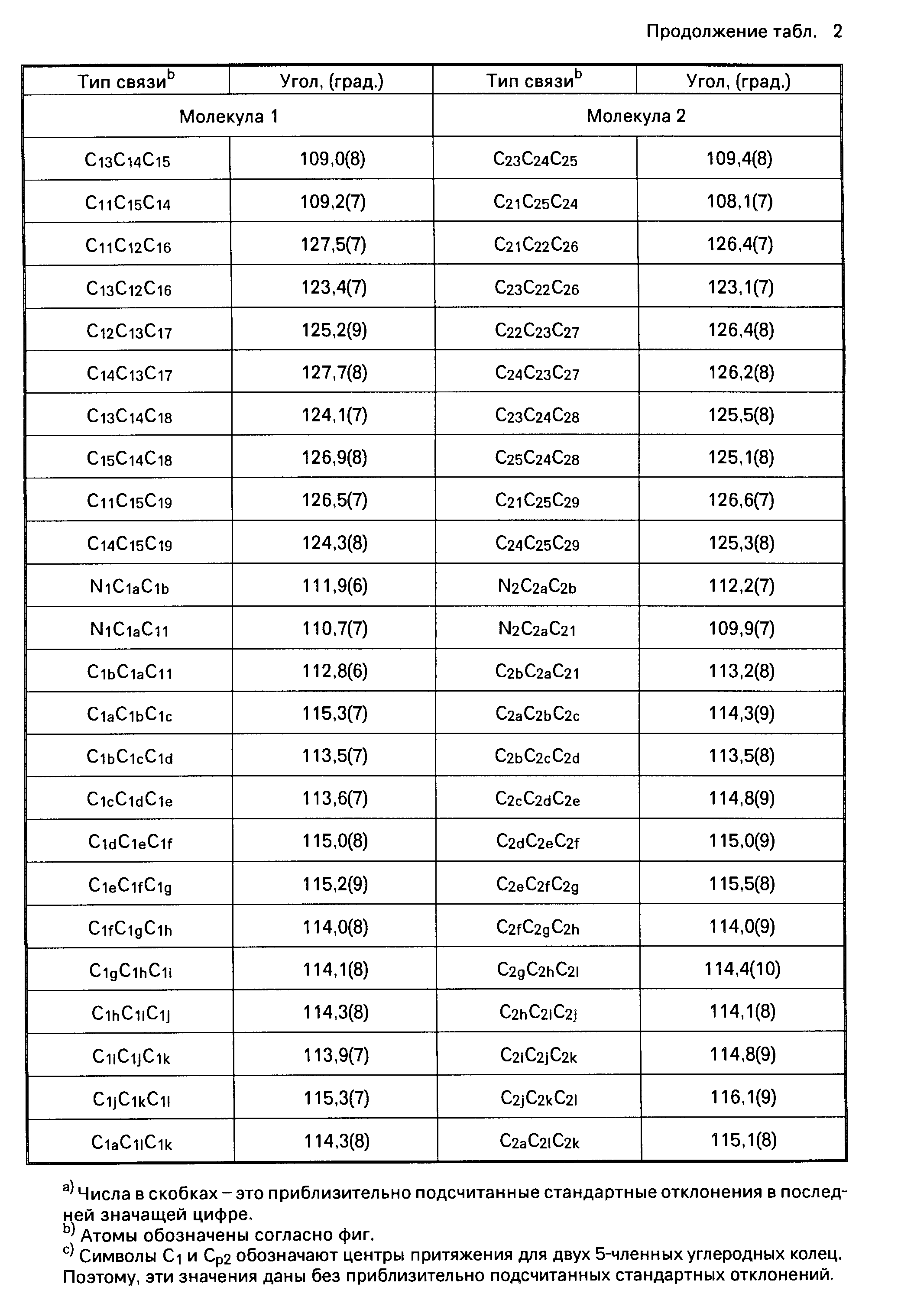
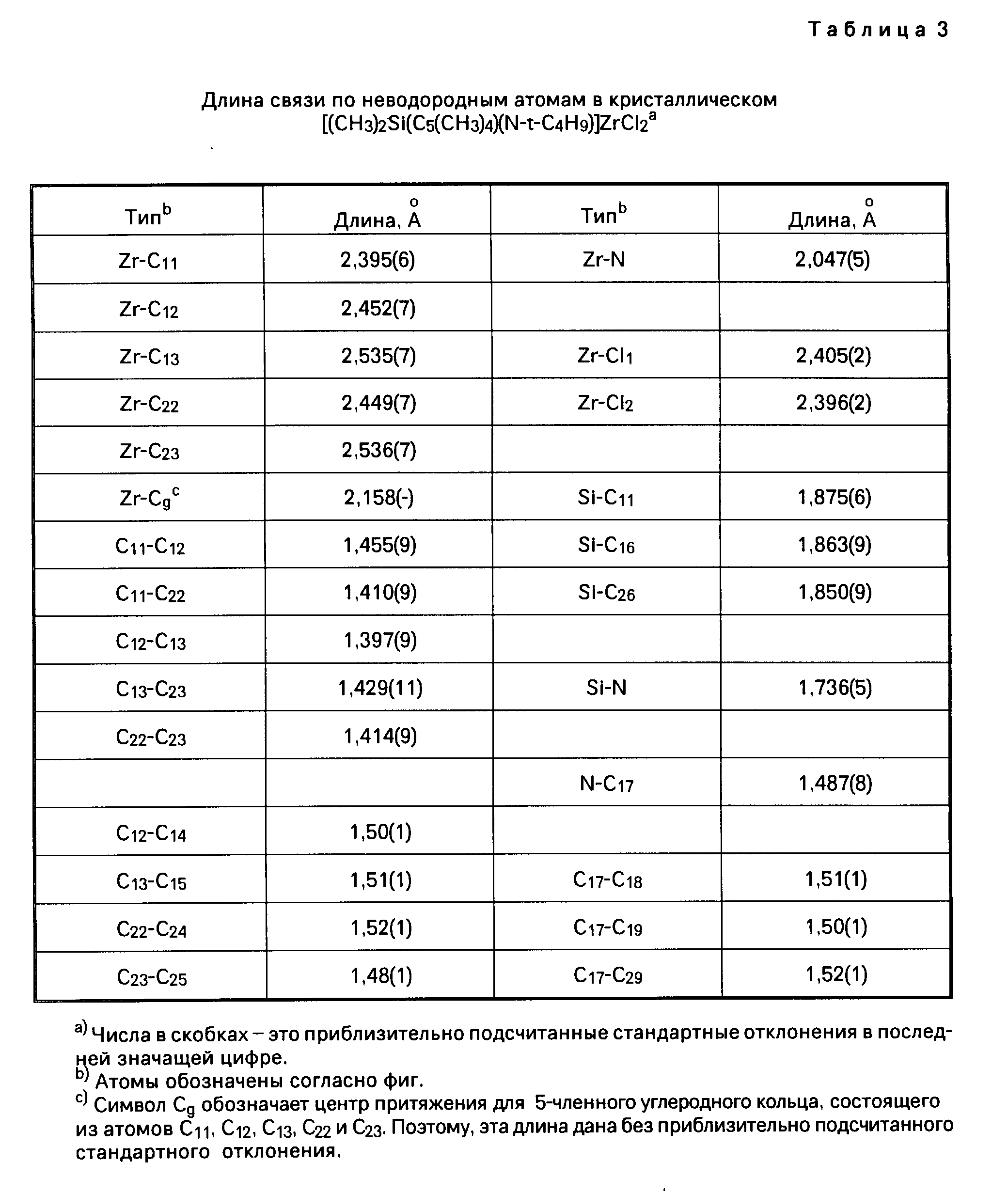
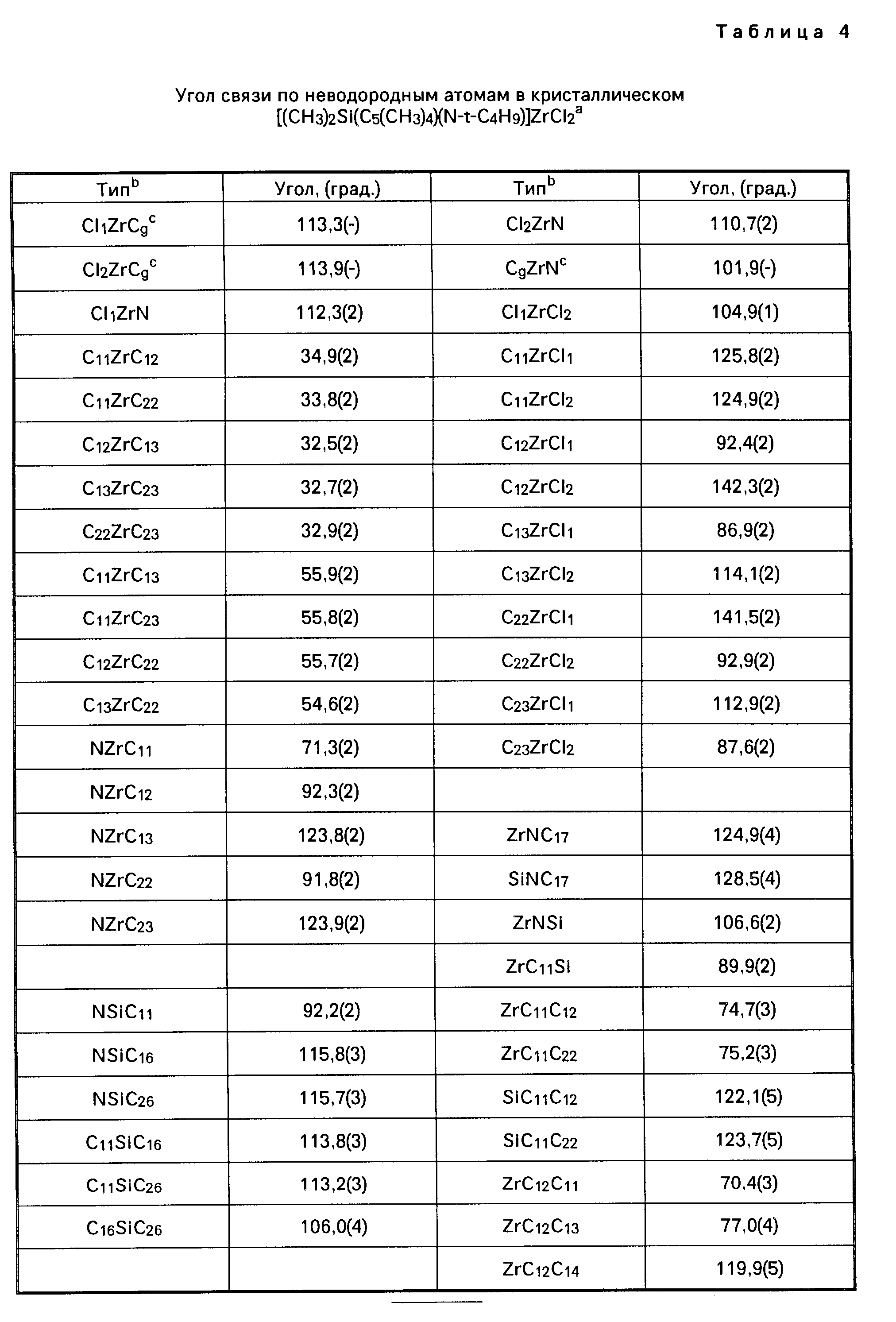
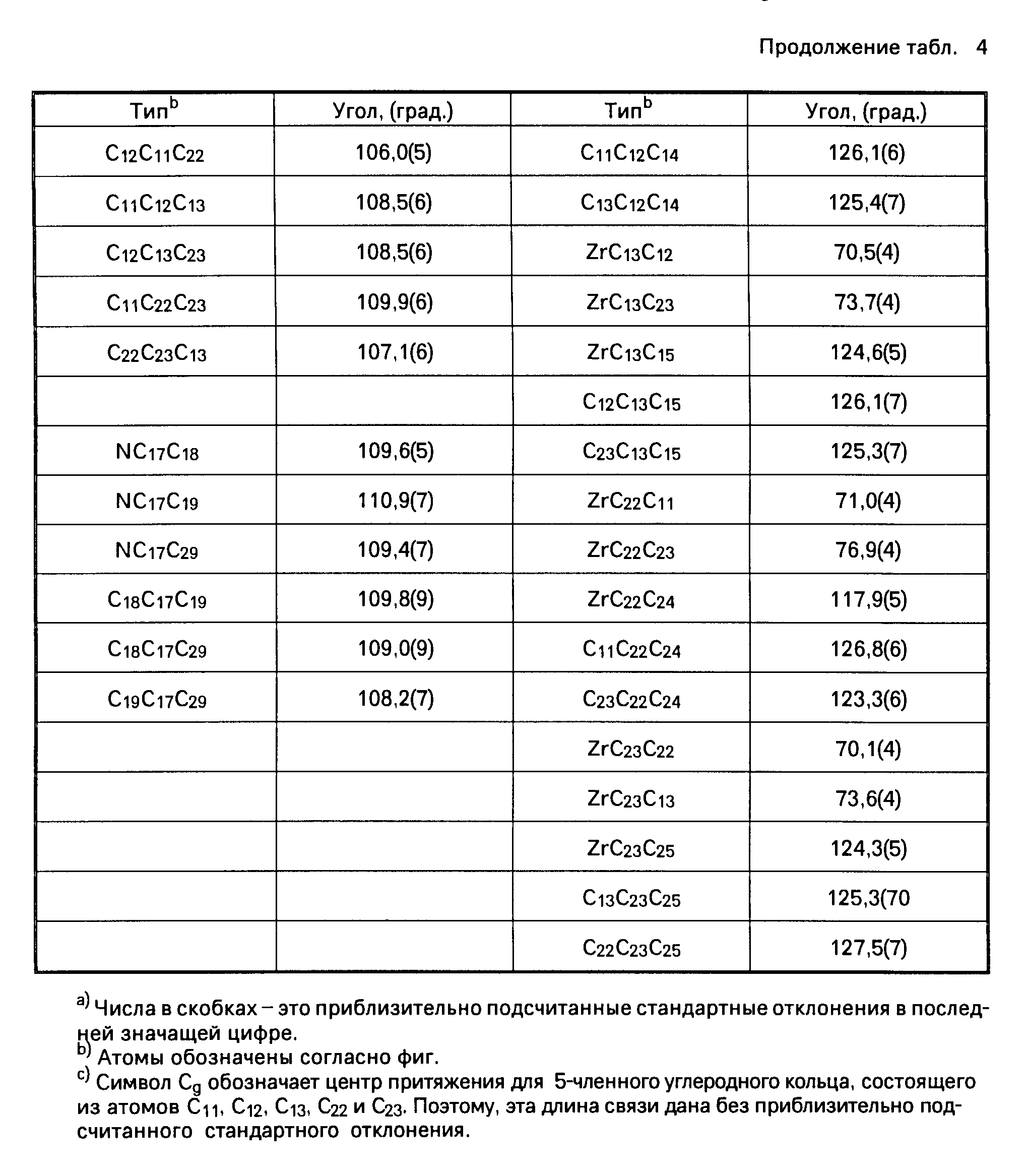
Координационные соединения моноциклопентадиенила с переходными металлами, способ их получения и каталитическая система для получения полиолефинов - RU2067981C1
Код документа: RU2067981C1
Чертежи





Описание
Настоящее изобретение относится к новым координационным соединениям моноциклопентадиенила с переходными металлами IVB группы Периодической системы элементов, способу их получения и каталитической системе, содержащей моноциклопентадиенил переходного металла IVB группы и алюмоксан для синтеза полиолефинов, например, полиэтилена, полипропилена, и α-олефиновых сополимеров, в частности, имеющих большую молекулярную массу сополимеров a-олефина с этиленом и пропиленом.
Предложенная каталитическая система обладает высокой активностью при небольших соотношениях между алюминием и переходным металлом группы IVB Периодической системы элементов и, таким образом, приводит к получению полиолефинового продукта с весьма низким содержанием остающегося в нем металла из каталитической системы. Каталитические системы, в которых в используется титан и те, которые используются в виде модификации без носителя, обладают стабильностью при высоких давлениях (в отличие от их титанорганических аналогов, а именно в отличие от бисциклопентадиенила титана) и проявляют способность катализировать (т.е. ускорять) процесс введения или внедрения более значительных количеств a-олефинового сомономера в структуру полимера при получении высокомолекулярных a-олефиновых сополимеров, чем аналогичные металлокомплексные (металлоорганические) каталитические системы, основанные на использовании моноциклопентадиенилов циркония и гафния.
В данной области техники хорошо известны различные способы и катализаторы для осуществления гомополимеризации или сополимеризации олефинов. Во многих случаях при практическом использовании весьма существенно, чтобы полученный полимерный продукт имел бы значительную среднемолекулярную массу и сравнительно узкое молекулярно-массовое распределение. Сочетание большой средней массы и молекулярной массы с узким молекулярно-массовым распределением позволяет получать полиолефин или сополимер этилена с a-олефином, обладающий высокими прочностными свойствами.
Хорошо известные каталитические системы Циглера-Натта, включающие в себя соединение переходного металла и алкилпроизводное алюминия (алюминийалкил), позволяют получать полиолефины большой молекулярной массы, но с широким молекулярно-массовым распределением.
Совсем недавно была разработана каталитическая система, в которой соединение переходного металла содержит два или большее число лигандов циклопентадиениловых колец. Такое соединение переходного металла получило название металлоцена и катализирует процесс получения полиолефинов из олефиновых мономеров. Подобные соединения переходного металла группы IVB Периодической системы элементов и, в частности, так называемые титаноцены и цирконоцены были использованы как содержащий переходный металл компонент в металлоценовой (т. е. содержащей металлоцен) каталитической системе для катализа процесса синтеза полиолефинов и сополимеров этилена с a-олефинами. Когда такие металлоцены используются в качестве сокатализаторов с алкилпроизводным алюминия, как это имеет место в хорошо известной каталитической системе Циглера-Натта, то каталитическая активность подобной, основанной на использовании металлоценов, каталитической системы, оказывается, как правило, слишком низкой и поэтому эти каталитические системы не нашли широкого промышленного применения.
Известно также, что указанные выше металлоцены могут применяться в качестве сокатализаторов в сочетании с так называемым алюмоксаном, а не с алюминийалкилом, для создания металлоценовой каталитической системы с высокой каталитической активностью для осуществления промышленного синтеза полиолефинов.
Так называемые цирконоцены в случае, если они используются в качестве сокатализаторов с алюмоксаном, как правило, являются более активными, чем их титановые или гафниевые аналоги, при полимеризации этилена, взятого отдельно или в сочетании с a-олефиновым сомономером. Когда цирконоценовые каталитические комплексы или системы используются без носителя, т.е. используются как цельная гомогенная или растворимая каталитическая система, то для обеспечения высокой производительности процесса полимеризации даже при применении самого активного цирконоценового катализатора приходится использовать такое количество алюмоксанового промотора, при котором отношение числа атомов алюминия к числу атомов переходного металла (в молекуле каталитического комплекса) по меньшей мере было бы больше 1000:1, а лучше больше чем 5000:1, и предпочтительно более 10000:1. При использовании таких больших количеств алюмоксанового промотора каталитической системы в полученном полимере остается нежелательная примесь в виде значительного количества остатка металла катализатора ("золы" или, другими словами, неулетучивающегося остатка металла катализатора). При осуществлении процессов полимеризации при высоких давлениях с использованием растворимых каталитических систем и, в частности, когда давление в полимеризационной установке или реакторе превышает 500 бар, можно применять только цирконоценовые или гафноценовые каталитические комплексы. Титаноценовые каталитические комплексы обычно нестабильны при таких высоких давлениях полимеризации, если они не нанесены на подложку или носитель.
В качестве компонентов промотированных алюмоксаном каталитических систем можно использовать весьма широкую гамму соединений переходных металлов группы IVB Периодической системы элементов. Хотя соединения переходных металлов группы IVB Периодической системы элементов с бис(циклопентадиенилом) были наиболее предпочтительными и широко исследовались на возможность их использования в промотированных (активированных) алюмоксаном каталитических системах для производства полиолефинов, были высказаны предположения, что практическое применение могут также найти и соединения переходных металлов группы IVB с моно- и трис(циклопентадиенилом) (см. патенты США 4522982, 4530914 и 4701431). В частности, среди подобных соединений переходных металлов с моно(циклопентадиенилом), которые можно было использовать в промотированной алюмоксаном каталитической системе, следовало бы назвать тригалоген- и триалкилпроизводные комплексов переходных металлов с моно(циклопентадиенилом).
В одной из самых последних международных публикаций, а именно в международной заявке 87/03887 говорится о возможности использования состава, содержащего переходный металл, который имеет координационную связь с по крайней мере одним циклопентадиениловым лигандом и по крайней мере одним гетероатомным лигандом, промотированный алюмоксаном для полимеризации a-олефинов. В качестве переходного металла предпочтительно использовать металл, входящий в IVB группу Периодической системы, связанный координационной связью с по крайней мере одним циклопентадиениловым лигандом и с 1-3 гетероатомными лигандами. При этом координация атома переходного металла каталитической системы обеспечивается циклопентадиениловыми или углеводородными лигандами. Каталитические системы, которые рассматриваются в указанной выше публикации, относятся к соединениям переходных металлов группы IVB Периодической системы элементов с бисциклопентадиенилом. Эти соединения переходных металлов называются, как указывалось выше, металлоценами.
На Третьем Химическом Конгрессе с участием специалистов стран Северной Америки, который проводился в г. Торонто в Канаде в июне 1988 года, докладчик Джон Беркоу говорил о предпринимаемых специалистами попытках практического использования соединения переходного металла группы IIIB Периодической системы, координационно-связанного с одним единственным лигандом, имеющим мостиковую связь с гетероатомом циклопентадиенилового кольца, в качестве каталитической системы для полимеризации олефинов. Хотя это соединение и проявляло некоторую каталитическую активность в практических условиях полимеризации, степень каталитической активности указанного соединения и свойства полученного полимерного продукта не подтвердили того мнения, что такой каталитический комплекс переходного металла с моноциклопентадиенилом может быть успешно использован для промышленного синтеза полимеров.
Таким образом, все еще существует необходимость в разработке каталитических систем, которые позволили бы производить высокомолекулярные полиолефины и желательно с узким молекулярно-массовым распределением. Кроме того, было бы желательно создать такой катализатор, который при практически разумных весовых соотношениях между мономерами, т.е. между этиленом и a-олефином, смог бы способствовать введению более значительных количеств a-олефиновых сомономеров при производстве сополимеров этилена с a-олефинами.
Согласно настоящему изобретению предлагаются новые
координационные соединения моноциклопентадиенила с переходными металлами общей структурной формулы I:
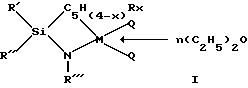
где: RI низший алкил,
RII низший алкил или фенил,
RIII низший алкил, незамещенный или замещенный низшим алкилом фенил или циклоалкил С6-C12, или низший алкоксил,
M цирконий, гафний, титан в их высшей степени окисления,
C5H(4-x)Rx циклопентадиениловое кольцо, которое может быть замещено 1-4 радикалами R, где R означает низший алкил или три(низший алкил)силил, либо в котором две или более групп R вместе с циклопентадиениловым кольцом образуют индениловую или флуорениловую систему,
x от 0 до 4,
n от 0 до 3,
Q галоид или низший алкил.
Объектом настоящего изобретения является также способ получения соединений
формулы I, заключающийся в том что галогенид соответствующего переходного металла или его диэфират подвергают взаимодействию с солью щелочного металла формулы:
[C5H(4-x)
RxSiR'R''(NR''')]Me2,
где R, R', R'', R''', x имеют вышеуказанные значения;
Me щелочной металл; при необходимости в присутствии (C2H5)2O.
Предпочтительно в качестве соли щелочного металла используют соль лития.
Предложенная в соответствии с настоящим изобретением каталитическая система содержит компонент на основе переходного металла, выбранного из группы IVB Периодической системы элементов (см. изданный фирмой "СиЭрСи" Справочник по химии и физике, 68-ое издание, 1987-1988 гг.), и алюмоксановый компонент. Эта каталитическая система может быть использована для осуществления процессов полимеризации в растворе, в суспензии или полимеризации в массе или объеме (блочной полимеризации) в целях производства полиолефинов со значительной среднемассовой молекулярной массой и со сравнительно узким молекулярно-массовым распределением.
Входящее в состав предложенной каталитической системы координационное соединение моноциклопентадиенила с переходным металлом IVB группы Периодической системы элементов имеет структурную формулу I, а в качестве алюмоксана содержит метилалюмоксан при молярном соотношении метилалюмоксана к соединению переходного металла, равном 465-16100.
Каталитические системы, предложенные в соответствии с настоящим изобретением, могут быть получены путем помещения указанного выше компонента на основе переходного металла группы IVB Периодической системы элементов и метилалюмоксана, находящимся в одном общем растворе, в жидкий (в нормальных условиях) алкановый или ароматический растворитель, предпочтительно в такой растворитель, который пригоден для осуществления жидкофазной полимеризации олефинового мономера. Указанные ранее типы компонентов на основе переходных металлов группы IVB Периодической системы, в которых в качестве переходного металла используется титан, придают, как это было установлено, весьма благоприятные положительные свойства каталитической системе. Появление таких свойств оказалось совершенно неожиданным с точки зрения того, что уже ранее было известно о свойствах соединений титана с бисциклопентадиенилом, используемых в качестве сокатализаторов в сочетании с алюмоксанами. Хотя и известно, что титаноцены в растворимой форме обычно являются нестабильными в присутствии алкилпроизводных алюминия, предлагаемые в соответствии с настоящим изобретением соединения типа с моноциклопентадиенилом и в которых гетероатомом является атом азота, как правило, обладают более высокой стабильностью (устойчивостью) в присутствии алюминийалкилов и более высокой каталитической активностью и способствуют более интенсивному внедрению молекул α -олефинового сомономера в структуру каталитического комплекса.
Кроме того, предложенный в соответствии с настоящим изобретением катализатор на основе соединения переходного металла группы IVB Периодической системы элементов, а именно титана, обычно обладает более высокой каталитической активностью, обеспечивает получение полимеров с большей молекулярной массой (т. е. более высокомолекулярных полимеров) и с более высоким содержанием a-олефинового сомономера в макромолекуле полимера, чем каталитические системы, полученные на основе соединений, входящих в группу IVB Периодической системы элементов других переходных металлов, а именно циркония или гафния.
Типичный процесс полимеризации, осуществляемый с помощью каталитических систем, предлагаемых в соответствии с настоящим изобретением, например, такой процесс, как полимеризация или сополимеризация этилена, включает в себя следующие две стадии: стадию ввода этилена или a-олефинов ряда C3-C20, взятых отдельно или в сочетании с другими ненасыщенными мономерами, включая a-олефины ряда C2-C20, диолефины ряда C5-C20 и/или ацетиленненасыщенные (ненасыщенные по ацетиленовым связям) мономеры, взятые отдельно или в сочетании с другими олефинами и/или другими ненасыщенными мономерами, в контакт с катализатором, содержащим находящиеся в пригодном для осуществления полимеризации растворителе (так называемом полимеризационном растворителе) следующие два компонента: указанное выше металлорганическое соединения переходного металла группы IVB Периодической системы и метилалюмоксан, взятые в таких количествах, при которых молярное соотношение между алюминием и переходным металлом находится в пределах между примерно 465 и 16100; и стадию осуществления реакции полимеризации указанного выше мономера в присутствии этой каталитической системы при температуре в пределах от около -100 до 300oC в течение интервала времени от 1 сек до примерно 10 часов. В результате реакции полимеризации получается полиолефин со среднемассовой молекулярной массой в пределах от примерно 1000 или менее до примерно 5000000 или более единиц и с молекулярно-массовым распределением в пределах от 1,5 до 15,0.
Низшими алкилами, используемыми в качестве лиганда "Q", могут быть, например, такие радикалы как метил, этил, пропил, бутил, амил, изоамил, гексил, изобутил, гептил. Наиболее предпочтительным радикалом-лигандом является, однако, метил. В качестве галогенов (атомов галогенов), входящих в состав лиганда "Q", можно назвать, например, хлор, бром, фтор и йод. При этом наиболее предпочтительным галогеном является хлор.
