Способ получения 2,4-дигидроксимасляной кислоты - RU2626531C2
Код документа: RU2626531C2
Чертежи








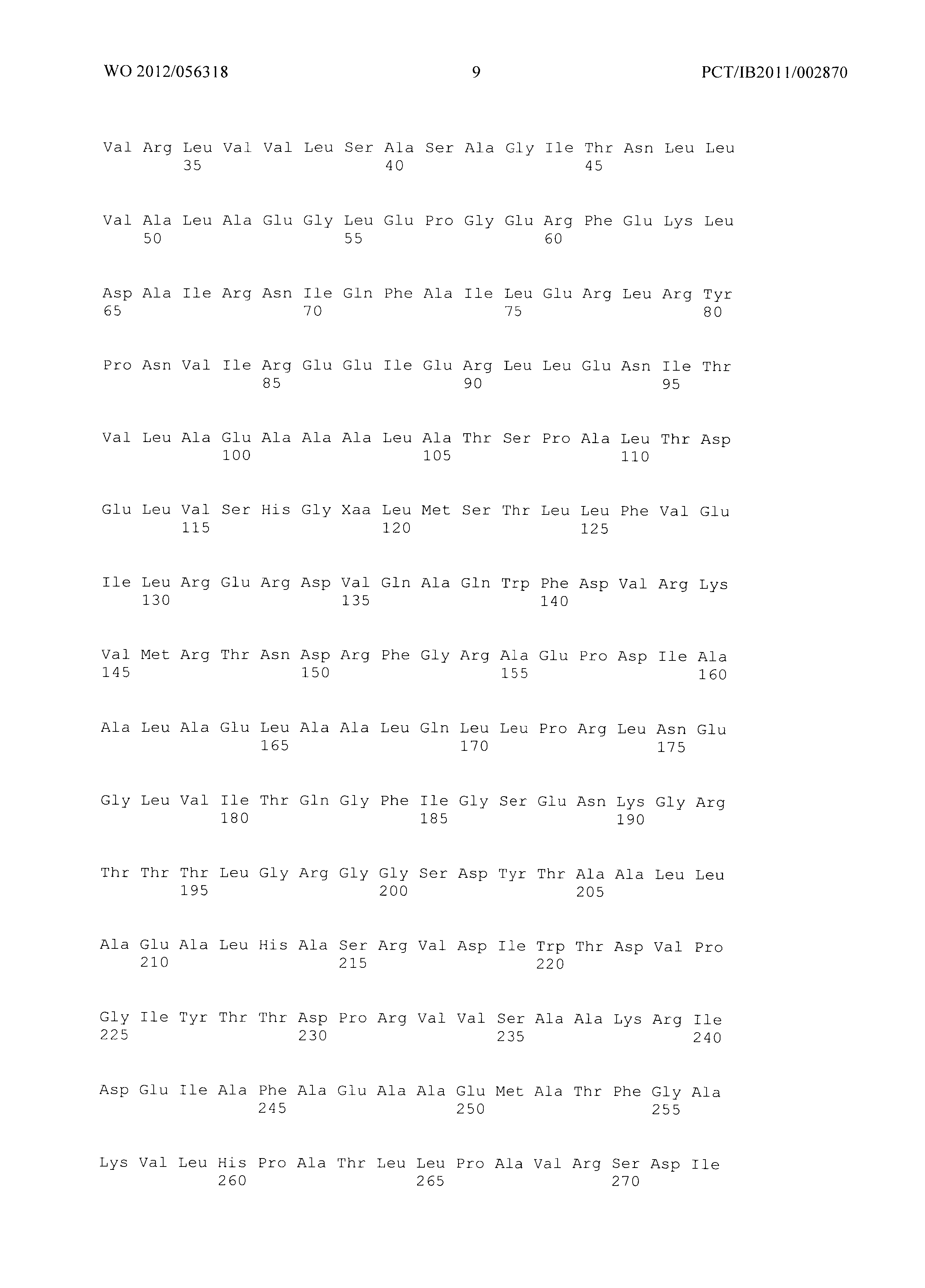



















































































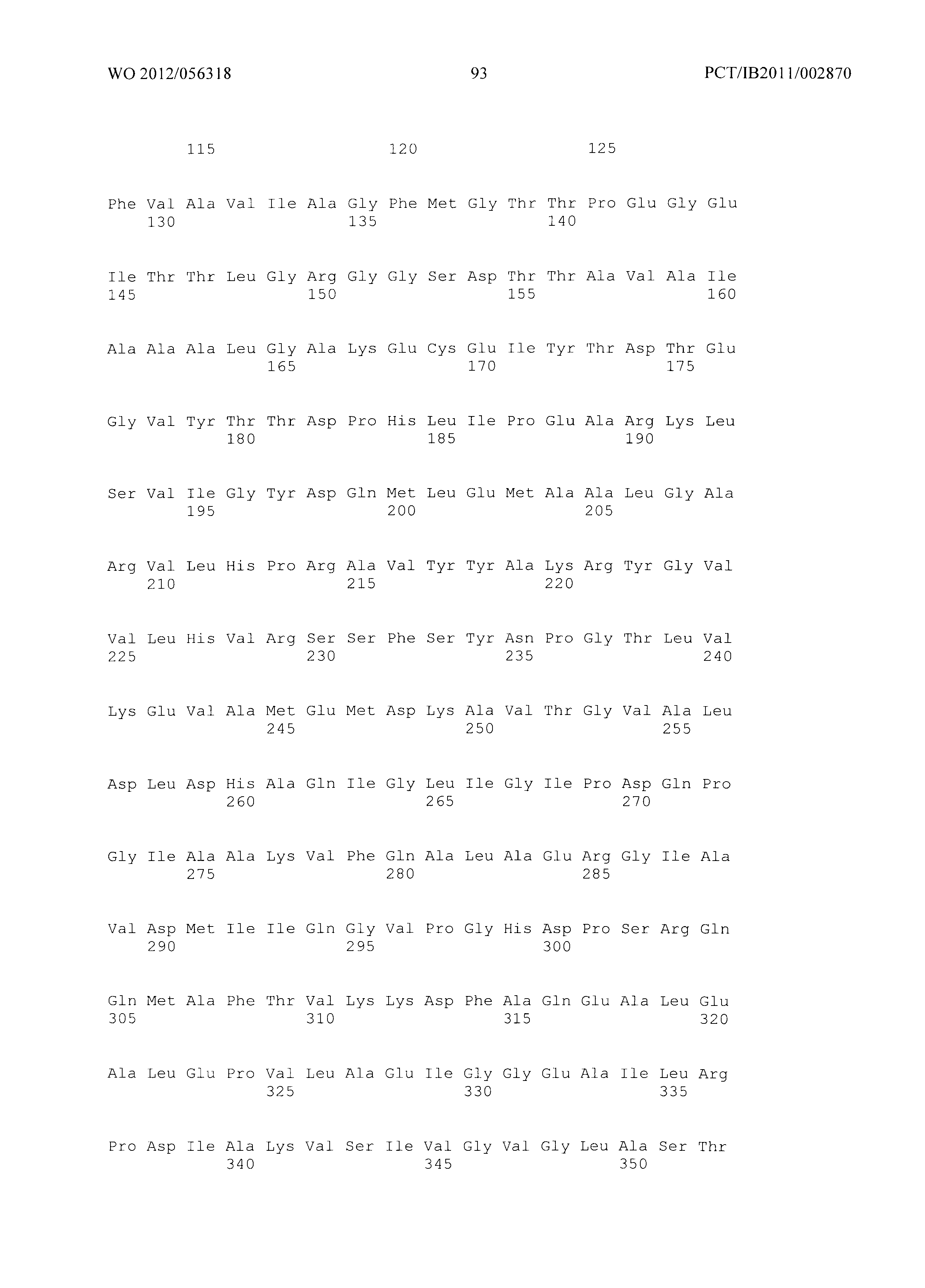
















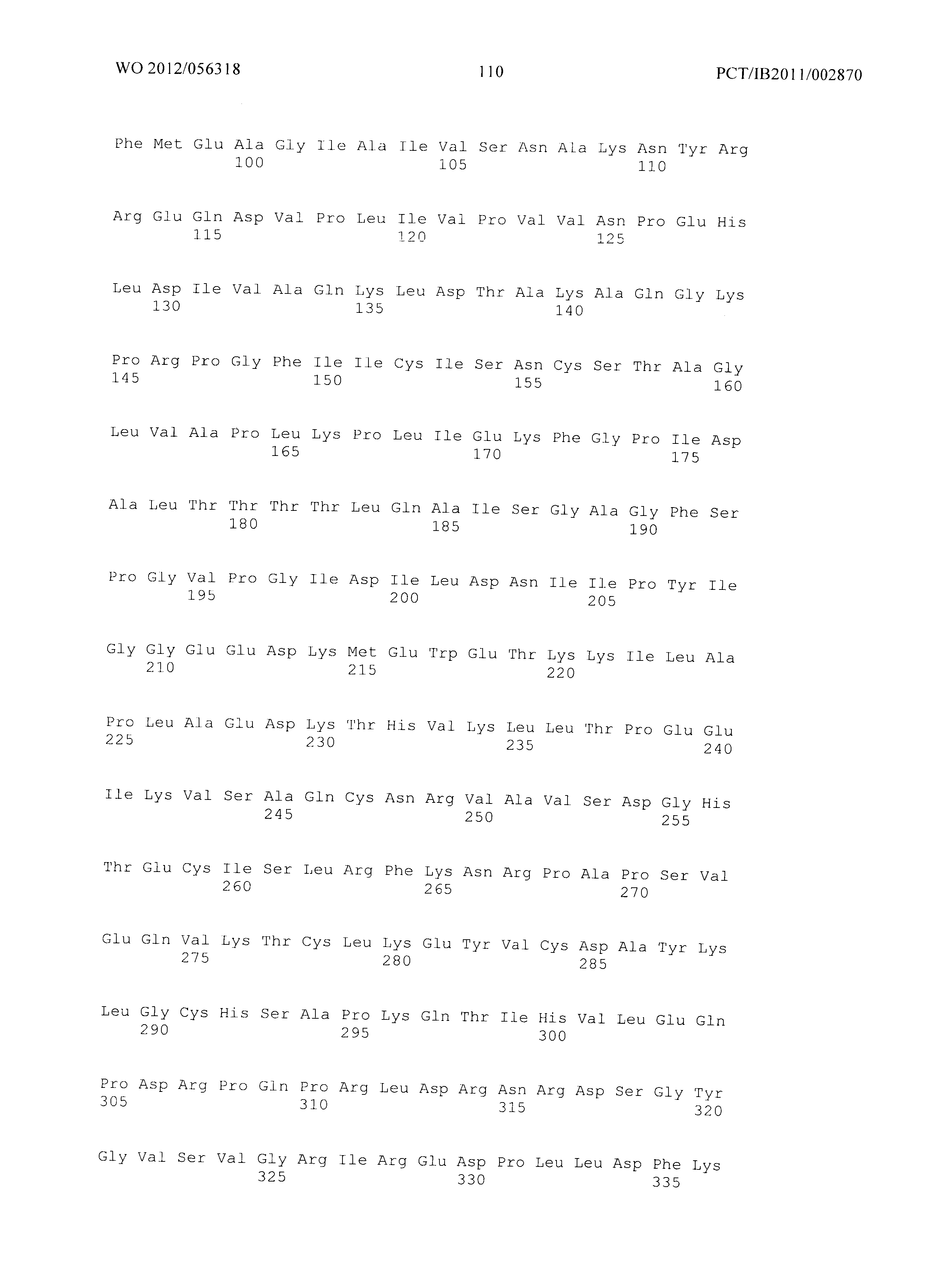













































Описание
Настоящее изобретение относится к новому способу получения 2,4-дигидроксимасляной кислоты из малата при помощи синтетического пути, который включает ферменты, обладающие, соответственно, малаткиназной, малат полуальдегиддегидрогеназной и 2,4-дигидроксибутиратдегидрогеназной активностью.
Карбоновые кислоты, цитированные в настоящей заявке на изобретение, в равной степени названы в соответствии со своей солевой (например, 2,4-дигидроксибутират) или кислотной формой (например, 2,4-дигидроксимасляная кислота).
2,4-Дигидроксимасляная кислота (в равной степени 2,4-ДГМ или ДГМ) представляет собой соединение, представляющее значительный экономический интерес. ДГМ может быть легко превращен в α-гидрокси-γ-бутиролактон в водных средах путем поддержания подходящего рН. α-Гидрокси-γ-бутиролактон представляет собой значимый предшественник для продукции метионинового заместителя 2-гидрокси-4-(метилтио)-бутирата (ГМТБ) (Deck et al., 2008), который занимает значительный рынок в кормлении животных. В настоящее время, α-гидрокси-γ-бутиролактон происходит из γ-бутиролактона при помощи многостадийного способа, который обеспечивает галогенирование γ-бутиролактона в позиции α, и последующее замещение атома галогена гидроксильной группой в щелочной среде (Deck et al., 2008).
Вследствие растущих цен на масло возрастает потребность в продукции ДГМ из возобновляемых источников. Микроорганизмы способны превращать происходящий из биомассы сырьевой материал, например сахара или органические кислоты, в огромное разнообразие различных химических соединений (Werpy & Petersen, 2004). Параллельно с растущей массой биохимической и генетической информации возможно модифицировать микроорганизмы, такие что они сверхпродуцируют встречающиеся в природе метаболические промежуточные соединения с высоким выходом и продуктивностью (Bailey, 1991). Оптимизация продукции микроорганизмов часто требует рационального конструирования метаболических сетей, которые обеспечивают, кроме того, сверхэкспрессию ферментов, требующихся для биосинтеза интересующего метаболита, и устранение ингибирования продукта по механизму обратной связи. Еще одна возможность заключается в применении новых ферментативных систем, которые катализируют продукцию интересующего метаболита.
