Комплексы металлов с пентадентантными (n3o2) лигандами, способ их получения, каталитические системы для проведения реакций диоксида углерода с эпоксидами, способ получения циклических карбонатов или алифатических поликарбонатов - RU2740944C1
Код документа: RU2740944C1
Чертежи
Описание
Изобретение относится к области получения новых катализаторов для реакции диоксида углерода с эпоксидами и способу проведения реакции с применением этих катализаторов.
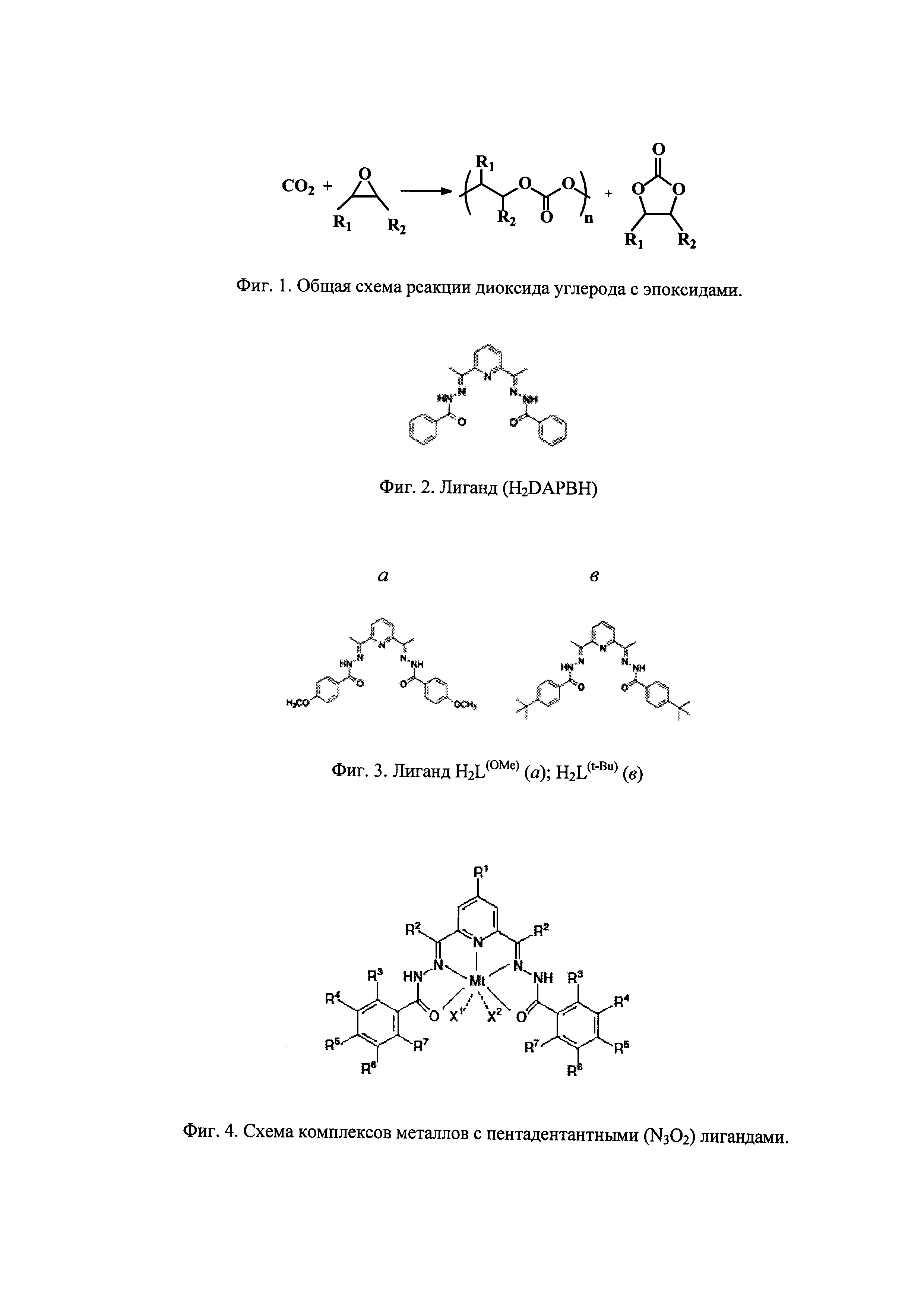
С 70-х годов XX века известно, что реакция диоксида углерода с эпоксидами может протекать в присутствии катализаторов. В последние десятилетия во всем мире проводились исследования, направленные на поиск активных каталитических систем. В 2000-х годах было показано, что к наиболее активным катализаторам относятся комплексы различных металлов (Cr, Со, Zn, Fe, Al и др.) с тетрадентантными основаниями Шиффа со связывающим узлом (N2O2) в качестве лигандов [A. Decortes, A.M. Costilla, A.W. Kleij // Angew. Chem. Int. Ed. 2010. V. 49, pp. 9822-9837; M.I. Childers, J.M. Longo, N.J. Van Zee, A.M. LaPointe, G.W. Coates // Chem. Rev. 2014. V. 114, pp. 8129-8152; C. Kozak, K. Ambrose, T.S. Anderson // Coord. Chem. Rev. 2018. V. 376, pp.565-587]. Реакция CO2 с эпоксидами протекает с образованием циклических карбонатов и алифатических поликарбонатов (Фиг. 1 Общая схема реакции диоксида углерода с эпоксидами). Состав продуктов реакции определяется в основном составом и строением каталитического комплекса и в некоторой степени условиями проведения реакции.
