Замещенные дигидропиразолоны в качестве ингибиторов hif-пролил-4-гидроксилазы - RU2509080C9
Код документа: RU2509080C9
Описание
Настоящая заявка касается новых замещенных производных дигидропиразолона, способов их получения, их применения для лечения и/или профилактики болезней, а также их применения для изготовления лекарственных средств для лечения и/или профилактики болезней, в частности, сердечно-сосудистых и гематологических заболеваний, заболеваний почек и для стимуляции заживления ран.
Недостаточное снабжение кислородом человеческого организма или его органов, которое из-за продолжительности и/или масштаба либо нарушает правильное функционирование организма или его органов, либо полностью парализует их функции, называется гипоксией. Гипоксия может быть вызвана уменьшением кислорода во вдыхаемом воздухе (например, при пребывании высоко в горах), нарушениями внешнего дыхания (например, в результате нарушенной функции легких или закупорки дыхательных путей), уменьшением минутного объема сердца (например, при сердечной недостаточности, при острой перегрузке правого желудочка сердца, при эмболии легочной артерии), малой кислородной транспортной емкостью крови (например, в результате малокровия (анемии) или интоксикации, например, угарным газом), может быть локализована недостаточным кровоснабжением в результате тромбоза сосудов (ишемические состояния, например, сердца, нижних конечностей или мозга, диабетическая макро- или микроангиопатия) или вызвана повышенной потребностью в кислороде ткани (например, в результате интенсивной мышечной работы или локальных воспалений) [Eder, Gedigk (Hrsg.), Allgemeine Pathologie und pathologische Anatomic, 33. Aufl., Springer Verlag, Berlin, 1990].
Человеческий организм относительно способен приспосабливаться к ситуациям пониженного снабжения кислородом в острых и хронических случаях. Кроме немедленного ответа, при котором вегетативно-нервные механизмы включают увеличение минутного объема сердца и временного объема дыхания, а также локальное расширение кровеносных сосудов, гипоксия приводит к изменению транскрипции многочисленных генов. При этом функция генных продуктов обеспечивает компенсацию нехватки кислорода. Таким образом, усиленно высвобождаются некоторые ферменты гликолиза и переносчики гликолиза 1, благодаря чему увеличивается выделение анаэробной АТФ и обеспечивается преодоление нехватки кислорода [Schmidt, Thews (Hrsg.), Physiologic des Menschen, 27. Aufl., Springer Verlag, Berlin, 1997; Loffler, Petrides (Hrsg.), Biochemie und Pathobiochemie, 7. Aufl., Springer Verlag, Berlin, 2003].
Кроме того, гипоксия приводит к усиленной экспрессии фактора роста сосудистых эндотелиальных клеток, VEGF, вследствие чего в испытывающих кислородное голодание тканях стимулируется новообразование кровеносных сосудов (ангиогенез). Этим самым улучшается долговременное кровоснабжение ишемической ткани. При различных заболеваниях, связанных с нарушением сердечного кровообращения, и заболеваниях, связанных с тромбозом сосудов, такой противоположно направленный регуляторный механизм осуществляется в недостаточной степени [обзор в работе: Simons und Ware, Therapeutic angio genesis in cardiovascular disease, Nat. Rev. Drug. Discov. 2 (11), 863-71 (2003)].
Далее, при систематической гипоксии усиленно высвобождается пептидный гормон эритропоэтин, образующийся преимущественно в интерстициальных фибропластах почек. Этим стимулируется образование красных клеток крови в костном мозге и вместе с тем кислородная транспортная емкость крови. Этот эффект использовался и используется спортсменами-рекордсменами при так называемых высотных тренировках. Снижение кислородной транспортной емкости крови, например, в результате постгеморрагической анемии, обычно вызывает рост образования эритропоэтинов в почках. При определенных видах анемии этот механизм регулирования может быть либо нарушен, либо продукция эритропоэтинов не достигает заданного параметра. Так, например, у страдающих почечной недостаточностью пациентов, хотя эритропоэтины и производятся в паренхиме почки, но в значительно меньшем количестве относительно кислородной транспортной емкости крови, результатом чего является так называемая почечная анемия. В частности, почечная анемия, а также анемии, обусловленные онкологией и ВИЧ-инфекцией, лечатся обычно назначением для парентерального приема рекомбинантных человеческих эритропоэтинов (rhEPO). В настоящее время для такого дорогостоящего лечения не существует альтернативной терапии принимаемым орально лекарством [обзоры в работах: Eckardt, The potential of erythropoietin and related strategies to stimulate erythropoiesis, Curr. Opin. Investig. Drugs 2(8), 1081-5 (2001); Bems, Should the target hemoglobin for patients with chronic kidney disease treated-with erythropoietic replacement therapy be changed?, Semin. Dial. 18 (1), 22-9 (2005)]. Последние исследования подтверждают, что эритропотеин кроме повышающего эритропоэз действия оказывает также независящее от него защитное (анти-апоптическое) действие на пораженные вследствие гипоксии ткани, в частности, на сердце и мозг. Далее по результатам последних исследований лечение эритропоэтином у пациентов с почечной недостаточностью в среднем уменьшает степень тяжести болезненного состояния [обзоры в работах: Caiola und Cheng, Use of erythropoietin in heart failure management, Ann. Pharmacother. 38 (12), 2145-9 (2004); Katz, Mechanisms and treatment of anemia in chronic heart failure. Congest. Heart. Fail. 10 (5), 243-7 (2004)].
Вместе с индуцированном гипоксией описанных выше генов, вызывается повышение их экспрессии при гипоксии путем так называемого индуцируемого гипоксией фактора транскрипции (HIF). Что касается фактора HIF, то речь идет о гетеродимерном факторе транскрипции, который состоит из одной альфа- и одной бета-субъединицы. Описаны три альфа-изоформы фактора HIF, из которых 1 альфа HIF и 2 альфа HIF представляют гомологические субъединицы и являются значимыми для индуцированной гипоксией экспрессии генов. В то время как бета-субъединица (описанная 2 изоформами), называемая также как ARNT (aryl hydrocarbon receptor nuclear translocator: арилгидрокарбоновый рецептор транслокатора ядра), является конститутивно выделенной, экспрессия альфа-субъединицы зависит от содержания кислорода в крови. При нормоксии альфа протеин фактора HIF поли-убиквитинируется и затем разлагается в протеазу. При гипоксии это разложение тормозится, так что HIF альфа образует димерное соединение с ARNT и может активизировать их целевые гены. При этом димер HIF прикрепляется к так называемым активным элементам гипоксии (HRE) в регуляторных последовательностях их целевых генов. Элементы HRE определяются смысловой последовательностью. Функциональные HRE были обнаружены в регуляторных элементах многочисленных генов, индуцированных гипоксией [обзоры в работах: Semenza, Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 7 (8), 345-50 (2001); Wenger und Gassmann, Oxygen(es) and the hypoxia-inducible factor-1: Biol. Chem. 378 (7), 609-16 (1997)].
Молекулярный механизм, лежащий в основе регуляции HIF-альфа, объяснен работами нескольких независимых исследовательских групп. В общем виде этот механизм представляется следующим: HIF альфа гидроксилируется обозначаемым как PHD или EGLN подклассом пролил-4-гидроксилаз, зависящих от кислорода, в два специфических остатка пролила (Р402 и Р564 человеческой субъединицы 1 альфа HIF). Что касается HIF пролил-4-гидроксилазы, то речь идет о зависящей от железа диоксигеназе, преобразующей оксоглутарат [Epstein et al., С.elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation, Cell 107 (1), 43-54 (2001); Bruick und McKnight, A conserved family of prolyl-4-hydroxylases that modify HIF, Science 294 (5545), 1337-40 (2001); Ivan et al., Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor, Proc. Natl. Acad. Sci. U.S.A. 99 (21), 13459-64 (2002)]. Ферменты были первые опубликованы в 2001 году как пролил-гидроксилазы [Aravind und Koonin, The DNA -repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate-and iron-dependent dioxygenases, Genome Biol. 2 (3), research0007.1-0007.8, Epub 2001 Feb 19].
К полигидроксилированной альфа субъединице HIF прикрепляется pVHL угнетающий опухоль протеин, который вместе с элонгином В и С образует так называемый комплекс VBC, который адаптирует альфа-субъединицу HIF к Е3 убиквитин-лигазе. Поскольку пролил-4-гидроксилирование альфа субъединицы HIF и ее последующее разложение осуществляется в зависимости от внутриклеточной концентрации кислорода, то HIF пролил-4-гидроксилазы называются также как клеточный датчик кислорода. Идентифицированы три изоформы этих ферментов: EGLN1/PHD2, EGLN2/PHD1 и EGLN3/PHD3. Из них два фермента (EGLN2/PHD1 и EGLN3/PHD3) индуцируются транскрипцией даже при гипоксии и могут быть ответственными за падение уровня HIF альфа, наблюдаемого при хронической гипоксии [см. Schofield und Ratcliffe, Oxygen sensing by HIF hydroxylases, Nat. Rev. Mol. Cell. Biol. 5 (5), 343-54 (2004)].
Селективное фармакологическое угнетение HIF пролил-4-гидроксилаз приводит к увеличению экспрессии зависящих от HIF целевых генов и поэтому используется для лечения многих болезней. В особенности, при заболеваниях сердечно-сосудистой системы можно ожидать улучшения течения болезни при индукции новых кровеносных сосудов, а также при изменении обмена веществ ишемических органов с аэробного до анаэробного выделения АТФ. Улучшение васкуляризации хронических ран способствует процессу заживления, в особенности, трудно излечиваемых язв Ulcera cruris и других хронических кожных ран. При определенных формах болезни, в частности, у пациентов с почечной анемией, индукция аутологичного эритропоэтина также представляет заслуживающую внимания терапевтическую цель.
Ингибиторы HIF пролил-4-гидроксилазы, описанные до настоящего времени в научной литературе, не удовлетворяют требованиям, предъявляемым к лекарственному средству. При этом речь идет либо о конкурентных аналогах оксоглутарата (как, например, N-оксалилглицин), которые отличаются очень низкой эффективностью и поэтому до сих пор не показали на образцах in vivo никакого действия в смысле индукции HIF целевых генов. Либо речь идет о железообразующих комплексах (комплексонах) как десферроксамин, которые, действуют как неспецифические ингибиторы содержащих железо диоксигеназ, и которые, хотя и вызывают индукцию целевых генов, как, например, эритропоэтин in vivo, но из-за комплексирования имеющегося железа подавляют эритропоэз.
2-Гетероарил-4-арил-дигидропиразолоны с бактерицидным и фунгицидным действием опубликованы в заявках ЕР 165 448 и ЕР 212281. Применение 2-гетероарил-4-арил-дигидропиразолонов в качестве ингибиторов липоксигеназы для лечения заболеваний дыхательных путей, болезней, связанных с нарушением сердечного кровообращения, и воспалительных заболеваний описано в EP 183 159. 2,4-дифенил-1,2-дигидропиразолоны с гербицидной активностью описаны в DE 2 651 008. О получении фармакологических свойств определенных 2-пиридил-дигидропиразолонов сообщается в работе Helv. Chim. Acta 49 (1), 272-280 (1966). В заявках WO 96/12706, WO 00/51989 и WO 03/074550 описаны соединения с парциальной структурой дигидропиразолонов для лечения различных заболеваний, а в WO 2006/101903 опубликованы замещенные гидрокси- или алкоксигруппами бипиразолы для лечения нервно-психических заболеваний. Далее, в WO 03/051833 и WO 2004/089303 описаны замещенные гетероарилом производные пиразола для лечения боли и различных заболеваний центральной нервной системы. В WO 2006/114213 были описаны 2,4-дипиридил-1,2-дигидропиразолоны в роли ингибиторов НГР-пролил-4-гидроксилаз.
О рентгеноструктурном анализе кристаллов соединения 3-метил-1-(пиридин-2-ил)-4-(1-пиридин-2-ил-3-метил-1H-пиразол-5-ил)-2H-3-пиразолин-5(1H)-он (другое название: 5,5'-диметил-2,2'-дипиридин-2-ил-1',2'-дигидро-2H,3'H-3,4'-бипиразол-3'-он) сообщается в работе Acta Crystallogr., Section E: Structure Reports Online E57 (11), oll26-oll27 (2001) [Chem. Abstr. 2001:796190]. Синтез определенных производных 3',5-диметил-2-фенил-1'-(1,3-тиазол-2-ил)-1H,2H-3,4'-бипиразол-5'-олов описан в работе Indian J. Heterocyclic Chem. 3. (1), 5-8 (1993) [Chem. Abstr. 1994:323362]. О получении и таутомерии отдельных производных 4-(пиразол-5-ил)-пиразолин-5-онов сообщается в работе J. Heterocyclic Chem. 27 (4), 865-870 (1990) [Chem. Abstr. 1991:428557]. Применение в терапевтических целях названных в этих публикациях соединений до настоящего времени не было описано. В заявке WO 2007/008541 приводится соединение 2-трети-бутил-1'-[4-(4-хлорфенил)-1,3-тиазол-2-ил]-3',5-диметил-1'H,2H-3,4'-бипиразол-5'-ола в качестве тестового примера.
Задачей настоящего изобретения является получение новых соединений, которые могут применяться для лечения заболеваний, в частности, сердечнососудистых и гематологических заболеваний.
В рамках настоящего изобретения описываются соединения, которые действуют как специфические ингибиторы HIF-пролил-4-гидроксилаз, и которые благодаря своему специфическому механизму действия in vivo после парентерального или орального введения приводят к индукции HIF целевых генов, как, например, эритропоэтин, и к вызванным ими биологическим процессам, как, например, эритропоэз.
Предметом изобретения являются соединения формулы
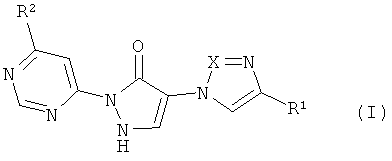
в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает насыщенный 4-7-членный остаток гетероциклил, связанный через атом азота,
причем гетероциклильный остаток может быть замещен одним заместителем, выбранным из группы, состоящей из гидрокси, гидроксикарбонила, C1-C3-алкила, C1-C3-алкиламино и C3-C6-пиююалкила, или причем гетеропиклильный остаток может быть замещен 1-4 заместителями фтор, и их соли, их сольваты и сольваты их солей.
Соединениями согласно изобретению являются соединения формулы (I) и их соли, сольваты и сольваты солей, а также охватываемые формулой (I) соединения, впоследствии названные как пример (примеры) исполнения, и их соли, сольваты и сольваты солей, если речь не идет о солях, сольватах и сольватах солей обоих видов охватываемых формулой (I), впоследствии названных соединений.
Соединения согласно изобретению в зависимости от их структуры могут существовать в стереоизомерных формах (энантиомеры, диастереомеры). Поэтому изобретение охватывает энантиомеры или диастереомеры и их соответствующие смеси. Из этих смесей энантиомеров и/или диастереомеров могут быть известными способами выделены стереоизомерные однородные компоненты.
Если соединения согласно изобретению могут быть представлены в таутомерных формах, то настоящее изобретение охватывает все таутомерные формы.
В качестве солей в рамках настоящего изобретения предпочтительны не опасные для физиологии соли соединений согласно изобретению. Охватываются также соли, которые сами не пригодны для фармацевтических применений, но которые могут использоваться, например, для выделения или очистки соединений согласно изобретению.
Не опасные для физиологии соли соединений согласно изобретению включают соли минеральных кислот, карбоновых кислот и сульфокислот, например, соли соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфокислоты, этансульфокислоты, толуолсульфокислоты, бензолсульфокислоты, нафталиндисульфокислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты и бензойной кислоты.
Не опасные для физиологии соли соединений согласно изобретению включают также соли обычных оснований, как, например, и преимущественно, соли щелочных металлов (соли натрия и калия), соли щелочноземельных металлов (например, соли кальция и магния) и соли аммония, полученные из аммиака или органических аминов с 1-16 атомами углерода, как, например, и преимущественно, этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метилморфолин, аргинин, лизин, этилендиамин и N-метилпиперидин.
В качестве сольватов в рамках настоящего изобретения называются такие виды соединений согласно изобретению, которые в твердом или жидком состоянии образуют комплекс путем координации с молекулами растворителей. Гидраты представляют специальную форму сольватов, при которой происходит координация с водой. В рамках настоящего изобретения в качестве сольватов предпочтительны гидраты.
Кроме того, настоящее изобретение охватывает также пролекарства соединений согласно изобретению. Понятие «пролекарства» включает соединения, которые сами могут быть биологически активными или неактивными, но которые в организме превращаются в соединения согласно изобретению (например, метаболическим или гидролитическим путем).
В рамках настоящего изобретения заместители имеют следующее значение, если нет другого определения:
Сам по себе алкил или «алкил» в группе алкиламино означает линейный или с разветвленной цепью алкильный остаток с 1-3 атомами углерода, например, и преимущественно, метил, этил, н-пропил, изопропил.
Алкиламино означает остаток алкиламино с одним или двумя (выбранными независимо один от другого) алкильными заместителями, например, и преимущественно, означает метиламине, этиламино, н-пропиламино, изопропиламино, N,N-диметиламино, N,N-диэтиламино, N-этил-N-метиламино, N-метил-N-н-пропиламино и N-изопропил-N-н-пропиламино. C1-C3-алкиламино означает, к примеру, остаток моноалкиламино с 1-3 атомами углерода или остаток диалкиламино соответственно с 1-3 атомами углерода в каждом алкильном заместителе.
Циклоалкил означает моноциклическую группу циклоалкила, как правило, с 3-6 атомами углерода, например, и преимущественно, означает циклоалкил под названием циклопропил, циклобутил, циклопентил и циклогексил.
