(-)-4-амино-5-фтор-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-(1h)-пиримидин- 2-он, смесь его энантиомеров, способы их получения, способ лечения - RU2126405C1
Код документа: RU2126405C1
Чертежи

Описание
Настоящее изобретение относится к нуклеозидным аналогам и их использованию в медицине. Более конкретно изобретение связано с нуклеозидными аналогами 1,3-оксатиолана, с включающими их фармацевтическими составами с их использованием при лечении вирусных инфекций.
Единственным соединением, которое в настоящее время одобрено для использования при лечении состояний, вызываемых ВИЧ, является 3'-азидо-3'-деоксимидин (AZT, зидовудин, BW 509U). Однако это соединение обладает значительной предрасположенностью к побочным эффектам, а потому не может использоваться или же после первых попыток использования дальнейшее его применение приходится прекращать для большого количества пациентов. Сохраняется настоятельная потребность в разработке соединений, которые были бы активными по отношению к ВИЧ, но имели бы значительно лучший сопутствующий терапевтический индекс.
Соединение по формуле (I)
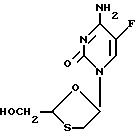
является рацемический смесью двух энантиомеров по формулам (I-1) и (I-2):
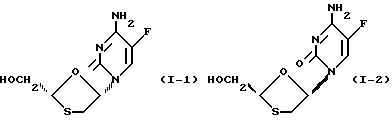
Мы неожиданно обнаружили, что (-)-энантиомер соединения (I) проявляет значительно большую активность, чем (+)-энантиомер, хотя оба энантиомера обладают неожиданно низкой цитотоксичностью. Таким образом, первой целью настоящего изобретения является (-) (или левовращающий) энантиомер соединения по формуле (I) и его фармацевтически приемлемые производные.
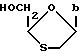
(-)-энантиомер имеет химическое название (-)-4-амино-5-фтор-1-(2-оксиметил-1,3-оксатиолан-5-ил)-(1H)-пиримидин-2-он (далее называется соединение (A)). Этот энантиомер имеет абсолютную стериохимическую структуру, представленную формулой (I-1).
Соединение (A) преимущественно должно быть свободно от соответствующего (+)-энантиомера, т. е. должно содержать не
более приблизительно 5 мас.% (+)-энантиомера, более предпочтительно не более приблизительно 1 мас.%
По "фармацевтически приемлемым производным" понимается фармацевтически приемлемая соль,
сложный эфир или соль этого эфира соединения (A) или другого соединения, которое при назначении пациенту способно выделять (прямо или косвенно) соединение (A), или метаболит, обладающий антивирусной
активностью, или его радикал.
Для специалиста очевидно, что соединение (A) может быть модифицировано, с целью получения его фармацевтически приемлемых производных, по функциональным группам как фрагмента основания, так и по оксиметильной группе оксатиоланового кольца. Модификации по всем этим функциональным группам включаются в объем притязаний по настоящему изобретению. Однако особый интерес представляют фармацевтически приемлемые производные, полученные модификацией 2-оксиметильной группы оксатиоланового кольца.
предпочтительными эфирами соединения (A) являются производные, в которых атом водорода в 2-оксиметильной группе замещается ацильной группой
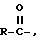
Что касается указанных выше эфиров, то, если особо не оговаривается, любой алкильный фрагмент в них преимущественно содержит от 1 до 16 атомов углерода, в частности от 1 до 4 атомов углерода. Любой арильный фрагмент в указанных соединениях преимущественно является фенильной группой.
В частности, эфирами могут быть эфиры, образованные (C1-C16)алкильными группами, незамещенные бензиловые эфиры или бензиловые эфиры, замещенные по крайней мере одним атомом галогена (брома, хлора, фтора или йода), одной (C1-C16)алкильной группой, (C1-C6)алкоксигруппой, нитрогруппой или трифторметильной группой.
Фармацевтически приемлемыми солями соединения (A) являются соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих кислот включают соляную кислоту, бромистоводородную кислоту, серную, азотную кислоту, хлорную кислоту, фумаровую кислоту, малеиновую кислоту, фосфорную кислоту, гликолевую кислоту, молочную кислоту, салициловую кислоту, янтарную кислоту, п-толуолсульфокислоту, винную кислоту, уксусную кислоту, лимонную кислоту, метансульфоновую кислоту, муравьиную кислоту, бензойную кислоту, малоновую кислоту, нафтен-2-сульфоновую кислоту и бензолсульфоновую кислоту. Другие кислоты, такие как щавелевая кислота, которые сами по себе не являются фармацевтически приемлемыми, могут использоваться для получения промежуточных соединений при синтезе соединений по настоящему изобретению и их фармацевтически приемлемых солей присоединения кислот.
Солями, образованными соответствующими основаниями, являются соли щелочных металлов (например, натрия), соли щелочноземельных металлов (например, магния), соли аммония и четвертичные аммониевые соли NR4+ (где R является (C1-C4) алкильной группой).
Ссылки в данном описании на соединение (A) по изобретению относятся как к собственно соединению (A), так и к его фармацевтически приемлемым производным.
Соединения по настоящему изобретению либо сами обладают антивирусной активностью, либо образуют такие соединения в процессе метаболизма. В частности, такие соединения эффективны при ингибировании репликации ретровирусов, в том числе ретровирусов человека, таких как вирус иммунодефицита человека (ВИЧ), который является причиной заболевания СПИДом.
Соединения по настоящему изобретению можно использовать также для лечения животных, в том числе человека, инфицированных вирусом гепатита B.
Следующей целью настоящем изобретения является использование соединения (A) или его фармацевтически приемлемого производного в качестве активного терапевтического средства, в частности антивиурсного средства, например, при лечении ретровирусных инфекций или инфекций, вызванных вирусом гепатита B.