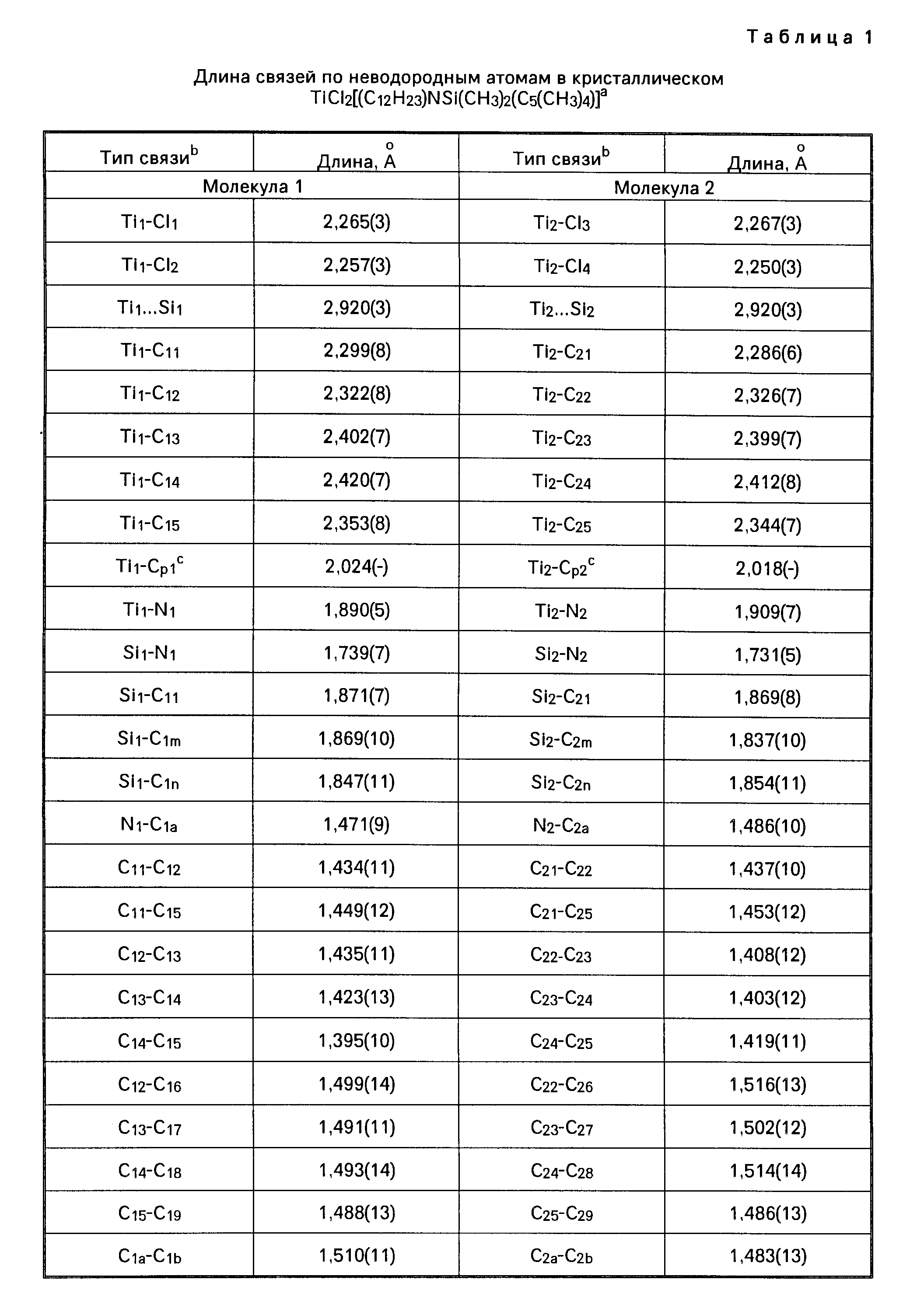
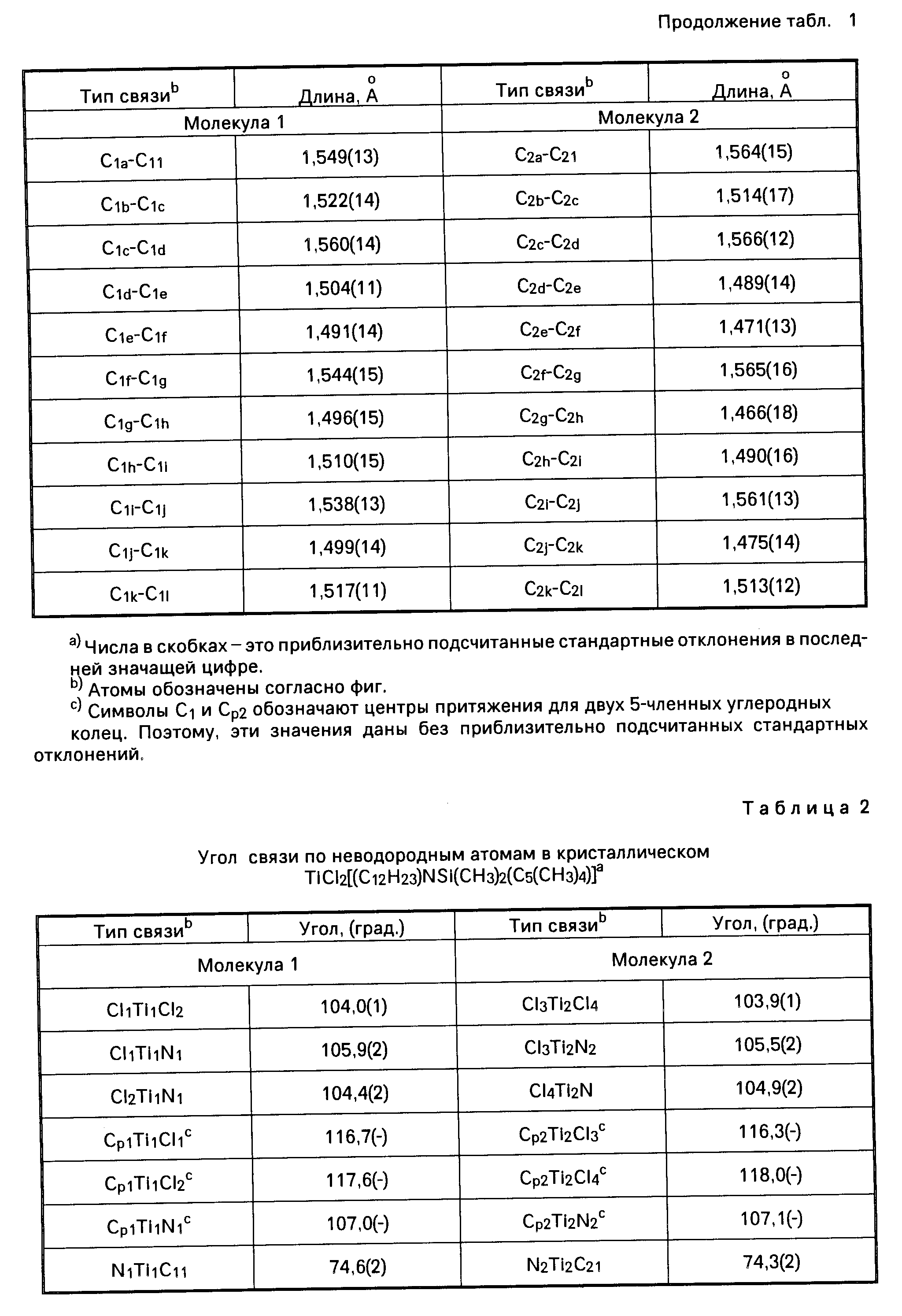
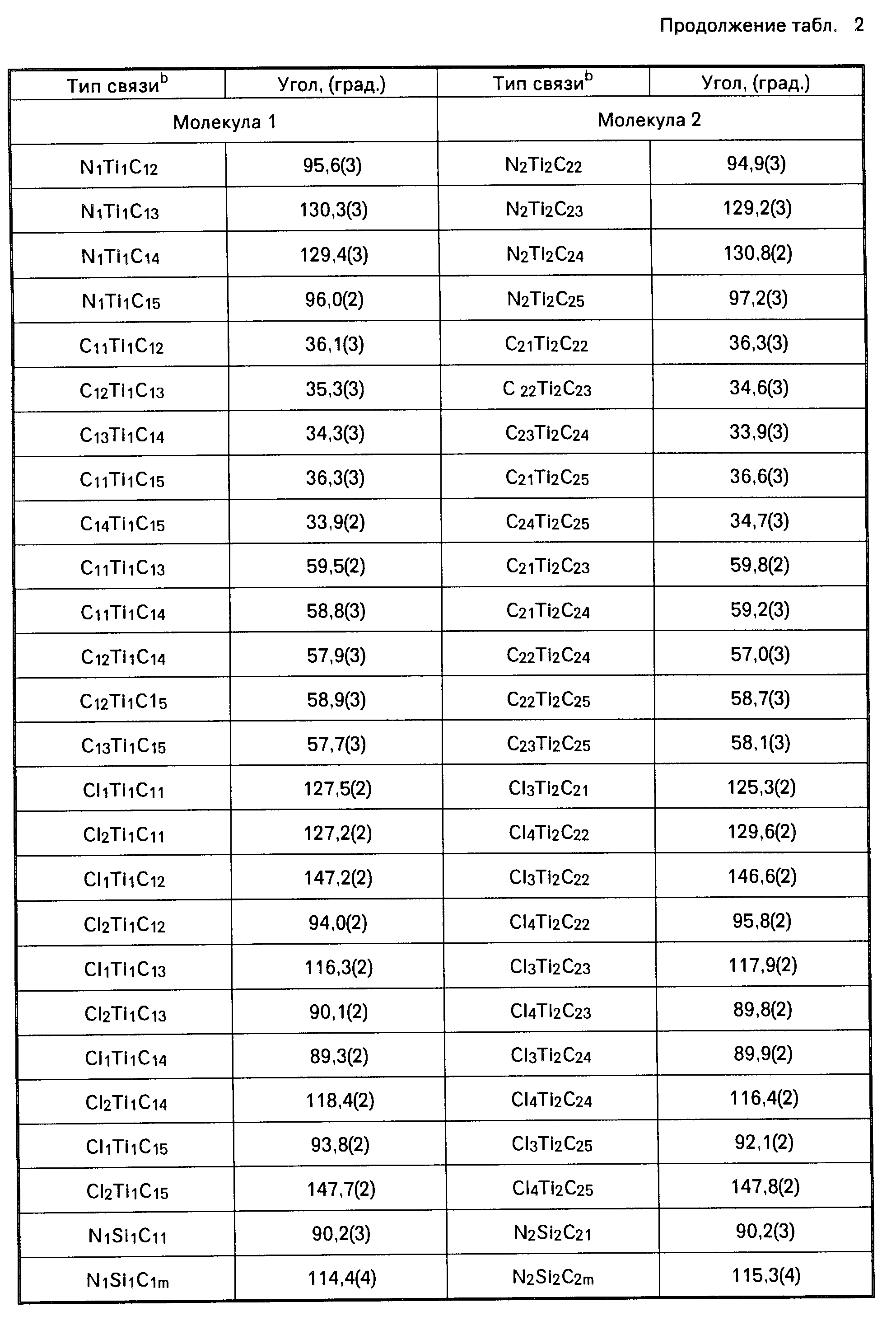
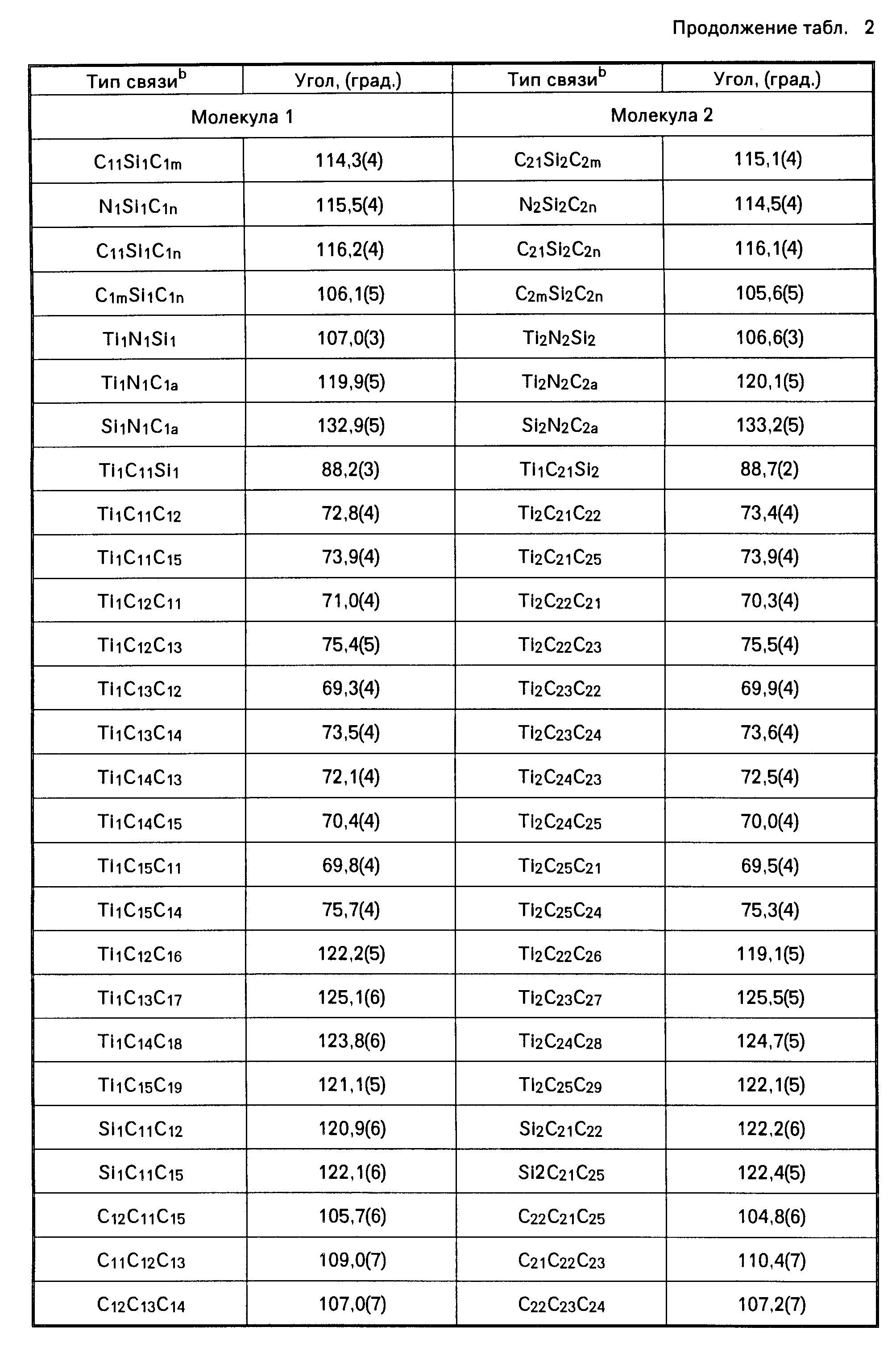
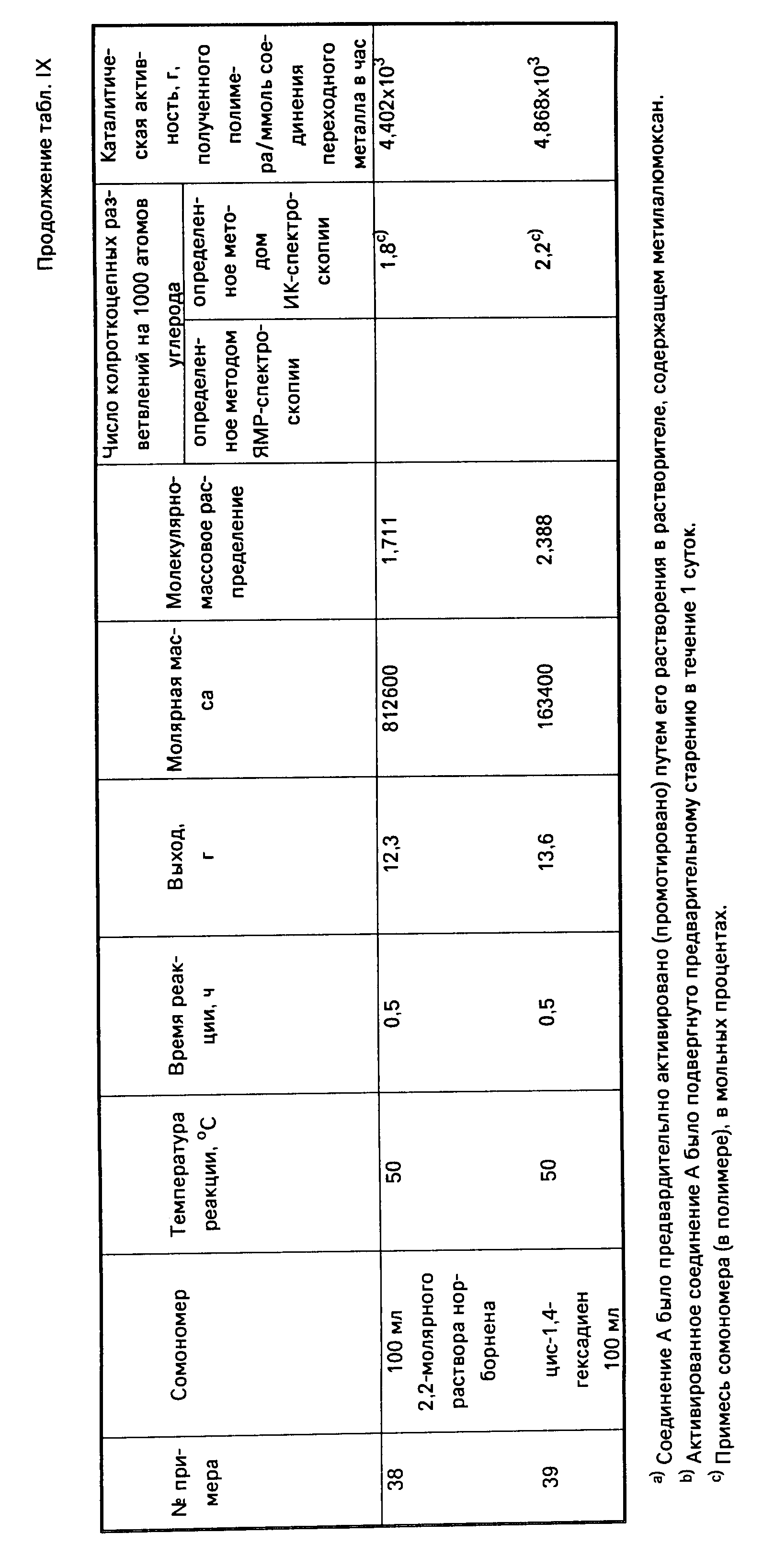
Амидами, используемыми в качестве лиганда "Q", могут быть, например, диметиламид, диэтиламид, метилэтиламид, ди-трет.-бутиламид, диизопропиламид и другие подобные амиды. В числе ариламидов следует назвать дифениламид или любой другой замещенный фениламид. Среди фосфидов, которые могут использоваться в качестве лиганда "Q", следует назвать, например, дифенилфосфид, дициклогексилфосфид, диэтилфосфид, диметилфосфид и другие подобные фосфиды. В качестве алкилиденовых радикалов в обоих лигандах "Q" могут использоваться, например, метилиден, этилиден и пропилиден. В приводимой далее в тексте описания настоящего изобретения таблице 1 (а именно в столбце 4, под заголовком "Q") указаны примеры групп или радикалов, используемых в качестве лиганда "Q" в соединении переходного металла группы IVB Периодической системы элементов, которое используется как одна из частей каталитической системы согласно настоящему изобретению.
Наиболее подходящие для осуществления целей настоящего изобретения углеводородные и замещенные радикалы, которые в виде R-группы могут замещать, как минимум, один атом водорода в циклопентадиениловом кольце, представляют собой прямые и разветвленные низшие алкильные радикалы, либо две или более группы R вместе с циклопентадиениловым кольцом образуют индениловую или флуорениловую группу.
Наиболее подходящие для осуществления целей настоящего изобретения металлорганические радикалы, которые в виде R-группы могут замещать, как минимум, один атом водорода в циклопентадиениловом кольце, включают в себя триметилсилил, триэтилсилил, этилдиметилсилил, метилдиэтилсилил.
В качестве возможных характерных примеров соединений можно назвать следующие: диметилсилилтетраметилциклопентадиенил-трет. -бутиламидоцирконийдихлорид, диметилсилилтетраметилциклопентадиенил-трет. -бутиламидо-гафнийдихлорид, диметилсилил-трет.-бутилциклопентадиенил-трет.-бутиламидо-цирконийдихлорид, диметилсилил-трет. -бутилциклопентадиенил-трет.-бутиламидо-гафнийхлорид, диметилсилилтриметилсилилциклопентадиенил-трет. -бутиламидо-гафнийхлорид, диметилсилилтетраметилциклопентадиенилфениламидо-цирконийдихлорид, диметилсилилтетраметилциклопентадиенилфениламидо-гафнийдихлорид, метилфенилсилилтетраметилциклопентадиенил-трет.-бутиламидо-цирконийдихлорид, метилфенилсилилтетраметилциклопентадиенил-трет.-бутиламидо-гафнийдихлорид, метилфенилсилилтетраметилциклопентадиенил-трет.-бутиламидо-гафнийдихлорид, диметилсилилтетраметилциклопентадиенил-n-н-бутилфениламидо-цирконийдихлорид и диметилсилилтетраметилциклопентадиенил-n-н-бутилфениламидо-гафнийдихлорид.
Как уже указывалось выше, соединения переходного металла (металлоорганическое комплексное соединение), а именно титана, дают возможность создавать каталитические системы, которые по сравнению с аналогичными соединениями циркония или гафния обладают более высокой каталитической активностью и обеспечивают более высокую способность или эффективность внедрения a-олефинового сомономера в структуру каталитического комплекса. В качестве этих характерных металлорганических соединений, а именно соединений титана, которые обладают такими превосходными каталитическими свойствами, можно назвать такие, далеко не единственные, комплексы титана, как, например: метилфенилсилилтетраметилциклопентадиенил-трет. -бутиламидо-титандихлорид, диметилсилилтетраметилциклопентадиенил-n-н-бутилфениламидо-титандихлорид, диметилсилилтетраметилциклопентадиенил-n-метоксифениламидо-титандихлорид, диметилсилил-трет.-бутилциклопентадиенил-2,5-ди-трет.-бутилфенил амидо-титандихлорид, диметилсилилинденил-трет. -бутиламидо-титандихлорид, диметилсилилтетраметилциклопентадиенилциклогексиламидо-титандихлорид, диметилсилилфлуоренилциклогексиламидо-титандихлорид, диметилсилилтетраметилциклопентадиенилфениламидо-титандихлорид, диметилсилилтетраметилциклопентадиенил-трет.-бутиламидо-титандихлорид.
