Способ и устройство для испарения и ингаляции выделенных веществ - RU2733642C2
Код документа: RU2733642C2
Чертежи

















Описание
Родственные заявки
В соответствии с настоящей заявкой испрашивается приоритет согласно предварительной заявке на выдачу патента США № 62/019225 (номер дела патентного поверенного 59426), поданной 30 июня 2014 г.; 62/035588 (номер дела патентного поверенного 59871), поданной 11 августа 2014 г.; 62/085772 (номер дела патентного поверенного 59424), поданной 1 декабря 2014 г.; 62/086208 (номер дела патентного поверенного 61145), поданной 2 декабря 2014 г., и 62/164710 (номер дела патентного поверенного 60652), поданной 21 мая 2015 г., содержание всех полностью включено в настоящий документ посредством ссылки, как если бы было полностью изложено в настоящем документе.
Область техники
Настоящее изобретение, согласно некоторым его вариантам осуществления, относится к области фармакологии и, более конкретно, но не исключительно, к способам и устройствам для контролируемой доставки посредством ингаляции испаряемых веществ.
Уровень техники
На протяжении многих лет многие способы и устройства были разработаны для достижения эффективной доставки биологически активного (фармацевтически активного) средства субъекту, требующему фармацевтического лечения. Пероральный прием внутрь, внутривенная доставка и подкожная инъекция представляют собой наиболее распространенные примеры современных техник доставки. Хотя эти техники, как правило, эффективны, они страдают от нескольких фармакокинетических ограничений, что дополнительно часто приводит к существенному несоблюдению пациентами. Например, терапевтическая польза от традиционных способов часто стирается в течение нескольких часов после первоначального введения дозы, в то время как дискомфорт, связанный с инъекциями, часто приводит к затруднениям во введении и поддержке. Даже пероральное введение может быть неэффективным в тех случаях, когда биологически активное средство обладает плохой биодоступностью и в случаях субъектов, не способных к проглатыванию биологически активного средства из-за тошноты и/или рвоты.
Одним из примеров высокоэффективного биологически активного средства является дронабинол - чистый изомер THC, или (-)-транс-Δ9-тетрагидроканнабинол, который представляет собой одно из основных биологически активных веществ, содержащихся в каннабисе. Дронабинол производится синтетически и продается под торговым наименованием Marinol®, однако, использование этого препарата весьма ограничено из-за присущих ему свойств, таких как вязкость и гидрофобность, которые выражаются в фармацевтически низкой биодоступности и неконтролируемой эффективности при доставке путем проглатывания. Например, у Marinol® занимает более одного часа достижение полного системного эффекта по сравнению с несколькими секундами или минутами для подвергаемого курению или испарению каннабиса. Некоторые пациенты, привыкшие к ингаляции достаточного количества дыма каннабиса для управления симптомами, жаловались на слишком интенсивную и несвоевременную запоздалую интоксикацию от предварительно определенных доз маринола. Многие пациенты говорили, что маринол производит более острый психоделический эффект, чем каннабис, и было предположено, что это несоответствие можно объяснить трудностью в регуляции количества биологически активного средства у субъекта в любой данный момент времени, поскольку это вязкое гидрофобное средство после того, как всасывается через желудочно-кишечный тракт, может временно сохраняться в жировой ткани до достижения рецепторов-мишений в центральной нервной системе.
В то время как курение, как правило, не рекомендуется из-за негативных последствий вдыхания дыма и низкой эффективности доставки несгоревшего биологически активного средства, испарение и вдыхание паров лекарственных средств, страдающих от низкой биодоступности, может представлять собой жизнеспособное решение проблем, связанных с их инъекцией и проглатыванием. Частичное решение обеспечивается некоторыми способами парообразования, направленными на доставку ингаляционных испаряемых биологически активных средств, избегая при этом опасности для дыхания курения. В то время как температура в центре горящей сигареты составляет 750-800°С, выпаривание можно проводить при любой заданной температуре, тем самым позволяя парам биологически активного средства образовываться ниже температуры горения, при которой образуются пиролитические токсичные соединения. Было показано, что способы испарения снижают образование окиси углерода и высоко канцерогенных соединений, таких как полициклические ароматические углеводороды (PAH), бензол и смола.
Однако ни один из известных в настоящее время бездымных устройств испарения не может быть использован для введения испаряемых биологически активных средств под общими фармацевтическими стандартами и практиками из-за невозможности точного и воспроизводимого управления количеством, которое вдыхает пациент. Легочная доставка испаряемых биологически активных средств в паровой фазе варьирует в пределах и между практически доставляемыми дозами из-за субъективной визуальной оценки дозированного количества, введенного пользователем, при неоднократных асинхронных ингаляциях из одной и той же вводимой дозы, непоследовательной динамики ингаляции и зависящей от времени конденсации паров на внутренней поверхности устройства. Как следствие, используемые в настоящее время испарители делают правильное фармацевтическое дозирование и контроль медицинского режима нереальным или нецелесообразным.
В публикации международной заявки на патент № WO 2008/116165 раскрыты системы и способы легочной доставки лекарственного средства в дыхательную систему пациента, причем лекарственное средство подает в очищенном воздухе при положительном давлении по отношению к атмосферному давлению, в то время как медикаментозное средство, доступное в разнообразных формах, вводят контролируемым образом в поток очищенного воздуха в аэрозольной, небулизированной или парообразной форме.
В публикации заявки на патент США № 20140238423 раскрыто электронное курительное изделие, которое включает наличие жидкого материала и нагревательного элемента-фитиля, выполненного с возможностью впитывать жидкий материал и нагревать жидкий материал до температуры, достаточной для испарения жидкого материала и образования аэрозоля. Нагревательный элемент-фитиль состоит из двух или более слоев электрически резистивного сетчатого материала. Это устройство не обеспечивает контролируемость и/или воспроизводимость доставляемого субъекту количества.
Rabinowitz J.D. с соавт. [J. Pharmacol. Exp. Ther., 2004, 309(2), p. 769-75] исследовали системную доставку чистых фармацевтических соединений без продуктов распада через соответствующий процесс, который также включает в себя вдыхание термически получаемого аэрозоля. Согласно Rabinowitz J.D. с соавт. лекарственное средство наносят в виде тонкой пленки на металлический нагревательный элемент и испаряют путем нагревания элемента; тонкая природа лекарственного покрытия минимизирует продолжительность времени, в течение которого лекарственное средство подвергается воздействию высоких температур, тем самым предотвращая его термический распад, и парообразное лекарственное средство быстро конденсируется и коагулирует в аэрозольные частицы микронного размера.
В международной заявке на патент № WO 2012/085919, которая включена в настоящее описание посредством ссылки, настоящими правопреемниками описаны, среди прочего, устройства для ингаляции отмеренных доз для контролируемого парообразования и легочной доставки биологически активных средств из растительного материала путем применения нагревания, причем устройство выполнено с возможностью испарять точное количество средства из растительного материала высоко воспроизводимым способом во время осуществления контроля воздушного потока, чтобы гарантировать полную легочную доставку предварительно определенной дозы.
Дополнительный предшествующий уровень техники включает международную заявку на патент № WO 2008/024490 и WO 2008/024408, патенты США № 6703418, 7169378, 7987846 и 8235037 и публикацию заявки на патент США № 20140100249, 20120252885, 20100168228, 20080181942, 20080176885, 20080078382, 20070072938, 20060258738 и 20060167084.
Сущность изобретения
В настоящем документе предусмотрена единица дозирования, содержащая по меньшей мере одно выделенное биологически активное средство, нанесенное на материал-носитель в термическом контакте с нагревательным элементом, выполненным с возможностью испарять предварительно определенное количество средства для легочной доставки, а также устройства для осуществления испарения и легочный доставки выделенного средства и способы получения единицы дозирования, контролируемого высвобождения средства из нее, способы их легочной доставки и способы лечения патологических состояний, поддающихся лечению посредством легочной доставки выделенного биологически активного средства.
В соответствии с аспектом некоторых вариантов осуществления настоящего раскрытия предусмотрена единица дозирования для легочной доставки по меньшей мере одного биологически активного средства пользователю, которая включает в себя:
пластину; а также
электрический резистивный нагревательный элемент в термоконтакте по меньшей мере с частью поверхности пластины или проходящий по ней,
причем по меньшей мере одно биологически активное средство включено в выделенное биологически активное средство, и пластина включает в себя твердый материал-носитель и биологически активное средство находится в материале-носителе и/или на нем.
В соответствии с некоторыми вариантами осуществления электрически резистивный нагревательный элемент проходит по меньшей мере через две противоположные поверхности пластины.
В соответствии с некоторыми вариантами осуществления материал-носитель по существу не реагирует с биологически активным средством, когда контактирует с биологически активным средством в температурном диапазоне, который находится в интервале от температуры хранения до температуры сгорания/распада биологически активного средства.
В соответствии с некоторыми вариантами осуществления материал-носитель по существу не реагирует с биологически активным средством, когда контактирует с биологически активным средством при температуре в интервале от температуры хранения до температуры на 50°С выше, чем температура испарения биологически активного средства.
В соответствии с некоторыми вариантами осуществления материал-носитель характеризуется температурой горения и/или распада, и/или плавления выше, чем температура испарения биологически активного средства.
В соответствии с некоторыми вариантами осуществления материал-носитель характеризуется температурой горения и/или распада, и/или плавления, которая выше, чем температура испарения биологически активного средства, по меньшей мере на 50°С.
В соответствии с некоторыми вариантами осуществления материал-носитель характеризуется удельным электрическим сопротивлением, составляющим по меньшей мере 10 мкОм⋅м.
В соответствии с некоторыми вариантами осуществления материал-носитель характеризуется удельной теплопроводностью, составляющей по меньшей мере 0,1 Вт/мK.
В соответствии с некоторыми вариантами осуществления материал-носитель включает в себя вещество, выбранное из группы, состоящей из стекла, кварца, керамического композита, карбида кремния, муллита, оксида алюминия, силикона и политетрафторэтилена.
В соответствии с некоторыми вариантами осуществления пластина характеризуется воздухопроницаемой структурой, которая делает возможным поток, составляющий по меньшей мере 0,5 литра газа в минуту, при вакууме, составляющем по меньшей мере 1-5 кПа.
В соответствии с некоторыми вариантами осуществления пластина представляет собой единую воздухопроницаемую матрицу.
В соответствии с некоторыми вариантами осуществления пластина представляет собой воздухопроницаемое множество упакованных частиц.
В соответствии с некоторыми вариантами осуществления частицы характеризуются диаметром более 10 микрон.
В соответствии с некоторыми вариантами осуществления выделенное биологически активное средство представляет собой жидкость, имеющую вязкость по меньшей мере 10 сантипуазов (сП).
В соответствии с некоторыми вариантами осуществления температура кипения выделенного биологически активного средства выше чем 80°С.
В соответствии с некоторыми вариантами осуществления коэффициент распределения октанол-вода (log P) выделенного биологически активного средства больше чем 5.
В соответствии с некоторыми вариантами осуществления коэффициент распределения октанол-вода (log P) выделенного биологически активного средства больше чем 1.
В соответствии с некоторыми вариантами осуществления выделенное биологически активное средство включает в себя синтетическое биологически активное средство.
В соответствии с некоторыми вариантами осуществления выделенное биологически активное средство включает в себя чистый экстракт растительного вещества.
В соответствии с некоторыми вариантами осуществления биологически активное средство выбирают из группы, состоящей из Δ9-тетрагидроканнабинола (ТНС), каннабидиола (CBD), каннабигеролов (CBG), каннабихроменов (CBC), каннабинола (CBN), каннабинодиола (CBDL), каннабициклола (CBL), каннабиэлсоина (CBE), каннабидиварина (CBDV), тетрагидроканнабиварина (THCV), каннабитриола (CBT), терпена, флавоноида и их комбинации.
В соответствии с некоторыми вариантами осуществления биологически активное средство выбирают из группы, состоящей из опиума, сальвинорина, катинона, пукатеина, туйона, дамианина, бульбокапнина, кавалактона, лагохилина, лактукария, глауцина, эргина, ибогаина, апорфина, леонурина, атропина, бупренорфина, буторфанола, фентанила, гидроморфона, метадона, мидазолама, нальбуфина, налоксона, налтрексона, оксикодона, фенитоина, ремифентанила, ризатриптана, силденафила, суфентанила и золпидема.
В соответствии с некоторыми вариантами осуществления биологически активное средство представляет собой (-)-транс-Δ9-тетрагидроканнабинол (дронабинол).
В соответствии с некоторыми вариантами осуществления биологически активное средство предусмотрено в материале-носителе и/или на нем в предварительно определенном количестве.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент представляет собой металлический нагревательный элемент.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент включает U-образную форму с двумя концами и отверстием, в котором располагается пластина, таким образом, что электрический ток протекает через обе по меньшей мере из двух противоположных поверхностей, когда напряжение прикладывается между двумя концами.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент закреплен на пластине, сохраняя пластину на единице дозирования.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент содержит часть, упакованную и проходящую внутри пластины.
В соответствии с некоторыми вариантами осуществления часть резистивного нагревательного элемента, проходящего через пластину, представляет собой воздухопроницаемый резистивный нагревательный элемент.
Согласно некоторым вариантам осуществления воздухопроницаемый резистивный нагревательный элемент обеспечивает поток, составляющий по меньшей мере 0,5 л газа в минуту, при вакууме, равном по меньшей мере 1-5 кПа.
Согласно некоторым вариантам осуществления резистивный нагревательный элемент включает в себя резистивную сетку.
Согласно некоторым вариантам осуществления резистивный нагревательный элемент включает в себя по меньшей мере одну ленту из протравленной металлической фольги.
В соответствии с некоторыми вариантами осуществления лента из протравленной металлической фольги обеспечивается полимерной подложкой, которая включает в себя множество перфораций, что делает ее воздухопроницаемой.
В соответствии с некоторыми вариантами осуществления лента из протравленной металлической фольги включает в себя суженную область, характеризующуюся повышенным сопротивлением, которая плавится, чтобы разорвать электрическую непрерывность вдоль ленты во время рассеивания электрической мощности, происходящей после выхода биологически активного средства.
В соответствии с некоторыми вариантами осуществления лента из протравленной металлической фольги прикрепляется к плавкому элементу, выполненному с возможностью размыкания электрической непрерывности вдоль ленты во время рассеивания электрической мощности, происходящей после выхода биологически активного средства.
В соответствии с некоторыми вариантами осуществления единица дозирования включает в себя воздухопроницаемую поддерживающую сетку, отделяющую пластину и нагревательный элемент, причем поддерживающая сетка в достаточной степени закрыта для удержания пластины в единице дозирования.
В соответствии с некоторыми вариантами осуществления воздухопроницаемая поддерживающая сетка позволяет поток, составляющий по меньшей мере 0,5 л газа в минуту, при вакууме, равном по меньшей мере 1-5 кПа.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент включает в себя принимающую контакт электрода область по обе стороны от области, проходящей по пластине.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент включает в себя блокировочную область транспортного рычага, приспособленную для крепления к транспортному рычагу приспособления для вытягивания дозы.
В соответствии с некоторыми вариантами осуществления единица дозирования содержит множество областей нагревательных элементов, причем каждая область отдельно выполнена с возможностью приема электрического тока.
В соответствии с некоторыми вариантами осуществления нагревательные элементы связаны с соответствующим множеством пластин.
В соответствии с некоторыми вариантами осуществления единица дозирования дополнительно включает в себя рамку, в отверстие которой пластина надлежащим образом вдавливается.
В соответствии с некоторыми вариантами осуществления рамка устойчива к нагреванию по меньшей мере до температуры, при которой биологически активное средство испаряется.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент находится в термальном контакте с пластиной и простирается по меньшей мере через отверстие.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент частично встроен в рамку, окружающую края отверстия.
В соответствии с некоторыми вариантами осуществления рамка включает в себя область в стороне от отверстия, к которому прикреплен резистивный нагревательный элемент.
В соответствии с некоторыми вариантами осуществления резистивный нагревательный элемент прикрепляется к области посредством по меньшей мере частичного плавление рамки в области таким образом, что материал рамки втекает в одно или несколько отверстий в резистивном нагревательном элементе.
В соответствии с некоторыми вариантами осуществления рамка включает в себя область блокировки транспортного рычага, приспособленного для крепления к транспортному рычагу приспособления для вытягивания дозы.
В соответствии с аспектом некоторых вариантов осуществления настоящего раскрытия предусмотрена активирующая единица для единицы дозирования в соответствии с любым из представленных в настоящем документе вариантов осуществления, которая включает в себя:
приспособление для вытягивания дозы, выполненное с возможностью перемещения единицы дозирования из положения хранения в положение использования;
держатель, выполненный с возможностью удержания единицы дозирования таким образом, что биологически активное средство находится в герметичном выравнивании с воздушным каналом активирующей единицы; а также
электроды, расположенные таким образом, чтобы быть в электрическом контакте по меньшей мере с двумя областями приема электрических контактов резистивного нагревательного элемента единицы дозирования, когда находятся в активирующей единице.
В соответствии с некоторыми вариантами осуществления приспособление для вытягивания дозы включает в себя рычаг для вытягивания дозы, приспособленный для сцепления с приемной областью единицы дозирования, так что перемещение рычага для вытягивания дозы передвигает единицу дозирования в положение использования или из него.
В соответствии с некоторыми вариантами осуществления герметизированное выравнивание определяет путь через пластину внутри просвета, вдоль которого воздух, проходящий через пластину, продолжается до достижения выходного отверстия.
В соответствии с некоторыми вариантами осуществления держатель включает в себя механизм, выполненный с возможностью перемещения единицы дозирования.
В соответствии с аспектом некоторых вариантов осуществления настоящего раскрытия предусмотрено ингаляционное устройство, которое включает в себя активирующую единицу согласно любому из представленных в настоящем документе вариантов осуществления.
В соответствии с некоторыми вариантами осуществления ингаляционное устройство включает в себя аппарат распыления единицы дозирования, который включает в себя множество единиц дозирования в замкнутом контейнере.
В соответствии с некоторыми вариантами осуществления замкнутый контейнер содержит блокиратор, который, после распыления первой единицы дозирования из контейнера, предотвращает распыление второй единицы дозирования из контейнера до тех пор, пока первая единица дозирования не вернется к аппарату распыления.
В соответствии с некоторыми вариантами осуществления единицу дозирования распыляют в испарительный аппарат, и работа блокиратора включает помещение испарительного аппарата в аппарат распыления единицы дозирования.
В соответствии с некоторыми вариантами осуществления устройство включает в себя аппарат зажимной камеры, который включает в себя:
компартмент, выполненный такого размера, чтобы получать единицу дозирования из контейнера единиц дозирования, в то время как аппарат зажимной камеры устанавливается в контейнер единиц дозирования, и
блок питания, выполненный чтобы, в то время как аппарат зажимной камеры удаляется из контейнера единиц дозирования, доставить ток на резистивный нагревательный элемент подходяще полученной единицы дозирования, для испарения биологически активного средства, содержащегося в единице дозирования.
В соответствии с некоторыми вариантами осуществления контейнер единиц дозирования содержит множество единиц дозирования.
В соответствии с некоторыми вариантами осуществления устройство выполнено с возможностью высвобождения по меньшей мере одного предварительно определенного парообразного количества биологически активного средства при контролируемом нагревании пластины, включающей в себя биологически активное средство.
В соответствии с некоторыми вариантами осуществления устройство включает в себя датчик температуры для измерения температуры в одной или нескольких из единиц дозирования и на единице дозирования.
В соответствии с аспектом некоторых вариантов осуществления настоящего раскрытия предусмотрен способ изготовления единицы дозирования в соответствии с любым из представленных в настоящем документе вариантов осуществления, который включает:
контактирование материала носителя с выделенным биологически активным средством;
образование пластины, которая включает в себя материал-носитель, содержащий биологически активное средство, внесенное в него и/или нанесенное на него; а также
покрытие пластины по меньшей мере на части одной стороны электрически резистивным нагревательным элементом.
В соответствии с некоторыми вариантами осуществления формирование пластины включает в себя:
размещение множества частиц материала-носителя, содержащего биологически активное средство, внесенное в него и/или нанесенное на него, в камеру дозирования на плоской поверхности;
вибрирование плоской поверхности до тех пор, пока множество частиц не выровняется; а также
прессование выровненного множества частиц с образованием пластины.
В соответствии с некоторыми вариантами осуществления образование пластины включает в себя отрезание участка от материала-носителя, чтобы образовать единую воздухопроницаемую матрицу.
В соответствии с некоторыми вариантами осуществления отрезание участка от материала-носителя осуществляется до контактирования материала-носителя с выделенным биологически активным средством.
В соответствии с аспектом некоторых вариантов осуществления настоящего раскрытия предусмотрен способ легочной доставки по меньшей мере одного биологически активного средства пациенту, который включает:
загрузку единицы дозирования в активирующую единицу ингаляционного устройства в соответствии с любым из представленных в настоящем документе вариантов осуществления;
приложение тока к резистивному нагревательному элементу единицы дозирования, чтобы, таким образом, выпарить предварительно определенное парообразное количество биологически активного средства, тем самым контролируемо высвобождая предварительно определенное парообразное количество.
В соответствии с некоторыми вариантами осуществления способ включает, после подведения тока, вдыхание окружающего воздуха через пластину, таким образом производя легочную доставку предварительно определенного парообразного количества к легким пациента.
Согласно некоторым вариантам осуществления предварительно определенное парообразное количество выбирают таким образом, чтобы проявлять по меньшей мере один предварительно выбранный фармакокинетический профиль и/или по меньшей мере один предварительно выбранный фармакодинамический профиль биологически активного средства у пациента.
В соответствии с некоторыми вариантами осуществления способ дополнительно включает:
определение по меньшей мере одного фармакокинетического параметра и/или по меньшей мере одной фармакокинетической переменной, и/или по меньшей мере одного фармакодинамического параметра, индуцированного легочной доставкой выделенного биологически активного средства у пациента из устройства; на основании фармакокинетического параметра и/или фармакокинетической переменной, и/или фармакодинамического параметра определение предварительно определенного парообразного количества, которое проявляет предварительно выбранный фармакокинетический профиль и/или предварительно выбранный фармакодинамический профиль биологически активного средства у пациента; а также
корректировку устройства для доставки по меньшей мере одного предварительно определенного парообразного количества биологически активного средства.
В соответствии с некоторыми вариантами осуществления каждый из фармакокинетического параметра и/или фармакокинетической переменной, и/или фармакодинамического параметра определяется для отдельного пациента таким образом, что предварительно определенное парообразное количество определяется персонально для пациента.
В соответствии с некоторыми вариантами осуществления предварительно выбранный фармакодинамический профиль варьирует в диапазоне от минимального уровня желаемого эффекта до уровня нежелательного эффекта.
В соответствии с некоторыми вариантами осуществления фармакодинамический профиль варьирует в диапазоне от минимального уровня желаемого эффекта до минимального уровня нежелательного эффекта.
В соответствии с некоторыми вариантами осуществления фармакодинамический профиль варьирует в диапазоне от минимального уровня желаемого эффекта до уровня выше, чем минимальный уровень нежелательного эффекта.
В соответствии с некоторыми вариантами осуществления определение по меньшей мере одного из желаемого эффекта и/или нежелательного эффекта включает получение инструкций от пациента и/или врача.
В соответствии с некоторыми вариантами осуществления предварительно выбранный фармакодинамический профиль выбран из группы, состоящей из:
фармакодинамического профиля в пределах уровня ниже минимального уровня терапевтического эффекта;
фармакодинамического профиля в пределах от минимального уровня терапевтического эффекта до максимального уровня терапевтического эффекта, где неблагоприятный эффект не проявляется или не ощущается, и
фармакодинамического профиля в пределах уровня выше минимального уровня неблагоприятного воздействия.
В соответствии с некоторыми вариантами осуществления фармакодинамический профиль варьирует в диапазоне от минимального уровня терапевтического эффекта до максимального уровня терапевтического эффекта, где неблагоприятный эффект не проявляется или не ощущается.
В соответствии с аспектом некоторых вариантов осуществления настоящего раскрытия предусмотрен способ лечения патологического состояния, подвергаемого лечению посредством ингаляции по меньшей мере одного предварительно определенного парообразного количества по меньшей мере одного биологически активного средства, осуществляемой с помощью способа согласно любому из представленных в настоящем документе вариантов осуществления.
В соответствии с некоторыми вариантами осуществления патологическое состояние выбрано из группы, состоящей из злоупотребления алкоголем, бокового амиотрофического склероза, нервной анорексии, тревожных расстройств, колебаний аппетита, бронхиальной астмы, атеросклероза, биполярного расстройства, нарушения функции мочевого пузыря, хронической обструктивной болезни легких (ХОБЛ), коллаген-индуцированного артрита, колоректальной злокачественной опухоли, болезни Крона, делирия, заболеваний органов пищеварения, синдрома Драве, наркотической зависимости и зависимости, дистонии, эпилепсии, фибромиалгии, генерализованной эпилепсии с фебрильными судорогами плюс (GEFS+), глаукомы, глиомы, гепатита С, ВИЧ-ассоциированной сенсорной невропатийной депрессии, болезни Хантингтона, повышенного давления, повышенного внутриглазного давления, воспалительного заболевание кишечника (IBD), бессонницы, синдрома раздраженного кишечника (IBS), отсутствия аппетита, лейкоза, мигрени, двигательных расстройств, рассеянного склероза (MS), тошноты, нейрогенной боли, невропатической боли, ноцицептивной боли, болезни Паркинсона, фантомной боли, посттравматического стрессового расстройства (PTSD), предменструального синдрома, кожного зуда, психических расстройств, психогенной боли (психалгии или соматоформной боли), судорог, септического и кардиогенного шока, сексуальной дисфункции, опухолей кожи, апноэ сна, спастичности, повреждения спинного мозга, тиков, симптомов Туретта, тремора, непреднамеренной потери веса и рвоты.
В соответствии с аспектом некоторых вариантов осуществления настоящего раскрытия предусмотрена единица дозирования для легочной доставки по меньшей мере одного биологически активного средства пользователю, которая включает в себя:
рамку, содержащую отверстие, а также
пластину, состоящую из твердого материала-носителя и подходящим образом вдавливаемую в отверстие;
причем пластина достаточно проницаема для воздуха, чтобы обеспечить поток, составляющий по меньшей мере 0,5 л газа в минуту, при вакууме, равном по меньшей мере 1-5 кПа, через пластину.
В соответствии с некоторыми вариантами осуществления единица дозирования включает в себя резистивный нагревательный элемент в термальном контакте по меньшей мере с двумя противоположными поверхностями пластины и проходящий по ним, причем пластина вместе с резистивным нагревательным элементом достаточно проницаема для воздуха, чтобы обеспечить поток по меньшей мере 0,5 литра газа в минуту при вакууме, равном по меньшей мере 1-5 кПа, через пластину между по меньшей мере двумя противоположными поверхностями.
В соответствии с некоторыми вариантами осуществления пластина достаточно проницаема для воздуха, чтобы обеспечить поток, составляющий по меньшей мере 0,5 л газа в минуту, при вакууме, равном по меньшей мере 1-5 кПа, через пластину между по меньшей мере двумя противоположными поверхностями.
В соответствии с некоторыми вариантами осуществления материал-носитель характеризуется удельным электрическим сопротивлением, составляющим по меньшей мере 10 мкОм⋅м.
В соответствии с некоторыми вариантами осуществления материал-носитель характеризуется удельной теплопроводностью, составляющей по меньшей мере 0,1 Вт/мK.
В соответствии с некоторыми вариантами осуществления материал-носитель выбирают из группы, состоящей из стекла, кварца, керамического композита, карбида кремния, муллита, оксида алюминия, силикона и политетрафторэтилена.
В соответствии с некоторыми вариантами осуществления пластина представляет собой единую воздухопроницаемую матрицу.
В соответствии с некоторыми вариантами осуществления пластина представляет собой воздухопроницаемое множество упакованных частиц.
В соответствии с некоторыми вариантами осуществления частицы характеризуются диаметром более 10 микрон.
Если не указано иное, все используемые в настоящем документе технические и/или научные термины характеризуются таким же значением, которое обычно понимает обычный специалист в настоящей области техники, к которой относится настоящее изобретение. Хотя способы и материалы, подобные или эквивалентные описанным в настоящем документе, могут быть использованы в практике или испытании вариантов осуществления настоящего изобретения, некоторые способы и/или материалы описаны ниже. В случае возникновения конфликта, патентное описание, включая определения, будут контролировать. Кроме того, материалы, способы и примеры являются только иллюстративными и не предназначены для обязательного ограничения.
Как будет понятно специалистам в настоящей области техники, аспекты изобретения могут быть реализованы в виде системы, способа или компьютерного программного продукта. Соответственно, аспекты изобретения могут принимать форму полностью аппаратного варианта осуществления, полностью программного варианта осуществления (включая в себя встроенное программное обеспечение, резидентное программное обеспечение, микро-код и т.п.) или варианта осуществления, сочетающего программные и аппаратные аспекты, которые могут все в целом называться в настоящем документе как «электронный блок», «модуль» или «система». Кроме того, некоторые аспекты могут принимать форму компьютерного программного продукта, воплощенного в один или несколько считываемых компьютером носителей, имеющего считываемый компьютером программный код, осуществленный на нем. Осуществление способа и/или системы вариантов осуществления может включать выполнение или завершение выбранных задач вручную, автоматически или их сочетание. Кроме того, в соответствии с реальной измерительной аппаратурой и оборудованием вариантов осуществления способа и/или системы настоящего раскрытия, несколько выбранных задач может быть реализовано аппаратными средствами, программным обеспечением или программно-аппаратными средствами или их комбинацией с использованием операционной системы.
Например, аппаратные средства для выполнения выбранных задач в соответствии с некоторыми вариантами осуществления могут быть реализованы в виде микросхемы или цепи. В виде программного обеспечения выбранные задачи в соответствии с некоторыми вариантами осуществления могут быть реализованы в виде множества программных инструкций, исполняемых компьютером с использованием любой подходящей операционной системы. Согласно некоторым вариантам осуществления одна или несколько задач в соответствии с некоторыми вариантами осуществления способа и/или системы, как описано в настоящем документе, выполняются процессором данных, таким как вычислительная платформа для выполнения множества команд. Необязательно, процессор данных включает в себя энергонезависимое запоминающее устройство для хранения команд и/или данных и/или энергонезависимое запоминающее устройство, например, магнитный жесткий диск, и/или съемные носители для хранения команд и/или данных. Необязательно, также предусмотрено сетевое подключение. Необязательно, также предусмотрен дисплей и/или пользовательское устройство ввода, такое как клавиатура или мышь.
Краткое описание чертежей
Некоторые варианты осуществления настоящего изобретения описаны в настоящем документе только в качестве примера со ссылкой на прилагаемые графические материалы. На конкретном примере подробных графических материалов следует подчеркнуть, что данные показаны в качестве примера и в целях иллюстративного обсуждения вариантов осуществления. В связи с этим, описание вместе с графическими материалами делает очевидным для специалистов в настоящей области техники то, как варианты осуществления настоящего изобретения могут быть осуществлены.
