Улучшенный способ получения меропенема с применением цинкового порошка - RU2490270C2
Код документа: RU2490270C2
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
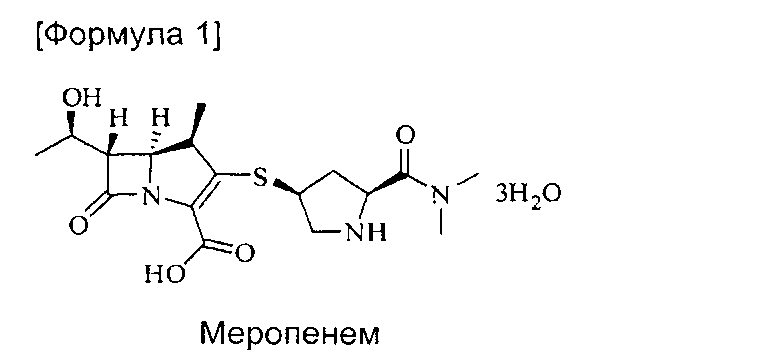
Настоящее изобретение относится к новому способу получения тригидрата меропенема, который был признан самым лучшим антибиотиком класса карбапанемов с отличной эффективностью и безопасностью. Конкретно, настоящее изобретение относится к способу получения тригидрата меропенема с высокой чистотой и выходом путем проведения реакции удаления защиты в мягких условиях с применением цинкового порошка.
УРОВЕНЬ ТЕХНИКИ
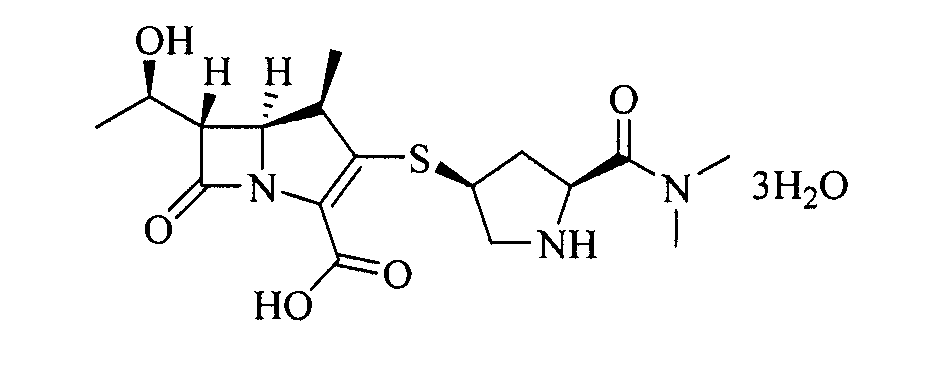
Тригидрат меропенем (меропенем·3Н2О) [Химическое название: (4R,5S,6S)-3-((3S,5S)-5-(диметилкарбамоил)пирролидин-3-илтио)-6-((R)-1-гидроксиэтил)-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты тригидрат] представляет собой соединение, имеющее структуру следующей формулы (1):
Формула 1
Что касается предшествующего уровня техники в отношении синтеза меропенема, в патенте US 4943569 раскрыт способ получения аморфного меропемена путем реакции сочетания MAP с несущим боковую цепь веществом, как представлено на схеме 1 ниже, с получением ПНБ-меропенема, карбоксильная группа которого защищена п-метоксибензильной группой или п-нитробензильной группой, растворения его в подходящем количестве растворителя, представляющего собой смесь тетрагидрофурана и этанола, гидрирования при комнатной температуре в течение 3 часов в буферном растворе морфолинопропансульфоновой кислоты в присутствии 10% палладия на угле с 120% массового соотношения, фильтрования катализатора, упаривания тетрагидрофурана и этанола в вакууме, промывания остаточного раствора этилацетатом, упаривания растворителя в водном растворе в вакууме, выделения с помощью колоночной хроматографии с применением CHP-20P и сублимационной сушкой.
