Производные 4-(тио- или селеноксантен-9-илиден)-пиперидина или акридина, фармацевтическая композиция на их основе и применение - RU2314305C2
Код документа: RU2314305C2
Описание
Настоящее изобретение относится к производным 4-(тио-или селеноксантен-9-илидена)-пиперидина и акридина и фармацевтически приемлемым солям этих соединений, обладающим полезными фармакологическими свойствами, включая возможность их применения в качестве селективных антагонистов рецептора 5-НТ2в для лечения болезненных состояний, которые могут быть облегчены лечением антагонистом рецептора 5-НТ2B.
Серотонин, нейромедиатор со смешанными и сложными фармакологическими свойствами, был открыт в 1948 году, и с тех пор является предметом фундаментальных исследований. Серотонин, называемый также как 5-гидрокситриптамин (5-НТ), действует как центрально, так и периферически на отдельные рецепторы 5-НТ. В настоящее время известно 14 подтипов серотониновых рецепторов, подразделяемых на 7 семейств, от 5-HT1 до 5-НТ7. Внутри семейства 5-НТ2 известны подтипы 5-НТ2А, 5-НТ2В и 5-НТ2C. Для этих подтипов характерна гомология последовательностей и сходство в специфичности к широкому спектру лигандов. Обзор номенклатуры и классификации рецепторов 5-НТ можно найти в работах (Martin and Humphrey, Neuropharm. 1994, 33, 261-273 и Hoyer et al., Pharm. Rev. 1994, 46, 157-203).
Рецептор 5-НТ2в, первоначально обозначенный как 5-HT2F, или рецептор, подобный серотониновому (SRL), впервые был охарактеризован в дне желудка, полученного от крыс (см. Clineschmidt et al., J. Pharmacol. Exp. Ther. 1985, 235, 696-708; Cohen and Wittenauer, J. Cardiovasc. Pharmacol. 1987, 10, 176-181) и сначала был клонирован из крысы (см. Foguet et al., EMBO 1992, 11, 3481-3487), а затем был клонирован рецептор 5-HT2B человека (см. Schmuck et al., FEBS Lett. 1994, 342, 85-90; Kursar et al., Mol. Pharmacol. 1994, 46, 227-234). Близкородственный рецептор 5-НТ2С, широко распространенный в мозге человека, впервые был охарактеризован как подтип 5-HT1С (см. Pazos et al., Eur. J. Pharmacol. 1984, 106, 539-546), а затем был отнесен к семейству рецепторов 5-НТ2 (см. Pritchett et al., EMBO J. 1988, 7, 4135-4140).
Благодаря сходству фармакологии лигандных взаимодействий в рецепторах 5-НТ2В и 5-НТ2С многие терапевтические мишени, которые были предложены для антагонистов рецептора 5-НТ2С, также являются мишенями для антагонистов рецептора 5-НТ2В. Современные данные свидетельствуют о терапевтической роли антагонистов рецепторов 5-НТ2В/2С в лечении беспокойства (например, общего беспокойства, панического страха и навязчивого невроза), алкоголизма и пристрастия к другим наркотикам, депрессии, мигрени, нарушений сна, нарушений питания (например, нервной анорексии) и приапизма. Кроме того, современные данные свидетельствуют о терапевтической роли селективного антагониста рецептора 5-НТ2В, обладающего рядом преимуществ: эффективностью, быстротой начала действия и отсутствием побочных эффектов. Ожидается, что такие агенты будут полезны при лечении гипертензии, нарушений со стороны желудочно-кишечного тракта (например, синдрома повышенной раздражимости кишечника, гипертонуса нижнего сфинктера пищевода, нарушений перистальтики), рестеноза, астмы и обструктивного заболевания дыхательных путей и гиперплазии простаты (например, доброкачественной гиперплазии простаты).
В US-A-3275640 описаны в общем замещенные 1-гидрокарбил-4-(9Н-тиоксантен-9-илиден)-пиперидины и их получение. Также описано, что эти соединения могут применяться в качестве терапевтических агентов ввиду их антигистаминных и/или антисеротониновых свойств.
US-A-3557287 относится к комбинированному препарату для применения в лечении головной боли сосудистого происхождения, содержащему в качестве активных составляющих: а) вазотоническую лизергиновую кислоту, выбираемую из ряда: эргостин, эрготамин, дигидроэргостин, дигидроэрготамин, эрговалин, 5'-метилэргоаланин; б) кофеин и в) 9-(1-метил-4-пиперидилиден)тиоксантен (=1-метил-4-(9Н-тиоксантен-9-илиден)-пипервдин.
В патенте DE-A-2256392 описаны производные 4-(9Н-тиоксантен-9-илиден)-пиперидина, в которых атом азота пиперидинового кольца связан с алкильным радикалом, замещенным циано-группой, -COR или -COOR. Эти производные обладают свойством вызывать сон.
Патент JP-A-61106573 относится к применению замещенных 4-(9Н-тиоксантен-9-илиден)-пиперидинов в качестве пестицидов.
Задачей настоящего изобретения является получение соединения, действующего как селективный антагонист рецептора 5-НТ2В.
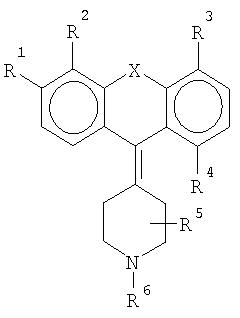
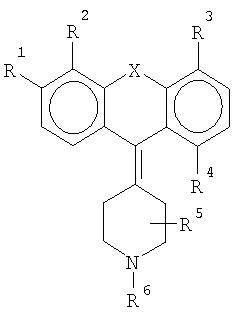
Этой задаче соответствует соединение формулы
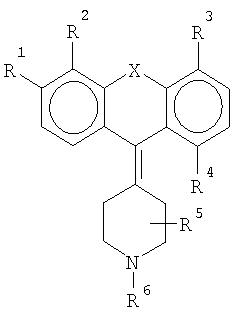
где R1 означает метил, этил, пропил, изопропил, бутил, изобутил, пентил (разветвленный или неразветвленный), гексил (разветвленный или неразветвленный), метокси-, этокси-, пропокси-, изопропокси-, бутокси-, изобутокси-, фенокси-, трифторметил-, трифторметокси-, амино-, диметиламино-, фтор, хлор, бром, - CON(CH3)2 или -CON(С2Н5)2, R2 означает метил, этил, пропил, изопропил, бутил, изобутил, пентил, гексил, гидрокси- или водород, или R1 и R2 образуют гетероцикл, R3 означает метил, этил, пропил, изопропил, бутил, изобутил, пентил, гексил, гидрокси- или водород, R4 означает гидрокси-, метокси-, этокси-, пропокси-, изопропокси-, бутокси-, изобутокси-, трифторметил, амино-, диметиламино-, диэтиламино-, фтор, хлор или бром, метил, этил, пропил, изопропил, бутил или водород, R5 означает метил или водород, R6 означает метил или этил, а Х означает S, N или Se, или фармацевтически приемлемая соль этого соединения, с условием что, если R1 означает этокси-, а Х означает S, то, по крайней мере, один из R2, R3, R4 и R5 не является атомом водорода.
Остатки R1 и R4 представляют особый интерес в рамках настоящего изобретения. R1 участвует в регулировании селективности соединения по отношению к рецептору 5-НТ2В. R4 участвует в регулировании аффинности к рецептору H1. Предпочтительно R1 означает низшую алкильную или алкокси-группу, или галогенированную алкильную или алкокси-группу, стерически способствующую, но не препятствующую связыванию с рецептором. R4 предпочтительно означает полярную группу, в частности небольшую полярную группу, однако подходит любая группа, снижающая аффинность H1. R2, R3 и R5 независимо могут быть любым остатком, если не затрудняется связывание с рецептором 5-НТ2В.
Настоящее изобретение также направлено на использование такого соединения для получения лекарства для лечения болезненных состояний, которые могут быть облегчены лечением антагонистом рецептора 5-НТ2В.
Далее настоящее изобретение относится к фармацевтической композиции, содержащей такое соединение или его фармацевтически приемлемую соль в сочетании с одним или более фармацевтически приемлемыми носителями.
Далее настоящее изобретение направлено на получение таких соединений.
Термин "фармацевтически приемлемая соль" относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований и которые не являются биологически или иначе нежелательными, эти соли образованы с неорганическими кислотами, такими как соляная, бромоводородная, серная, азотная, фосфорная и подобные кислоты, и с органическими кислотами, такими как уксусная, пропионовая, гликолевая, пировиноградная, щавелевая, яблочная, малоновая, янтарная, малеиновая, фумаровая, винная, лимонная, бензойная, коричная, миндальная, метансульфоновая, этансульфоновая, п-толуолсульфоновая, салициловая, аскорбиновая и подобные кислоты.
Термин "лечение" в настоящем документе подразумевает любое лечение заболевания млекопитающего, в частности человека, и включает:
i) предотвращение возникновения заболевания у пациента, который может быть предрасположен к заболеванию, но оно у него к настоящему моменту не диагностировано;
ii) ингибирование заболевания, то есть остановка его развития; или
iii) ослабление заболевания, то есть вызов его регрессии.
