Соединения пенема и фармацевтическая композиция на их основе - RU2051919C1
Код документа: RU2051919C1
Чертежи


Описание
Изобретение относится к соединениям пенема и, более точно, к соединениям пенема, которые могут найти клиническое применение в качестве перспективных антибиотиков.
Обнаружено, что группа соединений пенема, представленная формулой (V):

-А- кислородная или метиленовая группа;
-В- метиленовая, этиленовая или карбонильная групп, и их соли обладают превосходным антибактериальным действием как в отношении грам-позитивных, так и грам-негативных, аэробных или анаэробных бактерий (патент Японии N 203387/1988).
Высокая степень безопасности данных соединений была подтверждена испытанием на безопасность с использованием лабораторных животных. Ожидается их применение в качестве лекарственного средства.
В то же время в результате изучения взаимосвязи между структурой и антибактериальным действием данных соединений [J.Antibiotics Антибиотики, 41, 1685 (1988)] обнаружено, что среди 2 заместителей пенема (R)-2-тетрагидрофурильная группа обеспечивает наиболее сильное антибактериальное действие, в то время как (S)-2-тетрагидрофурильная группа, диастереомер в ее группе с двумя боковыми цепями, и (R) или (S)-3-тетрагидрофурильная группа, позиционный изомер, обеспечивают наиболее слабое действие конкретно против грам-негативных бактерий.
В
этой связи соединения, представленные следующей формулой (VI):

Биологическая доступность вышеописанных соединений самих по себе для лабораторных животных (крыс) оказалась ничуть не ниже биологической доступности промышленно выпускаемых препаратов, применяемых клинически.
С точки зрения безопасности и экономически очевидно более выгодным является дальнейшее повышение их биологической доступности. Что касается указанных соединений, существует возможность для дальнейшего усовершенствования.
Что касается улучшения всасывания при пероральном применении, были проведены активные исследования антибиотиков пенициллиновой и цефалоспориновой групп, в результате чего многие из данных антибиотиков находят применение как эффективные препараты. Тем не менее существует всего лишь несколько отчетов об исследованиях такого рода в отношении пенемсодержащих и карбапенемсодержащих антибиотиков. [J.Antibiotics Антибиотики, 36, 938 (1983); патент Японии N 67287(1990)] Вследствие этого важно выяснить применимы ли подходы к антибиотикам пенициллиновой и цефалоспориновой групп также и в отношении соединений пенема.
Исследованы соединения (VI) на предмет возможности совершенствования их биологической доступности. Обнаружено, что защита их карбоксильной группы конкретной эфирообразующей группой может значительно улучшить их биологическую доступность, что приведет к доработке настоящего изобретения.
Заявленное в
настоящем изобретении соединение пенем представлено следующей формулой (I):

-

R2 C1-C6 алкильная или алкенильная группа, С6-С10 арильная группа, С7-С11 аралкильная группа или указанная R2 группа, замещенная одним или несколькими заместителями, выбранными на С1-С6 алкильных групп, С6-С10 арильных групп, С7-С11 аралкильных групп, гидроксильных групп, С1-С6 алкоксильных групп и атомов галогена; а n целое число 1-6,
-

