Производные 3-трифторметил-1-карба-1-детиа-3-цефем-4-карбоновой кислоты и промежуточный продукт для их получения - RU2089551C1
Код документа: RU2089551C1
Чертежи

Описание
Изобретение относится к 1-карба(1-детиа)-цефалоспориновым антибиотикам, промежуточным соединениям для их получения, фармацевтическим составам, включающим антибиотики, и к способу лечения инфекционных заболеваний у человека и других животных.
1-Карба(1-детиа)- цефалоспориновые антибиотики имеют бициклическую кольцевую систему, представленную следующей формулой, в которой система нумерации соответствует общепринятой в произвольной системе номенклатуры цефема.
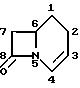
1-Карба(1-детиа)-цефалоспорины упоминаются для удобства в данном описании как 1-карбацефалоспорины или как 1-карба-(1-детиа)-3-цефем-4-карбоновые кислоты или их производные.
Получение 1-карбацефалоспоринов и C-3- замещенных метил-производных широко раскрывается в патенте США N 4226866 на имя Кристинсена и др. Хирата и др. в заявке на патент Великобритании N 2041923 раскрывают способ получения 3-Н и 3-гало-1-карбацефалоспоринов, тогда как Хатанака и др. Tetrahedron letters 24, N 44, стр.4837-4838 (1983), предлагают способ получения 3-гидрокси-(±)-1-карбоцефалоспоринов.
Хотя многие безопасные и сильнодействующие антибиотики класса β-лактамов известны и используются клинически, исследования в данном классе продолжаются в попытках обнаружить антибиотики с повышенной эффективностью, в частности против микроорганизмов, нечувствительных или резистентных относительно известных антибиотиков.
Изобретение предлагает антибиотики на основе 7-b-ациламино-1-карба(детиа)-3-цефем-4-карбоновой кислоты, замещенные в положении 3
трифторметилом.
Положение 7 этих антибиотиков замещено ациламино частью, такой как D-арилглицидиламино, или гетероциклически замещенной оксииминоацетиламиногруппой. Также предлагают фармацевтические
составы,
включающие антибиотики, промежуточные соединения и способы их получения. Примеры таких антибиотиков включают
7-b-D-фенилглицидиламино-1-карба(1-детиа)-3-трифторметил-3-цефем-4-карбоновую
кислоту,
7-b-D-p-гидроксифенилглициламидо-1-карба(1-детиа)-3-трифторметил-3-цефем-4-карбоновую кислоту,
7-b-D-m-метилсульфониламинофенилглициламино-1-карба(1-детиа)-3-трифторметил-3-цефем-4- карбоновую кислоту, а также
7-b-[(2-аминотиазол-4-ил)-(Z)- метоксииминоацетил]амино-1-карба(1-детиа)-3-трифторметил-3-цефем-4-карбоновую кислоту.
Изобретение предлагает соединения формулы (I):
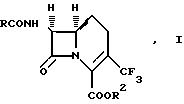
где
R2 означает водород или карбоксизащитную группу;
R означает замещенную метильную группу формулы:
R4 фенил или замещенный фенил формулы:
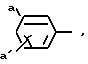
где
a и а' означают водород, галоген, гидрокси или (C1-C4) алкилсульфониламиногруппу; или
R4 обозначает тиазолил, замещенный аминогруппой; или
R является группой формулы
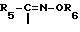
где
R5 обозначает тиазолил, замещенный аминогруппой,
R6 водород или (C1-C4) алкил, или
когда R2 обозначает водород, их фармацевтически приемлемые соли.
1-Карба(1-детиа)цефемы, представленные вышеприведенной формулой (I), где R2 обозначает водород, или их фармацевтически приемлемые соли ингибируют рост микроорганизмов, болезнетворных для человека и животных, и могут быть использованы для борьбы с инфекционными заболеваниями. Соединения изобретения получают в предлагаемом здесь способе в той же самой стехиометрической форме, в которой получают полусинтетические цефалоспориновые антибиотики.
Термин "карбоксизащитная группа", используемый в описании, относится к одному из сложноэфирных производных карбоновокислотной группа, обычно используемых для блокирования или защиты карбоновокислотной группы, тогда как реакции осуществляют в отношении других функциональных групп на соединении. Примерами таких защитных групп карбоновой кислоты являются 4-нитробензил, 4-метилбензил, 3, 4-диметоксибензил, 2, 4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, пентаметилбензил, 3,4-метилендиоксибензил, бензгидрил, 4,4'-диметоксибензгидрил, 2,2', 4, 4'-тетраметоксибензгидрил, трет-бутил, трет-амил, тритил, 4-метокситритил, 4,4'-диметокситритил, 4,4', 4''-триметокситритил, 2-фенилпроп-2-ил, триметилсилил, трет-бутилметилсилил, фенацил, 2,2, 2-трихлорэтил, β -(ди(н-бутил)метилсилил)этил, п-толуолсульфонилэтил, 4-нитробензилсульфонилэтил, аллил, циннамил, 1-(триметилсилилметил)проп-1-ен-3-ил и т.д. Вид используемой карбоксизащитной группы не является решающим, пока дериватизированная карбоновая кислота устойчива к условиям последующей реакции при других положениях кольцевой системы и ее можно отщепить в соответствующее время без разрыва остальной части молекулы. Предпочтительной защитной группой карбоновой кислоты является аллильная группа. Аналогичные карбоксизащитные группы, используемые в области цефалоспоринов, пенициллинов и пептидов, также можно использовать для защиты заместителей карбоксильной группы азетидинона. Другие примеры этих групп рассматриваются в работах Е.Хаслама "Защитные группы в органической химии", Дж. Г. У. МакОмие, ред. Пленум Пресс, Нью-Йорк, 1973, глава 5; Т.У. Грина "Защитные группы в органическом синтезе", Джон Уилли энд Санз, Нью-Йорк, 1981, глава 5. Родственный термин "защитный карбокси" означает, что карбоксильная группа замещена одной из вышеупомянутых карбоксизащитных групп.
В вышеприведенном определении соединений, представленных формулой (I), (C1-C4 )алкил относится к прямоцепочечным и разветвленным алкильным группам, таким как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил.
Когда в формуле (I) R обозначает замещенную фенильную группу, в которой заместитель(и) представлен(ы) в виде а и а', примерами таких групп являются галофенил, например 4-хлорфенил, 3-бромфенил, 2-фторфенил, 2,2-дихлорфенил и 3,5-дихлорфенил; гидроксифенил, например 2-гидроксифенил, 3-гидроксифенил, 4-гидроксифенил, 2,4-дигидроксифенил и 3,4-дигидроксифенил; алкилсульфониламино, например 3-метилсульфониламино, 4-метилсульфониламино, 3, 5-(диметилсульфониламино)фенил, 4-н-бутилсульфониламинофенил и 3-этилсульфониламинофенил.
Примеры соединений, представленных формулой
(I), когда R обозначает оксииминозамещенную группу,
представленную формулой:
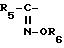
представляют собой, например, 2-(2-аминотиазол-4-ил)-2-метоксииминоацетил.
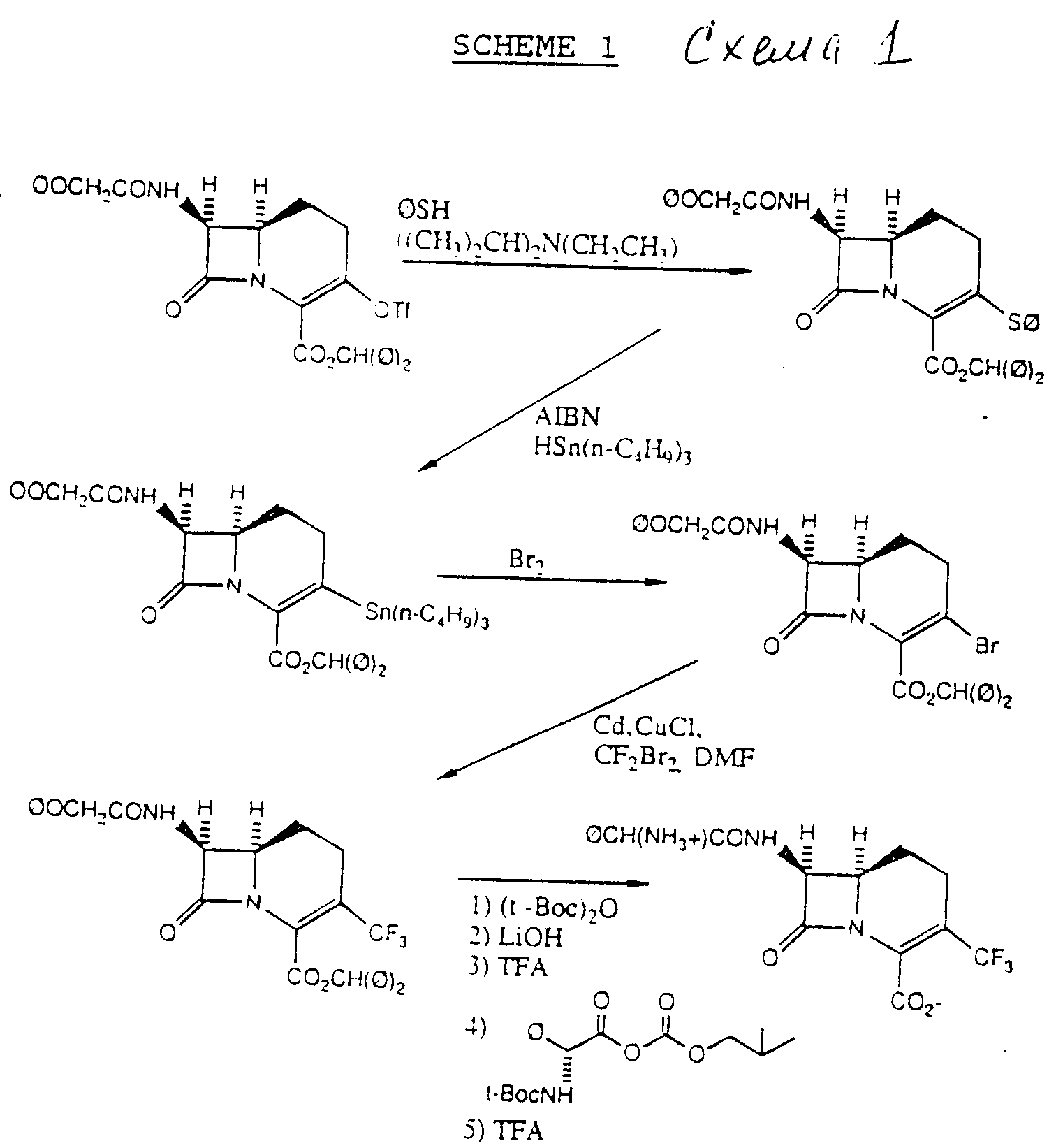
3-Трифторметил-карбацефемы формулы (I) могут быть получены в соответствии с приведенными в конце описания схемами.
3-Трифторметансульфонилокси ("трифлат" или "ТФ") карбацефем исходное вещество схемы 1 может быть получено способом Эванса и др. патент США N 4673737, который введен в данное описание в
качестве
ссылки. Это 3-трифлат - исходное вещество вначале подвергают взаимодействию с тиофенолом в атмосфере азота в присутствии амина, и предпочтительно третичного амина, такого как триэтиламин или
диизопропилэтиламин, с получением бензгидрил 7-β-феноксиацетиламино-1-карба(1-детиа)-3-фенилтио-3-цефем-4-карбоксилата. Промежуточный 3-фенилтио затем подвергают взаимодействию с гидридом
трибутилолова в атмосфере азота в присутствии свободно-радикального ингибитора, такого как 2,2'-азо(бис)изобутирилнитрил (AIBN), и нагревают, предпочтительно до температуры около 120oC, с
получением промежуточного 3-(три-н- бутил)станнана, как изображено на схеме 1. Это промежуточное соединение олова объединяют с положительным галогенирующим агентом, и предпочтительно бромом, с
получением бензгидрил-7 b-феноксиацетиламино-1-карба(1-детиа)-3-бром-3-цефем -4- карбоксилата. Промежуточное 3-бромсоединение затем нагревают (предпочтительно до температуры около 70oC) и
объединяют со смесью кадмия, хлористой меди, дибромдифторметана и ДМФ, получая бензгидрил-7 b-феноксиацетиламино-1-карба(1-детиа)-3-трифторметил-3-цефем-4-карбоксилат. Несмотря на то что ДМФ является
предпочтительным, другие пригодные соединения, включая соединения группы формулы:
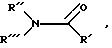
в которой
R' обозначает водород или (C1-C4) алкил;
R'' и R''' самостоятельно обозначают (C1-C4)алкил, или вместе с атомом азота образуют насыщенное кольцо, например пирролидин или пиперидин.
Это промежуточное соединение затем ацилируют с использованием ди-трет-бутилдикарбоната ((трет-BOC)2O) с последующей обработкой основанием, таким как LiOH, получая 7β-(трет-бутилоксикарбониламино) промежуточное соединение (не показано). (Дополнительные подробности данного взаимообмена трет-бутоксикарбонильной защитной группы на феноксиацетил-защитную группу можно найти в заявке на европатент N 8836996.5, публ. N 0301877 Докет N X-6913).
Группу трет-бутоксикарбонила (трет-BOC) и сложный бензгидрильный эфир можно отщепить с использованием известной методики, например с помощью трифторуксусной кислоты (TFA) в присутствии анизола. Полученное нуклеарное промежуточное соединение 7-амино-3-трифторметил затем N-ацилируют активированной формой желательной 7-ацильной группы. В схеме 1 D-фенилглициловую группу вводят путем взаимодействия 7-амино-нуклеарного промежуточного соединения с трет-BOC-защищенным ангидридом D-фенилглицина, образованным с использованием трет-BOC-защищенного фенилглицина и изобутилхлорформиата, как показано ниже.
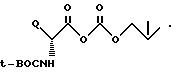
Любые оставшиеся трет-бутоксикарбонильные защитные группы затем можно отщепить путем обработки трифторуксусной кислотой. Несмотря на то что феноксиацетиловые и трет-бутоксикарбонильные (трет-BOC) группы используют в качестве аминозащитных групп, и бензгидрильную группу в качестве карбоксизащитной группы, специалист в данной области техники, относящейся к химии β-лактамов, поймет, что другие карбоксильные и аминозащитные группы служат в качестве функциональных эквивалентов.
Способ по схеме 1 осуществляют в основном в безводных условиях, которые представляют собой условия проведения реакции, свободные от присутствия воды. В соответствии с этим растворители сушат перед их использованием в способе. Пригодные органические растворители включают метиленхлорид, хлороформ, метиловый спирт, ди- или трихлорэтан, тетрагидрофуран (ТГФ), диметилпропилен-мочевину (DMPU), гексаметилфосфорный триамид (НMPA), диметилацетамид, тетрагидропиран, диоксан, ацетонитрил, простой диэтиловый эфир, диметилацетамид, диметилсульфоксид, диметоксиэтан и их смеси.
