Ковалентные конъюгаты липидов с фосфонокарбоновойкислотой, их получение и применение в качестве антивирусных лекарственных средств - RU2178418C2
Код документа: RU2178418C2
Чертежи
Описание
Изобретение относится к новым липидным производным фосфонокарбоновых кислот и их сложным эфирам общей формулы I
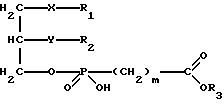
в которой R1 обозначает группировку -(CH2)e-Cycl, в которой (CH2)e обозначает линейную или разветвленную насыщенную алкиленовую цепь.
e равно целому числу от 4 до 16, причем один из атомов углерода, начиная с позиции 3, может быть
заменен гетероатомом (кислородом,
азотом или серой),
R2 обозначает водород, линейную или разветвленную, насыщенную или ненасыщенную алкильную цепь с 1-20 атомами углерода,
R3 обозначает H,
линейную или разветвленную алкильную цепь с 1-6 атомами углерода, в частности, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, гексил, неопентил,
тексил или фенил, холин,
этаноламин, карнитин, C5-C7-циклоалкил, бензил или одну из следующих групп
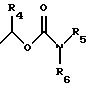
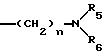
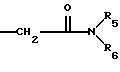
причем R4 обозначает C1-C6-алкил, бензил или фенил и R5, и R6 обозначают C1-C6 -алкил и n равно 1, 2 или 3,
X обозначает валентную связь, кислород, серу, оксикарбонил, карбонилокси, карбониламидо, амидокарбонил, сульфинильную или сульфонильную группу,
Y обозначает валентную связь, кислород, серу, оксикарбонил, карбонилокси, карбониламидо, амидокарбонил, сульфинильную или сульфонильную группу,
Cycl обозначает циклический алкильный остаток с 5-7 C-атомами или фенил, причем атом углерода в кольце может быть заменен азотом, и насыщенные или ароматические кольца могут быть одно или многократно замещены C1-C10-алкилом, C1-C10-алкокси, C1-C10-алкилмеркапто или галогеном,
m обозначает 0, 1, 2 или 3,
при условии, что R1 может быть равен R2, если R2 одновременно принимает значение R1, т. е. R1 и R2 могут быть взаимозаменяемы в своих значениях,
к их таутомерам и их физиологически приемлемым солям неорганических и органических оснований, а также к способу их получения и к содержащим эти соединения лекарственным средствам.
Так как соединения общей формулы I содержат асимметричные атомы углерода, то все оптически активные формы и рацемические смеси этих соединений являются предметом настоящего изобретения.
Терапия злокачественных неоплазий (карциномы, саркомы, гематологические неоплазии), воспалительных заболеваний или аутоиммунных заболеваний, а также заболеваний, вызываемых вирусами или ретровирусами, таких как, например, СПИД, ARC (родственный СПИД'у комплекс), инфекции цитомегалии и герпеса или гепатит, наряду с недостаточной эффективностью применяемых терапевтических активных веществ, часто связана также с их крайне нежелательными побочными действиями. Этот эффект объясняется слишком малой селективностью in vivo, соответственно ограниченным терапевтическим спектром применяемых фармакологически активных веществ. Оптимальные фармакологические свойства фармакологически активных веществ, проявляемые ими in vitro, часто не могут быть перенесены на условия in vivo.
Поэтому в течение многих лет пытаются путем модификации химической структуры фармакологически активных веществ создать новые вещества, обладающие улучшенным терапевтическим спектром действия. Кроме того, часто получают новые лекарственные формы фармацевтических препаратов с той целью, чтобы целенаправленно транспортировать активные вещества именно в нужное место, где они должны осуществлять свое терапевтическое действие. При этом, в частности, необходимо предотвратить нежелательное взаимодействие со здоровыми клетками. В случае опухолевых клеток, обладающих соответствующими поверхностными антигенами, были получены, например, антитела, которые узнают эти специальные поверхностные антигены и тем самым прицельно связываются с раковой клеткой. Антитела модифицированы подходящими токсинами таким образом, что после связывания с раковой клеткой токсин высвобождается, а раковая клетка погибает. Другая возможность улучшения терапевтического спектра состоит в том, что путем незначительной модификации фармакологически активного вещества, например, путем получения соли присоединения кислоты или основания, или путем получения фармакологически приемлемых сложных эфиров [например, эфиров жирных кислот: J. Pharm. Sci. 79, 531 (1990)] физические свойства активного вещества изменяют таким образом, чтобы была улучшена растворимость или переносимость активного вещества. Эти незначительно химически модифицированные соединения часто обозначаются также как "пролекарства", так как они при контакте с жидкостями организма или в печени (метаболизм первого прохода) почти непосредственно могут быть превращены в терапевтически активные агенты. Такие "пролекарства" соединений общей формулы I также включают настоящее изобретение.
Для улучшения катаболической стабильности
осуществляют химическое связывание нуклеозидов, например, ara-C
и ara-A, с фосфолипидами. Соответствующие производные проявляют меньшую токсичность и более высокую стабильность in vivo в сравнении с
немодифицированными нуклеозидами. Однако это не оказывало
практически никакого влияния на абсорбцию и проницаемость клетки [J. Med. Chem. 32, 367 (1989), Cancer Res. 37, 1640 (1977) и 41, 2707
(1981)] . Другие фосфолипидные производные нуклеозидов описаны,
например, в следующих литературных источниках:
В J. Biol. Chem. 265, 6112 (1990) описано получение и применение
липонуклеотидов в качестве антивирусных лекарственных средств. Однако в этом
случае были исследованы и синтезированы лишь связанные с известными нуклеозидами, например, AZT и ddC,
димиристоилфосфатидильные и дипальмитоилфосфатидильные остатки с их структурой сложных эфиров
жирных кислот.
В J. Med. Chem, 33, 1380 (1990) описаны нуклеозидные конъюгаты тиоэфирлипидов с цитидиндифосфатом, которые обладают противоопухолевым действием и могут найти применение в онкологии.
В Chem. Pharm. Bull. 36, 209 (1988) описаны 5'-(3-SN-фосфатидил)-нуклеозиды с противолейкемической активностью, а также их ферментативный синтез из соответствующих нуклеозидов и фосфохолинов в присутствии фосфолипазы D с трансферазной активностью.
Ферментативный синтез липонуклеотидов также описан, в частности, в Tetrahedron Lett; 28, 199 (1987) и Chem. Pharm. Bull, 36, 5020 (1988).
В WO 94/13324 описаны орально применяемые активные вещества с 1-O-алкил-, 1-O-ацил-, 1-S-ацил- и 1-S-алкил-sn-глицеро-3-фосфатами в качестве носителей липида.
В заявке EP 418814, а также в J. Med. Chem. 34, 1912 (1991) описаны изопреноидфосфинилформиаты в качестве ингибиторов скваленсинтетазы.
В Biochem. Biophys. Res. Commun. 171, 458 (1990) описан липидный конъюгат противоретровирусного фоскарнета с пальмитилфосфоноформиатом, а в J. Med . Chem. 20, 660 (1977) показана атни-ВИЧ-активность (гексилокси)-гидроксифосфинилуксусной кислоты.
В принципе большую помощь оказало бы нахождение эффективных путей транспортировки терапевтических концентраций лекарственных средств в соответствующие органы-мишени или в клетки-мишени, в случае СПИДа, например, в клетки иммунной системы и лимфатической системы, считающиеся главным резервуаром вирусной репликации.
Фосфономуравьиная кислота (PFA = phosphonoformic acid) и фосфоноуксусная кислота (PAA = phosphonoacenic acid) показывают высокую антивирусную активность против вируса простого герпеса (HSV) 1 и 2, гриппа, вируса гепатита В (HBV), вируса ветряной оспы (VZV), вируса Эпштейна-Барра (EVB), а также ретровирусных инфекций.
Фосфономуравьиная и фосфоноуксусная кислоты и их производные представляют собой в некоторых случаях эффективную альтернативу, соответственно дополнение для нуклеозидов, так как они тормозят широкий спектр ДНК- и РНК-полимераз, а также RT ретровирусов с достаточной селективностью.
Сами фосфономуравьиная и фосфоноуксусная кислоты благодаря их сходству с пирофосфатом проявляют токсичность из-за накопления в костях.
Соединения согласно настоящему изобретению также обладают ценными фармакологическими свойствами. В частности, они пригодны для терапии и профилактики инфекций, вызываемых ДНК-вирусами, например, такими, как вирус простого герпеса, вирус цитомегалии, папова-вирусы, вирус ветряной оспы, вирусы гепатита или вирус Эпштейна-Барра, или РНК-вирусами, такими как вирусы Тога, или, в частности, ретровирусами, такими как онковирусы HTLV-I и II (вирус человеческого Т- клеточного лейкоза), а также лентивирусами Visna и вирус иммунодефицита человека ВИЧ-1 и 2.
Особенно пригодны соединения формулы I для лечения клинических проявлений ретровирусной ВИЧ-инфекции у человека, такой как длительная генерализованная лимфаденопатия (PGL), далеко зашедшая стадия родственного СПИДу комплекса (ARC) и полная клиническая картина СПИДа, а также ассоциированные вирусные инфекции цитомегалии (CMV) и простого герпеса (HSV).
В J. Infect. Dis. 172, 225 (1995) описано антивирусное/антиретровирусное действие фоскарнета (тринатриевая соль фосфономуравьиной кислоты/фосфономуравьиная кислота) против ВИЧ у пациентов с CMV-ретинитом.
Антивирусный эффект в мышином вирусе цитомегалии (CMV) описан в Antiviral Res. 26, 1 (1995).
Далее, в JAMA 273, 1457 (1995) описано лечение CMV-ретинита с использованием фосфономуравьиной кислоты.
Конъюгаты фосфономуравьиной и фосфоноуксусной кислот с 2', 3'-дидезокси-3'-тиацитидином, ингибирующие ВИЧ-1-репликацию, описаны в J. Med. Chem. 37, 2216 (1994), а в J. Pharm. Sci. 83, 1269 (1994) описаны сложные ацилоксиалкильные эфиры фоскарнета.
