Соединения для предотвращения и лечения нейродегенеративных заболеваний и боли - RU2695358C2
Код документа: RU2695358C2
Чертежи




Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям для применения для предотвращения и/или лечения нейродегенеративных заболеваний или боли, или и того, и другого.
УРОВЕНЬ ТЕХНИКИ
В публикации заявки на патент США US 20120295863 описаны соединения двойного действия, нацеленные на аденозиновый рецептор А2А и транспортер аденозина, для предотвращения и лечения нейродегенеративных заболеваний. Было показано, что селективный агонист аденозинового рецептора А2А (A2AR), названный CGS21680 (кратко CGS), способен ослаблять симптомы болезни Хантингтона (БХ) в трансгенной модели на мышах и избавлять от нарушения цикла мочевины при БХ за счет увеличения активности системы убиквитин-протеасома (Chiang et al., 2009 Hum Mol Genet. 18:2929-2942; Chou et al., 2005 J Neurochem. 93:310-320). Однако известно, что CGS оказывает сильный иммуносупрессорный эффект и другие побочные эффекты и поэтому не подходит для клинического применения.
Было высказано предположение, что N6-(4-гидроксибензил)аденозин, обозначенный в документе US 20120295863 как Т1-11 и также являющийся агонистом A2AR, имеет терапевтический потенциал для лечения нейродегенеративных заболеваний. Однако Т1-11 до сих пор сложно получить в виде доступного для перорального введения лекарственного средства вследствие его низкой биодоступности (F<5%). Биодоступность при пероральном введении является важным свойством при разработке лекарственных средств, поскольку она представляет собой долю вещества в процентах, которая достигает системного кровотока после всасывания и метаболических превращений.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте настоящее изобретение относится к соединению формулы (I) или (IA):

или его фармацевтически приемлемой соли, где X представляет собой галоген.
Галоген (F, Cl, Br или I), может находиться в положении 3, 4 или 5 формулы (I) или в положении 2,4 или 5 формулы (IA).
В одном варианте реализации изобретения соединение выбрано из группы, состоящей из N6-[(3-галотиен-2-ил)метил]аденозина, N6-[(4-галотиен-2-ил)метил]аденозина и N6-[(5-галотиен-2-ил)метил]аденозина.
В другом варианте реализации изобретения соединение выбрано из группы, состоящей из N6-[(2-галотиен-3-ил)метил]аденозина, N6-[(4-галотиен-3-ил)метил]аденозина и N6-[(5-галотиен-3-ил)метил] аденозина.
Кроме того, в другом варианте реализации изобретения соединение выбрано из группы, состоящей из N6-[(5-бромтиен-2-ил)метил]аденозина, N6-[(4-бромтиен-2-ил)метил] аденозина, N6-[(3-бромтиен-2-ил)метил]аденозина, N6-[(5-хлортиен-2-ил)метил]аденозина, N6-[(4-хлортиен-2-ил)метил]аденозина и N6-[(3-хлортиен-2-ил)метил] аденозина.
В другом варианте реализации изобретения соединение выбрано из группы, состоящей из N6-[(2-бромтиен-3 -ил)метил] аденозина, N6-[(4-бромтиен-3-ил)метил]аденозина, N6-[(5-бромтиен-3-ил)метил]аденозина, N6-[(2-хлортиен-3-ил)метил]аденозина, N6-[(4-хлортиен-3-ил)метил] аденозина и N6-[(3-хлортиен-3-ил)метил]аденозина.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (I) или формулы (IA), упомянутого выше, включающему стадию (I) или (II):
(I):
(а) введение в реакцию 6-хлорпуринрибофуранозида в присутствии основания с (тиенил)метанамином, который замещен фтором, хлором, бромом или йодом и имеет формулу 3 или 3А:
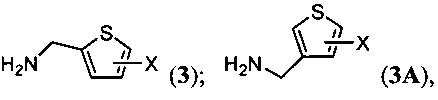
с получением соединения формулы (I) или (IA); или
(II):
(a1) введение в реакцию (2',3'-O-изопропилиден)аденина в присутствии основания с агентом, защищающим гидроксильную группу, с получением производного (2',3'-O-изопропилиден)аденина, содержащего гидроксил-защитную группу;
(b) введение в реакцию производного (2',3'-O-изопропилиден)аденина, содержащего гидроксил-защитную группу, с агентом, защищающим аминогруппу, с получением производного (2',3'-O-изопропилиден)аденина, содержащего гидроксил-защитную группу и амино-защитную группу;
(c) проведение реакции сочетания путем введения в реакцию производного (2',3'-O-изопропилиден)аденина, содержащего гидроксил- и аминозащищающие группы, с соединением формулы 7 или 7А, содержащим замещенную (тиенил)метильную группу:

где X представляет собой F, Cl, Br или I и Y представляет собой X, ОН, метансульфонат (OSO2CH3, OMs), n-толуолсульфонат (OSO2C6H4-n-CH3, OTs) или трифторметансульфонат (OSO2CF3, OTf),
с получением продукта, содержащего защитные группы и замещенную (тиенил)метильную группу; и
(d) удаление защитных групп из продукта стадии (с) в кислых условиях с получением соединения формулы (I) или (IA).
В одном варианте реализации изобретения основание на стадии (а) может представлять собой диизопропилэтиламин. Агент, защищающий гидроксильную группу, может представлять собой трет-бутилдиметилсилилхлорид. Агент, защищающий аминогруппу, может представлять собой ди-трет-бутилдикарбонат. Основание на стадии (a1) может представляет собой имидазол.
В другом варианте реализации изобретения на стадии (I)(а) основание представляет собой диизопропилэтиламин, а замещенный (тиенил)метанамин представляет собой: (i) (5-бромтиен-2-ил)метанамин с получением N6-[(5-бромтиен-2-ил)метил]аденозина; или (ii) (5-хлортиен-2-ил)метанамин с получением N6-[(5-хлортиен-2-ил)метил]аденозина.
Кроме того, в другом варианте реализации изобретения реакцию сочетания проводят в присутствии трифенилфосфина и диизопропилазодикарбоксилата. Соединение, содержащее замещенную (тиенил)метильную группу, может представлять собой (5-бромтиен-2-ил)метанол.
Кроме того, в другом аспекте настоящее изобретение относится к композиции, содержащей:
(a) терапевтически эффективное количество вышеупомянутого соединения или его фармацевтически приемлемой соли; и
(b) фармацевтически приемлемый носитель, вспомогательное вещество или переносящую среду.
В еще одном аспекте настоящее изобретение относится к применению вышеупомянутого соединения для получения лекарственного средства для лечения нейродегенеративного заболевания и/или боли у субъекта, нуждающегося в этом. Альтернативно, настоящее изобретение относится к вышеупомянутому соединению для применения для лечения нейродегенеративного заболевания и/или боли у субъекта, нуждающегося в этом. Нейродегенеративное заболевание может представлять собой заболевание, ассоциированное с неправильной укладкой белка.
В одном варианте реализации изобретения нейродегенеративное заболевание выбрано из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, прионного заболевания, болезни Хантингтона и спиноцеребеллярных атаксий. Спиноцеребеллярные атаксии могут быть выбраны из группы, состоящей из спиноцеребеллярных атаксий 2, спиноцеребеллярных атаксий 3 и спиноцеребеллярных атаксий 7. Боль может представлять собой боль, индуцированную кислотой. Боль, индуцированная кислотой, может представлять собой мышечную боль, индуцированную кислотой. Мышечная боль, индуцированная кислотой, может представлять собой хроническую мышечную боль, индуцированную кислотой.
В одном варианте реализации изобретения боль выбрана из группы, состоящей из боли при воспалении, боли при раке, боли в груди, боли в спине, боли в шее, боли в плече, мигрени, головной боли, миофасциальной боли, боли в суставах, мышечных болевых синдромов, невропатической боли, периферической боли, симпатической боли, послеоперационной боли, посттравматической боли и боли при рассеянном склерозе.
В другом варианте реализации изобретения боль может представлять собой дисфункциональную боль. Дисфункциональная боль может быть выбрана из группы, состоящей из фибромиалгии, миофасциальной боли, болевого синдрома мочевого пузыря и боли, вызванной синдромом раздраженного кишечника.
Эти и другие аспекты станут очевидными из следующего описания предпочтительного варианта реализации, рассматриваемого совместно со следующими чертежами, хотя могут быть затронуты его варианты и модификации без выхода за рамки новых идей настоящего описания.
Прилагаемые чертежи иллюстрируют один или более вариантов реализации настоящего изобретения и, вместе с описанием, служат для объяснения принципов настоящего изобретения. Везде, где это возможно, одни и те же номера ссылок использованы на всех чертежах для обозначения одинаковых или подобных элементов варианта реализации.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На ФИГ. 1 показаны концентрации в крови Т1-11 (А) и JMF3464 (В) у мышей ICR, получавших внутривенно (1 мг/кг) или перорально (10 мг/кг) Т1-11 или JMF3464. Биодоступность при пероральном введении Т1-11 и JMF3464, по оценкам, составляла 2,8% и 17,4%, соответственно.