Свободно-радиальные ингибиторы описаны в статье Лейрда и Йоргенсена "Радиальная цепная реакция", Журнал органической химии, т.55, N 1, стр. 9-27 (1990), раскрытие которой введено в данное описание в качестве ссылки. Пригодные свободно-радикальные ингибиторы включают AIBN, Bu3SnH, Cl3CBr, бензоил пероксид и др. приведенные в вышеупомянутой статье на стр.19 под таблицей VI. Другие ингибиторы включают фотолиз X2, где X Cl, Br и I (Шлекер, Хенкель, Сибах. Chem.Ber. 110, 2880 (1977)), O2 (Руссел, Кауп y.A.C.S. 91, 3851 (1967)), соли металлов (Тунг, Ято. J.Chem, Soc. Chem.Commun, 205 (1978)) и сложные эфиры тиогидроксамовых кислот (Бартон, Крич, Кретзахман, Tetra, Letters, 25, 1955 (1984)).
Определенный выше термин "положительный галогенирующий агент" относится к соединению, имеющему электрофильный галоген, или, другими словами, соединению, приводящему к получению галогена, имеющего положительный заряд. Примерами таких агентов являются, например, SbF5, F2, IF5, BrF3, SF4, Cl2, HOCl, (CH2CO)2 NCl, N-хлорсукцинамид, Me3COCl, NO2Cl, SO2Cl2, Br2, 1,3-дибромгидрантоин, N,N-дибромбензолсульфонамид, HOBr, N-бромсукцинамид, C4 H8O2Br2, ICl, IBr, I2,N -иодосукцинамид и 1,3-дииодо-5,5-диметилгидантоин.
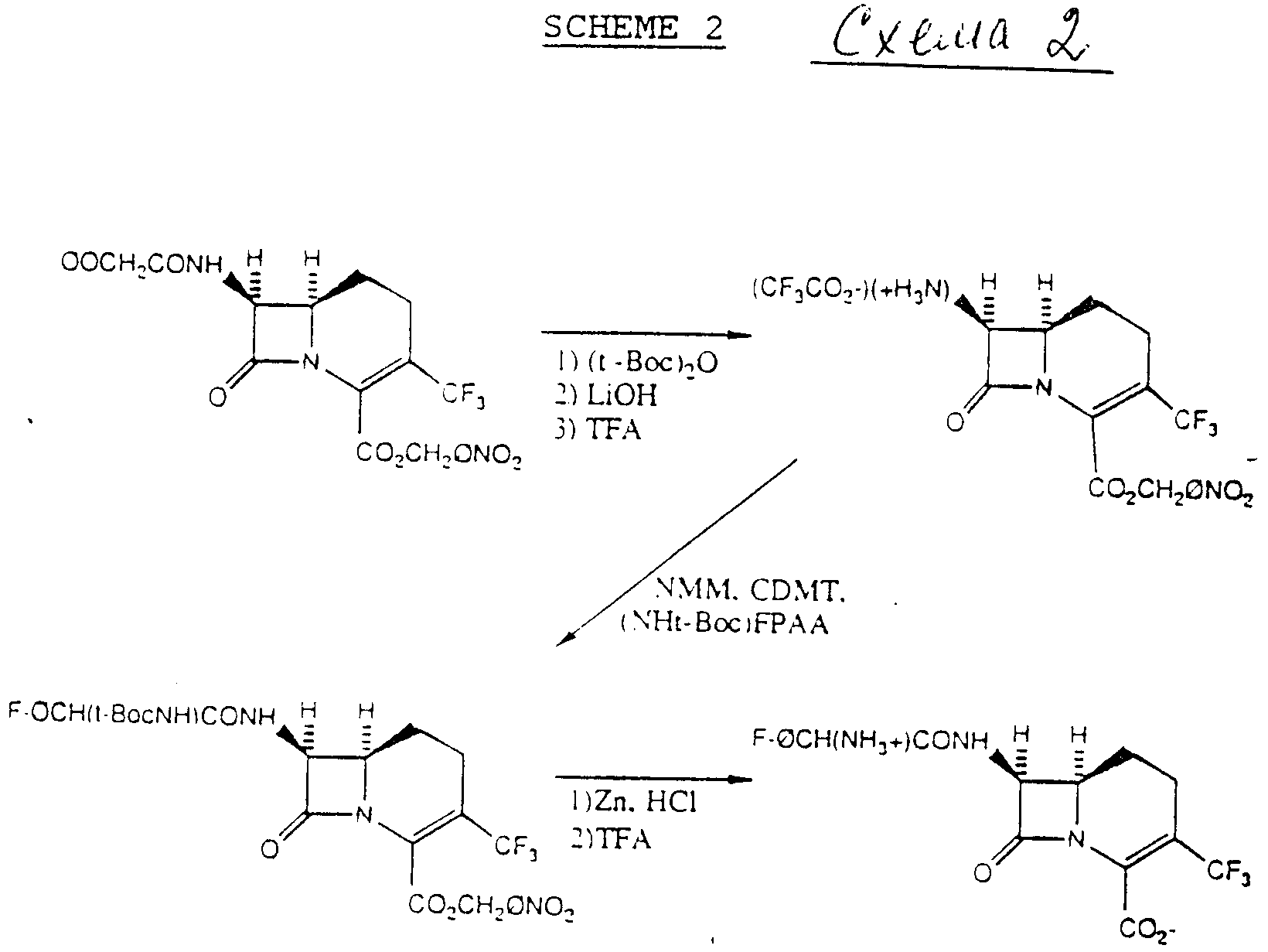
Схема 2 изображает синтез соединений формулы (I) с использованием способа по схеме 1 с применением п-нитробензилкарбоксизащитной группы вместо бензгидрила.
Промежуточную соль трифторуксусной кислоты затем обрабатывают смесью N-метилморфолина (NMM), (трет-бутоксикарбониламино)-3-фторфенилуксусной кислоты (N Hтрет-BOC-FPAA) и 1-хлор-3,5- диметокситриазина (CDMT) с получением трет-BOC-защищенного промежуточного соединения. Это промежуточное соединение затем обрабатывают цинком и HCl с последующей обработкой трифторуксусной кислотой.
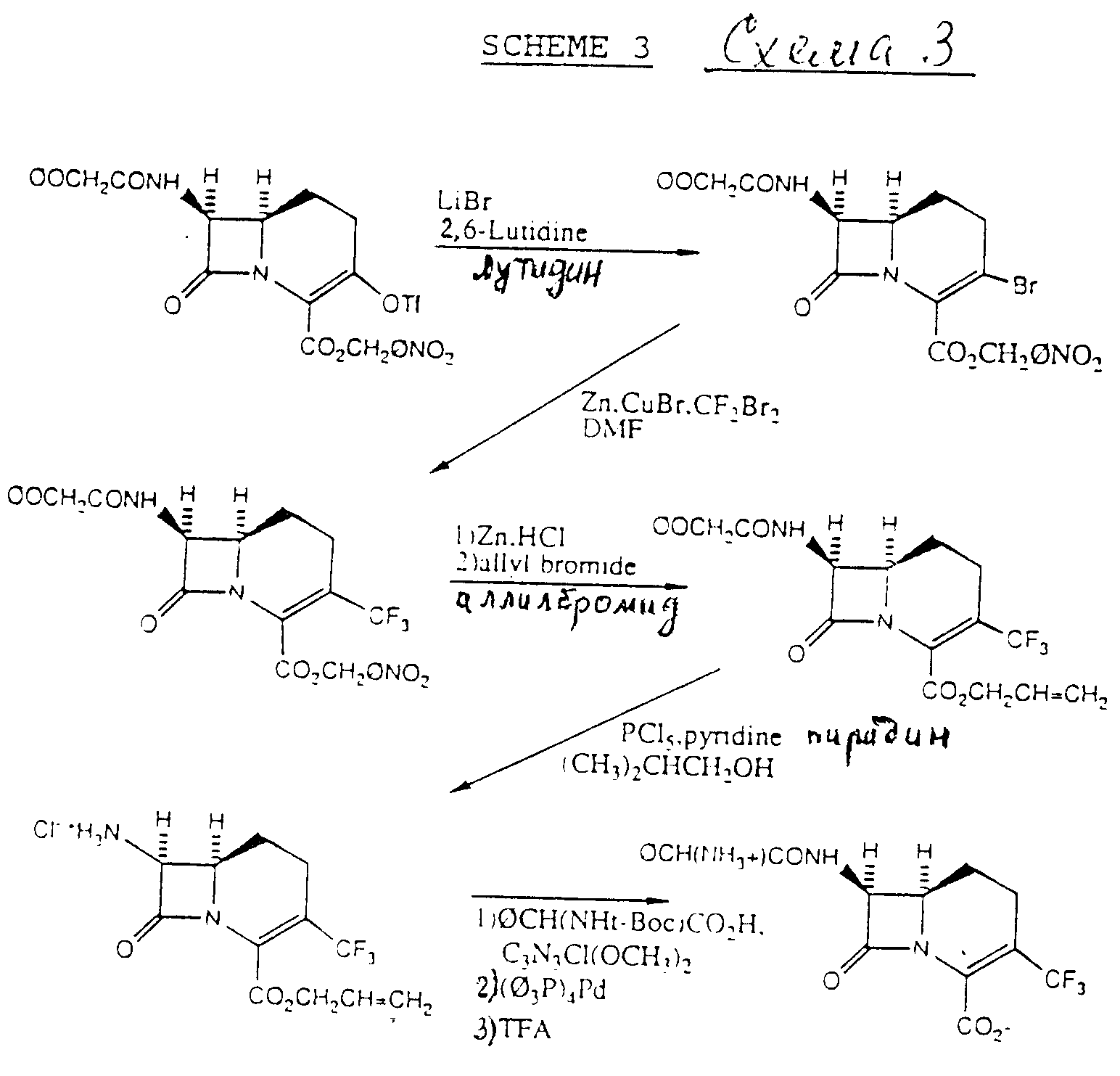
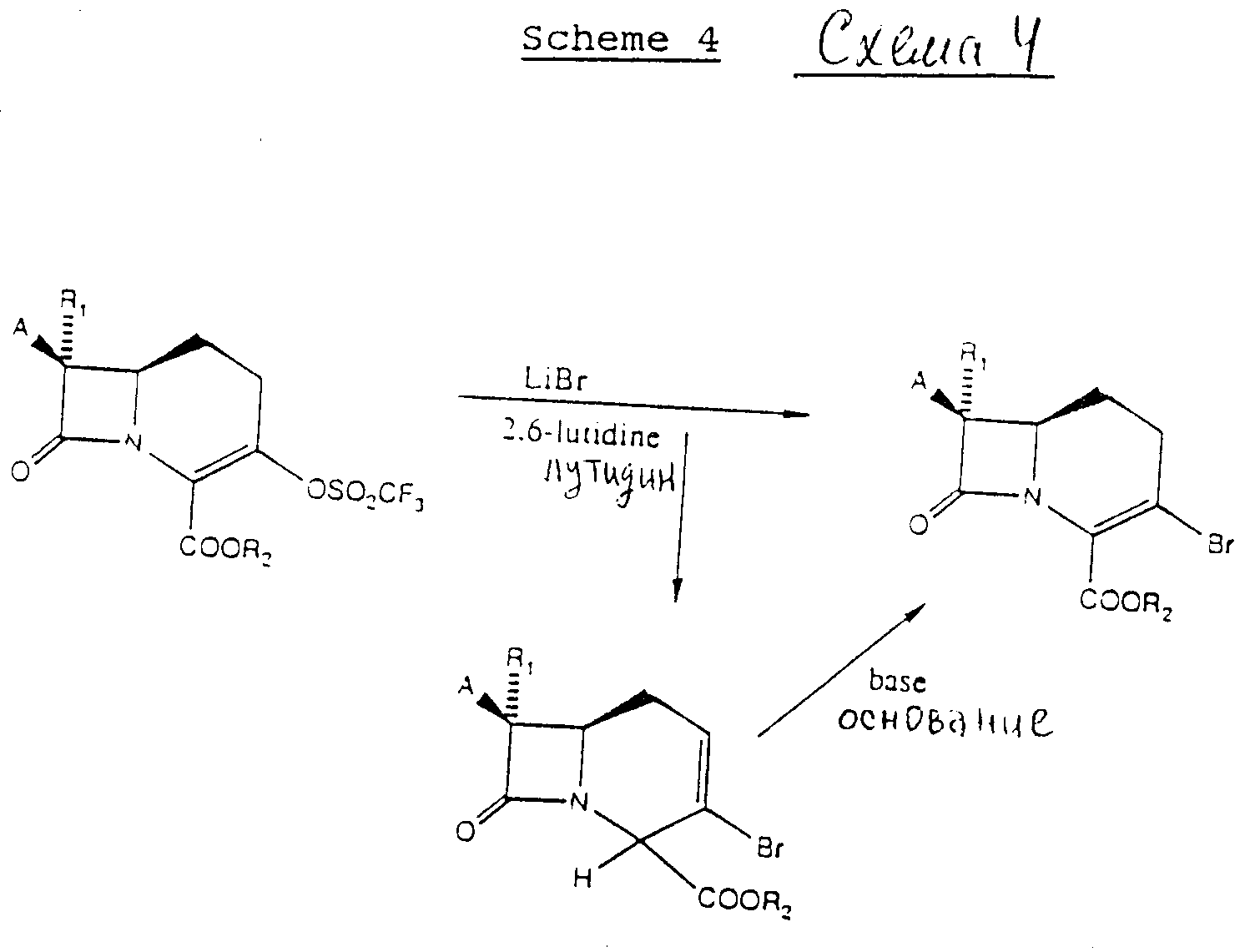
Альтернативно 3-трифторметилкарбацефемы формулы (I) могут быть получены в соответствии со схемой 3.
В схеме 3 3-трифлат-промежуточное соединение в безводном органическом растворителе превращают в 3-бромо-промежуточное соединение с использованием реакции замещения, применяя бромидсодержащую соль, предварительно LiBr, в присутствии затрудненного амин-основания. Бромидсодержащие соли включают бромид щелочного металла, тетраалкиламмоний бромид и тетраалкилфосфоний бромид. Предпочтительным затрудненным амином является 2,6-лутидин. Это 3-бромпромежуточное соединение затем превращают непосредственно в 3-трифторметиловое промежуточное соединение путем обработки смесью цинка и дибромдифторметана в присутствии бромистой меди и ДМФ. Промежуточный p-нитробензил 7β-феноксиацетиламино-1-карба(1-детиа)-3-трифторметил-3-цефем-4-карбоксилат обрабатывают смесью Zn/HCl с удалением группы сложного эфира p-нитробензила и затем повторно этерифицируют аллилбромидом. Феноксиацетильную группу отщепляют с использованием известной методики, т.е. применяя PCl5 /пиридин, изобутиловый спирт, с получением хлоргидрата аллил-7β-амино-1-карба(1-детиа)3-трифторметил-3-цефем-4-карбоксилата. Это промежуточное соединение затем можно обрабатывать активированной формой желательной карбоновой кислоты с получением 7-ациального заместителя, в основном в соответствии с изобретением на схеме 1. 4-Аллил карбоксизащитную группу можно затем отщепить с использованием известной методики освобождения от защиты, т.е. применяя тетракистрифенил-фосфинпалладий(0), с последующим отщеплением трет-BOC-защищенной группы трифторуксусной кислотой, получая соединения формулы (I). Как на схеме 1, используемые аминокислотные карбоксизащитные группы являются показательными, и специалист в области b-лактамов поймет, что можно использовать и другие защитные группы.