-СН2-Z, (IV) в которой Z-это 5-замещенная 2-окcо-1,3-диоксоленил-4-группа, причем указанные 5 заместителей являются С1-С6 алкильной группой, С6-С10 арильной группой, С7-С11 аралкильной группой или указанная 5-замещенная группа, замещенная одним или несколькими заместителями, выбранными из С1-С6 алкильных групп, С6-С10 арильных групп, С7-С11 аралкильных групп, гидроксильных групп, С1-С6 алкоксильных групп и атомов галогена.
Оптимальный способ осуществления изобретения.
Соединение
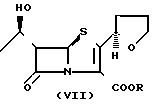
пенем, заявленное в настоящем изобретении, может быть синтезировано, например, введением в реакцию галогенированного алкильного соединения (VII)
с соединением
пенема (VI') согласно следующей
формуле:
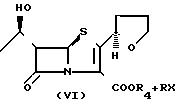
В случае, когда R4 в соединении (IV') является атомом щелочного металла или аминоостатком, согласно данному изобретению целевое соединение может быть получено перемешиванием соединения (VI'), галогенированного алкильного соединения (VII) в органическом растворителе.
В случае, когда R4 в соединении (VI') является атомом водорода, напротив, его сначала вводят в реакцию с гидроксидом щелочного металла, солью щелочного металла или аминосоединением в органическом растворителе для получения соли. Галогенированное алкильное соединение (VII) затем вводят в реакцию с реакционной смесью, в результате чего синтезируют целевое соединение.
Галогенированное алкильное соединение, представленное формулой (VII), служит для эффективной эстерификации карбоксильной группы соединения (VI') при взаимодействии с группой R таким образом, что получают целевое соединение формулы (I). Примеры соединения (VII) включают такие, в которых в качестве группы R выступают тетрагидрофурил-карбонилоксиметильная группа, тетрагидропиранилкарбонилоксиметильная группа, 1, 3-диоксоланилкарбонилоксиметильная группа, 2-оксо-1, 3-диок- соланилкарбонилоксиметильная группа, 1, 4-диоксанилкарбонилоксиметильная группа или тому подобные или ее оптически активный изомер, такой как (R)-2-тетрагидрофурил-карбонилоксиметильная группа, (S)-2-тетрагидрофурил-карбонилоксимети-льная группа, или (5-метил-2-оксо-1,3-диоксоленил-4) метильная группа и, в качестве галогена Х атом хлора, брома или йода.
На щелочной металл не существует конкретных ограничений до тех пор, пока он образует соль с соединением (VI'). Примеры щелочного металла включают литий, натрий и калий. Примеры их гидроксидов и солей включают гидроксид натрия, гидроксид калия, бикарбонат натрия, карбоксилат натрия, бикарбонат калия и карбонат калия. Примерные аминосоединения включают аммиак, триэтиламин и диизопропилэтиламин.
На растворитель реакции не существуют конкретных ограничений. Примеры растворителя реакции включают аромати- ческие углеводороды, такие как толуол и ксилол, алифатические углеводороды, такие как пентан и гексан, галогенированные алкилы, такие как метиленхлорид и хлороформ, галогенированные арилы, такие как хлорбензолы, кетоны, такие как ацетон и метилэтилкетон, нитрилы, такие как ацетонитрилы и пропионнитрилы, амиды, такие как диметилформамид, сульфоксиды, такие как диметилсульфоксид, простые эфиры, такие как простой диэтиловый эфир и тетрагидрофуран, и спирты, такие как изопропанол и t-бутанол. Они могут быть применены как по отдельности, так и в сочетании.
Реакция может быть осуществлена при комнатной температуре и, при необходимости, с нагреванием при температуре ниже 80оС. Время протекания реакции в основном 1-48 ч, хотя оно варьируется в зависимости от применяемого соединения галогенированного алкила.
В качестве
альтернативного способа получения соединения, заявленного в
настоящем изобретении, соединение может быть также синтезировано преобразованием
соединения формулы (VI) в его сложный эфир хлорметила или
йодметила в соответствии со способом, предложенным.
Б.Бальцером и др. [J.Antibiotics Антибиотики, 33, 1183 (1980)] и затем введением его в
реакцию с карбоксилатом, представленным следующей формулой
(VII):
R5-COOM, где R5
имеет то же значение, что и Y, а М щелочной металл или аминоостаток.
Данная реакция протекает при температуре в интервале от -40оС до комнатной температуры и обычно завершается в течение 1-48 ч.
Соединение пенем (I), полученное описанным выше способом, может быть применено в том виде, в каком оно существует, но, обычно, его при необходимости очищают способом колоночной хроматографии или перекристаллизацией для использования в качестве медицинского препарата.
Для перорального, парентерального или наружного применения соединения могут быть приготовлены в виде антибиотиков известным способом.
Несмотря на то, что дозировка каждого производного пенем, заявленного в настоящем изобретении, варьируется в зависимости от многих факторов, обычная дневная доза находится в пределах от 50 мг до 3 г для стандартного взрослого индивидуума при предпочтительном приеме от 100 мг до 2 г небольшими дозами через определенные интервалы времени. В целом, вышеуказанная доза будет введена в виде стандартной дозы, содержащей соответствующее количество активного ингредиента и подходящий физиологически приемлемый носитель или наполнитель.
При пероральном приеме могут применяться таблетки или капсулы. Они могут содержать наряду с активными ингредиентами наполнитель, например, лактозу, глюкозу, сахарозу, маннит, сорбит или целлюлозу и смазывающее вещество, например, тальк, стеариновая кислота или соль стеариновой кислоты. Таблетки могут дополнительно содержать связывающее вещество, например, гидроксипропилцеллюлозу или крахмал.
П р и м е р 1. Хлорметил (5R, 6S)-6-[(R)-1-гидроксиэтил]-2-[(R)-2-тетрагидрофурил] пенем-3-карбоксилат (соединение 6).