Еще одной целью настоящего изобретения является способ лечения вирусной инфекции, в частности инфекции, вызванной вирусом гепатита B или ретровирусами, такими как ВИЧ, у животных, в том числе у человека, который состоит в назначении эффективного количества соединения (A) или его фармацевтически приемлемого производного.
Еще одной или альтернативной целью изобретения является использование соединения (A) или его фармацевтически приемлемого производного в производстве медицинских препаратов для лечения вирусных инфекций.
Соединения по настоящему изобретению могут использоваться также при лечении связанных со СПИДом состояний, таких как связанных со СПИДом комплекс прогрессирующего общего поражения лимфатических узлов, связанных со СПИДом нейрологических состояний (таких как слабоумие и трофический парапарез), положительных анти-ВИЧ-антитело и ВИЧ-положительных состояний, саркомы Капози, тромботической пурпуры и заболеваний, вызываемых условно-патогенными микроорганизмами, например, pneumocystis carinii.
Соединения по настоящему изобретению полезны также для предотвращения развития заболевания в клиническую форму у пациентов, у которых обнаруживается положительная реакция на анти-ВИЧ-антитело и ВИЧ-антиген, и для профилактики после воздействия ВИЧ.
Соединение (A) или его фармацевтически приемлемые производные могут использоваться также для предотвращения вирусного загрязнения физиологических жидкостей, таких как кровь и семенная жидкость в условиях in vitro.
Соединения по настоящему изобретению могут использоваться также при лечении животных, в том числе человека, инфицированных вирусом гепатита B.
Для специалистов очевидно, что указанный способ лечения распространяется и на профилактику заболевания, а также и на лечение уже выявленных инфекций или заболеваний с выраженными симптомами.
Следует подчеркнуть, что количество соединения по настоящему изобретению, которое необходимо для лечения, будет меняться в зависимости не только от конкретно выбранного соединения, но и от пути назначения, природы заболевания, возраста и общего состояния пациента и устанавливается по усмотрению лечащего врача или ветеринара. Однако в общем случае приемлемая доза составляет приблизительно от 0,1 до приблизительно 750 мг/кг веса тела в день, предпочтительно от 0,5 до 60 мг/кг в день, и наиболее предпочтительно от 1 до 20 мг/кг в день.
Требуемая доза может представлять собой одну дозу или же разделена на несколько доз, которые назначаются через определенные промежутки времени, например, в виде двух, трех, четырех или более дозировок в день.
Соединение по настоящему изобретению удобно назначать в виде стандартной дозы, содержащей, например, от 10 до 1500 мг, предпочтительно от 20 до 1000 мг, еще более предпочтительно от 50 до 700 мг активного ингредиента на стандартную дозу.
В идеале, количество назначаемого активного ингредиента должно быть таким, чтобы максимальная его концентрация в плазме составляла приблизительно от 1 до приблизительно 75 мкМ, предпочтительно приблизительно от 2 до 50 мкМ, наиболее предпочтительно от 3 до приблизительно 30 мкМ. Это достигается, например, внутривенной инъекцией, в частности, солевого раствора с концентрацией от 0,1 до 5% активного ингредиента, или назначением орально шариков, содержащих приблизительно от 1 до приблизительно 100 мг активного ингредиента. Требуемый уровень содержания лекарства в крови можно поддерживать с помощью непрерывного вливания в количестве приблизительно от 0,01 до приблизительно 5,0 мг/кг в час или попеременных вливаний, содержащих приблизительно от 0,4 до приблизительно 15 мг/кг активного ингредиента.
Несмотря на то, что в терапевтических целях соединение по настоящему изобретению может назначаться в виде чистого вещества, предпочтительнее вводить активные ингредиенты в виде фармацевтических составов.
Таким образом, в настоящем изобретении далее заявляется фармацевтический состав, включающий соединение (A) или его фармацевтически приемлемое производное вместе с одним или большим количеством фармацевтически приемлемых носителей, а также, по выбору, с другими терапевтическими и/или профилактическими средствами. Носители должны быть "приемлемыми" в том смысле, что они должны совмещаться друг с другом в составе композиции и не оказывать вредное воздействие на пациента, который использует этот состав.
Фармацевтические составы включают такие фармацевтические композиции, которые пригодны для орального, ректального, внутриносового, местного (в том числе буккального или подъязычного), вагинального или парентерального (в том числе внутримышечного, подкожного или внутривенного) назначения или в форме, пригодной для ингаляции или вдувания порошкообразных лекарств. Составы, если это возможно, удобнее готовить в виде дискретных доз, которые могут быть получены любыми способами, хорошо известными в фармации. Все способы включают смешение активного вещества с жидким носителем или тщательно растертым твердым носителем или с обоими указанными носителями, а затем придание продукту требуемой формы.
Фармацевтические составы, удобные для орального назначения, могут быть в виде стандартных доз, таких как капсулы, крахмальные капсулы или таблетки, каждая из которых содержит определенное количество активного ингредиента; в виде порошков или гранул; в виде раствора, суспензии или эмульсии. Активный ингредиент может также быть в виде шарика, лекарственной кашки или пасты.
Таблетки и капсулы для орального назначения могут содержать обычные наполнители, такие как связующие вещества, смазывающие средства, разрыхлители или смачивающие вещества. Таблетки могут быть покрыты способами, хорошо известными из данной области техники. Жидкие препараты для орального назначения могут быть в форме, например, суспензий в воде или масле, растворов, эмульсий, сиропов или эликсиров, или могут представлять собой твердые продукты, которые необходимы перед употреблением смешать с водой или другим подходящим жидким носителем. Такие жидкие препараты могут включать обычные добавки, такие как суспендирующие агенты, эмульгаторы, неводные жидкие носители (которые могут включать употребляемые в пище масла) или консерванты.
