Альфа-амино-1-(фосфонометил)-1н-бензимидазол-2-пропановая кислота и способ ее получения - RU2073004C1
Код документа: RU2073004C1
Чертежи

Описание
α-глютамат и a-аспартат, эндогенные кислые аминокислоты, были однозначно установлены в качестве основных возбуждающих нейротрансмиттеров. Действие этих возбуждающих аминокислот передается через несколько подтипов различных рецепторов, наиболее изученным из которых является рецептор N-метил-D-аспартата (НМДА). Избыточное активирование комплекса НМДА - рецепторов может послужить причиной нейтронного перевозбуждения с патологическими последствиями. Экспериментальные исследования показывают, что продолжительная, индуцированная агонистом проводимость НМДА блокированного ионного канала допускает патологическое увеличение поступления кальция и образующиеся в результате увеличенные концентрации внутриклеточного кальция играют наиболее важную и неблагоприятную роль при эксцитотоксичном нейронном нарушении, нейродегенерации и замедленной гибели нейронов.
Возбуждающие аминокислоты образуются при нейропатологиях травматического, эндогенного генетического и окружающего происхождения. Нарушение работы головного мозга, связанного с гипоксией, гипогликемией, травматическими нарушениями, ударом, эпилепсией, специфическими метаболическими нарушениями и некоторыми хроническими нейродегенеративными заболеваниями, в значительной степени вызвано эксцитотоксичными механизмами.
Некоторые из этих исследований показали, что блокада рецепторов НМДА - подкласса существенно снижает нейронные нарушения и потери, которые имеют место в животных моделях, моделирующих самые разнообразные нейропатологические ситуации. Эти наблюдения однозначно указывают на то, что НМДА антагонисты обеспечивают эффективную нейропротекцию в нескольких клинических случаях. Таким образом, агенты антагонизирующие эксцитотоксичные эффекты, передающиеся НМДА рецептором, оказываются эффективны и при лечении ишемических заболеваний, ударов, нарушений работы головного или спинного мозга, и, в общем случае, у пациентов с возрастающими концентрациями возбуждающих трансмиттеров. Конкретные приложения включают также терапию старческого слабоумия типа Алзхаймера, комплекса слабоумия Паркинсона, хореи Хантингтона, и других доминантных или рецессивных спинномозговых и мозжечковых дегенераций, когда НМДА-антагонисты предотвращают или замедляют развитие заболевания.
Соединения
настоящего изобретения являются конкурирующими НМДА - антагонистами, которые имеют следующую формулу:
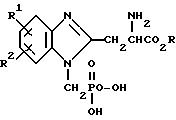
в которой
R является водородом, низшим алкилом, бензилом или пивалоилоксиметилом,
R1 и R2 являются, независимо друг от друга, водородом, низшим алкилом, низшим алкокси, трифторметилом, трифторметокси, метансульфониламино, ацетиламино, или гало, или, взятые вместе, R1 и R2 представляют метилендиокси-группы; и их приемлемые с фармакологической точки зрения соли.
Термин "низший алкил" и "низший алкокси" относится к составляющим, содержащим от 1 до 4 атомов углерода в углеродной цепи. Термин "гало" относится к фтору, хлору, брому и йоду.
Соединения настоящего изобретения проявляют хиральность и, таким образом, соединение настоящего изобретения включают не только рацемические смеси, но также отдельные энантиомеры. Энантиомеры строятся в соответствии с R/S системой, используя правило последовательности.
Соединения
настоящего изобретения могут
быть получены при помощи нескольких синтетических процедур. В соответствии с предпочтительной схемой, защищенное предшествующее соединение бензимидазолил-D-аланина
взаимодействует со сложным эфиром
алкилфосфоната с последующим снятием защиты, чтобы получить целевые финальные продукты:

в которой в приведенной выше последовательности R3 и R4 представляют известные защищающие аминокислоты группы такие, как низший алкил и бензил для R3 и третичн.-бутилоксикарбонил или бензилоксикарбонил для R4. Защищающие группы алкилфосфиновой кислоты R5 это низший алкил, бензил или 4-нитробензил.
Замещаемой группой Х реагента, алкил алкилфосфиновой кислоты, может быть гало, метилсульфонил, толилсульфонил, или трифторсульфонил, где трифторсульфонил является особенно предпочтительным. Защищенное предшествующее соединение бензимидазолилаланина может быть получено в оптически чистой форме при помощи энантиоселективного синтеза из R или S производного N-защищенного сложного эфира аспарагиновой кислоты при помощи процедуры Нестора и др. J. Med. Chem. т. 270 с. 320 (1984). Снятие защиты с алкилфосфинилированного промежуточного соединения может быть осуществлено с использованием кислотного или щелочного гидролиза, гидрогенолиза и/или обработки триметилсилилбромидом в зависимости от защищающей группы, подлежащей удалению. Эти стадии могут быть переставлены или соединены, когда это необходимо или уместно. Когда R3 является бензилом R4 является бензилоксикарбонилом, а R5 является 4-нитробензилом, можно осуществить одностадийную общую гидрогенолитическую депротекцию.