На ФИГ. 2 показано действие агонистов A2AR (CGS21680, T1-11, JMF3464 и JMF3818) на гибель клеток, индуцированную сывороточной депривацией. Лишенные сыворотки клетки PC12 обрабатывали в присутствии или в отсутствие указанного(-ых) реагента(-ов) в течение 24 ч. Жизнеспособность клеток выражали в процентах от результатов МТТ-тестов по сравнению со средним значением для контрольной группы, содержащей сыворотку. Точки данных представляют собой средние значения ±СО (n=3-6).
На ФИГ. 3А-В показано действие JMF3464 на двигательные дефекты и продолжительность жизни. JMF3464 (0,11 мкг/мышь/день), JMF1907 (0,11 мг/мышь/день) или переносящую среду (КОН) вводили подкожно указанным мышам в возрасте 7 недель с помощью осмотических минипомп ALZET в течение 6 недель. Оценивали способность удерживаться на вращающемся стержне (А) и продолжительность жизни (В) этих мышей. *p<0,05. ***p<0,005.
На ФИГ. 4А-В показаны эффекты Т1-11 по предотвращению агрегации белка, индуцированной гиперэкспрессией мутантного гена SCA2, и поведенческая активность трансгенных мышей SCA2. (А) Клетки, трансфицированные pSCA2-22Q-EGFP или pSCA2-104Q-EGFP, обрабатывали 10 мкМ Т1-11 или 10 мкМ JMF1907 (агониста A2AR, являющегося производным Т1-11) в течение 24 ч. Клетки собирали и подвергали анализу удержания на фильтре или получали их изображения. (В) Мыши дикого типа (ДТ) или трансгенные мыши (SCA2) пили воду, содержащую или не содержащую Т1-11. Способность удерживаться на вращающемся стержне использовали для измерения поведенческой функции мышей.
На ФИГ. 5А-С показано, что T1-11 уменьшает двигательную дисфункцию и гибель нейронов варолиевого моста у трансгенных мышей SCA3. (А) Трансгенные мыши SCA3 получали питьевую воду, содержащую переносящую среду (0,2% ДМСО) или Т1-11 (0,1 мг/мл) начиная с возраста 4 недель. Тест вращающегося стержня показал, что, по сравнению с мышами дикого типа (ДТ), обработанные переносящей средой трансгенные мыши в возрасте 4 месяцев, экспрессирующие атаксин-3-Q79, демонстрировали значительно более короткое время удерживания до падения и двигательную дискоординацию. Способность удерживаться на вращающемся стержне у трансгенных мышей SCA3 в возрасте 4 месяцев, обработанных T1-11 (1 мг в день), была значительно лучше, чем у обработанных переносящей средой мышей того же возраста, экспрессирующих атаксин-3-Q79. Каждая точка соответствует среднему значению ±С.О. для 7-8 мышей. (В-С) Иммуногистохимическое окрашивание нейронального маркера NeuN показало, что ежедневное пероральное введение T1-11 (1 мг в день) значительно уменьшало гибель нейронов в ядрах варолиевого моста трансгенной мыши SCA3 в возрасте 4 месяцев (SCA3+Т1-11). Масштабная полоса составляет 50 мкм. Каждый столбец соответствует среднему значению ±С.О. для 7-8 мышей. *Р<0,01 по сравнению с трансгенными мышами SCA3.
На ФИГ. 6А-С показано, что JMF1907 уменьшает атаксический симптом и гибель нейронов варолиевого моста у трансгенных мышей SCA3. (А) Тест вращающегося стержня показал, что, по сравнению с обработанными переносящей средой трансгенными мышами SCA3 в возрасте 4 месяцев, способность удерживаться на вращающемся стержне у трансгенных мышей SCA3 в возрасте 4 месяцев, обработанных JMF1907 (1 мг в день), была значительно улучшена. Каждая точка соответствует среднему значению ±С.О. для 6 мышей. (В-С) Иммуноцитохимическое окрашивание NeuN показало, что пероральное введение JMF1907 (1 мг в день) значительно предотвращало гибель нейронов в ядрах варолиевого моста мыши SCA3 в возрасте 4 месяцев (SCA3 + JMF1907). Масштабная полоса составляет 50 мкм. Каждый столбец соответствует среднему значению ±С.О. для 6 мышей. *Р<0,01 по сравнению с мышами SCA3.
На ФИГ. 7А-С показано, что JMF3464 уменьшает атаксию и гибель нейронов варолиевого моста у трансгенных мышей SCA3. (А) Трансгенные мыши SCA3 демонстрировали нарушенную способность удерживаться на вращающемся стержне. Ежедневное введение JMF3464 (0,3 мг в день) значительно улучшало способность удерживаться на вращающемся стержне у мышей SCA3 в возрасте 4 месяцев. (В-С) Иммуноцитохимическое окрашивание нейронального маркера NeuN показало, что ежедневное лечение путем перорального введения JMF3464 (0,3 мг в день) значительно уменьшало гибель нейронов в ядрах варолиевого моста трансгенной мыши SCA3 в возрасте 4 месяцев. Каждый столбец соответствует среднему значению ±С.О. для 6 мышей. # Р<0,01 по сравнению с трансгенными мышами SCA3.
На ФИГ. 8А-В показано, что лечение с помощью JMF1907 улучшало двигательные функции трансгенных мышей TDP-43. Были исследованы 4 различные дозы JMF1907 (3,7, 1,25, 0,5, 0,1 мг/кг). КТЛ: трансгенные мыши, обработанные ДМСО. ДТ: мыши дикого типа, обработанные ДМСО. Статистическая обработка была сделана посредством двухфакторного дисперсионного анализа (ANOVA). Для теста вращающегося стержня статистическая значимость была достигнута во всех точках для 1,25 и 0,5 мг/кг, от 12-21 недель для 0,1 мг/кг и 10-12 недель для 3,7 мг/кг. Для теста на силу захвата статистическая значимость была достигнута во всех точках для всех исследованных доз. N=18 (КТЛ), 15 (ДТ), 15 (0,1), 15 (0,5), 15 (1,25), 5 (3,7).
На ФИГ. 9 показано, что лечение с помощью JMF3464 уменьшало нарушение локализации TDP-43 в клетках NSC34. Клетки предварительно обрабатывали JMF3464 (30 мкМ) в течение одного часа и затем обрабатывали препаратом AICAR (1 мМ, AI) в присутствии JMF3464 дополнительно в течение 24 ч. Локализацию TDP-43 (красный) анализировали посредством иммуноокрашивания. Локализацию ядер отмечали с помощью красителя Hochest (синий).
На ФИГ. 10А-С показан обезболивающий эффект JMF3464 на модели фибромиалгии на мышах. (А-1 и А-2) Пероральное введение Т1-11 не показало обезболивающего эффекта на модели фибромиалгии на мышах, в которой у мышей развивалась хроническая мышечная боль после внутримышечной инъекции кислоты и обработки генистеином. Белые стрелки указывают на момент времени, в который мыши получали инъекцию кислоты. Черные стрелки указывают на момент времени, в который мыши получали Т1-11 (п.о.). (В) JMF3464 демонстрирует обезболивающий эффект на модели фибромиалгии на мышах, в которой у мышей развивалась хроническая генерализованная боль после подвергания периодическому холодовому стрессу в течение 2 дней. Обезболивающий эффект JMF3464 зависит от дозы. Эффективная доза начиналась от 100 мкг/кг (в.б.). (С) Пероральное введение JMF3464 (1 мг/кг) проявляло обезболивающий эффект на модели периодического холодового стресса.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ОПРЕДЕЛЕНИЯ
Если не определено иное, то все технические и научные термины, используемые в настоящем описании, имеют такие же значения, как и обычно понимаемые специалистом в области техники, к которой относится настоящее изобретение. В случае противоречия указанных терминов с общеизвестными, настоящий документ, включая определения, будет определяющим.
Термин "обработка" или "лечение" относится к введению эффективного количества терапевтического агента субъекту, нуждающемуся в этом, который страдает от нейродегенеративного заболевания и/или боли или симптома или предрасположенности к такому заболеванию и/или боли, с целью лечения, смягчения, облегчения, излечения, уменьшения проявлений или предотвращения заболевания и/или боли, его симптомов или предрасположенности к нему. Такой субъект может быть идентифицирован специалистом в области медицины на основании результатов любого подходящего способа диагностики.
"Эффективное количество" относится к количеству активного соединения, которое требуется для оказания терапевтического эффекта на подвергаемого лечению субъекта. Эффективные дозы будут изменяться, как признано специалистами в данной области техники, в зависимости от пути введения, применения вспомогательных веществ и возможности совместного применения с другими видами терапевтического лечения.
