Эрголиновые производные, способ их получения, фармацевтическая композиция - RU2191776C2
Код документа: RU2191776C2
Чертежи




Описание
Изобретение относится к новым эрголиновым производным, их получению и применению в качестве лекарственных средств, а также к содержащим их фармацевтическим композициям.
Более конкретно настоящее изобретение относится к соединению формулы I

где R1 обозначает водород, С1-С4 алкил или группу формулы

в которой n равно 1-4 и R7 обозначает фтор, хлор, гидрокси, С1-С4 алкил или С1-С4 алкокси;
R2 обозначает водород, хлор, бром, йод, С1-С4 алкил или С1-С4 алкилтио;
R3 обозначает водород или С1-С4 алкил; R4 обозначает (1) группу формулы
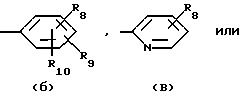

где R8 обозначает фтор, хлор, нитро или циано;
R9 и R10 независимо друг от друга обозначают водород, фтор, хлор, нитро или циано и
R11 обозначает С1-С4 алкил или С2-С8 алкенил, или (2) группу формулы

где радикалы Х независимо друг от друга обозначают О или S и
R5 и R6 каждый обозначает водород или вместе образуют дополнительную связь между двумя атомами углерода, к которым они присоединены,
при условии, что R8 не обозначает 4-NO2, если R1, R2, R9 и R10 обозначают водород, R3 обозначает метил и R5 и R6 вместе образуют дополнительную связь между двумя атомами углерода, к которым они присоединены, и что R8 не обозначает хлор, если R1, R2, R9 и R10 обозначают водород, и R3 обозначает метил,
в форме свободного основания или в форме кислотно-аддитивной соли.
Изобретение включает энантиомеры, а также их смеси, например, эпимерные или рацемицеские смеси, наличие которых обусловлено асимметричными атомами углерода в положениях 5, 8 и 10. Предпочтительной является [5R,10R]-конфигурация (5R соответствует 5β-водороду). Также предпочтительной является 8β -конфигурация.
Указанные выше алкильные, алкокси- и алкилтиогруппы предпочтительно представляют собой метил, метокси- и метилтиогруппу.
Еще одним объектом изобретения
является способ получения соединений формулы I и их кислотно-аддитивных солей, согласно которому соединение формулы II

где R1, R2, R3, R5 и R6 имеют указанные выше значения и М обозначает Н или щелочной металл,
подвергают взаимодействию с соединением формулы III