Графические материалы представляют собой:
Фиг. 1А-М представляют собой схематичные виды единицы дозирования (картриджа), разобранной и собранной, а также некоторые ее альтернативные конструкции, в соответствии с некоторыми вариантами осуществления.
На фиг. 2А-Е схематично показана система доставки дозы карусельного типа для использования в ингаляционном устройстве или в качестве его самого, в соответствии с некоторыми вариантами осуществления.
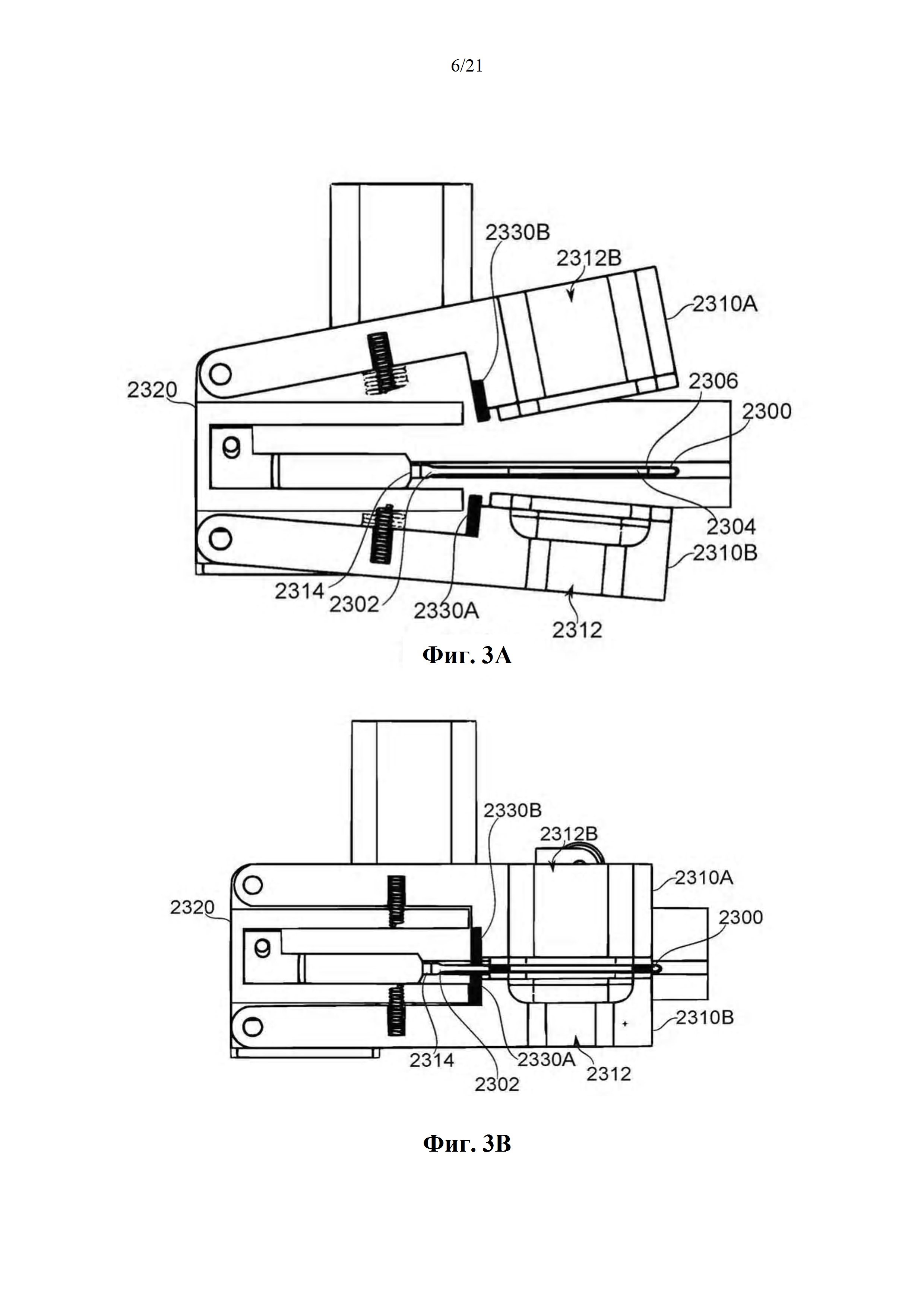
На фиг. 3A-B схематично показан аппарат зажимной камеры для испарения и доставки биологически активного средства из единицы дозирования, в соответствии с некоторыми вариантами осуществления.
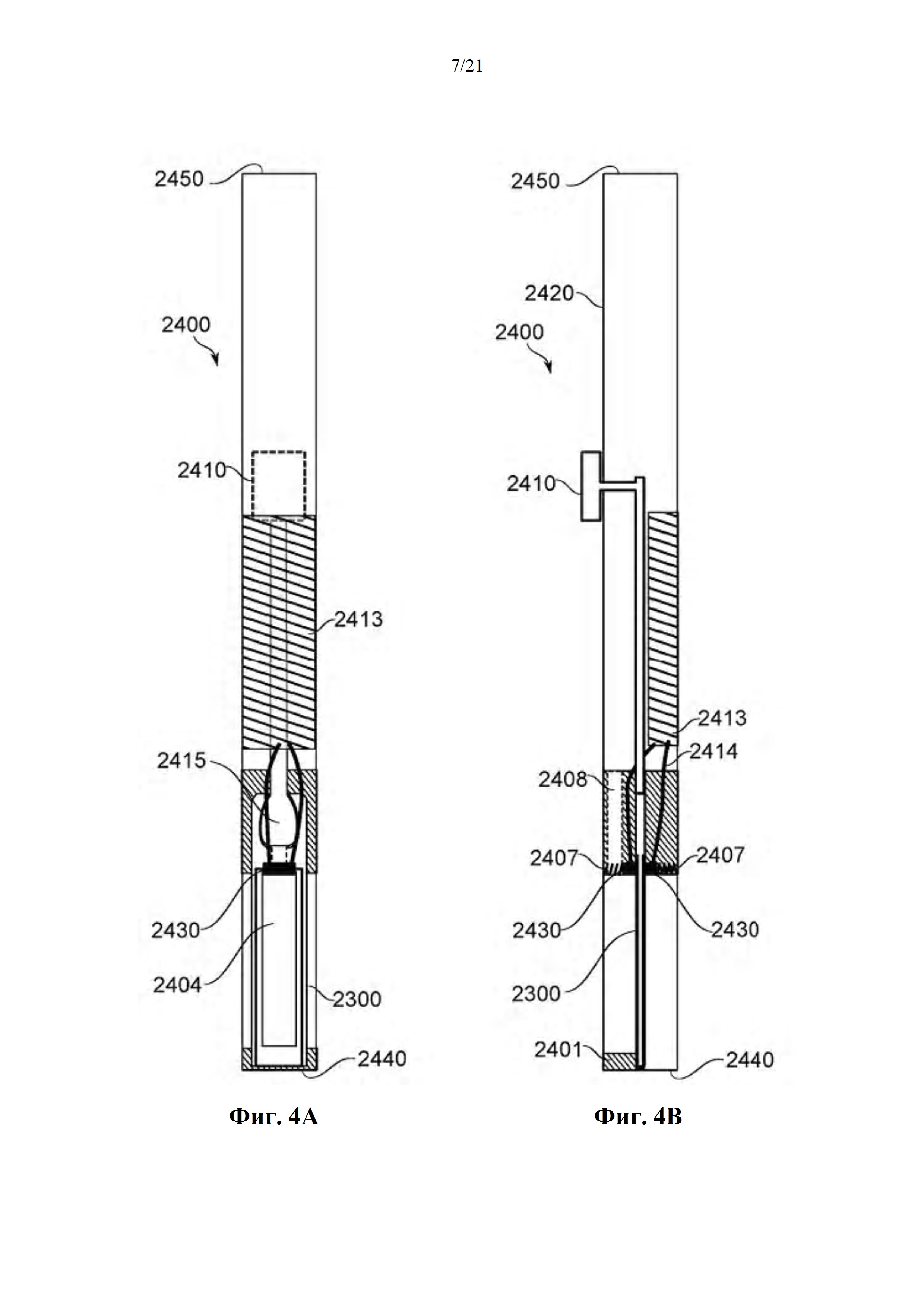
На фиг. 4A-B схематично показано устройство для загрузки из карусели и не зависимое от карусели для испарения и доставки выделенного биологически активного средства из единицы дозирования, в соответствии с некоторыми вариантами осуществления.
На фиг. 5 схематично показан аппарат распыления защищенной блокиратором дозы, вместе со съемным узлом введения дозы, в соответствии с некоторыми вариантами осуществления.
Фиг. 6 представляет собой схематическую диаграмму системы, содержащей устройство для ингаляции, интерфейс врача и/или интерфейс пациента, в соответствии с некоторыми вариантами осуществления настоящего изобретения.
На фиг. 7 показана блок-схема способа назначения персонифицированной схемы лечения пациенту в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Фиг. 8A-D представляют собой схематическую диаграмму (фиг. 8А) и изображения с экранов (фиг. 8B-D) интерфейса врача для выбора и назначения схемы лечения пациенту в соответствии с некоторыми вариантами осуществления настоящего раскрытия.
На фиг. 9 показана блок-схема способа получения персонального фармакодинамического параметра (PD) от пациента и модификации схемы лечения, соответственно, в соответствии с некоторыми вариантами осуществления настоящего изобретения.
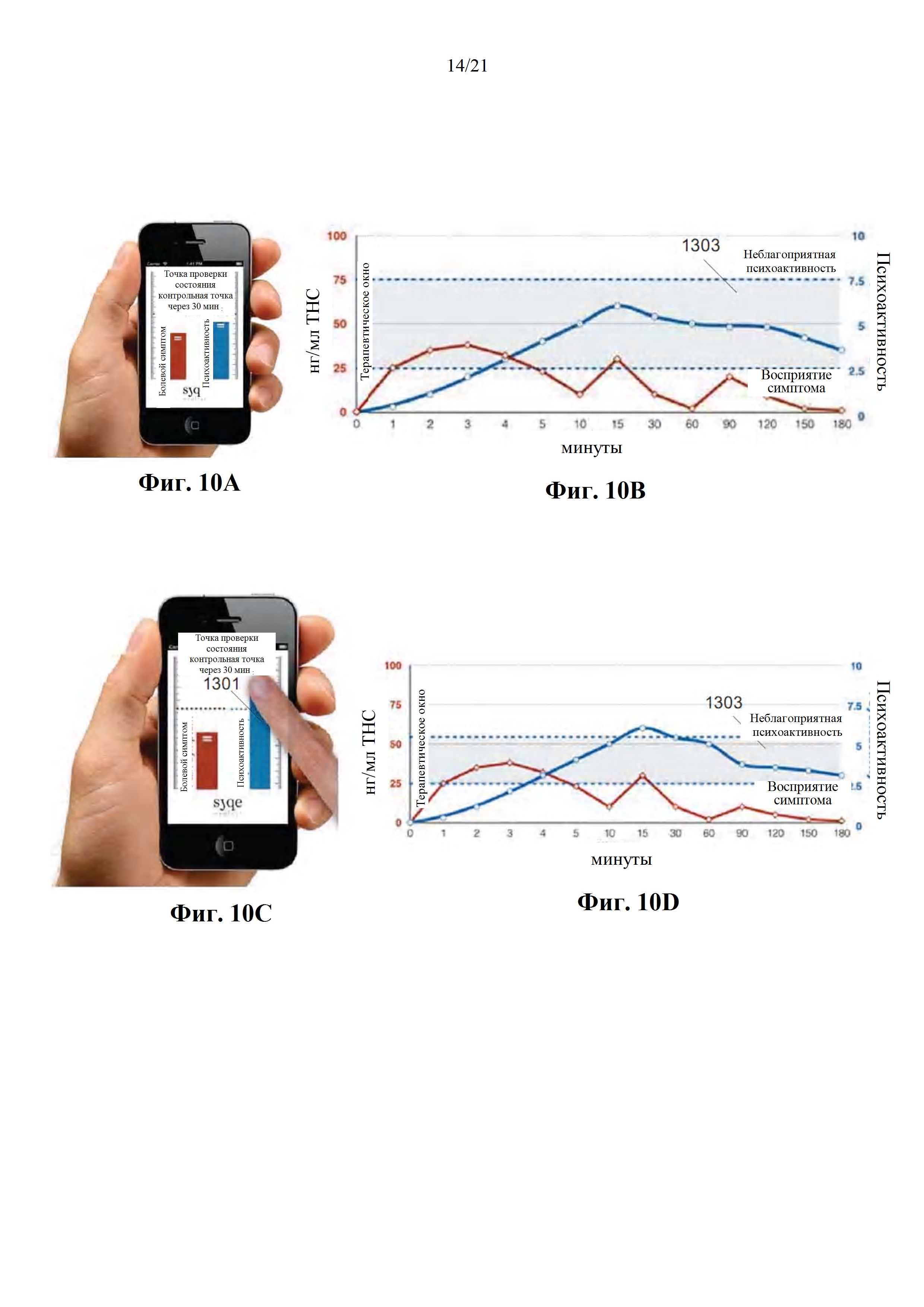
Фиг. 10А-Е представляют собой изображения с экранов интерфейса пациента (фиг. 10А, 10С, 10Е), а также графические представления ожидаемого фармакодинамического и фармакокинетического профилей пациента до и после получения персонального PD эффекта (фиг. 10В и 10D, соответственно), в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Фиг. 11 представляет собой блок-схему способа получения одного или нескольких биомаркеров с использованием персонального портативного устройства и/или с использованием ингаляционного устройства, и при необходимости модификации дозы и/или схемы лечения соответствующим образом, в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Фиг. 12A-C представляют собой изображения с экранов интерфейса пациента, содержащего различные приложения для получения биомаркеров и/или для оказания помощи пациенту в определении воспринимаемого терапевтического и/или неблагоприятного эффекта, в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Фиг. 13 представляет собой схематическую диаграмму ингаляционного устройства, выполненного с возможностью обеспечить автоматизированную контролируемую легочную доставку одного или нескольких активных средств, в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Фиг. 14A-B представляют собой схематическое изображение конфигурации ингаляционного устройства (фиг. 14А) и единицы дозирования ингаляционного устройства, содержащего дискретные пластины (фиг. 14В), в соответствии с некоторыми вариантами осуществления; а также
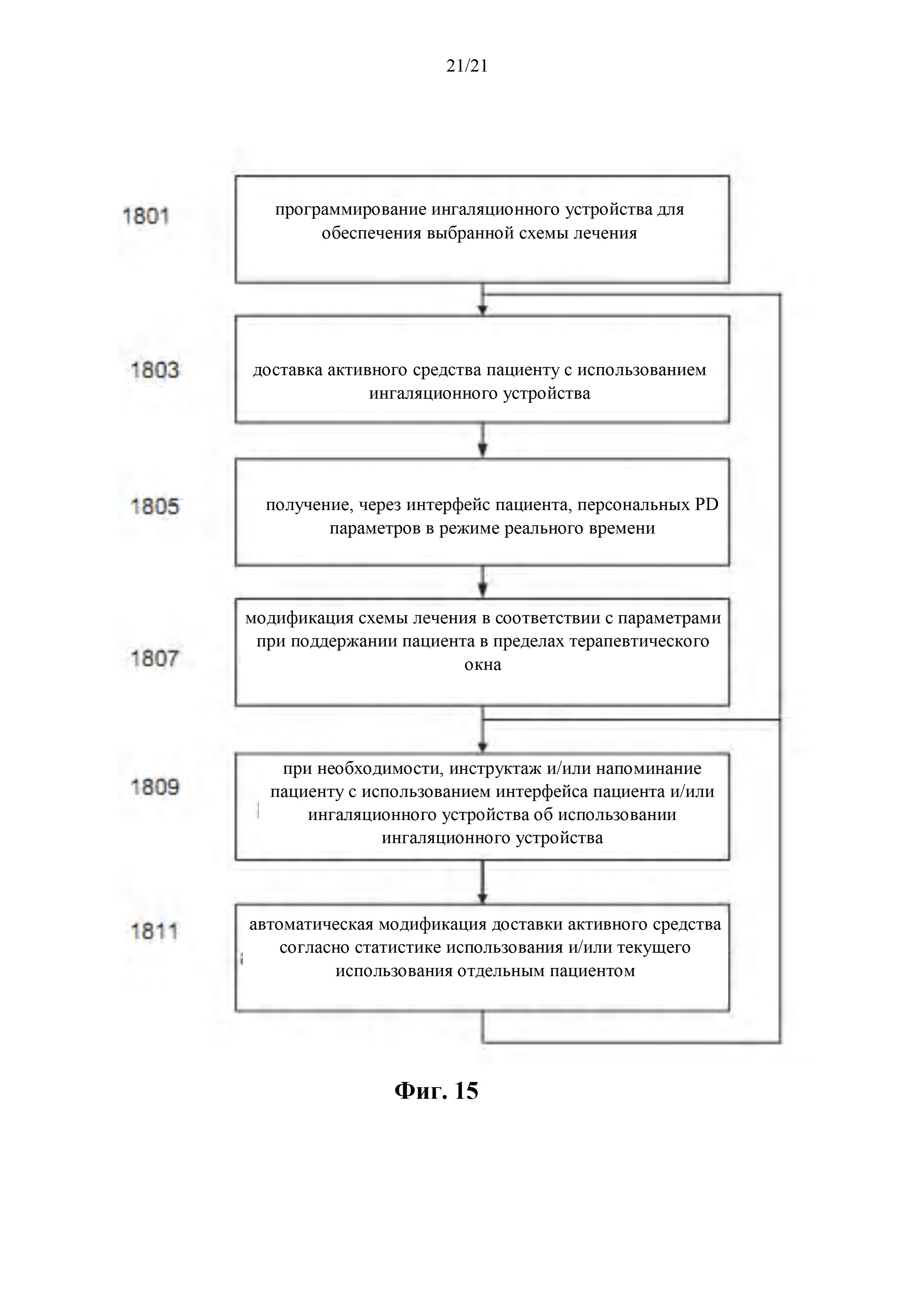
Фиг. 15 представляет собой блок-схему способа лечения отдельного пациента с использованием системы в соответствии с фиг. 9, поддерживая при этом пациента в персонифицированном терапевтическом окне, в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Подробное описание изобретения
Настоящее изобретение, согласно его некоторым вариантам осуществления, относится к области фармакологии и, более конкретно, но не исключительно, к способам и устройствам для контролируемой доставки посредством ингаляции испаряемых веществ.
Перед объяснением по меньшей мере одного из вариантов осуществления настоящего изобретения подробно следует понимать, что настоящее раскрытие не обязательно ограничивается в своем применении деталями конструкции и компоновки компонентов и/или способов, изложенных в приведенном ниже описании и/или проиллюстрированных в графических материалах и/или на примере примеров.
Трудности, связанные с контролируемой доставкой путем инъекции и/или проглатывания биологически активных (фармацевтически активных) средств, которые характеризуются низкой растворимостью в воде и/или высокой вязкостью, и/или высокой температурой кипения, приводят авторов настоящего изобретения к рассмотрению доставки таких биологически активных средство посредством испарения и ингаляции. Как уже указывалось выше, было показано, что способы и устройства для легочной доставки (ингаляции) испаряемых биологически активных средств из содержащих их растительных веществ весьма эффективны и способствуют широко приемлемым фармацевтическим стандартам и практикам. Однако эти способы и устройства не были разработаны для доставки выделенных биологически активных средств, а именно средств, которые больше не образуют часть растительного вещества.
В процессе поиска всеобъемлющего решения проблемы управляемой и воспроизводимой доставки предварительно определенного количества выделенного испаряемого биологически активного средства путем ингаляции, авторы настоящего изобретения рассматривали единицу дозирования, которая включает в себя пластину, содержащую по меньшей мере одно выделенное биологически активное средство в материале-носителе и/или на нем, и нагревательный элемент, находящийся в термическом контакте и проходящий по пластине, таким образом, что биологически активное средство испаряется с пластины при приложении тока к нагревательному элементу. Единица дозирования включает в себя количество биологически активного средства, которое соответствует одному или нескольким циклам (дозам) применения, и может быть использовано в ингаляционном устройстве, которое контролирует интенсивность и продолжительность нагревания и/или поток воздуха через единицу дозирования во время ингаляции, тем самым доставляя управляемое и воспроизводимое предварительно определенное парообразное количество биологически активного средства субъекту.
Способ испарения и ингаляции выделенного биологически активного средства:
В соответствии с аспектом некоторых вариантов осуществления предусмотрен способ легочной доставки путем ингаляции по меньшей мере одного биологически активного средства пациенту с использованием ингаляционного устройства, который выполнен с возможностью контролируемого высвобождения посредством испарения одного или нескольких биологически активных средств из единицы дозирования, содержащей выделенное биологически активное средство(а).
Этот способ представляет собой способ введения путем ингаляции парообразного биологически активного средства, которое иначе трудно вводить посредством проглатывания и/или инъекции по практическим соображениям, требованию пациента, а также внутренним свойствам некоторых выделенных биологически активных средств, что делает их непригодными или иным образом непредпочтительными для введения посредством проглатывания и/или инъекции. Необязательно, способ легочной доставки путем ингаляции по меньшей мере одного биологически активного средства пациенту включает устройство для ингаляции отмеренных доз (устройство MDI), которое представляет собой ингаляционное устройство, выполненное для контролируемого высвобождения посредством испарения по меньшей мере одного предварительно определенного парообразного количества одного или нескольких биологически активных средств.
Согласно некоторым вариантам термин «ингаляция» относится к действию, осуществляемому пользователем/пациентом в качестве добровольного и преднамеренного вдыхания окружающего воздуха через устройство, таким образом, чтобы внести парообразное средство в легкие. Следует отметить, что в соответствии с некоторыми вариантами осуществления спонтанное дыхание может также нести парообразное средство в легкие, а также непроизвольное дыхание, произведенное с помощью механического устройства для вентиляции/дыхания, так как такие устройства известны в настоящей области техники.
Согласно некоторым вариантам осуществления единица дозирования предназначена для содержания предварительно определенного и предварительно отмеренного количества биологически активного средства или выделенного биологически активного средства. Согласно некоторым вариантам осуществления количество соответствует однократной дозе, принимаемой время от времени или принимаемой в рамках схемы лечения. Согласно некоторым вариантам осуществления единица дозирования предназначена для включения некоторого количества выделенного биологически активного средства, которое соответствует более чем одной дозе, принимаемой время от времени или принимаемой в рамках схемы лечения. Поэтому единица дозирования может включать в себя несколько разовых доз, содержащихся отдельно в единице дозирования или содержащихся в комбинации и испаряемых в предварительно определенных аликвотах. Количество выделенного биологически активного средства в одной дозе может быть рассчитано с учетом эффективности испарения биологически активного средства.
Используемый в настоящем документе термин «выпаривание» во всех его склонениях относится к негорючему процессу, при котором вещество оказывается транспортабельным в виде газа (паров), тумана, капель, суспендированных во вдыхаемой атмосфере, или аэрозоля. Согласно некоторым вариантам осуществления «выпаривание» означает, что вещество оказывает транспортабельным в виде газа (паров) при нагревании. Согласно некоторым вариантам осуществления во время доставки путем ингаляции пары могут охлаждаться и конденсироваться с образованием тумана, а именно капелек вещества во взвешенном состоянии во вдыхаемой атмосфере или аэрозоля из них. В контексте некоторых вариантов осуществления термин «выпаривание» охватывает фазовый переход из жидкости в газ (испарение и кипение), а также фазовый переход из твердого состояния в газообразное (сублимация). Согласно некоторым вариантам осуществления термин «выпаривание» также включает в себя промежуточное состояние частично конденсированных паров, которые образуют маленькие капли, которые суспендированы во вдыхаемой атмосфере с образованием тумана или аэрозоля. В соответствии с некоторыми вариантами осуществления термин «выпаривание» относится к процессу, при котором выделенное вещество оказывается транспортабельным в качестве газа или его капель, суспендированных во вдыхаемой атмосфере, а именно, что предполагаемое вещество является по существу единственным веществом, которое испаряется, лишенное носителя или любого другого примечательного компонента, кроме вдыхаемой атмосферы. В соответствии с некоторыми вариантами осуществления термин «выпаривание» исключает пульверизацию (превращение жидкостей в мелкие брызги мелких капель, содержащих множество вещества в жидком состоянии, или превращение жидкостей в аэрозоль или туман, называемый также мелкодисперсным разбрызгиванием), а также другие виды транспорта веществ в виде мелких твердых частиц, содержащих множество вещества (порошок).
Согласно некоторым вариантам термин «выпаривание» исключает процессы, в которых вещество растворяют, суспендируют, эмульгируют или иным образом смешивают с жидким носителем, а затем приводят в транспортабельную форму в виде тумана, который включает в себя жидкий носитель или аэрозоль, который включает в себя жидкий носитель.
В соответствии с некоторыми вариантами осуществления выпаривание осуществляют путем нагревания вещества до температуры, которая достаточна для повышения парциального давления испаряемого вещества, при этом не вызывая горение вещества (ниже температуры сгорания). Как правило, выпаривание осуществляют путем нагревания вещества до температуры чуть ниже, равной или выше его нормальной точки кипения при атмосферном давлении. В соответствии с некоторыми вариантами осуществления выпаривание осуществляют путем повышения температуры вещества и понижения давления окружающей среды (с применением отрицательного давления или вакуума). Понижение давления окружающей среды, как правило, осуществляют под действием ингаляции, которая производит отрицательное давление в атмосфере, окружающей вещество, как правило, в пределах 5-50 мбар ниже по отношению к атмосферному давлению (отрицательные значения давления или от -5 до -50 мбар).
Используемый в настоящем документе термин «парообразное количество» относится к количеству средства, которое находится в форме пара, при этом количество парообразной формы получают с помощью нагревательных элементов в устройстве, при необходимости, принимая во внимание удаление паров путем воздушного потока. Следует отметить, что в настоящем документе согласно некоторым вариантам осуществления количество парообразного средства в контексте настоящего раскрытия не представляет собой расчетное количество, а представляет собой фактическое количество, выпаренное при указанном нагревании, как измерено непосредственно с помощью стандартных лабораторных способов.
Термин «предварительно определенное парообразное количество» относится к количеству, которое преднамеренно высвобождается устройством MDI из единицы дозирования, величина которого определяется конструкцией единицы дозирования, настройками устройства и/или протоколом схемы лечения, как описано в настоящем документе.
Термины «биоактивное средство», «фармацевтически активное средство», «биологически активное средство» и «средство» используются в настоящем документе взаимозаменяемо и относятся к соединению, полимеру, лекарственному средству, конъюгату или комплексу, или любой их комбинации, которая оказывает соматический и/или психотропный эффект при введении субъекту. Как правило, биологически активное средство оказывает желаемый и/или благотворный, и/или терапевтический эффект при его легочной доставке, а затем через системный путь (например, кровь, лимфу) в орган-мишень(и) и/или систему-мишень(и). Средство может быть природного или синтетического происхождения. Неограничивающие примеры активных средств включают в себя активные средства центральной нервной системы, химиотерапевтические средства, седативные средства, анальгетики и психотропные средства.
Используемый в настоящем документе термин «выделенное биологически активное средство» относится к биологически активному средству, которое получают синтетически, либо биологически активное средство, который экстрагируют из природного продукта.
Согласно некоторым вариантам осуществления настоящего изобретения термин «выделенное биологически активное средство» относится к по существу очищенному веществу, в противоположность, например, природному продукту, такому как растительное вещество, которое также содержит твердые нерастворимые компоненты, такие как целлюлозные материалы.
Термин «выделенное биологически активное средство» предназначено для охвата всей экстракции или селективной экстракции одного или нескольких веществ, извлеченных из природного продукта в виде растворимой фракции.
Согласно некоторым вариантам осуществления термин «выделенное биологически активное средство» относится к растворимой фракции экстрагированного препарата, который по существу смешивается с одним или несколькими растворителями и/или смесями растворителей и/или может по существу растворяться в нем. Под термином «по существу растворяется» подразумевается то, что по меньшей мере 90% по массе от общей массы выделенного биологически активного средства растворяют в одном или нескольких растворителях без распада биологически активного средства, в то время как менее чем 10%, менее чем 8%, менее чем 5%, менее чем 3% или менее чем 1% нерастворимой твердой массы остается нерастворенным во фракции. «По существу способен к смешиванию» подразумевает, что по меньшей мере 90% по массе от общей массы выделенного биологически активного средства находится в любой форме (например, жидкость, смола или растворимый порошок), которые могут объединяться с одним или несколькими растворителями без распада биологически активного средства для образования прозрачной жидкости, в то время как менее чем 10%, менее чем 8%, менее чем 5%, менее чем 3% или менее чем 1% нерастворимой твердой массы остается нерастворенным во фракции. В контексте некоторых вариантах осуществления выделенное биологически активное средство по существу лишено либо содержит менее чем 10%, менее чем 8%, менее чем 5%, менее чем 3% или менее чем 1% по массе нерастворимого вещества, нерастворимой фракции или нерастворимого компонента. Термин «нерастворимый» относится к веществу, которое не растворяется в растворителе или смеси растворителей, где растворяется выделенное биологически активное средство.
Согласно некоторым вариантам осуществления количество выделенного биологически активного средства, которое способно растворяться в одном или нескольких растворителях, составляет по меньшей мере 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97 %, 98% или 99% по массе от общей массы выделенного биологически активного средства.
Согласно некоторым вариантам осуществления «выделенное биологически активное средство» относится к биологически активному средству, которое может быть по существу выпарено, не оставляя существенного остатка, или включает в себя его. Под термином «по существу выпарено, не оставляя существенного остатка» подразумевается, что по меньшей мере 50% по массе от общей массы биологически активного средства испаряется без распада, в то время как менее 50% по массе остается неиспарившимся. Согласно некоторым вариантам осуществления количество биологически активного средства, которое способно испаряться по существу без распада и не оставляя существенного остатка, составляет по меньшей мере 50%, 60%, 70%, 80%, 90%, 95% или 99% по массе от общей массы биологически активного средства.
В контексте некоторых вариантов осуществления выделенное биологически активное средство по существу лишено неиспаряемого вещества, неиспаряемой фракции или неиспаряемого компонента. Термин «неиспаряемый» относится к веществу или смеси соединений, которые по существу не испаряются при условиях (например, температуре), используемых для испарения по меньшей мере 50% выделенного биологически активного средства и/или которые разрушаются или сгорают до кипячения или иным образом с образованием их паров, и/или характеризуются температурой кипения выше, чем температура, используемая для испарения по меньшей мере 50% выделенного биологически активного средства.
