Способ получения цефалоспориновых антибиотиков - RU2070200C1
Код документа: RU2070200C1
Описание
Изобретение относится к способу получения цефемовых, карба(детиа)цефемовых и окса(детиа)цефемовых соединений.
Но Shi и др. в патенте США N 4520022 (28 мая 1985 г.) описаны цефалоспориновые антибиотики, имеющие 1-пропениловую группу в
3-положении и описывающиеся следующей формулой, в которой R1 и R2 означают Н, ОН, ОСН3 или Сl:
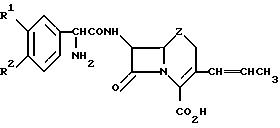
1-пропениловая группа описанных Но Shi и др. соединений предпочтительно имеет (Z)-конфигурацию, поскольку она проявляет более высокую антибактериальную активность, чем (Е)-конфигурация. Указанные соединения получают путем взаимодействия 3-галогенметилцефалоспорина с триарилфосфином с образованием в результате фосфоранилового промежуточного соединения, которое затем обрабатывают альдегидом для получения пропениловой или замещенной пропениловой группы.
При осуществлении вышеуказанного способа образуется смесь цис(Z)- и транс(Е)-изомеров. Поэтому для получения предпочтительного, обладающего более высокой антибактериальной активностью цис(Z)-изомера необходимо осуществлять дорогостоящее разделение или создавать такие условия процесса, при которых образуется целевой изомер. Общий выход целевого цис(Z)-изомера в расчете на исходный материал снижается за счет образования транс(Е)-изомера.
Сравнительно недавно в целях предупреждения образования нежелательного изомера и повышении таким образом выхода было предложено путем катализируемого палладием сочетания вводить алкениловые заместители в 3-положение цефалоспоринового кольца. Аллильное сочетание 3-галогенметицефемов с винилстаннанами, инициируемое соединениями Pd (0), галоидами метиллов и трис-(2-фурил)фосфином, приводит к образованию новых цефалоспоринов (S.R.Baker и др. патент США N 4847373, выданный 11 июля 1989 г.). Способ W.J.Scott и др. J. Amer. Chem. Soc. 106, 46304632 (1984), катализируемого смесью соединения Pd (0) и LiCi сочетания циклогексенилтрифлата с винилтрибутилстаннаном был применен для получения различных 3((Z)-1-пропенил)цеф-3-емов путем взаимодействия 3-трифлоксицеф-3-ема с (Z)-1-пропенилбутилстаннаном (S.R.Baker и др. патент США N 4870168, выданный 26 сентября 1989 г.).
О других исследованиях по расширению круга реакций и оптимизации условий проведения процесса для катализируемого палладием сочетания винилтрифлатов и винилстаннанов сообщается в работах Scott и Stille, J.Amer. Chem. Soc. 108, 3033-3040 (1986) и Stille и Groh. J.Amer.Chem, Soc. 109, 813-817 (1987).
Сочетание 3-трифлоксицев-3-ема с трибутилстаннаном в присутствии PdCi2(CH3CN)2 без фосфина или LiCi, необходимых в случае вышеописанного способа W.J.Scott и др. описано G.K.Cook и J.H.McDonald III (196th American Chemical Society National Meeting, Los Angeles, CA Sept 25-30, 1988 Division of Organic Chemistry, Abstract N 32).
Предметом изобретения является способ получения цефемов, оксацефемов и карбацефемов, имеющих алкенильную, алкинильную, арильную или гетероциклическую группу в 3-положении цефемового ядра. Предлагаемый способ включает взаимодействие 3-сульфонилоксизамещенного цефема, оксацефема или карбацефема с органостанном в присутствии Pd (II) или Pd (0)-катализатора, причем указанная 3-сульфонилокси-группа выбрана из фторсульфонилокси-, 4-нитробензолсульфонилокси- и 4-бромбензолсульфонилокси-групп. Предлагаемый способ представляет собой усовершенствованный вариант способа, описанного в нашем патенте США N 4870168. Усовершенствование это состоит в том, что взамен описанного в указанном патенте дорогого реагента 3-трифлоксицеф-3-ема в случае способа в соответствии с настоящим изобретением используются сравнительно дешевые реагенты.