где R4 имеет указанные выше значения,
и полученное таким образом соединение формулы I восстанавливают в форме свободного основания или в форме кислотно-аддитивной соли.
Реакция может быть осуществлена согласно известным методам получения амидов, например согласно способу, описанному в примере 1. В формуле II М обозначает щелочной металл, например натрий.
Обработку реакционных смесей, полученных согласно вышеуказанному процессу, и очистку полученных соединений можно проводить по известным методам.
Известным способом из свободных оснований могут быть получены кислотно-аддитивные соли и наоборот. Приемлемые кислотно-аддитивные соли для применения по настоящему изобретению включают, например, гидрохлорид. Исходные соединения формулы III могут быть получены взаимодействием пиперазина с соединением формулы R4-C1, например, как это описано в примере 1.
Соединения формулы II и соединения формулы R4-C1 являются известными или могут быть получены методом, аналогичным известным способам.
Соединения по изобретению, например соединения формулы I и их фармацевтически приемлемые кислотно-аддитивные соли (далее в контексте настоящего описания обозначенные как агенты по изобретению) обладают важными фармакологическим свойствами при тестировании in vitro с использованием клеточных культур, экспрессирующих SRIF-рецептор (рецептор соматотропин-рилизинг-ингибирующего фактора) и на животных моделях, и, следовательно, пригодны в качестве лекарственных средств.
В частности, агенты по изобретению связываются с рецепторами соматостатина. Более конкретно они являются избирательными антагонистами sst1 -рецепторов соматостатина, ранее названных SSTR-1-рецепторами (см. Ноуеr и др., TiPS, 1995, 16:86-88), что определено по связыванию радиолигандов и в исследованиях с использованием вторичных мессенджеров [см., например, у К. Kaupmann и др., FEBS LETTERS, 1993, 331: 53-59].
Таким образом, агенты по изобретению пригодны для лечения состояния тревоги, депрессии, шизофрении, нейродегенеративных заболеваний, таких как деменция, для лечения опухолей и для лечения сосудистых нарушений и иммунологических заболеваний.
Целесообразность применения агентов по изобретению при таких симптомах может быть подтверждена с помощью различных тестов, рассмотренных ниже.
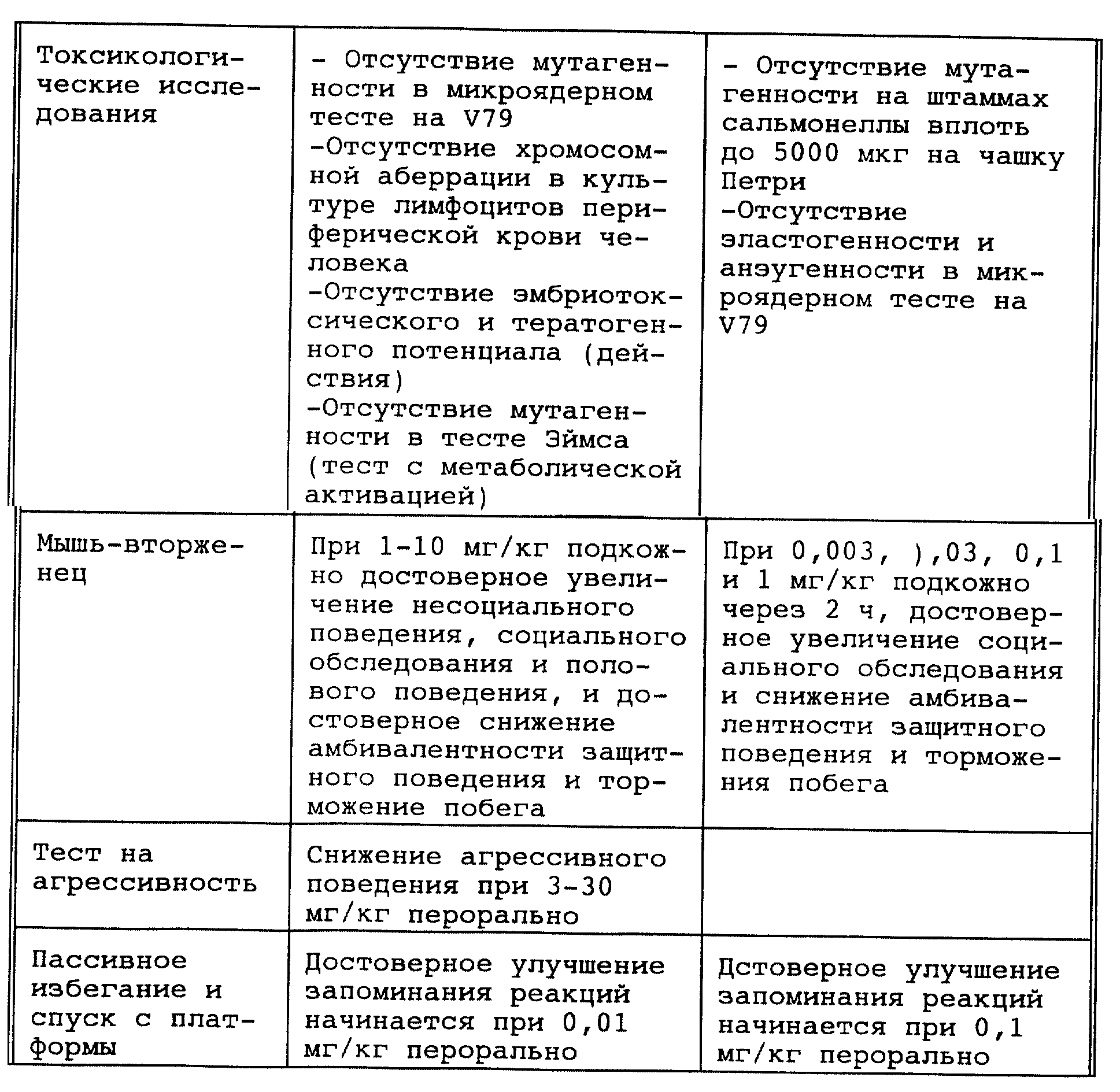
В дозах примерно от 0,3 до 3 мг/кг, вводимых перорально, агенты по изобретению усиливают исследовательское поведение мышей на платформе, у которой одна половина открыта, а другая закрыта, и такой тест позволяет смоделировать условия для выявления анксиолитической активности (Psychopharmacology, 1986, 89:31-37).
С использованием в качестве модели такой же наполовину закрытой платформы установлено, что агенты по изобретению в приведенных выше дозах также увеличивают внимательность мышей. Следовательно, соединения показаны для лечения депрессии, шизофрении и деменции, в частности старческого слабоумия типа болезни Альцгеймера.
В тесте с внедрением мышей [Triangle, 1982, 21: 95-105; J. Clin. Psychiatry, 1994, 55: 9 (дополнение В) 4-7] установлено, что агенты по изобретению усиливают необщественное поведение, общественное исследовательское поведение и половую активность и уменьшают защитную амбивалентность у внедренных мышей, которым подкожно было введено тестируемое соединение в дозах от примерно 1 до примерно 10 мг/кг, что позволяет предположить наличие антидепрессантного профиля, аналогичного таковому для карбамазепина и лития, нейролептического профиля, подобного таковому для клозапина, и анксиолического профиля, аналогичного таковому для диазепама.
Кроме того, в дозах от примерно 3 до примерно 30 мг/кг, вводимых перорально, агенты по изобретению снижают агрессивное поведение (нападения, преследования, укусы) в тесте на мышах, называемом ситуационным, тест по подбору пар [Dixon и др., J. Clin. Psychatry 55:(9) (прил. В) 4-7 (1994)]. С учетом указанного выше снижения защитного поведения в тесте с внедрением мышей соединения по изобретению обладают этофармакологическим профилем, очень сходным с таковым карбамазепина, хлорида лития и клозапина. Следовательно, они показаны для лечения нарушений, сопровождаемых аффектом, включая биполярные нарушения, например маниакально-депрессивный психоз, экстремальные психотические состояния, например маниакальный синдром, шизофрению и частые перепады настроения, при которых необходима стабилизация поведения. Кроме того, соединения показаны при состояниях тревоги, общего чувства страха, а также страха общества и агорафобии, а также при таких состояниях поведения, для которых характерен отказ от общества, например при отрицательных симптомах.