Возможность применения комплексов металлов с пентадентантными (N3O2) лигандами в реакции СО2 с эпоксидами до настоящего времени не исследовалась, хотя в литературе были описаны комплексы некоторых металлов (Co,Ni, редкоземельные элементы) с одним из подобных лигандов, (Лиганд H2DAPBH представлен на Фиг. 2) ((1,1'-(pyridine-2,6-diyl)bis(ethan-1-yl-1-ylidene))dibenzohydrazine, пентадентантным основанием Шиффа, не содержащим заместителей в фенильном кольце. [Thomas J. Giordano, Gus J. Palenik, Ruth C. Palenik, Douglas A. Sullivan // Inorganic Chemistry. 1979. V. 18. No. 9, pp. 2445-2450; Celine Pichon, Bahjat Elrez, Virginie Bereau, Carine Duhayon, and Jean-Pascal Sutter // Eur. J. Inorg. Chem. 2018. pp.340-348]
В 2019 году сотрудниками ИПХФ РАН были получены ряд новых комплексов хрома, кобальта, цинка, ванадия и др. металлов с неописанными ранее родственными (H2DAPBH) лигандами с метоксидными и трет-бутильными заместителями в фенильных кольцах лиганда (Фиг. 3).
Были начаты работы по систематическому изучению влияния природы металла в комплексе, состава и строения лиганда и комплекса на каталитическую активность комплексов в реакции СО2 с эпоксидами. Первые опыты показали, что комплексы хрома с незамещенным лигандом (H2DAPBH) не проявляют активности в реакции, однако, комплексы с родственными лигандами с заместителями в фенильных кольцах могут быть использованы как катализаторы в реакции СО2 с эпоксидами.
Задачей изобретения является получение новых катализаторов с пентадентантными (N3O2) лигандами, активных в реакции диоксида углерода с эпоксидами.
Задачей изобретения является также осуществление реакции диоксида углерода с эпоксидами с применением этих катализаторов.
Поставленные задачи решаются описываемым способом синтеза комплексов, имеющих общее строение согласно Фиг. 4,
где R1 может быть Н, алкильный заместитель из ряда С1-С4 или арильный заместитель, R2- R7 могут быть одинаковыми или различными и состоять из Н, алкильных заместителей ряда С1-С20, арильных заместителей, фтор-углеродных алкильных или арильных заместителей, алкоксидных или феноксидных заместителей с амино-, нитро-, гидроксо-группами; X1, X2 могут быть любыми нуклеофилами, способствующими раскрытию эпоксидной связи, например галоген-ионами, метокси- или фенокси-ионами, а также любыми анионами. Mt может быть CrIII, CoIII, ZnII, AlIII, VIV, MnIII, MoIV а также переходным металлом IV группы.
Комплексы, когда R1 = R2 = R3 = R4 = R5 = R6 = R7 = Н исключаются, так как они неактивны в реакции диоксида углерода с эпоксидами.
Кроме того задача решается способом проведения реакции диоксида углерода с эпоксидами с использованием синтезированного катализатора при температуре 20-90°С, давлении СО2 0.2-2 МПа, в присутствии сокатализаторов из ряда оснований или солей четвертичного аммония, отличающийся тем, что процесс проводят с предварительным перемешиванием раствора катализатора, сокатализатора и эпоксида в течение 0.5-1.5 часа при комнатной температуре в отсутствии СО2. Данные о скорости процесса получают, измеряя поглощение СО2 со временем на оригинальной установке высокого давления.
Сущность изобретения заключается в следующем.
Синтез комплексов с замещенными пентадентантными (N3O2) лигандами проводили по описанным ниже методикам.
