Карбамидсодержащие силаны, способ их получения и их применение - RU2678320C2
Код документа: RU2678320C2
Описание
Настоящее изобретение относится к карбамидсодержащим силанам, к способу их получения, а также к их применению.
Из CAS 1184961-62-3, 442527-46-0 и 498553-03-0 известны соединения формул
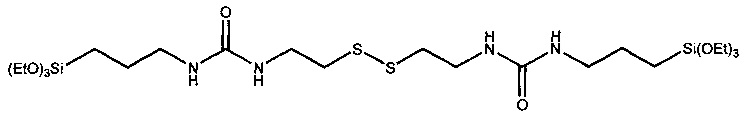

Помимо этого из US 2003/0191270 А1 известны силаны формулы
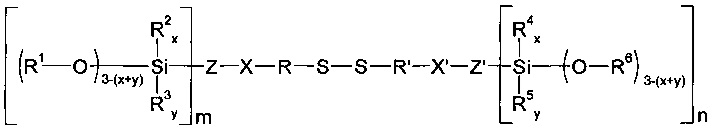
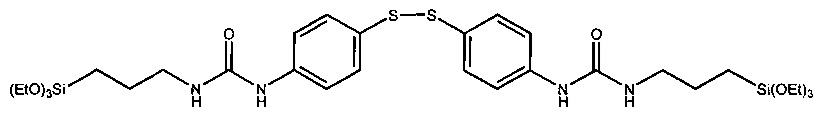
Из JP 2002-201312 А известны модификаторы каучука формулы

Из J. Mat. Chem. 2009, 19, сс. 4746-4752, известны далее золотые наночастицы, присутствующие в функционализированных SH-группами скелетных структурах из мезопористых кремниевых кислот, и получение карбамидсодержащих силанов. При получении силанов этим известным способом используют органические растворители.
Недостатки известных карбамидсодержащих дисульфидсиланов состоят в плохой перерабатываемости, неудовлетворительном сцеплении с мокрой дорогой и низкой динамической жесткости.
В основу настоящего изобретения была положена задача предложить карбамидсодержащие силаны, которые по сравнению с известными из уровня техники карбамидсодержащими силанами обладали бы улучшенной перерабатываемостью, лучшим сцеплением с мокрой дорогой и большей динамической жесткостью в резиновых смесях.
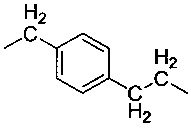
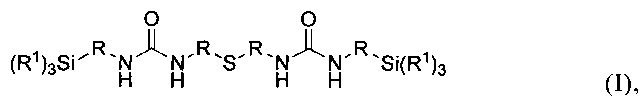
Объектом изобретения является карбамидсодержащий силан формулы I
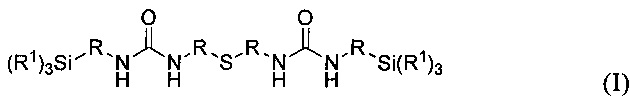
где R1 имеют одинаковые или разные значения и представляют собой C1-С10алкоксигруппы, предпочтительно метокси- или этоксигруппу, циклические С2-С10диалкоксигруппы, фенокигруппу, С4-С10циклоалкоксигруппы, С6-С20арильные группы, предпочтительно фенил, C1-С10алкильные группы, предпочтительно метил или этил, С2-С20алкенильную группу, С7-С20аралкильную группу или галоген, предпочтительно Cl, a R имеют одинаковые или разные значения и представляют собой разветвленную либо неразветвленную, насыщенную либо ненасыщенную, алифатическую, ароматическую либо смешанно алифатическую/ароматическую двухвалентную углеводородную группу с С1-С30, предпочтительно с С1-С20, более предпочтительно с C1-С10, особенно предпочтительно с С1-С7, наиболее предпочтительно с С2 и С3, которая необязательно замещена F-, Сl-, Br-, I-, -CN или HS-.
Карбамидсодержащие силаны могут представлять собой смеси карбамидсодержащих силанов формулы I.
Продукт осуществления способа может содержать олигомеры, которые образуются в результате гидролиза и конденсации алкоксисилановых функциональных групп карбамидсодержащих силанов формулы I.
Карбамидсодержащие силаны формулы I могут быть нанесены на носитель, например воск, полимер или сажу (технический углерод). Карбамидсодержащие силаны формулы I могут быть нанесены на кремниевую кислоту, с которой они при этом могут быть связаны физически или химически.
В предпочтительном варианте R может обозначать -СН2-, -СН2СН2-, -СН2СН2СН2-, -СН2СН2СН2СН2-, -СН(СН3)-, -СН2СН(СН3)-, -СН(СН3)СН2-, -С(СН3)2-, -СН(С2Н5)-, -СН2СН2СН(СН3)-, -СН(СН3)СН2СН2-, -СН2СН(СН3)СН2-, -СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2- или