Насыщенный 4-7-членный гетероциклильный остаток, связанный через атом азота, означает моноциклический, насыщенный, гетероциклический остаток с 4-7 кольцевыми атомами, содержащий атом азота, через который он связан, и не более 2, преимущественно не более одного дополнительного гетероатома и/или гетерогруппы из ряда N, O, S, SO, SO2, причем атом азота может образовывать оксид азота. Предпочтительны 4-7-членные, моноциклические насыщенные гетероциклильные остатки с максимум одним дополнительным гетероатомом из ряда O, N и S, например, и преимущественно, ацетидин-1-ил, пирролин-1-ил, пиперидин-1-ил, морфолин-4-ил, тиоморфолин-4-ил, пиперазин-1-ил, 1,2-оксазинан-2-ил, 1,4-оксазепан-4-ил, 1,4-тиазепан-4-ил.
Предпочтительны соединения формулы (I), в которой
Х означает N или CH,
R1 означает водород или циано,
R2 означает насыщенный 4-7-членный гетероциклильный остаток, связанный через атом азота,
причем гетероциклильный остаток замещен 1-4 заместителями фтор, или R2 означает пиперазин-1-ил,
причем пиперазин-1-ил замещен одним заместителем, выбранным из группы, состоящей из C3-C6-циклоалкила, или R2 означает ацетидин-1-ил,
причем ацетидин-1-ил замещен одним заместителем, выбранным из группы, состоящей из гидроксикарбонила, C1-C3-алкила, C1-C3-алкиламино и C3-C6-циклоалкила, или
R2 означает 1,2-оксазинан-2-ил или 1,4-оксазепан-4-ил,
и их соли, их сольваты и сольваты их солей.
Предпочтительны соединения формулы (I), в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает ацетидин-1-ил, пирролин-1-ил или пиперидин-1-ил,
причем ацетидин-1-ил, пирролин-1-ил и пиперидин-1-ил замещены
1-4 заместителями фтор,
или
R2 означает пиперазин-1-ил,
причем пиперазин-1-ил в 4-й позиции замещен одним заместителем, выбранным из группы, состоящей из C3-C6-циклоалкила,
или
R2 означает ацетидин-1-ил,
причем ацетидин-1-ил в 3-й позиции замещен одним заместителем,
выбранным из группы, состоящей из гидроксикарбонила, метила и
диметиламино,
или
R2 означает 1,2-оксазинан-2-ил или 1,4-оксазепан-4-ил,
и их соли, их сольваты и сольваты их солей.
Предпочтительны также соединения формулы (I), в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает насыщенный 4-7-членный гетероциклильный остаток,
связанный через атом азота,
причем гетероциклильный остаток замещен 1-4 заместителями фтор, и их соли, их сольваты и сольваты их солей.
Предпочтительны также соединения формулы (I), в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает ацетидин-1-ил, пирролин-1-ил или пиперидин-1-ил,
причем ацетидин-1-ил, пирролин-1-ил и пиперидин-1-ил замещены
1-4 заместителями фтор,
и их соли, их сольваты и сольваты их солей.
Предпочтительны также соединения формулы (I), в которой
Х означает N или CH,
R1 означает водород или циано,
R2 означает ацетидин-1-ил, пирролин-1-ил или пиперидин-1-ил,
причем ацетидин-1-ил, пирролин-1-ил и пиперидин-1-ил замещены 2 заместителями фтор, и эти заместители привязаны к тому же атому углерода,
и их соли, их сольваты и сольваты их солей. Предпочтительны также соединения формулы (I), в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает пиперазин-1-ил,
причем пиперазин-1-ил замещен одним заместителем, выбранным из
группы, состоящей из C3-C6-циклоалкила, и их соли, их сольваты и сольваты их солей.
Предпочтительны также соединения формулы (I), в которой
Х означает N или CH,
R1 означает водород или циано,
R2 означает пиперазин-1-ил,
причем пиперазин-1-ил в 4-й позиции замещен одним заместителем,
выбранным из группы, состоящей из C3-C6-циклоалкила, и их соли, их сольваты и сольваты их солей. Предпочтительны также соединения формулы (I), в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает ацетидин-1-ил,
причем ацетидин-1-ил замещен одним заместителем, выбранным из группы, состоящей из гидроксикарбонила, C1-C3-алкила, C1-C3-алкиламино и C3-C6-циклоалкила,
и их соли, их сольваты и сольваты их солей.
Предпочтительны также соединения формулы (I), в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает ацетидин-1-ил,
причем ацетидин-1-ил в замещен одним заместителем, выбранным из группы, состоящей из гидроксикарбонила, метила и диметиламино,
и их соли, их сольваты и сольваты их солей.
Предпочтительны также соединения формулы (I), в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает апетидин-1-ил,
причем апетидин-1-ил в 3-й позиции замещен одним заместителем, выбранным из группы, состоящей из гадроксикарбонила, метила и диметиламино,
и их соли, их сольваты и сольваты их солей.
Предпочтительны также соединения формулы (I), в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает 1,2-оксазинан-2-ил или 1,4-оксазепан-4-ил.
Предпочтительны также соединения формулы (I), в которой X означает N.
Предпочтительны также соединения формулы (I), в которой R1 означает водород.
Предпочтительны также соединения формулы (I), в которой R1 означает циано.
Предпочтительны также соединения формулы (I), в которой R2 означает 4-циклобутил-пиперазин-1-ил.
Предпочтительны также соединения формулы (I), в которой
X означает N или CH,
R1 означает циано,
R2 означает насыщенный 4-7-членный гетероциклильный остаток, связанный через атом азота,
причем гетероциклильный остаток замещен одним заместителем, выбранным из группы, состоящей из гидрокси, гидроксикарбонила, C1-C3-алкила, C1-C3-алкиламино и C3-C6-циклоалкила, или
причем гетероциклильный остаток замещен 1-4 заместителями фтор,
и их соли, их сольваты и сольваты их солей.
Указанные в частности определения остатков в соответствующих и в предпочтительных комбинациях остатков заменяются произвольно также определениями остатков других комбинаций независимо от соответствующих указанных комбинаций остатков.
Наиболее предпочтительны комбинации двух или нескольких из названной выше сферы предпочтений.
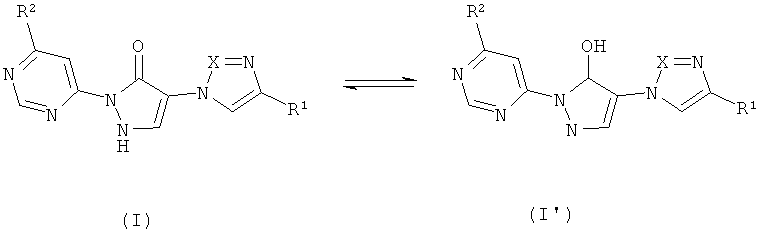
Согласно изобретению производные 1,2-дигидропиразол-3-она формулы (I) могут быть также представлены в таутомерной форме 1H-пиразол-5-ола (I') (см ниже схему 1); обе таутомерные формы охватываются настоящим изобретением.
Схема 1
Далее, предметом изобретения является способ получения соединений формулы (I), или их солей, их сольватов или сольватов их солей, причем согласно способу
[А] соединения формулы
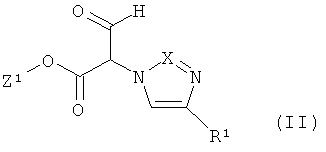
в которой R1 имеет указанное выше значение, и
Z1 означает метил или этил,
в инертном растворителе, при необходимости, в присутствии одной из кислот с соединением формулы
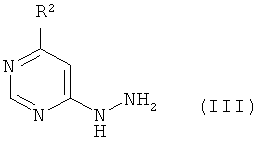
в которой R2 имеет указанное выше значение,
превращают в соединения формулы
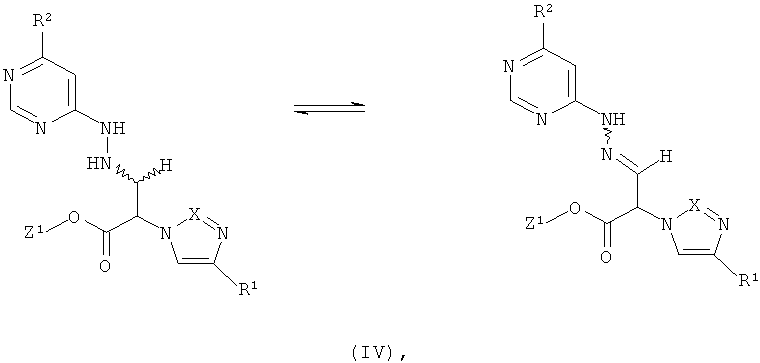
в которой Z1, R1 и R2 имеют указанные выше значения,
которые уже при этих условиях реакции или в последующей реакции под влиянием основания преобразуют в циклические соединения формулы (I),
и соединения формулы (I), при необходимости, с помощью соответствующих (i) растворителей и/или (ii) оснований или кислот преобразуют в их соли, их сольваты или сольваты их солей,
или
[В] соединения формулы
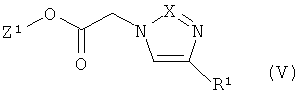
в которой Z1 и R1 имеют указанные выше значения,
с соединением формулы
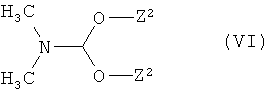
в которой
Z2 означает метил или этил,
конденсируют в соединения формулы
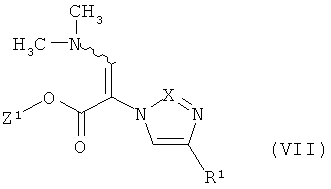
в которой Z1 и R1 имеют указанные выше значения,
и затем в присутствии одной из кислот с соединением формулы (III) превращают в соединения формулы (IV), которые уже при этих условиях реакции или в последующей реакции под действием основания преобразуют в циклические соединения формулы (I),
и соединения формулы (I), при необходимости, с помощью соответствующих (i) растворителей и/или (ii) оснований или кислот преобразуют в их соли, их сольваты или сольваты их солей,
или
[С] соединение формулы
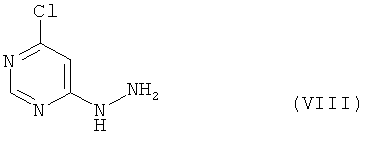
в воде в качестве растворителя методом айнтопф («одной пробирки») подвергают реакции сначала с соединениями формулы
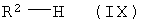
которой R2 имеет указанное выше значение,
и затем с соединениями формулы (VII) с превращением в соединения формулы (I),
и соединения формулы (I), при необходимости, с помощью соответствующих (i) растворителей и/или (ii) оснований или кислот преобразуют в их соли, их сольваты или сольваты их солей.
Свободное основание солей может быть получено реакцией обмена солей соединений или сольватов солей соединений с основанием.
В качестве оснований пригодны гидроксиды щелочных металлов, как, например, гидроксид натрия или калия, карбонаты щелочных или щелочноземельных металлов, как карбонат натрия, калия, кальция или цезия, или
водный раствор аммиака.
В альтернативном способе свободное основание солей может быть получено, например, с помощью хроматографии в колонке с обращенной фазой с градиентом ацетонитрил-вода при добавлении основания, в частности, с использованием колонки RP18 Phenomenex Luna С 18 (2) и диэтиламина в качестве основания.
Следующим предметом изобретения является способ получения соединений формулы (I) и их сольватов, по которому соли соединений или сольваты солей соединений реакцией обмена с основанием или с помощью хроматографии при добавлении основания преобразуют в соединения.
Далее, соединения согласно изобретению могут быть получены, при известных условиях, путем превращения функциональных групп отдельных заместителей, в частности, указанных символами R1 и R2, исходя из соединений формулы (I), полученных описанными выше способами. Эти превращения выполняют стандартными, известными специалистам способами и включают, например, такие реакции, как нуклеофильное или электрофильное замещение, оксидирование, восстановление, гидрогенизация, катализируемые переходными металлами реакции сочетания, алкилирование, ацилирование, аминирование, образование сложного эфира, расщепление сложного эфира, образование простого эфира, расщепление простого эфира, образование карбонамидов, сульфамидов, карбаматов и карбамидов, а также введение и удаление временных концевых групп.
В качестве инертных растворителей для стадий способа (II)+(III)→(IV), (VII)+(III)→(IV) и (IV)→(I) годятся, в частности, простые эфиры как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран и диоксан, или спирты как метанол, этанол, н-пропанол, изопропанол, н-бутанол и трет-бутанол, или вода, или смеси из растворителей, или смесь из растворителя с водой. Предпочтительно использование метанола, этанола, тетрагидрофурана или воды.
Стадию способа (V)+(VI)→(VII) проводят предпочтительно в диметилформамиде в качестве растворителя, или в присутствии избытка соединения (VI) без дополнительного растворителя. В некоторых случаях реакция может также выполняться преимущественно при микроволновом облучении. Реакцию обмена проводят в общем случае при температуре от +20°C до +150°C, предпочтительно при температуре от+80°C до 120°C [см. также J.P. Bazureau et aL, Synthesis 1998, 967; ibid. 2001 (4), 581].
В некоторых случаях стадии способа (II)+(III)→(IV) и (VII)+(III)→(IV) могут выполняться преимущественно с добавлением кислоты. Для этого пригодны обычные неорганические или органические кислоты, как, например, хлористый водород, уксусная кислота, трифторуксусная кислота, метансульфокислота, р-толуолсульфокислота или камфер-10-сульфокислота. Предпочтительно использование уксусных кислот, в особенности, трифторуксусной кислоты или р-толуолсульфокислоты.
Реакцию обмена (II)+(III)→(IV) в общем случае выполняют при температуре от 0°C до +100°C, предпочтительно от+10°C до+50°C. Реакцию (VII)+(III)→(IV) выполняют в общем случае при температуре от +20°C до +120°C, предпочтительно от +50°C до +100°C.
Последовательности способа (II)+(III)→(IV)→(I) и (VIII)+(III)→(IV)→(I) могут выполняться в два этапа или также как реакция «айнтопф» без выделения промежуточного этапа (IV). Для последнего варианта, в частности, пригодно превращение компонентов при микроволновом облучении; реакцию выполняют в общем случае при температуре от +50°C до +200°C, предпочтительно при температуре от +100°C до +180°C.
Отчасти замыкание цикла, приближаясь к соединению (I), начинается уже при получении соединения (IV); циклизация может быть завершена обработкой in situ реакционной смеси каким-либо основанием.
В качестве основания для такой выделенной стадии циклизации (IV)→(I) пригодны обычные неорганические или органические основания. К ним относятся, в частности, гидроксиды щелочных металлов, как, например, гидроксид натрия или калия, карбонаты щелочных или щелочноземельных металлов, как карбонат натрия, калия, кальция или цезия, алкоголяты щелочных металлов, как метанолат натрия или калия, этанолат натрия или калия, или трет-бутилат натрия или калия, или гидриды щелочных металлов как гидрид натрия. Предпочтительно применение метанолата или этанолата натрия.
Индуцированную основанием реакцию обмена (IV)→(I) выполняют в общем случае при температуре от 0°C до +60°C, предпочтительно от 0°C до +30°C.
Реакцию обмена (VIII)+(IX)+(VII)→(I) осуществляют при использовании 1,1-2,0 эквивалентов соединения формулы (IX) относительно 1 эквивалента соединения формулы (VIII), при необходимости, в присутствии от 1,1 до 2,0 эквивалентов основания. Предпочтительна реакция обмена с 1,1-1,5 эквивалентами соединения формулы (IX).
В качестве оснований для реакции обмена (VIII)+(IX)(+(VIII)→(I) пригодны обычные неорганические или органические основания. К ним относятся, в частности, гидроксиды щелочных металлов, как, например, гидроксид натрия или калия, или аминные основания, как, например, N-этил-N-(пропан-2-ил)пропан-2-амин. Предпочтителен N-этил-N-(пропан-2-ил)пропан-2-амин.
Реакцию обмена (VIII)+(IX)+(VII)→(I) осуществляют в общем случае при температуре от +20°C до +100°C, предпочтительно при температуре от +70°C до +100°C.
Все стадии способа могут выполняться при нормальном, повышенном или пониженном давлении (например, от 0,5 до 5 бар). Как правило, работы проводят при нормальном давлении.
Соединения формулы (II) могут быть получены описанными в литературе способами для C-ацилирования сложных эфиров сульфокислот из соединений формулы (V). Соединения формул (III), (V), (VI), (VIII) и (IX) имеются в продаже, известны из литературных источников или могут быть получены по аналогии со способами, описанными в литературе.