Кроме того, агенты по изобретению улучшают у мышей способность к понижающей пассивной парадигме избегания при их введении как до опыта (перорально), так и после опыта (внутрибрюшинно) в дозе от примерно 0,01 до примерно 10 мг/кг, усиливая восстановление способности к избеганию в тесте ступенчатого пассивного избегания (перорально) [Mondadori и др., Behav. Neural. Biol. 60:62-68 (1993)], и частично противодействуют индуцированной электрошоком амнезии (перорально) [Mondadori и др., Physiol. & Behav., 18: 1103-1109 (1997)]. И, наконец, агенты по изобретению, вводимые перорально в дозах от примерно 0,03 до примерно 3 мг/кг, специфично усиливают социальное распознавание знакомых молодых крыс и отличие их от незнакомых. Эти данные свидетельствуют о том, что агенты в низких дозах облегчают обучаемость и память. Эти особенности, которые все вместе характеризуются как антиагрессивные свойства и социотропные действия, позволяют предположить, что агенты по изобретению эффективны для лечения дефицита внимания и нарушений, связанных с гиперактивностью (ДВНГ).
Агенты по изобретению также эффективны для лечения различных типов опухолей, в частности опухолей, несущих sst1-рецептор, что выявлено в тестах по оценке пролиферации с использованием различных линий раковых клеток и в экспериментах по определению роста опухоли у лишенных волосяного покрова мышей, имеющих гормонально зависимые опухоли [см., например, G. Weckbecker и др., Cancer Research 1994, 54:6334-6337]. Таким образом, соединения показаны для лечения, например, рака молочной железы, предстательной железы, толстой кишки, поджелудочной железы, головного мозга и легкого (мелкоклеточный рак легких).
Для всех указанных выше показаний соответствующая доза, как очевидно, должна зависеть, например, от конкретно применяемого соединения, хозяина, пути введения, природы и серьезности болезненного состояния, подлежащего лечению. Однако, как правило, удовлетворительные результаты у животных получают при использовании суточной дозы от примерно 0,1 до примерно 10 мг/кг веса тела животного. Для более крупных млекопитающих, например людей, рекомендованы суточные дозы в диапазоне от примерно 5 до примерно 200 мг, предпочтительно примерно от 10 до примерно 100 мг соединения, предпочтительно при вводимых в виде разделенных доз до 4 раз в день или в форме с постоянным высвобождением.
Агенты по изобретению могут применяться в свободной форме или в форме фармацевтически приемлемой соли. Такие соли могут быть получены общепринятым методом и проявляют такой же уровень активности, что и свободные соединения.
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей агент по изобретению в сочетании по крайней мере с одним фармацевтически приемлемым растворителем или носителем. Такие композиции могут быть приготовлены общепринятым способом. Стандартные дозируемые формы содержат, например, от примерно 0,25 до примерно 50 мг агента по изобретению.
Агенты по изобретению можно вводить в организм любым приемлемым методом, например парентерально, в частности в форме инъецируемых растворов или суспензий, или энтерально, предпочтительно орально, например, в форме таблеток или капсул, или назально.
Для указанных выше показаний предпочтительным соединением является соединение из примера 1 (см. ниже). Указанное соединение обладает высоким сродством к нативным sst1-рецепторам крысы (pIC50=9,7) и к нативным и рекомбинантным sst1-рецепторам человека (pIC50=9,0 и 8,8 соответственно), не обладая при этом заметной активностью в отношении широкого спектра рецепторов нейромедиаторов. При введении перорально в дозе 3-30 мг/кг соединение в значительной степени понижает агрессивное поведение в указанном выше ситуационном тесте по подбору пар, а в вышеуказанном тесте с внедрением мышей меняет тип общественного поведения, характеризующийся уходом от общества. Эти действия также наблюдались при использовании стандартных лекарств против маниакального синдрома, таких как литий и карбамазепин, в дозе 3-30 мг/кг подкожно, что свидетельствует об аналогичных терапевтических воздействиях на человека. Однако обнаружено, что литий и карбамазепин являются менее активными и, кроме того, известно, что они обладают существенными недостатками, такими как узкий спектр терапевтической активности и медленное начало действия.
