Пентасахаридный конъюгат, способ его получения и фармацевтическая композиция на его основе - RU2266913C2
Код документа: RU2266913C2
Описание
Изобретение относится к новому антитромботическому соединению, фармацевтической композиции, содержащей соединение в качестве активного ингредиента, а также к применению названного соединения для производства лекарственных средств.
Сериновые протеазы являются ферментами, которые играют важную роль в каскаде свертывания крови. Важной сериновой протеазой является фактор Ха, который катализирует превращение протромбина в тромбин. Тромбин является конечным сериновым протеолитическим ферментом в каскаде свертывания. Основная функция тромбина заключается в расщеплении фибриногена с образованием фибриновых мономеров, которые поперечно сшиваются, формируя нерастворимый гель. Кроме того, тромбин регулирует свою собственную продукцию посредством активации факторов V и VIII на более ранней стадии каскада. Тромбин также осуществляет важные функции на клеточном уровне, где он воздействует на специфические рецепторы, чтобы вызвать агрегацию тромбоцитов, активацию эндотелиальных клеток и пролиферацию фибробластов. Таким образом, тромбин осуществляет центральную регуляторную роль в гемостазе и образовании тромба.
Недавно при разработке синтетических ингибиторов сериновых протеаз синтетический NAPAP-пентасахаридный конъюгат был описан как антитромботический агент, оказывающий двойное действие - как непосредственно антитромбиновую активность, так и ATIII-опосредованную анти-Ха активность (ATIII: антитромбин III) (Bioorg. Med. Chem. Lett. 1999, 9 (14), 2013-8). Хотя описанное антитромботическое средство может представлять интерес вследствие высокого сульфатного содержания в пентасахаридном остатке, с этим соединением связаны HIT-перекрестная реакционная способность и нейтрализация PF4 (Thromb. Haem. Suppl. 1997, p.363, PD1485).
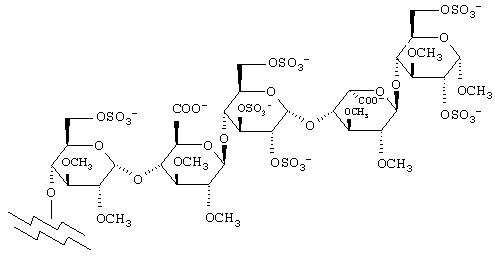
В настоящее время установлено, что соединения формулы (I) являются антитромботическими агентами, характеризующимися превосходным и благоприятным двойным профилем. Соединения формулы (I) имеют фармакологически удобный период полувыведения, допускающий прием лекарства один раз в день, и они с трудом нейтрализуются PF4. Кроме того, снижается риск кровотечения. В целом, соединения формулы (I) характеризуются привлекательным сочетанием фармакологических свойств.
Соединение формулы (I):

в которой R независимо означает SO3- или СН3;
вставка представляет собой гибкую вставку длиной 13-25 атомов, предпочтительно 16-22 и наиболее предпочтительно 19 атомов;
заряд пентасахаридного остатка уравновешивается положительно заряженными противоионами; а общее количество сульфатных групп в пентасахаридном остатке составляет 4, 5 или 6;
или его фармацевтически подходящая соль, пролекарство или сольват.
Соединения данного изобретения используются для лечения и профилактики заболеваний, опосредованных тромбином и связанных с тромбином. Заболевания включают в себя ряд тромботических и протромботических состояний, при которых активируется каскад свертывания, которые включают тромбоз глубоких вен, эмболию легких, тромбофлебиты, закупорку артерии в результате тромбоза или эмболии, повторную закупорку артерии во время или после ангиопластики или тромболизиса, рестеноз после повреждения артерии или инвазивных кардиологических процедур, послеоперационный тромбоз или эмболию вен, острый или хронический атеросклероз, инсульт, инфаркт миокарда, злокачественные опухоли и метастазы и нейродегенеративные заболевания, не ограничиваясь вышеперечисленным. Соединения изобретения также можно использовать как антикоагулянты в экстракорпоральном кровообращении, что необходимо при диализе и оперативном вмешательстве. Соединения изобретения также можно использовать как антикоагулянты in vitro.
В частности, соединения формулы (I) используются как антитромботические средства по артериальным показаниям.
Согласно данному изобретению, предпочтительными соединениями являются соединения, в которых пентасахаридный остаток имеет структуру:

Химическая природа вставки имеет второстепенное значение для антитромботической активности соединений изобретения. Однако вставка соединений изобретения является гибкой, что означает, что вставка не содержит жестких элементов, таких как ненасыщенные связи или циклические структуры. Подходящие вставки легко могут быть сконструированы специалистами в данной области. Предпочтительные вставки содержат по крайней мере один -(CH2CH2O)- элемент. Более предпочтительные вставки содержат три -(CH2CH2 O)- элемента. Наиболее предпочтительной вставкой является *-(CH2CH2O)3-(CH2)2-NH-C(О)-(CH2)3-NH-C(О)-СН2-, конец, обозначенный *, присоединяется к пентасахаридному остатку.
Предпочтительные соединения формулы (I) представляют собой соединения формулы (Ia), в которой р означает 1-5, n равно 1-5, и m равно 1 или 2. Наиболее предпочтительным соединением является соединение формулы (Ia), в которой р равно 3, n равно 3, и m равно 1.

Термин «положительно заряженный противоион» означает H+, Na+, K+, Ca2+ и тому подобное. Предпочтительно соединения формулы (I) находятся в форме их натриевой соли.
Термин «пролекарство» подразумевает соединение изобретения, в котором аминогруппа амино-фрагмента защищена, например, гидрокси или (1-6С)алкоксикарбонильной группой. Согласно данному изобретению сольваты включают в себя гидраты.