В "Практическом руководстве для экспертов в области оценки безопасной начальной дозы в клинических испытаниях для терапевтических средств на взрослых здоровых добровольцах", опубликованном Управлением по контролю за пищевыми продуктами и лекарственными средствами Департамента здравоохранения и социальных служб США, раскрыт термин "терапевтически эффективное количество", которое может быть получено с помощью расчетов с применением следующей формулы:
HED (эквивалентная доза для человека) = доза для животного в мг/кг × (масса животного в кг/масса человека в кг)0,33.
JMF 1907 обозначает соединение N6-[2-(индол-3-ил)этил]аденозин, и он был описан в публикации патента США №20120295863 А1 как соединение 6.
Химический синтез
Согласно одному подходу, 6-хлорпуринрибофуранозид (2) обрабатывали необязательно замещенным (тиенил)метанамином (3) в присутствии основания с получением целевого соединения формулы (I).
Например, диизопропилэтиламин применяли в качестве основания, и реакцию замещения 6-хлорпуринрибофуранозида с (5-бромтиен-2-ил)метанамином проводили путем нагревания в растворителе 1-пропаноле с получением N6-[(5-бромтиен-2-ил)метил]аденозина (JMF3464, структура 1).


Согласно другому подходу, (2',3'-O-изопропилиден)аденозин (4) обрабатывали трет-бутилдиметилсилилхлоридом (TBDMSCl) в присутствии основания имидазола с получением производного силилового простого эфира (5). 6-аминогруппу защищали в виде карбамата 6, содержащего трет-бутоксикарбонильный (Boc) заместитель. Вводили необязательно замещенную (тиенил)метильную группу, и получали целевое соединение формулы (I) после удаления всех защитных групп в кислых условиях.
Например, реакцию сочетания соединения 6 с (5-бромтиен-2-ил)метанолом (соединение 7, где X=5-Br, Y=ОН) осуществляли путем применения трифенилфосфина и диизопропилазодикарбоксилата (DIAD) с получением соединения 8. Общего снятия защитных силильной, ацетонидной и Вос-групп в соединении 8 достигали в кислых условиях с получением соединения 1.
Используя тот же химический подход, получали соединение (IA).

Биодоступность при пероральном введении
Для измерения биодоступности при пероральном введении исследуемого соединения забирали образцы крови мышей-самцов ICR (возраст 6 недель; 20-25 г) после перорального (10 мг/кг) или внутривенного (1 мг/кг) введений исследуемого соединения. Образцы крови, забранные в указанные периоды, экстрагировали метанолом, содержащим 0,1% муравьиной кислоты, и затем 10 мкл каждого экстрагированного образца вводили в систему для СЭЖХ-МСМС для количественного определения. Результаты (ФИГ. 1А-В) показали, что биодоступность при пероральном введении T1-11 и JMF3464 у мышей ICR составляла 2,8% и 17,4%, соответственно, что указывает на то, что всасывание при пероральном введении JMF3464 более чем в 6 раз превышало таковое для Т1-11.
Связывание JMF3464 с A2AR и ENT1 и защита нейрональных клеток от апоптоза
Сначала авторы настоящего изобретения охарактеризовали фармакологические свойства JMF3464 с применением анализов связывания радиолиганда. В таблице 1 представлены фармакологические свойства Т1-11, JMF1907 и JMF3464. Связывание указанных соединений с аденозиновым рецептором A2A (A2AR) и переносчиком аденозина (ENT1) характеризовали с применением стандартных протоколов связывания. Как показано в таблице 1, JMF3464 связывался с A2AR и транспортером аденозина - равновесным нуклеозидным транспортером 1. Аффинность JMF3464 по отношению к A2AR была близкой к таковой для Т1-11 и JMF1907 (аналога Т1-11), тогда как его аффинность по отношению к ENT1 была намного лучшей, чем аффинность Т1-11 и JMF1907. Настоящее исследование также показало, что N6-[(5-хлортиен-2-ил)метил]аденозин (JMF3818) также ингибировал апоптоз клеток PC12, вызванный лишением сыворотки. JMF3818 в концентрации 10 мкМ ингибировал рецептор A2A и транспортер аденозина (ENT1) на 54% и 96%, соответственно (ФИГ. 2).

JMF3464 оказывает благоприятное воздействие на основные симптомы болезни Хантингтона (БХ) в модели БХ на трансгенных мышах
Поскольку A2AR и ENT1 расположены в полосатом теле и вовлечены в функционирование полосатого тела, авторы настоящего изобретения предположили, что хроническая обработка JMF3464 будет модулировать прогрессирование БХ. Авторы настоящего изобретения проверили эффект JMF3464 в модели БХ на трансгенных мышах (R6/2), в которой агонисты A2AR оказывают благоприятные эффекты. Добавление JMF3464 (0,11 мг/кг/день) мышам в возрасте от 7 недель противодействовало прогрессирующему ухудшению двигательной координации, как оценивали по способности удерживаться на вращающемся стержне (ФИГ. 3А). Сокращенная продолжительность жизни мышей R6/2 также была увеличена путем пролонгированного (долгосрочного) лечения JMF3464.
Благоприятное воздействие JMF3464 при спиноцеребеллярных атаксиях 2 (SCA2)
Поскольку активация протеасомной активности может представлять собой механизм сигнального пути рецептора A2AR, участвующего в предотвращении агрегации мутантного хантингтина (Htt) или поведенческих характеристиках трансгенных мышей R6/2 (Huang et al., 2011 PLoS One. 6:e20934; Lee et al., 2012 PLoS One. 7:e38865), возможно, что агонист A2AR также может оказывать благоприятное воздействие по облегчению других полиглутаминовых заболеваний, таких как SCA2. Действительно, данные авторов настоящего изобретения показали, что Т1-11 является эффективным с точки зрения предотвращения агрегации мутантного ATXN2 (ФИГ. 4А) и поведенческой активности трансгенных мышей SCA2 (ATAXN2Q127) (ФИГ. 4В), что поддерживает вышеприведенную гипотезу. С учетом того, что биодоступность JMF3464 значительно лучше, чем у Т1-11, авторы настоящего изобретения сделали вывод, что JMF3464 также будет оказывать благоприятное воздействие при SCA2.
Благоприятное воздействие Т1-11 и его аналогов при спиноцеребеллярных атаксиях 3 (SCA3)
По сравнению с мышами дикого типа, обработанные переносящей средой трансгенные мыши SCA3, экспрессирующие атаксин-3-Q79 с удлиненной полиглутаминовой последовательностью, демонстрировали различные симптомы атаксии, включая нарушенную способность удерживаться на вращающемся стержне (ФИГ. 5А и 6А) начиная примерно с 3-4 месяцев. Как показано на ФИГ. 5А и 6А, трансгенные мыши SCA3 в возрасте 4 месяцев, подвергнутые лечению путем ежедневного перорального введения T1-11 (1 мг в день) или JMF1907 (1 мг в день), демонстрировали значительно улучшенную способность удерживаться на вращающемся стержне. Аналогично пациентам, страдающим SCA3, выраженная гибель нейронов была обнаружена в ядрах варолиевого моста трансгенных мышей SCA3 (ФИГ. 5В и 6В). В соответствии с результатами тестов вращающегося стержня, ежедневное лечение путем перорального введения Т1-11 (ФИГ. 5В и 5С) или JMF1907 (ФИГ. 6В и ФИГ. 6С) значительно уменьшала гибель нейронов варолиевого моста у трансгенных мышей SCA3 (ФИГ. 5В, 5С, 6В и ФИГ. 6С). Эти результаты свидетельствуют о том, что пролонгированное лечение путем перорального введения Т1-11 или JMF1907 улучшает неврологические и нейропатологические фенотипы трансгенных мышей SCA3.
JMF3464 имеет намного более высокую биодоступность. Поэтому также ожидалось, что JMF3464 будет оказывать благоприятный эффект при атаксии, индуцированной мутантным атаксином-3-Q79, и нейродегенерации у трансгенных мышей SCA3. Как и ожидалось, ежедневное применение JMF3464 (0,3 мг в день) уменьшало атаксию (ФИГ. 7А) и гибель нейронов варолиевого моста (ФИГ. 7В и 7С) у трансгенных мышей SCA3.
Благоприятное воздействие JMF1907 и JMF3464 при боковом амиотрофическом склерозе (БАС)
Как показано на ФИГ. 8А-В, трансгенные мыши, получавшие JMF1907 в 4 различных дозах (3,7, 1,25, 0,5 и 0,1 мг/кг/день) показывали значительно лучшие результаты в тесте вращающегося стержня и тесте на силу захвата, чем контрольные мыши. Доза 1,25 мг/кг показала наибольший благоприятный эффект. Эти данные указывали на явное улучшение двигательной функции. JMF3464, аналог T1-11 и JMF1907, имеет намного более высокую биодоступность. Поэтому ожидается, что JMF3464 будет оказывать благоприятное воздействие на мышей ALS. Обработка клеток NSC34 с помощью JMF3464 нормализовала нарушение локализации TDP-43, вызванное активацией AMPK (ФИГ. 9), что поддерживает представление о том, что JMF3464 способен предотвращать начальную стадию патогенеза БАС. Эти данные подтверждают, что JMF3464 проявляет благоприятное воздействие при БАС.