В предпочтительном варианте R1 может представлять собой метокси- или этоксигруппу.
Карбамидсодержащие силаны формулы I в предпочтительном варианте могут представлять собой ((EtO)3Si-CH2-NH-CO-NH-CH2)2S, ((EtO)3Si-CH2CH2-NH-CO-NH-CH2)2S, ((EtO)3Si-CH2-NH-CO-NH-CH2CH2)2S, ((EtO)3Si-CH2CH2-NH-CO-NH-CH2CH2)2S, ((EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2)2S, ((EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2)2S, ((EtO)3Si-CH2-NH-CO-NH-CH2CH2CH2)2S, ((EtO)3Si-CH2CH2-NH-CO-NH-CH2CH2CH2)2S, ((EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2CH2)2S, ((MeO)3Si-CH2-NH-CO-NH-CH2)2S, ((MeO)3Si-CH2CH2-NH-CO-NH-CH2)2S, ((MeO)3Si-CH2-NH-CO-NH-CH2CH2)2S, ((MeO)3Si-CH2CH2-NH-CO-NH-CH2CH2)2S, ((MeO)3Si-CH2CH2CH2-NH-CO-NH-CH2)2S, ((MeO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2)2S, ((MeO)3Si-CH2-NH-CO-NH-CH2CH2CH2)2S, ((MeO)3Si-CH2CH2-NH-CO-NH-CH2CH2CH2)2S или ((MeO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2CH2)2S.
Особенно предпочтительно соединение формулы (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-S-CH2CH2-NH-CO-NH-CH2CH2CH2-Si(OEt)3.
Еще одним объектом изобретения является первый способ получения предлагаемых в изобретении карбамидсодержащих силанов формулы I
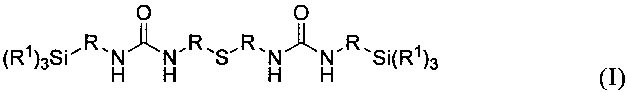
где R1 и R имеют указанные выше значения, отличающийся тем, что на первой стадии аминосилан формулы II
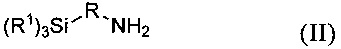
подвергают взаимодействию с изоцианатом формулы III

где R и R1 имеют указанные выше значения, a Hal обозначает F, Cl, Br или I, предпочтительно О, и на второй стадии продукт, полученный на первой стадии, подвергают взаимодействию с сульфидом натрия формулы (IV)

Соединения формулы II в предпочтительном варианте могут представлять собой (C2H5O)3Si-CH2-NH2, (C2H5O)3Si-CH2CH2-NH2, (C2H5O)3Si-CH2CH2CH2-NH2, (CH3O)3Si-CH2-NH2, (CH3O)3Si-CH2CH2-NH2 или (CH3O)3Si-CH2CH2CH2-NH2.
Соединения формулы III в предпочтительном варианте могут представлять собой OCN-CH2-Cl, OCN-CH2CH2-Cl или OCN-CH2CH2CH2-Cl.
При осуществлении первого предлагаемого в изобретении способа его первую и вторую стадии можно проводить в реакционном сосуде путем добавления всех исходных веществ (эдуктов).
На первой стадии первого предлагаемого в изобретении способа аминосилан формулы II можно дозировать к изоцианату формулы III.
На первой стадии первого предлагаемого в изобретении способа в предпочтительном варианте изоцианат формулы III можно дозировать к аминосилану формулы II.
На первой стадии первого предлагаемого в изобретении способа аминосиланы формулы II и изоцианаты формулы III можно использовать в молярном соотношении между ними от 0,85:1 до 1,15:1, предпочтительно от 0,90:1 до 1,10:1, особенно предпочтительно от 0,95:1 до 1,05:1.
Реакцию на первой стадии первого предлагаемого в изобретении способа можно проводить в условиях, исключающих доступ воздуха.
Реакцию на первой стадии первого предлагаемого в изобретении способа можно проводить в атмосфере защитного газа, например аргона или азота, предпочтительно в атмосфере азота.
Первую стадию первого предлагаемого в изобретении способа можно проводить при нормальном давлении, повышенном давлении или пониженном давлении. В предпочтительном варианте предлагаемый в изобретении способ можно осуществлять при нормальном давлении.
При проведении первой стадии при повышенном давлении оно может составлять от 1,1 до 100 бар, предпочтительно от 1,5 до 50 бар, особенно предпочтительно от 2 до 20 бар, наиболее предпочтительно от 2 до 10 бар.
При проведении первой стадии при пониженном давлении оно может составлять от 1 до 1000 мбар, предпочтительно от 1 до 500 мбар, особенно предпочтительно от 1 до 250 мбар, наиболее предпочтительно от 5 до 100 мбар.
Первую стадию первого предлагаемого в изобретении способа можно проводить при температуре в пределах от -78 до 100°С, предпочтительно от -70 до 50°С, особенно предпочтительно от -65 до 25°С.
Взаимодействие на первой стадии первого предлагаемого в изобретении способа можно проводить в отсутствие растворителя или в растворителе, например метаноле, этаноле, пропаноле, бутаноле, циклогексаноле, N,N-диметилформамиде, диметилсульфоксиде, пентане, гексане, циклогексане, гептане, октане, декане, толуоле, ксилоле, ацетоне, ацетонитриле, тетрахлорметане, хлороформе, дихлорметане, 1,2-дихлорметане, тетрахлорэтилене, диэтиловом эфире, метил-трет-бутиловом эфире, метилэтилкетоне, тетрагидрофуране, диоксане, пиридине или этилацетате. В предпочтительном варианте растворителем может служить дихлорметан, этанол, метил-трет-бутиловый эфир, толуол, этилацетат, пентан или гексан.
Взаимодействие на первой стадии первого предлагаемого в изобретении способа можно проводить без применения органического растворителя. В этом случае растворителем может служить вода.
Растворитель можно удалять по завершении первой стадии первого предлагаемого в изобретении способа, предпочтительно отгонять.
Продукт реакции, полученный на первой стадии первого предлагаемого в изобретении способа, можно затем отфильтровывать и промывать органическим растворителем. В предпочтительном варианте продукт реакции можно промывать алканом, особенно предпочтительно гексаном.
После фильтрации продукт реакции, полученный на первой стадии первого предлагаемого в изобретении способа, можно сушить. Сушку можно проводить при температуре в пределах от 20 до 100°С, предпочтительно от 25 до 50°С. Сушку можно проводить под пониженным давлением в пределах от 1 до 500 мбар.
Карбамидсодержащий галогенсилан формулы V
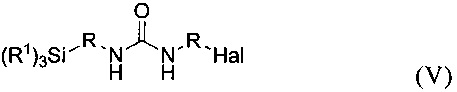
можно получать на первой стадии первого предлагаемого в изобретении способа с выходом более 50%, предпочтительно более 60%, особенно предпочтительно более 70%.
Реакцию на второй стадии первого предлагаемого в изобретении способа можно проводить в условиях, исключающих доступ воздуха.
Реакцию на второй стадии первого предлагаемого в изобретении способа можно проводить в атмосфере защитного газа, например аргона или азота, предпочтительно в атмосфере азота.
Вторую стадию первого предлагаемого в изобретении способа можно проводить при нормальном давлении, повышенном давлении или пониженном давлении. В предпочтительном варианте предлагаемый в изобретении способ можно осуществлять при нормальном давлении.
При проведении второй стадии при повышенном давлении оно может составлять от 1,1 до 100 бар, предпочтительно от 1,5 до 50 бар, особенно предпочтительно от 2 до 20 бар, наиболее предпочтительно от 2 до 10 бар.
При проведении второй стадии при пониженном давлении оно может составлять от 1 до 1000 мбар, предпочтительно от 1 до 500 мбар, особенно предпочтительно от 1 до 250 мбар, наиболее предпочтительно от 5 до 100 мбар.
Вторую стадию первого предлагаемого в изобретении способа можно проводить при температуре в пределах от 20 до 150°С, предпочтительно от 40 до 100°С, особенно предпочтительно от 45 до 80°С.
Взаимодействие на второй стадии первого предлагаемого в изобретении способа можно проводить в отсутствие растворителя или в растворителе, например метаноле, этаноле, пропаноле, бутаноле, циклогексаноле, N,N-диметилформамиде, диметилсульфоксиде, пентане, гексане, циклогексане, гептане, октане, декане, толуоле, ксилоле, ацетоне, ацетонитриле, тетрахлорметане, хлороформе, дихлорметане, 1,2-дихлорметане, тетрахлорэтилене, диэтиловом эфире, метил-трет-бутиловом эфире, метилэтилкетоне, тетрагидрофуране, диоксане, пиридине или этилацетате. В предпочтительном варианте растворителем может служить этанол.
Взаимодействие на второй стадии первого предлагаемого в изобретении способа можно проводить без применения органического растворителя. В этом случае растворителем может служить вода.
Продукт реакции, полученный на второй стадии первого предлагаемого в изобретении способа, можно отфильтровывать и промывать фильтровальный осадок органическим растворителем. В предпочтительном варианте продукт реакции можно промывать спиртом, особенно предпочтительно этанолом, или алканом, особенно предпочтительно гексаном.
Растворитель можно удалять по завершении второй стадии первого предлагаемого в изобретении способа, предпочтительно отгонять.
После фильтрации и удаления растворителя продукт реакции, полученный на второй стадии первого предлагаемого в изобретении способа, можно сушить. Сушку можно проводить при температуре в пределах от 20 до 100°С, предпочтительно от 25 до 50°С. Сушку можно проводить под пониженным давлением в пределах от 1 до 500 мбар.
Карбамидсодержащий силан формулы I

