Соединения, способ их получения и фармацевтическая композиция на их основе - RU2317985C1
Код документа: RU2317985C1
Описание
Область техники
Настоящее изобретение относится к эффективному ингибитору р-гликопротеина и к его фармацевтически приемлемой соли, способу получения указанного ингибитора и фармацевтической композиции, содержащей данный ингибитор в качестве активного ингредиента.
Уровень техники
Известно, что многие противораковые агенты, например винкаалкалоиды, антрациклин, эпиподофилотоксин, паклитаксел и доцетаксел, оказывают эффективное действие при введении больному, обладающему резистентностью ко многим лекарственным средствам (MDR), возникающей у больного за счет повышенной экспрессии р-гликопротеина. р-Гликопротеин ингибирует внутриклеточное накопление введенного противоракового агента, способствуя выведению его из клетки (D.W.Shen, et al., Science (1986), 232, 643-645; и Schinkel, et al., Cell (1994), 77, 491-502). Таким образом, многочисленные попытки были предприняты для повышения биодоступности вышеупомянутых агентов посредством объединения их с ингибитором р-гликопротеина.
Поскольку традиционные ингибиторы р-гликопротеина, такие как верапамил и циклоспорин А, вызывают серьезные неблагоприятные эффекты, например, снижение кровяного давления и подавление иммунитета, был разработан ряд новых ингибиторов п-гликопротеина, таких как пиперидин-2-карбоксилат, акридин, пиперазин-2,5-дион, антраниловая кислота и производные метандибензосуберана. Однако исследователями было сообщено, что подобные, вновь открытые ингибиторы п-гликопротеина проявляют токсичность и другие неблагоприятные свойства (смотри международные публикации РСТ № WO 94/07858; WO 92/12132; WO 96/20180 и 98/17648, и WO 98/22112).
Таким образом, авторы настоящего изобретения предприняли попытку разработать ингибитор п-гликопротеина, лишенный вышеуказанных проблем, и обнаружили новое соединение, которое в значительной степени повышает биодоступность противораковых агентов, подавляя активность п-гликопротеина.
Сущность изобретения
Таким образом, целью настоящего изобретения является предоставление соединения, которое может быть использовано в качестве эффективного ингибитора п-гликопротеина для увеличения биодоступности противоракового агента с минимальными неблагоприятными эффектами.
Другой целью настоящего изобретения является предоставление способа получения такого соединения.
Еще другой целью настоящего изобретения является предоставление фармацевтической композиции, содержащей такое соединение.
Подробное описание изобретения
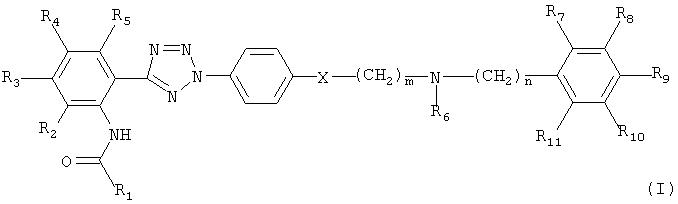
В соответствии с одним аспектом настоящего изобретения, изобретение обеспечивает соединение формулы (I) и его фармацевтически приемлемую соль:
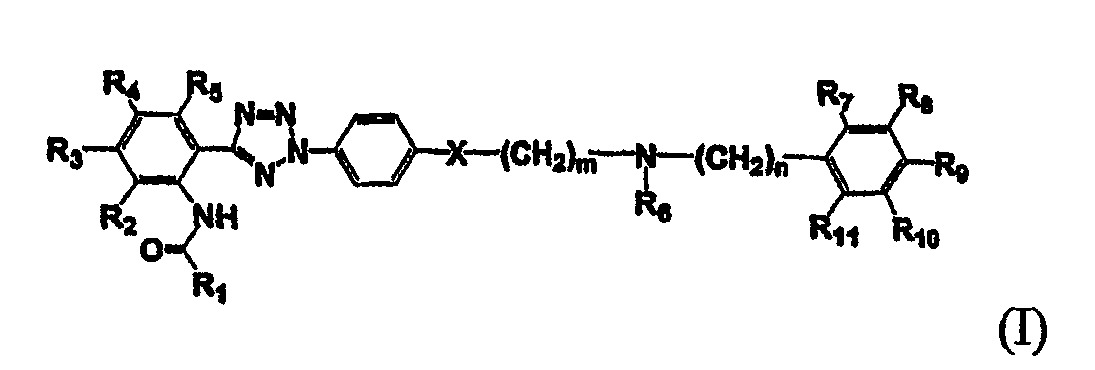
где R1 означает арил, гетероарил, акриларил, акрилгетероарил, гетероциклоалкенил или карбоцикло, который, необязательно, замещен одним или более заместителями, выбранными из С1-5 алкила, гидрокси, С1-5 алкокси, галогена, трифторметила, нитро- и амино;
R2, R3, R4, R5, R6, R7, R8, R9, R10 и R11 каждый независимо означает водород, гидрокси, галоген, нитро, С1-5 алкил или алкокси, причем R6 и R11, необязательно, объединены друг с другом с образованием 4-8-членного кольца;
m и n каждый независимо является целым числом, колеблющимся от 0 до 4, и
Х является СН2, О или S.
В отличие от традиционных ингибиторов п-гликопротеина, например, циклоспорина А, цинхонина и верапамила, соединение формулы (I) само по себе не обладает фармакологической активностью, и, следовательно, не вызывает побочных эффектов, в то же время оно повышает биодоступность противораковых агентов путем ингибирования активности п-гликопротеина.
У соединения формулы (I) согласно настоящему изобретению, предпочтительным радикалом R1 является незамещенный или замещенный фенил, пиридин, пиразин, хинолин, изохинолин, хиназолин, хиноксалин, пиразол, имидазол, триазол, оксазол, тиазол, оксадиазол, тиадиазол, бензотиазол, бензоксазол, хромон, хинолон, циннамил или хинолинакрил.
Характерные примеры соединения формулы (I) включают
хинолин-3-карбоновой кислоты
[2-(2-4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
хинолин-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
изохинолин-3-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
хинолин-8-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
изохинолин-1-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
хинолин-4-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
4-метоксихинолин-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
хиноксалин-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
пиридин-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4, 5-диметоксифенил]никотинамид;
N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]изоникотинамид;
пиразин-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
N-[2-(2-{4-[2-(6, 7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]бензамид;
нафталин-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4, 5-диметоксифенил]-2-фторбензамид;
N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-3-фторбензамид;
N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-4-фторбензамид;
N-[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-3,4-дифторбензамид;
тиофен-3-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
фуран-3-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
4-оксо-4Н-хромен-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
6-метил-4-оксо-4Н-хромен-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
5-гидрокси-4-оксо-4Н-хромен-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
5-метокси-4-оксо-4Н-хромен-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
6-фтор-4-оксо-4Н-хромен-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
6-бром-4-оксо-4Н-хромен-2-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
цинолин-4-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
4-оксо-4Н-хромен-3-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
хинолин-3-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-дифторфенил]амид;
хинолин-3-карбоновой кислоты
[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этилсульфанил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амид;
хинолин-3-карбоновой кислоты
2-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-илэтил)-2Н-тетразол-5-ил]-4,5-диметоксифениламид;
N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4, 5-диметоксифенил]-3-фенилакриламид;
N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-3-хинолин-3-илакриламид; и
4-оксо-4Н-хромен-2-карбоновой кислоты
(2-{2-[4-(2-{[2-(3,4-диметоксифенил)этил]метиламино}этил)фенил]-2Н-тетразол-5-ил}-4,5-диметоксифенил)амид.
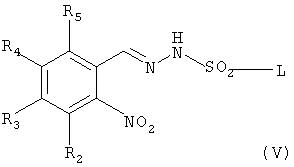
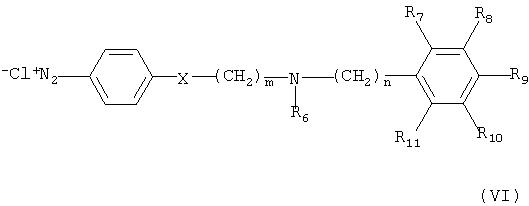
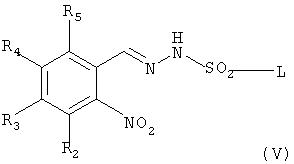
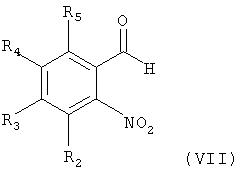
Соединение формулы (I) может быть получено по следующей реакционной схеме А:
Реакционная схема А
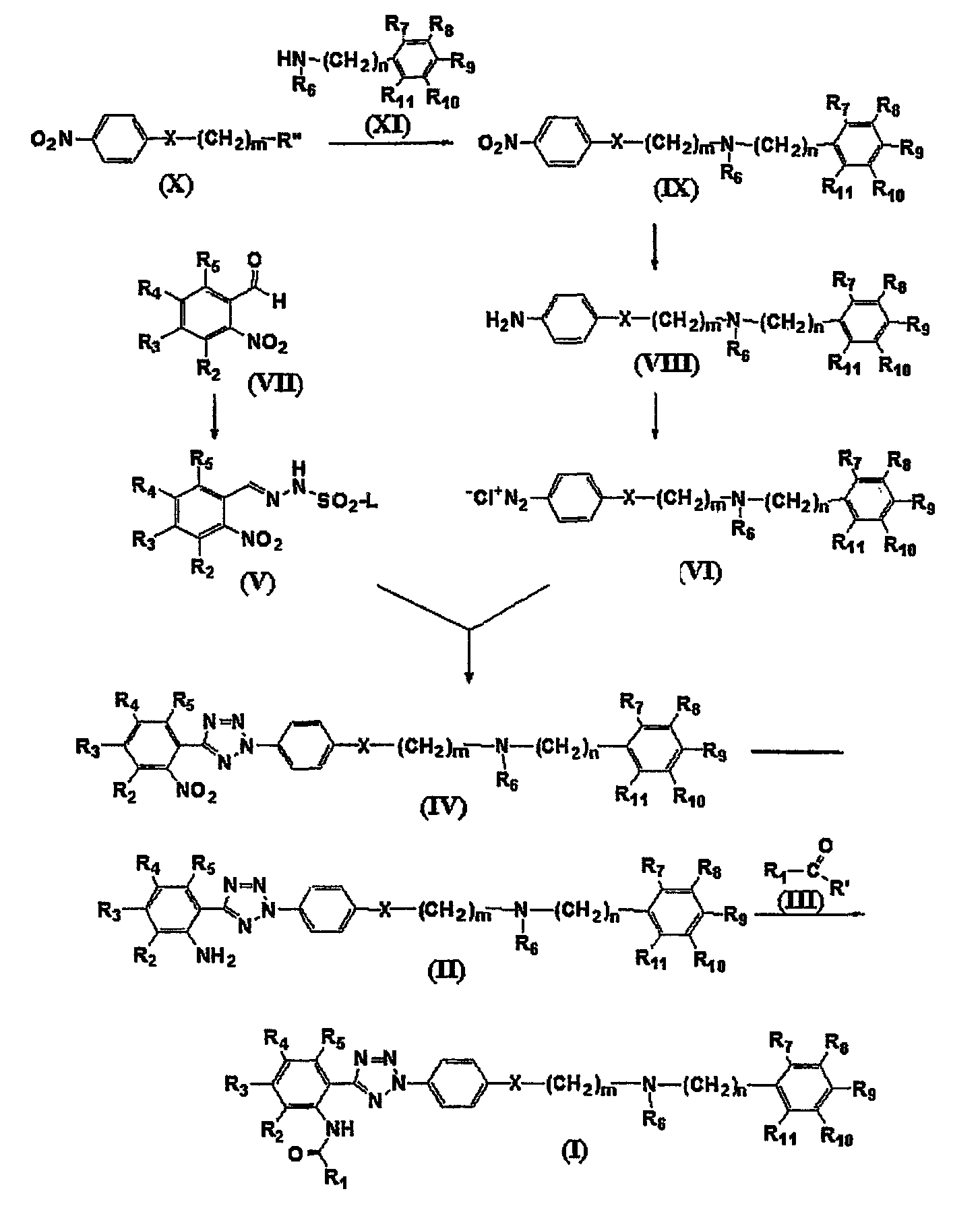
где R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, m, n и X имеют такие же значения, как определены в формуле (I);
R' и R" каждый в зависимости друг от друга означает OH, Cl или Br, и
L означает бензил или толил.