Подходы метаболической инженерии и ферментативные катализы требуют подробного знания биохимии и регуляции метаболического пути, приводящего к интересуемому метаболиту. В случае продукции ДГМ данная информация отсутствует. Только в нескольких исследованиях сообщается о появлении ДГМ у пациентов, страдающих от дефицита дегидрогеназы янтарного полуальдегида (Shinka et al., 2002), тем не менее без идентификации ферментативных реакций, вовлеченных в продукцию ДГМ. Таким образом, для ферментативной продукции ДГМ требуется (1) идентификация термодинамически реализуемого пути, который превращает доступный предшественник в ДГМ, (2) идентификация или конструирование ферментов, которые способны катализировать отдельные реакционные стадии в пути и (3) функциональная экспрессия ферментов в пути в соответствующем продуцирующем организме.
Задача настоящего изобретения заключается в том, чтобы удовлетворять этим потребностям.
Соответственно, одной из задач настоящего изобретения является способ получения 2,4-ДГМ, при котором осуществляют первую стадию превращения малата в 4-фосфомалат с использованием малаткиназы, вторую стадию превращения 4-фосфомалата в малат-4-полуальдегид с использованием малат полуальдегид дегидрогеназы, третью стадию превращения малат-4-полуальдегида в 2,4-ДГМ с использованием ДГМ дегидрогеназы.
В первой реакции (смотри фиг.1 (i)) малат (1) превращается в 4-фосфомалат (2) путем действия фермента, который обладает малаткиназной активностью (А). Во второй реакции (В) 4-фосфомалат превращается в малат-4-полуальдегид (3) путем действия фермента, который обладает малат полуальдегид дегидрогеназной активностью. Точнее, реакцию (В) катализируют при помощи фермента, обладающего дефосфорилирующей 4-фосфомалат редуктазной активностью в биосинтетическом пути. В третьей реакции (С) малат-4-полуальдегид превращается в ДГМ (4) путем действия фермента, который обладает ДГМ дегидрогеназной активностью. Точнее реакцию (С) катализируют при помощи фермента, обладающего малат-4-полуальдегид редуктазной активностью в биосинтетическом пути.
Ни один из вышеописанных ферментов и промежуточных продуктов не был описан и идентифицирован в живых клетках. Сами малаткиназа, малат полуальдегид дегидрогеназа, ДГМ дегидрогеназа и 4-фосфомалат представляют собой дополнительные задачи изобретения.
В еще одном из аспектов изобретения первая стадия в способе получения 2,4-ДГМ включает малаткиназу, которая характеризуется тем, что превращает малат в 4-фосфомалат. Указанный фермент получают при помощи по меньшей мере одной мутации фермента, где указанная(ые) мутация(и) улучшает(ют) активность и/или субстратную аффинность мутантного фермента в отношении малата.
В настоящем изобретении выражение "улучшают активность и/или субстратную аффинность" означает, что фермент до мутации был или не способен использовать субстрат (малат, 4-фосфомалат или малат-4-полуальдегид) и/или синтезировал продукт реакции (4-фосфомалат или малат-4-полуальдегид или ДГМ) с максимальной специфической скоростью, которая была по меньшей мере в три раза меньше, и/или
обладал аффинностью в отношении малата, 4-фосфомалата или малат-4-полуальдегида, которая была по меньшей мере в три раза меньше, и/или
обладал аффинностью в отношении природного субстрата (аспартат, 4-фосфо-аспартат, аспартат-4-полуальдегид), которая была по меньшей мере в 3 раза выше.
В еще одном из своих аспектов изобретение относится к применению малаткиназы для превращения малата в 4-фосфомалат.
Малаткиназная активность может быть измерена при помощи ферментативного теста, описанного в примере 1 (смотри "Ферментативный анализ").
В соответствии с еще одним аспектом изобретения малаткиназа может быть получена путем мутации аспартаткиназы.
Фиг.2 демонстрирует совмещение аминокислотных последовательностей аспартаткиназ различного биологического происхождения. Все ссылки на позицию аминокислот сделаны на основе аминокислотной последовательности аспартаткиназы, кодируемой геном LysC E.coli (представленной в SEQ ID No. 4). Относительные позиции соответствующих консервативных областей в других аспартаткиназах из различных организмов легко могут быть найдены специалистом в данной области техники путем простого совмещения последовательности, как представлено на Фиг.2, с ферментами, перечисленными ниже:
- AKIN - аспартаткиназа III из E. Coli (SEQ ID No. 4),
- AKI (SEQ ID No. 87) аспартаткиназа I из E.coli,,
- AKII (SEQ ID No. 88) - аспартаткиназа II из E.coli,
- MJ - Methanococcus jannaschii (SEQ ID No. 89),
- TT - Thermus thermophilus (SEQ ID No. 90),
- CG - Corynebacterium glutamicum (SEQ ID No. 91),
- AT - Arabidopsis thaliana (SEQ ID No. 92),
- SC - Saccharomyces cerevisiae. (SEQ ID No. 93).
Указанное совмещение может быть осуществлено при помощи программного обеспечения ClustaIW2. Например, остаток Е119 аспартаткиназы, представленной в SEQ ID No. 4, соответствует остатку Е207 аспартаткиназы А.thaliana (SEQ ID No. 50) или остатку Е147 аспартаткиназы S.cerevisiae (SEQ ID No. 51).
Мутантная аспартаткиназа в соответствии с изобретением содержит по меньшей мере одну мутацию по сравнению с ферментом дикого типа в по меньшей мере одной из следующих позиций: S39, Т45, V115, Е119, F184 и/или S201, где встречающаяся в природе аминокислота в указанных позициях заменена на любую из других 19 существующих в природе белокобразующих аминокислот, то есть на аланин, аргинин, аспарагин, аспарагиновую кислоту, цистеин, глутаминовую кислоту, глутамин, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин или валин.
В неисключительном примере конструирование малаткиназы путем сайтнаправленного мутагенеза продемонстрировано с использованием аспартаткиназы Lys С из Escherichia coli в качестве матрицы. В соответствии с одним из аспектов изобретения субстратная специфичность LysC меняется в сторону малата путем замены глутаминовой кислоты в позиции 119 на аспарагин, глутамин, цистеин, пролин, серин, треонин, валин или глицин.
В еще одном аспекте изобретения малаткиназа представлена в SEQ ID No. 9 и конкретней в SEQ ID No. 12, SEQ ID No. 14, SEQ ID No. 16, SEQ ID No. 18, SEQ ID No. 20, SEQ ID No. 22, SEQ ID No. 24 или SEQ ID No. 26.
Аспартаткиназы типично ингибируются метионином, треонином или лизином. Таким образом, малаткиназы, конструируемые путем случайного или сайтнаправленного мутагенеза аспартаткиназ также могут ингибироваться указанными аминокислотами. В еще одном аспекте изобретения ингибирование малаткиназы метионином, лизином или треонином уменьшается путем мутагенеза малаткиназы.
В специфическом аспекте изобретения вышеописанная мутантная LysC (малаткиназа) становится нечувствительной к ингибированию лизином путем мутации по меньшей мере по одной из следующих аминокислот Е250, М318, S321, V339, S338, F324, L.325, V339, S345, Е346, D340, Т344 и/или Т352 (смотри пример 3).
Настоящее изобретение также охватывает такие модифицированные ферменты и конкретней ферменты, представленные в SEQ ID No. 39, SEQ ID No. 41, SEQ ID No. 43 или SEQ ID No. 45.
В еще одном аспекте вторая стадия способа получения 2,4-ДГМ в соответствии с изобретением включает малат полуальдегид дегидрогеназу, отличающуюся тем, что она превращает 4-фосфомалат в малат-4-полуальдегид, где указанный фермент обладает активностью, дефосфорилирующей 4-фосфомалат редуктазу, в пути биосинтеза.
Малат полуальдегид дегидрогеназная активность может быть измерена при помощи ферментативного теста, описанного в примере 4 (смотри "Ферментативный анализ"),
Указанный фермент получают при помощи по меньшей мере одной мутации фермента, где указанная(ые) мутация(и) улучшает(ют) активность и/или субстратную аффинность мутантного фермента в отношении 4-фосфомалата.
В соответствии с еще одним аспектом малат полуальдегид дегидрогеназа по изобретению может быть получена путем мутации фермента, обладающего известной полуальдегид дегидрогеназной активностью, конкретней обладающего дефосфорилирующей активностью в восстановительном аспекте реакции, конкретней действующего на органические молекулы, которые состоят из 3, 4 или 5 углеродных молекул. В конкретном аспекте изобретения указанную малат полуальдегид дегидрогеназу получают путем мутации аспартат полуальдегид дегидрогеназы.
Аспартат полуальдегид дегидрогеназа, Asd из Е.coli и Hom2 из Saccharomyces cerevisiae в природе демонстрируют дегидрогеназную активность в отношении 4-фосфомалата 2.
В соответствии с еще одним аспектом изобретения малат полуальдегид дегидрогеназа может быть улучшена путем мутации аспартат полуальдегид дегидрогеназы.
Фиг.3 демонстрирует совмещение аминокислотных последовательностей аспартат полуальдегид дегидрогеназ различного биологического происхождения. Все ссылки на аминокислоты сделаны на основе аспартат полуальдегид дегидрогеназы, кодируемой геном Asd из Е.coli (как представлено в SEQ ID No. 20). Относительные позиции соответствующих консервативных областей в других аспартат полуальдегид дегидрогеназ из различных организмов легко могут быть найдены специалистом в данной области техники путем простого совмещения последовательностей, представленных на Фиг.4, с ферментами, перечисленными ниже:
- ЕС - Е.Coli (SEQ ID No. 49),
- MJ - Methanococcus jannaschii (SEQ ID No. 94),
- TT - Thermus thermophilus (SEQ ID No. 95),
- BS - Bacillus subtilis (SEQ ID No. 96),
- CG - Corynebacterium glutamicum (SEQ ID No. 97),
- AT - Arabidopsis thaliana (SEQ ID No. 98),
- SC - Saccharomyces cerevisiae. (SEQ ID No. 99)
Указанное совмещение легко может быть осуществлено с использованием программного обеспечения ClustaIW2.
Конструирование ферментов, обладающих улучшенной малат полуальдегид дегидрогеназной активностью, может быть осуществлено следующим образом.
Малат полуальдегид дегидрогеназа в соответствии с изобретением соответствует в специфическом аспекте аспартат полуальдегид дегидрогеназе, содержащей, по меньшей мере, одну мутацию по сравнению с ферментом дикого типа в по меньшей мере одной из позиций Т136, Q162, I230, Е241 и/или Н274, где встречающаяся в природе аминокислота в указанных позициях заменена на любую из других 19 существующих в природе белокобразующих аминокислот, то есть на аланин, аргинин, аспарагин, аспарагиновую кислоту, цистеин, глутаминовую кислоту, глутамин, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин или валин.
Как продемонстрировано в примере 5, сайтнаправленный мутагенез asd из Е.coli может улучшить активность и субстратную аффинность мутантного фермента в отношении 4-фосфомалата, в то же самое время уменьшая предпочтение фермента в отношении своего природного субстрата 4-фосфо-аспартата.
Для улучшения активности Asd в отношении 4-фосфомалата, и в соответствии с одним из аспектов изобретения Е241 заменен на остаток глутамина, аланина, цистеина, глицина, гистидина, изолейцина или метионина путем сайтнаправленного мутагенеза (Пример 5).
В еще одном аспекте изобретения малат полуальдегид дегидрогеназа представлена в SEQ ID No. 68 и конкретней в SEQ ID No. 54, SEQ ID No. 56, SEQ ID No. 58, SEQ ID No. 60, SEQ ID No. 62, SEQ ID No. 64 или SEQ ID No. 66.
В еще одном аспекте изобретение относится к применению малат полуальдегид дегидрогеназы для превращения 4-фосфомалата в малат-4-полуальдегид.
В еще одном аспекте третья стадия способа получения 2,4-ДГМ в соответствии с изобретением включает ДГМ дегидрогеназу, отличающуюся тем, что она превращает малат-4-полуальдегид в 2,4-ДГМ, где указанный фермент обладает малат-4-полуальдегид редуктазной активностью в биосинтетическом пути.
Ферменты-кандидаты ДГМ дегидрогеназы, которые потенциально уже обладают ДГМ дегидрогеназной активностью, могут быть выбраны из класса β-гидроксикислот дегидрогеназ, которые действуют на С3, С4 или С5 соединения.
В соответствии с еще одним аспектом изобретения указанные ферменты ДГМ дегидрогеназы могут быть структурно и механистически отнесены к гидроксикислот дегидрогеназам, таким как редуктазы тартронат полуальдегида, редуктазы янтарного полуальдегида, редуктазы малонат полуальдегида, редуктазы метилбутиральдегида, алкогольдегидрогеназы цинкового типа, L-треонин-3-дегидрогеназы или редуктазы гомосерина.
Настоящее изобретение также относится к применению редуктазы метилбутиральдегида или редуктазы янтарного полуальдегида для превращения малат-4-полуальдегида в 2,4-ДГМ.В конкретных воплощениях указанная редуктаза метилбутиральдегида представлена в SEQ ID No. 74 и указанная редуктаза янтарного полуальдегида представлена в SEQ ID No. 76. ДГМ дегидрогеназная активность может быть измерена при помощи ферментативного теста, описанного в примере 6 (смотри "Ферментативный анализ").
Аффинность ДГМ дегидрогеназы в отношении малат-4-полуальдегида может быть увеличена при помощи по меньшей мере одной мутации фермента, где указанная(ые) мутация(и) увеличивают активность и/или субстратную аффинность мутантного фермента в отношении малат-4-полуальдегида и/или уменьшает(ют) активность или аффинность в отношении своего природного субстрата по меньшей мере вдвое.
ДГМ дегидрогеназа в соответствии с изобретением соответствует в специфическом аспекте редуктазе янтарного полуальдегида М.sedula (SEQ ID No 76), содержащей по меньшей мере одну мутацию по сравнению с ферментом дикого типа в по меньшей мере одной из позиций S40, N43, Н39 Т49, F85, Q108, L281 и N305, где встречающаяся в природе аминокислота в указанных позициях заменена на любую из других 19 существующих в природе белокобразующих аминокислот, то есть на аланин, аргинин, аспарагин, аспарагиновую кислоту, цистеин, глутаминовую кислоту, глутамин, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин или валин.