На схемах 1 и 3 3-бромопромежуточное соединение подвергают взаимодействию со смесью галогенида меди (Br, Cl, I) дигалодифторметана, либо кадмия, либо цинка, и
ДМФ или группы формулы:
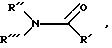
где R', R'' и R''' имеют вышеприведенные значения.
Полагают, что данная смесь приводит к получению in situ трифторметилмеди, которая взаимодействует с 3-бромопромежуточным соединением с образованием 3-фторметилового промежуточного соединения. Предпочтительно по крайней мере один молярный эквивалент трифторметилкадмия или трифторметилцинка присутствует с медью с тем, чтобы по крайней мере теоретически получить по крайней мере один молярный эквивалент трифторметилмеди.
Реакция замещения, в которой 3-трифлат превращают в 3-бромопромежуточное соединение, более полно представлена на схеме 4.
На схеме 4 R1 означает водород и R2 имеет вышеприведенное значение, тогда ка к А обозначает защищенный амино или ациламино формулы:
где R имеет вышеприведенное значение.
Реакции осуществляют в апротонном растворителе, таком как, например, представленный выше в описании. 3-Трифлат объединяют с бромидом лития и предпочтительно 2,6-лутидином. Смесь нагревают до температуры около 60-70oC, предпочтительно около 65oC, и сохраняют в течение периода времени, достаточного для получения 3-бромосоединения. Время нагревания предпочтительно составляет около 16 ч, и более предпочтительно около 48 ч. Обычно полученные продукты включают смесь Δ2/Δ3 изомеров. Δ2-Изомер можно затем изомеризовать в Δ3-изомер путем взаимодействия Δ2-изомера с сильным основанием, таким как 1,8-диазабицикло [5.4.0] ундец-7-ен (DBU) или 1,5-диазабицикло [4.3.0]нон-5-ен (DBN). Как раскрывается, 3-бромосоединение является пригодным промежуточным соединением для получения соединений, представленных формулой (I). Следует понять, однако, что сами по себе 3-бромосоединения являются ценными антибиотиками. Поэтому 3-бромопромежуточное соединение можно превратить, как 3-трифторметил, с образованием 1-карба(1-детиа)-3-цефем-3-бромо-4-карбоновых кислот или их фармацевтически приемлемых солей. Например, на схеме 3 стадию превращения 3-бромо в 3-трифторметил можно опустить с образованием (3-бромо)цефемов.
Термин "затрудненное аминоснование" включает как ароматические, так и алифатические затрудненные амины. Ароматические затрудненные амины включают амины с алкильным заместителем, связанным с ароматическим атомом углерода, примыкающим к атому азота. Предпочтительные заместители могут быть более крупными, нежели метил, например этил, изопропил, трет-бутил и арил. Более предпочтительными ароматическими аминами могут быть затрудненные амины по крайней мере с обоими ароматическими атомами углерода, примыкающими к азотному заместителю, с такими заместителями, как (C1-C6)алкилы и (C1-C4)алкокси. Кроме того, бициклические и полициклические амины могут быть использованы в том случае, если по крайней мере один атом углерода, примыкающий к атому азота, содержит подходящий заместитель. Алифатические затрудненные амины также могут быть использованы, и они включают третичные амины, такие как диизопропилэтиламин, этилдифениламин и т.д.
В одном аспекте
изобретение предлагает 7β-амино-1-карбоцефалоспориновые соединения, а также их соли и сложные эфиры, пригодные в качестве промежуточных
соединений при получении антибиотиков, представленных
формулой (I). Эти промежуточные соединения представлены формулой (II):
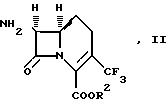
где R2 обозначает водород или карбоксизащитную группу, или их кислотно-аддитивные соли.
7-Аминосоединения формулы (II) образуют соли с общепринятыми кислотами, такими как минеральные кислоты, например хлористоводородная кислота, бромистоводородная кислота, серная кислота и фосфорная кислота, и сульфоорганические кислоты, например метансульфокислота, н-бутансульфокислота, бензолсульфокислота, p-толуолсульфокислота и нафталинсульфокислота. Такие соли используют для выделения и очистки 7-аминокислот и их сложных эфиров. Соединение формулы (II) также можно образовывать соли с гидроксидами, карбонатами и бикарбонатами щелочных или щелочно-земельных металлов. Примерами таких солей являются натриевые, калиевые, кальциевые и магниевые соли. Соли могут быть образованы с аминами, такими как дибензиламин, циклогенсиламин, триэтиламин, этаноламин, диэтаноламин и т.п.
Соединения 7β -амино-1-карба-3-цефема формулы (II) N-ацилируют карбоновой кислотой RCOOH или ее реакционноспособными производными для получения соединения формулы (I). N-ацилирование можно осуществлять с использованием общих способов ацилирования, используемых для N-ацилирования ядер цефалоспорина, например 7-ACA и 7-АDCА. Например, ядро (2) объединяют с кислотой RCOOH в присутствии осушителя, такого как карбодиимид, например дициклогексилкарбодиимид. Альтернативно карбоновую кислоту можно превратить в реакционноспособное производное карбоксильной группы и реакционноспособное производное, используемое в N-ацилировании. Реакционноспособными производными карбоксильной группы являются галогенангидриды, азиды кислот и ангидриды кислот, такие как активные сложные эфиры, образованные с использованием этилхлорформиата и изобутилхлорформиата; фенилкарбоматы; N-гидроксиимиды, такие как образованные с помощью N-гидроксисукцинимида и N-гидроксифталимида; а также образованные с помощью гидроксибенгзтриазола (HBT); и аналогичные активные карбоксипроизводные. Во время N-ацилирования любые свободные амино- или карбоксигруппы, присутствующие в карбоновой кислоте RCOOH, желательно защитить.
В другом аспекте изобретения предлагается предпочтительный
вариант соединения формулы:
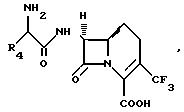
где R4 имеет вышеприведенные значения.
В другом аспекте изобретения предлагается способ получения соединения формулы:
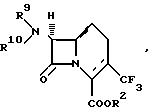
который заключается во взаимодействии соединения формулы:
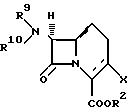
со смесью галогенида меди, кадмия или цинка, дигалодифторметана и либо ДВФ, либо группы формулы:
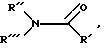
гдe R', R'' и R''' имеют вышеприведенные значения,
или с трифторметилмедью, по существу в безводном инертном органическом растворителе, где X обозначает галоген. R2 имеет вышеприведенные значения и группа R9 R10N обозначает защищенную аминогруппу или R9 обозначает водород и R10 обозначает ацильную группу, имеющую карбоновокислотное происхождение Способ может происходить при температуре в диапазоне около 10-90oC, предпочтительно около 60oC.
Соединения изобретения предлагаются для способа лечения инфекционных заболеваний у человека и других животных и для фармацевтических составов, пригодных для лечения. Терапевтический способ заключается во введении человеку или животному антибиотически эффективной нетоксичной дозы соединения, представленного формулой (I), или его фармацевтически приемлемой соли, когда R2 означает водород.
1-Карбацефалоспорины, предлагаемые изобретением, образуют соли с пригодными основаниями, в частности, фармацевтически приемлемые нетоксичные соли. Карбоксильная группа 1-карбацефалоспорина может образовать соли с гидроксидами, карбонатами и бикарбонатами щелочных и щелочноземельных металлов. Примерами таких фармацевтически приемлемых солей является натриевые, калиевые и магниевые соли. Соли также могут быть образованы с помощью аминов, таких как дибензиламин, дициклогексиламин, триэтиламин, этаноламин, диэтаноламин и аналогичные амины. Таким же образом, когда 1-карбацекфалоспорин замещен двумя или более карбоксильными группами, ди- и три-соли получают традиционными солеобразующими способами.
Соединения 1-карбацефалоспорина, представленные в соответствии с изобретением с заместителем аминогруппы в положении 7 боковой цепи, образуют соли с пригодными кислотами с получением антибиотиков в качестве фармацевтически приемлемых солей. Примерами пригодных кислот являются хлористоводородная, бромистоводородная, серная и фосфорная кислоты.
1-Карбацефалоспорины, представленные в соответствии с изобретением, в случае когда R2 обозначает водород, могут образовывать цвиттерионовую (внутрисолевую) форму соединения, когда заместитель в положении 7 включает свободный амино.
Антибиотически эффективным количеством является количество между 25 мг и 2 г приблизительно. Соединение, соль или сложный эфир можно вводить в разовой дозе или в многократных дозах в течение суток. Обработку (лечение) можно продолжать в течение периода времени от одной недели до десяти суток, или более длительно, в зависимости от продолжительности инфекции. Конкретная доза и схема приема лекарственного средства могут зависеть от таких факторов, как вес и возраст пациента, состояние конкретного организма, тяжесть инфекции, общее состояние здоровья пациента и толерантность пациента к антибиотику.
1-Карба(1-детиа)цефем можно вводить парентерально, перорально, подкожно или ректально. Как и в отношении с другими антибиотиками на основе β-лактама, способ изобретения может быть использован профилактически с целью предотвращения инфекций после экспонирования или перед возможным экспонированием, например предоперационно. Антибиотический 1-карба(1-детиа)цефем может быть введен общепринятыми способами, например в капсулах, таблетках суппозиториях, шприцом или путем внутривенного капельного вливания.
Фармацевтические составы изобретения включают антибиотически нетоксичное количество 1-карба(1-детиа)-3-цефема, представленного формулой (I) или формулой (III), где R2 обозначает водород, или его фармацевтически приемлемой соли, а также фармацевтический носитель.
Фармацевтически приемлемые соли представляют собой полезные формы антибиотиков для получения антибиотических составов. Составы для перорального введения включают капсулы, таблетки, лепешки и жидкие суспензии. Антибиотик или его соль или сложный эфир в форме сухого порошка включают в желатиновые капсулы для перорального применения. Антибиотик также может быть смешан с наполнителем, например стабилизатором, перед наполнением. Капсулы могут содержать около 100-5000 мг для получения составов унифицированной дозы.
Таблетки, содержащие около 100-500 мг антибиотика или его соли или сложного эфира, получают традиционными средствами, и они могут содержать вдобавок связующее вещество, дезинтегратор, стабилизатор, антиокислитель и т.д.
Жидкие препараты антибиотика могут быть получены для младенческого и гериатрического применения. Педиатрические суспензии составляют с антибиотическими пероральными наполнителями, такими как суспендирующие средства, ароматизаторы, стабилизаторы и т.д. Растворы антибиотиков аналогичным образом можно сформулировать с солюбилизаторами, ароматизаторами, сахаром, водой и т.д.
Парентеральные составы антибиотиков для инъекцирования формулируют с водой для инъекции, раствором Рингера, физиологическим раствором или раствором глюкозы. Антибиотик также можно вводить как внутривенную жидкость капельным методом.
Для парентерального применения антибиотик или его производное получают предпочтительно в форме сухого кристаллического порошка или в виде лиофилизированного порошка и вводят в ампулы. Такие ампулы содержат приблизительно от 100 мг до 2 г антибиотика на ампулу.
Следующие сокращения имеют указанные значения:
трет-ВОС трет-бутоксикарбонил или трет-бутилоксикарбонил,
ЖХВР жидкостная хроматография
высокого разрешения,
ТГФ тетрагидрофуран,
Y константа взаимодействия для спектров ЯМР в Гц,
ДМФ N,N-диметилформамид,
DMPU диметилпропиленмочевина,
BSU
бис(триметилсилил)мочевина,
BSA бис(триметилсилил)ацетамид,
DMAP диметиламинопиридин,
DBU 1,8-диазабицикло[5.4.0]ундец-7-ен,
AIBN азо(бис)изобутирилнитрил,
DCC дициклогексилкарбодиимид,
TFA трифторуксусная кислота,
HMPA гексаметилфосфорный триамид,
DMSO диметилсульфоксид.
Экспериментальная часть.
Препарат 1. Дименилметил (7S,6R)-7- феноксиацетамидо-3-фенилтио-1-карба-(1-детиа)-3-цефем-4-карбоксилат.
Раствор диметил [7S, 6R]-7-феноксиацетамидо-3- трифторметилсульфонилокси-1-карба(1-детиа)-3-цефем-4-карбоксилата (40,0 г, 63,00 ммоль) в 180 мл безводного ацетонитрила обрабатывают под N2 диизопропилэтиламином (15,4 мл, 88,0 ммоль) и тиофенолом (7,1 мл, 69,0 ммоль). Реакционную смесь перемешивают в течение ночи при температуре окружающей среды. Растворитель удаляют в вакууме и остаток очищают хроматографией на силикагеле (элюирование с помощью 35:65 этилацетат/гексан), получая 33,45 г (90%) светло-желтого твердого тела.
1H ЯМР (CDCl3), d 7.2-7.6 (м, 19 Н), 7.08 (т, Y 7 Гц, 1 Н), 7.05 (с, 1 Н), 6.90 (д, Y 8 Гц, 2 Н), 5.44 (м, 1 Н), 4.54 (с, 2Н), 3.81 (дт, Y 5, 12 Гц, 1 Н), 2.0-2.3 (м, 2 Н), 1.7-1.8 (м, 1 Н), 1.3-1.5 (м, 1 Н).
Препарат 2. Дифенилметил (7S, 6R)-7- феноксиацетамидо-3-бромо-1-карба(1-детиа)-3-цефем-4-карбоксилат.
Дифенилметил [7S, 6R]-7-феноксиацетамино-3- трифторметилсульфонилокси-1-карба
(1-детиа)-3-цефем-4-карбоксилат (150 г, 0.238 моль) растворяют в 1600 мл безводного ДМФ и обрабатывают 2,6-лутидином (63,8 г, 0,595 моль) и литийбромидом (121,1 г, 1,43 моль). Реакционную смесь
нагревают до 65oC в течение 30 мин и сохраняют при данной температуре в течение 64 ч. Реакционную смесь охлаждают до комнатной температуры и 75% растворителя удаляют при пониженном
давлении
при температуре 50oC. Суспензию разбавляют этилацетатом/простым эфиром, промывают раствором NaHCO3 (3 раза), 1 н. раствором HCl (3 раза) и солевым раствором, сушат в
присутствии
сульфата магния, фильтруют через силикагель с 10% этилацетата/CH2Cl2 и упаривают при пониженном давлении до осаждения твердого тела. Твердое тело бежевого цвета (34,
4 г) собирают
и после дальнейшего упаривания маточных растворов
собирают дополнительно 10,6 г твердого тела. Эти две партии твердых тел объединяют, поскольку обе они представляют собой желательные
D3-изомеры
(указанное в заголовке соединение). Оставшуюся жидкость отгоняют досуха, получая 64,7 г смеси Δ2/Δ3-изомеров. Смесь олефин-изомеров уравновешивают следующим образом.