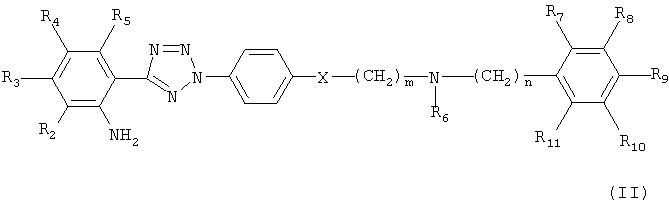
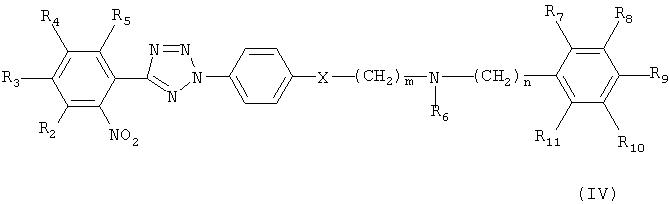
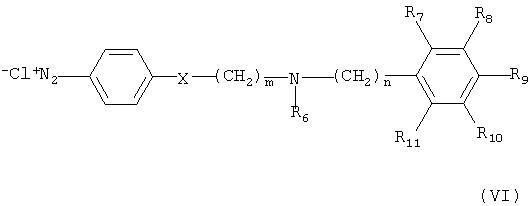
На реакционной схеме А, соединение формулы (I) может быть получено (i) циклизацией соединения формулы (V) c соединением формулы (VI) в присутствии основания с получением соединения формулы (IV); (ii) гидрированием соединения формулы (IV) в присутствии катализатора с получением соединения формулы (II), и (iii) ацилированием соединения формулы (II), полученного на стадии (ii), соединением формулы (III) в присутствии основания или конденсирующего реагента.
Основание, используемое на стадии (i), может быть выбрано из группы, состоящей из пиридина, триэтиламина и диизопропилэтиламина. Реакцию на стадии (i) можно проводить в таком растворителе, как метанол, этанол, хлороформ, дихлорметан, тетрагидрофуран, этиловый эфир, гексан и толуол, и соединение формулы (VI) может быть использовано в количестве в интервале от 1 до 2 эквивалентов, исходя из 1 эквивалента соединения формулы (V).
Реакцию на стадии (ii) можно проводить в таком растворителе, как метанол, этанол, хлороформ, дихлорметан, тетрагидрофуран, этиловый эфир, гексан и толуол, при температуре в интервале от 0 до 50°С и катализатор для стадии (ii) может быть выбран из группы, состоящей из палладиевого, платинового и цинкового катализаторов.
Для реакции на стадии (iii) соединение формулы (III) может быть использовано в количестве, в интервале от 1 до 1,5 эквивалентов относительно 1 эквивалента соединения формулы (II). Основание для стадии (iii) может быть использовано в количестве в интервале от 1 до 2 эквивалентов на 1 эквивалент соединения формулы (II), в то время как конденсирующий реагент может присутствовать в количестве в интервале от 1 до 5 эквивалентов, предпочтительно от 1 до 2 эквивалентов на 1 эквивалент соединения формулы (II). Основание для стадии (iii) включает триэтиламин, дипропилэтиламин и пиридин, и конденсирующий реагент для стадии (iii) выбирают из группы, состоящей из 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимида, N,N'-диизопропилкарбодиимида и 1-циклогексил-3-(2-(морфолинэтил)карбодиимид)метил-п-толуолсульфоната, предпочтительно 1-(3-диметиламинопропил)-3-этилкарбодиимида. В случае использования конденсирующего реагента на стадии (iii), 4-(диметиламино)пиридин может быть добавлен в качестве катализатора в количестве в интервале от 0,05 до 0,3 эквивалента, исходя из 1 эквивалента соединения формулы (II). Ацилирование соединения формулы (II) может быть выполнено в растворителе, выбранном из группы, состоящей из дихлорметана, хлороформа, N,N-диметилформамида, тетрагидрофурана и 1,4-диоксана, предпочтительно дихлорметана и хлороформа, при температуре в интервале от -20°С до температуры кипения используемого растворителя, предпочтительно от 10 до 40°С.
На реакционной схеме А показано, что соединение формулы (V) может быть получено взаимодействием соединения формулы (VII) с толуолсульфонилхлоридом или бензолсульфонилхлоридом способом, описанным в Bulletin of the Chemical Society of Japan Vol. 49(7), 1920-1923 (1976). В данной реакции толуолсульфонилхлорид или бензолсульфонилхлорид может быть использован в количестве в интервале от 0,5 до 5 эквивалентов, предпочтительно от 1 до 2 эквивалентов, на 1 эквивалент соединения формулы (VII), и реакцию можно проводить в растворителе, выбранном из группы, состоящей из хлороформа, тетрагидрофурана, этанола, метанола и воды, при температуре в интервале от -10 до 20°С, предпочтительно от 0 до 5° С.
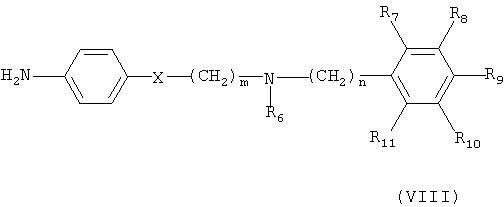
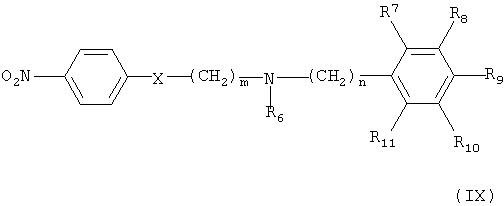
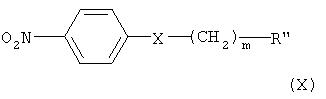
Кроме того, соединение формулы (VI) может быть получено взаимодействием соединения формулы (Х) с соединением формулы (XI) в присутствии основания, например, пиридина, триэтиламина или диизопропилэтиламина, с получением соединения формулы (IX), гидрированием соединения формулы (IX) в присутствии катализатора с образованием соединения формулы (VIII), и взаимодействием соединения формулы (VIII) с нитритом натрия и HCl (смотри публикацию в Bulletin of the Chemical Society of Japan Vol. 49(7), 1920-1923 (1976)).
В данном синтезе реакцию между соединением формулы (Х) и соединением формулы (XI) можно проводить в растворителе, выбранном из группы, состоящей из метанола, этанола, хлороформа, дихлорметана, тетрагидрофурана, этилового эфира, гексана и толуола, при температуре в интервале от 0 до 50°С.
Катализатор, подходящий для данного синтеза, представляет собой металлический катализатор, такой как палладиевый, платиновый или цинковый катализатор, и гидрирование соединения формулы (IX) можно проводить в растворителе, таком как метанол, этанол, хлороформ, дихлорметан, тетрагидрофуран, этиловый эфир, гексан или толуол, при температуре в интервале от 0 до 50°С.
Количество нитрита натрия, используемого в данном синтезе, может составлять от 1 до 5 эквивалентов, предпочтительно от 1 до 3 эквивалентов, на 1 эквивалент соединения формулы (VIII), в то время как кислота HCl может быть использована в количестве в интервале от 0,5 до 1 эквивалента, исходя из 1 эквивалента соединения формулы (VIII). Реакция превращения соединения формулы (VIII) в соединение формулы (VI) может быть проведена в растворителе, таком как этанол, метанол или вода, при температуре в интервале от -10 до 20°С предпочтительно от 0 до 5°С.
Кроме того, настоящее изобретение в его объеме охватывает фармацевтически приемлемую соль ингибитора п-гликопротеина формулы (I), образованную с неорганической или органической кислотой. Предпочтительная неорганическая или органическая кислота может быть выбрана из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты, уксусной кислоты, гликолевой кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаминовой кислоты, фумаровой кислоты, яблочной кислоты, миндальной кислоты, винной кислоты, лимонной кислоты, аскорбиновой кислоты, пальмитиновой кислоты, малеиновой кислоты, гидроксималеиновой кислоты, бензойной кислоты, гидроксибензойной кислоты, фенилуксусной кислоты, коричной кислоты, салициловой кислоты, метансульфокислоты, бензолсульфокислоты и толуолсульфокислоты.
Ингибитор п-гликопротеина согласно настоящему изобретению может быть введен в комбинации с противораковым агентом, который трудно всасывается в пищеварительном тракте вследствие ингибирующего действия п-гликопротеина. Таким образом, в следующем аспекте, настоящее изобретение предоставляет композицию, содержащую ингибитор п-гликопротеина формулы (I) или его фармацевтически приемлемую соль вместе с противораковым агентом, которая способствует:
(а) улучшению или повышению эффективности противоракового агента;
(b) повышению или восстановлению чувствительности опухоли к противораковому агенту, или
(с) снижению или уничтожению MDR резистентности опухоли к действию противоракового агента, независимо от того, является ли MDR приобретенной, индуцированной или врожденной.