Особый интерес представляют заявка США 5194654, соответственно заявка PCT WO 94/13682. В них описаны липидные производные фосфонокарбоновых кислот и их применение в липосомах с образованием особенно стабильного липосомального комплекса. Наряду с исключительно широкой формулой изобретения, в качестве объекта заявки описаны 1-O-алкил-sn-глицеро-3-фосфонокарбоновые кислоты, которые особенно хорошо встраиваются в двойной липидный слой липосом. Заявленные алкильные остатки могут включать от 2 до 24 атомов углерода, однако дополнительно они не замещены.
В качестве примера описано и подтверждено данными об антивирусном действии лишь соединение 1-O-октацедил-sn-глицеро-3-фосфоноформиат (батил-фосфоноформиат). В проведенных исследованиях и при получении это соединение оказалось нестойким. В противоположность названным патентным заявкам это соединение применялось в качестве чистого вещества в растворе/суспензии, а не в липосомах.
Предлагаемые согласно настоящему изобретению соединения общей формулы I в тех же условиях устойчивы и обладают как in vitro, так и in vivo (модель в мыши) очевидными преимуществами.
Сложные эфиры липидфосфонокарбоновых кислот in vitro имеют такую же эффективность, что и соответствующие свободные карбоновые кислоты. In vivo они, напротив, проявляют однозначные преимущества, в частности, при оральном введении. Сложные эфиры карбоновых кислот формулы I подвержены меньшему разложению в кислой среде путем декарбоксилирования и, следовательно, обеспечивают связанную с этим лучшую биодоступность. В соответствии с этим назначаемая доза может быть еще уменьшена в сравнении со свободной карбоновой кислотой. Кроме того, улучшается проходимость через мембрану, например, при преодолении гематоэнцефалического барьера и при прохождении через клеточную мембрану в клетку-мишень. Так как сложный эфир карбоновой кислоты in vivo должен быть расщеплен эстеразами, то период полураспада в сыворотке удлиняется.
Соединения, заявляемые в настоящей заявке, представляют собой интересное дополнение к публикациям WO 94/13682 и US 5194654, при этом они не описаны в указанных заявках.
Соединения формулы I являются новыми. Наряду с более высокой устойчивостью (в субстанции и в растворе) заявляемых соединений они проявляют еще и лучшее действие в сравнении с известными липидными производными.
Неожиданным образом было установлено, что фармацевтически активные вещества формулы I обладают в сравнении с фармакологически активными свободными, соответственно немодифицированными веществами более широким терапевтическим спектром. Кроме того, они улучшают их время пребывания в организме, биодоступность и часто известную в качестве критически важного фактора проходимость через мембрану (например, гематоэнцефалический барьер, клеточную мембрану и т. д. ). Соединения формулы I служат таким образом в качестве системы-носителя для фармакологически активных веществ. Конъюгаты формулы I могут быть обозначены с точки зрения их функции как внутриклеточная система депонирования лекарства, система целеуказания лекарства и система доставки лекарства. Они приводят к тому, что фармакологически активное вещество после орального приема высвобождается внутри клетки, причем это высвобождение, предпочтительно, происходит не во всех клетках, органах или тканях организма, но лишь прицельно в таких клетках, которые содержат определенный фермент. Однако, особенно поразительно то, что расщепление происходит уже не во время транспорта субстрата в жидкостях организма, таких как кровь, сыворотка или лимфатическая жидкость, или в печени, но лишь на или в соответствующих клетках-мишенях. Таким образом предотвращается нежелательное выделение фосфонокарбоновой кислоты почкой или расщепление конъюгата в печени, благодаря чему значительно большая часть активного вещества транспортируется на или в соответствующие клетки-мишени. Такого рода клетками, как уже упомянуто выше, являются, в частности, физиологически или патофизиологически активированные клетки, которые могут применяться в качестве целевого объекта для доставки фармакологически активных веществ, например, лейкоциты крови, лимфоциты, макрофаги и другие клеточные популяции иммунологически лимфатической системы. При этом речь, в частности, идет об активированных клетках (например, макрофагах, гранулоцитах, лимфоцитах, лейкоцитах, тромбоцитах, моноцитах и т. д. ), которые играют патофизиологическую или симптоматическую роль в конкретном патологическом процессе. Кроме того, имеются в виду также клетки, инфицированные вирусами, бактериями, грибками или другими микроорганизмами.
Неожиданно было также установлено, что терапевтический спектр фармакологически активной фосфонокарбоновой кислоты и ее сложных эфиров значительно улучшается, если вещество связывается с очень специальной липидообразной молекулой-носителем. Полученный таким образом конъюгат служит новым активным веществом для получения лекарственных форм фармацевтического препарата. Общим итогом такого связывания является усиление действия фармацевтически активной фосфонокарбоновой кислоты in vivo, так как образовавшаяся система депонирования, доставки и транспорта лекарства вызывает локализацию фармакологически активного вещества в клетках-мишенях и тем самым улучшает эффективность и переносимость фармакологически активного вещества. Это означает, что, с одной стороны, назначаемая дозировка фармакологически активной фосфонокарбоновой кислоты может быть снижена, а с другой стороны, при одном и том же эффективном качестве достигается усиление фармакологического действия.
Фармакологически активная фосфонокарбоновая кислота высвобождается из конъюгата в результате ферментативного гидролиза конъюгата.
Конъюгаты формулы I имеют явные преимущества в сравнении с неконъюгированной фармацевтически активной фосфонокарбоновой кислотой, соответственно с ее сложными эфирами. Специфический, ковалентно связанный с фармацевтически активным веществом носитель улучшает биодоступность плохо ресорбируемых фармацевтически активных веществ, переносимость потенциально токсичных активных молекул, время пребывания быстро выводимых или метаболизируемых лекарственных средств и проникновение через мембрану соединений с плохой мембранной проходимостью (например, через гематоэнцефалический порог, клетки и т. д. ).
Ферментативное расщепление липидного компонента in vivo происходит, как правило, не в сыворотке, а лишь внутри клетки. Кроме того, компонент носителя улучшает своей лецитинообразной структурой, которая существенна для обсуждаемого эффекта, проникновение через мембрану фармацевтически активного вещества и способствует эффекту депонирования. Кроме того, желудочно-кишечная переносимость липидных конъюгатов многократно лучше в сравнении с чистой фармацевтически активной фосфонокарбоновой кислотой. И при резорбции липидный конъюгат лучше проникает через мембранные структуры и тем самым лучше преодолевает резорбционные барьеры. То же самое относится к проникновению, например, через гематоэнцефалический барьер.
Далее, благодаря лучшему связыванию конъюгата с плазменными и тканевыми белками улучшается распределение in vivo. В результате нормальной биотрансформации конъюгат первично окисляется из тиоэфира в сульфоксид, что, однако, в силу равноценного действия сульфоксида в сравнении с тиоэфиром не является недостатком. Медленное высвобождение фармацевтически активной фосфонокарбоновой кислоты из конъюгата обеспечивает низкий, однако, постоянный в течение длительного периода времени активный уровень вещества и тем самым улучшает его действие и/или устраняет побочные токсические эффекты. Высвободившееся фармацевтически активное вещество в форме монофосфата в силу своей большой гидрофильности уже больше не покидает клетку.
Периоды полураспада фармацевтически активного вещества как в организме в целом, так и в клетке и в органах благодаря конъюгации значительно увеличиваются в результате более продолжительного времени пребывания конъюгата в организме. Так как отсутствует расщепляющая активность в сыворотке и в различных органах, то почти не наблюдается или наблюдается лишь очень незначительная токсичность для костного мозга и органов. В особенности преимуществом является то, что конъюгаты формулы I специфически накапливаются в различных органах-мишенях, тканях или клетках.
Соединения формулы I могут применяться в качестве активных веществ для получения лекарственных средств, применяемых для лечения всех тех заболеваний, при которых требуются или полезны высокие уровни фармацевтически активных веществ в клетках, органах или тканях. Существенной предпосылкой для этой системы, обозначаемой как система депонирования, доставки и целеуказания лекарства, является то, что интересные в смысле планируемой терапии клетки имеют фермент расщепления, так что на первой стадии активное вещество связывается и затем транспортируется через клеточную мембрану внутрь клетки, причем расщепление активного вещества в физиологически активную фосфонокарбоновую кислоту происходит либо в основном одновременно с транспортом через клеточную мембрану, либо позже отчасти внутри клетки. Внутриклеточное расщепление происходит, в частности, в тех случаях, когда фермент расщепления локализован также и внутри клетки.
Подходящими клетками-мишенями являются, в частности, клетки иммунологически лимфатической системы (например, лейкоциты крови, моноциты, макрофаги, лимфоциты) или инфицированные клетки.
Неожиданно было также установлено, что соединения общей формулы I ингибируют размножение ДНК-, соответственно РНК-вирусов на стадии вирус-специфической ДНК-, соответственно РНК-транскрипции. Эти вещества могут оказывать, через ингибирование фермента обратной транскриптазы, влияние на размножение ретровирусов (ср. Proc. Natl. Acad. Sci. USA 83, 1911, 1986 или Nature 325, 773, 1987). Особый терапевтический интерес представляет ингибирующее действие на ВИЧ-вирус, вызывающий заболевание человека иммунодефицитом (СПИДом). Для лечения СПИДа в настоящее время допущен 3'-азидо-3'-дезокситимидин (DE-A-3608606). Однако токсичные побочные действия 3'-азидо-3'-дезокситимидина на костный мозг у приблизительно 50% пациентов требуют переливания крови. Соединения общей формулы I не имеют этих недостатков. Их противовирусное действие не сопряжено с цитотоксичностью в фармакологически релевантных дозах.