Благоприятное воздействие JMF3464 при боли
Несмотря на то, Т1-11 (в.б.) продемонстрировал превосходное обезболивающее действие в 2 моделях фибромиалгии на мышах (моделях индуцированной кислотой хронической генерализованной боли и периодического холодового стресса), его биодоступность очень низка. Пероральное введение Т1-11 (до 2 мг/кг) не показало обезболивающего эффекта в модели индуцированной кислотой хронической генерализованной боли, в которой у мышей развивалась боль, подобная фибромиалгии, после внутримышечной инъекции кислоты и обработки генистеином (ФИГ. 10А). JMF3464, аналог Т1-11, продемонстрировал обезболивающее действие в модели фибромиалгии на мышах, в которой у мышей развивалась хроническая генерализованная боль после подвергания периодическому холодовому стрессу в течение 2 дней (ФИГ. 10В). В соответствии с хорошей биодоступностью JMF3464, его пероральное введение (1 мг/кг) продемонстрировало обезболивающее действие в модели периодического холодового стресса (ФИГ. 10С).
ПРИМЕР 1
Все реагенты и растворители имели высокую степень чистоты и были использованы без дополнительной очистки, если не указано иное. Тетрагидрофуран и диэтиловый эфир перегоняли над Na/бензофеноном, и CH2Cl2 перегоняли над CaH2. Все эксперименты, чувствительные к присутствию воздуха или влаги, проводили в атмосфере аргона. Всю стеклянную посуду сушили в печи в течение более 2 часов и использовали после охлаждения до комнатной температуры в эксикаторах. Реакции с применением микроволнового излучения проводили с применением фокусируемого однорежимного микроволнового реактора (СЕМ Discover). Данный прибор состоит из непрерывно фокусируемой системы подачи энергии в СВЧ-диапазоне с выходной мощностью по выбору оператора.
Точки плавления регистрировали на микроприборе Yanaco. Оптическое вращение измеряли на цифровом поляриметре DIP-1000 от компании JASCO Co., Япония. Значения [α]D приведены в единицах 10-1 град см2 г-1. Инфракрасные (ИК) спектры регистрировали на приборе Nicolet Magna 550-II. ЯМР-спектры получали на приборе Varian Unity Plus-400 (400 МГц), и химические сдвиги (δ) регистрировали в миллионных долях (м.д.) относительно δH 7,24/δС 77,0 (центральная линия t) для CHCl3/CDCl3, δH 2,05/δC 29,92 для (CH3)2CO/(CD3)2CO, δH 3,31/δC 49,0 для CH3OH/CD3OD, и δH 2,49 (m)/δС 39,5 (m) для (CH3)2SO/(CD3)2SO. Типы расщепления сигналов обозначены как s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет) и уш (уширенный). Константы спин-спинового взаимодействия (J) приведены в Гц. Эксперименты ИЭР-МС проводили на масс-спектрометре высокого разрешения Bruker Daltonics BioTOF III. Аналитическую тонкослойную хроматографию (ТСХ) проводили на пластинах силикагеля Е. Merck 60 F254 (0,25 мм). Соединения визуализировали с помощью УФ, распыления анисового альдегида или нингидрина. Колоночную хроматографию проводили на колонках, заполненных силикагелем с размером частиц 70-230 меш.
Чистота соединений по оценке посредством ВЭЖХ (Agilent HP-1100) составляла ≥95% при детектировании на длине волны 280 нм.
2',3'-O-Изопропилиден-5'-O-(трет-бутилдиметилсилил)аденозин (5)
К раствору 2',3'-(O-изопропилиден)аденозина (4, 614 мг, 2,00 ммоль) и имидазола (408 мг, 6 ммоль) в безводном CH2Cl2 (12 мл), охлажденного на ледяной бане, добавляли трет-бутилдиметилсилилхлорид (TBDMSCl, 452 мг, 3 ммоль) при 0°C в атмосфере N2. Ледяную баню удаляли, и полученную смесь перемешивали в течение 12 ч при комнатной температуре. Добавляли метанол (4 мл), и смесь перемешивали в течение еще 15 мин и затем концентрировали при пониженном давлении на роторном испарителе. Твердый остаток растворяли в CH2Cl2 и последовательно промывали 1 М HCl, деионизированной водой и солевым раствором. Органический слой собирали, сушили над MgSO4 и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 5 (819 мг, 1,57 ммоль, выход 78%) в виде белого твердого вещества.
6-N-трет-Бутоксикарбонил-2',3'-O-изопропилиден-5'-O-(трет-бутилдиметилсилил)аденозин (6)
Раствор соединения 5 (819 мг, 1,57 ммоль) и 4-(диметиламино)пиридина (ДМАП, каталитическое количество) в безводном ТГФ (10 мл) перемешивали в течение 2 мин в атмосфере N2. Раствор охлаждали на ледяной бане и добавляли по каплям ди-трет-бутилдикарбонат ((Вос)2О, 1,08 мл, 4,71 ммоль). Ледяную баню удаляли, и смесь перемешивали в течение 12 ч при комнатной температуре. После завершения реакции смесь концентрировали при пониженном давлении на роторном испарителе. Неочищенный продукт растворяли в CH2Cl2 и последовательно промывали 1 М HCl, деионизированной водой и солевым раствором. Органический слой собирали, сушили над MgSO4 и фильтровали. Фильтрат концентрировали при пониженном давлении с получением бис-Boc-соединения (859 мг, 1,38 ммоль, выход 87%) в виде бледно-желтой пены.
К раствору бис-Boc-соединения (400 мг, 0,64 ммоль) в метаноле (8 мл) добавляли метиламин (0,25 мл 40% раствора в метаноле, 2,54 ммоль). Смесь перемешивали при комнатной температуре в течение 20 ч до тех пор, пока анализ посредством ТСХ не показал полное расходование исходного вещества. Смесь концентрировали при пониженном давлении на роторном испарителе. Неочищенный продукт очищали посредством хроматографии на колонке с силикагелем с элюированием смесью EtOAc/гексан (градиенты от 0:1 до 2:1) с получением соединения 6 (300 мг, 0,57 ммоль, выход 88%) в виде бледно-желтого масла.
6-N-(5-Бромтиен-2-ил)метил-6-N-трет-бутоксикарбонил-2',3'-O-изопропилиден-5'-O-(трет-бутилдиметилсилил)аденозин (8, X=5-Br)
Раствор соединения 6 (300 мг, 0,57 ммоль), (5-бромтиен-2-ил)метанола (164 мг, 0,85 ммоль) и трифенилфосфина (226 мг, 0,85 ммоль) в безводном ТГФ (9 мл) перемешивали при 45°C в течение 2 мин в атмосфере N2. Добавляли по каплям диизопропилазодикарбоксилат (DIAD, 0,168 мл, 0,85 ммоль). Смесь перемешивали в течение еще 45 мин до тех пор, пока ТСХ-анализ не показал исчезновение соединения 6. Смесь концентрировали при пониженном давлении на роторном испарителе. Неочищенный продукт очищали посредством флэш-хроматографии (EtOAc/гексан = 1:9) с получением соединения 8 (X=5-Br) в виде бледно-желтого масла.
N6-[(5-Бромтиен-2-ил)метил]аденозин (JMF3464)
Способ А: Суспензию соединения 8 (X=5-Br 138 мг, 0,19 ммоль) в деионизированной воде (5 мл) и ТГФ (1 мл) перемешивали и охлаждали на ледяной бане. Добавляли по каплям при 0°C трифторуксусную кислоту (5 мл). Смесь перемешивали в течение 10 мин, ледяную баню удаляли, и смесь перемешивали в течение еще 30 мин. Смесь концентрировали при пониженном давлении на роторном испарителе. Неочищенный продукт очищали посредством флэш-хроматографии (MeOH/EtOAc=1:49) с получением JMF3464 (соединение 1) в виде белого порошка.
Способ В: Смесь (5-бромтиен-2-ил)метанамина (768 мг, 4 ммоль), 6-хлорпуринрибофуранозида (143 мг, 0,5 ммоль) и диизопропилэтиламина (2 мл, 12 ммоль) в 1-пропаноле (20 мл) нагревали при 70°C в течение 7 ч. Смесь, содержащую целевой продукт и не вступивший в реакцию (5-бромтиен-2-ил)метанамин, обрабатывали ди-трет-бутилдикарбонатом (0,92 мл, 4 ммоль) в ТГФ (6 мл) и NaHCO3 (672 мг, 8 ммоль) при комнатной температуре в течение 2 ч. Смесь концентрировали на роторном испарителе и очищали посредством флэш-хроматографии (силикагель, MeOH/EtOAc=1:9). Целевой продукт JMF3464 (59 мг, выход 27%) получали путем перекристаллизации из МеОН. Чистота продукта составляла 96%, как было показано с помощью ВЭЖХ на колонке НС-С18 (Agilent, 4,6×250 мм, 5 мкм) с элюированием градиентами 50% водного МеОН.