Предпочтительные примеры противоракового агента включают таксан (например, паклитаксел или доцетаксел), винкаалкалоид (например, винкристин, винбластин и винорельбин), антрациклин (например, дауномицин, даунорубицин, доксорубицин и акларубицин), камптотецин (например, топотекан и иринотекан), подофиллотоксин (например, этопозид и VP16), митоксантрон, актиномицин, колхицин, грамицидин D и амсакрин.
В другом аспекте, настоящее изобретение предоставляет фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль в качестве активного ингредиента вместе с фармацевтически приемлемыми носителями, наполнителями или другими добавками, для лечения млекопитающего, страдающего от рака:
(а) с целью улучшения или повышения эффективности противоракового агента;
(b) с целью повышения или восстановления чувствительности опухоли к действию противоракового агента, или
(с) с целью снижения или уничтожения MDR резистентности опухоли к действию противоракового агента, независимо от того, является ли MDR приобретенной, индуцированной или врожденной.
Фармацевтическая композиция согласно настоящему изобретению может быть приготовлена для перорального введения или парентерального введения, такого как внутримышечное, внутривенное или трансдермальное введение.
Для перорального введения фармацевтическая композиция согласно настоящему изобретению может быть приготовлена в виде таблетки, покрытой оболочкой таблетки, порошка, твердой или мягкой желатиновой капсулы, раствора, эмульсии, микроэмульсионной или водной дисперсии, обычным способом вместе, по крайней мере, с одним фармацевтически приемлемым носителем, таким как связывающие реагенты (например, прежелатинизированный кукурузный крахмал, поливинилпирролидон и гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза и вторичный кислый фосфат кальция); смазывающие реагенты (например, стеарат магния, тальк и двуокись кремния); дезинтегрирующие реагенты (например, лаурилсульфат натрия и натриевая соль крахмалгликолевой кислоты). Такие таблетки могут быть покрыты оболочками способами, хорошо известными в данной области. Жидкие препараты для перорального введения могут быть приготовлены в виде, например, растворов, сиропов или суспензий, или они могут быть представлены в виде сухого продукта, предназначенного для смешивания с водой или другим подходящим носителем до использования. Такие жидкие препараты могут быть приготовлены обычными способами вместе, по крайней мере, с одной фармацевтически приемлемой добавкой, такой как суспендирующий реагент (например, сироп сорбита, производное целлюлозы и гидрогенизованный пищевой жир); эмульгирующий реагент (например, лецитин и гуммиарабик); неводный носитель (например, миндальное масло, сложный эфир масла, этиловый спирт и фракционированное растительное масло) и консервант (например, метил- или пропил-п-гидроксибензоат и сорбиновая кислота). Указанные препараты также могут содержать, по крайней мере, одну буферную соль или, по крайней мере, один ароматизатор, краситель или подсластитель, что потребуется.
Фармацевтическая композиция согласно настоящему изобретению может быть приготовлена для парентерального введения посредством болюсного вливания или непрерывной инфузии. Препараты для инъекции могут быть представлены в виде стандартной лекарственной формы, например, в виде ампул или в виде контейнеров со множеством доз, с добавленным консервантом. Фармацевтические композиции могут быть представлены в виде суспензий, растворов или эмульсий в масляных, водных или спиртовых носителях, и могут содержать поверхностно-активные вещества, суспендирующие реагенты или эмульгаторы, которые могут быть выбраны из воды, солевого раствора, раствора глюкозы, сахароподобного раствора, спирта, гликоля, простого эфира (например, полиэтиленгликоля 400), масла, жирной кислоты, сложного эфира жирной кислоты и глицерида.
Фармацевтическая композиция согласно настоящему изобретению может быть введена отдельно, до или после введения противоракового агента, или в комбинации с противораковым агентом.
Предполагаемая суточная доза соединения согласно настоящему изобретению для введения человеку (приблизительно 70 кг веса тела) составляет приблизительно от 0,1 мг/кг до 100 мг/кг, более предпочтительно приблизительно от 1 мг/кг до 20 мг/кг. Следует признать, что суточная доза должна быть определена, исходя из существующих факторов, включая состояние, которое подвергается лечению, тяжесть симптомов у больного, способ введения или физиологическую форму противоракового агента, и поэтому, предложенная выше дозировка не должна рассматриваться, как ограничивающая объем изобретения каким бы то ни было образом.
Следующие примеры предназначены проиллюстрировать настоящее изобретение, не ограничивая его объема.
Пример 1: Синтез хинолин-3-карбоновой кислоты [2-(2-4-[2-(6, 7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Стадия 1: Получение 4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фениламина
2-(4-Нитрофенил)этанбромид в количестве 2,30 г и 2,29 г гидрохлорида 6,7-диметокси-1,2,3,4-тетрагидроизохинолина растворяли в 150 мл N, N'-диметилформамида, затем добавляли 4,15 г карбоната калия и 1,80 г йодида натрия, и смесь оставляли для проведения реакции при 100°С в течение 12 часов. После смешивания со 150 мл воды, реакционную смесь экстрагировали три раза 200 мл порциями этилацетата, и объединенные органические слои промывали насыщенным раствором NaCl и сушили над сульфатом магния. Из полученного раствора удаляли растворитель при пониженном давлении, и остаток перекристаллизовывали, используя этилацетат, с получением 2,40 г нитропроизводного. Нитропроизводное добавляли к смеси 150 мл тетрагидрофурана и 150 мл метанола, затем добавляли 0,24 г Pd/C и восстановление проводили в атмосфере водорода в течение 18 часов. Полученный раствор фильтровали и концентрировали при пониженном давлении с получением остатка, который привел к 2,03 г указанного в заголовке соединения (выход 65%).
1Н ЯМР (CDCl3)δ: 6,97 (д, 2Н), 6,57 (д, 2Н), 6,53(с, 1Н), 6,47 (с, 1Н), 3, 77 (с, 6Н), 3,57 (с, 2Н), 3,50 (с, 2Н), 2,71 (м, 8Н)
Стадия 2: Получение 4,5-диметокси-2-нитро-п-толуолсульфонилгидразона
п-Толуолсульфонилгидразид в количестве 6,90 г растворяли в 40 мл этанола, и добавляли 7,90 г альдегида 6-нитровератровой кислоты, растворенного в малом количестве этанола. Смесь перемешивали при 80°С течение 30 мин, охлаждали до комнатной температуры и смешивали со 100 мл воды. Образовавшееся твердое вещество фильтровали, промывали 100 мл этанола и сушили при пониженном давлении с получением 12,0 г указанного в заголовке соединения (выход 85%).
1Н ЯМР (CDCl3)δ: 8,47 (с, 1Н), 8,00 (с, 1Н), 7,87 (д, 2Н), 7,61 (с, 1Н), 7,41 (с, 1Н), 7, 32 (д, 2Н), 3,99 (д, 6Н), 2,42 (с, 3Н)
Стадия 3: Получение 2-(2-4-[5-(4,5-диметокси-2-нитрофенил)тетразол-2-ил]фенилэтил)-6,7-диметокси-1,2,3, 4-тетрагидроизохинолина
Полученное на стадии 1 соединение в количестве 7,4 г добавляли к 40 мл 50% этанола и охлаждали до 5°С. Затем добавляли 6,32 мл 35% HCl и раствор, полученный растворением 1,8 г нитрата натрия в 10 мл воды, и смесь охлаждали до -15°С. Полученное на стадии 2 соединение в количестве 9 г растворяли в 140 мл пиридина и медленно добавляли к вышеуказанной смеси. Образовавшийся раствор перемешивали в течение 14 часов и промывали посредством 1 н. HCl. Органический слой отделяли, сушили над сульфатом магния, фильтровали и подвергали дистилляции при пониженном давлении. Остаток очищали колоночной хроматографией с получением 9,0 г указанного в заголовке соединения (выход 70%).
1Н ЯМР (CDCl3 )δ: 8,08 (д, 2Н), 7,66 (с, 1Н), 7,45 (д, 2Н), 7,32 (с, 1Н), 6,59 (д, 2Н), 4,03 (с, 6Н), 3,85 (с, 6Н), 3,68 (с, 2Н), 3,01 (м, 2Н), 2,84 (м, 6Н)
Стадия 4: Получение 2-(2-4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил-2Н-тетразол-5-ил)-4,5-диметоксифениламина
Полученное на стадии 3 соединение в количестве 0,25 г смешивали с 3 мл этанола, 3 мл дихлорметана и 0,07 г Pd/C и оставляли в атмосфере водорода в течение 12 часов. Реакционную смесь фильтровали через слой целлита, слой промывали этанолом, и фильтрат и промывной раствор объединяли и подвергали дистилляции при пониженном давлении с получением 0,2 г указанного в заголовке соединения (выход 85%).
1Н ЯМР (CDCl3)δ: 8,21 (д, 2Н), 7,81 (с, 1Н), 7,58 (д, 2Н), 6,71 (д, 2Н), 6,48 (с, 1Н), 4,74 (ушир.с, 2Н), 4,02 (д, 6Н), 3,96 (д, 6Н), 3,79 (м, 2Н), 3,51 (м, 8Н)
Стадия 5: Получение хинолин-3-карбонилхлорида
3-Хинолинкарбоновую кислоту в количестве 10 г смешивали с 8,5 мл тионилхлорида и 150 мл толуола и оставляли для осуществления реакции при 100°С в течение 12 часов. Реакционную смесь конденсировали при пониженном давлении с получением остатка, который привел к 10 г указанного в заголовке соединения (выход 90%).
1Н ЯМР (CDCl3)δ: 9,64 (с, 1Н), 9,36 (с, 1Н), 8,85 (д, 1Н), 8,17 (м, 2Н), 7,92 (т, 1Н)
Стадия 6: Получение хинолин-3-карбоновой кислоты [2-(2-4-[2-(6, 7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Полученное на стадии 4 соединение в количестве 0,2 добавляли к 5 мл дихлорметана, к этому добавляли 0,07 г полученного на стадии 5 соединения и 0,1 мл триэтиламина, и смесь оставляли при комнатной температуре в течение 12 часов. После промывания посредством 50 мл дистиллированной воды, органический слой сушили над сульфатом магния, фильтровали и подвергали дистилляции при пониженном давлении. Остаток подвергали колоночной хроматографии с получением 0,18 г указанного в заголовке соединения (выход 69%).