Получение соединений согласно изобретению наглядно показано на следующей схеме реакций 2:
Схема 2
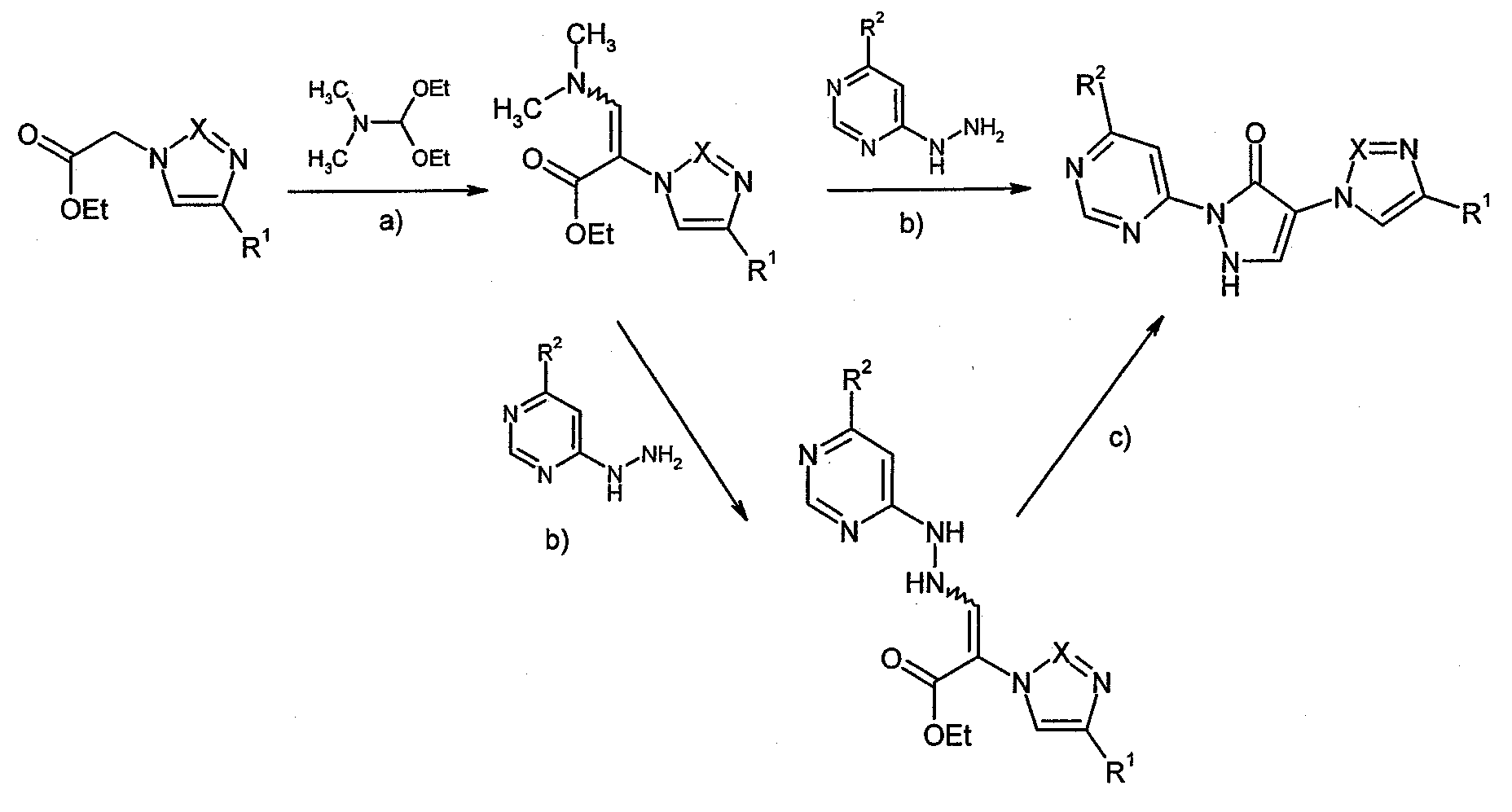
[а): диметилформамид, 16 часов,+100°C; b): этанол, трифторуксусная кислота, +78°C; с): NaOEt, этанол, 1 час, комнатная температура].
Соединения согласно изобретению показывают неожиданно полноценный фармакологический спектр действия.
Поэтому они пригодны для использования в качестве лекарственных средств для лечения и/или профилактики болезней у людей и животных.
Соединения согласно изобретению отличаются как специфические ингибиторы НIF-пролил-4-гидроксилазы.
Соединения согласно изобретению, благодаря их фармакологическим свойствам, могут применяться для лечения и/или профилактики заболеваний сердечно-сосудистой системы, в частности, сердечной недостаточности, коронарной болезни сердца, стенокардии, инфаркта миокарда, апоплексического удара, артериосклероза, эссенциальной, легочной и злокачественной гипертонии, а также периферийного облитерирующего заболевания
Соединения согласно изобретению пригодны, кроме того, для лечения и/или профилактики нарушений кроветворения, как, например, идиопатической анемии, почечной анемии и анемий, сопровождаемых онкологическим заболеванием (в частности, анемии, вызванной химиотерапией), инфекции (в частности, ВИЧ-инфекции) или других воспалительных заболеваний, как, например, ревматоидного артрита. Кроме того, соединения согласно изобретению пригодны для поддерживающего лечения анемий вследствие потери крови, железодефицитной анемии, авитаминозной анемии (например, вследствие недостатка витамина В-12 или недостатка фолиевой кислоты), гипопластической и апластической анемии, гемолитической анемии, или для поддерживающего лечения анемий в результате нарушения усвоения железа (железорефрактерная анемия) или анемий вследствие других эндокринных нарушений (например, гипотиреоз).
Далее, соединения пригодны для повышения гематокрита с целью получения крови для восполнения собственной крови перед операциями.
Кроме того, соединения согласно изобретению могут применяться для лечения и/или профилактики обусловленных операциями ишемических состояний и их последствий после хирургического вмешательства, в частности, после операций на сердце с применением искусственного сердца и легких (например, шунтирование, имплантации сердечных клапанов), операций на сонной артерии, операций на аорте и операций с инструментальным вскрытием или пенетрацией крыши черепа. Далее, соединения пригодны для общего лечения и/или профилактики при хирургических вмешательствах с целью ускорения заживления раны и сокращения времени выздоровления.
Кроме того, соединения пригодны для лечения и профилактики последствий острых и затяжных ишемических состояний мозга (например, апоплексический удар, родовая асфиксия).
Далее, соединения могут применяться для лечения и/или профилактики рака и для лечения и/или профилактики наступающего в ходе лечения рака ухудшения здоровья, в частности, после терапии цитостатическими средствами, антибиотиками и облучением.
Соединения пригодны также для лечения и/или профилактики ревматических заболеваний и других форм болезней, причисляемых к аутоиммунным заболеваниям и, в частности, для лечения и/или профилактики ухудшения здоровья, наступающего в ходе медикаментозного лечения такого рода болезней.
Кроме того, соединения согласно изобретению могут применяться для лечения и/или профилактики заболеваний глаз (например, глаукома), мозга (например, болезнь Паркинсона, болезнь Альцгеймера, слабоумие, хронические боли), хронических заболеваний почек, почечной недостаточности и острого отказа почек, а также для стимуляции заживления ран.
Далее, соединения согласно изобретению пригодны для лечения и/или профилактики общей физической слабости, часто встречающейся в глубоко преклонном возрасте вплоть до истощения.
Далее, соединения пригодны для лечения и/или профилактики сексуальной дисфункции.
Кроме того, соединения пригодны для лечения и/или профилактики сахарного диабета и его осложнений, как, например, диабетическая макро- и микроангиопатия, диабетическая нефропатия и невропатия.
Далее, соединения согласно изобретению пригодны для лечения и/или профилактики фиброзных заболеваний, например, сердца, легких и печени.
В частности, соединения согласно изобретению пригодны также для профилактики и лечения ретинопатии недоношенных детей (Retinopathia praematurorum).
Следующим предметом настоящего изобретения является применение соединений согласно изобретению для лечения и/или предупреждения заболеваний, в частности, названных выше заболеваний.
Следующим предметом настоящего изобретения является применение соединений согласно изобретению для изготовления лекарственного средства для лечения и/или предупреждения заболеваний, в частности, названных выше заболеваний.
Следующим предметом настоящего изобретения является способ лечения и/или предупреждения заболеваний, в частности, названных выше заболеваний, с использованием действующего количества, по крайней мере, одного из соединений согласно изобретению.
Соединения согласно изобретению могут применяться самостоятельно или, в случае необходимости, в комбинации с другими биологически активными веществами. Следующим предметом настоящего изобретения являются лекарственные средства, содержащие, по крайней мере, одно из соединений согласно изобретению и одно или несколько активных веществ, в частности, для лечения и/или предупреждения названных выше заболеваний. В качестве пригодных для комбинаций активных веществ, например, и преимущественно, могут быть названы следующие: ингибиторы ACE, антагонисты рецептора ангиотензин II, блокаторы бета-рецепторов, антагонисты кальция, ингибиторы фосфодиэстеразы (PDE), антагонисты минералокортикоидных рецепторов, мочегонные средства, аспирин, добавки железа, добавки витамина В 12 и фолиевой кислоты, статины, производные дигиталиса (дигоксин), онкологические химиотерапевтические препараты и антибиотики.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо ингибитором ACE, как, например, и преимущественно, эналаприл, каптоприл, лизиноприл, рамиприл, делаприл, фозиноприл, квиноприл, периндоприл или трандоприл.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо антагонистом ангиотензина II, как, например, и преимущественно, лозартан, кандезартан, валзартан, телмизартан или эмбузартан.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо блокатором бета-рецепторов, как, например, и преимущественно, пропранолол, атенолол, тимолол, пирдолол, альпренолол, окспренолол, пенбутолол, бупранолол, метипранолол, надолол, мепиндолол, каразалол, соталол, метопролол, бетаксолол, целипролол, бизопролол, картеолол, эсмолол, лабеталол, карведилол, адапролол, ландиолол, небиволол, эпанолол или буциндолол.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо антагонистом кальция, как, например, и преимущественно, нифедипин, амлодипин, верапамил или дилтиазем.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо ингибитором фосфодиэстеразы (PDE), как, например, и преимущественно, милринон, амринон, пимопендан, цилостазол, силденафил, варденафил или тадалафил.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо антагонистом минералокортикоидных рецепторов, как, например, и преимущественно, спиронолактон, эплеренон, канренон или канреноат калия.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо мочегонным средством, как, например, и преимущественно, фуроземид, буметанид, торземид, бендрофлуметиазид, хлортиазид, гидрохлортиазид, гидрофлуметиазид, метиклотиазид, поитиазид, трихлорметиазид, хлорталидон, индапамид, метолазон, квинетазон, ацетазоламид, дихлорфенамид, метазоламид, глицерин, изосорбид, маннитол, амилорид или триамтерен.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо ингибитором HMG-CoA-редуктазы из класса статинов, как, например, и преимущественно, ловастатин, симвастатин, правастатин, флувастатин, аторвастатин, розувастатин, церивастатин или питавастатин.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо онкологическим химиотерапевтическим средством, например, и преимущественно, из группы платиновых комплексов, как, например, цисплатин и карбоплатин, из группы алкилантов, как, например, циклофосфамид и хлорамбуцил, из группы антиметаболитов, как, например, 5-фторурацил и метотрексат, из группы ингибиторов топоизомеразы, как, например, этопозид и камтотецин, из группы антибиотиков, как, например, доксоробицин и даунорубицин, или из грауппы ингибиторов киназы, как, например, сорафениб и сунитиниб.
В предпочтительной форме исполнения изобретения соединения согласно изобретению выпускаются в комбинации с каким-либо антибиотиком, например, и преимущественно, из группы пенициллинов, цефалоспоринов или хинолонов, как, например, ципрофлоксацин и моксифлоксацин.
Следующим предметом настоящего изобретения являются лекарственные средства, содержащие, по крайней мере, одно соединение согласно изобретению, обычно вместе с одним или несколькими инертными, не токсичными, принятыми в фармакологии вспомогательными веществами, а также их применение для названных ранее целей.
Соединения согласно изобретению могут действовать системно или локально. Для этой цели они могут приниматься надлежащим способом, как, например, орально, парентерально, пульмонально, назально, под язык, на язык, трансбуккально, ректально, дермально, трансдермально, конъюктивально, через уши или в виде имплантата и стента.
Для этих путей приема соединения согласно изобретению могут быть приготовлены в соответствующих формах применения.
Для орального приема пригодны действующие по состоянию техники формы применения, быстро и/или в модифицированном виде отдающие соединения согласно изобретению, которые содержат соединения согласно изобретению в кристаллическом и/или аморфном и/или растворенном виде, как, например, таблетки (таблетки с покрытием или без покрытия, к примеру, устойчивыми к действию желудочного сока оболочками, растворимыми с задержкой или нерастворимыми оболочками, которые контролируют высвобождение соединения согласно изобретению), быстро распадающиеся в ротовой полости таблетки или пластинки/облатки, пластинки/лиофилизаты, капсулы (например, капсулы в твердом или мягком желатине), драже, грануляты, гранулы, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральный прием может осуществляться в обход стадии всасывания (например, внутривенно, внутриартериально, интракардиально, интраспинально или интралюмбально) или с включением всасывания (например, внутримышечно, подкожно, внутрикожно, чрезкожно или внутрибрюшинно). Для парентерального приема пригодны, среди прочего, препараты для инъекций и инфузий в виде растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для других путей приема пригодны, например, лекарственные формы для ингаляции (среди прочего, порошковые ингаляторы, распылители), капли, растворы или спреи для носа, таблетки, принимаемые на язык, под язык или за щеку, пластинки/облатки или капсулы, суппозитории, ушные или глазные препараты, вагинальные капсулы, водные суспензии (лосьоны, болтушки), жирорастворимые суспензии, мази, кремы, действующие через кожу системы (например, пластыри), молочко, пасты, пенки, присыпки, имплататы и стенты.
Предпочтителен оральный или парентеральный прием, в особенности оральный и внутривенный прием.
Соединения согласно изобретению могут быть переведены в указанные формы применения. Это может осуществляться известным способом с помощью инертных, нетоксичных, принятых в фармакологии вспомогательных веществ. К этим вспомогательным веществам относятся, среди прочего, носители (например, микрокристаллическая целлюлоза, лактоза, маннитол), растворители (например, жидкий полиэтиленгликоль) эмульгаторы и диспергаторы или смачивающие агенты (например, лаурилсульфат натрия, полиоксисорбитанолеат), связывающие вещества (например, поливинилпирролидон), искусственные и природные полимеры (например, альбумин), стабилизаторы (к примеру, антиоксиданты, как, например, аскорбиновая кислота), красители (к примеру, неорганические пигменты, как, например, оксиды железа) и вещества, улучшающие вкус и/или запах.
Для достижения ощутимого результата при парентеральном приеме следует назначать количества, в общем случае, от 0,001 до 1 мг/кг, преимущественно от 0,01 до 0,5 мг/кг веса тела. При оральном приеме дозировка составляет 0,01-100 мг/кг, преимущественно 0,01-20 мг/кг и особенно предпочтительно 0,1-10 мг/кг веса тела.
Тем не менее, в некоторых случаях следует отклоняться от названных количеств, а именно в зависимости от веса тела, пути приема, индивидуальной переносимости активного вещества, вида препарата и времени или интервала приема. Так, в некоторых случаях дозировка может быть достаточной в меньших количествах, чем указанные минимальные дозы, в то время как в других случаях должны быть превышены верхние границы. В случае назначения увеличенной дозы рекомендуется разделить ее на несколько приемов в день.
Следующие примеры исполнения объясняют изобретение. Изобретение не ограничивается приведенными примерами.
Данные, указанные в процентах в следующих тестах и примерах, если нет другого указания, являются весовыми процентами; доли являются весовыми частями. Соотношения растворителей, разбавителей и данные концентрации жидких/жидких растворов относятся к соответствующим объемам.
А. Примеры
Сокращения:
Методы LC-MS. GC-MS и HPLC:
Метод 1 (LC-MS): инструмент: Micromass Platform LCZ с HPLC Agilent Serie 1100; колонка: Thermo Hypersil GOLD 3µ, 20 мм × 4 мм; элюент А: 1 л воды+0.5 мл 50%-ной уксусной кислоты, элюент В: 1 л ацетонитрила+0.5 мл 50%-ной уксусной кислоты; градиент: 0.0 мин 100% А→0.2 мин 100% А→2.9 мин 30% А→3.1 мин 10% А→5.5 мин 10% А; печь: 50°C; поток: 0.8 мл/мин; УФ-детектирование: 210 нм.
Метод 2 (LC-MS): тип прибора MS: Micromass ZQ; тип прибора HPLC:
Waters Alliance 2795; колонка: Phenomenex Synergi 2µ Hydro-RP Mercury 20 мм × 4 мм; элюент A: 1 л воды+0.5 мл 50%-ной уксусной кислоты, элюент В: 1 л ацетонитрила+0.5 мл 50%-ной уксусной кислоты; градиент: 0.0 мин 90% А→2.5 мин 30% А→3.0 мин 5% А→4.5 мин 5% А; поток: 0.0 мин 1 мл/мин→2.5 мин/3.0 мин/4.5 мин 2 мл/мин; печь: 50°C; УФ-детектирование: 210 нм.
Метод 3 (LC-MS): тип прибора MS: Micromass ZQ; тип прибора HPLC:
Waters Alliance 2795; колонка: Phenomenex Synergi 2.5µ. MAX-RP 100A Mercury 20 мм × 4 мм; элюент A: 1 л воды+0.5 мл 50%-ной уксусной кислоты, элюент В: 1 л ацетонитрила +0.5 мл 50%-ной уксусной кислоты; градиент: 0.0 мин 90% А→0.1 мин 90% А→3.0 мин 5% А→4.0 мин 5% А→4.01 мин 90% А; поток: 2 мл/мин; печь: 50°C; УФ-детектирование: 210 нм.
Метод 4 (LC-MS): Инструмент: Micromass Quattro Micro MS c HPLC Agilent Serie 1100; колонка: Thermo Hypersil GOLD 3µ 20 мм × 4 мм; элюент А: 1 л воды+0.5 мл 50%-ной уксусной кислоты, элюент В: 1 л ацетонитрила+0.5 мл 50%-ной уксусной кислоты; градиент: 0.0 мин 100% А→3.0 мин 10% А→4.0 мин 10% А→4.01 мин 100% А (поток 2.5 мл/мин)→5.00 мин 100% А; печь: 50°C; поток: 2 мл/мин; УФ-детектирование: 210 нм.
Метод 5 (LC-MS): Инструмент: Micromass QuattroPremier с Waters UPLC Acquity; колонка: Thermo Hypersil GOLD 1.9µ 50 мм × 1 мм; элюент А: 1 л воды +0.5 мл 50%-ной уксусной кислоты, элюент В: 1 л ацетонитрила +0.5 мл 50%-ной уксусной кислоты; градиент: 0.0 мин 90% А→0.1 мин 90% А→1.5 мин 10% А→2.2 мин 10% А; поток: 0.33 мл/мин; печь: 50°C; УФ-детектирование: 210 нм.
Метод 6 (HPLC): Инструмент: HP 1100 с DAD-детектированием; колонка: Kromasil 100 RP-18, 60 мм × 2.1 мм, 3.5 мкм; элюент А: 5 мл перхлорной кислоты (70%-ной) / литр воды, элюент В: ацетонитрил; градиент: 0 мин 2% В→0.5 мин 2% В→4.5 мин 90% В→6.5 мин 90% В→6.7 мин 2% В→7.5 мин 2% В; поток: 0.75 мл/мин; температура колонки: 30°C; УФ-детектирование: 210 нм.
Метод 7 (GC-MS): Инструмент: Micromass GCT, GC6890; колонка: Restek RTX-35, 15 м × 200 мкм × 0.33 мкм; постоянный поток с гелием: 0.88 мл/мин; печь: 70°C; температура на входе: 250°C; градиент: 70°C, 30°C/мин→310°C (3 минуты выдерживается).