Как продемонстрировано в неисключительном примере аффинность редуктазы янтарного полуальдегида М sedula в отношении (L)-малат-4-полуальдегида увеличивалась путем введения двойной мутации H39R N43H путем сайтнаправленного мутагенеза, как представлено в SEQ ID No. 81. Простые мутанты H39R (SEQ ID No. 225) и N43H (SEQ ID No. 227) также охвачены настоящим изобретением (Пример 7).
ДГМ дегидрогеназа может быть использована для превращения малат-4-полуальдегида в 2,4-ДГМ, который составляет еще один аспект изобретения.
Последовательность нуклеиновой кислоты генов может быть адаптирована использованию кодонов организмом хозяином, таким образом, увеличивая продукцию гетерогенно экспрессируемых белков. Последнее составляет еще один аспект изобретения.
Синтез синтетического гена, кодирующего редуктазу янтарного полуальдегида М.sedula H39R N43H, нуклеотидная последовательность которого оптимизирована для экспрессии указанного фермента в Е.Coli, как представлено в SEQ ID No. 228, составляет еще один аспект изобретения.
В еще одном аспекте настоящее изобретение также относится к нуклеиновым кислотам, и конкретней к последовательностям выделенной нуклеиновой кислоты, кодирующим малаткиназу, как описано выше.
В еще одном аспекте указанная нуклеиновая кислота представлена в SEQ ID No. 13, SEQ ID No. 15, SEQ ID No. 17, SEQ ID No. 19, SEQ ID No. 21, SEQ ID No. 23, SEQ ID No. 25, SEQ ID No. 27, SEQ ID No. 38, SEQ ID No. 40, SEQ ID No. 42 или SEQ ID No. 44.
В еще одном аспекте настоящее изобретение также относится к последовательностям выделенной нуклеиновой кислоты, кодирующим малат полуальдегид дегидрогеназу, как описано выше.
Конкретней, указанная нуклеиновая кислота предпочтительно представлена в SEQ ID No. 55, SEQ ID No. 57, SEQ ID No. 59, SEQ ID No. 61, SEQ ID No. 63, SEQ ID No. 65 или SEQ ID No. 67.
В еще одном аспекте настоящее изобретение также относится к последовательностям выделенной нуклеиновой кислоты, кодирующим ДГМ дегидрогеназу, как описано выше.
В еще одном аспекте указанная нуклеиновая кислота представлена в SEQ ID No. 73 или SEQ ID No. 75, SEQ ID No. 224, SEQ ID No. 226 или SEQ ID No. 82
В соответствии с изобретением "последовательность нуклеиновой кислоты" относится к молекуле ДНК или РНК в одно- или двухцепочечной форме, предпочтительно молекуле ДНК. Использованный здесь термин "выделенная ДНК" относится к ДНК, которая не встречается в природе или больше не встречается в природном окружении, в котором она представлена исходно, например кодирующая последовательность ДНК, ассоциирующаяся с другими регуляторными элементами в химерном гене, ДНК, переносимая в другую клетку-хозяин, или искусственная синтетическая последовательность ДНК, имеющая нуклеотидную последовательность, отличающуюся от любой встречающейся в природе последовательности ДНК.
Настоящее изобретение также относится к химерному гену, содержащему, функционально связанные друг с другом, по меньшей мере один промотор, который является функциональным в организме хозяина, полинуклеотид, кодирующий любую из малаткиназы, малатполуальдегиддегидрогеназы или ДГМ дегидрогеназы в соответствии с изобретением, и терминаторный элемент, который является функциональным в том же самом организме хозяина. Различные элементы, которые может содержать химерный ген, представляют собой прежде всего элементы, регулирующие транскрипцию, трансляцию и созревание белков, таких как промотор, последовательность, кодирующую сигнальный пептид или транспортный пептид, или терминаторный элемент, составляющий сигнал полиаденилирования и, во вторых, полинуклеотид, кодирующий белок. Выражение "функционально связанные друг с другом" означает, что указанные элементы химерного гена связаны друг с другом таким образом, что на функцию одного из этих элементов влияет функция другого. В качестве примера промотор функционально связан с кодирующей последовательностью, где он способен влиять на экспрессию указанной кодирующей последовательности. Конструирование химерного гена в соответствии с изобретением и сборка его различных элементов может быть осуществлена с использованием способов, хорошо известных специалистам в данной области техники. Выбор регуляторных элементов, составляющих химерный ген, зависит по существу от организма-хозяина, в котором они должны функционировать, и специалисты в данной области техники способны выбирать регуляторные элементы, которые являются функциональными в заданном организме хозяина. Подразумевается, что термин "функциональный" обозначает способность функционировать в заданном организме хозяине.
Промоторы, которые может содержать химерный ген в соответствии с изобретением, являются конститутивными или индуцируемыми. В качестве примера, промоторы, используемые для экспрессии в бактериях, могут быть выбраны из промоторов, упомянутых ниже. Для экспрессии в Escherichia coli можно упомянуть lac, trp, Ipp, phoA, recA, araBAD, prou, cst-I, tetA, cadA, nar, tac, trc, Ipp-lac, Psyn, cspA, PL, PL-9G-50, PR-PL, T7, [lambda]PL-PT7, T3-lac, T5-lac, T4 ген 32, nprM-lac, VHb и промоторы белка А или еще промотор Ptrp (WO 99/64607). Для экспрессии в грамположительных бактериях, таких как Corynebacteria или Streptomyces, можно упомянуть промоторы PtipA или PS1 и PS2 (FR91/09870) или промоторы, описанные в заявке на изобретение ЕР 0629699А2. Для экспрессии в дрожжах и грибах можно упомянуть промоторы PLAC4 К.lactis или промотор Ppgk К.lactis (заявка на патент FR 91/05294), промотор tefl или cbhl Trichoderma (WO 94/04673), промотор his, csl или apf Penicillium (WO 00/68401) и промотор gla Aspergillus.
В соответствии с изобретением химерный ген может также содержать другие регуляторные последовательности, которые располагаются между промотором и кодирующей последовательностью, такие как активаторы транскрипции (энхансеры).
Сам по себе химерный ген по изобретению содержит в конкретном воплощении по меньшей мере в направлении транскрипции функционально связанные промоторную регуляторную последовательность, которая является функциональной в организме-хозяине, последовательность нуклеиновой кислоты, кодирующую малаткиназу малатполуальдегиддегидрогеназы по изобретению и терминаторную регуляторную последовательность, которая является функциональной в указанном организме-хозяине.
Настоящее изобретение также относится к клонированию и/или экспрессирующемуся вектору, содержащему химерный ген в соответствии с изобретением или последовательность нуклеиновой кислоты по изобретению. Вектор в соответствии с изобретением используют для трансформации организма-хозяина и экспрессии в этом организме любого из малаткиназы, малатполуальдегиддегидрогеназы и/или ДГМ дегидрогеназа. Этот вектор может представлять собой плазмиду, космиду, бактериофаг или вирус. Предпочтительно, трансформирующий вектор в соответствии с изобретением представляет собой плазмиду. Как правило, основные количества этого вектора должны обладать способностью поддерживать себя и реплицировать себя в клетках организма-хозяина, в частности за счет присутствия ориджина репликации, и для экспрессии в них любой из малаткиназы, малатполуальдегиддегидрогеназы и/или ДГМ дегидрогеназы. Для задачи стабильной трансформации организма-хозяина вектор также может интегрироваться в геном. Выбор такого вектора и также способов встраивания химерного гена в соответствии с изобретением в этот вектор и представляют собой часть общего знания специалистов в данной области техники. Благоприятно, вектор, используемый в настоящем изобретении, также содержит дополнительно к химерному гену в соответствии с изобретением химерный ген, кодирующий селективный маркер. Этот селективный маркер дает возможность отобрать организмы-хозяева, которые эффективно трансформируются, т.е. хозяева, которые включают вектор. В соответствии с конкретным воплощением изобретения организм-хозяин, который подвергают трансформации, представляет собой бактерию, дрожжи, гриб. Среди селективных маркеров, которые могут быть использованы, могут быть упомянуты маркеры, содержащие гены резистентности к антибиотикам, такие как, например ген гигромицин фосфотрансферазы. Другие маркеры могут представлять собой гены для дополнения ауксотрофности, такие как гены pyrA, pyrB, pyrG, pyr4, arg4, argB и trpC, ген молибдоптерин синтетазы или ген ацетамидазы. Также можно упомянуть гены, кодирующие легко идентифицируемые ферменты, такие как фермент GUS, или гены, кодирующие пигменты или ферменты, регулирующие продукцию пигментов в трансформированных клетках. Такие селективные маркерные гены в частности описаны в заявках на патенты WO 91/02071, WO 95/06128, WO 96/38567 и WO 97/04103.
Настоящее изобретение также относится к трансформированным организмам-хозяевам, содержащим по меньшей мере один химерный ген в соответствии с изобретением, интегрированный в их геном, или располагающийся на экстрахромосомальном генетическом элементе, например плазмиде. В более специфическом аспекте изобретения трансформированный организм-хозяин содержит нуклеиновую кислоту по изобретению, кодирующую малаткиназу, или химерный ген, содержащий нуклеиновую кислоту, кодирующую малаткиназу, или экспрессирующийся вектор, содержащий нуклеиновую кислоту, кодирующую малаткиназу, и/или нуклеиновую кислоту, кодирующую малатполуальдегиддегидрогеназу, или химерный ген, содержащий нуклеиновую кислоту, кодирующую малат полуальдегиддегидрогеназу, или экспрессирующийся вектор, содержащий нуклеиновую кислоту, кодирующую малатполуальдегиддегидрогеназу, и/или нуклеиновую кислоту, кодирующую ДГМ дегидрогеназу, химерный ген, содержащий нуклеиновую кислоту, кодирующую ДГМ дегидрогеназу, или экспрессирующийся вектор, содержащий нуклеиновую кислоту, кодирующую ДГМ дегидрогеназу.
В специфическом аспекте изобретения нуклеиновая кислота, кодирующая малаткиназу, представлена в SEQ ID No. 13, SEQ ID No. 15, SEQ ID No. 17, SEQ ID No. 19, SEQ ID No. 21, SEQ ID No. 23, SEQ ID No. 25, SEQ ID No. 27, SEQ ID No. 38, SEQ ID No. 40, SEQ ID No. 42 или SEQ ID No. 44, нуклеиновая кислота, кодирующая малат полуальдегид дегидрогеназу, представлена в SEQ ID 55, SEQ ID No. 57, SEQ ID No. 59, SEQ ID No. 61, SEQ ID No. 63, SEQ ID No. 65 или SEQ ID No. 67, и нуклеиновая кислота, кодирующая ДГМ дегидрогеназу, представлена в SEQ ID No. 73, SEQ ID No. 75, SEQ ID No. 224, SEQ ID No. 226 или SEQ ID No. 82.
Предполагается, что термин "организм-хозяин" обозначает любой низший одноклеточный организм, в который химерный(е) ген(ы), нуклеиновая(ые) кислота(ы) или вектор(ы) в соответствии с изобретением может(гут) быть введен(ы) для продукции 2,4-ДГМ.Предпочтительно, организм-хозяин представляет собой микроорганизм, в частности гриб, например рода Penicillium, Aspergillus и конкретней Aspergillus flavus, Chrysosporium или Trichoderma genus, дрожжи, в частности рода Saccharomyces, Kluyveromyces или Pichia, и конкретней Zygosaccharomyces rouxii, бактерию, например рода Escherichia, в частности Е.coli, или рода Corynebacterium, конкретней Corynebactenum glutamicum, или рода Streptomyces или бакуловируса.
Организм-хозяин может представлять собой организм-хозяин, который в природе сверхпродуцирует малат или сукцинат из сахаров, таких как глюкоза, или организм-хозяин, который сконструирован таким образом, чтобы сверхпродуцировать малат или сукцинат из сахаров, таких как глюкоза, и из которого удалены все потенциальные мембранные переносчики, которые облегчают экспорт органических кислот, таких как малат, пируват, сукцинат и фумарат. Организм-хозяин может представлять собой организм, который сконструирован для того, чтобы сверхпродуцировать ДГМ, и из которого удалены все мембранные переносчики, которые облегчают экспорт органических кислот, таких как малат, пируват, сукцинат и фумарат. Примеры пермеаз, которые облегчают экспорт малата и других органических кислот, представляют собой Мае1 из Schizosaccharomyces pombe (Camarasa et al., 2001; Grobler et al, 1995), DctA из Bacillus subtilis (Groeneveld et al., 2010), Dct 1-4 из Е.Coli, Jen1 из S.cerevisiae (Akita et al., 2000). Эксперт может идентифицировать кандидаты пермеаз в других микроорганизмах на основе гомологии последовательности. Эти конструкции служат для поддержания малата и других органических кислот внутри клетки для того, чтобы они были доступны для продукции ДГМ.
Подразумевается, что выражение "трансформированный организм-хозяин" обозначает организм-хозяин, который включает в свой геном или во внехромосомальный генетический элемент, например плазмиду, по меньшей мере один химерный ген в соответствии с изобретением и следовательно продуцирует любую из малаткиназы, малат полуальдегид дегидрогеназы и/или ДГМ дегидрогеназы в своих тканях, или в культуральной среде. Для получения организмов-хозяев в соответствии с изобретением специалисты в данной области техники могут использовать один из множества известных способов трансформации.
Один из этих способов заключается в том, чтобы привезти клетки организмов-хозяев, которые необходимо трансформировать, в контакт с полиэтиленгликолем (PEG) и с векторами в соответствии с изобретением. Электропорация представляет собой еще один способ, который заключается в том, чтобы подвергнуть клетки, которые необходимо трансформировать, и векторы по изобретению действию электрического поля. Еще один способ заключается в том, чтобы непосредственно инъецировать векторы в клетки или ткани путем микроинъекции. Может быть использован "биолистический" способ. Он состоит в бомбардировании клеток или тканей частицами, на которых адсорбированы векторы по изобретению (патент США No. 4945050).