Если это не оговорено или не вытекает из контекста, под использующимися в тексте настоящего описания выражениями "алкил", "алкенил", "алкинил",
"алкдиенил" и т.п. имеются в виду как прямые, так и разветвленные углеродные цепи. Соединения, содержащие фрагмент
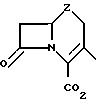
обозначаются термином "цефем", если Z означает атом серы, "оксацефем", если Z означает атом кислорода, и "карбоцефем", если Z означает метилен. Различные асимметрические атомы углерода азабициклокольцевой системы имеют такую же стереохимическую конфигурацию, как и асимметрические атомы углерода цефалоспориновых антибиотиков, широко использующихся в медицинской практике, которая соответствует продукту ферментации цефалоспорину С.
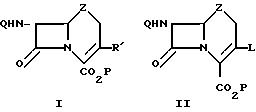
Согласно изобретению получают цефемы, оксацефемы и карбацефемы формулы I
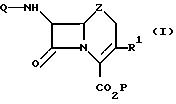
в которой Q, P и Z имеют те же определения, что и в формуле I. R1 выбран из группы, состоящей из Н, С2-6-алценила, С2-6-алкинила, С2-6-алкдиенила, С6-10-арила, замещенного С6-10-арила, незамещенного и замещенного гетероциклического остатка, причем указанные замещенные арил и гетероциклический остаток содержат 1-3 группы, выбранные из С1-3-алкила, гидрокси-, С1-3-алкоксигруппа, галогена, амино-, С1-3 -алкиламино-, ди-C1-3-алкиламино, нитрокарбоксильной, С1-3-алкоксикарбонильной и циано-групп. Примерами гетероциклического остатка являются придил, имидазолил, тиазолил, фурил, пирролил, тиенил и изоксазолил. Предлагаемый способ может использоваться, в частности, для получения соединений формулы I, у которых R1 означает Н, С2-6-алкенил или С2-6-алкинил, наиболее предпочтительно для получения соединений, у которых R1 означает С2-6-алкенил. В частности, предлагаемый способ может использоваться для получения цефпрозила, т. е. 77β-[D-2-амино-2-(4-гидрокси-фенил)-ацетамидо] -3-[(Z)-1-пропен -1-ил]цеф-3-ем-4-карбоновой кислоты.
Таким образом, соединения формулы I получают из
соединения
формулы II по следующей схеме:
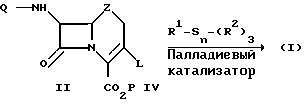
в которой Q, Z и Р имеют вышеприведенные определения, а L выбрано из группы, состоящей из фторсульфонилокси-, 4-нитробензолсульфонилокси- и 4-бромбензолсульфонилокси-групп. Соединение формулы II подвергают взаимодействию с органостаннаном формулы R1-Sn-(R2)3, в которой R1 имеет вышеприведенное определение, а R2 означает органическую группу, использующуюся обычно в реакциях сочетания органостаннанов (R2 может, например, означать С1-6-алкил, в частности бутил), в среде органического растворителя и в присутствии 1-10 мол. соединения Pd (II) или Pd (0) в качестве катализатора.
Использующиеся в вышеприведенной реакционной схеме в качестве исходного материала 3-(фторсульфонил)оксизамещенные соединения получают вышеописанным образом. Исходные 3-[(4-нитробензолсульфонил)окси] цефем и 3-[(4-бромбензол)сульфонил)окси] цефем получают с помощью модифицированного способа, описанного в патенте США N 3985737, выданного 12 октября 1976 г. Соответствующие оксацефемовые и карбацефемовые производные могут быть получены аналогичным образом.
Для получения предлагаемым способом соединений формулы I подбирают апротонный органический растворитель, в котором растворяются играющее роль катализатора соединение палладия и соединение формулы II. Катализирующее процесс соединение палладия берут в количестве 1-10 мол. в расчете на соединение формулы II. Чем меньше реакционная способность исходного материала, тем в большем количестве (в указанных пределах) берется катализатор. Реакцию проводят, осуществляя простое контактирование R1-замещенного органостаннана, палладиевого соединения и 3-сульфонилоксизамещенного реагента формулы II, растворенных или суспензированных в апротонном органическом растворителе. Реакция спонтанно протекает при комнатной температуре и затухает или полностью заканчивается в течение нескольких минут. Обычно продолжительность ее составляет от 10 мин до 1 ч. В неблагоприятных условиях реакция может протекать медленнее, до 2 или 3 дней. При проведении процесса в промышленных масштабах предпочтительно, чтобы продолжительность реакции составляла 1-4 ч. Продолжительность реакции может быть определена опытным путем на основании анализа реакционной смеси на отсутствие исходного соединения формулы II или по максимальному количеству образующегося продукта. Для анализа можно использовать тонкослойную хроматографию, высокопроизводительную жидкостную хроматографию, ядерный магнитный резонанс или спектрофотометрические методы.