Метод 8 (препаративная HPLC): колонка: Kromasil 100 С 18 5 мкм, 250 мм × 20 мм; элюент А: вода «Milli-Q», элюент В: водная 0.1%-ная трифторуксусная кислота, элюент С: ацетонитрил; градиент: 0.0 мин 76% А, 5% В, 19% С→15 мин 4% А, 95% В, 1% С→15.1 мин 76% А, 5% В, 19% С→20 мин 76% А, 5% В, 19% С; печь: 40°C; поток: 25 мл/мин; УФ-детектирование: 210 нм.
Метод 9 (препаративная HPLC): колонка: Sunfire С 18 5 мкм, 19 мм × 150 мм; элюент А: водная 0.2%-ная трифторуксусная кислота, элюент В: ацетонитрил; градиент: 0.0 мин 95% А→8 мин 50% А→8.01 мин 95% А→12 мин 95% А; КТ;
поток: 25 мл/мин; УФ-детектирование: 210 нм.
Метод 10 (препаративная HPLC): колонка: Sunfire С 18 5 мкм, 19 мм × 150 мм; элюент А: водная 0.2%-ная трифторуксусная кислота, элюент В: ацетонитрил; 0 мин 90% А→13 мин 90% А; печь: 40°C; поток: 25 мл/мин; УФ-детектирование: 210 нм.
Метод 11 (препаративная HPLC): колонка: XBridge С 18 5 мкм, 19 мм × 150 мм; элюент А: водная 0.2%-ная муравьиная кислота, элюент В: ацетонитрил; 0 мин 75% А→6 мин 75% А; КТ; поток: 25 мл/мин; УФ-детектирование: 210 нм.
Метод 12 (препаративная HPLC): колонка: XBridge С 18 5 мкм, 19 мм × 150 мм; элюент А: водная 0.2%-ная муравьиная кислота, элюент В: ацетонитрил; 0 мин 93% А→4 мин 93% А; КТ; поток: 25 мл/мин; УФ-детектирование: 210 нм.
Метод 13 (препаративная HPLC): колонка: XBridge С 18 5 мкм, 19 мм × 150 мм; элюент А: водная 0.2%-ная трифторуксусная кислота, элюент В: ацетонитрил;0 мин 90% А→12 мин 90% А; печь: 40°C; поток: 25 мл/мин; УФ-детектирование: 210 нм.
Исходные соединения
Пример 1А
(4-Циано-1H-имидазол-1-ил)этилацетат
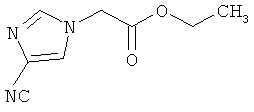
3,3 г (35,3 ммоль) 1H-имидазол-4-карбонитрила [Matthews et al., J. Org. Chem. 1986, 51, 3228-3231] вводят в 13,2 мл (11,5 г, 35.3 ммоль) 21%-ного раствора этилата натрия в этаноле, и добавляют 4,3 мл (6,5 г, 38,9 ммоль) бромэтилацетата. Реакционную смесь перемешивают 16 часов при комнатной температуре. Для переработки отфильтровывают выпавшее твердое вещество, фильтрационный осадок промывают этанолом, и фильтрат сгущают в вакууме. Остаток разводят диизопропиловым эфиром, еще раз фильтруют, фильтрат вновь сгущают в ротационном выпарном аппарате, и остаток высушивают в вакууме. Выход: 3,8 г (60% теор. возм.).
LC-MS (метод I): Rt=1.17 мин; MS (ESIpos): m/z=180 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.12 (s, 1H), 7.88 (s, 1H), 5.06 (s, 2H), 4.18 (q, 2H), 1.22 (t, 3H).
Пример 2А
2-(1 H-1,2,3 -триазол-1-ил)этилацетат
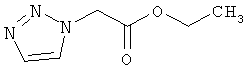
129.2 г (5.6 моль) натрия медленно добавляют к 4,0 литрам этанола. Затем добавляют 400,0 г (5,6 моль) 1,2,3-1H-триазола и прикалывают 623 мл (938,2 г, 5,6 моль) бромэтилацетата при внутренней температуре 20-25°C.Смесь перемешивают 48 часов при комнатной температуре. Выпавшее твердое вещество отфильтровывают, этанол удаляют в вакууме и снова фильтруют. Остаток разводят этилацетатом, фильтруют, вновь сгущают в вакууме и очищают дистилляцией в 30-ти сантиметровой колонке. Продукт получают при температуре бани 140°C, температуры верха 60-115°C и давлении 1 мбар. Выход:: 440,0 г (50% теор. возм.).
HPLC (метод б): Rt=1.58 мин;
LC-MS (метод I): Rt=0.71 мин; MS (ESIpos): m/z=156 [M+H]+.
Пример 3А
1H-Имидазол-1-илэтилацетат
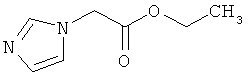
118,2 г (5,1 моль) натрия медленно добавляют к 2.5 литрам этанола. Затем добавляют 350,0 г (5,1 моль) имидазола и прикалывают 570 мл (858,6 г, 5,1 моль) бромэтилацетата при внутренней температуре 20-25°C. Смесь перемешивают 24 часа при комнатной температуре. Выпавшее твердое вещество отфильтровывают, этанол удаляют в вакууме и снова фильтруют. Остаток очищают в хроматографической колонке на силикагеле (растворитель: этилацетат). Выход: 639,0 г (81% теор. возм.).
GC-MS (метод 7): Rt=4.55 мин; MS (ESIpos): m/z=155 [M+H]+.
Пример 4А
(4-Циано-1H-1,2,3 -триазол-1-ил)этилацетат
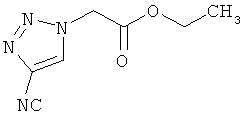
4,1 г (31,9 ммоль) ацидоэтилацетат и 2,8 г (31,9 ммоль) нитрила 2-хлоракриловой кислоты перемешиваются в 32 мл воды 16 часов при температуре бани 80°C. После охлаждения до комнатной температуры раствор подкисляют 1 N соляной кислотой и экстрагируют с помощью этилацетата. Органическую фазу высушивают над сульфатом натрия, фильтруют и сгущают в вакууме. Остаток разводят 50 мл этанола и 10 каплями концентрированной серной кислоты, и смесь перемешивают 16 часов при рефлюксе. Для переработки реакционную смесь концентрируют в вакууме, остаток разводят этилацетатом, суспензию промывают полуконцентрированным раствором гидрокарбонатом натрия, органическую фазу высушивают над сульфатом натрия. Растворитель полностью удаляют в ротационном выпарном аппарате, и остаток высушивают в вакууме. Выход: 1,5 г (25% теор. возм.).
LC-MS (метод 3): Rt=0.96 мин; MS (ESIpos): m/z=181 [M+H]+;
1Н-ЯМР (400 МГц, DMSOO: 5 - 9.06 (s, 1H), 5.57 (s, 2H), 4.19 (q, 2H), 1.22(t,3H).
Пример 5А
3-(N,N-Диметиламино)-2-(1H-имидазол-1-ил)акриловой кислоты этиловый эфир
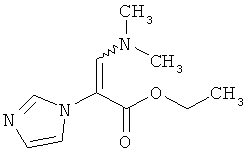
38,0 г (244,9 ммоль) соединения из примера ЗА перемешивают в 126 мл (108,1 г, 734,7 ммоль) N,N-диметилформамид-диэтилацетала 16 часов при температуре бани 90°C. После охлаждения смесь сгущают в вакууме, перемешивают с диизопропиловым эфиром, твердое вещество отфильтровывают, затем промывают диизопропиловым эфиром. Выход: 49,0 г (95% теор. возм.).
LC-MS (метод 2): Rt=2.42 мин; MS (ESIpos): m/z=211 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=7.52 (s, 1H), 7.49 (s, 1H), 7.05 (s, 1H), 6.91 (s, 1H), 4.02 (q, 2H), 2.63 (br. s, 6H), 1.12 (t, 3H).
Пример 6А
3-(N-диметиламино)-2-(4-циано-1H-имидазол-1-ил)акриловой кислоты этиловый эфир
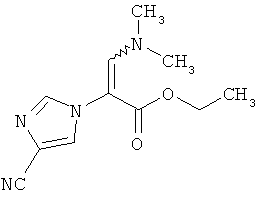
3,8 г (21,4 ммоль) соединения из примера 1А и 7,4 мл (6,3 г, 42,8 ммоль) N,N-диметилформамид-диэтилацетала перемешивают 16 часов при температуре бани 100°C. Для переработки охлажденный реакционный раствор сгущают в ротационном выпарном аппарате, и остаток высушивают в вакууме. Выход: 5,0 г (73% чистота, 73% теор. возм.).
LC-MS (метод I): Rt=2.69 мин; MS (ESIpos): m/z=235 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.13 (s. 1H), 7.85 (s, 1H), 7.58 (s, 1H), 4.03 (q, 2H), 2.69 (br. s, 6H), 1.12 (t, 3H).
Пример 7А
3-(Диметиламино)-2-(4-циано-1H-1,2,3-триазол-1-ил)акриловой кислоты этиловый эфир
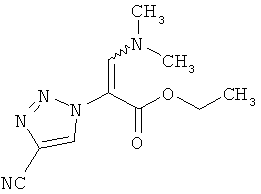
1,3 г (7,5 ммоль) соединения из примера 4А и 1,4 мл (1,2 г, 8,2 ммоль) N,N-диметилформамид-диэтилацетала перемешивают 16 часов при температуре бани 100°C.Для переработки охлажденный реакционный раствор сгущают в ротационном выпарном аппарате, и остаток высушивают в вакууме. Выход: 1,5 г (86% теор. возм.).
LC-MS (метод 4): Rt=1.55 мин; MS (ESIpos): m/z=236 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=9.14 (s, 1Н), 7.75 (s, 1H), 4.04 (q, 2H), 3.15 (br. s, 3H), 2.18 (br. s, 3H), 1.13 (t, 3h).
Пример 8А
3-(Диметиламино)-2-(1Н-1,2,3-триазол-1-ил)акриловой кислоты этиловый эфир
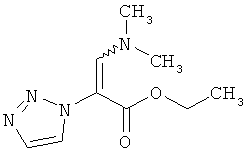
20,0 г (128,9 ммоль) соединения из примера 2А смешивают с 44,2 мл (38,0 г, 257,8 ммоль) N,N-диметилформамид-диэтилацетала, и смесь перемешивают 16 часов при 100°C. После охлаждения до комнатной температуры реакционную смесь сгущают в вакууме. Остаток перемешивают в диэтиловом эфире, фильтруют и промывают диэтиловым эфиром. Выход: 18,0 г (67% теор. возм.). LC-MS (метод 4): Rt=1.20 мин; MS (ESIpos): m/z=211 [M+H]+.
1H-ЯМР (400 МГц. DMSO-d6): δ=8.10 (d, 1H), 7.78 (d, 1H), 7.65 (s, 1H), 4.03 (q, 2H), 3.06 (br. s, 3H), 2.10 (br. s, 3H), 1.12 (t, 3H).
Пример 9А
4-(4-Циклобутилпиперазин-1-ил)-6-гидразинопиримидин
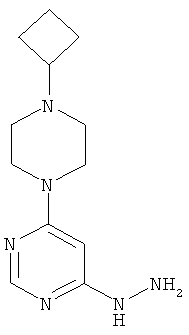
Этап а): 4-Хлор-6-(4-циклобугилпиперазин-1-ил)пиримидин
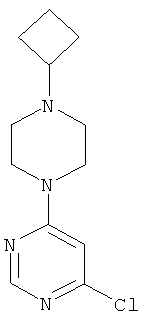
1,8 г (8,4 ммоль) 1-циклобутилпиперазиндигидрохлорида (Zaragoza et al., J. Med. Chem. 2004, 47, 2833) вводят в 18 мл воды и разбавляют 2,9 мл (2,1 г, 16,9 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина. Смесь перемешивают 30 минут при комнатной температуре, и к ней добавляют 1,3 г (8,4 ммоль) 4,6-дихлорпиримидина. Реакционную смесь перемешивают 1 час при 115°C, охлаждают до комнатной температуры, разбавляют 25 мл этилацетата и экстрагируют с помощью насыщенного водного раствора гидрокарбоната натрия. Органическую фазу отделяют, высушивают над сульфатом натрия, фильтруют и сгущают в вакууме. Исходный продукт очищают хроматографией на колонке на силикагеле (растворитель: дихлорметан/метанол 100/3). Выход: 1,9 г (89% теор. возм.)
HPLC (метод 6): Rt=2.79 мин; MS (DCI): m/z=254 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.32 (s, 1H), 6.95 (s, 1H), 3.65-3.58 (m, 4H), 2.70 (квинтет, 1H), 2.27 (t, 4H), 2.00-1.93 (m, 2H), 1.87-1.75 (m, 2H), 1.67-1.55 (m,2H).
Этап b): 4-(4-Циклобутилпиперазин-1-ил)-6-гидразинопиримидин
В раствор 1,9 г (7,5 ммоль) 4-6-(4-циклобутилпиперазин-1-ил)пиримидина в 28 мл этанола при размешивании каплями при комнатной температуре добавляют 4,4 мл (4,5 г, 89,7 ммоль) гидразингидрата. Реакционный раствор перемешивают 16 часов при 80°C.Для переработки раствор сгущают в вакууме, остаток несколько раз перемешивают в диэтиловом эфире, выпавшее твердое вещество отфильтровывают и высушивают в вакууме. Затем остаток очищают хроматографией на колонке на силикагеле (растворитель: дихлорметан/метанол 10/2). Выход: 1,5 г (80% теор. возм.).
LC-MS (метод 6): Rt=1.36 мин; MS (ESIpos): m/z=249 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=7.92 (s, 1H), 7.63 (s, 1H), 5.90 (NH), 4.09 (s, NH2), 3.45 (t, 4H), 2.69 (квинтет, 1H), 2.26 (t, 4H), 2.00-1.93 (m. 2H), 1.82-1.75 (m, 2H), 1.67-1.60 (m, 2H).
Пример 10А
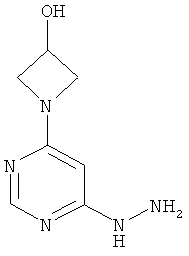
1-(6-Гидразинилпиримидин-4-ил)ацетидин-3-ол
Этап а): 1-(6-хлорпиримидин-4-ил)ацетидин-3-ол
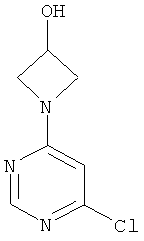
7,3 г (48,7 ммоль) 4,6-дихлорпиримидина в 140 мл превращают в суспензию и смешивают с 47 мл 1 N едкого натра. Добавляют 5,3 г (48,7 ммоль) 3-гидроксиацетидина, и реакционную смесь перемешивают 3 дня при 90°C. После охлаждения до комнатной температуры реакционную смесь сгущают в вакууме и используют в дальнейшем без очистки.
LC-MS (метод 5): Rt=0.36 мин; MS (ESIpos): m/z=1.87 [M+H]+.
Этап b): 1-(6-Гидразинилпиримидин-4-ил)ацетидин-3-ол
В раствор 10,4 г (55,8 ммоль) 1-(6-хлорпиримидин-4-ил)ацетидин-3-ола в 100 мл этанола при размешивании каплями добавляют 27,2 мл (27,9 г, 279,1 ммоль) гидразингидрата при комнатной температуре. Реакционный раствор перемешивают 16 часов при 80°C. Для переработки сгущают в вакууме, осадок отфильтровывают и дважды промывают этанолом (по 10 мл). Выход: 2,0 г (19% теор. возм.).
LC-MS (метод I): Rt=2.06 мин; MS (ESIpos): m/z=194 [M+H]+.
Пример 11А
4-Хлор-6-гидразинопиримидин
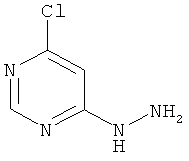
В раствор 20,0 г (134,3 ммоль) 4,6-дихлорпиримидина в 300 мл этанола при перемешивании каплями добавляют 11,8 мл (12,1 г, 241,6 ммоль) гидразингидрата при комнатной температуре. Во время добавления гидразингидрата происходит помутнение раствора, к нему добавляют еще этанол (около 400 мл). Реакционный раствор перемешивают 12 часов при комнатной температуре. Для переработки выпавшее твердое вещество отфильтровывают, фильтрационный осадок дважды промывают водой по 150 мл и дважды диэтиловым эфиром по 100 мл, и продукт высушивают в вакууме. Из сгущенного маточного раствора получают кристаллическую фракцию продукта. Выход: 16,8 г (87% теор. возм.).
LC-MS (метод 1): Rt=1.17 мин; MS (ESIpos): m/z=145 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.81 (s, 1H), 8.17 (br. s, 1H), 6.75 (s, 1H), 4.48 (br. s, 2H).
Пример 12А
2-(6-Хлорпиримидин-4-ил)-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
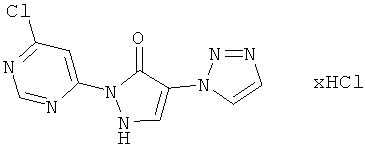
10,0 г (47,7 ммоль) соединения из примера 8А и 8,3 г (57,1 ммоль) соединения из примера ПА вводят в 100 мл этанола и разбавляют 1,5 мл (2,2 г, 19,0 ммоль) трифторуксусной кислоты. Раствор перемешивают 12 часов при рефлюксе. Затем к охлажденной реакционной смеси в избытке добавляют 4М раствора хлористого водорода в диоксане, смесь вымешивают около 1 часа, выпавшие кристаллы отфильтровывают, и фильтрационный осадок промывают диоксаном и этанолом. Полученный промежуточный продукт растворяют в 150 мл этанола, разбавляют 50 мл 25%-ного метанолового раствора метилата натрия, и смесь перемешивают 2 часа при комнатной температуре. Затем реакционную смесь подкисляют 1 N соляной кислотой до рН=5, вымешивают еще 2 часа при комнатной температуре, остаток отфильтровывают, фильтрационный осадок промывают этанолом, и продукт высушивают в вакууме. Выход: 7,0 г (49% теор. возм.).
