Некоторые аминоалкилглюкозаминидфосфатные производные и их применение - RU2389732C2
Код документа: RU2389732C2
Описание
Перекрестная ссылка на родственные заявки
Данная заявка претендует на приоритет предварительной заявки на патент США 60/438585, зарегистрированной 6 января 2003, включенной в данное описание в качестве ссылки.
Уровень техники
Toll-подобные рецепторы (TLR) связаны с возможной врожденной иммунной реакцией и распознают различные структурные компоненты, уникальные для патогенов; такое взаимодействие приводит иммунную систему в активированное состояние с кратковременными или долговременными последствиями. Существует значительный интерес к разработке агонистов и антагонистов TLR, так как фармакологическая манипуляция с врожденными иммунными реакциями может привести к более эффективным вакцинам и новым подходам к лечению аутоиммунных, атопических, злокачественных и инфекционных болезней. Первым разработанным микробным продуктом, представляющим собой агониста Toll-подобных рецепторов, стал LPS-компонент бактериальных мембран, характерный для грамотрицательных бактерий, который активирует Toll-подобный рецептор 4 (TLR-4). Хотя LPS является сильным иммуномодулирующим средством, его применение в медицине ограничивается из-за его исключительной токсичности, включая индукцию системного синдрома воспалительной реакции. Биологически активной эндотоксичной субструктурной частью LPS является липид-А - фосфорилированный полиацилированный жирной кислотой глюкозамин - дисахарид, служащий якорем всей структуры на внешней мембране грамотрицательных бактерий. Токсическое действие липида А может быть ослаблено селективной химической модификацией липида А, которая дает монофосфорилированные производные липида А (иммуностимулятор MPL™; Corixa Corporation; Seattle, WA). Описаны способы получения и применения иммуностимулятора MPL™ и сходных по структуре соединений в адъюванте вакцины и других применениях (см., например, патенты США №№4436727, 4877611, 4866034 и 4912094, 4987237; Johnson et al., J. Med. Chem., 42:4640-4649 (1999); Ulrich and Myers, Vaccine Design: The Subunit and Adjuvant Approach; Powell and Newman, Eds.; Plenum: New York, 495-524, 1995). В частности, в указанных и других ссылках демонстрируется, что иммуностимулятор MPL™ и родственные соединения обладают значительной адъювантной активностью, когда используются в композициях вакцин с белковыми и углеводными антигенами, усиливая гуморальный и/или клеточноопосредованный иммунитет к антигенам и взаимодействие с Toll-подобными рецепторами.
Исходя из экспериментов с иммуностимулятором MPL™ и другими компонентами стенки бактериальной клетки, разработано семейство новых синтетических соединений аминоалкилглюкозаминидфосфатов (AGP). Соединения AGP также взаимодействуют с TLR-4 как агонисты и антагонисты. AGP включают как ациклические, так и циклические соединения (патенты США №№6113918 и 6303347, WO 98/50399, опубл. 12 октября 1998, WO 01/34617, опубл. 17 мая 2001, WO 01/90129, опубл. 29 ноября 2001 и WO 02/12258, опубл. 14 февраля 2002). Показано, что подобно иммуностимулятору MPL™, такие соединения сохраняют существенные характеристики адъювантов при включении вместе с антигенами в композиции вакцин и, кроме того, имеют профили токсичности подобные или лучшие при сравнении с иммуностимулятором MPL™. AGP также демонстрируют активность адъювантов в слизистых оболочках и эффективны в отсутствие антигена, что делает их привлекательными соединениями для профилактического и/или лечебного применения.
Другим существенным преимуществом, предоставляемым AGP по сравнению с иммуностимулятором MPL™ и подобными средствами, является то, что AGP легко получать в коммерческом масштабе синтетическими способами. Так как AGP получают синтетически, они свободны от слабых биологических загрязнений, обнаруживаемых в MPL. Как таковые, AGP будут иметь преимущества перед MPL как адъюванты вакцин в некоторых случаях, таких как схемы иммунизации детей, где пирогенность адъювантов должна быть сведена к минимуму. Однако, поскольку AGP синтезируют химически, устойчивость ниже оптимальной может привести к накоплению продуктов разложения, что может отразиться в различной биологической активности и устойчивости от партии к партии. С точки зрения разработки способов GMP для получения веществ для клинических испытаний на человеке изменения устойчивости в партии и от партии к партии являются важнейшими проблемами. Поэтому нужны соединения, обладающие повышенной биологической активностью по сравнению с иммуностимулятором MPL™ и подобными средствами, взаимодействующие с toll-подобными рецепторами и/или оптимизированные для получения синтезом GPL в крупном масштабе. Настоящее изобретение обращается к таким потребностям и подобным и относится к соединениям, модифицированным для усиления биологической активности и устойчивости с повышенным сопротивлением ферментативному и химическому разложению и/или с улучшенным профилем безопасности.
Раскрытие изобретения
В одном аспекте данное изобретение относится к некоторым новым аминоалкилглюкозаминидфосфатам, определение которым дается в данном описании, и их фармацевтически приемлемым солям. Изобретение также относится к композициям, содержащим такие соединения и/или их соли, и способам применения соединений как адъювантов и их самих как фармацевтически эффективных соединений.
Осуществление изобретения
Соединения данного изобретения являются членами семейства аминоалкилглюкозаминид-4-фосфатов (AGP). Как описано ниже, соединения изобретения обладают различными модификациями по длинам шести ацильных цепей (первичных и вторичных), структурными модификациями алкильного плеча, включающего фосфатную часть, структурной модификацией для включения липида с группой первичного простого эфира в положении С-3 сахаров, а также трех липидов с вторичными простыми эфирными группами, и/или группой, блокирующей 6-гидроксил.
AGP, известные в химии как ω-аминоалкил-2-амино-2-дезокси-4-фосфоно-β-D-глюкопиранозиды, представляют собой класс синтетических миметиков липида А, которые структурно схожи с основным биологически активным компонентом компонента в монофосфориллипиде А. В AGP редуцирующий сахар заменен на N-[(R)-3-н-алканоилокситетрадеканоил]аминоалкилаглюконовое звено. Подобно другим дисахаридным производным липида А AGP содержат шесть остатков жирных кислот для максимальной биологической активности, но, в отличие от дисахаридных производных, AGP содержат конформационно гибкое агликоновое звено, присоединенное по β, которое создает возможность энергетически благоприятной плотной упаковки шести жирных ацильных цепей. Полагают, что компактная упаковка шести остатков жирных кислот в гексагональный мотив играет существенную роль в биологической активности молекул, подобных липиду A (Seydel et al., ImmunobioL, 187(3-5): 191-211, 1993).
Соединения настоящего изобретения рассматриваются как члены семейства AGP. Указанные соединения включают модификации по длинам шести ацильных цепей (первичных и вторичных).
В одном из самых широких аспектов отличительным признаком изобретения является соединение APG формулы (I)

где X выбирают из группы, состоящей из О и S в аксиальном или экваториальном положении; Y выбирают из группы, состоящей из О и NH; n, m, p и q равны целым числам от 0 до 6; R1, R2 и R3 являются одинаковыми или различными и представляют собой жирные ацильные остатки с 1 и до примерно 20 атомами углерода, и где один из R1, R2 или R3, необязательно, представляют собой водород; R4 и R5 являются одинаковыми или различными и выбраны из группы, состоящей из Н и метила; R6 и R7 являются одинаковыми или различными и выбраны из группы, состоящей из Н, гидрокси, алкокси, фосфоно, фосфоноокси, сульфо, сульфоокси, амино, меркапто, циано, нитро, формила и карбокси, и их сложных эфиров и амидов; R8 и R9 являются одинаковыми или различными и выбраны из группы, состоящей из фосфоно и Н, и, по меньшей мере, один из R8 и R9 представляет собой фосфоно; R10, R11 и R12 выбирают, независимо, из числа линейных незамещенных насыщенных алифатических групп с 1-10 атомами углерода;
или его фармацевтически приемлемые соли.
В предпочтительных воплощениях данного аспекта изобретения
- Х и Y оба представляют собой, предпочтительно, атомы кислорода;
- R1, R2 и R3 представляют собой, предпочтительно, нормальные ацильные группы, и наиболее предпочтительно, выбранные, независимо, из числа линейных (С6-С10)-ацильных групп (наиболее предпочтительно, насыщенные ацильные группы);
- R10, R11 и R12 представляют собой, предпочтительно, незамещенные насыщенные алифатические (т.е., алкильные) группы с 1-10, предпочтительно - 3-9, предпочтительнее - 3-7 атомами углерода и наиболее предпочтительно представляют собой идентичные незамещенные насыщенные алифатические группы с 3-7 атомами углерода.
Соединения 1, a, b и 2, а, b и их фармацевтически приемлемые соли являются примерами такого типа соединения (I)

Таким образом, соединения (I) данного изобретения имеют сходство с некоторыми известными AGP, за исключением того, что они содержат более короткие цепи первичных жирных кислот. Обнаружено, что изменение в длине цепи вторичной жирной кислоты воздействует на иммунностимулирующую способность AGP и гомологи вторичной жирной кислоты 3-D-MPL (Johnson et al., J. Med. Chem., 42:4640-4649, 1999). Низкая эндотоксичность некоторых природных вариантов липида А, таких как липид A R. sphaeroides, частично связана с наличием более коротких (С10) остатков первичных жирных кислот в таких молекулах (Qureshiet al., J. Biol. Chem., 266(10):6532-6538, 1991). Подобным образом, низкая токсичность LPS некоторых helicobacter и pseudomonas может иметь место из-за присутствия гексаацильного компонента, содержащего остатки первичных жирных кислот, отличающиеся по длине от остатков, обнаруженных в токсичном липиде A salmonella (Moran et al., J. Bacteriol, 179(20):6453-6463, 1997; Kulshin et al., Eur. J. Biochem., 198(3):697-704, 1991). Хотя соотношение между длинами первичных ацильных цепей исследовано в ограниченной степени с синтетическими аналогами субъединиц липида А, содержащими до трех остатков жирных кислот (Hasegawa et al., Biosci. Biotech. Biochem., 59(9): 1790-1792, 1995; и Ogawa et al., Carbohydr. Res., 220:155-164, 1991), и тетраацильными аналогами дисахаридов липида IVa (Fukase et al., Tetrahedron, 54:4033-4050, 1998), авторами никогда не проводились систематические исследования для сведения ни с основным гесаацилированным фармакофором липида А, ни с миметиком липида А.
К отличительным признакам данного изобретения также относятся некоторые глицил- и фосфонооксиэтильные (РЕ) соединения (L-серинолфосфаты). Они представляют собой соединения приведенной выше формулы (I), в которой R5 и R7 представляют собой водород, n, m и q равны 0, в которых р равен 1, R6 представляет собой СООН или в которых р равен 2, и R6 представляет собой ОРО3Н2. Таким образом, они имеют общую формулу (II)
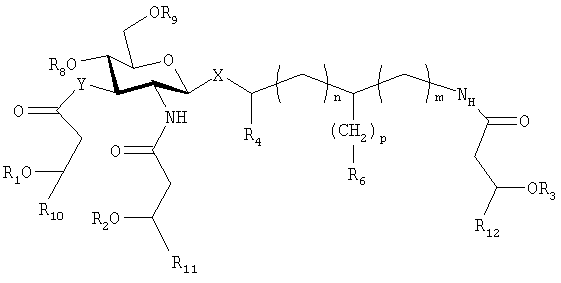
где Х выбирают из группы, состоящей из О и S, в аксиальном или экваториальном положении; Y выбирают из группы, состоящей из О и NH; n и m равны 0; R1, R2 и R3 являются одинаковыми или различными и представляют собой остатки жирных кислот с 1 и до примерно 20 атомами углерода, и где один из R1, R2 и R3, необязательно, представляет собой водород; R4 выбирают из группы, состоящей из Н и метила; р равен 1, и R6 представляет собой СООН, или р равен 2 и R6 представляет собой ОРО3Н2; R8 и R9 являются одинаковыми или различными и выбраны из группы, состоящей из фосфоно и Н, и, по меньшей мере, один из R8 и R9 представляет собой фосфоно; и R10, R11 и R12 выбирают, независимо, из числа линейных незамещенных насыщенных алифатических групп с 1-10 атомами углерода;
или представляют собой фармацевтически приемлемые соли соединений указанной формулы.
В предпочтительных воплощениях соединений (II) изобретения
- Х и Y оба представляют собой, предпочтительно, атомы кислорода;
- R1, R2 и R3 представляют собой, предпочтительно, нормальные ацильные группы и наиболее предпочтительно, выбранные, независимо, из числа линейных (С6-С10)-ацильных групп;
- группы R10, R11 и R12 представляют собой, предпочтительно, незамещенные насыщенные алифатические (т.е., алкильные) группы с 1-10, предпочтительно 3-9, предпочтительнее 3-7 атомами углерода и наиболее предпочтительно представляют собой идентичные незамещенные насыщенные алифатические группы с 3-7 атомами углерода.
Соединения 11а, b, и 12а, b, являются примерами соединения (II) такого типа
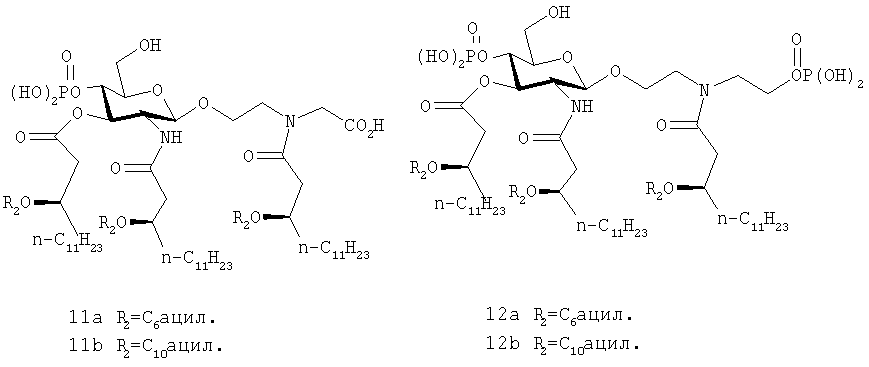
Соединения 12а и 12b содержат структурные модификации алкильного плеча, включающего фосфатную группу. Такие соединения считаются потенциально более устойчивыми, чем другие члены семейства. Такие соединения имеют преимущество перед серил/серинолфосфатными классами AGP в том, что они утратили стереогенный центр в агликоновом звене - особенность, которая может осложнить синтез и привести к затруднениям при отделении энантиомерных или диастереомерных примесей.
Другим типом соединения изобретения являются производные (R)-3-алкилокситетрадекановой кислоты. Они имеют такую же общую приведенную выше формулу (II), за исключением того, что R1, R2 и R3 не являются ацильными группами, но представляют собой линейные алкильные группы, что делает группы R1O-, R2O- и R3O- простыми эфирными, а не карбоксильными. В соединениях такого типа R1, R2 и R3 представляют собой, предпочтительно, (С6-С10)-алкильные группы. Они могут быть одинаковыми или различными группами, но наиболее предпочтительно являются идентичными.
Такие соединения имеют общую формулу (III)
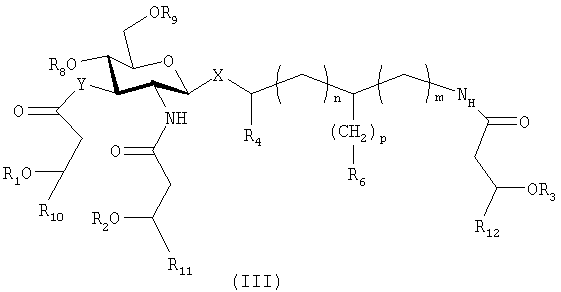
где Х выбирают из группы, состоящей из О и S, в аксиальном или экваториальном положении; Y выбирают из группы, состоящей из О и NH; n и m равны 0; R1, R2 и R3 являются одинаковыми или различными и представляют собой остатки жирных кислот с 1 и до примерно 20 атомами углерода, и где один из R1, R2 и R3, необязательно, представляет собой водород; R4 выбирают из группы, состоящей из Н и метила; р равен 1, R6 представляет собой СООН или р равен 2, и R6 представляет собой ОРО3Н2; R8 и R9 являются одинаковыми или различными и выбраны из группы, состоящей из фосфоно и Н, и, по меньшей мере, один из R8 и R9 представляет собой фосфоно; и R10, R11 и R12 выбирают, независимо, из числа линейных незамещенных насыщенных алифатических групп с 1-11 атомами углерода;
или представляют собой фармацевтически приемлемые соли соединений указанной формулы.