Катализируемое палладием сочетание 3-сульфонилоксизамещенного цефема, оксацефема или карбацефема с органостаннаном по способу в соответствии с изобретением предпочтительно проводить без помощи фосфинового лиганда или галоида металла. Хотя фосфиновый лиганд, например трифенилфосфин, и галоид металла, например хлорид цинка, и могут содержаться в реакционной среде, однако присутствие их не дает никакого преимущества в отношении выхода целевого продукта.
По предлагаемому способу предпочтительно использовать в качестве исходного материала 3-(фторсульфонил)оксизамещенное соединение формулы IV. Заместителем R2 органостаннана может быть этил, н-пропил, н-бутил, н-пентил, н-гексил и т.п.
Катализирующим процесс соединением палладия может быть соединение Pd (II) или Pd (0). Примерами подходящих соединений Pd (II) являются ацетат палладия, хлорид палладия, бромид палладия, иодид палладия, дихлорид бис-(ацетонитрил) палладия, дихлорид бис-(фенилацетонитрил)палладия, нитрат палладия, ацетоацетат палладия, сульфат и оксид палладия. Примерами подходящих соединений Pd (0) являются бис(дибензилиденацетон) палладий, трис(дибензилиденацетон)палладий и тетракис(трифенилфосфин)палладий. Предпочтительными каталитическими соединениями палладия являются ацетат палладия (II) и трис(дибензилиденацетон) палладий (0).
Апротонный растворитель, использующийся при осуществлении заявляемого способа, может быть выбран из 1-метил-2-пирролидинона, тетрагидрофурана, ацетонитрила, диметилсульфоксида, диметилформамида, простых эфиров, таких как диглим и диоксан, гексаметилфосфорамида, ацетона, нитрометана и нитробензола, а также галогенированных углеводородов, таких как метиленхлорид. Предпочтительными растворителями являются 1-метил-2-пирролидинон, тетрагидрофуран, ацетонитрил, диметилсульфоксид, метиленхлорид и диметилформамид. Наиболее предпочтительно использовать в качестве растворителя 1-метил-2-пирролидинон или метиленхлорид. Можно использовать также и смеси растворителей.
По предпочтительному варианту осуществления предлагаемого способа в качестве 3-сульфонилоксицефема используют 3-[(фторсульфонил)окси]цефем, в качестве органостаннана-R1трибутилстаннан, где R1 означает С2-6-алкенил, в качестве палладиевого катализатора ацетат палладия (II) или трис(дибензилиденацетон)-дипалладий (0); реакцию сочетания проводят в среде 1-метил-2-пирролидинона или метиленхлорида без добавок лигандов фосфина и галоидов металлов.
Предлагаемый способ, включающий катализируемую палладием реакцию сочетания 3-сульфонилоксицефема с органостаннаном, предпочтительно, в частности, использовать для получения цефпрозила. Использующийся для синтеза цефпрозила исходный станнан, Z-1-пропенилтриалкилстаннан, может быть получен из цис-1-бромпромена. Ниже описан разработанный эффективный способ получения чистого (> 99,О)цис-изомера 1-бромпропена.
Получение цис-1-бромпропена.