LC-MS (метод 5): Rt=1.20 мин; MS (ESIpos): m/z=264 [M+H]+
Пример 13А
2-(6-Хлорпиримидин-4-ил)-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
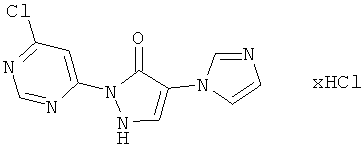
10,0 г (47,8 ммоль) соединения из примера 5А и 8,3 г (57,3 ммоль) соединения из примера 11А вводят в 100 мл этанола и разбавляют 1,5 мл (2,2 г, 19,0 ммоль) трифторуксусной кислоты. Раствор перемешивают 12 часов при рефлюксе. Выпавшие кристаллы отфильтровывают, фильтрационный осадок промывают этанолом, и промежуточный продукт высушивают в течение ночи в вакууме. Затем этот продукт превращают в суспензию в 20 мл, разбавляют 100 мл 4 М раствора хлористого водорода в диоксане, и смесь перемешивают 1 час при комнатной температуре. Твердое вещество отфильтровывают, фильтрационный осадок промывают диоксаном, этилацетатом и диизопропиловым эфиром, и продукт высушивают в вакууме. Выход: 4,6 г (32% теор. возм.).
HPLC (метод 6): Rt=2.81 мин; MS (ESIpos): m/z=263 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.46 (s, 1H), 8.96 (s, 1H), 8.56 (s, 1H), 8.51 (d, 1H), 8.07-8.04 (m, 1H), 7.85-7.82 (m, 1H).
Пример 14А
4-(6-Гидразинилпиримидин-4-ил)-1,4-оксазепан

Этап а): 4-(6-Хлорпиримидин-4-ил)-1,4-оксазепан
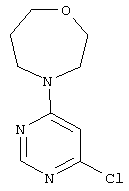
Смесь из 3,0 г (20,1 ммоль) 4,6-дихлорпиримидина, 2,8 г (20,1 ммоль) 1,4-оксазепан-гидрохлорида и 6,4 г (60,4 ммоль) карбоната натрия в 45 мл воды перемешивают 16 часов при рефлюксе. После охлаждения до комнатной температуры реакционную смесь экстрагируют с помощью этилацетата. Органическую фазу промывают насыщенным раствором хлорида натрия, высушивают над сульфатом натрия и фильтруют. Фильтрат сгущают в вакууме до сухого состояния. Продукт получают в виде масла. Выход: 3,9 г (86% теор. возм.).
LC-MS (метод 4): Rt=1.32 мин; MS (ESIpos): m/z=214 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.33 (s, 1H), 6.86 (s, 1H), 3.99-3.52 (m, 8H), 1.84 (m, 2H).
Этап b): 4-(6-Гидразинилпиримидин-4-ил)-1,4-оксазепан
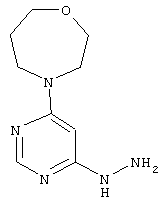
В раствор 3,9 г (18,0 ммоль) 4-(6-хлорпиримидин-4-ил)-1,4-оксазепана в 25 мл этанола при размешивании каплями добавляют 8,8 мл (9,0 г, 180,2 ммоль) гидразингидрата при комнатной температуре. После 16 часов перемешивания при 80°C реакционный раствор сгущают в вакууме. Остаток перемешивают в холодном этаноле, выпавшее твердое вещество отфильтровывают, и фильтрационный осадок промывают 25 мл диэтилового эфира. Продукт высушивают в вакууме. Выход: 1,4 г (36% теор. возм.).
HPLC (метод 11): Rt=2.48 мин; MS (ESIpos): m/z=210 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): 5=7.91 (s, 1H), 7.56 (br. s, 1H), 5.81 (s, 1H), 4.12 (br. s, 2H), 3.75-3.55 (m, 8H), 1.85 (квинтет, 2Н).
Примеры исполнения
Пример 1
2-[6-(4-Циклобутилпиперазин-1-ил)пиримидин-4-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
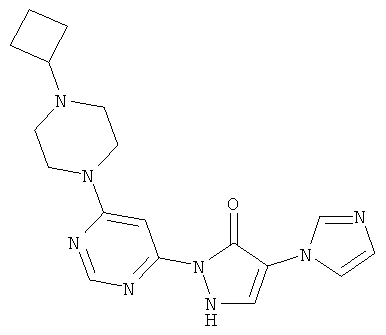
Смесь из 211 мг (1,0 ммоль) соединения из примера 5А и 250 мг (1,0 ммоль) соединения из примера 9А в 4 мл этилацетата разбавляют 16 мкл (23 мг, 0,2 ммоль) трифторуксусной кислоты и перемешивают 20 часов при 100°C. Реакционную смесь сгущают в вакууме, вновь разбавляют одинаковым количеством этилацетата и трифторуксусной кислоты и перемешивают еще 20 часов при 100°C. Реакционную смесь охлаждают до комнатной температуры, выпавшее твердое вещество отфильтровывают и промывают диэтиловым эфиром. Остаток очищают сначала хроматографией на колонке через силикагель (растворитель: дихлорметан/метанол/аммиак 10/2/0,2), а затем с помощью препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 137 мг (36% теор. возм.).
HPLC (метод 6): Rt=2.73 мин; MS (ESIpos): m/z=367 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.40 (s, 1H), 8.12 (s, 1H), 7.90 (s, 1H), 7.74 (s, 1H), 7.50 (s, 1H), 7.07 (s, 1H), 3.65-3.58 (m, 4H), 2.77 (квинтет, 1Н), 2.38-2.35 (m, 4H), 2.01-1.96 (m, 2H), 1.89-1.79 (m, 2H), 1.67-1.62 (m, 2H).
Пример 2
2-[6-(4-Циклобутилпиперазин-1-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
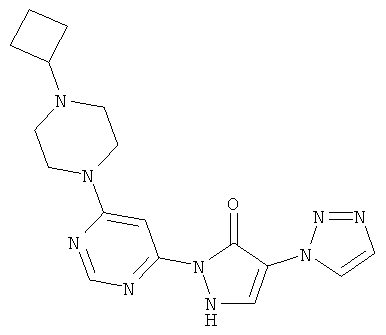
Смесь из 211 мг (1,0 ммоль) соединения из примера 8А и 250 мг (1,0 ммоль) соединения из примера 9А в 4 мл этилацетата разбавляют 16 мкл (23 мг, 0,2 ммоль) трифторуксусной кислоты и перемешивают 20 часов при 100°С.Реакционную смесь сгущают в вакууме, вновь разбавляют одинаковым количеством этилацетата и трифторуксусной кислоты и перемешивают 3 дня при 100°C. Реакционную смесь охлаждают до комнатной температуры, выпавшее твердое вещество отфильтровывают и промывают диэтиловым эфиром. Остаток превращают в суспензию в 2 мл воды, растворяют путем добавления водного 1 N едкого натра (рН=9-10) и очищают с помощью препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 195 мг (51% теор. возм.).
HPLC (метод 6): Rt=2.90 мин; MS (ESIpos): m/z=368 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.41 (s, 1H), 8.40 (s, 1H), 7.88 (s, 1H), 7.82 (s, 1H), 7.75 (s, 1H), 3.70-3.65 (m, 4H), 3.03-2.97 (m, 1H), 2.60-2.57 (m, 4H), 2.06-2.02 (m, 2H), 1.98-1.90 (m, 2H), 1.70-1.64 (m, 2H).
Пример 3
1-{2-[6-(4-Циклобутилпиперазин-1-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-имидазол-4-карбонитрил
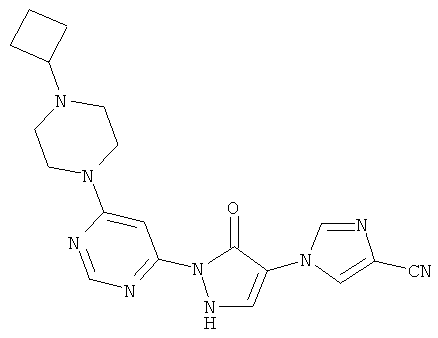
Смесь из 236 мг (1,0 ммоль) соединения из примера 6А и 250 мг (1,0 ммоль) соединения из примера 9А в 4 мл этилацетата разбавляют 16 мкл (23 мг, 0,2 ммоль) трифторуксусной кислоты и перемешивают 20 часов при 100°C. Реакционную смесь сгущают в вакууме, вновь разбавляют одинаковым количеством этилацетата и трифторуксусной кислоты и перемешивают 3 дня при 100°С. Реакционную смесь охлаждают до комнатной температуры, выпавшее твердое вещество отфильтровывают и промывают диэтиловым эфиром. Остаток превращают в суспензию в 2 мл воды, растворяют путем добавления водного 1 N едкого натра (рН=9-10) и очищают с помощью препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 219 мг (56% теор. возм.).
HPLC (метод 6): Rt=3.10 мин; MS (ESIpos): m/z=392 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.38 (d, 1H), 8.37 (d, 1H), 8.17 (d, 1H), 7.82 (s, 1H), 7.80 (s, 1H), 3.68-3.62 (m, 4H), 3.38-3.30 (m, 4H), 2.96-2.91 (m, 1H), 2.06-2.00 (m, 2H), 1.95-1.85 (m, 2H), 1.70-1.63 (m, 2H).
Пример 4
1-{2-[6-(3-Гидроксиацетидин-1-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-имидазол-4-карбонитрил
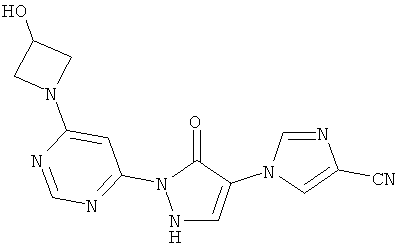
Смесь из 700 мг (3,0 ммоль) соединения из примера 6А и 541 мг (3,0 ммоль) соединения из примера 10А в 10 мл этилацетата разбавляют 46 мкл (68 мг, 0,6 ммоль) трифторуксусной кислоты и перемешивают 10 часов при 100°C. Реакционную смесь сгущают в вакууме, растворяют 5 мл этанола, и осадок отфильтровывают. Твердое вещество превращают в суспензию в 10 мл воды и разбавляют до растворения с помощью 1 N едкого натра (рН=9). Затем 1 N соляной кислотой устанавливается рН=7, раствор сгущают до объема порядка 5 мл, и образовавшийся осадок отфильтровывают. Остаток промывают водой и диизопропиловым эфиром и хроматографируется с помощью препаративной HPLC (метод 8). Затем твердое вещество превращают в суспензию в 10 мл воды и разбавляют до растворения 1 N едким натром (рН=9). Затем с помощью 1 N соляной кислоты устанавливается рН=7, раствор сгущают до объема порядка 2,5 мл, и образовавшийся осадок отфильтровывают. Фильтрат сгущают до объема около 2 мл и снова фильтруют. Оба остатка объединяют, промывают водой и этилацетатом и высушивают в вакууме. Выход: 78 мг (8% теор. возм.).
HPLC (метод 6): Rt=2.90 мин; MS (ESIpos): m/z=325 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.35 (s, 1H), 8.29 (s, 1H), 8.17 (s, 1H), 7.66 (s, 1H), 7.34 (s, 1H), 5.80-5.75 (m, 1H), 4.63-4.58 (m, 1H), 4.24-4.20 (m, 2H), 3.75-3.73 (m, 2H).
Пример 5
1-{6-[4-(1H-Имидазол-1-ил)-5-оксо-2,5-дигидро-1H-пиразол-1-ил]пиримидин-4-ил}ацетидин-3-карбоновая кислота
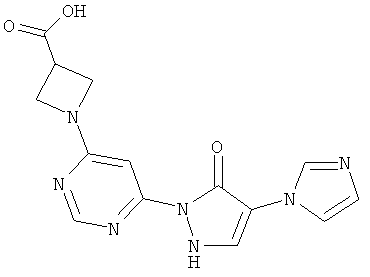
46 мг (0,3 ммоль) гидрохлорида ацетидин-3-карбоновой кислоты вводят в смесь из 1 мл воды и 0,3 мл этанола. Добавляют 100 мг (0,3 ммоль) соединения из примера 13А, и смесь перемешивают 1 час при 100°C. Затем реакционную смесь разводят водным 1 N едким натром до рН=7 и перемешивают 16 часов при 100°C. Вновь разводят 1 N едким натром до рН=7 и подвергают реакции обмена в течение 1 часа при 150°C в одномодовой микроволновой печи (single mode-Mikrowelle (Emrys Optimizer)). Реакционную смесь сгущают в вакууме и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 23 мг (21% теор. возм.).
LC-MS (метод 8): Rt=0.86 мин; MS (ESIpos): m/z=328 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.41 (s, 1H), 7.79 (s, 1H), 7.47 (s, 1H), 7.36 (s, 1H), 7.31 (s, 1H), 6.89 (s, 1H), 4.09 (t, 2H), 3.99 (t, 2H).
Пример 6
1-{6-[5-оксо-4-(177-1,2,3-триазол-1-ил)-2,5-дигидро-1H-пиразол-1-ил]пиримидин-4-ил}ацетидин-3-карбоновая кислота
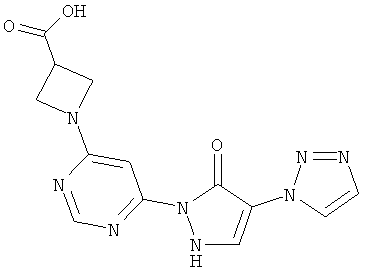
55 мг (0,4 ммоль) гидрохлорида ацетидин-3-карбоновой кислоты вводят в 2 мл воды. Добавляют 100 мг (0,3 ммоль) соединения из примера 12А, и смесь разводят водным 1 N едким натром до рН=7. Раствор в течение 1 часа при 150°C подвергают реакции обмена в одномодовой микроволновой печи (Emrys Optimizer). Реакционную смесь сгущают в вакууме и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 15 мг (13% теор. возм.).
HPLC (метод 6): Rt=2.81 мин; MS (ESIpos): m/z=329 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1H), 8.26 (s, 1H), 7.70 (s, 1H), 7.67 (s, 1H), 7.34 (s, 1H), 4.05-3.96 (m, 2H und 2H), 3.13-3.07 (m, 1H).
Пример 7
4-(1H-Имидазол-1-ил)-2-[6-(3-метилацетидин-1-ил)пиримидин-4-ил]-1,2-дигидро-3H-пиразол-3-он
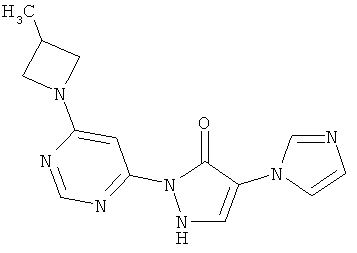
43 мг (0,4 ммоль) 3-метилацетидингидрохлорида, 100 мг (0,3 ммоль) соединения из примера 13А и 174 мкл (130 мг, 1,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина превращают в суспензию в 2 мл тетрагидрофурана, и смесь подвергают реакции обмена в течение 4,5 часов при 120°C в одномодовой микроволновой печи (Emrys Optimizer). Реакционную смесь сгущают в вакууме, разбавляют водой при добавлении водного 1 N едкого натра (до рН=9-10) и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 30 мг (30% теор. возм.).
HPLC (метод б): Rt=3.07 мин; MS (ESIpos): m/z=298 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.25 (s, 1H), 7.81 (s, 1H), 7.46 (s, 1H), 7.39 (s, 1H), 7.31 (s, 1H), 6.88 (s, 1H), 4.10 (t, 2H), 3.54 (dd, 2H), 2.86-2.75 (m, 1H), 1.25 (d, 3H).
Пример 8
2-[6-(3-Метилацетидин-1-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
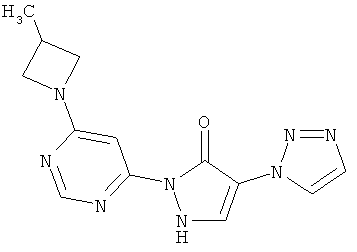
43 мг (0,4 ммоль) 3-метилацетидингидрохлорида, 100 мг (0,3 ммоль) соединения из примера 12А и 174 мкл (130 мг, 1,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина превращают в суспензию в 2 мл тетрагидрофурана, и смесь подвергают реакции обмена в течение 1,5 часов при 120°C в одномодовой микроволновой печи (Emrys Optimizer). Реакционную смесь сгущают в вакууме, разбавляют водой при добавлении водного 1 N едкого натра (рН=9-10), и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 25 мг (25% теор. возм.).
HPLC (метод 6): Rt=3.00 мин; MS (ESIpos): m/z=299 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.41 (s, 1H), 8.27 (s, 1H), 7.70 (s, 1H), 7.67 (s, 1H), 7.41 (s, 1H), 4.11 (t, 2H), 3.56 (dd, 2H), 2.86-2.78 (m, 1H), 1.25 (d, 3H).
Пример 9
2-{6-[3-(Диметиламино)ацетидин-1-ил]пиримидин-4-ил}-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-З -она дигидрохлорид
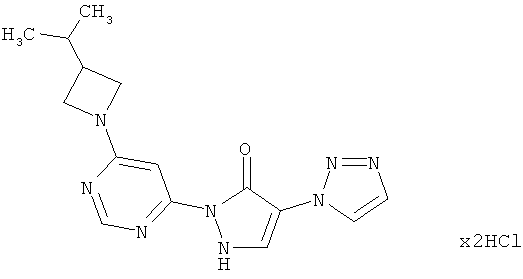
271 мг (1,5 ммоль) N,N-диметилацетидин-3-аминдигидрохлорида, 400 мг (1,5 ммоль) соединения из примера 12А и 847 мг (6.1 ммоль) карбоната калия превращают в суспензию в 8 мл N,N-диметилформамида, и смесь перемешивают 16 часов при 100°C. Реакционную смесь сгущают в вакууме и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода при добавлении 0,1%-ной трифторуксусной кислоты). Следующую очистку осуществляют посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода при добавлении 0,1%-ной муравьиной кислоты). Содержащие продукт фракции разбавляют 2 мл 1 N соляной кислоты и перемешивают 1 час при комнатной температуре. Твердое вещество отфильтровывают и высушивают в вакууме. Выход: 62 мг (17% теор. возм.).