Соединения согласно настоящему изобретению и их фармацевтические препараты могут применяться также в комбинации с другими лекарственными средствами для лечения и профилактики вышеназванных инфекций. Примеры этих других лекарств включают средства, которые могут быть использованы для лечения и профилактики ВИЧ-инфекций или заболеваний, сопровождающих эту болезнь, такие как 3'-азидо-3'-дезокситимидин, 2', 3'-дидезоксинуклеозиды, такие как 2', 3'-дидезокситидин, 2', 3'-дидезоксиаденозин и 2', 3'-дидезоксиинозин, ациклические нуклеозиды (например, Acyclovir), ненуклеозидные RT-ингибиторы, ингибиторы протеазы, такие как, например, инвиразы, интерфероны, такие как, например, интерферон α, β, γ, цитокины и интерлейкины (например, интерлейкин 16), хемокины, такие, как, например, MIP1 α, MIP1 β, CC1, ингибиторы почечных выделений, такие как, например, пробеницид, ингибиторы транспорта нуклеозидов, такие как, например, дипиридамол, а также иммуномодуляторы, такие как, например, интерлейкин II или стимулирующие факторы, такие как, например, факторы, стимулирующие колонию гранулоцитов-макрофагов (GM-CSF), факторы, стимулирующие колонию гранулоцитов (G-CSF, нейтропоэтин), тромбопоэтин и тромбопоэтино-подобные факторы. Соединения согласно настоящему изобретению и другое лекарственное средство могут применяться каждое отдельно, возможно одновременно, соответственно в одной единственной лекарственной форме или в двух различных лекарственных формах, или в разное время, так что достигается синергетический эффект.
В качестве возможных солей соединения общей формулы I могут рассматриваться прежде всего соли щелочных и щелочноземельных металлов и аммония с карбоксильной и фосфонатной группой. В качестве солей щелочных металлов предпочтительны соли лития, натрия и калия. В качестве солей щелочноземельных металлов могут рассматриваться, в частности, соли магния и кальция. Под солями аммония согласно изобретению подразумеваются соли, содержащие ион аммония, который может быть замещен вплоть до четырехкратно алкильными остатками с 1-4 атомами углерода и/или аралкильными остатками, предпочтительно, бензильными остатками. Заместители могут быть при этом одинаковыми или разными.
В формуле I остаток R1, предпочтительно, обозначает линейную, насыщенную алкиленовую цепь, в которой "e" обозначает 5-12 атомов углерода. Cycl, предпочтительно, представляет собой циклогексильный или циклопентильный остаток, соответственно фенил, который необязательно замещен C1-C4 -алкилом или галогеном. X и Y независимо один от другого, предпочтительно, являются серой, сульфинилом, сульфонилом, кислородом или валентной связью. Особенно предпочтительно, когда X обозначает серу, а Y - кислород. Остаток -(CH2)e-Cycl, предпочтительно, находится в положении 3 C3 -основы, "e", особенно предпочтительно, равно 6-10, (CH2)e-Cycl, особенно предпочтительно, обозначает фенилгексил или циклогексил-гексил. Предпочтительными алкильными остатками для R2 являются линейные или разветвленные, насыщенные или ненасыщенные алкильные цепи с 8-12 атомами углерода. Особенно предпочтительными алкильными остатками для R2 являются нонильная, децильная, ундецильная или додецильная группа.
Особенно
предпочтительными сопряженными фосфонокарбоновыми кислотами в предлагаемых конъюгатах общей формулы I являются:
- фосфономуравьиная кислота,
- фосфоноуксусная кислота,
- фосфонопропионовая кислота.
Соединения общей формулы I могут быть получены тем, что
1.
соединение общей формулы II
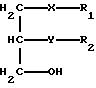
в которой R1, R2 и n имеют указанные значения, подвергают взаимодействию с соединением общей формулы III,
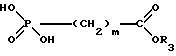
в которой m и R3 имеют вышеуказанное значение и R3, предпочтительно, представляет собой C1-C6-алкильный остаток, в присутствии, в случае необходимости замещенного хлорангидрида арилсульфоновой кислоты в органическом основании, соответственно в присутствии основания в инертном органическом растворителе, и затем сложный эфир карбоновой кислоты переводят щелочным омылением в производное формулы I, соответственно в его физиологически приемлемую соль,
или
2. получают смешанный ангидрид из соединения формулы III и хлорангидрида алкил- или арилсульфоновой кислоты и вводят его в реакцию со спиртом формулы II в присутствии основания в инертном органическом растворителе, соответственно непосредственно в основании, и затем, в случае необходимости подвергают сложный эфир карбоновой кислоты щелочному омылению,
или
3. подвергают фосфонокарбоновую кислоту формулы III, в которой R3 обозначает водород, со спиртом формулы II в присутствии основания и, в случае необходимости, замещенного хлорангидрида арилсульфоновой кислоты и, при желании, переводят в физиологически приемлемую соль или сложный эфир,
или
4. смешанный ангидрид из соединения формулы III, в которой R3 обозначает водород, и хлорангидрида алкил- или арилсульфоновой кислоты вводят в реакцию со спиртом формулы II в присутствии основания, соответственно в инертном органическом растворителе, и переводят конъюгат, в случае необходимости, в физиологически приемлемую соль,
или
5. дихлорангидриды фосфоновой кислоты общей формулы IV
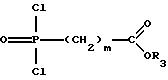
которые могут быть получены согласно Бонгле и сотр. (Synthetic Commun. 17, 1071 (1987)), исходя из сложного бис-триметилсилилового эфира фосфоновой кислоты путем последующего взаимодействия с оксалилхлоридом, подвергают взаимодействию со спиртом общей формулы II в присутствии основания в молярном соотношении 1: 1,
или
6. соединение формулы III переводят с помощью оксалилхлорида, как описано в Tetrahedron Letters 33, 7473 (1992), в соответствующий дихлорангидрид фосфоновой кислоты формулы IV, который затем подвергают взаимодействию со спиртом формулы II в присутствии основания в молярном соотношении 1: 1. Получаемый в качестве промежуточного продукта монохлорангидрид фосфоновой кислоты омыляют до полуэфира и переводят сложный эфир карбоновой кислоты путем щелочного омыления в производное формулы I, соответственно в его физиологически приемлемую соль.
Получение соединений общей формулы II описано в примерах в EP 0545966.
Содержащие соединения формулы I лекарственные средства для лечения, например, вирусных инфекций могут применяться в жидкой или твердой форме энтерально или парентерально. При этом приемлемы обычные формы применения, например, таблетки, капсулы, драже, сиропы, растворы или суспензии. В качестве инъекционной среды предпочтительно используют воду, содержащую обычные в инъекционных растворах добавки, такие как стабилизаторы, агенты растворения и буферные смеси. Такого рода добавками, например, являются тартратный и цитратный буфер, этанол, комплексообразователи, такие как этилендиаминтетрауксусная кислота и ее нетоксичные соли, высокомолекулярные полимеры, такие как жидкий полиэтиленоксид для регулирования вязкости.
Жидкие носители для инъекционных растворов должны быть стерильны, и предпочтительно, их разливают в ампулы. Твердыми носителями являются, например, крахмал, лактоза, маннит, метилцеллюлоза, тальк, высокодисперсные кремниевые кислоты, более высокомолекулярные жирные кислоты, такие как стеариновая кислота, желатин, агар-агар, фосфат кальция, стеарат магния, животные и растительные жиры, твердые высокомолекулярные полимеры, такие как полиэтиленгликоли, и т. д. Препараты, пригодные для орального применения, при необходимости, могут содержать вкусовые добавки и подсластители.
Дозировка может зависеть от различных факторов, таких как способ применения, вид заболевания, возраст или индивидуальное состояние пациента. Соединения согласно изобретению обычно применяют в количестве от 0,1 до 100 мг, предпочтительно, от 0,2 до 80 мг в день и на кг веса тела. Дневная доза, предпочтительно, распределяется на 2-5 приемов, причем при каждом приеме принимают по 1-2 таблетке с содержанием активного вещества от 0,5 до 500 мг. Таблетки могут быть также ретардированы, благодаря чему количество приемов лекарства в день уменьшается до 1-3. Содержание активного вещества в ретардированных таблетках может составлять от 2 до 1000 мг. Активное вещество может вводиться также путем продолжительного вливания, причем, как правило, достаточны количества от 5 до 5000 мг в день.
Помимо соединений, названных в примерах и
получающихся в результате комбинации всех указанных в формуле изобретения значений заместителей, в объеме настоящего изобретения могут
рассматриваться следующие соединения формулы I:
1.
((3-(4-Хлорфенил)гексилмеркапто)-2-децилокси)пропокси)- гидроксифосфинилмуравьиная кислота.
2. Изопропиловый эфир ((3-(6-циклогексилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты.
3. [3-(Фенил)окси-гексилмеркапто)-2-децилокси] пропокси)- гидроксифосфинилмуравьиная кислота.
4. [3-(Фенил)гептилмеркапто)-2-децилокси] пропокси)- гидроксифосфинилмуравьиная кислота.
5. Пентиловый эфир ((3-(6-циклогексилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты.
6. [3-(м-Этилфенил)децилмеркапто-2-децилокси] пропокси)- гидроксифосфинилмуравьиная кислота.
7. [3-(п-трет-Бутилфенил)октилмеркапто-2-децилокси] пропокси- гидроксифосфинилмуравьиная кислота.
8. [3-(Циклогексил)гептилмеркапто-2-децилокси] пропокси- гидроксифосфинилмуравьиная кислота.
9. [3-(Циклопентил)нонилмеркапто-2-децилокси] пропокси- гидроксифосфинилмуравьиная кислота.
10. [3-(Циклогептил)октилмеркапто-2-децилокси] пропокси- гидроксифосфинилмуравьиная кислота.
11. [3-(Циклогексил)окси-пентилмеркапто-2-децилокси] пропокси- гидроксифосфинилмуравьиная кислота.
12. [3-(Циклогексил)меркапто-пентилмеркапто-2-децилокси] пропокси- гидроксифосфинилмуравьиная кислота.
13. [3-(Фенил)ундецилмеркапто-2-децилокси] пропокси- гидроксифосфинилмуравьиная кислота.
14. [3-Додецилмеркапто-2-(фенил)гексилмеркапто] пропокси- гидроксифосфинилмуравьиная кислота.
15. [3-Децилокси-2-(циклогексил)гексилмеркапто] пропокси- гидроксифосфинилмуравьиная кислота.
16. [3-(п-Хлорфенил)гексилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
17. [3-(п-трет-Бутилфенил)окси-октилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
18. Бензиловый эфир ((3-(6-циклогексилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты.
19. [3-(Фенил)гептилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
20. [3-(п-Хлорфенил)окси-пентилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
21. [3-(м-Этилфенил)децилмеркапто-2-децилокси] пропокси)- гидроксифосфинилуксусная кислота.