Реакционная схема 1
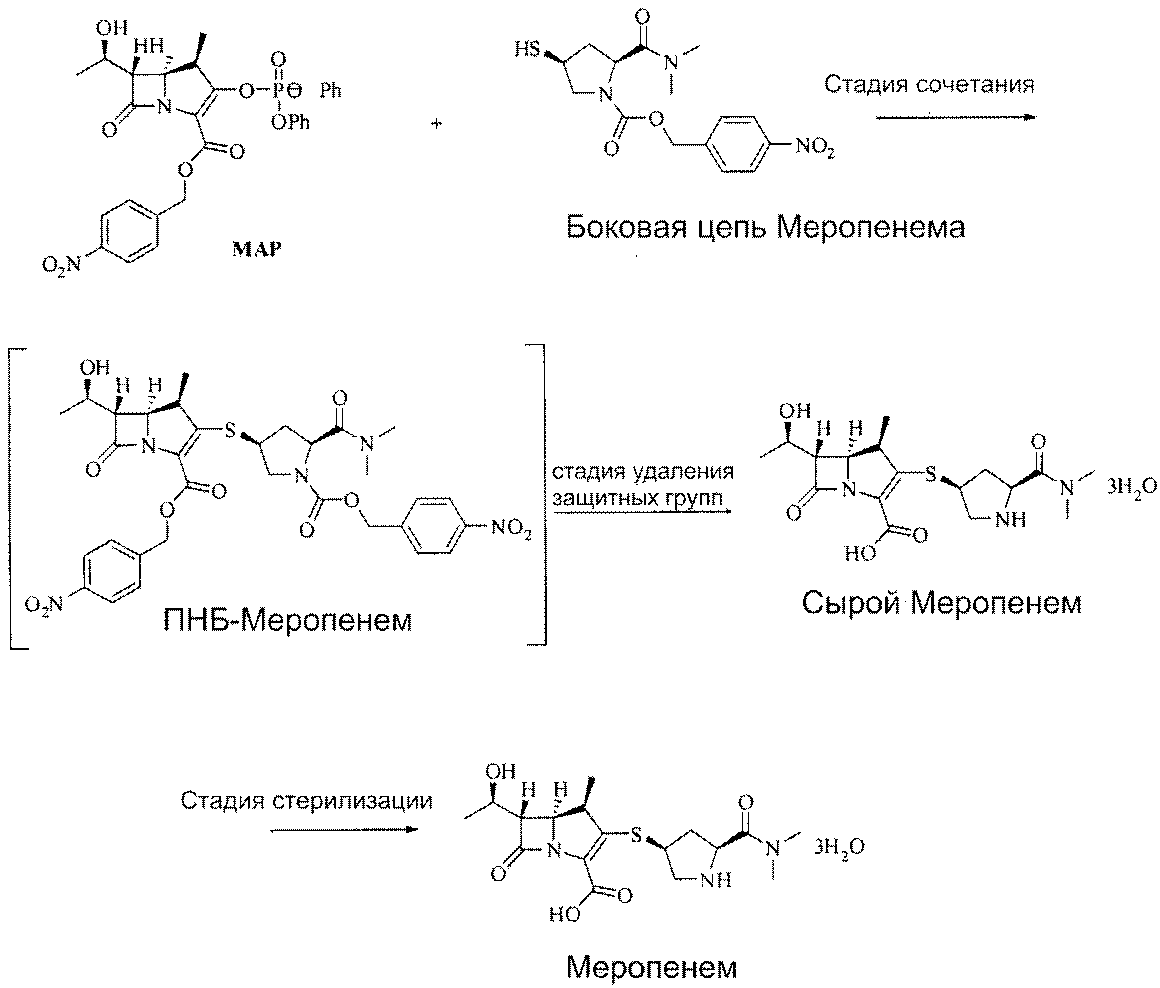
Помимо этого, в патенте US 4888344 представлен способ получения тригидрата меропенема путем растворения ПНБ-меропенема в растворителе, представляющем собой смесь тетрагидрофурана (ТГФ) и воды, добавления туда 10% палладия на угле и взаимодействия его в атмосфере водорода (4,8 атм. (486 кПа)) при комнатной температуре в течение 5 часов, фильтрования катализатора, упаривания тетрагидрофурана в вакууме, промывания остаточного раствора дихлорметаном, упаривания растворителя в водном растворе в вакууме, концентрирования с применением аппарата обратного осмоса и кристаллизации. По сравнению со способом патента US 4943569, учитывая, что в способе по патенту US 4888344 не используется буферный раствор морфолинопропансульфоновой кислоты и катализируемая реакция гидрирования проводится в растворителе, представляющем собой смесь воды и тетрагидрофурана, выгодно, что гидрат может быть получен непосредственно из водного концентрированного раствора без использования методов колоночной хроматографии, сублимационной сушки и извлечения.
Кроме того, в выложенной Корейской патентной публикации под номером 1994-14399 улучшается выход конечного целевого соединения путем введения нового способа синтеза ПНБ-меропенема, в котором может быть сокращено количество технологических операций и производство по сравнению с традиционным способом упрощается. Однако так как в данном способе также применяется процедура удаления защиты ПНБ-меропенема патента US 4943569, дополнительно проводится стадия кристаллизации после получения меропенема в аморфном состоянии с получением более стабильного тригидрата, приводящая к выходу тригидрата меропенема 55,3% (выход реакции удаления защиты: 69,1%; выход при кристаллизации: 80%).
Упомянутые выше способы включают сложные процессы и используют очень дорогое оборудование. В частности, для них требуется в большом количестве дорогого палладия на угле. Кроме того, поскольку следует использовать высоковзрывоопасный водород, их трудно внедрять в промышленность.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Задача изобретения
При получении меропенема в известном уровне техники существуют указанные выше проблемы. В частности, полагают, что условия удаления п-нитробензильной группы являются неподходящими для промышленного производства. Соответственно, авторы настоящего изобретения провели интенсивные исследования для разработки способа, который осуществляется в мягких условиях, является простым для использования в промышленном производстве и усовершенствован в отношении выхода и качества продукта. В результате этих исследований авторы настоящего изобретения обнаружили, что эти задачи могут быть решены путем применения цинкового порошка на стадии удаления защиты и таким образом создали настоящее изобретение.