1Н ЯМР (CDCl3)δ: 11,86 (с, 1Н), 9,69 (с, 1Н), 8,95 (с, 1Н), 8,75 (с, 1Н), 8,23 (д, 1Н), 8,12 (д, 1Н), 7,99 (д, 1Н), 7,86 (т, 2Н), 7,66 (м, 1Н), 7,46 (д, 2Н), 6,59 (д, 2Н), 4,06 (д, 6Н), 3,85 (с, 6Н), 3,69 (с, 2Н), 3,04 (м, 2Н), 2,83 (м, 6Н)
Пример 2: Синтез хинолин-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Полученное на стадии 4 примера 1 соединение в количестве 0,15 г и 0,05 г 2-хинолинкарбоновой кислоты добавляли к 5 мл дихлорметана, к этому добавляли 0,1 г гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида и 0,005 г 4-(диметиламино)пиридина, и смесь оставляли при комнатной температуре в течение 12 часов. После промывания 50 мл дистиллированной воды органический слой отделяли и сушили над сульфатом магния, фильтровали и подвергали дистилляции при пониженном давлении. Остаток подвергали колоночной хроматографии с получением 0,14 г указанного в заголовке соединения (выход 73%).
1Н ЯМР (CDCl3)δ: 12,60 (с, 1Н), 8,71 (с, 1Н), 8,40 (д, 2Н), 8,20 (д, 2Н), 8,13 (д, 1Н), 7,90 (с, 2Н), 7,65 (м, 2Н), 7,37 (д, 2Н), 6,58 (д, 2Н), 4,05 (д, 6Н), 3,85 (с, 6Н), 3,67 (с, 2Н), 3,01 (т, 2Н), 2,83 (м, 6Н)
Пример 3: Синтез изохинолин-3-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что гидрат 3-изохинолинкарбоновой кислоты использовали вместо 2-хинолинкарбоновой кислоты с получением 0,12 г указанного в заголовке соединения (выход 62%).
1Н ЯМР (CDCl3)δ: 12,67 (с, 1Н), 9,29 (с, 1Н), 8,83 (с, 1Н), 8,73 (с, 1Н), 8,41 (д, 2Н), 8,01 (д, 2Н), 7,93 (с, 1Н), 7,77 (м, 2Н), 7,53 (д, 2Н), 6,62 (с, 1Н), 6,57 (с, 1Н), 4,04 (д, 6Н), 3,85 (с, 6Н), 3,72 (с, 2Н), 3,07 (т, 2Н), 2,86 (м, 6Н)
Пример 4: Синтез хинолин-8-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 8-хинолинкарбоновую кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,13 г указанного в заголовке соединения (выход 67%).
1Н ЯМР (CDCl3 )δ: 13,69 (с, 1Н), 8,87 (д, 1Н), 8,77 (кв, 1Н), 8,37 (с, 1Н), 8,24 (д, 1Н), 8,06 (д, 1Н), 8,00 (д, 2Н), 7,38 (м, 1Н), 7,23 (с, 1Н), 6,58 (д, 2Н), 4,03 (д, 6Н), 3,85 (с, 6Н), 3,65 (с, 2Н), 2, 95(м, 2Н), 2,81(м, 6Н)
Пример 5: Синтез изохинолин-1-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4, 5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 1-изохинолинкарбоновую кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,12 г указанного в заголовке соединения (выход 62%).
1Н ЯМР (CDCl3)δ: 12,76 (с, 1Н), 9,76 (д, 1Н), 8,91 (с, 1Н), 8,73 (д, 1Н), 8,37 (д, 2Н), 8,05 (с, 1Н), 8,00 (м, 1Н), 7,93 (д, 1Н), 7,86 (м, 2Н), 7,47 (д, 2Н), 6,70 (д, 2Н), 4,17 (д, 6Н), 3,96 (с, 6Н), 3,80 (с, 2Н), 3,15 (т, 2Н), 2,94 (м, 6Н)
Пример 6: Синтез хинолин-4-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 4-хинолинкарбоновую кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,11 г указанного в заголовке соединения (выход 57%).
1Н ЯМР (CDCl3)δ: 11,38 (с, 1Н), 9,09 (д, 1Н), 8,74 (с, 1Н), 8,52 (д, 1Н), 8,23 (д, 1Н), 7,89 (с, 1Н), 7,79 (м, 4Н), 7,64 (т, 1Н), 7,36 (д, 2Н), 6,62 (с, 1Н), 6,65 (с, 1Н), 4,08 (с, 3Н), 4,01 (с, 3Н), 3,85 (с, 6Н), 3,67 (с, 2Н), 2,98 (т, 2Н), 2,82 (м, 6Н)
Пример 7: Синтез 4-метоксихинолин-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,06 г 4-метокси-2-хинолинкарбоновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,15 г указанного в заголовке соединения (выход 76%).
1Н ЯМР (CDCl3)δ: 12,58 (с, 1Н), 8,70 (с, 1Н), 8,22 (м, 3Н), 8,04 (д, 1Н), 7,90 (с, 1Н), 7,80 (с, 1Н), 7,66 (т, 1Н), 7,56 (т, 1Н), 7,36 (д, 2Н), 6,58 (д, 2Н), 4,16 (с, 3Н), 4,04 (д, 6Н), 3, 85 (с, 6Н), 3,00 (т, 2Н), 2,84 (м, 6Н)
Пример 8: Синтез хиноксалин-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 2-хиноксалинкарбоновую кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,14 г указанного в заголовке соединения (выход 73%).
1Н ЯМР (CDCl3)δ: 12,45 (с, 1Н), 9,75 (с, 1Н), 8,65 (с, 1Н), 8,14 (м, 4Н), 7,79 (м, 3Н), 7,37 (д, 2Н), 6,54 (д, 2Н), 4,00 (д, 2Н), 3,81 (с, 6Н), 3,64 (с, 2Н), 2,98 (т, 2Н), 2,79 (м, 6Н)
Пример 9: Синтез пиридин-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,04 г пиколиновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,13 г указанного в заголовке соединения (выход 73%).
1Н ЯМР (CDCl3)δ: 12,55 (с, 1Н), 8,77 (с, 1Н), 8,73 (д, 1Н), 8,35 (м, 3Н), 7,94 (т, 2Н), 7,50 (м, 3Н), 6,58 (д, 2Н), 4,03 (д, 6Н), 3,85 (д, 6Н), 3,69 (с, 2Н), 3,05 (т, 2Н), 2,84 (м, 6Н)
Пример 10: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]никотинамида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,04 г никотиновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,12 г указанного в заголовке соединения (выход 67%).
1Н ЯМР (CDCl3)δ: 11,77 (с, 1Н), 9,54 (с, 1Н), 8,92 (д, 1Н), 8,78 (с, 1Н), 8,55 (д, 1Н), 8,20 (д, 2Н), 7,93 (с, 1Н), 7,60 (м, 3Н), 6,69 (д, 2Н), 4,14 (д, 6Н), 3,96 (д, 6Н), 3,79 (с, 2Н), 3,14 (т, 2Н), 2,95 (м, 6Н)
Пример 11: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]изоникотинамида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,04 г изоникотиновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,12 г указанного в заголовке соединения (выход 67%).
1Н ЯМР (CDCl3)δ: 11,73 (с, 1Н), 8,86 (м, 2Н), 8,67 (с, 1Н), 8,10 (д, 2Н), 8,00 (д, 2Н), 7,83 (с, 1Н), 7,49 (д, 2Н), 6,58 (д, 2Н), 4,00 (д, 6Н), 3,85 (с, 6Н), 3,68 (с, 6Н), 3,03 (т, 2Н), 2,85 (м, 6Н)
Пример 12: Синтез пиразин-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,04 г 2-пиразинкарбоновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,14 г указанного в заголовке соединения (выход 78%).
1Н ЯМР (CDCl3)δ: 12,47 (с, 1Н), 9,56 (д, 1Н), 8,83 (д, 1Н), 8,73 (с, 1Н), 8,70 (м, 1Н), 8,30 (д, 2Н), 7,93 (с, 1Н), 7,52 (д, 2Н), 6,59 (д, 2Н), 4,05 (д, 6Н), 3,86 (д, 6Н), 3,70 (2Н), 3,06 (т, 2Н), 2,85 (м,6Н)
Пример 13: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]бензамида
Процедуру, описанную в примере 2, повторяли за исключением того, что бензойную кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,15 г указанного в заголовке соединения (выход 84%).
1Н ЯМР (CDCl3)δ: 11,39 (с, 1Н), 8,68 (с, 1Н), 8,15 (д, 2Н), 8,08 (д, 2Н), 7,78 (с, 1Н), 7,53 (м, 3Н), 7,42 (д, 2Н), 6,59 (с, 1Н), 6,52 (с, 1Н), 3,98 (д, 6Н), 3,82 (с, 6Н), 3,66 (с, 2Н), 2,98 (т, 2Н), 2,83 (м, 6Н)
Пример 14: Синтез нафталин-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,06 г 2-нафтойной кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,15 г указанного в заголовке соединения (выход 77%).
1Н ЯМР (CDCl3)δ: 11,65 (с, 1Н), 8,79 (с, 1Н), 8,69 (с, 1Н), 8,23 (д, 1Н), 8,11 (д, 2Н), 7,97 (м, 3Н), 7,60 (м, 2Н), 7,44 (м, 3Н), 6, 62 (с, 1Н), 6,56 (с, 1Н), 4,08 (с, 3Н), 4,03 (с, 3Н), 3,86 (с, 6Н), 3,69 (с, 2Н), 3,03 (т, 2Н), 2,85 (м, 6Н)
Пример 15: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-2-фторбензамида
Процедуру, описанную в примере 2, повторяли за исключением того, что 2-фторбензойную кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,12 г указанного в заголовке соединения (выход 66%).
1Н ЯМР (CDCl3)δ: 11,23 (с, 1Н), 8,58 (с, 1Н), 8,08 (м, 3Н), 7,84 (с, 1Н), 7,52 (м, 1Н), 7,44 (д, 2Н), 7,32 (т, 1Н), 7,23 (м, 1Н), 6,62 (с, 1Н), 6,55 (с, 1Н), 4,03 (д, 6Н), 3,85 (с, 6Н), 3,67 (с, 2Н), 3,01 (т, 2Н), 2,85 (м, 6Н)
Пример 16: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-3-фторбензамида
Процедуру, описанную в примере 2, повторяли за исключением того, что 3-фторбензойную кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,02 г указанного в заголовке соединения (выход 11%).