В альтернативной последовательности орто-фенилендиамин может взаимодействовать с реагентом, сложным эфиром алкилфосфоната, чтобы
получить промежуточное
соединение диамино, которое затем взаимодействует в соответствии с процедурой Нестора и др. см. выше, чтобы получить защищенное промежуточное соединение, которое затем
подвергают депротекции, как это
описано выше:

В тех случаях, когда структура замещения для R1 и R2 на кольце бензимидазола асимметрична, при этом в результате алкилирования при помощи сложного эфира алкилфосфоната в приведенной выше реакционной последовательности получали бы диастериометрическую смесь продуктов, поэтому должна быть необходима фракционная кристаллизация или хроматография, чтобы разделить продукты. Этого можно избежать при помощи использования соответствующих исходных материалов, которые дают целевые предшествующие материалы, Так соответствующим образом защищенный орто-нитроанилин или орто-галонитробензол взаимодействуют со сложным эфиром алкилфосфоната или сложным эфиром аминофосфоната, соответственно, чтобы получить соответствующее промежуточное соединение, которое затем взаимодействует в соответствии с процедурой Нестора и др. далее, защищу снимают и получают целевые финальные продукты:

Исходные материалы в приведенной выше последовательности либо производятся промышленностью, либо могут быть получены при помощи известных в этой области техники приемов и процедур.
Соединения, являющиеся предметом настоящего изобретения, могут образовывать фармакологически приемлемые соли из приемлемых с фармакологической точки зрения органических и неорганических кислот таких, как хлористоводородная, бромистоводородная, (моно) сульфо-, серная, фосфорная, азотная, малеиновая, фумаровая, бензойная, аскорбиновая, памоиновая, янтарная, метан (моно) сульфо-, ускусная, пропионовая, винная, лимонная, бензойная, молочная, яблочная, миндальная, коричная, пальмитиновая, итакконовая и бензол (моно) сульфокислота. Соединения настоящего изобретения такие, как фосфонокарбоновые кислоты, способы образовывать карбоксилаты щелочного и щелочно-земельного металлов, и карбоксилаты приемлемых с фармакологической точки зрения катионов, полученных из аммиака или щелочного амина. Примеры последних включают (но ими не исчерпывается полный список) катионы такие, как аммоний, моно-, ди и триметиламмоний, моно-, ди- и триэтиламмоний, моно-, ди и трипропиламмоний (изо и нормальный), этилдиметиламмоний, бензилдиметиламмоний, циклогексиламмоний, бензиламмоний, дибензиламмоний, пиперидиний, морфолиний, пирролидиний, пиперазиний, 1-метилпиперидиний, 4-этилморфолиний, 1-изопропилпирролидиний, 10,4-диметилпиперазиний, 1-н-бутил пиперидиний, 2-метилпиперидиний, 1-этил-2-метилпиперидиний, моно-, ди и триэтаноламмоний, этил диэтаноламмоний, н-бутилмоноэтаноламмоний, трис(оксиметил)метиламмоний, фенилмоноэтаноламмоний и т.п.
Соединения, являющиеся предметом настоящего изобретения, являются конкурирующими НМДА-антагонистами, используемыми при лечении судорог, церебральной ишемии, удара, поражений головного или спинного мозга, расстройств ЦНС (центральной нервной системы пер.) таких, как старческое слабоумие, заболевание Алзхеймера, хорея Хантингтона и других доминантных или рецессивных спинномозговых и мозжечковых дегенераций. Вышеупомянутые соединения могут быть особенно эффективными в качестве предварительных анестезирующих и нейрозащитных агентов во время хирургических вмешательств с высоким риском таких, как хирургия головного мозга и хирургия спинного мозга, или в результате травмы, когда риск ослабления сердечной деятельности или дыхания может вызвать частичное, временное или полное прекращение кровяного тока к головному мозгу. Дополнительные преимущества заключаются в использовании соединений настоящего изобретения в качестве предварительных анестезирующих агентов, так как они обладают слабыми транквилизаторными (седативными свойствами, свойством краткосрочной потери памяти) краткосрочная амнезия) и способностью усиливать эффект анестезирующих агентов так, что последние можно использовать в более низкой дозе.
Следовательно, в дополнение к новым соединениям предлагается способ предотвращения расстройств, вызванных сверхстимулированием возбудительных рецепторов аминокислоты в головном мозге и спинном мозге, который содержит применение к млекопитающему, страдающему таким заболеванием, НМДА - антагониста формулы, приведенной выше.
Как таковые, соединения настоящего изобретения могут быть применены чистыми или с фармакологическим носителем, и поэтому они могут быть изготовлены в форме стоматических доз таких, как таблетки, капсулы и т.п. Предлагаемые соединения могут быть применены при помощи комбинирования их с известными носителями такими, как карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, натрий карбоксиметилцеллюлоза, воск с низкой температурой плавления, кокосовые масло и т.п. Можно также использовать разбавители, ароматизирующие агенты, солюбилизаторы, смазочные агенты, суспендирующие агенты, связывающие материалы, агенты, разрыхляющие таблетки и т.п. Эти соединения могут быть также инъецированы парентерально, в этом случае они используются в форме стерильного раствора, содержащего друге растворенные вещества, например, достаточное количество соляного раствора или глюкозы, чтобы сделать раствор изотонным.