Техническое решение
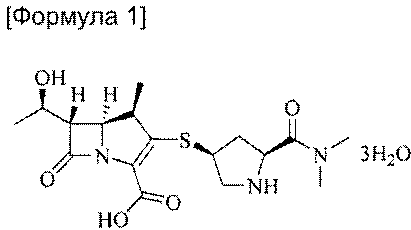
Таким образом, в настоящем изобретении обеспечивается способ получения тригидрата меропенема формулы (1)
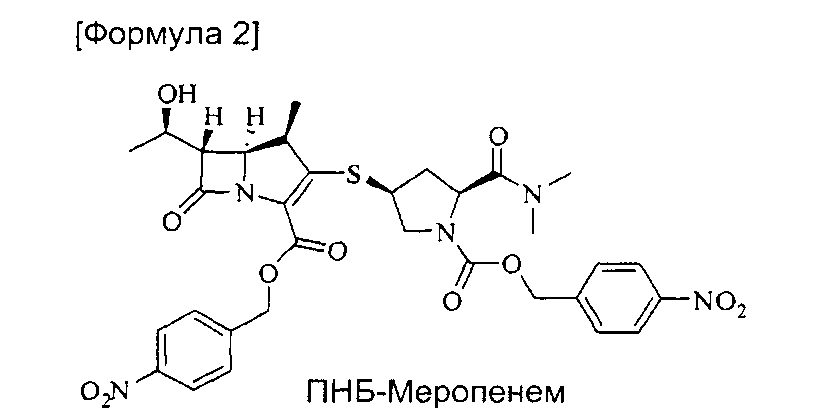
включающий реакцию ПНБ-меропенема формулы (2) [(4R,5S,6S)-4-нитробензил-3-((3S,5S)-5-(диметилкарбамоил)-1-((4-нитробензилокси)карбонил)пирролидин-3-илтио)-6-((R)-1-гидроксиэтил)-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат]
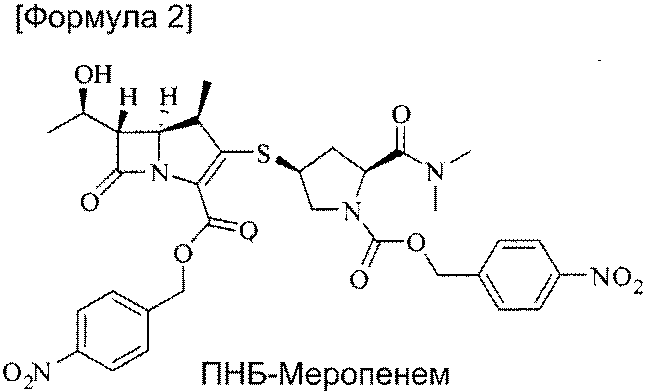
с цинковым порошком в растворителе, представляющем собой смесь органического растворителя и водного раствора фосфата, для удаления п-нитробензильной группы, удаление фосфата из полученной смеси и кристаллизацию тригидрата меропенема из растворителя, представляющего собой смесь для кристаллизации.
Эффект изобретения
Для преодоления проблемы предшествующего уровня техники - удаления п-нитробензильной группы посредством реакции с водородом при высоком давлении - в настоящем изобретении обеспечивается способ, который является промышленно безопасным, поскольку применяется удаление п-нитробензильной группы, которая является защитной группой карбоксильной группы в имино-(амино-)соединениях карбапенемового типа, в мягких условиях с применением цинкового порошка. Кроме того, в настоящем изобретении достигается эффект снижения стоимости за счет использования дешевых ионообменных смол и эффект улучшения качества путем эффективного удаления фосфатных примесей после реакции. Кроме того, в настоящем изобретении достигаются дополнительные эффекты легкости и повышения производительности путем проведения in situ реакции сочетания для синтеза ПНБ-меропенема и реакции удаления защиты для удаления п-нитробензильной группы.
Подробное описание изобретения
Способ получения тригидрата меропенема из ПНБ-меропенема формулы (2) представляет собой способ удаления п-нитробензильной группы, которая является защитной группой. Реакцию проводят в растворителе, представляющем собой смесь органического растворителя, способного растворять ПНБ-меропенем, и водного раствора фосфата при 20-50°С, в течение 0,5-5 часов, предпочтительно 1-1,5 часов.
Органический растворитель, способный растворять ПНБ-меропенем, выбирают из тетрагидрофурана, ацетонитрила, этилацетата, метиленхлорида, хлороформа и тому подобного. К раствору ПНБ-меропенема в органическом растворителе добавляют 0,5-1,6М водный раствор фосфата, предпочтительно 1,5М, доводят температуру до приблизительно 25°С и туда медленно добавляют цинковый порошок. Объемное соотношение органического растворителя и водного раствора фосфата в смеси составляет 5:5-15:30, предпочтительно 10:20, в расчете на массу ПНБ-меропенема. Фосфат может быть выбран из KH2PO4, K2HPO4, H3PO4, NaH2PO4 и Na2HPO4 и предпочтительным для использования является KH2PO4. Предпочтительно, чтобы его концентрация была близкой к насыщенной после добавления раствора ПНБ-меропенема. Если концентрация фосфата является низкой, реакция заканчивается на промежуточном соединении, в котором только нитрогруппа защитной группы восстановлена до амина, и таким образом целевое соединение не может быть получено с максимальным выходом. В случае если используется фосфатный буферный раствор, по сравнению с применением только фосфата, образуется большое количество примесей в результате значительного разложения целевого соединения, приводя к снижению его выхода и содержания. Цинковый порошок добавляется в количестве, превышающем в 4-8 раз по массе ПНБ-меропенем. При добавлении цинкового порошка порциями может быть минимизировано явление сильного выделения тепла, для того чтобы избежать разложения целевого соединения в результате значительного выделения тепла. Если температура реакции удаления защитной группы ниже 20°С, реакция протекает в основном только до промежуточного восстановленного амина, приводя к неполному протеканию реакции. Если температура составляет более 50°С, скорость реакции и полнота ее протекания повышаются, но целевое соединение разлагается в большей степени, приводя к снижению его выхода и чистоты.