22. [3-(п-трет-Бутилфенил)-октилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
23. [3-(Циклогексил)гептилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
24. [3-(Циклопентил)нонилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
25. [3-(Циклогептил)октилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
26. [3-(Циклогексил)окси-пентилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
27. [3-(Циклогексил)меркапто-пентилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
28. [3-(Фенил)ундецилмеркапто-2-децилокси] пропокси- гидроксифосфинилуксусная кислота.
29. [3-Додецилмеркапто-2-(фенил)гексилмеркапто] пропокси- гидроксифосфинилуксусная кислота.
30. [3-Децилокси-2-(циклогексил)гексилмеркапто] пропокси- гидроксифосфинилуксусная кислота.
31. [3-(п-Хлорфенил)гексилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
32. [3-(п-трет-Бутилфенил)окси-оксилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
33. [3-(Фенил)окси-гексилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
34. [3-(Фенил)гептилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
35. [3-(п-Хлорфенил)окси-пентилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
36. [3-(м-Этилфенил)децилмеркапто-2-децилокси] пропокси)- гидроксифосфинилпропионовая кислота.
37. [3-(п-трет-Бутилфенил)октилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
38. [3-(Циклогексил)гептилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
39. [3-(Циклопентил)нонилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
40. [3-(Циклогептил)октилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
41. [3-(Циклогексил)окси-пентилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
42. [3-(Циклогексил)меркапто-пентилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
43. [3-(Фенил)ундецилмеркапто-2-децилокси] пропокси- гидроксифосфинилпропионовая кислота.
44. [3-Додецилмеркапто-2-(фенил)гексилмеркапто] пропокси- фосфинилпропионовая кислота.
45. [3-Децилокси-2-(циклогексил)гексилмеркапто] пропокси- гидроксифосфинилпропионовая кислота.
46. Бутиловый эфир ((3-(6-циклогексилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты.
47. Этиловый эфир ((3-(6-фенилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты.
48. Пропиловый эфир ((3-(6-фенилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты.
49. трет-Бутиловый эфир ((3-(6-фенилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты.
50. (2-Диметиламино)этиловый эфир ((3-(6-фенилгексилмеркапто)-2-децилокси)- пропокси)- гидроксифосфинилмуравьиной кислоты.
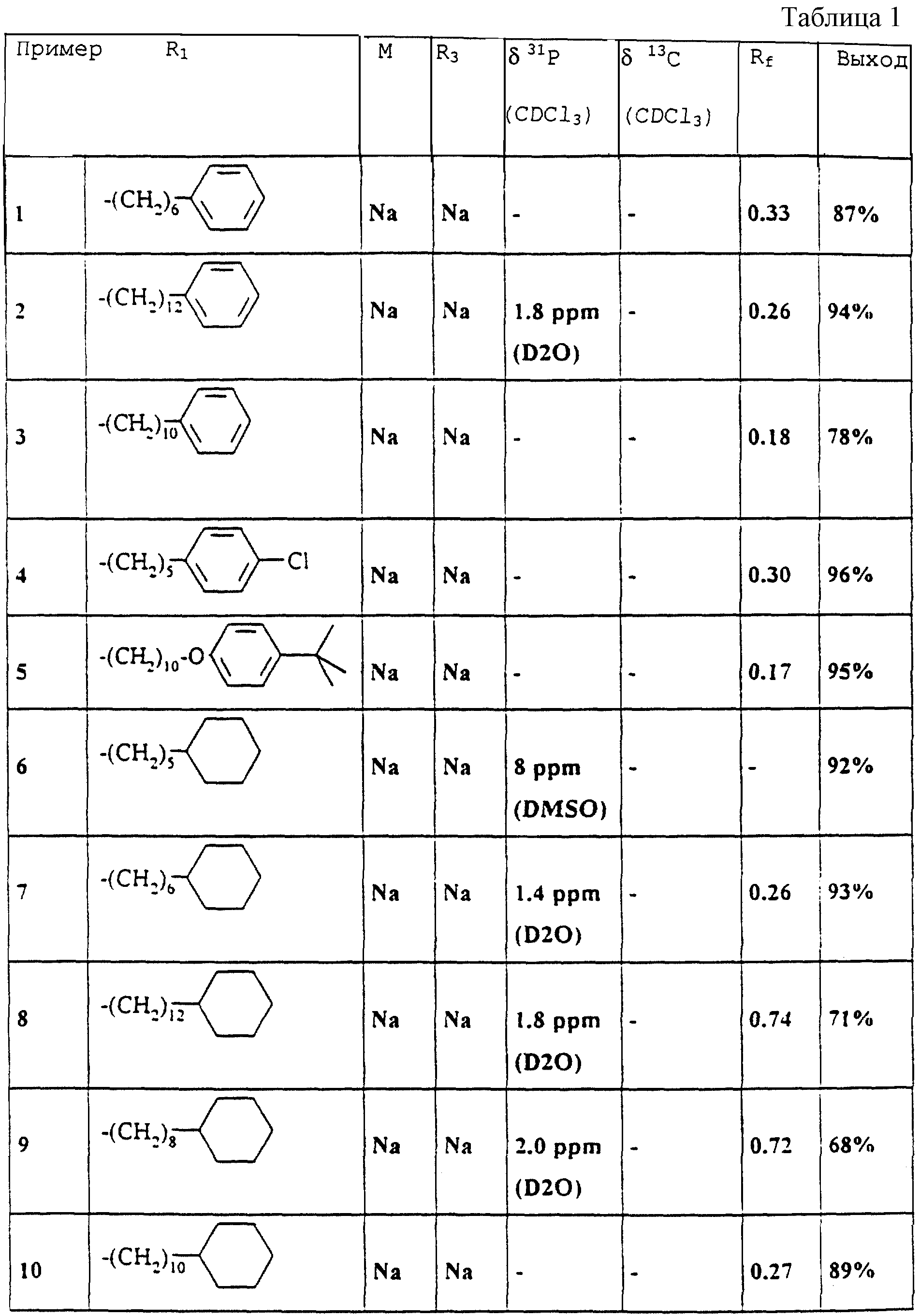
Пример 1
R, S-((3-(6-фенилгексилмеркапто)
-2-децилокси)-пропокси)- гидроксифосфинилмуравьиная кислота, двунатровая соль 1 (Ph6S10OP-PFA)
6-Фенил-1-гексантиол 13
В атмосфере азота добавляют 15,0 г (62,2 ммоля)
1-бром-6-фенилгексана (описан в описании к выложенной международной заявке PCT/EP95/04413), растворенного в 40 мл этанола, к раствору из 7,10 г (93,3 моля) тиомочевины в 30 мл этанола. После
выдерживания в течение 7 ч при температуре дефлегмации остужают до комнатной температуры, смешивают с 33 мл концентрированного аммиака и нагревают 4 ч с обратным холодильником. Затем подкисляют 15 мл
концентрированной HCl до pH 1. Экстрагируют трижды каждый раз с помощью 200 мл эфира, промывают водой и насыщенным раствором хлорида натрия, сушат над сульфатом магния и удаляют растворитель в
вакууме. Остаток растворяют в дихлорметане, твердое вещество отсасывают, дополнительно промывают дихлорметаном и фильтрат концентрируют в вакууме. Получают 9,80 г (82%) 13 в виде бесцветного
масла.
R, S-2-Децилокси-3- (6-фенилгексилмеркапто)-1-пропилбензоат 14
В атмосфере азота загружают 9,60 г (49,4 ммоля) 13 и 6,80 г (49,4 ммоля) карбоната калия в 100 мл
метилэтилкетона,
перемешивают 15 мин и смешивают затем с 19,7 г (49,4 ммоля) 3-бром-2-децилокси-1-пропил-бензоата 12 (EP 0545966) и с кристаллом иодида калия. После добавления 5 мл диметилформамида
перемешивают 48 ч
при комнатной температуре. Карбонат калия отсасывают, осадок промывают гептаном и фильтрат концентрируют в вакууме. Остаток растворяют в воде, экстрагируют гептаном и промывают
органическую фазу 0,5
н. NaOH, нейтрализуют водой, сушат над сульфатом магния и концентрируют. Получают 25,6 г (100%) 14, который применяют без дальнейшей очистки для синтеза 15.
R,
S-2-Децилокси-3- (6-фенилгексилмеркапто)-1- пропанол 15
В атмосфере азота смесь из 25,5 г (49,7 ммоля) 14, 30 мл этанола и 12 мл (60,0 ммоля) 5 н. NaOH перемешивают в общей сложности 48 ч
при
комнатной температуре. Концентрируют в вакууме, растворяют в воде, экстрагируют дихлорметаном, промывают 1 н. NaOH, водой, сушат над сульфатом магния и удаляют растворитель в вакууме. Получают 18,
9 г
(93%) продукта-сырца. Очистку производят посредством флэш-хроматографии на силикагеле (подвижная фаза: гептан/эфир уксусной кислоты 7: 1), причем выделяют 12,8 г (63%) 15 в виде бесцветного
масла.
Сложный метиловый эфир дихлорфосфинилмуравьиной кислоты 16
В атмосфере азота 28,2 г (99,2 ммоля) сложного метилового эфира
ди-(триметилсилилокси)-гидроксифосфинилмуравьиной
кислоты (Synthetic Commun. 17, 1071 (1987), Tetrahedron Lett. 33, 7473) растворяют в 150 мл дихлорметана и 5 каплях диметилформамида и добавляют по
каплям при 0oC в течение 30 мин 37,8 г (0,
297 ммоля) оксалилхлорида. После 2 ч перемешивания при комнатной температуре растворитель удаляют в вакууме и перегоняют в высоком вакууме.
Получают 12,1 г (69%) 16, Tкип = 42-45o
C/0,19 мбар.