Соединения по настоящему изобретению могут быть составлены для парентерального назначения (например, с помощью инъекции или непрерывного вливания) и могут быть представлены единичной дозой в виде ампул, заполненных шприцев, контейнеров для вливания небольшого количества лекарства или контейнерами, содержащими большое количество доз, в которые добавляют консерванты. Составы могут иметь форму суспензий, растворов или эмульсий в маслах или водных носителях, и могут содержать вспомогательные соединения, такие как суспендирующие средства, стабилизаторы и/или диспергаторы. Активный ингредиент может также быть в порошкообразной форме, получаемой асептическим выделением, стерилизацией или лиофилизацией из раствора, и его необходимо перед употреблением смешать с подходящим носителем, в частности со стерильной апирогенной водой.
Для местного нанесения на слой эпидерма соединение по настоящему изобретению может входить в состав мазей, кремов или лосьонов, а также в состав бляшек для нанесения на кожу. Мази и кремы могут, например, готовиться на основе воды или масла с добавлением подходящих загустителей и/или гелеобразующих средств. Лосьоны могут готовиться с использованием воды и масла, а также содержать в общем случае один или несколько эмульгаторов, стабилизаторов, диспергаторов, суспендирующих средств, загустителей или красителей.
Составы, пригодные для орального назначения, включают таблетки, состоящие из активного ингредиента в ароматизированной массе, обычно из сахарозы, камеди или смолы трагаканта; пастилки, состоящие из активных ингредиентов в инертном носителе, таком как желатин и глицерин или сахароза и камедь; жидкости для полоскания рта, состоящие из активных ингредиентов в подходящем жидком носителе.
Фармацевтические составы, удобные для ректального назначения, в которых носитель является твердым соединением, предпочтительно готовятся в виде суппозиторий, содержащий стандартную дозу. Подходящие носители включают шоколадное масло и другие вещества, обычно используемые для этих целей, а сами суппозитории удобно готовить смешением активных ингредиентов с размягченным или расплавленным носителем с последующим охлаждением и отливкой в форму.
Составы для вагинального назначения могут быть представлены вагинальными суппозиториями, тампонами, например, гелями, пастами, пенками или аэрозолями, которые в дополнение к активному ингредиенту содержат пригодные для этих целей известные носители.
Для внутриносового назначения соединения по настоящему изобретению могут использоваться в виде жидких аэрозолей или распыляемых порошков в форме капель.
Капли можно готовить с использованием воды и неводных жидкостей вместе с одним или несколькими диспергаторами, солюбилизаторами и суспендирующими агентами. Жидкие аэрозоли преимущественно подаются из баллончиков под давлением.
Для ингаляции соединения по настоящему изобретению обычно поступают из аппарата для вдувания, распылителя или из аэрозольной упаковки или подаются с помощью других подходящих средств доставки аэрозолей. В аэрозольных упаковках могут использоваться соответствующие летучие соединения, такие как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другие подходящие газы. В случае аэрозольной упаковки, стандартную дозу устанавливают с помощью клапана, который отмеряет определенное количество лекарства.
При назначении путем ингаляции или вдувания соединения по настоящему изобретению могут готовиться также в форме сухой порошкообразной композиции, например, смеси порошков соединения и нужной основы, такой как лактоза или крахмал. Стандартные дозы порошкообразных составов могут быть изготовлены, например, в виде капсул или картриджей или же, например, желатиновых или пластиковых упаковок, из которых порошок можно вводить с помощью ингалятора или аппарата для вдувания.
В случае необходимости описанные выше составы могут быть приспособлены для порционного выделения активного ингредиента в течение длительного периода времени.
Фармацевтические составы по настоящему изобретению могут содержать также другие активные ингредиенты, такие как антимикробные средства и консерванты.
Соединения по настоящему изобретению можно также использовать с другими терапевтическими средствами, например, с другими антиинфекционными средствами. В частности, соединения по настоящему изобретению можно применять совместно с известными антивирусными средствами.
Таким образом, еще одной целью настоящего изобретения является сложный состав, включающий соединение (A) или его физиологически приемлемое производное вместе с другим терапевтическим средством, в частности антивирусным средством.
Указанные сложные составы могут быть приготовлены для удобства в виде фармацевтических композиций, таким образом, еще одной целью изобретения являются фармацевтические композиции, состоящие из указанного выше сложного состава и фармацевтически приемлемого носителя.
Подходящими терапевтическими средствами для использования в таких сложных составах являются ациклические нуклеозиды, такие как ацикловир или ганцикловир, интерфероны, такие как альфа-, бета- или гамма-интерферон, ингибиторы почечных выделений, такие как пробенецид, ингибиторы транспорта нуклеозидов, такие как дипиридамол, 2',3'-дидеоксинуклеозиды, такие как AZT, 2', 3'-дидеокситидин, 2',3'-дидеоксиаденозин, 2',3'-дидеоксиинозин, 2', 3'-дидеокситимидин, 2',3'-дидеокси-2',3'-дидегидротимидин и 2',3'-дидеокси-2',3'-дидегидроцитидин, иммуномодуляторы, такие как интерлейкин-2 (IL-2) и гранулоцитарно-макрофагальный стимулирующий фактор (GM-CSF), эритропометин, амплиген, тимомодулин, тимопентин, фоскарнет, рибавирин и ингибиторы связывания ВИЧ с CD4 рецепторами, в частности растворимый CD4, фрагменты CD4, гибридные молекулы CD4, ингибиторы гликозилирования, такие как 2-деокси-D-глюкоза, кастаноспермин и 1-деоксинойиримицин.
Индивидуальные компоненты таких сложных составов могут назначаться как последовательно, так и одновременно в виде отдельных или совместных фармацевтических композиций.