Если реакция завершена, проводят фильтрование для удаления цинкового порошка. При фильтровании оставшийся на фильтре цинковый порошок промывают жидкостью, представляющей собой смесь воды и полярного органического растворителя, такого как тетрагидрофуран, спирт и так далее, для полного отделения целевого соединения, адсорбированного на цинковом порошке в большом количестве. Фильтрат затем разделяются на фазы, водный слой отделяют и промывают несколько раз неполярным органическим растворителем, например, дихлорметаном, хлороформом, тетрахлорметаном и тому подобным, чтобы отделить органический растворитель, используемый в качестве растворителя для реакции, или промывочную жидкость. Если оставшийся органический растворитель удалять путем концентрирования, целевое соединение постепенно разлагается в слабокислых условиях реакции, что приводит к потере на выходе и содержании. В частности, если гидрофильный органический растворитель остается в водном слое на протяжении процесса экстракции или концентрирования, это предотвращает адсорбцию целевого соединения на адсорбционной смоле и вызывает немедленное элюирование целевого соединения, что приводит к потере выхода. Соответственно, его удаление является предпочтительным.
Из результирующей смеси, полученной как указано выше, удаляют фосфат.
Первое удаление фосфата может быть осуществлено посредством кристаллизации. То есть большое количество фосфата сначала удаляют в кристаллической форме путем добавления растворителя, который растворяет меропенем, но не фосфат, например, смешиваемого с водой растворителя, такого как метанол, этанол, изопропиловый спирт, тетрагидрофуран, ацетон, ацетонитрил и тому подобное, предпочтительно метанол.
В соответствии с предпочтительным вариантом осуществления изобретения, после стадии экстракции содержащий целевое соединение водный слой охлаждают и сначала добавляют предварительно охлажденный метанол для удаления фосфата. Общее количество добавляемого метанола превышает в 10-80 по объему ПНБ-меропенем. В тоже время, если температура при добавлении охлажденного метанола слишком высока, образуется большое количество примесей и, таким образом, добавление предпочтительно осуществлять при 20°С или менее. После добавления позволяют кристаллам фосфата вырасти и отфильтровывают их, чтобы удалить.
Предпочтительно, продукт после первичного удаления фосфата путем кристаллизации и фильтрования как описано выше может быть пропущен через катионную смолу для дополнительного отделения фосфата.
В соответствии с предпочтительным вариантом осуществления изобретения, после завершения первичного удаления фосфата путем кристаллизации и фильтрования водный слой, содержащий целевое соединение, охлаждают и затем пропускают через предварительно промытую и охлажденную катионную смолу, такую как BCMB50, BC108, NM60G, Lewit up 1213, Lewit up 1243, IRC86RF, S8227 и тому подобное, предпочтительно BCMB50, для дополнительного удаления фосфата. То есть водный слой, содержащий целевое соединение, пропускают через катионную смолу, в результате чего дополнительно удаляют фосфат и целевое соединение очищают. Пропуская целевое соединение через смолу, значение рН повышают и элюированное целевое соединение полностью доводят до рН 5,0-7,0 и необязательно добавляют буферный раствор 1N-метилморфолина/уксусной кислоты (рН 6,5-7,0) и концентрируют до объема в 3-20 раз превышающего массу ПНБ-меропенема. Буферный раствор 1N-метилморфолина/уксусной кислоты подавляет разложение целевого соединения в течение процесса концентрирования, таким образом, повышая конечный выход дополнительно на 5% или выше. Во время процесса концентрирования элюата концентрирование следует проводить при пониженной температуре для минимизации разложения под действием тепла, и для этого концентрирование осуществляют с помощью аппарата обратного осмоса при пониженной температуре за короткий промежуток времени.
Альтернативно, первичное удаление фосфата может быть проведено путем адсорбции его на адсорбционной смоле.
В соответствии с еще одним вариантом осуществления изобретения, после завершения экстракции водный слой, содержащий целевое соединение, охлаждают и затем удаляют фосфат с помощью предварительно охлажденной адсорбционной смолы, такой как SP-207, Amberlite®, XAD4, XAD7, Diaion HP-20, HP-40 и тому подобной, предпочтительно SP-207. То есть, для удаления используемого для реакции фосфата и сильнополярных примесей водный слой, содержащий целевое соединение, адсорбируется на смолу и туда добавляется вода в количестве в 50-100 раз превышающем водный слой. После удаления фосфата целевое соединение извлекают, элюируя с помощью полярного растворителя, такого как метанол, этанол, изопропиловый спирт, тетрагидрофуран, ацетон, ацетонитрил и тому подобное, предпочтительно метанола или этанола, смешанного с водой при концентрации 20-80%.
При пропускании водного раствора, содержащего полярный органический растворитель, через адсорбционную смолу изменяется полярность и выделяется значительное количество тепла, в результате чего целевое соединение может разлагаться. Соответственно, используемый для элюирования раствор следует охлаждать так сильно, как это возможно, чтобы уменьшить разложение целевого соединения. Фракции элюата, содержащие целевое соединение, собирают и необязательно добавляют буферный раствор 1N-метилморфолина/уксусной кислоты (рН 6,5-7,0), и затем концентрируют до объема, 3-20-кратно превышающего массу ПНБ-меропенема. Буферный раствор 1N-метилморфолина/уксусной кислоты подавляет разложение целевого соединения в течение процесса концентрирования, таким образом, повышая конечный выход дополнительно на 5% или выше. Во время процесса концентрирования элюата концентрирование следует проводить при пониженной температуре для минимизации разложения под действием тепла, и для этого концентрирование осуществляют с помощью аппарата обратного осмоса при пониженной температуре за короткий промежуток времени.