В соответствии с некоторыми вариантами осуществления выделенное биологически активное средство представляет собой продукт процесса экстракции, который был отделен от других веществ без дополнительной очистки. Согласно некоторым вариантам осуществления содержание выделенного биологически активного средства в неочищенном экстракте по массе составляет по меньшей мере 20%, по меньшей мере 30%, по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60%, по меньшей мере 70%, по меньшей мере 80%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 98% по отношению к массе неочищенного экстракта, содержащего выделенное биологически активное средство.
В соответствии с некоторыми вариантами осуществления выделенное биологически активное средство представляет собой продукт процесса экстракции или продукт синтетического процесса, который был выделен из других веществ и очищен. Согласно некоторым вариантам осуществления чистота выделенного биологически активного средства составляет по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 98% чистоты по массе по отношению к массе образца, содержащего выделенное биологически активное средство.
В соответствии с некоторыми вариантами осуществления термин «выделенное биологически активное средство» относится к комбинации биологически активных средств, каждое из которых может оказывать различные или подобные эффекты и/или иметь синергетический эффект при комбинировании (кумулятивный эффект каждого в одиночку меньше, чем эффект комбинации).
В соответствии с некоторыми вариантами осуществления «полная экстракция» относится к процессу, при котором продукт природного происхождения обрабатывается таким образом, чтобы позволить растворимой фракции его компонентов раствориться в определенном растворителе, в то время как вода может экстрагировать водную фракцию, а органический растворитель или инертный газ может позволить себе органическую фракцию.
«Селективная экстракция» представляет собой процесс, в котором полная фракция или полная экстракция дополнительно обрабатывается на различных стадиях и различными растворителями, чтобы получить пасту, смолу или порошок, содержащий по существу одно или несколько веществ, которые выбраны в силу их растворимости в выбранные растворители, таким образом, получая селективную экстракцию, которая состоит в основном из нескольких выбранных основных компонентов (двух, трех, четырех, пяти, шести, семи, восьми, девяти или десяти веществ или соединений), называемых в настоящем документе «совместный экстракт».
Полный экстракт и/или совместный экстракт, и/или единственное экстрагированное и очищенное вещество может каждое быть превращено в выделенное биологически активное средства посредством существенного удаления (например, путем выпаривания) растворителя(ей), таким образом, получая выделенное биологически активное средство, возможно, в виде жидкой смолы или высушенного порошка, содержащего соответствующие растворимые в растворителе вещества.
Например, в то время как образец растения природного происхождения, культивируемого или разводимого, может содержать одно или несколько биологически активных средств, а также множество различных других веществ и нерастворимых веществ растительных происхождения, образец выделенного биологически активного средства может состоять в основном из одного вещества или соединения, а коэкстрагированный образец выделенных биологически активных средств может состоять в основном из нескольких (двух, трех, четырех, пяти, шести, семи, восьми, девяти или десяти) веществ или соединений, которые представляют собой основные компоненты образца, в то время как незначительные компоненты и примеси составляют менее 40%, менее 30%, менее 20%, менее 10%, менее 9%, менее 8%, менее 7%, менее 6%, менее 5%, менее 4%, менее 3%, менее 2% или менее 1% образца в единицах массы.
Согласно вариантам осуществления, в которых биологически активное средство получают синтетически, продукт реакции может содержать биологически активные средства, смешанные с множеством различных реагентов, продуктами побочной реакции, растворителем(растворителями) и другими веществами, и, таким образом, образец выделенного биологически активного средства дополнительно обрабатывают и очищают, чтобы оно по существу состояло из одного желаемого вещества или соединения, в то время как примеси составляли менее 10%, менее 9%, менее 8%, менее 7%, менее 6%, менее 5%, менее 4%, менее 3%, менее 2% или менее 1% образца в единицах массы.
В контексте некоторых вариантов осуществления выделенное биологически активное средство по существу лишено растворителя, нерастворимого вещества или носителя, а именно, оно находится не в растворе, эмульсии или суспензии и не смешано с другими веществами, если оно не комбинировано с другими выделенными биологически активными средствами, все из которых предназначены для совместной доставки, независимо от того, растворены ли некоторые из них или суспендированы, или найдены в эмульсии с любым другим выделенным биологически активными средствами.
Согласно вариантам осуществления, в которых объединены более чем одно выделенное биологически активное средство, комбинация охватывается термином «выделенное биологически активное средство», определенным в настоящем документе, причем каждое из биологически активных средств предназначено для его легочной совместной доставки пациенту. Используемый в настоящем документе термин «совместная доставка» означает, что два или более биологически активных средства доставляются пациенту на одной стадии ингаляции и/или присутствуют в одной единице дозирования и/или на ней.
Используемый в настоящем документе термин «чистый» относится к количеству одного идентифицированного и определенного вещества, по отношению к общему количеству смеси вещества с другими веществами, которое составляет более чем 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, более чем 99%, более чем 99,5%, более чем 99,9% или 100% от общей массы смеси.
Выделенные биологически активные средства, в соответствии с некоторыми из представленных в настоящем документе вариантов осуществления, включают в себя без ограничения:
синтетически полученное и очищенное (95% чистоты) биологически активное средство;
комбинацию двух или более отдельно синтезированных и очищенных (95% чистоты, каждый) биологически активных средств;
биологически активное вещество или комбинацию из более чем одного биологически активного вещества природного происхождения, которое экстрагируют из микроорганизма, растения или животного и дополнительно очищают до приблизительно 90% чистоты (очищенный экстракт);
целый/полный экстракт микроорганизма, растения или животного, который фракционируют в водном растворе или органическом растворителе (в зависимости от того, в чем биологически активное средство или комбинация биологически активных средств более растворима), сушат и используют без дополнительной очистки;
селективный экстракт микроорганизма, растения или животного, который фракционируют последовательно в различных водных и органических растворах для достижения дополнительного выделения одного или нескольких биологически активных средств из некоторых компонентов экстракта, сушат и используют без дополнительной очистки;
комбинацию более чем одного очищенного, целого или селективного экстракта, каждый из которых содержит одно или несколько биологически активных средств;
комбинацию одного или нескольких синтетически полученных и очищенных биологически активных средств с одним или несколькими очищенными, целыми или селективными экстрактами, каждый из которых содержит одно или несколько биологически активных средств.
Способ, описанный в настоящем документе, решает проблему управляемого и воспроизводимого введения некоторых типов биологически активных средств с использованием некоторых из наиболее преобладающих и приемлемых способов введения, таких как прием внутрь и внутривенная/подкожная инъекция. Например, гидрофобные биологически активные средства, которые по существу не смешиваются в водных средах и/или физиологических жидкостях, могут характеризоваться низкой абсорбцией, низким распределением и низкой биодоступностью при проглатывании или инъекции. Как известно в настоящей области техники, вероятность того, что соединение может быть подходящим в качестве лекарственного средства, возрастает, если соединение характеризуется некоторой степенью растворимости в водной среде; «правило пяти Липинского» относится к коэффициенту распределения октанол-вода (log P) и указывает на то, что соединение должно обладать log P менее чем 5, причем соединения, проявляющие log P более чем 5, считаются слишком гидрофобными для большинства режимов введения лекарственного средства, что приводит к плохой их абсорбции и распределению в организме. Следует отметить в настоящем документе, что варианты осуществления настоящего раскрытия не ограничиваются гидрофобными биологически активными средствами, и, следовательно, выделенные биологически активные средства, имеющие log P менее чем 5, также рассматриваются. Например, согласно некоторым вариантам осуществления выделенное биологически активное средство(а) характеризуется log P больше чем 1.
Устройства и способы, представленные в настоящем документе, применимы для введения любого выделенного биологически активного средства, которое может испаряться, независимо от его гидрофобности, в том числе средства, демонстрирующие высокие значения log P. В соответствии с некоторыми вариантами осуществления выделенное биологически активное средство характеризуется значением log P в интервале температур 20-37°С, составляющим более чем 3, более чем 4, более чем 5, более чем 6, более чем 7 или более чем 8.
Представленные в настоящем документе устройства и способы применимы для введения гидрофобных выделенных биологически активных средств в качестве средства, испаряемого и доставляемого путем ингаляции в виде газа (паров средства).
Другой фактор, который ограничивает использование некоторых выделенных биологически активных средств, представляет собой их физическую форму, а именно то, представляют ли они собой редкую или вязкую жидкость или твердое вещество в своем выделенном виде.
Предполагается, что представленные в настоящем документе устройства и способы применимы для введения любого выделенного биологически активного средства, которое может испаряться, независимо от его физической формы, включая в себя средства, которые представляют собой вязкие жидкости. В соответствии с некоторыми вариантами осуществления выделенное биологически активное средство характеризуется вязкостью по меньшей мере 10 сантипуазов (сП), по меньшей мере 20 сП, по меньшей мере 30 сП, по меньшей мере 40 сП, по меньшей мере 50 сП, по меньшей мере 60 сП, по меньшей мере 70 сП, по меньшей мере 80 сП, по меньшей мере 90 сП, по меньшей мере 100 сП, по меньшей мере 200 сП, по меньшей мере 300 сП, по меньшей мере 400 сП, по меньшей мере 500 сП, по меньшей мере 1000 сП, по меньшей мере 2000 сП, по меньшей мере 5000 сП или более чем 10000 сП.
Предусмотренные в настоящем документе способы и устройства подходят для испарения широкого спектра выделенных биологически активных средств, включая в себя и те, которые характеризуются относительно высокой температурой кипения. Согласно некоторым вариантам осуществления выделенное биологически активное средство характеризуется температурой кипения выше 80°С, выше 100°С, выше 150°С, выше 200°С, выше 250°С, выше 300°С, выше 350°С, выше 400°С, выше 450°С, выше 500°С, выше 550°С, выше 600°С, выше 650°С, выше 700°C или выше 750°С.
Предусмотренные в настоящем документе способы и устройства подходят для испарения широкого спектра испаряемых выделенных биологически активных средств независимо от их физической формы, гидрофобности, вязкости и/или температуры кипения, до тех пор, пока они испаряемые. Используемый в контексте некоторых вариантов осуществления термин «испаряемые» относится к свойству вещества, которое определяет его пригодность для легочной доставки посредством испарения и ингаляции. Это свойство соответствует температуре кипения или сублимации вещества, которое может быть в диапазоне от 80°С или даже 100°С до 750°С.
Согласно некоторым вариантам осуществления выделенное биологически активное средство, который эффективно доставляется пациенту в предварительно определенном и воспроизводимом терапевтическом количестве, является клейкой, густой, вязкой и жирной (гидрофобной с log P более чем 5) жидкостью, характеризующейся относительно высокой точкой кипения, в паровой фазе без совместного введения, преднамеренно или непреднамеренно, любого наполнителя, носителя или любого другого неактивного или нежелательного вещества с ним.
Как известно в настоящей области техники, представители семейства, относящегося к каннабису, содержат различные испаряемые биологически активные средства, которые, как было обнаружено, оказывают благотворное терапевтическое влияние на людей. Согласно некоторым вариантам осуществления биологически активное средство представляет собой или включает в себя каннабиноид, извлеченный и очищенный из каннабиса, или полученный синтетическим путем и очищенный каннабиноид. Согласно некоторым вариантам осуществления выделенное биологически активное средство представляет собой такой выделенный каннабиноид, как, например, Δ9-тетрагидроканнабинол (ТНС), каннабидиол (CBD), каннабигеролы (CBG), каннабихромены (CBC), каннабинол (CBN), каннабинодиол (CBDL), каннабициклол (CBL), каннабиэлсоин (CBE), каннабидиварин (CBDV), тетрагидроканнабиварин (THCV), каннабитриол (CBT) и любой их изомер и/или комбинацию.
Следует отметить, что другие испаряемые выделенные биологически активные средства включены в объем настоящего раскрытия, включая в себя без ограничения, природные биологически активные средства из экстрактов или синтетического происхождения, такие как сальвинорин, катионин, пукатеин, туйон, дамианин, бульбокапнин, кавалактоны, лагохилин, лактукария, глауцин, эргин, ибогаин, апорфин и леонурин, и синтетические испаряемые выделенные биологически активные средства, такие как атропин, бупренорфин, буторфанол, фентанил, гидроморфон, метадон, мидазолам, нальбуфин, налоксон, налтрексон, оксикодон, фенитоин, ремифентанил, ризатриптан, силденафил, суфентанил и золпидем.
Согласно некоторым вариантам осуществления выделенное биологически активное средство включает в себя совместные экстракты или синтетические комбинации биологически активных средств, содержащих терпены, флавоноиды, азотистые соединения и другие природные и синтетические соединения. Например, было показано, что некоторые комбинации каннабиноидов, терпенов и флавоноидов модулируют эффект каннабиноидов или даже оказывают синергическое действие по сравнению с действием каннабиноидов самих по себе.
Терпены и терпеноиды включают в себя без ограничения, Δ3-карен, β-селинен, β-пинен, β-фелландрен, β-фарнезен, β-кариофиллен, β-пинен, β-эудесмол, α-терпинолен, α-пинен, α-фелландрен, α-гумулен, α-бергамотен, α-терпинеол, α-терпинен, α-пинен, α-гумулен, α-гуайен (т), α-цедрен, α-бисаболол, валенсен (т), транс-оцимен, транс-оцимен, транс-кариофиллен, терпинолен, транс-2-пинанол (т), селина-3,7-(11)-диен, селина-(11)3,7-диен (т), сабинен гирдат, нерол, мирцен, мирцен, ментол, линалоол, лимонен, лимонен, изоборнеол, гуайол, гуайа-11,(10),1-диен (т), гермакрен В (т), гераниол, фарнезен (т), эудесм-7(11)-ен-4-ол (т), элемен (т), цис-оцимен, цис-оцимен, оксид кариофиллена, оксид кариофиллена, камфору, камфен, борнеол и (+)фенхол.
Флавиноиды включают в себя без ограничения каннфлавин A, каннфлавин B, каннфлавин C, витексин, изовитексин, апигенин, кемпферол, кверцетин, лютеолин и ориентин.
Согласно некоторым вариантам осуществления биологически активное средство представляет собой выделенный изомер любого из вышеуказанных каннабиноидов, таких как, например, (-)-транс-Δ9-тетрагидроканнабинол, также известный как дронабинол, который представляет собой изомер THC. Выделенный дронабинол, как и другие изомеры THC, проявляет растворимость в воде 0,0028 мг/мл при 23°С, значение log P 5,648, точка кипения 157°С и вязкость 85-140 сП.
Единица дозирования:
Из-за химических и физических свойств некоторых испаряемых выделенных биологически активных средств, способ введения путем ингаляции таких биологически активных средств осуществляют путем использования индивидуальной единицы дозирования, также называемой в настоящем документе взаимозаменяемо как картридж, который предназначен и настроен, чтобы позволить испарение и ингаляцию по меньшей мере одного биологически активного средства пользователю (например, пациенту). Как обсуждалось выше, биологически активное средство может представлять собой, согласно некоторым вариантам осуществления, выделенное биологически активное средство, характеризующееся одним или несколькими свойствами, которые делают его введение менее эффективным или даже неработоспособным при проглатывании и/или инъецировании пользователю.
В соответствии с аспектом некоторых вариантов осуществления единица дозирования предусмотрена для легочной доставки по меньшей мере одного биологически активного средства пользователю, она включает в себя пластину и электрорезистивный нагревательный элемент, также называемый в настоящем документе взаимозаменяемо как резистивный нагревательный элемент (например, металлический резистивный нагревательный элемент), в термальном контакте по меньшей мере с частью поверхности пластины и проходящий по ней, причем пластина содержит твердый материал-носитель и предварительно определенное количество биологически активного средства в материале-носителе и/или на нем.
Согласно некоторым вариантам осуществления резистивный нагревательный элемент проходит по меньшей мере по двум противоположным поверхностям пластины.
Фиг. 1A-B представляют собой схематические иллюстрации единицы дозирования (картридж испарения дозы вещества или картридж), в соответствии с некоторыми вариантами осуществления, показывая единицу дозирования 2300, содержащую пластину 2304, помещенную в отверстие 2303 в рамке 2308, которая образует часть корпуса 2301 (фиг. 1А), и резистивный нагревательный элемент 2306 в термальном контакте по меньшей мере с двумя противоположными поверхностями пластины и проходящий по ним 2304 (фиг. 1В). Фиг. 1С-М обеспечивают схематические иллюстрации альтернативных конструкций единиц дозирования, в соответствии с некоторыми вариантами осуществления.
Используемый в настоящем документе термин «пластина» относится к смеси химически связанных веществ, составляющих матрицу или платформу для обработки, поддержания, хранения, дозирования и доставки вещества, которое в противном случае представляет собой слишком диспергируемое для обработки, содержания, распределения и/или доставки само по себе (например, жидкость, паста, мелкодисперсный порошок, частицы или липкая смола). Пластина, например, позволяет распределение густой жидкости и в дальнейшем позволяет парообразование и последующую доставку ее с пластины.
Необязательно, жидкость, паста или липкая смола высушиваются и/или иным образом становятся твердыми после включения в пластину. Согласно некоторым вариантам осуществления пластина включает в себя любое вещество, которое остается и не доставляется пациенту при подведении к ней тепла.
В соответствии с вариантами осуществления пластина содержит твердый материал-носитель, который выбран и предназначен, чтобы позволять испарение и вдыхание из него выделенного биологически активного средства. Так как материал-носитель используют для переноса и дозирования испаряемого биологически активного средства, он определяется несколькими химическими и физическими критериями, которые включают в себя одно или несколько из следующего:
по существу не вступает в реакции (химически инертный) с биологически активным средством, когда контактирует с ним, по меньшей мере в интервале температур от такой низкой, как самая низкая ожидаемая температура хранения, и до такой высокой, как рабочая температура, возможно, с некоторым более широким диапазоном доверительного интервала (например, от 50°С ниже температуры хранения и до приблизительно 50°С выше рабочей температуры). Согласно некоторым вариантам осуществления температура хранения может быть ниже, чем приблизительно -80°С или приблизительно -40°С или -20°С, однако, более высокие и более низкие температуры находятся в пределах объема настоящего изобретения, включая в себя, например, комнатную температуру (например, 18-26°С). Материал-носитель является химически инертным при температуре до (по меньшей мере) максимальной температуры парообразования биологически активного средства (или немного выше, например, на 50°C) или до температуры сгорания и/или распада биологически активного средства, тем не менее, более высокие температуры находятся в пределах объема настоящего изобретения;
характеризуется температурой горения и/или распада, и/или плавления выше, чем горение/распад биологически активного средства, тем не менее, материалы-носители с более высокими температурами сгорания и/или распада, и/или плавления рассматриваются в пределах объема настоящего изобретения;
характеризуется удельной теплопроводностью, составляющей по меньшей мере 0,1 Вт/мК (что позволяет носителю легко распространять тепло по пластине); а также
характеризуется удельным электрическим сопротивлением, равным по меньшей мере 10 мкОм⋅м (снижение возможности носителя уменьшать ток, проходящий через резистивный нагревательного элемента).
В виде смеси химически связанных веществ, содержащей материал-носитель и биологически активное средство (загруженная пластина), загруженная пластина по существу представляет собой воздухопроницаемую. Другими словами, загруженная пластина характеризуется структурой, которая позволяет потоку вдыхаемого газа (как правило, окружающей атмосферы, несущей пары средства или нет) проходить сквозь нее. Согласно некоторым вариантам осуществления структура пластины характеризуется прохождением вдыхаемого газа через нее, в то время как проход определяется по меньшей мере 2 литрами газа в минуту (л/мин), по меньшей мере 1,5 л/мин, по меньшей мере 1 л/мин, по меньшей мере 0,8 л/мин или по меньшей мере 0,5 литрами газа в минуту, при вакууме, составляющем по меньшей мере 1-5 кПа, что соответствует силе тяги, соответствующей вхождению воздуха в легкие пользователя, тогда как средний легочный пик у здорового взрослого человека составляет приблизительно 25 мбар. В соответствии с некоторыми вариантами осуществления структура пластины характеризуется тем, что позволяет минимальный поток 0,5 литра в минуту и максимальное отрицательное (тяговое) давление 25-40 мбар или отрицательное давление 1-5 кПа (от -1 до - 5 кПа) вблизи пластины.
Для того чтобы пластина была воздухопроницаемой, она может, например, быть выполнена в виде единой пористой матрицы или содержать множество плотно или свободно упакованных отдельных пористых и/или непористых частиц. Согласно некоторым вариантам осуществления, при которых пластина выполнена из множества отдельных (неслитых) частиц, как правило, она окружена стенками, имеющими отверстия на двух противоположных поверхностях, чтобы позволить поток газа через них, следовательно, изделия больше, чем отверстия.
В соответствии с некоторыми вариантами осуществления структура пластины характеризуется отношением поверхность/масса, составляющим по меньшей мере 1000 квадратных метров на грамм (м2/г).
В соответствии с некоторыми вариантами осуществления структура пластины характеризуется отношением поверхность/масса, составляющим по меньшей мере 500 квадратных метров на грамм (м2/г).
Более конкретно, используемый в контексте некоторых вариантов осуществления термин «материал-носитель» представляет собой твердый материал пластины, который обеспечивает физическую поддержку для испаряемого биологически активного средства или термически испаряющегося вещества, которое включено в пластину.
В соответствии с некоторыми вариантами осуществления биологически активное средство не подается на электрически резистивный нагревательный элемент, а применяется в материале-носителе и/или на нем. Согласно некоторым вариантам осуществления биологически активное средство и резистивный нагревательный элемент не находятся в прямом физическом контакте, но находятся в термальном контакте через по меньшей мере пластину.
В соответствии с некоторыми вариантами осуществления материал-носитель или пластина, содержащая его, характеризуется по меньшей мере одним из следующих свойств:
химическая совместимость и соответствие;
относительно высокая температура горения/распала/плавления;
физическое единство, однородность и целостность;
пористость;
высокая теплопроводность; а также
низкая электропроводность.
В контексте некоторых вариантах осуществления химическая совместимость и соответствие может рассматриваться как требование к существенной химической стабильности и инертности, чистоте и отсутствию экстрагируемых и вымываемых веществ, и механической целостности.
Материал-носитель должен быть химически стабильным и инертным (не вступать в реакции) по отношению к биологически активному средству и компонентам, входящим в состав единицы дозирования ингалятора (картриджа), предусмотренной в настоящем документе, а также другим компонентам ингаляционного устройства, по меньшей мере в полном диапазоне между условия хранения и условиями эксплуатации устройства и картриджа. Согласно некоторым вариантам осуществления химическая инертность также требуется во время процесса изготовления пластины и/или единицы дозирования, такая как стабильность и химическая инертность при контакте с полярными и/или неполярными растворителями, которые могут быть использованы в процессе и/или в диапазоне температур или других условиях, применяемых в процессе производства. Химическая стабильность и инертность может быть определена по проценту материала-носителя или его составляющих, которые подвергаются химическому или физическому изменению в процессе, хранения и использования предусмотренного в настоящем документе картриджа, и/или по количеству полученных от материала-носителя веществ (упоминаемых в настоящем документе как «экстрагируемые и вымываемые»), которые вдыхаются во время использования предусмотренной в настоящем документе единицы дозирования, в соответствии с фармакопеей и другими широко используемыми стандартами и практиками, известными и доступными любому специалисту в настоящей области техники. Специалист в настоящей области техники сможет выполнить вышеизложенное, следуя обычно практикуемым руководящим принципам, как это предусмотрено, например, в публикациях, предусмотренных общедоступно Институтом качества продукции исследований (PQRI); в таких учебниках, как “Leachables and Extractables Handbook: Safety Evaluation, Qualification, and Best Practices Applied to Inhalation Drug Products”, 2008, Editors: Douglas J. Ball, Daniel L. Norwood, Cheryl L. M. Stults and Lee M. Nagao, Publisher: John Wiley & Sons, Inc., и в таких научных рецензируемых статьях, как “Best practices for extractables and leachables in orally inhaled and nasal drug products: an overview of the PQRI recommendations” by Norwood, D.L. et al., Pharm Res., 2008, 25(4), p. 727-39.
Материал-носитель дополнительно выбирают так, чтобы он был устойчивым к нагреванию при температуре, при которой испаряется биологически активное средство или несколько более высокой температуре. Другими словами, материал-носитель выбирают для проявления температуры горения, распада и/или плавления выше, чем температура, используемая при приготовлении единицы дозирования и выше, чем температура, при которой используется единица дозирования, чтобы выпарить биологически активное средство в процессе ингаляции. Например, согласно вариантам осуществления с использованием биологически активного средства, имеющего температуру кипения приблизительно 250°C, материал-носитель выбирают таким образом, чтобы он был химически и механически стабильным при нагревании до температуры, используемой для испарения биологически активного средства, таким образом, материал-носитель выбирают с температурой сгорания и/или температурой распада и/или температурой плавления выше 250°С, выше 270°С, выше 290°С, выше 300°С, выше 320°С, выше 350°C, выше 400°С, выше 450°С, выше 500°С, выше 600°С, выше 700°С или выше 750°С. Например, кварц, стекло, керамические материалы и некоторые органические и неорганические полимеры характеризуются температурой сгорания и/или температурой распада, и/или температурой плавления выше, чем точка кипения биологически активного средства приблизительно 250°С.
Согласно некоторым вариантам осуществления материал-носитель выполнен из одного или нескольких веществ, которые включают в себя без ограничения стекло, кварц, керамический композит, карбид кремния, муллит, оксид алюминия, виды углерода (такие как уголь, активированный уголь, графен, графит, фуллерены и подобные), силикон и политетрафторэтилен.
Требование физического единства, однородности и цельности соответствует химическому соответствию в том смысле, что материал-носитель выбирают таким образом, чтобы он поддерживал физическую и механическую целостность (не ломкий и не крошащийся) в той степени, чтобы его можно было транспортировать и использовать для подготовки пластины для предусмотренной единицы дозирования. Другими словами, материал-носитель выбирают таким образом, чтобы он не разрушался или не крошился до частиц, которые не являются однородными по размеру и форме, и, в частности, меньше, чем предполагаемый размер частиц материала-носителя (смотрите требование пористости ниже), при последующей переработке в пластину во время приготовления или во время использования единицы дозирования ингалятора. Материалы-носители могут, таким образом, быть выбраны в соответствии со свойствами хрупкости, пластичности и температуры вязко-хрупкого перехода, так как они известны и доступны любому специалисту в области науки о материалах, в то же время с учетом напряжения, которое применяется к материалу-носителю в процессе приготовления представленной в настоящем документе единицы дозирования, а также температур, которым подвергается материал-носитель в единице дозирования в процессе его использования, как описано в настоящем документе.
Воздухопроницаемость, пористость материала-носителя или характеристики содержащей его пластины определяются в соответствии с некоторыми вариантами осуществления с точки зрения потока воздуха, который может быть пропущен через пластину под давлением вдыхания, когда биологически активное вещество наносится на материал-носитель и/или вносится в него. Таким образом, материал-носитель выбирают подходящий для формирования пластин, который делает возможным поток воздуха по меньшей мере 0,5 литра газа в минуту (0,5/мин) или даже 1 л/мин при действии вакуума по меньшей мере 1-5 кПа, с предварительно определенным количеством биологически активного средства, нанесенным на него или внесенным в него. Согласно некоторым вариантам осуществления материал-носитель находится в форме множества частиц, имеющих форму, которая позволяет газу проходить между ними при упаковке пластины, как описано в настоящем документе. Согласно некоторым вариантам осуществления частицы материала-носителя находятся в форме шариков или сфероидов. Следует отметить, что шаровидными частицами легче манипулировать в процессе подготовки единицы дозирования, предусмотренной в настоящем документе.
Также предполагаются частицы материала-носителя в форме волокон, пленки или любой другой формы, которая может быть упакована в воздухопроницаемую пластину. Согласно некоторым вариантам осуществления частицы материала-носителя больше, чем размер отверстий в воздухопроницаемой удерживающей сетке или тканой сетке, содержащей электрорезистивный нагревательный элемент (например, металлический резистивный нагревательный элемент), образующей часть единицы дозирования, например, составляют более чем 10 мкм, 15 мкм, 20 мкм, 25 мкм, 30 мкм, 35 мкм, 40 мкм, 45 мкм или более чем 50 мкм.
Согласно некоторым вариантам осуществления материал-носитель находится в виде единой монолитной воздухопроницаемого матрицы, образующей пластину. Воздухопроницаемая матрица может быть образована путем слияния частиц материала-носителя в процессе, как правило, называемом спекание, и/или с помощью любой другой методики образования твердых вспененных материалов и других воздухопроницаемых матриц в пределах знаний в настоящей области техники.
Материал-носитель, из которого делают пластину, выбирают таким образом, чтобы он характеризовался теплопроводностью, которая способствует эффективному и однородному нагреванию и испарению биологически активного средства, нанесенного на него или внесенного в него. Теплопроводность материала-носителя, следовательно, выше, чем теплопроводность бумаги и другого высушенного материала растительного происхождения, такого как цветы каннабиса, и составляет приблизительно 0,01 Вт/мK. Например, теплопроводность материала-носителя составляет по меньшей мере 0,1 Вт/мK, по меньшей мере 0,2 Вт/мK, по меньшей мере 0,3 Вт/мK, по меньшей мере 0,4 Вт/мK, по меньшей мере 0,5 Вт/мK, по меньшей мере 0,6 Вт/мK, по меньшей мере 0,7 Вт/мK, по меньшей мере 0,8 Вт/мK, по меньшей мере 0,9 Вт/мK, по меньшей мере 1 Вт/мK, по меньшей мере 5 Вт/мK, по меньшей мере 10 Вт/мK, по меньшей мере 20 Вт/мK, по меньшей мере 50 Вт/мK или по меньшей мере 100 Вт/мK. Например, материал-носитель, содержащий силиконовую эпоксидную смолу, обладает теплопроводностью приблизительно 0,15-0,32 Вт/мK, материал-носитель, содержащий политетрафторэтилен (PTFE), обладает теплопроводностью приблизительно 0,25 Вт/мK, материал-носитель, содержащий стекло, обладает теплопроводностью приблизительно 1 Вт/мK, материал-носитель, содержащий кварц, обладает теплопроводностью приблизительно 3 Вт/мK, и материал-носитель, содержащий Cr/Ni сталь (18% Cr, 8% Ni), обладает теплопроводностью приблизительно 16,3 Вт/мK.
Согласно некоторым вариантам осуществления материал-носитель отличается (не представляет собой) от природного растительного материала, отличается от природного и химически необработанного растительного материала и отличается от природного и механического необработанного растительного материала. Согласно некоторым вариантам осуществления материал-носитель лишен природного химически необработанного и/или природного механически необработанного растительного материала. Например, бумага может быть определена как химически, так и механически обработанный растительный материал, в то время как кусочки растительного материала, прессуемые в пластину, рассматриваются в контексте настоящего раскрытия в качестве химически необработанного растительного материала.