Термин "болезненное состояние, которое может быть облегчено лечением антагонистом рецептора 5-НТ2В" в данном документе включает все болезненные состояния, которые, как известно в данной области техники, успешно лечатся соединениями, обладающими аффинностью к рецепторам 5-НТ2В, в общем, а также состояния, которые, как было обнаружено, успешно лечатся одним из соединений настоящего изобретения. Такие болезненные состояния включают, без ограничения, мигрень, боль (например, острую, хроническую, невропатическую, боль при воспалительных заболеваниях и раке), гипертензию, нарушения со стороны желудочно-кишечного тракта (например, синдром повышенной раздражимости кишечника, гипертонус нижнего сфинктера пищевода, нарушения перистальтики), рестеноз, астму и обструктивное заболевание дыхательных путей, гиперплазию простаты (например, доброкачественную гиперплазию простаты) и приапизм.
В предпочтительном способе осуществления изобретения остатки в общей формуле, приведенной выше, являются следующими: R1 означает изопропил, диметиламино-, метокси- или этокси-группу, R2 означает метил, этил или водород, или R1 и R2 образуют гетероцикл, R3 означает этил или водород, R4 означает метокси-, гидрокси-группу, этил, метил или водород, R5 означает метил или водород, R6 означает метил, а Х означает S, N или Se, или фармацевтически приемлемые соли этих соединений, при условии, что если R1 означает этокси-группу, а Х означает S, то, по крайней мере, один из остатков R2, R3, R4 и R5 не является атомом водорода.
В особенно предпочтительном способе осуществления настоящего изобретения соединение выбирается из группы, включающей: 4-(6-изопропил-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-этокси-1-этил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-этокси-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-метокси-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-диметиламино-1-метокси-тиоксантен-9-илиден)-1-метилпиперидин, 4-(6-диметиламино-1-гидрокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-селеноксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-1-метокси-селеноксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-1-гидрокси-селеноксантен-9-илиден)-1-метил-пиперидин, 3-этокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидроакридин, 6-этокси-1-метокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидроакридин, 6-этокси-1-гидрокси-9-(1-метил-пиперидин-4-илиден)-9, 10-дигидроакридин, 4-(3-этокси-5-этил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-4-метил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-4-этил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-тиоксантен-9-илиден)-1,3-диметил-пиперидин, 4-(3,4-(циклопент-3'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3, 4-(циклопент-4'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин и 4-(3,4-(циклопент-5'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин.
В особенно предпочтительном варианте осуществления настоящего изобретения соединение для применения в качестве медикамента и для получения фармацевтической композиции выбирается из группы, включающей: 4-(6-изопропил-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-изопропил-1-гидрокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-этокси-1-этил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-этокси-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(-этокси-1-гидрокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-метокси-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-диметиламино-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-диметиламино-1-гидрокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-селеноксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-1-метокси-селеноксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-1-гидрокси-селеноксантен-9-илиден)-1-метил-пиперидин, 3-этокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидроакридин, 6-этокси-1-метокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидроакридин, 6-этокси-1-гидрокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидроакридин, 4-(3-этокси-5-этил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-4-метил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-4-этил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-тиоксантен-9-илиден)-1,3-диметил-пиперидин, 4-(3, 4-(циклопент-3'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3,4-(циклопент-4'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин и 4-(3, 4-(циклопент-5'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин.
Хотя в US-A-3275640 в общем описаны антигистаминные и/или антисеротониновые свойства класса замещенных 1-гидрокарбил-4-(9Н-тиоксантен-9-илиден)-пиперидинов, в него не включены 4-(тио- или селеноксантен-9-илиден)-пиперидиновые или акридиновые производные настоящего изобретения, а также не указана связь выбранного производного со специальными антигистаминными и/или антисеротониновыми свойствами. Термин "антисеротониновое свойство" является очень широким и относится к 14 различным подтипам рецепторов. Более того, в US-A-3275640 не указано, что член класса замещенных 1-гидрокарбил-4-(9Н-тиоксантен-9-илиден)-пиперидинов обладает селективной аффинностью к одному из различных подтипов рецепторов 5-НТ, а именно к рецептору человека 5-НТ2В. Способность новых 4-(тио- или селеноксантен-9-илиден)-пиперидиновых или акридиновых производных функционировать в качестве антагониста рецептора 5-НТ2В обеспечивает возможность более специфического лечения указанных выше заболеваний и, в то же время, снижения нежелательных побочных эффектов.
Селективность по отношению к рецепторам 5-НТ2В достигается замещением в R1. Таким образом согласно настоящему изобретению для того, чтобы служить фармацевтическим соединением с селективной аффинностью к рецептору 5-НТ2В, в положение R1 должно быть замещено одним из остатков, указанных выше. Предпочтительным заместителем в положении R1 является низшая алкильная или алкокси-группа, или галогенированная алкильная или алкокси-группа. Эти группы особенно способствуют связыванию соединения с рецептором 5-НТ2В. Остаточная аффинность соединений к человеческим рецепторам H1 может быть снижена замещением в положении R4. Анти-гистаминергический эффект Р4-незамещенных 4-(тио- или селеноксантен-9-илиден)-пиперидиновых или акридиновых производных, вероятно, приведет к седативному действию. Для снижения этого нежелательного побочного эффекта согласно настоящему изобретению предпочтительно, чтобы в положении R4 был заместитель, предпочтительно выбираемый из небольших боковых групп, наиболее предпочтительными являются небольшие полярные боковые группы. Однако может применяться любая боковая группа, снижающая аффинность к рецептору H1.
Один из способов получения замещенных 1-гидрокарбил-4-(10-тиоксантилиден)-пиперидинов, не включающий новых соединений настоящего изобретения, описан в US-A-3275640.
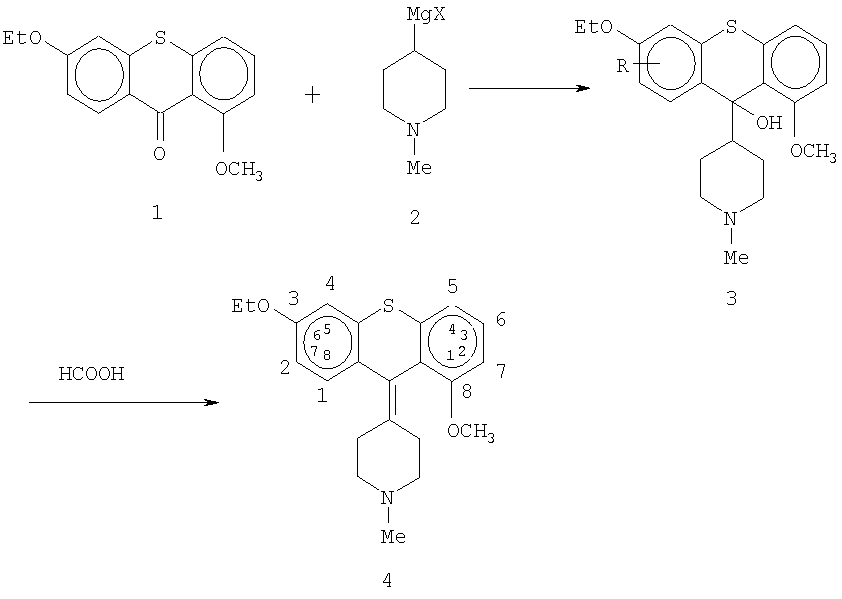
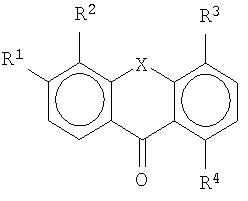
Способы, применяемые для синтеза новых соединений, состоят в следующем: "Верхний" кетон 1 вводят в реакцию с "нижним" реактивом Гриньяра 2, получают продукт присоединения - спирт 3. Спирт 3 дегидратируют муравьиной или другой кислотой, получают требуемый алкен 4.