Сложный метиловый эфир R, S-((3-(6-фенилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной
кислоты 17 (пример 12.21)
В атмосфере азота 1,50 г (8,
48 ммоля) сложного метилового эфира дихлорфосфинилмуравьиной кислоты 16 растворяют в 15 мл дихлорметана и охлаждают до 5oC. В
течение 15 мин добавляют по каплям смесь из 3,50 г (8,48 ммоля)
R, S-2-децилокси-3-(6-фенилгексилмеркапто)-1-пропанола 15 и 900 мг (8,48 ммоля) триэтиламина, растворенные в 20 мл дихлорметана, при
этом температура поднимается до 10oC. После 30 мин
перемешивания при 10oC перемешивают еще 3 ч при комнатной температуре и затем выливают в раствор из 7,85 мл 1 н. NaOH и 200 мл
ледяной воды. Дважды экстрагируют каждый раз с помощью 100 мл
дихлорметана, промывают комбинированные органические фазы водой и сушат над сульфатом магния. После удаления растворителя в вакууме
получают 4,3 г (95%) масла, которое очищают посредством
флэш-хроматографии на силикагеле. После элюирования неизрасходованного 15 (1,35 г, подвижная среда: эфир уксусной кислоты) и проявления
дихлорметан/метанолом 10: 1 получают в общей сложности 2,52 г
(56%) 17 (пример 21) в виде бесцветного масла.
В атмосфере азота смешивают смесь из 2,50 г (4,71 ммоля) 17, 20 мл этанола и 20 мл тетрагидрофурана с 4,7 мл (14,1 ммоля) 3 н. NaOH. Перемешивают 2 ч при комнатной температуре, удаляют растворитель на ротационном испарителе, остаток растворяют в 250 мл воды и дважды экстрагируют каждый раз с помощью 50 мл трет-бутилметилового эфира. Доводят pH водной фазы до значения 8,5 с помощью 1 н. NaOH и удаляют воду путем сушки вымораживанием. Получают 2,3 г (87%) 1, Tпл = 212-214oC.
Пример 2
Динатриевая соль R, S-((3-(12-фенилдодецилмеркапто)-2-децилокси)- пропокси)-гидроксифосфинилмуравьиной кислоты 2 (Ph12S10OP-PFA)
12-Фенил-1-додекантиол
18 Как при получении 13
(пример 1), 15,0 г (46,1 ммоля) 1-бром-12-фенил-додекана подвергают взаимодействию с 5,3 г (69,2 ммоля) тиомочевины. Получают 11,1 г
(87%) 18.
R,
S-2-Децилокси-3-(12-фенилдодецилтио)-пропил-бензоат 19
Из 10,8 г (38,8 ммоля) и 15,3 г (38,8 ммоля) 12 получают 20,0 г (92%) 19.
R,
S-2-Децилокси-3-(12-фенилдодецилтио)-пропанол 20
Гидролизом 4,40 г (7,37 ммоля) 19 с 3,0 мл (15 ммоля) 5 н. NaOH получают 3,08 г (85%) 20 в виде бесцветного масла.
Аналогично примеру 1 из 1,90 г (9,95 ммоля) 16 и 4,90 г (9,95 ммоля) R, S-2-децилокси-3- (12-фенилдодецилмеркапто)-1- пропанола 20 получают 3,39 г (52%) сложного метилового эфира R, S-((3-(12-фенилдодецилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты (пример 12.22) в виде бесцветного масла. После омыления раствором едкого натра (аналогично примеру 1) получают 2,90 г (94%) 2 с Tпл 224oC.
Пример 3
Динатриевая соль R, S-((3-(10-фенилдецилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной
кислоты 3 (Ph10S10OP-PFA)
Аналогично примеру 1 из 1,10 г (6,29 ммоля) 16 и 2,92 г (6,29 ммоля) R, S-2-децилокси-3- (10-фенилдецилмеркапто)-1- пропанола получают 0,85 г (23%) сложного
метилового эфира R, S-((3-(10-фенилдецилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты (пример 12.23) в виде бесцветной смолы. После омыления раствором едкого натра (аналогично
примеру 1) получают 0,71 г (79%) 3 с Tпл 219-220oC.
Пример 4
Динатриевая соль R, S-((3-(5-(4-хлорфенил)- пентилмеркапто)-2-децилокси)
-пропокси)- гидроксифосфинилмуравьиной кислоты 4 (CIPh5S10OP-PFA).
Аналогично примеру 1 из 1,10 г (6,29 ммоля) 16 и 2,70 г (6,20 ммоля) R, S-2-децилокси-3- (5-(4-хлорфенил)-пентилмеркапто)- 1-пропанола получают 3,30 г (97%) сложного метилового эфира R, S-((3-(5-(4-хлорфенил)-пентилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты (пример 12.24) в виде бесцветного масла. После омыления 2,80 г эфира раствором едкого натра (аналогично примеру 1) получают 2,90 г (96%) 4 с Tпл 170-172oC.
Пример 5
Динатриевая соль R,
S-((3-(10-(4-трет-бутилфенокси)-децилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиная кислота 5 (tBuPhO10S10OP-PFA).
Аналогично примеру 1 из 1,10 г (6,20 ммоля) 16 и 3,34 г (6,20 ммоля) R, S-2-децилокси-3- (5-(4-трет-бутилфенокси)-децилмеркапто)-1- пропанола получают 1,92 г (58%) сложного метилового эфира R, S-((3-(5-(4-трет-бутилфенокси) -децилмеркапто)-2-децилокси) -пропокси)- гидроксифосфинилмуравьиной кислоты (пример 12.25) в виде бесцветного масла. После омыления раствором едкого натра (аналогично примеру 1) получают 1,90 г (95%) 5.
Пример
6
Динатриевая соль R, S-((3-(5-циклогексилпентилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты 6 (CH5S10OP-PFA)
Аналогично примеру 1 из 1,10 г (6,20 ммоля) 16
и
2,48 г (6,20 ммоля) R, S-2-децилокси-3-(5-циклогексилпентилмеркапто)-1-пропанола получают 2,60 г (81%) сложного метилового эфира R,
S-((3-(5-циклогексилпентилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты (пример 12.26) в виде бесцветного масла. После омыления раствором едкого натра (аналогично примеру 1)
получают 1,50 г (92%) 6 с Tпл 217-219oC.
Пример 7
Динатриевая соль R,
S-((3-(6-циклогексилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты 7 (CH6S10OP-PFA)
Аналогично примеру 1 из 1,30 г (7,30 ммоля) 16 и 3,00 г (7,30 ммоля) R,
S-2-децилокси-3- (6-циклогексилгексилмеркапто)-1- пропанола получают 2,80 г (72%) сложного метилового эфира R, S-((3-(6-циклогексилгексилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной
кислоты (пример 12.27) в виде бесцветного масла. После омыления 2,02 г этого эфира раствором едкого натра (аналогично примеру 1) получают 2,00 г (93%) 7 с Tпл 199-202oC.
Пример 8
Динатриевая соль R, S-((3- (12-циклогексилдодецилмеркапто)-2-децилокси) -пропокси)- гидроксифосфинилмуравьиной кислоты 8 (CH12S10OP-PFA)
Аналогично примеру 1 из 0,
55 г (3,10 ммоля) 16 и 1,50 г (3,10 ммоля) R, S-2-децилокси-3-(12-циклогексилдодецилмеркапто)-1-пропанола получают 1,70 г (81%) сложного метилового эфира R, S-((3-(12-циклогексилдодецилмеркапто)
-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты (пример 12.28) в виде бесцветного масла. После омыления 1,50 г этого эфира раствором едкого натра (аналогично примеру 1) получают 1,10 г
(71%) 8 с Tпл 105-107oC.
Пример 9
Динатриевая соль R, S-((3-(8-циклогексилоктилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты 9
(CH8S10OP-PFA)
Аналогично примеру 1 из 1,10 г (6,29 ммоля) 16 и 2,75 г (6,29 ммоля) R, S-2-децилокси-3-(8-циклогексилоктилмеркапто)-1-пропанола получают 2,40 г (68%) сложного метилового
эфира
R, S-((3-(8-циклогексилоктилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты (пример 12.29) в виде бесцветного масла. После омыления 1,37 г этого эфира раствором едкого
натра
(аналогично примеру 1) получают 0,95 г (68%) 9, разложение при > 250oC.
Пример 10
Динатриевая соль R,
S-((3-(10-циклогексилдецилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты 10 (CH10S10OP-PFA)
Аналогично примеру 1 из 1,10 г (6,29 ммоля) 16 и 2,96 г (6,29 ммоля) R,
S-2-децилокси-3-(10-циклогексилдецилмеркапто)-1-пропанола получают 1,15 г (37%) сложного метилового эфира R, S-((3-(10-циклогексилдецилмеркапто)-2-децилокси)-пропокси)- гидроксифосфинилмуравьиной
кислоты (пример 12.30) в виде бесцветного масла. После омыления раствором едкого натра (аналогично примеру 1) получают 1,06 г (89%) 10 с Tпл 179-181oC.
Пример
11
Динатриевая соль R, S-((3-(5-(4-хлорфенокси) -пентилмеркапто)-2-децилокси) -пропокси)- гидроксифосфинилмуравьиной кислоты 11 (CIPhO5S10OP-PFA)
Аналогично примеру 1 из 1,10 г (6,
29
ммоля) 16 и 2,80 г (6,29 ммоля) R, S-2- децилокси-3-(5-(4-хлорфенокси)-пентилмеркапто)-1-пропанола получают 1,27 г (36%) сложного метилового эфира R, S-((3-(5-(4-хлорфенокси)
-пентилмеркапто)-2-децилокси)- пропокси)- гидроксифосфинилмуравьиной кислоты (пример 12.31) в виде бесцветного масла. После омыления раствором едкого натра (аналогично примеру 1) получают 1,34 г
(99%)
11 с консистенцией 7, Tпл 175-177oC.