В том случае, когда соединение (A) или его фармацевтически приемлемое производное используется совместно со вторым терапевтическим средством, которое активно по отношению к тому же самому вирусу, доза каждого соединения может быть такой же или же отличаться до дозы в случае использования каждого средства отдельно. Нужная доза устанавливается специалистом.
Соединение (A) и его фармацевтически приемлемое производное можно получить любым способом, который известен для синтеза соединений, обладающих аналогичной структурой, как это, например, описано в Европейской патентной публикации 0382526 A2.
Для специалиста очевидно, что в ряде описанных далее методов необходимая стерехимическая структура соединения (A) может быть получена либо при использовании оптически активного исходного соединения, либо путем расщепления рацемической смеси на одном из этапов синтеза. Во всех случаях оптически чистый целевой продукт может быть получен расщеплением конечного продукта каждой реакции.
В одном из таких способом 1,3-оксатиолан по формуле (VIII)
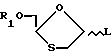
где аномерная группа L является уходящей группой, взаимодействует с соответствующим основанием.
Подходящими группами L являются группы OR, в которых R обозначает алкильную группу, в частности (C1-C6)алкильную группу, такую как метильная группа, либо же R обозначает (C1-C6)ацильную группу, в частности ацетильную группу, или атом галогена, например йода, брома или хлора.
Соединение по формуле (VIII) легко взаимодействует с 5-фторцитозином или соответствующим предшественником этого пиримидинового основания (предварительно силилированного силилирующим агентом, таким как гексаметилдисилазан) в подходящем растворителе, таком как хлористый метилен, в присутствии кислоты Льюиса, такой как тетрахлорид титана, триметилсилилтрифлат, йодтриметилсилан или соединение олова (V), например, SnCl4.
1,3-оксатиоланы по формуле (VIII) можно, например, синтезировать по реакции альдегида по формуле (VII) с меркаптоацеталем по формуле (VI) в подходящем органическом растворителе, таком как толуол, в присутствии кислотного катализатора, например, кислоты Льюиса, такой как хлорид цинка.
HSCH2CH(OC2H5)2; (VI)
C6H5CO2CH2CHO.
(VII)
Меркаптоацетали по формуле (VI) можно получить известными способами, например, G. Hesse and I. Jorder, Chem. Ber., Bd. 85, pp. 924-932 (1952).
Альдегиды по формуле (VII) можно получить известными способами, например, E. G. Halloquist and H. Hibbert, Can. J. Research, V. 8, pp. 129-136 (1933). Сырой альдегид (VII) удобно очищать, превращая его в кристаллический аддукт с бисульфитом с последующим выделением свободного альдегида.
Во втором способе соединение (A) получают интерконверсией в присутствии основания соединения по формуле (IX)
где B обозначает группу, которая превращается под действием основания в 5-фторцитозин.
Такая интерконверсия может быть осуществлена либо простыми химическими превращениями (в частности, преобразованием урацилового основания в цитозин), или ферментативными превращениями с использованием деоксирибозилтрансферазы. Указанные методы и условия интерконверсии хорошо известны в химии нуклеозидов.
В третьем способе по формуле (XI)
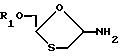
можно перевести в соединение (A) превращением аномерной аминогруппы в 5-фторцитозиновое основание методами, хорошо известными в химии нуклеозидов.
Многие из описанных здесь реакций в части синтеза нуклеозидов представлены в обзорах, например, Nucleoside Analogs-Chemistry, Biology and Medical Applikations, R.T. Walker et al., Eds., Plenum Press, N.Y. (1979), pp. 165-192 и T. Ueda, Chemistry of Nucleosides and Nucleotides, Vol. I, L.B. Towmnsend Ed., Plenum Press, N.Y. (1988) на стр. 165-192, которые приводятся здесь для справки.
Очевидно, что при осуществлении указанных выше реакций могут потребоваться исходные вещества, имеющие защищенные функциональные группы, так что на промежуточных или конечной стадиях для выделения целевого соединения может потребоваться проведение снятия указанной защиты. Защита и снятие защиты функциональных групп можно осуществить с использованием обычных способов. Так, например, аминогруппы можно защитить с помощью группы, выбранной из аралкильной группы (в частности, бензильной группы), ацильной группы, арильной группы (в частности, 2,4-динитрофенильной группы) или силильной группы; соответствующее удаление защитной группы проводят в нужный момент гидролизом или гидрогенолизом в стандартных условиях. Оксигруппы можно защитить любыми группами, которые используются для этой цели, как это описано, например, в Protective Groups in Organic Chemistry, J.F.W.McOmie, Ed., Plenum Press, N. Y. (1973) или T.W.Green, Protected Groups in Organic Synthesis, John Wiley and Sons, N.Y. (1989).
Примером подходящих групп для защиты оксигруппы являются группы, выбранные из алкильной группы (например, метильной группы, трет-бутильной группы или метоксиметильной группы), аралкильной группы (например, бензильной группы, дифенилметильной группы или трифенилметильной группы), гетероциклических групп, таких как тетрагипопиранильная группа, ацильной группы (например, ацетильной группы или бензоильной группы) и силильной группы, такой как триалкилсилильная группа (например, трет-бутилдиметилсилильная группа). Защита оксигруппы может быть снята обычными способами. Так, например, алкильную группу, ацильную группу и гетероциклическую группу можно удалить сольволизом, например, гидролизом в кислой или щелочной среде. Аралкильные группы, такие как трифенилметильная группа, можно также удалить сольволизом, например гидролизом в кислой среде. Аралкильные группы, такие как бензильная группа, можно снять на нужной стадии синтеза, например, обработкой эфиратом трехфтористого бора и уксусным ангидридом. Силильные группы также удобно удалять, используя доноры ионов фтора, такие как тетра-п-бутиламмонийфторид.