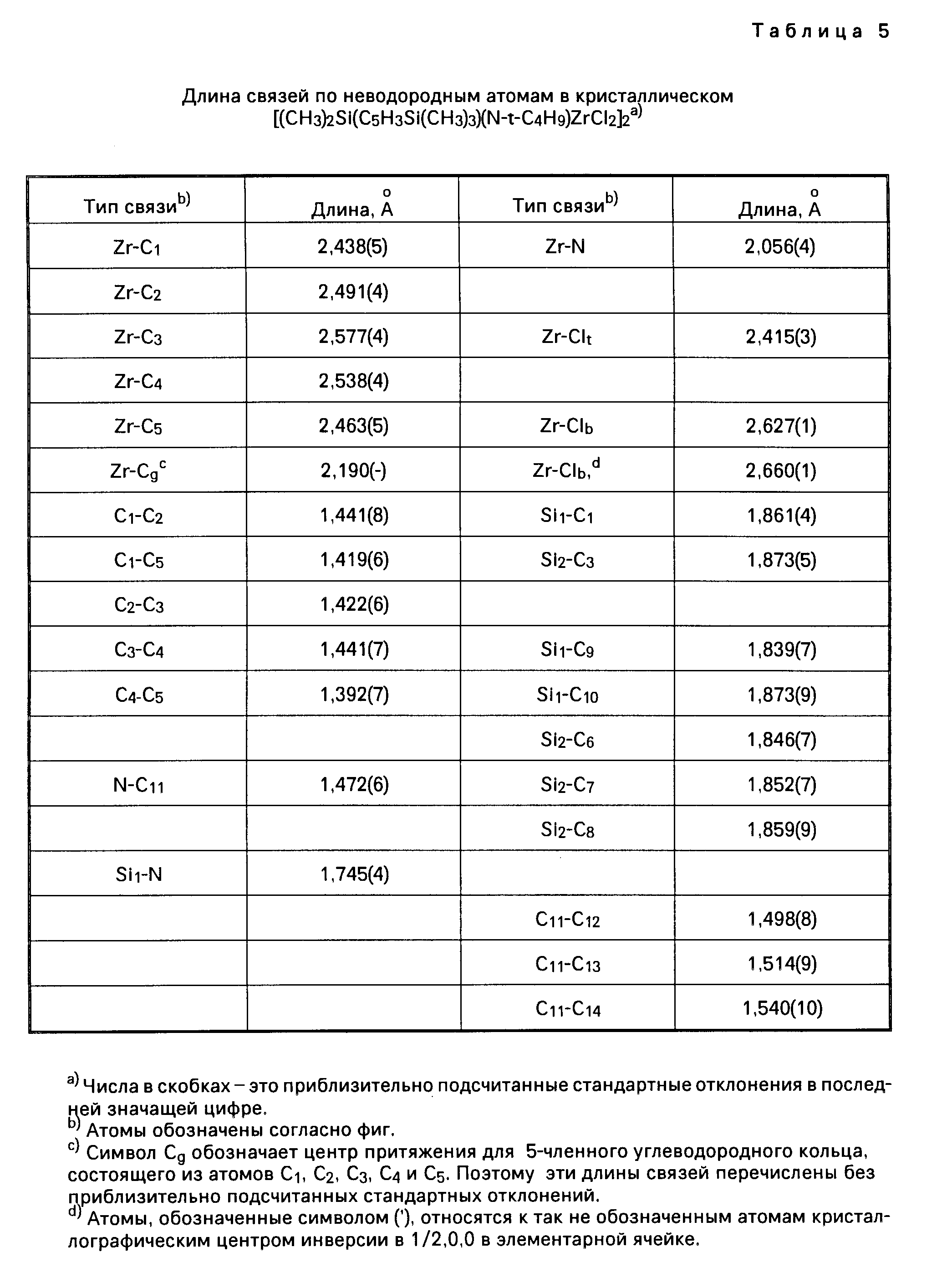
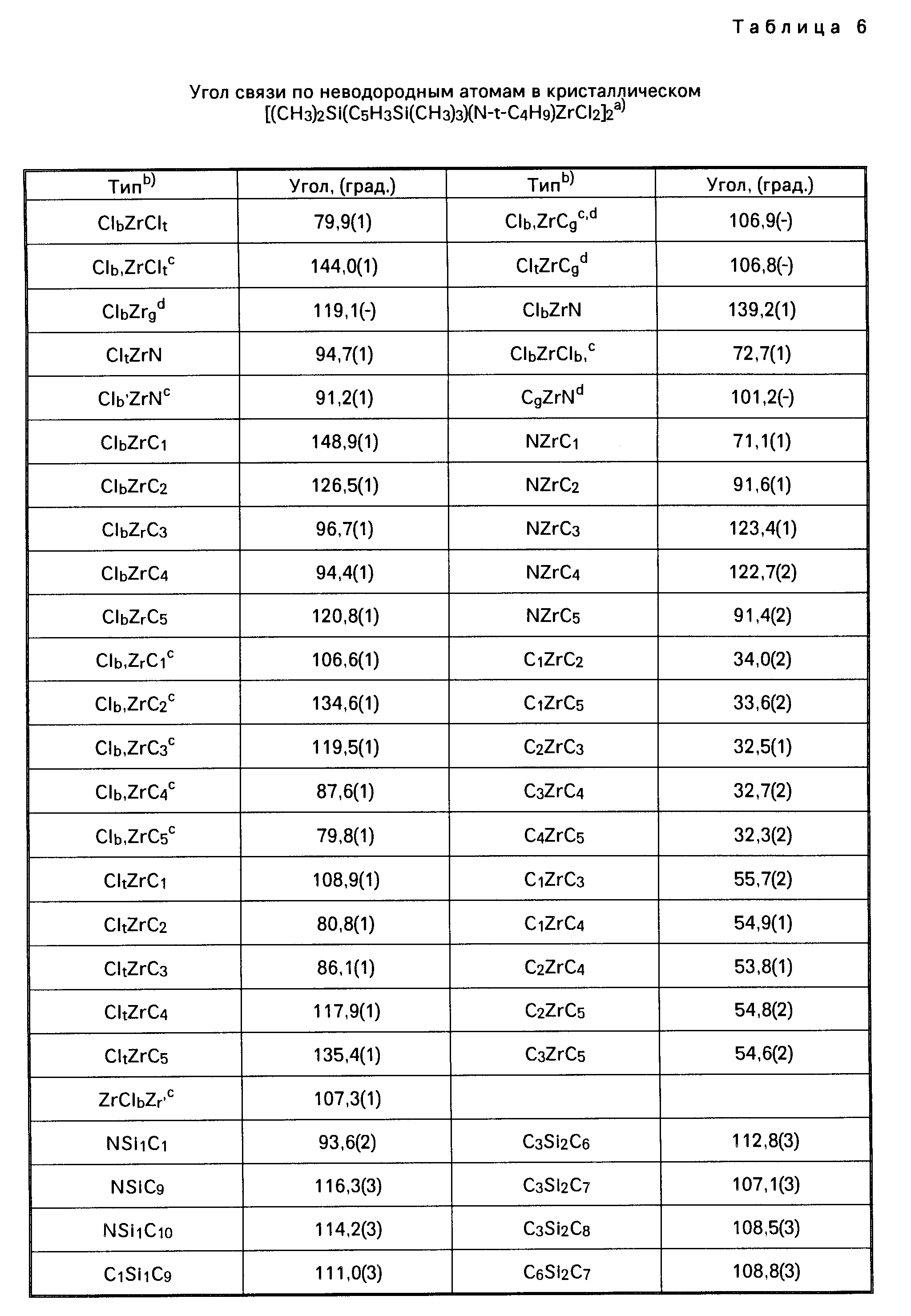
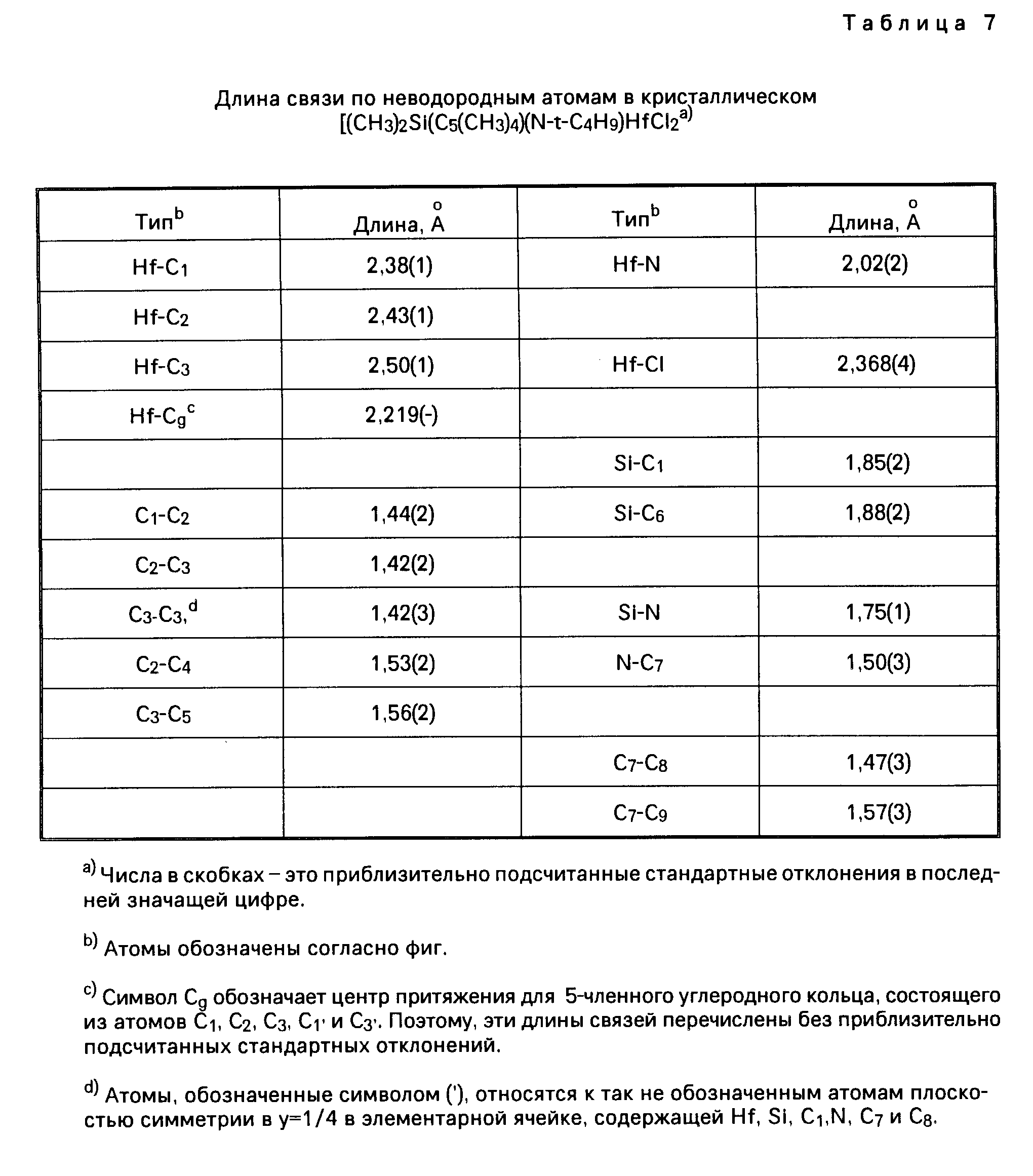
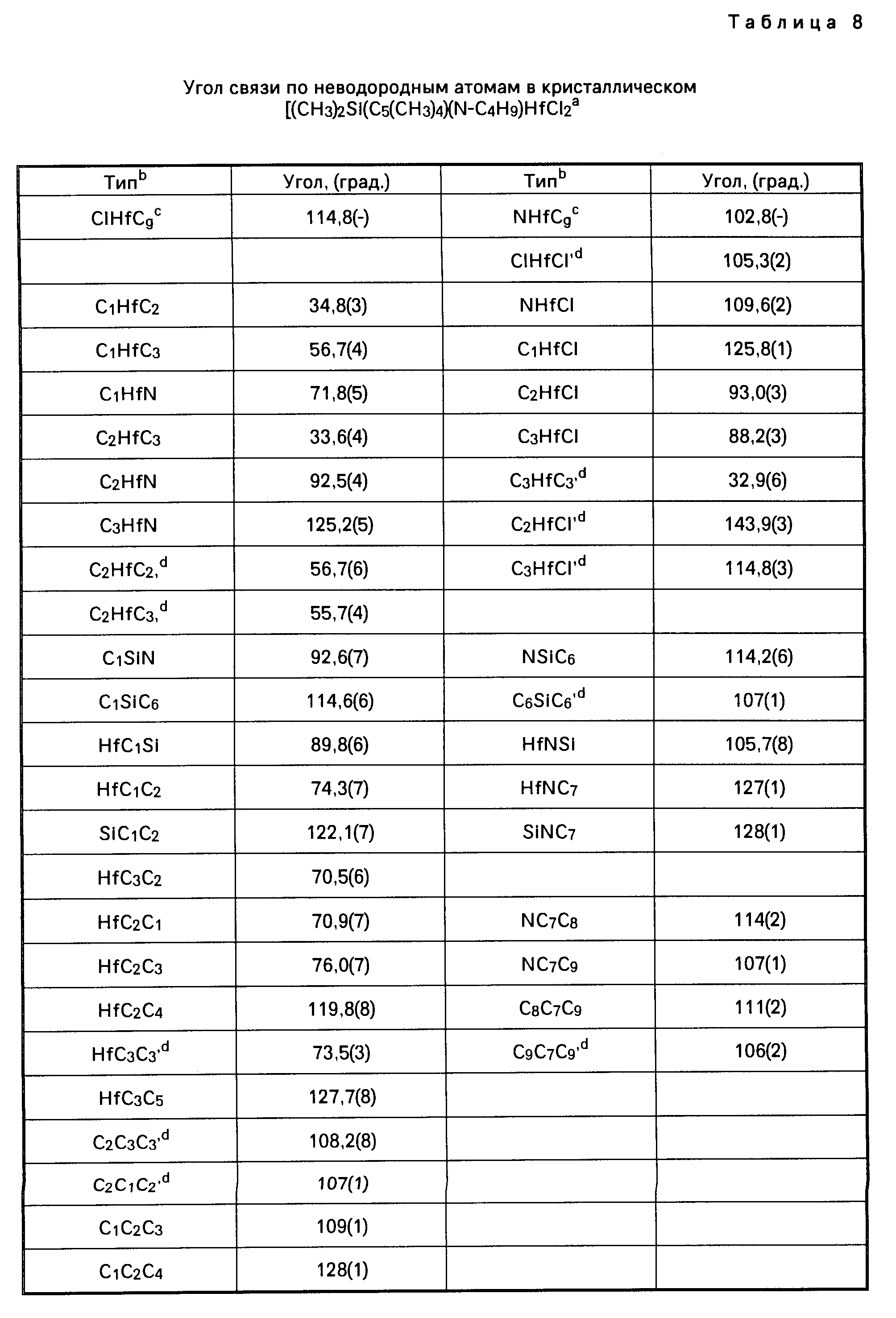
Для иллюстрации настоящего изобретения следует отметить, что указанные выше металлорганические соединения, получаемые перестановкой комбинаций соединений из таблицы 1, не включают в себя лиганд (L) в виде нейтрального основания Льюиса. Условия, при которых соединения, содержащие лиганды в виде нейтрального основания Льюиса, например, в виде простого эфира, образуют димерные соединения, определяются стерическим (т.е. пространственным) объемом лигандов, находящихся у активного "металлического" центра. Так, например, трет. -бутиловая группа в соединении Me2Si(Me4C5) (N-t-Bu)ZrCl2 имеет более значительный стерический объем, чем, скажем, фенильная группа в соединении Me2Si(Me4C5) (NPh)ZrCl2•Et2O, и, таким образом, эфир не координируется у атома циркония в указанном выше первом соединении. Аналогичным же образом, из-за меньшего стерического (пространственного) объема триметилсилилциклопентадиениловой группы соединения [Me2Si(Me3SiC5H3)(N-t-BuZrCl2]2 в сравнении со стерическим объемом тетраметилциклопентадиениловой группы в соединении [Me2 Si(Me4C5) (N-t-Bu)ZrCl2]2 первое из названных соединений является димерным, а второе им не является.
Предпочтительными для использования являются имеющие мостиковые связи соединения переходных металлов группы IVB Периодической системы элементов. Эти металлоорганические соединения могут быть получены путем осуществления реакции между литийорганическим соединением, а именно циклопентадиенил-литием и дигалогенсодержащим соединением. В результате этого взаимодействия выделяется соль в виде галогенида лития, а моногалоидный заместитель оказывается ковалентно связанным с циклопентадиенилом. Замещенный таким образом продукт реакции циклопентадиенила приводится затем в химическое взаимодействие с амидом лития. В результате этой реакции галоген, входящий в моногалоидную замещающую группу (в моногалоидный заместитель) в продукте реакции, образует галогенид лития, а содержащая аминогруппу составная часть амида лития оказывается ковалентно связанной с заместителем в продукте реакции с циклопентадиениловой группой. Полученное аминопроизводное циклопентадиенилсодержащего продукта вступает затем во взаимодействие с алкиллитиевым реагентом. При этом лабильные (т. е. приведенные в лабильное или неустойчивое состояние) атомы водорода при атоме углерода циклопентадиенилового комплекса и при атоме азота содержащей аминогруппу составной части, ковалентно связанной с заместителем, вступает во взаимодействие с алкильной группой алкиллитиевого реагента, в результате чего выделяется алкан и образуется циклопентадиенилпроизводное лития, а именно дилитийциклопентадиенил. После этого имеющие мостиковые связи модификациии металлоорганического соединения, а именно соединение переходного металла группы IVB Периодической системы элементов, получают путем осуществления реакции между дилитийциклопентадиениловым соединением и переходным металлом группы IVB, предпочтительно галогенидом переходного металла группы IVB.
Наиболее подходящими, но далеко не единственными металлоорганическими соединениями растворимого здесь типа, а именно соединений переходного металла группы IVB Периодической системы, которые могут быть использованы в каталитической системе, предлагаемой в соответствии с настоящим изобретением, являются те из имеющих мостиковые связи соединений ("Y" 1), в которых мостиковая группа Т представляет собой диалкилсилан, диарилсилан или алкиларилсилан. Примерами наиболее предпочтительных модификаций соединений (с мостиковыми связями) переходного металла группы IVB Периодической системы элементов являются соединения, которые имеют диметилсилиловые, метилфенилсилиловые, диэтилсилиловые, этилфенилсилиловые, дифенилсилиловые, этиленовые связи. Из них наиболее предпочтительными являются соединения с диметилсилиловыми, диэтилсилиловыми и метилфенилсилиловыми мостиковыми связями.
В приведенной выше таблице 1 указаны составные части соединения переходного металла группы IVB Периодической системы элементов. Эти составные части могут браться в различных комбинациях друг с другом. Примером металлоорганического соединения с мостиковыми связями может быть, в частности, такое соединение как диметилсилилциклопентадиенил-трет. -бутиламидодихлоридоцирконий.
В случае, когда требуется получить a-олефиновый
сополимер с высоким содержанием a-олефинов (a-олефиновых мономеров), преимущественно используют металлоорганические соединения (т. е. каталитические комплексы), в которых в качестве переходного
металла группы IVB Периодической системы элементов берут титан. Наиболее предпочтительные модификации титанорганических соединений могут быть представлены следующей общей формулой:
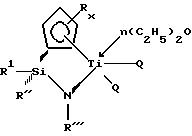
где символы Q, R, R', R'', R''', x и n обозначают то же самое, что и соответствующие символы в приведенных ранее структурных формулах.
В настоящее время общеизвестно, что алюмоксаны можно получать самыми различными способами. Алюмоксан можно получать, например, при взаимодействии триалкилалюминия с водой (вводимой в виде влажного инертного органического растворителя); или при взаимодействии (контактировании) триалкилалюминия с какой-либо солью в гидратной форме, например, с сульфатом меди в гидратной форме, находящимся во взвешенном состоянии в инертном органическом растворителе. Обычно, как бы тщательно алюмоксаны не получались, реакция триалкилалюминия с ограниченным количеством воды всегда приводит к образованию смеси линейной и циклической модификации алюмоксана.
Наиболее подходящими для использования в каталитических системах, предлагаемых согласно настоящему изобретению, алюмоксанами являются те из них, которые были получены гидролизом какого-либо триалкилалюминия.
Используемым в данной каталитической системе является так называемый метилалюмоксан (сокращенно МАО). Наиболее предпочтительными метилалюмоксанами считаются метилалюмоксаны со средней степенью олигомеризации в пределах от около 4 до около 25.
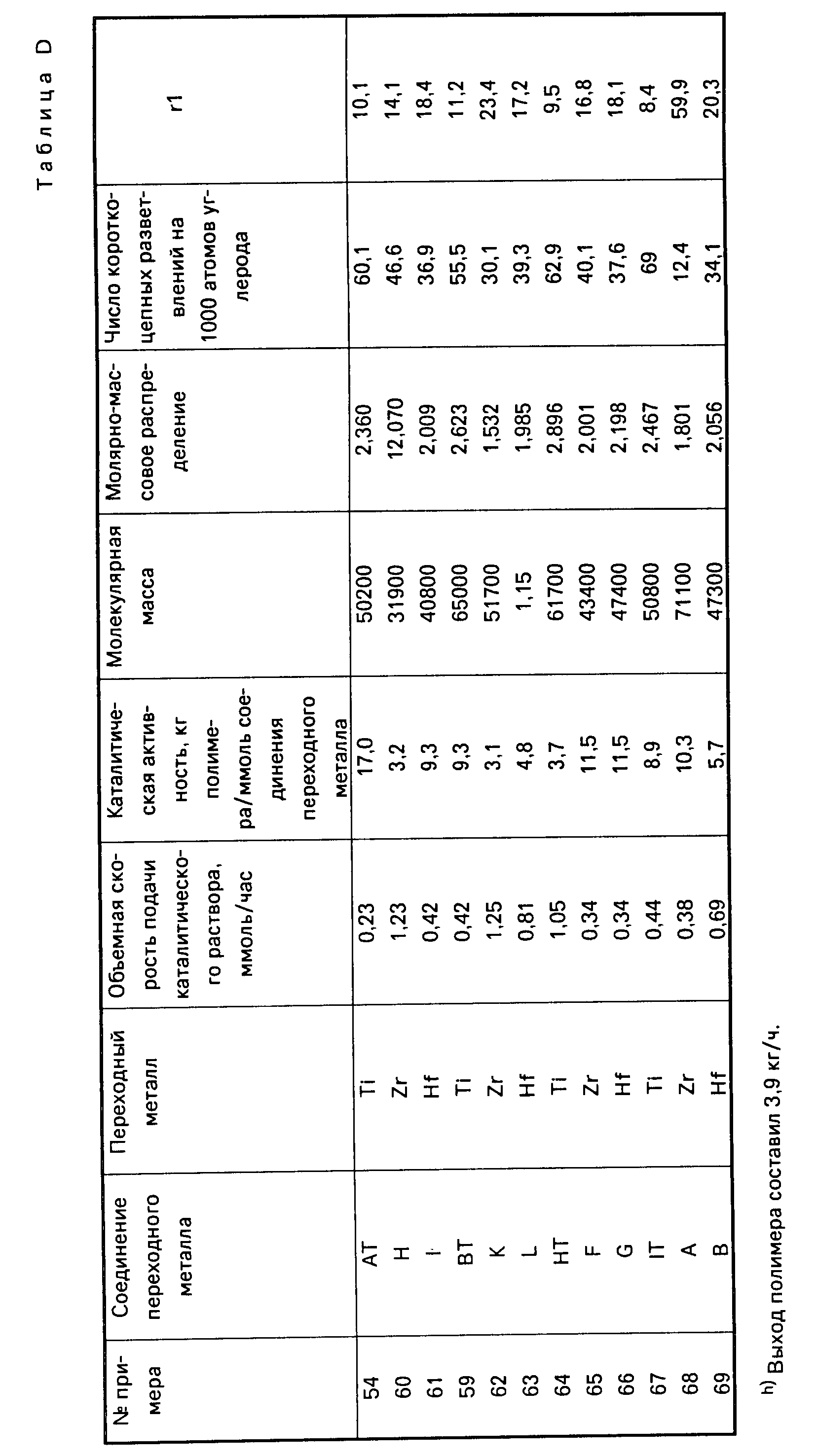
Каталитические системы
Каталитические системы, используемые при осуществлении процесса или способа, предлагаемого согласно настоящему изобретению, включают в себя каталитический
комплекс, получаемый путем смешивания металлоорганического соединения, а именно соединения переходного металла группы IVB Периодической системы элементов, с алюмоксановым компонентом. Каталитическую
систему этого типа можно получить посредством введения необходимых для осуществления данного конкретного процесса компонентов, а именно металлоорганического соединения переходного металла группы IVB
Периодической системы элементов, и алюмоксанового компонента в инертный растворитель такого типа, в котором полимеризация олефиновых мономеров может осуществляться способами полимеризации в растворе,
суспензии или в массе (или в блоке).
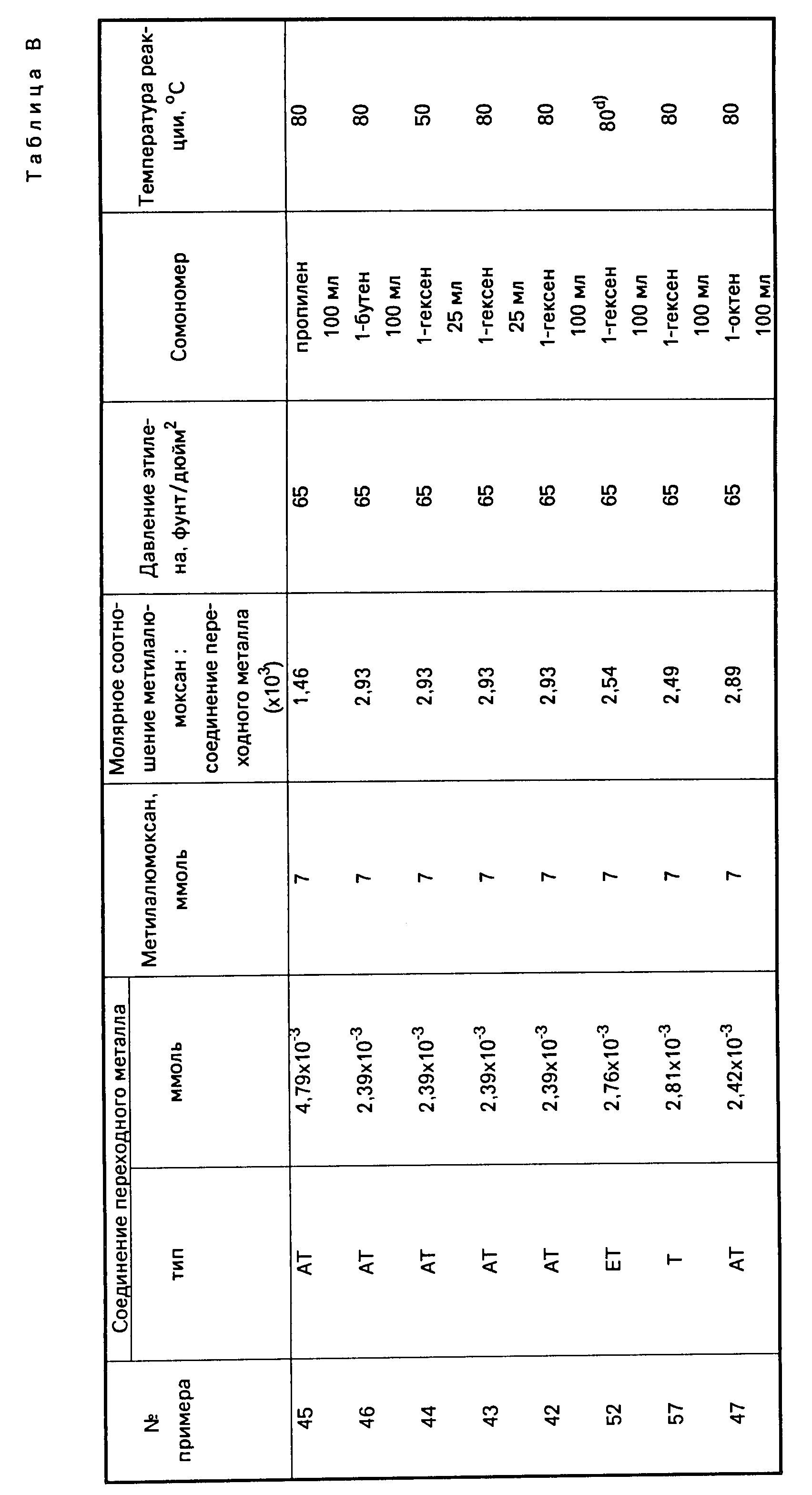
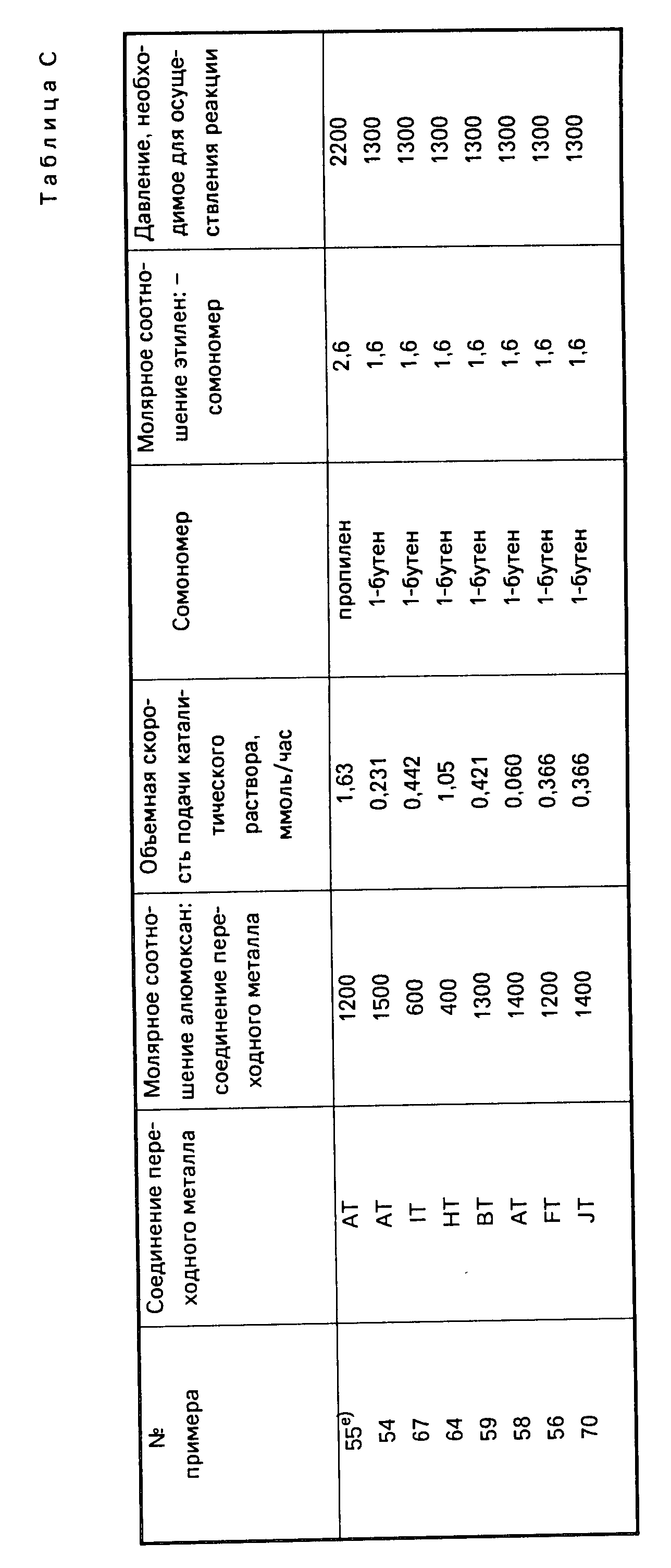
Каталитическую систему, предложенную согласно настоящему изобретению, можно весьма эффективно получать путем введения соединения переходного металла группы IVB Периодической системы элементов, и алюмоксанового компонента, в любой последовательности добавляя друг к другу, в алкановый или ароматический углеводородный растворитель, предпочтительно в такой растворитель, который одновременно годится и для использования в качестве растворителя (разбавителя) для проведения самого процесса полимеризации (в качестве полимеризационного растворителя). В тех случаях, когда применяемый углеводородный растворитель пригоден также и для использования в качестве полимеризационного растворителя, предлагаемую каталитическую систему можно получать непосредственно на месте проведения процесса полимеризации, т. е. в полимеризационном аппарате или реакторе (автоклаве). Кроме того, каталитическую систему можно приготавливать отдельно (т. е. вне реактора) в виде концентрата и затем ее можно вводить в полимеризационный растворитель, находящийся в реакторе. Или, при желании, указанные выше два составных компонента каталитической системы можно приготавливать в виде отдельных растворов, которые затем добавляются к полимеризационному растворителю в полимеризационном растворе в нужных весовых соотношениях или пропорциях, как это, например, бывает удобно для непрерывного осуществления процесса полимеризации в жидкой фазе. В качестве алкановых и ароматических углеводородов, пригодных для использования в качестве растворителей для приготовления предлагаемой каталитической системы и одновременно в качестве полимеризационного растворителя (т. е. растворителя для осуществления процесса полимеризации мономеров), можно, например, назвать следующие, далеко не единственные углеводороды с неразветвленными (прямыми) и разветвленными цепями: изобутан, бутан, пентан, гексан, гептан, октан и другие аналогичные углеводороды; циклические и алициклические углеводороды, например, такие как циклогексан, циклогептан, метилциклогексан, метилциклогептан и другие аналогичные углеводороды; ароматические и алкилзамещенные ароматические соединения, например, такие как бензол, толуол, ксилол и другие подобные соединения. Подходящими для достижения поставленной цели растворителями являются также жидкие олефины, которые могут одновременно использоваться как мономеры или сомономеры, например, этилен, пропилен, 1-бутен, 1-гексен и другие подобные олефины.