Схема I:
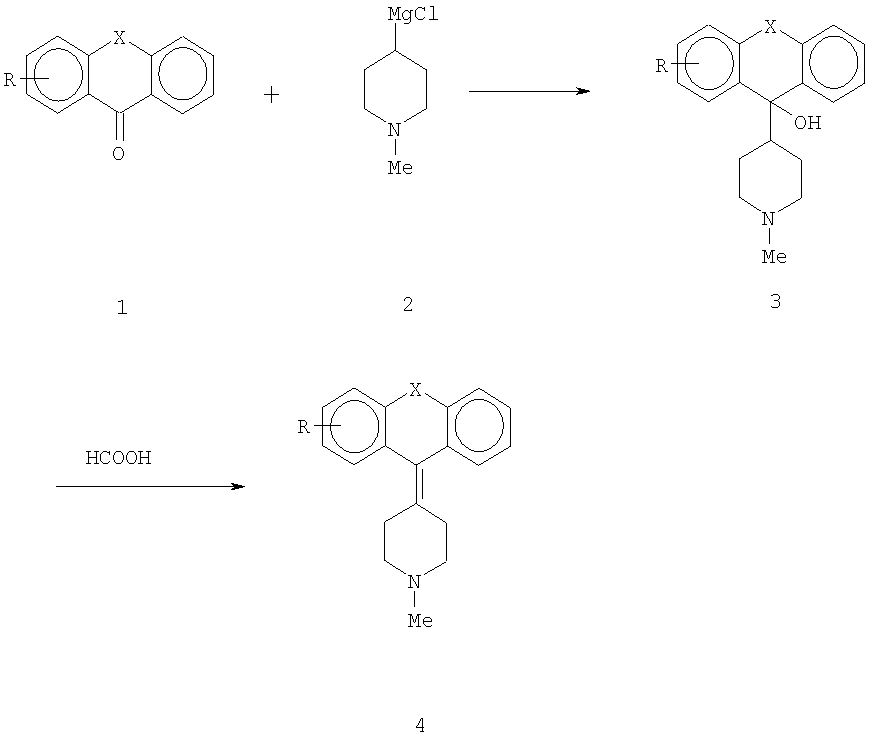
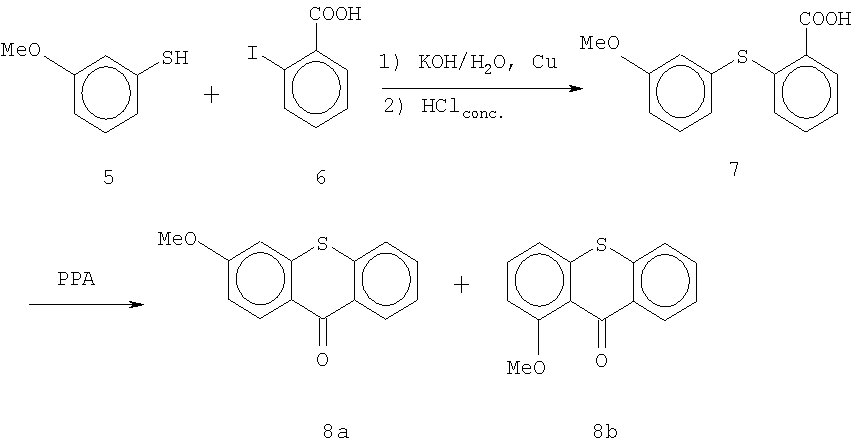
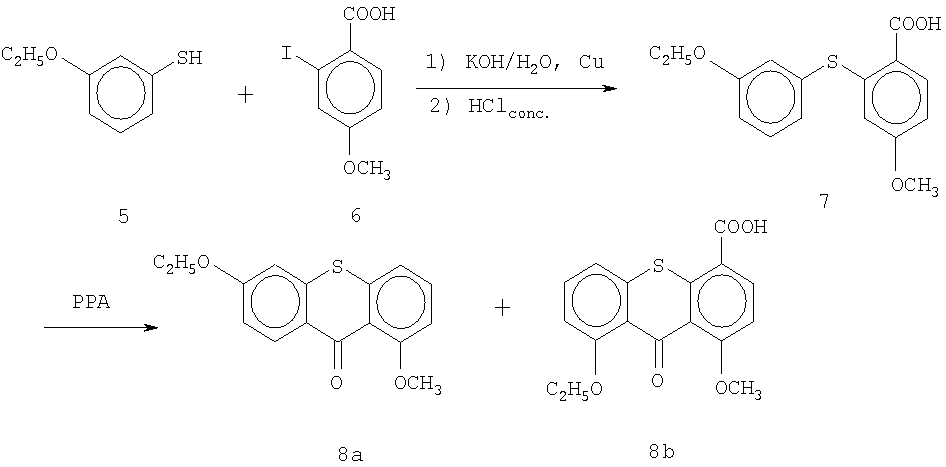
Один из способов синтеза исходного соединения 1, т.е. 3-этокситиоксантона, начинается с 3-метокситиоксантона 8а, который может быть получен методом, описанным в I.Cervena, J.Metysova, E.Svatek, В.Kakac, J.Holubek, M.Hrubantova and M. Protiva, Coll. Czech. Chem. Comm. 41, 881-904 (1978) (Схема II). 3-Метокситиофенол 5 вводят в реакцию с 2-йодбензойной кислотой 6 в кипящем растворе КОН в присутствии меди. После добавления соляной кислоты получают продукт присоединения - кислоту 7. Кислоту 7 циклизуют с полифосфорной кислотой, получают смесь изомеров 3-метокситиоксантона 8а и 1-метокситиоксантона 8b, которые можно разделить хроматографией, предпочтительно колоночной хроматографией. В промышленном масштабе для получения и разделения продуктов можно применять другие технологии разделения, включая, без ограничения, разделение на симулированном подвижном слое (simulated moving bed separations), капиллярный электрофорез, высокопроизводительные одно- или многоколоночные методы разделения при высоком давлении, с применением хиральных или нехиральных поддерживающих и разделительных сред. Также могут применяться методы ферментативной дегидратации в комбинации с любой из вышеупомянутых технологий, или методы разделения с ферментативным обогащением (enzyme enrichment separation), применяемые отдельно или в комбинации с вышеуказанными.
Схема II:
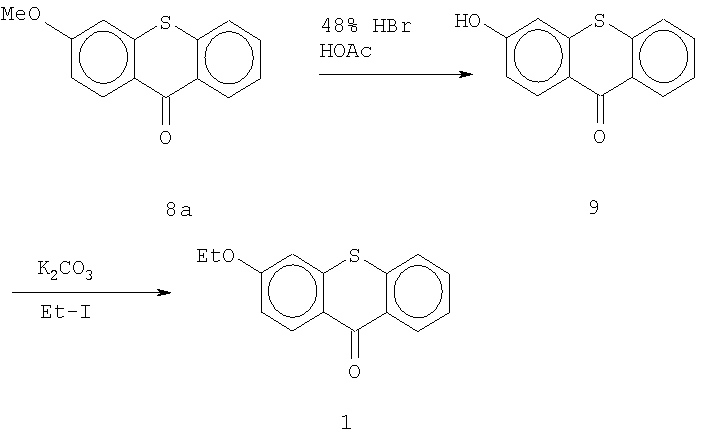
Для вариаций боковой цепи тиоксантена выделенный 3-метокситиоксантен 8а может быть превращен, например, в 3-гидрокситиоксантен 9 путем обработки бромоводородной и уксусной кислотами. 3-гидрокситиоксантен 9 можно ввести в реакцию с йодоэтаном в присутствии основания, предпочтительно К2СО3, и получить 3-этокситиоксантон 1 согласно Схеме III.
Схема III:
Таким образом, основываясь на методе Cervena, I. et al., описанном в Collection Czechoslov. Chem. Cumm. 41 (1978), 881-904, или на одном из методов Watanabe, М. et al., описанных в Chem. Pharm. Bull 32 (1984), 1264-1267, Chem. Pharm. Bull 34 (1986), 2810-2820 и Chem. Pharm. Bull 37 (1989), 36-41, можно синтезировать другие производные, например, 4-(6-изопропил-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-изопропил-1-гидрокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-этокси-1-этил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-этокси-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-этокси-1-гидрокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(6-диметиламино-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-селеноксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-1-метокси-селеноксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-1-гидрокси-селеноксантен-9-илиден)-1-метил-пиперидин, 3-этокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидро-акридин, 6-этокси-1-метокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидро-акридин, 6-этокси-1-гидрокси-9-(1-метил-пиперидин-4-илиден)-9, 10-дигидро-акридин, 4-(3-этокси-5-этил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-4-метил-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3-этокси-4-этил-тиоксантен-9-илиден)-1-метил-пиперидин и 4-(3-этокси-тиоксантен-9-илиден)-1,3-диметил-пиперидин,4-(3,4-(циклопент-3'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин, 4-(3, 4-(циклопент-4'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин и 4-(3,4-(циклопент-5'-окси-1'-ено)-тиоксантен-9-илиден)-1-метил-пиперидин.
Соединения настоящего изобретения являются антагонистами рецептора 5-НТ2B человека. Аффинность к рецепторам 5-НТ2B была показана в исследовании связывания in vitro при использовании клонированных рецепторов 5-НТ2B человека, радиоактивно меченных [3Н]-5НТ, как описано в примерах. Селективность по отношению к рецептору 5-HT2B человека была показана при исследовании против рецепторов человека 5-НТ2A и 5-НТ2C. Свойства антагониста по отношению к НТ2B были определены в продольных мускулах дна желудка крыс. Аффинность к рецептору человека H1 определяли в исследовании связывания in vitro при использовании клонированных рецепторов H1 человека, радиоактивно меченных [3H]-мепирамином, как описано в примерах. Свойства антагониста были определены при исследовании продукции фосфата [3H]-инозитола в транзиторно трансфицированных клетках НЕК-293.
Таким образом, соединения настоящего исследования применимы при лечении заболеваний, которые могут быть облегчены блокадой рецепторов 5-НТ2B. Из-за сходства в фармакологии лигандных взаимодействий в рецепторах 5-НТ2С и 5-НТ2B многие терапевтические мишени, предложенные для антагонистов рецептора 5-НТ2C, также являются мишенями для антагонистов рецептора 5-НТ2B. В частности, некоторые клинические наблюдения предполагают терапевтическую роль антагонистов рецептора 5-НТ2В в предотвращении мигрени, при этом полагается, что выброс 5-НТ в плазму является фактором, вызывающим мигрень. Кроме того, неселективные агонисты рецептора 5-НТ2В провоцируют приступы мигрени у чувствительных пациентов, а неселективные антагонисты рецептора 5-НТ2В эффективны в предотвращении приступа мигрени (см. Kalkman, Life Sciences 1994, 54, 641-644). Предполагается, что активация рецепторов 5-НТ2B, расположенных на эндотелиальных клетках менингеальных кровеносных сосудов, запускает приступы мигрени через образование оксида азота (см. Schmuck et al., Eur. J. Neurosci. 1996, 8, 959-967).