Растворение смеси
(64,7 г, 0,115 моль) в 675 мл безводного CH2Cl2 осуществляют путем обработки с помощью DBU (4,7 г, 0,031 моль). Через 3 ч при комнатной температуре реакционную
смесь фильтруют
через силикагель с 10% этилацетата/CH2Cl2 и упаривают. Остаток растворяют с небольшом количестве этилацетата, разбавляют гексаном и охлаждают до 0oC.
Это приводит к
получению 17,6 г чистого Δ3-изомера, и остальные 39,4 г (из маточного раствора) хроматографируют на силикагеле (элюируя градиентом толуола к 30/70 этилацетата в толуоле).
Получают 31,5 г смеси
Δ2/Δ3 --изомеров, которую растворяют в этилацетате, разбавляют гексаном, затравливают продуктом и охлаждают до 0oC. Получают 18,2 г Δ3-изомера,
оставляя 14,2 г смеси
Δ2/Δ3 -изомеров. Общее количество, равное 80,8 г, указанного в заголовке продукта выделяют при выходе 61%
Препарат 3. Аллил [7S,
6R]-7-феноксиацетил-амино-3-бромо-1-карба(1-детиа)-3-цефем-4- карбоксилат.
(a) Аллил [7S, 6R]-7-феноксиацетиламино-3- трифторметилсульфонилокси-1-карба(1-детиа)-3-цефем-4-карбоксилат
(1,0 г, 1,9826 ммоль) и высушенный литийбромид (0,689 г, 7,9302 ммоль) объединяют в ДМФ (3,0 мл). Смесь после растворения всех твердых тел медленно нагревают приблизительно до 67oC и
перемешивают в течение 16 ч. Смесь охлаждают до комнатной температуры и объединяют с 75 мл EtOAc, 50 мл 1:1 раствора H2O/насыщенный NaHCO3. Органические вещества отделяют и
промывают 50 мл 1 н. раствора HCl, сушат в присутствии сульфата натрия, фильтруют и концентрируют до красновато-коричневого масла. Флеш-хроматография при элюировании 7% EtOAc/CH2Cl2 приводит к получению 87 г продукта и с выходом около 10%
(b) 3-Трифлат из стадии (a) выше (200 мг, 0,3965 ммоль) объединяют с высушенным бромидом лития (138 мг, 1,586 ммоль) и DMPU
(2
мл) и смесь нагревают до 65-67oC в течение 6 ч. К смеси прибавляют 2,6-лутидин (0,4362 ммоль) и смесь перемешивают при температуре 67oC в течение ночи (14 ч). Смесь вливают в
50
мл EtOAc и 20 мл насыщенного раствора NaHCO3. Органические вещества промывают 25 мл 1 н. раствора HCl, отделяют и сушат в присутствии сульфата натрия, фильтруют и концентрируют до
коричневато-желтого масла. Флеш-хроматография с элюированием 7% EtOAc/CH2Cl2 позволяет получить 45 мг (26%) 60/40 смеси Δ2/Δ3-изомеров.
(c)
Повторяют методику, приведенную в стадии (b), за исключением того, что 2,6-лутидин помещают в смесь в начале методики, а не после начала нагрева. Используют следующие количества:
Исходное
вещество 3-трифталат 200 мг, 0,3965 ммоль
2,6-Лутидин 0,793 ммоль, 92 мкл
Бромид лития 138 мг, 1,586 ммоль
DMPU 2 мл
Данная методика позволяет получить 55 мг (31,
2%) смесиΔ2/Δ3- -изомеров в соотношении 60/40.
(d) Повторяют приведенную в стадии (c) методику за исключением того, что ДМФ используют вместо DMPU. Используют следующие
количества:
Исходное вещество 3-трифлат 145 мг, 0,2875 ммоль
2,6-Лутидин 0,5749 ммоль, 67 мкл
Бромид лития 100 мг, 1,15 ммоль
ДМФ 1,5 мл
Данная методика
приводит к получению 61,2 мг (49%) смеси Δ2/Δ3-изомеров в соотношении 70/30.
(e) Повторяют методику, приведенную в стадии (d), за исключением того, что смесь нагревают
приблизительно в 2,5 раза более длительно, или 48 ч. Используют следующие количества:
Исходное вещество 3-трифлат 11 г, 21,808 ммоль
2,6-Лутидин 43,616 ммоль, 5,08 мл
Бромид
лития 7,6 г, 87,232 ммоль
ДМФ 120 мл
Данный препарат позволяет получить 4,6 г указанного в заголовке соединения, или 48,5% выход смеси Δ2/Δ3-изомеров при
соотношении
свыше 95/5 соответственно. Данные относительно 2-изомера:
1H-ЯМР (300 МГц, CDCl3): δ 7.32 (т, J 1 Гц, 2 Н), 7.10 (д, J 9 Гц, 1 Н), 7.05 (т, J 6 Гц, 1
Н), 6.92 (д,
J 8 Гц, 2 Н), 6.50 (м, 1 Н), 5.95 (м, 1 Н), 5.35 (м, 3 Н), 4.90 (с, 1 Н), 4.63 (м, 2 Н), 4.55 (с, 2 Н), 4.15 (м, 1 Н), 2.35 (м, 1 Н), 2.15 (м, 1 Н); ИК (CHCl3): 3019, 1771,
1747, 1691, 1517,
1495, 1236 и 1182 см-1.
Масс-спектр, m/e 434 (M+), 436 (M+ + 2).
Анализ для C19H17N2
O5
Br:
Вычислено, C 52.43, H 4.40, N 6.44
Найдено, C 52.23, H 4.36, N 6.37
Пример 1. Дифенилметил [7S,
6R]-7- феноксиацетамидо-3-трибутилстаннил-1-карба(1-детиа)-3-цефем-карбоксилат.
Суспензию дифенилметил [7S, 6R]-7- феноксиацетамидо-3-фенилтио-1-карба(1-детиа)-3-цефем-карбоксилата (37, 0 г, 62,7 ммоль) в 32 мл безводного диглима обрабатывают под N2 гидридом трибутилолова (42,1 мл, 157,0 ммоль) и азо (бис) изобутилнитрилом (AIBN) (12,4 мл, 75,2 ммоль). Реакционную смесь нагревают до 120oC в течение 45 мин и затем охлаждают до температуры окружающей среды. Остаток очищают хроматографией на силикагеле (элюируя 40:60 этилацетат/гексан). Получают 37,1 г светлого вязкого масла (77%).
1H-ЯМР (CDCl3): d 7.55 (д,J=7Гц, 2H), 7.44 (д, J=7Гц, 2H), 7.2-7.4 (м, 7H), 7.0-7.1 (м, 2H), 6.9 (м, 3H), 5.48 (м,1H), 4.56 (с,2H), 3.87 (м, 1H), 2.6-2.7 (м, 1H), 2.3-2.45 (м,1H), 1.9-2.0 (м,1H), 1.2-1.4 (м,12H), 0.8-0.85 (м, 15H).
ИК (CHCl3): 2958.2, 2923.7, 1766.2, 1689.9, 1523.0, 1496.2, 1374.4 и 1239.5 см-1.
Масс-спектр (FAB): m/e (M+) 771.
УФ (EtOH): lmax275 нм (ε9960).
Анализ для C41H52
N2O5Sn: C, H, N,
Пример 2. Дифенилметил [7S, 6R]-7- феноксиацетамино-3-бромо-1-карба(1-детиа)-3-цефем -4-карбоксилат.
Станнан из примера 1 (8,58 г, 11, 13 ммоль) в 400 мл безводного ТГФ охлаждают в ванне, содержащей смесь ацетона с сухим льдом, и обрабатывают по каплям раствором Br2 (1,78 г, 11,13 ммоль) в 100 мл ТГФ в течение 30 мин. Растворитель удаляют при пониженном давлении и хроматографируют на силикагеле с 40:60 этилацетат/гексан, получая 5,02 г (80%) твердого тела белого цвета.
1H-ЯМР (CDCl3): δ 7.2-7.5 (м, 12H), 7.0-7.1 (м, 2H), 6.98 (с,1H), 6.9 (д, J= 8Гц, 2H), 5.40 (м,1H), 4.54(с, 2H), 3.95 (м, 1H), 2.7-2.8 (м, 2H), 1.9-2,0 (м,1H), 1.5-1.7 (м,1H), 1.3-1.4 (м, 1H).
ИК (CHCl3): 3025.2, 1787.0, 1742.2, 169.7, 1600.6, 1518.9, 1496.0, 1302.3 1242.7 см-1.
Масс-спектр (FAB)-m/e(M+) 561.
УФ
(EtOH): lmax275 нм (ε4870).
Анализ: C29H25BrN2O5: C,H,N.
Пример 3. Дифенилметил [7S, 6R]-7- феноксиацетамино-3-трифторметил-1-карба-(1-детиа)-3-цефем-4-карбоксилат.
Суспензию кадмия (20,0 г, 178 ммоль) в 100 мл безводного ДМФ охлаждают в ванне с ледяной водой под N2 и обрабатывают по каплям раствором CF2Br2. Периодическое удаление ледяной ванны необходимо для поддержания ровного продолжения реакции. После прибавления соединения ванну удаляют и реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. В отдельной колбе исходное промежуточное соединение 3-брома (10,0 г, 17,8 ммоль) растворяют в 18 мл безводного DMPU и нагревают до температуры 70oC. Раствор кадмиевого реагента обрабатывают раствором CuCl2 (17,6 г, 178 ммоль) и полученный красновато-бурый раствор прибавляют к 3-бромо-промежуточному соединению через канюлю в течение 1 ч. После охлаждения до комнатной температуры смесь разбавляют этилацетатом, фильтруют через броунмиллерит, промывают водой и солевым раствором, сушат в присутствии сульфата магния и восстанавливают до коричневого масла в вакууме. Это масло хроматографируют на силикагеле с 40:60 этилацетат/ гексан, получая 4,7 г (49%) указанного в заголовке продукта в виде пены.
1H-ЯМР (CDCl3) δ 7.3-7.4 (м, 12H), 7.0-7.2 (М, 3H), 6.9(д, J=8Гц, 2H), 5.49 (м, 1H), 4.58 (с,2H), 3.96 (м, 1H), 2.3-2.5 (м, 2H), 2.0-2.1(м,1H) и 1.4-1.5(м, 1H).
ИК (CHCl3): 1787.0, 1742.2, 1692.7, 1600.6, 1518.9, 1496.0, 1302.3 и 1242.7 см-1.
Масс-спектр (FAB) m/e (M++1) 517.
УФ (EtOH): lmax264 нм (ε8946).
Анализ: C30H25F3N2O5; C, H, N.
Пример 4. Дифенилметил [7S,6R]-7-трет бутоксикарбоксамидо -3-трифторметил-1-карба (1-детиа)-3-цефемкарбоксилат.
Дифенилметил [7S, 6R]-7-феноксиацетамидо-3- трифторметил-1-карба(1-детиа)-3-цефем-4-карбоксилат (4,7 г, 8,5 ммоль) растворяют в 85 мл безводного CH2Cl2 под N2, охлаждают в ванне с ледяной водой и обрабатывают ди-трет-бутилкарбонатом (2,04 г, 9,35 ммоль) и диметиламинопиридином (DMAP) (0,52 г, 4,3 ммоль). Через 45 мин реакционную смесь нагревают до температуры окружающей среды 4 ч. Раствор фильтруют через кремнезем с 20:80 этилацетат/CH2Cl2. После удаления растворителя в вакууме остаток берут в 85 мл безводного ТГФ и охлаждают в ванне со льдом. Прибавляют по каплям LiOH (9,7 мл 1 М раствора) и раствор нагревают до комнатной температуры. Реакционную смесь экстрагируют этилацетатом и данный экстракт промывают 1 н. раствором HCl, насыщенным раствором NaHCO3, солевым раствором, сушат в присутствии безводного сульфата магния и упаривают в вакууме досуха. Получают 4,2 г неочищенной пены оранжевого цвета.
1H-ЯМР (CDCl3) δ 7.3-7.4 (м, 10H), 6.95 (с,1H), 5.98 (шир.д, J=11 Гц, 1H), 5.22 (м, 1H), 3.87 (м, 1H), 2.3-2.5(м, 2H), 2.0-.1 (м, 1H) и 1.6-1.7 (м, 1H), 1.23 (с, 9H).
ИК (CHCl3): 1785.3, 1740.0, 1718.5, 1497.3, 1301.7 и 1243.3 см-1.
Масс-спектр (FAB):m/e (M++1) 517.
УФ (ЕтОН) lmax
264 нм (ε8946).
Анализ C27H27F3N2 O5; C, H, N.
Пример 5. Трифторацетатная соль [7S, 6R]-7- амино-3-трифторметил-1-карба-(1-детиа)-3-цефем-4- карбоновой кислоты.
Продукт из примера 4 (2,99 г, 3,87 ммоль) растворяют в холодной трифторуксусной кислоте (TFA) (40 мл) и триэтилсилане (13 мл) под N2 при 0oC. Реакцию поддерживают при 0oC в течение 30 мин в ванне со льдом, после чего ванну удаляют и смесь перемешивают 20 мин. Прибавляют безводный CH3CN и толуол, и объем уменьшают в вакууме. Данный процесс повторяют и колбу концентрируют досуха. Остаток затем суспендируют в CH3CN и фильтруют. Твердые тела промывают смесью CH3/простой диэтиловый эфир и затем дважды простым диэтиловым эфиром, получая 925 мг (66%) указанного в заголовке соединения в виде твердого тела белого цвета.
1H-ЯМР(DMSO-d6 /TFA) δ 8.0-8.2 (шир.с, 1H), 4,92 (д, J=6Гц, 1H), 3.97 (м, 1H), 2.0-2.1 (м, 2H), 1.7-1.8 (м, 2H).
Пример 6. [7S, 6R]-7-[D-альфа-(трет-бутокси-карбониламино)фенилацетиламино] -3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Трифторацетатную соль из примера 5 (0,90 г, 2,47 ммоль) суспендируют в 25 мл этилацетата под N2 и обрабатывают силилтрифторацетамидом (1,51 мл, 8,15 ммоль) и затем нагревают при 35oC до тех пор, пока все твердые тела будут в растворе. В отдельной колбе трест-ВОС-фенилглицин растворяют в 25 мл этилацетата под N2, охлаждают до -45oC и обрабатывают последовательно изобутилхлорформиатом (0,21 мл, 2,47 ммоль) и N-метилморфолином (0,326 мл, 2,96 ммоль). Через 30 мин раствор амина охлаждают до 0oC, а затем добавляют через канюлю в течение 5 мин и перемешивают 30 мин при температуре росы от -45 до -35oC и в течение 1,5 ч при температуре росы до 0oC. В это время прибавляют 2,0 мл метанола, ванну со льдом убирают и раствор перемешивают в течение 10 мин до его нагревания до комнатной температуры, упаривание и хроматография на кремнеземе с использованием 3%-ной уксусной кислоты в этилацетате к получению указанного в заголовке продукта в виде получистой пены бежевого цвета (1,03 г, 87%).