Предпочтительными показаниями являются депрессия, состояние тревоги, сопровождаемые аффектом нарушения, включая биполярные нарушения, например маниакальный синдром и ДВНГ.
Согласно вышеприведенному описанию настоящее изобретение также относится к агенту по изобретению, предназначенному для применения в качестве лекарственного средства, например, для лечения депрессии, состояния тревоги, биполярных нарушений и ДВНГ.
Кроме того, настоящее изобретение относится к агенту по изобретению, к приготовлению лекарственного средства для лечения любого указанного выше состояния, например депрессии, состояния тревоги, биполярных нарушений и ДВНГ.
Еще одним объектом изобретения является способ лечения любого указанного выше состояния, например депрессии, состояния тревоги, биполярных нарушений и ДВНГ, у пациента, нуждающегося в таком лечении. Этот способ предусматривает введение такому пациенту терапевтически эффективного количества агента по изобретению.
Ниже изобретение проиллюстрировано на примерах. Температура указана в градусах Цельсия и нескорректирована.
Пример 1. 4-(1-Метил-1Н-пирид-6-он-2-ил)пиперазинамид[5R, 8R,10R]-2-бром-9,10-дигидролизергиновой кислоты.
а) 2-Хлор-1-метилпирид-6-он.
51,82 г 2-хлорпирид-6-она (400 ммолей) и 12,7 г 83%-ной дисперсии гидрида натрия в минеральном масле (440 ммолей) подвергают взаимодействию в 400 мл абсолютного диметилформамида при комнатной температуре в атмосфере аргона в течение 15 мин. Добавляют по каплям 32,4 мл метилйодида (520 ммолей) и перемешивание продолжают при 50oС в течение 5 ч. Реакционную смесь упаривают на роторном испарителе до тех пор, пока растворитель не начнет отгоняться при температуре бани 60oС.
б) 4-(1-Метил-1Н-пирид-6-он-2-ил)пиперазин.
Неочищенный раствор 2-хлор-1-метилпирид-6-она, полученный на стадии а), смешивают с 103 г пиперазина (1,2 моля) и подвергают взаимодействию при 110oС в течение 6 ч. Растворитель выпаривают на роторном испарителе при 70oС при давлением 0,01 Торр. Удаление избытка пиперазина осуществляют распределением между 16%-ным раствором хлорида натрия в воде и смесью (50/50) толуол/1-пропанол. Указанное в заголовке соединение кристаллизуют в виде кислого оксалата из 2-пропанола и в виде свободного основания из толуола, т. пл. 108oС.
в) 4-(1-Метил-1Н-пирид-6-он-2-ил)пиперазинамид[5R,8R,10R]-2-бром-9,10-дигидролизергиновой кислоты.
2,794 г (8 ммолей) [5R,8R,10R]-2-бром-9,10-дигидролизергиновой кислоты суспендируют в 19,1 мл 38%-ного ангидрида пропанфосфиновой кислоты в диметилформамиде (24 ммоля) и 8 мл абсолютного пиридина и перемешивают в течение 15 мин при комнатной температуре. После добавления 1,546 г 4-(1-метил-1Н-пирид-6-он-2-ил)пиперазина (8 ммолей) перемешивание продолжают в течение ночи. Реакционную смесь распределяют между смесью (60/40) толуол/2-пропанол и 2М водным аммиаком, органический слой упаривают и хроматографируют на 24 г силикагеля с толуолом/этанолом/конц. водным аммиаком (90/0,9/0,1). Основной компонент перекристаллизовывают из этилацетата, т. пл. 250-253oС (разл.); [α] = -94,3° (0,5% в диметилформамиде).
Способом, аналогичным описанному в примере 1, получают приведенные ниже соединения формулы I, имеющие [5R, 8R,10R]-конфигурацию (см. табл. 1).
Состав покрытых пленкой таблеток (фильм-таблеток), каждая из которых содержит, например, 100 или 200 мг соединения из примеров 1 и 25.
Ниже приводится состав на каждую таблетку с вышеупомянутой дозой (см. табл. 2).
Активный ингредиент гранулируется с деминерализованной водой. Размолотая лактоза, маисовый крахмал, Avicel РН 102, цеплюлоза-НР-М-603 и лаурилсульфат натрия добавляют к вышеуказанной смеси и гранулируют с деминерализованной водой.
Влажное вещество высушивают и размалывают. После добавления оставшихся ингредиентов гомогенную смесь таблетируют с получением ядер таблеток, содержащих заданную дозу активного ингредиента. Эти ядра таблеток покрывают пленочным покрытием, которое получают из соответствующих подходящих ингредиентов, причем последние растворяют или суспендируют в воде или в небольшом количестве этанола с 5% изопропанола.
Реферат
Изобретение относится к новым эрголиновым производным формулы I, где R1 - H или группа