В предпочтительных воплощениях соединений (III) изобретения
- Х и Y оба представляют собой, предпочтительно, атомы кислорода;
- R1, R2 и R3, наиболее предпочтительно, выбирают, независимо, из числа незамещенных линейных (С6-С10)-алкильных групп;
- группы R10, R11 и R12 представляют собой, предпочтительно, незамещенные насыщенные алифатические (т.е., алкильные) группы с 1-11, предпочтительно 3-9, предпочтительнее 3-7 атомами углерода и, наиболее предпочтительно, представляют собой идентичные незамещенные насыщенные алифатические группы с 3-7 атомами углерода.
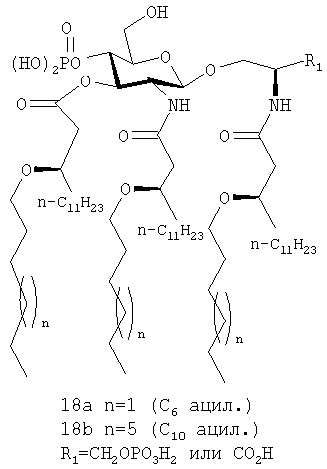
Соединения 18, а, b, являются примерами соединений данной группы, содержащих липид с остатком первичного простого эфира в положении С-3 сахара, а также три липида с остатками вторичных простых эфиров
Еще один тип соединения данного изобретения имеет формулу (IV)

где теперь Y определяется как кислород; Х выбирают из группы, состоящей из О и S, в аксиальном или экваториальном положении; n и m равны 0; R1, R3 и R3 являются одинаковыми или различными и представляют собой остатки жирных кислот с 1 и до примерно 20 атомами углерода, и где один из R1, R2 и R3, необязательно, представляет собой водород; R4 выбирают из группы, состоящей из Н и метила; р равен 1, и R6 представляет собой СООН, или р равен 2 и R6 представляет собой ОРО3Н2; R8 и R9 являются одинаковыми или различными и выбраны из группы, состоящей из фосфоно и Н, и, по меньшей мере, один из R8 и R9 представляет собой фосфоно; и R10, R11 и R12 выбирают, независимо, из числа линейных незамещенных насыщенных алифатических групп с 1-10 атомами углерода;
или представляет собой фармацевтически приемлемую соль соединения указанной формулы.
Таким образом, такие соединения содержат две ацилированные цепи и одну неацилированную цепь простого эфира.
В предпочтительных воплощениях соединений (IV) изобретения
- Х представляет собой, предпочтительно, атом кислорода;
- R1, R2 и R3, наиболее предпочтительно, выбирают, независимо, из числа незамещенных линейных (С6-С10)-алкильных групп;
- группы R10, R11 и R12 представляют собой, предпочтительно, незамещенные насыщенные алифатические (т.е., алкильные) группы с 1-10, предпочтительно 3-9, предпочтительнее 3-7 атомами углерода и, наиболее предпочтительно, представляют собой идентичные незамещенные насыщенные алифатические группы с 3-7 атомами углерода.
Соединения 20, а, b, являются примерами соединений такого класса
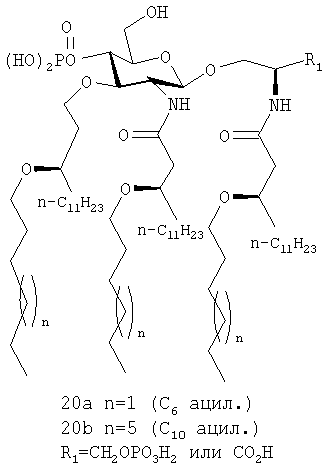
Такие соединения имеют свойства, позволяющие сопротивляться неблагоприятному метаболизму и/или водному гидролизу. Обусловлено, что селективное удаление остатков нормальных жирных кислот в структурно несхожих молекулах липида А человеческой ацилоксиацилгидролазой (АОАН), приводящее к антагонисту липиду IVа, развивается как защитный механизм для снижения токсичности липида A (Erwin and Munford, J. Biol. Chem., 265(27):16444-16449). Однако большая токсичность 3-D-MPL природного происхождения относительно токсичности основного гексаацильного компонента имеет место, вероятно, из-за присутствия высокоацилированных компонентов со структурами, отличными от липида IVa (Ulrich and Myers, Monophosphory lipid A as Adjuvant. Past experiences and new directions. In Vaccine Design: The Subunit and Adjuvant Approach. Ed. Powell M.F., Newman M.J. Plenum Press, New York, 1995, pp.495-524; Johnson et al., J. Med. Chem., 42:4640-4649, 1999). Структурная изменчивость в 3-D-MPL и других препаратах липида А проистекает, в своей основе, от сходного LPS, а также от расщепления сложного эфира во время полусинтетических процедур и извлечения. Действительно, сообщается, что при легком гидролитическом расщеплении связанных сложноэфирной связью ацильных групп во время химического синтеза предполагаемого липида A R.capsulatus - сильного антагониста, вызываемого LPS продуцирования TNF-α, образуются небольшие количества нежелательных побочных продуктов-агонистов (Christ et al., Science, 268:80-83, 1995). Таким образом, химическая и/или ферментативная неустойчивость может являться ахиллесовой пятой возможного лекарственного препарата на основе липида А, содержащего лабильные сложноэфирные связи. Химическая и метаболическая неустойчивость остатков жирных кислот, присоединяемых через сложноэфирную связь, присутствующих как в агонисте липида А, так и в молекулах антагониста, преодолевается гидролитически устойчивыми аналогами, содержащими простые эфирные связи вместо сложноэфирных связей, присоединяющих остатки первичных и/или вторичных жирных кислот (Christ et al., цит. выше; Lien et al., J. Biol. Chem, 276(3): 1873-1880, 2001).
Другие соединения настоящего изобретения содержат группу, блокирующую 6-гидроксил. Такие соединения имеют формулу (V)
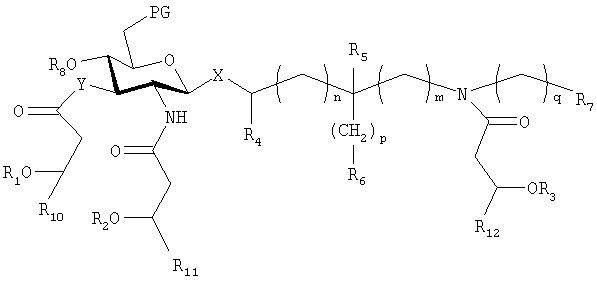
где Х выбирают из группы, состоящей из О и S, в аксиальном или экваториальном положении; Y выбирают из группы, состоящей из О и NH; n, m, p и q равны целым числам от 0 до 6; R1, R2 и R3 являются одинаковыми или различными и представляют собой остатки жирных кислот с 1 и до примерно 20 атомами углерода, и где один из R1, R2 и R3, необязательно, представляет собой водород; R4 и R5 являются одинаковыми или различными и выбраны из группы, состоящей из H и метила; R6 и R7 являются одинаковыми или различными и выбраны из группы, состоящей из Н, гидрокси, алкокси, фосфоно, фосфоноокси, сульфо, сульфоокси, амино, меркапто, циано, нитро, формила и карбокси и их сложных эфиров и амидов; R8 представляет собой фосфоно; PG представляет защитную группу для гидроксила, значения которой указаны ниже, и R10, R11 и R12 выбирают, независимо, из числа линейных незамещенных насыщенных алифатических групп с 1-10 атомами углерода;
или представляют собой фармацевтически приемлемые соли соединений указанной формулы.
Термин "защитная группа" (представленный в описании аббревиатурой "PG") относится к любой группе из большого числа групп, используемых для замены водорода гидроксигруппы с тем, чтобы блокировать, предупредить или ослабить реакционную способность группы. Примеры защитных групп (и перечень аббревиатур, обычно используемых для них) можно найти в T.W.Green and P.G.Futs, "Protective Groups in Organic Chemistry" (Wiley); Beacage and lyer. Tetrahedron, 48:2223 (1992), и в Harrison et al., Compendium of Synthetic Organic Methods, vols.1-8 (Wiley). Характерными защитными группами для гидроксигруппы являются группы, где гидроксигруппа или ацилируется, или алкилируется, например, путем образования простых или сложных эфиров, с использованием, например, метальной, ацетильной, бензильной, тритильной, алкильной, тетрагидропиранильной, аллильной и тризамещенных силильных групп или когда гидроксигруппа заменяется на фтор.
Выбор защитной группы для данного соединения, цели или набора условий находится в компетенции специалистов в данной области техники и делается таким образом, чтобы защитить, вообще или избирательно, реакционноспособную группу, представляющую интерес, в преобладающих условиях (присутствие других реакционноспособных соединений, рН, температура и т.д.). Защитные группы, которые можно использовать в данном изобретении, включают метальную, фталоильную, ацетильную (Ас), бензильную (Вn), 2,2,2-трихлорэтоксикарбонильную (Тrос), трет-бутилдиметилсилильную (TBS), трет-бутилдифенилсилильную (TBDPS) и 2,2,2-трихлор-1,1-диметилэтилхлорформильную (ТСВОС) группы. Также в качестве защитной группы можно использовать атом фтора. Как известно в технике, определенная защитная группа или тип группы могут быть более подходящими, чем другие, для применения с конкретным соединением или в данной ситуации, и при разработке процессов с участием соединений с реакционноспособными группами, такими как гидрокси, преимущество отдается таким подходящим группам.
В предпочтительных воплощениях таких соединений (V) изобретения
- Х и Y оба представляют собой, предпочтительно, атомы кислорода;
- R1, R2 и R3, предпочтительно, представляют собой нормальные ацильные группы, и, наиболее предпочтительно, выбираются, независимо, из числа линейных (С6-С10)-ацильных групп;
- группы R10, R11 и R12 представляют собой, предпочтительно, незамещенные насыщенные алифатические (т.е., алкильные) группы с 1-10, предпочтительно 3-9, предпочтительнее 3-7, атомами углерода и, наиболее предпочтительно, представляют собой идентичные незамещенные насыщенные алифатические группы с 3-7 атомами углерода.
Примерами членов такой группы являются соединения 25, a, b, содержащие простую метилэфирную группу, или соединения 26, a, b, содержащие группу фтора, используемые в сочетании с серил- или серинолфосфатсодержащими AGP
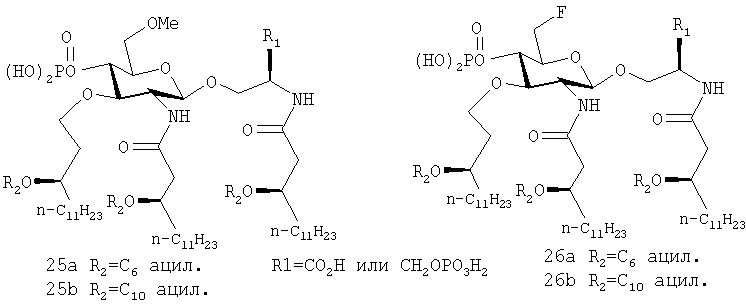
Незащищенная гидроксильная группа сахара С-6 может привести к образованию небольших количеств загрязнений во время синтеза производных липида А, которые могут удаляться с трудом (Chirst, цит. выше). Такие побочные продукты, вероятно, являются результатом образования сначала 4,6-циклического фосфата и последующей перегруппировки (Imoto et al., Tetrahedron Lett., 29(28):2227-2230, 1988).
В связи с обсуждением в данном описании термин "алифатический" в отношении заместителя самого по себе или части другого заместителя обозначает, если не указано иное, углеводородный радикал с линейной или разветвленной цепью, или циклический углеводородный радикал, или их сочетание, которые могут быть полностью насыщенными, моно- или полиненасыщенными и могут включать ди- и поливалентные радикалы с установленным числом атомов углерода (т.е., C1-С10 обозначает число атомов углерода от одного до десяти). Примерами насыщенных углеводородных радикалов являются такие группы, как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, циклогексилметил, циклопропилметил, гомологи и изомеры, например, н-пентила, н-гексила, н-гептила, н-окстила, и подобные группы. Ненасыщенная алифатическая группа представляет собой группу с одной или несколькими двойными связями или тройными связями. Примерами ненасыщенных алифатических групп являются винил, 2-пропенил, кротил, 2-изопентенил, 2-бутидиенил, 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1-и 3-пропинил, 3-бутинил и высшие гомологи и изомеры. Типично, алифатическая группа будет содержать от 1 до 24 атомов углерода. "Низшая алифатическая группа" представляет собой алифатическую группу с более короткой цепью, как правило, с восемью или меньшим числом атомов углерода.
Термин "ацил" относится к группе, образованной от органической кислоты путем удаления гидроксигруппы. Примерами ацильных групп являются ацетил, пропионил, додеканоил, тетрадеканоил, изобутирил и подобные группы. Соответственно, термин "ацил", используемый в данном описании, включает группу, иначе определяемую как -С(O)-алифатическая, где алифатическая группа представляет собой, предпочтительно, насыщенную алифатическую группу.
Термин "фармацевтически приемлемые соли" охватывает соли активных соединений, которые получают с относительно нетоксичными кислотами или основаниями, в зависимости от определенных заместителей в соединениях, описываемых в данном описании. Когда соединения настоящего изобретения содержат относительно кислотные функциональные группы, можно получить соли присоединения оснований путем приведения в контакт нейтральной формы таких соединений с достаточным количеством нужного основания или в чистом виде, или в подходящем инертном растворителе. Примерами фармацевтически приемлемых солей присоединения оснований являются натриевые, калиевые соли, соли аммония, органических аминов или магниевые соли, или подобные соли. Когда соединения настоящего изобретения содержат относительно основные функциональные группы, можно получить соли присоединения кислот путем приведения в контакт нейтральной формы таких соединений с достаточным количеством нужной кислоты или в чистом виде, или в подходящем инертном растворителе. Примерами фармацевтически приемлемых солей присоединения кислот являются соли, полученные с неорганическими кислотами, такими как хлороводородная, бромоводородная, азотная, угольная, моногидрокарбонаты, соли фосфорной кислоты, моногидрофосфаты, дигидрофосфаты, соли серной кислоты, моногидросульфаты, соли иодоводородной кислоты или фосфористой кислоты, и подобные соли, а также соли, образованные с относительно нетоксичными органическими кислотами, такими как уксусная, пропионовая, изомасляная, малеиновая, малоновая, бензойная, янтарная, субериновая, фумаровая, молочная, миндальная, фталевая, бензолсульфоновая, п-толуолсульфоновая, лимонная, винная, метансульфоновая и подобные кислоты. Также включаются соли аминокислот, такие как аргинат и подобные соли, и соли органических кислот, подобных глюкуроновой или галактуроновой кислотам, и подобные соли (см., например, Berge S.M. et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science, 66, 1-19, 1977). Некоторые конкретные соединения настоящего изобретения содержат как основные, так и кислотные функциональные группы, что позволяет превращать соединения в соли присоединения или оснований, или кислот.
Нейтральные формы соединений можно регенерировать путем приведения соли в контакт с основанием или кислотой и выделения исходного соединения обычным способом. Исходная форма соединения отличается от различных солевых форм некоторыми физическими свойствами, такими как растворимость в полярных растворителях, но по другим свойствам соли эквиваленты исходной форме соединения для целей настоящего изобретения.
Кроме солевых форм настоящее изобретение также относится к соединениям, которые существуют в форме пролекарств. Пролекарства соединений, описанных в данном изобретении, представляют собой соединения, которые легко претерпевают химические изменения в физиологических условиях с образованием соединений настоящего изобретения. Кроме того, пролекарства можно превращать в соединения настоящего изобретения химическими или биологическими способами в среде ex vivo. Например, пролекарства могут постепенно превращаться в соединения настоящего изобретения, когда они помещены в резервуар трансдермального пэтча с подходящим ферментом или химическим реагентом.