Пример 12
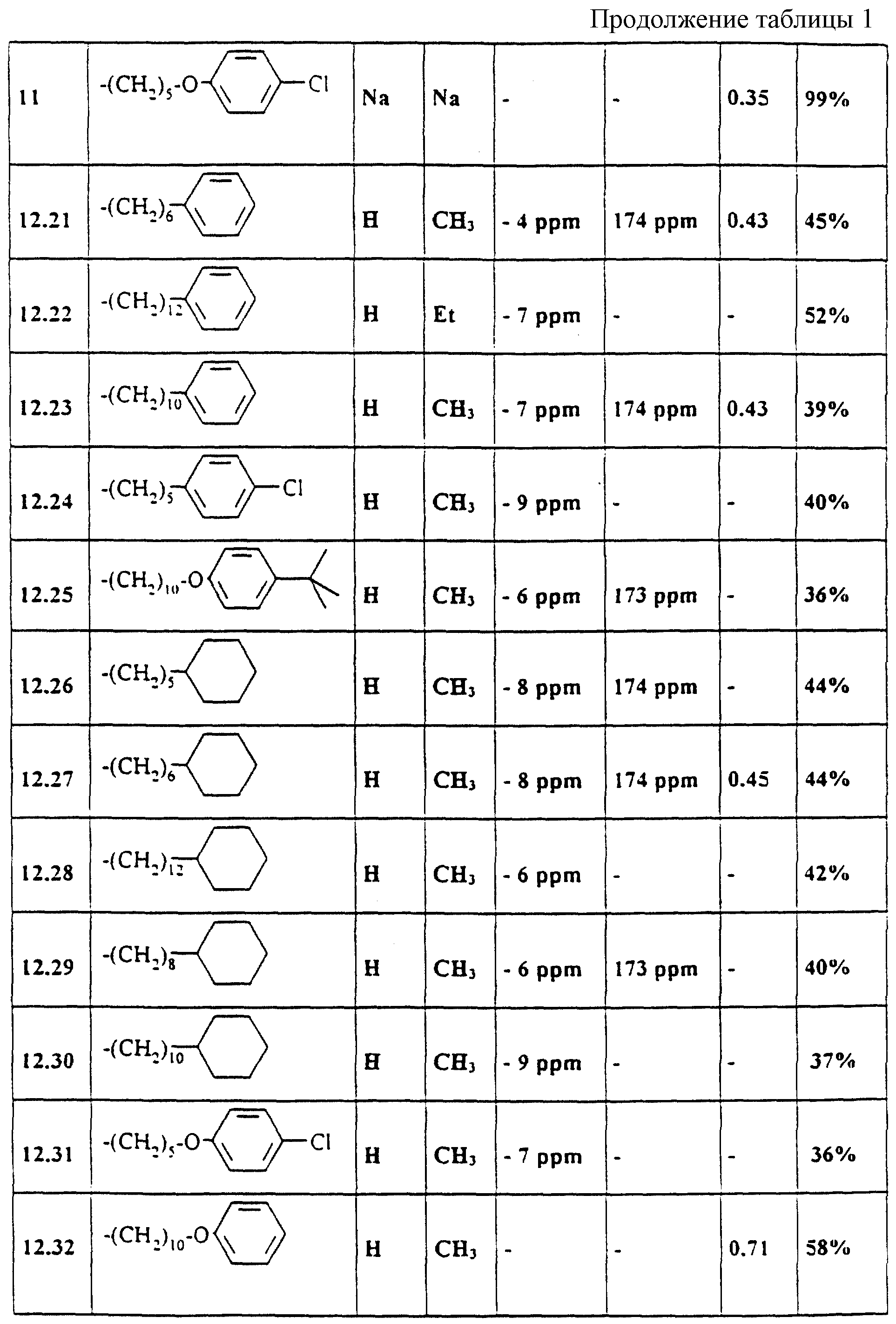
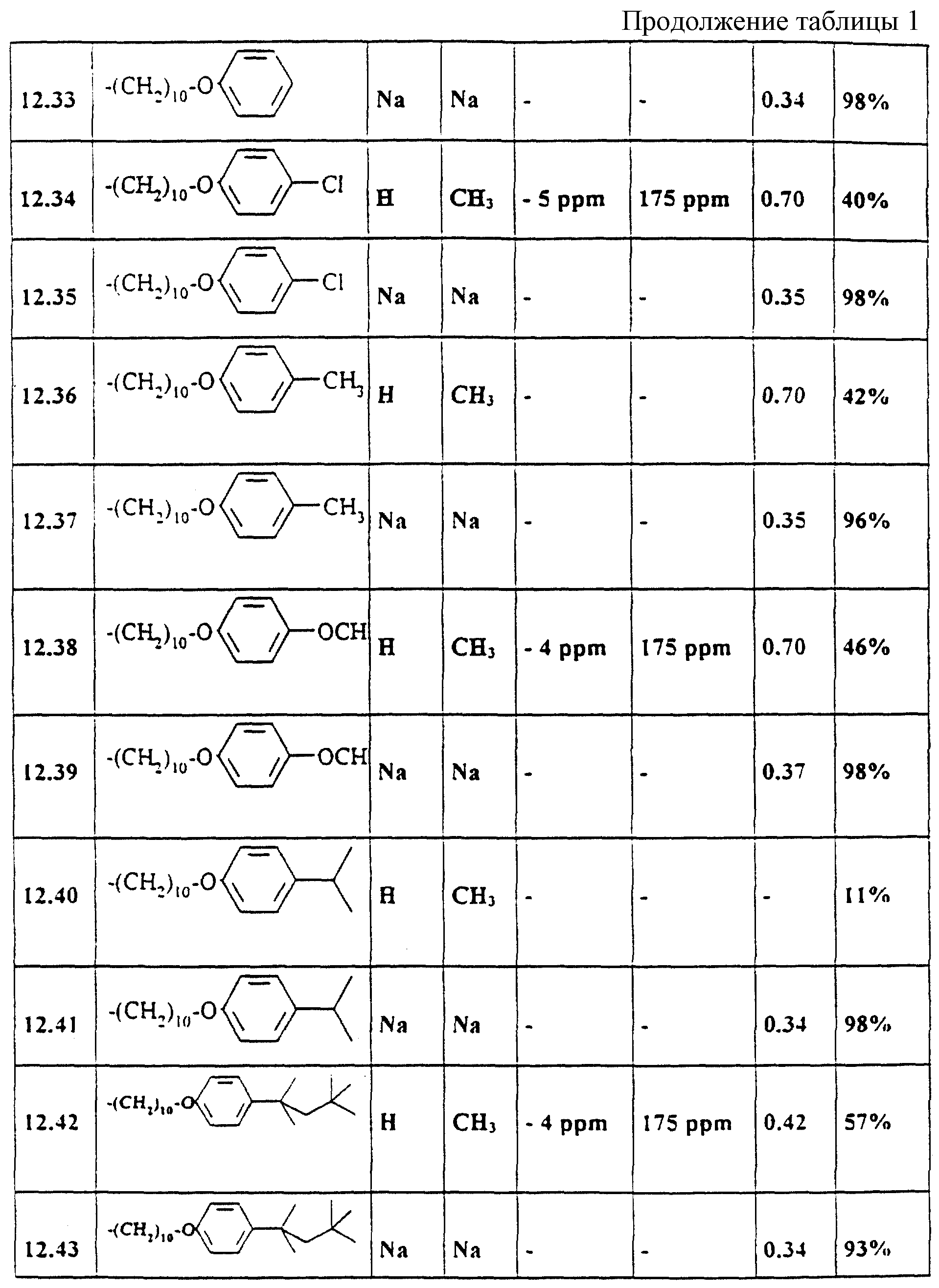
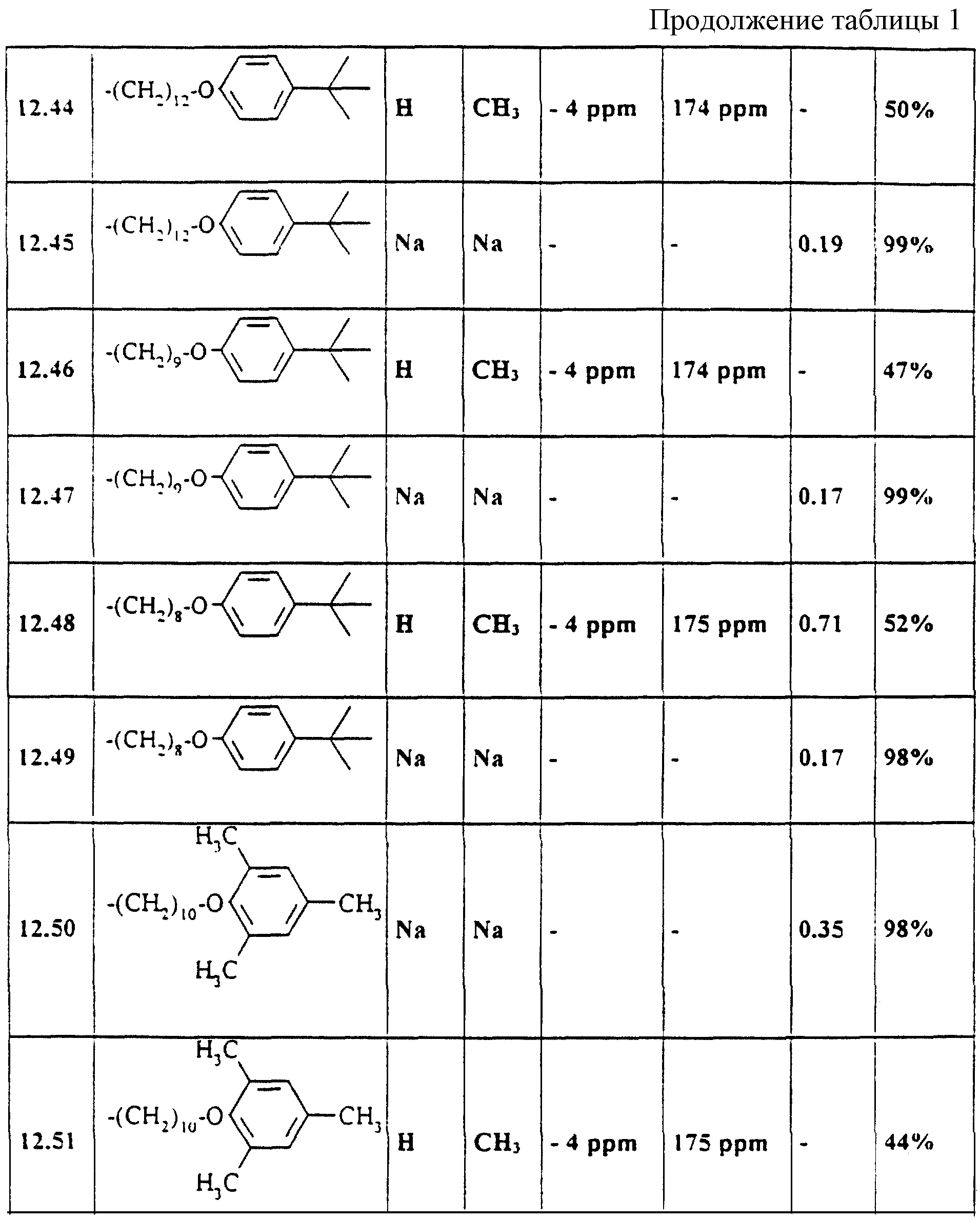
Аналогично примерам 1-11 получают содержащиеся в таблице 1 примеры по применению 21-51 (см. в
конце
описания).