Способ С: В запаянную пробирку (15 мл) добавляли (5-бромтиен-2-ил)метанамин (384 мг, 2 ммоль), 6-хлорпуринрибофуранозид (287 мг, 1 ммоль) и диизопропилэтиламин (3 мл, 17 ммоль) в EtOH (8 мл). Запаянную пробирку помещали в полость фокусируемого однорежимного микроволнового реактора (СЕМ Discover) и облучали при 100 Вт в течение 20 мин при 80°C. Растворитель удаляли на роторном испарителе. Остаток очищали посредством флэш-хроматографии (силикагель, MeOH/EtOAc=1:9) и перекристаллизовывали из МеОН с получением целевого продукта JMF3464 (159 мг, выход 36%).
C15H16BrN5O4S; желтый порошок; т.пл. 141,4-141,7°C; [α]25D=-43,0 (ДМСО, с=1,0); ТСХ (2-пропанол/гексан (2:3)) Rƒ=0,38;1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,55 (1Н, уш s), 8,40 (1Н, s), 8,29 (1Н, уш s), 7,03 (1Н, d, J=3,6 Гц), 6,78 (1Н, d, J=4,0 Гц), 5,90 (1Н, d, J=6,0 Гц), 5,45 (1Н, d, J=6,0 Гц), 5,34-5,32 (1Н, m), 5,19 (1Н, d, J=4,4 Гц), 4,76 (2Н, s), 4,63-4,59 (1Н, m), 4,15-4,14 (1Н, m), 3,96-3,95 (1Н, m), 3,69-3,65 (1Н, m), 3,57-3,52 (1Н, m),13С ЯМР (ДМСО-d6, 100 МГц) δ 153,9, 152,2, 148,5, 145,0, 140,2, 129,6, 126,3, 120,0, 109,7, 87,9, 85,8, 73,5, 70,6, 61,6, 42,9, ИЭР-МС рассчитано для C15H17BrN5O4S: 442,0185, найдено: m/z 442,0189 [М+Н]+.
N6-(Тиен-3-ил-метил)аденозин (JMF3461)
Смесь 3-(аминометил)тиофена (0,25 мл, 2,5 ммоль), 6-хлорпуринрибофуранозида (143 мг, 0,5 ммоль) и диизопропилэтиламина (2 мл, 12 ммоль) в 1-пропаноле (25 мл) нагревали при 80°C в течение 6 ч. Смесь концентрировали на роторном испарителе и перекристаллизовывали из МеОН с получением целевого продукта JMF3461 (136 мг, выход 75%). Чистота продукта составляла 99%, как было показано с помощью ВЭЖХ на колонке НС-С18 (Agilent, 4,6×250 мм, 5 мкм) с элюированием градиентами 50% водного МеОН. C15H17N5O4S; желтый порошок; т.пл. 134,3-135,1°C; [α]24D=-58,6 (ДМСО, с=1,0); ТСХ (2-пропанол/гексан, (2:3)) Rƒ=0,33;1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,36 (2Н, уш s), 8,22 (1Н, s), 7,43 (1Н, dd, J=3,5 Гц), 7,28 (1Н, d, J=1,6 Гц), 7,09 (1Н, d, J=4,8 Гц), 5,88 (1Н, d, J=6,4 Гц), 5,43 (1Н, d, J=6,0 Гц), 5,38 (1Н, q, J=4,6 Гц), 5,17 (1Н, d, J=4,4 Гц), 4,69 (2Н, s), 4,63-4,16 (1Н, m), 4,14-4,13 (1Н, m), 3,97-3,95 (1Н, m), 3,69-3,64 (1Н, m), 3,57-3,52 (1Н, m),13С ЯМР (ДМСО-d6, 100 МГц) δ 154,4, 152,3, 148,5, 140,8, 139,9, 127,9, 125,1, 120,0, 119,8, 87,9, 85,9, 73,5, 70,7, 61,7, 42,9; ИЭР-МС рассчитано для C15H18N5O4S: 364,1080, найдено: m/z 364,1079 [М+Н]+.
N6-(Тиен-2-ил-метил)аденозин (JMF3462)
Смесь 2-(аминометил)тиофена (0,25 мл, 2,5 ммоль), 6-хлорпуринрибофуранозида (143 мг, 0,5 ммоль) и диизопропилэтиламина (2 мл, 12 ммоль) в 1-пропаноле (25 мл) нагревали при 80°C в течение 7 ч. Смесь концентрировали на роторном испарителе и перекристаллизовывали из МеОН с получением целевого продукта JMF3462 (154 мг, выход 85%). Чистота продукта составляла 99%, как было показано с помощью ВЭЖХ на колонке НС-С18 (Agilent, 4,6×250 мм, 5 мкм) с элюированием градиентами 50% водного МеОН. C15H17N5O4S; белый порошок; т.пл. 149,2-149,7°C; [α]25D=-68,2 (ДМСО, с=1,0); ТСХ (2-пропанол/гексан, (2:3)) Rƒ=0,35;1Н ЯМР (ДМСО-d6, 400 МГц) δ 8,51 (1Н, уш s), 8,39 (1Н, s), 8,27 (1Н, уш s), 7,32 (1Н, d, J=5,2 Гц), 7,28 (1Н, d, J=3,2 Гц), 6,93 (1Н, dd, J=1,8, 2,6 Гц), 5,89 (1Н, d, J=6,0 Гц), 5,46-5,45 (1Н, m), 5,36 (1Н, q, J=4,6 Гц), 5,20-5,18 (1Н, m), 4,64 (2Н, s), 4,16-4,13 (1Н, m), 3,97-3,95 (1Н, m), 3,70-3,65 (1Н, m), 3,58-3,52 (1Н, m), 3,57-3,52 (1Н, m),13С ЯМР (ДМСО-d6 100 МГц) δ 154,1, 152,2, 148,5, 142,9, 140,0, 126,5, 125,3, 124,7, 120,0, 87,9, 85,9, 73,5, 70,6, 61,6, 42,9; ИЭР-МС рассчитано для C15H18N5O4S: 364,1080, найдено: m/z 364,1081 [М+Н]+.
N6-[(4-Бромтиен-2-ил)метил]аденозин
Смесь (4-бромтиен-2-ил)метанамина (1152 мг, 6 ммоль), 6-хлорпуринрибофуранозида (214 мг, 0,75 ммоль) и диизопропилэтиламина (3 мл, 18 ммоль) в 1-пропаноле (30 мл) нагревали при 70°C в течение 7 ч. Смесь, содержащую целевой продукт и не вступивший в реакцию (3-бромтиен-2-ил)метанамин, обрабатывали ди-трет-бутилдикарбонатом (1,4 мл, 6 ммоль) в ТГФ (8 мл) и NaHCO3 (1 г, 1,2 ммоль) при комнатной температуре в течение 2 ч. Смесь концентрировали на роторном испарителе и очищали посредством флэш-хроматографии (силикагель, MeOH/EtOAc=1:9) с получением N6-[(4-бромтиен-2-ил)метил]аденозина.
N6-[(3-Бромтиен-2-ил)метил] аденозин
В запаянную пробирку (15 мл) добавляли (3-бромтиен-2-ил)метанамин (576 мг, 3 ммоль), 6-хлорпуринрибофуранозид (430 мг, 1,5 ммоль) и диизопропилэтиламин (4,5 мл, 15,5 ммоль) в EtOH (10 мл). Запаянную пробирку помещали в полость фокусируемого однорежимного микроволнового реактора (СЕМ Discover) и облучали при 100 Вт в течение 20 мин при 80°C. Растворитель удаляли на роторном испарителе. Остаток очищали посредством флэш-хроматографии (силикагель, МеОН/EtOAc=1:9) с получением N6-[(3-бромтиен-2-ил)метил]аденозина.
N6-[(2-Бромтиен-3-ил)метил]аденозин
В запаянную пробирку (15 мл) добавляли (2-бромтиен-3-ил)метанамин (384 мг, 2 ммоль), 6-хлорпуринрибофуранозид (287 мг, 1 ммоль) и диизопропилэтиламин (3 мл, 17 ммоль) в EtOH (8 мл). Запаянную пробирку помещали в полость фокусируемого однорежимного микроволнового реактора (СЕМ Discover) и облучали при 100 Вт в течение 20 мин при 80°C. Растворитель удаляли на роторном испарителе. Остаток очищали посредством флэш-хроматографии (силикагель, МеОН/EtOAc=1:9) с получением N6-[(2-бромтиен-3-ил)метил]аденозина.