Некоторые соединения настоящего изобретения могут существовать в несольватированных формах, а также в сольватированных формах, в том числе в форме гидратов. Вообще, сольватированные формы эквивалентны несольватированным формам, и подразумевается, что они входят в объем настоящего изобретения. Некоторые соединения настоящего изобретения могут существовать в нескольких кристаллических или аморфных формах. Вообще, все физические формы эквивалентны для применений, предполагаемых настоящим изобретением, и подразумевается, что они входят в объем настоящего изобретения.
Некоторые соединения настоящего изобретения содержат асимметричные атомы углерода (оптические центры) или двойные связи; подразумевается, что все рацематы, диастереомеры, геометрические изомеры и отдельные изомеры входят в объем настоящего изобретения.
Соединения настоящего изобретения также могут содержать не встречающиеся в природе пропорции атомных изотопов одного или нескольких атомов, составляющих такие соединения. Например, соединения можно пометить радиоактивными изотопами, такими как, например, тритий (3H), иод-125 (125I) или углерод-14 (14С). Подразумевается, что все содержащие изотопы варианты соединений настоящего изобретения, являются ли они радиоактивными или нет, входят в объем настоящего изобретения.
Соединения настоящего изобретения можно получить любым подходящим способом (см. раздел примеров ниже), многие из которых описаны. Например, способы получения некоторых соединений, применимые для настоящего изобретения, описываются в патенте США №6113918, патенте США №6303347 и в PCT/US 98/09385 (WO 98/50300, 12 октября 1998). Также другие соединения можно получить с использованием способов, описанных в Johnson et al., J. Med. Chem., 42:4640-4649 (1999); Johnson et al., Bioorg. Med. Chem. Lett., 9:2273-2278(1999) и в PCT/US 98/50399 (WO 98/50399, 12 ноября 1998). Вообще, синтетические способы, описанные в указанных выше ссылках, и другие иные способы синтеза, хорошо известные в технике, можно широко применять для получения таких соединений. Например, при получении соединений с различными ацильными группами и заместителями специалисту в данной области техники следует иметь в виду, что описанные в указанных ссылках сходные способы можно модифицировать для использования других ацилирующих агентов или можно исходить из коммерчески доступных материалов с соответствующими присоединенными ацильными группами.
В композициях для вызывания или усиления иммунной реакции соединения данного изобретения вводят теплокровному животному, включая людей, с антигеном, таким как антиген белок или полипептид, или полинуклеотидом, экспрессирующим антиген белок или полипептид. Количество антигена, вводимого для вызывания или усиления иммунной реакции, может легко определить специалист в данной области техники, и оно будет изменяться в зависимости от типа вводимого антигена, способа введения и схемы иммунизации.
Соединения настоящего изобретения также можно вводить без антигена для того, чтобы вызвать немедленную защиту через эффект неспецифического сопротивления, как описано ниже (см. Persing et al., WIPO Publication WO 01/90129, 29 ноября 2001). Соединения, обладающие способностью стимулировать неспецифическое сопротивление и/или вызывать действие адъюванта, можно использовать в композиции быстрой вакцины. Введение соединений настоящего изобретения с антигеном приводит к приобретенной мышечной иммунной реакции в пределах трех-четырех недель. Еженедельное введение таких соединений, например интраназальным способом, на протяжении четырех недель обеспечивает быструю и надежную защиту за счет сочетания защиты, обеспечиваемой первоначальной врожденной иммунной реакцией с последующей приобретенной иммунной реакцией на антиген, представляющий интерес.
Соединения настоящего изобретения можно оценивать различными анализами для идентификации соединений, имеющих характеристики, наиболее подходящие для данного применения изобретения. Например, животные модели можно использовать для идентификации и оценки профилей высвобождения цитокинов в большой круг кровообращения после введения соединения настоящего изобретения. Кроме того, существуют различные модели in vitro и in vivo для проверки изменений в одном или нескольких аспектах иммунной реакции на различные антигенные компоненты для того, чтобы идентифицировать соединения, наиболее подходящие для вызывания специфической иммунной реакции, представляющей интерес. Например, соединение можно ввести в контакт in vivo с клетками-мишенями, такими как макрофаги, дендритные клетки или клетки Лангерганса, и можно определить выработанные цитокины. Кроме того, можно использовать массу событий экспрессии генов для идентификации специфических каскадов реакций, активированных или ингибированных определенным соединением, представляющим интерес.
Индукцию/продуцирование цитокинов можно определить с использованием обработки человеческой крови и/или клеток соединениями настоящего изобретения и измерения индукции методом ELISA (системы R & D). Такие методы также можно использовать для определения, если индукция зависит от Toll-рецепторов. Цитотоксическую реакцию Т-лимфоцитов после введения соединений настоящего изобретения определяют анализом на цитотоксичность на основе51Сr. При необходимости, действие соединений изобретения в этом отношении можно сравнить с действием других соединений, известных как функциональные в этом отношении, таких как липид A, MPL, AGP или подобных. Кроме того, соединения изобретения можно оценивать в сочетании с одним или несколькими адъювантами и/или иммуномодуляторами для идентификации синергических эффектов (см., например, патенты США №№6303347 и 6113918 и WO 01/90129, опубл. 29 ноября 2001).
Животные модели, такие как мышиная модель заражения гриппом и мышиная модель заражения Listeria monocytogenes, применимы для оценки активности адъюванта и иммуномодулятора. Коротко, вводят соединение с последующим заражением гриппом или L.monocytogenes. Контролируют показатель болезни (сильный налет на языке, сгорбленная осанка и одышка), потерю массы и смертность в случае гриппа или число колониеобразующих единиц в селезенке обработанных/необработанных мышей в случае L.monocytogenes, как показатель защиты, предоставляемой введением соединения изобретения (см., например, WO 01/90129, опубл. 29 ноября 2001).
Используемый в данном описании термин "полипептид" применяют в его обычном значении, т.е. как последовательность аминокислот. Полипептиды не ограничиваются конкретной длиной продукта; таким образом, в определение полипептид входят полипептиды, олигопептиды и белки, и указанные термины в данном случае можно использовать как взаимозаменяемые, если конкретно не указано иное. Данный термин также не относится к или исключает постэкспрессионные модификации, например гликозилирование, ацетилирование, фосфорилирование и т.п., а также другие модификации, известные в технике, как имеющие место в природе, так и не встречающиеся в природе. Полипептид может представлять собой полный белок или его последовательность. Конкретными полипептидами, представляющими интерес в контексте данного изобретения, являются аминокислотные последовательности, содержащие эпитопы, т.е. антигенные детерминанты, по существу, ответственные за иммуногенные свойства полипептида и способные вызывать иммунную реакцию.
Полипептиды, применимые в настоящем изобретении, иногда в данном описании называются опухолевыми белками или опухолевыми полипептидами как показатель того, что их идентификация основана, по меньшей мере частично, на повышенном уровне их экспрессии в опухолевых образцах. Таким образом, термины "опухолевый полипептид" или "опухолевый белок" относятся, главным образом, к полипептидной последовательности настоящего изобретения или полинуклеотидной последовательности, кодирующей такой полипептид, который экспрессируется в значительной части опухолевых образцов, например, предпочтительно, в более примерно 20%, предпочтительнее - в более примерно 30% и наиболее предпочтительно - в более примерно 50% или большей части испытываемых опухолевых образцов, на уровне, который, по меньшей мере, в два раза, а предпочтительно - по меньшей мере, в пять раз выше уровня экспрессии в здоровых тканях, что определяют с использованием типичного анализа, приведенного в данном описании.
В некоторых предпочтительных воплощениях полипептиды изобретения являются иммуногенными, т.е. во время иммуноанализа (такого как ELISA или анализ Т-клеточной стимуляции) можно обнаружить, что они реагируют с антисыворотками и/или Т-клетками от пациента с раковым заболеванием. Скрининг на иммуногенную активность можно осуществить с использованием методов, хорошо известных специалистам в данной области техники. Например, такие отборы можно осуществить с использованием таких способов, какие описаны в Harlow and Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988. Согласно одному пояснительному примеру полипептид можно иммобилизовать на твердом носителе и ввести в контакт с сыворотками пациентов для того, что создать возможность для связывания антител в сыворотках с иммобилизованным полипептидом. Затем несвязанную сыворотку можно удалить, а связанную сыворотку детектировать с использованием, например, меченного125! протеина А.
Как следует признать специалистам в данной области техники, иммуногенные части полипептидов, раскрываемых в данном описании, также входят в объем настоящего изобретения. Используемая в данном случае "иммуногенная часть" представляет собой фрагмент иммуногенного полипептида изобретения, которая сама является иммунологически реакционноспособной (т.е. специфически связывается) в отношении В-клеток и/или антигенных рецепторов на поверхности Т-клеток, распознающих полипептид. Иммуногенные части, как правило, можно идентифицировать с использованием хорошо известных методов, таких, какие сведены в Paul, Fundamental Immunology, 3rd ed., 243-247 (Raven Press, 1993), и ссылках, цитированных в указанной работе. Такие методы включают скрининг полипептидов на способность взаимодействовать с антигенспецифическими антителами, антисыворотками и/или Т-клеточными линиями или клонами. В данном случае антисыворотки и антитела являются "антигенспецифическими", если они специфически связываются с антигеном (т.е., они взаимодействуют с белком при ELISA или другом иммуноанализе и не взаимодействуют заметно с неродственными белками). Такие антисыворотки и антитела можно получить так, как описано в данном описании, и с использованием хорошо известных методов.
В предпочтительном воплощении иммуногенная часть полипептида настоящего изобретения представляет собой часть, которая взаимодействует с антисыворотками и/или Т-клетками на уровне, который, по существу, не ниже реакционной способности полноразмерного полипептида (например, в ELISA и/или анализе на реакционную способность Т-клеток). Предпочтительно, уровень иммуногенной активности иммуногенной части составляет, по меньшей мере, примерно 50%, предпочтительно - по меньшей мере, примерно 70% и наиболее предпочтительно - более примерно 90% от иммуногенности полноразмерного полипептида. В некоторых случаях предпочтительные иммуногенные части будут идентифицированы как имеющие уровень иммуногенной активности, превышающий активность соответствующего полноразмерного полипептида, т.е. имеющие уровень иммуногенной активности свыше примерно 100%, или 150%, или более.
В некоторых других воплощениях характерные иммуногенные части могут включать пептиды, в которых обнаруживаются N-концевая лидерная последовательность и/или трансмембранный домен. Другие характерные иммуногенные части будут содержать небольшую N- и/или С-концевую делецию (например, в 1-30 аминокислот, предпочтительно - 1-15 аминокислот) относительно зрелого белка.
В еще одном воплощении полипептидная композиция изобретения также может содержать один или несколько полипептидов, которые иммунологически реакционноспособны в отношении Т-клеток и/или антител, выработанных против полипептида изобретения, в частности полипептида с аминокислотной последовательностью, раскрываемой в данном описании, или его иммуногенного фрагмента, или варианта.
В другом воплощении изобретения полипептиды содержат один или несколько полипептидов, способных выявлять Т-клетки и/или антитела, иммунологически реакционноспособные в отношении одного или нескольких полипептидов, описанных в данном описании, одного или нескольких полипептидов, кодированных контигом нуклеотидных последовательностей, содержащимся в полинуклеотидах, описанных в данном описании, или их иммуногенных фрагментов, или вариантов.
Полипептиды могут содержать сигнальную (или лидерную) последовательность в N-конце белка, которая во время или после трансляции управляет переносом белка. Полипептид также может быть конъюгирован с линкерной или другой последовательностью для облегчения синтеза, очистки или идентификации полипептида (например, поли-His), или для усиления связывания полипептида с твердым носителем. Например, полипептид может быть конъюгирован с F-областью иммуноглобулина.
В других характерных воплощениях полипептид может представлять собой слитый полипептид, содержащий несколько полипептидов, описанных в данном описании, или полипептид, содержащий, по меньшей мере, один полипептид, описанный в данном описании, и неродственную последовательность, такую как известный опухолевый белок. Партнер по слиянию может, например, способствовать обеспечению Т-хелперных эпитопов (иммунологический партнер по слиянию), предпочтительно - Т-хелперных эпитопов, распознаваемых организмом человека, или может способствовать экспрессии белка (энхансер экспрессии) с более высокими выходами, чем выход нативного рекомбинантного белка. Некоторые предпочтительные партнеры по слиянию являются как иммунологическими партнерами, так и партнерами по слиянию, усиливающими экспрессию. Других партнеров по слиянию можно выбрать таким образом, чтобы повысить растворимость полипептида или придать полипептиду способность стать мишенью в нужных клеточных компартментах. Другие партнеры по слиянию включают аффинные метки, облегчающие очистку полипептида.
Слитые полипептиды, как правило, можно получить с использованием стандартных методов, в том числе химической конъюгации. Предпочтительно, слитый полипептид экспрессируется как рекомбинантный полипептид, причем в продуцирующей системе создается возможность продуцирования с повышенными уровнями относительно неслитого полипептида. Коротко, последовательности ДНК, кодирующие полипептидные компоненты, можно собрать по отдельности и лигировать в подходящий экспрессирующий вектор. Лигируют 3'-конец последовательности ДНК, кодирующей один полипептидный компонент, с помощью пептидного линкера или без него, с 5'-концом последовательности ДНК, кодирующей второй полипептидный компонент, так что рамки считывания последовательностей находятся в фазе. Это допускает трансляцию в один составной полипептид, который сохраняет биологическую активность обоих полипептидных компонентов.
Пептидную линкерную последовательность можно использовать для отдаления первого и второго полипептидных компонентов на расстояние, достаточное для уверенности в том, что каждый полипептид складывается в свои вторичные и третичные структуры. Такую пептидную линкерную последовательность вводят в слитый полипептид с использованием стандартных методов, хорошо известных в технике. Подходящие пептидные линкерные последовательности можно выбрать на основании следующих факторов: (1) их способность принимать гибкую растянутую конформацию; (2) их неспособность принимать вторичную структуру, которая может взаимодействовать с функциональными эпитопами на первом и втором полипептидах; и (3) отсутствие гидрофобных или заряженных остатков, которые могут взаимодействовать. с полипептидными функциональными эпитопами. Предпочтительные пептидные линкерные последовательности содержат остатки Gly, Asn и Ser. Другие почти нейтральные кислоты, такие как Thr и Аlа, также можно использовать в линкерной последовательности. Аминокислотные последовательности, которые можно использовать как линкеры, включают последовательности, раскрываемые в Maratea et al., Gene, 40:39-46, 1985; Murphy et al., Proc. Natl. Acad. Sci. USA, 83:8258-8262, 1986; патенте США №4935233 и патенте США №4751180. Линкерная последовательность, как правило, может иметь длину от 1 до примерно 50 аминокислот. Линкерные последовательности не требуются, когда первый и второй полипептиды имеют несущественные N-концевые аминокислотные участки, которые можно использовать для выделения функциональных доменов и предотвращения стерического влияния.
Лигированные последовательности ДНК операбельно соединяются с подходящими транскрипционными или транскрипционнорегуляторными элементами. Регуляторные элементы, ответственные за экспрессию ДНК, располагаются только 5' к последовательности ДНК, кодирующей первые полипептиды. Подобным образом, терминирующие кодоны, требуемые для окончания трансляции, и сигналы терминации транскрипции присутствуют только 3' к ДНК, кодирующей второй полипептид.
Слитый полипептид может содержать полипептид, описанный в данном описании, вместе с неродственным иммуногенным белком, таким как иммуногенная часть, способная вызывать отмененную реакцию. Примеры таких белков включают белки вирусов столбняка, туберкулеза и гепатита (см., например, Stoute et al., New Engl. J. Med., 336:86-91, 1997).