LC-MS (метод 5): Rt=0.19 мин; MS (ESIpos): m/z=328 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1H), 8.28 (s, 1H), 7.69 (s, 1H), 7.66 (s, 1H), 7.52 (s, 1H), 4.02 (t, 2H), 3.76 (dd, 2H), 3.24-3.18 (m, 1H), 2.12 (s, 6H).
Пример 10
2-[6-(4,4-дифторпиперидин-1-ил)пиримидин-4-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
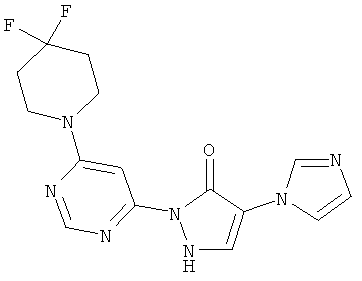
100 мг (0,3 ммоль) соединения из примера 13А, 63 мг (0,4 ммоль) 4,4-дифторпиперидин-гидрохлорида и 116 мкл (86 мг, 0,7 ммоль) N-этил-N-(пропан-2-ил)пропанп2-амина вводятся в 2 мл тетрагидрофурана, и смесь подвергают реакции обмена в течение 2,5 часов при 120°C в одномодовой микроволновой печи (Emrys Optimizer). После сгущения смеси в вакууме остаток разводят в ацетонитриле и воде и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 82 мг (71% теор. возм.).
HPLC (метод 6): Rt=3.37 мин; MS (ESIpos): m/z=348 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-dg): §=8.49 (s, 1H), 8.38 (s, 1H), 8.15 (s, 1H), 7.76 (s, 1H), 7.64 (s, 1H), 7.22 (s, 1H), 3.80 (t, 4H), 2.06 (гептет, 4Н).
Пример 11
2-[6-(4,4-дифторпиперидин-1-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
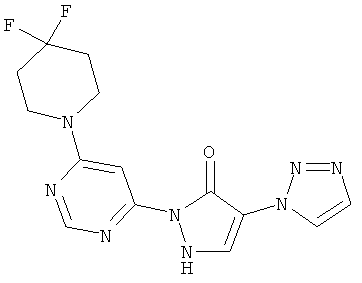
250 мг (0,8 ммоль) соединения из примера 12А, 158 мг (1,0 ммоль) 4,4-дифторпиперидин-гидрохлорида и 435 мкл (323 мг, 2,5 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина вводят в 5 мл тетрагидрофурана, и смесь подвергают реакции обмена в течение 30 минут при 120°C в одномодовой микроволновой печи (Emrys Optimizer). После предварительного очищения смеси посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода) продукт дополнительно очищают хроматографией на колонке через силикагель (растворитель: дихлорметан/метанол, 10/1). Выход: 29 мг (10% теор. возм.).
HPLC (метод 6): Rt=3.49 мин; MS (ESIpos): m/z=349 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.56 (s, 1H), 8.39 (s, 1H), 8.30 (s, 1H), 7.87 (s, 1H), 7.57 (s, 1H), 3.91-3.81 (m. 4H), 2.09 (гептет, 4H).
Пример 12
1-{2-[6-(4,4-дифторпиперидин-1-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-имидазол-4-карбонитрил
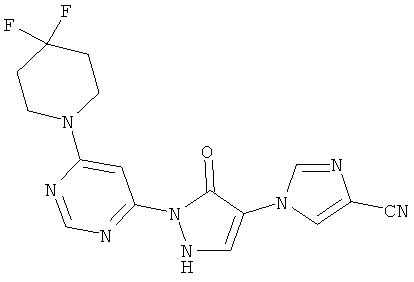
Смесь из 200 мг (1,4 ммоль) соединения из примера 11А, 262 мг (1,7 ммоль) 4,4-дифторпиперидингидрохлорида и 289 мкл (215 мг, 1,7 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина перемешивают в 3 мл воды 16 часов при 100°C. После добавления 53 мкл (79 мг, 0.7 ммоль) трифторуксусной кислоты и 324 мг (1,4 ммоль) соединения из примера 6А полученную реакционную смесь перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 111 мг (21% теор. возм.).
LC-MS (метод 4): Rt=1.69 мин; MS (ESIpos): m/z=373 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.55 (s, 1H), 8.44 (d, 1H), 8.33 (s, 1H), 8.22 (d, 1H), 7.60 (br. s, 1H), 3.84 (br. s, 4H), 2.09 (гептет, 4Н).
Пример 13
1-{2-[6-(4,4-дифторпиперидин-1-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-1,2,3-триазол-4-карбонитрил
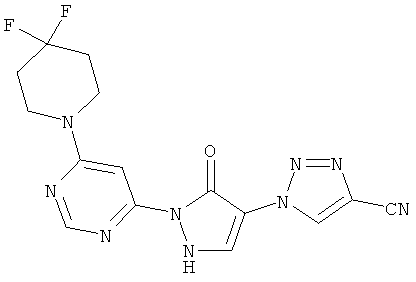
Смесь из 200 мг (1,4 ммоль) соединения из примера 11А, 262 мг (1,7 ммоль) 4,4-дифторпиперидингидрохлорида и 289 мкл (215 мг, 1,7 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина перемешивают в 3 мл воды 16 часов при 100°C. После добавления 53 мкл (79 мг, 0.7 ммоль) трифторуксусной кислоты и 325 мг (1,4 ммоль) соединения из примера 7А полученную реакционную смесь перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 34 мг (7% теор. возм.).
LC-MS (метод 4): Rt=1.77 мин; MS (ESIpos): m/z=374 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=9.24 (s, 1H), 8.59 (s, 1H), 8.27 (s, 1H), 7.54 (s, 1H), 3.90 (br. s, 4H), 2.12 (гептет, 4Н).
Пример 14
2-[6-(3,3-дифторпирролидин-1-ил)пиримидин-4-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
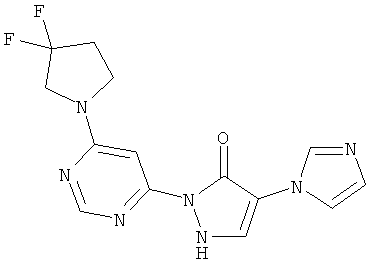
100 мг (0,3 ммоль) соединения из примера 13А, 58 мг (0,4 ммоль) 3,3-дифторпирролидин-гидрохлорида и 175 мкл (130 мг, 1,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина вводят в 2 мл тетрагидрофурана, и смесь подвергают реакции обмена в течение 1 часа при 120°C в одномодовой микроволновой печи (Emrys Optimizer). После сгущения в вакууме остаток разводят ацетонитрилом и водой и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 111 мг (99% теор. возм.).
HPLC (метод 6): Rt=3.18 мин; MS (ESIpos): m/z=334 [М+Н]+;
1Н-ЯМР (400 МГц, метанол-d4): δ=8.41 (s, 1H), 7.85 (s, 1H), 7.62 (s, 1H), 7.56 (s, 1H). 7.29 (s, 1H), 7.03 (s, 1H), 3.91 (t, 2H), 3.76 (t, 2H), 2.55 (гептет, 2Н).
Пример 15
2-[6-(3,3-дифторпирролидин-1-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
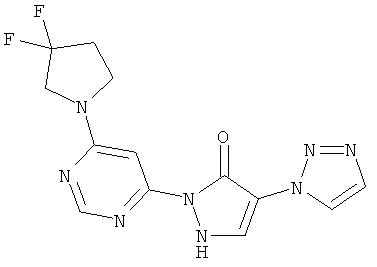
100 мг (0,8 ммоль) соединения из примера 12А, 57 мг (0,4 ммоль) 3,3-дифторпирролидин-гидрохлорида и 174 мкл (129 мг, 1,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина вводят в 2 мл тетрагидрофурана, и смесь подвергают реакции обмена в течение 30 минут при 120°C в одномодовой микроволновой печи (Emrys Optimizer). После сгущения в вакууме остаток разводят ацетонитрилом и водой и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 13 мг (12% теор. возм.).
HPLC (метод 6): Rt=3.33 мин; MS (ESIpos): m/z-335 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1H), 8.35 (s, 1H), 7.72-7.66 (m, 3H), 3.87 (t, 2H), 3.65 (t, 2H), 2.64-2.50 (m, частично при сигнале от DMSO, 2H).
Пример 16
1-{2-[6-(3,3-дифторпирролидин-1-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-имидазол-4-карбонитрил
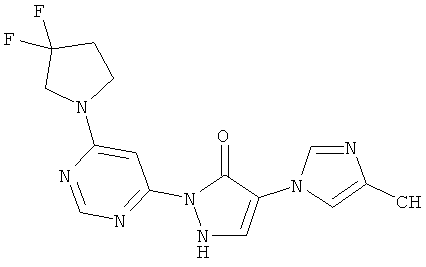
Смесь из 200 мг (1,4 ммоль) соединения из примера 11А, 238 мг (1,7 ммоль) 3,3-дифторпирролидингидрохлорида и 289 мкл (215 мг, 1,7 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина в 3 мл воды перемешивают 16 часов при 100°C. После добавления 53 мкл (79 мг, 0,7 ммоль) трифторуксусной кислоты и 324 мг (1,4 ммоль) соединения из примера 6А полученную реакционную смесь перемешивают 16 часов при 100°C.Реакционную смесь смешивается с 1 мл 1 N соляной кислоты. Выпавший при этом гидрохлорид исходного продукта отфильтровывают, промывают диэтиловым эфиром и высушивают. Промежуточный продукт содержит еще непрореагировавшее исходное соединение (соединение из примера 11А). Поэтому промежуточный продукт подвергают реакции обмена со 108 мкл (80 мг, 0,6 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина, 10 мг (0,1 ммоль) 3,3-дифторпирролидингидрохлорида и 2 мл воды в течение 15 минут при 170°C в одномодовой микроволновой печи (Emrys Optimizer). Реакционный раствор очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 30 мг (6% теор. возм.).
LC-MS (метод 4): Rt=1.58 мин; MS (ESIpos): m/z=359 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): 8=8.55 (s, 1H), 8.44 (d, 1H), 8.33 (s, 1H), 8.21 (d, 1H), 7.26 (br. s, 1H), 3.99 (t, 2H), 3.75 (br. s, 2H), 2.66-2.54 (m, частично при сигнале от DMSO, 2H).
Пример 17
1-{2-[6-(3,3-дифторпирролидин-1-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-1,2,3 -триазол-4-карбонитрил
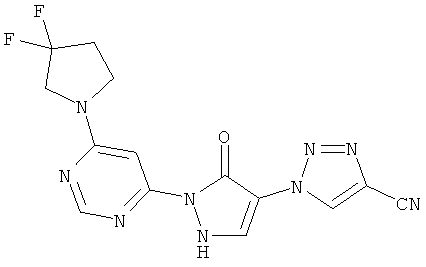
Смесь из 200 мг (1,4 ммоль) соединения из примера 11А, 238 мг (1,7 ммоль) 3,3-дифторпирролидингидрохлорида и 289 мкл (215 мг, 1,7 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина в 33 мл воды перемешивают 16 часов при 100°C. После добавления 53 мкл (79 мг, 0,7 ммоль) трифторуксусной кислоты и 325 мг (1,4 ммоль) соединения из примера 7А полученную реакционную смесь перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 137 мг (27% теор. возм.).
LC-MS (метод 4): Rt=1.66 мин; MS (ESIpos): m/z=360 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=9.25 (s, 1H), 8.61 (s, 1H), 8.29 (s, 1H), 7.21 (br. s, 1H), 4.06 (t, 2H), 3.82 (br. s, 2H), 2.69-2.55 (m, частично при сигнале от DMSO, 2H).
Пример 18
2-[6-(3,3-дифторацетидин-1-ил)пиримидин-4-ил]-4-(1H-имидазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
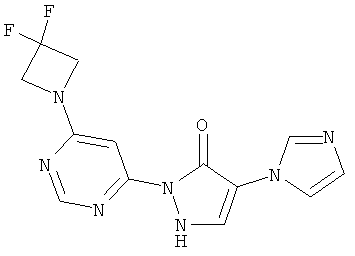
100 мг (0,3 ммоль) соединения из примера 13А, 52 мг (0,4 ммоль) 3,3-дифторацетидин-гидрохлорида и 175 мкл (130 мг, 1,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина вводят в 2 мл тетрагидрофурана, и смесь подвергают реакции обмена в течение 3 часов при 120°C в одномодовой микроволновой печи (Emrys Optimizer). Выпавшее твердое вещество отфильтровывают и фильтрат сгущают в вакууме. Остаток разводят ацетонитрилом и водой и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 36 мг (32% теор. возм.).
HPLC (метод 6): Rt=3.00 мин; MS (ESIpos): m/z=320 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1H), 8.16 (s, 1H), 7.79 (s, 1H), 7.56 (s, 1H), 7.50 (s, 1H), 7.07 (s, 1H), 4.49 (t, 4H).
Пример 19
2-[6-(3,3-дифторацетидин-1-ил)пиримидин-4-ил]-4-(1Н-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
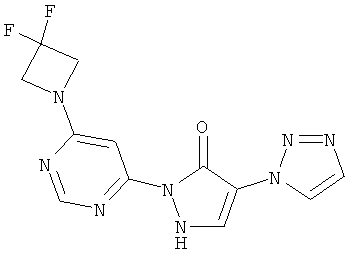
100 мг (0,3 ммоль) соединения из примера 12А, 52 мг (0,4 ммоль) 3,3-дифторацетидин-гидрохлорида и 174 мкл (130 мг, 1,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина вводят в 2 мл тетрагидрофурана, и смесь подвергают реакции обмена в течение 30 минут при 120°C в одномодовой микроволновой печи (Emrys Optimizer). После сгущения в вакууме остаток разводят ацетонитрилом и водой и очищают посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 36 mg (34% теор. возм.).
HPLC (метод 6): Rt=3.20 мин; MS (ESIpos): m/z=321 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.42 (s, 1H), 8.39 (s, 1H), 7.71 (s, 2H), 7.62 (s, 1H), 4.46 (t, 4H).
Пример 20
1-{2-[6-(3,3-дифторацетидин-1-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-имидазол-4-карбонитрил
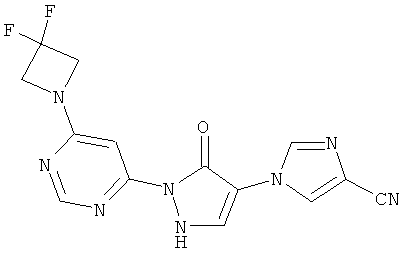
Смесь из 120 мг (0,8 ммоль) соединения из примера 11А, 129 мг (1,0 ммоль) 3,3-дифторацетидингидрохлорида и 174 мкл (129 мг, 1,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина перемешивают в 3 мл воды 16 часов при 100°C.
После добавления 32 мкл (47 мг, 0,4 ммоль) трифторуксусной кислоты и 194 мг (0,8 ммоль) соединения из примера 6А полученную реакционную смесь перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 32 мг (11% теор. возм.).
LC-MS (метод 4): Rt=1.48 мин; MS (ESIpos): m/z=345 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=8.80-8.08 (m, 4H), 7.25 (s, 1H), 4.61 (br. s, 4H).
Пример 21
1-{2-[6-(3,3-дифторацетидин-1-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-1,2,3-триазол-4-карбонитрил
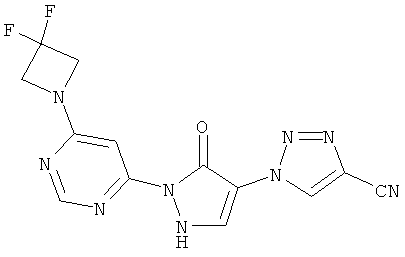
Смесь из 120 мг (0,8 ммоль) соединения из примера 11А, 129 мг (1,0 ммоль) 3,3-дифторацетидингидрохлорида и 174 мкл (129 мг, 1,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина в 3 мл воды перемешивают 16 часов при 100°C. После добавления 32 мкл (47 мг, 0,4 ммоль) трифторуксусной кислоты и 194 мг (0,8 ммоль) соединения из примера 7А полученную реакционную смесь перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 51 мг (18% теор. возм.).
LC-MS (метод 4): Rt=1.56 мин; MS (ESIpos): m/z=346 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=9.26 (s, 1H), 8.60 (s, 1H), 8.36 (s, 1H), 7.18(s,1H), 4.69 (t,4H).
Пример 22
4-(1H-Имидазол-1-ил)-2-[6-(1,4-оксазепан-4-ил)пиримидин-4-ил]-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
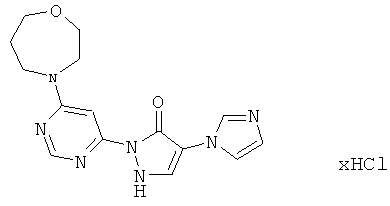
Смесь из 200 мг (1,0 ммоль) соединения из примера 5А, 200 мг (1,0 ммоль) соединения из примера 14А и 37 мкл (54 мг, 0,5 ммоль) трифторуксусной кислоты в 3 мл воды перемешивают 16 часов при 100°C. После предварительной очистки посредством препаративной HPLC (RР18-колонка; растворитель: градиент ацетонитрил/вода) промежуточный продукт перемешивают с 1 мл 1 N соляной кислоты, отфильтровывают и промывают диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 21 мг (6%. теор. возм.).
LC-MS (метод 4): Rt=0.95 мин; MS (ESIpos): m/z=364 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-d6): 5=9.46 (s, 1H), 8.56 (s, 1H), 8.30 (s, 1H), 8.06 (t, 1H), 7.83 (t, 1H), 7.50-7.33 (m, 1H), 4.05 (br. s, 2H), 3.91-3.70 (m, 4H), 3.68 (t, 2H), 2.04-1.73 (m,2H).