можно получать первым предлагаемым в изобретении способом на его второй стадии с выходом более 50%, предпочтительно более 60%, особенно предпочтительно более 70%.
Остаточное содержание карбамидсодержащего галогенсилана формулы V в продукте, полученном первым предлагаемым в изобретении способом, может составлять менее 25 мол. %, предпочтительно менее 10 мол. %, особенно предпочтительно менее 5 мол. %, наиболее предпочтительно менее 3 мол. %.
Выраженное в мол. % относительное содержание карбамидсодержащих галогенсиланов формулы V в продукте, полученном первым предлагаемым в изобретении способом, определяют с использованием1Н-ЯМР-метода путем интегрирования сигналов от Н-атомов группы -CH2CH2-Cl в карбамидсодержащих галогенсиланах формулы V по отношению к Н-атомам группы Si-CH2- в карбамидсодержащих силанах формулы I.
В целях определения такого относительного содержания для вещества формулы V (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-Cl используют, например, интеграл сигналов от Н-атомов группы -CH2CH2-Cl (δ=3,17 м.д.).
Остаточное содержание аминосилана формулы II в продукте, полученном первым предлагаемым в изобретении способом, может составлять менее 10 мол. %, предпочтительно менее 5 мол. %, особенно предпочтительно менее 1 мол. %, наиболее предпочтительно менее 0,1 мол. %.
Выраженное в мол. % относительное содержание аминосиланов формулы II в продукте, полученном первым предлагаемым в изобретении способом, определяют с использованием13С-ЯМР-метода путем интегрирования сигналов от С-атомов группы -CH2-NH2 в аминосиланах формулы II по отношению к С-атомам группы Si-CH2- в карбамидсодержащих силанах формулы I.
В целях определения такого относительного содержания для вещества формулы II (EtO)3Si-CH2-CH2-CH2-NH2 используют, например, интеграл сигналов от С-атомов группы -CH2-NH2 (δ=45,15 м.д.).
Остаточное содержание изоцианата формулы III в продукте, полученном первым предлагаемым в изобретении способом, может составлять менее 25 мол. %, предпочтительно менее 10 мол. %, особенно предпочтительно менее 5 мол. %, наиболее предпочтительно менее 3 мол. %.
Выраженное в мол. % относительное содержание изоцианатов формулы III в продукте, полученном первым предлагаемым в изобретении способом, определяют с использованием13С-ЯМР-метода путем интегрирования сигналов от С-атомов группы OCN-CH2- в изоцианатах формулы III по отношению к С-атомам группы Si-CH2- в карбамидсодержащих силанах формулы I.
В целях определения такого относительного содержания для вещества формулы III OCN-CH2-CH2-Cl используют, например, интеграл сигналов от С-атомов группы OCN-CH2- (δ=124,33 м.д.).
Еще одним объектом изобретения является второй способ получения предлагаемых в изобретении карбамидсодержащих силанов формулы I