В предпочтительном воплощении иммунологического партнера по слиянию получают из Mycobacterium sp., такого как полученный из Mycobacterium tuberculosis фрагмент Ra12. Композиции с Ra12 и способы их применения при усилении экспрессии и/или иммуногенности гетерологичных последовательностей полинуклеотидов/полипептидов описываются в заявке на патент США № 60/158585. Коротко, Ra12 называется участок полинуклеотида, представляющий собой последовательность нуклеиновой кислоты Mycobacterium tuberculosis MTB32A. МТВ32А представляет собой серинпротеазу с молекулярной массой 32 кД, кодированную геном, в вирулентных и авирулентных штаммах М.tuberculosis. Нуклеотидная последовательность и аминокислотная последовательность MTB32A описаны (например, в заявке на патент США № 60/158585; см. также Skeiky et al., Infection and Immun. (1999), 67:3998-4007). Последовательность, кодирующая MTB32A, экспрессирует С-концевые фрагменты на высоком уровне и сохраняется в виде растворимых полипептидов в процессе очистки. Кроме того, Ra12 может усиливать иммуногенность гетерологичных иммуногенных полипептидов, с которыми он сливается. Один из предпочтительных сливающихся с Ra12 полипептидов содержит С-концевой фрагмент в 14 кД, соответствующий аминокислотным остаткам 192-323 MTB32A. Другие предпочтительные полинуклеотиды Ra12, как правило, содержат, по меньшей мере, примерно 15 последовательных нуклеотидов, по меньшей мере, примерно 30 нуклеотидов, по меньшей мере, примерно 60 нуклеотидов, по меньшей мере, примерно 100 нуклеотидов, по меньшей мере, примерно 200 нуклеотидов или, по меньшей мере, примерно 300 нуклеотидов, которые кодируют часть полипептида Ra12. Полинуклеотиды Ra12 могут содержать нативную последовательность (т.е. эндогенную последовательность, кодирующую полипептид Ra12 или его часть) или могут содержать вариант такой последовательности. Варианты полинуклеотидов Ra12 могут содержать одну или несколько замен, добавлений, делеций и/или вставок, таких, что биологическая активность кодированного слитого полипептида, по существу, не уменьшается относительно слитого полипептида, содержащего нативный полипептид Ra12. Варианты, предпочтительно, показывают, по меньшей мере, примерно 70% идентичность, предпочтительнее, по меньшей мере, примерно 80% идентичность и наиболее предпочтительно, по меньшей мере, примерно 90% идентичность полинуклеотидной последовательности, кодирующей нативный полипептид Ra12 или его часть.
В других предпочтительных воплощениях иммунологичного партнера по слиянию получают из белка D - поверхностного белка грамотрицательной бактерии Haemophilus influenza В (WO 91/18926). Предпочтительно, производное белка D содержит приблизительно первую треть белка (например, первые 100-110 N-концевые аминокислоты), и производное белка D может быть липидизировано. В некоторых предпочтительных воплощениях первые 109 остатков партнера по слиянию белка D включают в N-конец для создания полипептида с дополнительными экзогенными Т-клеточными эпитопами и повышения уровня экспрессии в Е.coli (функционирующие, таким образом, как усилители экспрессии). Липидный хвост обеспечивает оптимальное представление антигена антиген-представляющим клеткам. К другим партнерам по слиянию относятся неструктурированный белок вируса гриппа NSI (гемаглутинин). Типично, используют 81 N-концевую аминокислоту, хотя можно использовать различные фрагменты, включающие Т-хелперные эпитопы.
В другом воплощении иммунологичный партнер по слиянию представляет собой белок, известный как LYTA, или его часть (предпочтительно, С-концевую часть). LYTA получают из Streptococcus pneumoniae, который синтезирует N-ацетил-L-аланинамидазу, известную как амидаза LYTA (кодированная геном LytA; Gene, 43:265-292, 1986). LYTA представляет собой аутолизин, который специфически разрушает некоторые связи в главной цепи пептидогликанов. С-Концевой домен белка LYTA ответственен за аффинность к холину или к некоторым аналогам холина, таким как DEAE. Такие свойства использовали для разработки экспрессирующих плазмид Е.coli C-LYTA, применимых для экспрессии слитых белков. Описана очистка гибридных белков, содержащих фрагмент С-LYTA в аминоконце (см. Biotechnology, 10:795-798, 1992). В предпочтительном воплощении в слитый полипептид можно включить часть повтора LYTA. Часть повтора найдена в С-концевом участке, начиная с остатка 178. Особенно предпочтительная часть повтора включает остатки 188-305.
Еще одно предпочтительное характерное воплощение включает слитые полипептиды и полинуклеотиды, кодирующие их, где партнер по слиянию содержит сигнал для мишени, способный направлять полипептид в эндосомальный/липосомальный компартмент, как описано в патенте США №5633234. Иммуногенный полипептид изобретения при слиянии с таким сигналом для мишени будет рациональнее ассоциироваться с молекулами класса II МНС и, таким образом, обеспечивает усиленную стимуляцию in vivo Т-клеток CD4+, специфических для полипептида.
Полипептиды изобретения получают с использованием любого из многочисленных хорошо известных в технике синтетических и/или рекомбинантных методов. Полипептиды, части и другие варианты, как правило, в менее примерно 150 аминокислот можно получить синтетическими способами с использованием методов, хорошо известных специалистам в данной области техники. Как один из примеров, такие полипептиды синтезируют с использованием любого из коммерчески доступных твердофазных методов, таких как способ твердофазного синтеза по Меррифилду, где аминокислоты последовательно добавляются к растущей аминокислотной цепи (см. Memfield, J. Am. Chem. Soc., 85:2149-2146, 1963). Оборудование для автоматического синтеза полипептидов коммерчески доступно от таких поставщиков, как Perkin Elmer/Applied BioSystems Division (Foster City, CA), и действия можно осуществлять по инструкциям изготовителя.
Вообще полипептидные композиции (включая слитые полипептиды) изобретения являются изолированными. "Изолированный" полипептид представляет собой полипептид, удаленный из его исходной окружающей среды. Например, встречающийся в природе белок или полипептид является изолированным, если он отделен от всех веществ, сосуществующих с ним в природе. Предпочтительно, такие полипептиды также являются очищенными, например, очищенными до, по меньшей мере, примерно 90% чистоты, предпочтительнее - до, по меньшей мере, примерно 95% чистоты, и наиболее предпочтительно - до, по меньшей мере, примерно 99% чистоты.
Настоящее изобретение в других аспектах относится к соединениям, содержащим один или несколько полинуклеотидов, кодирующих полипептидный антиген, как изложено выше в данном описании. Термины "ДНК" и "полинуклеотид" используются в данном описании, по существу, как взаимозаменяемые, и относятся к молекуле ДНК, изолированной в свободном виде от общей геномной ДНК определенных образцов. Используемый в данном описании термин "изолированный" означает, что полинуклеотид, по существу, отдален от других кодирующих последовательностей, и что молекула ДНК не содержит большие части неродственной кодирующей ДНК, такие как большие фрагменты хромосомы или другие функциональные гены или участки, кодирующие полипептид. Конечно, это относится к молекуле ДНК, которая изолирована изначально, и не исключает гены или кодирующие участки, позднее добавленные к сегменту в результате действий человека.
Полинуклеотиды могут содержать нативную последовательность (т.е. эндогенную последовательность, которая кодирует полипептид/белок изобретения или его часть) или могут содержать последовательность, кодирующую вариант или производное, предпочтительно, и иммуногенный вариант или производное, такой последовательности. Типично полинуклеотидные варианты будут содержать одну или несколько замен, добавлений, делеций и/или вставок, предпочтительно, таких, что иммуногенность полипептида, кодированного полинуклеотидом-вариантом, по существу, не уменьшается относительно полипептида, кодированного полинуклеотидной последовательностью, конкретно описанной в данном описании. Также следует иметь в виду, что термин "варианты" охватывает гомологичные гены ксеногенного происхождения.
В некоторых предпочтительных воплощениях полинуклеотиды, описанные выше, например полинуклеотидные варианты, фрагменты и гибридизирующие последовательности, кодируют полипептиды, которые являются иммунологически перекрестно-реакционноспособными в отношении антигенного или иммуногенного полипептида, описанного выше. В других предпочтительных воплощениях такие полинуклеотиды кодируют полипептиды с уровнем иммуногенной активности, составляющим, по меньшей мере, примерно 50%, предпочтительно - по меньшей мере, примерно 70% и предпочтительнее - по меньшей мере, примерно 90% от уровня для последовательности полипептида, конкретно описанной в данном описании.
Полинуклеотиды настоящего изобретения или их фрагменты, независимо от длины самой кодирующей последовательности, можно объединять с другими последовательностями ДНК, такими как промоторы, сигналы полиаденилирования, дополнительные сайты рестриктаз, сайты множественного клонирования, другие кодирующие сегменты и т.п., так что их общая длина может значительно изменяться. Поэтому предполагается, что можно использовать фрагмент нуклеиновой кислоты почти любой длины, причем общая длина, предпочтительно, ограничивается легкостью получения и применения в предполагаемой схеме метода рекомбинантных ДНК. Например, предполагается, что характерные полинуклеотидные сегменты с общей длиной в примерно 10000, примерно 5000, примерно, 3000, примерно 2000, примерно 1000, примерно 500, примерно 200, примерно 100, примерно 50 пар оснований (включая все промежуточные значения длины) применимы при многих осуществлениях данного изобретения.
Полинуклеотидные композиции настоящего изобретения можно идентифицировать, получать и/или манипулировать с ними с использованием любого из ряда хорошо известных методов (см. для общего представления Sambrook et al., Molecular Cloning: A Laboratory Mannual, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY, 1989 и другие подобные ссылки). Например, полинуклеотид можно идентифицировать так, как подробнее описано ниже, скринингом микроматрицы кДНК на экспрессию, ассоциируемую с опухолью (т.е. экспрессию, которая в опухоли, по меньшей мере, в два раза выше, чем в здоровой ткани, что определяют с использованием характерного анализа, приведенного в данном описании). Такой отбор можно осуществить, например, с использованием технологии микроматриц Affymetrix, Inc. (Santa Clara, CA) согласно инструкциям изготовителя (и, по существу, так, как описано в Schena et al., Proc. Natl. Acad. Sci. USA, 93:10614-10619, 1996 и в Heller et al., Proc. Natl. Acad. Sci. USA, 94:2150-2155, 1997). С другой стороны, полинуклеотиды можно амплифицировать из кДНК, полученной из клеток, экспрессирующих белки, описанные в данном описании, таких как опухолевые клетки.
Многие способы, зависящие от матрицы, доступны для амплификации матрицы-мишени в образце, представляющей интерес. Одним из наиболее известных способов амплификации является полимеразная цепная реакция (ПЦР™, PCR™), которая подробно описана в патентах США №№4683195, 4683202 и 4800159. Коротко, при ПЦР™ получают две праймерные последовательности, комплементарные областям на противоположно комплементарных цепях последовательности-мишени. В реакционную смесь добавляют избыток дезоксинуклеотидтрифосфатов вместе с ДНК-полимеразой (например, Taq-полимеразой). Если последовательность-мишень присутствует в образце, праймеры будут связываться с мишенью, и полимераза будет вызывать наращивание с праймеров вдоль последовательности-мишени путем присоединения нуклеотидов. За счет повышения и понижения температуры реакционной смеси достроенные праймеры будут диссоциировать от мишени с образованием продуктов реакции, избыточные праймеры будут связываться с мишенью и с продуктом реакции, и процесс повторяется. Предпочтительно, можно осуществить процедуру обратной транскрипции и амплификации ПЦР™ для того, чтобы определить количество амплифицированной мРНК. Методологии полимеразной цепной реакции хорошо известны в технике.
Целый ряд других способов, зависящих от матрицы, многие из которых являются вариациями метода амплификации ПЦР™, широко известны и доступны в технике. Как пример, некоторые такие способы включают лигазную цепную реакцию (обозначаемую LCR), описанную, например, в публ. заявке на евр. пат. №320308 и патенте США №4883750; способ Q-бета-репликазой, описанный в публ. заявки на междунар. пат. РСТ № PCT/US 87/00880; амплификацию с замещением цепи (Strand Displacement Amplification, SDA) и реакцию восстановления цепи (Repair Chain Reaction, RCR). Также другие способы амплификации описываются в заявке на пат. Великобритании №2202328 и в публ. заявки на междунар. пат. РСТ № PCT/US 89/01025. Другие процедуры амплификации нуклеиновых кислот включают системы амплификации, основанные на транскрипции (TAS) (публ. заявки на междунар. пат. РСТ WO 88/10315), включая амплификацию на основе нуклеотидной последовательности (NASBA) и 3SR. В публ. заявки на евр. пат. №329822 описывается способ амплификации нуклеиновых кислот, включающий циклический синтез одноцепочечной РНК ("оцRNА"), оцDNA и двухцепочечной ДНК (дцDNA). В публ. заявки на междунар. пат. РСТ WO 89/06700 описывается схема амплификации нуклеиновых кислот, основанная на гибридизации промоторной/праймерной последовательности с одноцепочечной ДНК-мишенью ("оцDNA") с последующей транскрипцией нескольких копий РНК последовательности. Специалистам в данной области техники также хорошо известны другие способы амплификации, такие как "RACE" (Frohman, 1990) и "односторонняя ПЦР" (Ohara, 1989).
Амплифицированную часть полинуклеотида настоящего изобретения можно использовать для выделения полноразмерного гена из подходящей библиотеки (например, библиотеки опухолевых кДНК) с использованием хорошо известных методов. В рамках таких методов библиотеку (кДНК или геномную) скринируют с использованием одного или нескольких полинуклеотидных зонодов или праймеров, подходящих для амплификации. Предпочтительно, библиотеку выбирают по размеру, включающую более крупные молекулы. Случайно премированные библиотеки также могут быть предпочтительны для идентификации 5' и предыдущих участков генов. Геномные библиотеки предпочтительны для получения интронов и достраивания 5'-последовательностей.
Для методов гибридизации неполную последовательность можно пометить (например, ник-трансляцией или32Р на конце) с использованием хорошо известных методов. Затем, как правило, бактериальную или бактериофаговую библиотеку скринируют методом гибридизации фильтров, содержащих денатурированные колонии бактерий (или "газоносодержащие" стерильные бляшки), с помощью меченого зонда (см. Sambrook et al., Molecular Cloning: A Laboratory Manuel, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY, 1989). Гибридизирующие колонии или бляшки отбирают и размножают, и выделяют ДНК для дальнейшего анализа. Клоны кДНК можно анализировать для определения количества дополнительной последовательности, например, с помощью ПЦР, с использованием праймера от неполной последовательности и праймера из вектора. Можно получить рестрикционные карты и неполные последовательности для идентификации одного или нескольких перекрывающихся клонов. Затем можно определить полную последовательность с использованием стандартных методов, которые могут включать получение ряда клонов делеции. Затем полученные перекрывающиеся последовательности можно собрать в единую последовательность контига. Молекулу полноразмерной кДНК можно получить путем лигирования подходящих фрагментов с использованием хорошо известных методов.
С другой стороны, методы амплификации, такие какие описаны выше, можно применить для получения полноразмерной кодирующей последовательности из неполной последовательности кДНК. Одним из таких методов амплификации является инверсная ПЦР (см. Triglia et al., Nucl. Acids Res., 16:8186, 1988), в которой используются рестриктазы для получения фрагмента в известном участке гена. Затем из фрагмента получают кольцевую молекулу путем внутримолекулярного лигирования и используют в качестве матрицы для ПЦР с дивергентными праймерами, полученными из известного участка. При другом подходе последовательности, прилегающие к неполной последовательности, можно восстановить амплификацией с праймером к линкерной последовательности и праймером, специфичным для известного участка. Амплифицированные последовательности типично подвергают второму циклу амплификации с тем же линкерным праймером и вторым праймером, специфичным для известного участка. Вариант такой процедуры, в котором используют два праймера, инициирующих достраивание в противоположных направлениях от известной последовательности, описывается в WO 96/38591. Другой такой метод известен как "быстрая амплификация концов кДНК" или RACE. Данный метод включает использование внутреннего праймера и внешнего праймера, который гибридизирует с областью поли-А или векторной последовательностью, для идентификации последовательностей, представляющих собой 5' и 3' известной последовательности. К другим методам относятся ПЦР с захватом (Lagerstrom et al., PCR Methods Applic., 1:111-19, 1991) и ПЦР с "прогулкой" (Parker et al., Nucl. Acids Res., 19:3055-60, 1991). Также можно использовать другие способы амплификации для получения полноразмерной последовательности кДНК.
В некоторых случаях возможно получение полноразмерной последовательности кДНК с помощью анализа последовательностей, полученных в базе данных о эспрессирующихся маркерах-последовательностях (EST), например, доступной от GenBank. Поиски перекрывающихся EST, как правило, можно осуществить с использованием хорошо известных программ (например, поиск NCBI BLAST), и такие EST можно использовать для получения полноразмерной последовательности контига. Полноразмерные последовательности ДНК также можно получить с помощью анализа геномных фрагментов.