Материал-носитель, из которого делают пластину, выбирают таким образом, чтобы он характеризовался низкой электрической проводимостью или высоким удельным сопротивлением с тем, чтобы избежать тока, проходящего через него, а не через встроенный резистивный нагревательный элемент или резистивную сетку. Например, материал-носитель выбирают с сопротивлением выше 1 мкОм⋅м при 20°C, что приблизительно соответствует удельному сопротивлению нихромового сплава, используемого в электрически резистивном нагревательном элементе. Следовательно, удельное сопротивление материала-носителя составляет по меньшей мере 10 мкОм⋅м, по меньшей мере 50 мкОм⋅м, по меньшей мере 100 мкОм⋅м, по меньшей мере 200 мкОм⋅м, по меньшей мере 400 мкОм⋅м, по меньшей мере 600 мкОм⋅м, по меньшей мере 800 мкОм⋅м или по меньшей мере 1000 мкОм⋅м (1 мОм⋅м). Например, материал-носитель, содержащий политетрафторэтилен (PTFE), характеризуется сопротивлением приблизительно 1023-1025 Ом⋅м, материал-носитель, содержащий стекло, обладает удельным сопротивлением приблизительно 1011-1015 Ом⋅м, и материал-носитель, содержащий плавленый кварц, обладает сопротивлением приблизительно 7,5×1017 Ом⋅м.
Согласно некоторым вариантам осуществления материал-носитель может быть образован из таких веществ, как, но без ограничения, стекло (в виде множества отдельных гранул или спеченной/приваренной воздухопроницаемой стеклянной матрицы или происходить из предшественника золя-геля), кварц (в виде множества отдельных гранул или плавленой воздухопроницаемого кварцевой матрицы), керамический композит, содержащий, например, карбид кремния (SiC), оксид алюминия (Al2O3) и/или муллит (Al2O3-SiO2) (в форме множества отдельных гранул или плавленой воздухопроницаемой керамической матрицы или воздухопроницаемой керамической композиционной матрицы), тугоплавкий полимер, например, PTFE или силиконовые смолы (в виде множества отдельных гранул полимера или воздухопроницаемой плавленой полимерной матрицы или полученной из эмульсионного шаблона полимерной вспененной матрицы). Согласно некоторым вариантам осуществления материал-носитель может быть выполнен из жидкокристаллического полимера (LCP), полиэфирэфиркетона (PEEK), Ultem, тефлона, торлона, Amodel, полифениленсульфида, фортона, Xydear, радела, Udel, полипропилена, Propylux, полисульфона или другого полимерного материала.
Следует отметить, что материал, образующий корпус, который обеспечивает механическую опору для пластины в соответствии с некоторыми вариантами осуществления (например, поддержку пластины 2304 посредством окружения отверстием 2303 в рамке 2308 корпуса 2301 на фиг. 1А; подробности смотрите ниже), выбирается с некоторыми критериями, которые следуют за выбором материала-носителя, такими как критерий химической приемлемости, критерий относительно высокой температуры сгорания/распада/плавления и критерий низкой электропроводности. Другие критерии, которые применяются для выбора материала-носителя, например, физическое единство, однородность и цельность, пористость и высокая теплопроводность, являются менее значимыми или не требуются для выбора материала корпуса.
Количество биологически активного вещества, которое применяется в материале-носителе и/или на нем в пластине, соответствует предварительно определенному парообразному количеству выделенного биологически активного средства(средств), которое должно быть доставлено в легкие пациента/пользователя, в соответствии с некоторыми вариантами осуществления, а именно, поскольку резервуар парообразного средства представляет собой пластину, она содержит средство в количестве, достаточном, чтобы обеспечить испарение и доставку требуемого парообразного и вдыхаемого количества. Количество средства в пластине может находиться в диапазоне от 20 до 500 мг, от 10 до 200 мг, от 9 до 150 мг, от 8 до 100 мг, от 7 до 50 мг, от 5 до 20 мг, от 1 до 10 мг, от 10 до 70 мг, от 10 до 60 мг, от 12 до 50 мг, от 12 до 40 мг, от 15 до 40 мг, от 12 до 30 мг или от 12 до 25 мг.
Узел сборки картриджа:
Согласно некоторым вариантам осуществления выделенное биологически активное средство испаряется при температуре, требующей существенного вклада экзогенного тепла, чтобы достичь температуры выше температуры окружающей среды. Согласно некоторым вариантам осуществления время до достижения температуры испарения находится, например, приблизительно в диапазоне от приблизительно 1000 мс до 5 сек, например, 250 мс, 500 мс, 1000 мс или составляет другое большее, меньшее или промежуточное значение.
Согласно некоторым вариантам осуществления нагревательный элемент, содержащий единицу дозирования, или картридж, представляет собой резистивный нагревательный элемент, содержащий металл, например, нихром, FeCrAl, мельхиор и/или нержавеющую сталь. Необязательно, нагревательный элемент упакован в термальном контакте с пластиной. Термальный контакт включает, например, нахождение в непосредственном контакте или в контакте через теплопередающий слой, позволяющий высокую скорость передачи тепла (например, состоящий из материала с высокой проводимостью тепла, такого как медь, алюминий, латунь или сталь; и/или имеющий тонкостенную конструкцию, толщиной менее чем приблизительно 10 мкм, 20 мкм, 25 мкм, 50 мкм или другой большей, меньшей или промежуточной толщиной). Согласно некоторым вариантам осуществления термальный контакт включает достаточно близкое расположение друг к другу пластины и нагревательного элемента таким образом, что пластина стягивает по существу весь теплоизлучающий угол части нагревательного элемента, покрывающей его; например, более чем 90%, 95%, 99% или другое большее, меньшее или промежуточное значение. Согласно некоторым вариантам осуществления пиковый ток, подаваемый на электрод, находится в диапазоне приблизительно от 1 до 10 ампер; например, составляет приблизительно 1 ампер, 2 ампера, 4 ампера, 6 ампер или другой более высокий, низкий или промежуточный ток.
Согласно некоторым вариантам осуществления термальный контакт содержит нагревательный элемент, проходящий через одну или несколько поверхностей пластины и контактирующий с одной или несколькими поверхностями пластины, например, с одной стороной или двумя противоположными, с наибольшей площадью, сторонами поверхности пластины. Согласно некоторым вариантам осуществления термальный контакт содержит нагревательный элемент, который по меньшей мере частично встроен в пластину.
Согласно некоторым вариантам осуществления нагревательный элемент является воздухопроницаемым (обеспечивает прохождение через него воздуха). Согласно некоторым вариантам осуществления пластина является воздухопроницаемой. Воздухопроницаемость обеспечивается в условиях, например, прохождения воздуха при температуре окружающей среды через нагретый узел сборки пластины и нагревательного элемента под давлением всасывания (сила тяги), таким как давление всасывания, производимое при вдохе, и/или положительное давление, производимое со стороны противоположной от стороны вдыхания картриджа.
Согласно некоторым вариантам осуществления приложенное давление находится в диапазоне 4-20 мм ртутного столба (приблизительно 5-30 мбар), 10-25 мм рт.ст., 5-30 мм рт.ст., 25-40 мм рт.ст., 30-50 рт.ст. или другом диапазоне, имеющем те же самые, более высокие, низкие и/или промежуточные границы.
Согласно некоторым вариантам осуществления нагревательный элемент содержит изгиб приблизительно на 180 градусов, так, что элемент выполнен в форме скобки и/или U-образной формы, покрывающей пластину по меньшей мере с двух сторон. При необходимости, каждая из двух сторон пластины находится в теплопроводном контакте с поверхностью нагревательного элемента. При необходимости, нагревательный элемент на картридже расположен таким образом, что нет никакого контакта между двумя сторонами U-образной формы. При необходимости, применение тока к нагревательному элементу с помощью нагревающего дозу узла сборки осуществляется вблизи двух концов (содержащих принимающие контакты области) U-образной формы, так что нагревание может иметь место на двух сторонах пластины сразу. Согласно некоторым вариантам осуществления применение тока к нагревательному элементу путем подключения к принимающей контакты области на любой стороне пластины на одной или обеих сторонах картриджа. Нагревательный элемент дополнительно разделен на две или более частей, каждая принимающая ток независимо друг от друга. Альтернативно, нагревательный элемент выполнен цельно (необязательно, кусок, который полностью окружает пластину); электроды применимы к принимающим контакты областям элемента таким образом, что потенциал напряжения генерируется на протяжении нагревательного элемента в термальном контакте с пластиной.
Аспект некоторых вариантов осуществления относится к предоставлению рамки вместе с пластиной и нагревательным элементом. Согласно некоторым вариантам осуществления рамка содержит отверстие для приема пластины (дозирующую камеру). Согласно некоторым вариантам осуществления площадь поверхности по всей ширине и длине дозирующей камеры находится в пределах приблизительно 20-100 мм2; например, составляет приблизительно 25 мм2, 50 мм2, 66 мм2, 80 мм2, 100 мм2 или другую большую, меньшую или промежуточную площадь поверхности. Согласно некоторым вариантам осуществления область отверстия открыта с одной стороны. Необязательно, открытая сторона области отверстия закрывается с применением U-образного нагревательного элемента.
Согласно некоторым вариантам осуществления размеры отверстия рамки составляют, например, приблизительно 6×10 мм, рамка, определяющая объем толщиной приблизительно 1 мм. Необязательно, площадь отверстия находится в пределах от приблизительно 20 до 100 мм2; например, 20 мм2, 40 мм2, 50 мм2, 60 мм2, 80 мм2, или оно может характеризоваться другой большей, меньшей или промежуточной площадью поверхности. Отверстие необязательно имеет квадратную форму или по существу квадратную форму (например, приблизительно 8×8 мм); необязательно отверстие имеет продолговатую форму с соотношением сторон, например, 1:2, 1:3, 1:4, 1:10 или характеризуется другим большим, меньшим или промежуточным соотношением длин сторон. Необязательно, отверстие, например, составляет приблизительно 30×2 мм по величине. Согласно некоторым вариантам осуществления отверстие является круглым, овальным или имеет любую форму и размеры в рамке.
Согласно некоторым вариантам осуществления рамка, содержащая отверстие, выполняет одну или несколько из следующих функций:
• располагает пластину в воспроизводимом положении относительно габаритных размеров картриджа;
• обеспечивает механическую устойчивость пластине (например, поддерживает по краям, обеспечивает жесткость, чтобы противостоять изгибу, и/или заякоривает);
• обеспечивает фиксируемые/якорные элементы и/или поверхности, позволяющие перенос картриджа механическими элементами, характеризующимися формой для взаимодействия с картриджем;
• обеспечивает изоляцию между двумя параллельными сторонами нагревательного элемента для предотвращения самоприкосновения и/или
• обеспечивает поверхностную область и/или объемную область для присоединения/заякоривания/погружения нагревательного элемента с картриджем.
Согласно некоторым вариантам осуществления общие функции отверстия включают в себя придание формы структуре дозы в процессе производства и/или содействие в манипулировании дозой для введения.
Согласно некоторым вариантам осуществления рамка содержит полимер или керамику, которые являются по существу термостойкими (например, негорючими, неплавящимися, не изменяющими размеры) при температуре возгонки. Согласно некоторым вариантам осуществления полимер содержит, например, жидкокристаллический полимер (LCP), полиэфирэфиркетон (PEEK), Ultem, тефлон, торлон, Amodel, полифениленсульфид, фортон, Xydear, радел, Udel, полипропилен, Propylux, полисульфон или другой полимерный материал.
Согласно некоторым вариантам осуществления фиксирующий/якорный элемент содержит блокировочную область транспортного рычага, характеризующуюся формой для прикрепления к транспортному рычагу приспособления для вытягивания дозы или другого механизма транспорта дозы.
Согласно некоторым вариантам осуществления пластина закрыта внутри в собранном картридже посредством нагревательного элемента, проходящего через отверстие. Согласно некоторым вариантам осуществления нагревательный элемент проходит через отверстие, не закрывая само отверстия (например, у ленточного нагревательного элемента предусмотрены зазоры между витками ленты). Согласно некоторым вариантам осуществления предусмотрен другой элемент, который действует в качестве защитного барьера (или стены). Необязательно, сдерживающий барьер расположен сразу над нагревательным элементом и пластиной вместе и/или между нагревательным элементом и пластиной.
Аспект некоторых вариантов осуществления относится к процессу изготовления единицы дозирования, как описано выше, в виде пластины и нагревательного элемента, расположенными в пределах рамки.
Согласно некоторым вариантам осуществления процесс включает контактирование материала-носителя с биологически активным средством;
формирование пластины, содержащей материал-носитель, содержащий биологически активное средство, внесенное в него и/или нанесенное на него; а также
покрытие пластины по меньшей мере с одной стороны электрорезистивным нагревательным элементом.
Как обсуждалось выше, пластина может быть в виде множества отдельных частиц или в виде единой воздухопроницаемой матрицы, и в каждом из этих вариантов материал-носитель контактирует с биологически активным средством(ами) путем погружения в материал-носитель, опрыскивания им и/или нанесения на материал-носитель биологически активного средства(средств), или иного нанесения биологически активного средства(средств) на материал-носитель.
Нанесение биологически активного средства(средств) может быть осуществлено до, во время и/или после того, как произведена пластина. Когда наносится множество выделенных биологически активных средств, они могут быть нанесены одновременно и/или последовательно.
Нанесение биологически активного средства(средств) может быть осуществлено, например, с использованием жидкой формы биологически активного средства, либо в виде выделенный (чистой) жидкости, либо раствора биологически активного средства, который может представлять собой растворенную жидкость или растворенное твердое вещество. При использовании раствора биологически активного средства(средств), процесс может дополнительно включать высушивание растворителя раствора таким образом, чтобы оставить покрытие выделенного биологически активного средства в материале-носителе и/или на нем или его части.
Согласно некоторым вариантам осуществления процесс образования пластины, сделанной из множества частиц, включает в себя размещение множества частиц материала-носителя, имеющего выделенное биологически активное средство(а), внесенное в него и/или нанесенное на него, в дозирующей камере на плоской поверхности;
вибрирование плоской поверхности до тех пор, пока множество частиц не выровняется; а также
прессование выровненного множества частиц, таким образом, чтобы сформировать пластину.
Согласно некоторым вариантам осуществления процесс образования пластины, выполненной из единой воздухопроницаемой матрицы, включает в себя отрезание секции от материала-носителя, чтобы образовать единую воздухопроницаемую матрицу. Согласно вариантам осуществления, где материал-носитель отрезают от большего куска материала, биологически активное средство(а) может быть нанесено на него до нарезания материала или после этого.
Согласно некоторым вариантам осуществления процесс образования пластины, сделанной из единой воздухопроницаемой матрицы, включает в себя прессование множества частиц материала-носителя в форме, имеющей обратную форму конечной пластины таким образом, чтобы объединить частицы в воздухопроницаемую матрицу, в то время как такой процесс может упоминаться как сплавление или спекание. В таких случаях нанесение биологически активного средства(средств) осуществляют после стадии прессования.
Согласно некоторым вариантам осуществления измеренное количество частиц материала-носителя, содержащего измеренное количество выделенного биологически активного средства, нанесенного на него, именуемого загруженным материалом-носителем, помещают в дозирующую камеру. Согласно некоторым вариантам осуществления измеренное количество загруженного материала-носителя находится в диапазоне, например, приблизительно 1-100 мг. Согласно некоторым вариантам осуществления дозирующая камера откалибрована таким образом, что размер пластины, при образовании, ограничен границами дозирующей камеры, например, границами ширины и длины пластины.
Согласно некоторым вариантам осуществления измеренное количество загруженного материала-носителя выравнивается вибрацией дозирующей камеры. При необходимости, вибрация происходит с амплитудой в диапазоне приблизительно 0,1-1,2 мм; например, 0,5 мм. Частота вибрации, например, находится в диапазоне приблизительно 20-300 Гц (например, 30 Гц, 45 Гц, 60 Гц, 75 Гц или составляет другую более высокую, низкую или промежуточную частоту). Продолжительность встряхивания, например, выбирают из диапазона 100-1100 мс (например, приблизительно 300 мс, 400 мс, 500 мс, 800 мс или другое более длинное, более короткое или промежуточное время). По желанию, камеру закрепляют до вибрации, чтобы предотвратить выход загруженного материала-носителя снизу из камеры.
Согласно некоторым вариантам осуществления пластину формируют из выровненного загруженного материала-носителя путем сжатия с помощью прессующего элемента. Согласно некоторым вариантам осуществления загруженный материал-носитель сжимается до пластины толщиной в диапазоне приблизительно 200-1500 мкм или толщиной в пределах другого диапазона, характеризующегося такими же, большими, меньшими и/или промежуточными границами.
«Пустая» единица дозирования:
Аспект некоторых вариантов осуществления относится к предоставлению рамки вместе с пластиной и, необязательно, также с нагревательным элементом без включенного в них биологически активного средства. Согласно некоторым вариантам осуществления единицу дозирования получают, по существу, как описано в настоящем документе отдельно для нанесения биологически активного средства в/на пластину единицы дозирования. После того, как биологически активное средство помещается в/на пластину такой «пустой» единицы дозирования, «заполненная» единица дозирования может быть использована для легочной доставки биологически активного средства, по существу, как описано в настоящем документе.
Согласно некоторым вариантам осуществления пустая единица дозирования может быть заполнена, например, посредством направления спрея или капания на пластину в единице дозирования. При необходимости, это выполняется при снятии нагревательного элемента или до его прикрепления. Согласно некоторым вариантам осуществления биологически активное средство наносится на протяжении единицы дозирования, включая в себя нагревательный элемент и/или рамку. При необходимости, в таких случаях рамка (и, возможно, также нагревательный элемент) выполнены из материалов или содержат такие, которые могут отталкивать биологически активное средство в мокром и/или сухом виде или иным образом имеют низкую тенденцию поддерживать с ним контакт.
Согласно некоторым вариантам осуществления пустая единица дозирования может быть заполнена путем удаления пластины из рамки единицы дозирования, нанося выделенное биологически активное средство на пластину и/или в нее, как описано в настоящем документе, и повторным прикреплением пластины к рамке. При необходимости, пластину отделяют от нагревательного элемента, чтобы не наносить биологически активное средство на нагревательный элемент, при нанесении биологически активного средства в/на пластину.
Устройство распыления единицы дозирования и активация:
Аспект некоторых вариантов осуществления относится к позиционированию и активации единицы дозирования, несущей пластину с интегрированным нагревательным элементом внутри камеры, которая активирует нагревательный элемент, при этом удерживая испаряемое выделенное биологически активное средство в канале доставки вещества, и, таким образом, относится к активирующей единице для позиционирования и активации единицы дозирования.
Согласно некоторым вариантам осуществления позиционирование осуществляется путем перемещения картриджа вдоль трека (например, с помощью механизма транспорта картриджа). Согласно некоторым вариантам осуществления камера содержит структуру, которая окружает картридж с обеих сторон, чтобы закрыть его в пределах определенного просвета, и обеспечивает электрический контакт с нагревательным элементом картриджа. Необязательно, электрический контакт находится с обеих сторон картриджа. Необязательно, электрический контакт делают по сторонам картриджа в точках, определенных позиционированием картриджа относительно электродов испаряющего аппарата. Необязательно, контактные площадки проходят от нагревательного элемента для получения электрического контакта с ним.
Аспект некоторых вариантов осуществления относится к контейнеру картриджа для использования с испарителем вещества, который попеременно:
• прикрепляется к контейнеру картриджа для приема единицы дозирования (картриджа) в испаритель вещества; а также
• отсоединяется от контейнера картриджа для введения дозы.
Согласно некоторым вариантам осуществления съемный испаритель вещества используется в качестве части механизма блокировки для контроля распыления единиц дозирования. Например, согласно некоторым вариантам осуществления испаритель вещества используется как часть активации блокировки, которая препятствует извлечению новой единицы дозирования до тех пор, пока ранее израсходованный картридж не вернется в распределительный контейнер.
Иллюстрация нескольких примеров:
В настоящем документе описаны различные примеры вариантов осуществления перечисленных элементов, а также примеры вариантов осуществления собранных единиц дозирования веществ, которые не содержат по меньшей мере один из этих элементов. Следует понимать, что различные варианты осуществления элементов необязательно объединены также в варианты осуществления собранных единиц дозирования в других комбинациях (например, любая конструкция нагревательного элемента предусмотрена с любой конструкцией рамки).
Фиг. 1А-В представляют собой схематичные изображения единицы дозирования 2300 (испарительный картридж дозирования вещества), разобранной и собранной, в соответствии с некоторыми вариантами осуществления. Ссылка также сделана на фиг. 1С-М, на которых схематически показаны альтернативные конструкции единиц дозирования 2350, 2360, 2370, 2380, 2390 и 2395, в соответствии с некоторыми вариантами осуществления. На фиг. 1C, 1E, 1G, 1J и 1L показаны разобранные единицы дозирования, в то время как на фиг. 1D, 1F, 1H, 1K и 1М показаны собранные единицы дозирования.
Согласно некоторым вариантам осуществления дозы выделенного биологически активного средства собраны на единице дозирования 2300 и/или в ней. Необязательно, единица дозирования 2300 содержит:
пластину 2304, при необходимости, образованную, например, путем уплощения для быстрого испарения;
механическую поддержку пластины 2304 (например, поддержку путем ограждения в пределах отверстия 2303 в рамке 2308 корпуса 2301, который необязательно имеет форму рамки);
средства для облегчения транспортировки единицы дозирования 2300 (например, защелку в виде челюстей 2302); и/или
средства для испарения пластины 2304 (например, резистивный нагревательный элемент 2306).
Необязательно, единица дозирования представляет собой одноразовую. Потенциальные преимущества одноразовой единицы дозирования включают в себя: удержание остатка активного средства для удаления; тесную интеграцию поддержки и транспортировки дозировки для надежного дозированного транспорта в пределах дозирующей аппаратуры; и/или сниженную необходимость поддержания и/или мониторинга частей системы дозирования (например, испаряющего нагревательного элемента), которые подвергаются условиям, которые могут привести к снижению производительности с течением времени.
Необязательно, единица дозирования применяется для использования в одной ингаляции. Потенциальные преимущества единицы дозирования однократного применения включают в себя повышение точности и/или надежности в регулировании парообразного количества биологически активного средства, в соответствии с настройками ингалятора.
Например, концентрацию и/или распыление выделенного биологически активного средства в загруженный материал-носитель можно контролировать в процессе производства в какой-то степени точности. В общем, степень изменения в выходе устройства (например, количество парообразного и вдыхаемого биологически активного средства) может поддерживаться в пределах допуска менее чем на +/- 15% от предполагаемого выхода. Другие факторы, которые могут оказать влияние на различия в выходе устройства, включают в себя условия окружающей среды, привычки использования пользователя и текущее состояние пользователя.
Согласно некоторым вариантам осуществления единица дозирования 2300 содержит корпус 2301, содержащий отверстие или приемную камеру 2303. Необязательно, корпус 2301 содержит уплощенную и вытянутую полосу, в то время как приемная камера 2303 содержит отверстие, обрамленное полосой (рамка 2308). Во время приготовления единицы дозирования 2300 пластина 2304 вставляется в приемную камеру 2303. Необязательно, пластину формируют до введения или во время него таким образом, чтобы она соответствовала сплющенной форме приемной камеры 2303. Потенциальным преимуществом для пластины 2304 является нахождение в уплощенной форме, так как большая площадь поверхности и/или равномерная толщина потенциально позволяет быстрее и/или более равномерно распределить нагревание и/или поток воздуха при испарении и доставке.
Согласно некоторым вариантам осуществления размеры пластины составляют, например, приблизительно 6×10 мм по всей площади открытой поверхности, а также приблизительно 1 мм толщиной. Необязательно, толщина пластины находится в диапазоне от приблизительно 0,1 до 1,0 мм или она может быть большей, меньшей или промежуточной толщины. Необязательно, площадь поверхности пластины находится в пределах от приблизительно 20 до 100 мм2; например, 20 мм2, 40 мм2, 50 мм2, 60 мм2, 80 мм2 или она может характеризоваться другой большей, меньшей или промежуточной площадью поверхности. Пластина необязательно имеет квадратную форму или по существу квадратную форму (например, приблизительно 8×8×1 мм); необязательно пластина имеет продолговатую форму с соотношением сторон, например, 1:2, 1:3, 1:4, 1:10 или характеризуется другим большим, меньшим или промежуточным соотношением длин сторон. Необязательно, пластина составляет, например, приблизительно 30×2×0,5 мм по величине. Соответствующая пластина по массе составляет приблизительно 15 мг согласно некоторым вариантам осуществления. Согласно некоторым вариантам осуществления массу пластины выбирают в диапазоне от приблизительно 1 до 100 мг или другом диапазоне, характеризующемся теми же, большими, меньшими и/или промежуточными границами.
Потенциальным преимуществом является окружение пластины 2304 корпусом с рамкой 2301 для большей механической стабильности. Например, пластина 2304 потенциально содержит отдельные частицы загруженного материала-носителя, таким образом, что пластина 2304 подвержена осыпанию частиц, особенно при перемещении или сгибании. Ограждение рамкой 2308 картриджа позволяет пластине 2304 перемещаться в системе без применения напряжений непосредственно самой пластине 2304. Согласно некоторым вариантам осуществления общая длина и ширина картриджа составляет приблизительно 20×10 мм или он может быть другого большего, меньшего или промежуточного размера. В процессе производства корпус с рамкой представляет собой потенциальное преимущество для формирования пластины нужного размера для подогнанной закупорки канала, через который с воздушным потоком подхватываются летучие вещества, выделившиеся во время нагревания пластины.
Следует понимать, что полное окружение пластины не требуется, согласно некоторым вариантам осуществления, для достижения достаточной механической стабильности. Например, согласно некоторым вариантам осуществления (смотрите, например, фиг. 1E-F) пластина 2304 помещается в открытую с одной стороны камеру 2363, определенную как «U»-образная часть рамки 2361. Потенциально, это позволяет упаковать пластину 2304 в единице дозирования 2360 с открытой стороны части рамки 2361. Потенциально, «U»-образная рамка упрощает и/или ускоряет формование и/или высвобождение самой рамки в процессе производства. Согласно некоторым вариантам осуществления открытая сторона закрыта, например, такой структурой, как резистивный нагревательный элемент 2306, проницаемая накладка 2375 (необязательно, удерживающая сетку; фиг. 1G) или другая структура.
Согласно некоторым вариантам осуществления предусмотрены другие виды поддержки пластины. Полностью безрамочный пример показан, например, в единице дозирования 2390A на фиг. 1L, где вся протяженность рамки 2391A (необязательно, включающая в себя даже защелки в виде челюсти 2392) обеспечивается материалом пластины. Согласно некоторым вариантам осуществления материал пластины достаточно стабилен после приготовления, таким образом, не требуется или требуется относительно мало дополнительной механической поддержки для использования (например, пластина сжимается так, что она остается неизменной во время транспортировки между магазином и зажимной камерой). Необязательно, по меньшей мере часть материала пластины смешивают со связующим веществом, чтобы добавить стабильность. Необязательно, пластина представляет собой цельную пластину, характеризующуюся достаточной стабильностью, и служит для удержания геля, жидкости или порошка, содержащего активное средство(а).
Согласно некоторым вариантам осуществления формируют цельную пластину/рамку, при необходимости, с множеством материалов пластины, например, материалом рамки для области рамки 2391A (который может содержать активное вещество или может не содержать его), а также материалом-носителем, содержащим активное средство для высвобождения в области пластины 2394A. Необязательно, формируют цельную пластину/рамку из одного материала, но активное средство(а) добавляют к ней только в области пластины 2394A. Дополнительно или альтернативно, условия формирования (например, степень компрессионной упаковки) различаются между рамочной частью пластины и биологически активным веществом, высвобождающим часть пластины. Согласно некоторым вариантам осуществления материал-носитель покрывает, например, приблизительно 60мм2 вблизи центра пластины 2393. Материал-носитель представляет собой содержащий активное средство материал, который расположен в положении в единице дозирования в сочетании с нагревательным элементом таким образом, что может быть нагрет. Согласно некоторым вариантам осуществления нагревательный элемент 2306 также обеспечивает механическую поддержку. Необязательно, узел сборки 2393 пластины/рамки, в свою очередь, обеспечивает электрическую изоляцию между частями нагревательного элемента 2306. Контакт между нагревательным элементом 2306 и пластиной 2393 осуществляется, например, с использованием любого способа, известного в настоящей области техники, который будет оставаться стабильным во время использования, включая в себя, например, одно или несколько из сварки, клея, холодного пресса, горячего пресса и/или штифтов.
Согласно некоторым вариантам осуществления (например, единица дозирования 2395 на фиг. 1М), пластина 2399 снабжена перфорациями 2398, которые увеличивают ее проницаемость для потока. Это представляет собой особенную потенциальную выгоду для безрамочных или почти безрамочных вариантов осуществления единиц дозирования. Пластина 2399 единицы дозирования 2395, например, ограничена только защелками в виде челюстей 2396 (которые могут быть выполнены в виде составной части пластины) и (прозрачно нарисованным) «U»-образным нагревательным элементом 2306. Потенциально, материал-носитель с достаточной плотностью для достижения механической собственной устойчивости уменьшает проницаемость для воздушного потока получаемой пластины, тем самым препятствуя улетучиванию лекарственных средств. Предусмотрены перфорации 2398, например, путем введения промежутков инструментом (пресс-форма, например), используемым в упаковке материала-носителя, путем перфорирования пластины после образования или другим способом.
Согласно некоторым вариантам осуществления (единица дозирования 2390 на фиг. 1J-К) предусмотрены защелки в виде челюсти 2391 в виде отдельной части (например, изготовленной из полимера или металла), прикрепленной к пластине 2394 материала-носителя, содержащего высвобождаемое активное средство. Согласно некоторым вариантам осуществления нагревательный элемент 2306 или другая оберточная структура обеспечивает дополнительную механическую поддержку. Необязательно, присоединение пластины 2394 к защелкам-челюстям 2391 включает использование адгезии. Необязательно, присоединение включает механическое присоединение; например, одну из защелок-челюстей 2391 и пластину 2394 формируют с язычком, а другую с прорезью, и/или защелки-челюсти 2391 снабжают выступами (например, гребнем шипов), вокруг которых формируется пластина 2394.
Согласно некоторым вариантам осуществления (например, поперечное сечение единицы дозирования 2380, показанное на фиг. 1I) нагревательный элемент 2386, который охватывает пластину 2304, приварен к узлу 2381, где сходятся две стороны нагревательного элемента. Потенциально, это дает преимущество для обеспечения дополнительной механической стабильности пластине 2304 (и, в частности, для одного из безрамочного или частично безрамочного вариантов осуществления). Так как сварной шов 2381 изменяет электропроводящую топологию нагревательного элемента 2386, электроды 2331 для обеспечения тепловой энергии к нагревательному элементу 2386 необязательно размещены на противоположных сторонах нагревательного элемента (при необходимости, но не обязательно в контакте с областью самого сварного шва 2381).