1H-ЯМР (CDCL3) d 7.1-7.4(м, 5H9,6.1 (шир.с, 1H), 5.4(шир.с, 1H),3.8 (шир.м,1H), 2.2-2.4 (шир.м,2H) и 1.2-1.5(шир.м,1H).
Пример 7. 7 b-[D-альфа-(амино)фенилацетил-амино]-3-трифторметил-1-карба(1-детиа)-3-цефем -4- карбоновая кислота.
Трет-ВОС-защищенную кислоту из примера 6 помещают в колбу при 0oC и обрабатывают смесью холодной трифторуксусной кислоты (TFA) (20 мл) и третилсилана (7 мл). После перемешивания в течение 15 мин при 0oC ванну со льдом убирают и реакционную смесь перемешивают в течение 10 мин при температуре окружающей среды, разбавляют раствором CH3CN и уменьшают в объеме. Дальнейшее разбавление раствором CH3CN сопровождают упариванием досуха, суспендированием остатка в EtO2 и фильтрацией. Полученное твердое тело промывают неоднократно раствором EtO2 и сушат в вакууме с получением 0,74 г неочищенного продукта со степенью чистоты около 85% Обращенно-фазовая хроматография на колонке C-18 со ступенчатым градиентом из 10% CH3CN/H2O в 30% CH3CN/H2O приводит к получению 0,39 г (50%) указанного в заголовке продукта в виде белого твердого тела.
1H-ЯМР (D1O)d 7.5-7.6 (м,5H), 5.44 (д, J=7 Гц, 1H), 5.21 (с,1H), 3.95 (м,1H), 2.25 (шир.м, 2H), 1.7-1.8 (м,1H), 1.0-1.1 (м,1H).
ИК (CHCl3): 1776.7, 1740.0, 1693.7, 1561.6, 1300.2 и 1166.1 см-1.
Масс-спектр (FAB): m/e (M++1) 384.
УФ (EtOH): lmax257 нм (ε9190).
Пример 8. [7S, 6R] -7-{ [2-(трет-бутокси- карбомил)амино-4-тиазолил]
(метоксиимино)ацетил}
амидо-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Трифторацетатную соль [7S, 6R] -7-амино-3- трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновой кислоты (0,10 г, 0,275 ммоль)обрабатывают под N2 бис(триметилсилил)мочевиной (0,281 г, 1,38 ммоль) и 1,4 мл безводного ДМФ. Твердые тела, растворенные после нагревания суспензии до температуры 45oC, сохраняют в растворе при данной температуре в течение 1 ч. В отдельной колбе [2-(трет-бутоксикарбонил)-амино-4-тиазолил] -(метоксиимино)уксусную кислоту (0,083 г, 0,275 ммоль) растворяют в 1,4 мл ДМФ, охлаждают до -5oC, обрабатывают оксалилхлоридом (0,035 г, 0,275 ммоль) и нагревают до температуры окружающей среды 30 мин. Раствор зародыша охлаждают до температуры охлаждающей среды, обрабатывают пиридином (0,067 г, 0,825 ммоль) и затем раствором хлорангидрида кислоты. После перемешивания в течение 2 ч раствор охлаждают в ванне со льдом и обрабатывают 2 мл воды, разбавляют этилацетатом и распределяют на фракции. Органический слой промывают два раза 1 н. раствором HCl, водой и солевым раствором, сушат в присутствии сульфата магния, фильтруют и упаривают досуха. Hеочищенное твердое тело (0,101 г) флеш-хроматографируют на кремнеземе с использованием 4% НОАс в этилацетате, получая 0,087 г (59%) указанного в заголовке продукта.
Пример 9. [7S, 6R] -7-([2-амино-4-тиазолил- (метоксиимино)ацетил]амино)-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Смесь трифторуксусной кислоты (1,5 мл) и триэтилсилана (0,5 мл) охлаждают в ванне со льдом и EtOH под N2 и обрабатывают дифенилметил [7S, 6R]-7-{ [2-(трет-бутоксикарбонил)-амино-4-тиазолил] (метоксиимино)ацетил} амидо-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоксилатом. Через 30 мин реакционную смесь обрабатывают раствором CH3CH и упаривают досуха. Этот неочищенный остаток (0,055 г) хроматографируют препаративной ЖХВР на обращенно-фазовой колонке с использованием ступенчатого градиента CH3CN:H2O:HOAc от 15:84:1 до 20:79:1, получая 0,021 г конечного продукта.
1Н-ЯМР (ДМСО-d6) δ 9,25 (д, J=11 Гц, 1Н), 7,18 (шир.с, 2Н), 6,70 (с, 1Н), 5,48 (м, 1Н), 3,9 (м, 1Н), 3,80 (с, 3Н), 2,37 (шир.м, 2Н), 1,95 (м, 1Н), 1,6 (м, 1Н).
Пример 10. [7S, 6R]-7-[D-альфа-(трет-бутоксикарбониламино)-4-гидроксифенилацетиламино] -3-трифторметил -1- карба-(1-детиа)-3-цефем-4-карбоновая кислота.
Раствор D-альфа-(трет-бутоксикарбониламино) -4- гидроксифенилуксусной кислоты (0,214 г, 0,799 мМ) в 2,4 мл безводного ТГФ обрабатывают 1-гидроксибензотриазолом (0,108 г, 0,799 мМ) и DCC (0,198 г, 0, 959 мМ) при 0oC. Осадок образуется через 5 мин, и суспензию нагревают до температуры окружающей среды и перемешивают в течение 2 ч, после чего раствор фильтруют и охлаждают до 0o C. Цвиттерион (амфотерный ион) карбацефема суспендируют в 2,4 мл безводного ТГФ и обрабатывают бистриметилсилилацетамидом (BSA) (0,8 мл), получая раствор, который концентрируют до густого масла, разбавляют 0,8 мл ТФГ, охлаждают до 0oC и обрабатывают холодным раствором сложного эфира. Через 30 мин ванну удаляют и реакционную смесь перемешивают в течение ночи. Растворитель упаривают и остаток распределяют между этилацетатом и водным раствором NaHCO3. Органический слой экстрагируют еще раз водным раствором NaHCO3 и объединенные органические слои разделяют этилацетатом и pH снижают до 2,5 с помощью 1 н. раствора HCl. Водный слой промывают этилацетатом и объединенные органические слои сушат в присутствии сульфата магния, фильтруют и концентрируют с получением 0,371 г (67%) указанного в заголовке продукта, который не подвергают дальнейшей очистке.
Пример 11. [6R, 7S] -7β-[D-a-(амино) -4-гидроксифенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Продукт из примера 10 помещают в колбу при температуре 0oC и обрабатывают смесью холодной трифторуксусной кислоты (TFA) (2,2 мл) и триэтилсилана (0,5 мл). Колбу сразу же помещают в ротационный испаритель и концентрируют до масла, которое растирают в порошок с помощью диэтилового эфира, получая твердое вещество, которое собирают фильтрацией и промывают раствором EtO2. Это твердое вещество сушат в вакууме в течение 3 ч с получением 0,132 г неочищенного продукта. Обращенно-фазовая хроматография на колонке C18 с использованием ступенчатого градиента от 99/1:H2O/HOAc до 5/94/1:CH2CN/H2O/ уксусная кислота после лиофилизации позволяет получить 0,055 г (25%) указанного в заголовке продукта в виде твердого тела белого цвета.
1Н-ЯМР (D2O) d7,35 (м, 2Н), 6,97 (м, 2Н), 5, 40 (д, J=7 Гц, 1Н), 5,13 (с, 1Н), 3,90 (с, 1Н), 2,1-2,3 (шир.м, 2Н), 1,73 (м, 1Н) и 1,10 (м, 1Н).
ИК (KBr):1768,0, 1688,9, 1613,6, 1519,1 1300,2 см-1.
Масс-спектр (FAB):m/e (M++1) 400.
УФ (этанол): lmax 234,257 нм (ε 1280, 9030).
Анализ:
Вычислено, C 51,13, H 4,04, N 10,52
Найдено, C 51,25, H 4,21, N 10,29
Пример 12. [6R, 7S]-7-b-[D-a-(амино)-4- фторфенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Раствор
рацемического трет-ВОС-защищенного 4-фторфенилглицина (0,216 г, 0,80 мМ) в 8,0 мл этилацетата охлаждают до температуры -45oC, обрабатывают изобутилхлороформиатом (0,104 мл, 0,80 мМ) и
N-метилморфолином (0,097 мл, 0,88 мМ), после чего перемешивают в течение 30 мин. 7-Амино-3-трифторметил-1-карба(1-детиа)-3-цефем-4- карбоновую кислоту (0,200 г, 0,80 мМ) растворяют в 8,0 мл
этилацетата после прибавления моносислилтрифторацетамида (0,341 мл, 1,84 мМ) и прибавляют к смешанному ангидриду при -45oC. Через 30 мин раствор нагревают до 0oC в течение 1 ч и
затем обрабатывают 0,5 мл MeOH, нагревают до температуры окружающей среды, фильтруют через броунмиллерит и концентрируют при температуре 30oC. Продукт хроматографируют на силикагеле и
использованием 3:87:10 НОАс/этилацетат/ гексан. Оба диастереомера видны при1Н-ЯМР вместе с незначительными примесями. Неочищенный продукт (0,228 г) обрабатывают холодным раствором TFA (1,
8
мл) и Et3SiH (0,6 мл), концентрируют, порошкуют с помощью EtO2, собирают фильтрацией и промывают EtO2. Соль неочищенной TFA (0,209 г) хроматографируют на колонке
C18 двумя ступенчатыми градиентами от 15% CH3/H2O до 20% CH3/H2O, получая чистые после лиофелизации 0,030 г (10%) продукта из L-аминокислоты и 0,
046 г
(14%) продукта, полученного из D-аминокислоты. Время удерживания при ЖХВР: C18 1/20/79: НОАс/CH3CN/H2O, 2 мл/мин, L=4,11 мин, D=5,38 мин. L-диастереомер:
1Н-ЯМР(CDCl) d 9,1 (м, 1Н), 7,50 (м, 2Н), 7,22 (т, J=10 Гц, 2 Н), 5,17 (шир.с, 1Н), 4,89 (шир.с, 1Н), 3,76 (м, 1Н), 2,17 (м, 2Н),1,73 (м, 1Н), 1,60 (м, 1Н).
ИК (CHCl3 ):см-1.
Масс-спектр (FAB):m/e (M+).
УФ (EtOH): lmax 257 нм (ε)
Пример 13. [7S,
6R]-7-{[2-(трифторметил)- амино-4-тиазолин] (трифенилметоксиимино)ацетил} амидо-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
7-Амино-3-трифторметил -1- карба(1-детиа)-3-цефем -4-карбоновую кислоту (0,300 г, 1,20 мМ) (цвиттерион карбацефема) растворяют в 6,0 мл сухого ДМФ с помощью бистриметилсилилмочевины (BSU)(1,23 г, 6,0 мМ) при 50o C в течение 1 ч и затем при комнатной температуре 30 мин. В отдельной колбе [2-тритиламино-4- тиазолил](тритилоксиимино)уксуную кислоту (0,806 г, 1,20 мМ) растворяют в 6,0 мл сухого ДМФ и охлаждают до 0oC, обрабатывают по каплям оксалилхлоридом (0,105 мл, 1,20 мМ) и нагревают до температуры окружающей среды. Ядро затем охлаждают до 0oC, обрабатывают пиридином (0,194 мл, 2, 4 мМ) и хлорангидридом кислоты, нагревают до температуры окружающей среды и перемешивают 2 ч. После прибавления 2,0 мл воды реакционную смесь обрабатывают 1 н. раствором HCl и экстрагируют этилацетатом. Этилацетатовый слой промывают водой, 1 н. раствором HCl, водой, солевым раствором, сушат в присутствии сульфата магния и концентрируют. Неочищенный продукт (1,12 г) хроматографируют фильтрацией через силикагель с помощью 3% НОАс/этилацетата, получая 0,95 г (88%) указанного в заголовке продукта в виде белого порошка.
Пример 14. [7S, 6R]-7-([2-амино-4-тиазолил-(оксиимино)ацетил]амидо)-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Раствор бис-тритилированного карбацефема из примера 13 в 4,5 мл ТГФ обрабатывают 7,6 мл 75% -ного водного раствора муравьиной кислоты и медленно нагревают до 40oC под азотом 2 ч. После охлаждения прибавляют 20 мл CH2CN с последующей концентрацией, и данный процесс повторяют с получением остатков коричневого цвета. Это твердое вещество промывают 3/1:EtO2/CH3CN и твердое вещество выделяют центрифугированием. После проведения дополнительных промывок с использованием EtO2 твердое вещество хроматографируют на колонке HP-20ss с помощью 1% HOAc/H2O после загрузки CH3CN.
Полученное твердое тело хроматографируют на колонке C18 с использованием 1/10/89:HOAc/CH3CN/H2O, получая 0,027 г твердого тела кремового цвета.
1H-ЯМР (ДМСО -d6) δ 9.13 (д, J=9 Гц, 1H), 7.07 (с, 2H), 6.64 (с, 1H), 5.50 (м, 1H), 3.89 (м, 1H), 2.25 (м, 2H), 1.95 (м, 1H), 1.60 (м, 1H).
ИК (CHCl3) см-1.
Масс-спектр (FAB): m/e (M+)
УФ (EtOH): lmax 257 нм.
Пример 15. Дифенилметил [7S, 6R]-7-фенокси-ацетамидо-3-трифторметил-1-карба-(1-детиа)-3-цефем-4-карбоксилат.
Суспензию промытого кислотой цинка (58,2 г, 0,89 моль) в 1780 мл безводного ДМФ под азотом обрабатывают по каплям 205,4 (0,98 ммоль) CF2Br2•CF2Br2 прибавляют с тем, чтобы поддержать температуру на уровне 45-50oC. После завершения прибавления реакционную смесь перемешивают при температуре окружающей среды 1,5 ч, и за это время смесь возвращается к комнатной температуре. Раствор цинкового реагента охлаждают до температуры -30oC и обрабатывают DMPU (100 мл) и CuCl (88,1 г, 0,89 ммоль). Через 15 мин реакционную смесь нагревают до 0oC в течение 30 мин. В отдельной колбе дифенилметил(6R, 7S)-7- феноксиацетамидо-3-бромо-1-карба(1-детиа)-3- цефем-4-карбксилат (50,0 г, 0,089 ммоль) растворяют в безводном DMPU (100 мл) и 100 мл ДМФ под азотом и нагревают до 65oC. Прибавляют медный раствор к бромиду через канюлю в течение 1 ч, поддерживая температуру на уровне 65oC. Через 1 ч реакционную смесь охлаждают до 50oC и перемешивают в течение 16 ч. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом, вливают в насыщенный раствор NaHCO3, фильтруют через броунмиллерит, промывают дважды NaHCO3, водой, 1 н. раствором HCl (два раза), водой и солевым раствором, сушат в присутствии сульфата магния и восстанавливают до бурого масла в вакууме. Это масло фильтруют через силикагель с помощью 60-40 этилацетата/гексана и перекристаллизовывают из этилацетата/гексана, получая 28,4 г (58%) твердого вещества не совсем белого цвета.