Несколько способов трансформации бактерий описаны в литературе для Escherichia coli и других грамотрицательных бактерий. Также может быть использована конъюгация. Для грамположительных бактерий может быть использована электропорация и также трансформация протопластов, в частности для бактерий рода Streptomyces.
Несколько способов трансформации грибов также описаны в литературе. Трансформация протопластов с использованием PEG описана для Aspergillus в ЕР 0260762, и адаптация этого способа для видов Penicillium funiculosum описана в WO 00/36120. Также известна трансформация путем интеграции, опосредованной рестрикционным ферментом, или REMI, как трансформация протопласта с использованием бактерий рода Agrobacterium. В литературе также описаны способы трансформации дрожжей.
В еще одном аспекте изобретение относится к способу продукции 2,4-ДГМ, при котором осуществляют стадию выращивания трансформированного микроорганизма по изобретению.
Для продукции ДГМ различные углеводы могут быть использованы индивидуально или в виде смеси, такие как глюкоза, фруктоза, сахароза, мелассы, мальтоза, тростниковые мелассы, крахмальный гидролизат (глюкоза, олигосахариды), лактоза, мальтоза, крахмал и крахмальные гидролизаты, целлюлоза, целлюлозный гидролизат, глицерин и определенные углеводороды, масла и жиры, такие как соевое масло, подсолнечное масло, арахисовое масло и кокосовое масло, а также жирные кислоты, такие как, например пальмитиновая кислота, стеариновая кислота и линолевая кислота. Эти вещества могут быть использованы индивидуально или в виде смесей.
Различные источники азота могут быть использованы индивидуально или в виде смесей для коммерческого и полупромышленного производства, включающие неорганические соединения, такие как газообразный и водный аммиак, аммониевые соли неорганических или органических кислот, такие как сульфат аммония, нитрат аммония, фосфат аммония, хлорид аммония, ацетат аммония и карбонат аммония. Альтернативно, также могут быть использованы природные азотсодержащие органические материалы, такие как соевый гидролизат, HCl-гидролизат соевого белка (общее содержание азота составляет приблизительно 7%), соевая мука, гидролизат соевого жмыха, жидкий кукурузный экстракт, казеиновый гидролизат, дрожжевой экстракт, мясной экстракт, солодовый экстракт, мочевина, пептоны и аминокислоты.
Производственный процесс может быть осуществлен в аэробных, анаэробных условиях и условиях с ограниченным содержанием кислорода. Он может быть осуществлен в виде подпитываемого процесса или периодического процесса.
Указанная продукция 2,4-ДГМ может быть осуществлена путем выращивания организма-хозяина в средах, в которые малат (или другую органическую кислоту, такую как пируват, сукцинат или фумарат) добавлены сами по себе или вместе с другим источником углерода, который обеспечивает рост. Малат (и другие органические кислоты) могут быть добавлены непосредственно или путем разработки двухстадийного способа ферментации, где малат (или другие органические кислоты) продуцирует на первой стадии способа микроорганизм, сверхпродуцирующий малат, а продукцию 2,4-ДГМ осуществляют на следующей стадии с использованием организма-хозяина по изобретению.
Разделение и очистка продукта представляет собой очень важный фактор, значительно влияющий на эффективность всего способа и стоимость продукта. Способы выделения продукта в общем содержат стадии отделения клеток, а также очистки, концентрирования и сушки продукта, соответственно.
Отделение клеток
Ультрафильтрация и центрифугирование могут быть использованы для отделения клеток из ферментационной среды. Отделение клеток от ферментационных сред часто осложняется высокой вязкостью среды. Таким образом, можно добавить добавки, такие как неорганическую кислоту или соли щелочных металлов, или нагревание культуральной среды для оптимизации клеточного разделения.
Выделение продукта
Различные способы ионообменной хроматографии могут быть применены для выделения ДГМ до или после удаления биомассы. Они включают применение первичных катионообменных смол, которые облегчают разделение продуктов в соответствии с их изоэлектрической точкой. Как правило, смола заряжается раствором, и оставшийся продукт элюируется отдельно в соответствии с увеличением рН (например, путем добавления гидроксида аммония) в элюенте. Еще одна возможность представляет собой применение ионообменной хроматографии с использованием смол с фиксированным или условным подвижным слоем. Различные хроматографические стадии могут быть комбинированы для достижения необходимой чистоты продукта. Эти способы очистки являются более экономными по сравнению с дорогостоящей стадией кристаллизации, также обеспечивая дополнительные преимущества и гибкость в отношении формы конечного продукта.
Концентрирование и сушка продукта
Способ очистки также может содержать стадию сушки, которая может включать любые подходящие средства сушки, такие как распылительный гранулятор, распылительная сушилка, барабанная сушилка, роторная сушилка и туннельная сушилка. Концентрированные растворы ДГМ могут быть приготовлены путем нагревания ферментационных бульонов при пониженном давлении паром при 130°С с использованием многофункционального концентратора или тонкослойного испарителя.
Эффективная продукция ДГМ может обеспечиваться путем оптимизации перераспределения углеродного потока в метаболической схеме организма-хозяина и путем обеспечения достаточного поступления НАДФН (восстановленный никотинамидадениндинуклеотидфосфат) и АТФ (аденозинтрифосфат) для трех ферментов в пути ДГМ.Направление углеродного потока в желаемом метаболическом пути и поступление кофактора NAD(P)H как правило облегчают путем устранения или уменьшения интенсивности конкурирующих природных ферментативных путей. Неисключительные примеры представляют собой
- оптимизацию продукции малата в S.cerevisiae путем блокирования образования этанола (путем устранения пируват декарбоксилаз (Zelle et al., 2008; Zelle et al., 2010).
- оптимизацию продукции сукцината или малата в Е.coli путем блокирования образования лактата (например, делеция ldhA), образования ацетата (например, делеция pta, ackA), образования этанола (например делеция adhE), образования формиата (например делеция pflB, focA), окисления пирувата (например делеция рохВ), разрушения малата (делеция maeB и scfA), образования сукцината (например делеция frdBC), образования метилглиоксаля (делеция mgsA) (Jantama et al., 2008a; Jantama et al., 2008b; Lin et al., 2005; Sanchez et al., 2005a; Zhang et al., 2011).
Еще одна возможность увеличения потока углерода и поступления АТФ для продукции органических кислот представляет собой конструирование фосфоенолпируват (РЕР)/пируват/оксалоацетатного пути (обзор в (Sauer & Eikmanns, 2005)). Неисключительные примеры метаболических сконструированных стратегий, которые обеспечивают увеличение потока углерода из фосфоенолпирувата в оксалоацетат, представляют собой
- оптимизацию продукции малата в S.cerevisiae путем блокирования функции пируваткиназы и увеличения активности PEP карбоксикиназы (Zelle et al., 2010).
- оптимизацию продукции сукцината в Е.coli путем увеличения активности природной или гетерологически экспрессируемой PEP карбоксилазы, PEP карбоксикиназы или пируват карбоксилазы (Millard et al., 1996; Sanchez et al., 2005b; Zhang et al., 2009).
Еще одна возможность для увеличения потока углерода и поступления АТФ для продукции органических кислот в Е.coli и другие бактерии, использующие фосфотрансферазную систему (PTS), потребляющую PEP (фосфоенолпируват), для исходной стадии фосфорилирования глюкозы заключается в устранении важных компонентов системы PTS (например ptsl или ptsG) (Lin et al., 2005; Zhang et al., 2009). Для обеспечения дополнительного захвата глюкозы в мутантах, несущих мутации, нарушающие систему PTS, должна быть обеспечена активность альтернативных систем захвата глюкозы (например GalP).
Еще одна возможность для увеличения потока углерода в желаемых путях продукции органических кислот заключается в конструировании цикла Кребса и глиоксилатного цикла. Неисключительные примеры представляют собой
- оптимизацию продукции янтарной кислоты в Е.coli путем увеличения активности изоцитратлиазы (устранение транскрипционного репрессора iclR) (Lin et al., 2005; Sanchez et al., 2005a).
- оптимизацию продукции янтарной кислоты путем устранения изоцитрат дегидрогеназы и/или сукцинат дегидрогеназы (Lin et al., 2005).
Еще одна возможность для увеличения потока углерода в желаемых путях продукции ДГМ представляет собой экспрессию соответствующих пируватдегидрогеназ и цитратсинтетаз в организме-продуценте. Природная пируватдегидрогеназа и цитратсинтетаза Е.coli ингибируются высокими внутриклеточными концентрациями НАДН, приводя к тому, что ферменты менее активны в анаэробных условиях. В Е coli экспрессия мутантной пируватдегидрогеназы, нечувствительной к НАДН, приводит в результате к сверхпродукции ацетил-СоА (ацетил-коэнзим А) в анаэробных условиях и модифицированному перераспределению потока углеродов между ферментативными конечными продуктами (ацетат, лактат, этанол, формиат и пируват) (Wang et al., 2010). Гетерологическая экспрессия цитратсинтетазы Bacillus subtilis, не чувствительной к НАДН, увеличивала продукцию янтарной кислоты в сконструированных штаммах Е.coli (Sanchez et al., 2005a). В комбинации с вышеописанными мутациями применение соответствующих пируват дегидрогеназ и цитратсинтетаз (чувствительных и нечувствительных к НАДН) дает возможность для переключения перераспределения потока углерода между реакциями глиоксилатного цикла и цикла Кребса и ферментативными путями в анаэробных и аэробных условиях.
Еще одна возможность увеличения потока углерода через ДГМ путь заключается в устранении ферментативных реакций, которые могут разрушать промежуточные соединения пути 4-фосфомалат, 4-малат полуальдегид. Ферменты-кандидаты, которые могут разрушать малат полуальдегид, представляют собой дегидрогеназы янтарного полуальдегида (sad, gabD), и другие дегидрогеназы, которые способны окислять С4 молекулы с концевыми альдегидными группами.
Еще одна возможность увеличить продуктивность ДГМ организмом-хозяином заключается в устранении метаболических реакций, которые разрушают ДГМ.ДГМ представляет собой конкурентный ингибитор малик-энзима, таким образом, обладая относительно высокой аффинностью в отношении активного сайта этого фермента (Rognstad & Katz, 1979). Таким образом, ДГМ может распознаваться другими ферментами и потенциально разрушаться. Эти ферменты могут быть идентифицированы и удалены из организма-хозяина.
Когда продукция 2,4-ДГМ основана на добавлении малата или других органических кислот, тогда микроорганизмы, продуцирующие 2,4-ДГМ, должны функционально экспрессировать белок мембранного транспорта, который облегчает захват малата (или других органических кислот, таких как пируват, сукцинат и т.д).
Следующие примеры иллюстрируют изобретение. Эти примеры приведены только для иллюстрации и их не следует рассматривать как ограничивающие каким-либо путем объем изобретения.
Краткое описание графических материалов
Фиг.1: (i) Реакционная схема, которая описывает превращение (L)-малата в (L)-2,4-дигидроксибутират (ДГМ), и (ii) аналогия с превращением (L)-аспартата в (L)-гомосерин.
Фиг.2: Совмещение аминокислотных последовательностей аспартаткиназ из различных организмов. (Ес_AKIII - аспартаткиназа III (SEQ ID No. 4), LysC из Е.coli, Ec_AKI (SEQ ID No. 87) - аспартаткиназа I, ThrA из Е.coli, Ec_AKII (SEQ ID No. 88 - аспартаткиназа II, MetL из Е.coli, Mj - Methanococcus jannaschii (SEQ ID No. 89), Tt - Thermus thermophilus (SEQ ID No. 90), Cg - Corynebacterium glutamicum (SEQ ID No. 91), At - Arabidopsis thaliana (SEQ ID No. 92), Sc - Saccharomyces cerevisiae. (SEQ ID No. 93)) Изображение получено с использованием ClustaIW2 (Larkin et al., 2007).
Фиг.3: Совмещение аминокислотных последовательностей аспартат полуальдегид дегидрогеназ из различных организмов. (Ec - Е.Coli (SEQ ID No. 49), Mj - Methanococcus jannaschii (SEQ ID No. 94), Tt - Thermus thermophilus (SEQ ID No. 95), Bs - Bacillus subtilis (SEQ ID No. 96), Cg - Corynebacterium glutamicum (SEQ ID No. 97), At - Arabidopsis thaliana (SEQ ID No. 98), Sc - Saccharomyces cerevisiae. (SEQ ID No. 99)). Изображение получено с использованием ClustaIW2 (Larkin et al., 2007).
Фиг.4: масштабирование GC хроматограмм области, соответствующей времени удерживания ДГМ, демонстрирующей: (А) стандарт ДГМ (концентрация = 1 мМ); (В) композиция реакционной смеси А, содержащей малаткиназу (МК), малатполуальдегиддегидрогеназу (MSA-Dh) и малатполуальдегидредуктазу (MSA-Red); (С) композиция контрольной реакционной смеси В (такой же как А, но без MSA-Red); (D) композиция контрольной реакционной смеси С (такой же как А, но без MSA-Dh).
Фиг.5: Относительные активности очищенных мутантов LysC E119G, LysC E119G Е250К, LysC E119G Т344М, LysC E119G S345L, LysC E119G T344M, и LysC E119G T352I в отношении концентрации лизина в реакционном буфере.
Примеры
Пример 1: Тестирование аспартаткиназ LysC и Hom3 из Escherichia coli и Saccharomyces cerevisiae, соответственно, в отношении аспартат- и малаткиназной активности
Конструирование плазмид, содержащих гены дикого типа аспартаткиназы: Плазмиду pLYSCwt конструировали путем амплификации гена IysC при помощи ПЦР (полимеразной цепной реакции) с использованием высокоточной полимеразы Phusion™ (Finnzymes) и прямых и обратных праймеров5’CACGAGGTACATATGTCTGAAATTGTTGTCTCC3’ (SEQ ID No. 1) и5’CTTCCAGGGGATCCAGT-ATTTACTCAAAC3’ (SEQ ID No. 2), которая вводит сайты рестрикции NdeI и SamHI выше стартового кодона и ниже стоп-кодона, соответственно. Геномную ДНК из Е.coli DH5α использовали в качестве матрицы. Продукт ПЦР расщепляли при помощи NdeI и BamHI, лигировали в соответствующие сайты экспрессирующегося вектора рЕТ28а (Novagen) с использованием Т4 ДНК лигазы (Biolabs), и трансформировали в клетки Е.coli DH5α. Получающуюся в результате плазмиду pAKIIIwt выделяли, и при помощи секвенирования ДНК показали, что она содержит полноразмерный ген lysC, имеющий правильную последовательность (SEQ ID No. 3). Соответствующий белок представлен в SEQ ID No. 4.