Выбранные ЯМР-данные и значения Rf примеров 1-11 и 12.21-12.51
Указанные значения Rf были получены на готовых силикагелевых пластинках 60F254DC фирмы Merck, Дармштадт (материал N 5715) при массе покрытия 10 мкг/мкл с текучей средой 36 (изопропанол/бутилацетат/вода/аммиак 50: 30: 15: 5, об. /об. ). Детектирование производилось с помощью распыляемого реагента HCl перхлорная кислота. Указанные13C-смещения относятся к карбонильному атому углерода (дуплет, J = 250 Гц).
Пример 13
Испытание эфирлипидо-фоскарнетных конъюгатов в модели мышиного вируса цитомегалии (MCMV) in vivo
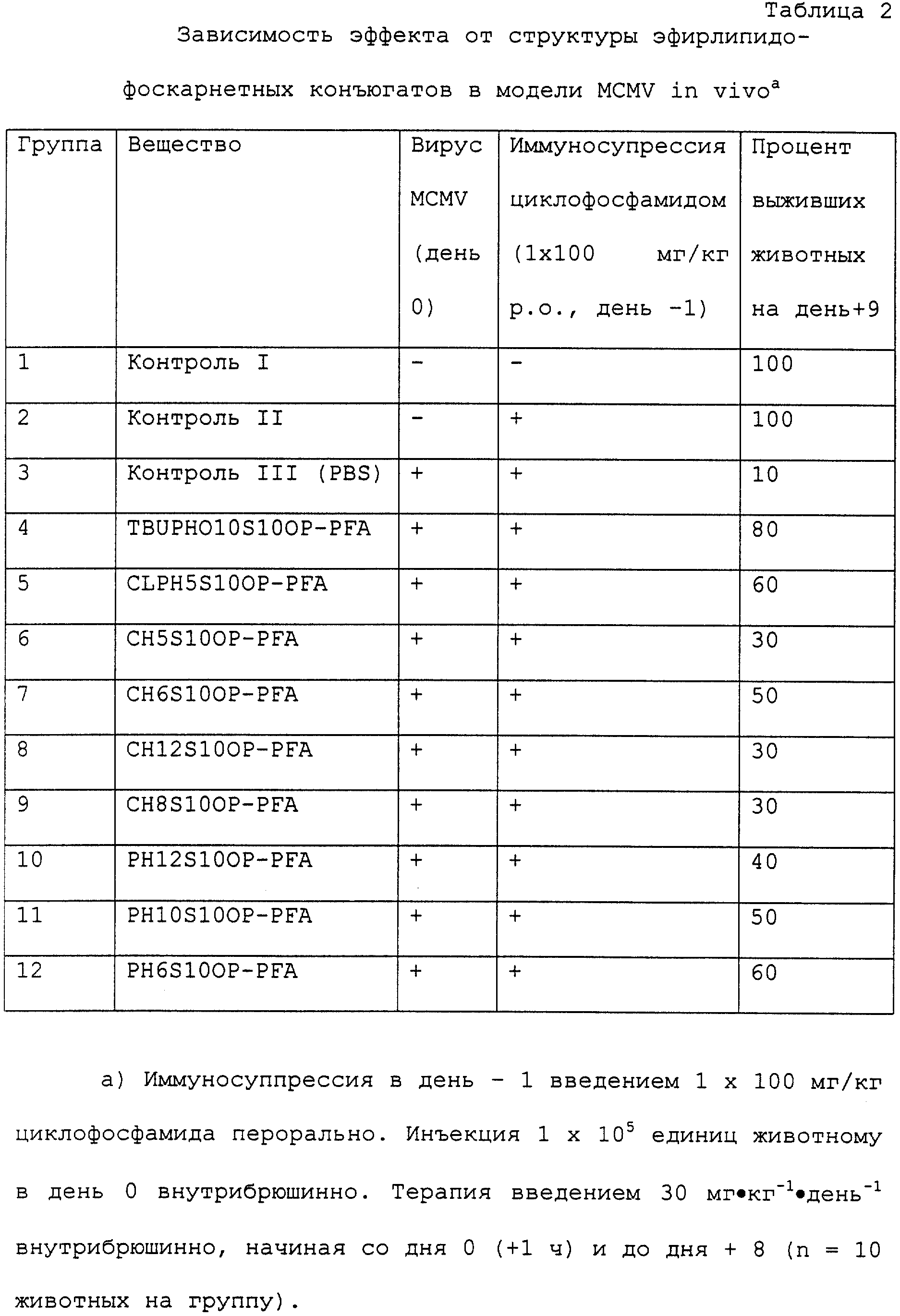
Испытывались различные эфирлипидо-фоскарнетные конъюгаты, различающиеся долей эфирлипида в молекуле, в модели MCMV in vivo. При этом определялась, в сравнении с контрольными животными, которым давали
плацебо, выживаемость после заражения вирусом MCMV на день + 9 после заражения (таблица 2).
Животных заражали (за исключением контроля I и II), вводя каждому животному по 2• 105 бляшкообразующих единиц (PFU) внутрибрюшинно в день 0. У всех животных (за исключением контроля I) в день - 1 подавляли иммунные реакции с помощью 100 мг/кг циклофосфамида (перорально). Всем подопытным животным ежедневно вводили внутрибрюшинно одной дозой в 30 мг•кг-1•день-1, начиная со дня 0 (+1 ч после заражения) и кончая днем +8. Из каждой группы брали по 10 животных. Число выживших животных определяли на день +9.
Как следует из табл. 2, в контрольной группе III (группа 3), которой давали PBS-плацебо (= фосфатносолевой буферный раствор), дожило до дня +9 лишь 1 животное из 10-ти. В этом тесте проявили эффективность все испытанные соединения. Что касается времени выживания, то для испытанных веществ было получено характерное соотношение между структурой и эффектом, причем наиболее активными соединениями были TBUPHO10S10OP-PFA, CLPH5S10OP-PFA и PH6S10OP-PFA.
Определение
антивирусной активности
конъюгатов производных фоскарнета
1. Материалы и методы
1.1 Анализ вирусных синцитиальных бляшек и определение концентрации ингибирования (IC50
)
HIV-1
Тест на вирисные синцитиальные бляшки использовался для оценки соединений на активность в отношении инфекции HIV-1, как описано Kucera 1990, AIDS Research & Human
Retroviruses 6, 491-501.
Монослой CEM-SS (Т-лимфоцитов) был приготовлен путем дозирования клеток в количестве 50.000 в 50 мкл среды RPMI-1640 в отсутствие сыворотки в каждую лунку, обработанную
поли-L-лизином, 96-луночного
микротитрационного планшета. Монослой клеток, сформированный после 30 минут инкубирования, при 37oC был инкулирован 30-60 единицами HIV-1, способствующими
образованию бляшек, в среде
RPMI-1640, способствующей росту. Через 120 минут адсорбции вируса, инфицированный монослой клеток был покрыт 100 мкл среды роста с добавлением серийных разведений
тестируемого соединения. Наибольшая
концентрация тестируемого соединения была вдвое или более ниже, чем IC50 для токсичности клеток. Контроль включал серийные разведения AZT. Все бляшки
были инкубированы при 37oC
во влажной атмосфере, содержащей 5% CO2 и 95% воздуха. После трех дней инкубирования добавляли второй слой сверху в количестве 100 мкл среды роста с
разбавлением тестируемого или
контрольного соединения и инкубировали при 37oC в течение 5 дней. Бляшки подсчитывали с помощью линз с 10-кратным увеличением и слабым микроскопом. Величину
IC50 для антивирусной
активности в отношении вируса HIV-1 рассчитывали с помощью компьютерной программы, анализирующей антивирусные данные в соответствии с методикой, описанной Chou с
сотр. и Piantadosi с сотр. в J. Med.
Chem. , 1991, 34, 1408-1414.
Вирус гепатита B (HBV)
Соединения проверяли на активность в отношении вируса HBV, используя линию клеток
гептобластомы человека, инфицированную
HBV (HepG2 2.2.15), как описано Korba и Gerin 1992, Antiviral Research, 55-70.
Клетки в количестве 5•104 в фазе логарифмического роста помещали в 96-луночный микротитрационный планшет, используя DMEM, 10%-ную эмбриональную бычью сыворотку, L-глютамин, пенициллин и стрептомицин (среда роста). Через 2 часа инкубирования при 37oC верхний слой среды был удален и помещен в свежую среду роста, содержащую серийные концентрации тестируемого соединения. Каждая концентрация тестируемого соединения определялась в трех лунках. Верхний слой среды перемещали ежедневно, исключая уикенд, в свежую среду, содержащую те же концентрации тестируемого соединения в общей сложности в течение 10 дней. Через 10 дней верхний слой среды, содержащей частицы HBV, анализировали на образование антигена "e" вируса гепатита B, используя коммерчески доступный EIA (INCSTAR Corp. Stilwater, MN). Монослои клеток были исследованы на IC50 с использованием теста нейтральности к красной краске.