Соединения изобретения, которые находятся в форме свободного основания, могут быть выделены из реакционной смеси в форме фармацевтически приемлемой соли. Фармацевтически приемлемые соли также можно получать в результате обработки свободного основания формулы (I) органической или неорганической кислотой, такой как хлористоводородная, бромистоводородная, йодистоводородная кислота, серная кислота, фосфорная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, метансульфоновая кислота, фумаровая кислота, янтарная кислота, винная кислота, лимонная кислота, бензойная кислота, аскорбиновая кислота и тому подобные.
Соединения данного изобретения содержат хиральные атомы углерода и, поэтому могут быть получены в виде чистого энантиомера или в виде смеси энантиомеров, или в виде смеси, содержащей диастереомеры. Способы получения чистых энантиомеров хорошо известны в данной области, например кристаллизация солей, которые получены из оптически активных кислот и рацемической смеси, или хроматография с использованием хиральных колонок. Для диастереомеров можно применять колонки с прямой или обращенной фазой.
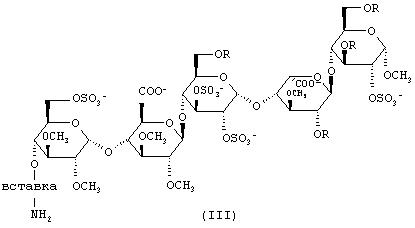
Соединения данного изобретения можно получать посредством сначала активации карбоксилатной группы аналога NAPAP формулы II, а впоследствии присоединения остатка пентасахарид-вставка, содержащего группу амина (формула III), возможно, с последующим снятием защиты с амидинового фрагмента.

Карбоксилатная группа в соединениях формулы II может быть активирована как смешанный ангидрид или, более предпочтительно, как активированный сложный эфир N-гидроксисукцинимида, пентафторфенол или 1-гидроксибензотриазол. На стадии связывания группа бензамидина в формуле II может быть незащищенной (R'=R''=H) или, возможно, может быть защищена с использованием группы карбамата, предпочтительно аллилоксикарбонила (R' и/или R'' соответствуют Н2С=СН-СН-С(О)О) или бензилоксикарбонила (R' и/или R'' соответствуют PhCH2-С(О)О). Аллилоксикарбонильная и бензилоксикарбонильная защитные группы могут быть удалены при относительно мягких условиях. Аллилоксикарбонильную группу можно удалить, используя Pd в присутствии слабого нуклеофила, такого как морфолин или малоновый эфир. Бензилоксикарбонильная группа может быть удалена при таких условиях, как водород / Pd(C). Альтернативно, можно применять синтетические предшественники бензамидина, такие как N-алкоксибензамидин или N-бензилоксибензамидин (R'=Н, R''=алкокси или бензилокси). Упомянутые синтетические предшественники могут быть превращены в бензамидин, используя восстанавливающие условия, такие как гидрирование (например, Fujii, Т et al., Chem. Pharm. Bull, 39, 301, 1991 и Fujii, T et al., Chem. Pharm. Bull, 42, 1231, 1994).
Предпочтительным предшественником бензамидина является 1,2,4-оксадиазолин-5-он (-R'-R''-=-С(О)О-). Указанный предшественник может быть превращен в бензамидин гидрированием (Bolton, R.E. et al. Tetrahedron Letters, vol 36, № 25, 1995, рр. 4471-4474).
Соединения формулы II могут быть получены различными путями при использовании способов, известных в данной области. Способ получения соединений формулы II, в которой R'=R''=Н; n равно 3 и m равно 1, описан в ЕР 0513543. Соединения формулы II, в которых амидин защищен, например, аллилоксикарбонильной или бензилоксикарбонильной группой, могут быть пролучены из соединений формулы IV, в которой амидин защищен аллилоксикарбонильной или бензилоксикарбонильной группой при использовании общеизвестных в данной области способов связывания пептидных фрагментов. Карбаматы формулы IV могут быть получены, например, из соответствующего амидина (формула IV, R'=R''=Н) как описано в литературе, например Weller, Т et al., J. Med. Chem. 39, 3119, 1996).

Соединения N-алкоксибензамидина и N-бензилоксибензамидина формулы II можно получить из соединения V (описано в ЕР 0513543) в результате обработки этого циано-соединения O-алкилгидроксиамином или O-бензилоксиамином с последующим удалением трет-бутилового эфира при кислых условиях.
Альтернативно, соединения N-алкоксибензамидина и N-бензилоксибензамидина формулы II могут быть получены посредством сначала удаления трет-бутилового эфира соединения V, используя кислые условия, чтобы получить соединение VI, и впоследствии реакции полученного циано-соединения с O-алкилгидроксиамином или O-бензилоксиамином.
Соединения формулы II, в которых -R'-R''-=-С(O)O-(группа 1,2,4-оксадиазолин-5-она) могут быть получены из соединений формулы IV, в которых -R'-R''-=-С(O)O-, используя способы связывания пептидных фрагментов, известные в данной области.
Синтез остатков амино-олигосахарид-вставка формулы III может быть осуществлен с использованием способов, описанных в ЕР 0649854. Сахаридные остатки соединений данного изобретения можно получать в соответствии со способами, известными в данной области, например из WO 99/25720.
Пептидное связывание, технологическая стадия вышеописанного способа получения соединений изобретения, может быть осуществлено посредством известных в данной области способов связывания или конденсации пептидных фрагментов, таких как азидный способ, способ смешанных ангидридов, способ активированных сложных эфиров или, предпочтительно, посредством карбодиимидного способа, особенно с присоединением каталитических или подавляющих рацемизацию соединений, подобных N-гидроксисукцинимиду и N-гидроксибензотриазолу. Обзор представлен в The Peptides, Analysis, Synthesis, Biology, vol 3, E. Gross and J. Meienhofer, eds. (Academic Press, New York, 1981) и Bodanszky, M.; Principles of peptide synthesis, Springer-Verlag, 1984.