В других воплощениях изобретения полинуклеотидные последовательности или их фрагменты, которые кодируют полипептиды, описанные выше в данном описании, или слитые белки, или их функциональные эквиваленты можно использовать в молекулах рекомбинантных ДНК для управления экспрессией полипептида в соответствующих клетках-хозяевах. Из-за свойственной вырожденности генетического кода можно получить другие последовательности ДНК, кодирующие, по существу, такую же или функционально эквивалентную аминокислотную последовательность, и такие последовательности можно использовать для клонирования и экспрессии данного полипептида.
Последовательности, кодирующие нужный полипептид, можно синтезировать, в целом или частично, с использованием химических способов, хорошо известных в технике (см. Caruthers M.H. et al. (1980), Nucl. Acids Res. Symp. Ser., 215-223; Horn T. et al. (1980), Nucl. Acids Res. Symp. Ser., 225-232).
Для того чтобы экспрессировать нужный полипептид, нуклеотидные последовательности, кодирующие полипептид, или их функциональные эквиваленты можно встроить в соответствующий экспрессирующий вектор, т.е. вектор, содержащий необходимые элементы для транскрипции и трансляции встроенной кодирующей последовательности. Для создания экспрессирующих векторов, содержащих последовательности, кодирующие полипептид, представляющий интерес, и соответствующие контролирующие транскрипцию и трансляцию элементы, можно использовать способы, известные специалистам в данной области техники. Такие способы включают методы in vitro рекомбинатных ДНК, синтетические методы и генетическую рекомбинацию in vivo. Такие методы описываются, например, в Sambrook J. et al., 1989, Molecular Cloning, A Laboratory Manuel, Cold Spring Harbor Press, Plainview N.Y., и Ausubel P.M. et al. (1989), Current Protocols in Molecular Biology, John Wiley & Sons, New York, NY.
"Контролирующие элементы" или "регуляторные последовательности", присутствующие в экспрессирующем векторе, представляют собой нетранслируемые участки вектора - энхансеры, промоторы, 5'- и 3'-нетранслируемые области - которые взаимодействуют с белками клеток-хозяев для осуществления транскрипции и трансляции. Такие элементы могут изменяться по их силе и специфичности. В зависимости от используемых векторной системы и хозяина можно использовать любое число подходящих элементов транскрипции и трансляции, включая конститутивные и индуцируемые промоторы.
Для клеток млекопитающих, как правило, пригоден ряд экспресирующих систем на основе вирусов. Например, в случаях, когда в качестве экспрессирующего вектора используют аденовирус, последовательности, кодирующие полипептид, представляющий интерес, можно лигировать в аденовирусный комплекс транскрипции/трансляции, состоящий из позднего промотора и лидерной последовательности из трех частей. Встраивание в несущественный участок Е1 или Е3 вирусного генома можно использовать для получения жизнеспособных вирусов, способных экспрессировать полипептид в инфицированных клетках-хозяевах (Logan J. and Sheak Т. (1984), Proc. Natl. Acad. Sci., 81:3655-3659). Кроме того, можно использовать энхансеры транскрипции, такие как вирус саркомы Рауса (RSV), для усиления экспрессии в клетках-хозяевах млекопитающих.
Также можно использовать специфические инициирующие сигналы для достижения более эффективной трансляции последовательностей, кодирующих полипептид, представляющий интерес. Такими сигналами являются инициирующий кодон ATG и прилегающие последовательности. В случаях, когда в соответствующий экспрессирующий вектор встраивают последовательности, кодирующие полипептид, его инициирующий кодон и предыдущие последовательности, дополнительные сигналы, контролирующие транскрипцию или трансляцию, могут не потребоваться. Однако в случаях, когда встраивают только кодирующую последовательность или ее часть, экзогенные сигналы, контролирующие трансляцию, включая инициирующий кодон ATG, должны предоставляться. Кроме того, инициирующий кодон должен находиться в правильной рамке считывания для обеспечения трансляции всей вставки. Экзогенные трансляционные элементы и инициирующие кодоны могут быть различного происхождения как природного, так и синтетического. Эффективность экспрессии можно усилить путем включения энхансеров, подходящих для определенной используемой клеточной системы, таких, какие описаны в литературе (Scharf D. et al. (1994), Results Probl. Cell Differ., 20:125-162).
В технике известны различные схемы детекции и измерения экспрессии продуктов, кодируемых полинуклеотидами, в которых используют поликлональные или моноклональные антитела, специфичные для данного продукта. Примерами являются твердофазный иммуноферментный анализ (ELISA), радиоиммуноанализ (RIA) и сортировка клеток, активированная флуоресценцией (FACS). Для некоторых применений может быть предпочтителен двухсайтовый иммуноанализ на основе моноклональных антител с использованием моноклональных антител, реакционноспособных в отношении двух неинтерферирующих эпитопов данного полипептида, но также можно использовать конкурентно-связывающий анализ. Указанные и другие способы анализа описаны, в числе прочих работ, в работе Hampton R. et al. (1990; Serological Methods, a Laboratory Manual, APS Press, St. Paul. Minn.) и Maddox D.E. et al. (1983; J. Exp. Med., 158:1211-1216).
Фармацевтические композиции и способы
Следует иметь в виду, что при необходимости соединения, раскрываемые в данной заявке, можно вводить в сочетании с другими лечебными способами воздействия, такими как антимикробные, противовирусные и противогрибковые соединения или терапии, различные лечебные средства на основе ДНК, лечебные средства на основе РНК, лечебные средства на основе полипептидов и/или другими иммуноэффекторами. Действительно, по существу, можно включить также любой другой компонент, при условии, что дополнительный(ые) компонент(ы) не оказывает(ют) существенного вредного действия при контакте с клетками-мишенями или тканями хозяина. Таким образом, композиции можно доставлять вместе с различными другими средствами, которые требуются или желательны для осуществляемого(ых) специфического(их) воплощения(й) изобретения. Как пример, фармацевтические композиции изобретения можно включать или использовать в сочетании с ДНК, кодирующей один или несколько лечебных белков, антисмысловыми РНК, рибосомами или подобными агентами.
В одном аспекте соединения изобретения и композиции, содержащие их, можно вводить вместе с антигеном для обеспечения адъювантного или усиливающего действия антигена, т.е. усиления иммунной реакции пациента или субъекта. В другом аспекте соединения и композиции изобретения вводят в отсутствие экзогенного антигена для лечебного воздействия самого соединения.
В другом аспекте, где соединения или композиции вводят без экзогенного антигена, настоящее изобретение относится к способам лечения, облегчения и/или, по существу, предотвращения инфекционных заболеваний у субъектов, в частности у животных, предпочтительно у людей. С учетом важности опосредуемых TLR сигналов при врожденной иммунной реакции на микробное заражение способность стимулировать такие каскады реакций селективно и с минимальной токсичностью представляет яркий подход для профилактических и/или исцеляющих способов воздействия против широкого ряда инфекционных факторов.
Способы, описанные в данном описании, применимы против, по существу, любого типа инфекционного фактора, в том числе бактерий, вирусов, паразитов и грибов. Как пример, изобретение применимо для профилактики и/или лечения бактериальных заражений видами Pseudomonas, Escherichia, Klebsiella, Enterobacter, Proteus, Serratia, Candida, Staphylococci, Streptococci, Chlamydia, Mycoplasma и многими другими. Примерами вирусных состояний, которые можно лечить согласно изобретению, являются состояния, вызванные, например, вирусами гриппа, аденовирусами, вирусами парагриппа, риновирусами, респираторно-синцитиальными вирусами (RSV), вирусами герпеса, цитомегаловирусами, вирусами гепатита, например вирусами гепатита В и С, и другими. Характерные примеры грибов включают, например, Aspergillis, Candida albicans, Cryptococcus neoformans, Coccidioides immitus и др.
В характерном воплощении изобретение относится к способам лечения субъектов, в частности субъектов с ослабленным иммунитетом, у которых развились, или у них существует опасность развития, инфекции, такие как нозокомиальная бактериальная и вирусная инфекции. Примерно у 2 миллионов из 40 миллионов индивидуумов, госпитализируемых каждый год, развивается нозокомиальная инфекция во время их пребывания, и примерно у 1% из них, или у примерно 400000 пациентов, развивается нозокомиальная пневмония, из которых более 7000 умирает. Это делает нозокомиальную пневмонию основной причиной смерти при внутрибольничных инфекциях. Таким образом, данное воплощение удовлетворяет существенной потребности в эффективных профилактических приемах при лечении нозокомиальных инфекций.
В родственном воплощении настоящее изобретение относится к профилактическому лечению пациентов с ослабленным иммунитетом, таких как ВИЧ-положительные пациенты, у которых развились, или у них существует опасность развития, пневмония или из-за инфекции, вызванной патогенно-условными микроорганизмами, или из-за реактивации подавленной или латентной инфекции. В 1992 г. сообщалось о примерно 20000 случаях инфекций Pneumocystis carini у пациентов со СПИДом только в США. Кроме того, в то же время 60-70% всех больных СПИДом приобрели Р.carini во время болезни. Таким образом, настоящее изобретение в данном воплощении относится к эффективным профилактическим способам для части населения с такой опасностью.
В другом родственном воплощении способы настоящего изобретения используют для лечения других групп пациентов, у которых может быть ослаблен иммунитет и/или существует опасность развития инфекционных болезней, включая, например, больных муковисцидозом, хронической обструктивной болезнью легких и других пациентов с ослабленным иммунитетом и/или помещенных в специальные лечебные учреждения.
В другом аспекте изобретения соединения и композиции изобретения используют (без экзогенного антигена) в способах лечения, облегчения или, по существу, предупреждения аллергических расстройств и состояний, таких как синусит, хронический риносинусит, астма, атопический дерматит и псориаз. Такой подход основан, по меньшей мере частично, на способности соединений активировать продуцирование цитокинов из клеток-мишеней, которые могут конкурировать с реакциями на цитокины типа стереотипичных аллергических реакций, характеризующихся продуцированном IL-4 или гиперчувствительностью к активности IL-4. Введение некоторых моно- и дисахаридов, раскрываемых в данной заявке, приводит к экспрессии IFN-гамма и IL-12 в антиген-процессирующих и представляющих клетках, а также других клетках, что приводит к отрицательной регуляции цитокинов, ассоциируемых с аллергическими реакциями, таких как IL-4, 5, 6, 10 и 13.
В другом аспекте изобретения соединения и композиции изобретения используют (без экзогенного антигена) в способах лечения аутоиммунных болезней и состояний. Соединения для применения в таком воплощении будут типично выбираться из числа соединений, способных антагонизировать, ингибировать или иначе отрицательно модулировать один или несколько Tool-подобных рецепторов, в частности Тlr2 и/или Тlr4, так что иммунная реакция, ассоциируемая с данным состоянием, ослабевает или, по существу, предотвращается. Как пример, способы по такому воплощению можно использовать при лечении состояний, таких как воспалительное заболевание кишечника, ревматоидный артрит, хронический артрит, рассеянный склероз и псориаз.
Соединения данного изобретения также можно использовать в качестве адъювантов и иммуноэффекторов, усиливающих образование антител у иммунизированных животных, стимулирующих продуцирование цитокинов и стимулирующих клеточноопосредованную иммунную реакцию, включая цитотоксичную Т-лимфоцитарную реакцию.
В способах по изобретению, например, для воздействия на иммунную реакцию индивидуума соединения и композиции данного изобретения можно ввести в композицию с фармацевтически приемлемым носителем для инъекции или для приема пищи. Используемый в данном описании термин "фармацевтически приемлемый носитель" обозначает среду, которая не влияет на иммуномодулирующую активность активного ингредиента и является нетоксичной для пациента, которому ее вводят. Фармацевтически приемлемые носители включают эмульсии типа масло в воде или вода в масле, водные композиции, липосомы, микрогранулы и микросомы. Например, носитель может представлять собой микросферу или микрочастицу с соединением данного изобретения внутри матрицы сферы или частицы или адсорбированным на поверхности сферы или частицы. Носитель также может представлять собой водный раствор или мицеллярную дисперсию, содержащую триэтиламин, триэтаноламин или другое вещество, по природе придающее композиции щелочные свойства, или суспензию, содержащую гидроксид алюминия, гидроксид кальция, фосфат кальция или тирозинадсорбат. Носителями также могут являться все растворители, дисперсионные среды, наполнители, образующие покрытия вещества, разбавители, антибактериальные и противогрибковые средства, придающие изотоничность и замедляющие поглощение вещества, буферы, растворы носителей, суспензии, коллоидные системы и подобные средства. Применение таких сред и средств для фармацевтически активных веществ хорошо известно в технике. Применение среды или средства в лечебных композициях рассматривается за исключением случаев, когда какая-либо среда или средство несовместимы с активным ингредиентом.
Композиции соединения данного изобретения, которые можно вводить парентерально, т.е. подкожно или внутримышечно, включают указанные далее предпочтительные носители. Примерами предпочтительных носителей для подкожного применения являются забуференный фосфатом физиологический раствор (PBS) и 0,01-0,1% раствор триэтаноламина в воде для инъекций, USP. Подходящими носителями для внутримышечных инъекций являются 10% этанол, USP, 40% раствор пропиленгликоля и соразмерное количество приемлемого изотонического раствора, такого как 5% декстроза.
Примерами предпочтительных носителей для внутривенного введения являются 10% этанол, USP, 40% раствор пропиленгликоля и соразмерное количество воды для инъекций, USP. Другой приемлемый носитель содержит 10% этанол, USP и воду для инъекций, USP; еще один приемлемый носитель представляет собой 0,01-0,1% раствор триэтаноламина в воде для инъекций, USP. Фармацевтически приемлемыми парентеральными растворителями являются растворители, раствор или дисперсию в которых можно фильтровать через 5-микронный фильтр без удаления активного ингредиента.
Предпочтительным способом введения композиций данного изобретения является назальное введение, в частности интраназальное введение, или введение путем ингаляции (легочное введение). Доставки лекарственного средства в легкие можно достичь с помощью нескольких разных средств, в том числе жидкостных ингаляторов, аэрозольных дозирующих ингаляторов (MDI) и устройств для дисперсии сухого порошка. Композиции для применения при введении такого типа представляют собой типично сухие порошки или аэрозоли. В случае введения аэрозолей, что является предпочтительным способом введения в данном изобретении, композиции доставляют с помощью ингаляторов, некоторые типы которых описываются ниже.
Сухие порошки содержат, кроме активного ингредиента, носитель, усилитель абсорбции и, необязательно, другие ингредиенты. Носитель представляет собой, например, моно-, ди- или полисахарид, спирт-сахар или другой полиол. Подходящими носителями являются лактоза, глюкоза, рафиноза, мелецитоза, лактит, мальтит, трегалоза, сахароза, маннит и крахмал. Лактоза является особенно предпочтительной, в особенности, в форме ее моногидрата. В композиции также включают усилители абсорбции, такие как полипептиды, поверхностно-активные вещества, алкилгликозиды, соли аминов и жирных кислот или фосфолипиды. Типично ингредиенты композиции должны находиться в тонко измельченной форме, т.е. средний диаметр их частиц должен, как правило, составлять от примерно 30 до примерно 200 микрон, при измерении лазерным дифрактометром или счетчиком coulter counter. Частицы нужного размера можно получить, используя способы, известные в технике, например размол, измельчение в микронайзере или прямое осаждение.
Интраназальный способ введения для соединений данного изобретения имеет ряд преимуществ перед многими другими формами введения. Система для инъекций требует стерилизации шприца для подкожных инъекций и ведет в лечебных учреждениях к беспокойству среди медицинского персонала в связи с опасностью заражения по причине случайного укалывания загрязненной иглой. Также в лечебном учреждении должны быть установлены строгие требования по безопасному обращению с использованными иглой и шприцем. Напротив, интраназальное введение требует небольшого времени со стороны пациента и обслуживающего медицинского персонала и является для учреждения значительно менее обременительным, чем инъекции.