В соответствии с настоящим изобретением оптимальные результаты достигаются, как правило, в тех случаях, когда соединение переходного металла группы IVB Периодической системы элементов содержится в полимеризационном растворителе в концентрациях от около 0,0001 до около 1 млмоль на 1 л полимеризационного растворителя и когда алюмоксановый компонент содержится в таком количестве, при котором молярное соотношение алюминий переходный металл находится в пределах от около 1:465 до около 16100:1. При этом растворитель рекомендуется использовать в количествах, достаточных для обеспечения надлежащих условий теплопередачи от компонентов каталитической системы в ходе реакции и для обеспечения хорошего перемешивания (компонентов в растворе).
Составные части предлагаемой каталитической системы, а именно соединение переходного металла группы IVB Периодической системы элементов и алюмоксан, и полимеризационный растворитель могут вводиться в реакционный аппарат быстро или постепенно. Температура, поддерживаемая во время контактирования компонентов каталитической системы друг с другом, может изменяться в довольно широких пределах, например, в интервале от 10 до 300oC. Можно также использовать более высокие и более низкие температуры. Желательно, чтобы реакция во время приготовления каталитической системы протекала при температуре в пределах от около 25 до 100oC, а предпочтительно при температуре около 25oC.
Следует особо отметить, что отдельные компоненты предлагаемой каталитической системы, а также уже приготовленная из этих компонентов каталитическая система должны быть всегда надежно защищены от воздействия на них кислорода и влаги. Это означает, другими словами, что реакции для получения каталитической системы, предложенной в соответствии с настоящим изобретением, следует проводить в бескислородной атмосфере, не содержащей влаги. Поэтому реакции следует осуществлять предпочтительно в присутствии сухого инертного газа, например, гелия или азота.
Процесс полимеризации
В предпочтительном варианте осуществления процесса полимеризации, основанного на использовании металлической
системы, предложенной согласно настоящему изобретению, эта каталитическая система предназначена для осуществления полимеризации олефинового мономера, причем полимеризация может проводиться в жидкой
фазе (например, в суспензии, растворе, жидкой массе или в смеси этих жидких сред), в текучей фазе под высоким давлением или в газовой фазе. Эти процессы могут использоваться в отдельности или в виде
последовательных ступеней. Так, например, при жидкофазной полимеризации олефиновый мономер приводится в контакт с каталитической системой в пригодном для осуществления полимеризации растворителе, а
сама реакция полимеризации этого мономера осуществляется в присутствии указанной выше каталитической системы в течение такого периода времени и при такой температуре, которые достаточны для
обеспечения получения полиолефина большой молекулярной массы.
В качестве мономера для осуществления жидкофазной полимеризации можно использовать только один этилен (в этом случае получают гомополимер) или этилен в сочетании с α-олефином с 3-20 атомами углерода. В этом последнем случае получают сополимер этилена с a-олефином. Можно также получать и гомополимеры высшего, т. е. более высокомолекулярного a-олефина, например, пропилена, бутена, стирола, а также сополимеры этого более высокомолекулярного a-олефина с этиленом и/или с a-олефинами и диолефинами с четырьмя или с большим числом атомов углерода.
Наиболее предпочтительным режимом проведения гомополимеризации или сополимеризации этилена является такой, при котором этилен подается в зону реакции при абсолютном давлении в пределах от около 0,019 фунтов/дюйм2 до около 50000 фунтов/дюйм2, а температура реакции находится в пределах от -100oC до 300oC. При этом целесообразно, чтобы молярное соотношение алюминий переходный металл находилось в пределах как указано выше. Продолжительность или время реакции составляет преимущественно от около 10 секунд до около 1 часа. В качестве одного из целого ряда возможных примеров осуществления процесса получения сополимера с использованием предлагаемой каталитической системы можно назвать вариант, когда жидкий a-олефиновый мономер, например, такой как 1-бутен подают в реактор, снабженный мешалкой, а каталитическая система в паровой или жидкой фазе вводится в реактор через соответствующие сопла. Исходное мономерное сырье, например, этилен в виде газа вводится в паровую фазу в реакторе или подается посредством барботажа в жидкую фазу, находящуюся в реакторе, способами, которые хорошо известны в данной области техники. В реакторе (реакционном аппарате) содержатся: жидкая фаза, состоящая, в основном, из жидкого a-олефинового сомономера вместе с растворенным в нем газообразном этилене, и паровая фаза, состоящая из паров всех мономеров. Температура и давление в реакционном аппарате могут регулироваться с помощью обратного холодильника, т. е. путем самоохлаждения обратного тока испаряющегося a-олефинового мономера, а также с помощью охлаждающих змеевиков, рубашек и так далее. Скорость полимеризации регулируется изменением концентрации катализатора. Содержание этилена в получаемом полимерном продукте определяется соотношением этилен: a-олефиновый сомономер в реакционном аппарате, а это соотношение регулируется изменением взаимных скоростей подачи этих компонентов, т. е. этилена и a-олефинового сомономера в реакционный аппарат.
Как уже указывалось выше, каталитическая система, в которой металлоорганическое соединение, а именно соединение переходного металла группы IVB Периодической системы элементов, является титанорганическим комплексом, способна обеспечивать внедрение значительных количеств a-олефиновых сомономеров. Таким образом, выбирая надлежащим образом модификацию соединения переходного металла группы IVB Периодической системы элементов, можно как бы контролировать и регулировать содержание этилена в получаемом сополимере в пределах практически разумного соотношения между этиленом и a-олефиновым сомономером.
Примеры
В приводимых далее примерах, которые иллюстрируют возможности практического
осуществления основных принципов настоящего изобретения, для анализа получаемых полиолефиновых продуктов использовались аналитические методы, которые рассматриваются в общих чертах ниже. Так, например,
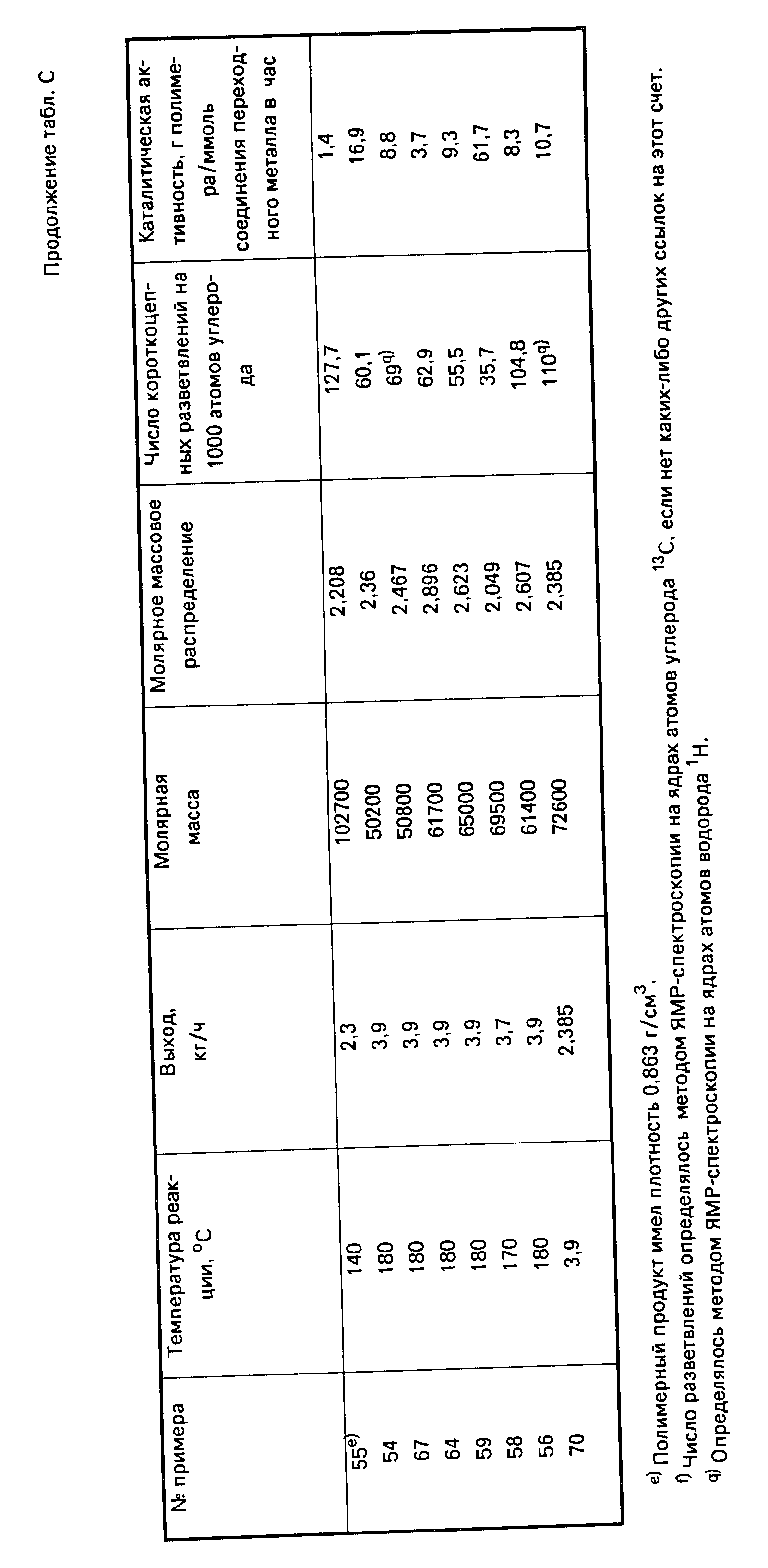
молекулярная масса получаемых полиолефиновых продуктов определялась с помощью гель-проникающей хроматографии следующим образом. Значения молекулярной массы и кривые молекулярно-массового
распределения замерялись и, соответственно, строились с помощью гель-проникающего хроматографа модели Уотерс 150, оборудованного дифференциальным детектором (датчиком) для определения показателя
преломления, и фотометром модели "Хроматикс КМХ-6" для измерения рассеянного света непосредственно в процессе работы реакционного аппарата для получения полимера. Эта хроматографическая система
использовалась для гель-проникающей хроматографии при температуре 135oC, а в качестве подвижной фазы в этом случае использовался 1,2,4-трихлорбензол. В качестве хроматографических колонок
использовались колонки производства фирмы "Седекс" (Сева Денко Америка, Инк.) моделей 802, 803, 804 и 805, заполненные полистироловым гелем. Этот метод аналитической хроматографии подробно
рассматривается в книге "Жидкостная хроматография полимеров и сопутствующих материалов", часть III, Издательство "Дж. Кейзес эдитор", Марсель Деккер, 1981 г. cтр. 207. Эта книга упоминается здесь в
порядке ссылки на справочную литературу по данному вопросу. Поправочные коэффициенты для учета разброса параметров хроматографических колонок не вводились. Однако данные, полученные по хроматограммам
общепринятых эталонов, т. е. известных стандартных веществ, а именно отвечающих требованиям национального Бюро Стандартов США эталонных полимеров как Полиэтилен 1484 и полученные анионной
полимеризацией гидрированные полиизопрены (так называемые альтернативные или чередующиеся сополимеры этилена с пропиленом) показали, что такие поправочные коэффициенты для значения отношения Mw/Mn, т.
е. соотношения между среднемассовой молекулярной массой и среднечисловой молекулярной массой (а это соотношение определяет молекулярно-массовое распределение, сокращенно ММР), не превышали 0,05.
Указанное выше отношение вычислялось по значению времени элюирования. Все количественные аналитические расчеты проводились с использованием широкодоступных средств программного обеспечения типа
"ЛАЛЛС" фирмы "Бекман" и электронной вычислительной машины фирмы "Хьюлетт-Паккард" модели НР 1000. Для гель-проникающей хроматографии использовалась стандартная насадка для гель-проникающей
хроматографии.
Приводимые ниже, далеко не единственные, примеры иллюстрируют конкретные варианты осуществления настоящего изобретения.
Все рассматриваемые здесь процессы осуществляются в инертной атмосфере, а именно в среде гелия или азота. Что касается выбора растворителя для осуществления процесса, то он часто не является строго обязательным. Так, например, в большинстве случаев можно использовать пентан или петролейный эфир с температурой кипения 30-60oC. При этом оба этих органических растворителя взаимозаменяемы. Амиды лития получали из соответствующих аминов и литийорганических соединений, как BuLi или MeLi. В журналах "Органометалликс" ("Металлоорганические соединения"), том 3, N 819 (1984 г.) в статье К.М.Фендрика и другие, а также в немецком журнале "Ц. Натурфоршунг" ("Изучение природы"), том 376, N 144 (1982 г.) в статье Ф.Г.Келера и К.Г. Долла рассматриваются, в частности, способы получения такого литийорганического соединения как LiHC5Me4. Другие литийорганические соединения, а именно литийсодержащие, замещенные соединения циклопентадиенила получают обычно из соответствующего циклопентадиенилового лиганда и н-бутиллития или метиллития. Указанные соединения можно также получать при взаимодействии метиллития с фульвеном (т.е.5-метиленциклопентадиеном). Тетрахлориды титана, циркония и гафния закупались у таких фирм как "Олдрич Кемикл Компани" или "Серак". Тетрахлорид титана TiCl4 использовался, как правильно, в форме эфирата. Эфират тетрахлорида титана, т.е. TiCl4•2Et2O может быть получен осторожным введением тетрахлорида титана в простой диэтиловый эфир. Амины, силаны и литийсодержащий реагенты закупались у таких фирм как "Олдрич Кемикл Компани" или "Петрарч Системз". Метилалюмоксан поставляется фирмами "Шерринг" или "Этил Корпорейшн".
Примеры A-L и A-IT соединения переходного металла группы IVB
Периодической системы элементов:
Пример А
Соединение А. Стадия 1. Литийорганическое соединение Me4HC5Li в количестве 10 г (0,078 моль) постепенно добавляли к
раствору соединения MeSiCl2 (11,5 мл, 0,095 моль) в 225 мл тетрагидрофурана. После этого раствор перемешивали 1 час для обеспечения завершения реакции. Органический растворитель, т.е.
тетрагидрофуран отгоняли в вакууме в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития добавляли пентан. Реакционную смесь пропускали затем через
цеолитовый фильтр, после чего из фильтрата удаляли растворитель. В результате было получено 15,34 г (0,071 моль) соединения MeHC5SiMeCl в виде жидкости бледно-желтого цвета.
Стадия 2. 10 г (0,047 моль) соединения MeHC5SiMe2Cl постепенно добавляли в суспензию 3,68 г (0,047 моль) LiHN-t-Bu в примерно 100 мл тетрагидрофурана. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Тетрагидрофуран отгоняли затем в вакууме в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития добавляли примерно 100 мл петролейного эфира, после чего реакционная смесь пропускалась через цеолитовый фильтр. Затем из фильтрата удаляли растворитель. В результате было выделено 11,14 г (0,044 моль) Me2Si(Me4HC5)(HN-t-Bu) в виде бледно-желтой жидкости.
Стадия 3. 11,14 г (0,044 моль) MeSi(Me4HC5)(HN-t-Bu) были разведены в 100 мл простого диэтилового эфира, после чего к полученному раствору постепенно добавляли 64 мл (0,09 моль) 1,4-молярного раствора литийорганического соединения, а именно метиллития (MeLi). По окончании смесь метиллития перемешивали в течение 30 минут. Полученный продукт (в виде выпавшего осадка) отфильтровывали после предварительно частичного удаления диэтилового эфира. Продукт в виде соединения, имеющего формулу [Me2Si(Me4C5)(N-t-Bu)]Li2, промывали несколькими порциями эфира и затем подвергали вакуумной сушке.
Стадия 4. 3 г (0,011 моль) соединения формулы [Me2Si(Me4C5)(N-t-Bu)]Li2 переводили во взвешенное состояние (суспендировали) в 150 мл простого диэтилового эфира. К полученной суспензии постепенно (медленно) добавляли 2,65 г (0,011 моль) тетрахлорида циркония, после чего получаемую смесь оставляли на ночь при непрерывном перемешивании. Эфир отгоняли с помощью вакуума в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития добавляли пентан. Полученную таким образом смесь пропускали дважды через цеолитовый фильтр. Пентан отгоняли, а выпавший осадок отфильтровывали. Было получено вещество бледно-желтого цвета в твердой фазе, которое было затем промыто растворителем. В результате было выделено 1,07 г (т. е. 0, 0026 моль) соединения формулы Me2Si(Me4C5)(N-t-Bu)ZrCl2.
Некоторое дополнительное количество этого же самого соединения циркония было выделено из фильтрата повторной перекристаллизацией. Общий выход целевого продукта составил 1,94 г (0,0047 моль).
Пример В
Соединение В. Повторяя указанную в примере А
методику получения соединения А и используя на стадии 4 тетрахлорид гафния вместо тетрахлорида циркония, в данном случае можно получить соединения гафния, аналогичное соединению циркония согласно
примеру А. Таким образом, при использовании 2,13 г (0,0081 моль) литийорганического соединения формулы [Me2S-i(Me4C5)(N-t-Bu)] Li2 и 2,59 г (0,0081 моль)
тетрахлорида гафния было получено 0,98 г (0,002 моль) соединения В формулы Me2Si(Me4C5)(N-t-Bu)HfCl2.
Пример С
Соединение С. Стадия
1. 7,5 мл (0,062 моль) Me2SiCl2 были разведены в примерно 30 мл тетрагидрофурана. К полученному таким образом раствору постепенно (медленно) добавляли раствор 7,29 г (0,056 моль)
t-Bu-H4C5Li в примерно 100 мл тетрагидрофурана. Полученную смесь растворов оставляли на ночь при непрерывном перемешивании. Органический растворитель, т.е. тетрагидрофуран,
удаляли с помощью вакуума в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития в реакционную смесь приливали пентан, после чего смесь пропускали
через цеолитовый фильтр. Затем из фильтрата удаляли пентан, в результате чего было получено 10,4 г (0,048 моль) соединения формулы t-Bu-H4C5SiMe2Cl в виде жидкости
бледно-желтого цвета.
Стадия 2. К раствору 3,83 г (0,048 моль) LiNH-t-Bu примерно в 125 мл тетрагидрофурана приливали по каплям 10,4 г (0,048 моль) соединения формулы t-Bu-H4 C5SiMe2Cl. Полученный таким образом раствор оставляли на ночь при непрерывном перемешивании. Тетрагидрофуран отгоняли с помощью вакуума в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития добавляли пентан, после чего реакционную смесь пропускали через цеолитовый фильтр. Из полученного фильтрата удаляли пентан, в результате чего осталось 11,4 г (0,045 моль) соединения формулы Me2Si(t-Bu-H4C5)(NH-t-Bu) в виде жидкости бледно-желтого цвета.
Стадия 3. 11,4 г (0,045 моль) Me2Si(t-Bu-H4C5)(NH-t-Bu) были разбавлены в примерно 100 мл простого диэтилового эфира, после чего в полученный раствор медленно добавляли 70 мл (0,098 моль) 1, 4-молярного раствора метиллития. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Эфир удаляли с помощью вакуума в охлаждаемую ловушку, в которой поддерживалась температура -196oC. После удаления эфира осталось 11,9 г (0,045 моль) литийсодержащего соединения формулы [Me2Si(t-Bu-H3C5)(N-t-Bu)] Li2 в виде твердой фазы бледно-желтого цвета.
Стадия 4. 3,39 г (0,015 моль) указанного выше соединения, а именно [Me2Si(t-Bu-H3C5)(N-t-Bu)] Li2 суспендировались в примерно 100 мл простого диэтилового эфира, после чего в полученную суспензию постепенно добавляли 3 г (0,013 моль) тетрахлорида циркония. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Эфир удаляли, а для осаждения хлорида лития добавляли пентан, после чего реакционную смесь пропускали через цеолитовый фильтр. После этого пентан отгоняли, и из раствора отфильтровывали твердый остаток бледного желтовато-коричневого цвета. Это вещество промывали несколько раз небольшими количествами пентана. Таким образом было выделено 2,43 г (0,0059 моль) продукта эмпирической формулы Me2Si(t-Bu-H3C5)(N-t-Bu)ZrCl2.
Пример D
Соединение D. Повторяя указанную в примере С методику получения соединения С и
используя на стадии 4 тетрахлорид гафния (вместо тетрахлорида циркония), можно в данном случае получать соединение гафния, аналогичное соединению циркония согласно примеру С. Так, например, при
использовании 3,29 г (0,012 моль) литийорганического соединения формулы [Me2Si(t-Bu-H3C5)(N-t-Bu)]Li2 и 4 г (0,012 моль) тетрахлорида гафния выделяли 1,86 г
(0,0037 моль) продукта, имевшего эмпирическую формулу Me2Si(t-Bu-H3C5)(N-t-Bu)HfCl2.
Пример Е
Соединение Е. Стадия 1. 7 г (0,054
моль) MeSiCl2 разводили в примерно 100 мл эфира, после чего в полученный раствор постепенно (медленно) добавляли 5,9 г (0,041 моль) литийорганического соединения формулы Me3
SiC5H4Li. Затем в раствор вводили около 75 мл тетрагидрофурана и смесь оставляли на ночь при непрерывном перемешивании. Растворитель удаляли с помощью вакуума в охлаждаемую
ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития в смесь добавляли пентан. Затем реакционную смесь пропускали через цеолитовый фильтр. Из полученного
фильтрата удаляли растворитель, в результате чего было выделено 8,1 г (0,035 моль) продукта формулы Me2Si(Me3SiC5H4)Cl в виде жидкости бледно-желтого
цвета.
Стадия 2. 3,96 г (0,017 моль) указанного выше продукта формулы Me2Si(Me3SiC5H4)Cl разводили в примерно 50 мл эфира, после чего к полученной смеси постепенно добавляли 1,36 г (0,017 моль) LiHN-трет.-бутила. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Эфир отгоняли с помощью вакуума, а для осаждения хлорида лития в смесь добавляли пентан. Полученную смесь пропускали через цеолитовый фильтр, после чего из фильтрата удаляли пентан. В результате было выделено 3,7 г (0,014 моль) продукта формулы Me2Si(Me3SiC5H4)(NH-t-Bu) в виде жидкости бледно-желтого цвета.
Стадия 3. 3,7 г (0,014 моль) продукта формулы Me2Si(Me3 SiC5H4)(NH-t-Bu) разводили в эфире, после чего в полученный раствор постепенно добавляли 25 мл (0,035 моль) 1,4-молярного раствора метиллития (MeLi) в эфире. После окончания добавления метиллития реакционную смесь перемешивали в течение 1,5 часов. Эфир отгоняли с помощью вакуума, в результате чего было выделено 4,6 г продукта белого цвета в твердой фазе, имевшего формулу Li2[Me2Si(Me3SiC5H3)(N-t-Bu)] 3/4Et2O. Непрореагировавший метиллитий не был удален из продукта в твердой фазе.
Стадия 4. 1,44 г (0,0045 моль) продукта формулы Li2[Me2Si(Me3S-iC5H3)(N-t-Bu)] 3/4Et2O переводились во взвешенное состояние в примерно 50 мл эфира. В приготовленную таким образом суспензию постепенно добавляли 1 г (0,0043 моль) тетрахлорида циркония. Полученную реакционную смесь перемешивали в течение нескольких часов. Растворитель удаляли с помощью вакуума, а для осаждения хлорида лития в смесь добавляли пентан. Реакционную смесь пропускали затем через цеолитовый фильтр. Затем был уменьшен объем фильтрата. Колбу помещали в морозильную камеру с температурой -40oC для осаждения максимального количества продукта. Выпавшая твердая фаза были отфильтрована, в результате чего было получено 0,273 г твердого вещества нестандартного белого цвета. Фильтрат снова концентрировали, а выпавший осадок был отфильтрован, в результате чего удалось получить дополнительные 0,345 г того же самого вещества. Всего было выделено таким образом 0,62 г соединения эмпирической формулы Me2Si(Me3SiC5H3)(N-t-Bu)ZrCl2. Данные рентгено-структурного анализа полученного кристаллического продукта показывают, что это соединение по характеру своего строения является димерным.
Пример F
Соединения F. Стадия 1. Продукт эмпирической формулы Me4HC5SiMeCl получали по методике, описанной в примере А, в котором говорилось о получении соединения А на стадии 1.
Стадия 2. 4,6 г (0,0462 моль) литийорганического соединения эмпирической формулы LiHNPh растворяли в примерно 100 мл тетрагидрофурана, после чего к приготовленному таким образом раствору постепенно добавляли 10 г (0,0466 моль) соединения эмпирической формулы Me4HC5S-iMe2Cl. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Тетрагидрофуран отгоняли с помощью вакуума, а для осаждения хлорида лития в смесь вводили петролейный эфир и толуол. После этого реакционную смесь пропускали через цеолитовый фильтр. Растворитель отгоняли, в результате чего осталось 10,5 г (0,0387 моль) продукта эмпирической формулы Me2Si(Me4HC5)(NHPh) в виде жидкости темно-желтого цвета.
Стадия 3. 10,5 г (0,0387 моль) продукта формулы Me2 Si(Me4HC5)(NHPh) разводили в примерно 60 мл эфира. После этого постепенно приливали 56 мл (0,0784 моль) 1,4-молярного раствора метиллития в эфире. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Выпавший осадок в виде твердой фазы белого цвета (11 г) продукта эмпирической формулы Li2[Me2Si(Me4Si(Me4C5 )(NPh)]3/4Et2O был отфильтрован и промыт эфиром.
Стадия 4. Навеска в 2,81 г (0,083 моль) указанного выше продукта формулы Li2[Me2Si(Me4C5)(NPh)]3/4Et2O переводилась во взвешенное состояние в примерно 40 мл эфира, после чего к полученной суспензии постепенно добавляли 1,92 г (0,0082 моль) тетрахлорида циркония. Затем реакционную смесь оставляли на ночь при непрерывном перемешивании. Эфир отгоняли с помощью вакуума, а для осаждения хлорида лития добавляли смесь петролейного эфира с толуолом. Затем реакционная смесь пропускалась через цеолитовый фильтр. Смесь растворителей удалялось с помощью вакуума, после чего в реакционную смесь добавляли пентан. Реакционную смесь помещали в морозильную камеру, в которой поддерживалась температура -40oC, для осаждения максимального количества целевого продукта. Целевой продукт отфильтровывали и промывали пентаном. В результате было выделено 1,89 г продукта эмпирической формулы Me2Si(Me4C5)NPh)ZrCl2•Et2O в виде твердой фазы бледно-желтого цвета.
Пример G
Соединение G.
Повторяя указанную в примере F методику получения соединения F и используя на стадии 4 тетрахлорид гафния вместо тетрахлорида циркония, можно получить соединения гафния, аналогичное соединению
циркония согласно примеру F. В частности, при использовании 2 г (0,0059 моль) литийорганического соединения формулы Li2[Me2Si(Me4C5)(NPh)] •3/4Et2O и 1,89 г (0,0059 моль) тетрахлорида гафния было получено 1,7 г соединения гафния эмпирической формулы Me2Si(Me4C5)(NPh)HfCl2•1/2Et2O.