Функциональные аминогруппы, присутствующие в соединениях, можно защитить N-защитной группой, которая подразумевает общеизвестную в пептидной химии группу для защиты α-амино-группы, подобную трет-бутилоксикарбонильной (Вое) группе, бензилоксикарбонильной (Z) группе, 9-фторенилметилокси-карбонильной (Fmoc) группе или группе фталоила (Phth). Удаление защитных групп может быть осуществлено различными способами в зависимости от природы этих защитных групп. Обычно снятие защиты происходит в кислых условиях и в присутствии акцепторов уходящих групп. Обзор защитных групп для аминогрупп соединений и способов их удаления представлен в вышеупомянутом The Peptides, Analysis, Synthesis, Biology, vol 3, и, кроме того, описано Greene, T.W. and Wuts, P.G.M. в Protective groups in organic synthesis, John Wiley & Sons Inc., 1991.
Соединения изобретения можно вводить энтерально или парентерально. Точная доза и схема приема названных соединений и их композиций обязательно будет зависеть от потребности индивидуального субъекта, которому вводится лекарственное средство, тяжести болезни или необходимости и заключения практикующего врача. Обычно, парентеральное применение требует более низких доз, чем другие способы введения, которые в большей степени зависят от всасывания. Однако для человека ежедневные дозы предпочтительно составляют 0,001-100 мг на кг массы тела, более предпочтительно 0,01-10 мг на кг массы тела.
Лекарства, приготовленные с соединениями данного изобретения, также можно использовать как адъювант в острой антикоагулянтной терапии. В таком случае, лекарственное средство вводят с другими соединениями, эффективными при лечении таких патологических состояний.
Смешанные с фармацевтически приемлемыми вспомогательными веществами, например как описано в стандартной рекомендации Gennaro et al., Remington's Pharmaceutical Sciences (18th ed., Mack Publishing Company, 1990, смотри особенно Part 8: Pharmaceutical Preparations and Their Manufacture) соединения могут быть спрессованы в твердые дозированные формы, такие как пилюли, таблетки, или приготовлены в виде капсул или суппозиторий. Вместе с фармацевтически приемлемыми жидкостями соединения также можно применять в виде раствора, суспензии, эмульсии, например для применения в виде инъекционного препарата или в виде спрея, например для использования в качестве назального спрея.
Для создания дозированных стандартных форм, например таблеток, предполагается использование общепринятых добавок, таких как наполнители, пигменты, полимерные связующие вещества и тому подобное. Вообще может быть использована любая фармацевтически приемлемая добавка, которая не препятствует действию активных соединений.
Приемлемые носители, с которыми могут быть введены композиции, включают в себя лактозу, крахмал, производные целлюлозы и тому подобное, или их смеси, используемые в соответствующих количествах.
Далее изобретение проиллюстрировано следующими примерами.
ПРИМЕР 1
Используемые сокращения:
Ас = ацетил
Bn = бензил
DBU = 1,8-диазабицикло[5.4.0]ундец-7-ен
DCC = дициклогексилкарбодиимид
DMF = N,N-диметилформамид
Su = сукцинимидил
Me = метил
TBTU = тетрафторборат 2-(1Н-бензотиазол-1-ил)-1,1,3, 3-тетраметилурония
TEA = триэтиламин
TFA = трифторуксусная кислота
Z = бензилоксикарбонил
Ряд соединений относится к соединениям, представленным в схемах 1-7.
Соединение 3
К перемешиваемому раствору соединения 1 (53,6 г, 143,6 ммоль) (R. Roy; W.K.C. Park; Q. Wu; S-N. Wang, Tetrahedron Lett., 1995, 36(25), 4377-80) и соединения 2 (27,9 г, 89,3 ммоль) (S.J.Danishefsky; M.P.DeNinno; G.B.Philips; R.E.Zeile, Tetrahedron, EN, 1986, 42, 11, 2809-2819) в 930 мл DMF добавляли гидрид натрия (7,7 г 60%-дисперсии, 192,2 ммоль) при 50°С. Через 1 час реакционную смесь нагревали до 120°С. После смешивания в течение 5 минут реакционную смесь охлаждали до 40°С и разбавляли водой и экстрагировали три раза дихлорметаном. Объединенные органические слои промывали водой и концентрировали в вакууме, получая неочищенный продукт 3 (54 г). TLC: Rf (фактор удержания)=0,23, простой эфир 100%.
Соединение 4
К перемешиваемому раствору соединения 3 (89,3 ммоль) в 800 мл сухого толуола и 800 мл уксусного ангидрида по каплям добавляли 361,5 мл охлажденного раствора серной кислоты в уксусном ангидриде (16,5 мл концентрированной серной кислоты и 345,0 мл уксусного ангидрида) при -30°С. Через 2 час реакционную смесь гасили 240 мл TEA и перемешивали при комнатной температуре. К раствору добавляли водный бикарбонат натрия (5%) и водный слой экстрагировали три раза этилацетатом. Объединенные органические слои дважды промывали водой и концентрировали в вакууме. Описанную процедуру повторяли, получая неочищенное соединение 4 (53 г). TCL: Rf=0,29, простой эфир 100%.
Соединение 5
К перемешиваемому раствору соединения 4 (89,3 ммоль) и этантиола (11,1 мл, 150,3 ммоль) в 370 мл сухого толуола по каплям добавляли раствор BF3-эфирата в толуоле (23,9 мл BF3-эфирата и 190 мл толуола) при 0°С. После перемешивания в течение 16 часов при комнатной температуре реакционную смесь гасили TEA и водным бикарбонатом натрия и три раза экстрагировали этилацетатом. Объединенные органические слои промывали водой и концентрировали в вакууме. Грубый продукт очищали методом колоночной хроматографии (толуол/этилацетат=1/1 до 0/1 об./об.), получая соединение 5 (21,4 г). TLC: Rf=0,31, толуол/этилацетат=4/6, об./об.