Дозировка будет меняться в зависимости от конкретной композиции, способа применения, степени серьезности симптомов и состояния пациента, подлежащего лечению. Лечение в общем случае начинают с небольших доз, меньших оптимальной дозы соединения. Далее, дозировку увеличивают до тех пор, пока при данных обстоятельствах не будет достигнут оптимальный эффект. В общем случае, соединения настоящего изобретения в наиболее предпочтительном варианте применяют в концентрации, которая будет в общем случае обеспечивать эффективные результаты без каких-либо опасных или неблагоприятных побочных эффектов, и они могут быть применены либо в виде одной дозы, либо, если это необходимо, эту дозу можно разделить на несколько поддоз, которые применяют в определенные моменты времени в течение дня.
Активность конкурирующих НМДА антагонистов соединений, являющихся предметом настоящего изобретения, может быть подтверждена при помощи стандартных фармакологических процедур, которые иллюируются их in vitro ингибирование (3H)ССР-связывания в ткани мозга крысы и их in viro антагонизм судорог у мышей, вызванных НМДА.
Приводимые ниже примеры показывают получение и фармакологическое испытание соединений, являющихся предметом настоящего изобретения.
Примеры
Энантиомерную чистоту примеров настоящего изобретения определяли при помощи модификации процедуры, предложенной Tapuh, Y. Miler, N. Karger, B. Journal of Chromatography, 1981, т. 205, стр.325 337.
Получение [бис-(4-нитрофенилметокси)фосфинил] метилового сложного эфира трифторметан(моно)сульфокислоты
Раствор ди-4-нитробунзил оксиметилфосфонаты (3,82 г, 10,0 ммолей)
[Hoffmonn, M. Synthesis, 1988 62] и пиридина (0,87 г, 11,0 ммолей) в дихлорметане (50 мл) обрабатывали при температуре -10 -20oC ангидридом трифторметан(моно)сульфокислоты (3,1 г, 11,0
ммолей) и перемешивали при -10oC в течение 1 часа. Раствор промывали холодным 1N раствором HCl (2 х 50 мл), холодной водой (3 х 50 мл) и сушили над сульфатом магния. Раствор фильтровали,
растворитель выпаривали, а оставшееся масло отверждали при отстаивании. Выход 4,18 г (80%). Материал был достаточно чистым, чтобы можно было использовать для последующих реакций. Аналитический
образец
получали с использованием хроматографии на сухой колонне на силикагеле Сорта II-III с этил ацетатом в качестве элюента. Продуктовые фракции выпаривали, а остаток кристаллизовали из
дихлорметана/гексана и сушили: температура плавления 73 - 75oC1H-ЯМР (400 МГц, CDCl3): δ 8,2 (дублет, 4Н), 7,5 (дублет, 4Н), 5,2 (мультиплет, 4Н), 4,8 (дублет,
2Н); МС (+ FAB) 515 (M + H)+.
Элементный анализ для C16H14N2O10PSF3.
Рассчитано: C 37,36, H 2,74 N 5, 45.
Найдено: C 37,27, H 3,03, N 5.52.
П р и м е р 1.
R-a-амино-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота.
Бензил N-(бензилоксикарбонил)-3-(2-бензимидазолил)-D-аланинат (30 г, 0,07 молей) (полученный в соответствии с процедурой Нестора и др. J. Med, Chem. 1984, т. 27, с. 320), (диэтоксифосфонил)метиловый сложный эфир трифторметан(моно)сульфокислоты (23,04 г, 0,076 молей) и порошкообразный безводный карбонат калия (37 г, 0,268 молей) перемешивали в ацетонитриле (500 мл) при комнатной температуре течение 20 ч. Смесь фильтровали, фильтрат выпаривали, а остаток растворяли в дихлорметане. Дихлорметан промывали водой (500 мл), 5% NHCO3 (2 х 300 мл), водой (300 мл) и сушили над сульфатом магния. Раствор фильтровали, а растворитель выпаривали до смолы. Выход 43 г. Смолу растворяли в дихлорметане (300 мл) и подвергали очистке на сухой колонне с силикагелем с градиентным элюированием (от метилен хлорида до этил ацетата). Фракции продукта [Rf (метилен хлорид/этил ацетат 4:1) 0,17] собирали и выпаривали до смолы. Выход 10,6 г (26%).1Н-ЯМР (400 МГц, CDCl3): d 7,7 (дублет, 1Н), 7,35 (синглет, 5Н), 7,3 (синглет, 5Н), 7,2 (синглет, 2Н)= 6,9 (дублет, 1Н), 5,2 5,0 (мультиплет 5Н), 4,4 4,2 (мультиплет, 2Н), 4,1 (мультиплет, 1Н), 3,9 (мультиплет, 4Н), 3,6 (двойной дублет, 1Н), 3,4 (двойной дублет, 1Н), 1,2 (триплет, 3Н), 1,1 (триплет, 3Н), МС (+ FAB) 580 (М + Н)+. Материал (10,6 г, 0,0183 молей) растворяли в уксусной кислоте (300 мл) и встряхивали с 10% Pd/C (1 г) при давлении 1 атм Н2 при комнатной температуре до тех пор, пока не прекратится поглощение Н2, фильтрат выпаривали, а остаток совместно выпаривали с диоксаном (4 х 100 мл), ИК- ЯМР- и масс-спектр остатка (8 г) указывали на удаление бензильных защищающих групп. 7,5 г остатка и триметилсилил бромид (15 г, 0,1 молей) дефлегмировали в N2 в дихлорэтане (110 мл) в течение 1,5 ч. Растворитель выпаривали, а остаток перемешивали в воде (50 мл) и простом эфире. Получали твердое вещество, которое фильтровали и хранили. Фильтрат отделяли, водный слой разбавляли этанолом (50 л) и обрабатывали окисью пропилена (10 мл) при перемешивании в течение 0,5 ч. Раствор выпаривали, чтобы удалить этанол, а твердое вещество, которое при этом образуется, соединяли с твердым веществом, полученным ранее. Продукт растворяли в 1 vN растворе HCl (100 мл) и фильтрованный раствор, выпаривали до сухого состояния. Выход 4 г (65%) в пересчете не полностью защищенное промежуточное соединение. Продукт кристаллизовали из теплой воды (50 мл) и этанола (100 мл), и сушили под вакуумом. Выход 2,27 г (37%). Соединение не плавилось при температуре ниже 310oC.1H-ЯМР (400 ДМСО-d6): d 7,5 (триплет, 2Н), 7,3 (мультиплет, 2Н), 4,35 (дублет, 2Н), 4,25 (триплет, 1Н), 3,5 (мультиплет, 2Н); МС (-FAB) 298 (M-H)-; [a] -49,5o (с 1,01 1 N HCl); ЖХВД анализ: энантиомерная чистота: 1S 99R.
Элементный
анализ для C11H14N3O5P • 2H2O:
Рассчитано: C 38,88, H 5,41, N 12,37
Найдено: С 38,95, Н 5,13, N 12,52.
Дигидрат хлоргидрата получали при помощи растворения свободной кислоты в 2 N HCl, выпаривали до сухого состояния, а затем сушили под вакуумом.1H-ЯМР (400 МГц, ДМСО-d6: δ 9,2 8,2 (мультиплет, 2Н), 7,9 (дублет, 1Н), 7,7 (дублет, 1Н), 7,5 (мультиплет, 2Н), 4,8 (мультиплет, 3Н), 3,8 (дублет, 2Н); [a] -41,6o (c 1,0 1 N HCl).
Элементный анализ для С11H14N3O5P • HCl •
2H2
O:
Рассчитано: C 32,37, H 4,93, N 10,29
Найдено: C 32,57, H 4,87, N 10,53
П р и м е р 2.
R-α-амино-5, 6-дихлор-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота.
При помощи той же процедуры, что использовали в Примере 1, но используя бензил N-(бензилоксикарбонил)-3-[(5, 6-дихлор)-2-бензимидазоил] -D-аланинат (полученный в соответствии с процедурой Нестора и др. J. Med. Chem. 1984, т. 27, стр. 320), получали соединение из заголовка примера (Выход 15,3%), температуру точки плавления не определяли.1Н-ЯМР (400 МГц, ДМСО-d6): d 8,55 (синглет, 3Н), 8,95 (синглет, 1Н), 8,85 (синглет, 1Н), 4,6 (дублет, 2Н), 4,55 (триплет, 1Н), 3,8 (мультиплет, 2Н); МС (-FAB) 366 (M-H)- [a] -50,0o (c 1,01; 1 N HCl); ЖХВД анализ (свободная кислота) энантиомерная чистота: 2,6 S 97,4 R.
Элементный анализ для С11H12N3O5PCl2 • 2 HCl:
Рассчитано: C 29,99;
H 3,20, N 9,53
Найдено: C 29,78, H 3,31, N 9,65.
П р и м е р 3.
R-α-амино-5, 6-диметил-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота.
Бензил N-(бензилоксикарбонил)-3-[/5,6-диметил)-2-бензимидазоил] -D-аланинат (Нестор и др. J. Med. Chem, 1984, т. 27, с.