В указанных выше способах соединение (A) обычно получают в виде смеси цис- и транс-изомеров, из которых соединением, представляющим интерес, является цис-изомер.
Эти изомеры можно разделить физическими методами, например, хроматографией на силикагеле или дробной перекристаллизацией непосредственно или в виде подходящего производного, в частности, в виде ацетатов (получаемых, например, с использованием уксусного ангидрида) с последующим (после разделения изомеров) обратным превращением в нужный продукт (в частности, деацетилированием метанольным раствором аммиака).
Фармацевтически приемлемые соли соединения по настоящему изобретению можно получить способом, описанным в Патенте США 4383114, который приводится здесь для справки. Так, например, когда требуется получить из соединения (A) соль присоединения кислоты, то продукт по одному из представленных выше способов можно обычным способом превратить в соль обработкой выделенного свободного основания соответствующей кислотой в присутствии, если необходимо, подходящего растворителя, такого как сложный эфир (например, этилацетат) или спирт (например, метанол, этанол или изо-пропанол). Соли неорганических оснований могут быть получены взаимодействием целевого соединения с подходящим основанием, таким как спирт (в частности, метанол). Фармацевтически приемлемые соли можно получать также из других солей, в том числе других фармацевтически приемлемых солей соединения (A), применяя обычные методы.
Соединение (A) можно превратить в фармацевтически приемлемые фосфаты или другие эфиры взаимодействием с фосфорилирующими агентами, такими как хлорокись фосфора, или подходящими этерифицирующими агентами, такими как ацилгалогенид или ангидрид. Эфир или соль соединения (A) можно, например, перевести в исходное соединение гидролизом.
Расщепление конечного продукта, промежуточного продукта или исходного вещества, можно провести любым известным способом: см., например, E.L.Elies, Stereochemistry of Carbon Compounds, McGraw Hill (1962) и S.H.Wilen, Tables of Resolving Agents.
Так, например, соединение (A) можно выделить с помощью хиральной жидкостной хроматографии высокого разрешения, используя подходящую стационарную фазу, например ацетилированный бета-циклодекстрин или триацетат целлюлозы и соответствующий растворитель, например спирт, такой как этанол, или водный раствор, например, ацетата триэтиламмония. Соединения можно расщепить также с помощью стимулируемой ферментом энантиоселективной диссимиляции подходящим ферментом, таким как цитидиндеаминаза или селективным ферментативным разложением подходящего производного 5'-нуклеотидазы. В том случае, если расщепление проводят ферментативно, фермент может содержаться в растворе или, что более удобно, присутствовать с иммобилизованной форме. Иммобилизация ферментов может быть осуществлена известными способами, например нанесением на смолу, такую как Eupergit S..
Изобретение далее поясняется следующими примерами, которые ни в коем случае не огранивают настоящее изобретение. Все температуры приведены в градусах Цельсия.
Промежуточное соединение 1.
(+)-цис-2-Оксиметил-5-(5'-фторцитозин-1'-ил)-1,3-оксатиолан.
(i) 2-Бензоилоксиметил-5-ацетокси-1,3-осатиолан.
Бензоилоксиацетальдегид (216,33 г, 1,32 мол) растворяют в пиридине (373 мл, 4,61 мол) и к полученному раствору прибавляют 1,4-дитиан-2, 5-диол (100,31 г, 9,66 мол). Гетерогенную смесь перемешивают в атмосфере азота при температуре 65oC в течение 1 часа. К этому времени растворение завершается. К реакционной смеси прибавляют дихлорметан (650 мл) и смесь охлаждают до 0oC в бане со льдом. По каплям при 0-5oC в течение 1,5-2 часов прибавляют хлористый ацетил (281 мл, 3,95 мол). Перемешивают при 0-5oC еще в течение 30 минут и осторожно выливают смесь в холодный (0oC) насыщенный раствор бикарбоната натрия. Органический слой отделяют. Водный слой экстрагируют дихлорметаном (3 • 200 мл). Органические вытяжки объединяют, моют насыщенным раствором бикарбоната натрия (3 • 200 мл) и соли (200 мл), сушат над сульфатом натрия и упаривают в вакууме. Остатки пиридина удаляют азеотропной перегонкой с бензолом. Получают 320,79 г сырого продукта, который очищают дистилляцией или фильтрованием через короткую колонку с силикагелем (элюент - гексан/этилацетат, 3/1).
(ii) цис- и транс-2-бензоилоксиметил-5-(N'-ацетил-5'- фторцитозин-1'-ил)-1,3-оксатиолан
5-фторцитозин (4,30 г, 33,3 ммол), гексаметилдисилазан (25 мл) и сульфат аммония (120 мг)
кипятят с обратным холодильником до полного растворения цитозина (3 часа), а затем кипятят еще в течение 2 часов. Гексаметилдисилазан отгоняют в вакууме и к остатку прибавляют толуол (100 мл), чтобы
упарить растворители. Полученный раствор бис(триметилсилил)фторцитозина в дихлорметане (40 мл) прибавляют в атмосфере аргона к раствору 2-бензилоксиметил-5-ацетокси-1,3-оксатиолана (8,537 г, 30,3
ммол) в сухом дихлорметане (100 мл), содержащем молекулярные сита (4A, 2 г), которые предварительно выдерживают в атмосфере аргона и охлаждают до 0oC в течение 20 минут.
К полученной смеси прибавляют при 0oC [(трифторметансульфонил) окси]триметилсилан (6 мл, 31 ммол) и полученный раствор перемешивают при комнатной температуре в течение 2 часов. Отфильтровывают, фильтрат промывают дважды по 300 мл насыщенным раствором соли и один раз дистиллированной водой. Органический слой сушат над сульфатом магния, отфильтровывают и упаривают досуха. Получают сырое производное 5-фторцитозина (10,1 г). Rf = 0,57 (EtOAc:MeOH, 9:1).