1Н ЯМР (CDCl3)δ: 11,57 (с, 1Н), 8,76 (с, 1Н), 8,17 (д, 2Н), 8,03 (д, 1Н), 7,94 (д, 2Н), 7,58 (м, 3Н), 7,37 (м, 1Н), 6,69 (с, 1Н), 6,62 (с, 1Н), 4,15 (д, 6Н), 3,92 (с, 6Н), 3,75 (с, 2Н), 3,10 (т, 2Н), 2,91 (м, 6Н)
Пример 17: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-4-фторбензамида
Процедуру, описанную в примере 2, повторяли за исключением того, что 4-фторбензойную кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,13 г указанного в заголовке соединения (выход 70%).
1Н ЯМР (CDCl3)δ: 11,41 (с, 1Н), 8,60 (с, 1Н), 8,12 (м, 2Н), 8,06 (д, 2Н), 7,76 (с, 1Н), 7,48 (д, 2Н), 7,19 (т, 2Н), 6,59 (с, 1Н), 6,51 (с, 1Н), 3,98 (д, 6Н), 3,82 (с, 6Н), 3,68 (с, 2Н), 3,03 (т, 2Н), 2,84 (м, 6Н)
Пример 18: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-3,4-дифторбензамида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,06 г 3,4-дифторбензойной кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,12 г указанного в заголовке соединения (выход 63%).
1Н ЯМР (CDCl3)δ: 11,53 (с, 1Н), 8,65 (с, 1Н), 8,10 (д, 2Н), 7,98 (м, 1Н), 7,90 (м, 1Н), 7,84 (с, 1Н), 7,49 (д, 2Н), 7,35 (д, 1Н), 6, 62 (с, 1Н), 6,55 (с, 1Н), 4,03 (д, 6Н), 3,85 (с, 6Н), 3,68 (с, 2Н), 3,04 (т, 2Н), 2,85 (м, 6Н)
Пример 19: Синтез тиофен-3-карбоновой кислоты [2-(2-{4-[2-(6, 7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 3-тиофенкарбоновую кислоту использовали вместо 2-хинолинкарбоновой кислоты с получением 0,10 г указанного в заголовке соединения (выход 55%).
1Н ЯМР (CDCl3 )δ: 11,43 (с, 1Н), 8,63 (с, 1Н), 8,21 (д, 1Н), 8,08 (д, 2Н), 7,76 (с, 1Н), 7,74 (с, 1Н), 7,48 (д, 2Н), 7,38 (м, 1Н), 6,61 (с, 1Н), 6,54 (с, 1Н), 3,99 (д, 6Н), 3,83 (с, 6Н), 3,67 (с, 2Н), 3,02 (т, 2Н), 2,83 (м, 6Н)
Пример 20: Синтез фуран-3-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4, 5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,04 г 3-фуранкарбоновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,11 г указанного в заголовке соединения (выход 62%).
1Н ЯМР (CDCl3)δ: 11,32 (с, 1Н), 8,64 (с, 1Н), 8,22 (с, 1Н), 8,11 (д, 2Н), 7,78 (с, 1Н), 7, 51 (м, 3Н), 7,03 (д, 1Н), 6,62 (с, 1Н), 6,55 (с, 1Н), 4,01 (д, 6Н), 3,85 (с, 6Н), 3,68 (с, 2Н), 3,04 (т, 2Н), 2,85 (м, 6Н)
Пример 21: Синтез 4-оксо-4Н-хромен-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,07 г хромон-2-карбоновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,16 г указанного в заголовке соединения (выход 80%).
1Н ЯМР (CDCl3)δ: 12,63 (с, 1Н), 8,76 (с, 1Н), 8,37 (д, 1Н), 8,27 (д, 2Н), 7,91 (м, 3Н), 7,60 (м, 3Н), 7,39 (с, 1Н), 6,70 (д, 2Н), 4,13 (д, 6Н), 3,98 (с, 6Н), 3, 81 (с, 2Н), 3,16 (т, 2Н), 2,97 (м, 6Н)
Пример 22: Синтез 6-метил-4-оксо-4Н-хромен-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,08 г 6-метилхромон-2-карбоновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,16 г указанного в заголовке соединения (выход 79%).
1Н ЯМР (CDCl3)δ: 12,49 (с, 1Н), 8,62 (с, 1Н), 8,14 (д, 2Н), 8,02 (с, 1Н), 7,78 (с, 1Н), 7,69 (д, 1Н), 7,57 (д, 1Н), 7,47 (д, 2Н), 6,58 (д, 2Н), 4,02 (д, 6Н), 3,85 (д, 6Н), 3,68 (с, 2Н), 3,04 (т, 2Н), 2,82 (м, 6Н), 2,49 (с, 3Н)
Пример 23: Синтез 5-метокси-4-оксо-4Н-хромен-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,3 г соединения, полученного на стадии 4 примера 1, и 0,19 г 5-метоксихромон-2-карбоновой кислоты использовали вместо 0,15 г соединения со стадии 4 примера 1 и 0,05 г 2-хинолинкарбоновой кислоты, соответственно, чтобы получить 0, 23 г указанного в заголовке соединения (выход 55%).
1Н ЯМР (CDCl3)δ: 12,39 (с, 1Н), 8,62 (с, 1Н), 8,15 (д, 2Н), 7,78 (с, 1Н), 7,64 (т, 1Н), 7,48 (д, 2Н), 7, 36 (д, 1Н), 7,15 (с, 1Н), 6,84 (д, 1Н), 6,63 (с, 1Н), 6,56 (с, 1Н), 4,02 (м, 9Н), 3,85 (с, 6Н), 3,76 (с, 2Н), 3,09 (м, 2Н), 2,91 (м, 6Н)
Пример 24: Синтез 6-фтор-4-оксо-4Н-хромен-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,3 г соединения, полученного на стадии 4 примера 1, и 0,16 г 6-фторхромон-2-карбоновой кислоты использовали вместо 0,15 г соединения со стадии 4 примера 1 и 0,05 г 2-хинолинкарбоновой кислоты, соответственно, чтобы получить 0,27 г указанного в заголовке соединения (выход 66%).
1Н ЯМР (CDCl3)δ: 12,60 (с, 1Н), 8,66 (с, 1Н), 8,17 (д, 2Н), 7,92 (дд, 1Н), 7,87 (дд, 1Н), 7,82 (с, 1Н), 7,56 (м, 3Н), 7,29 (с, 1Н), 6,65 (с, 1Н), 6,58 (с, 1Н), 4,06 (д, 6Н), 3,88 (с, 6Н), 3,72 (с, 2Н), 3,08 (м, 2Н), 2,88 (м, 6Н)
Пример 25: Синтез 6-бром-4-оксо-4Н-хромен-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4, 5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,25 г соединения, полученного на стадии 4 примера 1, и 0,20 г 6-бромхромон-2-карбоновой кислоты использовали вместо 0,15 г соединения со стадии 4 примера 1 и 0,05 г 2-хинолинкарбоновой кислоты, соответственно, чтобы получить 0,22 г указанного в заголовке соединения (выход 60%).
1Н ЯМР (CDCl3)δ: 12,55 (с, 1Н), 8,59 (с, 1Н), 8,35 (с, 1Н), 8,12 (д, 2Н), 7,86 (д, 1Н), 7,75 (с, 1Н), 7,67 (д, 1Н), 7,48 (д, 2Н), 7,26 (с, 1Н), 6,62 (с, 1Н), 6, 56 (с, 1Н), 4,01 (с, 6Н), 3,85 (с, 6Н), 3,70 (с, 2Н), 3,07 (м, 2Н), 2,86 (м, 6Н)
Пример 26: Синтез цинолин-4-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,3 г соединения, полученного на стадии 4 примера 1, и 0,13 г цинолин-4-карбоновой кислоты использовали вместо 0,15 г соединения со стадии 4 примера 1 и 0,05 г 2-хинолинкарбоновой кислоты, соответственно, чтобы получить 0,16 г указанного в заголовке соединения (выход 41%).
1Н ЯМР (CDCl3)δ: 11,64 (с, 1Н), 9,79 (с, 1Н), 8,73 (с, 1Н), 8,67 (дд, 2Н), 7,95 (м, 5Н), 7,44 (д, 2Н), 6,65 (с, 1Н), 6,58 (с, 1Н), 4,13 (с, 3Н), 4,07 (с, 3Н), 3,88 (с, 6Н), 3,70 (с, 2Н), 3,02 (м, 2Н), 2,86 (м, 6Н)
Пример 27: Синтез 4-оксо-4Н-хромен-3-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0, 06 г хромон-3-карбоновой кислоты использовали вместо 0,05 г 2-хинолинкарбоновой кислоты с получением 0,08 г указанного в заголовке соединения (выход 40%).
1Н ЯМР (CDCl3)δ: 12,15 (с, 1Н), 9,04 (с, 1Н), 8,89 (д, 1Н), 8,50 (д, 2Н), 7,60 (м, 3Н), 7,49 (м, 3Н), 7,04 (с, 1Н), 6,55 (с, 1Н), 6,54 (с, 1Н), 4,04 (д, 6Н), 3,84 (с, 6Н), 3,67 (с, 2Н), 3,03 (м, 2Н), 2,84 (м, 6Н)
Пример 28: Синтез хинолин-3-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4, 5-дифторфенил]амида
Стадия 1: Получение 4,5-дифтор-2-нитро-п-толуолсульфонгидразона
п-Толуолсульфонгидразид в количестве 17,7 г добавляли к 100 мл этанола, затем добавляли 17,7 г 4,5-дифтор-2-нитробензальдегида, растворенного в малом количестве этанола, и смесь перемешивали при 80°С в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и смешивали со 150 мл воды. Выпавшее в осадок твердое вещество фильтровали, промывали 100 мл этанола и сушили при пониженном давлении с получением 31,6 г указанного в заголовке соединения (выход 94%).
1Н ЯМР (CDCl3)δ: 11,78 (с, 1Н), 8,66 (с, 1Н), 7,81 (д, 2Н), 7,64 (с, 1Н), 7,45 (д, 2Н), 7,17 (с, 1Н), 2,54 (с, 3Н)
Стадия 2: Получение 2-(2-{4-[5-(4,5-дифтор-2-нитрофенил)тетразол-2-ил]фенил}этил)-6,7-диметокси-1,2,3,4-тетрагидроизохинолина
Полученное на стадии 1 примера 1 соединение в количестве 28,9 г добавляли к 100 мл 50% этанола и охлаждали до 0°С. Затем добавляли 25 мл 35% HCl и 6,6 г нитрата натрия, охлаждали до -15°С, и затем медленно добавляли 31, 6 г соединения, полученного на стадии 1, растворенного в 500 мл пиридина. Реакционную смесь перемешивали в течение 20 часов и промывали посредством 1 н. HCl. Органический слой отделяли, сушили над сульфатом магния, фильтровали, подвергали дистилляции при пониженном давлении и перекристаллизовывали, используя этилацетат, с получением 28 г указанного в заголовке соединения (выход 60%).