320 (3,7 н, 0,8 ммолей), [бис-(4-нитрофенилметокси/фосфинил] метиловый
сложный эфир трифторметан(моно)сульфокислоты (5,5 г, 10 ммолей) и порошкообразный безводный K2CO3 (5,5 г,
40 ммолей) перемешивали в ацетонитриле (100 мл) при комнатной
температуре в атмосфере N2 (20 ч). Ацетонитрил выпаривали, а остаток встряхивали с дихлорметаном (100 мл) и водой (2 х 100
мл). Слой дихлорметана промывали 5% NaHCO3 (2 x 100 мл),
соляным раствором (100 мл) и сушили над сульфатом магния, фильтрованный раствор выпаривали и оставшуюся смолу растворяли в уксусной
кислоте (100 мл) и встряхивали на аппарате гидрогенизации Парра с
10% Pd/C (1 г) и Н2 [38 фунтов на кв. дюйм (2,67 кг/см2) в начале] до тех пор, пока поглощение H2 не
прекратится (3 ч). Фильтрованный раствор выпаривали и совместно
выпаривали с диоксаном (3 х 50 мл) и остаток разбавляли водой (350 мл). рН смеси обеспечивали на уровне 3 при помощи 6 N раствора HCl,
быстро охлаждали и фильтровали. Высушенное воздухом твердое
вещество (2,8 г) растворяли в воде (50 мл) с 1 N раствором НСl (1 мл) и осаждали добавлением 1 N раствора NaOH (1 мл). Продукт промывали
водой, этанолом и простым эфиром. ИК-, ЯМР- и масс-спектры
согласовывались с фосфонометил аминокислотой. Этот материал растворяли с 1 N растворе НСl (20 мл), обрабатывали активированным углем и
быстро охлаждали. Соль хлордигидрата фильтровали, промывали
ледяной водой и сушили под вакуумом. Выход 1,23 г (37,4%1П-ЯМР (400 МГц, ДМСО-d6 + 20% DCl (D2 (2 капли):
d 7,75 (синглет, 1Н), 5,7 (cинглет, 1Н), 5 4,75 (мультиплет,
3Н), 3,85 (мультиплет, 1Н), 2,35 (синглет, 3Н); МС (-FAB) 326 (M-H)-; [a] 54,4o (c 1,01, 1 N HCl); ЖХВД анализ: энантиомерная чистота: 1S 99R
Элементный анализ для C13H18N3O5P • 2 HCI:
Рассчитано: C 39,
02, H 5,04 N 10,50.
Найдено: C 38,62, H 5,27, N 10,32.
П р и м е р 4.
R-(-)(-α -амино-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота.
В соответствии с процедурой из примера 3 и, используя бензил-N-(бензилоксикарбонил)-3-(2-бензимидазоил)-D-аланинат, получали соединение из заголовка примера. В результате кристаллизации из горячей воды и длительной сушки под вакуумом получали гидрат с 3,5 молями воды, выход (61%).
1H-ЯМР (400 Гц, ДМСО-d6 + DCl): d 7,95 (квартет, 1Н), 7,8 (квартет, 1Н), 7,55 (2Н), 4,8 (мультиплет, 3Н), 3,8 (дублет, 2Н); [a] 53,1o (c 1,03 I NHCl); ЖХВД анализ, энантиомерная чистота: 1,3S 98,7R.
Элементный анализ С11H14N3
O5P • 3,5 H2O
Рассчитано: C 36,46, H 5,84, N
11,59.
Найдено: C 36,67, H 5,64, N 11,74.
Хлордигидрат получали при помощи растворения в 2 N HCl (5 мл) и воде (15 мл), и выпаривания до сухого состояния. [a] -40,2 (c 1,0, 1 N HCl).
Элементный анализ для С11H14N3 O5P • HCl • H2O.
Рассчитано: C 33,86, H 4,65, N 10,77.
Найдено: C 33,77, H 4,59, N 10,67.
П р и м е р 5.
2-Амино-2-(оксиметил)-1,3-пропандиол, R-α-амино-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота (2:1).
Соединение свободной кислоты из примера 4 (1,86 г, 5 ммолей) трометаминовое основание (1,33 г, 11 ммолей), нагревали в воде (10 мл) и осаждали этанолом (100 мл). Материал фильтровали, промывали этанолом, затем простым эфиром и сушили воздухом в течение ночи. Продукт подвергали рекристаллизации из теплой оды (30 мл) и этанола (200 мл), фильтровали, промывали этанолом и сушили под вакуумом над P2O5. Выход 2,58 г (86,6%).
Элементный анализ для C11H14N3O5P • 2 C4H11NO3 • 3H2O.
Рассчитано: C 38, 35, H 7,11, N 11,77.
Найдено: C 38,39, H 6,76, N 11,86.
П р и м е р 6.
S-a-амино-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота.
Бензил N-(бензилоксикарбонил)-3-(2-бензимидазоил)-a-аланинат (2,85 г, 6,3 ммолей), [бис-(4-нитрофенилметокси)-фосфонил]метиловый сложный эфир трифторметан(моно)сульфокислоты (3,67 г, 8,9 ммолей) и порошкообразный безводный карбонат калия (4 г, 28,9 ммолей) перемешивали в ацетонитриле (100 мл) при комнатной температуре в течение ночи. Затем продолжали процедуру из Примера 1 и получали производные тетрабензила. Сырой продукт (4,3 г) подвергали очистке на ЖХВД типа Уотерз преп 500, используя градиентное элюирование от гексана (100% ) до этил ацетата (100%). Выход 1,0 г. Продукт растворяли в уксусной кислоте с 10% Bd/C, (0,5 г и подвергали гидрогенизации при давлении в 1 атм. Смесь фильтровали, выпаривали на установке Ротовапор (Rotovapor), быстро разбавляли дважды диоксаном и перемешивали в воде (10 мл). Продукт фильтровали и сушили. Выход 0,35 г (17, 6%); ЯМР идентичен продукту из примера 1 MC (+FAB) 300 (M + H)Б+ [a] +32,8o (c 1,45, метанол + HCl); ЖХВД анализ, энантиомерная чистота: 98,2S/1,8R.
Элементный анализ для C11H14N3O5P • H2O.
Рассчитано: C 41,65, H 5,08, N 13,25.
Найдено: C 41,60, H 5,04, N 13,09.
П р и м е р 7.