В колбу емкостью 500 мл, снабженную мешалкой, термометром и капельной воронкой, загружали кротоновую кислоту (51,68 г, 0,6 моля, Aldrich) и 320 мл гептана. Смесь нагревали на водяной бане до температуры реакции 30oC в защитной атмосфере сухого азота, после чего добавляли в колбу по каплям в течение примерно 45 минут, 34,4 мл (0,63 моля, 1,05 эквивалента) брома (Fisher), поддерживая с помощью холодной водяной бани температуру 30oC. В течение 4-5 мин после окончания добавления брома происходила кристаллизация образующегося продукта, эритро-2,3-диброммасляной кислоты. Для поддержания температуры реакции примерно 34oС колбу охлаждали на водяной бане. После этого смеси давали остыть до температуры окружающей среды, перемешивали ее в течение еще 16 ч и охлаждали на водно-ледяной бане в течение 30 мин. Бесцветные кристаллы отсасывали на фильтре, дважды промывали гептаном порциями до 75 мл и высушивали в вакууме до постоянного веса при температуре окружающей среды. В результате получали 130 г (88%) эритро-2, 3-диброммасляной кислоты с т.пл. 87-89oC. В 2-л колбу, снабженную мешалкой, термометром и обратным холодильником со смонтированным на нем берботером с минеральным маслом, загружали 717,5 мл (3,71 моля, 4,13 эквивалента) 89%-ного триэтиламина (Aldrich). При интенсивном перемешивании в колбу добавляли десятью порциями с интервалами 5 мин 221,33 г (0,90 моля) эритро-2,3-диброммасляной кислоты. В процессе добавления происходило выделение газа (через барботер пробулькивали пузырьки) и наблюдался разогрев смеси до 40oC. Реакционную смесь перемешивали в течение 3,4 ч при температуре окружающей среды, затем нагревали до 40oC и продолжали перемешивание при этой температуре в течение еще 3,5 ч (до прекращения выделения газа). Смесь затем охлаждали до температуры окружающей среды и добавляли к ней 321 мл воды. Твердый осадок промывали и растворяли, после чего в колбу добавляли 230 мл концентрированного раствора HCl (Fisher), поддерживая температуру реакции равной 0oC. При отделении нижней фазы и разделительной воронке получали 82,15 г (75%) сырого цис-1-бромпропена. Водную фазу сохраняли для извлечения из нее триэтиламина.
Сырой продукт промывали равным объемом насыщенного раствора NaHCO3 и перегоняли при атмосферном давлении, получая в результате чистый цис-изомер 1-бромпропена в виде бесцветной жидкости. Т.кип. 59-60o C.
Кислую водную фазу охлаждали до 0-5oC и добавляли к ней при интенсивном перемешивании 750 мл 25% -ного водного раствора NaCH. После отделения в разделительной воронке верхней фазы количественно извлекали триэтиламин.
Соединения формулы I, у которых Q означает карбоксиацильную группу известного 7-ациламиноцефалоспоринового антибиотика, сами являются антибиотиками, которые можно использовать для лечения инфекционных заболеваний, вызываемых бактериями и другими чувствительными микроорганизмами. Однако эти антибиотики не составляют объект изобретения, предметом которого являются способ и полупродукты.
Те из соединений формулы I, у которого Q означает Н или защитную группу, являются полупродуктами для получения вышеуказанных соединений-антибиотиков формулы I путем ацилирования или отщепления вначале защитной группы и затем ацилирования, осуществляемых известным способом.
Нижеследующими примерами иллюстрируются получение различных 3-R1-защищенных цеф-3-емов формулы I предлагаемым способом из соответствующих вышеупомянутых 3-сульфонилоксицеф-3-емов. Эти примеры, однако, никоим образом не ограничивают объем изобретения.
Пример 1. Дифенилметил-7-феноксиацетамидо-3 [(4-нитрофенилсульфонилокси] -3-цефем-4-карбоксилат.
Раствор 0,516 г (0, 001 моля) дифенилметил-7-феноксиацетамидо-3-окси-3-цефем-4-карбоксилата в 5 мл сухого тетрагидрофурана охлаждали до 0oC в атмосфере азота, после чего добавляли к нему 0,040 г (0,001 моля) гидрида натрия (в виде 60%-ного раствора в минеральном масле), в результате чего происходило выделение водорода. Реакционную смесь перемешивали в течение 5 мин при 0oC и добавляли к ней 0, 221 г (0,001 моля) 4-нитробензолсульфонилхлорида, перемешивали в течение 1 ч при 0oC и в течение еще 19 ч при комнатной температуре. Растворитель затем удаляли при пониженном давлении и остаток растворяли в 30 мл этилацетата. Полученный раствор трижды промывали водой и органическую фазу концентрировали в вакууме, получая пенистый остаток. Этот остаток подвергали очистке с помощью хроматографии на силикагеле. В результате получали 0,5 г (71%) целевого соединения.
Результаты анализа.1 H-ЯМР (СDCl3, 360 МГц): δ 8,15 (д, 2Н); 7,7 (д, 2Н); 7,4-6,9 (м, 16Н); 6,72 (с, 1Н); 5,95 (дв.д, 1Н); 5,3 (д, IH); 4,55 (с, 2Н); 3,9 (д, 1Н); 3,58 (д, 1Н).