Плазмиду pHOM3wt конструировали путем амплификации гена НОМ3 при помощи ПЦР с использованием высокоточной полимеразы Phusion™ (Finnzymes) и прямых и обратных праймеров5’TATAATGCTAGCATGCCAATGGATTTCCAACC3’ (SEQ ID No. 5) и5’TATAATGAATTCT-TAAATTCCAAGTCTTTTCAATTGTTC3’ (SEQ ID No. 6), которая вводит сайты рестрикции NheI и EcoRI выше стартового кодона и ниже стоп-кодона, соответственно. Геномную ДНК из S.cerevisiae BY4741wt использовали в качестве матрицы. Продукт ПЦР расщепляли при помощи NheI и EcoRI, и лигировали в соответствующие сайты экспрессирующегося вектора рЕТ28а (Novagen) с использованием Т4 ДНК лигазы (Biolabs), и трансформировали в клетки Е.coli DH5α. Получающуюся в результате плазмиду pHOM3wt выделяли, и при помощи секвенирования ДНК показали, что она содержит полноразмерный ген НОМЗ, имеющий правильную последовательность (SEQ ID No. 7). Соответствующий белок представлен в SEQ ID No. 8.
Экспрессия ферментов; Клетки Е.coli BL21 D3 star трансформировали соответствующими плазмидами. Ферменты с N-концевой последовательностью гекса-His экспрессировали в 250 мл культуры LB, которую инокулировали из ночной культуры при OD600 0,1 и выращивали до OD600 0,6, а затем экспрессию белка индуцировали путем добавления 1 мМ изопропил β-D-1-тиогалактопиранозида (IPTG) в культуральную среду. После 3 ч экспрессии белка клетки собирали путем центрифугирования при 1300 g в течение 10 мин и хранили при -20°С до последующего анализа. Рост и экспрессию белка осуществляли при 37°С. Культуральные среды содержали 50 мкг/л канамицина.
Очистка ферментов: Замороженные клеточные осадки экспрессирующихся культур ресуспендировали в 0,5 мл разрушающего буфера (50 мМ Hepes, 300 мМ NaCl, pH 7,5) и разрушали при помощи четырех последующих раундов ультразвуковой обработки (Bioblock Scientific, VibraCell™ 72437) с выходной мощностью, составляющей 30%. Клеточный дебрис удаляли путем центрифугирования неочищенных экстрактов в течение 15 мин при 4°С при 13000 g, и получали прозрачный супернатант. РНК и ДНК удаляли из экстрактов путем добавления 15 мг/мл стрептомицина (Sigma), центрифугирования образцов при 13000 g в течение 10 мин при 4°С и получения супернатанта. Прозрачный белковый экстракт инкубировали в течение 1 ч при 4°С с объемом слоя кобальтовой аффинной смолы Talon™ (Clontech) 0,75 мл. Суспензию центрифугировали при 700 g в настольной центрифуге, и супернатант удаляли. Смолу промывали 10 объемами слоя промывающего буфера (50 мМ Через, 300 мМ NaCl, 15 мМ Имидазол, рН 7,5), а затем аспартаткиназы элюировали 0,5 мл элюирующего буфера (50 мМ Hepes, 300 мМ NaCl, 500 мМ Имидазол, рН 7,5). Чистоту элюрованных ферментов проверяли при помощи анализа SDS-PAGE (электрофорез в полиакриламидном геле с додецилсульфатом натрия).
Ферментативный анализ. Активности аспартат- или малаткиназы измеряли путем сочетания продукции АДФ (аденозиндифосфата) в киназных реакциях с окислением НАДН в присутствии фосфоенолпирувата, пируваткиназы и лактатдегидрогеназы.
Схема реакции:
Аспартат (или малат) киназа
аспартат (или малат) + АТФ → 4-фосфо-(L)-аспартат (или 4-фосфо-(L)-малат) + АДФ
Пируваткиназа
АДФ + фосфоенолпируват → АТФ + пируват
Лактатдегидрогеназа
пируват + НАДН → NAD+ + лактат
Анализируемая смесь содержала 50 мМ Hepes (рН 7,5), 50 мМ KCl, 5 мМ MgCl2, 0,24 мМ НАДН, 0,96 мМ АТФ, 0,96 мМ ФЕП (фосфоенолпируват), 9 мкг/мл лактатдегидрогеназы (Sigma, L2500), 12,4 мкг/мл пируваткиназы (Sigma, P1506), и соответствующие количества очищенной аспартат (малат) киназы. Реакции инициировали путем добавления 50 мМ (L)-аспартата или (L)-малата. Ферментативные анализы осуществляли при 30°С в 96-луночных плоскодонных микротитровальных планшетах в конечном объеме 250 мкл. Реакции контролировали по характеристическому поглощению НАДН при 340 нм в микропланшетном ридере (BioRad 680XR).
Гидроксаматный анализ. Для проверки фосфорилирования субстрата, т.е. образования ацилфосфатного ангидрида аспартаткиназами дикого типа или мутантными аспартаткиназами продукт киназной реакции инкубировали с гидроксиламином с образованием соответствующего аспартат- или малатгидроксаматного производного. Анализируемая смесь содержала 120 мМ Hepes (рН 8), 200 мМ KCl, 10 мМ АТФ, 200 мМ гидроксиламин, 10 мМ аспартат или малат, и соответствующее количество очищенного белка. Реакцию останавливали через 30 мин путем добавления равного объема 1,7% (масс./об.) FeCl3 в 1 М соляной кислоты. Образование комплекса гидроксамата с железом подтверждали путем измерения его характеристического поглощения при 540 нм в микропланшетном ридере. Анализируемые смеси, содержащие все компоненты за исключением АТФ, использовали в качестве отрицательного контроля.
Результаты: Очищенные LysC (без His последовательности, SEQ ID No. 4) и Hom3 (без His последовательности, SEQ ID No. 7) ферменты демонстрировали аспартаткиназную активность, но не способны фосфорилировать малат, что подтверждается при помощи гидроксаматного анализа (Keng & Viola, 1996). Максимальные активности для LysC и Hom3 по аспартату составляли 4,5 мкмоль/(мин*мгбелка) и 1,6 мкмоль/(мин*мгбелка), соответственно. Значение Km для аспартата оценивали при помощи способа Eadie и Hofstee путем измерения исходных скоростей реакции (v) при различных концентрациях субстрата (с) и путем вычитания наклона v в зависимости от графика v/c. Оценивали, что Km очищенного LysC с His последовательностью составляла приблизительно 0,6 мМ, демонстрируя то, что белок с His последовательностью обладает той же самой субстратной аффинностью как и очищенный фермент без концевой последовательности, которая, как сообщается, составляет 0,6 мМ (Marco-Marin et al., 2003).
Пример 2: Сайтнаправленный мутагенез аспартаткиназы LysC из Escherichia coli и тестирование мутантных ферментов в отношении малаткиназной активности
Сайтнаправленный мутагенез осуществляли с использованием пар олигонуклеотидов, перечисленных в таблице 1, и плазмиды pLYSCwt (SEQ ID No. 3) в качестве матрицы. Точечные мутации для изменения аминокислотной последовательности вводили при помощи ПЦР (Phusion 1U, буфер HF 20% (об./об.), dNTP 2,5 мМ, прямые и обратные праймеры по 1 мкМ каждого, матричная плазмида 200 нг, вода) с использованием пары олигонуклеотидов, представленной в таблице 1. Плазмиды, образованные при помощи ПЦР, содержали новый сайт рестрикции для Nco1 (введенный с использованием молчащих мутаций) в дополнение к функциональной мутации для облегчения идентификации мутантных клонов. Продукты ПЦР расщепляли при помощи DpnI при 37°С в течение 1 ч для удаления матричной ДНК, и трансформировали в NEB 5-альфа компетентные клетки Е.coli (NEB). Мутантные плазмиды идентифицировали при помощи анализа по сайтам рестрикции и убеждались в том, что они несут желаемые мутации, при помощи секвенирования ДНК.

Последовательность, демонстрирующая мутацию в позиции 119, может быть представлена в SEQ ID No. 9, где остаток в позиции 119 представляет собой X, где Х представляет собой любую из 19 встречающихся в природе аминокислот (за исключением глутамина).
Мутантные ферменты экспрессировали, очищали и тестировали в отношении аспартат- и малаткиназной активности, как описано в примере 1. Результаты обобщены в таблице 2.
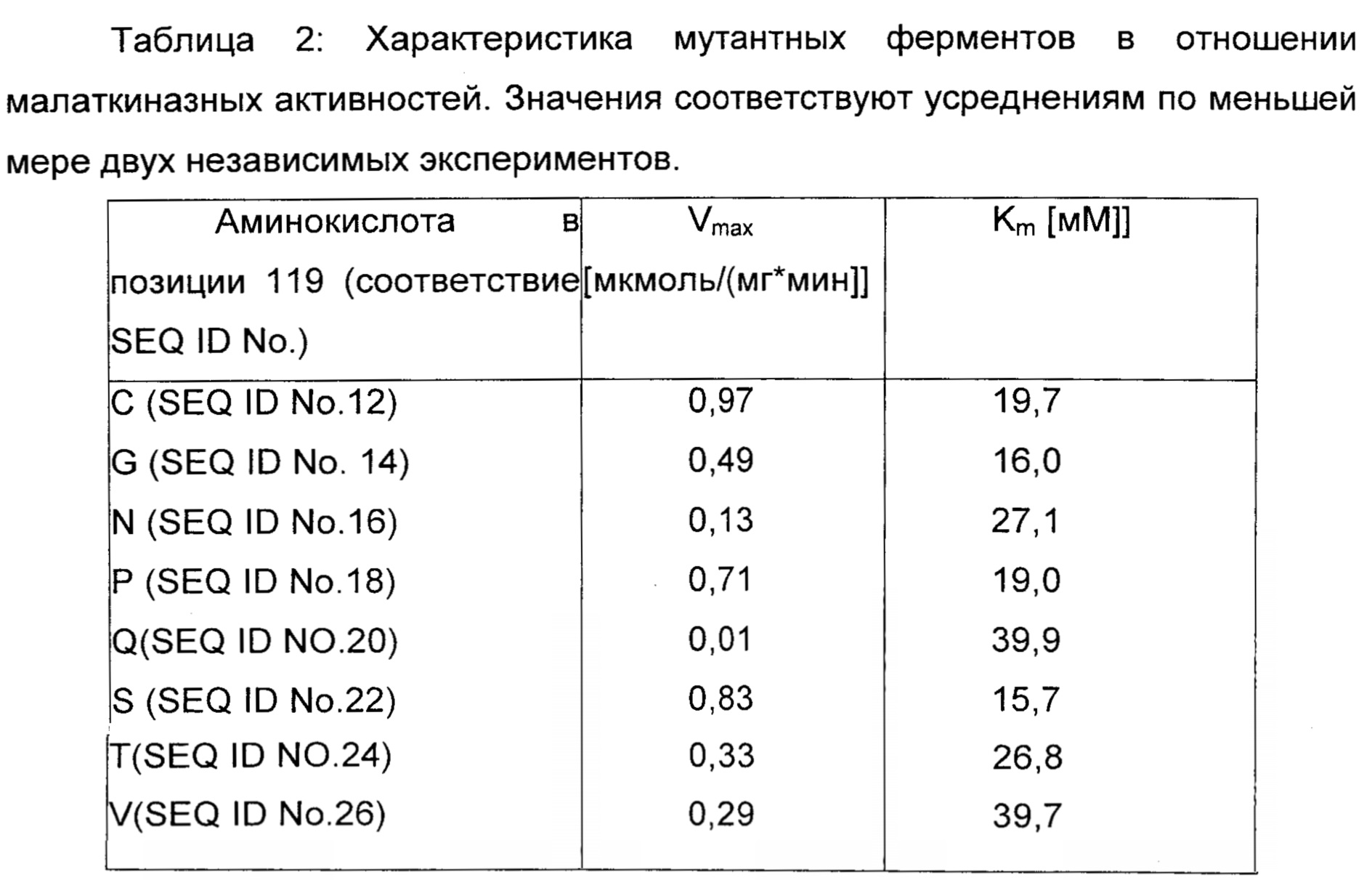
Ни один из мутантов, перечисленных в Таблице 2, не обладал активностью в отношении аспартата.
Результаты демонстрируют, что аспартаткиназа может быть превращена в малаткиназу путем замены консервативного глутамата в позиции 1.19 на цистеин, глицин, аспарагин, пролин, глутамин, серин, треонин или валин.
Соответствующие последовательности нуклеиновой кислоты фермента, представленного в таблице 2, представляют собой SEQ ID No. 13, SEQ ID No. 15, SEQ ID No. 17, SEQ ID No. 19, SEQ ID No. 21, SEQ ID No. 23, SEQ ID No. 25 и SEQ ID No. 27.
Пример 3: Конструирование малаткиназы с сильно уменьшенной чувствительностью для ингибирования лизином
Сайтнаправленный мутагенез осуществляли с использованием пар олигонуклеотидов, представленных в таблице 3, и плазмиды pLYSC_E119G в качестве матрицы (плазмида pLYSC_E119G получена, как описано в примере 2 путем осуществления следующих изменений в последовательности ДНК гена lysC: (SEQ ID No. 15). Точечные мутации для изменения аминокислотных последовательностей вводили при помощи ПЦР (Phusion 1U, буфер HF 20% (об./об.), dNTP 2,5 мМ, прямой и обратный праймеры 1 мкМ каждого, матричная плазмида 200 нг, вода) с использованием пар олигонуклеотидов, перечисленных в таблице 1. Когда возможно, плазмиды, созданные при помощи ПЦР, содержали новые сайты рестрикции (введенные с использованием молчащих мутаций) в дополнение к функциональной мутации для облегчения идентификации мутантных клонов. Продукты ПЦР расщепляли при помощи DpnI при 37°С в течение 1 ч для удаления матричной ДНК, и трансформировали в NEB 5-альфа компетентные клетки Е.coli (NEB). Мутантные плазмиды идентифицировали при помощи анализа сайтов рестрикции и убеждались в том, что они несут желаемые мутации, при помощи секвенирования ДНК.