Экспериментальные данные указывают на то, что соединения настоящего изобретения применимы в лечении боли, включая острую, хроническую, невропатическую боль при воспалительных заболеваниях и раке, особенно в лечении воспалительной боли. 5-НТ (серотонин) играет ключевую роль в регулировании передачи ноцицептивной информации на разных уровнях периферической и центральной нервной систем (см. Richardson В.Р., "Serotonin and Pain", Ann. N.Y. Acad. Sci., 1990, 600, 511-520). Кроме того, нейрональные системы, содержащие 5-НТ, участвуют не только в регуляции ноцицептивного сигнала на спинальном и супраспинальном уровне, но и в опосредовании ноцицептивного действия других анальгетиков, включая опиаты. 5-НТ является медиатором сенсибилизации ноцицепторов нервных окончаний, которые могут участвовать в происхождении боли, связанной с воспалением. Рецептор 5-НТ2B высоко чувствителен к активации 5-НТ, и специфическая блокада селективными антагонистами 5-НТ2B может обеспечить новый подход к обезболиванию.
Экспериментальные данные подтверждают терапевтическую роль антагонистов рецептора 5-HT2B в лечении гипертензии. При гипертензии наиболее значительное повышение чувствительности сосудов наблюдается для серотонина. Две линии свидетельств предполагают, что это является следствием переключения рецептора, опосредующего сужение сосудов, с преимущественно 5-НТ2А на преимущественно 5-НТ2B. Во-первых, индуцированные серотонином сокращения кровеносных сосудов, выделенных от животных с гипертензией, становятся устойчивыми к блокировке селективными антагонистами рецептора 5-НТ2А, но остаются чувствительными к неселективным антагонистам рецептора 5-НТ2B. Во-вторых, в сосудах животных с гипертензией наблюдается повышение уровня мРНК рецептора 5-НТ2B (см. Watts et al., J. Pharmacol. Exp. Ther. 1996, 277, 1103-13 и Watts et al., Hypertension 1995, 26, 1056-1059). Этот индуцированный гипертензией сдвиг в популяции подтипа рецептора, опосредующего сократительный ответ на 5-НТ, предполагает, что селективная блокировка сужающих сосуды рецепторов 5-НТ2B может иметь применение в лечении гипертензии.
Клинические и экспериментальные данные подтверждают терапевтическую роль антагонистов рецептора 5-НТ2B в лечении заболеваний желудочно-кишечного тракта, в частности синдрома раздраженного кишечника (IBS). Хотя патология, лежащая в основе IBS, остается неясной, существует хорошо подтвержденное предположение об участии в этом процессе серотонина. Так, пища с высоким содержанием серотонина может обострять симптомы у некоторых пациентов (см. Lessorf, Scand. J. Gastroenterology 1985, 109, 117-121), а в доклинических исследованиях было прямо показано, что серотонин повышает чувствительность висцеральных сенсорных нейронов, что приводит к повышенному болевому ответу, сходному с наблюдаемым при IBS (см. Christian et al., J. Applied Physiol. 1989, 67, 584-591 и Sanger et al., Neurogastroenterology and Motility 1996, 8, 319-331). Возможная ключевая роль рецепторов 5-НТ2B в сенсибилизирующем действии серотонина предполагается несколькими линиями фактов. Во-первых, рецепторы 5-НТ2B присутствуют в кишечнике человека (см. Borman et al., Brit. J. Pharmacol. 1995, 114, 1525-1527 и Borman et al., Ann. of the New York Acad. of Sciences 1997, 812, 222-223). Во-вторых, активация рецепторов 5-НТ2В может привести к продуцированию оксида азота, агента, способного сенсибилизировать сенсорные нервные волокна (см. Glusa et al., Naunyn-Schmied. Arch. Pharmacol. 1993, 347, 471-477 и Glusa et al., Brit. J. Pharmacol. 1996, 119, 330-334). В-третьих, слабоселективные лекарства, обладающие высокой аффинностью к рецептору 5-НТ2B, клинически эффективны в облегчении боли, ассоциированной с IBS и взаимосвязанными заболеваниями (см. Symon et al., Arch. Disease in Childhood 1995, 72, 48-50 и Tanum et al., Scand. J. Gastroenterol. 1996, 31, 318-325). Все эти факты вместе позволяют предположить, что селективный антагонист рецептора 5-НТ2B будет облегчать как боль в желудочно-кишечном тракте, так и ненормальную перистальтику, ассоциированную с IBS.
Клинические и экспериментальные данные подтверждают терапевтическую роль антагонистов рецептора 5-НТ2B в лечении рестеноза. Ангиопластика и автография при шунтировании связаны с рестенозом, который ограничивает эффективность этих процедур. Образование богатых тромбоцитами тромбов является основной причиной закупорки сосудов, в то время как предполагается, что серотонин, в ряду других происходящих из тромбоцитов медиаторов, вносит вклад в поздний рестеноз (см. Barradas et al., Clinica Chim. Acta 1994, 230, 157-167). В этом позднем рестенозе задействована пролиферация гладкой мускулатуры сосудов. Две линии фактов подтверждают роль рецепторов 5-НТ2B в этом процессе. Во-первых, серотонин обладает мощной митогенной активностью в культуре клеток гладкой мускулатуры и эндотелия через активацию рецепторов 5-НТ2 (см. Pakala et al., Circulation 1994, 90, 1919-1926). Во-вторых, эта митогенная активность опосредуется через активацию второго тирозинкиназного информационного пути, задействующего митогенно активированную протеинкиназу (МАРК) (см. Lee et al., Am. J. Physiol. 1997, 272 (1 pt 1), C223-230 и Kelleher et al., Am. J. Physiol. 1995, 268 (6 pt 1), L894-901). Недавнее обнаружение того, что рецепторы 5-НТ2B связываются с МАРК (см. Nebigil et al., Proc. Natl. Acad Sci. U.S.A. 2000, 97, 22591-2596), связанной с высокой аффинностью серотонина к рецепторам этого подтипа, указывает на то, что селективные антагонисты рецептора 5-НТ2B могут защищать от рестеноза сосудов при автографии или после ангиопластики.
Клинические и экспериментальные данные подтверждают терапевтическую роль антагонистов рецептора 5-НТ2В в лечении астмы и обструктивного заболевания дыхательных путей. Ненормальная пролиферация гладкой мускулатуры дыхательных путей вместе с гиперреактивностью гладкой мускулатуры в ответ на стимуляторы сжатия, включающие серотонин, играет значительную роль в патогенезе заболеваний дыхательных путей человека, таких как астма и бронхиально-легочная дисплазия (см. James et al., Am. Review of Respiratory Disease 1989, 139, 242-246 и Margrafet al., Am. Review of Respiratory Disease 1991, 143, 391-400). Наряду с другими подтипами серотонинового рецептора рецепторы 5-НТ2В присутствуют в бронхиальной гладкой мускулатуре (см. Choi et al., Febs Letters 1996, 391, 45-51), было показано, что они стимулируют митогенез гладких мышц в гладкой мускулатуре дыхательных путей (см. Lee et al., Am. J. Physiol. 1994, 266, L46-52). Так как повышенные концентрации циркулирующего свободного серотонина тесно связаны с тяжестью клинических проявлений и легочной функцией у симптоматических астматиков, серотонин может играть важную роль в патофизиологии острых приступов (см. Lechin et al., Ann. Allergy, Asthma, Immunol. 1996, 77, 245-253). Эти данные предполагают, что антагонист рецепторов 5-НТ2B в гладкой мускулатуре дыхательных путей может быть применим при предотвращении сужения дыхательных путей, случающегося при повышенном уровне циркулирующего серотонина и предотвращении пролиферации гладкой мускулатуры дыхательных путей, вносящей вклад в долгосрочную патологию этого заболевания.
Экспериментальные данные подтверждают терапевтическую роль антагонистов рецептора 5-НТ2В в лечении гиперплазии простаты. В результате гиперплазии простаты и избыточного простатического сужения уретры может происходить непроходимость мочевыводящих путей. В свою очередь, это ведет к уменьшению потока мочи и повышению частоты и необходимости мочеиспускания. Рецепторы 5-НТ2B присутствуют в простате человека (см. Kursar et al., Mol. Pharmacol. 1994, 46, 227-234), а рецептор с фармакологическими свойствами этого подтипа рецептора опосредует сокращение ткани (см. Killam et al., Eur. J. Pharmacol. 1995, 273, 7-14). Некоторые лекарства, эффективные в лечении доброкачественной гиперплазии простаты, блокируют опосредованные 5-НТ сокращения простаты (см. Noble et al., Brit. J. Pharmacol. 1997, 120, 231-238). Рецепторы 5-НТ2B опосредуют фиброзную гиперплазию и гиперплазию гладких мышц (см. Launay et al., J. Biol. Chem. 1996, 271, 3141-3147), а серотонин является митогенным в простате (см. Cockett et al., Urology 1993, 43, 512-519), таким образом, селективный антагонист рецептора 5-НТ2B может применяться не только при снижении избыточного сокращения простаты, но также и при предотвращении прогрессирования гиперплазии ткани.
Экспериментальные данные подтверждают терапевтическую роль антагонистов рецептора 5-НТ2C в лечении приапизма (см. Kennett, Curr. Opin. Invest. Drugs 1993, 2, 317-362). МСРР (м-хлорфенилпиперазин) вызывает эрекцию пениса у крыс, причем этот эффект блокируется неселективными антагонистами рецептора 5-НТ2C/2А/2B, но не селективными антагонистами рецептора 5-НТ2А (см. Hoyer, Peripheral actions of 5-HT 1989, Fozard J. (ed.), Oxford University Press, Oxford, 72-99). Эта терапевтическая мишень для антагонистов рецептора 5-НТ2С также является мишенью для антагонистов рецептора 5-НТ2В.