Так как сконцентрированная жидкость элюата с катионной смолы или адсорбционной смолы, как объясняется выше, уже содержит растворяющий меропенем растворитель, туда добавляют не растворяющий меропенем растворитель, такой как ацетон, изопропиловый спирт и тетрагидрофуран, предпочтительно ацетон, в объеме в 10-110 раз превышающем массу ПНБ-меропенема и через 1 час при комнатной температуре образуются кристаллы. Кристаллам дают вырасти при охлаждении до 0-5°С и перемешивании в течение 2 часов, отфильтровывают, промывают ацетоном и сушат в вакууме при 25°С. В примерах осуществления настоящего изобретения, в результате анализа с помощью ЯМР-спектра подтверждают, что целевое соединение, синтезированное в соответствие с настоящим способом, является веществом эквивалентным стандартному веществу меропенему фармакопеи США.
В соответствии с еще одним вариантом осуществления изобретения, полученная после первого удаления фосфата жидкость может быть сконцентрирована до объема, 3-20-кратно превышающего массу ПНБ-меропенема без пропускания ее через смолу. На данном этапе предпочтительно может использоваться буферный раствор 1N-метилморфолина/уксусной кислоты, как изложено выше. В указанной стадии концентрирования следует проводить концентрирование при пониженной температуре для минимизации разложения под действием тепла, и для этого концентрирование осуществляют с помощью аппарата обратного осмоса при пониженной температуре за короткий промежуток времени. Более того, поскольку данная сконцентрированная жидкость также уже содержит растворяющий меропенем растворитель, туда добавляют не растворяющий меропенем растворитель, такой как ацетон, изопропиловый спирт и тетрагидрофуран, предпочтительно ацетон, в объеме в 10-110 раз превышающем массу ПНБ-меропенема, и через 1 час при комнатной температуре образуются кристаллы. Первые кристаллы меропенема, содержащие небольшое количество фосфата, выращивают при охлаждении до 0-5°С и перемешивании в течение 2 часов, отфильтровывают, промывают ацетоном и сушат в вакууме при 25°С. Затем первые кристаллы меропенема, содержащие фосфат, суспендируют в предварительно охлажденном растворителе с низкой способностью растворять меропенем, таком как вода, для дополнительного отделения оставшегося фосфата. Для эффективного удаления оставшегося фосфата, и в тоже время минимизируя потери в выходе меропенема, кристаллы промывают охлажденным раствором, представляющим собой смесь изопропанол:вода с объемным соотношением 1:2-3:1, и полученные вторые кристаллы меропенема сушат в вакууме при 25°С.
Так дополнительное удаление фосфата посредством второй кристаллизации меропенема может применяться для кристаллов меропенема, полученных из сконцентрированной жидкости элюата с катионной смолы или адсорбционной смолы, как объясняется выше.
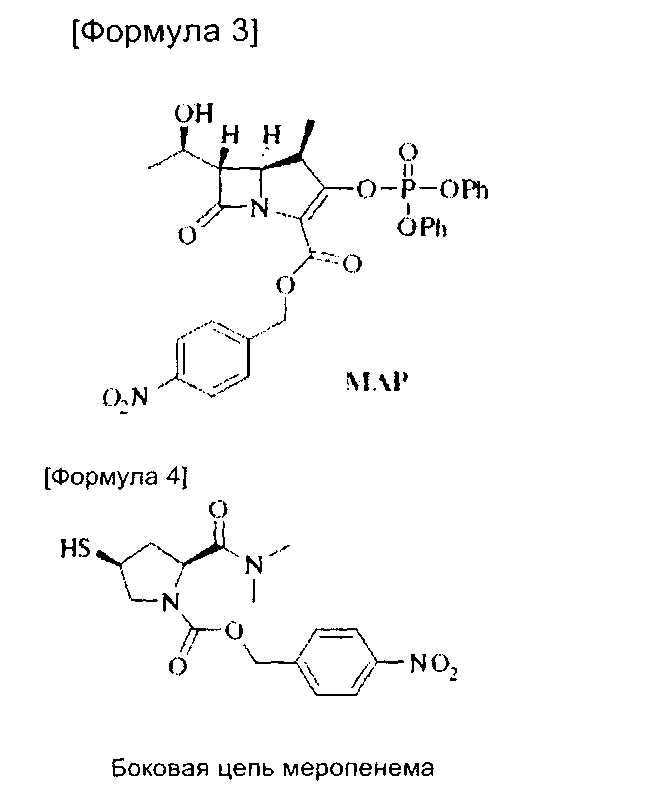
ПНБ-меропенем формулы (2), используемый в настоящем изобретении, может быть получен путем реакции сочетания MAP следующей формулы (3) [(4R,5S,6S)-4-нитробензил-3-(дифеноксифосфорилокси)-6-((R)-1-гидроксиэтил)-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат] с несущим боковую цепь соединением следующей формулы (4) [(2S,4S)-4-нитробензил-2-(диметилкарбамоил)-4-(меркаптопирролидин-1-карбоксилат].
