Способ взаимодействия и промежуточные продукты, пригодные для получения цефалоспоринов - RU2237670C1
Код документа: RU2237670C1
Описание
Данное изобретение относится к новому способу получения цефалоспоринов, 3-замещенных циклическим простым эфиром. Изобретение относится также к новым способам получения цвиттерионов, пара-нитробензиловых сложных эфиров и аллиловых сложных эфиров, пригодных при получении вышеуказанных цефалоспоринов. Изобретение относится также к цефалоспоринам, 3-замещенным циклическим простым эфиром. Эти соединения обладают некоторыми благоприятными свойствами, такими как кристаллическая форма и высокий энантиомерный избыток (е.е.).
3-Замещенные циклическим простым эфиром цефалоспорины, полученные способами настоящего изобретения, имеют пролонгированную антибактериальную активность и высокие уровни антибактериальной активности и обладают хорошей парентеральной абсорбцией у людей и животных. 3-Замещенные циклическим простым эфиром цефалоспорины, полученные способами настоящего изобретения, содержат в качестве заместителя циклический простой эфир у углерода 3 кольца цефалоспорина.
В патенте Великобритании 1405758 описаны альтернативные способы получения некоторых цефалоспоринов, 3-замещенных циклическим простым эфиром.
В J. Antibiotics (1994), vol. 47(2); page 253, и WO 92/01696 описаны также альтернативные способы получения соединений формулы I, как указано здесь ниже, и соединения, пригодные в указанных способах.
В патентах Соединенных Штатов № 6020329 и 6077952 описаны соли, полиморфы, сольваты и гидраты цефалоспоринов, 3-замещенных циклическим простым эфиром.
В патенте Соединенных Штатов № 6001997 описаны альтернативные способы получений цефалоспоринов, 3-замещенных циклическим простым эфиром.
Предварительная заявка на патент Соединенных Штатов, озаглавленная “Process and Ester Derivatives Useful For Preparation of Cephalosporins”, зарегистрированная 30 ноября 2000 г., относится к промежуточным продуктам и способам получения цефалоспоринов, 3-замещенных циклическим простым эфиром.
Каждое из вышеуказанных публикаций, пациентов и заявок на патенты, таким образом, включено в качестве ссылки во всей его полноте.
Авторы настоящего изобретения описали новое соединение формулы I, как определено здесь ниже. Авторы настоящего изобретения описали также способ получения указанных соединений формулы I с высоким выходом.
Настоящее изобретение относится к способу получения 3-замещенного циклическим простым эфиром цефалоспорина формулы I

или его фармацевтически приемлемых солей, где
группа СО2R1 представляет карбоновую кислоту или карбоксилатную соль и
R2 имеет формулу

где A1 представляет С6-10-арил, С1-10-гетероарил или С1-10-гетероциклил;
А2 представляет водород, С1-6-алкил, С3-10-циклоалкил, С6-10-арил, С1-6-алкил(СО)(С1-6 )алкил-О-, НО(СО)(С1-6)алкил, моно-(С6-10-арил)(С1-6-алкил), ди-(С6-10-арил)(С1-6-алкил) или три-(С6-10-арил)(С1-6-алкил);
который включает взаимодействие соединения формулы II

с соединением формулы III
R2L (III)
где R2 имеет указанные выше значения и L представляет уходящую группу, в присутствии растворителя и основания. Вышеуказанный способ, необязательно, можно проводить в присутствии конденсирующего агента и катализатора.
Группа ОА2 указанных соединений формулы III, предпочтительно, находится в цис-положении к амидной связи, т.е. предпочтительной является Z-конфигурация.
Подходящие растворители для вышеуказанного способа превращения соединений формулы II в соединения формулы I изобретения включают воду, ацетон, тетрагидрофуран, этилацетат, диметилацетамид, диметилформамид, ацетонитрил, метиленхлорид, 1,2-дихлорэтан или их смеси. В одном варианте осуществления изобретения растворителем является тетрагидрофуран. В другом варианте осуществления изобретения растворителем является этилацетат. Растворителем, предпочтительно, является вода, ацетон или их смеси. Более предпочтительно, растворителем является смесь ацетона и воды. Наиболее предпочтительно, растворителем является смесь 1,3:1 ацетона и воды.
Подходящие основания для вышеуказанного превращения по изобретению включают диизопропилэтиламин или гидроксид натрия. Предпочтительно, основанием является гидроксид натрия, наиболее предпочтительно, 15% водный гидроксид натрия.
Подходящие конденсирующие агенты для вышеуказанного превращения по изобретению включают N,N’-диэтилкарбодиимид, N,N’-дипропилкарбодиимид, N,N’-диизопропилкарбодиимид, N, N’-дициклогексилкарбодиимид, N-этил-N’-[3-(диметиламино)пропил]карбодиимид, N,N’-карбонилдиимидазол или N,N’-карбонилдитиазол. Предпочтительным конденсирующим агентом является N, N’-дициклогексилкарбодиимид. Вышеуказанное превращение, предпочтительно, проводят без использования любого связующего агента.
Подходящие катализаторы для вышеуказанного превращения по изобретению включают кислоты Льюиса. Подходящие кислоты Льюиса выбраны из группы, состоящей из тригалогенида бора, такого как трибромид бора, и галогенида алюминия, такого как хлорид алюминия. Вышеуказанное превращение, предпочтительно, проводят без использования каких-либо катализаторов.
Вышеуказанное превращение по изобретению можно проводить при температуре приблизительно от -40°С до приблизительно +30°С, предпочтительно, приблизительно от +20°С до приблизительно +30°С. Вышеуказанный способ можно проводить в течение периода приблизительно от 1 часа до приблизительно 24 часов; предпочтительно, приблизительно 3 часов.
Подходящие уходящие группы L вышеуказанного соединения формулы III вышеуказанного превращения включают гидрокси, галоген, азидо, моно-(С1-6-алкил)карбонат, (С1-6-алкил)карбоксилат, (С6-10-арил)карбоксилат, моно-(С6-10-арил)(С1-6-алкил)карбоксилат, ди-(С6-10 -арил)(С1-6-алкил)карбоксилат, ди-(С1-6-алкил)фосфоротиоат, (С1-6-алкил)сульфонил, моно-(С-1-6-алкил)(С6-10-арил)сульфонил, ди-(С1-6 -алкил)(С6-10-арил)сульфонил, (С1-6-алкил)-(СО)-S-, циано-С1-6-алкокси, С6-10-арилокси, 3-бензотиазолилокси, 8-хинолинилокси- или N-оксисукцинимидил.
В одном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из гидрокси, галогена и азидо.
В другом варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из моно(С1-6-алкил)карбоната, (С1-6 -алкил)карбоксилата, (С6-10-арил)карбоксилата, моно-(С6-10-арил)(С1-6-алкил)карбоксилата, ди-(С6-10-арил)(С1-6-алкил)карбоксилата и ди-(С1-6-алкил)фосфоротиоата.
Еще в одном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из (С1-6-алкил)сульфонила, моно-(С1-6-алкил)(С6-10-арил)сульфонила, ди-(С1-6-алкил)(С6-10-арил)сульфонила и (С1-6-алкил)-(СО)-S-.
Еще в одном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из циано-С1-6-алкокси, С6-10-арилокси, 3-бензотиазолилокси, 8-хинолинилокси- и N-оксисукцинимидила.
Еще в одном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из галогена, метансульфонила, диэтилфосфоротиоата и 3-бензтиазолилокси.
В предпочтительном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III представляет ди(С1-6-алкил)фосфоротиоат, более предпочтительно, диэтилфосфоротиоат.
Настоящее изобретение относится также к альтернативному способу получения вышеуказанного 3-замещенного циклическим простым эфиром цефалоспорина формулы I или его фармацевтически приемлемых солей, включающему взаимодействие соединения формулы V