Пример 2. Дифенилметил-7-феноксиацетамидо-3-(Z-1-пропенил)-3-цефем-4-карбоксилат.
К смеси 1,75 (0,0025 моля) продукта в соответствии с примером 1, 1,03 г (0,003125 моля) Z-1-пропенил-три-н-бутилстаннана и 0,06 г (0,00025 моля) 4-нитробензосульфонихлорида и 17,5 мл 1-метил-2-пирролидинона при комнатной температуре в атмосфере азота добавляли 0,06 г (0,00025 моля) ацетата палладия (II). Реакционную смесь перемешивали в течение 19 ч при комнатной температуре, разбавляя затем 125 мл этилацетата и органическую фазу трижды промывали водой. Этилацетатный раствор обрабатывали углем, который отфильтровывали затем на броунмиллерите. Растворитель удаляли при пониженном давлении, а остаток фильтровали через слой силикагеля с 50%-ным этилацетатом в н-гексане. Сырой продукт перекристаллизовывали из 2-пропанола, получая в результате 1,031 г (76%) целевого соединения.
Результаты анализа1H-ЯМР (СDCl3, 360 мГц); d 7,4-6,8 (м, 17Н), 6,1 (широкий д, 1Н); 5,85 (дв.д, 1Н); 5,55 (м, 1Н); 5,05 (д, 1Н); 4,58 (с, 2Н); 3,43 (д, 1Н); 3,25 (д, 1Н).
Пример 3. Дифенилметил-7-феноксиацетамидо-3-(фторсульфонил)окси -3-цефем-4-карбокстилат.
Раствор дифенилметил-7-феноксиацетамидо-3-окси-3-цефем -4-карбоксилата (2,0 г, 3,8 ммоля) в дихлорметане (20 мл) охлаждали до -78oC в инертной атмосфере и добавляли к нему по каплям в течение 2 мин N, N-диизопропилэтиламин (0,74 мл, 4,2 ммоля, 1,1-эквивалента). Образующийся в результате бледножелтый раствор перемешивали в течение 5 мин и обрабатывали затем фторсульфоновым ангидридом (0,77 г, 4,2 ммоля, 1,1 эквивалента). Реакционную смесь перемешивали в течение 30 мин и затем прекращали реакцию путем добавления к смеси воды (10 мл). После нагревания реакционной смеси до температуры окружающей среды органическую фазу высушивали над сульфатом магния и фильтровали раствор через тонкий слой силикагеля. Силикагель промывали этилацетатом (10 мл) и объединенные органические фракции концентрировали, получая в результате бледно-желтую пенистую массу. Сырой продукт перекристаллизовывали из диэтилового эфира и в результате получали 2,2 г (96%) целевого соединения в виде белых игольчатых кристаллов.
Т.пл. 131-132oC (с разложением).
Результаты анализа.
1 H-ЯМР (CDCl3, 360 мГц); d 7,42-7,24 (комплексы м, 13Н); 7,03 (видимый т, 2Н); 6,90 (д, 2Н, J 7,9 Гц); 5,97 (дв. д, 1Н, J 5,0, 9,2 Гц); 6,08 (д, 1Н, J 5,0 Гц); 4,55 (с, 2Н); 3,83 (А из АВ, 1Н, J 18,5 Гц); 3,51 (В из АВ, 1Н, J 18,5 Гц).
13C ЯМР (90,5 мГц, CDCl3): d 168,63; 164,26; 157,62; 156,73; 139,94; 138,46; 138,28; 129,89; 128,57; 128,51; 128,44; 128,34; 127,76; 127,25; 122,55; 114,71; 80,60; 66,98; 58,23; 57,33; 25,58.
Результаты анализа из расчета на формулу:
C28H23FN2O8
S2
Рассчитано, C 56,17; Н 3,87; N 4,68.
Найдено, С 55,88; Н 3,94; N 4,56.
Пример 4. Дифенилметил-7-феноксиацетамидо-3-винил-3-цефем-4-карбоксилат.