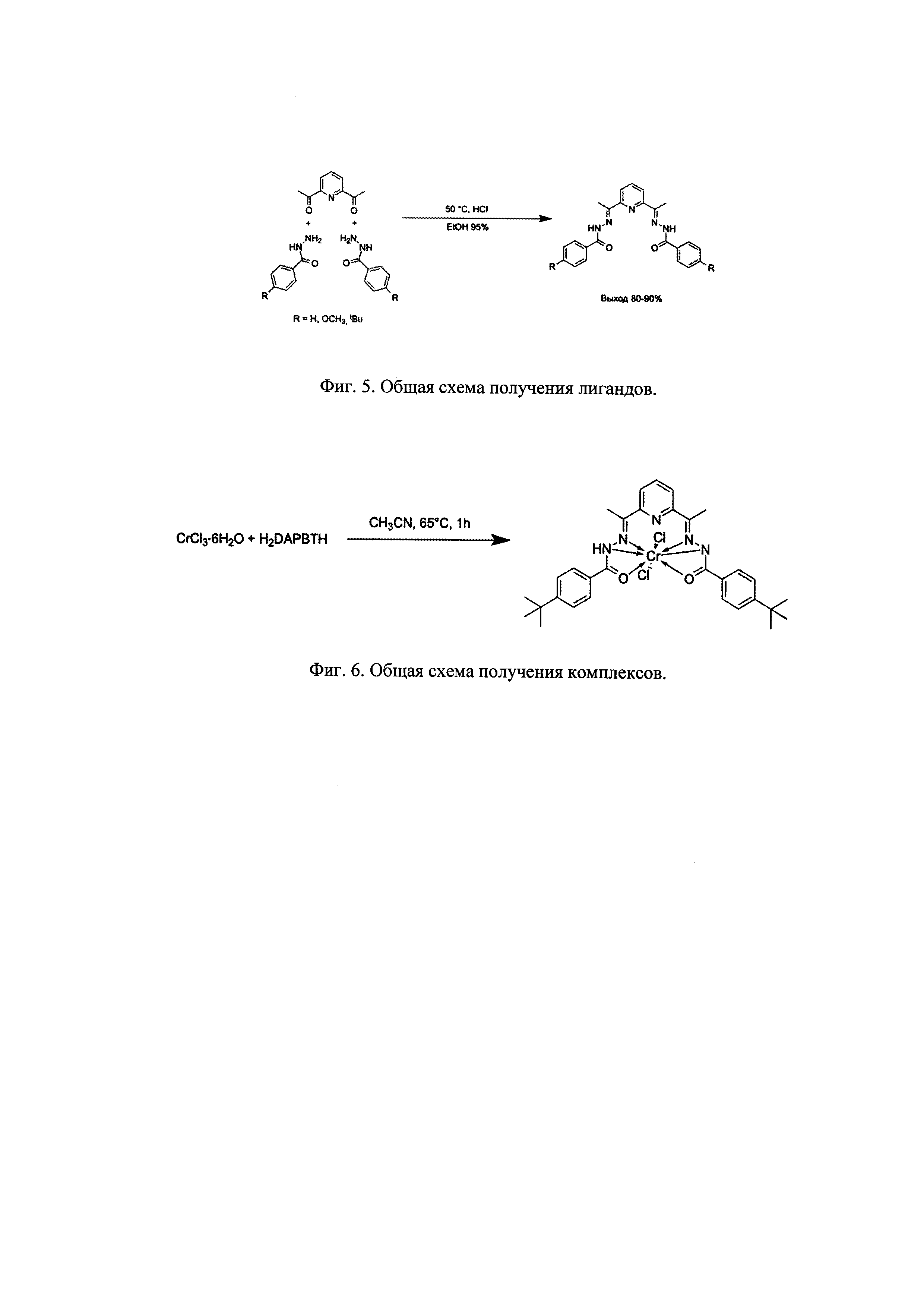
На начальном этапе были синтезированы лиганды с различными заместителями в фенильных кольцах. На первой стадии проводили реакцию конденсации диацетилпиридина (ДАП) с соответствующими бензоилгидразидами (Схема синтеза лигандов представлена на Фиг. 5).
Комплексы получали прямой реакцией хлорида соответствующего металла (CrCl2, CrCl3, CoCl2, ZnCl2 и др.) с лигандом в среде метилового спирта или ацетонитрила (Схема синтеза комплексов с пентадентантными лигандами представлена на Фиг. 6).
Для исследования влияния типа заместителей в лиганде на активность комплексов в реакции были использованы комплексы хрома с R5 = ОМе иtBu.
Влияние природы противоионов X на скорость реакции диоксида углерода с эпоксидами исследовали на примере комплексов хрома с R5 = ОМе, Х1 = Х2 = Cl; Х1 = Х2 = N3; Х1 = ОМе, X2 = МеОН.
Комплекс с Х = N3 получали по реакции LCrCl2 с NaN3; комплекс с Х1 = ОМе, Х2 = МеОН -по реакции LCrCl2 с раствором NaOMe в СН3ОН.
Изучение влияния природы металла на активность комплексов проводили, применяя комплексы Cr, Со, Zn, V с R5 =tBu. Все перечисленные выше комплексы являются новыми.
Реакцию диоксида углерода с эпоксидами проводили аналогично известным методикам, описанным при применении тетрадентантных (N2O2) комплексов [G. W. Coats, Z. Qin, С.Т. Cohen US 8,278,239 В2 (2012)]. Отличие применяемой нами методики от литературных методов заключалось в предварительном перемешивании растворов катализатора, сокатализатора и эпоксида в течение 0.5-1.5 часа при комнатной температуре в атмосфере аргона в присутствии комплексов с пентадентантными (N3O2) лигандами. Кинетика реакции впервые была изучена с использованием оригинальной установки высокого давления, разработанной в ИПХФ РАН, по поглощению СО2 в ходе реакции. Измерение скорости реакции проводили на стационарном участке кинетической кривой, длительность которого составляла 1 час. Далее в примерах скорость была рассчитана в единицах TOF, ч-1 как количество молей СО2 в расчете на моль каталитического комплекса за час.
Анализ состава продуктов реакции проводили методом ЯМР1Н по описанным литературным методикам [A. Decortes, A.M. Castilla, A. W. Kleij // Angew. Chem. Int. Ed. 2010. V. 49, pp. 9822-9837; M.I. Childers, J.M. Longo, N.J. Van Zee, A.M. LaPointe, G.W. Coates // Chem. Rev. 2014. V. 114, pp.8129-8152, и ссылки в обзорах].
Далее приводятся примеры для иллюстрации описанного способа синтеза новых катализаторов и проведения реакции СО2 с эпоксидами с использованием этих катализаторов.
В примерах 1, 2 описан постадийный синтез лигандов с заместителями, в примерах 3-6 представлено описание синтеза комплексов с металлами Cr, Со, Zn, V с этими лигандами.