где R2 имеет формулу

где А1 представляет С6-10-арил, С1-10-гетероарил или C1-10-гетероциклил;
А2 представляет водород, С1-6-алкил, С3-10-циклоалкил, С6-10-арил, С1-6-алкил(СО)(С1-6 )алкил-О-, НО(СО)(С1-6)алкил, моно-(С6-10-арил)(С1-6-алкил), ди-(С6-10-арил)(С1-6-алкил) или три-(С6-10-арил)(С1-6-алкил) и
R3 представляет пара-нитробензил или аллил, предпочтительно, аллил, с подходящим снимающим защиту агентом в присутствии растворителя.
Термин “алкил”, используемый здесь, если не оговорено особо, включает насыщенные одновалентные углеводородные радикалы, имеющие линейные разветвленные части или их комбинации. Алкильные группы, где бы они не находились, могут быть, необязательно, замещены подходящим заместителем.
Термин “циклоалкил”, используемый здесь, если не оговорено особо, включает моно- или бициклическое карбоциклическое кольцо (например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклопентенил, циклогексенил, бицикло[2.2.1]гептанил, бицикло[3.2.1]октанил и бицикло[5.2.0]нонанил и т.д.), необязательно, содержащее 1 или 2 двойные связи и, необязательно, замещенное 1-3 подходящими заместителями, как указано ниже, такими как фтор, хлор, трифторметил, (С1-4)алкокси, (С6-10)-арилокси, трифторметокси, дифторметокси или (С1-4)алкил, более предпочтительно, фтор, хлор, метил, этил и метокси.
Термин “алкокси”, используемый здесь, включает группы О-алкил, где “алкил” имеет указанные выше значения.
Термин “галоген”, используемый здесь, если не оговорено особо, включает фтор, хлор, бром или иод, предпочтительно, бром или хлор.
Термин “арил”, используемый здесь, если не оговорено особо, включает органический радикал, образованный из ароматического углеводорода удалением одного или нескольких водородов, такой как фенил или нафтил, необязательно замещенный 1-3 подходящими заместителями, такими как фтор, хлор, циано, нитро, трифторметил, (С1-6)алкокси, (С6-10)арилокси, (С3-8)циклоалкилокси, трифторметокси, дифторметокси или (С1-6)алкил.
Термин “гетероарил”, используемый здесь, если не оговорено особо, включает органический радикал, образованный из ароматического гетероциклического соединения удалением одного или нескольких водородов, такой как бензимидазолил, бензофуранил, бензофуразанил, 2Н-1-бензопиранил, бензотиадиазин, бензотиазинил, бензотиазолил, бензотиофенил, бензоксазолил, хроманил, циннолинил, фуразанил, фуропиридинил, фурил, имидазолил, индазолил, индолинил, индолизинил, индолил, 3Н-индолил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, нафтиридинил, оксадиазолил, оксазолил, фталазинил, птеридинил, пуринил, пиразинил, пиридазинил, пиридинил, пиримидинил, пиразолил, пирролил, хиназолинил, хинолинил, хиноксалинил, тетразолил, тиазолил, тиадиазолил, тиенил, триазинил и триазолил, где указанный (С1-10)гетероарил необязательно замещен у любого из атомов углерода кольца, способных образовывать дополнительную связь, одним или двумя заместителями, независимо выбранными из F, Cl, Br, CN, OH, (С1-4)алкила, (С1-4 )перфторалкила, (С1-4)перфторалкокси, (С1-4)алкокси и (С3-6)циклоалкилокси. Вышеуказанные группы, как образованные из соединений, перечисленных выше, могут быть C-присоединенными или N-присоединенными, где такое возможно. Например, группа, образованная из пиррола, может быть пиррол-1-илом (N-присоединенная) или пиррол-3-илом (С-присоединенная).
Термин “гетероциклил”, используемый здесь, если не оговорено особо, включает органический радикал, образованный из неароматического гетероциклического соединения удалением одного или нескольких водородов, такой как 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азетидинил, дигидрофуранил, дигидропиранил, дигидротиенил, диоксанил, 1,3-диоксоланил, 1,4-дитианил, гексагидроазепинил, гексагидропиримидин, имидазолидинил, имидазолинил, изоксазолидинил, морфолинил, оксазолидинил, пиперазинил, пиперидинил, 2Н-пиранил, 4Н-пиранил, пиразолидинил, пиразолинил, пирролидинил, 2-пирролинил, 3-пирролинил, хинолизинил, тетрагидрофуранил, тетрагидропиранил, 1,2,3,6-тетрагидропиридинил, тетрагидротиенил, тетрагидротиопиранил, тиоморфолинил, тиоксантил и тритианил. Вышеуказанные группы, как образованные из соединений, перечисленных выше, могут быть C-присоединенными или N-присоединенными, где такое возможно. Например, группа, образованная из пиперидина, может быть пиперидин-1-илом (N-присоединенная) или пипиридин-4-илом (С-присоединенная). Указанные выше группы, как образованные из соединений, перечисленных выше, могут быть, необязательно, замещены, где такое возможно, подходящим заместителем, таким как оксо, F, Cl, Br, CN, OH, (С1-4)алкил, (С1-4)перфторалкил, (С1-4)перфторалкокси, (С1-4)алкокси или (С3-6 )циклоалкилокси.
Фраза “подходящий заместитель” предназначена для обозначения химически и фармацевтически приемлемой функциональной группы, т.е. части, которая не сводит на нет ингибирующую активность соединений изобретения. Такие подходящие заместители могут быть стандартным образом выбраны специалистом в данной области. Иллюстративные примеры подходящих заместителей включают, но не ограничиваются перечисленным, галогенные группы, перфторалкильные группы, перфторалкоксигруппы, алкильные группы, гидроксигруппы, оксогруппы, меркаптогруппы, алкилтиогруппы, алкоксигруппы, арильные или гетероарильные группы, арилокси- или гетероарилоксигруппы, аралкильные или гетероаралкильные группы, аралкокси- или гетероаралкоксигруппы, карбоксигруппы, аминогруппы, алкил- и диалкиламиногруппы, карбамоильные группы, алкилкарбонильные группы, алкоксикарбонильные группы, алкиламинокарбонильные группы, диалкиламинокарбонильные группы, арилкарбонильные группы, арилоксикарбонильные группы, алкилсульфонильные группы, арилсульфонильные группы и тому подобное.
Термин “карбоксилатная соль”, используемый здесь, включает соли металлов (таких как алюминия, соли щелочных металлов, таких как натрия или калия, предпочтительно, натрия), соли щелочноземельных металлов (таких как кальция или магния) и соли аммония. Соли аммония могут быть замещены С1-6-алкиламинами (такими как триэтиламин), гидрокси-(С1-6)алкиламинами (такими как 2-гидроксиэтиламин, бис-(2-гидроксиэтил)амин или трис-(2-гидроксиэтил)амин), циклоалкиламинами (такими как дициклогексиламин), прокаином, дибензиламином, N,N-дибензилэтилендиамином, 1-эфенамином, N-метилморфолином, N-этилпиперидином, N-бензил-β-фенетиламином, дегидроабиетиламином, N, N’-бисдегидроабиетиламином, этилендиамином или основаниями типа пиридина (такими как пиридин, коллидин или хинолин) или другими аминами, которые используют для образования солей с известными пенициллинами и 3-замещенными циклическим простым эфиром цефалоспоринами. Другие пригодные соли включают соль лития и соль серебра. Соли соединений формулы I можно получить солеобменом общепринятым способом.
Термин “активное соединение”, используемый здесь, относится к соединениям формулы I.
Соединения формулы I содержат хиральные центры и, следовательно, существуют в различных энантиомерных формах. Данное изобретение относится ко всем оптическим изомерам, энантиомерам, диастереомерам и стереоизомерам соединений формулы I и их смесям. Соединения изобретения могут существовать также в различных таутомерных формах. Данное изобретение относится ко всем таутомерам формулы I. Специалисты в данной области хорошо знают, что ядро цефалоспорина существует в растворе в виде смеси таутомеров. Различные отношения таутомеров в твердой и жидкой форме зависят от различных заместителей на молекуле, а также конкретного способа кристаллизации, используемого для выделения соединения.
Группа ОА2 указанных соединений формулы III, предпочтительно, находится в цис-положении к амидной связи, т.е. предпочтительной является Z-конфигурация.
Подходящие агенты снятия защиты для вышеуказанного способа превращения соединений формулы V в соединения формулы I изобретения включают дитионит натрия или тетракистрифенилфосфинпалладий(0).
Подходящие растворители для вышеуказанного превращения включают ацетон, воду, тетрагидрофуран, метиленхлорид или их смеси. В одном варианте осуществления изобретения растворителем является метиленхлорид, тетрагидрофуран или их смеси. В другом варианте осуществления изобретения растворителем является тетрагидрофуран. В предпочтительном варианте осуществления вышеуказанного превращения по изобретению растворителем является метиленхлорид.
Вышеуказанное превращение можно проводить при температуре приблизительно от 0°С до приблизительно 45°С. Вышеуказанное превращение можно проводить в течение периода приблизительно от 1 часа до приблизительно 24 часов.
В одном варианте осуществления вышеуказанного превращения R3 представляет пара-нитробензил. В пределах данного варианта осуществления вышеуказанное превращение проводят подходящим образом при температуре приблизительно 40°С. В пределах данного варианта осуществления вышеуказанный способ проводят подходящим образом в течение приблизительно 4 часов.
В предпочтительном варианте осуществления вышеуказанного превращения R3 представляет аллил. В данном варианте осуществления предпочтительным агентом снятия защиты является тетракистрифенилфосфинпалладий(0). В данном варианте осуществления вышеуказанный способ проводят при температуре приблизительно от 20°С до приблизительно 35°С, предпочтительно, приблизительно от 27°С до приблизительно 30°С. В данном варианте осуществления вышеуказанный способ проводят, предпочтительно, в течение приблизительно 5 часов.
Настоящее изобретение включает также способ получения вышеуказанного соединения формулы II, включающий взаимодействие соединения формулы IV