Пример Н
Соединение Н. Стадия 1. 14,9 г (0,078 моль) соединения эмпирической формулы MePhSiCl2 были разведены в примерно 250 мл тетрагидрофурана, после
чего в полученный раствор постепенно добавляли 10 г (0,078 моль) литийорганического соединения формулы Me4C5HLi в виде твердой фазы. Реакционный раствор оставляли на ночь при
непрерывном перемешивании. Органический растворитель (тетрагидрофуран) удаляли с помощью вакуума в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида
лития в реакционную смесь добавляли петролейный эфир. Реакционную смесь пропускали через цеолитовый фильтр. Из фильтрата удаляли петролейный эфир. Было выделено 20,8 г (0,075 моль) продукта
эмпирической формулы MePhSi(Me4C5H)Cl в виде вязкой жидкости желтого цвета.
Стадия 2. 4,28 г (0,054 моль) LiHN-t-Bu растворяли в примерно 100 мл тетрагидрофурана, после чего в полученный раствор по каплям добавляли 15 г (0,054 моль) продукта формулы MePhSi(Me4C5H)Cl. Полученный таким образом реакционный раствор желтого цвета оставляли на ночь при непрерывном перемешивании. После этого растворитель (тетрагидрофуран) отгоняли с помощью вакуума. Для осаждения хлорида лития добавляли петролейный эфир. Реакционную смесь пропускали затем через цеолитовый фильтр. Фильтрат упаривали, в результате чего было получено 16,6 г (0,053 моль) продукта эмпирической формулы MePhSi(Me4C5H)(NH-t-Bu) в виде высоковязкой жидкости.
Стадия 3. 16,6 г (0,053 моль) полученного на стадии 2 продукта формулы MePhSi(Me4C5H)(NH-t-Bu) были разведены примерно 100 мл эфира. В полученный раствор постепенно добавляли 76 мл (0,106 моль) 1,4-молярного раствора метиллития MeLi, после чего полученную таким образом реакционную смесь перемешивали в течение 3 часов. После этого отгоняли эфир и отфильтровывали соль лития, которую промывали пентаном, в результате чего было получено 20 г продукта эмпирической формулы Li2[MePhSi(Me4C5)(N-t-Bu)] • 3/4Et2O в виде твердой фазы бледно-желтого цвета.
Стадия 4. 5 г (0,0131 моль) полученного на стадии 3 продукта формулы Li2[MePhSi(Me4C5 )(N-t-Bu)]•3/4Et2O суспендировали в примерно 100 мл диэтилового эфира (Et2O), после чего к полученной суспензии постепенно добавляли 3,06 г (0,0131 моль) тетрахлорида циркония. Реакционную смесь перемешивали при комнатной температуре примерно 1,5 часа. За это время реакционная смесь приобрела слегка темную окраску. Растворитель отгоняли с помощью вакуума, после чего добавляли смесь петролейного эфира с толуолом. Для удаления хлорида лития реакционную смесь пропускали через цеолитовый фильтр. Образовавшийся фильтрат упаривали почти досуха, а сухой остаток отделили фильтрованием. Полученное вещество в твердой фазе нестандартного белого цвета промывали петролейным эфиром. Выход целевого продукта эмпирической формулы MePhSi(Me4C5 )(N-t-Bu)ZrCl2 составил 3,82 г (0,0081 моль).
Пример I
Соединение 1. Продукт эмпирической формулы Li2[MePhSi(Me4C5)(N-t-BuO]
•3/4Et2O приготавливали по методике, описанной в примере Н получения соединения Н на стадии 3.
Стадия 4. 5 г (0,0131 моль) указанного выше продукта формулы Li2[MePhSi(Me4C5)(N-t-Bu)]•3/4Et2O суспендировали в примерно 100 мл диэтилового эфира, после чего к суспензии постепенно добавляли 4,2 г (0,0131 моль) тетрахлорида гафния. Приготовленную таким образом реакционную смесь оставляли на ночь при непрерывном перемешивании. Растворитель отгоняли с помощью вакуума, а для осаждения хлорида лития в смесь вводили петролейный эфир. Реакционную смесь пропускали через цеолитовый фильтр, а образовавшийся фильтрат упаривали почти досуха и отделяли фильтрованием. Оставшееся твердое вещество нестандартного белого цвета промывали петролейным эфиром. Было выделено 3,54 г (0,0058 моль) целевого продукта формулы MePhSi(Me4C5)(N-t-Bu)HfCl2.
Пример J
Соединение J. Продукт эмпирической формулы MePhSi(Me4C5)(N-t-Bu)HfMe2 получали, добавляя стехиометрическое количество метиллития (в виде 1,4-молярного раствора в
эфире) к соединению эмпирической формулы MePhSi(Me4C5)(N-t-Bu)HfCl2, находящемуся во взвешенном состоянии в эфире. При этом был выделен целевой продукт в виде твердого
вещества белого цвета. Выход продукта достаточно высокий.
Пример К
Соединение К. Стадия 1. Соединение эмпирической формулы Me4C5Si-Me2Cl
получали, используя методику примера А применительно к получению соединения А на стадии 1.
Стадия 2. 10 г (0,047 моль) продукта эмпирической формулы Me4C5SiMe2Cl разводили в примерно 25 мл диэтилового эфира (Et2O), после чего к полученному раствору постепенно добавляли 7,57 г (0,047 моль) литийорганического соединения (в эфиратной форме) формулы (LiHNC5H4-n-н-Bu)•1/10Et2O. Полученную таким образом реакционную смесь перемешивали в течение примерно 3 часов, после чего растворитель отгоняли в вакууме. Для осаждения хлорида лития в реакционную смесь добавляли петролейный эфир, а затем смесь пропускали через цеолитовый фильтр. После удаления растворителя получали 12,7 г (0,039 моль) продукта эмпирической формулы Me2Si(Me4C5H)(HNC6H4-n-н-Bu) в виде вязкой жидкости оранжевого цвета.
Стадия 3. 12,7 г (0,039 моль) полученного на стадии 2 продукта формулы Me2Si(Me4C5H)(HNC6H4-n-н-Bu) были разведены в примерно 50 мл диэтилового эфира, после чего к полученному раствору постепенно приливали 55 мл (0,077 моль) 1,4-молярного раствора MeLi (метиллития) в эфире. Реакционную смесь перемешивали около 3 часов. Продукт отфильтровывали и затем промывали диэтиловым эфиром, в результате чего было выделено 13,1 г (0,033 моль) литийорганического соединения эмпирической формулы Li[Me2Si(Me4C5)(NC6H4-n-н-Bu)] 3/4Et2O в виде твердой фазы белого цвета.
Стадия 4. 3,45 г (0,0087 моль) полученного на стадии 3 продукта эмпирической формулы Li2[Me2Si(Me4 C5)(NC6H4-n-н-Bu-)] •3/4Et2O переводили во взвешенное состояние в примерно 50 мл диэтилового эфира, после чего в полученную суспензию постепенно добавляли 2 г (0,0086 моль) тетрахлорида циркония. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Затем эфир отгоняли с помощью вакуума, а для осаждения хлорида лития в реакционную смесь вводили петролейный эфир, после чего реакционную смесь пропускали через цеолитовый фильтр. Фильтрат упаривали досуха, в результате чего осталась твердая фаза желтого цвета, которая затем была перекристаллизована из пентана. Выход целевого продукта эмпирической формулы Me2Si(Me4C5)(NC6H4-n-н-Bu)] ZrCl2•3Et2 O составил 4,2 г.
Пример L
Соединение L. Литийорганическое соединение формулы Li2[MeSi(Me4C5)(NC6H4-n-н-Bu)] 3/4Et2O было получено по методике, описанной в примере К при получении соединения К на стадии 3.
Стадия 4. 3,77 г (0,0095 моль) литийорганического соединения формулы Li2 [MeSi(Me4C5)(NC6H4-n-н-Bu)] 3/4Et2O переводили во взвешенное состояние в примерно 50 мл диэтилового эфира. К приготовленной таким образом суспензии постепенно добавляли 3 г (0,0094 моль) тетрахлорида гафния в твердой фазе, после чего реакционную смесь оставляли на ночь при непрерывном перемешивании. Эфир удаляли с помощью вакуума, а для осаждения хлорида лития в смесь добавляли петролейный эфир. Затем реакционную смесь пропускали через цеолитовый фильтр. Петролейный эфир отгоняли с помощью вакуума, а остаток в виде твердой фазы нестандартного белого цвета перекристаллизовывали из пентана. Выход целевого продукта, имевшего, как было установлено, эмпирическую формулу Me2Si(Me4C5)(N-C6 H4-n-н-Bu)HfCl2, составил 1,54 г (0,0027 моль).
Пример АТ
Соединение Ат. Стадия 1. 14,9 г (0,078 моль) соединения формулы MePhSiCl2 были
разведены в 250 мл тетрагидрофурана, после чего в полученный таким образом раствор постепенно добавляли 10 г (0,078 моль) литийорганического соединения формулы Me4HC5Li в твердой
фазе. Реакционный раствор оставляли на ночь при непрерывном перемешивании. После этого с помощью вакуума отгоняли растворитель в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития в реакционную смесь добавляли петролейный эфир. Затем реакционную смесь пропускали через цеолитовый фильтр, а из фильтрата удаляли пентан. В результате было
выделено 20,8 г (0,075 моль) соединения эмпирической формулы MePhSi(Me4C5H)Cl в виде вязкой жидкости желтого цвета.
Стадия 2. 4,28 г (0,054 моль) соединения формулы LiHN-t-Bu растворяли в примерно 100 мл тетрагидрофурана, после чего к приготовленному таким образом раствору по каплям добавляли 15 г (0,054 моль) соединения формулы MePhSi(C5Me4H)Cl. Полученный реакционный раствор желтого цвета оставляли на ночь при перемешивании. Растворитель (тетрагидрофуран) отгоняли в вакууме, а для осаждения хлорида лития добавляли петролейный эфир. Реакционную смесь пропускали затем через цеолитовый фильтр, а оставшийся фильтрат упаривали. В результате было выделено 16,6 г (0,053 моль) соединения эмпирической формулы MePhSi(C5 Me4H)NH-t-Bu) в виде жидкости очень высокой вязкости.
Стадия 3. 17,2 г (0,055 моль) соединения эмпирической формулы MePhSi(C5Me4H)(NH-t-Bu) разводили в
примерно 20 мл эфира, после чего к полученному раствору постепенно добавляли 60 мл (0,096 моль) 1,6-молярного раствора н-бутиллития в гексане. Полученную реакционную смесь перемешивали около 3 часов.
Затем растворитель отгоняли в вакууме. В результате было получено 15,5 г (30,48 моль) твердого вещества бледно-коричневого цвета. Полученное вещество имело эмпирическую формулу Li2
[MePhSi(C5Me4)(N-t-Bu)]
Стадия 4. 8,75 г (0,027 моль) полученного на стадии 3 соединения формулы Li2[MePhSi(C5Me4)(N-t-Bu)] переводили
во взвешенное состояние в примерно 125 мл холодного эфира, имевшего температуру -30oC. К полученной суспензии постепенно добавляли 9,1 г (0,027 моль) тетрахлорида титана в виде эфира, а
именно TiCl4•2Et2O. Реакционную смесь перемешивали в течение нескольких часов, а затем эфир отгоняли в вакууме. Затем для растворения продукта добавляли смесь толуола с
дихлорметаном. Растворитель удаляли в основном с помощью вакуума, а затем добавляли эфир. Полученную смесь охлаждали для обеспечения осаждения максимального количества продукта. Неочищенный продукт
отфильтровывали, а затем снова растворяли в толуоле. Нерастворимые в толуоле вещества были отфильтрованы. Затем толуол отгоняли и добавляли петролейный эфир. Для обеспечения осаждения максимального
количества целевого продукта реакционную смесь охлаждали. После этого было отфильтровано 3,34 г (7,76 моль) целевого продукта в виде твердого вещества желтого цвета. Продукт имел формулу MePhSi(C5Me4)(N-t-Bu)TiCl2.
Пример ВТ
Соединение ВТ. Стадия 1. 10 г (0,078 моль) литийорганического соединения формулы C5Me4HLi
постепенно добавляли к 11,5 мл (0,095 моль) раствора Me2SiCl2 в 225 мл тетрагидрофурана. Для обеспечения проведения полной реакции реакционный раствор перемешивали в течение 1
часа. После этого растворитель отгоняли в вакууме, а для осаждения хлорида лития в смесь добавляли пентан. Реакционную смесь пропускали затем через цеолитовый фильтр. Растворитель удаляли из фильтрата
с помощью вакуума. В результате было выделено 15,34 г (0,071 моль) продукта в виде жидкости бледно-желтого цвета. Продукт имел эмпирическую формулу (C5Me4H)SiMe2Cl.
Стадия 2. 10 г (0,047 моль) полученного на стадии 1 продукта формулы (C5Me4H)SiMe2Cl были разведены в примерно 25 мл диэтилового эфира. В полученный таким образом раствор постепенно вводили 7,75 г (0,048 моль) соединения формулы LiHNC5H4-n-н-бутил•1/10Et2O, после чего полученную реакционную смесь перемешивали около 3 часов. Растворитель отгоняли в вакууме, а для осаждения хлорида лития в смесь добавляли петролейный эфир. Затем реакционную смесь пропускали через цеолитовый фильтр. После удаления растворителя осталось 12,7 г (0,039 моль) вязкой жидкости оранжевого цвета. Этот продукт имел эмпирическую формулу Me2Si(C5Me4H)(HNC6H4 -n-н-бутил).
Стадия 3. 12,7 г (0,039 моль) полученного на стадии 2 продукта формулы Me2Si(C5Me4H)(HNC6H4-n-н-Bu) были разведены в
примерно 50 мл диэтилового эфира. К полученному таким образом раствору постепенно приливали 55 мл (0,077 моль) 1,4-молярного раствора метиллития в эфире. Реакционную смесь перемешивали около 3 часов.
Продукт был отфильтрован, промыт диэтиловым эфиром и просушен. Было выделено 13,1 г (0,033 моль) твердого продукта белого цвета. Продукт имел следующую эмпирическую формулу Li2[Me2Si(C5Me4)(NC6H4-n-н-Bu-)]
•3/4Et2O.
Стадия 4. 2,36 г (5,97 моль) полученного на стадии 3 продукта формулы Li2[Me2Si(C5Me4)(NC6H4-n-н-Bu-)] •3/4Et2O переводились во взвешенное состояние в холодном эфире, после чего в полученную таким образом суспензию постепенно добавляли 2 г (5,92 г) эфирата тетрахлорида титана TiCl4•2Et2O. Реакционную смесь оставляли на ночь при перемешивании. Растворитель отгоняли с помощью вакуума, после чего в смесь добавляли петролейный эфир и дихлорметан. Для удаления хлорида лития реакционную смесь пропускали через цеолитовый фильтр. Растворитель отгоняли с помощью вакуума, после чего добавляли толуол и петролейный эфир. После охлаждения смесь была отфильтрована, в результате чего был получен продукт нестандартного желтого цвета. Этот продукт был снова растворен в дихлорметане, а в полученный раствор добавили петролейный эфир. Полученную смесь охлаждали, после чего из нее отфильтровывали 0,83 г (1,87 ммоль) твердого продукта желтого цвета. Продукт имел формулу Me2Si(C5Me4)(NC6H4-n-н-Bu)TiCl2.
Пример СТ
Соединения СТ. Стадия 1. Соединение формулы
(C5Me4H)SiMe2Cl получали по методике, описанной в примере ВТ применительно к получению соединения на стадии 1.
Стадия 2. 8,14 г (0,038 моль) соединения формулы (C5Me4H)SiMe2Cl смешивали с примерно 100 мл тетрагидрофурана. К полученной смеси постепенно добавляли 4,89 г (0,038 моль) литийорганического соединения формулы LiHNC6H4-p-OMe, после чего смесь перемешивали в течение около 2 часов. Растворитель отгоняли с помощью вакуума, а для осаждения хлорида лития добавляли петролейный эфир. Осажденный хлорид лития отфильтровывали. Растворитель удаляли из фильтрата с помощью вакуума. В результате было выделено 9,8 г (0,033 моль) продукта в виде вязкой жидкости оранжево-желтого цвета. Продукт имел формулу Me2Si(C5Me4H)(NC6H4-p-OMe).
Стадия 3. 10 г (0,033 моль) продукта формулы Me2Si(C5 Me4H)(HNC6H4-p-OMe) были разведены в тетрагидрофуране, после чего к полученному раствору медленно приливали 47 мл (0,066 моль) 1,4-молярного раствора метиллития в эфире. Реакционную смесь перемешивали в течение нескольких часов. После этого растворитель отгоняли в вакууме, в результате чего был выделен твердый остаток белого цвета. Продукт имел тетрагидрофурановые координационные связи. Было получено 14,7 г (0,032 моль) продукта эмпирической формулы Li2[Me2Si(C5Me4)(NC6H4 -p-OMe)] •2ТГФ, где ТГФ тетрагидрофуран.
Стадия 4. 7 г (0,015 моль) Li2[Me2Si(C5Me4)(NC6H4-p-OMe)] •2ТГФ cуспендировали в примерно 125 мл холодного эфира, после чего к приготовленной таким образом суспензии постепенно добавляли 5,1 г (0,015 моль) эфирата тетрахлорида титана, а именно TiCl2 •2Et2O. Реакционную смесь оставляли на ночь при перемешивании. Растворитель удаляли в вакууме, после чего добавляли петролейный эфир, дихлорметан и толуол. Для удаления выпавшего осадка хлорида лития смесь пропускали через цеолитовый фильтр. Затем растворитель отгоняли и добавляли петролейный эфир. Реакционную смесь охлаждали, после чего было отфильтровано твердое вещество коричневого цвета. Затем многократной экстракцией и перекристаллизацией из толуола и петролейного эфира было получено 2,3 г (5,5 моль) целевого продукта, имевшего эмпирическую формулу Me2 Si(C5Me4)(NC6H4-p-OMe)TiCl2.
Пример DT
Соединение DT. Стадия 1. 7,5 мл (0,062 моль) соединения формулы Me2
SiCl2 разводили в примерно 30 мл тетрагидрофурана, после чего к полученному раствору медленно приливали 7,29 г (0,057 моль) раствора t-Bu-H4C5Li в примерно 100 мл
тетрагидрофурана. Приготовленную таким образом реакционную смесь оставляли на ночь при перемешивании. Тетрагидрофуран отгоняли в вакууме, а для осаждения хлорида лития добавляли пентан. После этого
смесь пропускали через цеолитовый фильтр. Пентан удаляли из фильтрата, в результате чего остался продукт в виде жидкости бледно-желтого цвета в количестве 10,4 г (0,048 моль). Продукт имел формулу
(t-Bu-C5H4)SiMe2Cl.
Стадия 2. 5 г (0,023 моль) полученного на стадии 1 продукта формулы (t-Bu-C5H4)SiMe2Cl добавляли к примерно 50 мл тетрагидрофурана, после чего к полученному раствору постепенно добавляли 4,94 г (0,023 моль) литийорганического соединения формулы LiHN-2,5-t-Bu2C6H3. Реакционную смесь перемешивали в течение 2 часов. Растворитель отгоняли с помощью вакуума, а для осаждения хлорида лития добавляли петролейный эфир. Выпавший в осадок хлорид лития отфильтровывали. Растворитель удаляли из фильтрата, в результате чего был выделен маслянистый продукт, имеющий формулу Me2Si(t-Bu-C5H4)(HN-2,5-t-Bu2C6H3 ).
Стадия 3. К полученному на стадии 2 продукту формулы Me2Si(t-Bu-C5H4)(HN-2,5-t-Bu2C6H3), взятому в количестве около
8 г (0,021 моля), постепенно добавляли 30 мл (0,042 моль) 1,4-молярного раствора метиллития в эфире. Реакционную смесь перемешивали затем в течение нескольких часов, после чего растворитель отгоняли в
вакууме. Оставшаяся после удаления растворителя твердая фаза слегка розоватого цвета была промыта эфиром, профильтрована и просушена. В результате было выделено 4,42 г (0,011 моль) литийорганического
соединения формулы Li2[Me2Si(t-BuC5H3) (N-2,5-t-Bu2C6H3)]
Стадия 4. 7,6 г (0,019 моль) полученного на стадии 3
литийорганического соединения формулы Li2[Me2Si(t-BuC5H3) (N-2,5-t-Bu2C6H3)] переводились во взвешенное состояние в
холодном эфире, после чего к полученной суспензии постепенно добавляли 6,5 г (0,019 моль) эфирата тетрахлорида титана TiCl4•2Et2O. Приготовленную таким образом реакционную
смесь оставляли на ночь при непрерывном перемешивании. Растворитель отгоняли в вакууме, после чего в реакционную смесь вводили толуол и дихлорметан. Для удаления хлорида лития смесь пропускали через
цеолитовый фильтр. Затем фильтрат упариванием концентрировали и добавляли петролейный эфир. Для наиболее полного осаждения реакционную смесь охлаждали. Выпавшая в осадок твердая фаза темно-желтого
цвета была отфильтрована и перекристаллизована из толуола и петролейного эфира. В результате была выделена твердая фаза светло-коричневого цвета. Всего было выделено 1,6 г (3,2 ммоль) соединения
формулы Me2Si(t-Bu-C5H3)(N-2,5-t-Bu2C6H3-) TiCl2.
Пример ЕТ
Соединение ЕТ. Стадия 1. 40 г (0,33
моль) литийорганического соединения эмпирической формулы LiC9H7 литийинден или сокращенно Li(ind)] постепенно добавляли к 60 мл (0,49 моль) раствора Me2SiCl2
в эфире и тетрагидрофуране. Реакционную смесь перемешивали в течение 1,5 часов перед отгонкой растворителя в вакууме, после чего добавляли петролейный эфир и отфильтровывали хлорид лития.
Растворитель отгоняли из фильтрата с помощью вакуума. Осталось 55,7 г (0,27 моль) жидкости бледно-желтого цвета. Этот жидкий продукт имел формулу (ind)Me2SiCl.
Стадия 2. 20 г (0,096 моль) полученного на стадии 1 продукта формулы (ind)Me2SiCl были разведены в эфире, после чего к этому раствору постепенно добавляли 7,6 г (0,096 моль) LiHN-t-Bu. Приготовленную таким образом реакционную смесь оставляли на ночь при непрерывном перемешивании. Затем отгоняли растворитель с помощью вакуума и добавляли петролейный эфир и толуол. Выпавший в осадок хлорид лития отфильтровывали, а растворитель удаляли с помощью вакуума. В результате был выделен продукт формулы Me2Si(ind)(HN-t-Bu).
Стадия 3. 21 г (0,086 моль) полученного на стадии 2 продукта формулы Me2Si(ind)(HN-t-Bu) был разведен в смеси петролейного эфира с диэтиловым эфиром, после чего к полученному раствору постепенно добавляли 108 мл (0,17 моль) 1,6-молярного раствора трет.-бутиллития в гексане. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Растворитель отгоняли в вакууме, а оставшаяся твердая фаза была промыта петролейным эфиром и отфильтрована. Было выделено 26 г (0,094 моль) продукта эмпирической формулы Li2[Me2Si(ind)(N-t-Bu)] •1/4Et2O, где сокращение "ind" обозначает инден. Продукт представлял собой твердый продукт бледно-желтого цвета.
Стадия 4. 10 г (0,036 моль) полученного на стадии 3 продукта формулы Li2[Me2Si(ind)(N-t-Bu)] • 1/4Et2O растворяли в эфире. К охлажденному раствору добавляли 12,1 г (0,036 моль) эфирата тетрахлорида титана TiCl4•2Et2O. Реакционную смесь оставляли на ночь при непрерывном перемешивании, после чего растворитель отгоняли в вакууме и добавляли смесь толуола с дихлорметаном. Для удаления хлорида лития реакционную смесь пропускали через цеолитовый фильтр. Затем отгоняли растворитель и добавляли горячий толуол. Нерастворимые вещества отфильтровывались, после чего растворитель отгоняли и приливали петролейный эфир. Реакционную смесь охлаждали, после чего был отфильтрован твердый продукт, который имел эмпирическую формулу Me2Si(ind)(N-t-Bu)TiCl2. Эта твердая фаза была затем несколько раз перекристаллизована. Выход целевого продукта составил 2,5 г (6,8 ммоль).
Пример FT
Соединение FT. Стадия 1. Соединение эмпирической формулы (C5Me4H)SiMe2Cl получали, используя
методику, описанную в примере ВТ применительно к получению соединения ВЕ на стадии 1.
Стадия 2. 5,19 г (0,024 моль) полученного на стадии 1 соединения формулы (C5Me4H)SiMe2Cl постепенно добавляли к раствору 2,52 г (0,024 моль) литийорганического соединения формулы LiHNC6H11 в примерно 125 мл тетрагидрофурана. Полученный таким образом реакционный раствор перемешивали в течение нескольких часов. Затем отгоняли тетрагидрофуран с помощью вакуума, а для осаждения хлорида лития в смесь добавляли петролейный эфир. Выпавший в осадок хлорид лития отфильтровывали, после чего растворитель удаляли из фильтрата с помощью вакуума. В результате было выделено 6,3 г (0,023 моль) продукта в виде желтой жидкости, имевшего эмпирическую формулу Me2Si(C5H4)(HNC6H11).
Стадия 3. 6,3 г (0,023 моль) полученного на стадии 2 продукта формулы Me2
Si(C5H4)(HNC6H11) были разведены в примерно 100 мл эфира, после чего к полученному раствору постепенно добавляли 33 мл (0,046 моль) 1,4-молярного раствора
метиллития в эфире. Реакционную смесь перемешивали в течение 0,5 часа, после чего была отфильтрована твердая фаза белого цвета, которая была затем промыта эфиром и просушена в вакууме. Таким способом
было выделено 5,4 г (0,019 моль) литийорганического соединения эмпирической формулы Li2[Me2Si(C5Me4)(NC6H11)]
Стадия 4. 2,57
г (8,9 ммоль) полученного на стадии 3 соединения формулы Li2[Me2Si(C5Me4)(NC6H11)] переводили во взвешенное состояние в примерно 50 мл
холодного эфира, после чего к полученной суспензии постепенно добавляли 3 г (8,9 ммоль) эфирата тетрахлорида титана TiCl4•2Et2O. Приготовленную таким образом реакционную
смесь оставляли на ночь при непрерывном перемешивании. Растворитель отгоняли в вакууме, после чего добавляли смесь толуола с дихлорметаном. Для удаления выпавшего в осадок побочного продукта, а именно
хлорида лития, реакционную смесь пропускали через цеолитовый фильтр. Из фильтрата удаляли растворитель. После этого добавляли сначала небольшое количество толуола, а затем петролейный эфир. Для
обеспечения наиболее полного осаждения реакционную смесь охлаждали. Была отфильтрована твердая фаза коричневого цвета, которая сначала была растворена в горячем толуоле, а затем пропущена через
цеолитовый фильтр. Растворитель отгоняли из фильтрата, а затем добавляли петролейный эфир. После охлаждения была отфильтрована твердая фаза оливково-зеленого цвета, которая затем дважды
перекристаллизовывалась из дихлорметана и петролейного эфира. Таким образом было выделено 0,94 г (2,4 ммоль) твердого продукта бледного оливково-зеленого цвета. Полученный целевой продукт имел
эмпирическую формулу Me2Si(C5Me4)(NC6H11)TiCl.
Пример GT
Соединение GT. Стадия 1. 150 мл (1,24 моль) соединения формулы
Me2SiCl2 разводили в примерно 200 мл диэтилового эфира, после чего в полученный раствор постепенно добавляли 28,2 г (0,11 моль) эфирата флуоренлития формулы Li(C13
H9)•Et2O. Реакционную смесь перемешивали в течение около 1 часа, а затем отгоняли растворитель с помощью вакуума и приливали толуол. Полученную смесь пропускали через
цеолитовый фильтр для удаления выпавшего в осадок хлорида лития, после чего из фильтрата удаляли растворитель. В результате осталось 25,4 г (0,096 моль) продукта в виде твердой фазы нестандартного
белого цвета. Продукт имел формулу Me2Si(C13H3)Cl.