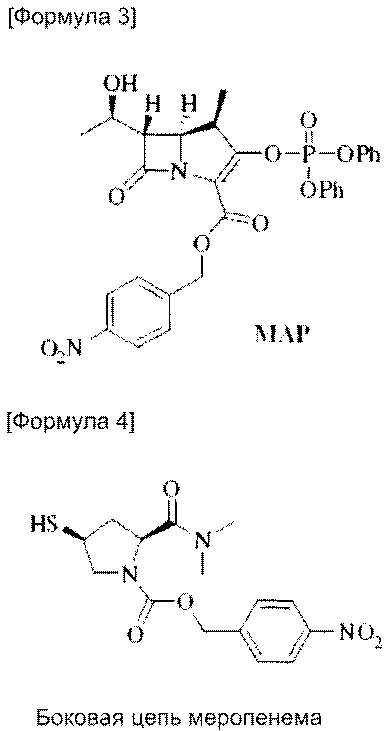
При проведении стадии удаления защитных групп для удаления п-нитробензильной группы, которая защищает карбоксильную группу, вслед за стадией сочетания с получением ПНБ-меропенема формулы (2), стадия удаления защитных групп может проводиться после кристаллизации ПНБ-меропенема после стадии сочетания или, без кристаллизации, она может проводиться in situ в растворителе для экстракции без выделения и очистки. Так, процесс in situ возможен, поскольку после реакции сочетания меропенем растворен в водном слое, а побочные продукты растворены в органическом слое, и, таким образом, первичная очистка возможна только путем экстракции. Если стадия удаления защитных групп протекает in situ, время, затрачиваемое на получение, сокращается и выход увеличивается и, таким образом, производительность может быть значительно повышена.
Более конкретно, реакция сочетания MAP формулы (3) и несущего боковую цепь соединения формулы (4) может проводиться с использованием высокополярного органического растворителя, такого как диметилацетамид, диметилфорамид и тому подобного, в качестве растворителя для реакции в присутствии основания, такого как диизопропилэтиламин. Реакцию проводят в течение 1-5 часов при -20°С - комнатной температуре. После завершения реакции реакционный раствор экстрагируют органическим растворителем, промывают 0,1-6н. раствором HCl, насыщенным раствором соли и обрабатывают безводным сульфатом магния и активированным углем. После фильтрования, минуя стадию концентрирования растворителя, сразу проводится кристаллизация в этилацетате для получения ПНБ-меропенема. Альтернативно, как описано выше, минуя проведение стадии кристаллизации может использоваться экстракция из жидкого состояния in situ в последующей реакции удаления защитных групп.
Вкратце, способ по настоящему изобретению имеет много преимуществ, указанных далее:
Во-первых, он является экономичным, поскольку в реакции удаления защитных групп используется дешевый цинковый порошок, и он легко применим в промышленности без риска взрыва, поскольку реакция проводится в мягких условиях при температуре и давлении окружающей среды.
Во-вторых, традиционные способы требуют использования очень дорогого оборудования для реакции удаления защитных групп с применением водорода. Однако в способе по настоящему изобретению могут применяться обычные реакционные установки. Кроме того, может быть достигнут эффект снижения стоимости, так как может применяться дешевая катионная смола и может беспрепятственно осуществляться масштабирование реакции.
В-третьих, потеря выхода может быть минимизирована путем проведения реакции удаления защитных групп in situ после получения ПНБ-меропенема, минуя выделению и очистку.
В-четвертых, поскольку удаление фосфата является эффективным, увеличивается содержание и улучшается качество соответственно уменьшению примесей.
В следующих далее примерах настоящее изобретение объясняется более подробно. Однако данные примеры предназначены только для иллюстрации и не ограничивают объем настоящего изобретения.
Пример 1
1-1) Получение ПНБ-меропенема
Растворяли 20 г MAP в 80 мл диметилацетамида и добавляли туда 12,5 г несущего боковую цепь соединения. После охлаждения до 0-5°С по каплям добавляли 6,5 мл диизопропилэтиламина. После перемешивания при 0-5°С в течение 5 часов добавляли 120 мл этилацетата и 200 мл воды и реакционную смесь перемешивали и затем фазы разделяли. К этилацетатному слою добавляли 80 мл 0,5н. HCl для удаления оставшегося основания и полученную смесь промывали 200 мл насыщенного водного раствора соли. Для удаления остатков воды и обесцвечивания использовали безводный сульфат магния и активированный уголь. Этилацетатный раствор, который представляет собой фильтрат, перемешивали для получения кристаллов. После перемешивания при комнатной температуре в течение 8 часов, охлаждения до 0-5°С и перемешивания в течение 2 часов и фильтрования получали 18,78 г ПНБ-меропенема.
1-2) Получение тригидрата меропенема
Растворяли 20 г ПНБ-меропенема в 200 мл тетрагидрофурана. Добавляли туда 60 г одноосновного фосфата калия (KH2PO4), растворенного в 400 мл воды и нагревали до 27°С. Медленно порциями добавляли 80 г цинкового порошка и перемешивали между 25 и 35°С в течение 1 часа. После подтверждения завершения реакции добавляли 220 мл метиленхлорида и перемешивали в течение 10 минут и затем фильтровали для удаления цинкового порошка. Водный слой отделяли и промывали два раза 100 мл метиленхлорида, в результате чего полностью удаляли присутствующий в водном слое тетрагидрофуран. Водный слой охлаждали и пропускали через колонку, заполненную адсорбционной смолой SP-207 для адсорбции. Колонку со смолой промывали 2 л воды для удаления соли фосфата калия, используемой в реакции, и примесей и элюировали 2 л 60% метанола для полного извлечения меропенема. Фракции, содержащие меропенем, собирали и добавляли туда буферный раствор N-метилморфолина/уксусной кислоты (рН 6,5), и проводили концентрирование до объема в 10 раз превышающего массу ПНБ-меропенема. Для образования кристаллов добавляли 1100 мл ацетона и перемешивали в течение 1 часа при комнатной температуре. После фильтрования и высушивания получали 9,78 г тригидрата меропенема.