N6-[(4-Бромтиен-3-ил)метил]аденозин
Смесь (4-бромтиен-3-ил)метанамина (768 мг, 4 ммоль), 6-хлорпуринрибофуранозида (143 мг, 0,5 ммоль) и диизопропилэтиламина (2 мл, 12 ммоль) в 1-пропаноле (20 мл) нагревали при 70°C в течение 7 ч. Смесь, содержащую целевой продукт и не вступивший в реакцию (4-бромтиен-3-ил)метанамин, обрабатывали ди-трет-бутилдикарбонатом (0,92 мл, 4 ммоль) в ТГФ (6 мл) и NaHCO3 (672 мг, 8 ммоль) при комнатной температуре в течение 2 ч. Смесь концентрировали на роторном испарителе и очищали посредством флэш-хроматографии (силикагель, МеОН/EtOAc=1:9) с получением N6-[(4-бромтиен-3-ил)метил]аденозина.
6-N-(5-Бромтиен-3-ил)метил-6-N-трет-бутоксикарбонил-2',3'-O-изопропилиден-5'-O-(трет-бутилдиметилсилил)аденозин
Раствор соединения 6 (360 мг, 0,68 ммоль), (5-бромтиен-3-ил)метанола (197 мг, 1,02 ммоль) и трифенилфосфина (271 мг, 1,02 ммоль) в безводном ТГФ (10 мл) перемешивали при 45°C в течение 2 мин в атмосфере N2. Добавляли по каплям диизопропилазодикарбоксилат (DIAD, 0,20 мл, 1,02 ммоль). Смесь перемешивали до тех пор, пока анализ посредством ТСХ не показал исчезновение соединения 6. Смесь концентрировали при пониженном давлении на роторном испарителе. Неочищенный продукт очищали посредством флэш-хроматографии (EtOAc/гексан=1:9) с получением 6-N-(5-бромтиен-3-ил)метил-6-N-трет-бутоксикарбонил-2',3'-O-изопропилиден-5'-O-(трет-бутилдиметилсилил)аденозина.
N6-[(5-Бромтиен-3-ил)метил]аденозин
Суспензию 6-N-(5-бромтиен-3-ил)метил-6-N-трет-бутоксикарбонил-2',3'-O-изопропилиден-5'-O-(трет-бутилдиметилсилил)аденозина (138 мг, 0,19 ммоль) в деионизированной воде (5 мл) и ТГФ (1 мл) перемешивали и охлаждали на ледяной бане. Добавляли по каплям при 0°C трифторуксусную кислоту (5 мл). Смесь перемешивали в течение 10 мин, ледяную баню удаляли, и смесь перемешивали в течение еще 30 мин. Смесь концентрировали при пониженном давлении на роторном испарителе. Неочищенный продукт очищали посредством флэш-хроматографии (MeOH/EtOAc=1:49) с получением N6-[(5-бромтиен-3-ил)метил]аденозина.
(5-Хлортиен-2-ил)метанамин
Смесь 2-(аминометил)тиофена (1 мл, 10 ммоль), ди-трет-бутилдикарбоната (2,5 мл, 11 ммоль) и NaHCO3 (840 мг, 10 ммоль) в ТГФ (13 мл) перемешивали при комнатной температуре в течение 3 ч с получением суспензии, содержащей бледно-желтое твердое вещество. Смесь концентрировали при пониженном давлении, и остаток экстрагировали CH2Cl2 и H2O. Органическую фазу сушили над MgSO4, фильтровали и очищали посредством флэш-хроматографии на колонке с силикагелем с элюированием смесью EtOAc/гексан (1:20) с получением трет-бутил(тиен-2-ил)метилкарбамата (C10H15NO2S, 1,62 г, выход 76%).
Смесь трет-бутил(тиен-2-ил)метилкарбамата (106 мг, 0,5 ммоль), N-хлорсукцинимида (73 мг, 0,55 ммоль) в бензоле (0,3 мл) перемешивали при 80°C. Через 2 ч добавляли уксусную кислоту (0,3 мл, 5 ммоль) и проводили реакцию в течение еще 21 ч. Смесь экстрагировали CH2Cl2 и H2O. Органическую фазу сушили над MgSO4, фильтровали и очищали посредством флэш-хроматографии на колонке с силикагелем с элюированием смесью EtOAc/гексан (1:20) с получением трет-бутил(5-хлортиен-2-ил)метилкарбамата (C10H14ClNO2S, 82,6 мг, выход 67%).
Смесь полученного выше соединения (65 мг, 0,26 ммоль) и ТФУ (1 мл, 13 ммоль) в CH2Cl2 (1 мл) перемешивали в течение 3 ч при комнатной температуре. Раствор концентрировали при пониженном давлении с получением (5-хлортиен-2-ил)метанамина (выход ~100%). C5H6NSCl; бледно-желтое твердое вещество;1Н ЯМР (400 МГц, CD3OD) δ 7,07 (1Н, d, J=4,0 Гц), 6,95 (1Н, d, J=3,6 Гц), 4,26 (2Н, s);13С ЯМР (100 МГц, CD3OD) δ 134,9, 132,8, 130,7, 128,0, 38,8; ИЭР-МСВР рассчитано для C5H7ClNS: 147,9988, найдено: m/z 147,9995 [М+Н]+.
N6-[(5-Хлортиен-2-ил)метил]аденозин (JMF3818)
Смесь (5-хлортиен-2-ил)метанамина (35,4 мг, 0,24 ммоль), 6-хлорпуринрибофуранозида (0,12 ммоль) и диизопропилэтиламина (0,36 мл, 2 ммоль) в EtOH (1 мл) перемешивали в запаянной пробирке при 80°C с помощью микроволнового излучения в течение 30 мин. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением бледно-желтого масла, которое последовательно промывали H2O и МеОН с получением указанного в заголовке соединения JMF3818. C15H16ClN5O4S; белое твердое вещество;1Н ЯМР (400 МГц, CD3OD) δ 8,29 (1Н, s), 8,27 (1Н, s), 6,88 (1Н, d, J=3,6 Гц), 6,79 (1Н, d, J=3,6 Гц), 5,96 (1Н, d, J=6,8 Гц), 4,74-4,77 (1Н, m), 4,32-4,34 (1Н, m), 4,17 (1Н, d, J=2,8 Гц), 3,89 (1Н, dd, J=12,4, 2,0 Гц), 3,75 (1Н, dd, J=12,4, 2,8 Гц);13С ЯМР (100 МГц, CD3OD) δ 156,0, 153,6, 142,8, 142,0, 129,9, 127,0, 126,6, 121,7, 91,5, 88,4, 75,6, 72,9, 63,7, 40,4; ИЭР-МСВР рассчитано для C15H17ClN5O4S: 398,0690, найдено: m/z 398,0692 [М+Н]+.
Фармакокинетическое исследование. Соединение вводили в виде водных растворов в обычном физиологическом растворе. Мыши-самцы ICR были приобретены в компании BioLASCO Taiwan Co., Ltd. Для измерения биодоступности при пероральном введении исследуемых соединений (Т1-11 и JMF3464) забирали образцы крови мышей-самцов ICR (возраст 6 недель; 20-25 г) после перорального (10 мг/кг) или внутривенного (1 мг/кг) введений исследуемых соединений. Для Т1-11 образцы крови забирали через 2, 10, 30, 60, 120, 360 минут после внутривенного введения и через 15, 30, 45, 60, 120, 360 минут после перорального введения. Для JMF3464 образцы крови забирали через 2, 10, 30, 60, 120, 240, 360, 480 минут после внутривенного введения и через 15, 30, 60, 90, 120, 240, 360, 480 минут после перорального введения. Образцы крови экстрагировали метанолом с 0,1% муравьиной кислотой, и затем 10 мкл экстрагированных образцов вводили в систему для СЭЖХ-МСМС для количественного определения.
Фармакокинетические параметры получали с применением программы для фармакокинетических расчетов WinNonlin с приведением данных в соответствие с некомпартментной моделью. Фармакокинетические параметры, включая площадь под кривой зависимости концентрации в плазме от времени (AUC) до последнего момента забора, (AUC0-120), до бесконечности (AUC0-∞), период полувыведения в конечной фазе (T1/2), максимальную концентрацию соединения в плазме (Cmax), время достижения Cmax (Tmax) и константу скорости первого порядка, связанную с конечным участком кривой (k), оценивали с помощью линейной регрессии зависимости времени от логарифма концентрации. Общий плазменный клиренс (CL) рассчитывали как доза/AUCв.в. Биодоступность при пероральном введении (F) исследуемого соединения путем перорального введения рассчитывали как значение AUC0-∞ для пероральной дозы, разделенное на AUC0-∞ для в.в. дозы.