Стадия 2. 8 г (0,031 моль) полученного на стадии 1 продукта формулы Me2Si(C13 H9)Cl суспендировали в смеси эфира и тетрагидрофурана, взятых в весовом соотношении 5: 1. К полученной таким образом суспензии постепенно добавляли 3,25 г (0,031 моль) соединения формулы LiHNC6H11. Реакционную смесь оставляли затем на ночь при перемешивании. После удаления растворителя в вакууме добавляли толуол. Для удаления выпавшего в осадок хлорида лития реакционную смесь пропускали через цеолитовый фильтр. После этого фильтрат упаривали, в результате чего осталась вязкая жидкость оранжевого цвета, которую затем разводили в диэтиловом эфире. К разведенной в эфире жидкости постепенно приливали 43 мл (0,06 мл) 1,4-молярного раствора метиллития в эфире. Реакционную смесь после этого оставляли на ночь при непрерывном перемешивании. Растворитель отгоняли под вакуумом. В результате было выделено 13 г (0,031 моль) эфирата литийорганического соединения эмпирической формулы Li2[Me2Si(C13H8)(NC6H11)] •1,25Et2O.
Стадия 3. 6,5 г (0,015 моль) полученного на стадии 2 соединения формулы Li2[Me2Si(C13H8 )(NC6H11)] •1,25Et2O растворили в холодном эфире, после чего в полученный таким образом раствор постепенно добавляли 5,16 г (0,015 моль) эфирата тетрагидрохлорида титана формулы TiCl4•2Et2O. Реакционную смесь оставляли на ночь при перемешивании. Растворитель отгоняли под вакуумом и затем добавляли метиленхлорид. После этого реакционную смесь пропускали через цеолитовый фильтр для удаления выпавшего в осадок хлорида лития. Фильтрат упаривали до меньшего объема и затем добавляли петролейный эфир. Смесь охлаждали для обеспечения осаждения максимального количества твердой фазы, которую затем отфильтровывали. Поскольку выделенная твердая фаза не растворялась полностью в толуоле, ее смешивали с толуолом и отфильтровывали. Фильтрат упариванием концентрировали и затем в него добавляли петролейный эфир для осаждения твердой фазы. Полученную смесь охлаждали, а затем фильтровали. В результате было выделено 2,3 г (5,2 моль) твердого продукта красновато-коричневого цвета. Продукт имел следующую эмпирическую формулу Me2Si(C13H8)(NC6H11 )TiCl2.
Пример НТ
Соединение НТ. Стадия 1. Соединение эмпирической формулы (C5Me4H)SiMe2Cl получали по методике, описанной в
примере ВТ при получении соединения ВТ на стадии 1.
Стадия 2. Литийорганическое соединение формулы LiHNPh в количестве 4,6 г (0,046 моль) растворяли в примерно 100 мл тетрагидрофурана, после чего к полученному раствору постепенно добавляли 10 г (0,047 моль) соединения формулы (C5Me4H)SiMe2Cl. Реакционную смесь оставляли на ночь при непрерывном перемешивании. Тетрагидрофуран отгоняли под вакуумом, а для осаждения хлорида лития добавляли петролейный эфир и толуол. Полученную смесь пропустили затем через цеолитовый фильтр. Из фильтрата удалили растворитель, в результате чего осталось 10,5 г (0,039 моль) жидкого продукта эмпирической формулы Me2Si(C5Me4H)(NHPh) темно-желтого цвета.
Стадия 3. 9,33 г (0,034 моль) полученного на стадии 2 продукта формулы Me2Si(C5Me4H)(NHPh) разводили в примерно 30 мл эфира, после чего в полученный раствор постепенно приливали 44 мл (0,062 моль) 1,4-молярного раствора метиллития в эфире. Реакционную смесь перемешивали в течение 2 часов. После удаления растворителя получали 9,7 г (0,03 моль) продукта в твердой фазе эмпирической формулы Li2[Me2Si(C5Me4)(NPh)]•1/2Et2O. Продукт был отфильтрован, промыт эфиром и просушен.
Стадия 4. 4,3 г (0,013 моль) полученного на стадии 3 продукта формулы Li2[Me2Si(C5Me4)(NPh)] •1/2Et2O суспендировали в примерно 50 мл охлажденного эфира, после чего к суспензии постепенно добавляли 4,1 г (0,012 моль) эфирата тетрахлорида титана формулы TiCl4•2Et2O. Реакционную смесь перемешивали в течение нескольких часов. Растворитель отгоняли под вакуумом, а для растворения продукта добавляли толуол и дихлорметан. Для удаления выпавшего в осадок хлорида лития смесь пропускали через цеолитовый фильтр. Растворитель отгоняли под вакуумом и добавляли небольшую порцию толуола вместе с петролейным эфиром. Для обеспечения осаждения максимального количества твердой фазы реакционную смесь охлаждали. Была получена твердая фаза светло-коричневого цвета, которая была отфильтрована и промыта небольшим количеством толуола. Оставшаяся твердая фаза была снова растворена в горячем толуоле и пропущена через цеолитовый фильтр для удаления нерастворимых в толуоле примесей. После отгонки растворителя было получено 2,32 г (5,98 моль) твердого продукта желтого цвета. Целевой продукт имел эмпирическую формулу Me2Si(C5Me4)(NPh)TiCl2.
Пример IT
Соединение IT. Стадия 1. Соединение формулы (C5Me4H)SiMe2Cl получали
по методике, описанной в примере ВТ при получении соединения ВТ на стадии 1.
Стадия 2. 10 г (0,047 моль) полученного на стадии 1 соединения формулы (C5Me4 H)SiMe2Cl постепенно добавляли к суспензии 3,68 г (0,047 моль) LiHN-t-Bu в примерно 100 мл тетрагидрофурана. Реакционную смесь оставляли на ночь при перемешивании, после чего тетрагидрофуран отгоняли под вакуумом в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида в смесь приливали петролейный эфир. Смесь пропускали затем через цеолитовый фильтр. Из фильтрата удаляли растворитель, в результате чего было выделено 11,14 г (0,044 моль) продукта формулы Me2Si(C5Me4H)(NH-t-Bu) в виде жидкости бледно-желтого цвета.
Стадия 3. 11,14 г (0,044 моль) полученного на стадии 2 жидкого продукта формулы Me2Si(C5Me4H)(NH-t-Bu) разбавляли в примерно 100 мл эфира, после чего к полученному разбавленному раствору медленно приливали 64 мл (0,09 мл) 1,4-молярного раствора метиллития в эфире. После окончания добавления метиллития реакционную смесь перемешивали в течение 0,5 часа. Эфир отгоняли, а затем отфильтровывали выпавший в осадок продукт формулы [Me2Si(C5Me4)(N-t-Bu)] Li2. Продукт был промыт несколькими порциями эфира, а затем был подвергнут вакуумной сушке.
Стадия 4. 6,6 г (0,025 моль) полученного на стадии 3 продукта формулы [Me2Si(C5Me4)(N-t-Bu)] Li2 суспендировали в охлажденном эфире, после чего к суспензии постепенно добавляли 8,4 г (0,025 моль) эфирата тетрахлорида титана формулы TiCl4•2Et2O. Полученную таким образом реакционную смесь оставляли на ночь при перемешивании. Затем эфир отгоняли под вакуумом в охлаждаемую ловушку, в которой поддерживалась температура -196o C. Для осаждения хлорида лития добавляли метиленхлорид, после чего реакционную смесь пропускали через цеолитовый фильтр. Растворитель упаривали, а для осаждения продукта приливали петролейный эфир. Полученную смесь перед фильтрованием охлаждали для обеспечения осаждения максимального количества целевого продукта. Было выделено 2,1 г (5,7 ммоль) целевого продукта формулы Me2Si(C5Me4)(N-t-Bu)TiCl2.
Пример JT
Соединение JT. Стадия 1. Соединение формулы (C5Me4H)SiMe2Cl получали, используя
методику, описанную в примере ВТ при получении соединения ВТ на стадии 1.
Стадия 2. 8 г (0,037 моль) полученного на стадии 1 соединения формулы (C5Me4H)SiMe2Cl постепенно вводили в суспензию 7 г (0,037 моль) литийорганического соединения формулы LiHNC12H23 (где С12H23 обозначает циклододецил) в примерно 80 мл тетрагидрофурана. Полученную таким образом реакционную смесь оставляли на ночь при перемешивании, после чего тетрагидрофуран отгоняли под вакуумом в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития в смесь приливали петролейный эфир и толуол, после чего смесь пропускали через цеолитовый фильтр. Из образовавшегося фильтрата удаляли растворитель. При этом было выделено 11,8 г (0,033 моль) продукта формулы Me2Si(C5Me4H)(NHC12H23) в виде бледно-желтой жидкости.
Стадия 3. 11,9 г (0,033 моль) полученного на стадии 2 жидкого продукта формулы Me2Si(C5Me4H)(NHC12H23) разбавляли в примерно 150 мл эфира, после чего к полученному разбавленному раствору медленно приливали 47 мл (0,066 моль) 1,4-молярного раствора метиллития в эфире. После окончания добавления метиллития реакционную смесь перемешивали 2 часа. Перед отфильтровыванием продукта эмпирической формулы [Me2Si(C5Me4)(NC12H23)]Li2 отгоняли эфир. Полученный таким образом продукт промывали несколькими небольшими порциями эфира и сушили в вакууме. Выход продукта составил 11,1 г (0,03 моль).
Стадия 4. 3 г (0,008 моль) полученного на стадии 3 литийорганического соединения формулы [Me2Si(C5Me4)(NC12H23)]Li2 суспендировали в охлаждаемом эфире, после чего к приготовленной таким образом суспензии постепенно добавляли эфират тетрахлорида титана формулы TiCl4•2Et2O в количестве 2,7 г (0,008 моль). Полученную реакционную смесь оставляли на ночь при перемешивании. Затем эфир отгоняли под вакуумом в охлаждаемую ловушку, в которой поддерживалась температура -196oC. Для осаждения хлорида лития добавляли метиленхлорид, после чего смесь пропускали через цеолитовый фильтр. Из образовавшегося фильтрата отгоняли растворитель, а для осаждения продукта приливали петролейный эфир. Перед фильтрованием смесь охлаждали для выпадения максимального количества продукта в осадок. Собранная твердая фаза была перекристаллизована из метиленхлорида, в результате чего был выделен 1 г (2,1 ммоль) целевого продукта формулы Me2Si(C5Me4)(NC12H23)TiCl2.
КРИСТАЛЛОГРАФИЧЕСКИЕ ДАННЫЕ
Нижеследующие кристаллографические данные касаются четырех моно-Ср соединений, одно из
которых имеет димерную структуру, заявленную в данной заявке. Включены только ORTEP-чертежи, а также таблицы данных, иллюстрирующие длину химической связи и угол связи в каждом перечисленном случае.
Также имеются дополнительные данные по кристаллографическим координатам, термопараметрам, вычислениям структурных факторов и т.д.
Краткое изложение содержания таблиц: 1 8.
1. Для немостиковых соединений, J=O и N. Для мостиковых соединений, все J= N. Информация о синтезе, ЯМР и полимеризации также имеется для немостиковых соединений, где J= O. Примерами таких соединений являются: CpZr(O-t-Bu)Cl2•xТГФ (Z2Z), CpTi(O-t-Bu)Cl2 (X2Z), Cp*Ti(O-t-Bu)Cl2 (B3Z), CP*Ti(OSiMe3)Cl2 (C3Z). Однако последнее соединение не входит в объем притязаний данной заявки.
2. В примере J, Q=Me. Вся работа была проведена с соединениями, где Q=Cl или Me. Большая же часть работы была проделана с последним.
3. Для мостиковых соединений, Lw является Et2O (примеры F, G и К) или второй молекулой для образования димера (пример Е). Для немостиковых соединений, имеется одно соединение, содержащее ТГФLw. Этим соединением является CpZr(O-t-Bu)Cl2•xТГФ (Z2Z). Данные по ЯМР, лабораторному синтезу и полимеризации, касающиеся последнего, имеются. На дату приоритета указанное соединение еще не было раскрыто ни в одной публикации.
4. Интервал соотношения A1/M, используемого в примерах, составляет 465-16100.
Анализ кристаллической структуры показан: для табл. 1 TiCl2[(C12H23)NSi(CH3)2 (C5(CH3)4)] на фиг.1; для табл. 3 [CH3)2Si(C5(CH3)4 (N-t-C4H9)] ZrCl2 на фиг. 2; для табл. 5 [(CH3)2Si(C5H3)3 (N-t-C4H9)ZrCl2]2 на фиг.3; для табл. 7 [(CH3)2Si(C5(CH3)4) (N-t-C4H9)]HfCl2 на фиг.4
Примеры 1-70 даны для иллюстрации вариантов практического осуществления процесса полимеризации олефинов с использованием
предложенной каталитической системы.
Пример 1
Полимеризация с использованием соединения А
Процесс полимеризации осуществляется в реакционном аппарате (реакторе) в
виде автоклава емкостью 1 л, оборудованного лопастной мешалкой, наружной водяной рубашкой для регулирования температуры, регулируемым устройством подвода азота (сухого), этилена, пропилена, 1-бутена и
гексана, а также элементом впуска (в виде мембраны) для ввода в реакционный аппарат растворителей, растворов комплексных соединений переходных металлов и растворов алюмоксанов. Перед началом работы
реакционный аппарат был подвергнут тщательной сушке и дегазации. Типичный процесс синтеза полимерного продукта включал в себя ввод в реакционный аппарат 400 мл толуола, 6 мл 1,5-молярного раствора
метилалюмоксана и 0,23 мг соединения А (0,2 мл раствора, 11,5 мг соединения А в 10 мл толуола). Реакционный аппарат нагревали затем до 80oC и в него вводили этилен (под давлением 60
фунтов/дюйм2). Растворитель отгоняли из полимера потоком азота. Было получено 9,2 г полиэтилена (с молекулярной массой 257200 и молекулярно-массовым распределением с преобладанием фракции 2,
275).
Пример 2
Полимеризация с использованием соединения А
Полимеризация осуществлялась как и в примере 1. При этом в реакционный аппарат вводили 300 мл толуола, 3
мл 1,5-молярного раствора метилалюмоксана и 0,115 мг соединения А (0,1 мл раствора, 11,5 мг соединения А в 10 мл толуола). Было синтезировано 3,8 г полиэтилена с молекулярной массой 359800 и
молекулярно-массовым распределением с преобладанием фракции 2,425.
Пример 3
Полимеризация с использованием соединения А
Полимеризацию проводили также, как и в
примере 2, используя те же самые концентрации исходных компонентов. Отличие состояло в основном в том, что реакцию осуществляли при температуре 40oC, а не при 80oC, как это имело
место в предыдущем примере. Было синтезировано 2,4 г полиэтилена с молекулярной массой 635000 и молекулярно-массовым распределением с преобладанием фракции 3,445.
Пример 4
Полимеризация с использованием соединения А
Полимеризацию проводили также, как и в примере 1, за исключением того, что в данном случае использовали 300 мл гексана, а не 400 мл толуола. Было
получено 5,4 г полиэтилена с молекулярной массой 212600 и молекулярно-массовым распределением с преобладанием фракции 2,849.
Пример 5
Полимеризация с использованием
соединения А
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику, что и в примере 1, в реакционный аппарат вводили: 300 мл толуола, 200 мл пропилена, 6 мл 1,
5-молярного раствора метилалюмоксана и 0,46 мг соединения А (0,4 мл раствора, 11,5 мг соединения А в 10 мл толуола). Реакционный аппарат нагревали до температуры 80oC, после чего в аппарат
вводили этилен (под давлением 60 фунтов/дюйм2). Реакцию полимеризации проводили в течение 30 минут, после чего производили быстрое охлаждение и продувку аппарата. После испарения
растворителя было получено 13,3 г сополимера этилена с пропиленом с молекулярной массой 24900, молекулярно-массовым распределением с преобладанием фракции 2,027 и с так называемой короткоцепной
разветвленностью макромолекул сополимера, соответствующей 73,5 разветвлений на 1000 атомов углерода (число разветвлений или боковых ответвлений от основной цепи определяли ИК-спектроскопией).
Пример 6
Полимеризация с использованием соединения А
Полимеризацию проводили точно так же, как и в примере 5, за исключением того, что брали 200 мл толуола и 0,92 мг
соединения А (0,8 мл раствора, 11,5 мг соединения А в 10 мл толуола). Температура полимеризации также была ниже, а именно 50oC. Было синтезировано 6 г сополимера этилена и пропилена.
Сополимер имел молекулярную массу 83100, молекулярно-массовое распределение с преобладанием фракции 2,370 и короткоцепную разветвленность, соответствующую 75,7 разветвлением на 1000 атомов углерода
(число разветвлений определялось методом ИК-спектроскопии).
Пример 7
Полимеризация с использованием соединения А
Используя реакционный аппарат той же самой
конструкции и ту же самую общую методику, что и в примере 1, в реакционный аппарат вводили следующие исходные компоненты: 150 мл толуола, 100 мл 1-бутена, 6 мл 1,5-молярного раствора метилалюмоксана и
2,3 мг соединения А (а именно 2 мл раствора, 11,5 мг соединения А в 10 мл толуола). Реакционный аппарат нагревали до температуры 50oC, после чего в аппарат вводили этилен (под давлением 65
фунтов/дюйм2). Полимеризацию проводили в течение 30 минут, после чего производилось быстрое охлаждение и продувка аппарата. После испарения толуола было получено 25,4 г сополимера этилена с
1-бутеном. Сополимер имел молекулярную массу 184500, молекулярно-массовое распределением с преобладанием фракции 3,424, а число короткоцепных боковых разветвлений на 1000 атомов углерода, определенное
методами спектроскопии ядерного магнитного резонанса (ЯМР), а именно резонанса на ядрах атомов углерода13C, и методами ИК-спектроскопии, составляло 23,5 и 21,5, соответственно.
Пример 8
Полимеризация с использованием соединения А
Полимеризация проводилась точно также, как и в примере 7, за исключением того, что толуол и 1-бутен брали в количестве
100 мл и 150 мл соответственно. Было синтезировано 30,2 г сополимера этилена и 1-бутена. Полученный сополимер имел молекулярную массу 143500 и молекулярно-массовое распределение с обладанием фракции 3,
097. Число короткоцепных боковых разветвлений на 1000 атомов углерода, определенное методом ЯМР на ядрах атомов углерода13C и методом ИК-спектроскопии, составляло 30,8 и 26,5
соответственно.
Пример 9
Полимеризация с использованием соединения А
Полимеризацию проводили точно также, как и в примере 7. При этом были изменены количества
вводимых в реакционный аппарат исходных компонентов, а именно брали 200 мл толуола, 8 мл 1-молярного раствора метилалюмоксана и 50 мл 1-бутена. Было синтезировано 24,9 г сополимера этилена с
1-бутеном. Полученный сополимер имел молекулярную массу 163200 и молекулярно-массовое распределение с преобладанием фракции 3,290. Число короткоцепных разветвлений на 1000 атомов углерода,
определенное методом ЯМР на ядрах атомов углерода13C и методом ИК-спектроскопии, составляло 23,3 и 18,9 соответственно.
Пример 10
Полимеризация с использованием
соединения А
Полимеризацию проводили точно также, как и в примере 9, за исключением того, что вместо 200 мл толуола брали 200 мл гексана. Было получено 19,5 г сополимера этилена и 1-бутена.
Полученный сополимер имел молекулярную массу 150600 и молекулярно-массовое распределение с преобладанием фракции 3,510. Число короткоцепных разветвлений на 1000 атомов углерода, определенное методом
ЯМР на ядрах атомов углерода13C и методом ИК-спектроскопии, составляло 12,1 и 12,7 соответственно.
Пример 11
Полимеризация с использованием соединения А
Полимеризацию осуществляли точно также, как и в примере 10, за исключением того, что были изменены количества вводимых в реакционный аппарат компонентов. В частности, в данном случае использовали 150
мл гексана и 100 мл 1-бутена. Было синтезировано 16 г сополимера этилена с 1-бутеном. Сополимер имел молекулярную массу 116200 и молекулярно-массовое распределение с преобладанием фракции 3,158. Число
короткоцепных разветвлений на 1000 атомов углерода, определенное методом спектроскопии ЯМР на ядрах атомов углерода13C и методом ИК-спектроскопии, составляло 19,2 и 19,4 соответственно.
Пример 12
Полимеризация с использованием соединения А
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику осуществления процесса, что и
в примере 1, в реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,2 мл раствора предварительно активированного соединения А (а
именно 11,5 мг соединения А, растворенного в 9 мл толуола, и 1 мл 1-молярного раствора метилалюмоксана). Реакционный аппарат нагревали до 80oC. Этилен вводили в аппарат под давлением 60
фунтов/дюйм2. Процесс проводили в течение 30 минут, после чего производили быстрое охлаждение и продувку аппарата. После испарения растворителя было получено 3,4 г полиэтилена с
молекулярной массой 285000 и молекулярно-массовым распределением с преобладанием фракции 2,808.
Пример 13
Полимеризация соединения А
Полимеризацию проводили точно
также, как и в примере 12, за исключением того, что раствор предварительно активированного соединения А подвергали старению в течение одних суток. Было синтезировано 2 г полиэтилена молекулярной массы
260700 с молекулярно-массовым распределением с преобладанием фракции 2,738.
Пример 14
Полимеризация с использованием соединения А
Используя реакционный аппарат той
же самой конструкции и ту же самую общую методику осуществления процесса, что и в примере 1, в реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола, 0,25 мл 1-молярного раствора
метилалюмоксана и 0,2 мл раствора предварительно активированного соединения А (11,5 мг соединения А, растворенного в 9,5 мл толуола и 0,5 мл 1-молярного раствора метилалюмоксана). Реакционный аппарат
нагревали до 80oC, а исходный мономер, а именно этил, вводили в аппарат под давлением 60 фунтов/дюйм2. Реакцию полимеризации проводили в течение 30 минут, после чего производили
быстрое охлаждение и продувку аппарата. После испарения растворителя было выделено 1,1 г полиэтилена с молекулярной массой 479600 и молекулярно-массовым распределением с преобладанием фракции 3,
130.
Пример 15
Полимеризация с использованием соединения А
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику проведения
полимеризации, что и в примере 1, в реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола и 2 мл раствора предварительно активированного соединения А (11,5 г соединения А,
растворенного в 9,5 мл толуола, и 0,5 мл 1-молярного раствора метилалюмоксана). Реакционный аппарат нагревали до 80oC, а этилен вводили в аппарат под давлением 60 фунтов/дюйм2.
Полимеризацию проводили в течение 30 минут, после чего производили быстрое охлаждение и продувку аппарата. После испарения растворителя было выделено 1,6 г полиэтилена с молекулярной массой 458800 и
молекулярно-массовым распределением с преобладанием фракции 2,037.
Пример 16
Полимеризация с использованием соединения А
Используя общую методику проведения процесса,
описанную в примере 1, в реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,23 г соединения А (0,2 мл раствора, 11,5 мг
соединения А в 10 мл толуола). Реакционный аппарат нагревали до 80oC, а исходный мономер (этилен) вводили в аппарат под давлением 400 фунтов/дюйм2. Полимеризацию проводили в
течение 30 минут, после чего производили быстрое охлаждение и продувку аппарата. После испарения растворителя было получено 19,4 г полиэтилена с молекулярной массой 343700 и молекулярно-массовым
распределением с преобладанием фракции 3,674.
Пример 17
Полимеризация с использованием соединения А
Полимеризацию проводили в автоклаве из нержавеющей стали емкостью
100 мл, снабженном мешалкой и рассчитанном на осуществление полимеризации при давлениях до 40000 фунтов/дюйм2 и температуре до 300oC. Реакционный аппарат (автоклав) продували
азотом и нагревали до 160oC. Растворы соединения А и алюмоксана приготавливали в отдельных сосудах (флаконах или ампулах). Исходный раствор приготавливали путем растворения 26 мг соединения
А в 100 мл толуола. Раствор соединения А приготавливали, разбавляли в 5 мл толуола 0,5 мл исходного раствора. Раствор алюмоксана состоял из 2 мл 4%-ного раствора метилалюмоксана и добавленных к нему 5
мл толуола. Раствор соединения А приливали к раствору алюмоксана, после чего 0,43 мл смешанных растворов нагнетали с помощью потока азота, находящегося под давлением, в инжекторную трубку постоянного
объема. В автоклаве создавали повышенное давление путем подачи в него этилена под давлением 1784 бар. Реакционная смесь в автоклаве перемешивалась путем вращения мешалки со скоростью (частотой
вращения) 1500 об/мин. Смешанные растворы инжектировались в автоклав (при одновременном перемешивании) с избыточным давлением. При этом наблюдалось повышение температуры на 4oC. Значения
температуры и давления непрерывно регистрировались в течение 120 секунд, после чего содержимое автоклава быстро выгружалось в приемный сосуд продувкой. Рабочее пространство в автоклаве промывали
ксилолом для сбора дополнительного количества оставшегося в автоклаве полимера. Эти промывочные порции ксилола с остатками полимера объединялись вместе с основным полимером, выгруженным из автоклава
продувкой. Выход полимера (полиэтилена) составил 0,7 г. Полиэтилен имел молекулярную массу 245500 и молекулярно-массовое распределение с преобладанием фракции 2,257.
Пример 18
Полимеризация с использованием соединения В
Используя общую методику проведения процесса полимеризации, описанную в примере 1, в реакционный аппарат вводили следующие исходные компоненты:
400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,278 мг соединения В (0,2 мл раствора, 13,9 мг соединения В в 10 мл толуола). Реакционный аппарат нагревали до 80oC, после чего
в него вводили этилен под давлением 60 фунтов/дюйм2. Реакцию проводили в течение ограниченного интервала времени (10 минут). Реакцию прервали быстрым охлаждением и продувкой аппарата.
Растворитель отгоняли из полимера потоком азота. Было синтезировано 9,6 г полиэтилена с молекулярной массой 241200 и молекулярно-массовым распределением с преобладанием фракции 2,268.
Пример 19
Полимеризация с использованием соединения С
Используя общую методику проведения процесса, описанную в примере 1, в реакционный аппарат вводили следующие исходные
компоненты: 300 мл толуола, 4 мл 1-молярного раствора метилалюмоксана и 0,46 г соединения С (0,4 мл раствора, 11,5 мг соединения С в 10 мл толуола). Реакционный аппарат нагревали до 80oC.
Этилен вводили в аппарат под давлением 60 фунтов/дюйм2. Реакцию полимеризации проводили в течение ограниченного периода времени (30 минут). Реакцию прерывали быстрым охлаждением и продувкой
(разгрузкой) аппарата. Растворитель отгоняли из полимера продувкой азотом. Было синтезировано 1,7 г полиэтилена с молекулярной массой 278400 и молекулярно-массовым распределением с преобладанием
фракции 2,142.
Пример 20
Полимеризация с использованием соединения D
Используя общую методику проведения процесса, описанную в примере 1, в реакционный аппарат
вводили следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,278 мг соединения D (0,2 мл раствора, 13,9 мг соединения D в 10 мл толуола). Реакционный аппарат
нагревали до 80oC и в него вводили исходный мономер, а именно этилен, под давлением 60 фунтов/дюйм2. Реакция полимеризации осуществлялась в течение ограниченного периода времени
(30 минут). Реакцию прерывали быстрым охлаждением и продувкой (разгрузкой) аппарата. Растворитель отгоняли из полимера продувкой азотом. Было синтезировано 1,9 г полиэтилена с молекулярной массой
229700 и молекулярно-массовым распределением с преобладанием фракции 2,618.
Пример 21
Полимеризация с использованием соединения Е
Используя общую методику
осуществления процесса, описанную в примере 1, в реакционный аппарат вводили следующие исходные компоненты: 300 мл гексана, 9 мл 1-молярного раствора метилалюмоксана и 0,24 мг соединения Е (0,2 мл
раствора, 12 мг соединения Е в 10 мл толуола). Реактор нагревали до 80oC, а этилен вводили в аппарат под давлением 60 фунтов/дюйм2. Продолжительность реакции полимеризации
ограничивали периодом времени в 30 минут. Реакцию прерывали быстрым охлаждением и продувкой (разгрузкой) аппарата. Растворитель отгоняли из полимера продувкой азотом. В результате было получено 2,2 г
полиэтилена с молекулярной массой 258200 и молекулярно-массовым распределением с преобладанием фракции 2,348.