Соединение 7
Раствор донора 5 (15,0 г, 30,3 ммоль) и акцептора 6 (23,0 г, 30,3 ммоль) (WO 99/25720) в смеси сухой простой эфир/дихлорметан (232 мл, 3/1, об./об.) перемешивали в течение 30 минут в потоке азота в присутствии активированных молекулярных сит 4А (7,6 г). Затем к реакционной смеси по каплям добавляли раствор 1,3-дибром-5,5-диметилгидантоина (5,5 г, 19,1 ммоль) и трифторметансульфокислоты (triflic acid) (0,49 мл, 5,6 ммоль) в смеси диоксан/дихлорметан (69,8 мл, 1/1, об./об.) в течение 75 минут при -20°С. Через 30 минут, TEA (5 мл) добавляли к реакционной смеси, которую перемешивали в течение 10 минут, а потом фильтровали. Фильтрат промывали водным тиосульфатом натрия (10%) и водным бикарбонатом (10%) и концентрировали в вакууме. Продукт очищали колоночной хроматографией (0 до 5% ацетона в дихлорметане), получая соединение 7 (19,6 г). TLC: Rf=0,1, простой эфир/гептан=8 /2, об./об.
Соединение 8
К перемешиваемому раствору соединения 7 (19,5 г, 16,4 ммоль) в смеси сухой толуол/уксусный ангидрид (442 мл, 1/1, об./об.) по каплям добавляли 131,5 мл охлажденного раствора серной кислоты в уксусном ангидриде (11,5 мл концентрированной серной кислоты и 120 мл уксусного ангидрида) при -26°С. Через 75 минут добавляли TEA (73,5 мл) при -20°С. Уксусный ангидрид разлагали, постепенно добавляя 330 мл воды, поддерживая температуру между 25°С и 30°С. После перемешивания в течение 16 часов смесь вливали в 800 мл воды и дважды экстрагировали толуолом. Объединенные органические слои промывали водой и концентрировали в вакууме. Грубый продукт очищали колоночной хроматографией (толуол/этилацетат/этанол=96/2/2, об./об./об.), получая соединение 8 в виде белой пены (13,2 г).
TLC: Rf=0,29, толуол/этанол=9/1, об./об.
Соединение 9
К раствору соединения 8 (13,2 г, 11,7 ммоль) в сухом толуоле (66 мл) добавляли морфолин (4,1 мл, 46,9 ммоль) при 32°С. После перемешивания в течение 42 час при 32°С реакционную смесь охлаждали до комнатной температуры и добавляли водную хлористоводородную кислоту (17,6 мл, 4 N). Смесь разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические слои дважды промывали водой, высушивали над сульфатом натрия и концентрировали в вакууме, получая неочищенное соединение 9 (12,6 г).
TLC: Rf=0,32, толуол/ацетон=7/3, об./об.
Соединение 12
К раствору соединения 9 (12,6 г, 11,6 ммоль) в дихлорметане (114 мл) добавляли трихлорацетонитрил (3,5 мл, 34,9 ммоль) и DBU (52, 2 мкп, 0,35 ммоль). После перемешивания в течение 2 час при комнатной температуре к реакционной смеси добавляли активированные молекулярные сита 4А (24 г) и акцептор 11 (8,9 г, 13,0 ммоль) (WO 99/25720) в дихлорметане (45 мл). После перемешивания в течение 30 минут при комнатной температуре, смесь охлаждали до -20°С и по каплям добавляли раствор триметилсилилтрифторметансульфоната (405 мкл, 2,1 ммоль) в дихлорметане (100 мл). После перемешивания в течение 30 минут -20°С добавляли бикарбонат натрия и реакционную смесь фильтровали. Фильтрат вливали в водный бикарбонат натрия и три раза экстрагировали дихлорметаном. Объединенные органические слои дважды промывали водой и концентрировали в вакууме. Продукт очищали посредством колоночной хроматографии (1: SiO2: 0-10% ацетон в простом эфире; 2: SiO2: толуол/ацетон=85/15 до 80/20, об./об.; 3: RP-18: вода/ацетонитрил=2/8 до 0/10, об./об.), получая чистое соединение 12 (8,9 г). TLC: Rf=0,37, толуол/ацетон=7/3, об./об.
Соединение 14
Суспензию соединения 12 (8,9 г, 5,1 ммоль) и 10% Pd/C (8,9 г) в 312 мл DMF и 45 мл воды перемешивали в непрерывном потоке водорода. Через 4,5 час катализатор Pd/C удаляли фильтрованием. Фильтрат концентрировали до объема 400 мл и обрабатывали 10% Pd/C (1,5 г) в непрерывном потоке водорода в течение 5,5 час. Катализатор удаляли фильтрованием. К фильтрату (900 мл) добавляли водную гидроокись натрия (32 мл, 4 N). После перемешивания в течение 4 час при комнатной температуре смесь подкисляли до рН 6,6 1 N хлористоводородной кислотой, а затем концентрировали в вакууме. Неочищенный продукт обессоливали на колонке с сефадексом G-25, который уравновешивали водой. Соответствующие фракции объединяли и лиофилизировали, получая соединение 14 (4,0 г).
Соединение 15
Пентасахарид 14 (700 мг, 0,61 ммоль) растворяли в воде (13,2 мл) и DMF (3,3 мл) и обрабатывали N-(бензилоксикарбонилокси)сукцинимидом (224 мг, 0,90 ммоль) и N-этилморфолином (233 мкл, 1,83 ммоль). После перемешивания в течение 15 минут реакционную смесь непосредственно наносили на колонку RP-18 и элюировали смесью вода/ацетонитрил 10/0 до 7/3. Соответствующие фракции объединяли и концентрировали до небольшого объема и наносили на ионообменную колонку с Dowex 50 WX4-H+ в воде. Элюат концентрировали в вакууме, чтобы получить чистое соединение 15 (482 мг).