Пример 16. Аллил [7S, 6R]-7- феноксиацетамидо-3-трифторметил-1-карба-(1-детиа)-3-цефем-4- карбоксилат.
После растворения продукта из примера 15 (26,00 г, 0,050 моль) в 375 мл N,N- диметилформамида и 750 мл тетрагидрофурана под азотом прибавляют 375 мл 1 н. раствора HCl с последующим прибавлением цинкового порошка (19,61 г, 0,30 ммоль). Раствор становится теплым, и его перемешивают 90 мин, а затем фильтруют через броунмиллерит. Затем ТГФ упаривают. После разбавления этилацетатом осуществляют четыре промывки 1 н. раствором HCl, водой, солевым раствором, сушат в присутствии сульфата натрия и упаривают при пониженном давлении до неочищенного твердого вещества оранжевого цвета. Твердое вещество растворяют в этилацетате, прибавляют 50%-ный водный раствор бикарбоната натрия, и двухфазный раствор обрабатывают кислой сернокислой соль тетрабутиламмония (17,83 г, 0,052 моль). После перемешивания в течение 15 мин слои разделяют, и этилацетатный слой сушат в присутствии сульфата натрия, фильтруют и упаривают при пониженном давлении до вязкого масла. Масло растворяют в 110 мл хлороформа и обрабатывают аллилбромидом (7,26 г, 0,060 моль), растворенным в 35 мл хлороформа, через капельную воронку в течение 30 мин. Раствор перемешивают при температуре окружающей среды в течение ночи, восстанавливают до твердого тела и хроматографируют на силикагеле с использованием 70% гексан/30% этилацетат, получая после упаривания при пониженном давлении 14,35 г указанного в заголовке соединения (68%).
1H-ЯМР (CDCl3) δ 7.45 (д, J=8 Гц, 1H), 7.30 (т, J=7 Гц, 2H), 7.00 (т, J= 7 Гц, 1H), 6.87 (д, J=10 Гц, 2H), 2.0-6.58 (м, J= Гц, 1H), 5.45 (дд, J=9 Гц, 1H), 5.4-5.2 (м, 2H), 5.72 (т, J=6 Гц, 2H), 5.43 (с, 2H), 3.95-3.85 (м, 1H), 2.55-2.4 (м, 1H), 2.4-2.25 (м, 1H), 2.1-1.95 (м, 1H), 1.6-1.4 (м, 1H).
Пример 17. Дифенилметил [7S, 6R]-7-фенокси-ацетамидо-3-трифторметил-1-карба-(1-детиа)-3-цефем-4-карбоксилат.
Суспензию только что промытого кислотой цинкового порошка (58,2 г, 0,89 моль) суспендируют в 1780 мл безводного ДМФ в трехлитровой трехгорлой колбе, снабженной перегородкой, термометром, трубопроводом для азота, капельной воронкой и конденсатором с сухим льдом. Приблизительно 20% охлажденного на льду дибромдифторметана (205,4 г, 0,98 моль) прибавляют по каплям в быстром темпе через капельную воронку, и через 15 мин начало экзотермической реакции вызывает повышение температуры до 50oC. Остальной дибромдифторметан прибавляют со скоростью, которая поддерживает внутреннюю температуру реакции на уровне 45-50oC, после чего дибромдифторметан перемешивают в течение 90 мин после прибавления всего соединения. Вслед за охлаждением раствора до температуры -30oC прибавляют 150 мл DMPU с последующим прибавлением CuCl (88,1 г, 0,89 моль), перемешивают 20 мин, затем ванну заменяют ледяной баней и перемешивание продолжают в течение 20 мин. В отдельной колбе исходный винилбромид (50,0 г, 0,089 моль) растворяют в 100 мл безводного DMPU и 100 мл безводного ДМФ и нагревают до температуры 65-70oC. К бромиду прибавляют холодный медный реагент через канюлю в течение приблизительно 1 ч, поддерживая температуру на уровне 65-70oC во время прибавления, и затем перемешивают в течение 1 ч при температуре 70oC. После охлаждения до температуры окружающей среды разбавляют этилацетатом, медленно вливают в 50%-ный водный раствор бикарбоната, отделяют, промывают два раза 50%-ным водным бикарбонатом, водой, два раза 1 н. раствором HCl, водой, солевым раствором, сушат в присутствии сульфата магния и восстанавливают до бурого масла. Масло хроматографируют на силикагеле с использованием 60:40 гексанэтилацетат, после чего кристаллизуют из этилацетата/гексана с получением 28,4 г твердого вещества бежевого цвета (58%).
Пример 18. п-Нитробензил[7S, 6R]-7- феноксиацетамидо-3-трифторметил-1-карба-(1-детиа)-3-цефем-4-карбоксилат.
Указанное в заголовке соединение получают в соответствии с методикой примера 17 (за исключением того,
что CuBr используют вместо CuCl), получая твердое вещество бежевого цвета с выходом 64%
1H-ЯМР (CDCl3) d 8.19 (д, J=9 Гц, 2H), 7.63 (д, J=9.0 Гц, 2H), 7.30 (т, J= 6 Гц,
3H), 7.03 (т, J=8 Гц, 1H), 6.86 (д, J=9 Гц, 2H), 5.45 (т, J=5 Гц, 1H), 5.37 (квартет, J=15 Гц, 2H), 4.57 (с, 2H), 4.0-3.9 (м, 1H), 2.6-2.35 (м, 1H), 2.4-2.3 (м, 1H), 2.1-2.0 (м, 1H), 2.6-2.4 (м,
1H).
Пример 19. Хлористоводородная смесь аллил-[7S- 6R]-7-амино-3-трифторметил-1-карба(1-детиа)-3-цефем-карбоксилата.
Продукт из примера 16 (10,53 г, 0,0246 моль) растворяют в 83 мл сухого метиленхлорида под азотом и охлаждают до 0oC. Раствор обрабатывают пиридином (2,41 г, 0,0305 моль) через шприц, после чего порционно в течение 20 мин прибавляют PCl5 (5,84 г, 0,0280 моль). Ледяную ванну удаляют через 30 мин и раствор перемешивают в течение 1,5 ч и за это время раствор переохлаждают до температуры ледяной бани и обрабатывают изобутиловым спиртом (18,4 г, 0,248 моль), растворенным в 20 мл диэтилового эфира, через шприц в течение 5 мин. Через 20 мин ледяную ванну удаляют и реакционную смесь перемешивают 1 ч. Образуется густое, ворсистое твердое вещество белого цвета, которое охлаждают до 0oC, собирают фильтрацией, промывают два раза 50/50 смесью метиленхлорида/простого диэтилового эфира и один раз диэтиловым эфиром. Это твердое вещество сушат в вакууме в течение 3 ч, получают 6,49 г указанного в заголовке продукта в виде хлопьеобразного твердого вещества белого цвета (77%).
1H-ЯМР (DMCO-d6) d 9,20 (c, 3H), 6,0-5,8 (м, 1H), 5,35 (д, J 1 Гц, 1H), 5,24 (д, J 1 Гц, 1H), 4,90 (д, J 1 Гц, 1H), 4,75 (т, 2H), 4,1- 3,9 (м, 1H), 3,5-3,2 (м, 2H), 2,2-2,1 (м, 1H), 1, 9-1,7 (м, 1H).
Пример 20. Аллил (7S, 6R)-7-[D-a- (третбутоксикарболамино)-3-фторфенилацетамино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоксилат.
Раствор (трет-бутоксикарбониламино)-3-фторфенилуксусной кислоты (1,05, 0,00321 моль ) в 21 мл безводного метиленхлорида обрабатывают N-метилморфолином (0,35 г, 0,00343 моль) и 1-хлор-3,5-диметокситриазином (0, 58 г, 0,00321 моль) при температуре 0oC под азотом и перемешивают в течение 45 мин. Продукт из примера 16 (1,00 г, 0,00306 моль) суспендируют в 8 мл метиленхлорида, обрабатывают N-метилморфолином (0,35 г, 0,00343 моль) и полученный раствор прибавляют по каплям в течение 10 мин к вышеуказанному раствору активированного сложного эфира. Через 20 мин при 0oC ванну удаляют и раствор перемешивают в течение 90 мин. После упаривания растворителя в вакууме остаток растворяют в этилацетате, нерастворимые вещества удаляют фильтрацией через броунмиллерит и упаривают до масла, после чего неочищенный продукт хроматографируют на силикагеле с использованием 75:25 гексан/этилацетат, затем 65: 45 гексан/этилацетат, получая 1,45 г (88%) указанного в заголовке продукта в виде бежевой пены.
1H-ЯМР (CDCl3) d 7,4-7,3 (м, 2H), 7,2-7,0 (м, 2H), 6,75 (д, 1H), 6,0-5,85 (м, 1H), 5,7-5,5 (м, 1H), 5,4-5,2 (м, 2H), 5,2-5,1 (м, 1H), 4,8- 4, 7 (м, 1H), 3,95-3,8 (м, 1H), 2,6-2 2,2 (М, 2H), 2,0-1,7 (м, 1H), 1,40 (с, 9H) и 1,3-1,1 (м, 1H).
Пример 21. (7S, 6R)-7[D, L-a-(трет-бутоксикарбониламино)-3- фторфенилацетамино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Продукт из примера 20 (1,45 г, 0,00268 моль) растворяют в 4,5 мл этилацетата и 1 мл метиленхлорида под азотом и дезоксигенируют барботированием азота в колбу в течение 5 мин. Раствор охлаждают в ванне со льдом и обрабатывают 0,070 г тетракистрифенилфосфинпалладия, 0,070 г тетрафенилфосфина и 6,4 мл (0,00322 моль) 0,5 М раствора 2-этилгексаноата натрия в этилацетате. Через 5 мин ванну удаляют и раствор перемешивают в течение 30 мин, вливая в 30 мл смеси 50/50 диэтилового эфира и гексана. Полученный осадок собирают фильтрацией с отсосом, растворяют в этилацетате и небольшом количестве метиленхлорида, промывают 1 н. раствором HCl, сушат в присутствии сульфата магния и упаривают с получением 1,20 г (89%) указанного в заголовке соединения, которое не подвергают дальнейшей очистке.
Пример 22. (6R, 7S)-7-b-[D-a-(амино)-3-фторфенилацетиламино]-3-трифторметил-1-карба (1-детиа)-3-цефем-4-карбоновая кислота.
Продукт из примера 21 (1,15 г, 0,00229 моль) прибавляют за одну порцию к охлажденному на льду раствору TFA (11,5 мл) и анизола (0,75 мл, 0,00687 моль) под азотом и перемешивают 30 мин. Реакционную смесь разбавляют ацетонитрилом и восстанавливают в вакууме, вновь разбавляют ацетонитрилом и упаривают при температуре 30oC досуха. Остаток порошкуют простым диэтиловым эфиром и полученное твердое тело собирают фильтрацией и промывают неоднократно простым диэтиловым эфиром. Неочищенное твердое вещество сушат в вакууме с получением 0,89 г смеси диастереомеров; 0,20 г продукта хроматографируют обращенно-фазовой хроматографией на колонке C-18 порционно (0,020 г) с использованием 10% ацетонитрил/1% AcOH/H2O, получая после лиофилизации 0,032 г искомого продукта.
Пример 23. Аллил (7S, 6R)-7-a-(трет-бутоксикарбониламино)-3-этилсульфонамидофенилацетиламино]-3-трифторметил-1-карба (1-детиа)-3-цефем-4-карбоксилат.
Указанное в заголовке соединение, полученное в соответствии с примером 20, имеет вид белой пены (88%).
1H-ЯМР (CDCl3) d 7,50 (с, 1H), 7,35-7,1 (м, 6H), 6,0-5,75 (м, 2H), 5,4-5,1 (м, 4H), 5,8-5,7 (м, 2H), 3, 95-3,8 (м, 1H), 3,10 (квартет, J 8 Гц, 2H), 2,6-2,0 (м, 3H), 1,40 (с, 9H), 1,30 (м, 1H).
Пример 24. (7S, 6R)-7-[D, L-a-(третбутоксикарбониламино)-3-этилсульфонамидофенилацетиламино] -3-трифторметил-1-карба (1-детиа)-3-цефем-4-карбоновая кислота.
Неочищенное соединение, указанное в заголовке, получают из продукта примера 23 в соответствии с методикой примера 21 (96%) и не подвергают дальнейшей очистке.
Пример 25. (6R, 7S)-7-b-[D-a-(амино)-3-этилсульфонамидофенилацетил-амино]-3-трифторметил-1карба-(1-детиа)-3-цефем-4-карбоновая кислота.
Указанный в заголовке продукт получают из неочищенного продукта примера 24 в соответствии с методикой примера 22 и хроматографируют на колонке C-18, используя 20% ацетонитрил/1% ацетат аммония/ H2O, с получением 14% продукта после лиофилизации.
1H-ЯМР (DMCO-d6) d 9,85 (шир.с, 1H), 9,3- 9,1 (м, 1H), 7,3-7,0 (м, 4H), 5,3-5,2 (м, 1H), 4, 72 (с, 1H), 3,7-3,6 (м, 1H), 3,1-2,9 (м, 1H), 2,2-1,9 (м, 2H), 1,4-1,3 (м, 1H), 1,25-0,95 (м, 4H).
Пример 26. Аллил (7S, 6R)-7-[a-(третбутоксикарбониламино)-3-бромфенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоксилат.
Указанное в заголовке соединение получают в соответствии с методикой в примере 20 и хроматографируют на силикагеле с использованием 70/30 гексан/этилацетат, получая не совсем белую пену (91%).
1H-ЯМР (CDCl3 d 7,5-7,2 (м, 5H), 6,0-5, 7 (м, 2H), 5,5-5,2 (м, 3H), 5,0-4,7 (м, 1H), 3,95-3,8 (м, 1H), 2,6-2,2 (м, 2H), 2,1-1,7 (м, 1H), 1,4 (с, 9H), 1,1-1,3 (м, 1H).
Пример 27. (7S, 6R)-7-[D, L-a- (третбутоксикарбониламино)-3-бромфенилацетиламино]-3-трифторметил-1-карба (1-детиа)-3-цефем-4-карбоновая кислота.
Указанный в заголовке продукт с выходом свыше 100% выделяют в результате реакции, аналогичной реакции примера 21. Исходным веществом является продукт из примера 26.
Пример 28. [6R, 7S]-7-b-[D-a-(амино)-3- бромфенилацетиламино]-3-трифторметил-1-карба(1- детиа)-3-цефем-4-карбоновая кислота.
Указанное в заголовке соединение получают в соответствии с методикой
примера 26 и хроматографируют с использованием 15% ацетонитрил /1% AcOH/H2O, получая продукт с выходом 10%
1H-ЯМР (ДМСО-d6) d 9.20 (шир.с, 1H), 7.65 (с, 1H),
7.65 (с, 1H), 7.50 (д, J 5 ГЦ, 1 H), 7.40 (д, J= 5 Гц, 1H), 7.30 (т, J 5 Гц, 1H), 5.3-5.2 (м, 1H), 4.75 (с, 1H), 3.7-3.6 (м, 1H), 2.20-2.0 (м, 2H), 1.4-1.2 (м, 2H).