Пример 22
Полимеризация с использованием соединения Е
Полимеризацию проводили точно также, как и в примере 1. При этом в реакционный аппарат вводили следующие исходные компоненты: 200 мл толуола, 100 мл 1-бутена, 9 мл 1-молярного раствора метилалюмоксана
и 2,4 мг соединения Е (2 мл раствора, 12 мг соединения Е в 10 мл толуола). Реакционный аппарат нагревали до 50oC. В аппарате создавали высокое давление, вводя в него этилен под давлением 65
фунтов/дюйм2. Реакцию полимеризации проводили в течение 30 минут, после чего аппарат быстро охлаждали и продували (разгружали). После удаления растворителя испарением было получено 1,8 г
сополимера этилена и 1-бутена. Сополимер имел молекулярную массу 323600 и молекулярно-массовое распределение с преобладанием фракции 2,463. Короткоцепная разветвленность макромолекулы сополимера
соответствовала 33,5 разветвлениям на 1000 атомов углерода (число разветвлений определялось с помощью ИК-спектроскопии).
Пример 23
Полимеризация с использованием соединения
F
Полимеризацию проводили точно также, как и в примере 1. При этом в реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,
242 мг соединения F (0,2 мг раствора, 12,1 мг соединения F в 10 мл толуола). Реакционный аппарат нагревали до 80oC, а этилен вводили под давлением 60 фунтов/дюйм2. Реакцию
полимеризации проводили в течение 30 минут. Было синтезировано 5,3 г полиэтилена с молекулярной массой 319900 и молекулярно-массовым распределением с преобладанием фракции 2,477.
Пример 24
Полимеризация с использованием соединения F
Реакция полимеризации проводилась точно также, как и в примере 1. При этом в реакционный аппарат вводили следующие исходные
компоненты: 150 мл толуола, 100 мл 1-бутена, 9 мл 1-молярного раствора метилалюмоксана и 2,42 мг соединения F (2 мл раствора, 12,1 мг соединения F в 10 мл толуола). Реакционный аппарат нагревали до
50oC, а этилен вводили под давлением 65 фунтов/дюйм2. Полимеризация продолжалась 30 минут. Было синтезировано 3,5 г сополимера этилена с 1-бутеном. Сополимер имел молекулярную
массу 251300 и молекулярно-массовое распределение с преобладанием фракции 3,341. Короткоцепная разветвленность макромолекулы сополимера 33,3 разветвления на 1000 атомов углерода. Число разветвлений
определялось с помощью ИК-спектроскопии.
Пример 25
Полимеризация с использованием соединения G
Реакция полимеризации проводилась точно также, как и в примере 1. При
этом в реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,29 мг соединения G (0,2 мл раствора, 14,5 мг соединения G в 10 мл
толуола). Реакционный аппарат нагревали до 80oC. Этилен вводился под давлением 60 фунтов/дюйм2. Полимеризацию проводили в течение 30 минут. Было синтезировано 3,5 г полиэтилена с
молекулярной массой 237300 и молекулярно-массовым распределением с преобладанием фракции 2,549.
Пример 26
Полимеризация с использованием соединения G
Реакция
полимеризации проводилась точно также, как и в примере 1. При этом в реакционный аппарат вводили следующие компоненты: 150 мл толуола, 100 мл 1-бутена, 7 мл 1-молярного раствора метилалюмоксана и 2,9
мг соединения G (2 мл раствора, 14,5 мг соединения G в 10 мл толуола). Реакционный аппарат нагревали до 50oC, а этилен вводили в него под давлением 65 фунтов/дюйм2. Реакцию
полимеризации проводили в течение 30 минут. Было синтезировано 7 г сополимера этилена и 1-бутена. Полученный сополимер имел молекулярную массу 425000 и молекулярно-массовое распределение с
преобладанием фракции 2,816. Короткоцепная разветвленность макромолекулы сополимера 27,1 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ИК-спектроскопии).
Пример 27
Полимеризация с использованием соединения Н
Реакция полимеризации проводилась точно также, как и в примере 1. При этом в реакционный аппарат вводили следующие
компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,266 мг соединения Н (0,2 мл раствора, 13,3 мг соединения Н в 10 мл толуола). Реакционный аппарат нагревали до 80oC,
а этилен вводили в него под давлением 60 фунтов/дюйм2. Полимеризация проводилась в течение 30 минут. Было синтезировано 11,1 г полиэтилена с молекулярной массой 299800 и
молекулярно-массовым распределением с преобладанием фракции 2,569.
Пример 28
Полимеризация с использованием соединения Н
Реакция полимеризации проводилась точно
также, как и в примере 1. При этом в реакционный аппарат вводили следующие компоненты: 150 мл толуола, 100 мл 1-бутена, 7 мл 1-молярного раствора метилалюмоксана и 2,66 мг соединения Н (2 мл раствора,
13,3 мг соединения Н в 10 мл толуола). Реакционный аппарат нагревали до 50oC, а этилен вводили под давлением 65 фунтов/дюйм2. Реакция полимеризации протекала 30 минут. Было
синтезировано 15,4 г сополимера этилена и 1-бутена. Сополимер имел молекулярную массу 286600 и молекулярно-массовое распределение с преобладанием фракции 2,980. Короткоцепная разветвленность
макромолекулы сополимера 45,4 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ИК-спектроскопии).
Пример 29
Полимеризация с использованием
соединения I
Реакция полимеризации проводилась точно также, как в примере 1. При этом в реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора
метилалюмоксана и 0,34 мг соединения I (0,2 мл раствора, 17 мг соединения I в 10 мл толуола). Реакционный аппарат нагревали до 80oC, а этилен вводили под давлением 60 фунтов/дюйм2
. Реакцию полимеризации проводили в течение 30 минут, после чего аппарат быстро охлаждали и продували (для выгрузки полимера ). После испарения растворителя было получено 0,9 г полиэтилена с
молекулярной массой 377000 и молекулярно-массовым распределением с преобладанием фракции 1,996.
Пример 30
Полимеризация с использованием соединения J
Реакция
полимеризации проводилась точно также, как и в примере 1. При этом в реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,318 мг
соединения J (0,2 мл раствора, 15,9 мг соединения J в 10 мл толуола). Реакционный аппарат нагревали до 80oC, а этилен вводили в него под давлением 60 фунтов/дюйм2. Полимеризацию
проводили в течение 30 минут. Было синтезировано 8,6 г полиэтилена с молекулярной массой 321000 и молекулярно-массовым распределением с преобладанием фракции 2,803.
Пример 31
Полимеризация с использованием соединения J
Реакция полимеризации проводилась точно также, как в примере 1. При этом в реакционный аппарат вводили следующие исходные компоненты: 150 мл
толуола, 100 мл 1-бутена, 7 мл 1-молярного раствора метилалюмоксана и 3,18 мг соединения J (2 мл раствора 15,9 мг соединения J в 10 мл толуола). Реакционный аппарат нагревали до 50oC, а
этилен вводили под давлением 65 фунтов/дюйм2. Реакцию проводили в течение 30 минут. Было синтезировано 11,2 мг сополимера этилена и 1-бутена. Полученный сополимер имел молекулярную массу
224800, молекулярно-массовое распределение с преобладанием фракции 2,512 и короткоцепную разветвленность, соответствующую 49,6 и 55,4 разветвлением на 1000 атомов углерода (первое из этих чисел было
определено с помощью ИК-спектроскопии, а второе с помощью спектроскопии ЯМР).
Пример 32
Полимеризация с использованием соединения К
Реакция полимеризации проводилась
точно также, как и в примере 1. При этом в реакционный аппарат вводились следующие исходные компоненты: 300 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,272 мг соединения К (0,2 мл
раствора, 13,6 мг соединения К в 10 мл толуола). Реакционный аппарат нагревали до 80oC, а этилен вводили под давлением 60 фунтов/дюйм2. Полимеризацию проводили в течение 30
минут. Было синтезировано 26,6 г полиэтилена с молекулярной массой 187300 и молекулярно-массовым распределением с преобладанием фракции 2,401.
Пример 33
Полимеризация с
использованием соединения К
Реакция полимеризации проводилась точно также, как и в примере 1. При этом в реакционный аппарат вводились следующие исходные компоненты: 150 мл толуола, 100 мл
1-бутена, 7 мл 1-молярного раствора метилалюмоксана и 2,72 мг соединения К (2 мл раствора, 13,6 мг соединения К в 10 мл толуола). Реакционный аппарат нагревали до 50oC, а этилен вводился
под давлением 65 фунтов/дюйм2. Реакцию полимеризации проводился в течение 30 минут. Было получено 3,9 г сополимера этилена и 1-бутена. Сополимер имел молекулярную массу 207600 и
молекулярно-массовое распределение с преобладанием фракции 2,394. Короткоцепная разветвленность макромолекулы сополимера 33,9 разветвлений на 1000 атомов углерода (число разветвлений определялось с
помощью ИК-спектроскопии).
Пример 34
Полимеризация с использованием соединения L
Полимеризация осуществлялась точно также, как и в примере 1. В данном случае в
реакционный аппарат вводили следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,322 мг соединения L (0,2 мл раствора, 16,1 мг соединения L в 10 мл толуола).
Реакционный аппарат нагревали до 80oC. Исходный мономер, а именно этилен, вводили в аппарат под давлением 60 фунтов/дюйм2. Реакция полимеризации проводилась 30 минут. Было
синтезировано 15,5 г полиэтилена с молекулярной массой 174300 и молекулярно-массовым распределением с преобладанием фракции 2,193.
Пример 35
Полимеризация с использованием
соединения А
Полимеризация осуществлялась точно также, как и в примере 1. При этом в реакционный аппарат загружались следующие исходные компоненты: 250 мл толуола, 150 мл 1-гексена, 7 мл
1-молярного раствора метилалюмоксана и 2,3 мг соединения А (2 мл раствора, 11,5 мг соединения А в 10 мл толуола). Реактор нагревали до 50oC, а этилен вводился в него под давлением 65
фунтов/дюйм2. Реакцию полимеризации проводили в течение 30 минут, после чего реактор быстро охлаждали и продували. После испарения растворителя было получено 26,5 г сополимера этилена и
1-гексена. Сополимер имел молекулярную массу 222800 и молекулярно-массовое распределение с преобладанием фракции 3,373. Короткоцепная разветвленность макромолекулы сополимера 39,1 разветвлений на 1000
атомов
углерода (число разветвлений определялось с помощью ИК-спектроскопии).
Пример 36
Полимеризация с использованием соединения А
Реакция полимеризации
осуществлялась точно также, как и в примере 1. В данном случае в реакционный аппарат были загружены следующие исходные компоненты: 300 мл толуола, 100 мл 1-октена, 7 мл 1-молярного раствора
метилалюмоксана и 2,3 мг соединения А (2 мл раствора, 11,5 мг соединения А в 10 мл толуола). Реакционный аппарат нагревали до 50oC, а исходный мономер (этилен) вводился под давлением 65
фунтов/дюйм2. Реакцию проводили в течение 30 минут, после чего аппарат быстро охлаждали и продували. После испарения растворителя было получено 19,7 г сополимера этилена и 1-октена.
Сополимер имел молекулярную массу 548600 и молекулярно-массовое распределение с преобладанием фракции 3,007. Короткоцепная разветвленность макромолекулы сополимера 16,5 разветвлений на 1000 атомов
углерода (число разветвлений определялось с помощью спектроскопии ЯМР на ядрах атомов углерода13C).
Пример 37
Полимеризация с использованием соединения А
Полимеризация осуществлялась точно также, как и в примере 1. В данном случае в реакционный аппарат были загружены следующие исходные компоненты: 300 мл толуола, 100 мл 4-метил-1-пентена, 7 мл
1-молярного раствора метилалюмоксана и 2,3 мг соединения А (2 мл раствора, 11,5 мг соединения А в 10 мл толуола). Реакционный аппарат нагревали до 50oC. Этилен вводили в реакционный аппарат
под давлением 65 фунтов/дюйм2. Реакцию полимеризации проводили в течение 30 минут, после чего реакционный аппарат быстро охлаждали и продували. После отгонки растворителя было получено 15,1
г сополимера этилена и 4-метил-1-пентена. Сополимер имел молекулярную массу 611800 и молекулярно-массовое распределение с преобладанием фракции 1,683. Было обнаружено наличие примеси сомономера в
количестве 1,8 мол. определение производилось с помощью спектроскопии ЯМР на ядрах атомов углерода13C.
Пример 38
Полимеризация с использованием соединения А
Полимеризация осуществлялась точно также, как и в примере 1. При этом в реактор загружали: 300 мл толуола, 100 мл 2,2-молярного раствора норборнена в толуоле, 7 мл 1-молярного раствора
метилалюмоксана и 2,3 мг соединения А (2 мл раствора, 11,5 мг соединения А в 10 мл толуола). Реакционный аппарат нагревали до 50oC. В аппарате создавали высокое давление (65 фунтов/дюйм2) подачей в него этилена под соответствующим давлением. Реакцию проводили в течение 30 минут, после чего реактор быстро охлаждали и продували (разгружали). После отгонки растворителя было
выделено 12,3 г сополимера этилена и норборнена. Сополимер имел молекулярную массу 812600, молекулярно-массовое распределение с преобладанием фракции 1,711. С помощью ЯМР-спектроскопии на ядрах атомов
углерода13 было обнаружено наличие примесей сомономера в количестве 0,3 мол.
Пример 39
Полимеризация с использованием соединения А
Полимеризацию
осуществляли точно также, как и в примере 1. В данном случае в реакционный аппарат были загружены следующие исходные компоненты: 300 мл толуола, 100 мл цис-1,4-гексадиена, 7 мл 1-молярного раствора
метилалюмоксана и 2,3 мг соединения А (2 мл раствора 11,5 мг соединения А в 10 мл толуола). Реакционный аппарат нагревали до 50oC. Этилен вводили под давлением 65 фунтов/дюйм2.
Реакцию полимеризации проводили в течение 30 минут, после чего реакционный аппарат быстро охлаждали и продували. После отгонки растворителя было получено 13,6 г сополимера этилена и цис-1,
4-гексадиена. Сополимер имел молекулярную массу 163400, молекулярно-массовое распределение с преобладанием фракции 2,388. Было обнаружено наличие примеси сомономера в количестве 2,2 мол. определение
производилось с помощью спектроскопии ЯМР на ядрах атомов углерода13C.
Пример 40
Полимеризация с использованием соединения АТ
Полимеризация проводилась в
реакционном аппарате в виде автоклава емкостью 12 л, оборудованного лопастной мешалкой, наружной водяной рубашкой для регулирования температуры реакции, регулируемым устройством подвода сухого азота,
этилена, пропилена, 1-бутена и гексана, а также впускным устройством мембранного типа для загрузки в реакционный аппарат других растворителей, сомономеров, раствора комплексного соединения переходного
металла и раствора алюмоксана. Перед началом работы реакционный аппарат был тщательно высушен и подвергнут дегазации. Для полимеризации в реакционный аппарат загружали исходные компоненты, а именно
400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,206 мг соединения АТ (0,2 мл раствора, 10,3 мг соединения АТ в 10 мл толуола), и в последующем нагревали аппарат до 80oC. Затем
в аппарат вводили этилен под давлением 60 фунтов/дюйм2. Реакцию полимеризации проводили в течение ограниченного периода времени 30 минут, после чего реакцию прерывали быстрым охлаждением и
продувкой аппарата. Растворитель испаряли (отгоняли) из полимера потоком азота. Было синтезировано 11,8 г полиэтилена с молекулярной массой 279700 и молекулярно-массовым распределением с преобладанием
фракции 2,676.
Пример 41
Полимеризация с использованием соединения АТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику ведения
процесса, что в примере 40, в реакционный аппарат загружали следующие исходные компоненты: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,2 мл раствора предварительно активированного
соединения АТ (10,3 мг соединения АТ, растворенного в 9,5 мл толуола и 0,5 мл 1-молярного раствора метилалюмоксана). Реакционный аппарат нагревали до 80oC, а этилен загружали под давлением
60 фунтов/дюйм2. Реакцию полимеризации проводили в течение 30 минут, после чего аппарат быстро охлаждали и продували. После удаления растворителя было получено 14,5 г полиэтилена с
молекулярной массой 406100 и молекулярно-массовым распределением с преобладанием фракции 2,486.
Пример 42
Полимеризация с использованием соединения АТ
Используя
реакционный аппарат той же самой конструкции и ту же самую общую методику ведения процесса, что и в примере 40, в реакционный аппарат загружали следующие исходные компоненты: 300 мл толуола, 100 мл
1-гексена, 7 мл 1-молярного раствора метилалюмоксана и 1,03 мг соединения АТ (1 мл раствора 10,3 мг соединения АТ в 10 мл толуола). Реакционный аппарат нагревали до 80oC, а этилен подавали
под давлением 65 фунтов/дюйм2. Реакцию полимеризации проводили в течение 10 минут, после чего аппарат быстро охлаждали и продували. После удаления толуола было выделено 48,6 г сополимера
этилена и 1-гексена. Сополимер имел молекулярную массу 98500 и молекулярно-массовое распределение с преобладанием фракции 1,745. Короткоцепная разветвленность макромолекулы сополимера 117 разветвлений
на 1000 атомов углерода (число разветвлений определялось с помощью спектроскопии ЯМР на ядрах атомов углерода13C).
Пример 43
Полимеризация с использованием
соединения АТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику ведения процесса, что и в примере 40, в реакционный аппарат загружали следующие компоненты:
375 мл толуола, 25 мл 1-гексена, 7 мл 1-молярного раствора метилалюмоксана и 1,03 мг соединения АТ (1 мл раствора, 10,3 мг соединения АТ в 10 мл толуола). Реакционный аппарат нагревали до 80oC, а этилен подавали в него под давлением 65 фунтов/дюйм2. Реакцию полимеризации проводили в течение 10 минут, после чего аппарат быстро охлаждали и продували. После удаления толуола
было выделено 29,2 г сополимера этилена и 1-гексена с молекулярной массой 129800 и молекулярно-массовым распределением с преобладанием фракции 2,557. Короткоцепная разветвленность макромолекулы
сополимера соответствовала 53 разветвлениям на 1000 атомов углерода (число разветвлений определялось с помощью спектроскопии ЯМР на ядрах атомов углерода13C).
Пример 14
Полимеризация с использованием соединения АТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику ведения процесса, что и в примере 40, в реакционный
аппарат загружали следующие исходные компоненты: 375 мл толуола, 25 мл 1-гексена, 7 мл 1-молярного раствора метилалюмоксана и 1,03 мг соединения АТ (1 мл раствора 10,3 мг соединения АТ в 10 мл
толуола). Реакционный аппарат нагревали до 50oC, а этилен подавали в него под давлением 65 фунтов/дюйм2. Реакцию проводили в течение 10 минут, после чего реакционный аппарат
быстро охлаждали и продували. После удаления толуола было выделено 15 г сополимера этилена и 1-гексена. Сополимер имел молекулярную массу 310000 и молекулярно-массовое распределение с преобладанием
фракции 2,579. Короткоцепная разветвленность макромолекулы сополимера соответствовала 47,2 разветвлениям на 1000 атомов углерода (число разветвлений определялось с помощью спектроскопии ЯМР на ядрах
атомов углерода13C).
Пример 45
Полимеризация с использованием соединения АТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую
методику ведения процесса, что и в примере 40, в реакционный аппарат загружали: 300 мл толуола, 100 мл пропилена, 7 мл 1-молярного раствора метилалюмоксана и 2,06 мг соединения АТ (2 мл раствора, 10,3
мг соединения AT в 10 мл толуола). Затем реакционный аппарат нагревали до 80oC, а этилен подавали в него под давлением 65 фунтов/дюйм2. Реакцию полимеризации проводили в течение
10 минут, после чего реакционный аппарат быстро охлаждали и продували. После испарения толуола было получено 46 г сополимера этилена и пропилена. Сополимер имел молекулярную массу 110200 и
молекулярно-массовое распределение с преобладанием фракции 5,489. С помощью ИК-спектроскопии было обнаружено наличие примеси вес. (мас.) этилена в полученном сополимере.
Пример 46
Полимеризация с использованием соединения АТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику осуществления процесса, что и в примере 40, в него
загружали: 300 мл толуола, 100 мл 1-бутена, 7 мл 1-молярного раствора метилалюмоксана и 1,03 мг соединения АТ (1 мл раствора, 10,3 мг соединения АТ в 10 мл толуола). Реакционный аппарат нагревали до
80oC, а этилен подавали в него под давлением 65 фунтов/дюйм2. Реакцию полимеризации проводили 10 минут, после чего аппарат быстро охлаждали и продували. После испарения толуола
было выделено 35,1 г сополимера этилена и 1-бутена. Сополимер имел молекулярную массу 94400 и молекулярно-массовое распределение с преобладанием фракции 2,405. Короткоцепная разветвленность
макромолекулы сополимера соответствовала 165 разветвлениям на 1000 атомов углерода (число разветвлений определялось с помощью спектроскопии ЯМР на ядрах атомов углерода13C).
Пример 47
Полимеризация с использованием соединения АТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику осуществления процесса, что и в
примере 40, в него загружали: 300 мл толуола, 100 мл 1-октена, 7 мл 1-молярного раствора метилалюмоксана и 1,04 мг соединения АТ (1 мл раствора, 10,4 мг соединения АТ в 10 мл толуола). Реакционный
аппарат нагревали до 80oC, а этилен подавали в него под давлением 65 фунтов/дюйм2. Реакцию полимеризации проводили в течение 10 минут, после чего аппарат быстро охлаждали и
продували. После отгонки толуола было выделено 30,6 г сополимера этилена и 1-октена. Сополимер имел молекулярную массу 73100 и молекулярно-массовое распределение с преобладанием фракции 2,552.
Короткоцепная разветвленность соответствовала 77,7 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью спектроскопии ЯМР на ядрах атомов углерода13C).
Пример 48
Полимеризация с использованием соединения ВТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику осуществления процесса, что и в
примере 40, в него загружали: 400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,248 мг соединения ВТ (0,2 мл раствора, 12,4 мг соединения ВТ в 10 мл толуола). Реакционный аппарат нагревали
до 80oC, а этилен подавали в него под давлением 60 фунтов/дюйм2. Реакцию полимеризации проводили в течение 10 минут, после чего аппарат быстро охлаждали и продували. После
отгонки растворителя (толуола) было выделено 3,8 г полиэтилена с молекулярной массой 451400 и молекулярно-массовым распределением с преобладанием фракции 3,692.
Пример 49
Полимеризация с использованием соединения СТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику осуществления процесса, что и в примере 40, в него загружали:
400 мл толуола, 5 мл 1-молярного раствора метилалюмоксана и 0,234 мг соединения СТ (0,2 мл раствора, 11,7 мг соединения СТ в 10 мл толуола). Реакционный аппарат нагревали до 80oC, а этилен
подавали в него под давлением 60 фунтов/дюйм2. Реакцию полимеризации проводили в течение 10 минут, после чего аппарат быстро охлаждали и продували. После отгонки (испарения) толуола было
выделено 2,7 г полиэтилена с молекулярной массой 529100 и молекулярно-массовым распределением с преобладанием фракции 3,665.
Пример 50
Полимеризация с использованием
соединения DT
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику осуществления полимеризации, что и в примере 40, в аппарат загружали: 400 мл толуола, 5 мл
1-молярного раствора метилалюмоксана и 0,28 мг соединения DT (0,2 мл раствора, 14 мг соединения DT в 10 мл толуола). Реакционный аппарат нагревали до 80oC. Этилен подавали под давлением 60
фунтов/дюйм2. Реакцию полимеризации осуществляли в течение 10 минут, после чего реакционный аппарат быстро охлаждали и продували. После отгонки толуола было выделено 9 г полиэтилена с
молекулярной массой 427800 и молекулярно-массовым распределением с преобладанием фракции 3,306.
Пример 51
Полимеризация с использованием соединения DT
Используя
реакционный аппарат той же самой конструкции и ту же самую общую методику проведения процесса, что и в примере 40, в аппарат загружали: 300 мл толуола, 100 мл пропилена, 7 мл 1-молярного раствора
метилалюмоксана и 1,4 мг соединения DT (1 мл раствора, 14 мг соединения DT в 10 мл толуола). Реакционный аппарат нагревали до 30oC. Реакцию полимеризации проводили в течение 1 часа, после
чего аппарат быстро охлаждали и продували. После отгонки толуола было выделено 15 г аморфного полипропилена с молекулярной массой 18600 и молекулярно-массовым распределением с преобладанием фракции 1,
657.
Пример 52
Полимеризация с использованием соединения ЕТ
Используя реакционный аппарат той же самой конструкции и ту же самую общую методику проведения
полимеризации, что и в примере 40, в аппарат загружали: 300 мл толуола, 100 мл 1-гексена, 70 мл 1-молярного раствора метилалюмоксана и 1 мг соединения ЕТ (1 мл раствора, 10 мг соединения ЕТ в 10 мл
толуола). Реакционный аппарат нагревали до 80oC. Этилен подавали под давлением 65 фунтов/дюйм2. Во время полимеризации температура в реакционном аппарате повысилась на 20oC. Спустя 10 минут реакционный аппарат был быстро охлажден и продукт. После отгонки толуола было выделено 106 г сополимера этилена и 1-гексена. Сополимер имел молекулярную массу 17900 и
молекулярно-массовое распределение с преобладанием фракции 2,275. Короткоцепная разветвленность макромолекулы сополимера соответствовала 39,1 разветвлениям на 1000 атомов углерода (число разветвлений
определялось с помощью спектроскопии ЯМР).
Пример 53
Полимеризация с использованием соединения АТ
Реакция полимеризации проводили в автоклаве емкостью 100 мл из
нержавеющей стали, оборудованном мешалкой и рассчитанном на проведении полимеризации при температуре до 300oC и давлении до 2500 бар. Реакционный аппарат сначала вакуумировали, продували
азотом, затем продували этиленом и нагревали до 200oC. Затем в реакционный аппарат вводили 75 мл 1-гексена под давлением, равным давлению этилена. Исходный раствор соединения АТ готовили
путем растворения 6,5 мг соединения АТ в 12,5 мл толуола. Далее добавляли 1 мл исходного раствора соединения АТ к 1,9 мл 1-молярного раствора метилалюмоксана. Затем к полученной таким образом смеси
растворов приливали 7,1 мл толуола. 0,43 мл полученного раствора подавали под напором азота в трубчатый дозатор инжекторного типа. В автоклаве создавали высокое давление (равное 1748 бар) путем подачи
в него этилена под соответствующим давлением. Реакционную смесь перемешивали при вращении мешалки со скоростью 1800 об/мин. Раствор соединения АТ с алюмоксаном впрыскивали в автоклав с избыточным
давлением. В этом момент наблюдалось повышение температуры в автоклаве на 16oC. Значения температуры и давления непрерывно регистрировались в течение 120 секунд. Сразу же после этого
содержимое автоклава быстро выгружалось (продувкой) в приемный сосуд (приемник). Затем автоклав промывали ксилолом с целью сбора дополнительного количества частично оставшегося в автоклаве полимера.
Эти промывочные порции были объединены затем с основной частью образовавшегося полимера, выгруженного из автоклава продувкой. Осаждением полимера из смеси путем добавления ацетона удалось выделить 2,7
г полимера с молекулярной массой 64000 и молекулярно-массовым распределением с преобладанием фракции 3,16. Короткоцепная разветвленность макромолекулы полученного таким образом полимера
соответствовала 14,7 разветвлениям на 1000 атомов углерода (число разветвлений определялось с помощью ИК-спектроскопии).