R-α
-амино-6-хлор-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота
A) В атмосфере сухого азота a-бензиловый сложный эфиp
N-Boc-D-аспарагиновой кислоты (13,4 ммолей, 4,34 г) растворяли в
сухом тетрагидрофуране (67 мл) и охлаждали до -10oC. Последовательно добавляли триэтиламин (13,4 ммолей, 1,28 мл) и
реакционную смесь перемешивали в течение 10 мин при температуре -10oC, после чего медленно добавляли раствор производимого промышленностью 4-хлор-1,2-фенилендиамина (14,7 ммолей, 2,1 г) в
сухом тетрагидрофуране (27 мл). Смеси давали возможность медленно
нагреваться до окружающей температуры. Затем ее сливали в ледяной соляной раствор (150 мл), экстрагировали этил ацетатом (2 х 100 мл).
Соединенный органический слой промывали последовательно
насыщенным льдом NaHCO3 (100 мл), затем соляным раствором (100 мл), а затем сушили над сульфатом магния, фильтровали и выпаривали до
сухого состояния под вакуумом. Остаток подвергали
хроматографии (ЖХВД). В результате элюирования этил ацетатом (гексаном получали 2,9 г N4-(2-амино-5-хлорфенил)-N2
-[(диметилэтокси/карбонил] - D-аспарагин фенилметилового
сложного эфира.
В) Раствор полученного выше масла (4,8 ммолей, 2,15 г) в ледяной уксусной кислоте (70 мл) нагревали до 70oC на 5 ч без доступа влажности. Затем смесь выпаривали под вакуумом, а остаток оперативно подвергали хроматографии на силикагеле (60 г). В результате элюирования 20% этилацетатом/гексаном получали 1,6 г фенилметилового сложного эфира 6-хлор-a-[1,1-диметилэтокси/карбонил]амино]-1Н-бензимидазол-2-пропановой кислоты в виде масла.
C) Раствор масла со Стадии В (3,7 ммолей, 1, 6 г) в ацетонитриле (50 мл) обрабатывали при температуре 25oC в сухом азоте [диметоксифосфинил] метиловым сложным эфиром трифторметан(моно)сульфокислоты (4,1 ммолей, 1,115 г) и безводным порошкообразным карбонатом калия (10 ммолей, 1, 38 г). Реакционную смесь перемешивали при температуре 25oC в течение ночи, фильтровали, промывали метиленхлоридом (20 мл), соединенные фильтраты выпаривали под вакуумом, а остаток разделяли между метилен хлоридом и водой. Органический слой отделяли, сушили над сульфатом магния, фильтровали и выпаривали под вакуумом. Остаток подвергали оперативной хроматографии на силикагеле (60 г). В результате элюирования хлороформом/этил ацетатом получали 1,4 г фенилметилового сложного эфира 6-хлор-1-[(диметоксифосфинил)метил] - a-[[(1, 1-диметилэтокси)карбонил]амино]-1Н-бензимидазол-2-пропановой кислоты в виде масла.
D) Раствор масла со Стадии С (2,5 ммолей, 1,4 г) в ледяной уксусной кислот (20 мл) обрабатывали 10% палладием на древесном угле (140 мг) и подвергали гидрогенизации в течение 3 часов при 25oC. Реакционную смесь продували азотом, фильтровали через фильтр Солка-флок (Solka-floc), лепешку промывали уксусной кислотой (10 мл) и фильтрат выпаривали до сухого состояния под вакуумом. Остаток отгоняли с толуолом (2 х 10 мл) и выпаривали под глубоким вакуумом, чтобы получить 1,15 г 6-хлор-1- [(диметоксифосфинил)-метил] -a-[[1,1-диметилэтокси/карбонил]амино]- 1Н-бензимидазол-2-пропановой кислоты в виде масла.
Е) Масло по Стадии D (1,9 ммолей, 0,9 г) подвергали дефлегмации в 6 N растворе HCl (20 мл в течение 45 мин. Реакционную смесь затем выпаривали до сухого состояния под вакуумом, остаток отгоняли с толуолом (2 х 20 мл), а затем подвергали кристаллизации из горячей воды/ацетонитрила, чтобы получить 330 мг соединения из заголовка примера; температура точки плавления 198 - 200oC.1H-ЯМР (ДМСО-d6 1 капля DCl, 400 МГц: d 3, 87 (двойной дублет, J1 5,5 Гц, J2 7,2 Гц, 2Н,
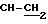

Элементный анализ для С11 H13ClN3O5P • 0,8 HCl • H2O.
Рассчитано: C 36,62, H 4,47, N 11,64
Найдено: C 36,32, H 4,55 N 11,35
П р и м е
р 8.
R-α-амино-5-хлор-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота.
А) Использовали процедуру, как в примере 7А), однако, полученный остаток не очищали, а использовали на следующей стадии.
В) Использовали процедуру, как в примере В), однако, полученный остаток не очищали, но использовали на следующей стадии.