где R1 и R имеют указанные выше значения, отличающийся тем, что на первой стадии изоцианатосилан формулы VI

подвергают взаимодействию с амином формулы VII

где R и R1 имеют указанные выше значения, a Hal обозначает F, Cl, Br или I, предпочтительно Cl, и на второй стадии продукт, полученный на первой стадии, подвергают взаимодействию с сульфидом натрия формулы IV

Соединения формулы VI в предпочтительном варианте могут представлять собой (C2H5O)3Si-CH2-NCO, (C2H5O)3Si-CH2CH2-NCO, (C2H5O)3Si-CH2CH2CH2-NCO, (CH3O)3Si-CH2-NCO, (CH3O)3Si-CH2CH2-NCO или (CH3O)3Si-CH2CH2CH2-NCO.
Соединения формулы VII в предпочтительном варианте могут представлять собой H2N-CH2-Cl, H2N-CH2CH2-Cl или H2N-CH2CH2CH2-Cl.
При осуществлении второго предлагаемого в изобретении способа его первую и вторую стадии можно проводить в реакционном сосуде путем добавления всех исходных веществ (эдуктов).
На первой стадии второго предлагаемого в изобретении способа амины формулы VII можно дозировать к изоцианатосиланам формулы VI.
На первой стадии второго предлагаемого в изобретении способа в предпочтительном варианте изоцианатосиланы формулы VI можно дозировать к аминам формулы VII.
На первой стадии второго предлагаемого в изобретении способа изоцианатосиланы формулы VI и амины формулы VII можно использовать в молярном соотношении между ними от 0,85:1 до 1,15:1, предпочтительно от 0,90:1 до 1,10:1, особенно предпочтительно от 0,95:1 до 1,05:1.
Реакцию на первой стадии второго предлагаемого в изобретении способа можно проводить в условиях, исключающих доступ воздуха.
Реакцию на первой стадии второго предлагаемого в изобретении способа можно проводить в атмосфере защитного газа, например аргона или азота, предпочтительно в атмосфере азота.
Первую стадию второго предлагаемого в изобретении способа можно проводить при нормальном давлении, повышенном давлении или пониженном давлении. В предпочтительном варианте предлагаемый в изобретении способ можно осуществлять при нормальном давлении.
При проведении первой стадии при повышенном давлении оно может составлять от 1,1 до 100 бар, предпочтительно от 1,5 до 50 бар, особенно предпочтительно от 2 до 20 бар, наиболее предпочтительно от 2 до 10 бар.
При проведении первой стадии при пониженном давлении оно может составлять от 1 до 1000 мбар, предпочтительно от 1 до 500 мбар, особенно предпочтительно от 1 до 250 мбар, наиболее предпочтительно от 5 до 100 мбар.
Первую стадию второго предлагаемого в изобретении способа можно проводить при температуре в пределах от -78 до 100°С, предпочтительно от -75 до 60°С, особенно предпочтительно от -70 до 40°С.
Взаимодействие на первой стадии можно проводить в отсутствие растворителя или в растворителе, например метаноле, этаноле, пропаноле, бутаноле, циклогексаноле, N,N-диметилформамиде, диметилсульфоксиде, пентане, гексане, циклогексане, гептане, октане, декане, толуоле, ксилоле, ацетоне, ацетонитриле, тетрахлорметане, хлороформе, дихлорметане, 1,2-дихлорметане, тетрахлорэтилене, диэтиловом эфире, метил-трет-бутиловом эфире, метилэтилкетоне, тетрагидрофуране, диоксане, пиридине или этил ацетате.
Взаимодействие на первой стадии второго предлагаемого в изобретении способа можно проводить без применения органического растворителя. В этом случае растворителем в предпочтительном варианте может служить вода.
Растворитель можно удалять по завершении первой стадии второго предлагаемого в изобретении способа, предпочтительно отгонять.
Продукт реакции, полученный на первой стадии второго предлагаемого в изобретении способа, можно затем отфильтровывать и промывать органическим растворителем. В предпочтительном варианте продукт реакции можно промывать алканом, особенно предпочтительно гексаном.
После фильтрации продукт реакции, полученный на первой стадии второго предлагаемого в изобретении способа, можно сушить. Сушку можно проводить при температуре в пределах от 20 до 100°С, предпочтительно от 25 до 50°С. Сушку можно проводить под пониженным давлением в пределах от 1 до 500 мбар.
Карбамидсодержащий галогенсилан формулы V