Для анализа влияния различных заместителей в лиганде, природы противоиона, природы металла на активность комплексов реакцию проводили в одинаковых условиях.
Влияние типа заместителей X на скорость реакции показано в примерах 7-9. Скорость реакции при прочих равных условиях изменяется в ряду заместителей X следующим образом: (X1 = X2 = Cl) > (X1 = X2 = N3) > (X1 = ОМе, X2 = МеОН) (Таблица, №7-9).
Влияние природы заместителя R5 на скорость реакции показано в примерах 7, 13. Скорость изменялась в ряду (R5 =tBu) > (R5 = ОМе) (Таблица, №7,13).
Изменение скорости реакции при применении изоструктурных комплексов с различными металлами показано в примерах 10, 13, 14, 15. Наибольшую активность в реакции демонстрируют комплексы хрома и цинка, а скорость изменяется в ряду (Mt = Cr) > (Mt = Zn) > (Mt = V) > (Mt = Co). Наблюдалось изменение состава продуктов при использовании комплексов с различными металлами. При низкой скорости реакции на комплексах Со образовывалось небольшого количества сополимера, смесь продуктов получена также в случае применения комплекса Zn (Таблица, №10,13-15). Изменение селективности процесса в сторону образования сополимера возможно при варьировании условий реакции, природы металла в комплексе.
Применение разных эпоксидов в реакции показано в примерах 10-12 (Таблица, №10-12). При прочих равных условиях скорость реакции изменялась в ряду: пропиленоксид > бутеноксид > циклогексеноксид. Различие в скоростях реакции в последних двух случаях было небольшим, в то время как с пропиленоксидом скорость реакции в два раза выше, чем с другими эпоксидами.
Синтез замещенных пентадентантных (N3O2) лигандов
Пример 1. Синтез H2L(OMe) (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 = OMe, Фиг. 4) 2,6-диацетилпиридин (26.78 ммоль) и п-метоксибензгидразид (53.56 ммоль) были растворены в 96% этиловом спирте. После этого, к раствору были добавлены 3 капли концентрированной соляной кислоты, раствор приобрел желтый цвет. Раствор нагревали до 50°С при перемешивании. Спустя 15 минут после добавления кислоты образовался объемный белый осадок. Нагревание продолжалось в течение 1 часа. Затем смесь была оставлена на ночь в холодильнике (+4°С), осадок отфильтрован и промыт холодным этанолом и диэтиловым эфиром, затем высушен под вакуумом. В результате был получен белый кристаллический порошок (21.71 ммоль, выход 81%). Расчетные значения содержания элементов для C25H25N5O4: С, 65.36; Н, 5.45; N, 15.25%. Найдено: С, 65.62; Н, 5.71; N, 15.35%. FT-IR (НПВО) νmax/см-1: 3411 cp., 3218 ср., 2296 ср., 1646 с., 1606 с., 1580 ср., 1547 с., 1502 с., 1455 ср., 1365 ср., 1285 с., 1250 с., 1176 с., 1145 ср., 1120 ср., 1025 с., 919 ср., 834 с., 758 ср.
Пример 2. Синтез H2L(t-Bu) (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 =tBu, Фиг. 4) Синтез проводили аналогично Примеру 1, за тем исключением, что использовали 2,6-диацетилпиридин (26.78 ммоль) и п-третбутилбензгидразид (53.56 ммоль). Получено 11,11 г, 21.71 ммоль лиганда, выход составил 81%). Расчетные значения содержания элементов для C31H37N5O2 (%) С, 72.77; Н, 7.29; 13.69. Найдено (%): С, 70.43; Н, 7.85; N, 12.22.
Синтез комплексов с пентадентантными лигандами.
Пример 3. Синтез комплексов Cr с замещенными пентадентантными лигандами L(R)CrCl2
К смеси CrCl3⋅6H2O (1.17 ммоль) и H2L(OMe) или H2L(t-Bu) (1.17 ммоль) был добавлен абсолютный ацетонитрил (30 мл). Раствор при этом приобретал темно-зеленую окраску за счет частичного растворения соли. Реакционную смесь нагревали до 70°С и перемешивали на магнитной мешалке в течение часа. Раствор приобретал коричневую окраску. Через час реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный фильтр Шотта от небольшого осадка. Маточный раствор оставляли при температуре -18°С на 12 часов. Из раствора выпадали светло-коричневые кристаллы L(R)CrCl2. Раствор декантировали, кристаллы промывали диэтиловым эфиром и сушили откачкой в вакууме. Выход составлял 45% (0.524 ммоль). Монокристаллы комплекса для проведения рентгеноструктурного анализа получали путем наслоения диэтилового эфира на раствор полученных кристаллов в хлористом метилене.
L(OMe)CrCl2 CH3CN Расчетные значения содержания элементов для CrC27H27N6O4Cl2 (%): С, 52.01; Н, 4.37; N, 13.50 Найдено (%): С, 52.55; Н, 4.84; N, 13.40. FT-IR (НПВО) νmax/см-1: 683 с, 698 с, 712 с, 727 с, 736 с, 762 ср, 808 с, 825 с, 834 с, 851 ср, 861 ср, 878 с, 884 с, 906 с, 916 с, 922 с, 930 с, 940 с, 957 с, 978 с, 987 с, 1079s, 1092 с, 1112 с, 1135 с, 1145 с, 1177 ср, 1256 ср, 1291 с, 1301 с, 1342 с, 1380 ср, 1402 с, 1498 ср, 1534 с, 1559 с, 1602 ср, 2251 с.