Согласно некоторым вариантам осуществления испарение выделенного биологически активного средства включает нагревание с помощью резистивного нагревательного элемента 2306 или другой формы резистивного нагревательного элемента. Резистивная сетка дополнительно содержит материал, который показывает существенное резистивное нагревание; например, нихром (типичное сопротивление составляет приблизительно 1-1,5 мкОм⋅м), FeCrAl (типичное сопротивление составляет приблизительно 1,45 мкОм⋅м), нержавеющая сталь (типичное сопротивление составляет приблизительно 10-100 мкОм⋅м) и/или мельхиор (типичное сопротивление составляет приблизительно 19-50 мкОм⋅м). В соответствии с выбором материала (например, металла) параметры, такие как длина и ширина, толщина, размер и/или рисунок отверстий нагревательного элемента корректируют так, чтобы характеризоваться общим сопротивлением через резистивный нагревательный элемент, который, например, находится в диапазоне приблизительно 0,05-1 Ом, 0,5-2 Ом, 0,1-3 Ом, 2-4 Ом или в другом диапазоне, имеющем такие же, выше, ниже и/или промежуточные границы.
Необязательно, во время сборки, резистивный нагревательный элемент 2306 прикреплен к корпусу 2301 в положении, перекрывающем пластину 2304 на одной или нескольких сторонах. Например, резистивный нагревательный элемент 2306 проходит по дорсальной поверхности 2309A, складывается на конце корпуса 2311 и проходит назад вдоль вентральной поверхности 2309B. Необязательно, резистивный нагревательный элемент 2306 проходит вокруг камеры 2303 таким образом, что пластина 2304, содержащаяся в камере 2303, загорожена нагревательным элементом 2306. Согласно некоторым вариантам осуществления резистивный нагревательный элемент 2306 содержит множество отдельных панелей, например, панели 2356 и 2356A на фиг. 1C-D, по одной на каждой стороне единицы дозирования 2350. Необязательно, панели электрически соединены одна с другим. Альтернативно, каждая получает отдельные электрические соединения. Потенциальное преимущество нескольких и отдельных панелей для резистивного нагревательного элемента заключается в возможности осуществления управляемого парообразования в предварительно выбранных областях пластины. Потенциальное преимущество двухстороннего ограждения корпуса пластины 2304 (используемого согласно некоторым вариантам осуществления) заключается в увеличении скорости и/или равномерности парообразования при приложении тока к нагревательному элементу 2306. Согласно некоторым вариантам осуществления только одна панель 2356 ограждения представляет собой электрически резистивный элемент, а другая панель 2356A, необязательно, представляет собой сетку или другую воздухопроницаемую структуру (например, пористую структуру), которая обеспечивает механическую поддержку.
Согласно некоторым вариантам осуществления электрические резистивные нагревательные элементы 2356, 2356A работают одновременно. Согласно некоторым вариантам осуществления резистивные нагревательные элементы работают раздельно. Это потенциальное преимущество, например, для возможности раздельного управления и/или высвобождения двух различных средств и/или одного средства в двух последовательных доставках. Например, первый нагревательный элемент (панель, например) работает с энергией, достаточной, чтобы испарить средство непосредственно под ним, но в течение достаточно короткого промежутка времени или с таким профилем нагревания, что тепло не проходит полностью через пластину. С некоторым смещением во времени (необязательно, перекрывающимся или полностью отделенным от первого нагревания) работает второй нагревательный элемент. Потенциально, это является преимуществом, когда должны быть высвобождены два вещества, имеющие различные свойства улетучивания в зависимости от времени или температуры (например, из двух различных материалов пластин). Необязательно, корректируют два профиля нагревания, чтобы привести к одновременному парообразованию. Дополнительно или альтернативно, выпаривание двух средств намеренно смещено во времени. Например, пластину, содержащую отдушку или маскирующее средство, помещают в пластину вблизи нагревательного элемента, где оно испаряется в первую очередь, а второе средство испаряется вскоре после этого (или наоборот). Это потенциальное преимущество, например, чтобы замаскировать потенциально неприятный вкус для сигнализации пользователю о том, что состояние испарения в процессе, и/или иным образом модифицировать сенсорный опыт ингаляции. Необязательно, каждый электрод нагревается на целой стороне пластины. Альтернативно, каждый нагревательный элемент выполнен таким образом, что нагревание для парообразования происходит только через часть пластины, возможно, в другой части для каждого электрода. Согласно некоторым вариантам осуществления один нагревательный элемент используется для «предварительного нагревания» пластины до порогового значения ниже высвобождения активного средства, а второй нагревательный элемент приводится в действие для достижения самого высвобождения. Потенциально, предварительное нагревание с последующим нагреванием для высвобождения укорачивает период парообразования средства и/или увеличивает концентрацию после высвобождения. Потенциально, это помогает увеличить количество средства, достигающего легких, и/или целевое высвобождение до более узкой выбранной респираторной глубины.
Согласно некоторым вариантам осуществления резистивная сетка 2306 характеризуется соотношением открытой (отверстие) к закрытой (сетчатый материал) площади поверхности, составляющей от приблизительно 1:1 (50%) до 1:3 (33%). Согласно некоторым вариантам осуществления соотношение находится в пределах приблизительно 10-20%, приблизительно 20-40%, приблизительно 30-50%, приблизительно 40-70%, приблизительно 60-80%, приблизительно 70-90% или характеризуется другим диапазоном соотношений, имеющим такие же, большие, меньшие и/или промежуточные границы. Согласно некоторым вариантам осуществления отверстия сетки находятся в диапазоне приблизительно 10 мкм, приблизительно 25 мкм, 32 мкм, 50 мкм, 75 мкм, 100 мкм, 200 мкм, 300-750 мкм, 700-1200 мкм или другом большем, меньшем или промежуточном диапазоне. Необязательно, по меньшей мере два отверстия имеют разные размеры и/или форму. Согласно некоторым вариантам осуществления сетка представляет собой сетку из нержавеющей стали 400/0,03 316, с отверстиями по 0,033 мм, 400 отверстий на квадратный дюйм, причем каждое отверстие составляет приблизительно 0,033 мм (33 мкм), толстая проволока 0,03 мм.
Согласно некоторым вариантам осуществления по меньшей мере один нагревательный элемент 2306 встроен полностью или частично в пластину 2304. Необязательно, нагревательный элемент 2306 встроен частично или полностью в рамку корпуса 2301. Например, корпус 2301 первоначально формуют с установленным на месте нагревательным элементом и/или нагревательный элемент 2306 вдавливается на место при высокой температуре на другой стадии изготовления. Необязательно, множество нагревательных элементов 2306 встроено полностью или частично в пластину 2304 таким образом, что они могут работать одновременно или по отдельности.
На фиг. 1G-Н показан другой вариант осуществления единицы дозирования 2370, содержащей погруженный нагревательный элемент 2376 в рамку 2371. Согласно некоторым вариантам осуществления нагревательный элемент 2376 содержит нагревательную секцию 2378, расположенную между множеством электродных накладок 2377. В собранной единице дозирования нагревательная секция 2378 проходит поперек или внутри камеры 2303 и поперек или через пластину 2304. Например, пластина 2304, необязательно, образуется вдавливанием сыпучего материала на место вокруг нагревательного элемента 2376, погружая его. Необязательно, рамка 2371 содержит одну или несколько выемок 2377A, которые получают накладки электрода 2377. Согласно некоторым вариантам осуществления дополнительная механическая поддержка для пластины обеспечивается проницаемым наложением 2375, простирающимся по меньшей мере на одну сторону рамки единицы дозирования 2371. Перекрытие 2375 необязательно содержит полимерную сетку или другую структуру, обеспечивающую поток газа.
Согласно некоторым вариантам осуществления нагревательная секция 2378 нагревательного элемента 2376 выполнена в виде проволоки, которая пересекает камеру 2303 один или несколько раз при подключении к электродным накладкам 2377. Согласно некоторым вариантам осуществления нагревательная секция 2378 содержит сетку, ленту или другую форму. Согласно некоторым вариантам осуществления нагревательная секция 2378 разделена на множество отдельных частей (ветвей, слоев или других подразделений). Согласно некоторым вариантам осуществления нагревательная секция 2378 проходит рядом (например, в пределах 1 мм, в пределах 2 мм или в пределах другого, большего или меньшего расстояния) по существу со всеми частями пластины, содержащей высвобождаемое лекарственное вещество. Это представляет собой потенциальное преимущество для получения более быстрого и/или равномерного высвобождения вещества при нагревании.
Следует понимать, что хотя электродные контакты 2377 электрически отделены друг от друга, за исключением того, как присоединились к нагревательной секции 2378, они не должны быть размещены на физическом удалении друг от друга, в зависимости, например, от курса(ов), проходящего нагревательной секцией 2378 самой по себе. Необязательно, электродные контакты размещены на той же или на разных сторонах камеры 2303, например.
Согласно некоторым вариантам осуществления резистивный нагревательный элемент 2306 содержит резистивную фольгу (например, фольгу, вытянутую в непрерывную ленту или другую форму, поддерживаемую полимером, таким как полиимид и/или силиконовый каучук). Необязательно, поддерживаемая резистивная фольга перфорирована через подложку, чтобы позволить выходить воздуху во время улетучивания вещества дозирования. Согласно некоторым вариантам осуществления к резистивной фольге добавляется запал, например, в качестве дополнительного компонента и/или в качестве области ленты, изготовленной преднамеренно тонкой, так, чтобы обеспечить способ уничтожения нагревательного элемента после использования (путем проведения соответствующего высокого тока через нагревательный элемент в течение достаточного периода времени).
Согласно некоторым вариантам осуществления резистивный нагревательный элемент 2306 прикрепляют к корпусу картриджа 2301, нажав на сетку на корпусе с использованием температуры, достаточно высокой, чтобы расплавить корпус и/или смягчить таким образом, чтобы сетка погрузилась в материал корпуса. Согласно некоторым вариантам осуществления корпус содержит инертный, термостойкий, подходящий материал. Согласно некоторым вариантам осуществления используемый материал корпуса содержит, например, жидкокристаллический полимер (LCP), полиэфирэфиркетон (PEEK), Ultem, тефлон, торлон, Amodel, полифениленсульфид, фортон, Xydear, радел, Udel, полипропилен, Propylux, полисульфон или другой полимерный материал.
Потенциальное преимущество LCP и/или PEEK заключается в хорошей устойчивости к температуре выше температуры, необходимой для испарения вещества, поддерживаемого в картридже. Согласно некоторым вариантам осуществления прикрепление сетки и корпуса происходит при температуре приблизительно 280°C (или при другой достаточно высокой температуре, чтобы расплавить и/или смягчить LCP или PEEK). LCP и PEEK обеспечивают потенциальное преимущество хорошей термической стабильности при более низких температурах, например, при температуре испарения приблизительно 230°С.
Потенциальное преимущество обеспечения нагревательным элементом, таким как резистивный нагревательный элемент 2306, каждой отдельной единицы дозирования, заключается в обеспечении равномерности производительности между использованиями. Потенциально, часть биологически активного средства, с которым нагревательный элемент вступает в контакт, остается прилипшей к нагревательному элементу после остывания. Это накопление может потенциально влиять на производительность парообразования. Дистанционное нагревание (посредством излучения и/или опосредованной проводимости, например) потенциально создает систему, имеющую относительно высокую тепловую инерцию (требуется больше мощности нагрева) по сравнению с прямым проводящим нагреванием с помощью контактного электрода; проблема загрязнения контактного электрода удаляется путем разработки его для одноразового использования. Пониженное требование к нагреванию потенциально повышает безопасность и/или долголетие устройства. Потенциально, пониженное требование к нагреванию также снижает требования к доставке мощности, позволяя варианты осуществления с повышенной портативностью, большей жизнью заряда и/или пониженной стоимостью (например, для систем с батарейным питанием нагревательных элементов).
Согласно некоторым вариантам осуществления единица дозирования 2300 содержит запирающий элемент для использования при транспортировке единицы дозирования. Запирающий элемент содержит, например, защелку в виде челюсти 2302. Запирание делает возможным сцепление одним или несколькими подходящими элементами механизма транспорта системы дозирования для закрепления и/или перемещения единицы дозирования. Перемещение и/или закрепление единицы дозирования от нежелательного перемещения может произойти во время цикла службы единицы дозирования, например: когда единица дозирования помещается в очередь единиц дозирования, содержащих множество единиц дозирования, организованных для использования, когда единица дозирования продвигается в очереди, когда единицу дозирования выбирают для использования, когда единица дозирования перемещается в положение для использования, когда единица дозирования действительно используется и/или когда единица дозирования отбрасывается или, альтернативно, перемещается в «использованное» положение в очереди единиц дозирования.
Карусель и испарительное устройство:
На фиг. 2А-Е схематически показана система доставки дозы карусельного типа 2340 для использования в качестве части ингаляционного устройства или даже устройства MDI, в соответствии с некоторыми вариантами осуществления.
Согласно некоторым вариантам осуществления система доставки дозы 2340 содержит карусель 2322, удерживающую множество единиц дозирования 2300, заключенную в корпусе 2324, и испарительный аппарат 2321, содержащий приспособление для вытягивания дозы 2314 и аппарат зажимной камеры 2320. Корпус карусельного типа 2324 и испарительный аппарат 2321 прикреплены друг к другу; карусель 2322 вращается, чтобы представлять единицы дозирования испарительному аппарату 2321 в порядке их загрузки или в другом порядке, по выбору работы карусели 2322.
Необязательно, корпус карусельного типа 2324 (и его содержимое) является сменным для новой сборки корпуса, например, когда некоторые или все единицы дозирования расходуются, истекают или их необходимо заменить, чтобы изменить композицию единицы дозирования. Число единиц дозирования, переносимых в корпусе составляет, например, приблизительно 100. При желании, количество единиц дозирования представляет собой другое число в диапазоне 10-200 (например, 10, 40, 80, 120, 180 или 200) или другое большее или меньшее число. Согласно некоторым вариантам осуществления диаметр карусели находится, например, в диапазоне приблизительно 7-10 см или составляет больший или меньший диаметр, в соответствии, например, с числом и размером единиц дозирования, которые будут размещены. При необходимости, карусель 2322 содержит одинаковые единицы дозирования или множество различных единиц дозирования (например, содержащих различное количество, концентрации и/или выделенные композиции биологически активных средств). Следует понимать, что карусель представляет собой не единственную форму устройства хранения картриджа, которая может использоваться с единицами дозирования. Например, единицы дозирования могут сохраняться в пружинной системе хранения магазинного типа. Потенциальное преимущество карусели заключается в свободном (а не строго последовательном) доступе во время загрузки и/или выгрузки к положениям единиц дозирования; например, для корректировки режима дозирования. Другие потенциальные преимущества использования карусели относятся к обеспеченному хранению, контролю злоупотреблений, безопасности и другим нормативным требованиям и реквизитам.
В примере рабочего цикла, приспособление для вытаскивания дозы 2314 простирается от испарительного аппарата в карусельный корпус 2324, где оно присоединяется к единице дозирования 2300, например, с помощью защелки в виде челюсти 2302. Согласно некоторым вариантам осуществления приспособление для вытаскивания дозы 2314 «защелкивает» на место в пределах защелок-челюстей 2302. Согласно некоторым вариантам осуществления приспособление для вытаскивания дозы 2314 содержит две части, которые перемещаются в поперечном направлении по противоположным сторонам, а затем близко друг к другу в пределах пространства, ограниченного челюстями 2302 (потенциальное применение более низкой силы к челюстям 2302 и/или единице дозирования 2300, чем способ защелкивания путем вставки). Дальнейшее действие тянет обратно в испарительный аппарат привод и прикрепленную к нему единицу дозирования 2300. Загрузка вещества дозирования 2304 единицы дозирования 2300 тянется, таким образом, вместе с воздухозаборником 2312. Следует понимать, что приспособление для вытаскивания дозы потенциально работает в режиме, отличном от приводимого в действие рукой транспорта: например, как «толкатель» дозы (содержащий, например, загружаемый посредством пружины элемент в самом карусельном объеме) и/или магнит (в режиме вытягивания) или магниты (в режиме толкания или вытягивания).
Согласно некоторым вариантам осуществления зажимные элементы 2310A и 2310B располагаются близко на картридже, приводя электроды на место для нагревания вещества дозирования для испарения из него летучих веществ.
На фиг. 3A-B схематически изображен аппарат зажимной камеры 2320 для испарения и доставки выделенного биологически активного средства из единицы дозирования 2300, в соответствии с некоторыми вариантами осуществления.
Единица дозирования 2300 транспортируется в аппарат зажимной камеры 2320, например, путем перемещения приспособления для вытягивания дозы 2314, одновременно с зацеплением с защелкой в виде челюсти 2302.
Согласно некоторым вариантам осуществления аппарат зажимной камеры 2320 (также называемый испарительный аппарат) состоит из двух зажимных элементов 2310A, 2310B, которые зацепляют единицу дозирования 2300 при испарении. Согласно некоторым вариантам осуществления каждый зажим 2310A, 2310B несет соответствующий электрод 2330A, 2330B, который располагается таким образом, чтобы контактировать с нажатием с резистивным нагревательным элементом 2306 или другим нагревательным элементом, которые образуют часть единицы дозирования 2300. Электроды 2330A, 2330B, в свою очередь находятся в электрическом контакте с источником питания. Нагревание осуществляется, согласно некоторым вариантам осуществления, за счет переключения тока через электроды 2330A, 2330B, через резистивный нагревательный элемент 2306. Электроды 2330A, 2330B расположены таким образом, что ток следует по пути, простирающемся по существу по всей по меньшей мере одной стороне (двум сторонам на изображенном примере) загрузки вещества дозирования, таким образом, что тепло может быть равномерно распределено по поверхности загрузки 2304 и подводится к ней.
Согласно некоторым вариантам осуществления поток воздуха проходит через воздухозаборник 2312, через нагретую пластину 2304 и выходит через выходное отверстие 2312B. Необязательно, выходное отверстие 2312B находится в сообщении по текучей среде с трубкой, которая направляется для доставки парообразного вещества пользователю. Необязательно, зажимные элементы 2310A, 2310B содержат части воздухозаборника 2312 и выхода 2312B. Потенциально это позволяет зажимным элементам 2310A, 2310B попеременно открываться для получения единицы дозирования 2300, а также закрываться для герметизации воздушного прохода вокруг единицы дозирования 2300, так что парообразное выделенное биологически активное средство(а) ограничивается определенным каналом.
После доставки дозы, выброс единицы дозирования включает разъединение приспособления для вытягивания дозы 2314 от защелки-челюсти 2302; например, путем перемещения одной из двух частей, при этом сдерживая другую и/или деформируя одну из двух частей. Например, приспособление для вытягивания 2314 дополнительно втягивается, в то время как следование за ним единицы дозирования 2300 предотвращается ограничением размера щели, через которую она движется. Согласно некоторым вариантам осуществления разъединение следует после выбрасывания: например, единица дозирования выпадает из гнезда, толкается возвращающимся действием приспособления для вытягивания дозы 2314 и/или иным образом транспортируется из устройства в целом. Согласно некоторым вариантам осуществления единица дозирования возвращается к карусели 2322 в качестве использованной дозы (в тот же или другой доступный слот, отличный от того, из которого она была извлечена). Необязательно, это выполняется через короткое время или сразу же по окончании использования. Альтернативно, единица дозирования выбрасывается в рамку следующего использования устройства, и в этом случае карусель также продвигает, представляя следующую единицу дозирования, которая будет использоваться.
Согласно некоторым вариантам осуществления доступ к дозам, загруженным в карусель, осуществляется последовательно в порядке их загрузки. Согласно некоторым вариантам осуществления порядок дозирования предопределен, но варьирует, например, дозирования различных количеств для введения в течение определенного периода времени расположены в таком порядке, как загружается карусель. Согласно некоторым вариантам осуществления движение карусели (продвижение) осуществляется по существу в соответствии с последовательностью действий, которые механически соединены с тянущими дозу и/или возвращающими дозу действиями. Согласно некоторым вариантам осуществления движение карусели находится под управлением контроллера, например, регулируемого микропроцессором шагового двигателя или другим механизмом продвижения. Необязательно, контроллер управляет той дозой, которая находится в слоте картриджа и/или ее статусом. Необязательно, контроллер автоматически и/или по команде выбирает соответствующую единицу дозирования и перемещает ее в положение посредством стольких шагов, сколько необходимо, чтобы сделать его доступным для вытягивания. Необязательно, этот выбор позволяет внеочередной доступ к единице дозирования в карусели. Необязательно, карусель продвигается в результате приведения в действие устройства пользователем.
Съемное устройство испарения и доставки:
На фиг. 4A-B схематически показано ингаляционное устройство для загрузки из карусели и отделимое от карусели для испарения и доставки выделенного биологически активного средства из единицы дозирования, в соответствии с некоторыми вариантами осуществления.
Согласно некоторым вариантам осуществления функции, выполняемые посредством аппарата зажимной камеры 2320, выполняются отделяемыми частями таким образом, что узел зажима/нагревания/введения отделим от частей узла вытягивания и транспортировки хранимой дозы по меньшей мере для введения дозы пользователю. Согласно некоторым вариантам осуществления узел зажима/нагревания/введения 2400 содержит по существу цилиндрический корпус (например, в форме сигареты, сигарильо, сигары и/или ручки), который вставляется в емкость узла вытягивания и транспортировки дозы. Узел 2400 содержит ползунковый механизм 2410 или другую структуру, которая включена в транспортный узел и/или активируется с помощью ручной или другой внешней операции.
Необязательно, ползунковый механизм 2410 выскальзывает из впускного конца 2440 узла 2400 для вовлечения единицы дозирования 2300 с соединительной частью 2415, как это описано, например, в связь с приспособлением для вытаскивания дозы 2314. Необязательно, единица дозирования 2300 (сформированная, например, с длинной и узкой пластиной 2404) втягивается в зажимный/нагревательный узел. Зажимный/нагревательный узел необязательно содержит электроды 2430, которые загружаются с пружинными элементами 2407 или другими средствами для прижимания резистивного нагревательного элемента 2306 для обеспечения электрического контакта с ним. Необязательно, мощность нагревания обеспечивается батареей 2413, соединенной с электродами 2430 по проводам 2414.
Необязательно, батарея 2413 является перезаряжаемой, например, батарея 2413 перезаряжается от источника, предоставленного основным корпусом, собранным вместе с каруселью. Необязательно, нагревание начинается после работы управления (например, кнопки) и/или подвергается одной или нескольким автоматическим активациям, модуляциям и/или средствам управления блокировкой, такими как нагревание при зондировании изменения давления и/или открытие шунта воздуха для управления скоростью и/или количеством доставки дозы. Во время доставки, воздух втягивается через тело 2420 (например, перорально путем ингаляции) путем применения всасывания к концу 2450. Воздух, втягиваемый во впускной конец 2440, принудительно направляется перегородками/трубопроводами 2401, 2408 для прохождения через нагретую пластину 2404, несущую парообразное биологически активное средство к концу 2450.
Потенциальное преимущество разъемной конструкции заключается в уменьшении усилия, необходимого пользователю для управления устройством дозирования во время введения дозы. Другим потенциальным преимуществом является разделение функций выбора доз, управления и контроля от самого дозирования. Существует потенциальный позитивный психологический эффект, обусловленный разделением акта дозирования, который приближается к нормальной электронной сигарете, от более клинических аспектов контроля дозирования.
Согласно некоторым вариантам осуществления съемная единица дозирования содержит множество отдельно нагреваемых областей; например, материал загружается в различные отверстия и/или отверстие пересекается множеством отдельно доступных нагревательных элементов. Необязательно, различные загрузки содержат различные выделенные биологически активные средства. Например, за загрузкой каннабиса необязательно следует одна или несколько загрузок выделенных каннабиноидов, таких как изолят ТНС, и/или загрузок различных каннабиноидов, таких как выделенный CBD.
Согласно некоторым вариантам осуществления логика аналоговой и/или цифровой схемы используется для управления тем, какая область нагревательного элемента получает ток. Например, каждый нагревательный элемент необязательно намеренно «сжигается» (посредством преломления предохранителя, например) после использования. Соответствующим образом организованная управляющая схема обнаруживает первую неиспользованную область дозирования и выбирает ее для следующей активации. Потенциальное преимущество этого заключается в том, чтобы позволить дозировке распространяться в несколько вдохов. Другим потенциальным преимуществом является возможность дозирования для одной цели (например, медицинского назначения) сочетаться с дозированиями для других целей (например, альтернативного медицинского назначения, или разрешить доступ дополнительным ингаляциям для рекреационных целей). Другим потенциальным преимуществом является возможность использования нескольких типов доз (например, композиций различных выделенных биологически активных средств) для целей придания разнообразия опыту пользователя.
На фиг. 5 схематически показан аппарат распыления защищенных блокировкой доз 2500 вместе со съемным узлом введения дозы 2400, в соответствии с некоторыми вариантами осуществления.
Согласно некоторым вариантам осуществления аппарат распыления 2500 включает в себя множество приемных отверстий 2501, 2502 для узла введения 2400. Согласно некоторым вариантам осуществления отверстие 2501 представляет собой отверстие, из которого неиспользованная единица дозирования 2300C, 2300A извлекается в узел введения 2400. Согласно некоторым вариантам осуществления после того, как единица дозирования 2300A извлекается из аппарата распыления 2500, следующая единица дозирования 2300C не продвигается в нужное положение до тех пор, пока условия, выполняемые блокировочным устройством, не будут выполнены. Согласно некоторым вариантам осуществления работа блокировочного устройства включает вставку узла введения 2500 в отверстие 2502. Необязательно, вставка запускает (например, с помощью активируемой механически и/или контроллером операции) перемещение карусели таким образом, что единица дозирования 2300C перемещается в положение. Согласно некоторым вариантам осуществления вставка (необязательно, вставка и удаление) узла введения 2400 извлекает единицу дозирования 2300A, которая теперь занимает прежнее положение использованной единицы дозирования 2400B. Потенциально, этот блокировочный механизм помогает гарантировать, что только одна единица дозирования в это же время удаляется из устройства распыления 2500. Согласно некоторым вариантам осуществления продвижение карусели не происходит, если единица дозирования 2300A не определяется внутри узла введения 2400 при вставке в отверстие 2502. Согласно некоторым вариантам осуществления единица дозирования 2300A вставляется в узел введения 2400 таким образом, что она не может быть удалена без разрушения единицы дозирования 2300A и/или узла введения 2400.
Ингаляционное устройство:
Единица дозирования, содержащая пластину одного или нескольких выделенных биологически активных средств, расположенных поверх материала-носителя, и выполненная с возможностью осуществления их испарения с помощью нагревательного элемента в соответствии с некоторыми вариантами осуществления, может быть использована в ингаляционном устройстве, выполненном с возможностью проведения электрического тока через нагревательный элемент и обеспечения прохождения воздуха через нагретую пластину для переноса парообразного средства в легкие пациента или пользователя. Ингаляционное устройство, использующее представленную в настоящем документе единицу дозирования, может представлять собой устройство для ингаляции отмеренных доз (MDI).
Согласно некоторым вариантам осуществления ингаляционное устройство выполнено с возможностью точной дозировки, пригодной для медицинских целей. Точная доза может быть осуществлена с использованием предварительно измеренного количества биологически активного средства(средств) и/или посредством управления профилем нагревания (например, температурой и/или скоростью нагревания, и/или продолжительностью), применяемого для парообразования и/или профиля потока (например, скорости потока и/или изменения скорости потока, и/или продолжительности). Согласно некоторым вариантам осуществления устройство выполнено с возможностью общего испарения биологически активного средства(средств) при слабых требованиях к точности.
Согласно некоторым вариантам осуществления ингаляционное устройство содержит по меньшей мере одну описанную в настоящем документе единицу дозирования и дополнительно содержит активирующую единицу, выполненную с возможностью перемещения единицы дозирования из положения хранения в рабочее положение и приведения в действие прохождения тока через нагревательный элемент, как описано выше.
В соответствии с некоторыми вариантами осуществления ингаляционное устройство дополнительно содержит аппарат распыления единицы дозирования, который содержит множество единиц дозирования.
В соответствии с некоторыми вариантами осуществления ингаляционное устройство дополнительно содержит аппарат зажимной камеры (также известный как испарительный аппарат), как описано выше.
В соответствии с некоторыми вариантами осуществления ингаляционное устройство представляет собой по существу устройство MDI, как описано в международной заявке на патент WO 2012/085919 и/или в любой из предварительных заявок на патент США № 62/035588, 62/085772 и 62/086208, включая в себя любой из описанных в нем вариантов осуществления и любые их комбинации.
В соответствии с одним аспектом некоторых вариантов осуществления предусмотрено устройство MDI, выполненное с возможностью легочной доставки предварительно определенного парообразного количества по меньшей мере одного биологически активного средства (фармакологически активного средства) пациенту, причем средство представляет собой выделенное биологически активное средство, а также:
устройство выполнено с возможностью доставки указанного предварительно определенного парообразного количества средства при контролируемом нагревании пластины, содержащей средство;
предварительно определенное парообразное количество выбирают таким образом, чтобы оно обеспечивало предварительно выбранный фармакокинетический профиль и/или предварительно выбранный фармакодинамический профиль средства у пациента; а также
предварительно определенное парообразное количество получают путем измерения по меньшей мере одного фармакокинетического (PK) параметра и/или по меньшей мере одного фармакодинамического (PD) параметра, индуцированного легочной доставкой средства пациенту из устройства MDI (PK/PD исследования). Такие PK/PD параметры, как правило, известны в настоящей области техники и обсуждаются в настоящем документе ниже, и могут быть измерены или оценены в соответствии с хорошо известными способами, называемыми в настоящем документе PK/PD исследованиями.
В соответствии с некоторыми вариантами осуществления устройство MDI выполнено с возможностью связи с электронным блоком интерфейса пациента и интеграции в систему, предназначенную для обеспечения сбора PK и/или PD (PK/PD) данных и ввода, хранения записей о пациенте, автоматической или ручной калибровки, корректировки, сброса и повторного определения начальной предварительной настройки устройства пациентом и/или практикующим врачом, как это будет подробно описано в настоящем документе ниже.
Вариабельность PK/PD среди когорты пациентов представляет собой особенно низкую для некоторых выделенных биологически активных средств, и может быть обеспечена за счет использования точного и последовательного устройства MDI, в соответствии с некоторыми вариантами осуществления настоящего раскрытия.
В соответствии с некоторыми вариантами осуществления представленные в настоящем документе способы и устройства также характеризуются высокой точностью, последовательностью, правильностью и воспроизводимостью результатов, которые выражаются минимальным отклонением между вдыхаемым пациентом фактическим парообразным количеством средства и предварительно определенным парообразным количеством средства.
В соответствии с некоторыми вариантами осуществления ингаляционное устройство для контролируемого испарения по меньшей мере одного активного фармацевтически активного средства по меньшей мере из одного вида вещества посредством применения нагревания включает в себя:
по меньшей мере одну единицу дозирования (картридж), содержащий пластину, которая содержит по меньшей мере одно выделенное биологически активное (фармацевтически активное) средство;
нагревательный элемент, приспособленный для применения нагревания к пластине для испарения фармацевтически активного средства; а также
механизм, приспособленный для перемещения картриджа относительно контроллера для питания нагревательного элемента.