При использовании соединений настоящего изобретения в лечении указанных выше состояний активное соединение и его соли, описанные здесь, могут быть введены любым принятым способом, включая оральный, парентеральный и другие системные пути введения. Для введения может быть использована любая фармацевтически приемлемая форма, включая твердую, полутвердую и жидкую дозировочные формы, например таблетки, суппозитории, пилюли, капсулы, порошки, жидкости, суспензии и подобные, предпочтительны одиночные формы дозировки для однократного введения точной дозы или формы для продолжительного или контролируемого высвобождения дозы для пролонгированного введения соединения с определенной скоростью. Композиции обычно содержат традиционный фармацевтический носитель или наполнитель и, по меньшей мере, одно из соединений настоящего изобретения или его фармацевтически приемлемую соль, и, кроме того, может содержать другие лекарственные и фармацевтические агенты, носители, адъюванты и т.п.
Количество вводимого производного настоящего изобретения зависит от заболевания, его тяжести, способа введения и мнения лечащего врача. Однако эффективная доза для орального, парентерального и другого системного пути введения находится в пределах от 0,01 до 20 мг/кг/день, предпочтительно - 0,1-10 мг/кг/день. Для среднего взрослого весом 70 кг эта величина составляет 0,7-1400 мг/день, предпочтительно - 7-700 мг/день.
Специалист в области лечения таких заболеваний сможет без ненужных экспериментов, полагаясь на собственные знания и описание настоящего изобретения, установить эффективное количество одного из пиперидиновых соединений настоящего изобретения для лечения конкретного заболевания.
Для твердых композиций обычные нетоксичные твердые носители включают, например, маннитол, лактозу, целлюлозу, производные целлюлозы, кросскармелозу натрия, крахмал, стеарат магния, сахарин натрия, тальк, глюкозу, сахарозу, карбонат магния и подобные соединения фармацевтического качества. Активное соединение, определенное выше, может быть приготовлено в форме суппозиториев при использовании в качестве носителей, например полиалкиленгликолей, например ПЭГ (полиэтиленгликоля) или производных ПЭГ, ацетилированных триглицеридов и подобных веществ. Фармацевтические композиции для введения в жидком виде могут быть приготовлены, например, путем растворения, диспергирования и т.п. активного соединения, определенного выше, и необязательных фармацевтических добавок в носителе, например в воде, солевом растворе, водном растворе декстрозы, глицерине, этаноле и подобных с образованием раствора или суспензии. При желании фармацевтическая композиция может также содержать минимальные количества нетоксичных вспомогательных веществ, таких как смачивающие и эмульгирующие агенты, агенты для поддержания буферного рН и подобные, например ацетат натрия, сорбитмонолаурат, триэтаноламин ацетат натрия, триэтаноламинолеат и т.д. Композиция или состав для введения в любом случае содержит количество активного соединения (шт), эффективное для облегчения симптомов заболевания.
Могут быть приготовлены дозировочные формы или композиции, содержащие одно из пиперидиновых соединений настоящего изобретения в количестве от 0,25 до 95 мас.%, остальное составляет нетоксичный носитель.
Для орального введения фармацевтически приемлемую нетоксичную композицию получают добавлением любого из обычных наполнителей, таких как, например, маннитол, лактоза, целлюлоза, производные целлюлозы, кросскармелоза натрия, крахмал, стеарат магния, сахарин натрия, тальк, глюкоза, сахароза, карбонат магния и подобные соединения фармацевтического качества. Такие композиции могут быть в форме растворов, суспензий, таблеток, пилюль, капсул, порошков, составов для замедленного высвобождения и т.п. Такие композиции могут содержать от 1 до 95 мас.% одного из соединений настоящего изобретения, более предпочтительно - от 2 до 50 мас.%, наиболее предпочтительно - от 5 до 8 мас.%.
Парентеральное введение обычно осуществляется путем инъекции: подкожной, внутримышечной или внутривенной. Составы для инъекции могут быть приготовлены в виде обычных форм, либо в виде жидких растворов или суспензий, либо в виде твердых форм, пригодных для растворения или суспендирования в жидкости перед введением, либо в виде эмульсий. Подходящими наполнителями являются, например, вода, солевой раствор, декстроза, глицерин, этанол или подобные. Кроме того, по желанию фармацевтические композиции могут содержать также минимальные количества нетоксичных вспомогательных веществ, таких как смачивающие и эмульгирующие агенты, агенты для поддержания буферного рН и подобные, например ацетат натрия, сорбитмонолаурат, триэтаноламинолеат, триэтаноламин ацетат натрия и т.д.
Трансдермальное или "импульсное" трансдермальное введение могут осуществляться посредством кремов, гелей, дисперсий и подобного.
Недавно разработанный подход к парентеральному введению заключается в имплантации систем для медленного или продолжительного высвобождения, таким образом, что поддерживается постоянный уровень дозировки (см., например, US-A-3710795).
Процент активных соединений, содержащихся в таких парентеральных композициях, в высокой степени зависит как от их специфической природы, так и от активности соединения и потребностей пациента. Пиперидиновые соединения настоящего изобретения применяются в концентрациях от 0,1 до 10 мас.% в растворе, эти концентрации выше в случае твердой композиции, которую затем разводят до указанных концентраций. Предпочтительно композиция содержит от 0,2 до 2 мас.% одного из пиперидиновых соединений в растворе.
При применении соединений настоящего изобретения в лечении заболеваний или нарушений со стороны глаз, связанных с ненормально высоким внутриглазным давлением, введение соединения осуществляется любым фармацевтически приемлемым способом, обеспечивающим соответствующие местные концентрации для формирования требуемого ответа. К этим способам относятся как прямое введение соединения в глаз посредством капель и вставок или имплантатов с контролируемым высвобождением, так и системное введение, описанное выше.
Капли и растворы, вносимые прямо в глаз, обычно представляют собой стерилизованные водные растворы, содержащие от 0,1 до 10 мас.%, более предпочтительно - от 0,5 до 1 мас.% одного из новых пиперидиновых соединений, а также подходящий буфер, стабилизатор и консервант. Общая концентрация растворенных веществ по возможности должна быть такой, чтобы полученный раствор был изотоническим со слезной жидкостью (хотя это не является абсолютно необходимым) и имел эквивалентный уровень рН в пределах рН 6-8. Типичными консервантами являются ацетат фенилртути, тимеросал, хлорбутанол и хлорид бензалкония. Типичные буферные системы и соли основаны на, например, цитрате, борате или фосфате; к подходящим стабилизаторам относятся глицерин и полисорбат 80. Для получения водных растворов просто растворяют вещества в подходящем количестве воды, доводят рН до приблизительного значения 6,8-8,0, доводят водой до конечного объема и стерилизуют препараты методами, известными в данной области техники.
Дозировка полученной композиции зависит от концентрации капель, состояния пациента и индивидуального ответа на лечение. Однако типичную композицию для глаз вводят в количестве 2-10 капель в день в глаз 0,5 мас.% раствора одного из пиперидиновых соединений.
Композиции по настоящему изобретению могут также быть приготовлены для введения любым удобным способом по аналогии с другими композициями для местного применения у млекопитающих. Эти композиции могут быть представлены для применения любым удобным способом при использовании любого из разнообразных фармацевтических носителей. Для такого местного применения фармацевтически приемлемый нетоксичный состав может быть в полутвердой, жидкой или твердой форме, например в форме гелей, кремов, лосьонов, растворов, суспензий, мазей порошков или подобных. Например, активный компонент может быть приготовлен в форме сиропа или геля при использовании этанола, пропиленгликоля, пропиленкарбоната, полиэтиленгликолей, диизопропиладипата, глицерина, воды и т.п. с применением соответствующих гелеобразующих агентов, таких как карбомеры, гидроксипропилцеллюлозы Klucel и т.д. При желании состав может также содержать минимальные количества нетоксичных вспомогательных веществ, таких как консерванты, антиоксиданты, агенты для поддержания буферного рН, поверхностно-активные вещества и подобные. Методы, применяемые для получения таких дозировочных форм, хорошо известны специалистам в данной области техники, см. Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., 19th Edition, 1995.
Предпочтительно, чтобы фармацевтическая композиция вводилась одной дозировочной единицей для продолжительного лечения или одной дозировочной единицей по необходимости, если особенно требуется облегчение симптомов.
Примеры
Пример 1:
6-этокси-1-метокситиоксантон 1 вводят в реакцию с реактивом Гриньяра 2, полученным из 1-метил-4-галопиперидина, предпочтительно -1-метил-4-хлорпиперидина, согласно Схеме I. Выделяют спирт 3 и дегидратируют его кислотой, предпочтительно соляной или муравьиной кислотой, получают 1-метил-4-(3-этокси-9Н-тиоксантен-9-илиден)-пиперидин (4).
Схема I:
Один из способов синтеза 6-этокси-1-метокситиоксантона 1 описан в работе I.Cervena, J.Metysova, E.Svatek, В.Kakac, J.Holubek, M. Hmbantova and M. Protiva, in Coll. Czech. Chem. Comm. 41, 881-904 (1978) (Схема II). 3-этокситиол 5 вводят в реакцию с 2-йод-4-метоксибензойной кислотой 6 в кипящем водном растворе КОН в присутствии меди. После добавления соляной кислоты получают кислоту присоединения 7. Кислоту 7 циклизуют полифосфорной кислотой, получают смесь изомеров 1-метокси-6-этокситиоксантона 8а и 1-метокси-8-этокситиоксантона 8b, которые можно разделить методом хроматографии, предпочтительно колоночной хроматографии.