Раствор ацетат палладия (II) (3,6 мг, 0,0165 ммоля, 0,1 эксивалента) в 1-метил-2-пирролидиноне (2 мл) обрабатывали винил-три-н-бутилстаннаном (58,4 мл, 0,2 ммоля, 1,2 эквивалента) в инертной атмосфере и перемешивали смесь в течение 3 мин. Образующуюся темную суспензию обрабатывали затем дифенилметил-7-феноксиацетамидо-3-[(фторсульфонил)окси] -3-цефем-4-карбоксилатом (100,0 мг, 0,16 ммоля, 1,0 эвивалент) и реакционную смесь перемешивали в течение 10 мин. Затем ее разбавляли этилацетамом и трижды промывали водой порциями по 20 мл. Органическую фракцию высушивали над сульфатом магния и концентрировали. Сырой остаток коричневого цвета подвергали очистке с помощью флеш-хроматографии на силикагеле (W.R.Grace, 951W), используя вначале дихлорметан (50 мл) для удаления остатка станнана, а затем 1%-ный этилацетат в дихлорметане (75 мл). После концентрирования получали 74,8 мг (85%) целевого соединения в виде твердого вещества белого цвета.
Результаты анализа.
1H ЯМР (360 мГц, CDCl3: d 7,42-7,23) комплексн. м, 12Н); 7,04-6,92 (комплексн. м, 2Н); 6,91 (д, 1Н, J 7,82 Гц); 5,90 (дв. д. 1Н, J 4,9, 9,2 Гц); 5,42 (д, 1Н, J 17,6 Гц);5,26 (д, 1Н, J 11,2 Гц); 5,02 (д, 1Н, J 4,9 Гц); 4,55 (с, 2Н); 3,62 (а из АВ, 1Н, J 17,7 Гц); 3,46 (В из АВ, 1Н, J 17,7 Гц).13C ЯМР (90,5 мГц, CDCl3): d 168,66; 164,19; 161,00; 156,82; 139,28; 139,01; 131,79; 129,77; 128,51; 128,39; 128,16; 128, 04; 127,72; 127,54; 127,01; 126,48; 122,32; 118,07; 114,69; 79,37; 67,01; 58,47; 57,27; 24,09.
Пример 5. Дифенилметил-7-феноксиацетамидо-3-(Z-1-пропенил)-3-цефем-4-карбоксилат.
Раствор ацетата палладия (II) (3,6 мг, 0,016 ммоля, 0,1 эквивалента) в дихлорметане (2 мл) обрабатывали Z-1-пропенил-три-н-бутилстаннаном (66,2 мг, 0,2 ммоля, 1,2 эквивалента) в инертной атмосфере и перемешивали смесью в течение 3 мин. Образующуюся темную суспензию обрабатывали затем добавляемым в один прием дифенилметил-7-феноксиацетамидо-3-[(фторсульфонил)окси] -3-цефем-4-карбоксилатом (100,0 мг, 0,16 ммоля, 1,0 эквивалент) и реакционную смесь перемешивали в течение 10 мин. Затем ее разбавляли дихлорметаном и промывали 10 мл воды. Органическую фракцию высушивали над сульфатом магния и подвергали очистке с помощью флеш-хроматографии на силикагеле (W.R.Grace, 951W), используя вначале дихлорметан (50 мл) для удаления остатков станнана, а затем 10%-ный этилацетат в дихлорметане (50 мл). После концентрирования получали 80,4 мг (89%) целевого соединения в виде бледно-желтого твердого вещества, которое перекристаллизовывали из изопропилового спирта. В результате получали 62,3 мг (69%) белого твердого продукта. Т.пл. 103-104oC.
Результаты анализа.
1H ЯМР (360 мГц, CDCl3): d 7, 41-6,90 (комплексн. м, 17Н); 6,10 (д, 1Н, J 11,7 Гц); 5,90 (дв, д, J 4,5, 9,8 Гц); 5,56 (м, 1Н); 5,07 (д, 1Н, J 4,5 Гц); 4,58 (с, 2Н); 3,47 (А из АВ, 1Н, J 17,5 Гц); 3,28 (В ил АВ, 1Н, J 17,5 Гц); 1, 41 (дв, д, 3Н, J 1,7, 7,1 Гц).
13C ЯМР (90,5 мГц, CDCl3): d 169,63; 164,28; 161,28; 156,83; 139.42; 139,10; 130,33; 130,33; 129,80; 129,74; 128,45; 128,28; 128,08; 127,79; 127,81; 127,18; 125,88; 122,37; 114,74; 78,99; 67,06; 58,45; 57,55; 28,50.