В соответствии с некоторыми вариантами осуществления ингаляционное устройство выполнено с возможностью контролируемого испарения по меньшей мере одного активного фармацевтически активного средства по меньшей мере из одного вида вещества путем применения нагревания и воздушного потока и, соответственно, также содержит механизм, приспособленный для управления потоком воздуха через пластину.
Согласно некоторым вариантам осуществления устройство дополнительно содержит множество единиц дозирования, размещенных в ленте, гирлянде (карусели) или магазине. При необходимости или дополнительно, пластина организована с предварительно определенным количеством фармацевтически активного средства на единицу площади пластины в каждой единице дозирования в ленте, гирлянде или магазине.
При желании или дополнительно, толщина единицы дозирования варьирует в диапазоне от приблизительно 0,1 мм до приблизительно 2,0 мм. Необязательно или дополнительно, лента, карусель или магазин содержит в общей сложности от приблизительно 5 граммов до приблизительно 100 граммов загруженных пластин. Необязательно или дополнительно, лента, карусель или магазин содержит достаточное количество активного фармацевтически активного средства по меньшей мере для двух воздействий. Необязательно или дополнительно, картридж содержит первый слой материала, соединенный с пластиной, первый слой, содержащий отверстия, достаточно большие, чтобы позволить удаление газов, но достаточно малые, чтобы содержать подогреваемый материал пластины. Необязательно или дополнительно, диаметр отверстий составляет от 25 мкм до 500 мкм. Необязательно или дополнительно, картридж содержит второй слой материала, соединенный с пластиной, а второй слой выполнен с возможностью передачи тепла к пластине без существенного распределения тепла по второму слою. Необязательно или дополнительно, нагревательный элемент и пластина удерживаются между первым и вторым слоями.
Согласно некоторым вариантам осуществления устройство дополнительно содержит ингаляционную единицу, содержащую мундштук для ингаляции фармацевтически активного средства, причем мундштук сообщается по текучей среде с испарительной камерой устройства, причем испарительная камера содержит парообразное активное фармацевтически активное средство.
Необязательно, мундштук содержит односторонний клапан для регулирования потока жидкости от испарительной камеры. Необязательно или дополнительно, устройство дополнительно содержит датчик, сообщающийся по текучей среде с мундштуком, при этом датчик приспособлен для оценки скорости потока воздуха и посылает сигнал на контроллер, приспособленный для испарения фармацевтически активного средства в соответствии со скоростью воздушного потока.
Согласно некоторым вариантам осуществления устройство дополнительно содержит контроллер, выполненный с возможностью синхронизировать приложение тепла с движением картриджа и/или со скоростью воздушного потока, осуществляемого путем ингаляции.
Согласно некоторым вариантам осуществления устройство дополнительно содержит электронный блок для управления (контроллер) активацией нагревательного элемента.
Согласно некоторым вариантам осуществления устройство дополнительно содержит интерфейс связи для связи с одним или несколькими внешними компьютерами и/или системами и/или интерфейсами пациента/врача.
Согласно некоторым вариантам осуществления устройство дополнительно содержит или связано с индикатором дозы для обеспечения визуального вывода испарения фармацевтически активного средства.
Согласно некоторым вариантам осуществления устройство является портативным и весит менее чем приблизительно 300 г.
Согласно некоторым вариантам осуществления устройство дополнительно содержит или связано с запоминающим устройством, приспособленным для хранения по меньшей мере одних из данных о назначении и данных об использовании, при этом запоминающее устройство соединено с контроллером, приспособленным к управлению по меньшей мере одним из нагревательного элемента, воздушного потока и транспортного механизма в соответствии с дозой и/или данными о схеме лечения.
Согласно некоторым вариантам осуществления устройство дополнительно содержит уникальный идентификатор, приспособленный для отслеживания использования устройства связанным с ним пациентом.
Согласно некоторым вариантам осуществления устройство дополнительно содержит датчик, приспособленный для обнаружения физического нарушения устройства.
В соответствии с некоторыми вариантами осуществления предусмотрен способ контролируемого испарения активного фармацевтически активного средства с пластины, организованной в виде картриджа (единицы дозирования), причем указанный способ включает:
подвод тепла к области картриджа для испарения предварительно определенного количества активного фармацевтически активного средства и
перемещение картриджа относительно источника тепла.
Альтернативно, нагревательный элемент содержится внутри картриджа, и картридж перемещается относительно электрических контактов для питания нагревательного элемента.
Согласно некоторым вариантам осуществления способ дополнительно включает корректировку по меньшей мере одного из времени и скорости перемещения для испарения активного фармацевтически активного средства в соответствии с профилем доставки.
Согласно некоторым вариантам осуществления испарение включает испарение во время легочной доставки.
Согласно некоторым вариантам осуществления применение нагревания включает применение нагревания, чтобы достичь заданной температуры менее чем за 500 миллисекунд после сигнала запуска.
В соответствии с некоторыми вариантами осуществления предусмотрен способ контролируемого испарения по меньшей мере одного выделенного активного фармацевтически активного средства по меньшей мере из одного типа пластины путем применения нагревания, причем способ включает:
нагревание одной или нескольких площадей одной или нескольких пластин, организованных в одном или нескольких картриджах с одним пользовательским включателем для высвобождения по меньшей мере одного активного фармацевтически активного средства. Необязательно, площади содержат различные выделенные активные фармацевтически активные средства.
В соответствии с некоторыми вариантами осуществления предусмотрен способ изготовления картриджа, содержащего пластину, содержащую выделенное активное фармацевтически активное средство, причем картридж приспособлен для использования с устройством для автоматического применения локализованного нагревания для испарения фармацевтически активного средства, причем способ включает:
нанесение по меньшей мере одного, необязательно, предварительно отмеренного количества выделенного фармацевтически активного средства в материал пластины и/или на него;
при необходимости, измерение количества фармацевтически активных средств, присутствующих в единице массы загруженного материала пластины и/или на ней, а также
прессование пластины в картридж.
Необязательно, измерение количества фармацевтически активного средства включает в себя одно или несколько из непосредственного измерения фармацевтически активного средства и взвешивания количества материала, содержащего фармацевтически активное средство.
Согласно некоторым вариантам осуществления прессование материала пластины с загруженными частицами выполняют в картридже с отверстиями с размером меньшим, чем размер частиц.
Согласно некоторым вариантам осуществления способ дополнительно включает маркировку картриджа с предварительно определенным количеством активного фармацевтически активного средства.
В соответствии с некоторыми вариантами осуществления предусмотрен картридж для терапевтической доставки лекарственного средства, содержащий пластину с загруженным выделенным активным фармацевтически активным средством, причем указанную пластину загружают предварительно определенным количеством фармацевтически активного средства на единицу площади поверхности картриджа, и нагревательный элемент.
Согласно некоторым вариантам осуществления множество картриджей организовано в виде рулона ленты, карусели (ромашки) или магазина.
Контролируемое высвобождение и доставка:
В соответствии с аспектом некоторых вариантов осуществления предусмотрен способ контролируемого высвобождения по меньшей мере одного выделенного биологически активного средства с использованием описанного в настоящем документе ингаляционного устройства.
В соответствии с некоторыми вариантами осуществления способ осуществляют с использованием MDI, который способен доставлять воспроизводимо и точно, путем легочной ингаляции, некоторое количество по меньшей мере одного испаряемого средства посредством нагревания пластины, содержащей испаряемое средство, в соответствии с некоторыми вариантами осуществления, причем испарение средства эффективно, и указанное парообразное средство вдыхается пользователем. Такие требования к MDI удовлетворяются, для неограничивающего примера, таким MDI, который описан в международной заявке на патент № WO 2012/085919 и в любой из предварительных заявок на патент США № 62/035588, 62/085772 и/или 62/086208, которые полностью включены в настоящий документ посредством ссылки, как если бы были полностью изложены в настоящем документе.
Регулируемость достигается посредством одного или нескольких из регулирования количества выделенного биологически активного средства(средств) в единице дозирования, регулирования уровня нагрева подаваемого на единицу дозирования, регулирования протекающего через нагревательный элемент тока и/или его продолжительности и регулирования конфигурации и/или потока воздуха через воздушные каналы в устройстве, которое может иногда обеспечить полное вдыхание всего объема, который включает в себя парообразное количество биологически активного средства.
Регулируемость парообразного количества биологически активного средства в его выделенной форме обеспечивает, например, способы для использования биологически активного средства в качестве фармацевтического средства (лекарственного средства; медикамента) с известными и по существу предсказуемыми и воспроизводимыми фармакологическими параметрами, такими как фармакокинетический (PK), фармакодинамический (PD) профиль, что позволяет достигать желаемой схемы лечения, чтобы соответствовать известному и по существу предсказуемому и воспроизводимому терапевтическому окну. Таким образом, в соответствии с некоторыми вариантами осуществления способ контролируемого высвобождения посредством испарения и легочной доставки путем ингаляции предварительно определенного парообразного количества по меньшей мере одного представленного в настоящем документе выделенного биологически активного средства осуществляется таким образом, что предварительно определенное парообразное количество выбирают таким образом, чтобы проявлять предварительно выбранный фармакокинетический профиль и/или предварительно выбранный фармакодинамический профиль средства в организме пациента.
Используемые в настоящем документе термины «терапевтическое окно» и «фармацевтическое окно» являются взаимозаменяемыми и относятся к диапазону фармакодинамических эффектов, вызванных диапазоном доз одного или нескольких фармацевтически активных средств, обеспечивая баланс между одним или несколькими желательными (положительными) эффектами и одним или несколькими неблагоприятными (отрицательными) эффектами. В соответствии с некоторыми вариантами осуществления фармацевтическое/терапевтическое окно относится к фармакодинамическому профилю. Окно может иметь отношение к данному моменту времени или может охватывать период времени любой продолжительности, включая в себя, например, минуты, часы, дни или более длинные, более короткие или любой промежуточный период времени. Желательность и нежелательность эффекта может быть определена на основе различных критериев и включать в себя без ограничения медицинскую практику, правила и нормы, культурные и демографические нормы, генетические факторы и личные предпочтения и допуски. Например, желательность и нежелательность эффекта может быть определена, основываясь на цели лечения и основываясь на общепринятых значениях и, возможно, может принимать во внимание другие параметры, такие как предпочтение, способность и активность пациента. Следует отметить, что данный эффект можно рассматривать как желаемый в некоторых случаях, но можно рассматривать как нежелательный в других случаях, и наоборот.
Следует отметить в настоящем документе, что согласно некоторым вариантам осуществления путем проявления предварительно выбранного фармакокинетического и/или фармакодинамического профиля, это означает, что парообразное количество выделенного биологически активного средства было предварительно определено на основе фармакокинетических/фармакодинамических (PK/PD) исследований, проводимых в соответствии с хорошо установленной практикой и приемлемыми стандартами по меньшей мере у одного субъекта, посредством легочной доставки субъекту средства с использованием устройства MDI, которое выполнено с возможностью высвобождать точное и адекватное парообразное количество средства при нагревании содержащей его пластины, как описано в настоящем документе. Также в настоящем документе отмечено, что в соответствии с некоторыми вариантами осуществления путем проявления предварительно выбранного фармакокинетического профиля, это означает, что был идентифицирован по меньшей мере один желаемый фармакокинетический профиль, и что было показано, что по меньшей мере одно предварительно определенное парообразное количество выделенного биологически активного средства вызывает желаемый фармакокинетический профиль у субъекта. Также в настоящем документе отмечено, что в соответствии с некоторыми вариантами осуществления путем проявления предварительно выбранного фармакокинетического профиля, это означает, что по меньшей мере один желаемый фармакодинамический профиль был идентифицирован и что было показано, что по меньшей мере одно предварительно определенное парообразное количество выделенного биологически активного средства вызывает желаемый фармакодинамический профиль у субъекта воспроизводимым образом. Следует также отметить, что для некоторых выделенных биологически активные средств их проглатывание и/или его инъецирование случайным субъектом приводит к непредсказуемым и/или непоследовательным, и/или нерабочим значениям фармакокинетических параметров, для которых предусмотренный в настоящем документе способ может обеспечить весьма выгодное решение.
Согласно некоторым вариантам осуществления термины «предварительно выбранный» и «предварительно определенный» относятся к или используются взаимозаменяемо с терминами «предназначенный», « целенаправленный», «желаемый» или «желательный» или с терминами «эффективный», «необходимый» и «терапевтический».
Следует также отметить, что в настоящем документе определение желаемого фармакокинетического профиля и/или желаемого фармакодинамического профиля, как правило, выполняют путем проведения PK/PD исследования для конкретного фармацевтически активного средства у данного субъекта или их группы. Следует также отметить в настоящем документе, что способность проводить стандартные и широко признанные исследования PK/PD у определенного субъекта или их группы для фармацевтически активного средства, которое доставляется посредством ингаляции (легочной доставки) при управляемом и воспроизводимом высвобождении парообразного количества выделенного биологически активного средства путем нагревания содержащей его пластины, становится возможным, например, посредством устройства MDI, например, как раскрыто в настоящем документе и в международной заявке на патент № WO 2012/085919 и в любой из предварительных заявок на патент США № 62/035588, 62/085772 и/или 62/086208, все из которых полностью включены в настоящий документ посредством ссылки.
Согласно некоторым вариантам осуществления термин «предварительно определенное парообразное количество» также используется в настоящем документе для описания количества средства, которое определяется на основании данных, указывающих на фармакокинетический параметр и/или фармакодинамический параметр, а именно, парообразного количества, которое было определено путем мониторинга и/или записи, и/или приема, и/или анализа, и/или определения по меньшей мере одного PK параметра и/или PD параметра, который/которые индуцируются данным средством у одного или нескольких субъектов/пациентов.
Согласно некоторым вариантам осуществления настройка устройства MDI для высвобождения предварительно определенного количества означает калибровку устройства для проявления предварительно выбранного PK и/или предварительно выбранного PD профиля. Регулируемое, точное и воспроизводимое высвобождение выделенного биологически активного средства из единицы дозирования, представленной в настоящем документе, может позволить калибровку устройства MDI, как это предусмотрено в настоящем документе.
В соответствии с некоторыми вариантами осуществления способ осуществляют путем определения по меньшей мере одного фармакокинетического параметра (также называемого взаимозаменяемо в настоящем документе как фармакокинетический эффект) и/или по меньшей мере одной фармакокинетической переменной и/или по меньшей мере одного фармакодинамического параметра (также взаимозаменяемо называемого в настоящем документе как фармакодинамический эффект), поскольку эти термины известны в настоящей области техники, которые индуцируются легочной доставкой парообразного количества биологически активного средства пациенту с использованием устройства MDI:
на основе фармакокинетического параметра и/или фармакокинетической переменной, и/или фармакодинамического параметра, определение предварительно определенного парообразного количества, которое проявляет предварительно выбранный фармакокинетический профиль и/или предварительно выбранный фармакодинамический профиль средства у пациента, а также
корректировку/повторную корректировку/конфигурацию устройства MDI для доставки предварительно определенного парообразного количества средства.
Используемая в настоящем документе фраза «фармакокинетический профиль» относится к концентрации в организме фармацевтически активного средства или его метаболита (например, активного метаболита), а именно, концентрации средства или его метаболита в физиологической системе организма (всем организме, крови, плазме, лимфе, ткани, органе и т.п.), в который вводилось соединение, как функция времени. Как правило, фармакокинетический (РК) профиль рассматривается с момента времени введения соединения до момента времени, в который соединение больше не обнаруживается в организме, или до любого промежуточного периода времени; следовательно, PK профиль может описывать концентрацию в организме в конкретной физиологической системе конкретного соединения между введением и исчезновением под влиянием механизмов высвобождения, поглощения, распределения, метаболизма и выведения/секреции соединения. Так как каждый организм и каждый отдельный организм в пределах рода организма по-разному реагирует на введение средства, PK профиль может быть различным, а в некоторых случаях сильно изменяющимся от субъекта к субъекту, и может отличаться в пределах отдельного субъекта на основе текущего физиологического состояния, состояния здоровья, условий окружающей среды и даже от времени суток.
В соответствии с некоторыми вариантами осуществления фармакокинетический профиль достигается путем обеспечения субъекта одним или несколькими из следующего:
Доза - одно количество соединения или средства, которое ему вводится;
Дозирование - множество предварительно определенных доз, которые могут быть различными или аналогичными по количествам и/или
Схема лечения - дозирование, вводимое в различные интервалы времени, которые могут быть разными или схожими с точки зрения продолжительности. Согласно некоторым вариантам осуществления схема лечения также охватывает время периода доставки (например, период введения средства или период лечения).
Альтернативно, схема лечения представляет собой множество предварительно определенного множества предварительно определенных парообразных количеств, даваемых в предварительно определенные промежутки времени.
Следует отметить, что PK профиль может быть определен в соответствии с изменением PK параметра в зависимости от времени или комбинации PK параметров в зависимости от функции времени. PK профиль, как правило, оценивают по концентрации на временной шкале, используя непосредственные и/или опосредованные измеряемые PK эффекты. Например, PK профиль может представлять собой концентрацию в плазме данного фармацевтически активного средства у субъекта, как функцию времени.
Используемый в настоящем документе термин «предварительно выбранный фармакокинетический профиль» относится к PK профилю, который был выбран в качестве желательного. Предварительно выбранный PK профиль может быть выбран, так как было установлено что он, эффективен в достижении желаемого фармакодинамического эффекта у субъекта, как описано в любом из соответствующих вариантов осуществления (например, для поддержания субъекта в пределах терапевтического окна, как описано в настоящем документе).
PK параметры, как правило, включают в себя без ограничения:
Ct, что представляет собой концентрацию средства, как определено, измерено или оценено в определенной физиологической системе (например, в плазме) после его введения (доставки, например, легочной доставки) дозы или схемы лечения субъекту;
Cmax, что представляет собой пиковую концентрацию средства, как определено, измерено или оценено в определенной физиологической системе (как правило, в плазме), после его введения субъекту;
Tmax, что представляет собой время, прошедшее между введением и достижением Cmax;
Площадь под кривой (AUC0→∞; от нуля до бесконечности), которая представляет собой интеграл кривой концентрации в зависимости от времени, как правило, после однократной дозы или в стационарном состоянии;
Cmin, что представляет собой самую низкую концентрацию средства в организме до очередной вводимой дозы;
Tmin, что представляет собой время, прошедшее до обнаружения Cmin или до введения следующей дозы;
Cпоследняя, что представляет собой последнюю наблюдаемую количественную концентрацию;
λz, что представляет собой конечную константу фазы скорости;
Период полувыведения (t½), который представляет собой время, необходимое для того, чтобы концентрация средства достигла половины любого выбранного значения;
Константа скорости элиминации (kE), которая представляет собой скорость, при которой средство удаляется из организма;
Скорость введения (kin), которая представляет собой скорость введения необходимого баланса элиминации;
Клиренс, который представляет собой объем плазмы, очищенный от средства, на единицу времени;
Биодоступность, которая представляет собой системно доступную фракцию средства; а также
Колебание, которое представляет собой пик колебания в пределах одного интервала дозирования в стационарном состоянии.
В качестве инструмента оценки PK профиля у представителя популяции подобных отдельных субъектов (сходных в биологическом смысле, как и в группе людей), PK переменные, которые, как было обнаружено, коррелируют с PK профилем в подмножестве популяции, могут быть использованы для обобщения (экстраполирования) PK профиля для каждого из индивидуумов, входящих в состав целой популяции. Фармакокинетические переменные, как правило, включают в себя без ограничения массу тела, рост, индекс массы тела (ИМТ), индекс талия-бедро, безжировую массу тела (LBM), возраст, расу, сопутствующие заболевания, историю болезни пациента, сопутствующие лекарственные средства и пол. Следует понимать, что PK переменные зависят от генетической и эпигенетической композиции каждого отдельного субъекта и, следовательно, могут быть использованы для прогнозирования PK/PD профилей у отдельного субъекта с определенной степенью точности; тем не менее, персонализация/индивидуализация лечения, основанная на введении фармацевтически активного средства, как правило, основана на персональных PK/PD параметрах (данных), определенных для отдельного субъекта. В целом, отклонение отдельных параметров от средних параметров, установленных для широкой популяции, заметно мала.
В контексте некоторых вариантов осуществления термин «лечение» относится к одному легочному введению средства в данной дозе; фиксированной и ограниченной серии легочных введений средства (дозированию), получаемым при одних и тех же или разных дозах в одни и те же или различных дозовые интервалы (схемы лечения); или хроническому лечению, которое вводят в виде ограниченных серий, но без запланированного прекращения лечения (непрерывное лечение). Как правило, серия предварительно определенных доз, вводимых через предварительно определенные промежутки времени, называется в настоящем документе как схема лечения или схема.
В соответствии с некоторыми вариантами осуществления предусмотренная в настоящем документе единица дозирования представляет собой физический вариант осуществления разовой дозы, которая используется в одном сеансе ингаляции.
В соответствии с некоторыми вариантами осуществления представленного в настоящем документе способа легочная доставка выделенного биологически активного средства включает одну дозу, доставленную как одно предварительно определенное парообразное количество, высвобожденное устройством MDI в одном сеансе ингаляции, или дозу можно вводить пациенту в виде нескольких сопутствующих ингаляций. Альтернативно, серия доз, каждая вводимая в виде одной или нескольких предварительно определенных парообразных количеств, которая называется в настоящем документе как дозирование и вводится в предварительно определенные промежутки времени, относится в настоящем документе к схеме лечения. Схема лечения, следовательно, определяется одной или несколькими дозами, вводимыми в одном или нескольких предварительно определенных парообразных количествах (дозирование) в предварительно определенные интервалы времени, причем каждое из предварительно определенных парообразных количеств, доз и интервалов дозирования может быть одинаковым или различным.
В контексте вариантов осуществления PK профиль данного фармацевтически активного средства представляет собой результат дозы, дозирования и/или схемы лечения, с помощью которой средство вводят пациенту, или, альтернативно, в соответствии с некоторыми вариантами осуществления PK профиль представляет собой средство для достижения конкретного, предварительно выбранного или иного желаемого фармакодинамического профиля средства у пациента.
Используемый в настоящем документе термин «фармакодинамический профиль» относится к эффекту фармацевтически активного средства у субъекта, как функция от времени. Соответственно, термин «фармакодинамический профиль» относится к сумме всех биологических выражений и реакций организма, как функции от времени, при введении фармацевтически активного средства. Фармакодинамическим профиль, как правило, представляет собой прямой или косвенный результат фармакокинетического эффекта(ов) в любой данный момент времени либо фармакокинетический профиль средства в организме пациента в течение любого заданного периода времени.
Фармакодинамический профиль представляет собой изменение/вариацию прямо и/или косвенно определяемого фармакодинамических эффектов как функция от времени.
Фармакодинамические эффекты, как правило, могут быть определены без ограничения посредством желательного (терапевтического) эффекта (например, субъективно воспринимаемого терапевтического эффекта), нежелательного (неблагоприятного) эффекта (например, субъективно воспринимаемого неблагоприятного эффекта), а также с помощью определения содержания биомаркера (который представляет собой показатель желательного и/или нежелательного эффекта), так как эти термины описаны ниже. Фармакодинамический профиль, который может представлять собой предварительно выбранный (желательный) фармакодинамический профиль, в соответствии с некоторыми вариантами осуществления, определяется терапевтическим окном данного средства у данного субъекта, как этот термин определен в настоящем документе.
Фармакодинамический (PD) профиль, как правило, представляет собой зависимую от времени оценку и/или измерение по шкале, идущей от отсутствия ответа, через начало желаемого терапевтического эффекта (ниже порогового значения терапевтического эффекта), через терапевтическое окно, через начало неблагоприятного эффекта (выше порогового значения неблагоприятного эффект) и вплоть до токсического эффекта. Потенциальное преимущество единицы дозирования, устройства и способов, представленных в настоящем документе, заключается в реализации возможности практического введения посредством ингаляции определенных выделенных биологически активных средств, что способствует более точной и воспроизводимой оценке PD параметров у любого данного субъекта, по сравнению с оценкой параметров PD при введении одного и того же средства путем проглатывания и/или инъекции, в связи с низкой биодоступностью, связанной с гидрофобностью, вязкостью и другими специфическими для средства свойствами, как обсуждалось выше.
Результаты такого PK/PD исследования, проведенные с использованием единицы дозирования, устройств и способов, предусмотренных в настоящем документе, у одного или нескольких субъектов, могут быть поэтому использованы для определения начального предварительно определенного парообразного количества по меньшей мере одного фармакологически активного средства, которое сразу при введении с помощью устройства MDI, настроенного для его легочной доставки, приводило бы к начальному предварительно выбранному фармакокинетическому профилю и/или начальному предварительно выбранному фармакодинамическому профилю средства у конкретного пациента, и в дальнейшем могло быть использовано для калибровки и предварительной установки аналогичных устройств MDI таким образом, чтобы доставить начальное предварительно определенное парообразное количество для достижения аналогичных последовательных первоначальных результатов.
Следует отметить, что в соответствии с некоторыми вариантами осуществления, в то время как пациент/пользователь может начать легочную доставку с использованием начального предварительно определенного парообразного количества, которое не было определено на основании личных/индивидуальных параметров и переменных пациента, предусмотренный в настоящем документе способ включает дополнительный шаг, на котором личные параметры и переменные пациента рассматриваются в определении предварительно определенного парообразного количества. Таким образом, в соответствии с некоторыми вариантами осуществления способ может включать персонализацию предварительно определенного парообразного количества, которая приводит к получению предварительно выбранного PK/PD профиля. Представленная ниже стадия персонализации может заменить предварительную калибровку устройства MDI или выступать в качестве дополнительной стадии после калибровки устройства MDI.
Способ лечения:
Предусмотренная в настоящем документе единица дозирования, используемая в устройстве MDI, выполненного с возможностью воспроизводимой и точной доставки терапевтического количества выделенного биологически активного средства или их комбинации, может быть использована для лечения патологических состояний, которые поддаются лечению с помощью биологически активного средства, которое предпочтительно вводят с помощью легочной доставки (ингаляции). Такой способ лечения может быть предпочтительным по сравнению с другими способами, особенно когда лечение проводится с использованием выделенного биологически активного средства, которое трудно вводить с помощью других режимов введения, или когда их введение с помощью других режимов является неэффективным.
В соответствии с аспектом некоторых вариантов осуществления предусмотрен способ лечения пациента, страдающего от патологического состояния, которое поддается лечению путем легочной доставки (ингаляции) по меньшей мере одного предварительно определенного парообразного количества по меньшей мере одного выделенного биологически активного средства.
Способ, в соответствии с некоторыми вариантами осуществления, выполняют посредством легочной доставки пациенту свободно вдыхаемых паров выделенного биологически активного средства из устройства для ингаляции отмеренных доз, выполненного с возможностью контролируемого высвобождения по меньшей мере одного предварительно определенного парообразного количество средства при контролируемом нагревании содержащей средство пластины.
Согласно некоторым вариантам осуществления предварительно определенное парообразное количество средства выбирают таким образом, чтобы проявлять по меньшей мере один предварительно выбранный фармакокинетический профиль и/или по меньшей мере один предварительно выбранный фармакодинамический профиль средства у пациента.
Согласно некоторым вариантам осуществления выделенное биологически активное средство представляет собой такой выделенный каннабиноид, как без ограничения Δ9-тетрагидроканнабинол (ТНС), дронабинол ((-)-транс-ТНС), каннабидиол (CBD), каннабигеролы (CBG), каннабихромены (CBC), каннабинол (CBN), каннабинодиол (CBDL), каннабициклол (CBL), каннабиэлсоин (CBE), каннабидиварин (CBDV), тетрагидроканнабиварин (THCV), каннабитриол (CBT) и любая их комбинация. Другие выделенные и испаряемые биологически активные средства рассматриваются в контексте некоторых аспектов и вариантов осуществления настоящего раскрытия.
Согласно некоторым вариантам осуществления выделенное биологически активное средство содержит каннабиноид и терпен и/или флавоноид.
Неограничивающие репрезентативные патологические состояния, подвергаемые лечению посредством легочной доставки испаряемого выделенного биологически активного средства, такого как каннабиноиды, необязательно, с терпенами и/или, необязательно, флавиноидами включают в себя без ограничения злоупотребление алкоголем, боковой амиотрофический склероз, нервную анорексию, тревожные расстройства, колебания аппетита, бронхиальную астму, атеросклероз, биполярное расстройство, нарушение функции мочевого пузыря, хроническую обструктивную болезнь легких (COPD), коллаген-индуцированный артрит, колоректальную злокачественную опухоль, болезнь Крона, делирию, заболевания органов пищеварения, синдром Драве, наркотическую зависимость и зависимость, дистонию, эпилепсию, фибромиалгию, генерализованную эпилепсию с фебрильными судорогами плюс (GEFS+), глаукому, глиому, гепатит С, ВИЧ-ассоциированную сенсорную невропатийную депрессию, болезнь Хантингтона, повышенное давление, повышенное внутриглазное давление, воспалительное заболевание кишечника (IBD), бессонницу, синдром раздраженного кишечника (IBS), отсутствие аппетита, лейкоз, мигрень, двигательные расстройства, рассеянный склероз (MS), тошноту, нейрогенную боль, невропатическую боль, ноцицептивную боль, болезнь Паркинсона, фантомную боль, посттравматическое стрессовое расстройство (PTSD), предменструальный синдром, кожный зуд, психические расстройства, психогенную боль (психалгию или соматоформную боль), судороги, септический и кардиогенный шок, сексуальную дисфункцию, опухоли кожи, апноэ сна, спастичность, повреждение спинного мозга, тики, симптомы Туретта, тремор, непреднамеренную потерю веса и рвоту.
В соответствии с некоторыми вариантами осуществления способ выполняется путем использования устройства MDI, которое выполнено с возможностью высвобождения предварительно определенного парообразного количества таким образом, что отклонение фактического парообразного количества выделенного биологически активного средства от предварительно определенного парообразного количества средства составляет 20% или менее, 15% или менее, 10% или менее или 5% или менее предварительно определенного парообразного количества.
В соответствии с некоторыми вариантами осуществления способ осуществляют таким образом, что отклонение фактического фармакокинетического профиля от предварительно выбранного фармакокинетического профиля составляет 40% или менее, чем предварительно выбранный фармакокинетический профиль. Альтернативно, отклонение составляет 35% или менее, 30% или менее, 25% или менее или 20% или менее. Следует отметить, что отклонение может быть в фармакокинетическом профиле или в одном или нескольких составляющих профиль фармакокинетических параметрах, например, Ct или Cmax. Такие отклонения, как ожидается, будут низкими даже для выделенных биологически активных средств, для которых было обнаружено, что их проглатывание и инъекция неблагоприятны, из-за низкой вариабельности между PK параметрами, полученными при использовании предусмотренной в настоящем документе единицы дозирования в правильном, последовательном и точном устройстве MDI.