Схема II:
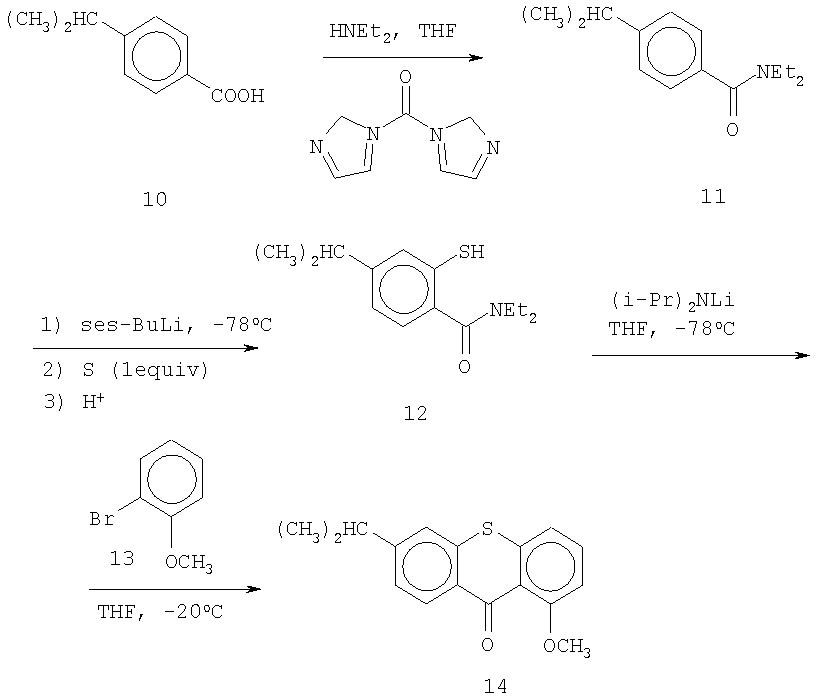
Второй способ получения исходного кетона основан на методе М. Watanabe et al., Chem. Pharm Bull. 37, 36-41 (1989). Исходную 4-изопропилбензойную кислоту 10 превращают в амид 11, который обрабатывают сильным основанием для отщепления протона, смежного с амидной группой. И после введения молекулярной серы и обработки кислотой вводят тиольную группу, как в соединении 12. Обработка бромоанизола 13 сильным основанием превращает его в бензин, который реагирует с соединением 12 с образованием требуемого кетона 14.
Способ, который подходит для различных соединений настоящего изобретения, зависит от доступности исходных веществ.
Пример 2:
Этапы получения 5 соединений: 4-(6-этокси-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидина, 4-(6-изопропил-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидина, 4-(6-этокси-1-этил-тиоксантен-9-илиден)-1-метил-пиперидина, 4-(3-этокси-селеноксантен-9-илиден)-1-метил-пиперидина, 3-этокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидроакридина.
Все реакции, описанные в последующих разделах, должны проводиться в защитной атмосфере (сухой аргон или сухой N2). Анализ проводят совместным методом ВЭЖХ-МС (214 нм, положительная ESI (ионизация электрораспылением), +30 В). Этапы экстракции тиолов следует проводить с растворителями, дегазированными или насыщенными аргоном, для предотвращения повторного окисления до дисульфида.
2.1 Синтез тиоксантенов/селеноксантена
2.1.1 Синтез диэтиламида 4-этоксибензойной кислоты
4-этоксибензойную кислоту (20,0 г, 120 ммоль) растворяют в тионилхлориде (14 мл) и нагревают с обратным холодильником в течение 3 часов. После охлаждения реакционной смеси до комнатной температуры выпаривают избыток тионилхлорида. Полученный неочищенный кислотный хлорид сразу растворяют в безводном DCM (дихлорметане) (125 мл) и охлаждают раствор ледяной водой. По каплям добавляют 125 мл диэтиламина. После добавления амина реакционную смесь оставляют нагреться до комнатной температуры. После перемешивания смеси при комнатной температуре в течение ночи (16 ч) раствор промывают 1,0 н раствором соляной кислоты (3×100 мл), насыщенным раствором NaHCO3 (3×100 мл) и солевым раствором (3×100 мл). Органический слой сушат над Na2SO4, фильтруют и упаривают досуха.
Чистота: 98%.
Выход: 25 г, 94%.
2.1.2 Синтез диэтиламида 4-изопропилбензойной кислоты
4-изопропилбензойную кислоту (5 г, 30,5 ммоль) растворяют в тионилхлориде (10 мл) и нагревают с обратным холодильником в течение 3 часов. После охлаждения реакционной смеси до комнатной температуры выпаривают избыток тионилхлорида. Полученный неочищенный кислотный хлорид сразу растворяют в безводном DCM (50 мл) и охлаждают раствор ледяной водой. По каплям добавляют диэтиламин (32 мл, 0,3 моль). После добавления амина реакционную смесь оставляют нагреться до комнатной температуры. После перемешивания смеси при комнатной температуре в течение ночи (12-14 ч) раствор промывают 1,0 н раствором соляной кислоты (3×50 мл), насыщенным раствором NaHCO3 (3×50 мл) и солевым раствором (3×50 мл). Органический слой сушат над Na2SO4, фильтруют и упаривают досуха.
Чистота: 96%.
Выход: 6,0 г, 90%.
2.1.3 Синтез диэтиламида 4-этокси-2-тиобензойной кислоты
Диэтиламид 4-этоксибензойной кислоты (4,43 г, 20 ммоль) и N,N,N,N-тетраметилэтилендиамин (5,1 мл, 23,8 ммоль) растворяют в безводном тетрагидрофуране (ТГФ) (100 мл). Раствор охлаждают до -78°С. Затем по каплям добавляют втор-бутиллитий (1,3 М раствор в н-гексане/ТГФ, 13,73 мл, 23,8 ммоль) таким образом, что температура не превышает -70°С. Раствор перемешивают при -78°С в течение 1 часа, затем в один прием добавляют порошок серы (1,36 г, 40 ммоль).
Убирают холодную баню, оставляют раствор нагреться до комнатной температуры и перемешивают в течение ночи (16 ч). Добавляют насыщенный раствор NH4Cl (100 мл), затем - 10% водный раствор HCl (50 мл). Смесь упаривают досуха. Остаток экстрагируют метиленхлоридом (100 мл), сушат над Na2SO4, фильтруют и выпаривают растворитель.
Неочищенный продукт растворяют в уксусной кислоте (100 мл). Добавляют воду (100 мл) и цинк (5 г) и перемешивают смесь на магнитной мешалке при 65°С в течение 24 часов. Раствор экстрагируют метиленхлоридом (1×100 мл). Отделяют органический слой и промывают водой (3×100 мл). Затем удаляют растворитель. Продукт очищают на системе флэш-хроматографии ISCO при использовании метиленхлорида и метанола (градиент).
Чистота: 75%.
Выход: 3,5 г, 70%.
2.1.4 Синтез диэтиламида 4-изопропил-2-тиобензойной кислоты
Диэтиламид 4-изопропилбензойной кислоты (2,19 г, 10 ммоль) и N,N,N,N-тетраметилэтилендиамин (2,6 мл, 11,9 ммоль) растворяют в безводном ТГФ (33 мл). Раствор охлаждают до -78°С. Затем по каплям добавляют втор-бутиллитий (1,3 М раствор в н-гексане/ТГФ, 9,15 мл, 11,9 ммоль) таким образом, что температура не превышает -70°С. Раствор перемешивают при -78°С в течение 1 часа, затем в один прием добавляют порошок серы (0,64 г, 30 ммоль).
Убирают холодную баню, оставляют раствор нагреться до комнатной температуры и перемешивают в течение ночи (16 ч). Добавляют насыщенный раствор NH4Cl (100 мл), затем - 10% водный раствор HCl (50 мл). Смесь упаривают досуха. Остаток экстрагируют метиленхлоридом (100 мл), сушат над Na2SO4, фильтруют и выпаривают растворитель.
Неочищенный продукт растворяют в уксусной кислоте (50 мл). Добавляют воду (50 мл) и цинк (2,5 г) и перемешивают смесь на магнитной мешалке при 65°С в течение 24 часов. Раствор экстрагируют метиленхлоридом (1×100 мл). Отделяют органический слой и промывают водой (3×100 мл) и солевым раствором (1×100 мл). Раствор сушат над Na2SO4 и затем фильтруют. Затем удаляют растворитель. Продукт очищают на системе флэш-хроматографии ISCO при использовании метиленхлорида и метанола (градиент).
Чистота: 76%.
Выход: 950 мг, 38%.
2.1.5 Синтез ди-[4-этокси-N,N-диэтил-2-селено-бензамида]
Диэтиламид 4-этоксибензойной кислоты (1,32 г, 6 ммоль) и N,N,N,N-тетраметилэтилендиамин (1,63 мл, 7,6 ммоль) растворяют в безводном ТГФ (40 мл). Раствор охлаждают до -78°С. Затем по каплям добавляют втор-бутиллитий (1,3 М раствор в н-гексане/ТГФ, 5,85 мл, 7,6 ммоль) таким образом, что температура не превышает -70°С. Раствор перемешивают при -78°С в течение 1 часа, затем в один прием добавляют порошок селена (0,95 г, 12 ммоль).
Убирают холодную баню, оставляют раствор нагреться до комнатной температуры и перемешивают в течение ночи (16 ч). Добавляют насыщенный раствор NH4Cl (100 мл), затем - 10% водный раствор HCl (50 мл). Смесь упаривают досуха. Остаток экстрагируют метиленхлоридом (100 мл), сушат над Na2SO4, фильтруют и выпаривают растворитель.