L(t-Bu)CrCl2 Расчетные значения содержания элементов для CrCl2C31H36O2N5 С, 58.77; Н, 5.73; N, 11.05%. Найдено: С, 58.05; Н, 5.81; N, 10.68%. FT-IR (НПВО) νmax/см-1: 371 с., 524 с., 551 с., 682 с., 846 ср., 921 ср., 1075 ср., 1127 с., 1166 с., 1190 с., 1269 с., 1314 с., 1386 с., 1435 ср., 1458 ср., 1491 ср., 1529 с., 1557 с., 1590 ср., 1605 с., 2867 с., 2902 с., 2960 с., 3068 сл.
Синтез L(OMe)CrCl2 МеОН.
К суспензии H2L(OMe) (300 мг, 0.65 ммоль) в абсолютном СН3ОН (19 мл) в атмосфере Ar при перемешивании добавили CrCl2⋅4H2O (127 мг, 0.65 ммоль). Цвет раствора изменился на зелено-коричневый. Смесь перемешивали 1.5 часа при 60-70°С. Через 30 минут после начала нагревания выпал желто-коричневый осадок. Смесь оставили на ночь при комнатной температуре, после чего осадок был отфильтрован через стеклянный фильтр Шотта, промыт абсолютным СН3ОН и Et2O, и высушен в вакууме.
Получен L(OMe)CrCl2 МеОН (60 мг, 0.05 ммоль, выход 16%). Расчетные значения содержания элементов для CrC26H29N5O5Cl2 (%): С, 50.81; Н, 4.72; N, 11.4. Найдено (%) С, 51.0; Н, 4.74; N, 11.14. FT-IR (НПВО) νmax/см-1: 682 ср, 744 с, 761 с, 804 ср, 849 с, 1021 с, 1051 ср, 1171 с, 1259 с, 1291 ср, 1309 ср, 1380 с, 1439 ср, 1499 с, 1602 с, 1626 ср, 2995 ср, 3132 ср.
Синтез L(OMe)Cr(N3)2
К метанольному раствору азида натрия NaN3 (56 мг, 0.86 ммоль, 16 мл СН3ОН) добавляли навеску комплекса L(OMe)CrCl2 МеОН (50 мг, 0.086 ммоль). Реакционную смесь нагревали и перемешивали в течение 10 минут при +60°С. Реакционную смесь красно-коричневого цвета отфильтровывали через стеклянный фильтр Шотта и оставляли для кристаллизации при комнатной температуре. Через 12 часов наблюдали образование темных кристаллов. Маточный раствор декантировали, кристаллы промывали метанолом (1.5 мл) и диэтиловым эфиром (2 мл) и сушили на воздухе. Получили черное кристаллическое вещество L(OMe)Cr(N3)2 (34 мг, выход 68%). Расчетные значения содержания элементов для C26H28N11O5Cr (%): С, 49.84; Н, 4.50; N, 24.59. Найдено (%): С, 49.85; Н, 4.36; N, 25.01. FT-IR (НПВО) νmax/см-1: 688 с., 696 с., 717 с., 736 с., 759 ср., 800 с., 807 с., 851 с., 859 с., 907 с., 997 с., 1026 ср., 1050 с., 1110 с., 1120 с., 1137 с., 1171 ср., 1160 ср., 1288 ср., 1308 с., 1338 ср., 1376 ср., 1433 с., 1454 с., 1490 с., 1506 ср., 1542 с., 1556 с., 1568 с., 1584 с., 1607 ср., 2048 ср., 2348 с., 2357 с., 2371 с.
Синтез L(OMe)Cr(OMe)(MeOH)
К навеске комплекса L(OMe)CrCl2 МеОН (350 мг, 0.6 ммоль) добавили МеОН (22 мл) и 0.44 М раствор NaOMe в МеОН (3.9 мл, 92 мг, 1.7 ммоль). Раствор приобрел красно-коричневую окраску. После нескольких дней медленного испарения растворителя при комнатной температуре были выделены красно-коричневые кристаллы. Кристаллы промыты H2O (5 мл), EtOH (5 мл), Et2O (5 мл) и высушены в вакууме. Получен L(OMe)Cr(OMe)(MeOH) МеОН (210. mg, 0.35 mmol, выход 61%). Расчетные значения содержания элементов для C28H34N5O7Cr (%): С, 55.62; Н, 5.67; N, 11.58. Найдено (%): С, 55.31; Н, 5.81; N, 11.62. FT-IR (НПВО) νmax/см-1: 683s, 698 с., 714 с., 744 с., 761 ср., 781 с., 788 с., 813 с., 828 с., 842 с., 864 с., 882 с., 889 с., 906 с., 920 с., 993 с., 1028 ср., 1043 ср., 1072 с., 1091 с., 1102 с., 1119 с., 1135 с., 1171 ср., 1156 ср., 1308 с., 1337 с., 1379 ср., 1462 с., 1505 ср., 1559 с., 1582 с., 1607 с.