где R3 представляет пара-нитробензил или аллил, предпочтительно, пара-нитробензил, и Х представляет галоген, предпочтительно, хлор, с подходящим удаляющим защитную группу агентом в присутствии растворителя.
Подходящие растворители для способа превращения соединений формулы IV в соединения формулы II изобретения включают ацетон, воду, тетрагидрофуран, метиленхлорид или их смеси. В одном варианте осуществления изобретения растворителем является ацетон, вода, тетрагидрофуран или их смеси. Растворителем, предпочтительно, является смесь ацетона и воды. Более предпочтительно, растворителем является смесь 3:1 ацетона и воды.
Подходящие снимающие защиту агенты для вышеуказанного превращения включают дитионит натрия, каталитический гидрирующий агент (такой как газообразный водород над 10% палладием на угле) или тетракистрифенилфосфинпалладий(0).
Вышеуказанное превращение можно проводить при температуре приблизительно от 0°С до приблизительно 45°С. Вышеуказанное превращение можно проводить в течение периода приблизительно от 1 часа до приблизительно 24 часов.
В предпочтительном варианте осуществления вышеуказанного превращения R3 представляет пара-нитробензил. В данном варианте осуществления предпочтительным агентом снятия защиты является дитионит натрия. Вышеуказанный способ, предпочтительно, проводят при температуре приблизительно 45°С. Вышеуказанный способ, предпочтительно, проводят в течение приблизительно 1 часа.
В другом варианте осуществления изобретения R3 представляет аллил. В данном варианте осуществления подходящим снимающим защиту агентом является тетракистрифенилфосфинпалладий(0). Подходящие растворители включают метиленхлорид и тетрагидрофуран. Вышеуказанный способ можно проводить при температуре приблизительно от 20°С до приблизительно 35°С.
Настоящее изобретение относится также к способу получения вышеуказанного соединения формулы V, включающему взаимодействие вышеуказанного соединения формулы IV, где R3 представляет пара-нитробензил или аллил, предпочтительно, аллил, и Х представляет галоген, предпочтительно, хлор, с соединением формулы III, как определено выше, в присутствии растворителя. Вышеуказанный способ, необязательно, можно проводить в присутствии необязательного конденсирующего агента или необязательного катализатора.
Подходящие растворители для вышеуказанного превращения соединений формулы IV в соединения формулы V включают метиленхлорид, тетрагидрофуран или их смеси.
В одном варианте осуществления вышеуказанного превращения по изобретению используют конденсирующий агент. В данном варианте осуществления подходящие конденсирующие агенты включают N,N’-диэтилкарбодиимид, N,N’-дипропилкарбодиимид, N, N’-диизопропилкарбодиимид, N,N’-дициклогексилкарбодиимид, N-этил-N’-[3-(диметиламино)пропил]карбодиимид, N,N’-карбонилдиимидазол или N,N’-карбонилдитиазол. Предпочтительным конденсирующим агентом является N,N’-дициклогексилкарбодиимид. Вышеуказанное превращение, предпочтительно, проводят в отсутствие любых конденсирующих агентов.
В другом предпочтительном варианте осуществления вышеуказанного превращения по изобретению используют катализатор. В данном варианте осуществления катализатором может быть кислота Льюиса. Подходящими кислотами Льюиса являются тригалогенид бора, такой как трибромид бора, или галогенид алюминия, такой как хлорид алюминия. Вышеуказанное превращение, предпочтительно, проводят в отсутствие любых катализаторов.
Вышеуказанное превращение можно проводить при температуре приблизительно от -40°С до приблизительно +40°С. Вышеуказанное превращение можно проводить в течение периода приблизительно от 1 часа до приблизительно 24 часов.
В одном варианте осуществления вышеуказанного превращения по изобретению R3 представляет пара-нитробензил. В указанном варианте осуществления вышеуказанное превращение подходящим образом проводят при температуре приблизительно от +20°С до приблизительно +30°С. В указанном варианте осуществления вышеуказанное превращение подходящим образом проводят в течение приблизительно 3 часов.
В предпочтительном варианте осуществления вышеуказанного превращения по изобретению R3 представляет аллил. В данном варианте осуществления растворителем, предпочтительно, является метиленхлорид. В предпочтительном варианте осуществления вышеуказанное превращение, предпочтительно, проводят при температуре приблизительно от 20°С до приблизительно 40°С. В предпочтительном варианте осуществления вышеуказанное превращение, предпочтительно, проводят в течение приблизительно 24 часов.
Подходящие уходящие группы L соединения формулы III в вышеуказанном превращении изобретения включают гидрокси, галоген, азидо, моно-(С1-6-алкил)карбонат, (С1-6-алкил)карбоксилат, (С6-10 -арил)карбоксилат, моно-(С6-10-арил)(С1-6-алкил)карбоксилат, ди-(С6-10-арил)(С1-6-алкил)карбоксилат, ди-(С1-6-алкил)фосфоротиоат, (С1-6 -алкил)сульфонил, моно-(С1-6-алкил)(С6-10-арил)сульфонил, ди-(С1-6-алкил)(С6-10-арил)сульфонил, (С1-6-алкил)-(СО)-S-, циано-С1-6 -алкокси, С6-10-арилокси, 3-бензтиазолилокси, 8-хинолинилокси- или N-оксисукцинимидил. В одном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из гидрокси, галогена и азидо.
В другом варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из моно-(С1-6-алкил)карбоната, (С1-6-алкил)карбоксилата, (С6-10-арил)карбоксилата, моно-(С6-10-арил)(С1-6 -алкил)карбоксилата, ди-(С6-10-арил)(С1-6-алкил)карбоксилата и ди-(С1-6-алкил)фосфоротиоата.
Еще в одном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из (С1-6-алкил)сульфонила, моно-(С1-6-алкил)(С6-10-арил)сульфонила, ди-(С1-6-алкил)(С6-10-арил)сульфонила и (С1-6-алкил)-(СО)-S-.
Еще в одном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из циано-С1-6-алкокси, С6-10-арилокси, 3-бензтиазолилокси, 8-хинолинилокси- и N-оксисукцинимидила.
Еще в одном варианте осуществления вышеуказанного превращения по изобретению уходящая группа L соединения формулы III выбрана из группы, состоящей из галогена, метансульфонила, диэтилфосфоротиоата и 3-бензтиазолилокси.
В предпочтительном варианте осуществления вышеуказанного превращения по изобретению уходящей группой L соединения формулы III является моно(С1-6 -алкил)карбонат, более предпочтительно, ацетат. Настоящее изобретение относится также к соединению формулы II