где n= 1-4, R7- F, Cl, C1-C4 алкил, R2- Н, Cl, Br, I, C1-C4 алкил, C1-C4 алкилтио, R3- Н, C1-C4 алкил, R4 - (i) группа формул (в), (г), где R8- Н, NO2, и R11- C1-C4 алкил, С2-С8 алкенил или (ii) группа формул (д), (е), R5 и R6 - H или вместе образуют дополнительную связь. Соединения I являются селективными антагонистами соматостатинового sst1 -рецептора и могут найти применение в качестве лекарственных средств для лечения депрессий, состояний тревоги, биполярных нарушений и дефицита внимания и нарушений, связанных с гиперактивностью. 3 с. и 4 з.п.ф-лы, 3 табл.



Формула

где R1 обозначает водород, или группу формулы

в которой n равно 1-4 и R7 обозначает фтор, хлор или С1-C4алкил;
R2 обозначает водород, хлор, бром, йод, С1-С4алкил или С1-С4алкилтио;
R3 обозначает водород или С1-С4алкил;
R4 обозначает i группу формулы

где R8 обозначает водород или нитро;
R11 обозначает С1-С4алкил или С2-С8алкенил,
или (ii) группу формулы

R5 и R6 каждый обозначает водород или вместе образуют дополнительную связь между двумя атомами углерода, к которым они присоединены,
в форме свободного основания или в форме кислотно-аддитивной соли.

где R1, R2, R3, R5 и R6 имеют значения, указанные в п.1, а М обозначает Н или щелочной металл,
с соединением формулы III

где R4 имеет значения, указанные в п.1,
и выделение полученного таким путем соединения формулы I в форме свободного основания или в форме кислотно-аддитивной соли.
Комментарии