2. Результаты
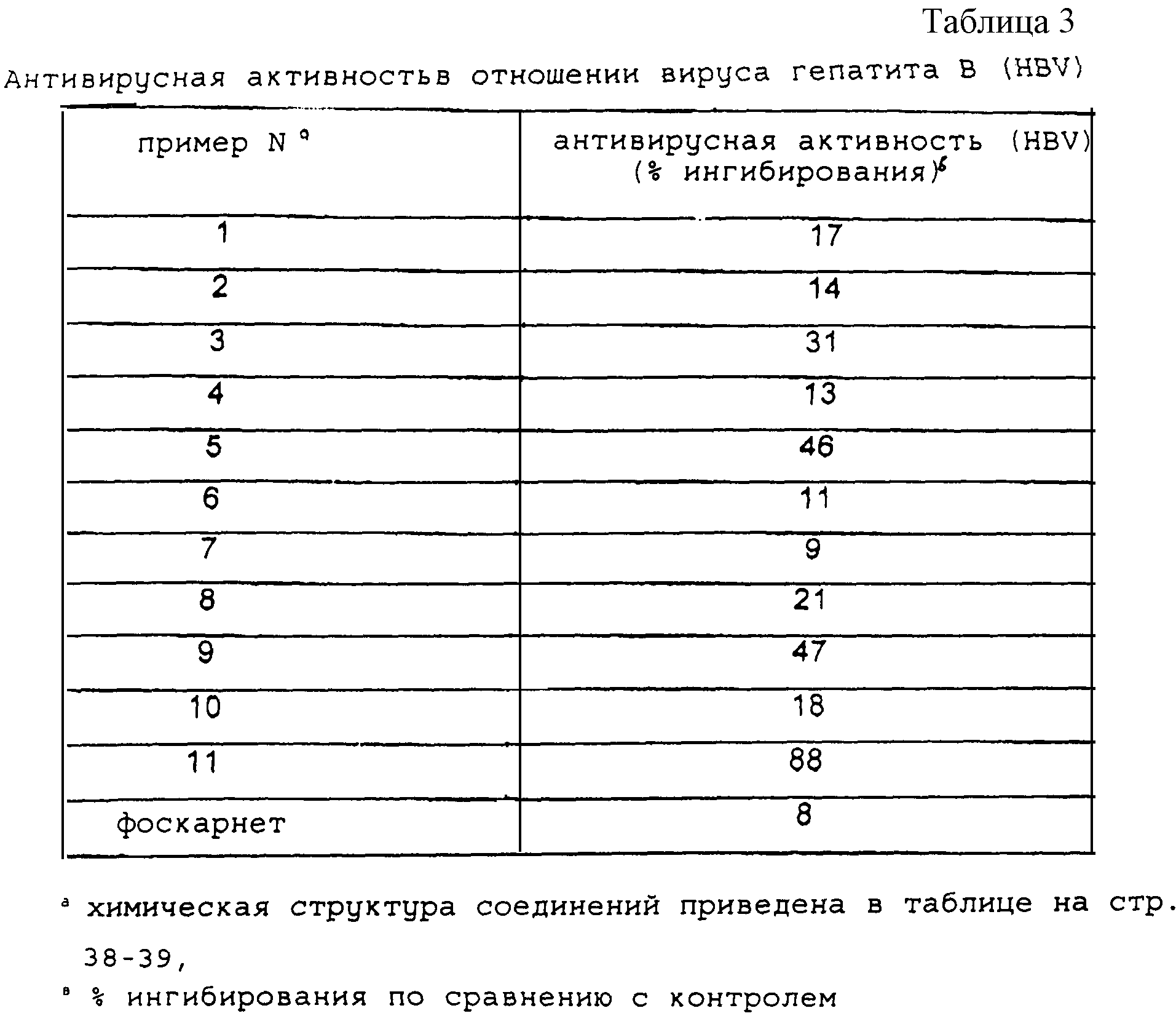
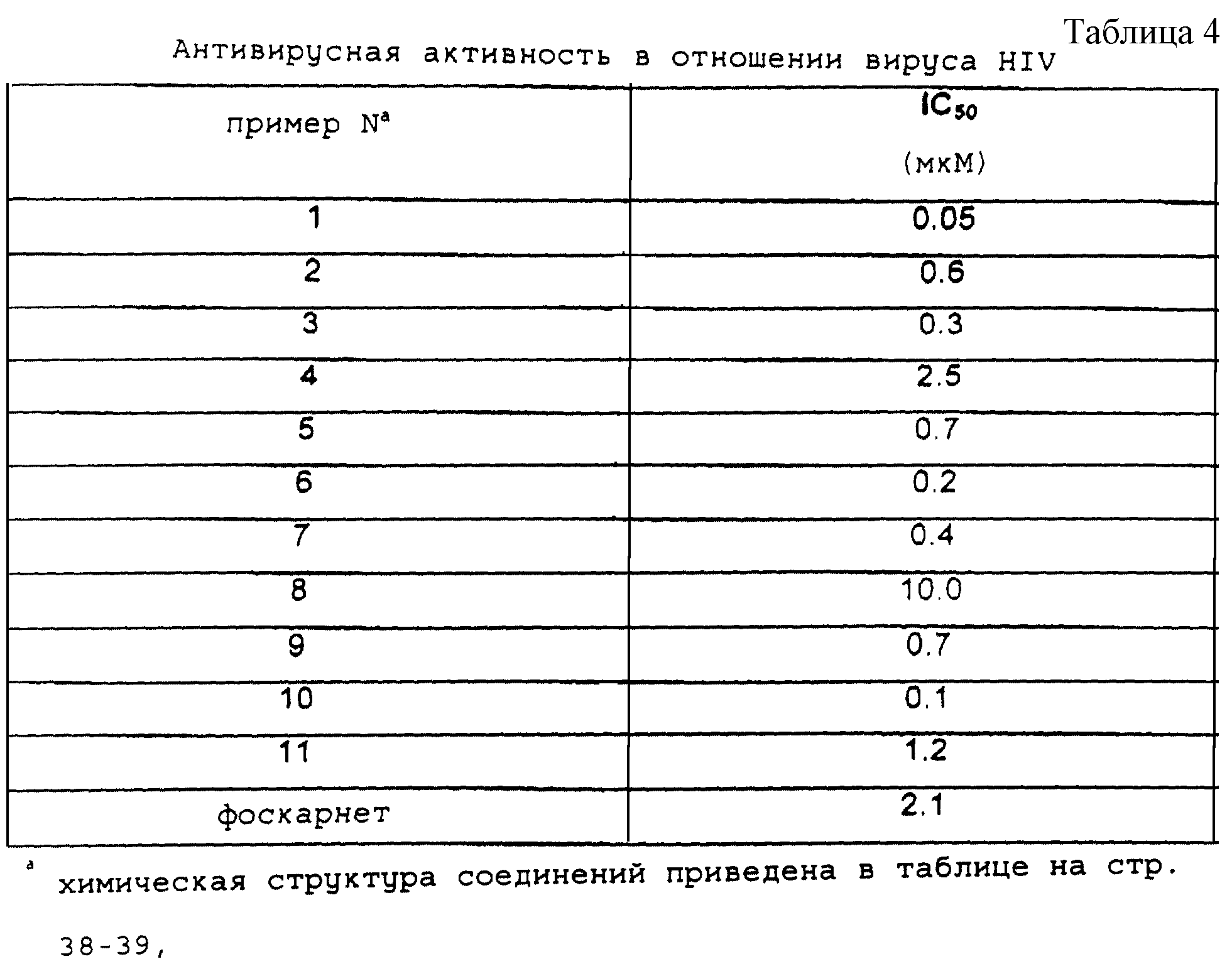
Антивирусная активность в отношении вирусов HBV и HIV для ряда
различных конъюгатов производных фоскарнета
приведены в таблицах 3 и 4 соответственно. Приведенные данные демонстрируют прямую связь структуры соединения и его активности.
Пример
14
Кальциевая соль метилового эфира R,
S- ((3-(8-циклогексил-октилмеркапто) -2-децилокси)-пропокси)- гидроксифосфинилмуравьиной кислоты
Свободная кислота была получена согласно
примеру 12.29 и растворена в метаноле. Путем
добавления водного раствора ацетата кальция выпадала кальциевая соль и в течение часа при комнатной температуре проводилось перемешивание.
Твердое вещество отсасывали, промывали метанолом и ацетоном и сушили в сушильном шкафу под вакуумом при температуре 40oC.
Выход: 89%, Т. пл. 198-199oC, Rf = 0,63 (Polygram SIL G/UV254, Macharey & Nagel, 805021, 0,2 мм; элюент хлороформ/метанол/конц. аммиак при соотношении 70/30/4).
Реферат
Изобретение относится к фосфолипидным производным фосфонокарбоновых кислот формулы I (см. формулу), в которой R1 обозначает группировку -(СН2)еСус1, в которой -(СН2)е обозначает линейную или разветвленную, насыщенную алкиленовую цепь, где е равно 4-16; R2 обозначает линейную насыщенную алкильную цепь с 1-20 атомами углерода; R3 обозначает Н или линейную алкильную цепь с 1-6 атомами углерода; Х обозначает серу, Y обозначает кислород, Сус1 обозначает циклический алкильный остаток с 5-7 С-атомами или фенил, одно- или многократно замещенный С1-С10-алкилом, С1-С10-алкокси или галогеном, m равно 0,1-3, при условии, что R1 может быть равен R2, если R2 одновременно обозначает R1, и их физиологически приемлемым солям. Также описывается лекарственное средство, содержащее указанное соединение в качестве активного вещества, для лечения вирусных, в том числе ретровирусных, заболеваний. 2 с. и 9 з. п. ф-лы, 4 табл.
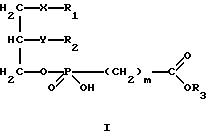
Формула
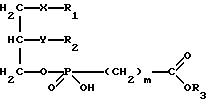
в которой R1 обозначает группировку -(СН2)еCycl, в которой - (СН2)е обозначает линейную или разветвленную насыщенную алкиленовую цепь, где е равно целому числу от 4 до 16, причем один из атомов углерода, начиная с позиции 3, может быть заменен атомом кислорода;
R2 обозначает линейную насыщенную алкильную цепь с 1-20 атомами углерода;
R3 обозначает Н или линейную алкильную цепь с 1-6 атомами углерода;
Y обозначает кислород;
X обозначает серу;
Сусl обозначает циклический алкильный остаток с 5-7 С-атомами или фенил, причем насыщенные или ароматические кольца могут быть одно- или многократно замещены С1-С10-акилом, С1-С10 -алкокси или галогеном;
m обозначает 0, 1-3, при условии, что R1 может быть равен R2, если R2 одновременно обозначает R1,
их физиологически приемлемые соли неорганических и органических оснований.
Комментарии