В одном варианте осуществления изобретения соединение формулы II имеет энантиомерную или диастереомерную чистоту от 96% до 100%, предпочтительно, 97%.
Настоящее изобретение относится также к соединению формулы V

где R2 имеет указанные выше значения и R3 представляет пара-нитробензил или аллил, предпочтительно, аллил.
В одном варианте осуществления изобретения соединение формулы V имеет энантиомерную или диастереомерную чистоту от 96% до 100%, предпочтительно 97%.
В видовых (общих) или подвидовых вариантах осуществления каждого из вышеуказанных вариантов осуществления часть А1 указанного R2 представляет С6-10-арил, такой как фенил. В других видовых или подвидовых вариантах осуществления изобретения часть А1 указанного R2 представляет С1-10-гетероарил, выбранный из группы, состоящей из фурила, тиенила, пиридила, аминотиазолила и аминотиадиазолила, где аминогруппа указанного аминотиазолила или аминотиадиазолила, необязательно, защищена. В других видовых или подвидовых вариантах осуществления изобретения часть А1 указанного R2 представляет С1-10-гетероциклил, такой как 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азетидинил, дигидрофуранил, дигидропиранил, дигидротиенил, диоксанил, 1,3-диоксоланил, 1,4-дитианил, гексагидроазепинил, гексагидропиримидин, имидазолидинил, имидазолинил, изоксазолидинил, морфолинил, оксазолидинил, пиперазинил, пиперидинил, 2Н-пиранил, 4Н-пиранил, пиразолидинил, пиразолинил, пирролидинил, 2-пирролинил, 3-пирролинил, хинолизинил, тетрагидрофуранил, тетрагидропиранил, 1,2,3,6-тетрагидропиридинил, тетрагидротиенил, тетрагидротиопиранил, тиоморфолинил, тиоксанил и тритианил. Часть А1 указанного R2 предпочтительно представляет аминотиазолил.
В других видовых или подвидовых вариантах осуществления изобретения часть А2 указанного R2 представляет водород или С1-6-алкил. Предпочтительный вариант осуществления изобретения включает каждый из вышеуказанных видовых или подвидовых вариантов осуществления, у которых часть А2 указанного R2 представляет С1-6-алкил, более предпочтительно, метил.
В предпочтительном варианте осуществления каждого из видовых или подвидовых вариантов осуществления изобретения соединение формулы III имеет формулу IIIa

где L представляет уходящую группу, такую как галоген, метансульфонил, диалкилфосфоротиоат, такой как диэтилфосфоротиоат или 3-бензтиазолилокси.
В наиболее предпочтительном варианте осуществления каждого из вышеуказанных вариантов осуществления изобретения соединение формулы III имеют формулу IIIa, как указано выше, где L представляет диэтилфосфоротиоат или ацетат.
Необязательное превращение R2 в другой R2 и необязательное образование фармацевтически приемлемой соли можно проводить с использованием способов, хорошо известных в данной области.
В способах, описанных здесь выше и ниже, может быть необходимо удалить защитные группы. Удаление защитных групп можно проводить любым пригодным способом, известным в данной области, так чтобы минимизировать нежелательные побочные реакции. Отделение нежелательных побочных продуктов можно проводить с использованием стандартных способов, известных специалисту в данной области (см., например, “Protection of the Amino Group”, in Protective Groups in Organic Synthesis, 2nd Edition, T.W.Greene and P.G.M.Wuts, Ed., Wiley and Sons, Inc. 1991, pp. 309-405).
Настоящее изобретение относится также к способу использования цвиттерионного промежуточного продукта для получения 3-замещенных циклическим простым эфиром цефалоспоринов.
Способ настоящего изобретения и получение соединения настоящего изобретения иллюстрируется следующими реакционными схемами. За исключением случаев, в которых указывается иначе, в схемах реакций и обсуждении, которые следуют ниже, заместители R1, R2, R3, L, A1, A2 и Х имеют указанные выше значения, если не описано особо.
СХЕМА 1