ЯМР1H (CDCl3, 400 МГц) д 5,5 (1H), 5,20 (2H), 4,75 (1H), 4,26 (2H), 3,4-3,8 (4H), 3,3 (2H), 3,0 (6H), 2,62 (1H), 1,2-1,3 (8H).
Пример 2
Получение тригидрата меропенема
Растворяли 20 г ПНБ-меропенема, полученного в соответствии с примером 1-1), в 200 мл этилацетата. Добавляли туда 60 г одноосновного фосфата калия (KH2PO4), растворенного в 400 мл воды и нагревали до 30°С. Добавляли 80 г цинкового порошка к раствору этилацетата/одноосновного фосфата калия и перемешивали между 25 и 35°С в течение 1 часа. После завершения реакции отфильтровывали цинковый порошок, и фильтрат промывали 60 мл раствора, представляющего собой смесь тетрагидрофуран/вода. Водный слой отделяли, промывали 200 мл метиленхлорида и пропускали через колонку, заполненную адсорбционной смолой SP-207 для адсорбции. Колонку со смолой промывали 2 л воды для удаления одноосновного фосфата калия, используемой в реакции, и примесей и элюировали 2 л 60% метанола для полного извлечения меропенема. Фракции, содержащие меропенем, собирали и добавляли туда буферный раствор N-метилморфолина/уксусной кислоты (рН 6,5), и проводили концентрирование до объема в 10 раз превышающего массу ПНБ-меропенема. Для образования кристаллов добавляли 1100 мл ацетона и перемешивали в течение 1 часа при комнатной температуре. После фильтрования и высушивания получали 9,66 г тригидрата меропенема.
Пример 3
Получение тригидрата меропенема
Растворяли 20 г ПНБ-меропенема, полученного в соответствии с примером 1-1), в 200 мл метиленхлорида. Добавляли туда 60 г одноосновного фосфата калия (KH2PO4), растворенного в 400 мл воды, и нагревали до 30°С. Добавляли 80 г цинкового порошка к раствору метиленхлорида/одноосновного фосфата калия и перемешивали между 25 и 35°С в течение 1 часа. После завершения реакции отфильтровывали цинковый порошок, и фильтрат промывали 60 мл раствора, представляющего собой смесь тетрагидрофуран/вода. Водный слой отделяли, промывали 200 мл метиленхлорида и пропускали через колонку, заполненную адсорбционной смолой SP-207 для адсорбции. Колонку со смолой промывали 2 л воды для удаления одноосновного фосфата калия, используемого в реакции, и примесей и элюировали 2 л 60% метанола для полного извлечения меропенема. Фракции, содержащие меропенем, собирали и добавляли туда буферный раствор N-метилморфолина/уксусной кислоты (рН 6,5), и проводили концентрирование до объема в 10 раз превышающего массу ПНБ-меропенема. Для образования кристаллов добавляли 1100 мл ацетона и перемешивали в течение 1 часа при комнатной температуре. После фильтрования и высушивания получали 9,84 г тригидрата меропенема.
Пример 4
Получение тригидрата меропенема (способ in situ)
Растворяли 20 г MAP в 80 мл диметилацетамида и добавляли туда 12,5 г несущего боковую цепь соединения. После охлаждения до 0-5°С по каплям добавляли 6,5 мл диизопропилэтиламина. После перемешивания в течение 5 часов добавляли 120 мл этилацетата и 200 мл воды и реакционную смесь перемешивали и затем фазы разделяли. К этилацетатному слою добавляли 80 мл 0,5н. HCl для удаления оставшегося основания и полученную смесь промывали 200 мл насыщенного водного раствора соли. Для удаления остатков воды и обесцвечивания использовали безводный сульфат магния и активированный уголь. Минуя стадию концентрирования растворителя после фильтрования, следующую стадию проводили непосредственно в этилалцетатном растворе. К этилацетатному раствору добавляли 60г одноосновного фосфата калия (KH2PO4), растворенного в 400 мл воды, и нагревали до 30°С или выше. Добавляли туда 80 г цинкового порошка и перемешивали между 25 и 35°С в течение 1 часа. После завершения реакции отфильтровывали цинковый порошок, и фильтрат промывали раствором, представляющим собой смесь тетрагидрофуран/вода. Водный слой отделяли, промывали 4 раза 100 мл метиленхлорида и пропускали через колонку, заполненную адсорбционной смолой SP-207 для адсорбции. Колонку со смолой промывали 2 л воды для удаления одноосновного фосфата калия, используемого в реакции, и примесей и элюировали 2 л 60% метанола для полного извлечения меропенема. Фракции, содержащие меропенем, собирали и добавляли туда буферный раствор N-метилморфолина/уксусной кислоты (рН 6,5), и проводили концентрирование до объема в 10 раз превышающего массу ПНБ-меропенема. Для образования кристаллов добавляли 1100 мл ацетона и перемешивали в течение 1 часа при комнатной температуре. После фильтрования и высушивания получали 10,30 г тригидрата меропенема.