Пример 6. Дифенилметил-7-феноксиацетамидо-3-[(4-бромфенилсульфонил)окси] -3-цефем-4-карбоксилат.
Указанное целевое cоединение получали путем модификации способа, описанного в патенте США N 3985737. Раствор 0,51 г (0,001 моля) дифенилметил-7-феноксиацетамидо-3-окси-3-цефем-4-карбоксилата в 5 мл ацетонитрила охлаждали до 0oC в атмосфере азота и добавляли к нему 0,030 г (0,001 моля) гидрида натрия (в виде 80%-ного раствора в минеральном масле), в результате чего происходило выделение водорода. Реакционную смесь перемешивали при 0oC в течение 5 мин и добавляли к ней 0,229 г (0,009 моля) 4-бромбензолсульфонилхлорида. Охлаждающую баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение 19 ч. Затем ее фильтровали и фильтрат обрабатывали углем. Угол отфильтровывали и затем удаляли растворитель, получая остаток в виде пены, который перекристаллизовывали из 1-пропанола. В результате получали 0,398 г (54%) целевого соединения.
Результаты анализа.
1H ЯМР (СDCl3, 360 мГц): d 7,5 (д, 2Н); 7,4 (д, 2НО); 7,4-609 (м, 16Н); 6,79 (с, 1Н); 5,9 (дв. д, 1Н); 5,1 (д, 1Н); 4,55 (с, 2Н); 3,85 (д, 1Н); 3,5 (д, 1Н).
Пример 7. Дифенилметил-7-феноксиацетамидо-3-(z-1-пропенил)-3-цефем-4-карбоксилат.
К смеси 0,184 г (0,00025 моля) продукта в соответствии с примером 6, 0,
103 г (0,0003215 моля)
Z-1-пропенил-три-н-бутилстаннана и 0,0064 г (0,000025 моля) 4-бромбензолсульфонилхлорида в 2,5 мл 1-метил-2-пирролидинона в атмосфере азота, при комнатной температуре,
добавляли 0,006 г (0,000025
моля) ацетата палладия (II). Реакционную смесь перемешивали при комнатной температуре в течение 20 ч. На хроматограмме высокопроизводительной жидкостной хроматографии
реакционной смеси наблюдался пик
целевого соединения с таким же временем удерживания, как и время удерживания идентичного образца целевого соединения. Площадь этого пика равнялась 21,2%
Пример 8.
Дифенилметил-7-феноксиацетамидо-3-(z-1-пропенил)-3-цефем-4-карбоксилат, получаемый через трис(дибензилиденацетон)дипалладий (О).
Раствор трис(дибензилиденацетон)дипалладия (14,6 мг, 0, 016 ммоля, 0,1 эквивалента) в дихлорметане (2 мл) или 1-метил-2-пирролидиноне (2 мл) обрабатывали Z-1-пропенил-три-н-бутилстаннаном (66,2 мг, 0,2 ммоля, 1,2 эквивалента) в инертной атмосфере. Полученный раствор обрабатывали затем добавляемым в один прием дифенилметил-7-феноксиацетамидо-3- -[(фторсульфонил)окси]-3-цефем-4-карбоксилатом (100,0 мг, 0,16 ммоля, 1,0 эквивалент). Протекание реакции контролировали с помощью высокопроизводительной жидкостной хроматографии. При использовании в качестве растворителя дихлорметана реакция заканчивалась через 3 ч, в случае же 1-метил-2-пирролидинона продолжительность ее составляла 8 ч. Выход целевого продукта, определенный с помощью высокопроизводительной жидкостной хроматографии составлял более 98% Идентичность полученных при использовании обеих растворителей продуктов с целевым соединением была подтверждена с помощью ЯМР при 360 мГц.
Пример 9. Получение дифенилметил-7-феноксиацетамидо-3-(Z-1-пропенил) -3-цефем-4-карбоксилата через трис(дибензилиденацетон)дипалладий (О).
К смеси 0,175 г (0,00025 моля) продукта в соответствии с примером 1, 0,099 г, (0,0003 моля) Z-1-пропенил-три-н-бутилстаннана в 1,0 мл 1-метил-2-пирролидинона в атмосфере азота, при комнатной температуре, добавляли 0,014 г (0,000025 моля) трис(дибензилиденацетон)дипалладия (О). Реакционную смесь перемешивали при комнатной температуре в течение 17 ч, затем разбавляли ее этилацетатом и органическую фазу дважды промывали водой. Этилацетатный раствор обрабатывали углем, который затем отфильтровывали, пропуская смесь через слой броунмиллерита. Растворитель удаляли из фильтра в вакууме, получая пенообразный остаток. Остаток подвергали очистке с помощью хроматографии на силикагеле. В результате получали 0,060 г (44%) целевого соединения, идентичность которого была подтверждена с помощью ЯМР.