C) Масло, полученное со Стадии В (21,8 ммолей, 9,4 г, смесь 5- и 6-хлорорегиозомеров), в ацетонитриле (250 мл) обрабатывали один раз в сухом азоте и при перемешивании [диметоксифосфинил]метиловым сложным эфиром трифторметан/моно(сульфокислоты) 24 ммолей, 6,528 г) и безводным порошкообразным K2CO3 (65 ммолей, 8,97 г). Реакционную смесь перемешивали при 25oC в течение ночи, фильтровали и промывали ацетонитрилом. Фильтрат выпаривали, а остаток разделяли между водой и метилен хлоридом. Отделенный органический слой сушили, а затем выпаривали под вакуумом до сухого состояния. Остаток подвергали хроматографией (ЖХВД), элюировали этил ацетатом/гексаном, в результате чего получали 4 г фенилметилового сложного эфира 5-хлор-1-[(диметоксифосфинил/метил] альфа-[[(1, 1-диметоксиэтокси/карбонил]-амино]-1Н-бензимидазол-2-пропа- новой кислоты в виде масла.
D) Раствор масла со Стадии С (7,25 ммолей, 4 г) в ледяной уксусной кислоте (60 мл) обрабатывали 10% палладием (древесный уголь (400 мг) и подвергали гидрогенизации при температуре 25oC при атмосферном давлении в течение 4 ч. Затем смесь продували азотом, фильтровали через фильтр Solka-floc, промывали уксусной кислотой (20 мл), а фильтрат выпаривали до сухого состояния под вакуумом. Остаток отгоняли с толуолом (2 х 20 мл), и, наконец, выпаривали под глубоким вакуумом, чтобы получить 3,43 г 5-хлор-1-[(диметоксифосфинил)метил] -a-[[(1,1- диметилэтокси/карбонил]амино]-1Н-бензимидазхол-2-пропановой кислоты в виде масла.
Е) Масло по Стадии D (7,2 ммолей, 3,43 г) подвергали дефлегмации в 6 N растворе хлористоводородной кислоты (60 мл) в течение 50 мин. Затем смесь выпаривали до сухого состояния под вакуумом, а остаток еще один раз выпаривали с водой (15 мл). Остаток сушили под высоким вакуумом, а затем кристаллизовали из горячей воды (ацетонитрила; Соединение фильтровали, промывали простым эфиром (10 мл) и сушили при давлении 1 мм рт.ст. 60oC (над P2O5), чтобы получить 1,4 г целевого продукта. Температура точки плавления 113 116oC (разлож.).1H-ЯМР (ДМСО-d6 + 1 капля DCl, 400 МГц): d 3,86 (триплет, J 6,4 Гц, 2Н,

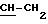
Элементный анализ для С11H13CIN3O5P • HCI.
Рассчитано: C,
35,69, H 3,81 N 11,35
Найдено: C, 35,87, H 3,94, N 11,26
П р и м е р 9.
S-α-Амино-5,6-дихлор-1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота.
Соединение получали в соответствии с процедурой из Примера 7; температура точки плавления 230oC (разлож.).1H-ЯМР (ДМСО-d6 + капля DCl/: d 3,78 (дублет, J 7 Гц, 2Н,


Элементный анализ для С11H12N3O5P
• 2H2O
Рассчитано: C, 32,69, H 3,99, N 10,39
Найдено: C, 32,66, H 4,13, N 10,34
П р и м е р 10.
Соединения, являющиеся предметом настоящего изобретения, испытывали на их НМДА-конкурирующую антагонистическую активность через их способность вытеснить насыщенную тритием 3-(2-карбоксипиперазин-4-ил)пропил-1-алкилфосфиновую кислоту (КПП), известного конкурирующего антагониста НМДА, в гомогенатах передней лобной доли крысы в in vitro анализе на3H/КПП-связывание.
Этот анализ осуществляли следующим образом.
Крыс обезглавливали и их головной мозг немедленно удаляли, взвешивали и помещали в приблизительно 15 объемов охлаждаемой льдом 10% сахарозы. Каждый мозг гомогенизировали, используя стеклянный гомогенизатор типа Поттер Эльвехьем (12 ходов при скорости 840 об/мин), снабженный пестиком из Тефлона. Эатем гомогенат подвергали центрифугированию со скоростью 1 000 х g течение 10 мин. Полученную в результате таблетку сбрасывали, а верхний слой центрифугировали со скоростью 20 000 x g в течение 20 мин. Сырую митохондриальную таблетку снова суспендировали в охлаждаемую льдом дистиллированную воду и диспергировали с использованием устройства типа Политрон Бринкманн (РТ-10 с установленным режимом 6 в течение 30 с). Суспензию центрифугировали со скоростью 8000 х g в течение 20 мин. Полученный в результате верхний слой и темно-желтое покрытие центрифугировали со скоростью 48 000 х g в течение 20 мин. Финальную неочищенную таблетку синапсных мембран снова суспендировали в охлажденную льдом дистиллированную воду и центрифугировали со скоростью 48 000 x g в течение 20 мин.
Чтобы облегчить удаление эндогенного глютамата, мембраны снова суспендировали в 15 объемов охлаждаемого льдом 50 мМ ТРИС/рН 7,6), содержащего 0,04% Тритона Х-100. Суспензию инкубировали при температуре 37oC в течение 15 минут, а затем центрифугировали со скоростью 20 000 x g в течение 20 мин. Таблетку промывали (т.е. снова суспендировали в охлаждаемый льдом ТРИС-буфер и центрифугировали со скоростью 20 000 х g течение 20 мин дважды. Таблетку мембран, наконец, снова суспендировали в 15 объемов охлаждаемого льдом 50 мМ ТРИС, распределяли по нескольким приборкам для центрифуги и подвергали центрифугированию со скоростью 20 000 х g в течение 20 мин, и таблетки замораживали (-70oC) для последующего использования с целью анализа на связывание.