Пример 23
2-[6-(1,4-Оксазепан-4-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он-гидрохлорид
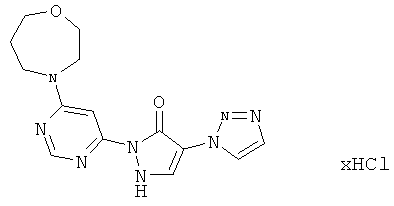
Смесь из 400 мг (1,3 ммоль) соединения из примера 12А и 220 мг (1,6 ммоль) 1,4-оксазепангидрохлорида подвергают реакции обмена в 4 мл пропан-2-ола в течение 30 минут при 115°C в одномодовой микроволновой печи (Emrys Optimizer). Добавляют 232 мкл (172 мг, 1,3 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина, и реакционную смесь подвергают реакции обмена в течение 20 минут при 115°C в одномодовой микроволновой печи (Emrys Optimizer). После сгущения в вакууме остаток разводят ацетонитрилом/водой/трифторуксусной кислотой и обрабатывают препаративной HPLC (метод 9). Полученную путем разделения хроматографией HPLC, лиофилизированную соль трифторацетата с помощью 2 мл 1 N соляной кислоты переводят в гидрохлорид. Выход: 7 мг (1% теор. возм.).
LC-MS (метод 4): Rt=1.20 мин; MS (ESIpos): m/z=329 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ-8.54 (s, 1H), 8.38 (s, 1H), 8.25 (s, 1H), 7.86 (d, 1H), 7.35 (br. s, 1H), 4.14-3.69 (m, 6H), 3.67 (t, 2H), 2.03-1.77 (m, 2H).
Пример 24
2-[6-(1,4-Оксазепан-4-ил)пиримидин-4-ил]-4-(1Н-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
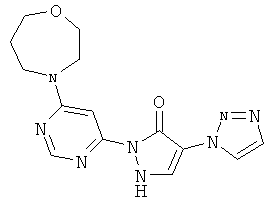
Смесь из 500 мг (1,7 ммоль) соединения из примера 12А, 688 мг (5,0 ммоль) 1,4-оксазепангидрохлорида и 1,4 мл (1077 мг, 8,3 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина подвергают реакции обмена в 10 мл тетрагидрофурана и этанола (1/1) в течение 30 минут при 140°C в одномодовой микроволновой печи (Emrys Optimizer). После предварительной очистки посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода) промежуточный продукт разводят ацетонитрилом и трифторуксусной кислотой и обрабатывают препаративной HPLC (метод 10). В полученной путем разделения хроматографией HPLC, лиофилизированной соли трифторацетата с помощью 1 N едкого натра устанавливают рН=7-8. Продукт выделяют посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 176 мг (31% теор. возм.).
LC-MS (метод 4): Rt=1.19 мин; MS (ESIpos): m/z=329 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): 6=8.41 (d, 1H), 8.33 (d, 1H), 7.78 (br. s, 1H), 7.72-7.70 (m, 2H), 3.91-3.65 (m, 6H), 3.62 (t, 2H), 1.95-1.83 (m, 2H).
Пример 25
1-{2-[6-(1,4-Оксазепан-4-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-имидазол-4-карбонитрил
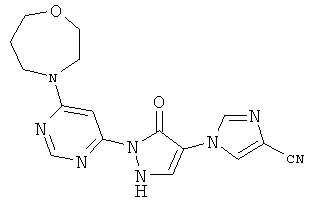
Смесь из 200 мг (0,9 ммоль) соединения из примера 6А, 178 мг (0,9 ммоль) соединения из примера 14А и 33 мкл (49 мг, 0,4 ммоль) трифторуксусной кислоты в 3 мл воды перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 120 мг (40% теор. возм.).
HPLC (метод 6): Rt=3.27 мин; MS (ESIpos): m/z=353 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.52 (s, 1H), 8.42 (d, 1H), 8.25 (s, 1H), 8.19 (d, 1H), 7.39 (br. s, 1H), 4.14-3.70 (m, 6H), 3.66 (t, 2H), 2.05-1.68 (m, 2H).
Пример 26
1-{2-[6-(1,4-Оксазепан-4-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-1,2,3-триазол-4-карбонитрил
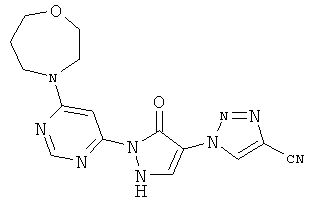
Смесь из 200 мг (0,9 ммоль) соединения из примера 7А, 178 мг (0,9 ммоль) соединения из примера 14А и 33 мкл (49 мг, 0,4 ммоль) трифторуксусной кислоты в 3 мл воды перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 80 мг (27% теор. возм.).
LC-MS (метод 4): Rt=1.40 мин; MS (ESIpos): m/z=354 [M+H]+;
1H-ЯМР (400 МГц, DMSO-d6): δ=9.22 (s, 1H), 8.56 (s, 1H), 8.21 (s, 1H), 7.45-7.26 (m, 1H), 4.06 (br. s, 2H), 3.92-3.71 (m, 4H), 3.68 (t, 2H), 2.06-1.74 (m, 2H).
Пример 27
2-[6-(1,2-Оксазинан-2-ил)пиримидин-4-ил]-4-(1H-1,2,3-триазол-1-ил)-1,2-дигидро-3H-пиразол-3-он
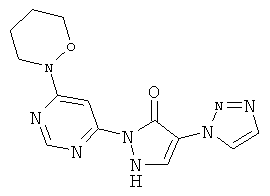
Смесь из 200 мг (0,7 ммоль) соединения из примера 12А, 99 мг (0,8 ммоль) 1,2-оксазинангидрохлорида [Bhat et al., J. Chem. Soc. Perkin Trans. 2 2000, 7, 1435-1446] и 348 мкл (258 мг, 2,0 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина подвергают реакции обмена в 4 мл тетрагидрофурана в течение 30 минут при 140°C в одномодовой микроволновой печи (single mode-Mikrowelle (Emrys Optimizer)). После предварительной очистки посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода) промежуточный продукт разводят ацетонитрилом/водой/метанолом и обрабатывают препаративной HPLC (метод 11). В полученной путем разделения хроматографией HPLC, лиофилизированной соли формиата с помощью 1 N едкого натра устанавливают рН=7-8. Продукт выделяют посредством препаративной HPLC (RP18-колонка; растворитель: градиент ацетонитрил/вода). Выход: 23 мг (11% теор. возм.).
LC-MS (метод 4): Rt=1.38 мин; MS (ESIpos): m/z=315 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ=8.58 (d, 1H), 8.42 (d, 1H), 8.40 (br. s, 1H), 7.88 (d, 1H), 7.66 (br. s, 1H), 4.09 (t, 2H), 3.98 (t, 2H), 1.86-1.68 (m, 4H).
Пример 28
1-{2-[6-(1,2-Оксазинан-2-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1H-имидазол-4-карбонитрил
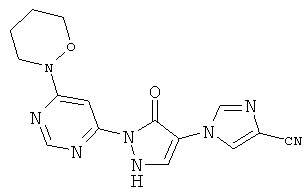
Смесь из 200 мг (1,4 ммоль) соединения из примера 11А, 205 мг (1,7 ммоль) 1,2-оксазинангидрохлорида [Bhat et al., J. Chem. Soc. Perkin Trans. 2 2000, 7, 1435-1446] и 289 мкл (215 мг, 1,7 ммоль) N-этил-N-(пропан-2-ил)пропан-2 -амина в 3 мл воды перемешивают 1,5 часа при 100°C. После добавления 59 мкл (49 мг, 0,4 ммоль) трифторуксусной кислоты и 324 мг (1,4 ммоль) соединения из примера 6А полученную реакционную смесь перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 199 мг (39% теор. возм.).
LC-MS (метод 5): Rt=0.87 мин; MS (ESIpos): m/z=339 [M+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): 8=8.57 (d, 1H), 8.45 (d, 1H), 8.40 (br. s, 1H), 8.23 (d, 1H), 7.70 (br. s, 1H), 4.07 (t, 2H), 3.96 (t, 2H), 1.85-1.65 (m, 4H).
Пример 29
1-{2-[6-(1,2-Оксазинан-2-ил)пиримидин-4-ил]-3-оксо-2,3-дигидро-1H-пиразол-4-ил}-1Н-1,2,3-триазол-4-карбонитрил
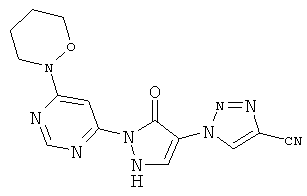
Смесь из 200 мг (1,4 ммоль) соединения из примера 11А, 205 мг (1,7 ммоль) 1,2-оксазинангидрохлорида [Bhat et al., J. Chem. Soc. Perkin Trans. 2 2000, 7, 1435-1446] и 289 мкл (215 мг, 1,7 ммоль) N-этил-N-(пропан-2-ил)пропан-2-амина в 3 мл воды перемешивают 1,5 часа при 100°C. После добавления 59 мкл (49 мг, 0,4 ммоль) трифторуксусной кислоты и 325 мг (1,4 ммоль) соединения из примера 7А полученную реакционную смесь перемешивают 16 часов при 100°C. Выпавшее твердое вещество отфильтровывают и промывают сначала водой, а затем диэтиловым эфиром. Продукт высушивают в вакууме. Выход: 122 мг (24% теор. возм.).
LC-MS (метод 4): Rt=1.66 мин; MS (ESIpos): m/z=340 [М+Н]+;
1H-ЯМР (400 МГц, DMSO-d6) δ=9.28 (s, 1H), 8.58 (s, 1H), 8.36 (s, 1H), 7.60 (br. s, 1H), 4.12 (t, 2H), 4.03 (t, 2H), 1.88-1.70 (m, 4H).
В. Оценка фармакологической эффективности
Фармакологические свойства соединений согласно изобретению могут быть показаны в следующих испытаниях:
Сокращения:
DMEM Среда Dulbecco's Modified Eagle
FCS Фетальная телячья сыворотка
ТМВ 3,3',5,5'-тетраметилбензидин
Iris Трис(гидроксиметил)-аминометан
1. Испытания in vitro для определения активности и селективности ингибиторов НIF-пролил-4-гидроксидазы
1.а) Угнетение активности HIF-пролилгидроксилазы:
Гидроксилированный HIF особо привязан к комплексу протеин Гиппеля-Линдау - элонгин В - элонгин С (VBC-комплекс). Такое взаимодействие происходит только в том случае, если HIF гидроксилирован в консервированном остатке пролила. Это является основой для биохимического определения активности HIF-пролилгидроксилазы. Испытание проводят по способу, описанному в статье [Oehme F., Jonghaus W., Narouz-Ott L., Huetter J., Flamme I., Anal. Biochem. 330(1), 74-80 (2004)]:
Прозрачный, покрытый NeutrAvidin НВС 96-луночный микротитрационный планшет (фирма Pierce) на 30 минут инкубируют блокатором-казеином. Затем планшет промывают трижды по 200 мкл моечным буферным раствором (50 мМ Tris, pH 7.5, 100 мМ Nad, 10% (об/об) блокатор-казеин, 0.05% (об/об) Tween 20) на лунку. Добавляют пептид биотин-DLDLEMLAPYIPMDDDFQL (фирма Eurogentec, 4102 Seraing, Бельгия) в концентрации 400 нМ на 100 мкл моечного буферного раствора. Этот пептид служит в качестве питательной среды для пролилгидроксилирования и привязан к микротитрационному планшету. Через 60 минут инкубации планшет трижды промывают моечным буферным раствором, 30 минут инкубирует биотин в блокаторе-казеине, и затем снова трижды промывают моечным буферным раствором.
Для выполнения реакции пролилгидроксилазы привязанную к планшету пептидную питательную среду инкубируют в течение 1 - 60 минут клеточным лизатом, содержащим пролилгидроксилазу. Реакцию проводят в 100 мкл реакционного буферного раствора (20 мМ Tris, pH 7.5, 5 мМ KCl,1.5 мМ MgCl2, 1 мкМ-1 мМ 2-оксоглютарата, 10 мкМ FeSO4, 2 мМ аскорбата) при комнатной температуре. Реакционная смесь кроме предназначенного для испытания ингибитора содержит пролилгидроксилазу в различных концентрациях. Испытуемое вещество является предпочтительным, но только при концентрациях между 1 нМ и 100 мкМ. Путем трехкратного промывания планшета моечным буферным раствором реакцию останавливают.
Для количественного определения пролилгидроксилирования добавляют составной белок, который содержит как тиоредоксин из Е. coli (из микрофлоры Escherichia coli), так и VBC-комплекс, в 80 мкл связующего буферного раствора (50 мМ Tris, pH 7.5, 120 мМ NaCl). Через 15 минут добавляют 10 мкл раствора поликлональных анти-тиоредоксин-антител из кролика в связующий буферный раствор. Через 30 минут инкубации при комнатной температуре планшет трижды промывают моечным буферным раствором, чтобы удалить несвязанные антитела и VBC-комплекс. Для определения количества связанного VBC-комплекса осуществляют инкубацию в течение 15 минут с ТМВ. Окрашиваемую реакцию останавливают путем добавления 100 мкл 1М серной кислоты. Количество связанного VBC-комплекса определяют измерением оптической плотности на волне 450 нм. Она пропорциональна количеству гидроксилированного пролила в пептидной питательной среде.
Для индикации пролилгидроксилирования альтернативно может использоваться VBC-комплекс, связанный с европием (фирма Perkin Elmer). В этом случае количество связанного VBC-комплекса определяют по времени флуоресценции. Кроме того, возможно применение VBC-комплекса, маркированного радиоактивным [35S]метионином. Для этого VBC-комплекс с радиоактивной меткой может быть изготовлен в лабораторных условиях путем транскрипции-трансляции в лизат ретикулоцита.
Примеры исполнения угнетают активность HIF-пролилгидроксилазы в этом испытании с показателем IC50 порядка ≤30 мкМ. В приведенной ниже таблице 1 показаны характерные показатели IС50 для примеров исполнения.
1.b) Клеточный, функциональный тест in vitro:
Эффективность соединений согласно изобретению количественно оценивают с помощью рекомбинантной клеточной линии. Клетки первоначально получены от человеческой раковой клеточной линии легких (А549, АТСС: American Type Culture Collection, Manassas, VA 20108, USA). Испытуемая клеточная линия стабильно переносится вектором, который содержит репортерный ген люциферазы Photinus pyralis (называемый в дальнейшем люцифераза) под контролем искусственного минипромотора. Минипромотор состоит из двух чувствительных к гипоксии элементов против последовательности ТАТА-блока [Oehme F., Ellinghaus P., Kolkhof P., Smith T.J., Ramakrishnan S., Htitter J., Schramm M., Flamme I., Biochem. Biophys. Res. Commun. 296 (2), 343-9 (2002)]. При воздействии гипоксии (например, культивирование в присутствии 1% кислорода в течение 24 часов) или при воздействии неселективных ингибиторов диоксигеназы (например, десферроксамин в концентрации 100 мкМ, хлорид кобальта в концентрации 100 мкМ или диэтиловый эфир iV-оксалилглицина в концентрации 1 мМ) испытуемая клеточная линия продуцирует люциферазу, которая может быть обнаружена и количественно определена с помощью подходящих реагентов биолюминисценции (например, Steady-Glo® Luciferase Assay System, Promega Corporation, Мэдисон, штат Висконсин 53711, США) и подходящего люминометра.
Ход испытания: Клетки за день до испытания вводят в точно отмеренную питательную среду (DMEM, 10% FCS, 2 mМ глутамина) в микротитрационном планшете с 384 или 1536 лунками и помещают в клеточный инкубатор (96% влажность воздуха, 5% об/об CO2, 37°C). В день испытания к питательной среде добавляют испытуемые вещества в ступенчатых концентрациях. В приставки, служащие в качестве негативного контроля, к клеткам испытуемое вещество не добавляют. В качестве позитивного контроля для определения чувствительности клеток к ингибиторам добавляют, например, десферроксамин в конечной концентрации 100 мкМ. Через 6-24 часов после нанесения испытуемых веществ в лунки микротитрационного планшета измеряют результирующий световой сигнал в люминометре. По измеренным величинам устанавливают отношение дозы действия, которое служит основанием для определения полумаксимальной концентрации действия (обозначаемой как EC50).
1.с) Клеточный, функциональный тест in vitro для изменения экспрессии генов:
Чтобы исследовать изменение экспрессии специфических мРНК в человеческих клеточных линиях после обработки испытуемыми веществами, культивируют следующие клеточные линии на планшетах с 6 или 24 лунками:
человеческие гепатомные клетки (HUH, JCRB Cell Bank, Япония), человеческий эмбриональный почечный фибропласт (НЕК/293, АТСС, Манассас, штат Вирджиния 20108, США), человеческие раковые клетки шейки (HeLa, АТСС, Манассас, штат Вирджиния 20108, США), человеческие венозные эндотелиальные клетки пуповины (HUVEC, Cambrex, East Rutherford, Нью-Джерси 07073, США). Через 24 часа после добавления испытуемых веществ клетки промывают физиологическим раствором с фосфатом, и из них соответствующим методом получают общую РНК (например, Trizol®-Reagenz, Invitrogen GmbH, 76131 Карлсруэ, Германия).
Для обычного аналитического эксперимента каждый 1 мкг полученной таким образом общей РНК усваивается Dnase 1, и подходящей реакцией реверса транскриптазы (ImProm-II Reverse Transcription System, Promega Corporation, Мэдисон, WI 53711, США) преобразуется в дополнительную ДНК (кДНК). 2,5% полученной таким образом композиции кДНК используют для цепной реакции полимеразы. Уровень экспрессии исследуемых генов мРНК устанавливают с помощью количественного анализа цепной реакции полимеразы в реальном времени [TaqMan-PCR; Heid C.A, Stevens J., Livak K.J, Williams P.M., Genome Res. 6 (10), 986-94 (1996)] с применением прибора последовательной индикации ABI Prism 7700 (фирма Applied Biosystems, Inc.). Используемые при этом комбинации затравка-зонд вырабатываются программой Primer Express 1.5 (фирма Applied Biosystems, Inc.). В частности, исследуют мРНК эритропоэтина, карбоангидразы IX, лактатдегидрогеназы А и фактора роста сосудистых эндотелиальных клеток.