Пример 4. Синтез L(t-Bu)CoCl2
К суспензии H2L(t-Bu) (250 мг, 0.49 ммоль) в ректификате С2Н5ОН (15 мл) добавили при перемешивании раствор безводного CoCl2 в абсолютном метаноле (0.05 ммоль, 1 мл 0.5М раствора). Цвет раствора изменился на ярко-коричневый, весь лиганд растворился. Затем к раствору добавили 0.14 мл триэтиламина Et3N и смесь перемешивали в течение 1 часа при 60°С. После охлаждения до комнатной температуры к реакционной смеси добавили избыток раствора HCl в СН3ОН, удалили часть растворителя откачкой в вакууме, а выпавшие при этом светло-зеленые кристаллы промыли этиловым спиртом и эфиром и высушили в вакууме. Получен L(t-Bu))CoCl2 (0.32 ммоль, выход 65%). Расчетные значения содержания элементов для CoCl2C31H37O2N5 С, 56.88; Н, 5.66; N, 10.70%. Найдено: С, 57.05; Н, 5.81; N, 11.35%. FT-IR (НПВО) νmax/см-1: 682c., 846 ср., 921 ср., 1075 ср., 1127 с., 1166 с., 1190 с., 1269 с., 1314 с., 1386 с., 1435 ср., 1458 ср., 1491 ср., 1529 с., 1557 с., 1590 ср., 1605 с., 2867 с., 2902 с., 2960 с., 3068 сл.
Пример 5. Синтез L(t-Bu)ZnCl2
К суспензии H2L(t-Bu) (385 мг, 0.75 ммоль) в ректификате C2H5OH (17 мл) при перемешивании добавили навеску ZnCl2 (102 мг, 0.75 ммоль). Цвет раствора изменился на светло-лимонно-желтый, соль и лиганд растворились. Реакционную смесь перемешивали в течение 15 минут при 60°С, затем фильтровали. После нескольких дней медленного испарения растворителя при комнатной температуре в растворе образовались светло-желтые кристаллы, которые были отделены декантацией маточного раствора, промыты эфиром и высушены в вакууме. Получен L(t-Bu))ZnCl2 МеОН (0.51 ммоль, выход 68%). Расчетные значения содержания элементов для ZnCl2C32H40O3N5 С, 56.64; Н, 5.90; N, 10.32%. Найдено: С, 57.05; Н, 5.88; N, 10.58%. FT-IR (НПВО) νmax/см-1: 551 с., 682 с, 846 ср., 921 ср., 1075 ср., 1127 с, 1166 с, 1190 с, 1269 с, 1314 с, 1386 с, 1435 ср., 1458 ср., 1491 ср., 1529 с, 1557 с, 1590 ср., 1605 с, 2867 с, 2902 с, 2960 с, 3068 сл.
Пример 6 Синтез L(t-Bu)V(O)(SO4)
К суспензии H2L(t-Bu) (213 мг, 0.42 ммоль) в абсолютном СН3ОН (15 мл) в атмосфере Ar при перемешивании добавили твердый кристаллический VOSO4⋅3H2O (90 мг, 0.42 ммоль). Цвет раствора изменился на яркий красно-коричневый. Смесь перемешивали 1 час при 60°С, затем охлаждали до комнатной температуры, фильтровали от небольшого осадка, затем удаляли часть растворителя откачкой в вакууме, а выпавший при этом кристаллический осадок промывали эфиром и сушили откачкой в вакууме. Получен L(t-Bu)V(O)(SO4) (0.27 ммоль, выход 64%). Расчетные значения содержания элементов для L(t-Bu)V(O)(SO4): С, 55.27; Н, 5.35; N, 10.40; S, 4.75%. Найдено: С, 56.05; Н, 5.88; N, 10.68; S, 5.15%. FT-IR (НПВО) νmax/см-1: 554 с., 636 с., 846 ср., 916 ср., 1089 ср., 1144 с., 1165 с., 1190 с., 1268 с., 1363 с., 1435 ср., 1452 ср., 1497 ср., 1533 с., 1567 с., 1590 ср., 1605 с., 2867 с., 2901 ср., 2960 ср., 3225 ср.
Реакция СО2 с эпоксидами
Пример 7. Реакция СО2 с пропиленоксидом (РО) в присутствии L(OMe)CrCl2 (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 = OMe, Х1 = Х2 = Cl, Фиг. 4).
Комплекс хрома (0.027 ммоль) и сокатализатор бис-(трифенилфосфин)иминия хлорид (PPNCl, 0.054 ммоль) растворяли в 4 мл РО в атмосфере аргона при перемешивании в течение 40 минут. Раствор переливали в предварительно высушенный реактор объемом 0.1 л в атмосфере СО2. В реакторе поднимали давление в подачей СО2 и подключали к термостату, нагретому до 70°С. В ходе реакции поддерживали постоянное давление 0.7 МПа. Кинетику регистрировали, измеряя изменение давления СО2 в калиброванной мерной емкости. В течение часа реакция протекала при постоянной скорости. На стационарном участке кинетической кривой рассчитывали скорость реакции в единицах TOF, ч-1 (мольное количество диоксида углерода, вступившего в реакцию, в расчете на моль хрома за час). Реакцию останавливали, сбрасывая давление и охлаждая реактор до комнатной температуры. Небольшую часть раствора отбирали для регистрации1Н ЯМР спектра.