Соединение 16
К раствору соединения 15 (471 мг, 0,37 ммоль) в DMF (4,7 мл) добавляли комплекс трехокись серы-пиридин (1,1 г, 6,6 ммоль) и смесь перемешивали в течение 16 часов при 30°С. Смесь охлаждали до комнатной температуры и по каплям добавляли к охлажденному 10% раствору бикарбоната натрия (16,7 мл, 19,9 ммоль) и перемешивали в течение 1 часа при комнатной температуре. Смесь концентрировали до небольшого объема и наносили на колонку с сефадексом G-25 и элюировали водой. Соответствующие фракции объединяли и концентрировали до небольшого объема, который впоследствии пропускали через колонку с Dowex Na+ HCRW2, элюируя водой. Элюат концентрировали и повторно растворяли в 8,3 мл 0,2 N хлористоводородной кислоты и оставляли стоять в течение 16 часов при 4°С. Реакционную смесь нейтролизовали 8 мл 0,2 N гидроокиси натрия и обессоливали на колонке с сефадексом G-25, элюируя водой. Соответствующие фракции объединяли и концентрировали в вакууме, получая чистое соединение 16 (840 мг).
Соединение 17
Суспензию соединения 16 (0,37 ммоль) и 10% Pd/C (820 мг) в трет-бутаноле (85 мл) и воде (79 мл) и в присутствии нескольких капель уксусной кислоты перемешивали в непрерывном потоке водорода. Через 3 часа катализатор Pd/C удаляли фильтрованием, а фильтрат концентрировали и лиофилизировали, получая чистое соединение 17 (675 мг).
Гидрохлорид бензилового эфира 4-[{4-[[(1R)-[[4-(аминоиминометил)фенил]метил]-2-оксо-2-(1-пиперидинил)этил]амино]-3-[[(4-метокси-2,3, 6-триметилфенил)сульфонил}амино]-1,4-(S)-диоксобутил]амино]бутановой кислоты (18)
К раствору гидрохлорида 4-[[(1R)-1[{4-(аминоиминометил)фенил]метил]-2-оксо-2-(1-пиперидинил)этил]амино]-3-[[(4-метокси-2,3,6-триметилфенил)сульфонил]амино]-4-оксо-(3S)бутановой кислоты, (2,38 г, 3,96 ммоль) (Tetrahedron 51, 12047-12068, 1995) и бензил-(4-аминомасляная кислота)бензолсульфоната (1,52 г, 3,96 ммоль) (J. Am. Chem. Soc. 105, 5278-5284, 1983) в DMF (40 мл) в атмосфере азота добавляли N, N-диизопропилэтиламин (0,689 мл, 3,96 ммоль) и тетрафторборат тетраметилбензотриазолилурония (1,91 г, 5,94 ммоль). рН реакционной смеси поддерживали при 6, используя N,N-диизопропилэтиламин. Реакционную смесь перемешивали в течение 4 дней при комнатной температуре, концентрировали, растворяли в этилацетате, промывали 5% карбонатом натрия и 0,1 N хлористоводородной кислотой, высушивали над сульфатом магния и концентрировали. Остаток растворяли в сухом этаноле (5 мл), осаждали сухим диизопропиловым эфиром, фильтровали, чтобы получить 2,47 г указанного в заголовке соединения 18.
Rf=0,8, этилацетат/пиридин/уксусная кислота/вода=88/31/18/7, об./об./об./об.; Mass (ESI+): 777,4 [М+Н]+
Гидрохлорид 4-[{4-[[(1R)-1-[[4-(аминоиминометил)фенил]метил]-2-оксо-2-(1-пиперидинил)этил]амино]-3-[[(4-метокси-2,3,6-триметилфенил)сульфонил}амино]-1,4-(S)-диоксобутил]амино]бутановой кислоты (19)
Суспензию соединения 18 (2,42 г, 3,11 ммоль) и 10% Pd/C (400 мг) в смеси метанол/вода (40 мл, 3/1, об./об.) перемешивали в непрерывном потоке водорода. Через 8 часов реакционную смесь фильтровали, фильтрат концентрировали и три раза выпаривали вместе со смесью метанол/толуол (1/10, об./об.). Остаток растворяли в сухом этаноле (5 мл), осаждали сухим диэтиловым эфиром, фильтровали и высушивали. Остаток растворяли в воде и наносили непосредственно на препаративную колонку DeltaPak Rp-C18 для ВЭЖХ, используя систему градиентной элюции 20% А/60% В/20% С до 20% А/14% В/66% С в течение 60 мин при скорости течения 40 мл/мин (А: 0,5 М фосфатный буфер, рН 2,1; В: вода; С: ацетонитрил/вода=6/4). Выход 598 мг.