Пример 10. Третбутил-7-(бензилоксикарбониламино)-3-фторсульфонилокси-1 -карба(1-детиа)-3-цефем-4-карбоксилат.
1,35 М раствор фторсульфонилового ангидрида в метиленхлориде (665 мкл, 0,906 ммоля) добавляли по каплям к перемешиваемому, охлаждаемому на бане со смесью сухого льда и ацетона раствору третбутил-7-(бензилоксикарбониламино)-3-окси-1-карба(1 -детиа(-3-цефем-4-карбоксилата (158 мкл, 0, 906 ммоля) в метиленхлориде (5 мл). Раствор перемешивали при -78oC в течение 0,25 ч, после чего охлаждающую баню удаляли и продолжали перемешивать при температуре окружающей среды в течение еще 0,25 часа. Раствор затем трижды промывали водой и высушивали над сульфатом натрия. После удаления растворителя в вакууме получали вязкую смолу, которую подвергали очистке с помощью хроматографии на SiO2 (20 г), используя в качестве элюента смесь метиленхлорида и этилацетата в соотношении 95:5. В результате получали 139 мг (выход 30%) целевого соединения в виде вязкой смолы, которую перекристаллизовывали из смеси этилацетата и гексана, получая бесцветные кристаллы (56 мг) с т.пл. 118oC (с разложением).
Результаты анализа.1H ЯМР (CDCl3, 300 мГц): d 7,35 (с, 5Н); 5,37 (м, 1Н); 5,22 (м, 1Н); 5,11 (с, 2Н); 3,87 (м, 1Н); 2,65 (м, 2Н); 2,13 (м, 1Н,); 1,68 (м, 1Н); 1,53 (с, 9Н).
Масс-спектр: (положительный ион FAБ, N ОВА) m/z 471 (М+1).
Реферат
Использование: в химико-фармацевтической промышленности. Сущность изобретения: способ получения цефалоспориновых антибиотиков формулы I, где Р - Н, t-Bu-, gu(Ph)CH-карбоксизащитная группа, Q-H, PhOCH2CO или PHCH2OCO-аминозащитная группа Z-S или-СН2-, R'-(C2-C3)-алкенил. Реагент 1:3-сульфонилоксисодержащее производное цефема формулы II, где Q и P имеют указанные выше значения, Z-фторсульфонилокси, 4-нитробензолсульфонилокси и 4-бромфенилсульфонилокси. Реагент II: органостаниан R'Sn(R2)3 где R' имеет указанные выше значения, R2 - низший алкил. Условия: среда апротонного растворителя, катализатор соединение Pd (II) или Рd (0) в количестве 1-10 моль.% на реагент 1. 2 з.п. ф-лы, Структура соединений формулы I и II.
Формула
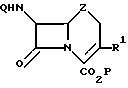
где P водород, трет-бутильная или дифенилметильная карбоксизащитная группа;
Q водород, феноксиацетильная или бензилоксикарбонильная аминозащитная группа;
Z сера или метилен;
R1 C2 C3-алкенил,
путем взаимодействия 9-сульфонилоксисодержащего производного цефема с органостаннаном формулы
R1Sn(R2 )3,
где R1 имеет указанные значения;
R2 низший алкил,
в среде апротонного растворителя в присутствии в качестве катализатора соединения Pd (II) или Pd (0) в количестве 1 10 мол. в расчете на 3-сульфонилоксисодержащее производное цефема, отличающийся тем, что в качестве производного 3-сульфонилоксисодержащего производного цефема используют соединение формулы II
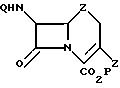
где Q и P имеют указанные значения;
Z фторсульфонилокси, 4-нитробензолсульфонилокси и 4-бромфенилсульфонилокси.
21.11.89 при Z сера или метиленовая группа, Q водород, феноксиацетильная группа, P водород, дифенилметильная группа;
31.10.90 при Z сера или метиленовая группа, Q бензилоксикарбонильная группа, P трет-бутильная группа.
Комментарии