1H ЯМР (CDCl3, 500 МГц) м.д.: пропиленкарбонат 1.49, 1.51 (д, 3Н, СН3) 4.02, 4.03, 4.05 (дд, 1Н, СН2); 4.55, 4.56, 4.58 (дд, 1Н, СН2); 4.86 (м, 1Н, СН); поли(пропилен)карбонат 1.33, 1.35 (д, 3Н, СН3), 4.11-4.30 (м, 2Н, СН2), 5.01 (м, 1H, СН).
Продукты реакции взвешивали после откачивания эпоксида в вакууме. За 100 мин. выход составил 1.23 г, скорость была равна 360 ч-1 (Таблица). Расход CO2 в реакции соответствовал количеству полученного продукта с точностью ±10%. По данным ЯМР, получен в основном циклический пропиленкарбонат. Селективность реакции по циклическому карбонату (ƒ) рассчитывали как долю эпоксида в циклическом карбонате в расчете на суммарное количество эпоксида, вступившего в реакцию. В этом опыте ƒ составило 98%.
Пример 8. Реакция СО2 с пропиленоксидом в присутствии L(OMe)Cr(N3)2 (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 = OMe, X1 = X2 = N3, Фиг. 4)
Реакцию проводили аналогично методике примера 7, за исключением того, что количество комплекса хрома было 0.036 ммоль, сокатализатора PPNCl 0.072 ммоль. Смесь перемешивали в течение 50 мин до полного растворения реагентов. Реакцию проводили в течение 120 мин, выход продуктов составил 0.62 г., скорость была равна 140 ч-1 (Таблица). В продуктах реакции присутствует только циклических пропиленкарбонат.
Пример 9. Реакция CO2 с пропиленоксидом в присутствии L(OMe)Cr(OMe)(MeOH) (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 = OMe, X1 = ОМе, X2 = MeOH, Фиг. 4) Реакцию проводили аналогично методике примера 7, за исключением того, что количество комплекса хрома было 0.035 ммоль, сокатализатора PPNCl 0.07 ммоль. Смесь перемешивали в течение 40 мин до полного растворения реагентов. Реакцию проводили в течение 100 мин, выход продуктов составил 0.77 г., скорость была равна 170 ч-1 (Таблица). В продуктах реакции присутствует только циклических пропиленкарбонат.
Пример 10. Реакция CO2 с пропиленоксидом в присутствии L(t"Bu)CrCl2 (R1 = r3 = R4 = R6 = R7 = H, R2 = Me, R5 =tBu, X1 = X2 = Cl, Фиг. 4)
Реакцию проводили аналогично методике примера 7, за исключением того, что количество комплекса хрома было 0.025 ммоль, сокатализатора PPNCl 0.05 ммоль. Смесь перемешивали в течение 30 мин. до полного растворения компонентов. Реакцию проводили в течение 120 мин, выход продуктов составил 1.51 г., скорость была равна 490 ч-1 (Таблица). Селективность по циклическому карбонату f = 98%.
Пример 11. Реакция CO2 с бутеноксидом в присутствии L(t-Bu)CrCl2 (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 =tBu, X1 = X2 = Cl, Фиг. 4)
Реакцию проводили аналогично методике примера 7, за исключением того, что количество комплекса хрома было 0.028 ммоль, сокатализатора PPNCl 0.056 ммоль. Смесь перемешивали в течение 40 мин. Реакцию проводили в течение 120 мин, выход продуктов составил 0.8 г., скорость была равна 250 ч-1 (Таблица). Получен циклический бутенкарбонат с селективностью 100%.1Н ЯМР (CDCl3, 500 МГц) м.д.: бутенкарбонат 0.98 (т, 3Н, CH3), 1.81-1.70 (м, 2Н, CH2), 4.05 (т, 1Н, CH2), 4.51 (т, 1Н, CH2), 4.60-4.65 (м, 1Н, СН); поли(бутен)карбонат 0.94 (м, 3Н, CH3), 1.67 (м, 2Н, CH2), 4.07-4.34 (м, 2Н, CH2), 4.83 (м, 1Н, СН).
Пример 12. Реакция CO2 с циклогексеноксидом в присутствии L(t-Bu)CrCl2 (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 =tBu, Х1 = Х2 = Cl, Фиг. 4)
Реакцию проводили аналогично методике примера 7, за исключением того, что количество комплекса хрома было 0.029 ммоль, сокатализатора PPNCl 0.058 ммоль. Смесь перемешивали в течение 50 мин до полного растворения компонентов. Реакцию проводили в течение 120 мин, выход продуктов составил 0.7 г., скорость была равна 220 ч-1 (Таблица). Получен циклический циклогексенкарбонат с селективностью 100%.1Н ЯМР (CDCl3, 500 МГц) м.д.: циклогексенкарбонат 1.30-1.40 (м, 2Н, CH2), 1.55-1.60 (м, 2Н, CH2), 1.85-2.10 (м, 4Н, CH2), 4.62-4.66 (м, 2Н, СН); поли(циклогексен)карбонат 1.30 (м, 2Н, CH2), 1.44 (м, 2Н, CH2), 1.69 (м, 2Н, CH2), 2.10 (м, 2Н, CH2), 4.62 (м, 2Н, СН).