Схема 1 относится к получению соединений формулы I. Cоединение формулы I согласно схеме 1 можно получить взаимодействием соединения формулы II с соединение формулы III
R2-L (III),
где L представляет уходящую группу, в присутствии основания и растворителя.
Подходящие уходящие группы включают гидрокси, галоген, азидо, моно(С1-6-алкил)карбонат, (С1-6-алкил)карбоксилат, (С6-10-арил)карбоксилат, моно-(С6-10-арил)(С1-6-алкил)карбоксилат, ди-(С6-10-арил)(С1-6-алкил)карбоксилат, ди-(С1-6-алкил)фосфоротиоат, (С1-6-алкил)сульфонил, моно-(С1-6-алкил)(С6-10-арил)сульфонил, ди-(С1-6-алкил)(С6-10-арил)сульфонил, (С1-6-алкил)-(СО)-S-, циано-С1-6-алкокси, С6-10-арилокси, 3-бензтиазолилокси, 8-хинолинилокси- или N-оксисукцинимидил. Уходящая группа, предпочтительно, представляет ди-(С1-6-алкил)фосфоротиоат, такой как диэтилфосфоротиоат.
Подходящие основания включают диизопропилэтиламин или гидроксид натрия, предпочтительно, гидроксид натрия, наиболее предпочтительно, 15% водный гидроксид натрия.
Подходящие растворители включают воду, ацетон, тетрагидрофуран, этилацетат, диметилацетамид, диметилформамид, ацетонитрил, метиленхлорид, 1,2-дихлорэтан или их смеси, предпочтительно, смесь воды и ацетона, наиболее предпочтительно, смесь 1:1,3 воды и ацетона.
Вышеуказанное взаимодействие можно проводить при температуре приблизительно от -40°С до приблизительно 30°С, предпочтительно, приблизительно от 20°С до приблизительно 30°С. Вышеуказанную реакцию можно проводить в течение периода приблизительно от 1 часа до приблизительно 24 часов, предпочтительно, в течение приблизительно 3 часов.
Вышеуказанное взаимодействие, необязательно, можно проводить в присутствии связывающего кислоту агента, например, третичного амина (такого как триэтиламин), пиридина (такого как 2,6-лутидин или 4-диметиламинопиридин) или диметиланилина. Вышеуказанное взаимодействие, необязательно, можно также проводить в присутствии молекулярных сит, неорганического основания (такого как карбонат кальция или бикарбонат натрия) или оксирана, который связывает газообразный водород, выделяемый при вышеуказанном взаимодействии. Оксираном, предпочтительно, является оксид С1-6-алкил-1,2-алкилена, такой как оксид этилена или оксид пропилена.
Вышеуказанное взаимодействие, необязательно, можно проводить в присутствии конденсирующего агента. Подходящие конденсирующие агенты включают N,N’-диэтилкарбодиимид, N,N’-дипропилкарбодиимид, N,N’-диизопропилкарбодиимид, N, N’-дициклогексилкарбодиимид, N-этил-N’-[3-(диметиламино)пропил]-карбодиимид, N,N’-карбонилдиимидазол и N,N’-карбонилдитиазол. Предпочтительно, конденсирующим агентом является N,N’-диэтилкарбодиимид. Вышеуказанное превращение, предпочтительно, проводят в отсутствие любых конденсирующих агентов.
Вышеуказанное взаимодействие, необязательно, можно проводить в присутствии катализатора. Подходящие катализаторы включают кислоту Льюиса, такую как тригалогенид бора или галогенид алюминия. Взаимодействие, предпочтительно, проводят в отсутствие любых катализаторов.
Соединение формулы III можно получить способами, известными в данной области. Подходящие способы включают способы, описанные, например, в патенте Великобритании № 2107307, описании изобретения к патенту Великобритании № 1536281 и описании изобретения к патенту Великобритании № 1508064. Соединение формулы III (т.е. R2L), где R2 имеет формулу:

где А1 представляет 2-аминотиазол-4-ил, А2 представляет метил и L представляет (С1-6-алкил)сульфонил, такой как метилсульфонил, или ди(С1-6-алкил)фосфоротиоат, такой как диэтилфосфоротиоат, можно получить, предпочтительно, взаимодействием соединения формулы IIIb