В соответствии с некоторыми вариантами осуществления способ осуществляют таким образом, что отклонение между воспринимаемым PD профилем и предварительно выбранным PD профилем в любой данный момент времени составляет 25% или менее, 20% или менее, 15% или менее, 10% или менее или 5% или менее. Отклонение между воспринимаемым PD профилем и предварительно выбранным PD профилем в любой данный момент времени может быть оценено путем определения PD параметра, как описано выше. Отклонение, как ожидается, будет низким также из-за низкой вариабельности между описанными выше PK параметрами.
Так как устройство может быть выполнено с возможностью последовательной доставки любого точного количества таким образом, чтобы проявлять любой предварительно выбранный PD профиль у пациента, предусмотренные в настоящем документе устройство и способ могут осуществлять предварительно выбранный PD профиль, который можно тонко регулировать таким образом, чтобы быть:
в пределах уровня ниже, чем минимальный уровень терапевтического эффекта (ниже терапевтического окна);
в пределах от минимального уровня указанного терапевтического эффекта до максимального уровня указанного терапевтического эффекта, в котором неблагоприятный эффект терпимый и/или приемлемый, а именно, по существу низкий или не проявляемый или не воспринимаемый (в пределах терапевтического окна); а также
в пределах уровня выше, чем минимальный уровень нежелательного эффекта (выше терапевтического окна).
Как обсуждалось выше, в соответствии с некоторыми вариантами осуществления предварительно выбранный PD профиль соответствует терапевтическому окну средства у пациента, а именно, находится в диапазоне в пределах от минимального уровня желательного эффекта до уровня нежелательного эффекта.
Согласно некоторым вариантам осуществления предварительно выбранный PD профиль варьирует в диапазоне от минимального уровня желательного эффекта до минимального уровня нежелательного эффекта.
Согласно некоторым вариантам осуществления предварительно выбранный PD профиль варьирует в диапазоне от минимального уровня желательного эффекта до уровня выше, чем минимальный уровень нежелательного эффекта.
Согласно некоторым вариантам осуществления предварительно выбранный PD профиль варьирует в диапазоне от минимального уровня терапевтического эффекта до максимального уровня терапевтического эффекта, в котором неблагоприятный эффект является приемлемым.
В любом предварительно выбранном PD профиле, способ и устройство обеспечивают высокую точность и воспроизводимость результатов; следовательно, в соответствии с некоторыми вариантами осуществления отклонение воспринимаемого фармакодинамического профиля от предварительно выбранного фармакодинамического профиля в любой данный момент времени составляет 25% или менее, 20% или менее, 10% или менее или 5% или менее ниже предварительно выбранного PD профиля, и/или 25% или менее, 20% или менее, 10% или менее или 5% или менее выше указанного предварительно выбранного PD профиля.
Не ограничивающий пример патологического состояния, которое можно лечить легочной доставкой испаряемого фармацевтически активного средства, представляют собой боль, которая поддается лечению с помощью легочной доставки дронабинола, испаряемого из представленной в настоящем документе единицы дозирования.
Интерфейс и система:
Предусмотренные в настоящем документе единица дозирования (картридж), ингаляционное устройство и способы весьма подходят для персонализации, самостоятельного подбора дозы, механизации и автоматизации в противном случае сложного и затруднительного способа введения и лечения различных патологических состояний, которые поддаются лечению при ингаляции одного или нескольких биологически активных средств; в то время как любой персонализированный протокол лечения в соответствии с фармацевтическими правилами и требованиями представляет собой проблемы, воспроизводимое и контролируемое лечение, основанное на легочной доставке активных средств, выпаренных под воздействием тепла, представляет собой нетривиальную задачу по любым меркам.
После того, как проблема точности, согласованности и воспроизводимости результатов была решены, как это было сделано, например, в раскрытом в настоящем документе устройстве MDI и в WO/2012/085919, и так как была выполнена необходимость в калибровке и предварительной установке устройства, чтобы оставаться в пределах желаемого терапевтического окна, основанного на широко принятых экспериментальных PK/PD параметрах, авторы настоящего изобретения задумали интегрированную систему, которая может управлять устройством для легочной доставки выделенных биологически активных средств с использованием вводных данных, собранных из различных источников, с тем, чтобы обеспечить персонализированное и эффективное лечение для любого данного пациента, а также в режиме реального времени.
Фиг. 9 представляет собой принципиальную схему системы, содержащей устройство MDI, интерфейс врача и/или интерфейс пациента, в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Согласно некоторым вариантам осуществления устройство MDI 901 выполнено с возможностью связи с интерфейсом врача 903 и/или с интерфейсом пациента 905. Согласно некоторым вариантам осуществления устройство MDI 901 выполнено с возможностью приема входных данных от одного или обоих интерфейсов 903 и/или 905. Дополнительно или альтернативно, устройство MDI 901 выполнено с возможностью посылать выходные данные одному или обоим интерфейсам 903 и/или 907.
Согласно некоторым вариантам осуществления связь между компонентами системы осуществляется с помощью одного или нескольких средств передачи данных, таких как соединение USB, кабельное соединение, беспроводное соединение и/или любой подходящий проводной и/или беспроводной протокол связи.
Согласно некоторым вариантам осуществления связь между компонентами системы осуществляется через один или несколько коммуникационных модулей, таких как модуль связи 907 устройства MDI 901, модуль связи 909 интерфейса врача 903 и/или модуль связи 911 интерфейса пациента 905.
Согласно некоторым вариантам осуществления устройство MDI 901 содержит контроллер 913, выполненный с возможностью, например, активации нагревания пластины, тем самым испаряя активное средство, регулировки профиля нагревания и/или активации нагревания, регулировки механизма подачи картриджа устройства MDI, считывания данных из запоминающего устройства 919 устройства MDI 901, управления потреблением энергии и/или другими функциями. Согласно некоторым вариантам осуществления контроллер 913 осуществляет связь с запоминающим устройством 919. Дополнительно, запоминающее устройство 919 выполнено с возможностью хранения рецептурных данных, персональных данных об использовании, данных о пациенте, персональных PD параметров, полученных от пациента, модификаций дозы, дозирования и/или схемы лечения, параметров, полученных от пациента в ответ на изменение в дозе и/или схеме лечения и/или других ценностей или информации. Согласно некоторым вариантам осуществления контроллер 913 активирует легочную доставку активного средства в соответствии с данными о дозе, дозировании и/или схеме лечения, хранимыми в запоминающем устройстве 919.
Согласно некоторым вариантам осуществления запоминающее устройство 919 выполнено с возможностью хранения данных об использовании и/или данных об обратной связи от пациента относительно конкретной дозы и/или схемы лечения и/или по отношению к предварительно выбранному (желательному) PD профилю активного средства в организме пациента.
Согласно некоторым вариантам осуществления интерфейс врача 903, содержащий, например, одно или несколько из контроллера 915, запоминающего устройства 921 и/или модуля связи 909, настроен на персональный компьютер (планшетный компьютер, портативный компьютер, настольный компьютер или другие), мобильное устройство, такое как смартфон, портативное устройство, носимое устройства, наручное устройство или встроенное в защитные очки устройство, монитор клиники или больницы и/или любое другое подходящее устройство. При необходимости, врачу предоставляется удаленный доступ к устройству MDI 901. Дополнительно или альтернативно, врач непосредственно активизирует устройство MDI 901. Согласно некоторым вариантам осуществления врач предварительно программирует (предварительно калибрует или устанавливает) устройство MDI 901 с предварительно определенным парообразным количеством (дозой, дозированием и/или схемой введения), подходящим для конкретного пациента. Согласно некоторым вариантам осуществления данные посылаются от интерфейса врача 903 к интерфейсу пациента 905, например для инструктирования пациента или для осуществления предварительно настроенных корректировок.
Согласно некоторым вариантам осуществления интерфейс пациента 905, содержащий, например, одно или несколько из следующего: контроллер 917, запоминающее устройство 923 и/или модуль связи 911, настроен на персональный компьютер (планшетный компьютер, портативный компьютер, настольный компьютер или другие), мобильное устройство, такое как смартфон, и/или на само устройство MDI 901.
Согласно некоторым вариантам осуществления интерфейс пациента 905 принимает входные данные 929. Входные данные могут быть получены от одного или нескольких из следующего: пациент, интерфейс врача, сервер базы данных, устройство MDI. Примеры различных типов входных данных могут включать в себя дозу и/или схему лечения, определенную врачом и полученную через интерфейс врача, текущий персональный PD параметр пациента, внесенный пациентом и/или полученный от пациента, статистические данные личного пользования, записанные, например, на сервере базы данных и/или в запоминающем устройстве устройства MDI, указание продолжительности ингаляции и/или объема ингаляции, считанное устройством MDI, и/или другие типы вводных данных.
Согласно некоторым вариантам осуществления интерфейс пациента 905 содержит дисплей 927.
Необязательно, дисплей представляет собой интерактивный дисплей, например, сенсорный экран смартфона, портативного устройства, носимого устройства, наручного устройства или встроенного в защитные очки устройства.
В некоторых случаях, некоторые функции, такие как передача данных врачу, получение доступа к базе данных, чтобы получить такую информацию, как инструкции пользователя/пациента, и/или другие функции активируются с помощью интерфейса пациента 905, в то время как другие функции, такие как изменение предварительно определенного парообразного количества (дозы), дозирования и/или схемы лечения, просмотр протоколов других пациентов и/или другие функции не разрешены посредством интерфейса пациента 905. При необходимости, врач устанавливает определения доступа интерфейса пациента на отдельного пациента.
Согласно некоторым вариантам осуществления интерфейс пациента 905 и/или устройство MDI 901 выполнены с возможностью уведомлять пациента каждый раз о необходимости легочной доставки (ингаляции). При необходимости, уведомление осуществляется автоматически на основании запланированного дозирования (схемы лечения), хранящейся в запоминающем устройстве. Дополнительно или альтернативно, уведомление устанавливается пациентом. Дополнительно или альтернативно, уведомление выдается лечащим врачом.
Согласно некоторым вариантам осуществления один или несколько компонентов системы осуществляет связь с сервером базы данных 925 путем приема вводимых данных из базы данных и/или отправки информации в базу данных. Согласно некоторым вариантам осуществления база данных содержит индивидуальные данные пациента, например, включая в себя историю болезни пациента, данные, передаваемые с помощью устройства MDI 901, входные данные от врача, входные данные от пациента и/или другую информацию. Необязательно, сервер базы данных выполнен с возможностью выполнения вычислений над данными. Согласно некоторым вариантам осуществления сервер базы данных 925 содержит коллективные данные, включающие в себя, например, одно или несколько из следующего: клинические результаты экспериментов, результаты других пациентов, данные исследований и/или другие данные. Необязательно, сервер базы данных 925 осуществляет связь со множеством систем обработки, используемых различными пациентами. Данные различных взаимодействий между пациентами и устройством MDI собирают в центральной базе данных, непрерывно изучая индивидуальные профили использования пациентов и рекомендуя дозу, дозирование и/или схему лечения, соответственно. Использование коллективной базы данных пользователей может улучшить создание точной предсказательной дозы, дозирования и/или схемы лечения для существующих и новых пациентов, улучшение общего терапевтического показателя успешности лечения.
Согласно некоторым вариантам осуществления в соответствии с персональными данными обратной связи, полученными от пациента с использованием устройства MDI 901 и/или с помощью интерфейса пациента 905, предварительно определенное парообразное количество (доза, дозирование и/или схема лечения) автоматически изменяется с помощью контроллера 917 интерфейса пациента и/или контроллера 913 устройства MDI для компенсации неадекватных настроек или неправильного использования устройства MDI, например, в ситуации, в которой пациент не использует устройство MDI, когда предписано, и/или использование устройства MDI осуществляется в отличное от заданной схемы лечения время. В ответ может быть принято одно или несколько действий, например, отсрочка следующей дозы, увеличение или уменьшение следующей дозы (и/или следующих доз) и/или иное изменение схемы лечения.
Согласно некоторым вариантам осуществления пациент с использованием устройства MDI 901 может пожелать запланировать свои дозу, дозирование и/или схему лечения таким образом, при котором возможные неблагоприятные эффекты меньше мешают повседневной деятельности пациента. В то время как некоторые неблагоприятные эффекты переносимы в домашних условиях или в определенное время суток и являются приемлемым компромиссом для облегчения симптомов, эти неблагоприятные эффекты могут быть нежелательны, когда пациент занимается такой деятельностью, как вождение, участие в совещании и/или другие виды деятельности. По желанию, используя интерфейс пациента 905 и/или путем прямой активации устройства MDI 901, пациент настраивает дозу, дозирование и/или схему лечения таким образом, чтобы меньше препятствовать своей планируемой деятельности.
Дополнительно или альтернативно, устройство MDI 901 и/или интерфейс пациента 905 выполняют с возможностью активно навязывать определенную дозу и/или схему лечения, например, на основе входных данных от пациента. Например, пациент вносит свои запланированные ежедневные мероприятия и сроки проведения этих мероприятий, а доза, дозирование и/или схема лечения автоматически изменяется соответствующим образом. Необязательно, доза, дозирование и/или схема лечения автоматически изменяется, чтобы гарантировать, что пациент находится в подходящем состоянии для выполнения намечаемой деятельности, например, гарантируя, что во время вождения уровень неблагоприятного эффекта является относительно низким или не воспринимается.
Согласно некоторым вариантам осуществления пациент может добровольно изменить дозу, дозирование и/или схему лечения, например, с использованием интерфейса пациента 905. При необходимости, степень модификации представляет собой ограниченную, чтобы предотвратить состояние, при котором пациент находится в опасности, например, предотвратить передозировку.
Согласно некоторым вариантам осуществления пациент может просто использовать устройство MDI 901, даже если специально не предписано. В таком случае, следующая доза и/или схема лечения могут быть автоматически модифицированы в ответ на использование. По желанию, пациент получает уведомление о модификациях в дозе и/или схеме введения через интерфейс пациента 905.
Дополнительно или альтернативно, врач получает уведомление о таких модификациях, например, через интерфейс врача 903.
Фиг. 7 представляет собой блок-схему способа назначения схемы лечения пациенту с использованием устройства MDI для доставки по меньшей мере одного активного средства, в соответствии с некоторыми вариантами осуществления.
Согласно некоторым вариантам осуществления врач может принять решение лечить пациента путем осуществления легочной доставки одного или нескольких активных средств с помощью устройства MDI (1001).
Согласно некоторым вариантам осуществления данные о пациенте, такие как одно или несколько, например, из следующего: PK переменные (например, возраст, пол, индекс массы тела и т.д.), патофизиологический статус, фармакогенетические и/или фармакогеномические переменные и/или другие параметры, вносимые в систему (1003), например, врачом и/или другим клиническим персоналом. Необязательно, параметры пациента и персональные переменные вносят с помощью интерфейса врача.
Согласно некоторым вариантам осуществления создают предполагаемую дозу, дозирование и/или схему лечения (1005). Необязательно, дозу, дозирование и/или схему лечения создают автоматически, например, с помощью программного обеспечения интерфейса врача. Дополнительно или альтернативно, доза, дозирование и/или схема лечения планируется врачом. Согласно некоторым вариантам осуществления дозу, дозирование и/или схему лечения создают путем сопоставления внесенных данных пациента с предварительно определенной дозой и/или схемой лечения с использованием данных из базы данных или в соответствии с персональными данными обратной связи, или, например, в соответствии со справочной таблицей.
Согласно некоторым вариантам осуществления производится (1007) моделирование ожидаемого PK/PD профиля пациента для выбранной дозы, дозирования и/или схемы лечения. Согласно некоторым вариантам осуществления моделируется ожидаемый PK/PD профиль, включая в себя, например, терапевтические эффекты и/или неблагоприятные эффекты. Согласно некоторым вариантам осуществления путем корреляции между фармакодинамическим профилем и/или фармакокинетическим профилем и личными данными пациента выбирается терапевтическое окно. Необязательно, моделирования PK/PD профиля и/или предварительно выбранное терапевтическое окно графически отображаются у врача, например, на дисплее интерфейса врача. При рассмотрении моделирования, врач может принять решение об изменении дозы, дозирования и/или схемы лечения для более точного соответствия (персонифицированного) пациенту (1009). В некоторых случаях, врач может принять решение об изменении параметров, предложенной дозы и/или схемы лечения, таких как одна или несколько доз, дозирование, схема лечения или общая продолжительность лечения, и/или других параметров лечения.
В некоторых случаях лечение включает введение двух или более биологически активных средств из одной, двух или более пластин, одновременно или последовательно, для получения желаемого терапевтического эффекта у пациента. Система, в соответствии с некоторыми из любых вариантов осуществления, включает использование MDI для доставки более чем одного фармацевтически активного средства (из одной или нескольких пластин) в любом соотношении или в любых предварительно определенных парообразных количествах, чтобы проявлять предварительно выбранный PD профиль (например, поддержание отдельного пациента в пределах терапевтического окна, рассчитанного для пациента). Согласно некоторым вариантам осуществления различные дозы выборочно вводят в соответствии со схемой введения с тем, чтобы не допустить неблагоприятных эффектов при сохранении облегчения симптомов.
Согласно некоторым вариантам осуществления пациенту назначают выбранную (и, возможно, улучшенную) дозу, дозирование и/или схему лечения (1011).
Согласно некоторым вариантам осуществления в виде проверки и в течение определенного периода времени, в течение которого пациент подвергается лечению (например, в течение нескольких часов, в течение дня, в течение недели, в течение месяца и/или в течение промежуточных, более длинных или более коротких, периодов), врач получает один или несколько показателей, таких как показатели в отношении общего использования устройства пациентом, показатели в отношении дозы, дозирования и/или схемы лечения пациента, потребляемых пациентом единиц дозирования, одного или нескольких персональный PD параметров пациента, например, в связи с наличием неблагоприятных эффектов, таких как психотропный уровень, и/или показателей, относящихся к интенсивности симптомов, таких как уровень боли и/или содержание одного или нескольких биомаркеров, и/или другие показатели (1013). При необходимости, один или несколько показателей предоставляются в режиме реального времени. Дополнительно или альтернативно, показатели предусмотрены в конце легочной доставки средства. Дополнительно или альтернативно, показатели предусмотрены по требованию врача.
Дополнительно или альтернативно, пациент принимает решение, когда посылать показатели врачу.
Согласно некоторым вариантам осуществления настоящего изобретения показатели передаются врачу с помощью устройства MDI и/или с помощью интерфейса пациента, автоматически и/или в ответ на инструкцию от врача и/или пациента. Необязательно, один или несколько показателей хранятся в базе данных для дальнейшего использования.
Согласно некоторым вариантам осуществления на основе предоставленных показателей, дозу, дозирование и/или схему лечения корректируют или иным образом модифицируют (1015). При необходимости, модификацию выполняют в режиме реального времени. Согласно некоторым вариантам осуществления конкретную дозу, дозирование и/или схему лечения модифицируют, при необходимости, в режиме реального времени. Согласно некоторым вариантам осуществления дозу и/или схему лечения модифицируют с учетом верхнего и нижнего пределов PD параметра, определенных отдельно для каждого пациента. Верхний предел может позволить дозу, дозирование и/или схему лечения, выше которой присутствуют существенные неблагоприятные эффекты. Нижний предел может позволить дозу и/или схему лечения, ниже которой симптом, который должен был подвергаться лечению путем доставки активного средства, не достаточно смягчается.
Фиг. 8A-D представляют собой схематическую диаграмму (фиг. 8А) и изображения с экранов (фиг. 8B-D) интерфейса врача для выбора и назначения дозы, дозирования и/или схемы лечения пациенту, в соответствии с некоторыми вариантами осуществления.
На фиг. 8А показан общий дисплей 1107 интерфейса врача. Согласно некоторым вариантам осуществления данные пациента вводятся врачом через вход 1109.
Согласно некоторым вариантам осуществления графическое отображение ожидаемого и/или предварительно выбранного фармакокинетического профиля 1111 и/или ожидаемого и/или предварительно выбранного фармакодинамического профиля 1113 представляется врачу. По желанию, показываются один или несколько из профилей, по отдельности или вместе, в отношении временного ряда 1115, в том числе, например, продолжительности (например, почасовой шкале), в течение которой пациент подвергается лечению. Согласно некоторым вариантам осуществления определяется терапевтическое окно 1117, устанавливая верхний предел 1119 и нижний предел 1121. Согласно некоторым вариантам осуществления дозу, дозирование и/или схему лечения выбирают таким образом, чтобы получить ожидаемые и/или предварительно выбранные PK/PD профили, вписывающиеся в диапазон терапевтического окна 1117.
Согласно некоторым вариантам осуществления предел определяют как постоянное значение, представленное в виде прямой линии, например, как показано на фиг. 8А. Альтернативно, предел может содержать изменяющийся набор значений и быть представлен в виде кривой линии. Например, нижний предел 1121 представляет собой желательный терапевтический эффект, верхний предел 1119 представляет собой приемлемый неблагоприятный эффект, и более высокий порог Cmax фармакокинетического профиля может быть установлен для начальной части воздействия, например, для ускорения облегчение симптомов, а порог Cmax может уменьшаться по мере продолжения лечения по желанию. Согласно некоторым вариантам осуществления дозу и/или схему лечения выбирают и/или корректируют для достижения первоначального накопления активного средства у пациента, например, в начальной части лечения, а затем, чтобы обеспечить постоянное дозирование для поддержания пациента в стационарном состоянии (поддержание дозирования). В общем, первоначальное накопление средства основано на сравнительно большом количестве средства по сравнению с количествами, приведенными при поддерживающем дозировании.
Согласно некоторым вариантам осуществления, например, при улучшении предварительно определенного парообразного количества средства (дозы, дозирования и/или схемы лечения) для отдельного пациента, врач может выполнить одно или несколько из повышения и/или понижения предела 1119 и/или предела 1121, повышения и/или понижения пиков профиля 1113 и/или профиля 1111, расширения и/или укорачивания продолжительности лечения по оси времени и/или другие модификации.
Следует отметить, что графическое представление показано в настоящем документе в качестве примера и что могут быть использованы различные графические представления, такие как гистограммы. Согласно некоторым вариантам осуществления профиль 1111 и/или профиль 1113 может быть представлен в прерывистом виде, например, в виде множества точек.
На фиг. 8В показано моделирование ожидаемого фармакокинетического профиля пациента с использованием предварительно определенного парообразного количества, доставленного в соответствии с предварительно определенной схемой введения, в соответствии с некоторыми вариантами осуществления. В этом примере для экрана интерфейса врача, врач может заполнить данные пациента 1101 (например, пол, массу, рост, вводимое лекарственное средство, ID пациента и/или другие данные), а также получить экстраполяцию фармакокинетического профиля отдельного пациента, как и показано, например, с помощью графика 1103, имитируя концентрацию активного средства в плазме пациента с течением времени.
Аналогичным образом, на фиг. 8C показана экстраполяция 1105 ожидаемого фармакодинамического профиля отдельного пациента, показывающая уровень неблагоприятного эффекта на пациента с течением времени.
На фиг. 8D показано изображение с экрана интерфейса врача, в соответствии с некоторыми вариантами осуществления настоящего изобретения. Смоделированный фармакокинетический профиль представлен графом 1103 и смоделированный фармакодинамический профиль представлен графом 1105, которые отображаются на оси динамических рядов 1115, в этом примере представляющую собой 8-часовой период. Шкала фармакодинамического параметра пациента визуально разделена на секции, указывающие, например, на состояние «боли» (ниже терапевтического уровня эффекта), указывающие на «оптимальное» состояние (в пределах терапевтического окна), указывающие, например, на «психоактивное» состояние (выше уровня неблагоприятного эффекта), как определено у отдельного пациента, а смоделированные профили PK/PD в виде графов приведены по отношению к этим разделам. В этом моделировании, первая доза предоставляется в 8:00, приводя в результате к изменению, как фармакодинамических, так и фармакокинетических параметров, переходя из раздела «боли» в раздел «оптимальный». Показано, что вторая доза, предоставляемая в 11:00, поддерживает пациента в пределах «оптимального» (терапевтическое окно).
На фиг. 9 показана блок-схема способа получения данных обратной связи от пациента и модификации/корректировки дозы и/или схемы лечения соответствующим образом, в соответствии с некоторыми вариантами осуществления.
Согласно некоторым вариантам осуществления получают персональный PD параметр пациента (1201). Согласно некоторым вариантам осуществления PD параметр относится к неблагоприятному эффекту, такому как психотропный уровень, терапевтическому эффекту, такому как уровень боли, и/или изменению в любом из этих уровней. PD параметр может включать в себя абсолютное количественное определение уровня и/или относительное количественное определение уровня, оцениваемое, например, по отношению к уровню, измеренному до доставки единицы дозирования и/или до доставки дозирования и/или схемы лечения. PD параметр может быть получен до, во время и/или после доставки единицы дозирования и/или до, во время и/или после доставки дозирования и/или схемы лечения, и/или до, во время и/или после общего периода времени, в течение которого пациенту предоставляется лечение.
Согласно некоторым вариантам осуществления PD параметр предоставляется непосредственно пациентом, например, с помощью интерфейса пациента. Согласно некоторым вариантам осуществления пациент может вручную откорректировать визуальное представление PD параметра, основанное на персональном определении уровня PD параметра. В качестве примера, пациент может повысить или понизить столбик на графике, указывающий на уровень боли, например, на сенсорном экране сотового телефона и/или любого другого персонального устройства, на котором настроен интерфейс пациента.
Согласно некоторым вариантам осуществления пациенты, которые не в состоянии сформулировать уровни PD параметра, могут использовать интерактивный набор инструментов, чтобы помочь им в определении их текущего уровня PD параметра, например, как описано далее.
Дополнительно или альтернативно, к сознательному, субъективно воспринимаемому PD параметру, указанному пациентом, персональный PD параметр, такой как биомаркер, получают с помощью интерфейса пациента и/или системы, например, с использованием датчика. Согласно некоторым вариантам осуществления один или несколько стандартных компонентов сотового телефона и/или персонального компьютера, на котором интерфейс пациента выполнен с возможностью действовать как датчик для получения параметра. Некоторые компоненты, которые могут быть использованы в качестве датчиков для получения PD параметров у пациента, могут включать в себя: сенсорный экран, который может быть использован, например, для оценки ловкости, координации глаз и руки и/или памяти и умственного состояния; гироскоп, акселерометр, датчик близости и/или датчик жестов, например, ИК-датчик может быть использован, например, для оценки моторики; камера и/или источник света может быть использован, например, для обнаружения визуального слежения, саккадической дисперсии, расширения сосудов глаз, расширения и/или пульсации зрачков; RGB-подсветка может использоваться, например, для оценки восприятия окружающей среды; магнитометр и/или GPS могут быть использованы, например, для оценки ориентации; динамик и/или микрофон может быть использован, например, для оценки слуховых и/или голосовых навыков; датчик температуры и/или влажности может быть использован, например, для оценки температуры тела.
Согласно некоторым вариантам осуществления устройство MDI выполнено с возможностью получения персональных данных обратной связи. В качестве примера, устройство MDI содержит датчик потока и/или датчик давления. Необязательно, связанный с дыханием показатель пациента получают с использованием датчика потока и/или давления. Согласно некоторым вариантам осуществления датчик адаптирован для обнаружения объема ингаляции. Поскольку может существовать корреляция между объемом ингаляции и PD параметром, такая как уровень боли, согласно некоторым вариантам осуществления инициируют измерение потока и/или давления для определения PD параметра у пациента.
После того, как получен один или несколько персональных PD параметров, доза и/или схема лечения может быть модифицирована соответствующим образом (1203). Согласно некоторым вариантам осуществления дозу и/или режим модифицируют, с одной стороны, чтобы улучшить или иным образом изменить состояние пациента на основании предоставленной индикации, а с другой стороны, для достижения предварительно выбранного фармакодинамического профиля, такого как поддержание пациента в пределах терапевтического окна - между нижним пределом терапевтического эффекта, который обеспечивает облегчение симптомов, и верхним пределом неблагоприятного эффекта, в котором уровень неблагоприятного эффекта все еще терпим. Согласно некоторым вариантам осуществления устройство MDI может быть выполнено таким образом, что, когда оно ниже минимального терапевтического эффекта, входные данные пациента могут увеличивать дозу и/или корректировать схему лечения по частоте и/или количеству. Необязательно, дозу и/или схему лечения модифицируют для получения уровня выше минимального терапевтического эффекта.
Дополнительно или альтернативно, дозу и/или схему лечения модифицируют на столько, на сколько позволяет максимальный уровень неблагоприятного эффекта.
Фиг. 10A-Е представляют собой изображения с экранов интерфейса пациента (фиг. 10А, фиг. 10С, фиг. 10Е), а также графические изображения ожидаемого фармакодинамического и фармакокинетического профилей пациента до и после получения входных данных персонального PD параметра пациента (10В и 10D, соответственно), в соответствии с некоторыми вариантами осуществления.
Фиг. 10B представляет собой пример для рассчитанной 3-часовой схемы лечения для конкретного пациента (пациент X, 35 лет и характеризуется ИМТ 22). В соответствии с примером для расчетной схемы лечения, чтобы поддерживать пациента X в пределах терапевтического окна в течение 3-х часов, осуществляемого PK профилем, представленным красной кривой на фиг. 10В, пациент Х обязан подвергаться легочной доставке активного средства с использованием устройства MDI в соответствии с некоторыми вариантами осуществления в следующие моменты времени и с использованием следующих доз: 00 минут - 1,2 мг; 10 минут - 1,0 мг; 60 минут - 0,5 мг. Синяя кривая представляет собой пример для расчетного PD профиля в указанных дозах. Как видно, расчетная схема лечения поддерживает пациента X в пределах уровней, а именно, ниже уровня неблагоприятного эффекта и выше уровня терапевтического эффекта, а именно, в терапевтическом окне 1303 в диапазоне от 2,5 до 7,5 по шкале представленного в примере неблагоприятного психоактивного эффекта.
На фиг. 10С, во время и/или после лечения, пациент Х указывает на желание изменить предел неблагоприятного эффекта, например, за счет повышения уровня столбика психоактивности 1301 на экране интерфейса пациента. Поднимая столбик, пациент может указать, что он готов увеличить допустимый уровень неблагоприятного психоактивного эффекта. Терапевтическое окно, как показано на фиг. 10D, затем пересматривают на основе входных данных пациента - например, окно сужается до диапазона от 2,5 до 5 по шкале психоактивности.
Текущая вводимая доза и/или схема лечения затем может быть модифицирована соответствующим образом. Например, предварительно определенное парообразное количество, которое запланировано для легочной доставки, например, через 60 минут после начальной легочной доставки, снижают от 0,5 мг до 0,3 мг, в попытке снизить уровень испытываемого пациентом неблагоприятного эффекта (психоактивного эффекта).
Согласно некоторым вариантам осуществления дозу и/или схему лечения автоматически модифицируют на основе входных данных пациента. Дополнительно или альтернативно, входные данные пациента и/или смоделированные профили передаются, автоматически и/или по желанию пациента, врачу, и врач изменяет схему лечения.