Другим важным преимуществом интраназального введения является принятие пациентом системы доставки лекарственного средства. Интраназальное введение ощущается как неинвазивное, не вызывающее боли, не имеет существенных последствий и вызывает у пациента, испытывающего симптомы, удовлетворение от быстрого облегчения. Это является особенно важным преимуществом, когда пациентом является ребенок. Другим важным обстоятельством является то, что пациент может сам вводить предписанную(ые) дозировку(и) назального спрея.
Для интраназального введения композиции данного изобретения могут иметь форму жидкостей или твердых веществ. Такие композиции могут содержать один или несколько адъювантов, веществ для усиления абсорбции активных ингредиентов путем проникания через носовую слизистую оболочку и (в случае жидких композиций) водный разбавитель, например воду. С другой стороны, разбавитель может содержать водный буфер, такой как фосфатный буфер. Композиция также может включать, необязательно, один или несколько многоатомных спиртов и один или несколько консервантов, таких как, например, гентамицин, бацитрацин (0,005%) или крезол. Композиции можно вводить в носовую полость в форме спрея, используя аэрозольный ингалятор, жидкостной ингалятор, распылитель, капельницу или другое устройство, которое обеспечивает контакт раствора со слизистой оболочкой носа. Устройство может быть простым, таким как назальный распылитель, который может использовать пациент, или оно может быть более совершенным для более точного распределения композиций, которое можно использовать во врачебном кабинете или в лечебном учреждении.
Назальные порошковые композиции можно получить, смешивая активный ингредиент и эксципиент, имеющие обе частицы нужного размера. Сначала получают раствор активного ингредиента и циклодекстриновых эксципиентов, после чего проводят осаждение, фильтрацию и пульверизацию. Растворитель также можно удалить сушкой вымораживанием с последующей пульверизацией порошка до частиц нужного размера, используя обычные методы, известные из фармацевтической литературы. Конечной стадией является классификация по размеру, например просеиванием, для получения частиц, имеющих, предпочтительно, диаметр от 30 до 200 микрон. Порошки можно вводить с использованием назального инсуффлятора или их можно поместить в ряд капсул в устройстве для ингаляции или инсуффляции. Через капсулу протыкают иглу для того, чтобы получить отверстия в верхней и нижней части капсулы, и пропускают воздух, выдувающий частицы порошка. Порошковые композиции также можно вводить в струе инертного газа или суспендированными в жидких органических средах.
В специфическом воплощении фармацевтическую композицию можно доставлять в системе с регулируемым или отсроченным высвобождением (см. Langer, Science, 249:1527-1533 (1990); Sefton, 1987, CRC Crit. Ref. Biomed. Eng., 14:10; Buschwald et al., 1980, Surgery, 88:507; Saudek et al., 1989, N. Engl. J. Med., 321:574). В другом воплощении можно использовать полимерные материалы для достижения регулируемого или отсроченного высвобождения агониста κ-опиоидных рецепторов и/или опиоидного антагониста (см., например. Medical Applications of Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Florida, 1974; Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, 1983, J. Macromol. Sci. Rev. Macrol. Chem., 23:61; см. также Leavy et al., 1985, Science, 228:190; During et al., 1989, Ann. Neurol, 25:351; Howard et al., J. Neurosurg., 7 1:105; патент США №5679377; патент США №5916597; патент США №5912015; патент США №5989463; патент США №5128326; публикация РСТ WO 99/12154 и публикация РСТ WO 99/20253). Примерами полимеров, используемых в композициях с отсроченным высвобождением, являются, но не ограничиваются перечисленным, поли(2-гидроксиэтилметилакрилат), полиметилметакрилат, полиакриловая кислота, сополимер этилена и винилацетата, полиметакриловая кислота, полигликолиды (PLG), полиангидриды, поли-N-винилпирролидон, поливиниловый спирт, полиакриламид, полиэтиленгликоль, полиактиды (PLA), сополимеры лактида и гликолида (PLGA) и сложные полиортоэфиры. В предпочтительном воплощении полимер, используемый в композиции с отсроченным высвобождением, является инертным, свободным от выщелачиваемых примесей, устойчивым при хранении, стерильным и поддающимся биологическому разложению. В еще одном воплощении систему с регулируемым или отсроченным высвобождением можно поместить вблизи лечебной мишени, причем тогда требуется только часть системной дозы (см., например, Goodson, в Medical Applications of Controlled Release, цит. выше, vol.2, pp.115-138 (1984)).
Носители для применения с такими фармацевтическими композициями являются биосовместимыми и также могут быть поддающимися биологическому разложению. В некоторых воплощениях композиция, предпочтительно, обеспечивает постоянный уровень высвобождения активного компонента. Однако в других воплощениях может быть желательной более быстрая скорость высвобождения непосредственно после введения. Получение таких композиций, с использованием известных методов, входит в компетенцию специалиста в данной области техники. Характерными носителями в таком случае являются микрочастицы сополимера лактида и гликолида, полиакрилата, латекса, крахмала, целлюлозы, декстрана и подобных веществ. Другими характерными носителями в случае отсроченного высвобождения являются надмолекулярные биологические векторы, содержащие нежидкую гидрофильную сердцевину (например, полисахарид или олигосахарид с поперечными связями) и, необязательно, наружный слой, содержащий амфифильное соединение, такое как фосфолипид (см., например, патент США №5151254 и заявки РСТ WO 94/20078, WO 94/23701 и WO 96/06638). Количество активного соединения, содержащееся в композиции с отсроченным высвобождением, зависит от места имплантации, скорости и ожидаемой длительности высвобождения и характера состояния, от которого лечат или которое предупреждают.
Соединения данного изобретения вводят индивидууму в эффективном количестве или фармацевтически эффективном количестве для выявления или усиления иммунной реакции индивидуума. Используемый в данном описании термин "эффективное количество" или "фармацевтически эффективное количество" обозначает такое количество, которое показывает реакцию, превышающую реакцию на носитель или отрицательный контроль. "Адъювантэффективное количество" означает такое количество соединения в данном случае, которое при введении в сочетании с антигеном показывает реакцию, превышающую реакцию, вызываемую одним антигеном. Точная дозировка соединений данного изобретения, вводимая пациенту, будет зависеть от конкретного используемого соединения, способа введения, фармацевтической композиции и пациента. Например, при подкожном введении для усиления реакции на антитела количество используемого соединения составляет от 1 до примерно 250 мкг, предпочтительно - от примерно 25 до примерно 50 мкг, для введения в расчете на типичного взрослого пациента с массой тела 70 кг.
В другом воплощении характерные иммуногенные композиции настоящего изобретения, например иммуногенные композиции и/или вакцины, содержат ДНК, кодирующую один или несколько полипептидов, описанных выше, так что полипептид генерируется in situ. Как указано выше, полинуклеотид можно вводить в любой из разнообразных систем доставки, известных специалистам в данной области техники. Действительно, в технике хорошо известны многие методы доставки генов, например, описанные в Rolland, Crit. Rev. Therap. Drug Carrier Systems, 15:143-198, 1998, и в ссылках, цитированных в указанной работе. Подходящие системы экспрессии полинуклеотидов, конечно, будут содержать необходимые регуляторные последовательности регуляторной ДНК для экспрессии в организме пациента (такие, как подходящий промотор и сигнал терминации).
Следовательно, в некоторых воплощениях полинуклеотиды, кодирующие иммуногенные полипептиды, описанные в данном описании, вводят в подходящие клетки-хозяев млекопитающих для экспрессии с использованием любой из многих известных систем на основе вирусов. В одном характерном воплощении удобную и эффективную основу для систем доставки генов обеспечивают ретровирусы. Выбранную нуклеотидную последовательность, кодирующую полипептид настоящего изобретения, можно встроить в вектор и упаковать в частицы ретровирусов с использованием методов, известных в технике. Затем такие рекомбинантные вирусы можно извлечь и доставить субъекту. Описаны многие характерные ретровирусные системы (например, в пат. США №5219740; Miller and Rosman (1989), BioTechniques, 7:980-990; Miller A.D. (1990), Human Gene Therapy, 1:5-14; Scarpa et al. (1991), Virology, 180:849-852; Burns et al. (1993), Proc. Natl. Acad. Sci. USA, 90:8033-8037; и в Boris-Lawrie and Temin (1993), Cur. Opin. Genet. Develop., 3:102-109.
Кроме того, также описан ряд характерных систем на основе аденовирусов. В отличие от ретровирусов, которые интегрируются в геном хозяина, аденовирусы остаются вне хромосомы, причем, таким образом, сводятся к минимуму опасности, связанные с инсерционным мутагенезом (Haj-Ahmad and Graham (1986), J.Virol., 57:267-274; Bett et al. (1993), J.Virol., 67:5911-5921; Mittereder et al. (1994), Human Gene Therapy, 5:717-729; Seth et al. (1994), J.Virol., 68:933-940; Barr et al. (1994), Gene Therapy, 1:51-58; Berkner K.L. (1988), BioTechniques, 6:616-629; и Rich et al. (1993), Human Gene Therapy, 4:461-476).
Для доставки полинуклеотидов также разработаны различные векторные системы на основе аденоассоциированных вирусов (AAV). Векторы AAV можно легко сконструировать с использованием методов, хорошо известных в технике (см., например, пат. США №№5173414 и 5139941, заявки на международный патент WO 92/01070 и WO 93/03769; Lebkowski et al. (1988), Molec. Cell. Biol, 8:3988-3996; Vincent et al. (1990), Vaccines 90 (Cold Spring Harbor Laboratory Press); Carter B.J. (1992), Current Opinion in Biotechnology, 3:533-539; Muzyczka N. (1992), Current Topics in Microbiol. and Immunol, 158:97-129; Kotin R.M. (1994), Human Gene Therapy, 5:793-801; Shelling and Smith (1994), Gene Therapy, 1:165-169; и Zhou et al. (1994), J. Exp. Med., 179:1867-1875.
Другие вирусные векторы, применимые для доставки методом переноса генов полинуклеотидов, кодирующих полипептиды настоящего изобретения, включают векторы, образованные от вирусов семейства покс, таких как вирус коровьей оспы и птичий поксвирус. Как пример, рекомбинанты вируса коровьей оспы, экспрессирующие новые молекулы, можно сконструировать следующим образом. Сначала ДНК, кодирующую полипептид, встраивают в соответствующий вектор так, чтобы она соседствовала с промотором коровьей оспы и фланкирующими последовательностями ДНК коровьей оспы, такими как последовательность, кодирующая тимидинкиназу (ТК). Затем такой вектор используют для трансфекции клеток, которые одновременно инфицируют вирусами коровьей оспы. Гомологичная рекомбинация служит для встраивания в вирусный геном промотора коровьей оспы вместе с геном, кодирующим нужный полипептид. Полученный рекомбинант TK.sup.(-) можно отобрать, культивируя клетки в присутствии 5-бромдезоксиуридина и собирая вирусные бляшки, устойчивые к нему.
Систему инфицирования/трансфекции на основе вирусов коровьей оспы можно удобно использовать для обеспечения индуцируемой транзиторной экспрессии или коэкспрессии одного или нескольких полипептидов, приведенных в данном описании, в клетках-хозяевах организма. В такой определенной системе клетки сначала инфицируют in vitro рекомбинантом вируса коровьей оспы, кодирующим РНК-полимеразу бактериофага Т7. Данная полимераза проявляет совершенную специфичность в том, что она транскрибирует только матрицы, содержащие промоторы Т7. После инфицирования клетки трансфицируют полинуклеотидом или полинуклеотидами, представляющими интерес, управляемыми промотором Т7. Полимераза, экспрессированная в цитоплазме от рекомбинанта вируса коровьей оспы, транскрибирует трансфицированную ДНК в РНК, которая затем транслируется в полипептид трансляционным механизмом хозяина. Способ обеспечивает на высоком уровне транзиторное цитоплазматическое продуцирование больших количеств РНК и продуктов ее трансляции (см., например, Elroy-Stein and Moss, Proc. Natl. Acad. Sci. USA (1990), 87:6743-6747; Fuerst et al., Proc. Natl. Acad. Sci. USA (1986), 83:8122-8126).
С другой стороны, для доставки нужных кодирующих последовательностей также можно использовать птичьи поксвирусы, такие как вирусы fowlpox и canarypox. Известно, что рекомбинантные птичьи поксвирусы, экспрессирующие иммуногены из патогенов млекопитающих, придают защитный иммунитет при введении видам, не относящимся к птицам. Использование вектора авипокс особенно желательно для человека и других видов млекопитающих, так как члены рода Avipox могут продуктивно реплицировать только в восприимчивых видах птиц и поэтому являются незаразными в клетках млекопитающих. Способы получения рекомбинантных птичьих поксвирусов известны в технике, и в них используют генную рекомбинацию, как описано выше, в отношении получения вирусов коровьей оспы (см., например, WO 91/12882, WO 89/03429 и WO 92/03545).
Также для доставки полинуклеотидных композиций настоящего изобретения можно использовать любой вектор из числа альфавирусных векторов, таких как векторы, описанные в патентах США №№5843723, 6015686, 6008035 и 6015694. Также можно использовать некоторые векторы на основе вирусов венесуэльского энцефалита у лошадей (VEE), характерные примеры которых можно найти в патентах США №№5505947 и 5643576.
Кроме того, для доставки генов по изобретению также можно использовать молекулярные конъюгированные векторы, такие как аденовирусные химерные векторы, описанные в Michael et al., J. Biol. Chem. (1993), 268:6866-6869, и в Wagner et al., Proc. Natl. Acad. Sci. USA (1992), 89:6099-6103.
Другую пояснительную информацию об указанных и других системах доставки на основе вирусов можно найти, например, в Fisher-Hoch et al., Proc. Natl. Acad. Sci. USA, 86:317-321, 1989; Flexner et al., Ann. N. Y. Acad. Sci., 569:86-103, 1989; Flexner et al., Vaccine, 8:17-21, 1990; патентах США №№4603112, 4769330 и 5017487; WO 89/01973; патенте США №4777127; GB 2200651; ЕР 0345242; WO 91/02805; Berkner, Biotechniques, 6:616-627, 1988; Rosenfeld et al., Science, 252:431-434, 1991; Kolls et al., Proc. Natl. Acad. Sci. USA, 91:215-219, 1994; Kass-Eisler et al., Proc. Natl. Acad. Sci. USA, 90:11498-11502, 1993; Guzman et al., Circulation, 88:2838-2848, 1993; и Guzman et al., Cir. Res., 73:1202-1207, 1993.
В некоторых воплощениях полинуклеотид можно интегрировать в геном клетки-мишени. Такая интеграция может происходить в определенном месте и ориентации путем гомологичной рекомбинации (замена гена) или его можно интегрировать в случайное неопределенное место (усиление гена). В других воплощениях полинуклеотид может устойчиво сохраняться в клетке в виде отдельного эписомального сегмента ДНК. Такие полинуклеотидные сегменты или "эписомы" кодируют последовательности, которых достаточно для возможности сохранения и репликации, независимой от или синхронной с циклом клетки-хозяина. Способ, которым экспрессирующая конструкция доставляется в клетку, и где в клетке остается полинуклеотид, зависит от типа используемой экспрессирующаей конструкции.
В другом воплощении изобретения полинуклеотид вводят/доставляют в виде "голой" ДНК, например, как описано в Ulmer et al., Science, 259:1745-1749, 1993, и в обзоре Cohen, Science, 259:1691-1692, 1993. Поглощение голой ДНК можно усилить путем нанесения ДНК на биологически разлагаемые гранулы, которые эффективно транспортируются в клетки.
В еще одном воплощении композицию настоящего изобретения можно доставлять способами с бомбардировкой частицами, многие из которых описаны. В одном характерном примере ускорения частиц, образованных газом, можно достичь с помощью таких устройств, как приборы, изготовляемые Powderject Pharmaceuticals PLC (Oxford, UK) и Powderject Vaccines Inc. (Madison, WI), примеры которых описаны в патентах США №№5846796, 6010478, 5865796, 5584807 и в патенте ЕР №0500799. Такой подход представляет собой способы доставки без иглы, где микроскопические частицы сухого порошкового препарата, такие как частицы полинуклеотида или полипептида, ускоряются до высокой скорости в струе гелия, генерированной портативным прибором, продвигающей частицы к ткани-мишени, представляющей интерес.