можно получать на первой стадии второго предлагаемого в изобретении способа с выходом более 50%, предпочтительно более 60%, особенно предпочтительно более 70%.
Реакцию на второй стадии второго предлагаемого в изобретении способа можно проводить в условиях, исключающих доступ воздуха.
Реакцию на второй стадии второго предлагаемого в изобретении способа можно проводить в атмосфере защитного газа, например аргона или азота, предпочтительно в атмосфере азота.
Вторую стадию второго предлагаемого в изобретении способа можно проводить при нормальном давлении, повышенном давлении или пониженном давлении. В предпочтительном варианте предлагаемый в изобретении способ можно осуществлять при нормальном давлении.
При проведении второй стадии при повышенном давлении оно может составлять от 1,1 до 100 бар, предпочтительно от 1,5 до 50 бар, особенно предпочтительно от 2 до 20 бар, наиболее предпочтительно от 2 до 10 бар.
При проведении второй стадии при пониженном давлении оно может составлять от 1 до 1000 мбар, предпочтительно от 1 до 500 мбар, особенно предпочтительно от 1 до 250 мбар, наиболее предпочтительно от 5 до 100 мбар.
Вторую стадию второго предлагаемого в изобретении способа можно проводить при температуре в пределах от 20 до 150°С, предпочтительно от 40 до 100°С, особенно предпочтительно от 45 до 80°С.
Взаимодействие на второй стадии второго предлагаемого в изобретении способа можно проводить в отсутствие растворителя или в растворителе, например метаноле, этаноле, пропаноле, бутаноле, циклогексаноле, N,N-диметилформамиде, диметилсульфоксиде, пентане, гексане, циклогексане, гептане, октане, декане, толуоле, ксилоле, ацетоне, ацетонитриле, тетрахлорметане, хлороформе, дихлорметане, 1,2-дихлорметане, тетрахлорэтилене, диэтиловом эфире, метил-трет-бутиловом эфире, метилэтилкетоне, тетрагидрофуране, диоксане, пиридине или этилацетате. В предпочтительном варианте растворителем может служить этанол.
Взаимодействие на второй стадии второго предлагаемого в изобретении способа можно проводить без применения органического растворителя. В этом случае растворителем может служить вода.
Продукт реакции, полученный на второй стадии второго предлагаемого в изобретении способа, можно отфильтровывать и промывать фильтровальный осадок органическим растворителем. В предпочтительном варианте продукт реакции можно промывать спиртом, особенно предпочтительно этанолом, или алканом, особенно предпочтительно гексаном.
Растворитель можно удалять по завершении второй стадии второго предлагаемого в изобретении способа, предпочтительно отгонять.
После фильтрации и удаления растворителя продукт реакции, полученный на второй стадии второго предлагаемого в изобретении способа, можно сушить. Сушку можно проводить при температуре в пределах от 20 до 100°С, предпочтительно от 25 до 50°С. Сушку можно проводить под пониженным давлением в пределах от 1 до 500 мбар.
Карбамидсодержащий силан формулы I
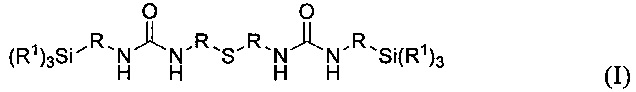
можно получать вторым предлагаемым в изобретении способом на его второй стадии с выходом более 50%, предпочтительно более 60%, особенно предпочтительно более 70%.
Остаточное содержание карбамидсодержащего галогенсилана формулы V в продукте, полученном вторым предлагаемым в изобретении способом, может составлять менее 25 мол. %, предпочтительно менее 10 мол. %, особенно предпочтительно менее 5 мол. %, наиболее предпочтительно менее 3 мол. %.
Выраженное в мол. % относительное содержание карбамидсодержащих галогенсиланов формулы V в продукте, полученном вторым предлагаемым в изобретении способом, определяют с использованием1H-ЯМР-метода путем интегрирования сигналов от Н-атомов группы -СН2СН2-Cl в карбамидсодержащих галогенсиланах формулы V по отношению к Н-атомам группы Si-CH2- в карбамидсодержащих силанах формулы I.
В целях определения такого относительного содержания для вещества формулы V (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-Cl используют, например, интеграл сигналов от Н-атомов группы -CH2CH2-Cl (δ=3,17 м.д.).
Остаточное содержание изоцианатосиланов формулы VI в продукте, полученном вторым предлагаемым в изобретении способом, может составлять менее 10 мол. %, предпочтительно менее 5 мол. %, особенно предпочтительно менее 1 мол. %, наиболее предпочтительно менее 0,1 мол. %.
Выраженное в мол. % относительное содержание изоцианатсиланов формулы VI в продукте, полученном вторым предлагаемым в изобретении способом, в интервале значений более 1 мол.% определяют с использованием13С-ЯМР-метода путем интегрирования сигналов от С-атомов группы -NCO в изоцианатсиланах формулы VI по отношению к С-атомам группы Si-CH2- в карбамидсодержащих силанах формулы I.
В целях определения такого относительного содержания в интервале значений более 1 мол. % для вещества формулы VI (EtO)3Si-CH2-CH2-CH2-NCO используют, например, интеграл сигналов от С-атомов группы -NCO (δ=122,22 м.д.).
Выраженное в мол. % относительное содержание изоцианатсиланов формулы VI в продукте, полученном вторым предлагаемым в изобретении способом, в интервале значений менее 1 мол. % определяют известным методом количественной инфракрасной спектроскопии с фурье-преобразованием. Для калибровки при анализе этим методом используют калибровочные растворы пригодной для этого концентрации (например, в С2Сl4). Для проведения измерений навеску пробы массой около 1 г помещают в 25-миллилитровую склянку с закатанным краем и добавляют 25 г C2Cl4. Пробу в течение 1-2 ч встряхивают в машине для встряхивания. Затем нижнюю жидкую фазу осторожно дозируют в 20-миллиметровую кювету для инфракрасной спектроскопии и анализируют инфракрасной спектроскопией с фурье-преобразованием (4000-1200 см-1, разрешение 2 см-1). В тех же условиях для вычитания регистрируют спектр растворителя.
В целях определения такого относительного содержания в интервале значений менее 1 мол. % для вещества формулы VI (EtO)3Si-CH2-CH2-CH2-NCO используют, например, длину волны валентного колебания группы -NCO при 2270 см-1.
Остаточное содержание амина формулы VII в продукте, полученном вторым предлагаемым в изобретении способом, может составлять менее 25 мол. %, предпочтительно менее 10 мол. %, особенно предпочтительно менее 5 мол. %, наиболее предпочтительно менее 3 мол. %.
Выраженное в мол. % относительное содержание аминов формулы VII в продукте, полученном вторым предлагаемым в изобретении способом, определяют с использованием13С-ЯМР-метода путем интегрирования сигналов от С-атомов группы -CH2-NH2 в аминах формулы VII по отношению к С-атомам группы Si-CH2- в карбамидсодержащих силанах формулы I.
В целях определения такого относительного содержания для вещества формулы VII H2N-CH2-CH2-Cl используют, например, интеграл сигналов от С-атомов группы H2N-CH2-CH2-Cl (δ=39,47 м.д.) или группы H2N-CH2-CH2-Cl (δ=37,95 м.д.).
Амины формулы VII можно перед их взаимодействием с изоцианатсиланами формулы VI получать из гидрохлоридных солей аминов формулы VIII