Пример 5
Получение тригидрата меропенема
Растворяли 20 г ПНБ-меропенема, полученного в соответствии с примером 1-1), в 200 мл тетрагидрофурана. Добавляли туда 400 мл 1,6М раствора одноосновного фосфата натрия (NaH2PO4) и нагревали до 27°С. Добавляли медленно 80 г цинкового порошка и перемешивали между 25 и 35°С в течение 1 часа. После завершения реакции добавляли 220 мл метиленхлорида и перемешивали в течение 10 минут, а затем фильтровали для удаления цинкового порошка. Водный слой отделяли, промывали 2 раза по 100 мл метиленхлорида, при этом полностью удаляли присутствующий в водном слое тетрагидрофуран. Водный слой охлаждали и пропускали через колонку, заполненную адсорбционной смолой SP-207, для адсорбции. Колонку со смолой промывали 2 л воды для удаления одноосновного фосфата натрия, используемого в реакции, и примесей и элюировали 2 л 60% метанола для полного извлечения меропенема. Фракции, содержащие меропенем, собирали и добавляли туда буферный раствор N-метилморфолина/уксусной кислоты (рН 6,5), и проводили концентрирование до объема в 10 раз превышающего массу ПНБ-меропенема. Для образования кристаллов добавляли 1100 мл ацетона и перемешивали в течение 1 часа при комнатной температуре. После фильтрования и высушивания получали 8,53 г тригидрата меропенема.
Пример 6
Получение тригидрата меропенема
Растворяли 20 г ПНБ-меропенема в 200 мл тетрагидрофурана. Добавляли туда 60 г одноосновного фосфата калия (KH2PO4), растворенного в 400 мл воды, и нагревали до 27°С. Медленно порциями добавляли 80 г цинкового порошка и перемешивали между 25 и 35°С в течение 1 часа. После завершения реакции цинковый порошок удаляли фильтрованием. Добавляли 220 мл метиленхлорида и перемешивали, и затем фазы разделяли. Водный слой отделяли и промывали 2 раза 100 мл метиленхлорида, в результате чего полностью удаляли присутствующий в водном слое тетрагидрофуран. Водный слой охлаждали и добавляли по каплям 800 мл охлажденного метанола для осаждения кристаллов фосфата калия. После кристаллы отфильтровывали, фильтрат пропускали через колонку, заполненную промытой катионной смолой BCMB50, для удаления оставшегося фосфата. Добавляли туда буферный раствор N-метилморфолина/уксусной кислоты (рН 6,5), и проводили концентрирование до объема в 10 раз превышающего массу ПНБ-меропенема. Для образования кристаллов добавляли 1100 мл ацетона и перемешивали в течение 1 часа при комнатной температуре. После фильтрования и высушивания получали 10,15 г тригидрата меропенема.
В результате анализа с помощью ЯМР-спектра было подтверждено, что целевое соединение, синтезированное в соответствие с настоящим примером, является веществом эквивалентным стандартному веществу меропенему фармакопеи США.
Пример 7
Получение тригидрата меропенема
Растворяли 20 г ПНБ-меропенема в 200 мл тетрагидрофурана. Добавляли туда 60 г одноосновного фосфата калия (KH2PO4), растворенного в 400 мл воды, и нагревали до 27°С. Медленно порциями добавляли 80 г цинкового порошка и перемешивали между 25 и 35°С в течение 1 часа. После завершения реакции цинковый порошок удаляли фильтрованием. Добавляли 220 мл метиленхлорида, перемешивали и фазы разделяли. Водный слой отделяли и промывали 2 раза с помощью 100 мл метиленхлорида, в результате чего полностью удаляли присутствующий в водном слое тетрагидрофуран. После охлаждения водного слоя добавляли по каплям 800 мл охлажденного метанола для осаждения кристаллов фосфата калия. После отделения кристаллов фильтрованием фильтрат концентрировали до объема в 10 раз превышающего массу ПНБ-меропенема. Для образования кристаллов добавляли 1100 мл ацетона и перемешивали в течение 1 часа при комнатной температуре. После фильтрования получали 12,2 г кристаллов, содержащих тригидрат меропенем и фосфат. 12,2 г данных кристаллов добавляли в 48 мл воды и перемешивали в течение 30 минут. После фильтрования и высушивания получали 9,9 г тригидрата меропенема.
В результате анализа с помощью ЯМР-спектра было подтверждено, что целевое соединение, синтезированное в соответствие с настоящим Примером, является веществом эквивалентным стандартному веществу меропенему фармакопеи США.
Реферат
Изобретение относится к усовершенствованному способу получения тригидрата меропенема, химическое наименование, [(1R,5S,6S)-2-[((2'S,4'S)-2-(диметиламинокарбозил)пирролидин-4'-илтио)-6-[(R)-1-гидроксиэтил]-1-метилкарбапен-2-ем-3-карбоновой кислоты тригидрат], формулы (1):включающий реакцию ПНБ-меропенема формулы (2):с цинковым порошком в растворителе, представляющем собой смесь органического растворителя и водного раствора фосфата, для удаления п-нитробензильной группы, последующее удаление фосфата из полученной смеси и проведения кристаллизации полученного тригидрата меропенема из смеси растворителей. Способ позволяет получить целевой продукт с выходом 78%, исключить взрывоопасный водород и дорогой катализатор Pd/C. 18 з.п. ф-лы, 1 схема, 7 пр.
Формула
включающий реакции ПНБ-меропенема формулы (2)
с цинковым порошком в растворителе, представляющем собой смесь органического растворителя и водного раствора фосфата, для удаления п-нитробензильной группы,
удаление фосфата из полученной смеси и
кристаллизацию тригидрата меропенема из смеси растворителей, для кристаллизации.
Документы, цитированные в отчёте о поиске
Соединение карбапенема
Комментарии