Пример 54
Полимеризация с использованием соединения
АТ
Для проведения реакции полимеризации согласно данному примеру использовали реакционный аппарат в виде стального автоклава емкостью 1 л, оборудованного мешалкой и рассчитанного на
проведение непрерывных реакций полимеризации на катализаторах Циглера (Циглера-Натта) при давлении до 2500 бар и температуре до 300oC. Реакционный аппарат был снабжен, кроме того,
термопарой и датчиком давления для непрерывного измерения температуры и давления, соответственно, а также устройствами для непрерывной подачи в аппарат сжатого очищенного этилена и 1-бутена (или
пропилена). В комплект системы для проведения реакции полимеризации входили также: оборудование (устройства) для непрерывной подачи в аппарат дозированных количеств раствора катализаторов и
оборудование для быстрой продувки (разгрузки) аппарата и быстрого прекращения реакции полимеризации резким охлаждением. В комплект оборудования для проведения полимеризации были включены также
устройства для сбора синтезируемого полимерного продукта. Реакцию полимеризации осуществляли при молярном соотношении этилен:1-бутен равном 1,6:1, без добавления растворителя. Температура очищенного
реакционного аппарата, в который были загружены этилен и 1-бутен, поддерживалась на уровне, соответствующем желаемой температуре реакции, а именно была равна 180oC. Раствор катализатора
готовили путем смешивания 0,888 г соединения АТ в твердой фазе с 0,67 л 30%-ного (по массе) раствора метилалюмоксана в 4,3 л толуола в инертной атмосфере. Этот каталитический раствор (раствор
катализатора) непрерывно подавался в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 0,56 л/ч, благодаря чему в реакционном аппарате создавалась температура в
180oC. В процессе полимеризации исходные мономеры, а именно этилен и 1-бутен непрерывно подавались в автоклав под общим давлением 1300 бар. Содержимое реакционного аппарата (автоклава)
перемешивалось со скоростью вращения мешалки 1000 об/мин. Выход целевого полимерного продукта, т.е. сополимера этилена и 1-бутена составил 3,9 кг/ч. Полученный сополимер имел среднемассовую
молекулярную массу 50200 и молекулярно-массовое распределение с преобладанием фракции 2,36. Короткоцепная разветвленность макромолекулы сополимера соответствовала 60,1 разветвлениям на 1000 атомов
углерода. Число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C.
Пример 55
Полимеризация с использованием соединения АТ
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а этилен и пропилен вводились в молярном соотношении 2,6: 1, соответственно. При этом, не добавляя
растворитель, температуру очищенного реакционного аппарата, в которой были загружены этилен и пропилен, устанавливали на уровне, соответствующем желаемой температуре реакции 140oC.
Каталитический раствор (раствор катализатора) приготавливали смешиванием 0,779 г соединения АТ в твердой фазе с 0,5 л 30%-ного (по массе) раствора метилалюмоксана в 24,5 л толуола в инертной
атмосфере. Этот каталитический раствор непрерывно подавали в реакционный аппарат (автоклав) насосом высокого давления с объемной скоростью потока раствора 0,9 л/ч, в результате чего в реакционном
аппарате создавалась температура 140oC. Во время проведения полимеризации исходные мономеры, а именно этилен и пропилен, непрерывно подавались в автоклав (реакционный аппарат) под общим
давлением 2200 бар. Содержимое реакционного аппарата перемешивалось со скоростью вращения мешалки 1000 об/мин. Выход целевого полимерного продукта, а именно сополимера этилена и пропилена, составил 2,
3 кг/ч. Полученный сополимерв имел среднемассовую молекулярную массу 102700, молекулярно-массовое распределение с преобладанием фракции 2,208 и плотность 0,863 г/см3.
Пример 56
Полимеризация с использованием соединения FT
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а этилен и 1-бутен вводились в
молярном соотношении 1,6: 1, соответственно. При этом, не добавляя растворитель, температуру очищенного реакционного аппарата, загруженного этиленом и 1-бутеном, устанавливали на уровне,
соответствующем желаемой температуре реакции 180oC. Каталитический раствор готовили смешиванием 0,859 г соединения FT в твердой фазе с 30%-ным (по массе) раствором метилалюмоксана и
толуолом в такой пропорции, чтобы концентрация катализатора составляла 0,162 г/л, а молярное отношение между алюминием и переходным металлом было бы равно 1200:1. Этот каталитический раствор готовили
в инертной атмосфере и подавали непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 1,15 л/ч, в результате чего в аппарате устанавливалась температура
180oC. Во время полимеризации исходные материалы, а именно этилен и 1-бутен, непрерывно подавались в автоклав под общим давлением 1300 бар. Реакционная смесь в автоклаве перемешивалась при
скорости вращения мешалки 1000 об/мин. Выход целевого полимерного продукта, т.е. сополимера этилена и 1-бутена, составил 3,9 кг/ч. Полученный сополимер имел среднемассовую молекулярную массу 61400 и
молекулярно-массовое распределением с преобладанием фракции 2,607. Короткоцепная разветвленность макромолекулы сополимера соответствовала 104,8 разветвлениям на 1000 атомов углерода (число
разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 57
Полимеризация с использованием соединения GT
Используя
реакционный аппарат той же самой конструкции и ту же самую общую методику ведения полимеризации, что и в примере 40, в аппарат загружали: 300 мл толуола, 100 мл 1-гексана, 7 мл 1-молярного раствора
метилалюмоксана и 1,23 мг соединения GT (1 мл раствора, 12,3 мг соединения в 10 мл толуола). Реакционный аппарат нагревали до температуры 80oC, а этилен загружался в него под давлением 65
фунтов/дюйм2. Реакцию полимеризации проводили в течение 30 минут, после чего аппарат быстро охлаждали и продували. После упаривания толуола было выделено 47,2 г сополимера этилена и
1-гексана. Сополимера имел молекулярную массу 313000 и молекулярно-массовое распределение с преобладанием фракции 3,497. Короткоцепная разветвленность макромолекулы сополимера 41 разветвление на 1000
атомов углерода. Число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C.
Пример 58
Полимеризация с использованием соединения
АТ
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а молярное отношение этилен 1-бутен брали равным 1,6 1. Растворитель не добавляли.
Температура очищенного реакционного аппарата, в который были загружены этилен и 1-бутен, устанавливалась на уровне, соответствующем желаемой температуре реакции 170oC. Каталитический
раствор готовили путем смешивания 0,925 г соединения АТ в твердой фазе с 2 л 10%-ного (по массе) раствора метилалюмоксана в 8 л толуола в инертной атмосфере. Этот каталитический раствор непрерывно
подавали в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 0,28 л/ч, в результате чего в аппарате устанавливалась температура 170oC. Во время проведения
полимеризации этилен и 1-бутен непрерывно подавались в аппарат при общем давлении 1300 бар. Реакционная смесь в реакционном аппарате перемешивалась при скорости вращения мешалки 1000 об/мин. Выход
целевого полимерного продукта, а именно сополимера этилена и 1-бутена, составил 3,7 кг/ч. Сополимер имел среднемассовую молекулярную массу 69500 и молекулярно-массовое распределение с преобладанием
фракции 2,049. Короткоцепная разветвленность 35,7 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 59
Полимеризация с использованием соединения ВТ
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а молярное отношение
этилен 1-бутен составляло 1,6 1. Растворитель не добавляли. Температура очищенного реакционного аппарата, загруженного этиленом и 1-бутеном, устанавливалась на уровне, соответствующем желаемой
температуре реакции 180oC. Каталитический раствор готовили путем смешивания 0,995 г соединения ВТ в твердой фазе с 30%-ным (по массе) раствором метилалюмоксана и толуолом в таком
соотношении, чтобы концентрация катализатора составила 0,187 г/л, а молярное отношение алюминий:переходный металл было бы равно 1300:1. Этот каталитический раствор готовили в инертной атмосфере и
подавали непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 1 л/ч, в результате чего в аппарате устанавливалась температура 180oC. Во время
полимеризации этилен и 1-бутен подавались в реакционный аппарат при общем давлении 1300 бар. Реакционная смесь в реакционном аппарате перемешивалась при скорости вращения мешалки 1000 об/мин. Выход
целевого полимерного продукта, а именно сополимера этилена и 1-бутена, составил 3,9 кг/ч. Сополимер имел среднемассовую молекулярную массу 65000, молекулярно-массовое распределение с преобладанием
фракции 2,623. Короткоцепная разветвленность 55,5 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 60
Полимеризация с использованием соединения Н
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а этилен и 1-бутен
брали в молярном соотношении 1,6: 1. При этом, не добавляя растворитель, температуру в очищенном реакционном аппарате, загруженном этиленом и 1-бутеном, устанавливали на уровне, соответствующем
желаемой температуре реакции 180oC. Каталитический раствор готовили путем смешивания 1,94 г соединения Н в твердой фазе с 2 л 10%-ного (по массе) раствора метилалюмоксана в 3 л толуола в
инертной атмосфере. Этот каталитический раствор подавали непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 1,5 л/ч, в результате чего в реакционном
аппарате создавалась температура 180oC. Во время этого процесса полимеризации этилен и 1-бутен подавались в аппарат при общем давлении (в аппарате) 1300 бар. Реакционная смесь в аппарате
перемешивалась при скорости вращения 1000 об/мин. Выход целевого полимерного продукта, а именно сополимера этилена и 1-бутена, составил 3,9 кг/ч. Сополимер имел среднемассовую молекулярную массу
31900. Короткоцепная разветвленность 46,5 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 61
Полимеризация с использованием соединения I
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а этилен и 1-бутен брали в
молярном соотношении 1,6: 1, соответственно. При этом, не добавляя растворитель, температуру в очищенном реакционном аппарате, загруженном этиленом и 1-бутеном, устанавливали на уровне,
соответствующем желаемой температуре реакции 180oC. Каталитический раствор готовили, смешивая 1,92 г соединения I в твердой фазе с 2 л 10% -ного (по массе) раствора метилалюмоксана в 3 л
толуола в инертной атмосфере. Приготовленный каталитический раствор подавали непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 0,67 л/ч, в результате чего
в реакторе установилась температура 180oC. Во время полимеризации этилен и 1-бутен подавались в аппарат при общем давлении (в аппарате) 1300 бар. Реакционная смесь в аппарате перемешивалась
при скорости вращения 1000 об/мин. Выход сополимера этилена и 1-бутена составил 3,9 кг/ч. Сополимер имел среднемассовую молекулярную массу 40800 и молекулярно-массовое распределение с преобладающей
фракцией 2,009. Короткоцепная разветвленность макромолекулы сополимера 36,9 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 62
Полимеризация с использованием соединения К
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54,
а этилен и 1-бутен брали в молярном соотношении 1,6: 1, соответственно. Не добавляя растворитель, температуру в очищенном реакционном аппарате, загруженном этиленом и 1-бутеном, устанавливали на
уровне, соответствующем желаемой температуре реакции 180oC. Каталитический раствор готовили, смешивая 1,8 г соединения К в твердой фазе с 2 л 10%-ного (по массе) раствора метилалюмоксана в
3 л толуола, в инертной среде. Готовый каталитический раствор подавали непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 1,7 л/ч, в результате чего в
реакционном аппарате установилась температура 180oC. Во время полимеризации этилен и 1-бутен подавались в аппарат при общем давлении 1300 бар. Реакционная смесь в аппарате перемешивалась
при скорости вращения мешалки 1000 об/мин. Выход сополимера этилена и 1-бутена составил 3,9 кг/ч. Сополимер имел среднемассовую молекулярную массу 51700 и молекулярно-массовое распределение с
преобладанием фракции 1,532. Короткоцепная разветвленность 30,1 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13
C).
Пример 63
Полимеризация с использованием соединения L
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а этилен и
1-бутен брали в молярном соотношении 1,6: 1, соответственно. Не добавляя растворителя, температуру в очищенном реакционном аппарате, загруженном этиленом и
1-бутеном, устанавливали на уровне,
соответствующем желаемой температуре реакции 180oC. Каталитический раствор готовили, смешивая 1,95 г соединения L в твердой фазе с 2 л 10%-ного (по массе) раствора метилалюмоксана в 3 л
толуола, в инертной среде. Готовый каталитический раствор подавали непрерывно в реакционный аппарат насосом высокого давления в объемной скоростью потока раствора 1,2 л/ч, в результате чего в аппарате
установилась температура 180oC. Во время полимеризации этилен и 1-бутен подавали в аппарат при общем давлении 1300 бар. Содержимое реакционного аппарата перемешивали при скорости вращения
мешалки 1000 об/мин. Выход сополимера этилена и 1-бутена составил 3,9 кг/ч. Сополимер имел среднемассовую молекулярную массу 38800 и молекулярно-массовое распределение с преобладанием фракции 1,985.
Короткоцепная разветвленность макромолекулы этого сополимера 39,3 разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 64
Полимеризация с использованием соединения НТ
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а
этилен и 1-бутен брали в молярном соотношении 1,6: 1. Не вводя растворитель, температуру в очищенном реакционном аппарате, загруженном этиленом и 1-бутеном, устанавливали на уровне, соответствующем
желаемой температуре реакции 180oC. Каталитический раствор готовили, смешивая 2,01 г соединения НТ в твердой фазе с 30%-ным (по массе) раствором метилалюмоксана и толуолом с таким расчетом,
чтобы концентрация катализатора составила 0,354 г/л и молярное отношение алюминий:переходный металл было бы равно 400:1. Каталитический раствор готовили в инертной среде и подавали непрерывно в
реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 1,15 г/л, в результате чего в аппарате установилась температура 180oC. Во время полимеризации этилен и
1-бутен находились в реакционном аппарате под общим давлением 1300 бар. Реакционная смесь в аппарате перемешивалась при скорости вращения мешалки 1000 об/мин. Выход сополимера этилена и 1-бутена
составил 3,9 кг/ч. Полученный сополимер имел среднемассовую молекулярную массу 61700 и молекулярно-массовое распределение с преобладанием фракции 2,896. Макромолекула сополимера имела 62,9
разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 65
Полимеризация с
использованием соединения F
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а этилен и 1-бутен брали в молярном соотношении 1,6: 1,
соответственно. Не вводя растворитель, температуру в очищенном реакционном аппарате, загруженном этиленом и 1-бутеном, устанавливали на уровне, соответствующем желаемой температуре реакции 180oC. Каталитический раствор готовили, смешивая 1,31 г соединения F в твердой фазе с 2 л 10%-ного (по массе) раствора метилалюмоксана в 3 л толуола, в инертной среде. Готовый каталитический
раствор вводили непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 0,56 л/ч, в результате чего в аппарате создавалась температура 180oC. Во
время этого процесса полимеризации этилен и 1-бутен находились в реакционном аппарате под общим давлением 1300 бар. Содержимое реакционного аппарата перемешивали при скорости вращения мешалки 1000
об/мин. Выход целевого полимерного продукта, а именно сополимера этилена и 1-бутена, составил 3,9 кг/ч. Синтезированный таким образом сополимер имел среднемассовую молекулярную массу 43400 и
молекулярно-массовое распределение с преобладанием фракции 2,001. Макромолекула этого сополимера имела короткоцепную разветвленность, соответствующую 40,1 разветвлениям на 1000 атомов углерода (число
разветвлений от основной цепи определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 66
Полимеризация с использованием соединения G
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а этилен и 1-бутен брали в том же самом молярном соотношении 1,6: 1, соответственно. Не вводя
растворитель, температуру в очищенном реакционном аппарате, загруженном этиленом и 1-бутеном, устанавливали на уровне, соответствующем желаемой температуре реакции 180oC. Каталитический
раствор готовили, смешивая 1,53 г соединения G в твердой фазе с 0,5 л 30% -ного (по массе) раствора метилалюмоксана в 4,5 л толуола, в инертной среде. Готовый каталитический раствор подавали
непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 0,58 л/ч, в результате чего в аппарате устанавливалась температура 180oC. Во время
полимеризации этилен и 1-бутен находились в реакционном аппарате под общим давлением 1300 бар. Реакционная смесь в аппарате перемешивалась при скорости вращения мешалки 1000 об/мин. Выход целевого
полимерного продукта, а именно сополимера этилена и 1-бутена, составил 3,9 кг/ч. Полученный сополимер имел среднемассовую молекулярную массу 74700 и молекулярно-массовое распределение с преобладанием
фракции 2,198. Макромолекула этого сополимера имела короткоцепную разветвленность, соответствующую 37,6 разветвлениям на 1000 атомов углерода (число разветвлений определялось с помощью
ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 67
Полимеризация с использованием соединения IT
В данном случае использовали реакционный аппарат
той же самой конструкции, что и в примере 54, а этилен и 1-бутен брали в том же самом молярном соотношении 1,6:1, что и в примере 54. Не добавляя к смеси мономеров растворитель, температуру в
очищенном реакционном аппарате, в который были загружены этилен и 1-бутен, устанавливали на уровне, соответствующем желаемой температуре реакции 180oC. Каталитический раствор готовили путем
смешивания 1,94 г соединения IT c 30%-ным (по массе) раствором метилалюмоксана и толуолом в таком соотношении, чтобы концентрация катализатора составила 0,388 г/л и молярное отношение
алюминий:переходный металл было бы равно 600:1. Каталитический раствор готовили в инертной среде и подавали непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока
раствора 0,42 л/ч, в результате чего в аппарате устанавливалась температура 180oC. Во время проведения полимеризации этилен и 1-бутен в аппарате находились под общим давлением 1300 бар.
Содержимое реакционного аппарата перемешивалось при скорости вращения мешалки 1000 об/мин. Выход целевого полимерного продукта, а именно этилена и 1-бутена, составил 3,9 кг/ч. Синтезированный таким
образом сополимер имел среднемассовую молекулярную массу 50800 и молекулярно-массовое распределение с преобладанием фракции 2,467. Макромолекула этого сополимера имела 69 короткоцепных разветвлений на
1000 атомов углерода (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов водорода1H).
Пример 68
Полимеризация с использованием соединения
А
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а исходные мономеры, а именно этилен и 1-бутен, брали в том же самом молярном соотношении 1,
6:1, что и в примере 54. Не добавляя растворитель, температуру в очищенном реакционном аппарате, загруженном этиленом и 1-бутеном, устанавливали на уровне, соответствующем желаемой температуре реакции
180oC. Каталитический раствор (т.е. раствор катализатора) готовили путем смешивания 1,95 г соединения А в твердой фазе с 0,67 л 30%-ного (по массе) раствора метилалюмоксана в 4,3 л толуола,
в инертной среде. Готовый каталитический раствор подавали непрерывно в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 0,4 л/ч, в результате чего в реакционном
аппарате устанавливалась температура 180oC. Во время проведения полимеризации этилен и 1-бутен находились в реакционном аппарате под общим давлением 1300 бар. Реакционная смесь в аппарате
перемешивалась при скорости вращения мешалки 1000 об/мин. Выход целевого полимерного продукта, а именно сополимера этилена и 1-бутена, составил 3,9 кг/ч. Сополимер имел среднемассовую молекулярную
массу 71100 и молекулярно-массовое распределение с преобладанием фракции 1,801. Макромолекула сополимера имела 12,4 короткоцепных разветвлений на 1000 атомов углерода (число разветвлений определялось
с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 69
Полимеризация с использованием соединения В
В данном случае использовали реакционный
аппарат той же самой конструкции, что и в примере 54, а исходные мономеры этилен и 1-бутен брали в том же самом молярном соотношении 1,6:1, что и в примере 54. Не добавляя растворитель, температуру в
очищенном реакционном аппарате, загруженном этиленом и 1-бутеном, устанавливали на уровне, соответствующем желаемой температуре реакции 180oC. Каталитический раствор готовили путем
смешивания 1,97 г соединения В в твердой фазе с 0,67 л 30%-ного (по массе) раствора метилалюмоксана в 4,3 л толуола, в инертной среде. Готовый каталитический раствор закачивали непрерывно в
реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 0,35 л/ч, в результате чего в аппарате устанавливалась температура 180oC. При проведении полимеризации
этилен и 1-бутен находились в реакционном аппарате под общим давлением 1300 бар. Содержимое аппарата перемешивалось при скорости вращения мешалки 1000 об/мин. Выход целевого полимерного продукта в
виде сополимера этилена и 1-бутена составлял 3,9 кг/ч. Сополимер имел среднемассовую молекулярную массу 47300 и молекулярно-массовое распределение с преобладанием фракции 2,056. Макромолекула
сополимера имела 34,1 короткоцепных разветвлений (число разветвлений определялось с помощью ЯМР-спектроскопии на ядрах атомов углерода13C).
Пример 70
Полимеризация с использованием соединения JT
В данном случае использовали реакционный аппарат той же самой конструкции, что и в примере 54, а исходные мономеры этилен и 1-бутен брали в том же
самом молярном соотношении 1,6:1, что и в примере 54. Не добавляя растворитель, температуру в очищенном реакционном аппарате, в который были загружены этилен и 1-бутен, устанавливали на уровне,
соответствующем желаемой температуре реакции 180oC. Каталитический раствор готовили, смешивая 1,78 г соединения JT в твердой фазе с 30%-ным (по массе) раствором метилалюмоксана и толуолом с
таким расчетом, чтобы концентрация катализатора составляла 0,318 г/л и молярное отношение алюминий:переходный металл было бы равно 1400:1. Каталитический раствор готовили в инертной атмосфере.
Приготовленный таким образом каталитический раствор непрерывно закачивали в реакционный аппарат насосом высокого давления с объемной скоростью потока раствора 0,55 л/ч, в результате чего в аппарате
устанавливалась температура 180oC. При проведении полимеризации этилен и 1-бутен находились в реакционном аппарате под общим давлением 1300 бар. Содержимое аппарата перемешивалось при
скорости вращения мешалки 1000 об/мин. Выход целевого полимерного продукта в виде сополимера этилена и 1-бутена составил 3,9 кг/ч. Cополимер имел среднемассовую молекулярную массу 72600 и
молекулярно-массовое распределение с преобладанием фракции 2,385. Макромолекула полимера имела 110 короткоцепных разветвлений на 1000 атомов углерода (число разветвлений определялось с помощью
ЯМР-спектроскопии на ядрах атомов водорода1H).
В приводимой ниже таблице IX указаны используемые режимы полимеризации, а также основные свойства полимерных продуктов, полученных согласно примерам 1-39, рассмотренным выше.
В приведенных ниже таблицах А, В и С, D указаны используемые режимы полимеризации и основные свойства полимерных продуктов, полученных согласно примерам 40-50, 52, 54-59, 64, 67 и 70. Во всех этих примерах полимеризация проводилась на каталитической системе, основанной на таком соединении переходного металла группы IVB Периодической системы элементов с циклопентадиениловой группой, в котором в качестве переходного металла использовался титан.
В таблице указаны используемые режимы полимеризации и основные свойства полимерных продуктов, полученных с помощью каталитических систем, основанных на использовании соединения каждого из переходных металлов группы IVB Периодической системы, а именно титана, гафния или циркония с моноциклопентадиениловой группой. Эти соединения по своей структуре аналогичны друг другу, за исключением того, что в них переходным металлом является либо титан, либо гафний, либо цирконий.
Таким образом, можно заметить, что требования, предъявляемые к алюмоксановому компоненту каталитической системы, могут быть значительно снижены, если перед инициированием полимеризации предварительно смешать катализатор с алюмоксаном (см. примеры 12-15).
Благодаря правильному выбору: 1) переходного металла группы IVB Периодической системы элементов, входящего в состав каталитической системы; 2) типа используемого алюмоксана и его количества в каталитической системе; 3) типа растворителя (разбавителя), используемого для полимеризации, а также его объема; 4) температуры реакции; и 5) давления, при котором протекает полимеризация, можно обеспечить получение полимерного продукта с весьма значительной среднемассовой молекулярной массой и с желаемым узким молекулярно-массовым распределением, определяемым величиной отношения менее 4:1. Для практического осуществления предлагаемого согласно настоящему изобретению способа (процесса) полимеризации рекомендуется, предпочтительно, использовать такие ароматические растворители или разбавители как толуол, или же алканы, например, такой как гексан.
Из приведенных выше примеров становится очевидным, что применительно к каталитической системе, в которой используется соединение такого переходного металла группы IVB Периодической системы элементов,
как титан, и в которой это соединение титана имеет указанную ниже структуру:
природа R'-группы может существенно повлиять на каталитические свойства системы. Так, например, для получения сополимеров этилена с α-олефином, которые (сополимеры) имеют максимальное содержание сомономера, рекомендуется при выбранном соотношении между мономерами, т.е. между этиленом и a-олефином, использовать в качестве R'-группы предпочтительно неароматический заместитель, например, такой, как алкильный или циклоалкильный заместитель, который в качестве первичного или вторичного атома углерода предпочтительно содержит атом углерода, связанный с атомом азота.
Кроме того, из изложенного выше можно заметить, что природа структуры циклопентадиенилового лиганда в соединении титана также влияет на свойства каталитической системы. Рекомендуется преимущественно использовать те циклопентадиениловые лиганды, которые не слишком сильно блокированы стерически и которые содержат хорошие (активные) электронодонорные группы, например, Me4C5-лиганд.
Смолы, получаемые предложенными согласно настоящему изобретению способом, могут быть использованы для производства широкого ассортимента изделий, например, полимерной пленки и искусственного (полимерного) волокна.
Настоящее изобретение было рассмотрено здесь со ссылкой на предпочтительные варианты его осуществления. Вполне очевидно, что специалисты, работающие в данной области техники, могут вносить всевозможные изменения в варианты практического осуществления, не отходя от основного принципа изобретения, изложенного в приводимой ниже патентной формуле. ТТТ1 ТТТ2 ТТТ3 ТТТ4 ТТТ5 ТТТ6 ТТТ7 ТТТ8 ТТТ9 ТТТ10 ТТТ11 ТТТ12 ТТТ13 ТТТ14 ТТТ15 ТТТ16 ТТТ17 ТТТ18 ТТТ19 ТТТ20 ТТТ21 ТТТ22 ТТТ23 ТТТ24 ТТТ25 ТТТ26 ТТТ27 ТТТ28 ТТТ29 ТТТ30 ЫЫЫ2
Реферат
Согласно настоящему изобретению предлагается каталитическая система, включающая в себя соединение переходного металла группы IVB Периодической системы элементов, и алюмоксан. Предложенная каталитическая система может быть использована для полимеризации олефинов с целью получения высокомолекулярного полимера. 3 с. и 1 з.п.ф-лы, 13 табл., 4 ил.

Формула
где R' низший алкил;
R'' низший алкил или фенил;
R''' низший алкил, незамещенный или замещенный низшим алкилом фенил, или C6 C12-циклоалкил, или низший алкоксил;
М цирконий, гафний, титан в их высшей степени окисления;
С5Н(4-x)Rx циклопентадиениловое кольцо, которое может быть замещено 1 4 радикалами R, где R низший алкил или три(низший алкил)силил, либо в котором две или более группы R вместе с циклопентадиениловым кольцом образуют индениловую или флуорениловую систему;
x 0 4;
n 0 3;
Q галоид или низший алкил.
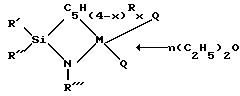
где R, R', R'', R''', М, Q, n, х имеют указанные значения,
отличающийся тем, что галогенид соответствующего переходного металла или его диэфират подвергают взаимодействию с солью щелочного металла общей формулы
[C5H(4-x)RxSiR'R''(NR''')]Me2
где Ме щелочной металл;
R', R'', R''' и x имеют указанное значение,
при необходимости, в присутствии (С2Н5)2O.
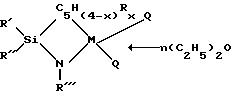
где R' низший алкил;
R'' низший алкил или фенил;
R''' низший алкил, незамещенный или замещенный низшим алкилом фенил, или C6 C12-циклоалкил, или низший алкокси;
М цирконий, гафний, титан в их высшей степени окисления;
С5Н(4-x)Rx циклопентадиениловое кольцо, которое может быть замещено 1 4 радикалами R, где R низший алкил или три(низший алкил)силил, либо в котором две или более группы R вместе с циклопентадиениловым кольцом образуют индениловую или флуорениловую систему;
x 0 4;
n 0 3;
Q галоид или низший алкил,
в качестве алюмоксана система содержит метилалюмоксан при молярном отношении метилалюмоксана к соединению переходного металла 465 16100.
Комментарии