добавлением основания, предпочтительно NaOEt. Основание можно при этом добавлять до тех пор, пока величина рН не установится на значение в пределах 7 до 14.
В одном из предпочтительных вариантов второй способ получения карбамидсодержащих силанов формулы I

где R и R1 имеют указанные выше значения, может отличаться тем, что гидрохлоридную соль амина формулы VIII

растворяют в этаноле и подвергают взаимодействию с основанием, после чего добавляют изоцианатсилан формулы VI

а затем добавляют сульфид натрия формулы IV

фильтруют и удаляют растворитель, предпочтительно отгоняют.
Остаточное содержание гидрохлоридной соли амина формулы VIII в продукте, полученном вторым предлагаемым в изобретении способом, может составлять менее 25 мол. %, предпочтительно менее 10 мол. %, особенно предпочтительно менее 5 мол. %, наиболее предпочтительно менее 3 мол. %.
Выраженное в мол. % относительное содержание гидрохлоридной соли аминов формулы VIII в продукте, полученном вторым предлагаемым в изобретении способом, определяют с использованием13С-ЯМР-метода путем интегрирования сигналов от С-атомов группы -CH2-NF2⋅HCl в гидрохлоридной соли аминов формулы VIII по отношению к С-атомам группы Si-CH2- в соединениях формулы I.
В целях определения такого относительного содержания для вещества формулы VIII HCl⋅H2N-CH2-CH2-Cl используют, например, интеграл сигналов от С-атомов группы HCl⋅H2N-CH2-CH2-Cl (δ=41,25 м.д.) или группы HCl⋅H2N-CH2-CH2-Cl (δ=40,79 м.д.).
Карбамидсодержащие силаны формулы I, полученные предлагаемыми в изобретении способами, можно характеризовать путем анализа известным1Н-,13С- или29Si-ЯМР-методом.
Растворимый в ДМСО-d6 или CDCl3 компонент карбамидсодержащих силанов формулы I в полученных предлагаемыми в изобретении способами продуктах определяют путем добавления внутреннего стандарта, такого, например, как трифенилфосфиноксид (ТФФО), в ДМСО-d6 или в CDCl3 и путем анализа известным1H-ЯМР-методом.
Карбамидсодержащие силаны формулы I можно использовать в качестве усилителей (промоторов) адгезии между неорганическими материалами, например стеклянными шариками, стеклянной крошкой, стеклянными поверхностями, стекловолокнами или оксидными наполнителями, предпочтительно кремниевыми кислотами, такими как осажденные кремниевые кислоты и пирогенные кремниевые кислоты, и органическими полимерами, например термореактопластами, термопластами или эластомерами, соответственно в качестве сшивающих агентов и модификаторов оксидных поверхностей.
Карбамидсодержащие силаны формулы I можно далее использовать в качестве аппретов в наполненных резиновых смесях, например в резиновых смесях для изготовления протекторов шин, резинотехнических изделий или обувных подошв.
Преимущества предлагаемых в изобретении карбамидсодержащих силанов формулы I состоят в их улучшенной перерабатываемости, в лучшем сцеплении с мокрой дорогой и в большей динамической жесткости в резиновых смесях.
Примеры
Сравнительный пример 1: Получение [(EtO)3Si-(CH2)3-NH-C(=O)-NH-(CH2)2-S-]2 в воде
В продутую азотом (N2) 1-литровую четырехгорлую колбу с двойной рубашкой, оснащенную мешалкой KPG, обратным холодильником, внутренним термометром и капельной воронкой, помещают дигидрохлорид цистамина (108,39 г, 0,47 моля, 1,00 экв.) и растворяют в полностью обессоленной воде (382 мл). Далее с помощью капельной воронки при температуре в пределах от 15 до 23°С дозируют 50%-ный раствор КОН (92,31 г, 0,82 моля, 1,75 экв.) и перемешивают в течение 30 мин. Затем дозируют 3-изоцианатопропил-триэтоксисилан (221,05 г, 0,85 моля, 1,8 экв.) таким образом, чтобы внутренняя температура не превышала 30°С. После этого перемешивают в течение часа при 24°С. Белую суспензию фильтруют под давлением, промывают тремя порциями полностью обессоленной воды (в общей сложности 340 мл) и сушат в течение 2 ч сухим N2. Фильтровальный осадок сушат на роторном испарителе в токе N2 в течение 7 ч при температуре 35°С и давлении 166 мбар, в течение 10 ч при температуре 35°С и давлении 150 мбар и в течение 9 ч при температуре 35°С и давлении 100 мбар. Таким путем в качестве продукта получают [(EtO)3Si-(CH2)3-NH-C(=O)-NH-(CH2)2-S-]2 в виде мелкого белого порошка (246,38 г, 90,7% от теории).
1Н-ЯМР (δ в м.д., 500 МГц, ДМСО-d6): 0,52 (4Н, t), 1,14 (18Н, t), 1,42 (4Н, m), 2,74 (4Н, m), 2,96 (4Н, m), 3,29 (4Н, m), 3,74 (12Н, q), 6,05 (4Н, m).
13С-ЯМР (δ в м.д., 125 МГц, ДМСО-d6): 7,3 (2С), 18,2 (6С), 23,5 (2С), 38,5 (2С), 39,6 (2С), 42,0 (2С), 57,7 (6С), 157,9 (2С).
29Si-ЯМР (δ в м.д., 100 МГц, ДМСО-d6): -45,3 (100% силана).
Содержание растворимых в ДМСО-d6 компонентов с использованием ТФФО в качестве внутреннего стандарта: 86,0%.
Водосодержание (согласно DIN 51777): 0,7%.
Температура начала плавления: 97°С.
Остаточное содержание изоцианата: 0,08%.
Пример 1: Получение (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-S-CH2CH2-NH-CO-NH-CH2CH2CH2-Si(OEt)3 из (EtO)3Si-CH2CH2CH2-NH2, OCN-CH2CH2-Cl и Na2S (аналогично первому предлагаемому в изобретении способу)