Неочищенный продукт растворяют в уксусной кислоте (50 мл). Добавляют воду (25 мл) и цинк (2,5 г) и перемешивают смесь на магнитной мешалке при 65°С в течение 24 часов. Раствор экстрагируют метиленхлоридом (1×100 мл). Отделяют органический слой и промывают водой (3×100 мл) и солевым раствором (1×100 мл). Раствор сушат над Na2SO4 и затем фильтруют. Затем удаляют растворитель. Чистота полученного продукта достаточно высока, так что дополнительной очистки не требуется.
Чистота: 93%.
Выход: 1,05 г, 58%.
2.1.6 Синтез 6-этокси-1-метокси-тиоксантен-9-она
Диэтиламид 4-этокси-2-тиобензойной кислоты (1 г, 6,98 ммоль) растворяют в безводном ТГФ (105 мл) и охлаждают до -78°С. По каплям добавляют ЛДА (литий-диизопропиламид, 1М раствор, 27,92 мл, 13,96 ммоль). Раствор перемешивают 1 час при -78°С. Раствор оставляют нагреться до -20°С и по каплям добавляют 2-метоксибромбензол (2, 176 мл, 17,45 ммоль в 30 мл безводного ТГФ). Раствор оставляют нагреться до комнатной температуры и перемешивают в течение ночи (16 ч).
К реакционной смеси добавляют насыщенный раствор NH4Cl (50 мл), а затем - 10% раствор HCl (50 мл). Смесь упаривают досуха, а остаток экстрагируют СН2Cl2 (150 мл). Органический слой промывают солевым раствором (3×100 мл), сушат над Na2SO4, фильтруют и выпаривают растворитель. Продукт дважды очищают методом флэш-хроматографии (градиент СН2Cl2/метанол).
Чистота (ВЭЖХ, 214 нм): 84,4%.
Выход: 0,5 г (25%).
2.1.7 Синтез 6-этокси-1-этил-тиоксантен-9-она
Синтез проводят согласно этапу 2.1.6, используя 2-этилбромбензол вместо 2-метоксибромбензола.
Очистку продукта проводят один раз методом флэш-хроматографии.
Чистота: 86% (ВЭЖХ, 214 нм), выход: 220 мг, 10%.
2.1.8 Синтез 6-изопропил-1-метокси-тиоксантен-9-она
Синтез проводят согласно этапу 2.1.6, используя в качестве исходного соединения диэтиламид 4-изопропил-2-тиобензойной кислоты.
Очистку продукта проводят один раз методом флэш-хроматографии.
Чистота: 79% (ВЭЖХ, 214 нм), выход: 250 мг, 14%.
2.1.9 Синтез 3-этоксиселеноксантен-9-она
Синтез проводят согласно этапу 2.1.6, используя в качестве исходного соединения диселенид, полученный на этапе 2.1.5 (1,0 г, 4,3 ммоль селенида), и бромбензол (1,146 мл, 10,9 ммоль) в качестве реагента.
Очистку продукта проводят один раз методом флэш-хроматографии.
Чистота: 86% (ВЭЖХ, 214 нм), выход: 300 мг, 22%.
2.2 Синтез конечных соединений 4-(6-этокси-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидина, 4-(6-изопропил-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидина, 4-(6-этокси-1-этил-тиоксантен-9-илиден)-1-метил-пиперидина, 4-(3-этокси-селеноксантен-9-илиден)-1-метил-пиперидина.
2.2.1 Синтез реактива Гриньяра 1-метил-пиперидин-4-ил-магний хлорида
4-хлор-1-метилпиперидин*HCl (20 г) превращают в свободный амин путем растворения в 25% растворе NH3/H2O с последующей экстракцией эфиром. Органический слой дважды промывают солевым раствором, сушат над Na2SO4 и упаривают. Чистый амин держат в атмосфере аргона.
2 г амина (около 15 ммоль) растворяют в безводном ТГФ (7 мл). К гранулам магния (0,434 г, 16,5 ммоль) и безводному ТГФ (1,4 мл) добавляют один кристалл йода и этилйодид (60,4 мкл, 0,75 мкмоль). Когда начинается реакция Гриньяра, к раствору по каплям добавляют раствор амина. Раствор слегка перемешивают и иногда подогревают тепловентилятором для поддержания кипения ТГФ. После добавления раствора амина для завершения реакции реакционную смесь нагревают с обратным холодильником в течение 2 часов.
Раствор следует держать в атмосфере аргона.
2.2.2 Синтез 4-(6-этокси-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидина
6-этокси-1-метокси-тиоксантен-9-он (100 мг, 0,349 ммоль) растворяют в безводном ТГФ (5 мл). По каплям добавляют 1-метил-пиперидин-4-ил-магний хлорид (3 экв., с этапа 2.2.1). После завершения добавления реактива Гриньяра раствор оставляют при перемешивании на ночь. Контроль реакции проводят методом тонкослойной хроматографии (ТСХ) (1% метанол/CH2Cl2). После завершения реакции добавляют сначала уксусную кислоту (5 мл), затем воду (5 мл). Выпаривают растворители, а полученный карбинол дегидратируют уксусным ангидридом (5 мл, нагревание с обратным холодильником, 4 ч).
Выпаривают уксусный ангидрид, а остаток растворяют в 1 н растворе КОН (5 мл). Свободный амин экстрагируют один раз метиленхлоридом (10 мл). Раствор сушат над Na2SO4 и выпаривают растворитель.
Продукт очищают методом флэш-хроматографии и затем переводят в его гидрохлорид путем нагревания в водном растворе HCl (50 экв. HCl, 5 мл, 4 ч, 50°С). Выпаривают растворитель, а продукт растворяют в смеси трет-бутилового спирта и воды (2 мл, 4:1, об/об) и лиофилизируют.
Чистота: 92%.
Выход: 20 мг, 16%.
2.2.3 Синтез 4-(6-изопропил-1-метокси-тиоксантен-9-илиден)-1-метил-пиперидина
6-изопропил-1-метокси-тиоксантен-9-он (100 мг, 0,35 ммоль) растворяют в безводном ТГФ (5 мл). По каплям добавляют 1-метил-пиперидин-4-ил-магнийхлорид (3 экв., с этапа 2.2.1). После завершения добавления реактива Гриньяра раствор оставляют при перемешивании на ночь. Контроль реакции проводят методом ТСХ (1% метанол/СН2Cl2. После завершения реакции добавляют сначала уксусную кислоту (5 мл), затем воду (5 мл). Выпаривают растворители, а полученный карбинол дегидратируют уксусным ангидридом (5 мл, нагревание с обратным холодильником, 4 ч).
Выпаривают уксусный ангидрид, а остаток растворяют в 1 н растворе КОН (5 мл). Свободный амин экстрагируют метиленхлоридом, сушат над Na2SO4 и выпаривают растворитель.
Продукт очищают методом флэш-хроматографии и затем переводят в его гидрохлорид путем нагревания в водном растворе HCl (50 экв. HCl, 5 мл, 4 ч, 50°С). Выпаривают растворитель, а продукт растворяют в смеси трет-бутилового спирта и воды (2 мл, 4:1, об/об) и лиофилизируют.
Чистота: >98%.
Выход: 25 мг, 20%.
2.2.4 Синтез 4-(6-этокси-1-этил-тиоксантен-9-илиден)-1-метил-пиперидина 6-этокси-1-этил-тиоксантен-9-он (50 мг, 0,18 ммоль) растворяют в безводном ТГФ (5 мл). По каплям добавляют 1-метил-пиперидин-4-ил-магнийхлорид (3 экв., с этапа 2.2.1). После добавления первой капли раствор меняет цвет со светло-желтого на темно-коричневый. После завершения добавления реактива Гриньяра раствор оставляют при перемешивании на ночь. Контроль реакции проводят методом ТСХ (1% метанол/СН2Cl2). После завершения реакции добавляют сначала уксусную кислоту (5 мл), затем воду (5 мл). Выпаривают растворители, а полученный карбинол дегидратируют уксусным ангидридом (5 мл, нагревание с обратным холодильником, 4 ч).
Выпаривают уксусный ангидрид, а остаток растворяют в 1 н растворе КОН (5 мл). Свободный амин экстрагируют метиленхлоридом, сушат над Na2SO4 и выпаривают растворитель.
Продукт очищают методом флэш-хроматографии и затем переводят в его гидрохлорид путем нагревания в водном растворе HCl (50 экв. HCl, 5 мл, 4 ч, 50°С). Выпаривают растворитель, а продукт растворяют в смеси трет-бутилового спирта и воды (2 мл, 4:1, об/об) и лиофилизируют.
Чистота: >97%.
Выход: 10,8 мг, 16%.
2.2.5 Синтез 4-(4-этокси-селеноксантен-9-илиден)-1-метил-пиперидина
4-этокси-1-селеноксантен-9-он (50 мг, 0,16 ммоль) растворяют в безводном ТГФ (5 мл). По каплям добавляют 1-метил-пиперидин-4-ил-магнийхлорид (3 экв., с этапа 2.2.1). После добавления первой капли раствор меняет цвет со светло-желтого на темно-коричневый. После завершения добавления реактива Гриньяра раствор оставляют при перемешивании на ночь. Контроль реакции проводят методом ТСХ (1% метанол/СН2Cl2). После завершения реакции добавляют сначала уксусную кислоту (5 мл), затем воду (5 мл). Выпаривают растворители, а полученный карбинол дегидратируют уксусным ангидридом (5 мл, нагревание с обратным холодильником, 4 ч).