с (С1-6-алкил)сульфонилгалогенидом, таким как метансульфонилхлорид, или ди-(С1-6-алкил)тиофосфоновой кислотой, такой как диэтилтиофосфоновая кислота.
Наиболее предпочтительно, соединением формулы III является диэтилтиофосфорил-[Z]-2-аминотиазол-4-илметоксиамин (DAMA), который можно получить по способам, описанным в патенте США № 5567813 и Европейском патенте 628561. Схема 2 относится к получению соединения формулы II. Соединение формулы II согласно схеме 2 можно получить взаимодействием соединения формулы IV, где R3, предпочтительно, представляет пара-нитробензиловый сложный эфир и Х, предпочтительно, представляет хлор, с подходящим агентом удаления защитной группы в растворителе.
Подходящие агенты удаления защитной группы включают дитионит натрия или каталитический гидрирующий агент, такой как газообразный водород над 10% палладия на угле.
Подходящие растворители включают ацетон, воду, тетрагидрофуран, метиленхлорид или их смеси. Растворителем, предпочтительно, является смесь 3:1 ацетона и воды.
Вышеуказанное взаимодействие можно проводить при температуре приблизительно от 0°С до приблизительно 45°С, предпочтительно, приблизительно при 45°С. Вышеуказанное взаимодействие можно проводить в течение периода приблизительно от 1 часа до приблизительно 24 часов, предпочтительно, приблизительно в течение 1 часа.
Соединение формулы IV можно получить различными синтетическими способами, таким как способы, описанные в предварительной заявке на патент Соединенных Штатов, озаглавленной “Process and Ester Derivatives Useful For Preparation of Cefalosporins”, поданной 30 ноября 2000 г.
Схема 3 относится к альтернативному способу получения соединения формулы I. Что касается схемы 3, соединение формулы I можно получить взаимодействием соединения формулы V, где R3 представляет, предпочтительно, аллил, с подходящим агентом удаления защитной группы в растворителе.
Подходящие агенты удаления защитной группы включают дитионит натрия или тетракистрифенилфосфинпалладий(0).
Подходящие растворители включают ацетон, воду, тетрагидрофуран, метиленхлорид или их смеси. Растворителем, предпочтительно, является метиленхлорид.
Вышеуказанное взаимодействие проводят при температуре приблизительно от 0°С до приблизительно 45°С. Вышеуказанное взаимодействие можно проводить в течение периода приблизительно от 1 часа до приблизительно 24 часов.
Соединение формулы V можно получить взаимодействием соединения формулы IV, где R3 представляет, предпочтительно, аллил и Х представляет, предпочтительно, хлор, с соединением формулы III
R2-L (III),
в растворителе.
Подходящие растворители для вышеуказанного взаимодействия включают метиленхлорид, тетрагидрофуран или их смеси. Растворителем, предпочтительно, является метиленхлорид.
Вышеуказанное взаимодействие можно проводить, необязательно, в присутствии конденсирующего агента. Подходящие конденсирующие агенты включают N,N’-диэтилкарбодиимид, N, N’-дипропилкарбодиимид, N,N’-диизопропилкарбодиимид, N,N’-дициклогексилкарбодиимид, N-этил-N’-[3-(диметиламино)пропил]карбодиимид, N,N’-карбонилдиимидазол или N,N’-карбонилдитиазол. Предпочтительно, конденсирующим агентом является N,N’-диэтилкарбодиимид. Вышеуказанное превращение, предпочтительно, проводят без любых конденсирующих агентов.
Вышеуказанное взаимодействие, необязательно, можно проводить в присутствии катализатора. Подходящие катализаторы включают кислоту Льюиса, такую как тригалогенид бора или галогенид алюминия. Вышеуказанное взаимодействие, предпочтительно, проводят без любых катализаторов.
Вышеуказанное взаимодействие можно проводить при температуре приблизительно от -40°С до приблизительно +40°С, предпочтительно, приблизительно от +20°С до приблизительно +40°С. Вышеуказанное взаимодействие можно проводить в течение периода приблизительно от 1 часа до приблизительно 24 часов, предпочтительно, в течение приблизительно 24 часов.
Соединение формулы IV можно получить, как указано выше в описании для схемы 2.
Соединения данного изобретения можно кристаллизовать или перекристаллизовать из растворителей, таких как органические растворители. В таких случаях могут образовываться сольваты. Данное изобретение включает в свой объем стехиометрические сольваты, в том числе гидраты, а также соединения, содержащие изменяемые количества воды, которые можно получить такими способами, как лиофилизация.
Соединения формулы (I) являются пригодными для получения 3-замещенного циклическим простым эфиром цефалоспорина, т.е. активного соединения. Активное соединение обладает активностью против грамположительных и грамотрицательных бактерий. Способы анализа активности и способы получения препаратов активных соединений и введения их описаны в патенте Соединенных Штатов № 6020329, выданном 1 февраля 2000 г.. В вышеуказанном патенте описаны также способы лечения.
Нижеследующие примеры иллюстрируют получение соединений настоящего изобретения. Точки плавления являются некорректированными. Данные ЯМР приводятся в частях на миллион (м.д.) и указываются со ссылкой на запирающий сигнал растворителя образца (дейтериохлороформ, если не указано иначе). Коммерческие реагенты используют без дальнейшей очистки. Комнатная температура или температура окружающей среды относится к температуре от 20°С до 25°С. Для удобства и получения максимальных выходов продуктов все неводные реакции проводят в атмосфере азота. Концентрирование при пониженном давлении означает, что использовали роторный испаритель. ТСХ означает тонкослойную хроматографию. ВЭЖХ означает высокоэффективную жидкостную хроматографию. ГХ означает газовую хроматографию.
Пример 1
7-(2-(2-Аминотиазол-4-ил)-2-метоксиимино-3-(тетрагидрофуран-2-ил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат натрия
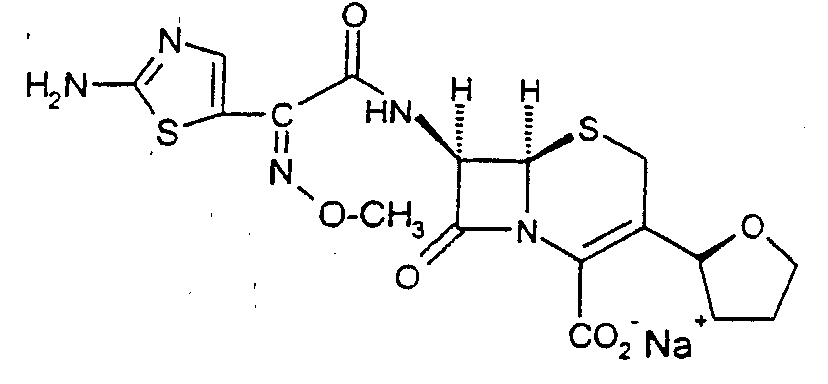
СПОСОБ А: ИЗ 7-АМИНО-8-ОКСО-3-(ТЕТРАГИДРОФУРАН-2-ИЛ)-5-ТИА-1-АЗАБИЦИКЛО[4.2.0]ОКТА-1(6),2,4-ТРИЕН-2-КАРБОНОВОЙ КИСЛОТЫ7-Амино-8-оксо-3-(тетрагидрофуран-2-ил)-5-тиа-1-азабицикло[4.2.0]окта-1(6),2,4-триен-2-карбоновую кислоту (20 г, 75 ммоль), воду (300 мл), ацетон (400 мл) и смесь ангидрида (Z)-2-амино-α -(метоксиимино)-4-тиазолуксусной кислоты и О,О-диэтилгидрофосфоротиоата (27 г, 1,06 эквивалента) смешивают с образованием суспензии. рН суспензии регулируют до величины между 7 и 7,5 с использованием водного гидроксида натрия. После достижения полного растворения реакционную смесь перемешивают в течение 3 часов. Продукт осаждают добавлением ацетона (3200 мл). образовавшуюся суспензию гранулируют, фильтруют и сушат в вакууме, получая при этом указанное в заголовке соединение (29,0 г, 80%).
СПОСОБ В: ИЗ СОЛИ БЕНЗОЛСУЛЬФИНОВОЙ КИСЛОТЫ АЛЛИЛ-7-(2-(2-АМИНОТИАЗОЛ-4-ИЛ)2-МЕТОКСИИМИНО)-3-(ТЕТРАГИДРОФУРАН-2-ИЛ)-8-ОКСО-5-ТИА-1-АЗАБИЦИКЛО[4.2.0]ОКТ-2-ЕН-2-КАРБОКСИЛАТА
В стеклянный сосуд на 10 литров загружают метиленхлорид (4,50 литр), затем тетракис(трифенилфосфин)палладий (9,0 г, 7,8 ммоль) в атмосфере азота. Добавляют трифенилфосфин (1,0 г, 3,8 ммоль) и перемешивают в растворе. Загружают соль бензолсульфиновой кислоты аллил-7-(2-(2-аминотиазол-4-ил)-2-метоксиимино)-3-тетрагидрофуран-2-ил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилата (225,0 г, 354 ммоль) и смесь нагревают до 27-30°С. Мониторинг реакции проводят ВЭЖХ и, если требуется, добавляют дополнительный катализатор. После завершения реакции твердый продукт отфильтровывают и промывают два раза метиленхлоридом (всего 700 мл). Продукт цвета от желтого до рыжевато-коричневого затем сушат на воздухе до достижения постоянной массы перед хранением в холодильнике. Выходы составляют 49-110,1%.
Пример 2
7-Амино-8-оксо-3-(тетрагидрофуран-2-ил)-5-тиа-1-азабицикло-[4.2.0]окта-1(6),2,4-триен-2-карбоновая кислота