На первой стадии реакции в 4-литровую трехгорлую колбу, оснащенную мешалкой KPG, внутренним термометром, капельной воронкой и обратным холодильником, помещают 3-аминопропилтриэтоксисилан (73,05 г, 0,33 моля, 1,00 экв.) в пентане (2,5 л) и охлаждают до -78°С. Далее в течение 4,5 ч при температуре в пределах от -78 до -70°С по каплям добавляют 2-хлорэтилизоцианат (34,82 г, 0,33 моля, 1,00 экв.) и затем нагревают до комнатной температуры. Белую суспензию фильтруют, фильтровальный осадок промывают пентаном и оставляют сушиться на ночь в токе N2. Таким путем в качестве промежуточного продукта получают (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-Cl (100,52 г, количественный выход) в виде белого, хлопьевидного порошка.
На второй стадии реакции (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-Cl (100,00 г, 0,31 моля, 2,00 экв.) в этаноле (75 мл) помещают в 500-миллилитровую трехгорлую колбу, оснащенную мешалкой, обратным холодильником и внутренним термометром. Далее добавляют сухой сульфид натрия (Na2S, 14,47 г, 0,18 моля, 1,17 экв.) и смесь нагревают с обратным холодильником. После протекания реакции в течение 4,5 ч реакционную смесь охлаждают до комнатной температуры и суспензию фильтруют. От фильтрата на роторном испарителе удаляют растворитель и фильтрат сушат в вакууме. Таким путем в качестве продукта получают (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-S-CH2CH2-NH-CO-NH-CH2CH2CH2-Si(OEt)3 (93,34 г, 99,2% от теории) в виде воскоподобного светло-зеленого твердого вещества.
1Н-ЯМР (δ в м.д., 500 МГц, CDCl3): 0,64 (4Н, t), 1,22 (18Н, t), 1,60 (4Н, m), 2.66 (4Н, t), 3,15 (4Н, m), 3,39 (4Н, m), 3,82 (12Н, q), 5,2-6,4 (4Н, шир.).
13С-ЯМР (δ в м.д., 125 МГц, CDCl3): 7,8 (2С), 18,3 (6С), 23,8 (2С), 32,8 (2С), 40,2 (2С), 42,8 (2С), 58,4 (6С), 159,2 (2С).
Пример 2: Получение (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-S-CH2CH2-NH-CO-NH-CH2CH2CH2-Si(OEt)3 из (EtO)3Si-CH2CH2CH2-NH2, OCN-CH2CH2-Cl и Na2S (аналогично первому предлагаемому в изобретении способу)
На первой стадии реакции в 4-литровую трехгорлую колбу, оснащенную мешалкой KPG, внутренним термометром, капельной воронкой и обратным холодильником, помещают 3-аминопропилтриэтоксисилан (159,39 г, 0,72 моля, 1,00 экв.) в этаноле (3,0 л) и охлаждают до -78°С. Далее в течение 2,25 ч при температуре в пределах от -78 до -69°С по каплям добавляют 2-хлорэтилизоцианат (75,92 г, 0,72 моля, 1,00 экв.) и затем нагревают до 50°С. После этого пятью порциями добавляют сухой сульфид натрия (Na2S, 28,09 г, 0,36 моля, 0,50 экв.) и смесь нагревают с обратным холодильником. После протекания реакции в течение 4,5 ч реакционную смесь охлаждают до комнатной температуры и суспензию фильтруют. От фильтрата на роторном испарителе удаляют растворитель и фильтрат сушат в вакууме. Таким путем в качестве продукта получают (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-S-CH2CH2-NH-CO-NH-CH2CH2CH2-Si(OEt)3 (214,09 г, 96,7% от теории) в виде воскоподобного белого твердого вещества.
1Н-ЯМР (δ в м.д., 500 МГц, CDCl3): 0,64 (4Н, t), 1,22 (18Н, t), 1,61 (4Н, m), 2.67 (4Н, t), 3,15 (4Н, m), 3,40 (4Н, m), 3,82 (12Н, q), 5,16 (2Н, шир.), 5,43 (2Н, шир.).
13С-ЯМР (δ в м.д., 125 МГц, CDCl3): 7,8 (2С), 18,3 (6С), 23,8 (2С), 33,0 (2С), 40,3 (2С), 42,9 (2С), 58,4 (6С), 159,3 (2С).
Пример 3: Получение (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-S-CH2CH2-NH-CO-NH-CH2CH2CH2-Si(OEt)3 из (EtO)3Si-CH2CH2CH2-NCO, HCl⋅H2N-CH2CH2-Cl и Na2S (аналогично второму предлагаемому в изобретении способу)
На первой стадии реакции в 4-литровую трехгорлую колбу, оснащенную мешалкой KPG, внутренним термометром, капельной воронкой и обратным холодильником, помещают гидрохлорид 3-хлорэтиламина (73,86 г, 0,70 моля, 1,00 экв.) в этаноле (3,0 л), охлаждают до -78°С и добавляют этанолят натрия (226,83 г, 0,70 моля, 1,00 экв., 21%-ный в этаноле). Далее в течение 3 ч при температуре в пределах от -78 до -70°С по каплям добавляют 3-изоцианатопропил(триэтоксисилан) (173,15 г, 0,70 моля, 1,00 экв.) и затем нагревают до 50°С. После этого пятью порциями добавляют сухой сульфид натрия (Na2S, 27,31 г, 0,35 моля, 0,50 экв.) и смесь нагревают с обратным холодильником. После протекания реакции в течение 4 ч реакционную смесь охлаждают до комнатной температуры и суспензию фильтруют. От фильтрата на роторном испарителе удаляют растворитель и фильтрат сушат в вакууме. Таким путем в качестве продукта получают (EtO)3Si-CH2CH2CH2-NH-CO-NH-CH2CH2-S-CH2CH2-NH-CO-NH-CH2CH2CH2-Si(OEt)3 (212,68 г, 98,8% от теории) в виде желтого масла.
Пример 4: Резиновые смеси
Рецептура резиновых смесей приведена ниже в таблице 1. При этом величина "част./100 част, каучука" представляет собой массовую долю соответствующего ингредиента в пересчете на 100 частей используемого сырого каучука. Предлагаемый в изобретении силан используют в изомолярных количествах по отношению к сравнительному силану.