На следующей стадии полученный продукт без дальнейшей очистки ацетилируют. Сырой продукт растворяют в атмосфере аргона в дихлорметане (120 мл) в круглодонной колбе емкостью 500 мл. К полученному раствору прибавляют триэтиламин (12,7 мл, 91,1 мол) и диметиламинопиримидин (111 мг, 0,9 ммол). Колбу под аргоном помещают на 1 час в баню со льдом. В охлажденную колбу впрыскивают уксусный ангидрид (4,5 мл, 45 ммол), перегнанный над ацетатом натрия. Оставляют смесь при перемешивании на ночь, а затем осторожно декантируют в колбу Эрленмейра, содержащую насыщенный раствор бикарбоната натрия. Продукт промывают дистиллированной водой, а затем насыщенным раствором соли. Порции раствора в хлористом метилене сушат и упаривают досуха в глубоком вакууме, получая ацетилированную смесь альфа/бета изомеров в виде бесцветной пенки, вес которой после осушки составляет 9,6 г. Тонкослойной испарительной хроматографией из этого вещества, используя в качестве элюента этилацетат:метанол (9:1), получают 3,1 г (7,8 ммол, 46%) чистого транс- и 3,5 г (8,9 ммол, 30%) чистого цис-изомера целевого продукта.
транс-изомер: Rf = 0,65 (этилацетат:метанол, 9:1)
УФ-спектр (MeOH) λmax: 309 нм
Спектр ЯМР ( δ м.д. в CDCl3): 8,77 (1H, шир., C'4)-N


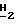
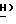
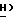

цис-изомер: Rf = 0,58 (этилацетат:метанол, 9:1)
УФ-спектр (MeOH) λmax: 309 нм
Спектр ЯМР ( δ м.д. в CDCl3): 8,72 (1H, шир., C'4-N



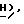

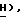
(iii) (+)-цис-оксиметил-5-(5'-фторцитозин-1'-ил)-1,3-оксатиолан
1,2 г (3,05 ммол) цис-2-бензоилоксиметил-5-(N'-ацетил-5'-фторцитозин-1'-ил)- 1,3-оксатиолана перемешивают в течение 2 часов при 0oC в 30 мл метанольного раствора аммиака и оставляют перемешиваться на ночь при комнатной температуре. Смесь упаривают при пониженном давлении и остаток растирают дважды (2 х 30 мл) с абсолютным эфиром. Твердый остаток перекристаллизовывают из абсолютного спирта и получают 655 мг (2,65 ммол, 87%) чистого цис-изомера целевого соединения:
т.пл 204 - 206oC; Rf = 0, 21 (этилацетат:метанол, 9:1). Целевое соединение охарактеризовано методами H и C-ЯМР спектроскопии и УФ спектроскопии
УФ-спектр (вода): λmax = 280,9 нм
Спектр1H-ЯМР ( δ м.д. в ДMCO-d6: 8,22 (1H, дубл., C'6-H, JCF = 7,2 Гц), 7,84 (2H, дубл. , C'4)-N


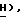
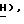
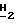
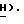
Спектр13C-ЯМР (ДМСО-d6): 153,46 (C'6), 158,14 (C'2,2JCF = 14,0 Гц), 134,63 (C'4, JCF = 24,1 Гц), 126,32 (C'5, JCF = 32,5 Гц), 86,82 (C5), 86,80 (C4), 86, 77 (C2), 62,32 (CH2OH).
Пример 1. (-)-4-Амино-5-фтор-1-(2-оксиметил-1,3-оксатиолан-5-ил)-(1H)-пиримидин-2-он.
(i) Монофосфат (+)-цис-2-оксиметил-5-(5'-фторцитозин-1'-ил)-1,3-оксатиолана.
К перемешиваемой смеси Промежуточного соединения 1 (500 мг, 2,024 ммол) в сухом триметилфосфате (10 мл), охлажденной до 0oC, прибавляют по каплям хлорокись фосфора (1,22 мл, 13,1 ммол). Реакционную смесь перемешивают при указанной температуре в течение часа и реакцию прерывают, прибавляя ледяную волу. Доводят pH охлажденной смеси до 3 прибавлением 1N водного раствора едкого натра, переносят на колонку с активированным древесным углем (5 г, Darco ) и элюируют сначала водой, а затем этанолом и водным раствором аммиака (соотношение 10:10:1). Фракции, которые содержат монофосфат, отделяют и упаривают, а затем переносят на колонку, наполненную 15 г DEAE Sephadex A25 (форма HCO3). Проводят градиентное элюирование водой (300 мл), 0,1 M раствором бикарбоната аммония (300 мл) и 0,2 M раствором бикарбоната аммония (100 мл). Упаривание соответствующих фракций после разбавления водой (30 мл) позволяет выделить монофосфат (+)-цис-2-оксиметил-5-(5'-фторцитозин-1'-ил)-1,3-оксатиолана в виде бесцветного вещества, Rf = 0,5 (n-PrOH: NHOH, 6: 4), выходом 612 г (1,77 ммол, 87,9%).
Спектр1H-ЯМР ( δ м.д. в D2O): 8,27 (1H, дубл., C'6-H, JCf = 6,47 Гц), 7,33 (1H, дубл. дублетов, C5-H ), 5,47 (1H, трипл., C2-H), 4,84 (2H, мульт. C2- CH2-OH), 3,63 (1H, дубл. дублетов, C4-H), 3,30 (1H, дубл. дублетов, C4-H).