4-Нитробензиловый эфир 7-амино-8-оксо-3-(тетрагидрофуран-2-ил)-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты (20 г, 54 ммоль), воду (30 мл) и ацетон (90 мл) смешивают с образованием суспензии. Значение рН суспензии регулируют до 7 с использованием водного раствора аммиака (15%). К образовавшемуся раствору добавляют раствор гидросульфита натрия (32 г, 3,8 эквив.) в воде (40 мл). Значение рН образовавшегося раствора регулируют до 7 с использованием водного аммиака (15%) при поддержании температуры между 40°С и 45°С. После перемешивания в течение 1 часа при 45°С рН устанавливают до 3,5 водным раствором (15%) хлористоводородной кислоты. Образовавшуюся суспензию гранулируют, фильтруют и сушат, получая при этом указанное в заголовке соединение (11,3 г, 80%).
Получение 1: 4-нитробензиловый эфир (3-бензил-7-оксо-4-тиа-2,6-диазабицикло[3.2.0]гепт-2-ен-6-ил)гидроксиуксусной кислоты
Изопропанол (500 мл), метиленхлорид (1800 мл) и (1R)-(4-нитрофенил)метиловый эфир (α,1-метилэтилиден)-7-оксо-3- (фенилметил)-4-тиа-2,6-диазабицикло[3.2.0]гепт-2-ен-6-уксусной кислоты (250 г) смешивают и реакционную смесь охлаждают до -70°C. Через охлажденную реакционную смесь барботируют озон до завершения озонолиза. К образовавшемуся раствору добавляют смесь ледяной уксусной кислоты (625 мл) и изопропанола (750 мл), затем смесь изопропанола (100 мл), воды (100 мл) и борогидрида натрия (22 г). После завершения реакции добавляют метабисульфит натрия в воде с последующим регулированием рН до значения 1,5-2,5 хлористоводородной кислотой (15%). Слои разделяют и органический слой промывают два раза водным хлоридом натрия (1000 мл). Органический слой концентрируют в вакууме и образовавшуюся суспензию гранулируют, фильтруют и осадок на фильтре промывают изопропанолом. Продукт сушат в вакууме.
Получение 2: 4-Нитробензиловый эфир гидрокси-{2-оксо-4-[2-оксо-2-(тетрагидрофуран-2-ил)этилсульфанил]-3-фенилацетиламино-азетидин-1-ил}уксусной кислоты
Бром (51 г) и метанол (270 мл) смешивают с последующим добавлением по каплям 1-(тетрагидро-2-фуранил)этанона (30 г) в растворе метанола (30 мл) при 30°С. Затем добавляют водный раствор тиосульфата натрия с последующим добавлением метиленхлорида (300 мл). Слои разделяют и органический слой промывают два раза водным раствором бикарбоната натрия (300 мл). Образовавшийся органический слой концентрируют с последующим добавлением ацетона (600 мл) и пара-толуолсульфоновой кислоты (6 г) После нагревания для кипячения с обратным холодильником в течение 2 часов реакционную смесь охлаждают и загружают 4-нитробензиловый эфир (3-бензил-7-оксо-4-тиа-2, 6-диазабицикло[3.2.0]гепт-2-ен-6-ил)гидроксиуксусной кислоты (100 г) и дополнительную пара-толуолсульфоновую кислоту (6 г). Образовавшийся раствор перемешивают в течение 2 часов с последующим регулированием рН до значения между 3 и 4 с использованием пиридина. Реакционную смесь концентрируют с последующим добавлением воды (180 мл), метиленхлорида (600 мл) и хлористоводородной кислоты (9 мл, 15%) для регулирования значения рН между 1 и 2. Слои разделяют и метиленхлорид заменяют метанолом (600 мл). Для завершения осаждения добавляют изопропанол (300 мл) и образовавшуюся суспензию гранулируют, фильтруют и осадок фильтрования промывают изопропанолом. Продукт сушат в вакууме.
Получение 3: 4-Нитробензиловый эфир 7-амино-8-оксо-3-(тетрагидрофуран-2-ил)-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновой кислоты
Тионилхлорид (45 мл, 0,615 моль) добавляют по каплям к раствору 4-нитробензилового эфира гидрокси-{2-оксо-4-[2-оксо-2-(тетрагидрофуран-2-ил)этилсульфанил]-3-фенилацетиламиноазетидин-1-ил}уксусной кислоты (202 г, 0,362 моль) и 2,6-лутидина (58 мл, 0,500 моль) в дихлорметане (4 литра) при -20°С. После перемешивания в течение 1 часа раствор промывают два раза насыщенным хлоридом натрия (1 литр) и концентрируют. К концентрированному раствору добавляют триметилфосфин в растворе тетрагидрофурана (110 мл, 3 М, 330 ммоль), раствор перемешивают в течение 1 часа, промывают разбавленным гидрокарбонатом натрия и насыщенным хлоридом натрия. После перемешивания при кипячении с обратным холодильником в течение 16 часов раствор промывают водой и насыщенным хлоридом натрия. Раствор концентрируют и охлаждают до -40°С с последующим добавлением по каплям пентахлорида фосфора (104 г, 0,5 моль). Добавляют α-пиколин (92 мл) в растворе дихлорметана (60 мл) при поддержании температуры между -40°С и 30°С. Смесь перемешивают в течение 1 часа с последующим добавлением изопропанола (660 мл). Реакционную смесь нагревают до 22°С, гранулируют, фильтруют и сушат, получая при этом указанное в заголовке соединение (250 г, 45%).
Пример 3
Соль бензолсульфиновой кислоты аллил-7-(2-(2-аминотиазол-4-ил)-2-метоксиимино)-3-тетрагидрофуран-2-ил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилата

Получение 1: Аллил-7-фенилацетамидо-3-(тетрагидрофуран-2-ил)-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат
В стеклянный сосуд на 100 литров загружают толуол (47 литров) и аллил-2-три-N-метилфосфоранилиден-2-(3-фенилацетамидо-4-(тетрагидрофуран-2-илкарбонилметилтио)азетидинон-1-ил)ацетат (1990 г). Раствор продувают азотом и доводят до кипячения с обратным холодильником. Любую присутствующую воду собирают и раствор кипятят с обратным холодильником в течение 20 часов. После взятия образца для анализа ТСХ/ВЭЖХ раствор охлаждают до температуры окружающей среды. Раствор затем пропускают через силикагель 60 (4,5 кг), причем силикагель далее элюируют дополнительным толуолом (33 литра). Толуол затем выпаривают в вакууме при максимальной температуре 60°С. Затем добавляют этилацетат и выпаривают его в вакууме при максимальной температуре 60°С. К полутвердому маслу добавляют трет-бутилметиловый эфир (2,5 литра) и раствор перемешивают на протяжении ночи. Кристаллический продукт отфильтровывают и промывают дополнительным трет-бутилметиловым эфиром (0,3 литра). Маточные растворы концентрируют и снова подвергают хроматографии на диоксиде кремния (растворяют в 5 литрах толуола, вводят в диоксид кремния, элюируют 15 литрами толуола) и кристаллизуют таким же образом, получая при этом вторую порцию продукта. Продукт выделяют в виде белого кристаллического твердого вещества. Выход составляет от 70% до 80%.
Получение 2: Аллил-2-три-N-метилфосфоранилиден-2-(3-фенилацетамидо-4-(тетрагидрофуран-2-илкарбонилметилтио)-азетидинон-1-ил)ацетат
Раствор аллил-2-гидрокси-2-(3-фенилацетамидо-4-(тетрагидрофуран-2-илкарбонилметилтио)азетидинон-1-ил)ацетата в тетрагидрофуране, который был получен в получении 1 примера 3, далее разбавляют дополнительным тетрагидрофураном (общее количество тетрагидрофурана 12 литров). Раствор снова охлаждают до -20°С в атмосфере азота и добавляют 2,6-лутидин (654,0 г, 6,09 моль) с последующим добавлением по каплям тионилхлорида (724,0 г, 6,09 моль) при максимальной температуре -20°С. После перемешивания в течение тридцати минут раствору дают возможность нагреться до -10°С и отбирают образец для ТСХ. ТСХ показывает, что исходный материал полностью превратился в аллил-2-хлор-3-(3-фенилацетамидо-4-(тетрагидрофуран-2-илкарбонилметилтио)-азетидинон-1-ил)ацетат. Осажденные соединения затем отфильтровывают и промывают далее тетрагидрофураном. Раствор в тетрагидрофуране затем концентрируют в вакууме при максимальной температуре 30°С, снова растворяют в свежем тетрагидрофуране (6 литров) и снова охлаждают до -10°С. После перемешивания на протяжении ночи при температуре окружающей среды отбирают образец раствора на завершение превращения, разбавляют этилацетатом (35 литров) и промывают 5% бикарбонатом натрия (20 литров) и 20% насыщенным хлоридом натрия (20 литров) Этилацетат затем выпаривают в вакууме при максимальной температуре 40°С, получая при этом густое темное масло. Выход составляет от 88% до 90%.
Получение 3: Аллил-2-гидрокси-2-(3-фенилацетамидо-4-(тетрагидрофуран-2-илкарбонилметилтио)азетидинон-1-ил)ацетат
В сосуд на 20 литров добавляют метиленхлорид (10,0 литров), тетрагидрофуран (1,0 литр) и аллил 2-гидрокси-2-(3-бензил-4-тиа-2,6-диазабицикло[3.2.0]гепт-2-ен-7-он)ацетат (2016 г, 6,05 моль). К этому раствору добавляют 45% водный раствор (500,0 г) пара-толуолсульфоновой кислоты. После перемешивания раствора в течение трех часов отбирают образец на завершение превращения ТСХ. Раствор затем переносят в стеклянную делительную воронку на 50 литров и добавляют метиленхлорид (5 литров), затем воду (2 литра). Отделенную органическую фазу затем промывают водой (4 литра). Фазу метиленхлорида затем сушат над сульфатом натрия, получая при этом сухой раствор аллил-2-гидрокси-2-(3-фенилацетамидо-4-меркаптоазетидинон-1-ил)ацетата в метиленхлориде, который затем без задержки используют. К вышеуказанному раствору добавляют 86% раствор 2-бромацетилтетрагидрофурана в метиленхлориде (6,3 моль). Образовавшийся раствор упаривают в вакууме при максимальной температуре 30°С до 50% его объема. При максимальной температуре 10°С добавляют пиридин (503,1 г, 6,36 моль). Раствор перемешивают на протяжении ночи, разбавляют метиленхлоридом (10 литров) и промывают два раза водой (всего 10 литров), затем один раз насыщенным хлоридом натрия (10%, 10 литров). После сушки над сульфатом натрия раствор концентрируют в вакууме при максимальной температуре 40°С до гарантии высушивания. Раствор снова растворяют в тетрагидрофуране (5 литров) для использования в следующей стадии. Если требуется хранение, раствор в тетрагидрофуране сохраняют и сушат перед использованием.
Получение 4: 2-Бромацетилтетрагидрофуран
В стеклянный сосуд на 20 литров добавляют метиленхлорид (10,0 литров), затем ацетилтетрагидрофуран (838,0 г, 7,34 моль). Раствор затем охлаждают до -10°С и добавляют триэтиламин (854,0 г, 8,44 моль). Сосуд продувают азотом и при максимальной температуре -8°С по каплям добавляют триметилсилилтрифторметансульфонат (1713,0 г, 7,71 моль). Добавление обычно завершается через 45 минут. После 15 минут перемешивания отбирают образец для анализа ТСХ и ГХ, которые показывают, что реакция завершилась. К раствору при максимальной температуре -5°С добавляют N-бромсукцинимид (1340 г, 7,53 моль) на протяжении периода приблизительно 45 минут в виде шести порций. После 30 минут перемешивания отбирают образец раствора для анализа ГХ и ТСХ, который показывает, что реакция завершилась. Раствор затем переносят в делительную воронку на 50 литров и осторожно добавляют 5% бикарбонат натрия (5 литров). Раствор перемешивают и разделяют. Нижнюю водную фазу выгружают и фазу метиленхлорида промывают водой, сушат над сульфатом натрия, фильтруют и хранят в холодильнике перед использованием в следующей стадии.
Получение 5: Аллил-2-гидрокси-2-(3-бензил-4-тиа-2,6-диазабицикло[3.2.0]гепт-2-ен-7-он)ацетат
В стеклянный сосуд на 50 литров добавляют метиленхлорид (20,6 литра), затем 3-бензил-4-тиа-2, 6-диазабицикло[3.2.0]гепт-2-ен-7-он (1700 г, 7,79 моль). К данной суспензии добавляют моногидрат аллилглиоксилата (1285 г, 9,74 моль), затем достаточное количество триэтиламина (приблизительно 175 г) для установления рН раствора 7,5-7,9. После перемешивания 1 час отбирают образец для анализа ТСХ/ВЭЖХ. После завершения реакции раствор гасят 0,1 М хлористоводородной кислотой (2,75 литра) до рН 4, 50-5,00. Нижнюю водную фазу выгружают и фазу метиленхлорида промывают водой (8 литров) и насыщенным хлоридом натрия (8 литров). Раствор сушат над сульфатом натрия и концентрируют до густого масла. Масло диспергируют в гексане (5 литров), фильтруют и снова диспергируют в трет-бутилметиловом эфире (5 литров) перед фильтрованием и промыванием дополнительным трет-бутилметиловым эфиром. Сушка на воздухе дает не совсем белый кристаллический продукт. Выход составляет 72-99%.
Несмотря на то, что изобретение было описано и иллюстрировано со ссылкой на некоторые конкретные варианты осуществления его, специалист в данной области должен понимать, что могут быть сделаны различные адаптации, изменения, модификации, замены, исключения или дополнения к процедурам и протоколам без выхода за пределы сущности и объема изобретения. Следовательно, подразумевается, что изобретение определяется объемом формулы изобретения, которая следует за этим, и что такая формула изобретения интерпретируется так широко, как может быть приемлемо.
Реферат
Изобретение относится к новому способу получения 3-замещенных циклическим простым эфиром цефалоспоринов формулы I:

где группа CO2R1 представляет карбоновую кислоту или карбоксилатную соль, а R2 имеет формулу:

где A1 выбран из группы, состоящей из С6-10-арила, С1-10-гетероарила или С1-10 -гетероциклила и А2 имеет значения, указанные в формуле изобретения, который включает: а) взаимодействие соединения формулы (IV)

где R3 представляет пара-нитробензил или аллил и Х представляет галоген, с подходящим снимающим защиту агентом в присутствии растворителя с получением соединения формулы (II) и в) взаимодействие соединения формулы II

с соединением формулы III

где R2 имеет указанные выше значения и L имеет значения, указанные в формуле изобретения, в присутствии растворителя, основания, возможного конденсирующего агента и возможного катализатора с получением соединения формулы I. Также описаны соединения формулы II и формулы V. Технический результат - полученные соединения обладают такими благоприятными свойствами как кристаллическая форма и высокий энантиомерный избыток (е.е.). 4 н. и 9 з. п. ф-лы, 1 ил.

Формула











Комментарии