ПРИМЕР 2
Анализы связывания радиолиганда. Анализы связывания радиолиганда были проведены в компании MDS Pharma Services Taiwan (Taipei, Taiwan) с применением стандартных протоколов связывания. Для анализов связывания A2AR мембранные белки, полученные из клеток НЕК293, избыточно экспрессирующих A2AR человека, инкубировали в буфере для реакции [50 мМ Трис-HCl (pH 7,4), 10 мМ MgCl2, 1 мМ ЭДТА и 2 Ед/мл аденозиндезаминазы], содержащем3H-CGS21680 (50 нМ) в течение 90 мин при 25°C. Неспецифическое связывание оценивали в присутствии 50 мкМ аденозин-5'-N-этилкарбоксамида. Для измерения аффинности связывания Т1-11 по отношению к A3R мембранные белки, полученные из клеток яичника китайского хомячка (CHO)-K1, избыточно экспрессирующих A3R человека, инкубировали с3Н-АВ-МЕСА (0,5 нМ) в течение 60 мин при 25°C буфере для реакции, содержащем 25 мМ HEPES (pH 7,4), 5 мМ MgCl2, 1 мМ СаС12 и 0,1% бычьего сывороточного альбумина. Неспецифическое связывание оценивали в присутствии 1 мкМ IB-MECA (Tocris Bioscience, Ellisville, МО, USA). Анализы связывания для транспортеров аденозина проводили, как описано ранее. Мембранные фракции, полученные из коры головного мозга морских свинок Duncan Hartley, инкубировали с3Н-меченным 6-[(4-нитробензил)тио]-9-β-D-рибофуранозилпурином (NBTI, 0,5 нМ) в течение 30 мин при 25°C в буфере для инкубации, содержащем 50 мМ Трис-HCl (pH 7,4). Неспецифическое связывание оценивали в присутствии 5 мкМ NBTI, эффективного ингибитора равновесных нуклеозидных транспортеров. Необходимо отметить, что NBTI представляет собой высокоаффинный ингибитор ENT1 и ингибирует только (h)ENT1 человека в конценрации 0,5 нМ. Реакции завершали путем фильтрования через фильтры из стекловолокна GF/B и промывания буфером для реакции.
Культура клеток и временная трансфекция
Клетки крысы PC12, приобретенные в Американской коллекции типовых культур (АТСС; Manassas, VA, USA) поддерживали в среде DMEM, содержащей 10% лошадиной сыворотки и 5% ФБС, и инкубировали в CO2-инкубаторе (5%) при 37°C. LIPOFECTAMINE™ 2000 (Invitrogen) применяли в качестве переносящей среды для переноса плазмид в клетки в соответствии с протоколом производителя. Плазмиды были любезно предоставлены Dr. Pulst (Факультет неврологии, Университет штата Юта, США). Как правило, 5 мкг ДНК вместе с 5 мкл LIPOFECTAMINE™ 2000 вносили в каждую лунку 6-луночных планшетов. Число посеянных клеток составляло (1~1,5)×106клеток/лунка. После трансфекций в течение 6 ч клетки обрабатывали реагентами в течение еще 24 ч. Изображения регистрировали с помощью инвертированного флуоресцентного микроскопа Zeiss Axiovert 200М (
MTT-тест. Выживаемость оценивали посредством анализа метаболизма 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия бромида (МТТ). Вкратце, после обработки, МТТ добавляли в среду (0,5 мг/мл) и инкубировали при 37°C в течение 2~3 ч. Число посеянных клеток составляло 1×104 клеток/лунка в 96-луночном планшете. После удаления среды в лунки вносили ДМСО для растворения кристаллов формазана, и в каждой лунке измеряли поглощение при 570 и 630 нм на устройстве для прочтения микропланшетов для иммуноферментного анализа (ИФА).
ПРИМЕР 3
Животные и введение лекарственных средств. Мыши-самцы R6/2 (Mangiarini et al., 1996 Cell. 87:493-506) и однопометные контрольные мыши первоначально были получены от Jackson Laboratories (Bar Harbor, ME, USA) и спарены с контрольными мышами-самками (B6CBAFI/J). Потомство идентифицировали с помощью метода генотипирования с применением полимеразной цепной реакции (ПЦР) по геномной ДНК, выделенной из тканей хвоста, с использованием праймеров, расположенных в трансгене (5'-CCGCTCAGGTTCTGCTTTTA-3'; SEQ ID NO: 1 и 5'-GGCTGAGGAAGCTGAGGAG-3'; SEQ ID NO: 2), для обеспечения того, что количество СAG повторов оставалось приблизительно равным 150. Животных содержали в Лаборатории по уходу за животными Института биомедицинских наук в условиях цикла 12/12-ч свет/темнота. Массу тела мышей регистрировали один раз в день. Эксперименты на животных проводили в соответствии с протоколами, одобренными Институциональным комитетом по содержанию и использованию животных Академии Синика, Тайбэй, Тайвань.
Способность удерживаться на вращающемся стержне. Координацию движений оценивали с применением устройства с вращающимся стержнем (UGO BASILE, Comerio, Italy) при постоянной скорости (12 об/мин) в течение периода времени 2 мин (Carter et al., 1999 J Neurosci. 19:3248-3257). Всех мышей в возрасте 4 недель обучали в течение 2 дней для обеспечения возможности их ознакомления с устройством с вращающимся стержнем. Затем животных подвергали тесту три раза в неделю в возрасте 4~12 недель. Для каждого теста животных помещали в устройство до начала вращения. Время удерживания до падения регистрировали автоматически. Каждую мышь подвергали трем испытаниям в течение максимум 2 мин для каждого испытания.
ПРИМЕР 4
Животные. Мыши C57BL/6 были приобретены в Национальном центре лабораторных животных (Тайвань). Трансгенные мыши (ATAXN2Q127) были предоставлены доктором Пульстом (Dr. Pulst). Мышей содержали в звуконепроницаемом помещении в условиях цикла 12/12-ч свет/темнота и при контролируемой температуре (22±2°C). Пища и вода были доступны без ограничения. Были предприняты все усилия для минимизации количества используемых животных и их боли и дискомфорта в соответствии с принципами и директивами Руководства по содержанию и использованию лабораторных животных Национального института здравоохранения (NIH). Эти эксперименты также были рассмотрены и одобрены Институциональным комитетом по содержанию и использованию животных в Национальном исследовательском институте китайской медицины (№ свидетельства: 100-15).
Анализ удержания на фильтре. Этот метод соответствовал описанному Wanker et al. (1999 Methods Enzymol. 309:375-386) с некоторыми изменениями. Вкратце, собранные клетки повторно суспендировали в буфере для лизиса (50 мМ Трис-HCl (pH 8,8), 100 мМ NaCl, 5,0 мМ MgCl2, 1 мМ ЭДТА и 0,5% (масс./об.) IPGEAL, содержащий 1х смесь ингибиторов протеаз (Roche Diagnostics, Indianapolis, IN, USA)) и обрабатывали ультразвуком в течение 10 с (1 импульс/с). Образцы с равными концентрациями белка (15-20 мкг/лунка) в каждой группе фильтровали через ацетатцеллюлозную мембрану, предварительно уравновешенную 2% додецилсульфатом натрия (ДСН) (0,2 мкм; Whatman, Maidstone, Kent, UK) с использованием прибора Bio-Dot SF (Bio-Rad, Hercules, CA, USA). Во время отсасывания каждую лунку дважды промывали 200 мкл 0,1% SDS. Блот блокировали в ТСБ (100 мМ Трис-HCl и 150 мМ NaCl; pH 7,4), содержащем 3% обезжиренное сухое молоко, в течение 1 ч при комнатной температуре и затем инкубировали с антителами против полиглутамина (1:5000; МАВ1574) в 3% бычьем сывороточном альбумине (БСА) с 0,02% NaN3 (4°C в течение ночи) для детекции нормального и мутантного ATAXN2. Последующие методы являлись такими же, как описанные выше.
Способность удерживаться на вращающемся стержне. В данном исследовании использовали мышей дикого типа и трансгенных мышей в возрасте 5 недель. Координацию движений оценивали с применением устройства с вращающимся стержнем (UGO BASILE, Comerio, Italy) при увеличивающейся скорости (от 10 до 28 об/мин) в течение периода времени 5 мин. Всех мышей в возрасте 5 недель обучали в течение 2 дней для обеспечения возможности их ознакомления с устройством с вращающимся стержнем. Кроме того, на этой неделе часть трансгенных мышей начинала получать Т1-11, растворенный в их повседневной питьевой воде. Затем животных подвергали тесту согласно протоколу 2 раза в неделю начиная с возраста 6 недель. Для каждого теста животных помещали в устройство до начала вращения. Время удерживания до падения регистрировали автоматически. Каждую мышь подвергали 2 испытаниям за период времени. Мышам обеспечивали отдых в течение 20~30 минут между испытаниями.
ПРИМЕР 5
Поведенческий тест. Функции равновесия и координации у мышей определяли путем проведения теста вращающегося стержня. Мышей помещали на движущийся барабан устройства с вращающимся стержнем (модель с ускорением, Ugo Basile Biological Research Apparatus), который затем ускоряли до тех пор, пока мышь не падала с барабана на пластину с остановкой таймера. Время удерживания до падения измеряли в четырех ежедневных испытаниях в течение 4 дней.