Следует отметить, что чувствительность пациента к терапевтическому и/или неблагоприятному эффекту может меняться в течение дня, например, демонстрируя более высокую болевую чувствительность в вечернее время, снижение когнитивных способностей утром, таким образом, пациент может быть менее восприимчивым к терапевтическому эффекту в вечернее время или более восприимчивым к неблагоприятному эффекту утром.
Дополнительно или альтернативно к уровню неблагоприятного эффекта, пациент может указывать на уровень терапевтического эффекта и/или другие условия, и доза и/или схема лечения будет модифицироваться соответствующим образом.
На фиг. 10E показано приложение интерфейса пациента, включающее регулируемый ползунок 1305, перемещаемый пациентом. Согласно некоторым вариантам осуществления приложение представляет пациенту оценку текущего состояния, рассчитанного на основе одного или нескольких из следующего: предварительно определенной дозы, дозирования и/или схемы лечения, предыдущих входных данных, полученных от пациента, например, включая в себя биологические маркеры и/или другие прямые и/или опосредованные персональные PD параметры, лечение и историю эффектов для конкретного пациента, использование записи пациента, медицинского состояния пациента, информации из коллективной базы данных и/или другую информацию.
Во время лечения и/или после лечения пациент может перетащить ползунок, чтобы отразить свой воспринимаемый PD профиль. Например, если пациент испытывает полный терапевтический эффект (например, у пациент больше нет боли), пациент может передвинуть ползунок на «оптимальное» состояние (например, к «психоактивному» состоянию).
Используя материалы, полученные от пациента, интерфейс пациента может автоматически модифицировать следующую дозу и/или схему лечения. Согласно некоторым вариантам осуществления указатель на модификацию 1307 отображается для пациента, например, уведомление пациента о том, что следующая доза увеличена в количестве. По желанию, приложение настроено для запроса подтверждения 1309 от пациента изменить дозу, дозирование и/или схему лечения.
Согласно некоторым вариантам осуществления вводные данные от пациента и/или модифицированные настройки автоматически передаются на интерфейс врача. В некоторых случаях врач может принять решение изменить вручную заново определенные настройки дозы и/или схемы лечения.
Фиг. 11 представляет собой блок-схему способа измерения одного или нескольких биомаркеров с использованием персонального портативного устройства и/или с использованием устройства MDI, и соответствующей модификации дозы и/или схемы лечения, в соответствии с некоторыми вариантами осуществления.
Согласно некоторым вариантам осуществления измеряют один или несколько биомаркеров (1401). Согласно некоторым вариантам осуществления биомаркеры указывают на существование и/или степень неблагоприятных эффектов у подвергнутого лечению больного. Необязательно, измерения биомаркеров используются для определения терапевтического окна для отдельного пациента и/или для контроля дозы и/или схемы лечения для поддержания пациента в пределах терапевтического окна.
Неблагоприятные эффекты, такие как когнитивные нарушения и другие психоактивные эффекты, могут отличаться среди пациентов с различными генетическими и биологическими чертами. Таким образом, согласно некоторым вариантам осуществления отдельные биомаркеры, такие как биомаркеры ЦНС, получают от пациента, используя, например, один или несколько датчиков в системе, и/или один или несколько датчиков, установленных в интерфейсе устройства пациента, такие как датчики сотовых телефонов, например, как описано выше.
Некоторые неинвазивные способы оценки биомаркеров могут включать в себя одно или несколько из следующего: оценка саккадического движения глаз (например, саккадическое движение), тестирование памяти, адаптивное слежение, оценка пальцевого теппинга, оценка равновесия тела, соответствие визуально-аналоговой шкале и/или другие способы оценки.
Согласно некоторым вариантам осуществления могут быть выполнены различные известные в настоящей области техники неинвазивные тесты биомаркеров, таких как когнитивные задачи, включая в себя, например, время реакции, внимание, зрительно-пространственную ориентацию, воспроизведение названия, воспроизведение повествования, воспроизведение лиц, ассоциации имя-лицо, формирование, беглость речи, присвоение имен объектам, имплицитную память, логическое мышление и/или другие когнитивные задачи.
Согласно некоторым вариантам осуществления измерения биомаркеров сообщаются врачу (1403). Необязательно, измерения PD параметра сохраняются в памяти устройства MDI и/или памяти интерфейса пациента. Дополнительно или альтернативно, измерения PD параметра загружаются в базу данных. Необязательно, измерения PD параметра сравнивают с измерениями PD параметра, хранящимися в базе данных, включая в себя, например, предыдущие измерения PD параметра конкретного пациента, измерения PD параметра других пациентов, измерения PD параметра из литературы и т.д.
Согласно некоторым вариантам осуществления дозу и/или схему лечения модифицируют в соответствии с измерениями PD параметра (1405).
Фиг. 12A-C представляют собой изображения с экранов интерфейса пациента, содержащие различные приложения для получения данных PD параметра и/или для оказания помощи пациенту в определении парообразного количества средства (дозы и/или схемы лечения), в соответствии с некоторыми вариантами осуществления.
В приложении, показанном в настоящем документе в качестве примера, которое может быть установлено на персональном портативном устройстве, таком как сотовый телефон и/или планшетный компьютер, пациент в интерактивном режиме выполняет одну или несколько задач, которые могут быть включены как часть игры или тому подобного, основанных на персональном PD параметре, который может быть оценен на основании задачи. Согласно некоторым вариантам осуществления уровень неблагоприятного эффекта, такой как психоактивный уровень пациента, автоматически выводится приложением. Дополнительно или альтернативно, приложение помогает пациенту в озвучивании своего воспринимаемого терапевтического и/или неблагоприятного эффекта, которое затем может быть предусмотрено как вход в систему.
Предусмотренные в настоящем документе задачи, например, включают в себя слежение за мишенью пальцем (фиг. 12А), визуальное слежение за мишенью (фиг. 12В), выравнивание мишени (фиг. 12С).
Другие приложения могут включать в себя различные измерения персонального PD параметра с использованием действий и способов, известных в настоящей области техники, такие как моделируемое вождение, сортировка карт, тестирование арифметических навыков, оценка времени, копирование символов, зрительно-двигательная координация, время реакции, навыки изображения и/или буквенного написания, а также/или другие приложения, например, как описано выше.
Фиг. 13 представляет собой схематическое изображение ингаляционного устройства, выполненное с возможностью обеспечения автоматизированной контролируемой легочной доставки одного или нескольких активных средств, в соответствии с некоторыми вариантами осуществления.
Согласно некоторым вариантам осуществления устройство 1601 включает в себя распылитель единицы дозирования 1603, например, распылитель для пластины, которая содержит фармацевтически активное средство и позволяет фармацевтически активному средству испаряться из него. Согласно некоторым вариантам осуществления распылитель единицы дозирования содержит или сообщается с источником по меньшей мере одной пластины, из которой берет свое начало активное средство, и механизмом обработки единицы дозирования для получения доставляемого активного средства, например, как описано выше.
Пластина может содержать различные формы, такие как, например, твердую массу, твердые частицы или порошок. При необходимости, пластина содержится в картридже, капсуле и/или других контейнерах. Согласно некоторым вариантам осуществления механизм обработки включает в себя одно или несколько из, например, нагревания (например, для испарения), превращения в аэрозоль, вызывая химическую реакцию, например, путем смешивания с другими материалами, высвобождения биологически активного средства из контейнера, например, путем разламывания капсулы, создания давления, мобилизации и/или других видов обработки. Альтернативно, активное средство уже находится в готовой к применению форме и не требует какой-либо обработки перед доставкой пользователю посредством нагревания пластины.
Согласно некоторым вариантам осуществления ингаляционное устройство 1601 представляет собой устройство MDI, которое содержит вход 1605. Необязательно, модуль ввода 1605 выполнен с возможностью приема данных, относящихся к дозе и/или схеме введения, в соответствии с которыми активное средство будет доставлено пациенту. Дополнительно или альтернативно, вход 1605 выполнен с возможностью приема одного или нескольких показаний от датчика (не показан на фиг. 13), находящегося в устройстве 1601 и/или сконфигурирован снаружи устройства 1601.
Согласно некоторым вариантам осуществления ингаляционное устройство 1601 содержит контроллер 1607, выполненный с возможностью инициировать и/или модифицировать и/или прекратить легочную доставку фармацевтически активного средства. Согласно некоторым вариантам осуществления контроллер 1607 управляет распылителем единицы дозирования 1603, например, активируя нагревание пластины с помощью нагревательного элемента, такого как резистивный нагревательный элемент Согласно некоторым вариантам осуществления контроллер 1607 активизирует доставку предварительно определенного парообразного количества средства, такого как доза и/или схема лечения, полученные в качестве входных данных. Согласно некоторым вариантам осуществления контроллер 1607 управляет потоком активного средства, например, путем активации одного или нескольких клапанов. Согласно некоторым вариантам осуществления контроллер приспособлен для высвобождения средства на основании текущей скорости потока.
Согласно некоторым вариантам осуществления ингаляционное устройство 1601 содержит выход 1609.
Необязательно, выход 1609 выполнен в виде мундштука для взаимодействия с пациентом. Альтернативно мундштуку, выход 1609 может быть выполнен в виде маски для дыхания, соски, как приспособления для грудных детей, и/или других структур, пригодных для доставки потока паров пациенту.
Согласно некоторым вариантам осуществления компоненты устройства 1601, такие как распылитель единицы дозирования и/или контроллер и/или другие компоненты, содержатся внутри корпуса 1611. Необязательно, корпус имеет форму и размеры для использования в качестве портативного устройства.
Согласно некоторым вариантам осуществления устройство MDI 1601 содержит механизм управления потоком.
Необязательно, поток паров контролируется с использованием одного или нескольких клапанов. Согласно некоторым вариантам осуществления поток выбирают и/или модифицируют для отдельного пациента, например, по времени доставки и возможности потока активного средства к пациенту только при вдохе пациента, показанного, например, с помощью датчика, встроенного в устройство. Согласно некоторым вариантам осуществления устройство выполнено с возможностью модификации потока, чтобы позволить пациенту инстинктивно определить, когда следует прекратить ингаляцию, вдыхать глубже и/или иным образом изменить ритм и/или интенсивность дыхания. В качестве примера, устройством подается импульс увеличенного объема потока, чтобы указать пациенту прекратить ингаляцию.
Согласно некоторым вариантам осуществления поток выбирают и/или модифицируют, чтобы уменьшить количество активного средства, которое остается захваченным внутри выходного тракта устройства и не доставляется пациенту. В некоторых случаях количество скопившегося активного средства снижается до известного, предварительно определенного количества путем регулирования потока.
Согласно некоторым вариантам осуществления поток регулируется контроллером 1607. Необязательно, поток регулируется в соответствии с данными, полученными на входе 1605, данными, полученными с помощью датчика, и/или другими показателями.
Потенциальное преимущество устройства, содержащего механизм управления потоком, который предназначен для конкретного пациента, может включать в себя улучшенную точность доставки пациенту в отношении сроков и/или предварительно определенного парообразного количества активного средства, поставляемого устройством, улучшая производительность системы/устройства MDI.
Фиг. 14А представляет собой схематическое изображение конфигурации ингаляционного устройства 1701, в соответствии с некоторыми вариантами осуществления.
В этой конфигурации, распылитель единицы дозирования 1703 содержит единицу дозирования (картридж) 1705, нагревательный элемент 1707 и подложку 1709, которая перемещает единицу дозирования относительно нагревательного элемента 1707, например, чтобы находиться в контакте или в непосредственной близости от нагревательного элемента.
Согласно некоторым вариантам осуществления нагревательный элемент выполнен с возможностью обеспечения локализованного нагревания, например, за счет кондукции, конвекции и/или излучения. Согласно некоторым вариантам осуществления пластину нагревают достаточно быстро до температуры, подходящей для образования паров испаряемого фармацевтически активного средства, содержащегося в нем. Согласно некоторым вариантам осуществления пластина организована в виде подвижного элемента, который может быть выборочно и/или локально активирован. Необязательно, пластина организована в компактные формы. Необязательно, каждая форма представляет собой предварительно определенное парообразное количество.
Согласно некоторым вариантам осуществления пары, высвобождаемые из пластины, собирают внутри испарительной камеры 1711, из которой они переносятся пациенту через выходной тракт.
Необязательно, клапан 1713 расположен вдоль тракта для управления скоростью потока.
Согласно некоторым вариантам осуществления устройство 1701 содержит мундштук 1715, из которого пары доставляются пациенту в ответ на вдыхание. Альтернативно, мундштук 1715 может быть присоединен к другим элементам, например, к маске и/или носовой канюле, необязательно с дополнительным кислородом, например, для доставки средства лечения ослабленным больным. По желанию, мундштук находится в жидкостном сообщении с клапаном 1713.
Согласно некоторым вариантам осуществления устройство 1701 включает в себя источник питания 1717, например, батарею, закрученную вручную пружину и/или штепсель.
Согласно некоторым вариантам осуществления устройство 1701 включает в себя контроллер 1719, например, как описано в настоящем документе выше, выполненный с возможностью управления одним или несколькими из клапана 1713, источника питания 1717 и/или распылителя единицы дозирования 1703 в виде полностью и/или отдельно контролируемых компонентов распылителя единицы дозирования. Согласно некоторым вариантам осуществления контроллер 1719 проверяет, чтобы единицу дозирования было разрешено использовать.
Согласно некоторым вариантам осуществления контроллер 1719 сообщается с запоминающим устройством 1721, которое может быть прочитано с помощью контроллера и/или на которое может быть произведена запись.
На фиг. 14В показана единица дозирования 1723, содержащая множество дискретных пластин 1725. Каждая пластина 1725 содержит одну или несколько секций или площадей поверхности 1727, предназначенных для испарения одного или нескольких выделенных биологически активных средств, заключенных в нагревательный элемент 1729, который функционирует в качестве корпуса пластины. Согласно некоторым вариантам осуществления нагревательный элемент 1729 имеет форму подобной клетке сети проводов, которые упаковывают пластину. Согласно некоторым вариантам осуществления, чтобы выпарить активное средство, электрический ток проходит через нагревательный элемент 1729, нагревая загруженную пластину, содержащуюся в пределах конкретной отдельной единицы дозирования. Производимые пары, необязательно, собираются в испарительной камере и доставляются пациенту.
Потенциальное преимущество индивидуально нагреваемых единиц дозирования может включать в себя более точный контроль над предварительно определенными парообразными количествами биологически активного средства, доставляемого пациенту, например, по сравнению с движущейся лентой единиц дозирования, нагреваемых стационарным нагревательным элементом. Индивидуальная загрузка и нагрев конкретной единицы дозирования в определенный момент времени может повысить точность устройства MDI.
Фиг. 15 представляет собой блок-схему способа лечения отдельного пациента с использованием системы в соответствии с фиг. 6 при сохранении пациента в пределах терапевтического окна, в соответствии с некоторыми вариантами осуществления.
Согласно некоторым вариантам осуществления устройство MDI программируют на предварительно определенное парообразное количество (дозы и/или схемы лечения) (1801). Необязательно, дозу и/или схему лечения устанавливают в ингаляционном устройстве с помощью врача, вручную (например, путем активации кнопок на самом устройстве) и/или с использованием интерфейса врача. Дополнительно или альтернативно, дозу и/или схему лечения устанавливают в устройстве MDI в соответствии с инструкциями, посланными из интерфейса пациента.
Согласно некоторым вариантам осуществления устройство активируется для доставки активного средства пациенту (1803). Согласно некоторым вариантам осуществления прямые и/или опосредованные данные обратной связи от пациента получают в режиме реального времени (1805). Необязательно, данные обратной связи получают в ходе легочной доставки (ингаляционной сессии). Лечение, как правило, может начинаться с легочной доставки и заканчивается через 5-20 минут после этого, например, когда предварительно выбранный фармакодинамический профиль проявится в полной мере для активного средства и/или в более позднее время. Дополнительно или альтернативно, данные обратной связи получают во время серии легочных доставок, например, в течение периода времени, составляющего 1 час, 3 часа, 5 часов, 9 часов, 12 часов или промежуточные, более длинные или более короткие периоды времени. Протокол может включать в себя 5-10 легочных доставок в день, через определенные интервалы времени в диапазоне от 15 до 180 минут между последовательными легочными доставками.
Согласно некоторым вариантам осуществления данные обратной связи, которые получают от пациента, включают в себя персональные PD параметры, такие как терапевтические эффекты, например, интенсивность симптомов, и/или неблагоприятные эффекты, например, психоактивное состояния пациента.
Согласно некоторым вариантам осуществления интерфейс пациента взаимодействует с пациентом для получения данных обратной связи. Согласно некоторым вариантам осуществления вопросы к пациенту, касающиеся его текущего состояния, отображаются на экране, и пациент отвечает на вопросы. Такой вопрос может быть представлен, например, в форме столбика, указывающего на уровень боли, например, который пациент поднимает и/или понижает. Дополнительно или альтернативно, данные обратной связи получают с помощью одного или нескольких приложений, таких как игры, с которыми пациент взаимодействует. Необязательно, содержание неинвазивных биомаркеров оценивают путем анализа входных данных пациента при взаимодействии с пользовательским интерфейсом. Дополнительно или альтернативно, данные обратной связи от пациента получают путем измерения различных биомаркеров с использованием одного или нескольких датчиков, например, за счет использования компонентов смартфона, карманного устройства, носимого устройства, наручного устройства или встроенного в защитные очки устройства, чтобы действовать в качестве датчиков неинвазивных биомаркеров.
Согласно некоторым вариантам осуществления персональные PD параметры получают периодически, например, полусуточно, ежедневно, еженедельно, ежемесячно, согласно требованию, например, перед дозой и/или серией доз, до и/или после изменения в дозировке и/или схеме введения или иным образом.
Согласно некоторым вариантам осуществления в ответ на PD параметры, дозу и/или схему лечения модифицируют (1809). Необязательно, дозу и/или схему лечения модифицируют для достижения желаемого эффекта, например, для снижения уровня боли у пациента, при этом поддерживая пациента в пределах терапевтического окна. Согласно некоторым вариантам осуществления дозу и/или схему лечения итеративно модифицируют посредством интерфейса пациента. Модификации могут происходить множество раз, например, во время, между или после одной или нескольких легочных доставок и/или в течение общего времени периода лечения (дни, недели, месяцы, годы), которому подвергают пациента. Модификация ограничена отсечками безопасности, например, дозами, которые могут подвергнуть пациента риску.
Согласно некоторым вариантам осуществления интерфейс пациента и/или ингаляционное устройство (MDI) напоминает пациенту выполнять одну или несколько легочных доставок (1811). Такое напоминание может быть предусмотрено в виде визуального сигнала (например, световой индикации), звука, вибрации, уведомления на портативном/карманном устройстве, например смартфоне, карманном устройстве, носимом устройстве, наручном устройстве или встроенном в защитные очки устройстве или их сочетании.
Согласно некоторым вариантам осуществления данные пациента об использовании регистрируются и сохраняются в памяти устройства MDI и/или в памяти интерфейса пациента. При необходимости, доставку активного средства модифицируют, потенциально в режиме реального времени, в соответствии с данными об использовании. Например, в случае, когда пациент пропустил одну или несколько легочных доставок, доза и/или схема лечения может быть автоматически модифицирована, чтобы установить доставку, например, увеличенного количества активного средства в следующие одну или несколько легочных доставок.
Согласно некоторым вариантам осуществления одно или несколько действий, описанных в 1801-1811, могут повторяться. Предпочтительно, получение персональных PD параметров и/или данных об использовании от пациента повторно могут включать постоянную корректировку дозы и/или схемы лечения, обеспечивая гибкое, правильное и точное индивидуальное лечение пациента на основе фактического эффекта лечения на отдельном пациенте.
Ожидается, что в течение срока действия патента, являющегося следствием этой заявки, будет разработано много соответствующих единиц дозирования для выпаривания и доставки посредством ингаляции выделенных биологически активных средств, и объем термина единицы дозирования предназначен для включения всех таких новые технологий априори.
Размеры и значения, раскрытые в настоящем документе, не следует понимать как строго ограниченные точными числовыми значениями. Вместо этого, если не указано иное, каждый такой размер должен обозначать как указанное значение, так и функциональный эквивалентный интервал, окружающий это значение. Например, раскрытый размер «10 мкм» предполагается для обозначения «приблизительно 10 мкм».
Используемые в настоящем документе числовые диапазоны, которым предшествует термин «приблизительно», не следует рассматривать как ограниченные указанным диапазоном. Скорее, числовые диапазоны, которым предшествует термин «приблизительно», следует понимать как включающие диапазон, принятый специалистами в настоящей области техники для любого данного элемента в микрокапсулах или составах согласно настоящему изобретению.
Используемый в настоящем документе термин «приблизительно» означает нахождение в пределах приемлемого диапазона ошибок для конкретного значения, как определено обычным специалистом в настоящей области техники, который будет частично зависеть от того, как измеряется или определяется значение, то есть ограничениями системы измерения. Например, «приблизительно» может означать диапазон до 10%, более предпочтительно до 5% и еще более предпочтительно до 1% заданного значения. Там, где конкретные значения описаны в заявке и в формуле изобретения, если не указано иное, значение термина «приблизительно» находится в пределах приемлемого диапазона ошибок для конкретного значения.
Термины «содержит», «содержащий», «включает в себя», «включающий», «имеющий» и родственные им по значению слова означают «включающий в себя без ограничения».
Термин «состоящий из» означает «включая в себя и ограничиваясь этим».
Термин «состоящий по существу из» означает, что композиция, способ или структура может включать в себя дополнительные ингредиенты, стадии и/или части, но только в том случае, если дополнительные ингредиенты, стадии и/или их части не оказывают значительного влияния на основные и новые характеристики заявляемой композиции, способа или структуры.
Используемая в настоящем документе форма единственного числа включает в себя ссылки на множественное число, если из контекста явно не следует иное. Например, термин «соединение» или «по меньшей мере одно соединение» может включать в себя множество соединений, включая в себя их смеси.
Слова «пример» и «иллюстративный» используется в настоящем документе для обозначения «служащий в качестве примера, отдельного случая или иллюстрации». Любой вариант осуществления, описанный как «пример» или «иллюстративный», не должен обязательно рассматриваться как предпочтительный или имеющий преимущество перед другими вариантами осуществления и/или исключать включение элементов из других вариантов осуществления.
Слово «необязательно» используется в настоящем документе для обозначения «предусмотрено согласно некоторым вариантам осуществления и не предусмотрено согласно другим вариантам осуществления». Любой конкретный вариант осуществления может включать множество «необязательных» признаков, за исключением тех случаев, когда такие признаки конфликтуют.
В настоящей заявке различные варианты осуществления могут быть представлены в формате диапазона. Следует понимать, что описание в формате диапазон представлено просто для удобства и краткости и не должно быть истолковано в качестве негибкого ограничения объема изобретения. Соответственно, описание диапазона следует рассматривать, чтобы конкретно раскрыть все возможные поддиапазоны, а также отдельные числовые значения в пределах этого диапазона. Например, описание такого диапазона, как от 1 до 6, следует рассматривать как диапазон, специфически раскрывающий все возможные поддиапазоны, такие как от 1 до 3, от 1 до 4, от 1 до 5, от 2 до 4, от 2 до 6, от 3 до 6 и т.д., а также отдельные значения в пределах этого диапазона, например, 1, 2, 3, 4, 5 и 6. Это применяется независимо от ширины диапазона.
Всякий раз, когда в настоящем документе указывается диапазон числовых значений, это означает включение любого цитируемого числового значения (дробного или целого) в пределах указанного диапазона. Фразы «варьируя в диапазоне/варьирует в диапазоне между» первым указывающим числом и вторым указывающим числом и «варьируя в диапазоне/варьирует в диапазоне от» первого указывающего числа «до» второго указывающего числа используются в настоящем документе взаимозаменяемо и предназначены для включения первого и второго указывающих чисел и всех дробных и целых чисел между ними.
Используемый в настоящем документе термин «способ» относится к подходам средствам, технологиям и процедурам для выполнения данной задачи, включая в себя без ограничения подходы, средства, технологии и процедуры либо известные, либо легко разработанные из известных подходов, средств, технологий и процедур практиками химических, фармакологических, биологических, биохимических и медицинских областей техник.
Используемый в настоящем документе термин «лечение» включает в себя отмену, по существу ингибирование, замедление или обращение прогрессирования состояния, по существу, облегчающие клинические или эстетические симптомы состояния или по существу предотвращающие появления клинических или эстетических симптомов состояния.
Предполагается, что все значения измеряемых параметров измеряют при стандартных условиях температуры и давления или т.п., если не указано иное.
Следует понимать, что определенные признаки изобретения, которые для ясности описаны в контексте отдельных вариантов осуществления, также могут быть представлены в комбинации в одном варианте осуществления. Наоборот, различные признаки изобретения, которые для краткости описаны в контексте одного варианта осуществления, могут также быть представлены отдельно или в любой подходящей подкомбинации или в качестве пригодных в любом другом описанном варианте осуществления. Некоторые функции, описанные в контексте различных вариантов осуществления, не должны рассматриваться как существенные признаки этих вариантов осуществления, если вариант осуществления не находится в нерабочем состоянии без этих элементов.
Различные варианты осуществления и аспекты настоящего изобретения, которые очерчены выше и заявлены в формуле изобретения, находят экспериментальное подтверждение в следующих примерах.
ПРИМЕРЫ
Следующие примеры, вместе с приведенными выше описаниями, иллюстрируют некоторые варианты осуществления настоящего изобретения неограничивающим образом.
Пример 1
Кусок спеченного стекла с пористостью 30 (лабораторный стандарт), имеющий размеры пластины, подходящей для установки в рамку или корпус пластины, использовали в качестве единой воздухопроницаемой матрицы.
Готовили раствор из 50 мг выделенного и очищенного CBD в 50 микролитрах этанола. Раствор выливали на воздухопроницаемую матрицу таким образом, чтобы раствор оставался охваченным и пропитанным в матрице без выщелачивания.
Загруженную воздухопроницаемую матрицу, т.е. пластину, помещали в сушилку для испарения этанола при температуре ниже точки кипения CBD, такой как 100°С.
После того, как этанол выпаривали, что оценивали по достижению постоянной массы пластины, пластина была готова продолжить монтаж единицы дозирования. После того, как пластину устанавливали в рамку единицы дозирования, сетку сплавляли с рамкой единицы дозирования с помощью теплового пресса (плавлением рамки и перекрывающей сетки), ультразвуковой сварки или, при необходимости, любым биосовместимым клеем.
CBD характеризуется температурой кипения, равной 180°С. Для испарения и ингаляции лекарственного средства предусмотрено короткое время (приблизительно всего 3 секунды), так что большая часть, если не все лекарственное средство, может быть выпарено и ингалировано в одной ингаляции большинством предполагаемых пользователей. Пластину быстро нагревают до температуры выше 180°С, но ниже температуры горения материала воздухопроницаемой матрицы, материала рамы и CBD.
Пример 2
Кусок керамики с пористостью 30 (лабораторный стандарт), имеющий размеры пластины, подходящей для установки в раму или корпус пластина, использовали в качестве единой воздухопроницаемой матрицы.
Готовят раствор 20 мг чистого дронабинола в 50 микролитрах этанола.
Раствор выливают на воздухопроницаемую матрицу таким образом, чтобы раствор оставался охваченным и пропитанным в матрице без выщелачивания.
Загруженную воздухопроницаемую матрицу, т.е. пластину, помещают в сушилку для испарения этанола при температуре ниже точки кипения дронабинола, такой как 100°С.
После того, как этанол испаряется, что оценивают по достижению постоянной массы пластины, пластина готова продолжить монтаж единицы дозирования. После того, как пластину устанавливают в рамку единицы дозирования, сетку сплавляют с рамкой единицы дозирования с помощью теплового пресса (плавлением рамки и перекрывающей сети), ультразвуковой сварки или, при необходимости, любым биосовместимым клеем.
Дронабинол характеризуется температурой кипения, равной 250°С. Для испарения и ингаляции лекарственного средства предусмотрено короткое время (приблизительно всего 3 секунды). Пластину быстро нагревают до температуры выше 250°С, но ниже температуры горения материала воздухопроницаемой матрицы, материала рамки и дронабинола.
Пример 3
Измеренное количество (например, 30м3) промытых кислотой/силанизированных стеклянных гранул, имеющих средний размер 75 мкм (таких как, например, SUPELCO 59201), размещают в дозовой рамке. Необязательно, гранулы распределяют в дозовой рамке, помещая их при этом в горизонтальной плоскости по отношению к поддерживающей поверхности и встряхивая дозовую рамку с гранулами внутри вертикально (например, с помощью вибрации ее самой и/или поверхности, на которой она расположена), до тех пор, пока не образуется выровненная плоскость гранул в рамке. При необходимости, дозовую рамку прикрепляют перед вибрацией, чтобы предотвратить выкатывание гранул из рамки снизу.
Готовят раствор 5 мг Δ9-тетрагидроканнабинола (дронабинол ТНС-10015S) и 1 мг лимонена (Sigma-Aldrich 62118-1 мл) в 50 мкл этанола.
Раствор осторожно выливают на стеклянные гранулы, и дозу помещают в сушилку для того, чтобы испарить этанол. При необходимости, вместо этого гранулы погружают в раствор, а затем удаляют, чтобы высушить, прежде чем поместить в рамку, как это описано выше.
После того, как этанол выпаривают, что может быть оценено, например, путем достижения постоянной массы гранул или пластины (если уже сформирована), количество ТНС и лимонена может быть измерено или оценено, например, путем сравнения масс высушенных покрытых гранул с промытыми гранулами перед воздействием раствора THC-лимонена.
Хотя настоящее изобретение было описано в связи с конкретными вариантами осуществления, понятно, что многие альтернативы, модификации и вариации будут очевидны специалистам в настоящей области техники. Соответственно, предполагается охватить все такие альтернативы, модификации и вариации, которые попадают в пределы сущности и широкого объема прилагаемой формулы изобретения.
Все публикации, патенты и патентные заявки, упомянутые в настоящем документе, полностью включены посредством ссылки в описание, в той же степени, как если бы конкретно и отдельно было указано, что каждая отдельная публикация, патент или патентная заявка включена в настоящий документ посредством ссылки. Кроме того, цитирование или идентификация любой ссылки в данной заявке не должно быть истолковано как признание того, что такая ссылка доступна в качестве предшествующего уровня техники для настоящего изобретения. В той степени, в которой используются заголовки разделов, они не должны быть истолкованы как обязательное ограничение.
Реферат
Изобретение относится к медицинской технике. Описан элемент дозирования, содержащий рамку с отверстием и воздухопроницаемую пластину внутри отверстия. Пластина имеет отношение площади поверхности к массе по меньшей мере 1000 м/г и/или отношение площади поверхности к объему по меньшей мере 500 м/мл. 24 з.п. ф-лы, 15 ил.
Комментарии