К смеси натрий (5R, 6S)-6-[(R)-1-гидроксиэтил]-2- [(R)-2-тетрагидрофурил] пенем-3-карбоксилата 2,5 гидрата (соединена 1, 24, 7 г), тетрабутиламмонийсульфата (2,38 г) и бикарбоната калия (21,0 г), добавили воду (70 мл) и метиленхлорид (70 мл). К полученной смеси по каплям добавляли разбавленный в метиленхлориде (280 мл) раствор хлорметил хлорсульфата (11,5 г) в течение 10 мин при охлаждении льдом и перемешивании. Реакционную смесь затем дополнительно перемешивали в течение 2,5 ч при комнатной температуре. Органический слой собирали и промывали водой. Органический слой сушили и концентрировали. Полученный осадок очищали, пропуская через колонку с силикагелем. Получали 9,92 г соединения, указанного в названии примера.
П р и м е р 2. Иодометил (5R, 6S)-6-[(R)-1-гидроксиэтил]-2- [(R)-2-тетрагидрофурил] пенем-3-карбоксилат (соединение 7). Смесь хлорметил (5R, 6S)-6-[(R)-1-гидроксиэтил] -2-[(R)-2-тетрагидрофурил] пенем-3-карбоксилата (соединение 6, 334 мг), иодида натрия (225 мг) и ацетона (2 мл) перемешивали в течение ночи при комнатной температуре и реакционную смесь концентрировали. К остатку добавили этилацетат. Полученный раствор промыли водой, сушили и затем концентрировали. Получили 359 мг целевого соединения.
П р и м е р 3. [5-(аллилокси)глутарил]оксиметил (5R, 6S)-6-[(R)-1-гидроксиэтил] -2-[(R)-2-тетрагидрофурил] -пенем-3- карбоксилат (соединение 2). Смесь иодометил (5R, 6S)-6-[(R)-1-гидроксиэтил]-2-[(R)-2-тетрагидрофурил] пенем-3-карбоксилата (соединение 7; 3,4 г), натрий моно аллил-глутарата (3,83 г) и N, N-диметилформамида (20 мл) была выдержана в течение ночи при -20оС. Затем реакционную смесь разбавили простым этиловым эфиром и промыли водой. Органический слой был высушен и затем концентрирован. Остаток был очищен подачей через колонну с силикагельной насадкой, в результате чего было получено 0,553 г заявленного соединения.
П р и м е р 4. (R)-2-тетрагидрофурилкарбонилоксиметил (5R, 6S)-6-[(R)-1-гидроксиэтил] -2-[(R)-2-тетрагидрофурил] -пенем-3-карбоксилат (соединение 3).
Смесь йодметил (5R, 6S)-6-[(R)-1-гидроксиэтил]-2-[(R)-2-тетрагидрофурил] пенем-3-карбоксилата (соединение 7, 3,4 г), натриевой соли (R)-тетрагидро-2-фуранкарбоновой кислоты (1,38 г) и N,N-диметилформамида (20 мл) перемешивалось в течение 2 ч при комнатной температуре. Реакционная смесь была разбавлена простым этиловым эфиром и затем промыта водой. Органический слой был высушен и затем концентрирован. Остаток был очищен подачей через колонну с силикагельной насадкой, в результате чего было получено 0,72 г заявленного соединения.
П р и м е р 5. (S)-2-тетрагидрофурилкарбонилоксиметил (5R, 6S)-6-[(R)-1-гидроксиэтил] -2-[(R)-2-тетрагидрофурил] пенем-3-карбоксилат (соединение 4).
Смесь йодметил (5R, 6S)-6-[(R)-1-гидроксиэтил]-2-[(R)-2-тетрагидрофурил] пенем-3-карбоксилата (соединение 7, 3,4 г), натриевой соли (S)-тетрагидро-2-фуранкарбоновой кислоты (1,38 г) и N, N-диметилформамида (20 мл) перемешивалась в течение 2 ч при комнатной температуре. Реакционная смесь была разбавлена простым этиловым эфиром и затем промыта водой. Органический слой был высушен и затем концентрирован. Остаток был очищен подачей через колонну с силикагельной насадкой, в результате чего было получено 0,92 г заявленного соединения.
П р и м е р 6. (5-метил-2-оксо-1,3-диоксоленил-4)метил (5R, 6S)-6-[(R)-1-гидроксиэтил] -2-[(R)-2-тетрагидрофурил] пенем- 3-карбоксилат (соединение 5).
Смесь 2,5 гидрата натрия (5R, 6S)-6-[(R)-1-гидроксиэтил]-2-[(R)-2-тетрагидрофурил] пенем-3-карбоксилата (соединение 1, 3,4 г), 4-йодметил-5-метил-2-оксо-1, 3-диоксолена (1,38 г) и N,N-диметилформамида (20 г) перемешивалась в течение 5 ч при комнатной температуре. В реакционную смесь была добавлена вода. Реакционная смесь была разбавлена простым этиловым эфиром и затем промыта водой. Органический слой был высушен и концентрирован. Остаток был очищен подачей через колонну с силикагельной насадкой, в результате чего было получено 0,72 г заявленного соединения.
Физические свойства полученных соединений по примерам 1-6 приведены в табл.1.
П р и м е р 7. Биологическая активность некоторых соединений (I) настоящего изобретения была испытана в зависимости от скорости их регенерации в моче.
Каждое испытавшееся соединение (30 μмоль/кг) было перорально введено крысам линии SD (трем самцам крысы в каждой группе). Моча была взята по истечении 6 ч после введения соединений и с помощью анализа на биологическую активность была определена скорость регенерации соответствующего компонента, присутствующего в моче. Результаты показаны в табл. 2.
Как очевидно следует из результатов, соединения (I) настоящего изобретения продемонстрировали более высокую скорость регенерации в моче, другими словами, более высокую биологическую доступность по сравнению с соединением пенем (VI).
В каждом из нижеследующих примеров препаратов активными ингредиентами могут быть, например, соединение 5 или эквивалентное качество любого из остальных соединений, заявленных в настоящем изобретении (см. табл.3 и 4).
Технология производства.
Ингредиенты 1 и 2 были соединены в подходящем смесителе, куда был добавлен ингредиент 3 с последующим дополнительным перемешиванием. Полученная смесь была введена в капсулировочную машину.
Ингредиенты 1-3 были соединены в подходящем смесителе, куда был добавлен ингредиент 4 с последующим дополнительным перемешиванием в течение нескольких минут. Полученной смеси под давлением была придана заданная форма и масса с помощью таблетирующей машины.
Соединения настоящего изобретения демонстрируют превосходную биологическую доступность, таким образом они могут успешно применяться в качестве антибиотиков для перорального приема.
Реферат
Сущность изобретения: соединения пенема ф-лы I, где
R- группа ф-лы II: -СН2-ОС(О)-(СН2)n
-С(О)-ОR1 при R1-(С1 - С6
)-алкенил, n - целое число 1 - 6; или R - группа ф-лы III:
(-СН2-ОС(О)-Y), где Y - (R) - или (S)-2-тетрагидрофурильная
группа; или R - группа ф-лы IV: (-СН2-Z), где Z - группа
ф-лы V. Соединения ф-лы I обладают антибиотическим действием
и входят в состав фармацевтической композиции. 2 с. и 5 з. п. ф-лы, 2
табл. Структура ф-лы I и V:

Формула
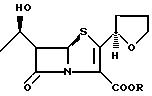
где R представляет собой группу общей формулы II
-CH2-OCO(CH2)n COOR1,
где R1 - C1 - C6-алкенил;
n = 1 - 6 - целое число,
или R представляет собой группу общей формулы III
-CH2OCO-Y,
где Y - группа формулы

или R представляет собой группу общей формулы IV
-CH2-Z,
где Z - группа формулы

где А - С1 - С6-алкил
2. Соединения по п.1 общей формулы I, где Y представляет собой (S)-2-тетрагидрофурильную группу.
4. Соединения по п.1 общей формулы I, где R1 - аллил,
5. Соединение по п.1 общей формулы I, выбранное из группы, содержащей { [5-(аллилокси)глутарил] окси}метил (5R, 6S)-6-[(R)-1-гидроксиэтил]-2-[(R)-2-тетрагидрофурил] -пенем-3-карбоксилат, (R)-2- тетрагидрофурилкарбонилоксиметил(5r,6S)-6-[(R)-1-гидроксиэтил] -2-[(R)-2-тетрагидрофурил]пенем-3-карбоксилат, (S)-2-тетрагидрофурилкарбонилоксиметил(5r,6S)-6-[(R)-1-гидроксиэтил] -2-[(R)-2-тетрагидрофурил]-пенем-3-карбоксилат, (5-метил-2-оксо-1, 3-диоксоленил-4)метил-(5R, 6S)-6-[(R)-1-гидроксиэтил] -2-[(R)-2-тетрагидрофурил]-пенем-3-карбоксилат.
Комментарии