Иммуногистохимическое окрашивание. Мышей дикого типа или трансгенных мышей SCA3 подвергали анестезии и осуществляли транскардиальную перфузию 4% параформальдегидом в ФСБ. Ткань мозга уравновешивали в заключающей среде Tissue-Tek и замораживали в жидком азоте. Коронарные срезы (20 мкм), полученные с помощью резки в криостате, пермеабилизовали в 0,1% Тритон Х-100 и инкубировали при 4°C в течение 48 часов с разбавленной антисывороткой, содержащей моноклональные антитела против NeuN (Chemicon). Далее срезы промывали и инкубировали с биотинилированным IgG лошади против антител мыши с последующей инкубацией с комплексом авидин-биотин-пероксидаза хрена. Затем срезы промывали и обрабатывали раствором диаминобензидина. NeuN-положительные нейроны визуализировали и подсчитывали с помощью микроскопа Leica DM2500, оснащенного ПЗС-камерой Retiga-2000R (QImaging) и трехосевой платформой с приводом MAC 600 (Ludl Electronics) с компьютерным управлением, с применением программного обеспечения StereoInvestigator (MBF Bioscience). Каждый анализ включал обработку 15 срезов мозга для каждой мыши.
ПРИМЕР 6
Животные и доставка лекарственных средств. Трансгенных мышей B6SJL-Tg(Prnp-TARDBP)4Jlel/J приобретали в Jackson Laboratory (Bar Harbor, Maine USA) и разводили в Национальном центре лабораторных животных в Тайнане. Трансгенных мышей подвергали скринингу с помощью ПЦР с прямым праймером 5'-GGTGGTGGGATGAACTTTGG-3' (SEQ ID NO: 3) и обратным праймером 5'-GTGGATAACCCCTCCCCCAGCCTAGAC-3' (SEQ ID NO: 4). Мыши дикого типа являлись нетрансгенными однопометниками. Мышей в возрасте 6 недель подвергали операции по установке микроосмотической помпы ALZET, модель 1004 (DURECT Corporation, Cupertino, СА, USA), содержащей, как указано, ДМСО или JMF1907, помещенной подкожно в переднюю боковую область брюшной полости. Помпу заменяли каждые 28 дней.
Сила захвата. Силу захвата измеряли с помощью измерителя силы захвата (TSE Systems, Inc., МО, USA). Вкратце, мышь брали рукой за хвост и позволяли ей схватить регулируемую по высоте рукоятку, установленную на датчике силы. Растягивающее усилие прикладывали к мыши за хвост. Максимальная сила была показана на цифровом дисплее панели подключенного блока управления, когда мышь отпускала свой захват. Каждую мышь повторно подвергали тесту 3 раза.
Способность удерживаться на вращающемся стержне. Координацию движений мышей дикого типа и трансгенных мышей оценивали с применением устройства (UGO BASILE, Comerio, Italy) при постоянной скорости (40 об/мин) в течение 120 сек. Всех мышей обучали в течение 2 дней в неделю в течение 2 недель. Затем мышей подвергали тесту согласно протоколу 2 раза в неделю начиная с возраста 9 недель. Для каждого теста животных помещали в устройство до начала вращения. Время удерживания до падения регистрировали автоматически. Каждую мышь подвергали 3 испытаниям за период времени. Мышам обеспечивали отдых в течение 20 минут между испытаниями.
Культура клеток и трансфекция. Линия клеток двигательных нейронов (NSC34) была щедрым даром от Dr. Neil Cashman (Brain Research Centre, The University of British Columbia, Canada), и ее культивировали в модифицированной по методу Дульбекко среде Игла (DMEM) с высоким содержанием глюкозы, содержащей 10% фетальной телячьей сыворотки (ФТС), 2 мМ L-глутамина и 1% пенициллина/стрептомицина (Invitrogen GibcoBRL, Carlsbad, СА, USA) при 37°C в атмосфере 5% CO2.
ПРИМЕР 7
Мыши. Мыши-самки C57BL6N в возрасте 8-12 недель были приобретены в компании BioLASCO (Yi-Lan, Taiwan).
Модель индуцированной кислотой хронической генерализованной боли. Модель фибромиалгии являлась модификацией модели индуцированной кислотой хронической боли, созданной группой Sluka (Sluka et al., 2003 Pain. 106:229-239). Мыши были подвергнуты короткой анестезии с помощью 2% газообразного изофлурана и получали в.м. инъекцию 20 мкл генистеина (1 мкМ) в левую икроножную мышцу. Через 3 минуты осуществяли инъекцию 20 мкл физиологического раствора, содержащего кислоту (pH 4,0), в то же место. Затем у мышей развивалась длительная механическая гипералгезия в течение более 2 недель. Обезболивающее эффекты Т1-11 (п.о. с применением зонда 0,9 мм/7 см) исследовали через 4 дня после развития у мышей механической гипералгезии. Механическую гипералгезию анализировали путем проверки реакции отдергивания задних лап мыши при стимуляции нитью фон Фрея с усилием 0,2 мН.
Модель периодического холодового стресса. Модель фибромиалгии была разработана группой Ueda, при этом мышей подвергали периодическому холодовому стрессу в течение 2 дней (Nishiyori and Ueda, 2008 Mol Pain. 4:52). У мышей, подвергнутых периодическому холодовому стрессу, развивалась длительная (>2 недель) механическая и термическая гипералгезия. Обезболивающее эффекты JMF3464 (в.б. или п.о.) исследовали на этих мышах через 5 дней после периодического холодового стресса. Механическую гипералгезию анализировали путем проверки реакции отдергивания задних лап мыши при стимуляции нитью фон Фрея с усилием 0,2 мН.
Предшествующее описание типовых вариантов реализации настоящего изобретения было представлено исключительно в целях иллюстрации и описания и не является исчерпывающим и не ограничивает настоящее изобретение до точных раскрытых форм. Возможны многие модификации и варианты в свете вышеприведенной идеи.
Варианты реализации и примеры были выбраны и описаны с целью объяснения принципов настоящего изобретения и их практического применения с тем, чтобы обеспечить другим специалистам в данной области техники возможность использования настоящего изобретения и различных вариантов реализации с различными модификациями, подходящими для конкретного рассматриваемого применения. Альтернативные варианты реализации будут очевидными для специалистов в данной области техники, к которой относится настоящее изобретение, без выхода за его рамки.
Полное содержание всех ссылок, приведенных и обсужденных в настоящем описании, включено в настоящий документ посредством ссылки в той же степени, как если бы каждая ссылка была индивидуально включена посредством ссылки.
Реферат
Настоящее изобретение относится к соединению для лечения нейродегенеративных заболеваний и боли, способу его получения и композициям на его основе. Предложено соединение, имеющее следующую структуру:или его фармацевтически приемлемая соль, где Х представляет собой Cl или Br. Предложенный способ включает стадии: (a) взаимодействия (2',3'-О-изопропилиден)аденозина в присутствии основания с агентом, защищающим гидроксильную группу, с получением производного (2',3'-О-изопропилиден)аденозина, содержащего гидроксил-защитную группу; (b) взаимодействия указанного производного (2',3'-О-изопропилиден)аденозина, содержащего гидроксилзащитную группу, с агентом, защищающим аминогруппу, с получением производного (2',3'-О-изопропилиден)аденозина, содержащего гидроксил защитную группу и аминозащитную группу; (c) проведения реакции сочетания путем взаимодействия указанного производного (2',3'-О-изопропилиден)аденозина, содержащего гидроксилзащитную и аминозащитную группы, с соединением формулы 7, содержащим замещенную (тиенил)метильную группу:,где Х представляет собой Cl или Br и Y представляет собой Х, ОН, метансульфонат (OSOCH, OMs), п-толуолсульфонат (OSOCH-п-СН, OTs) или трифторметансульфонат (OSOCF, OTf), с получением продукта, содержащего указанные защитные группы и замещенную (тиенил)метильную группу; и (d) удаления защитных групп из продукта со стадии (c) в кислой среде. Предложены новые соединения, эффективные для лечения нейродегенеративных заболеваний и боли, эффективные способы их получения и композиции на их основе. 5 н. и 12 з.п. ф-лы, 7 пр., 10 ил.
Формула

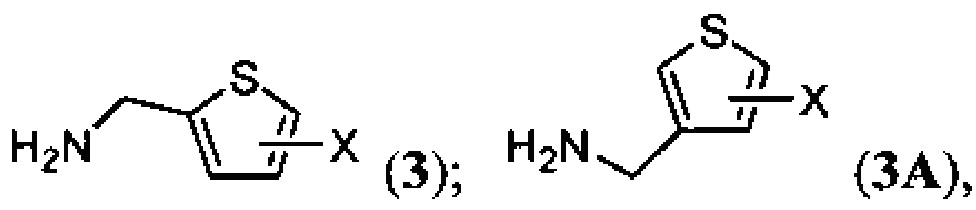

Комментарии