Вещества согласно настоящему изобретению приводят к значимому, зависящему от дозы увеличению мРНК индуцированных гипоксией генов в клетках человеческого происхождения.
2. Тесты in vivo для доказательства действия в сердечно-сосудистой системе
2.а) Тест т vivo для изменения экспрессии генов:
Мышам или крысам вводят испытуемые соединения, растворенные в соответствующих растворителях, орально или через желудочный зонд, внутрибрюшинно или внутривенно. Типичными дозами являются 0,1, 0,5, 1, 5, 10, 20, 50, 100 и 300 мг вещества на кг веса тела и прием. Контрольные животные получают только растворитель. Через 4, 8 или 24 часа после приема испытуемого вещества животных умертвляли сверхдозой изофлурана с последующим переломом шейного позвоночника, и извлекали предназначенные для исследования органы. Фрагменты органов мгновенно замораживали в жидком азоте. Из фрагментов органов полученная общая РНК, как описано в пункте B.I. а), преобразуется в кДНК. Уровень экспрессии исследуемых генов мРНК устанавливают с помощью количественного анализа цепной реакции полимеразы в реальном времени [TaqMan-PCR; Heid C.A., Stevens J., Livak K.J., Williams P.M., Genome Res. 6 (10), 986-94 (1996)] с применением прибора последовательной индикации ABI Prism 7700 (фирма Applied Biosystems, Inc.).
Вещества согласно настоящему изобретению после орального или парентерального приема приводят к значимому, зависящему от дозы увеличению мРНК эритропоэтина в почках по сравнению с контрольными образцами, получившими плацебо.
2.b) Определение уровня эритропоэтина в сыворотке:
Мышам или крысам вводят испытуемое вещество в соответствующем растворителе либо внутрибрюшинно либо орально один или два раза ежедневно. Типичными дозами являются 0,1, 0,5, 1, 5, 10, 20, 50, 100 и 300 мг вещества на кг веса тела и прием. Контрольные животные получают только растворитель. Перед приемом и через четыре часа после последнего введения лекарства у животных под кратковременным наркозом берут 50 мкл крови из ретроорбитального венного сплетения или из хвостовой вены. Свертывание крови предотвращают добавлением литий-гепарина. Путем центрифугирования получают плазму крови. В плазме крови определяют содержание эритропоэтина с помощью прибора Erythropoetin-ELISA (Quantikine® mouse Epo Immunoassay, R&D Systems, Inc., Миннеаполис, США) в соответствии с инструкцией изготовителя. Измеренные величины пересчитывают в пг/мл на основе контрольного измерения эритропоэтина у мыши.
Вещества согласно настоящему изобретению после орального или парентерального приема приводят к значимому, зависящему от дозы увеличению эритропотеина по сравнению с исходным показателем и с контрольными образцами, получившими плацебо.
2.с) Определение клеточного состава периферийной крови:
Мышам или крысам вводят испытуемое вещество в соответствующем растворителе либо внутрибрюшинно, либо орально один или два раза в течение нескольких дней. Типичными дозами являются, например, 0,1, 0,5, 1, 5, 10, 20, 50, 100 и 300 мг вещества на кг веса тела и прием. Контрольные животные получают только растворитель. В конце испытания у животных под кратковременным наркозом берут кровь из венного сплетения угла глазной щели или из хвостовой вены, и свертывание крови предотвращают добавлением цитрата натрия. В подходящем электронном измерительном приборе определяют в пробе крови концентрации эритроцитов, лейкоцитов и тромбоцитов. Концентрацию етикулоцитов определяют по мазкам крови, которые для этого окрашивают соответствующим красящим раствором (фирма КАВЕ Labortechnik, Нюмбрехт) путем просмотра через микроскоп каждой 1000 эритроцитов. Для определения гематокрита кровь из ретроорбитального сплетения вен берут с помощью гематокритного капилляра, и показатель гематокрита считывают вручную после обработки капилляра в пригодной для этого центрифуге.
Вещества согласно настоящему изобретению после орального или парентерального приема приводят к значимому, зависящему от дозы увеличению гематокрита, числа эритроцитов и лейкоцитов по сравнению с исходным показателем и с контрольными образцами, получившими плацебо.
3. Определение растворимости
Изготовление исходного раствора (первичного раствора):
Не менее 1,5 мг испытуемого вещества точно отвешивают в Wide Mouth 10 mm Screw V-Vial (фирма Glastechnik Grafenroda GmbH, Art.-Nr. 8004-WM-Н/V15 мк) с подходящим навинчивающимся колпачком и перегородкой, вещество разбавляют соединением DMSO до концентрации 50 мг/мл и в течение 30 минут взбалтывают с помощью вихревой мешалки.
Изготовление калибровочных растворов:
Необходимые ступени дозирования осуществляют в планшете Deep Well Plate (DWP) с 96 лунками 1,2 мл с помощью роботов манипулирования жидкостями. В качестве растворителя используют смесь из ацетонитрила и воды 8:2.
Изготовление исходного раствора для калибровочных растворов (концентрированный раствор): 10 мкл первичного раствора разводят 833 мкл смеси растворов (концентрация=600 мкг/мл) и смесь гомогенизируют. Каждое испытуемое вещество разбавляют до концентрации 1:100 в отдельных планшетах DWP и снова гомогенизируют.
Калибровочный раствор 5 (600 нг/мл): 30 мкл концентрированного раствора разбавляют 270 мкл смеси растворов и гомогенизируют.
Калибровочный раствор 4 (60 нг/мл): 30 мкл калибровочного раствора 5 разбавляют 270 мкл смеси растворов и гомогенизируют.
Калибровочный раствор 3 (12 нг/мл): 100 мкл калибровочного раствора 4 разбавляют 400 мкл смеси растворов и гомогенизируют.
Калибровочный раствор 2 (1.2 нг/мл): 30 мкл калибровочного раствора 3 разбавляют 270 мкл смеси растворов и гомогенизируют.
Калибровочный раствор 1 (0.6 нг/мл): 150 мкл калибровочного раствора 2 разбавляют 150 мкл смеси растворов и гомогенизируют.
Изготовление пробных растворов:
Необходимые ступени дозирования осуществляют в планшете DWP с 96 лунками 1,2 мл с помощью роботов манипулирования жидкостями. 10,1 мкл концентрированного раствора разводят 1000 мкл буферного раствора PBS рН 6,5. (Буферный раствор PBS рН 6,5: 61,86 г хлорида натрия, 39,54 г дигидрофосфата натрия и 83,35 г 1 N едкого натра взвешивают в мерной колбе на 1 литр, дополняют водой, и смесь перемешивают около 1 часа. 500 мл этого раствора заливают в мерную колбу на 5 литров, и доливают воду. С помощью 1 N едкого натра устанавливают рН 6,5.)
Выполнение:
Необходимые ступени дозирования осуществляют в планшете DWP с 96 лунками 1,2 мл с помощью роботов манипулирования жидкостями. Приготовленные таким образом пробные растворы взбалтывают 24 часа при 1400 об/мин с помощью термостатированной вихревой мешалки при 20°C.Из этих растворов берут пробы по 180 мкл и помещают в полиалломерные центрибужные пробирки Бекмана. Эти растворы центрифугируют 1 час с силой 223.000 × г. От каждого пробного раствора отбирают по 100 мкл жидкой фракции, которые разводят буферным раствором PBS 6,5 в концентрации 1:10 и 1:1000.
Методика анализа:
Анализ проб осуществляют с помощью HPLC/MS-MS. Количественную оценку получают по пятиточечной калибровочной кривой испытуемого соединения. Растворимость выражается в мг/л. Последовательность анализа: 1) контрольная проба (смесь растворителей); 2) калибровочный раствор 0,6 нг/мл; 3) калибровочный раствор 1,2 нг/мл; 4) калибровочный раствор 12 нг/мл; 5) калибровочный раствор 60 нг/мл; 6) калибровочный раствор 600 нг/мл; 7) контрольная проба (смесь растворов); 8) пробный раствор 1:1000; 7) пробный раствор 1:10.
Метод HPLC/MS-MS
HPLC: Agilent 1100, четырехкомпонентный насос (G1311A), автоматический пробоотборник СТС HTS PAL, дегазатор (G1322A) и термостат колонки (G1316A); колонка: Oasis HLB 20 мм × 2,1 мм, 25 мк; температура: 40°C; растворитель А: вода+0.5 мл муравьиной кислоты/л; растворитель В: ацетонитрил+0,5 мл муравьиной кислоты/л; скорость потока: 2,5 мл/мин; время останова 1,5 мин; градиент: 0 мин 95% А, 5% В; изменение: 0-0,5 мин 5% А, 95% В; 0,5-0,84 мин 5% А, 95% В; изменение: 0,84-0,85 мин 95% А, 5% В; 0,85-1,5 мин 95% А, 5% В.
MS/MS: WATERS Quattro Micro Tandem MS/MS; Z-Spray API-Interface; HPLC-MS-делитель на входе 1:20; измерение в режиме ESI.
С. Примеры исполнения для фармацевтических составов
Соединения согласно изобретению могут быть переведены в лекарственные препараты следующим образом:
Таблетки:
Состав:
100 мг соединения согласно изобретению, 50 мг лактозы (моногидрат), 50 мг кукурузного крахмала (природного), 10 мг поливинилпирролидона (PVP 25) (фирма BASF, Людвигсгафен, Германия) и 2 мг стеарата магния.
Вес таблетки 212 мг. Диаметр 8 мм, радиус выпуклости 12 мм.
Изготовление:
Смесь из соединения согласно изобретению, лактозы и крахмала с 5%-ным раствором (масса/масса) ПВП гранулируют в воде. После сушки гранулированный продукт 5 минут перемешивают со стеаратом магния. Эту смесь прессуют стандартным таблеточным прессом (формат таблетки см. выше). Ориентировочной величиной для прессования является усилие пресса 15 кН.
Суспензии для орального применения:
Состав:
1000 мг соединения согласно изобретению, 1000 мг этанола (96%), 400 мг Rhodigel® (ксантановая смола фирмы FMC, Пенсильвания, США) и 99 г воды.
Разовая доза 100 мг соединения согласно изобретению соответствует 10 мл оральной суспензии.
Изготовление:
Родигель превращают в этаноле в суспензию, к суспензии добавляют соединение согласно изобретению. При размешивании добавляют воду. Размешивание продолжают около 6 часов до окончательного набухания родигеля.
Раствор для орального применения:
Состав:
500 мг соединения согласно изобретению, 2,5 г полисорбата и 97 г полиэтиленгликоля 400. Разовая доза 100 мг соединения согласно изобретению соответствует 20 г орального раствора.
Изготовление:
Соединение согласно изобретению при размешивании в смеси из полиэтиленгликоля и полисорбата превращают в суспензию. Процесс размешивания продолжают до полного растворения соединения согласно изобретению.
Раствор для внутривенного вливания:
Соединение согласно изобретению с концентрацией, ниже насыщенной растворимости, растворяют в физиологически приемлемом растворителе (например, изотонический раствор хлорида натрия, раствор глюкозы 5% и/или раствор PEG 400 30%). Раствор фильтруют в стерильных условиях и разливают в стерильные и апирогенные емкости для инъекций.
Реферат
Изобретение относится к новым замещенным дигидропиразолонам формулы (I) и его фармацевтически приемлемым солям, обладающим свойствами ингибитора HIF-пролил-4-гидрооксилазы. Соединения могут найти применение для получения лекарственного средства, пригодного для применения в способе лечения и/или профилактики сердечно-сосудистых заболеваний, сердечной недостаточности, анемии, хронических заболеваний почек и почечной недостаточности. В соединении формулы (I)X означает N или CH, Rозначает водород или циано, Rозначает насыщенный 4-7-членный остаток гетероциклила, связанный через атом азота, который содержит от 1 до 2 гетероатомов, выбранных из N и O. Причем гетероциклильный остаток может быть замещен одним заместителем, выбранным из группы, состоящей из C-C-циклоалкила, или 1-4 атомами фтора. Изобретение также относится к способу получения соединений и лекарственному средству на их основе. 5 н. и 11 з.п. ф-лы, 1 табл., 29 пр.
Формула
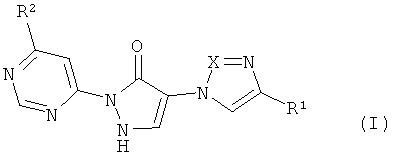
в которой
X означает N или CH,
R1 означает водород или циано,
R2 означает насыщенный 4-7-членный остаток гетероциклил, связанный через атом азота, который содержит от 1 до 2 гетероатома, выбранных из N и O,
причем гетероциклильный остаток может быть замещен одним заместителем, выбранным из группы, состоящей из C3-C6-циклоалкила,
или
причем гетероциклильный остаток замещен 1-4 заместителями фтор, или одна из его фармацевтически приемлемых солей.
R1 означает водород или циано,
R2 означает насыщенный 4-7-членный остаток гетероциклил, связанный через атом азота, который содержит от 1 до 2 гетероатома, выбранных из N или O, причем гетероциклильный остаток замещен 1-4 заместителями фтор,
или
R2 означает пиперазин-1-ил,
причем пиперазин-1-ил замещен одним заместителем, выбранным из группы, состоящей из C3-C6-циклоалкила,
причем пиперазин-1-ил замещен одним заместителем, выбранным из группы, состоящей из C3-C6-циклоалкила,
или
R2 означает азетидин-1-ил,
причем азетидин-1-ил замещен одним заместителем, выбранным из группы, состоящей из C3-C6-циклоалкила,
или одна из его фармацевтически приемлемых солей.
X означает N или CH,
R1 означает водород или циано,
R2 означает азетидин-1-ил, пирролин-1-ил или пиперидин-1-ил,
причем азетидин-1-ил, пирролин-1-ил и пиперидин-1-ил замещены 1-4 заместителями фтор,
или
R2 означает пиперазин-1-ил,
причем пиперазин-1-ил в 4-й позиции замещен одним заместителем, выбранным из группы, состоящей из C3-C6-циклоалкила,
или одна из его фармацевтически приемлемых солей.
X означает N или CH,
R1 означает водород или циано,
R2 означает пиперазин-1-ил,
причем пиперазин-1-ил замещен одним заместителем, при этом заместитель выбирают из группы, состоящей из C3-C6-циклоалкил,
или одна из его фармацевтически приемлемых солей.
X означает N или CH,
R1 означает водород или циано,
R2 означает пиперазин-1-ил,
причем пиперазин-1-ил в 4-й позиции замещен одним заместителем, при этом заместитель выбирают из группы, состоящей из C3-C6-циклоалкил,
или одна из его фармацевтически приемлемых солей.
X означает N или CH,
R1 означает водород или циано,
R2 означает азетидин-1-ил,
причем азетидин-1-ил замещен одним заместителем, выбранным из группы, состоящей из C3-C6-циклоалкил,
или одна из его фармацевтически приемлемых солей.
R2 означает 4-циклобутил-пиперазин-1-ил.
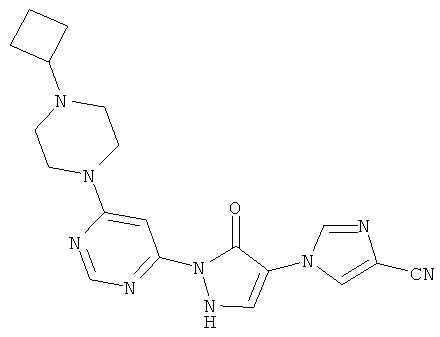
и его фармацевтически приемлемые соли.
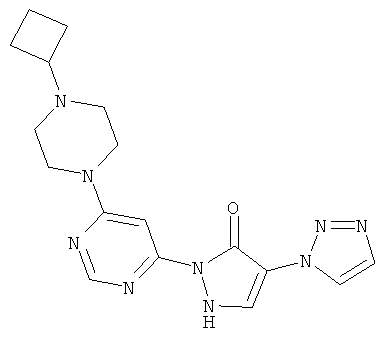
или одна из его фармацевтически приемлемых солей
в которой остатки X, R1 и R2 имеют указанные в одном из пп.1-11 значения, и которое обладает свойствами селективных ингибиторов HIF-пролил-4-гидрооксилазы для лечения и/или профилактики болезней, опосредованных активностью HIF-пролил-4-гидрооксилазы.
в которой остатки X, R1 и R2 имеют указанные в одном из пп.1-11 значения, для получения лекарственного средства, пригодного для применения в способе лечения и/или профилактики сердечно-сосудистых заболеваний, сердечной недостаточности, анемии, хронических заболеваний почек и почечной недостаточности.
в которой X, Z1 и R1 имеют указанные в п.1 значения, с соединением формулы
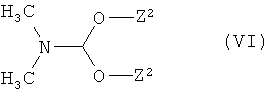
в которой
Z2 означает метил или этил,
конденсируют в соединение формулы
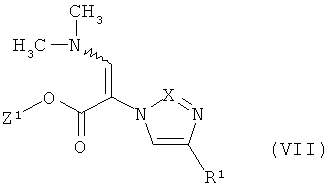
в которой X, Z1 и R1 имеют указанные в п.1 значения,
и затем в присутствии одной из кислот подвергают взаимодействию с соединением формулы (III)
в которой R2 имеет указанное в п.1 значение,
и полученное при этом соединение при этих условиях реакции взаимодействия соединения формулы (VII) с соединением формулы (III) или под влиянием основания преобразуют в циклическое соединение формулы (I),
и соединение формулы (I), при необходимости, с помощью соответствующего (i) растворителя и/или (ii) основания или кислоты преобразуют в одну из его солей.
Документы, цитированные в отчёте о поиске
Замещенные дигидропиразолоны для лечения кардиоваскулярных и гематологических заболеваний
Комментарии