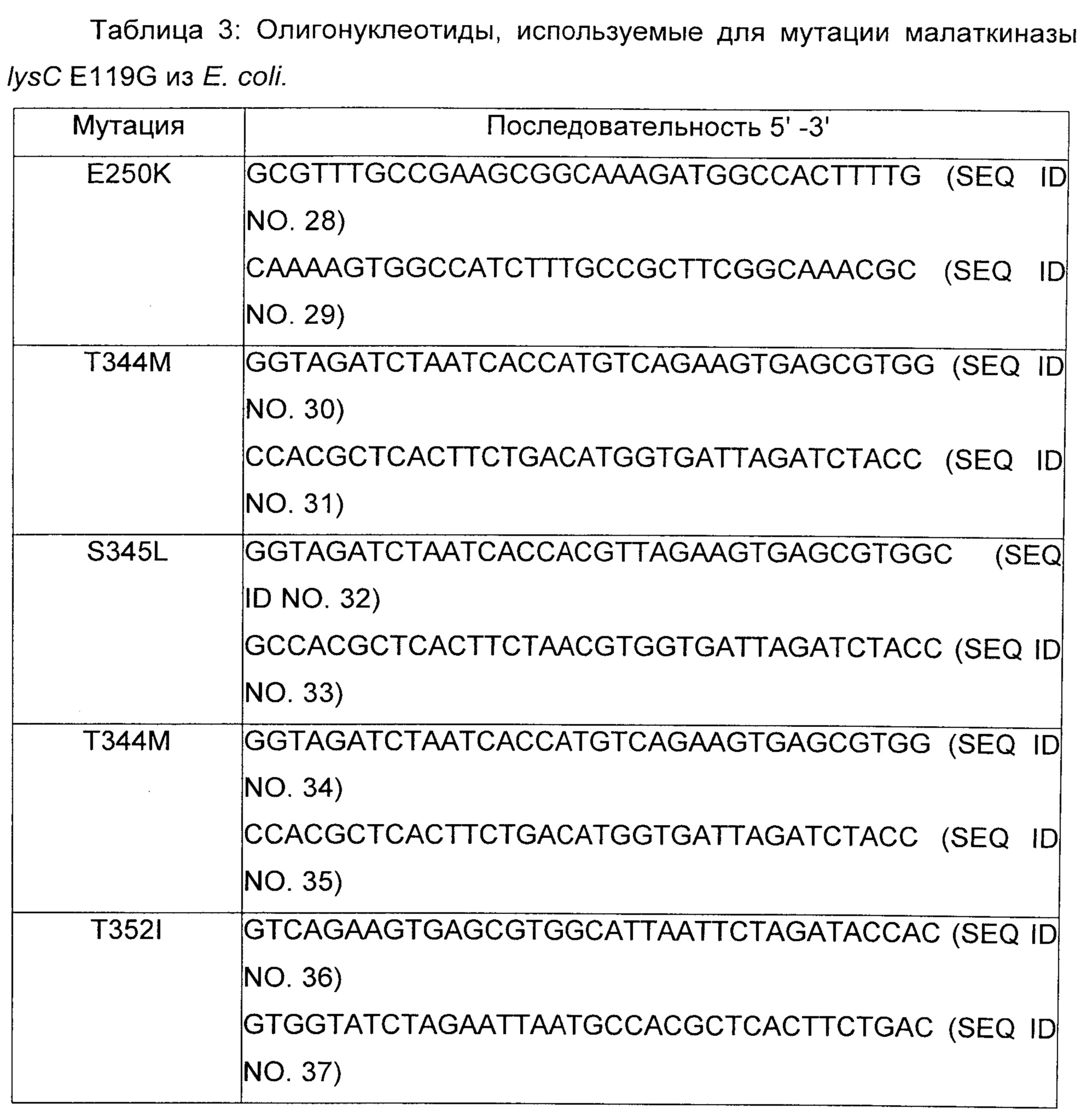
Последовательность нуклеиновой кислоты белка LysC E119G, содержащая дополнительную мутацию, соответствующую (2) замене глутаминовой кислоты в позиции 250 на лизин, представлена в SEQ ID No. 38; ее соответствующая аминокислотная последовательность представлена в SEQ ID No. 39; (2) замена треонина в позиции 344 на метионин представлена в SEQ ID No. 40; ее соответствующая аминокислотная последовательность представлена в SEQ ID No. 41; (3) замена треонина в позиции 352 на изолейцин представлена в SEQ ID No. 42; ее соответствующая аминокислотная последовательность представлена в SEQ ID No. 43, (4) замена серина в позиции 345 на лейцин представлена в SEQ ID No. 44; ее соответствующая аминокислотная последовательность представлена в SEQ ID No. 45.
Экспрессия и очистка ферментов; Экспрессию белка для His-маркированных ферментов LysC E119G, LysC E119G Е250К, LysC E119G Т344М, LysC E119G S345L, LysC E119G T352I осуществляли, как описано в примере 1.
Ферментативный анализ: Малаткиназные активности измеряли как описано в примере 1. Концентрация лизина в реакционном буфере варьировала.
Результаты: Введение мутаций Е250К, Т344М или S345L в LysC E119G приводит к тому, что малаткиназная активность становится в значительной степени нечувствительной к повышенным концентрациям лизина (смотри Фиг.4).
Пример 4: Тестирование аспартат полуальдегид дегидрогеназ Asd из Escherichia coli в отношении аспартат- и малат полуальдегид дегидрогеназной активности
Конструирование плазмид, содержащих гены дикого типа аспартат полуальдегид дегидрогеназы; Плазмиду pASDwt конструировали путем амплификации гена asd из Е.coli при помощи ПЦР с использованием высокоточной полимеразы Phusion™ (Finnzymes) и прямых и обратных праймеров5’TATAATGCTAGCATGAAAAATGTTGGTTTTATCGG3’ (SEQ ID No. 46) и5’TATAATGGATCCTTACGCCAGTTGACGAAGC3’ (SEQ ID No. 47), которая вводит сайт рестрикции NheI и BamHI выше стартового кодона и ниже стоп-кодона, соответственно. Геномную ДНК из Е.coli DH5α использовали в качестве матрицы. Продукт ПЦР расщепляли при помощи NheI и BamHI, лигировали в соответствующие сайты экспрессирующегося вектора рЕТ28а (Novagen) с использованием Т4 ДНК лигазы (Biolabs), и трансформировали в клетки Е.coli DH5α. Получающуюся в результате плазмиду pASDwt выделяли и при помощи секвенирования ДНК показали, что она содержит полноразмерный ген asd, имеющий правильную последовательность (SEQ ID No. 48). Соответствующая аминокислотная последовательность указанного фермента представлена в SEQ ID No. 49.
Экспрессия и очистка ферментов: Экспрессию белка для His-маркированных ферментов Asd осуществляли, как описано в примере 1.
Ферментативный анализ: Аспартат или малат полуальдегид дегидрогеназные активности измеряли в обратном направлении биосинтеза по восстановлению НАДФ во время окисления аспартат или малат полуальдегида до 4-фосфо-(L)-аспартата или 4-фосфо-(L)-малата, соответственно (Roberts et al., 2003).
(L)-аспартат полуальдегид (или (L)-малат полуальдегид) + НАДФ + Pi → 4-фосфо-(L)-аспартат (или 4-фосфо-(L)-малат) + НАДФН
Анализируемая смесь содержала 200 мМ Hepes (pH 9), 50 мМ K2HPO4, 0,25 мМ НАДФ. Реакции инициировали путем добавления (L)-аспартат полуальдегида или (L)-малатполуальдегида. (L)-аспартатполуальдегид добавляли в форме L-аспарагиновой кислоты β-полуальдегида гидрата трифторацетата (поддерживаемого при pH 3 для предупреждения разрушения), который представлял собой подходящий субстрат для ферментативных тестов гомосерин дегидрогеназы и аспартат полуальдегид дегидрогеназы (Roberts et al., 2003). Нестабильный малат полуальдегид готовили свежим перед ферментативными тестами путем удаления защиты со стабильного малат полуальдегидного производного 2-[(4S)-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]ацетальдегида (DMODA). Малат полуальдегид получали путем инкубации DMODA в 2М соляной кислоте в течение 15 мин при 25°С и выпаривания высвободившегося ацетона (35°С, 50 мбар). pH малат полуальдегидного раствора удерживали на уровне 3 с использованием бикарбоната натрия.
Ферментативные анализы осуществляли в 96-луночных плоскодонных микротитровальных планшетах в конечном объеме 250 мкл при 30°С. Реакции контролировали при помощи при помощи характеристического поглощения НАДФН при 340 нм в микропланшетном ридере (BioRad 680XR).
Результаты: His-маркированная аспартат полуальдегид дегидрогеназа дикого типа, Asd, окисляла (L)-аспартат полуальдегид до 4-фосфо-(L)-аспартата с максимальной специфической активностью 160 мкмоль/(мин*мгбелка). В отношении (L)-малат полуальдегида фермент обладал активностью 0,01 мкмоль/(мин*мгбелка).
Пример 5: Сайтнаправленный мутагенез аспартат полуальдегид дегидрогеназы Asd из Escherichia coli и тестирование мутантных ферментов в отношении малат полуальдегид дегидрогеназной активности
Точечные мутации в аминокислотной последовательности Asd вводили с использованием плазмиды pASDwt в качестве матрицы и в соответствии с протоколом, изложенном в примере 2. Пары олигонуклеотидов, перечисленные в таблице 4, использовали для мутации глутаматного остатка в позиции 241 или остатка треонина в позиции 136. Мутантные плазмиды идентифицировали при помощи анализа сайтов рестрикции и убеждались в том, что они несут желаемые мутации, при помощи секвенирования ДНК
Белок Asd, мутантный в позиции 241, может быть представлен в SEQ ID No. 68, где остаток в позиции 241 представляет собой X, где Х представляет собой любую из других 19 биологически встречающихся аминокислот (за исключением глутамина).

Результаты: Активности и значения Km Asd, мутантной в позиции Е241, обобщены в таблице 5. Мутанты Asd, где глутамат 241 был замещен на аланин, цистеин, глицин, гистидин, изолейцин, метионин или глутамин, окисляли (L)-аспартат-4-полуальдегид до 4-фосфо-(L)-аспартата со значительно более высокой максимальной специфической активностью, чем фермент дикого типа. Двойной мутант Asd E241Q T136N (SEQ ID No. 231) обладал максимальной специфической активностью 0,25 мкмоль/(мин*мгбелка) и Km 0,25 мМ.

Соответствующие нуклеиновые кислоты представлены в SEQ ID No. 55, SEQ ID No. 57, SEQ ID No. 48, SEQ ID No. 59, SEQ ID No. 61, SEQ ID No. 63, SEQ ID No. 65 и SEQ ID No. 67.
Двойной мутант Asd E241Q T136N имел последовательность нуклеиновой кислоты, представленную на SEQ ID No. 230.
Пример 6: Идентификация 2,4 ДГМ дегидрогеназы
Для идентификации подходящей 2,4 ДГМ дегидрогеназы дегидрогеназы бета-гидрокискислоты из различных биологических источников тестировали в отношении их способности восстанавливать малат полуальдегид. Среди тестируемых ферментов были метилбутиральдегид редуктаза, Ypr1 (Ford & Ellis, 2002))(SEQ ID No. 73 и SEQ ID No. 74) из Saccharomyces cerevisiae; и редуктаза янтарного полуальдегида, Ms-Ssr из Metallosphaera sedula (Kockelkorn & Fuchs, 2009)(SEQ ID No. 75 и SEQ ID No. 76). Гены YPR1 и Ms-SSR амплифицировали с использованием праймеров, перечисленных в таблице 6 и клонированных в вектор рЕТ28 (ферменты рестрикции смотри в таблице 3) с получением плазмид pYPRt и pMs-SSR, соответственно. Белки экспрессировали и очищали как описано в примере 1.

Тестирование активности малат полуальдегид редуктазы
Реакция:
(L)-Малат полуальдегид + НАД(Ф)Н → (L)-2,4-дигидроксимасляная кислота + НАД(Ф)
Анализируемая смесь содержала 200 мМ Hepes (pH 7,5), 50 мМ KCl, 5 мМ MgCl2, 0,24 мМ НАДН или НАДФН и подходящие количества очищенного фермента. Реакции запускали путем добавления 10 мМ (L)-малат полуальдегида (малат полуальдегид готовили свежим для каждого теста, смотри Пример 4). Ферментативные анализы осуществляли при 30°С в 96-луночных плоскодонных микротитровальных планшетах в конечном объеме 250 мкл. За реакциями следили при помощи характеристического поглощения NAD(P)H при 340 нм в микропланшетном ридере (BioRad 680XR). Результаты представлены в таблице 7.

Дегидрогеназа янтарного полуальдегида M.sedula и метилбутиральдегид редуктаза S.cerevisiae обладают малат полуальдегид редуктазной активностью. Km Ms-SSR для малат полуальдегида составляла 1,1 мМ.
Пример 7: Сайтнаправленный мутагенез редуктазы янтарного полуальдегида M.sedula
Сайтнаправленный мутагенез осуществляли с использованием пар олигонуклеотидов, перечисленных в таблице 8, и плазмиды pMs-SSR в качестве матрицы. Точечные мутации для изменения аминокислотных последовательностей вводили при помощи ПЦР (Phusion 1U, буфер HF 20% (об./об.), dNTP (дезоксирибонуклеотидтрифосфаты) 2,5 мМ, прямой и обратный праймеры 1 мкМ каждого, матричная плазмида 200 нг, вода). При возможности плазмиды, созданные при помощи ПЦР, содержали новые сайты рестрикции (введенные с использованием молчащих мутаций) в дополнение к функциональной мутации для облегчения идентификации мутантных клонов. Продукты ПЦР расщепляли при помощи DpnI при 37°С в течение 1 ч для удаления матричной ДНК, и трансформировали в NEB 5-альфа компетентные клетки Е.coli (NEB). Мутантные плазмиды идентифицировали при помощи анализа сайтов рестрикции и подтверждали, что они несут желаемые мутации, путем секвенирования ДНК. В таблице 9 обобщены кинетические параметры мутантов. Результаты демонстрируют, что двойной мутант Ms-SSR H39R N43H (SEQ ID No. 81, SEQ ID No. 82) обладают улучшенной аффинностью в отношении малат полуальдегида по сравнению с ферментом дикого типа.