1Н ЯМР (CDCl3)δ: 8,10 (д, 2Н), 7,67 (с, 1Н), 7,48 (д, 2Н), 7,42 (с, 1Н), 6,61 (д, 2Н), 3,98 (с, 6Н), 3,77 (с, 2Н), 3,00 (м, 2Н), 2,85 (м, 6Н)
Стадия 3: Получение 2-(2-4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил-2Н-тетразол-5-ил)-4,5-дифторфениламина
Полученное на стадии 2 соединение в количестве 28 г смешивали со смесью 360 мл этанола и 360 мл дихлорметана, добавляли 8,4 г Pd/C, и смесь оставляли в атмосфере водорода в течение 18 часов. Смесь после реакции восстановления фильтровали через слой целлита, слой промывали этанолом, фильтрат и промывной раствор объединяли и конденсировали при пониженном давлении с получением остатка, который привел к 22 г указанного в заголовке соединения (выход 84%).
1Н ЯМР (CDCl3)°С: 8,24 (д, 2Н), 7,88 (с, 1Н), 7,61 (д, 2Н), 6,75 (д, 2Н), 6,49 (с, 1Н), 4,79 (ушир, 2Н), 3,99 (д, 6Н), 3, 81 (м, 2Н), 3,54 (м, 8Н)
Стадия 4: Получение хинолин-3-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-дифторфенил]амида
Полученное на стадии 3 соединение в количестве 1,0 г смешивали с 15 мл дихлорметана, добавляли 0,47 г соединения, полученного на стадии 5 примера 1, и 0,4 мл триэтиламина, и смесь перемешивали при комнатной температуре в течение 20 часов. После промывания 100 мл дистиллированной воды, образовавшийся органический слой сушили над сульфатом магния, фильтровали и подвергали дистилляции при пониженном давлении. Полученный таким образом остаток подвергали колоночной хроматографии с получением 0,8 г указанного в заголовке соединения (выход 61%).
1Н ЯМР (CDCl3)°С: 11,84 (с, 1Н), 9,70 (с, 1Н), 8,97 (с, 1Н), 8,81 (с, 1Н), 8,22 (д, 1Н), 8,19 (д, 1Н), 7,97 (д, 1Н), 7,89 (т, 2Н), 7,68 (м, 1Н), 7,48 (д, 2Н), 6,61 (д, 2Н), 4,01 (д, 6Н), 3,68 (с, 2Н), 3,08 (м, 2Н), 2,85 (м, 6Н)
Пример 29: Синтез хинолин-3-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этилсульфанил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида
Стадия 1: Получение 1-(2-бромэтилсульфанил)-4-нитробензола
1,2-Дибромэтан в количестве 6,94 мл разбавляли 100 мл ацетонитрила, смешивали с 11,2 г карбоната калия и 5,0 г 4-нитробензолтиола и перемешивали при 80°С в течение 18 часов. После промывания посредством 300 мл дистиллированной воды и 300 мл водного раствора NaCl, образовавшийся органический слой сушили над сульфатом магния, фильтровали и подвергали дистилляции при пониженном давлении. Полученный остаток подвергали колоночной хроматографии с получением 6,9 г указанного в заголовке соединения (выход 82%).
1Н ЯМР (CDCl3)δ: 7,60 (с, 2Н), 7,42 (с, 2Н), 2,92 (м, 4Н)
Стадия 2: Получение 4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этилсульфанил]фениламина
Полученное на стадии 1 соединение в количестве 6,9 г, 6,1 г гидрохлорид 6,7-диметокси-1,2,3,4-тетрагидроизохинолина и 7,7 г карбоната калия добавляли к 80 мл ацетонитрила и перемешивали при 80°С в течение 14 часов. После промывания посредством 250 мл дистиллированной воды и 300 мл водного раствора NaCl, образовавшийся органический слой сушили над MgSO4, фильтровали и подвергали дистилляции при пониженном давлении с получением 5,3 г 6,7-диметокси-2-[2-(4-нитрофенилсульфанил)этил]-1,2,3, 4-тетрагидроизохинолина. Соединение перемешивали с 3,4 г железа, 6,74 мл 35% HCl и 35 мл метанола в атмосфере азота при давлении, составляющем 1 атмосферу, в течение 20 часов. Реакционную смесь фильтровали через слой целлита, слой промывали метанолом, и фильтрат и промывной раствор объединяли и подвергали дистилляции при пониженном давлении с получением остатка, который приводил к 3,0 г указанного в заголовке соединения (выход 33%).
1Н ЯМР (CDCl3)δ: 7,58 (с, 2Н), 7,32 (с, 2Н), 6,61 (с, 1Н), 6,55 (с, 1Н), 4,01 (с, 6Н), 3,84 (д, 6Н), 3,68 (с, 2Н), 3,26 (м, 2Н), 2,83 (м, 6Н)
Стадия 3: Получение 2-(2-{4-[5-(4,5-диметокси-2-нитрофенил)тетразол-2-ил]фенилсульфанил}этил)-6,7-диметокси-1,2,3, 4-тетрагидроизохинолина
Полученное на стадии 2 соединение в количестве 0,8 г добавляли к 4 мл 50% этанола. После охлаждения до 00С добавляли 0,6 мл 35% HCl и 0,16 г нитрата натрия, и после охлаждения до -15°С медленно добавляли раствор 0,9 г соединения, полученного на стадии 2 примера 1, растворенного в 14 мл пиридина. Смесь перемешивали в течение 20 часов, промывали посредством 1 н. HCl, сушили над сульфатом магния, фильтровали и подвергали дистилляции при пониженном давлении. Полученный остаток подвергали колоночной хроматографии с получением 0,7 г указанного в заголовке соединения (выход 52%).
1Н ЯМР (CDCl3)δ: 8,08 (д, 2Н), 7,58 (с, 1Н), 7,50 (д, 2Н), 7,32 (с, 1Н), 6,60 (с, 1Н), 6,53 (с, 1Н), 4,04 (с, 6Н), 3,85 (д, 6Н), 3,66 (с, 2Н), 3,25 (м, 2Н), 2,85 (м, 6Н)
Стадия 4: Получение 2-(2-{4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этилсульфанил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифениламина
Соединение, полученное на стадии 3, в количестве 0,2 г, 0,1 г железа и 0,15 мл концентрированной HCl добавляли к 3 мл метанола и перемешивали в атмосфере водорода при давлении, равном 1 атмосфере, в течение 18 часов. Реакционную смесь фильтровали через слой целлита, слой промывали метанолом, фильтрат и промывной раствор объединяли и перегоняли при пониженном давлении с получением остатка, который приводил к 0,15 г указанного в заголовке соединения (79%).
1Н ЯМР (CDCl3)δ: 8,14 (д, 2Н), 7,74 (с, 1Н), 7,56 (д, 2Н), 6,65 (с, 1Н), 6,56 (с, 1Н), 6,40 (с, 1Н), 3,96 (д, 6Н), 3,88 (с, 6Н), 3,69 (с, 2Н), 3,30 (м, 2Н), 2,88 (м, 6Н)
Стадия 5: Получение хинолин-3-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этилсульфанил]фенил}-2Н-тетразол-5-ил)-4, 5-диметоксифенил]амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,15 г соединения, полученного на стадии 4, и 0,06 г гидрата 3-изохинолинкарбоновой кислоты использовали в качестве исходных материалов, чтобы получить 0,10 г указанного в заголовке соединения (выход 52%).
1Н ЯМР (CDCl3)δ: 11,84 (с, 1Н), 9,70 (д, 1Н), 8,96 (д, 1Н), 8,75 (с, 1Н), 8,24 (д, 1Н), 8,12 (д, 2Н), 8,01 (д, 1Н), 7,88 (м, 2Н), 7,70 (т, 1Н), 7,51 (д, 2Н), 6,64 (с, 1Н), 6,56 (с, 1Н), 4,08 (д, 3Н), 4,04 (д, 3Н), 3,86 (с, 6Н), 3,69 (с, 2Н), 3,29 (м, 2Н), 2,88 (м, 6Н)
Пример 30: Синтез хинолин-3-карбоновой кислоты 2-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил-этил)-2Н-тетразол-5-ил]-4, 5-диметоксифениламида
Стадия 1: Получение 2-(2-бромэтил)-6,7-диметокси-1,2,3,4-тетрагидроизохинолина
Гидрохлорид 6,7-диметокси-1,2,3, 4-тетрагидроизохинолина в количестве 1 г смешивали с 15 мл N,N-диметилформамида, затем добавляли 1,05 г 1,3-дибромэтана, 1,80 г карбоната калия и 0,6 г йодида калия, и смесь перемешивали при 100°С в течение 6 часов. Реакционную смесь экстрагировали посредством 250 мл этилацетата, и экстракт промывали 250 мл дистиллированной воды. Образовавшийся органический слой сушили над сульфатом магния и фильтровали при пониженном давлении для удаления растворителя. Полученный остаток подвергали колоночной хроматографии с получением 1 г указанного в заголовке соединения (выход 60%).
1Н ЯМР (CDCl3): 6,52 (д, 2Н), 3,85 (с, 6Н), 3,59 (с, 2Н), 2,93-2,81 (м, 4Н), 2,77-2,68 (м, 2Н), 2,64-2,56 (м, 2Н)
Стадия 2: Получение 5-(4,5-диметокси-2-нитрофенил)-2Н-тетразола
4,5-Диметокси-2-нитробензонитрил в количестве 2,33 г добавляли к 15 мл толуола, затем добавляли 0,28 г дибутилоловооксида и 2,58 г триметилсилилазида, и смесь перемешивали при 100°С в течение 16 часов. Реакционную смесь перегоняли при пониженном давлении для удаления растворителя, и образовавшееся твердое вещество промывали 250 мл дихлорметана с получением 2,0 г указанного в заголовке соединения в виде твердого вещества серого цвета (выход 71%).