Применяемые ингредиенты:
аНК ТСК: натуральный каучук типа SIR 20 SED, фирма Aneka Bumi Pratama (сокращение "ТСК" означает "технически специфицированный каучук", сокращение "SIR" от англ. "Standard Indonesian Rubber" означает "стандартный индонезийский каучук"),
бСКД: полибутадиен Europrene Neocis BR 40, фирма Polimeri,
вР-СКС: полимеризованный в растворе СКС Sprintan® SLR-4601, фирма Styron,
гкремниевая кислота: ULTRASIL® VN3 GR, фирма Evonik Industries AG,
д6ПФД: N-(1,3-диметилбутил)-N'-фенил-n-фенилендиамин,
еДФГ: дифенилгуанидин,
жЦБС: N-циклогексил-2-бензотиазолсульфенамид.
Резиновые смеси приготавливали в обычных условиях в две стадии в лабораторном резиносмесителе (объем от 300 мл до 3 л), при этом сначала на первой стадии смешения (стадия приготовления маточной смеси) все ингредиенты за исключением вулканизующей системы (сера и влияющие на вулканизацию вещества) перемешивают в течение 200-600 секунд при 145-165°С (целевая температура 152-157°С). Добавлением вулканизующей системы на второй стадии получают окончательную смесь (стадия приготовления окончательной смеси), при этом перемешивают в течение 180-300 секунд при 90-120°С.
Общий способ приготовления резиновых смесей и получения их вулканизатов описан в справочнике "Rubber Technology Handbook", W. Hofmann, изд-во Hanser Verlag, 1994.
Резинотехнические свойства исследуют по методам, представленным в таблице 2.

Из всех смесей приготавливали образцы для испытаний путем вулканизации до момента t95 (по данным измерения на вискозиметре с пуансоном согласно стандарту ISO 6502/ASTM D5289-12), проводимой под давлением при 160°С. Результаты исследования резинотехнических свойств невулканизованных резиновых смесей и полученных из них вулканизатов представлены ниже в таблице 3.

Резиновая смесь II, которая содержит предлагаемый в изобретении карбамидсодержащий силан из примера 2, обладает улучшенной перерабатываемостью (меньший минимальный крутящий момент после 2-й стадии смешения), лучшим сцеплением с мокрой дорогой (эластичность по отскоку при 23°С), а также большей динамической жесткостью (модуль Е' при удлинении на 0,15% и разность между модулем Е' при удлинении на 8% и модулем Е' при удлинении на 0,15%) по сравнению со сравнительной резиновой смесью I, которая содержит используемый в изомолярном количестве силан из сравнительного примера 1 (дисульфидсилан).
Реферат
Изобретение относится к карбамидсодержащим силанам. Предложен карбамидсодержащий силан формулы (I), где Rимеют одинаковые или разные значения и представляют собой С-Салкоксигруппы, а R имеют одинаковые или разные значения и представляют собой неразветвленную насыщенную алифатическую двухвалентную углеводородную группу с С-С. Предложены также варианты способа получения указанных силанов. Технический результат – предложенные карбамидсодержащие силаны обладают улучшенной перерабатываемостью и придают содержащим их резиновым смесям улучшенное сцепление с мокрой дорогой и динамическую жесткость. 3 н. и 12 з.п. ф-лы, 3 табл., 4 пр.
Формула







Комментарии