В родственном воплощении другие устройства и способы, которые можно применять для проводимой без иглы с помощью газа инъекции композиций настоящего изобретения, включают устройства и способы, предлагаемые Bioject, Inc. (Portland, OR), примеры которых описаны в патентах США №№4790824, 5064413, 5312335, 5383851, 5399163, 5520639 и 5993412.
В некоторых воплощениях изобретения фармацевтическая композиция представляет собой, предпочтительно, композицию, вызывающую иммунную реакцию преимущественно типа Тhl. Высокое содержание цитокинов типа Th-1 (например, IFN-γ, TNF-α, IL-2 и IL-12) благоприятствует индукции клеточноопосредованных иммунных реакций на вводимый антиген. Напротив, высокие уровни цитокинов типа Тh-2 (например, IL-4, IL-5, IL-6 и IL-10) благоприятствуют индукции гуморальных иммунных реакций. После применения иммуногенной композиции, описанной в данном описании, у пациента будет поддерживаться иммунная реакция, включающая реакции типа Тh1 и Th2. В предпочтительном воплощении, где реакция преимущественно представляет тип Тh1, содержание цитокинов типа Тh1 будет повышаться в большей степени, чем содержание цитокинов типа Th2. Уровни указанных цитокинов можно легко оценить с использованием стандартных анализов. С другой стороны, или, кроме того, высокие уровни цитокинов типа Th2 (например, IL-4, IL-5, IL-6 и IL-10) могут быть желательны в случае некоторых лечебных применений. Уровни таких цитокинов можно легко оценить с использованием стандартных анализов. Для общего представления о семействах цитокинов см. Monsmann and Coggman, Ann. Rev. Immunol., 7:145-173, 1989.
Характерные композиции для применения в индукции цитокинов типа Тh1 включают, например, сочетание CpG-содержащих олигонуклеотидов (в которых динуклеотид CpG является неметилированным), описанных, например, в WO 96/02555, WO 99/33488 и в патентах США №№6008200 и 5856462. Иммуномодулирующие последовательности ДНК также описаны, например, в Sato et al., Science, 273:352, 1996. Другие подходящие иммуностимуляторы содержат сапонины, как, например, QS21 (Aquila Biopharmaceuticals Inc., Framingham, MA), и родственные производные сапонинов и их миметики.
Любой из многочисленных иммуностимуляторов можно включать в композиции данного изобретения. Например, цитокины, такие как GM-CSF, интерфероны или интерлейкины также модулируют нужную иммунную реакцию. Кроме того, также можно использовать Montanide ISA 720 (Seppic, Франция), SAF (Chiron, California, Соединенные Штаты), ISCOMS (CSL), MF-59 (Chiron), адъюванты группы SBAS (например, SBAS-2 или SBAS-4, доступные от SmithKline Beecham, Rixensart, Бельгия) и иммуномодулятор Enhanzyn™ (Corixa, Hamilton, MT). Иммуностимуляторы на основе простых эфиров полиоксиэтилена описаны в WO 99/52549 A1 и также могут использоваться.
Фармацевтические композиции изобретения часто также будут содержать один или несколько буферов (например, нейтральный забуференный физиологический раствор или забуференный фосфатом физиологический раствор), углеводы (например, глюкозу, маннозу, сахарозу или декстраны), маннит, белки, полипептиды или аминокислоты, такие как глицин, антиоксиданты, бактериостаты, комплексообразователи, такие как ЭДТК или глутатион, адъюванты (например, гидроксид алюминия), растворенные вещества, придающие препарату изотоничность, гипотоничность или слабую гипертоничность с кровью реципиента, суспендирующие вещества, загустители и/или консерванты. С другой стороны, композиции настоящего изобретения можно получить в виде лиофилизатов.
Фармацевтические композиции, приведенные в данном описании, можно представить в однодозовых или многодозовых емкостях, таких как запаянные ампулы или флаконы. Такие емкости типично плотно закрывают, чтобы таким способом сохранить стерильность и устойчивость препарата до применения. Вообще препараты можно хранить в виде суспензий, растворов или эмульсий в масляных или водных носителях. С другой стороны, фармацевтическую композицию можно хранить как высушенную вымораживанием, причем требуется только добавление стерильного жидкого носителя непосредственно перед применением.
Разработки подходящих схем дозирования и лечения для применения с определенными композициями, описанными в данном описании, в разных схемах лечения, в том числе, в случае перорального, парентерального, внутривенного, интраназального и внутримышечного введения, и препарата, хорошо известны в технике, и ряд из них коротко обсуждается ниже в целях пояснения общих положений.
В некоторых применениях фармацевтические композиции, описанные в данном описании, можно доставлять животному пероральным введением. В таком случае такие композиции можно сочетать в препарате с инертным разбавителем или с усвояемым съедобным носителем, или их можно заключать в капсулы с твердой или мягкой желатиновой оболочкой, или их можно прессовать в таблетки, или их можно включать непосредственно в пищевые продукты рациона.
Активные соединения даже можно смешивать с эксципиентами и использовать в форме таблеток для проглатывания, таблеток для медленного рассасывания в щечном кармане, пастилок, капсул, эликсиров, суспензий, сиропов, облаток и т.п. (см., например, Mathiowitz et al., Nature, 1997, Mar 27, 386 (6623):410-4; Hwang et al., Crit. Rev. Ther. Drug Carrier Sits, 1998, 15(3):243-84; патент США №5641515; патент США №5580579 и патент США №5792451). Таблетки, пастилки, пилюли, капсулы и т.п. также могут содержать любой из различных дополнительных компонентов, например связующее вещество, такое как трагакантовая смола, аравийская камедь, кукурузный крахмал или желатин; эксципиенты, такие как дикальцийфосфат; вещество, способствующее рассыпанию, такое как кукурузный крахмал, картофельный крахмал, альгиновая кислота и т.п.; смазывающее вещество, такое как стеарат магния; и может быть добавлено подслащивающее вещество, такое как сахароза, лактоза или сахарин, или корригент, такой как перечная мята, винтергеновое масло или вишневая отдушка. Когда стандартная лекарственная форма представляет собой капсулу, она может содержать, кроме веществ указанных выше типов, жидкий носитель. Различные другие материалы могут присутствовать в качестве покрытий или для иной модификации физической формы лекарственной единицы. Например, на таблетки, пилюли или капсулы может быть нанесено покрытие из шеллака, сахара или того и другого. Конечно, любое вещество, используемое при получении любой стандартной формы, должно являться фармацевтически чистым и, по существу, нетоксичным в используемых количествах. Кроме того, активные соединения могут быть включены в препарат и составы с отсроченным высвобождением лекарственного средства.
Типично такие препараты будут содержать, по меньшей мере, примерно 0,1% или более активного соединения, хотя процентное содержание активного(ых) ингредиента(ов) может, конечно, изменяться и обычно может составлять от примерно 1 или 2% до примерно 60 или 70% от массы или объема всего препарата. Естественно, количество активного(ых) соединения(й) в каждой терапевтически применимой композиции, которую можно получить, является таким, что подходящую дозировку можно будет получить в любой данной единичной дозе соединения. Факторы, такие как растворимость, биодоступность, биологический полупериод существования, способ введения, срок хранения продукта, а также другие фармакологические обстоятельства, будут учитываться специалистами в области получения таких фармацевтических препаратов, и если так, могут быть желательны различные схемы дозировки и лечения.
Для перорального введения композиции настоящего изобретения можно, с другой стороны, смешивать с одним или несколькими эксципиентами в форме жидкости для полоскания рта, средства для чистки зубов, таблетки для медленного рассасывания в щечном кармане, перорального спрея или вводимого подъязычно перорального препарата. С другой стороны, активный ингредиент можно включить в пероральный раствор, например, содержащий борат натрия, глицерин и бикарбонат натрия, или диспергировать в средстве для чистки зубов или добавить в терапевтически эффективном количестве в композицию, которая может содержать воду, связующие вещества, абразивы, отдушки, пенообразователи и смачивающие вещества. С другой стороны, композициям можно придать формы таблеток или раствора, которые можно поместить под язык или иным способом растворить во рту.
В некоторых случаях будет желательна доставка фармацевтических композиций, описанных в данном описании, парентеральным, внутривенным, внутримышечным или даже интраперитонеальным способом. Такие подходы хорошо известны специалистам в данной области техники, и некоторые из них описаны, например, в патенте США №5543158, патенте США №5641515 и патента США №5399363. В некоторых воплощениях растворы активных соединений в виде свободных оснований или фармакологически приемлемых солей можно получить в воде в смеси с подходящим поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Также можно получить дисперсии в глицерине, жидких полиэтиленгликолях и их смесях и маслах. В обычных условиях хранения и применения такие препараты, как правило, будут содержать консервант для предотвращения размножения микроорганизмов.
Характерными фармацевтическими формами, подходящими для инъекций, являются стерильные водные растворы или дисперсии и стерильные порошки для приготовления стерильных растворов или дисперсий для немедленного применения (например, см. патент США №5466468). Во всех случаях форма должна быть стерильной и должна быть жидкой до такой степени, чтобы ее можно было легко вводить шприцем. Она должна быть устойчивой в условиях изготовления и хранения, и ее следует предохранять от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащий, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль, и т.п.), их подходящие смеси и/или растительные масла. Подходящую текучесть можно сохранить, например, используя покрытие, такое как лецитин, поддерживая требуемый размер частиц в случае дисперсии и/или используя поверхностно-активные вещества. Предотвращение действия микроорганизмов можно облегчить, например, с помощью различных антибактериальных и противогрибковых средств, например парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимеросаля и т.п. Во многих случаях будет предпочтительно включать вещества для придания изотоничности, например сахара или хлорида натрия. Длительного всасывания инъецируемых композиций можно добиться, используя в композициях вещества, замедляющие поглощение, например моностеарат алюминия и желатин.
В одном воплощении в случае парентерального введения в водном растворе раствор следует, при необходимости, соответствующим образом забуферировать, и жидкому разбавителю в первую очередь придают изотоничность достаточным количеством физиологического раствора или глюкозы. Такие определенные водные растворы особенно подходят для внутривенного, внутримышечного, подкожного и интраперитонеального введения. В этой связи стерильная водная среда, которую можно использовать в свете данного раскрытия, будет известна специалистам в данной области техники. Например, одну дозировку можно растворить в 1 мл изотонического раствора NaCl и или добавить к 1000 мл жидкости, вводимой в подкожную клетчатку, или инъецировать в нужное место инфузии (см., например, "Remington's Pharmaceutical Sciences", 15th Edition, pages 1035-1038, 1570-1580). Некоторые изменения в дозировке обязательно будут иметь место в зависимости от состояния субъекта, которого лечат. Кроме того, в случае введения человеку препараты, конечно, будут, предпочтительно, удовлетворять требованиям стерильности, пирогенности и общей безопасности и стандартам чистоты, которые требуют стандарты FDA Комитета по биологическим стандартам.
В другом воплощении изобретения композиции, раскрываемые в данном описании, могут содержаться в нейтральной или солевой форме. Характерными фармацевтически приемлемыми солями являются соли присоединения кислот (образованные по свободным аминогруппам белка) и образованные с неорганическими кислотами, такими как, например, хлороводородная или фосфорная кислота, или такими органическими кислотами как уксусная, щавелевая, винная, миндальная или подобная кислота. Также можно получить соли, образованные по свободным карбоксильным группам с неорганическими основаниями, такими как, например, гидроксиды натрия, калия, аммония, кальция или железа, и такими органическими основаниями, как изопропиламин, триметиламин, гистидин, прокаин и т.п. После получения растворы будут вводиться способом, совместимым с лекарственным препаратом, и в таком количестве, которое является терапевтически эффективным.
В некоторых воплощениях фармацевтические композиции можно доставлять с помощью интраназальных спреев, ингаляции и/или других аэрозолей для доставки. Способы доставки композиций с генами, нуклеиновыми кислотами и пептидами назальными аэрозолями непосредственно в легкие описаны, например, в патенте США №5756353 и патенте США №5804212. Подобным образом, также в фармацевтической технике хорошо известна доставка лекарственных средств с использованием интраназального введения микрочастиц смолы (Takenaga et al., J. Controlled Release, 1998, Mar 2, 52(1-2):81-7) и лизофосфатидилглицеринов (патент США №5725871). Также в патенте США №5780045 описаны примеры трансмукозной доставки лекарственных средств в форме несущей матрицы из политетрафторэтилена.
В некоторых воплощениях используют липосомы, нанокапсулы, микрочастицы, липидные частицы, везикулы и т.п. для внедрения композиций настоящего изобретения в подходящие хозяйские клетки/организмы. В частности, можно получить композиции настоящего изобретения для доставки или инкапсулированными в липидные частицы, липосомы, везикулы, наносферы, или наночастицы, или подобным образом. С другой стороны, композиции настоящего изобретения можно связать, или ковалентно, или нековалентно с поверхностью таких носителей-наполнителей.
Получение и применение липосомоподобных препаратов как потенциальных носителей лекарственных средств, вообще, известно специалистам в данной области техники (см., например, Lasic, Trends Biotechnol., 1998, Jul, 16(7):307-21; Takakura, Nippon Rinsho, 1998, Mar, 56(3):691-5; Chandran et al., Indian. J. Exp. Biol., 1997, Aug, 35(8):801-9; Margalit, Crit. Rev. Ther. Drug Carrier Syst, 1995, 12(2-3):233-61; патент США №5567434; патент США №5552157; патент США №5565213; патент США №5738868 и патент США №5795587).
Липосомы успешно используются с рядом типов клеток, которые обычно трудно трансфицировать другими процедурами, в том числе суспензиями Т-клеток, первичными культурами гепатоцитов и клетками PC 12 (Renneisen et al., J. Biol. Chem., 1990, Sep 25, 265(27): 16337-42; Muller et al., DNA Cell Biol., 1990, Apr, 9(3):221-9). Кроме того, липосомы свободны от ограничений по длине ДНК, типичных для систем доставки на основе вирусов. Липосомы эффективно используют для введения генов, различных лекарственных средств, средств для лучевой терапии, ферментов, вирусов, факторов транскрипции, аллостерических эффектронов и т.п., в различные культивированные клеточные линии и животным. Кроме того, оказывается, что использование липосом не связано с аутоиммунными реакциями или неприемлемой токсичностью после системной доставки.
В некоторых воплощениях липосомы получают из фосфолипидов, диспергированных в водной среде, с самопроизвольным образованием мультиламеллярных концентрических двухслойных везикул (также называемых мультиламеллярными везикулами (MLV)).
С другой стороны, в других воплощениях изобретение относится к фармацевтически приемлемым нанокапсулярным препаратам с композициями настоящего изобретения. Нанокапсулы, как правило, могут захватывать соединения прочно и воспроизводимо (см., например, Quintanar-Guerrero et al., Drug Dev. Ind. Pharm., 1998, Dec, 24(12):1113-28). Для того чтобы избежать побочного действия из-за перегрузок клеток полимерами, такие ультрамелкие частицы (размером примерно 0,1 мкм) можно создать с использованием полимеров, способных разлагаться in vivo. Такие частицы можно получить так, как описано, например, в Couvrer et al., Crit. Rev. Ther. Drug Carrier Syst., 1988, 5(1):1-20; zur Muchlen et al., Eur. J. Pharm. Biopharm., 1998, Mar, 45(2):149-55; Zambaux et al., J. Controlled Release, 1998, Jan 2, 50(1-3):31-40; и патенте США 5145684.
Лечение раковых заболеваний
Иммунологические подходы к лечению рака основаны на знании того, что раковые клетки часто могут не поддаваться действию защитных сил организма против аберрантных или чужеродных клеток и молекул, и что такие защитные силы можно терапевтически стимулировать для возвращения утраченных позиций (см., например, pgs. 623-648 в Klein, Immunology (Wiley-Interscience, New York, 1982)). Многочисленные последние наблюдения того, что различные иммунные эффекторы могут прямо или косвенно ингибировать рост опухолей, привели к возвращению интереса к такому подходу к лечению рака (см., например, Jager et al., Oncology, 2001, 60(1):1-7; Renner et al., Ann. Hematol., 2000, Dec, 79(12):651-9).