Чистота по данным жидкостной хроматографии высокого разрешения (ЖХВР) составляет не менее 99%.
(ii) (+)-цис-2-Оксиметил-5-(5'-фторцитозин-1'-ил)-1,3-оксатиолан.
К раствору монофосфата (+)-цис-2-оксиметил-5-(5'-фторцитозин-1'-ил)-1,3-оксатиолана (100 мг, 0,29 ммол) в 3 мл буферного раствора на основе глицина [глицин (52,6 мг) и хлорид магния (19 мг) в воде (10 мл)] быстро прибавляют 5'-нуклеотидазу ("Sigma", 3,5 мг с активностью 29 ед/мг). Полученную смесь выдерживают в термостате при перемешивании при 37oC. За ходом реакции следят с помощью ЖХВР (хиральная колонка, содержащая гликопротеин альфа-кислоты, элюент - 0,2M раствор фосфата натрия с pH 7, скорость подачи 0,15 мл/мин) через различные промежутки времени. Через 2,5 часа наблюдается присутствие лишь (+)-энантиомера. Прибавляют еще некоторое количество (2 мг) фермента и смесь выдерживают в термостате еще в течение 3 часов. Анализ по данным ЖХВР ясно указывает на селективный и полный гидролиз (+)-энантиомера. Полученную смесь переносят на колонку, наполненную DEAE Sephadex A25 (форма HCO). Проводят градиентное элюирование водой (155 мл), а затем 0,1 M и 0,2 M раствором бикарбоната аммония (по 100 мл каждого). Соответствующие фракции, содержащие первый элюированный нуклеозид, объединяют и упаривают. Оставшееся твердое вещество очищают на короткой колонке с силикагелем, используя в качестве элюента смесь этилацетата и метанола (4,5:0,5), а затем разделяют с помощью ЖХВР (в тех же условиях, что и приведенные ранее). Получают чистый (+)-цис-2-оксиметил-5-(5'-фторцитозин-1'-ил)-1, 3-оксатиолан (23 мг, 0,093 ммол, 32%) в виде белого твердого вещества.
[α] +123o (С, 1,00, MeOH); т.пл. 185oC.
Спектр1Н-ЯМH ( δ м.д. в ДМСО): 8,26 (1H, дубл., C'6-H, JHF в 5,22 Гц), 7,87 (1H, синг., NH2, D2O, обмен), 7,63 (1H, сингл., NH2, D2O, обмен), 6,20 (1H, дубл. дублетов, C5-H), 5,48 (1H, трипл. C2-H), 5,24 (1H, трипл., CH2-O
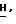
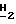
(iii) (-)-цис-2-Оксиметил-5-(5'-фторцитозин-1'-ил)-1,3-оксатиолан. Соответствующие фракции после колонки с сефадексом, содержащие второй элюированный нуклеозид, получение которого описано на стадии (ii), объединяют и упаривают при пониженном давлении. Остаток растворяют в 2 мл воды и обрабатывают алкалинфосфатазой ("Sigma", 1 мл с активностью 60 ед/мг) с последующим термостатированием в течение 1,5 часов при температуре 37oC. Растворитель упаривают и остаток очищают колоночной хроматографией на силикагеле, элюируя смесью этилацетатметанол (4:1) с последующей ЖХВР (в тех же условиях, что и указанные ранее). Таким образом получают чистый (-)-цис-2-оксиметил-5-(5'-фторцитозин-1'-ил)-1,3-оксатиолан (20 мг, 0,081 ммол, 28%) с т.пл. 190oC (с разл.): Rf = 0,21 (EtAc:MeOH, 4:1).
УФ-спектр (вода): λmax = 279,1 мм
Спектр1H-ЯМH ( δ м.д. в ДМСО-d6): 8,25 (1H, дубл., C'6-H, JHF = 7,26 Гц), 7,88 (1H, шир. , С4'-NH2, D2O, обмен) 7,85 (1H, шир., C4'-NH2; D2O, обмен), 5,24 (1H, трипл. , C2-H),
3,83 (2H, мульт., C2C

Промежуточное соединение 2 и Пример 2 поясняют альтернативный способ получения соединения (А).
Промежуточное соединение 2. (1'R, 2'S, %R)-Ментил-5R-(5'-фторцитозин-1''-ил)-1,3-оксатиолан-2S-карбоксилат.
К суспензии 5-фторцитозина (155 мг, 1,2 ммол) в хлористом метилене (1 мл) при комнатной температуре в атмосфере аргона прибавляют последовательно 2,4,6-коллидин (0,317 мл, 2,4 ммол) и трет-бутилметилсилил-трифторметансульфонат (0,551 мл, 2,4 ммол). Полученную смесь перемешивают в течение 15 минут до получения прозрачного раствора. Прибавляют раствор (1'R, 2'S, 5'R)-ментил-5R-ацетокси-1,3-оксатиолан-2S-карбоксилата (330 мг, 1 ммол) в хлористом метилене (0,5 мл), а затем йодтриметилсилан (0,156 мл, 1,1 ммол). Перемешивание продолжают в течение 3 часов. Разбавляют смесь хлористым метиленом (20 мл) и промывают последовательно водным раствором бисульфита натрия, водой, насыщенным раствором соли и упаривают. Остаток переносят в смесь эфир-гексан (1: 1, 10 мл) и насыщенный раствор бикарбоната натрия (2 мл) и перемешивают при комнатной температуре в течение 15 минут. Водный слой отделяют, а органический слой подвергают центрифугированию и получают твердое вещество белого цвета, которое промывают гексаном (3х5 мл) и сушат в вакууме. Полученный таким способом (+)-(1'R, 2'S, 5'R)-ментил-5R-(5''-фторцитозин-1''-ил)-1,3-оксатиолан-29-карбоксилат (350 мг, 88%) содержит около 6% (1'R, 2'S, 5'R)-ментил-5S-(5''-фторцитозин-1''-ил)-1,3-оксатиолан-2S-карбоксилата (ЯМР). Полученное вещество перекристаллизовывают из смеси метанол-хлористый метилен и получают кристаллический продукт.