Для анализа на связывание таблетки мембран оттаивали и снова суспендировали в 15 объемов охлаждаемого льдом 50 мМ ТРИС (рН 7,6) буфера. Всего три образца (1000μл) суспензии мембран, содержащей от 0,2 до 0,4 мг протеина/мл, инкубировали при температуре 23oC в течение 15 мин с 8 нМ (3H/КПП) фирма New England Nuclear), одним из различных испытываемых растворов, и буфер в финальном объеме инкубирования 2 мл, используя пластиковые минипробирки (фирма Skatron). Затем образцы центрифугировали со скоростью 48 000 х g в течение 20 мин, а верхние слои сбрасывали. Таблетки переваривали солюбилизатором тканей (фирма Amersham, NCS; 500 μл образец) в течение 1 ч. В каждый образец добавляли хлористоводородную кислоту /100μл, 4N/, чтобы восстановить хемолюминисценцию во время последующего подсчета. В каждую из минипробирок добавляли коктейл сцинтилляций /Agasol, фирма Du Pont; 3,2 мл/, затем их накрывали и встряхивали в течение 15 минут перед подсчетом. Пробирки помещали в счетчик типа Packard 460 CD /или его эквивалент/ c целью определения радиоактивности.
Общее специфическое связывание определяли как общее связывание минус связывание в присутствии 1 мМ НМДА. Специфическое связывание в присутствии испытываемого препарата выражали в процентах от общего специфического связывания, когда такого препарата нет. Когда испытываемые соединения анализировали на соотношение доза-реакция, результаты наносили на плоскость в виде log связывания относительно log концентрации испытываемого препарата. Анализ линейных регрессий дает тогда прямую линию, из которой можно вычислить IC50 с 95% доверительными пределами.
Стандартные соединения:
Лиганд IC50 + ст. ош. М./мМ/ iμ
Л. Глютаминовая кислота 64,3
≈ 4,4 (3)
AP7 639,2
≈ 128,6 (3)
НМДА 1 882,6 ≈ 612,2 (5)
При испытании в этом анализе соединения, являющиеся предметом настоящего изобретения, давали
следующие результаты.
Соединение из Примера N IC50, нМ
1 59
2 18,2
3 123
6 63% c 10 M
7 21,7
8 7
9 1010
П р и м е р 11.
Соединения настоящего изобретения, далее, испытывали на их in vivo антагонистическую способность по отношению к НМДА в опытах на мышах в судорожном состоянии вызванном НМДА.
Этот
анализ осуществляли следующим образом:
Самцов мышей Swiss-albino (штамм СD-I, Charles River) весом 18 22 гр, спустя 18 ч лишения пищи, помещали в
наблюдательную камеру на 30 мин. Мышей
предварительно обрабатывали представителем испытываемых соединений, затем через тридцать минут при помощи НМДА, в дозе 195 мг/кг внутрибрюшинным способом,
причем эта доза, в общем случае, в 90%
случаев приводит к гибели, наступающей в результате паралича двигательной функции, включающего неконтролирумое царапание задними лапами или подергивание мышц
конечностей и/или спиральной мышцы с
утратой правильного рефлекса с последующей гибелью животного в течение периода наблюдения в 30 мин после применения НМДА. Из последних определяли ЕД50
для живых мышей.
Данные анализировали с использованием программы независимого анализа PS-NONLIN (Версия Естественной Скорости Реакции). Выход из этой программы содержит статистическую значимость наклона кривой доза-реакция с ЕД с 50% и 95% доверительными пределами для живых мышей.
Контрольные соединения приведены в таблице.
При испытании в этом
анализе соединения, являющиеся
предметом настоящего изобретения, давали следующие результаты:
Соединение из Примера N ED50, мг/кг, внутрибрюш. или живых
1 2,0
2
2,7
3 10% при 3
мг/кг
7 <5
8 0,13
9 > 10
Реферат
Использование: в медицине в качестве конкурирующих НМ ДА - антагонистов. Сущность изобретения: продукт α-амино 1-(фосфонометил)-1Н-бензимидазол-2-пропановая кислота ф-лы I, где R1 и R2 - водород, низший алкил или галоген, или фармакологически приемлемые соли. Реагент I. Соединение ф-лы 2, где R3 - R5 - защищающие группы. Условия реакции: осуществляют снятие защиты с соединения ф-лы 2 с использованием кислотного или щелочного гидролиза, или гидрогенолиза.


соединение 1 - соединение 2 1табл.
Формула

где R1 и R2 водород, низший алкил или галоген,
или их фармакологически приемлемые соли.

где R1 и R2 водород, низший алкил или галоген,
R3 и R4 защищающие аминокислотные группы, R5 - защищающие алкилфосфоновую кислоту группы,
с последующим выделением целевого продукта в виде кислоты или переводом ее в фармакологически приемлемую соль.
Комментарии