Rf=26,4 мин (3-10 мин: 20-43% С + 20% А; 10-50 мин: 43-66% С + 20% А), (А: фосфатный буфер, рН 2,1; В: вода; С:ацетонитрил/вода=6/4, об./об.), аналитическая колонка supelcosil LC-18-DB для ВЭЖХ; Mass (ESI+): 687,2 [M+H]+, (ESI-): 685,2 [M-H]
Соединение 21 из соединения 17 и соединения 19
К раствору соединения 19 (40 мг, 58,3 мкмоль) в DMF (800 мкл) добавляли N-гидроксисукцинимид (9,0 мг, 78,1 мкмоль), DCC (18,5 мг, 89,7 мкмоль) и 1-гидроксибензотиазол (8,8 мг, 65,1 мкмоль). Реакционную смесь перемешивали в течение 40 часов при комнатной температуре. Реакционную смесь фильтровали через дикалит и дикалит четыре раза промывали DMF (284 мкл). К фильтрату добавляли 0,1 М Na2HPO4-буфер (1936 мкл, рН 7,5) и пентасахарид 17 (94,7 мг, 52,6 мкмоль). После перемешивания в течение 30 минут, смесь фильтровали через дикалит, концентрировали и наносили на колонку с сефадексом G-25 и элюировали смесью ацетонитрил/вода (1/1, об./об.). Соответствующие фракции объединяли, концентрировали и дважды обессоливали посредством колоночной хроматографии через сефадекс G-50 (вода). Соответствующие фракции объединяли и лиофилизировали, получая конъюгат 21 в виде белого твердого вещества (95,8 мг). Mass (ESI+)=2469, ВЭЖХ: Rt=8,3 мин (20-80% В в течение 15 минут; А=вода/ацетонитрил 8/2; В=2 М NaCl/ацетонитрил 8/2, об./об.), аналитическая колонка MonoQ HR 5 ВЭЖХ.
Соединение 22
Раствор (R)-N-Вос(4-цианофенил)аланина (25,0 г, 86,1 ммоль), пиперидина (21,3 мл, 215,3 ммоль) и TBTU (41,5 г, 129,2 ммоль) в сухом СН2Cl2 (500 мл) перемешивали при комнатной температуре в потоке азота в течение 2 часов. Реакционную смесь промывали последовательно 0,2 N хлористоводородной кислотой, водой, водным бикарбонатом натрия (насыщенный) и водой. Органический слой высушивали над MgSO4, фильтровали и концентрировали в вакууме. Продукт растворяли в горячем этилацетате (35 мл), осаждали гептаном (190 мл) и фильтровали, чтобы получить соединение 22 (27,75 г).
TLC: Rf=0,58, гептан/этилацетат=3/7, об./об.
Соединение 23
Раствор соединения 22 (25,6 г, 71,7 ммоль), гидроксиламин·HCl (7,1 г, 101,8 ммоль) и триэтиламина (16,8 мл, 120,5 ммоль) в абсолютном этаноле (307 мл) перемешивали при 80°С в течение 4 часов. При охлаждении смеси до комнатной температуры формировались кристаллы. Кристаллы отфильтровывали, промывали этанолом и простым эфиром и высушивали в эксикаторе, чтобы получить соединение 23 (24,5 г).
TLC: Rf=0,15, гептан/этилацетат=3/7, об./об.
Соединение 24
Раствор соединения 23 (24,5 г, 62,7 ммоль) и этилхлорформиата (7,2 мл, 75 ммоль) в сухом пиридине (245 мл) перемешивали при 115°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и вливали в воду (1250 мл) и три раза экстрагировали этилацетатом (500 мл). Объединенный органический экстракт высушивали над MgSO4, фильтровали и концентрировали в вакууме, чтобы получить соединение 24 (24,3 г).
TLC: Rf=0,42, CH2 Cl2/MeOH 95/5, об./об.
Соединение 25
Раствор соединения 24 (24,3 г, 58,4 ммоль) в сухом CH2Cl2 (122 мл) и TFA (122 мл) перемешивали при комнатной температуре в течение 2 часов и концентрировали в вакууме в присутствии толуола, чтобы получить соединение 25 (37,6 г).
TLC: Rf=0,35, CH2Cl2/MeOH 8/2, об./об.
Соединение 26
Суспензию H-Asp-(Отрет-Bu)-ОН (39 г, 206,35 ммоль), 4-метокси-2,3,6-триметилбензолсульфонилхлорида (62 г, 249,3 ммоль) и диизопропиламина (89 мл, 635 ммоль) в DMF (950 мл) и воде (450 мл) перемешивали при 0°С в течение 3 часов. Реакционную смесь вливали в смесь лед/вода (5 л) и дважды промывали диэтиловым эфиром, подкисляли водной хлористоводородной кислотой (4 N, 72 мл) и 3 раза экстрагировали этилацетатом. Объединенные этилацетатные слои высушивали над MgSO4, фильтровали и концентрировали в вакууме, чтобы получить соединение 26 (97,7 г).
TLC: Rf=0,67, CH2Cl2/MeOH 8/2, об./об.
Соединение 27
Раствор соединения 25 (33,5 г), соединения 26 (24,7 г), TBTU (36,8 г, 114,6 ммоль) и диизопропиламина (27,2 мл, 194,1 ммоль) в сухом DMF (670 мл) перемешивали в течение 2 часов и концентрировали в вакууме. Остаток растворяли в этилацетате (750 мл), промывали водным бикарбонатом натрия (5%, 1250 мл) и водной хлористоводородной кислотой (0,1 N, 1250 мл), высушивали над MgSO4, фильтровали и концентрировали в вакууме, чтобы получить соединение 27 (33,8 г).
TLC: Rf=0,88, CH2Cl2/MeOH 8/2, об./об.
Соединение 28
Раствор соединения 27 (33,8 г, 48,3 ммоль) в сухом СН2Cl2 (170 мл) и TFA (170 мл) перемешивали при комнатной температуре в течение 2 часов и концентрировали в вакууме в присутствии толуола, чтобы получить соединение 28 (32,3 г).
TLC: Rf=0,73, СН2Cl2/МеОН 8/2, об./об.