1Н ЯМР (CD3OD): 7,90 (c, 1Н), 7,30 (с, 1Н), 4,04 (c, 3Н), 3,99 (с, 3Н)
Стадия 3: Получение 2-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил-этил)-2Н-тетразол-5-ил]-4, 5-диметоксифениламина
Полученное на стадии 2 соединение в количестве 0,25 г, 0,3 г соединения, полученного на стадии 1, и 0,17 мл триэтиламина добавляли к 10 мл дихлорметана и перемешивали в течение 16 часов. Реакционную смесь экстрагировали 250 мл этилацетата, и экстракт промывали 250 мл дистиллированной воды. Образовавшийся органический слой сушили над сульфатом магния и фильтровали при пониженном давлении, затем подвергали колоночной хроматографии с получением 0,1 г нитропроизводного. Нитропроизводное смешивали с 30 мл дихлорметана, 30 мл этанола и 0,10 г Pd/C и оставляли в атмосфере водорода при давлении, равном 1 атмосфере, в течение 18 часов. После проведения реакции восстановления смесь фильтровали через слой целлита при пониженном давлении, слой промывали метанолом, фильтрат и промывной раствор объединяли и подвергали дистилляции при пониженном давлении с получением остатка, который привел к 0,40 г указанного в заголовке соединения (выход 91%).
1Н ЯМР (CD3OD): 7,37 (c, 1Н), 6,59 (с, 1Н), 6,52 (c, 1Н), 6,47 (с, 1Н), 4,00 (c, 2Н), 3,80 (с, 3Н), 3,79 (c, 3Н), 3,77 (с, 3Н), 3,76 (с, 3Н), 2,80-2,70 (м, 2Н), 2,65-3,1 (м, 6Н)
Стадия 4: Получение хинолин-3-карбоновой кислоты 2-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил-этил)-2Н-тетразол-5-ил]-4, 5-диметоксифениламида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,40 г соединения, полученного на стадии 3, и 0,22 г 3-хинолинкарбоновой кислоты использовали в качестве исходных материалов для получения 0,30 г указанного в заголовке соединения (выход 56%).
1Н ЯМР (CDCl3): 9,30 (с, 1Н), 8,80 (с, 1Н), 8,20 (м, 1Н), 8,15 (д, 1Н), 7,90 (т, 2Н), 7,80 (т, 1Н), 7,53 (с, 1Н), 6,60 (с, 1Н), 6,55 (с, 1Н), 4,01 (с, 2Н), 3,80 (с, 6Н), 3,70 (с, 6Н), 3,7-3,5 (м, 8Н)
Пример 31: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-3-фенилакриламида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,15 г соединения, полученного на стадии 4 примера 1, и 0,05 г транс-изомера коричной кислоты использовали в качестве исходных материалов с получением 0,11 г указанного в заголовке соединения (выход 59%).
1Н ЯМР (CDCl3)δ: 11,07 (с, 1Н), 8,82 (с, 1Н), 8,10 (м, 4Н), 7,83 (с, 1Н), 7,79 (с, 1Н), 7,58 (с, 1Н), 7,50 (с, 1Н), 7,41 (с, 1Н), 7,31 (д, 1Н), 6,71 (с, 1Н), 6,68 (с, 1Н), 6,62 (с, 1Н), 6,55 (с, 1Н), 3,98 (д, 6Н), 3,85 (с, 6Н), 3,69 (с, 2Н), 3,01 (т, 2Н), 2,83 (м, 6Н)
Пример 32: Синтез N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]-3-хинолин-3-ил-акриламида
Стадия 1: Получение 3-хинолин-3-ил-акриловой кислоты
3-Хинолинкарбоксальдегид в количестве 80 г, 85 г малоновой кислоты и 6,50 г пиперидина добавляли к 350 мл пиридина и перемешивали при 100°С в течение 3 часов. После смешивания с 1000 мл дистиллированной воды добавляли концентрированную кислоту HCl до тех пор, пока рН раствора не становился равным рН 4,8, и перемешивали в течение 1 часа. Полученное твердое вещество фильтровали при пониженном давлении, промывали посредством 1500 мл дистиллированной воды и сушили от 15 часов до 40 часов с получением 96 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 95%).
1Н ЯМР (DMSO-d6): 9,23 (с, 1Н), 8,67 (с, 1Н), 8,04-7,98 (м, 2Н), 7,82-7,75 (м, 2Н), 7,64 (т, 1Н), 6, 85 (д, 1Н)
Стадия 2: Получение N-[2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4, 5-диметоксифенил]-3-хинолин-3-ил-акриламида
Процедуру, описанную в примере 2, повторяли за исключением того, что 0,22 г соединения, полученного на стадии 4 примера 1, и 0,10 г соединения, полученного на стадии 1, использовали в качестве исходных материалов и получали 0,18 г указанного в заголовке соединения (выход 61%).
1Н ЯМР (CDCl3 )δ: 9,09 (с, 1Н), 8,22 (с, 1Н), 7,89-7,84 (м, 2Н), 7,77-7,71 (м, 3Н), 7,60-7,56 (м, 3Н), 7,26-7,19 (м, 3Н), 6,81 (м, 2Н), 6,61 (с, 1Н), 6,55 (с, 1Н), 3,96 (с, 6Н), 3,88 (с, 6Н), 3,68 (с, 2Н), 2, 99-2,77 (м, 8Н)
Пример 33: Синтез 4-оксо-4Н-хромен-2-карбоновой кислоты
(2-{2-[4-(2-{[2-(3, 4-диметоксифенил)этил]метиламино}этил)фенил]-2Н-тетразол-5-ил}-4,5-диметоксифенил)амида
Стадия 1: Получение 4-(2-{[2-(3, 4-диметоксифенил)этил]метиламино}этил)фениламина
2-(4-Нитрофенил)этилбромид в количестве 7,0 г, 5,94 г [2-(3,4-диметоксифенил)этил]метиламина, 8,41 г карбоната калия и 4,56 г йодида натрия добавляли к 70 мл N,N-диметилформамида и оставляли для проведения реакции при 100°С в течение 6 часов. После смешивания со 100 мл дистиллированной воды реакционную смесь экстрагировали три раза 200 мл порцией этилацетата, и объединенный органический слой промывали насыщенным раствором NaCl, сушили над сульфатом магния, фильтровали при пониженном давлении и перегоняли для удаления растворителя. Полученный остаток кристаллизовали из этилацетата с получением 7,86 г нитропроизводного. Нитропроизводное смешивали с 200 мл тетрагидрофурана и 200 мл метанола, добавляли 0,5 г Pd/C, и смесь оставляли в атмосфере водорода при давлении, равном 1 атмосфере, в течение 18 часов. Смесь после реакции восстановления фильтровали через слой целлита при пониженном давлении, слой промывали метанолом, фильтрат и промывной раствор объединяли и подвергали дистилляции при пониженном давлении с получением остатка, который привел к 6,52 г указанного в заголовке соединения (выход 68%).
1Н ЯМР (CDCl3)δ: 7,35 (д, 1Н), 6,90 (д, 2Н), 6,67 (д, 2Н), 6,60 (д, 1Н), 6,54 (с, 1Н), 3,90 (с, 6Н), 3,86 (с, 6Н), 2,95-2,71 (м, 8Н), 2,35 (с, 3Н)
Стадия 2: Получение 2-{2-[4-(2-{[2-(3,4-диметоксифенил)этил]метиламино}этил)фенил]-2Н-тетразол-5-ил}-4,5-диметоксифениламина
Процедуру, применяемую на стадии 3 и 4 примера 1, повторяли за исключением того, что 1,2 г соединения, полученного на стадии 2 примера 1, и 1 г соединения, полученного на стадии 1, использовали в качестве исходных материалов и получали 0,98 г указанного в заголовке соединения (выход 70%).
1Н ЯМР (CDCl3)δ: 7,70-7,66 (м, 1Н), 7,62-7,52 (м, 3Н), 7,45-7,20 (м, 2Н), 6,93 (д, 1Н), 6,70 (с, 1Н), 6,55 (с, 1Н), 3,98 (с, 6Н), 3,85 (с, 6Н), 2,93-2,73 (м, 8Н), 2,43 (с, 3Н)
Стадия 3: Получение 4-оксо-4Н-хромен-2-карбоновой кислоты (2-{2-[4-(2-{[2-(3, 4-диметоксифенил)этил]метиламино}этил)фенил]-2Н-тетразол-5-ил}-4,5-диметоксифенил)амида
Процедуру, описанную в примере 2, повторяли за исключением того, что 1 г соединения, полученного на стадии 2, и 0,97 г хромон-2-карбоновой кислоты использовали в качестве исходных материалов и получали 0,99 г указанного в заголовке соединения (выход 75%).
1Н ЯМР (CDCl3)δ: 7,70-7,63 (м, 1Н), 7,62-7,51 (м, 3Н), 7,45-7,18 (м, 2Н), 6,93 (д, 1Н), 6,68 (с, 1Н), 6,53 (с, 1Н), 3,97 (с, 6Н), 3,86 (с, 6Н), 2,95-2,75 (м, 8Н), 2,44 (с, 3Н)
Соединения, полученные в примерах 1-33, перечислены в таблице I.
Пример 34: Синтез хинолин-3-карбоновой кислоты [2-(2-4-[2-(6,7-диметокси-3, 4-дигидро-1Н-изохинолин-2-ил)этил]фенил-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида метансульфоната
Соединение, полученное в примере 1, в количестве 1 г перемешивали с 70 мл метанола в течение 30 мин, и смесь 0,1 мл метансульфокислоты и 5 мл метанола добавляли по каплям при 0°С. Смесь нагревали до комнатной температуры в течение 10 мин и перемешивали в течение 6 часов и получали 0,95 г указанного в заголовке соединения (выход 83%).
1Н ЯМР (CD3OD)δ: 9,71 (c, 1Н), 9,55 (с, 1Н), 8,33 (д, 1Н), 8,31 (д, 1Н), 8,24-8,20 (м, 3Н), 8,10 (т, 1Н), 7,89 (с, 1Н), 7,65 (м, 3Н), 6,86 (с, 1Н), 6,84 (с, 1Н), 4,00 (д, 6Н), 3,86 (д, 6Н), 3,65-3,55 (м, 4Н), 3,37-3,26 (м, 6Н), 2,19 (с, 3Н)
Пример 35: Синтез 4-оксо-4Н-хромен-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида метансульфоната
Процедуру, описанную в примере 33, повторяли за исключением того, что 1,2 г соединения, полученного в примере 21, и 0,12 мл метансульфокислоты использовали в качестве исходных материалов и получали 1,1 г указанного в заголовке соединения (выход 80%).