Пример 13. Реакция СО2 с пропиленоксидом в присутствии L(t-Bu)CoCl2 (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 =tBu, X1 = X2 = Cl, Фиг. 4)
Реакцию проводили аналогично методике примера 7, за исключением того, что количество комплекса кобальта было 0.029 ммоль, сокатализатора PPNCl 0.058 ммоль. Смесь перемешивали в течение 50 мин. Реакцию проводили в течение 130 мин, выход продуктов составил 0.23 г., скорость была равна 60 ч"1 (Таблица). Получен циклический пропиленкарбонат с селективностью 90%, а также небольшое количество поли(пропилен)карбоната.
Пример 14. Реакция CO2 с пропиленоксидом в присутствии L(t-Bu)ZnCl2 (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 =tBu, X1 = X2 = Cl, Фиг. 4)
Реакцию проводили аналогично методике примера 7, за исключением того, что количество комплекса цинка было 0.024 ммоль, сокатализатора PPNCl 0.048 ммоль. Смесь перемешивали в течение 40 мин до растворения компонентов. Реакцию проводили в течение 120 мин, выход продуктов составил 0.47 г., скорость была равна 150 ч-1 (Таблица). Получен циклический пропиленкарбонат с селективностью 85%, а также небольшое количество поли(пропилен)карбоната.
Пример 15. Реакция CO2 с пропиленоксидом в присутствии L(t-Bu)V(O)(SO4) (R1 = R3 = R4 = R6 = R7 = H, R2 = Me, R5 =tBu, X1 = SO4, X2 = О, Фиг. 4)
Реакцию проводили аналогично методике примера 7, за исключением того, что количество комплекса цинка было 0.025 ммоль, сокатализатора PPNCl 0.05 ммоль. Смесь перемешивали в течение 40 мин до растворения компонентов. Реакцию проводили в течение 100 мин, выход продуктов составил 0.26 г., скорость была равна 90 ч-1 (Таблица). Получен циклический пропиленкарбонат с селективностью 100%.

Описанные примеры показывают, что состав лигандного окружения, природа металла, природа заместителей X оказывают большое влияние на скорость и селективность реакции диоксида углерода с эпоксидами.
Введение различных заместителей в лиганд приводит к значительному изменению скорости реакции. Например, почти в 1.5 раза выросла скорость при замене ОМе -заместителя наtBu (примеры 7,10).
Примеры 7-9 показывают, что природа противоиона X также влияет на скорость реакции. Наиболее активными из представленных в таблице являются комплексы с X = Cl при прочих равных условиях.
Различные эпоксиды могут быть использованы в реакции, как линейного, так и циклического строения (примеры 10-12). С более высокой скоростью протекает реакция с пропиленоксидом.
Природа металла в комплексе оказывает влияние как на скорость процесса, так и на его селективность. С большей скоростью протекает реакция в присутствии комплексов хрома. По данным, представленным в таблице, скорость изменяется в ряду Cr > Zn > V > Со при прочих равных условиях (примеры 7, 13, 14, 15). В присутствии катализаторов Zn и Со продукты реакции представляли собой смесь циклического карбоната и сополимера. При изменении условий реакции в присутствии этих комплексов выход полимерного продукта можно увеличить. Сравнение полученных данных с литературными довольно сложно провести, так как строение лигандного окружения, природа металла и сокатализатора, а также условия проведения реакции оказывают большое влияние на ее скорость. Для примера можно сопоставить данные, полученные с использованием одного из известных комплексов хрома с тетрадентантным (N2O2) лигандом [D.J. Darensbourg // Chem. Rev. 2007. V. 107, p.2401, Fig. 28]. Реакция диоксида углерода с пропиленоксидом при 60°С, 3.4 МПа, в присутствии сокатализатора PPNCl протекала со скоростью 150 ч-1. Однако при формировании комплексов со сложными заместителями в лиганде можно получить более активные системы.
Итак, представленные в таблице результаты показывают, что комплексы различных металлов активны в реакции диоксида углерода с эпоксидами. Как природа заместителей в лиганде, так и природа металла или противоиона X оказывают большое влияние как на скорость, так и на селективность процессов. В реакции можно использовать различные эпоксиды. Продуктами реакции являются циклический карбонат и сополимер диоксида углерода с эпоксидом. И тот, и другой продукт представляют большой интерес для современной химии.
Реферат
Изобретение относится к области получения новых комплексов, а именно к комплексам металлов с пентадентантными (N3O2) лигандами общей формулы,где R1-R7могут быть одинаковыми или различными и представляют собой водород, алкильный заместитель ряда С1-С20или алкоксидный заместитель, X1и Х2являются нуклеофилами или анионами, способствующими раскрытию эпоксидной связи, Mt представляет металл, выбранный из группы, включающей CrIII, CoIII, ZnII, VII, при условии, что R1-R7не могут одновременно представлять водород. Также предложены способ получения указанных выше комплексов, каталитические системы для проведения реакций диоксида углерода с эпоксидами и способ получения циклических карбонатов или алифатических поликарбонатов. Технический результат изобретения заключается в получении нового типа каталитических комплексов, обеспечивающих более эффективную координацию металлоцентра в каталитически активном комплексе и возможность гибкого регулирования скорости и селективности процесса за счет модификации строения лиганда или природы металла. Предложенные комплексы могут применяться для получения циклических карбонатов и алифатических поликарбонатов из диоксида углерода и эпоксидов линейного или циклического строения. 4 н. и 3 з.п. ф-лы, 6 ил., 1 табл., 15 пр.
Формула

Документы, цитированные в отчёте о поиске
Способ каталитического связывания двуокиси углерода
Комментарии