Соединение 29
Раствор соединения 28 (32,3 г), H-GABA-Отрет-Bu·HCl (9,5 г, 48,4 ммоль), TBTU (29,0 г, 90,5 ммоль) и диизопропиламина (25,2 мл, 179,8 ммоль) в сухом DMF (622 мл) перемешивали при комнатной температуре в течение 2 часов и концентрировали в вакууме. Остаток растворяли в этилацетате (840 мл), промывали водным бикарбонатом натрия (5%, 1400 мл) и водной хлористоводородной кислотой (0,1 N, 1400 мл), высушивали над MgSO4, фильтровали и концентрировали в вакууме. Остаток растворяли в этаноле (75 мл) и медленно добавляли к перемешиваемому диизопропиловому эфиру (2990 мл), получая соединение 29 в виде не совсем белых кристаллов (32,0 г).
TLC: Rf=0,56, CH2Cl2/MeOH 9/1, об./об.
Соединение 30
Раствор соединения 29 (3,0 г, 3,82 ммоль) в сухом CH2Cl2(15 мл) и TFA (15 мл) перемешивали при комнатной температуре в течение 2 часов и концентрировали в вакууме в присутствии толуола. Остаток очищали на силикагеле, используя смесь СН2 Cl2/МеОН (0%-6% МеОН), получая чистое соединение 30 (1,98 г).
TLC: Rf=0,56, СН2Cl2/МеОН 8/2, об./об.
Соединение 31
Раствор соединения 30 (900 мг, 1,23 ммоль), TBTU (396 мг, 1,2 ммоль) и диизопропиламина (215 мкл, 1,53 ммоль) в DMF (45 мл) перемешивали в течение 2 часов при комнатной температуре. Добавляли соединение 17 (2,0 г, 1,11 ммоль) и после перемешивания в течение 4 часов смесь концентрировали в вакууме, получая соединение 31 (4,17 г).
Соединение 21 из соединения 31
Суспензию соединения 31 (4,17 г) и 10% Pd/C (2,8 г) в трет-бутиловом спирте (28 мл) и воде (56 мл) перемешивали в течение ночи в непрерывном потоке водорода. Катализатор Pd/C удаляли фильтрованием, а фильтрат концентрировали в вакууме. Остаток растворяли в воде и очищали на колонке с Q-сефарозой. Соответствующие фракции объединяли, концентрировали и обессоливали с помощью колоночной хроматографии через сефадекс G-25 (вода). Соответствующие фракции объединяли и лиофилизировали, чтобы получить конъюгат 21 в виде белого твердого вещества (1,74 г).







Биологические активности соединений данного изобретения определяли с помощью следующих тест-способов.
I Исследование антитромбиновой активности
Тромбин (Фактор IIa) является фактором каскада свертывания крови.
Антитромбиновую активность соединений данного изобретения оценивали посредством спектрофотометрического измерения скорости гидролиза хромогенного субстрата s-2238, расщепляемого тромбином. Описанный анализ антитромбиновой активности в буферной системе использовали, чтобы определить величину IC50 испытываемого соединения.
Исследуемая среда: Буфер - трометамин-NaCl-полиэтиленгликоль 6000 (TNP)
Стандартное соединение: 12581 (Kabi)
Наполнитель: TNP-буфер.
Растворимость может быть установлена при использовании диметилсульфоксида, метанола, этанола, ацетонитрила или трет-бутилового спирта, которые не проявляют побочных эффектов в концентрациях вплоть до 2,5% в конечной реакционной среде.
Методика:
Реагенты* 1. Трометамин-NaCl (TN) буфер; состав буфера:
Трометамин (трис) 6,057 г (50 ммоль), NaCl 5,844 г (100 ммоль), вода до 1 л. рН раствора доводили HCl до 7,4 при 37°С (10 ммоль·л-1). 2. TNP буфер: Полиэтиленгликоль 6000 растворяли в TN-буфере для получения концентрации 3 г·л-1 3. Раствор S-2238: S-2238 из одной ампулы (25 мг Chromogenix; Sweden) растворяли в 20 мл TN-буфера для получения концентрации 1,25 мг·мл-1 (2 ммоль·л-1). 4. Раствор тромбина: Тромбин человека (1000 NIH единиц/ампулу. Enzyme Res. Lab. Inc., USA) растворяли в TNP-буфере, чтобы получить исходный раствор 50 NIH единиц·мл-1. Непосредственно перед использованием приготовленный раствор разбавляли TNP-буфером до получения концентрации 30,2 NIH единиц· мл-1.
- Все использованные ингредиенты были аналитически чистыми.
- Для водных растворов использовали ультрачистую воду (качество - Milli-Q).
Приготовление растворов испытываемых и стандарных соединений
Испытываемые и стандартные соединения растворяли в Milli-Q-воде, чтобы получить исходные концентрации 10-2 моль.л-1. Каждую концентрацию ступенчато разбавляли наполнителем до получения концентраций 10-3, 10-4 и 10-5 моль·л-1. Разведения, включая исходный раствор, использовали в исследованиях (конечные концентрации в реакционной смеси: 3·10-4; 10-4; 3·10-5; 10-5; 3·10-6 ; 10-6; 3·10-7 и 10-7 моль·л-1, соответственно).
Способ
При комнатной температуре 0,075 мл и 0,025 мл растворов испытываемого соединения или стандартного соединения или наполнителя попеременно пипетировали в лунки титрационного микропланшета и эти растворы разбавляли 0,115 мл и 0,0165 мл TNP-буфера, соответственно. В каждую лунку добавляли аликвоты по 0,030 мл раствора S-2238 и планшет предварительно нагревали и проинкубировали, встряхивая, в термостате (Amersham) в течение 10 минут при 37° С. После преинкубации гидролиз S-2238 начинали добавлением 0,030 мл раствора тромбина в каждую лунку. Планшет инкубировали (со встряхиванием в течение 30 сек) при 37°С. Начиная после 1 минуты инкубации, измеряли оптическую плотность каждого образца при 405 нм каждые 2 минуты в течение периода 90 минут, используя кинетический микропланшетридер (Twinreder plus, Flow Laboratories).