Четырьмя основными типами клеток, функции которых связаны с противоопухолевым клеточным иммунитетом и удалением опухолевых клеток из организма, являются: i) В-лимфоциты, секретирующие иммуноглобулины в плазму крови для идентификации и мечения чужих вторгшихся клеток; ii) моноциты, секретирующие белки комплемента, ответственные за лизис и процессинг меченных иммуноглобулинами вторгшихся клеток; iii) природные лимфоциты-киллеры, имеющие два механизма разрушения опухолевых клеток - антителозависимую клеточноопосредованную цитотоксичность и естественный лизис; и iv) Т-лимфоциты, обладающие антигенспецифическими рецепторами и способностью узнавать опухолевые клетки, несущие комплементарные маркерные молекулы (Schreiber H., 1989, в Fundamental Immunology (ed.) W.F.Paul, pp.923-955).
Противораковая иммунотерапия, как правило, сосредоточена на индуцировании гуморальных иммунных реакций, клеточных иммунных реакций или тех и других. Кроме того, твердо установлено, что индукция хелперных Т-клеток CD4+ необходима для того, чтобы вторично индуцировать или антитела, или цитотоксичные Т-клетки CD8+. Полипептидные антигены, селективные или идеально специфические в отношении раковых клеток, представляют сильный подход для индукции иммунных реакций против рака и составляют важный аспект настоящего изобретения.
Поэтому в других аспектах настоящего изобретения фармацевтические композиции, описанные в данном описании, могут использоваться для стимуляции иммунной реакции против рака. В рамках таких способов фармацевтические композиции, описанные в данном описании, вводят пациенту, типично, теплокровному животному, предпочтительно - человеку. Пациент может быть поражен или не поражен раком. Фармацевтические композиции или вакцины можно вводить или перед, или после хирургического удаления первичных опухолей и/или лечения, такого как лучевая терапия или обычные химиотерапевтические лекарственные средства. Как описано выше, введение фармацевтических композиций можно осуществлять любым подходящим способом, в том числе внутривенным, интраперитонеальным, внутримышечным, подкожным, интраназальным, интрадермальным, анальным, вагинальным, местным и пероральным путями.
В некоторых воплощениях иммунотерапия может представлять собой активную иммунотерапию, при которой лечение основано на стимуляции in vivo эндогенной иммунной системы хозяина для реакции против опухолей с помощью введения средств, модифицирующих иммунные реакции (таких как полипептиды и полинуклеотиды, описанные в данном описании).
Способы и частота введения лечебных композиций, описанных в данном описании, а также дозировка, будут изменяться от индивидуума к индивидууму и могут быть легко установлены с использованием стандартных методов. Вообще фармацевтические композиции и вакцины можно вводить инъекцией (например, интрадермальной, внутримышечной, внутривенной или подкожной), интраназально (например, путем аспирации) или перорально. Предпочтительно, можно вводить от 1 до 10 доз на протяжении 52 недель. Предпочтительно, вводят 6 доз с интервалом в 1 месяц, и затем можно периодически осуществлять бустер-вакцинацию. Для отдельных пациентов могут подходить другие схемы. Подходящей дозой является количество соединения, которое, при таком введении, как описано выше, способно промотировать противоопухолевую иммунную реакцию, по меньшей мере, на 10-50% превышающую базовый уровень (т.е., в отсутствие обработки). Такую реакцию можно контролировать, определяя у пациента антитела против опухоли, или с помощью генерации вакцинозависимых цитолитических эффекторных клеток, способных убивать опухолевые клетки пациента in vitro. Такие вакцины также должны быть способны вызывать иммунную реакцию, ведущую к улучшенному клиническому результату (например, более частым ремиссиям, продолжительному существованию при полном, или частичном, или более длительном отсутствии заболевания) у вакцинированных пациентов по сравнению с невакцинированными пациентами. Вообще, в случае фармацевтических композиций и вакцин, содержащих один или несколько полипептидов, количество каждого полипептида, присутствующего в дозе, колеблется от примерно 25 мкг до 5 мг на кг массы тела хозяина. Величины подходящих доз могут изменяться с массой пациента, но типично, будут колебаться от примерно 0,1 мл до примерно 5 мл.
Вообще, подходящие дозировка и схема лечения предоставляют активное(ые) соединение(я) в количестве, достаточном для обеспечения лечебного и/или профилактического благоприятного результата. Такую реакцию можно контролировать по улучшенному клиническому результату (например, более частым ремиссиям, продолжительному существованию при полном, или частичном, или более длительном отсутствии заболевания) у пациентов, прошедших лечение, по сравнению с пациентами, не получавшими лечение. Усиление прежних иммунных реакций на белок опухоли, как правило, коррелирует с улучшенным клиническим результатом. Такие иммунные реакции, как правило, можно оценить с использованием стандартных анализов на пролиферацию, цитотоксичность или цитокины, которые можно осуществлять с использованием образцов, взятых у пациента до и после лечения.
Настоящее изобретение далее описывается с помощью примеров, не являющихся ограничительными, и примеров испытаний, которые приводятся только в целях пояснения. Все ссылки, указанные в описании, включены в него в качестве ссылок.
Примеры
Пример 1
Модификации первичных жирных ацильных цепей
В данном примере описывается получение производных первичных жирных кислот, имеющих ацильные цепи разной длины одни или в сочетании с различными цепями вторичных жирных кислот. Например, соединения 1, а-с, и 2, а-с являются соединениями, в которых первичные жирные кислоты с короткой (С6) и средней (С10) цепями сочетаются с вторичными жирными кислотами с короткими, средними или длинными цепями.
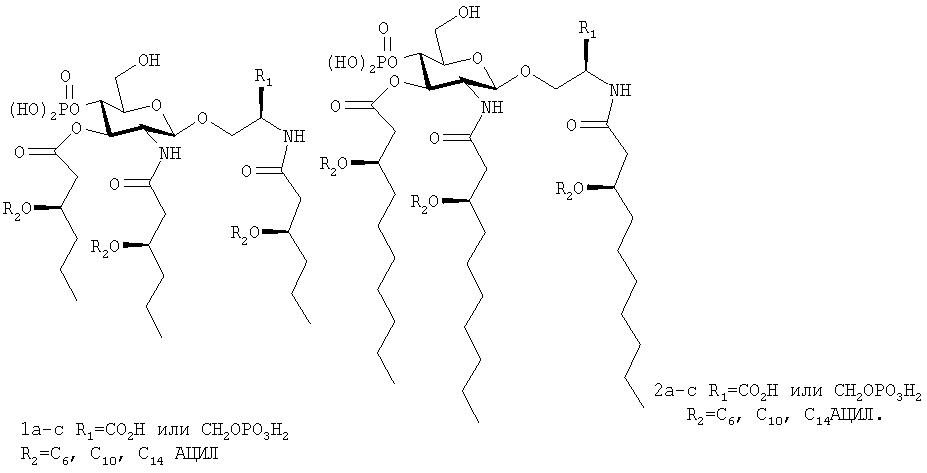
Такие соединения получают или с использованием хорошо известного серинагликона (R1=CO2H) или, с другой стороны, с использованием химически более устойчивого и способного к ионизации звена серинолфосфатагликона (R1=СН2OРО3Н2). Выбор серина/серинолфосфата будет основываться на сравнении биологических активностей известных серилпроизводных 3, a, b с новыми серинолфосфатами 4, а, b.

Производные с модифицированными первичными цепями синтезируют модификацией ранее описанного способа (В1 в Johnson et al., патент США №6355251) с использованием общего специального промежуточного соединения, позволяющего вводить остатки ацилоксикислот, присоединяемые через амидные или сложноэфирные связи, ближе к окончанию синтеза (схема I). Начальной стадией синтеза является гликозилирование акцептора 6 известным тетраацетатом 5 (полученным в 4 стадии из глюкозамина) с образованием β-гликозида 7 и конверсией 7 в общее специальное промежуточное соединение (CAI) 8, которое оптимизируют для R1=СО2Вn. Селективное 4-O-ацилирование и удаление N-защитной группы/ацилирование приводит к гексаацильному производному 9, которое превращают в 3-4 стадии в 1, а-с или 2, а-с, фосфорилированием и удалением блокирующей группы.

Требуемые (R)-3-н-алканоилоксиалкановые кислоты получают согласно Keegman et al., Tetrahedron: Asymmetry, 7(12):3559-3564, 1996, исходя из соответствующих 3-оксометиловых эфиров. Высокой химической и диастереомерной чистоты продуктов 1 и 2 достигают с помощью или хроматографии с нормальной фазой на силикагеле или, с другой стороны, хроматографией на целлюлозе, или жидкость-жидкостной распределительной хроматографией на геле сефадекс LH-20. Чистоту выделенных солей триэтиламмония определяют спектроскопией (ИК,1H и13С ЯМР) и физическими способами (анализ сжиганием, FAB-MS), а также ВЭЖХ.
Пример 2
Глицил- и фосфонооксиэтильные (РЕ) производные
В данном примере описывается синтез глицилсодержащих соединений 11, a, b и фосфонооксиэтильных (РЕ) производных 12, a, b, которые являются близкими региоизомерами 3, a, b и 4, a, b.

Такие соединения легче всего получить более конвергентным синтезом, чем описанный на схеме I, где общий С6- или С10-гликозильный донор 13 вводят в сочетание с соответствующим N-ацилированным (или, с другой стороны, N-Troc-защищенным (не показано)) акцепторным элементом 14 или 15 в присутствии ионов серебра с образованием β-гликозидов 16 (схема II). Акцептор глицин 14 получают согласно Bulusu et al., J. Med. Chem., 35(19):3463-3469, 1992, из этаноламина и бензил(или трет-бутил)бромацетата с последующим N-ацилированием или введением защитной группы. Фосфат 15 получают монофосфорилированием N-ацилированного (или защищенного) диэтаноламина. Ожидается, что удаление N-защитной группы/ацилирование или N,N-диацилирование в случае Тroc-защищенного агликона (Jiang et al., Tetrahedron, 58(43):8833-8842, 2002) β-гликозидов 16 и отщепление фенильной и других защитных групп полученного гексаацилированного производного 17 даст нужные соединения 11, a, b и 12, a, b, которые выделяют и характеризуют в виде их триэтиламмониевых солей после хроматографической очистки на диоксиде кремния или геле LH-20 или целлюлозе DEAE.
Соединения 16 являются новыми и составляют еще один аспект данного изобретения.

Пример 3
Липиды с группами вторичных простых эфиров
В данном примере описывается синтез производных (R)-3-алкилокситетрадекановой кислоты (18, a, b), которые устойчивы к неблагоприятному метаболизму и/или водному гидролизу. Для того чтобы синтезировать соединения 18, а, b - аналоги липидов с группами простых эфиров, сначала надо получить соединения вторичных жирных кислот 3, a, b или соответствующие серинолфосфаты 5, a, b. Как видно на схеме III обратного синтеза, синтеза целевых молекул 18, a, b, можно достичь заменой (R)-3-гексилокситетрадекановой кислоты или (R)-3-децилокситетрадекановой кислоты на соответствующие ацилоксикислоты, начиная с селективного 3-O-ацилирования общего специального промежуточного соединения 8 на схеме I и затем через промежуточное соединение 9 (R2=С6- или С9-алкил, n=9). Требуемые алкилоксикислоты 19 синтезируют из (R)-3-гексилокситетрадекановой кислоты или ее фенилового эфира - промежуточных соединений при синтезе ацилоксикислот, известными способами с общим выходом >50% (Keegman et al., Tetrahedron: Asymmetry, 7(12):3559-3564, 1996; Watanabe et al., Carbohydr. Res., 332(3):257-277, 2001; Jiang, Bioorg. Med. Chem. Lett., 12(16):2193-2196, 2002; Chirst et al., патент США №5530113, 1996).

Пример 4
Липиды с группами первичного и вторичного простых эфиров
В данном примере описываются соединения (20, a, b), содержащие липид с группой первичного простого эфира в положении С-3 сахара, а также три липида с группами вторичного простого эфира. Такие соединения синтезируют путем алкилирования ацетонида 21 - промежуточного соединения при синтезе гликозильного донора 13 (схема II), сульфонатом 22, который, в свою очередь, получают в одну стадию из предшественника спирта 19, и получают простой диэфир 23 (схема IV). 4,6-Функционализация и аномерная активация дают гликозилхлорид 24, который затем обрабатывают согласно схеме II с использованием соответствующих алкилоксикислот на стадиях N-ацилирования. По другой схеме на стадии 3-O-алкилирования можно использовать 2-азидо42- или 2-тpифтopaцeтaмидo45-пpoизвoднoe.
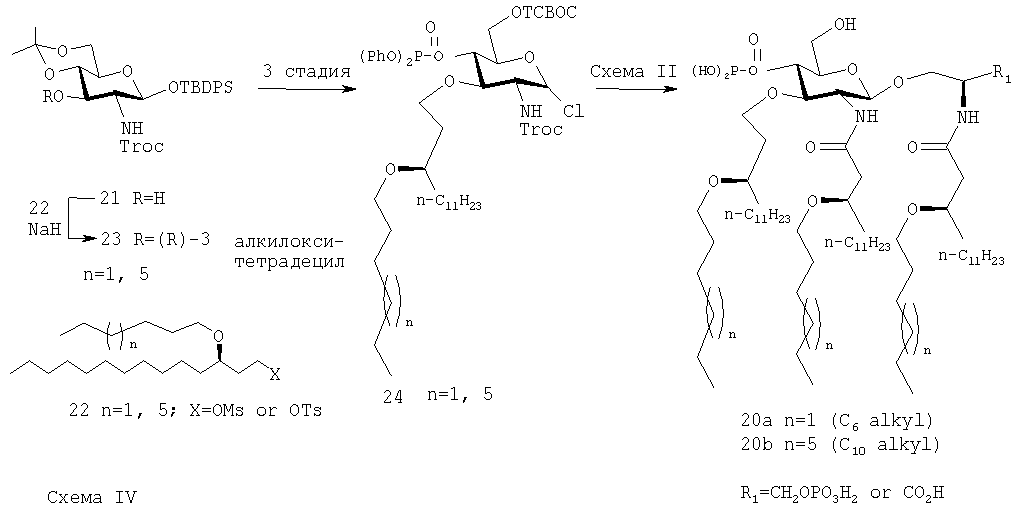
Пример 5
Соединения, модифицированные по С-6
В данном примере описываются соединения с блокированным 6-гидроксилом. В данном примере используют простую метилэфирную или фторгруппу в конъюгации с серилсодержащими соединениями или серинолфосфатами 25, a, b и 26, a, b. Как указано выше, такие соединения также составляют аспект изобретения.

Соединения получают из диола 27, как показано на схеме V.
Промежуточное соединение 27, полученное в две стадии из ацетонида 21, функционализируют в положении 6 известными способами (Chirst et al., патент США №5530113, 1996; Watanabe et al., Carbohydr. Res., 333(3):203-231, 2001) и получают спирт 28. Конверсия 28 в хлориды 29 в две стадии и дополнительная обработка согласно схеме II дают целевые молекулы 25, a, b и 26, a, b. Соединения с группами первичных и/или вторичных простых эфиров, описанные выше в примере 4, можно модифицировать так, как описано в данном примере, для дополнительной защиты молекул от химического и ферментативного разложения.

Следует иметь в виду, что приведенные выше примеры являются только пояснениями настоящего изобретения. Определенные изменения композиций и/или используемых способов можно осуществить с достижением, все еще, целей изобретения. Такие модификации рассматриваются как входящие в объем заявляемого изобретения.
Реферат
Настоящее изобретение относится к соединению приведенной общей структурной формулы, в которой n представляет собой целое число 1 или 5, а R6 представляет собой СООН или СН2ОРО3Н2, или к его фармацевтически приемлемой соли. Изобретение относится также к фармацевтической композиции на основе данных соединений или их фармацевтически приемлемых солей, предназначенной для вызывания или усиления иммунной реакции у субъекта. Кроме того, изобретение относится к способу вызывания, или усиления иммунной реакции у субъекта, а также к способу облегчения или, по существу, предупреждения инфекционной болезни, включающим введение указанному субъекту эффективного количества указанного выше соединения. 4 н. и 3 з.п. ф-лы.
Формула

где n представляет собой целое число 1 или 5 и R6 представляет собой СООН или
СН2ОРО3Н2 или его фармацевтически приемлемая соль.
Комментарии