1Н ЯМР (CD3OD)δ: 8,35 (c, 1Н), 8,18-8,16 (м, 3Н), 7,95 (т, 1Н), 7,79 (д, 1Н), 7,71-7,64 (м, 4Н), 7,08 (с, 1Н), 7,05 (с, 1Н), 7,01 (с, 1Н), 4,05 (с, 6Н), 4,01 (с, 3Н), 3,90 (с, 3Н), 3,90-3,73 (м, 4Н), 3,51-3,41 (м, 6Н), 2,18 (с, 3Н)
Пример 36: Синтез 4-оксо-4Н-хромен-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амида гидрохлорида.
Процедуру, описанную в примере 33, повторяли за исключением того, что 1,5 г соединения, полученного в примере 21, и 0,1 мл хлористоводородной кислоты использовали в качестве исходных материалов и получали 1,4 г указанного в заголовке соединения (выход 89%).
1Н ЯМР (CD3OD)δ: 8,37 (c, 1Н), 8,19-8,15 (м, 3Н), 7,97 (т, 1Н), 7,82 (д,1Н), 7,73-7,65 (м, 4Н), 7,18 (с, 1Н), 7,15 (с, 1Н), 7,11 (с, 1Н), 4,07 (с, 6Н), 4,02 (с, 3Н), 3,98 (с, 3Н), 3,94-3,75 (м, 2Н), 3,52-3,43 (м, 8Н)
Препаративный пример 1: Приготовление препарата для перорального введения
Таблетку приготовляли с использованием следующих ингредиентов, где активным ингредиентом было соединение примера 21:
Другие таблетки также были приготовлены таким же способом, используя в качестве активного ингредиента любое из тетразольных производных согласно изобретению из примеров 1-20 и примеров 22-36. В случае соединения из примера 35, используемое количество составило 114 мг.
Препаративный пример 2: Приготовление препарата для перорального введения
Твердую желатиновую капсулу приготовляли с использованием следующих ингредиентов, где активным ингредиентом было соединение примера 21:
Другие капсулы также были приготовлены таким же способом, используя в качестве активного ингредиента любое из тетразольных производных согласно изобретению из примеров 1-20 и примеров 22-36. В случае соединения из примера 35, используемое количество составило 114 мг.
Препаративный пример 3: Приготовление препарата для инъекции
Инъецируемый препарат приготовляли с использованием следующих ингредиентов, где активным ингредиентом было соединение из примера 21:
Другие инъецируемые препараты также были приготовлены таким же способом, используя в качестве активного ингредиента любое из тетразольных производных согласно изобретению из примеров 1-20 и примеров 22-36. В случае соединения из примера 35, используемое количество составило 23 мг, и HCl не использовали.
Препаративный пример 4: Приготовление препарата для инъекции
Инъецируемый препарат приготовляли с использованием следующих ингредиентов, где активным ингредиентом было соединение из примера 21:
Другие инъецируемые препараты также были приготовлены таким же способом, используя в качестве активного ингредиента любое из тетразольных производных согласно изобретению из примеров 1-20 и примеров 22-36. В случае соединения из примера 35, используемое количество составило 23 мг.
Пример тестирования 1: Ингибирующее действие соединения согласно изобретению по отношению к п-гликопротеину
Для того, чтобы исследовать биодоступность любого из соединений согласно изобретению как ингибитора п-гликопротеина, его клеточную токсичность измеряли, используя линию клеток MCF-7 и линию клеток MCF-7/Dox, которые являются MCF-7-клеточными линиями, экспрессирующей п-гликопротеин.
Клетки субкультивировали в среде 5% FBS (сыворотка плода коровы)/RPMI1640, дополненной глутамином 2 ммоль, бикарбонатом натрия 3,7 г/л и гентамицином 10 мг/л при 37°С в инкубаторе с 5% СО2 при 100%-ной влажности, и собирали, используя 0, 25%-ный раствор трипсина, содержащий 3 мМ 1,2-циклогександиаминтетрауксусной кислоты.
Собранные клетки помещали на 96-луночный планшет с плоскодонными лунками при плотности клеток 2х103 клеток/лунку и инкубировали в такой же среде в течение 24 часов. Паклитаксел, противораковый агент, разбавляли такой же средой с получением тестируемых растворов с концентрацией 10-11˜10-6 М. После удаления культуральных сред каждую лунку обрабатывали 100 мкл тестируемого раствора, отдельно или в комбинации с 50 нМ любого из тестируемых соединений из примеров 1-30. После инкубации в течение 72 часов, культуральную среду удаляли, каждую лунку обрабатывали 10%-ной трихлоруксусной кислотой в течение 1 часа для фиксации клеток, промывали водой и сушили при комнатной температуре. После добавления окрашивающего раствора, содержащего 1% уксусной кислоты с 0,4% SRB (сульфородамин В), лунки оставляли при комнатной температуре в течение 30 мин и промывали 1%-ной уксусной кислотой для удаления остатков SRB. Затем добавляли 10 мл основного раствора trisma, имеющего рН 10,3˜10,5, и измеряли поглощение каждой лунки при 520 нм, используя аппарат для чтения микропланшетов, чтобы оценить ED50 концентрацию лекарственного средства, при которой рост раковых клеток ингибируется на 50%. Кроме того, увеличение противораковой активности паклитаксела относительно клеточной линии MCF7/Dox (устойчивые раковые клетки) измеряли путем определения отношения EDPAC50/ED50, где EDPAC50 является величиной, определенной только для паклитаксела. Результаты приведены в таблице II.
Как показано в таблице II, паклитаксел проявляет цитотоксичность относительно клеток MCF-7/Dox в значительно большей степени в случае обработки им в комбинации с соединениями примеров, чем когда их обрабатывают только паклитакселом, и можно увидеть, что соединения согласно изобретению формулы (I) эффективно подавляют активность р-гликопротеина даже при низкой концентрации, составляющей 50 нМ.
Пример тестирования 2: In vivo всасывание перорально введенного паклитаксела
Для того чтобы исследовать активность соединений согласно изобретению, синтезированных в примерах, проверки всасывания in vivo осуществляли следующим образом.
Двадцать пять 14-15-недельных крыс линии Sprague-Dawley держали голодными в течение 24 часов, в то же время крысы имели свободный доступ к воде, и затем крыс разделяли на 4 группы от 5 до 8 крыс в каждой группе. Трем группам из упомянутых выше групп перорально вводили 20 мг/кг веса тела паклитаксела (6 мг паклитаксела/0,5 мл кремофора EL + 0,5 мл этанола) и любое из соединений примера 1, 21 и 22 (12 мг соединений примеров/4 мл 5%-ной декстрозы + 1,2 мкг метансульфокислоты), и контрольной группе вводили носитель (4 мл 5%-ной декстрозы + 1,2 мкг метансульфокислоты) и 20 мг паклитаксела (композиция: 6 мг паклитаксела в 0,5 мл кремофора EL + 0,5 мл этанола). Образцы крови брали прямо из сердца каждой крысы до введения и через 1, 2, 4, 6, 8 и 24 часа после введения препаратов.
Каждый из образцов крови центрифугировали при 12000 об./мин с получением образца сыворотки, 200 мкл которой смешивали с 400 мкл ацетонитрила (внутренний стандарт), и смесь встряхивали для получения экстракта. Экстракт центрифугировали при 12000 об./мин, 4°С в течение 5 мин и получали супернатант. Супернатант в количестве 50 мкл подвергали полумикроанализу ВЭЖХ при следующих условиях:
- полумикросистема ВЭЖХ: модель SI-1 (Shiseido),
- колонка для анализа: Capcell Pak C18 UG120 (5 мкм, 1,5×250 мм, Shiseido),
- пре-колонка: Capcell Pak C18 MF Ph-1 (4,6×10 мм, Shiseido),
- концентрационная колонка: Capcell Pak C18 UG120 (5 мкм, 1,5×35 мм, Shiseido),
- подвижная фаза для пре-колонки: 20% ацетонитрил,
- подвижная фаза для колонки для анализа: 55% ацетонитрил,
- объем впрыскиваемого раствора: 5 мкл,
- скорость потока: 5 мкл/мин,
- детектор: 227 нм.
Зависимые от времени изменения концентрации паклитаксела в крови представлены в таблице III.
Представленные в таблице III результаты свидетельствуют о том, что соединения согласно изобретению можно с успехом использовать для повышения биодоступности паклитаксела, который сам по себе трудно всасывается в пищеварительном тракте.
Хотя изобретение было описано с признанием вышеприведенных специфических аспектов, следует отметить, что специалисты в данной области могут внести модификации и изменения к изобретению, которые также попадают в объем изобретения, определенный приложенной формулой изобретения.
Реферат
Описываются новые соединения общей формулы (I)
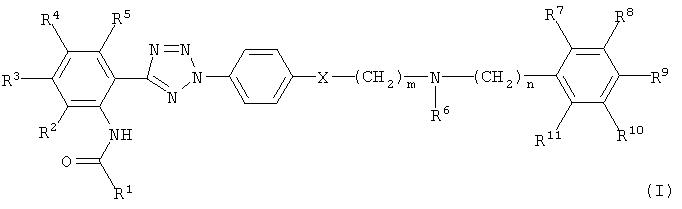
где R1 - хинолинил, возможно замещенный С1-5 алкокси, изохинолинил, хиноксалинил, пиридинил, пиразинил, бензил, возможно замещенный галогеном, нафталинил, тиофенил, фуранил, циннолил, фенилвинил, хинолилвинил или 4-оксо-4Н-хроменил, возможно замещенный галогеном, С1-5 алкилом или С1-5 алкокси;
R2, R5, R8 и R11 - водород;
R3 и R4 - галоген, C1-5 алкокси;
R6 и R7 - Н или C1-5 алкил или вместе образуют радикал -СН2-СН2-;
R9 и R10 - C1-5 алкокси;
m и n - независимо целое число от 0 до 4.
Х означает CH2 или S, их фармацевтически приемлемые соли, способ их получения и фармацевтическая композиция на их основе. Данные соединения являются ингибиторами р-гликопротеина, усиливают биодоступность противоракового агента и могут найти применение в медицине. 3 н. и 4 з.п. ф-лы, 3 табл.
Формула
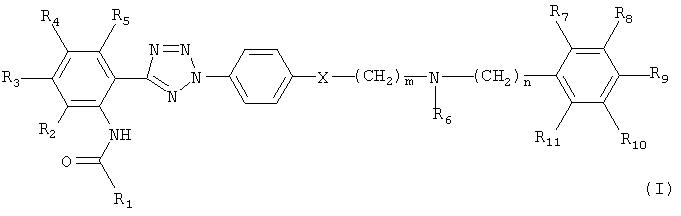
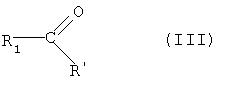

Комментарии