Все данные сосредотачивали в персональном компьютере, используя программу обработки данных (Biolise). Для каждой концентрации соединения (выраженной в моль·л-1 реакционной смеси) и для контроля строили график: оптическая плотность в зависимости от времени реакции в мин.
Оценка реакций: Для каждой конечной концентрации максимальную оптическую плотность рассчитывали по графику анализа. Величину IC50 (конечная концентрация, выраженная в мкмоль·л-1, вызывающая 50% ингибирование максимальной оптической плотности контроля) рассчитывали, используя способ логарифмического преобразования согласно Hafner et al. (Arzneim.-Forsch./Drug Res. 1977; 27(II): 1871-3).
Антитромбиновая активность соединения примера 1: величина IC50: 17 нМ
II Исследование анти-фактор Ха-активности
Активированный Фактор Х (Ха) является фактором каскада свертывания крови. Анти-Ха-активность соединений данного изобретения оценивали посредством спектрофотометрического измерения скорости гидролиза хромогенного субстрата s-2222, расщепляемого Ха. Упомянутое исследование анти-Ха-активности в буферной системе использовали для определения значения IC50испытываемого соединения.
Стандартное соединение: пентасахарид Org 31540
Наполнитель: TNP-буфер.
Растворимость может быть определена с диметилсульфоксидом, метанолом, этанолом, ацетонитрилом или трет-бутиловым спиртом, которые не вызывают побочных эффектов в концентрациях вплоть до 1% (для DMSO) и 2,5% (для других растворителей) в конечной реакционной смеси.
Методика
Реагенты* 1. Трометамин-NaCl (TN)-буфер; состав буфера: Трометамин (трис) 6,11 г (50,4 ммоль), NaCl 10,17 г (174 ммоль), полиэтиленгликоль 6000 3 г·л-1, вода до 1 л. рН раствора доводили HCl (10 ммоль·л-1) до 7,4 при 37°С. 3. Раствор S-2222: S-2222 из одной ампулы (25 мг; Chromogenix, Sweden) растворяли в воде, чтобы получить концентрацию 0,375 мг/мл-1 (0,5 ммоль·л-1). 4. Раствор Ха: Бычий фактор Ха (71 нКат/ампулу-1; Chromogenix) растворяли в 10 мл TNP-буфера, а затем далее разбавляли TNP-буфером, чтобы получить концентрацию 0,75 нКат/(1,5 Ед.)/мл-1. Разведение должно быть свежеприготовленным. 5. Раствор ATIII: ATIII человека (Chromogenix) растворяли в воде для получения концентрации 1 Ед./мл-1, после этого раствор далее разбавляли 3 объемами TNP-буфера до концентрации 0,25 Ед./мл-1. 6. Стандартный раствор: исходный раствор Org 31540 5,7 анти-Ха Ед./мл-1 разбавляли TNP-буфером до 0,05 Ед./мл-1. 7. Исследуемые образцы: Каждый препарат растворяли в воде и разбавляли TNP-буфером до концентрации 0,05 нмоль·мл-1. Для каждого препарата была приготовлена серия из 9 разведений (фактор разведения 1,5).
Определение активности Ха
Каждый исследуемый образец (0,05 мл) вносили пипеткой в лунку титрационного микропланшета при комнатной температуре. Раствор AT-III (0,05 мл) добавляли к каждому образцу и планшет встряхивали, используя Vari-вибратор. Аликвоты раствора Ха (0,05 мл) вносили пипеткой в каждую лунку через 10 минут после добавления раствора AT-III и планшет снова встряхивали. Строго через 2 мин после добавления раствора Ха 0,1 мл раствора S-2222 пипеткой вносили в каждую лунку и планшет снова встряхивали. Для всех добавлений использовали 12-канальную пипетку. Оставшееся количество Ха катализировало гидролиз S-2222, скорость которого измеряли фотометрически после периодов инкубации 2 и 22 минуты, соответственно, при комнатной температуре. Оптическую плотность каждого образца измеряли при 405 нм, используя аппарат для считывания Microelisa, модель 310С (Organon Teknika, Oss, The Netherlands) и рассчитывали увеличение в оптической плотности (ΔOD). Для каждого исследуемого образца дублировали определение. В серию из каждых 10 образцов включали контроль (0,05 мл TNP-буфер).
Калибровочная кривая
Из аликвоты стандартного раствора калибровочного образца готовили ряд разведений (фактор разведения 1,4 для образцов Org 31540). Полученные стандартные образцы (приблизительно 12 образцов) должны содержать 0,01-0,05 анти-Ха Ед./мл. В пределах каждой серии по 0,05 мл каждого стандартного образца тестировали по крайней мере 3 раза, как описано "Determination of Xa activity". Калибровочная кривая получена посредством построения прямой линии к log среднее ΔOD (контроль) - среднее ΔOD (стандартный образец) против log анти-Ха среднее ΔOD (стандартный образец) величинам Ед./мл, используя метод наименьших квадратов.
Оценка реакций: Для каждого образца определяли среднюю анти-Ха-активность в Ед./мл, используя калибровочную кривую.
Анти-фактор Ха-активность соединения примера 1: 1012 Ед./мкмоль.
Реферат
Описывается новый пентасахаридный конъюгат формулы (I);

в которой R независимо представляет собой SO3-или СН3; вставка представляет гибкую вставку длиной 13-25 атомов; заряд пентасахаридного остатка уравновешен положительно заряженными противоионами; и общее количество сульфатных групп в пентасахаридном остатке составляет 4, 5 или 6. Способ его получения и фармацевтическая композиция на его основе для лечения заболеваний, опосредованных или связанных с тромбином. 3 н. и 6 з.п. ф-лы.
Формула



Комментарии