Пример 29. Аллил [7S, 6R]-7-[a-трет- бутоксикарбониламино)-3-трифторметилфенилацетиламино]-3-трифторметил-1- карба(1-детиа)-3-цефем-4-карбоксилат.
Указанное в заголовке соединение получают в соответствии с примером 20 в виде белой пены (91%).
1H-ЯМР (CDCl3) d 7.4-7.2 (м, 4H), 6.9-6.8 (м, 1H), 6.0-5.85 (м, 1H), 5.7-5.55 (м, 1H), 5.4-5.2 (м, 4H), 4.75 (т, J 3 Гц), 4.0-3.8 (м, 1H), 2.5-2.2 (м, 2H), 2.1-1.7 (м, 1H), 1.7-1.5 (м, 1H), 1.40 (с, 1 H).
Пример 30. [7S, 6R)-7-(D, L-a-(трет - бутокискарбониламино)-3-трифторометилфенилацетиламино] -3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Указанное в заголовке соединение получают в соответствии с методикой примера 21, получая свыше 100% неочищенного твердого тела светло-желтого цвета. Исходным веществом является продукт из примера 29.
Пример 31. [6R, 7S]-7-b-[D-a-(амино)-3-трифторметилфенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Указанное в заголовке соединение получают в соответствии с
методикой
примера 22, используя продукт из примера 30 в качестве исходного вещества, и хроматографируют на колонке C18 порционно (0,020 г), применяя 15% ацетонитрил /1% AcOH/H2O, с
получением
продукта, имеющего выход 12%
1ЯМР (ДМСО-d6)d 9.5-9.3 (м, 1H), 8.6 (шир.с, 3H), 7.9-7.6 (м, 4H), 5.56-5.35 (м, 1H), 5.16 (с, 1H), 3.85-3.75 (м, 1H), 2.3-2.1 (м,
2H),
1.35-1.25 (м, 1H), 1.25-1.1 (м, 1H).
Пример 32. [6R, 7S)-7-b-[D-a-(амино)-3,4 -дихлорфенилацетиламино]-3-трифторметил-1-карба(1 -детиа)-3-цефем-4-карбоновая кислота.
Рацемический раствор трет-ВОС-защищенного 3,4-дихлорфенилглицина (0,550 г, 0,00173 моль) в 21 N,N-диметилформамида охлаждают до -45oC под азотом, прибавляют изобутилхлорформиат 80,22 мл, 0, 00173 моль) с последующим прибавлением N-метилморфолина (0,19 мл, 0,00173 моль) и перемешиванием полученного раствора 30 минут. Моносилилтрифторацетамид (1,53 г, 0,00824 моль) прибавляют к перемешанной суспензии 7-амино-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновой кислоты (0,600 г, 0,00165 моль) в 15 мл N,N- диметилформамида и раствор нагревают до 40oC в течение 30 мин. Раствор силилированного ядра хлаждают и прибавляют к смешанному ангидриду со скоростью, которая поддерживает внутреннюю температуру реакции на уровне -40oC. Раствор медленно нагревают до 0oC через 2 ч, прибавляют 1, 2 мл MeOH, и ванну со льдом удаляют. После достижения температуры окружающей среды раствор разбавляют этилацетатом, фильтруют через броунмиллерит, промывают четыре раза 1 н. раствором HCl, водой, солевым раствором, сушат в присутствии сульфата магния и концентрируют при пониженном давлении. Остаток хроматографируют на силикагеле с использованием 2/80/ 18:HOAc/этилацетат/гексан. Неочищенный продукт прибавляют к перемешанному раствору TFA (4,0 мл) и Et3SiH (1,2 мл) в ванне со льдом и через 30 мин раствор концентрируют, порошкуют простым диэтиловым эфиром, собирают фильтрацией и промывают простым диэтиловым эфиром. Хроматография неочищенной соли TFA (0,114 ) на колонке C18 с использованием градиента к 25% CH3 CN/H2O позволяет получить после лиофилизации 0,021 г (3%) смеси диастереомеров (90% D-боковой цепи и 10% L боковой цепи при анализе ЖХВР).
1H-ЯМР (ДМСО d6) d 9.15 (шир.с, 1H), 7.7-7.6 (м, 2H), 7.38 (д, J 8 Гц, 1H), 5.3-5.25 (м, 1H), 4.75 (с, 1H), 3.8-3.7 (м, 1H), 2.2-2.1 (м, 2H), 1.4-1.2 (м, 2H).
Пример 33. [6R, 7S]-7b-[D-a-(амино)-3-хлор-4-гидроксифенилацетиламино] -3-трифторметил-1-карба-(1-детиа)-3-цефем-4-карбоновая кислота.
Указанный в заголовке продукт получают в соответствии с условиями проведения реакции примера 32, за исключением того, что чистый трет-ВОС-защищенный D-3-хлор-4-гидроксифенилглицин используют для ацилирования, получая 41% -ный выход твердого вещества белого цвета.
1H-ЯМР (ДМСО-d6) d 7.40 (с, 1H), 7.15 (д, J 9 Гц, 1H), 6.95 (д, J 10 Гц, 1H), 5.17 (д, J 5 Гц, 1H), 4.75 (с, 1H), 3.7-3.6 (м, 1H), 2.2 1.9 (м, 2H), 1.45-1.3 (м, 1H), 1.15-1.0 (м, 1H).
Пример 34. Аллил [7S, 6R] -7-[[2- (трифенилметил)амино-4-тиазолил] (третбутокси-карбонилметоксиимино) -ацетил] амидо-3- трифторметил-1-карба(1-детиа)-3-цефем-4-карбоксилат.
Указанное в заголовке соединение получают в соответствии с методикой примера 20 с выходом 41% твердого тела белого цвета.
1H-ЯМР (CDCl3) d 8.68 (д, 1H), 7.34 (c, 16H), 7.02 (с, 1H), 6.80 (с, 1H), 6.0-5.9 (м, 1H), 5.57 (д, 1H), 5.4-5.2 (м, 2H), 4.8-4.7 (м, 4H), 4.0-3.9 (м, 1H), 2.5-2.4 (м, 1H), 2.4-2.3 (м, 1H), 2.2-2.1 (м, 1H), 1.65-1.5 (м, 1H), 1.44 (с, 9H).
Пример 35. [7S, 6R]-7-[2-амино-4- тиазолил(карбоксиметоксиимино)ацетил] -3-трифторметил-1-карба(1-детиа)-3-цефем-4- карбоновая кислота.
200 мг (0,245 ммоль) вышеуказанного продукта примера 34 обрабатывают 2,5 мл холодной трифторуксусной кислоты и 0,8 мл холодного триэтилсилата под азотом в ванне со льдом 1 ч. Затем ванну удаляют и раствор нагревают до комнатной температуры и перешивают в течение ночи. Раствор разбавляют ацетонитрилом и упаривают до небольшого объема в вакууме при 30oC, затем разбавляют еще 2 раза ацетонитрилом и упаривают досуха. Остаток порошкуют простым диэтиловым эфиром и 73 мг неочищенного твердого тела бежевого цвета собирают фильтрацией с отсосом (57%-ный выход).
65 мг (0,126 ммоль) вышеуказанного неочищенного продукта растворяют в 3 мл безводного ацетонитрила под азотом с последующим прибавлением 3 мг (0,0126 ммоль) трифенилфосфина и 1 мг (0,00252 ммоль) палладиевого ацетата. Раствор охлаждают в ванне со льдом при одновременном прибавлении через шприц 0,036 мл (0,132 ммоль) гидрида трибутилолова. Через 1 ч раствор обрабатывают 0,014 мл (0,252 ммоль) уксусной кислоты и перемешивают в течение 30 мин. Осадок собирают и промывают простым диэтиловым эфиром и затем перекристаллизовывают из метанола/этилацетата/диэтилового эфира с получением 21 мг светло-желтого твердого тела (выход 35%).
1Н-ЯМР (ДМСО-d6) d 9,45-9,3 (м, 1H)
7,
14 (c, 2H), 6,76 (c, 1H), 5,5-5,4 (м, 1H), 4,56 (c, 2H), 3,9-3,8 (м, 1H), 2,4-2,2 (м, 2H), 1,95-1,85 (м, 1H), 1,6-1,45 (м, 1H)
Пример 36. Аллил (7S, 6R)-7-[D,
L-a- (третбутоксикарбониламино)-3-тиенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоксилат.
Указанное в заголовке соединение, полученное в соответствии с методикой примера 20, имеет вид твердого вещества не совсем белого цвета (91%).
Пример 37. [7S, 6R]-7-[D, L-a- (третбутоксикарбониламино)-3-тиенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Указанное в заголовке соединение получают из продукта примера 36 в соответствии с методикой примера 21 без дальнейшей очистки.
Пример 38. [6R,7S]-7-b-[D-a-(амино)-3-тиенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Указанный в заголовке продукт получают из неочищенного продукта примера 37 в соответствии с методикой, приведенной для примера 22, и хроматографируют на колонке C18 с использованием метанола/уксусной кислоты, воды, получая 18%-ный выход лиофилизации.
1Н-ЯМР (ДМСО-d6) d 9,3-9,2 (м, 1H), 7,6-7,5 (м, 2H), 7,15 (д, 1H), 5,3-5,2 (м, 1H), 4,90 (с, 1H), 3,85-3,6 (м, 1H), 2,2-2,0 (м, 2H), 1,45-1,2 (м, 2H).
Пример 39. Аллил [7S, 6R]-7-b-[D-a-(третбутоксикарбониламино)-3-бензиенилацетиламино]-3-трифторметил-1-карба-(1-детиа)-3-цефем-4-карбоксилат.
Указанное в заголовке соединение, полученное в соответствии с методикой примера 20, имеет вид пены белого цвета (выход 60%).
Пример 40. [7S, 6R]-7-b-[D-a-(третбутоксикарбониламино)-3-бензтиенилацетиламино]-3-трифторметил-1- карба(1-детиа)-3-цефем-4-карбоновая кислота.
Указанное в заголовке соединение получают из продукта примера 39 в соответствии с методикой примера 21, и не подвергают очистке.
Пример 41. [6R, 7S)-7-b-[D-a-(амино)-3-бензтиенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Указанное в заготовке соединение получают в соответствии с методикой
примера 22 из неочищенного продукта примера 40 и хроматографируют на кнопке C18 с использованием метанола/уксусной кислоты/воды, после чего лиофилизуют с получением твердого вещества белого цвета с
выходом 32%
1Н-ЯМР (ДМСО-d6) d 9,35 (д, 1H), 9,1-8,5 (шир.с, 3H), 8,1-7,9 (м, 2H), 7,9 (с, 1H), 7,5-7,3 (м, 2H), 5,5-5,3 (м, 2H), 3,9-3,7 (м, 1H), 2,2-2,1 (м, 2H), 1,4-1,
3 (м, 1H), 1,2-1,1 (м, 1H).
Пример 42. Аллил 7-[(N-трет- бутилоксикарбонил-N-феноксиацетил)-амино] -3-бромо-1-карба(1-детиа)-2,3-цефем-4-карбоксилат
Смесь 60/40 D2/Δ3
соединения 3-бромо из препарата 3 (100 мг, 0,2297 ммоль) объединяют с ДМАР (29 мг, 0,2343 ммоль) и ди-трет-бутилдикарбонатом (0,2412 ммоль, 84 мкл). Смесь перемешивают приблизительно 1 ч при
комнатной
температуре. Смесь непосредственно хроматографируют на силикагеле с элюированием 20% EtOAc/CH2Cl2, получая 104 мг (85%-ный выход) указанного в заголовке соединения в
виде смеси
60/40 Δ2/Δ3 изомеров соответственно. Данные относительно Δ2-изомера.
1Н-ЯМР (300 МГц, CDCl3) δ 7,30 (т, J=8 Гц, 2H), 7,0 (т, J 6 Гц, 1H), 6,90 (д, J 8 Гц, 2 H), 6,30 (м, 1H), 5,95 (м, 1H), 5,65 (м, 1H), 5,35 (м, 2H), 5,10 (м, 2H), 4,88 (с, 1H), 4,75 (м, 2H), 4,10 (м, 1H), 2,25 (м, 2H), 1,50 (с, 9H).
Пример 43. Аллил-7-[(N-трет-бутоксикарбонил-N-феноксиацетил)амино]-3-бромо-1-карба(1-детиа)-2-цефем-4-карбоксилат.
Чистый D2-изомер 3-бромосоединения из препарата 3 (4,6 г, 10,57 ммоль) объединяют с ди-трет-бутилдикарбонатом (2,54 г, 11,6241 ммоль, 2,65 мл) и DMAР (1,33 г, 10,88 ммоль), после чего перемешивают. Смесь обрабатывают, как в примере 34, получая 5,3 г (93,6%-ный выход) указанного в заголовке продукта.
Пример 44. Аллил [7S, 6R]-7-трет-бутилоксикарбониламино-3-бромо-1-карба-(1-детиа)-2-цефем-4-карбоксилат.
Продукт из примера
42 (104
мг, 0,1947 ммоль) объединяют с ТГФ (2 мл) и первой порцией LiOH (0,1652 мл) и перемешивают в течение 40 мин. Затем прибавляют вторую порцию LiOH (0,0295 мл) и смесь перемешивают 1 ч. Смесь
выливают в
40 мл EtOAc и затем смешивают с 10 мл воды и 10 мл насыщенного раствора NaHCO3. Органические вещества отделяют, сушат в присутствии сульфата натрия, фильтруют и концентрируют с
получением
79 мг (выход свыше 100%) неочищенного продукта. Данные относительно Δ2-изомера:
1Н-ЯМР (300 МГц, CDCl3) δ 6,35 (м, 1H), 5,95 (м, 1H), 5,35 (м,
2H), 5,15 (м,
2H), 4,88 (с, 1H), 4,68 (м, 2H), 4,05 (м, 1H), 2,35 (м, 2H) и 1,42 (с, 9H).
Пример 45. Аллил [7S, 6R]-7- бутилоксикарбониламино-3-бромо-1-карба(1-детиа)-2-цефем-4-карбоксилат.
Продукт из примера 43 (5,25 г, 9,8076 ммоль) объединяют с ТГФ (95 мл) и первой порцией LiOH (8,75 мл) и перемешивают приблизительно 40 мин. Прибавляют вторую порцию LiOH (1,55 мл) и смесь перемешивают в течение 1 ч. Смесь обрабатывают в соответствии с примером 36, получая 4,02 г (выход свыше 100%) указанного в заголовке продукта.
Пример 46. Аллил [7S, 6R]-7- бутилоксикарбониламино-3-бромо-1-карба(1-детиа)-3-цефем-4-карбоксилат.
Продукт из примера 44 (16 мг, 0, 0399 ммоль) объединяют с 0,3 мл CH2Cl2 и DBU (0, 0064 ммоль, 1,0 мкл). Смесь перемешивают 60 мин при комнатной температуре. Смесь отмеривают пипеткой через 1 ч кремнезема и затем элюируют с использованием 10% EtOAc/CH2Cl2. Указанный в заголовке продукт пропускают через DBU на кремнеземе. Смесь концентрируют до пены, получая 15,1 мл продукта при соотношении 15/85D2/Δ3- изомеров, что представлятет 94,9% восстановления на основе теоретического минимума от 16 г.