Соответствующие последовательности нуклеиновой кислоты представлены в SEQ ID No. 224, SEQ ID No. 226 и SEQ ID No. 82.
Пример 8: Продукция ДГМ in vitro
Ферменты малаткиназу (LysC E119G, SEQ ID No. 15), малат полуальдегид дегидрогеназу (Asd E241Q; SEQ ID No. 67) и малат полуальдегид редуктазу (Ms SSrR, SEQ ID No. 76) экспрессировали и очищали как описано в примере 1. Продукцию ДГМ продемонстрировали in vitro путем добавления 50 мМ малата к реакционной смеси, содержащей 50 мМ Hepes (рН 7,5), 50 мМ KCl, 5 мМ MgCl2, 1 мМ НАДФН, 180 мкг/мл малаткиназы (Lys E119G), 325 мкг/мл малат полуальдегид дегидрогеназы (Asd E241Q) и 130 мкг/мл малат полуальдегид редуктазы (Ms_Ssr) (Реакция А). Контрольные реакционные смеси содержали все компоненты, но были лишены малат полуальдегид редуктазы (Реакция В) или малат полуальдегид дегидрогеназы (Реакция С). После 30 мин инкубации при 30°С реакционную смесь анализировали при помощи газовой хроматографии [CPG Varian Series 430; оборудованный FID (пламенно-ионизационным) детектором; автосэмплером СР8400; инжектором без деления потока 1177 (230°С); колонка: CP-WAX58/FFAP, 30 м × 0,53 мм, df 0,50 мкм; и вкладыш: вводная втулка, колено 6,5×78,5×4 мм GWOL (Varian). Газ-носитель представлял собой азот при скорости потока 25 мл/мин. Пламенную ионизацию осуществляли с использованием смеси воздух-водород (скорости потока составляли 300 мл/мин и 30 мл/мин, соответственно). Температура детектора составляла 240°С. Инжектируемый объем составлял 1 мкл. Температурная программа приведена в таблице 10.
Продукцию ДГМ обнаружили в реакции А (присутствие всех ферментов), но она отсутствовала в контрольных реакциях В и С (Фиг.5).
Пример 9: Оптимизация кодирующей последовательности редуктазы янтарного полуальдегида М.sedula в отношении его экспрессии в Е.coll.
Кодирующую последовательность редуктазы янтарного полуальдегида М.sedula, включающую мутации H39R и N43H, оптимизировали в отношении максимальной экспрессии в Е.coli с использованием программного обеспечения GeneOptimizer®. Синтетический ген получали при помощи GeneArt® Gene Synthesis (Invitrogen Life Technologie). Сайты рестрикции NheI и EcoRI вводили выше стартового кодона и ниже стоп-кодона, соответственно, давая возможность для прямого клонирования в рЕТ28а+ (Novagen).
Получающуюся в результате плазмиду pSSR-H39RN43H-opt выделяли и при помощи секвенирования ДНК показали, что она содержит полноразмерный ген SSR H39R N43H М sedula, имеющий правильную последовательность (SEQ ID No. 228).
Пример 10: Конструирование плазмиды, которая облегчает одновременную экспрессию малаткиназы (мутант гена lysC из Е.coli), малат полуальдегид дегидрогеназы (мутант гена asd из Е.coli), и ДГМ дегидрогеназы (мутант гена редуктазы янтарного полуальдегида М.sedula) с использованием Е.coli в качестве организма-хозяина
Плазмиду pLYSC-E119G E250K (SEQ ID No. 38) использовали в качестве скелета для конструирования оперона. Фрагмент ДНК, содержащий сайт связывания с рибосомой рЕТ28 (Novagen) (rbs) и кодирующую область ASD-E241Q, получали при помощи ПЦР (высокоточная полимераза Phusion™ (Finnzymes)) с использованием pASD-E241Q (SEQ ID No. 55 в качестве матрицы, и прямых и обратных праймеров5’TATAAGGATCCGTTTAACTTTAAGAAGGAGATATACCATGGG3’ (SEQ ID No. 83) и5’TATAAGAATTCTTACGCCAGTTGACGAAG3’ (SEQ ID No. 84), которая вводит сайт рестрикции BamHI и EcoRI выше rbs и ниже стоп-кодона, соответственно. Продукты ПЦР расщепляли при помощи BamHI и EcoRI, лигировали в соответствующие сайты pLYSC-E119G E250K с использованием Т4 ДНК лигазы (Biolabs), и трансформировали в клетки Е.coli DH5α. Получающуюся в результате плазмиду pLYSC-E119G-E250K_ASD-E241Q выделяли, и при помощи секвенирования ДНК показали, что она обладает правильной последовательностью.
Фрагмент ДНК, содержащий сайт связывания с рибосомой рЕТ28 (rbs) и кодирующую область оптимизированной по кодонам Ms-SSR-H39RN43H-opt, получали при помощи ПЦР с использованием pSSR-H39RN43H-opt в качестве матрицы, и прямых и обратных праймеров5’TATAAGCGGCCGCGTTTAACTTTAAGAAGGAGATAT3’ (SEQ ID No. 85) и5’TATAAACTCGAGCTTACGGAATAATCAGG3’ (SEQ ID No. 86), которая вводит сайт рестрикции NotI и PspXI выше rbs и ниже стоп-кодона, соответственно. Продукты ПЦР расщепляли при помощи NotI и PspXI, лигировали в соответствующие сайты pLYSC-E119G-E250K_ASD-E241Q с использованием Т4 ДНК лигазы (Biolabs) и трансформировали в клетки Е.coli DH5α. Получающуюся в результате плазмиду рЕТ28-ДГМ (SEQ ID No. 229) выделяли и при помощи секвенирования ДНК показали, что она обладает правильной последовательностью.
Промоторная область выше 5’, одновременно регулирующая экспрессию трех генов (т.е. промотор Т7 в рЕТ28-ДГМ), может быть заменена на любой другой промотор, как индуцируемый, так и конститутивный, путем расщепления рЕТ28-ДГМ при помощи SphI и XbaI и клонирования еще одной промоторной области подходящими сайтами рестрикции. В качестве примера применения индуцируемого промотора, промотор Т7 скелета рЕТ28-ДГМ заменяли на промотор tac, характеристики которого дают возможность для экспрессии белка в присутствии глюкозы (de Boer et al., 1983). Промотор tac получали из плазмиды рЕХТ20 (Dykxhoorn et al., 1996) путем расщепления плазмиды при помощи SphI и XbaI. Фрагмент ДНК, содержащий промотор, очищали и клонировали в плазмиду рЕТ28-ДГМ, расщепленную SphI и XbaI. Получающуюся в результате плазмиду рТАС-ДГМ выделяли, и при помощи секвенирования ДНК показали, что она обладает правильной последовательностью.

Пример 11: Конструирование штаммов Е.coli для оптимизации перераспределения потока углерода и поступления кофактора НАДФН для ферментативной продукции ДГМ
Несколько генов нарушали в штамме Е.coli MG1655 для оптимизации перераспределения потока углерода и поступления кофактора для продукции ДГМ. Генные делеции осуществляли с использованием способа с рекомбиназой lambda red в соответствии с Datsenko et al. (Datsenko & Wanner, 2000).
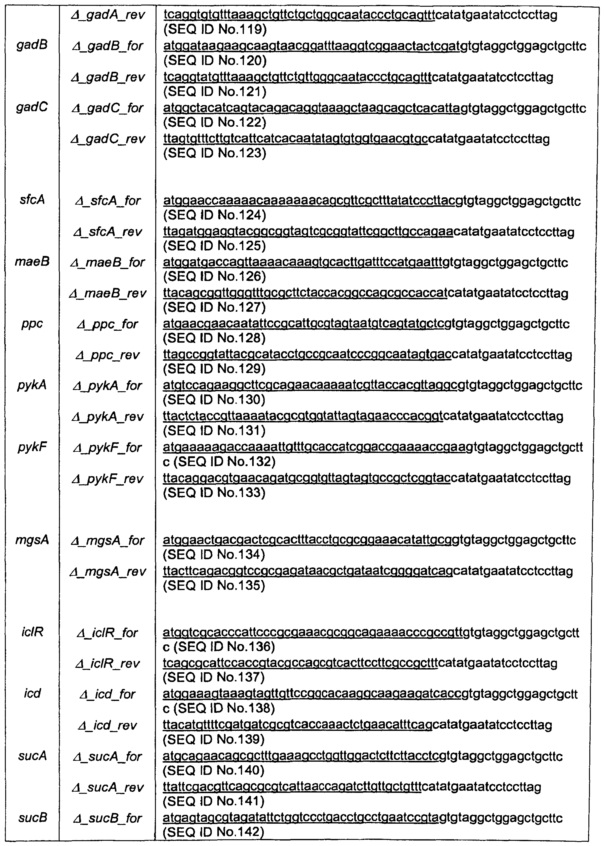
Кассеты с делениями готовили при помощи ПЦР с использованием высокоточной полимеразы Phusion™ (Finnzymes) и FRT-фланкированным геном резистентности к канамицину (kan) плазмиды pKD4 в качестве матрицы (Datsenko & Wanner, 2000). Смысловые праймеры содержали последовательности, соответствующие 5’ концу каждого гена-мишени (подчеркнутому) с последующими 20 п.о., соответствующими кассете FRT-kan-FRT из pKD4. Антисмысловые праймеры содержали последовательности, соответствующие 3’ концевой области каждого гена-мишени (подчеркнутой) с последующими 20 п.о., соответствующими кассете. Праймеры описаны в таблице 12. Продукты ПЦР расщепляли при помощи DpnI и очищали перед трансформацией.
Штам Е.coli MG1655 делали электрокомпетентным путем выращивания клеток до OD600 0,6 в жидкой среде LB (Луриа-Бертани) при 37°С, 100-кратного концентрирования клеток и двойного промывания охлажденным на льду 10% глицерином. Клетки трансформировали плазмидой pKD46 (Datsenko & Wanner, 2000) путем электропорации (2,5 кВ, 200 Ω, 25 мкФ, в кюветах с 2 мм щелью). Трансформантов отбирали при 30°С на ампициллине (100 мкг/мл) на твердой среде LB.
Нарушающие кассеты трансформировали в электрокомпетентные штаммы Е.Coli, несущие плазмиду pKD46, экспрессирующую рекомбиназу lambda Red. Клетки выращивали при 30°С в жидкой среде SOB, содержащей ампициллин (100 мкг/мл). Систему рекомбиназы lambda red индуцировали путем добавления 10 мМ арабинозы, когда OD600 в культурах достигала 0,1. Клетки дополнительно выращивали до OD600 0,6, а затем их собирали путем центрифугирования, дважды промывали охлажденным на льду 10% глицерином, и трансформировали нарушающей кассетой путем электропорации. После фенотипической экспрессии в течение ночи при 30°С в среде LB клетки высевали на твердую среду LB, содержащую 25 мкг/мл канамицина. Трансформанты отбирали путем выращивания при 30°С.
Генную замену проверяли при помощи ПЦР из колонии с использованием Taq полимеразы Crimson (NEB). Первую реакцию осуществляли с фланкирующими локус-специфическими праймерами (смотри таблицу 13) для проверки одновременной утраты родительского фрагмента и получения нового специфического для мутантов фрагмента. Две дополнительные реакции осуществляли с использованием соседних локус-специфических праймеров с соответствующим общим тестовым праймером k1rev или k2for (смотри таблицу 13) в кассете FRT-резистентность к канамицину (смысловой праймер локуса/k1 rev и k2for/o6paтный праймер локуса).
Ген резистентности (FRT-kan-FRT) затем вырезали из хромосомы с использованием плазмиды рСР20, несущей FLP рекомбиназу (Cherepanov & Wackernagel, 1995), оставляя область, содержащую один сайт FRT. рСР20 представляет собой плазмиду, резистентную к амипициллину и CmR, которая демонстрирует температурочувствительную репликацию и температурную индукцию синтеза FLP рекомбиназы. Мутанты, резистентные к канамицину, трансформировали рСР20, и трансформанты, резистентные к ампициллину, отбирали при 30°С. Трансформанты затем выращивали на твердой среде LB при 37°С и тестировали в отношении утраты резистентности к антибиотикам. Вырезание кассеты FRT-канамицин анализировали при помощи ПЦР колоний с использованием taq-полимеразы crimson и фланкирующих локус-специфических праймеров (таблица 13). Множественные делеции получали путем повторения вышеописанных стадий.
Штаммы, несущие единичную или множественные делеции, делали электрокомпетентными, как описано выше, трансформировали плазмидой рТАС-ДГМ, которая давала возможность для индуцируемой IPTG (изопропилтиогалактозид) экспрессии ферментов пути ДГМ (смотри пример 10), и отбирали на твердой среде LB, содержащей 50 мкг/мл канамицина.
Плазмида рАСТ3-pck, несущая ген pck Е.coli, кодирующий PEP карбоксикиназу, конструировали путем амплификации последовательности, кодирующей pck, с использованием геномной ДНК из Е.coli MG1655 в качестве матрицы и прямого и обратного праймеров, соответственно,5’TATAATCCCGGGATGCGCGTTAACAATGGTTTGACC3’ (SEQ ID No. 100) и5’TATAATTCTAGATTACAGTTTCGGACCAGCCG3’ (SEQ ID No. 101). Фрагмент ДНК расщепляли при помощи XmaI и XbaI, лигировали в соответствующие сайты экспрессирующегося вектора рАСТ3 (Dykxhoorn etal., 1996) с использованием Т4 ДНК лигазы (Biolabs), и трансформировали в клетки Е.coli DH5α. Трансформанты отбирали на твердой среде LB, содержащей хлорамфеникол (25 мкг/мл). Получающуюся в результате плазмиду выделяли, и правильность встраивания гена pck подтверждали путем секвенирования. Плазмиды рАСТ3-асеА, рАСТ3-ррс, рАСТ3-galP, рАСТ3-pykA и рАСТ3-рус, несущие, соответственно, асеА, ррс, galP, или pykA (все Е.coli) или русА из Lactococcus lactis, конструировали по аналогии с использованием праймеров, перечисленных в таблице 14.