[α] +22o (С, 0,19, MeOH); т.пл. 216-218oC
Спектр1H-ЯМH ( δ м.д. в CDCl3): 0,78 (3H, дубл., J
= 7 Гц), 0,91 (6H, трипл. , J = 7,3 Гц), 1,00 (2H, мульт.), 1,39 - 2,04 (7H, мульт.), 3,12 (1H, дубл. дублетов, J = 6,6 Гц, 6,1 Гц), 3,52 (1H, дубл. дублетов, J = 4,7 Гц, 6,1 Гц), 4,79 (1H, дубл.
дублетов, J = 4,6 Гц, 4,3 Гц), 5,46 (1H, сингл.), 5,75 (1H, шир. сингл., обмен), 6,42 (1H, трипл., J = 5,0 Гц), 8,10 (1H, шир. сингл., обмен), 8,48 (1H, дубл., J = 6,6 Гц).
Пример 2. 2S-оксиметил-5R-(5'-фторцитозин-1'-ил)-1,3-оксатиолан.
К суспензии литийалюмогидрида (10 мг, 0,54 ммол) в ТГФ (1 мл) медленно прибавляют в атмосфере аргона при комнатной температуре раствор (+) - (1'R, 2'S, 5'R)-ментил-5R-(5''-фторицтозин-1''-ил)-1,3-оксатиолан-2S-карбоксилата (54 мг, 0,135 ммол) в ТГФ (2 мл). Перемешивают реакционную смесь в течение 30 минут и прерывают реакцию, прибавляя избыток метанола (2 мл), а затем прибавляют силикагель (3 г). Образовавшуюся массу очищают колоночной хроматографией на силикагеле (этилацетат-гексан-метанол, 1:1:1) и получают смолоподобное твердое вещество, которое высушивают азеотропной перегонкой с толуолом и выделяют 20,7 мг (63%) твердого белого вещества.
[α] +114o (С, 0,12, MeOH)
Спектр1H-ЯМР ( δ м.д. в ДМСО-d6): 3,14 (1H, дубл. дублетов, J = 4,3 Гц, 11,9
Гц), 2,42 (1H, дубл. дублетов, J = 5,3 Гц, 11,9 Гц), 3,76 (2H, мульт.), 5,18 (1H, мульт. ), 5,42 (1H, трипл., J = 4,8 Гц), 6,14 (1H, мульт), 7,59 (1H, нир. мульт., обмен), 7,83 (1H, шир. мульт.,
обмен), 8,29 (1H, дубл., J = 7,66 Гц).
Пример 3. Биологическая активность.
(i) Антивирусная активность.
Антивирусную активность соединения по примеру 1 определяют по отношению к ВИЧ-1 в следующих клеточных линиях.
Клетки С8166, Т-лимфобластоидная клеточная линия человека, инфицированные ВИЧ-1, штамм RF.
Клетки МТ-4, клеточная линия T-клеток лейкемии, инфицированные ВИЧ-1, штамм RF.
Антивирусную активность в клетках С8166 определяют по ингибированию образования соклетий (Tochikura et al. , Virology, Vol. 164, 542-546), а в клетках МТ-4 по ингибированию превращения формазана [Baba et al., Biochem. Biophys. Res. Commun., Vol, 65, pp. 128-134 (1987); Mossman, J. Immun. Meth. , Vol. 65, pp. 55-57 (1983)]. Антивирусную активность определяют также путем анализа количества антигенов ВИЧ p24, синтезируемых в присутствии и в отсутствие энантиомеров.
Полученные результаты представлены в таблицах 1 и 2.
(ii) Цитоксичность
Цитоксичность соединения по примеру 1 и рацемической смеси (Промежуточное соединение 1) определяют в двух клеточных линиях
CD4: H9, и CEM.
Исследуемые соединения последовательно разбавляют от исходной концентрации 100 мкг/мл до 0,3 мкг/мл (окончательная концентрация) в 96 ячеек для микротитрования. В каждую ячейку помещают по 3,6•104 клеток, в том числе в контрольные ячейки, которые не содержат лекарство. Термостатируют в течение 5 дней при температуре 37oC и подсчитывают количество жизнеспособных клеток путем отделения суспензии клеток и подсчета количества клеток, за исключением трипаносом, в гемоцитометре.
Полученные результаты представлены в таблице 3.
Реферат
Изобретение относится к новым нуклеозидным аналогам 1,3-оксатиолана и их использованию при лечении вирусных инфекций, против Вич-инфекции, гепатита В, более конкретно к (-)-4-амино-5-фтор-1-(2-оксиметил-1,3-оксатиолан-5-ил)-(1Н )-пиримидин-2-ону (I) и его фармацевтически приемлемым производным и содержащим иx фармацевтическим составам. Описывается новая смесь из (-)-4-амино-5-фтор-1-(2-гидроксиметил-1,3-оксатиолан-5-ил )-(1H) -пиримидин-2-oнa и (+)-4-амино-5-фтор-1-(2-гидроксиметил)-1,3-оксатиолан-5-ил)-(1H)-пиримидин-2-она или их фармацевтически приемлемых производных, где (+)-энантиомер присутствует в количестве не более 5 мас.%. Способ получения соединения I, заключаются в том, что проводят выделение (-)-энантиомера из смеси (-)- и (+)-энантиомеров. Способ лечения заключается в ведении эффективного количества соединения (±). 5 с. и 19 з.п.ф-лы, 3 табл.
Комментарии