Пример 47. Аллил [7S, 6R]-7- бутилоксикарбониламино-3-бромо-1-карба-(1-детиа)-3-цефем-4-карбоксилат.
Продукт
из примера 45 (4,0 г, 9,97 ммоль) обрабатывают в соответствии с примером 46 за исключением того,
что 6% etAOc/CH2Cl2 используют вместо 10% раствора, получая чистый Δ
3-изомер. Используют следующие реакционные условия:
DBU 2,5 ммоль, 0,373 мл
CH2Cl2 70 мл
Неочищенную смесь вносят непосредственно в хроматографическую
колонку глубиной 8 дюймов (20,32 см) и диаметром 4 дюйма (10,16 см) (кремнезем) и элюируют с помощью
6% EtOAc/CH2Cl2, получая 1,98 г желательного Δ3-изомера и 610 мг смеси
Δ2/Δ3 изомеров, повторно хроматографируют, как описано выше с получением
дополнительных 250 мл чистого Δ3-изомера и 360 мг чистого Δ2-изомера. Чистый Δ3-изомер
прибавляют с получением общего количества 2,23 г (66%-ный выход) чистого Δ
3- изомера.
1Н-ЯМР (300 МГц, CHCl3)δ 5,95 (м, 1H), 5,35 (м, 3H), 5,15 (м, 1H), 4,75 (м, 2H), 3,85 (м, 1H), 2,75 (м, 2H), 2,0 (м, 1H), 1,70 (м, 1H) и 1,42 (с, 9H).
ИК (CHCl3): 3019, 1775, 1718, 1504, 1369, 1206 1055 см-1.
Macc-cпектр: m/e 400 (М+).
Анализ для C16H21N2O2Br:
Вычислено, C 47,89, H 5,28, N 6,78, Br 19,91
Найдено, C 47,85, H 5,05, N 6,77, Br 19,76
Пример 48. [7S,
6R]-7-(аминофенилацетил)-амино-3-бромо -1-карба(1-детиа)-3-цефем-4- карбоновая кислота.
A) В 25-мл колбу прибавляют при температуре 30oC 78 мг (0,1944 ммоль) аллил [7S, 6R)-7- бутилоксикарбониламино-3-бромо-1-карба(1-детиа)-3-цефем-4-карбоновой кислоты в 4 мл EtOH и 37 мг (0,1944 ммоль) моногидрата n-толуолсульфокислоты, после чего смесь концентрируют досуха. Прибавляют EtOH (4 мл) и смесь вновь концентрируют досуха. В отдельной колбе 46 мг (0,1944 ммоль) (2-пропенилокси)карбониламино-D-фенилглицина и 34 мг (0,1944 ммоль) хлордиметокситриазина в 1,5 мл CH2Cl2 объединяют и охлаждают до 0oC. Прибавляют N-метилморфолин (NMM) и смесь перемешивают 40 минут. В это время прибавляют другой эквивалент NMM, после чего прибавляют смесь в первой колбе. Смесь нагревают до комнатной температу ры 2-5 ч. Смесь концентрируют почти досуха, после чего прибавляют 1 мл CH2Cl2 и 1 мл 50/50 смеси EtOAc/гексана. Затем смесь нагружают на 15 г (кремнезем) хроматографической колонки и элюируют с использованием 10% MeOH/EtOAc, получая 74 мг аллил-7-(фенил(2-пропенилокси)карбониламино)ацетиламино-3-бромо-1-карба(1-детиа)-3-цефем-4-карбоксилата, физические данные которого приведены ниже.
1Н-ЯМР (300 МГц, CDCl3) d 7,30 (с, 5H), 7,08 (м, 1H), 5,95 (м, 3H), 5,25 (м, 6H), 4,72 (д, J 6 Гц, 2H), 4,52 (м, 2H), 3,82 (м, 1H), 2,62 (м, 2H), 1,58 (м, 1H), 1,25 (м, 1H).
ИК (CHCl3): 3019, 1775, 1727, 1689, 1496, 1241 и 1052 см-1.
Масс-спектр: m/e 517.
Анализ для C23H24
N3O6Br:
Вычислено, C 53,29 H 4,67, N 8,11, Br 15,4
Найдено, C 53,51, H 4,46, N 7,93, Br 15,4
B) В 25-мл колбе объединяют 73 мг (0,1408 ммоль) продукта
из части (A) в 2,0 мл CH3CH
и 1,0 мл Et2O, 7,4 мг (0,0282 ммоль) трифенилфосфина и 1,4 мг (0,0056 ммоль) ацетата палладия. Смесь охлаждают до 0oC через 5 мин и через
микрошприц прибавляют 76 мкл раствора
Bu3SnH. Смесь нагревают до комнатной температуры и перемешивают 45 мин. К смеси прибавляют 4 мкл Bu3SnH и 2 мл Et2O и
перемешивают дополнительно 30 мин. Прибавляют
концентрированный раствора HCl (23,5 мкл, 12 М), и в это время наблюдается преципитация. Прибавляют 10 мл Et2O и смесь переносят в 40-мл
центрифужную пробирку. Затем прибавляют 20 мл
Et2O и смесь центирифугируют. Твердое вещество декантируют и промывают (2х10 мл CH3CN/10 мл Et2O), затем промывают дважды
15 мл Et2O. Смесь сушат в вакууме
с получением 51,5 мг твердого вещества бурого цвета, которое растворяют в 1,2 мл CH3CN и 0,13 мл 1 н. раствора HCl, центрифугируют и декантируют
в 15-мл центрифужную пробирку, образуя
завихрения, прибавляют несколько выше, чем 1 эквивалент, 1,5 М раствора NH4OH с тем, чтобы установить pH на уровне около 4,0, и в это время осаждается
твердое вещество белого цвета,
которое затем центрифугируют и декантируют, промывая один раз 8 мл Et2O и дважды 8 мл 1/1 Et2O/гексан, после чего сушат с получением 46,5 кг
указанного в заголовке продукта.
Физические данные этого продукта приведены ниже.
1H-ЯМР (300 МГц, D2O) d 7.55 (м, 5H), 5.40 (д, J=12Гц, 1H), 5.20 (с, 1H), 3.85 (м,1H), 2.50 (м, 2H), 1.55 (м, 1H), 1.25 (м, 1H).
ИК(КВ) 3150, 3050, 1770, 1750, 1625, 1550, 145 и 1325 см-1.
Масс-спектр: m/e 394(M++1).
Пример 49. 7-(((2-Амино-4-тиазолил)-метоксииминоацетил)амино)-3-бромо-1-карба(1-дтиа)-3-цефем-4-карбоновая кислота.
А) В 50-мл колбе объединяют 240 мг (0,5981 ммоль) аллил-7-бутоксиамино-3-бромо-1-карба(1-детиа)-3-цефем-4-карбоксилата в 8 мл EtOH и 114 мг (0,5891 ммоль) моногидрата п-толуолсульфокислоты, после чего смесь разрушают ультразвуком до растворения всех твердых веществ. Смесь концентрируют досуха, прибавляют 8 мл EtOH и смесь вновь концентрируют досуха. Во второй колбе объемом 50 мл объединяют 181 мг (0,5891 ммоль)[2-(трет-бутоксикарбонил)амино-4-тиазолил](метоксиимино)-уксусной кислоты и 105 мг (0,5891 ммоль) хлордиметокситриазина в 4,5 мл CH2Cl2 и охлаждают до 0oC. Во вторую колбу прибавляют N-метилморфолин (NMM) (69 мкл, 0,628 ммоль) и содержимое перемешивают 45 мин. После перемешивания прибавляют другой эквивалент NMM и содержимое первой колбы в CH2 Cl2 через пипетку. Температуру во второй колбе доводят до комнатной и содержимое перемешивают 2,5 ч. Смесь концентрируют почти досуха, после чего прибавляют 2,5 мл CH2Cl2/EtOAc. Затем смесь загружают в колбу хроматографической колонки (50 г, кремнезем) и элюируют с помощью 80/20 CH2Cl2/EtOAc. Желательное вещество концентрируют с получением 270 мг указанного в заголовке продукта.
1H= ЯМР (300 МГц, CDCl3) d 9,45 (с, 1H), 8.25 (д, J=8 Гц, 1H), 6.98 (с, 1H), 5.95 (м, 1H), 5.68 (м, 1H), 5.35 (м, 2H), 4.75 (м, 1H), 4.0 (м, 1H), 3.95 (с, 3H), 2.80 (м, 2H), 2.10 (м,1H), 1.90 (м, 1H) и 1.52 (с, 9H).
Масс-спектр: m/e 583 (M++1).
Анализ для C22
H25N5O7SBr:
Вычислено, C 45.29, H 4.32, N 12.00
Найдено, C 45.09, H 4.52, N 11.76
В) Продукт из части (А) выше (270 мг, 0,4623
ммоль) в 3 мл
CH2Cl2 помещают в 50-мл колбу с 54 мг (0,5085 ммоль) этилгексаноата натрия в 3 мл EtOAs. В колбу помещают 3,1 мг (0,0116 ммоль) трифенилфосфина и 13,4 мг (0,0116
ммоль)
тетракистрифенилфосфинпалладия (0) и смесь перемешивают 1,5 ч. Для осаждения твердого тела прибавляют 30 мл Et2O и смесь перемешивают 20 мин, после чего вливают в 200 мл CH2
Cl2 и 75 мл 1 н. раствора HCl. Твердое вещество выделяют, сушат в присутствии сульфата натрия, фильтруют и концентрируют досуха с получением 250 мг.
С) В колбу емкостью 50 мл помещают 251 мг (0,4591 ммоль) продукта из части (В) выше и 3 мл CH2Cl2 и смесь охлаждают до 0oC. В колбу прибавляют 0,22 мл (1,36 ммоль) Et3 SiH и 3 мл трифторуксусной кислоты, и смесь нагревают до комнатной температуры и перемешивают в течение 35-40 мин. Смесь разбавляют 25 мл CH3CN и концентрируют до 1 мл. Три раза прибавляют по 10 мл CH3CN и по 10 мл толуола и смесь концентрируют досуха с получением желтовато-коричневого твердого вещества. Твердое вещество хроматографируют на 75 г (кремнезем) и элюируют с помощью 0,5% AcOH/4,5% изопропанол/20% CH3CN/75% EtOAc до выхода всего искомого твердого тела. Искомые фракции концентрируют до 1/2 мл и преципицируют с помощью Et2O. Смесь центрифугируют и сушат с получением 60 мг указанного в заголовке продукта. Твердое тело хроматографируют повторно на 10 г NP20SS, элюируя с помощью 0,25% AcON, 10% CH3CN, 89,75% воды и собирают фракции по 10 мл.
1H-ЯМР(300 МГц, D2O) d 7.08 (с.1H), 5.46 (д, J=12Гц, 1H), 4.10 (м, 1H), 4.05 (с, 3H), 2.78 (м, 2H),05 (м, 1H) и 1.80 (м, 1H).
Масс-спектр: m/e 444 (M++1).
Соединения формулы (I) ингибируют рост некоторых болезнетворных организмов, как продемонстрировано методом разведения в агаре, в
котором испытуемые
соединения разводят до приемлемого диапазона концентраций в 0,1 М фосфат-забуференном растворе, pH 7.0, вводят в агар Мюллера-Хинтона (Дифко), пополненный 1% Бакто-Сапплементом С
(Дифко), при 50oC и отверждают в чашках Петри. Свежие ночные культуры исследуемых бактерий разбавляют приблизительно до 1 x (10)4 клеток/мкл и наносят в объеме 1 мкл на
поверхности агаровых пластин.
Иннокулированные пластины инкубируют в течение ночи при температуре 35oC в окружающем воздухе. Конечные значения минимальной ингибирующей концентрации (МИК)
регистрируют как наименьшие
концентрации антибиотика в микрограммах на миллилитр, которые ингибируют развитие видимого роста на пластинах. Ниже суммированы результаты таких испытаний с использованием
соединений приведенных выше
примеров. Следующие номера соединений присвоены тем соединениям, которые упоминаются в таблице:
1.
7b-[D-a-(амино)фенилацетиламино]-3- трифторметил-1-карба-(1-детиа)-3-цефем-4- карбоновая
кислота
2. [6R, 7S]-7β
-[D-a-(амино)-4- гидроксифенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота
3. [6R,
7S]-7-b-[D-a-(амино)-4- фторфенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота
4. [7S,
6R]-7-([2-амино-4- тиазолил(метоксиимино)ацетил]амидо)-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота
5. [7S,
6R]-7-([2-амино-4- тиазолил(оксиимино)ацетил]амидо)-3-трифторметил-1-карба-(1-Детиа)-3-цефем-4-карбоновая кислота
6. [6R, 7S]
-7-b-[D-a-(амино)-3- этилсульфонамидофенилацетиламино]-3-трифторметил-1-карба (1-детиа)-3- цефем-4-карбоновая кислота
7. [6R,
7S]-7-b-[D-a-(амино)-3- бромфенилацетиламино]-3-трифторметил-1-карба (1-детиа)-3-цефем-4-карбоновая кислота
8. [6R,
7S]-7-b-[D-a-(амино)-3- фторфенилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота
9. [6R,
7S]-7-b-[-a-(амино-3- трифторметилацетиламино]-3-трифторметил-1-карба(1-детиа)-3-цефем-4-карбоновая кислота.
Реферат
Производные 3-трифторметил-1-карба-1-детиа-3-цефем-4-карбоновой кислоты и промежуточный продукт для их получения. Сущность изобретения: предлагаются в качестве антибиотиков 7-β -ациламино-1-карба(1-детиа)-3-трифторметил-3-цефем-4-карбоновые кислоты и их производные. Также предлагаются фармацевтические составы, включающие антибиотики, промежуточные соединения и способ их получения. 3 с. и 8 з.п. ф-лы, 1 табл.
Формула
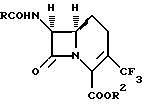
где R2 водород или карбоксизащитная группа;
R замещенная метильная группа общей формулы
R4-CH-Q
где Q NH2, R4 фенил или замещенный фенил общей формулы
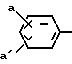
где а и а' водород, галоген, гидрокси или С1 - С4 -алкилсульфониламиногруппа, или R4 тиазолил, замещенный аминогруппой;
или R группа общей формулы
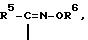
где R5 тиазолил, замещенный аминогруппой, R6 водород или С1 С4-алкил, или, когда R2 водород,
их фармацевтически приемлемые соли.
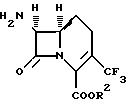
где R2 водород или карбоксизащитная группа,
или их кислые аддитивные соли.
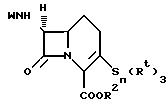
где R2 водород или карбоксизащитная группа;
Rt С1 С6-алкил;
W аминозащитная группа.
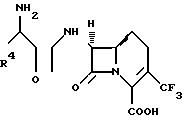
где R4 фенил или замещенный фенил общей формулы
где а и а' имеют указанные значения.
где а и а' имеют указанные значения.
Комментарии