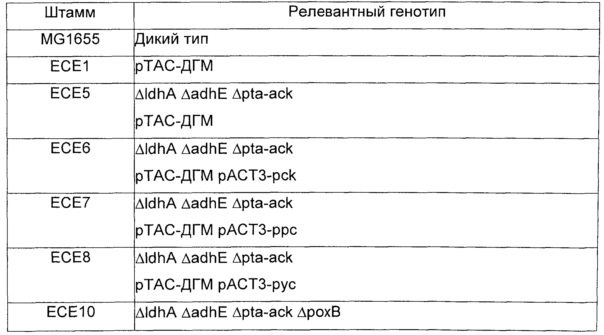
Вышеупомянутые плазмиды, полученные из рАСТ3, и плазмиду рТАС-ДГМ трансформировали мутантам Е.coli MG1655, несущим комбинации делеций, перечисленных в таблице 12. Трансформанты, содержащие обе плазмиды, отбирали на твердой среде LB, содержащей хлорамфеникол (25 мкг/мл) и канамицин (50 мкг/мл). Примеры конструированных штаммов перечислены в таблице 15.







Пример 12: Продукция 2,4-дигидроксимасляной кислоты путем ферментации глюкозы
Штаммы и условия выращивания: Эксперименты осуществляли со штаммом Е.coli ЕСЕ1, соэкспрессирующим малаткиназу, малат полуальдегид дегидрогеназу и ДГМ дегидрогеназу из плазмиды рТАС-ДГМ (смотри пример 11), и изогенным контрольным штаммом, содержащим только пустую плазмиду (т.е. скелет рТАС без кодирующих последовательностей вышеупомянутых ферментов). 1 литр культуральной среды содержал 20 г глюкозы, 18 г Na2HPO4 * 12 H2O, 3 г ⋅ KH2PO4, 0,5 г NaCl, 2 г NH4Cl, 0,5 г MgSO4 * 7 Н2О, 0,015 CaCl2 * 2 H2O, 1 мл 0,06 моль/л концентрированного раствора FeCl3, приготовленного в разбавленной в 100 раз концентрированной HCl, 2 мл 10 мМ концентрированного раствора тиамина HCl, 20 г MOPS, 50 мкг канамицин сульфата и 1 мл раствора следовых элементов (содержащего на литр: 0,04 г Na2ЭДTA * 2H2O, 0,18 г CoCl2 * 6 H2O, ZnSO4 * 7 H2O, 0,04 г Na2MoO4 * 2 H2O, 0,01 г Н3ВО3, 0,12 г MnSO4 * H2O, 0,12 г CuCl2 * H2O). pH доводили до 7 и среду стерилизовали путем фильтрования. Все культивирования осуществляли при 37°С в роторном шейкере Infors при скорости 170 об./мин. Ночные культуры (3 мл среды в тестируемой пробирке) инокулировали из глицериновых концентрированных растворов и использовали для доведения исходной OD600 до 0,05 в 100 мл ростовых культур, выращиваемых в колбах для перемешивания объемом 500 мл. IPTG добавляли в концентрации 1 ммоль/л, пока OD600 в ростовых культурах не достигала 0,2.
Оценка концентрации ДГМ при помощи анализов LC-MS/MS (жидкостная хроматография/тандемная масс-спектрометрия)
Культуральную среду отделяли от клеток путем центрифугирования (Beckmann-Coulter Allegra 21R, Ротор Beckmann S4180, 10 мин, 4800 об./мин). Осветленный супернатант хранили при -20°С до последующего анализа. Содержание ДГМ количественно оценивали с использованием HPLC (Waters) (высокоэффективная жидкостная хроматография), оборудованной колонкой ACQUITY UPLC ВЕН (С18, 1,7 мкм, 100×2,1 мм, Waters), соединенной с масс-спектрометрическим детектором (TQ, Waters, режим ESI (ионизация путем распыления электронов), напряжение на капилляре: 2,5 кВ, напряжение на конусе 25 В, напряжение на экстракторе: 3 В, температура источника: 150°С, температура десольватации: 450°С, поток газа на конусе: 50 л/ч, поток газа для десольватации: 750 л/л). Температуру колонки поддерживали при 30°С. Подвижная фаза представляла собой смесь 88% 0,08% раствора тетра-н-бутиламмония гидрокисда и 12% ацетонитрила. Скорость потока фиксировали на уровне 0,4 мл/мин. Объем инжекции образцов составлял 5 мкл.
Результаты:
Содержание ДГМ в культуральной среде штамма Е.coli ЕСЕ1 и контрольного штамма оценивали через 8 ч и 24 ч после индукции экспрессии малаткиназы, аспартат полуальдегид дегидрогеназы и ДГМ дегидрогеназы путем добавления IPTG. Как можно видеть в таблице 16, штамм ЕСЕ1, который экспрессировал ферменты пути ДГМ, продуцировал значительно более высокие количества ДГМ по сравнению с контрольным штаммом, демонстрируя возможность ферментативной продукции ДГМ при помощи метаболического пути, представленного на Фиг.1 (i).

Ссылки:
Akita, O., Nishimori, C,, Shimamoto, T., Fujii, T. & lefuji, H. (2000). Transport of pyruvate in Saccharomyces cerevisiae and cloning of the gene encoded pyruvate permease. Biosci Biotechnol Biochem 64, 980-984.
Bailey, J.E. (1991). Toward a science of metabolic engineering. Science 252, 1668-1675.
Camarasa, C., Bidard, F., Bony, M., Barre, P. & Dequin, S. (2001). Characterization of Schizosaccharomyces pombe malate permease by expression in Saccharomyces cerevisiae. AppI Environ Microbiol 67, 4144-4151.
Cherepanov, P.P. & Wackernagel, W. (1995). Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9-14.
Datsenko, K.A. & Wanner, B.L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97, 6640-6645.
Deck, P., Exner, K. & Buschhaus, B. (2008). Method for the production of D,L-hydroxy-4-alkylthio butyric acid. Edited by B. AG.
Dykxhoorn, D.M., St Pierre, R. & Linn, T. (1996). A set of compatible tac promoter expression vectors. Gene 177, 133-136.
Ford, G. & Ellis, E.M. (2002). Characterization of Yprlp from Saccharomyces cerevisiae as a 2-methylbutyraldehyde reductase. Yeast 19, 1087-1096.
Grobler, J., Bauer, F., Subden, R.E. & Van Vuuren, H.J. (1995). The mae1 gene of Schizosaccharomyces pombe encodes a permease for malate and other C4 dicarboxylic acids. Yeast 11, 1485-1491.
Groeneveld, M., Weme, R.G., Duurkens, R.H. & Slotboom, D.J. (2010). Biochemical characterization of the C4-dicarboxylate transporter DctA from Bacillus subtilis. J Bacteriol 192, 2900-2907.
James, C.L. & Viola, R.E. (2002). Production and characterization of bifunctional enzymes. Domain swapping to produce new bifunctional enzymes in the aspartate pathway. Biochemistry 41, 3720-3725.
Jantama, K., Haupt, M.J., Svoronos, S.A., Zhang, X., Moore, J.C., Shanmugam, K.T. & Ingram, L.O. (2008a). Combining metabolic engineering and metabolic evolution to develop nonrecombinant strains of Escherichia coli C that produce succinate and malate. Biotechnol Bioeng 99, 1140-1153.
Jantama, K., Zhang, X., Moore, J.C., Shanmugam, K.T., Svoronos, S.A. & Ingram, L.O. (2008b). Eliminating side products and increasing succinate yields in engineered strains of Escherichia coli C. Biotechnol Bioeng 101, 881-893.
Keng, Y.F. & Viola, R.E. (1996). Specificity of aspartokinase III from Escherichia coli and an examination of important catalytic residues. Arch Biochem Biophys 325, 73-81.
Kockelkorn, D. & Fuchs, G. (2009). Malonic semialdehyde reductase, succinic semialdehyde reductase, and succinyl-coenzyme A reductase from Metallosphaera sedula: enzymes of the autotrophic 3-hydroxypropionate/4-hydroxybutyrate cycle in Sulfolobales. J Bacteriol 191, 6352-6362.
Larkin, M.A., Blackshields, G., Brown, N.P. & other authors (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947-2948.
Lin, H., Bennett, G.N. & San, K.Y. (2005). Metabolic engineering of aerobic succinate production systems in Escherichia coli to improve process productivity and achieve the maximum theoretical succinate yield. Metab Eng 7, 116-127.
Marco-Marin, C., Ramon-Maiques, S., Tavarez, S. & Rubio, V. (2003). Site-directed mutagenesis of Eschehchia coli acetylglutamate kinase and aspartokinase III probes the catalytic and substrate-binding mechanisms of these amino acid kinase family enzymes and allows three-dimensional modelling of aspartokinase. J Mol Biol 334, 459-476.
Millard, C.S., Chao, Y.P., Liao, J.C. & Donnelly, M.I. (1996). Enhanced production of succinic acid by overexpression of phosphoenolpyruvate carboxylase in Escherichia coli. Appl Environ Microbiol 62, 1808-1810.
Roberts, S.J., Morris, J.C., Dobson, R.C. & Gerrard, J.A. (2003). The preparation of (S)-aspartate semi-aldehyde appropriate for use in biochemical studies. Bioorg Med Chem Lett 13, 265-267.
Rognstad, R. & Katz, J. (1979). Effects of 2,4-dihydroxybutyrate on lipogenesis in rat hepatocytes. J Biol Chem 254, 11969-11972.
Sanchez, A.M., Bennett, G.N. & San, K.Y. (2005a). Novel pathway engineering design of the anaerobic central metabolic pathway in Escherichia coli to increase succinate yield and productivity. Metab Eng 7, 229-239.
Sanchez, A.M., Bennett, G.N. & San, K.Y. (2005b). Efficient succinic acid production from glucose through overexpression of pyruvate carboxylase in an Escherichia coli alcohol dehydrogenase and lactate dehydrogenase mutant. Biotechnol Prog 21, 358-365.
Sauer, U. & Eikmanns, B.J. (2005). The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol Rev 29, 765-794.
Shinka, T., Inoue, Y., Ohse, M., Ito, A., Ohfu, M., Hirose, S. & Kuhara, T. (2002). Rapid and sensitive detection of urinary 4-hydroxybutyric acid and its related compounds by gas chromatography-mass spectrometry in a patient with succinic semialdehyde dehydrogenase deficiency. J Chromatogr B Analyt Technol Biomed Life Sci 776, 57-63.
Wang, Q., Ou, M.S., Kirn, Y., Ingram, L.O. & Shanmugam, K.T. (2010). Metabolic flux control at the pyruvate node in an anaerobic Escherichia coli strain with an active pyruvate dehydrogenase. Appl Environ Microbiol 76, 2107-2114.
Werpy, T. & Petersen, G. (2004). Top value added chemicals from biomass. Results of screening for potential candidates from sugars and synthesis gas. Washington, DC: http://dx.doi.org/10.2172/15008859.
Zelle, R.M., de Hulster, E., van Winden, W.A. & другие авторы (2008). Malic acid production by Saccharomyces cerevisiae: engineering of pyruvate carboxylation, oxaloacetate reduction, and malate export. Appl Environ Microbiol 74, 2766-2777.
Zeile, R.M., Trueheart, J., Harrison, J.C., Pronk, J.T. & van Maris, A.J. (2010). Phosphoenolpyruvate carboxykinase as the sole anaplerotic enzyme in Saccharomyces cerevisiae. Appl Environ Microbiol 76, 5383-5389.
Zhang, X., Jantama, K., Moore, J.C., Jarboe, L.R., Shanmugam, K.T. & Ingram, L.O. (2009). Metabolic evolution of energy-conserving pathways for succinate production in Escherichia coli. Proc Natl Acad Sci USA 106, 20180-20185.
Zhang, X., Wang, X., Shanmugam, K.T. & Ingram, L.O. (2011). L-malate production by metabolically engineered Escherichia coli. Appl Environ Microbiol 77, 427-434
Реферат
Группа изобретений относится к получению 2,4-дигидроксимасляной кислоты (2,4-ДГМ). Предложен способ получения 2,4-ДГМ, включающий первую стадию превращения малата в 4-фосфомалат с использованием фермента, способного осуществить подобное превращение, вторую стадию превращения 4-фосфомалата в малат-4-полуальдегид с использованием фермента, способного осуществить подобное превращение, и третью стадию превращения малат-4-полуальдегида в 2,4-ДГМ с использованием фермента, способного осуществить подобное превращение. При этом на первой стадии используют фермент, который имеет последовательность, выбранную из группы, состоящей из SEQ ID No. 12, SEQ ID No. 14, SEQ ID No. 16, SEQ ID No. 18, SEQ ID No. 20, SEQ ID No. 22, SEQ ID No. 24, SEQ ID No. 26, SEQ ID No. 39, SEQ ID No. 41, SEQ ID No. 43 и SEQ ID No. 45. На второй стадии используют фермент, который имеет последовательность, выбранную из группы, состоящей из SEQ ID No. 54, SEQ ID No. 56, SEQ ID No. 58, SEQ ID No. 60, SEQ ID No. 62, SEQ ID No. 64, SEQ ID No. 66 и SEQ ID No. 231. На третьей стадии используют фермент, который имеет последовательность, выбранную из группы, состоящей из SEQ ID No. 74, SEQ ID No. 76, SEQ ID No. 81, SEQ ID No. 225 и SEQ ID No. 227. Предложены также варианты ферментов, используемых в способе, варианты нуклеиновых кислот, кодирующих соответствующие ферменты, варианты химерных генов для трансформации микроорганизма-хозяина и экспрессии в этом микроорганизме соответствующего фермента, варианты вектора экспрессии, варианты микроорганизма-хозяина, экспрессирующего соответствующий фермент. 19 н. и 12 з.п. ф-лы, 5 ил., 16 табл.,12 пр.
Формула
Документы, цитированные в отчёте о поиске
Способ ферментативного получения 2-гидрокси-2-метилкарбоновых кислот
Комментарии