Выпаривают уксусный ангидрид, а остаток растворяют в 1 н растворе КОН (5 мл). Свободный амин экстрагируют метиленхлоридом, сушат над Na2SO4 и выпаривают растворитель.
Продукт очищают методом флэш-хроматографии и затем переводят в его гидрохлорид путем нагревания в водном растворе HCl (50 экв. HCl, 5 мл, 4 ч, 50°С). Выпаривают растворитель, а продукт растворяют в смеси трет-бутилового спирта и воды (2 мл, 4:1, об/об) и лиофилизируют.
Чистота: >97%.
Выход: 28 мг, 46%.
2.3 Синтез соединения 3-этокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидроакридина
Только этап 2.3.3 должен проводиться в защитной атмосфере (аргон).
2.3.1 Синтез 2-(3-этокси-фениламино)-бензойной кислоты
К раствору КОН (7,385 г, 131,6 ммоль), воды (77 мл) и 2-йодбензойной кислоты (9,6 г, 38,7 ммоль) добавляют м-фенетидин (5 мл, 38,7 ммоль). Добавляют порошок меди (77 мг, 1,22 мкмоль) и нагревают смесь с обратным холодильником при перемешивании в течение ночи (16 ч).
Раствор фильтруют горячим и подкисляют концентрированной соляной кислотой для осаждения требуемого продукта. Продукт очищают методом флэш-хроматографии.
Чистота: 95%.
Выход: 3,0 г, 30%.
2.3.2 Синтез 3-этокси-10Н-акридин-9-она
К 2-(3-этоксифениламино)-бензойной кислоте (1,5 г, 4,2 ммоль) добавляют полифосфорную кислоту (20 г) и смесь нагревают при перемешивании до 120°С.
Через 4 часа раствор вливают в ледяную крошку. Полученный водный раствор экстрагируют метиленхлоридом. Органический слой снова промывают водой (2 х) и солевым раствором (1 х), сушат над Na2SO4 и упаривают досуха.
Продукт очищают методом флэш-хроматографии.
Чистота: >95%.
Выход: 200 мг, 14%.
2.3.3 Синтез 3-этокси-9-(1-метил-пиперидин-4-илиден)-9,10-дигидроакридина
3-этокси-10Н-акридин-9-он (50 мг, 0,21 ммоль) растворяют в безводном ТГФ (5 мл). По каплям добавляют 1-метил-пиперидин-4-ил-магнийхлорид (3 экв., с этапа 2.2.1). После завершения добавления реактива Гриньяра раствор оставляют при перемешивании на ночь. Контроль реакции проводят методом ТСХ (1% метанол/СН2Cl2). После завершения реакции добавляют сначала уксусную кислоту (5 мл), затем воду (5 мл). Выпаривают растворители, а полученный карбинол дегидратируют уксусным ангидридом (5 мл, нагревание с обратным холодильником, 4 ч).
Выпаривают уксусный ангидрид, а остаток растворяют в 1 н растворе КОН. Свободный амин экстрагируют метиленхлоридом, сушат над Na2SO4 и выпаривают растворитель.
Продукт очищают методом флэш-хроматографии и затем переводят в его гидрохлорид путем нагревания в водном растворе HCl (50 экв. HCl, 5 мл, 4 ч, 50°С). Выпаривают растворитель, а продукт растворяют в смеси трет-бутилового спирта и воды (2 мл, 4:1, об/об) и лиофилизируют.
Чистота: 99%.
Выход: 22 мг, 32%.
Пример 3:
Исследование связывания с клонированным рецептором 5-НТ2B человека
Далее описано исследование связывания in vitro с применением клонированных рецепторов 5-НТ2B, меченных [3H]-5HT.
Исследование связывания с рецептором
Клетки НЕК 293 транзиторно трансфицированные экспрессионной плазмидой pXMD1-hu2B, кодирующей рецептор 5-НТ2B человека (см. Schmuck et al., FEBS Lett., 1994, 342, 85-90), используют так, как описано ранее (Schmuck et al., Eur. J. Pharmacol, 1996, 8, 959-967). Через два дня после трансфекции клетки собирают, осаждают при 500 g в течение 5 минут при 4°С, аккуратно ресуспендируют в ледяном буфере 1 (50 мМ трис рН 7,7, 4 мМ CaCl2) и гомогенизируют при помощи гомогенизатора для тканей Polytron PT 1200 (положение 6, 30 с). Клетки осаждают при 50000 g в течение 10 мин при 4°С, промывают буфером 1 и снова осаждают. Конечный осадок ресуспендируют в буфере для инкубации (50 мМ трис рН 7,7, 4 мМ CaCl2, 10 мкМ паргилина и 0,1 мас.% аскорбиновой кислоты). Для исследования связывания берут 300 мкл суспензии мембран (концентрация белка - от 0,3 до 0,5 мг/мл), 150 мкл конкурирующего лекарства и 50 мкл [3H]-5НТ в конечной концентрации от 4 до 5 нМ. Смесь инкубируют при 37°С в течение 30 минут и останавливают реакцию быстрым фильтрованием и двумя промывками 5 мл холодного 20 мМ трис-HCl рН 7,5 и 154 мМ NaCl через фильтры Whatman GFB. Фильтры учитывали методом жидкой сцинтилляции. Неспецифическое связывание определяли в присутствии избытка 5-НТ (100 мкМ). Связанный радиолиганд составлял менее 1% свободного радиолиганда. В экспериментах по конкуренции специфическое связывание составляло около 60% общего связывания. Результаты, выраженные как величина рКi, приведены в Таблице.
В результате эксперимента, описанного в вышеприведенном примере, было установлено, что соединения настоящего изобретения обладают селективной аффинностью к рецептору 5-НТ2В.
Пример 4:
Методы связывания с рецепторами 5-НТ2А, 5-НТ2В, 5-НТ2C
Ниже описаны методы связывания с рецепторами, в которых лиганды с высокой аффинностью к рецепторам 5-НТ2B используют в сравнительном исследовании с рецепторами 5-НТ2А и 5-HT2C для демонстрации селективности.
Рецепторы 5-НТ2А метят [3H]-кетансерином в коре головного мозга человека, в клетках НЕК293, экспрессирующих клонированный рецептор 5-НТ2А человека и в клетках НЕК293, экспрессирующих рецептор 5-НТ2А крыс. Для исследования конкурентного связывания концентрация лиганда составляет приблизительно 0,1 нМ. Для исследования насыщения связывания концентрации лиганда находятся в пределах от 0,01 нМ до 2,0 нМ. Исследования проводят в 0,5 мл буфера для исследования (50 мМ трис-HCl, 4 мМ хлорида кальция, 0,1 мас.% аскорбиновой кислоты) (рН 7,4 при 4°С). Неспецифическое связывание определяют при 10 мМ немеченого кетансерина. После инкубации в течение 60 минут при 32°С мембраны собирают на фильтры, обработанные 0,1% по массе полиэтиленимином и определяют связанную радиоактивность.
Рецепторы 5-НТ2B человека метят в клетках НЕК293, как описано выше, за исключением того, что радиолигандом является [3Н]-5-НТ и буфер для исследования содержит паргилин в концентрации 10 мМ и 0,1 мас.% аскорбиновой кислоты. Для исследования конкурентного связывания концентрация радиолиганда составляет приблизительно 0,4 нМ, а для исследования насыщения связывания концентрация [3H]-5НТ находится в пределах от 0,05 до 8 нМ. Неспецифическое связывание определяют при 10 мМ 5-НТ. Инкубации проводят при 4°С в течение 120 минут.
Рецепторы 5-НТ2C метят в хориоидном сплетении, клетках Cos-7, экспрессирующих рецептор 5-HT2C человека, и NIH-3T3, экспрессирующих рецептор 5-HT2C крыс.
Исследования проводят как описано для рецептора 5-НТ2А, за исключением того, что радиолигандом является [3H]-мезулергин. Для исследования конкуренции концентрация радиолиганда составляет приблизительно 0,2 нМ, а для исследования насыщения связывания концентрация находится в пределах от 0,1 до 18 нМ. Неспецифическое связывание определяют при 10 мкМ немеченого мезулергина.
Данные конкурентного связывания радиолиганда анализируют при использовании логистического уравнения с четырьмя параметрами и итерационного метода подбора кривой, получают оценочные значения IC50 и коэффициент Хилла. Значения Kd, определенные в исследовании насыщения связывания, затем используют для вычисления констант диссоциации ингибирования (Кi).
По результатам исследований, проводимых, как описано в вышеприведенном примере, было обнаружено, что соединения настоящего изобретения обладают селективной аффинностью к рецептору 5-НТ2B. Результаты приведены в Таблице.
Пример 5:
Функциональный анализ, основанный на ткани с рецептором 5-НТ2B
Ниже описан функциональный анализ in vitro, характеризующий рецепторы 5-НТ (предположительно 5-НТ2B) в продольных мышцах дна желудка крыс (Baxter et al., Brit. J. Pharmacol. 1994, 112, 323-331).
Полоски продольных мышц получают из дна желудка самцов крыс Sprague Dawley. Удаляют слизистую оболочку и суспендируют полоски со средним напряжением 1 g в кислородсодержащем (95% O2 / 5% CO2) растворе Тайрода при 37°С. Состав раствора Тайрода (мМ): NaCl 136,9; KCl 2,7; NaH2PO4 0,4; MgCl2 1, 0; глюкоза 5,6; NaHCO3 11,9; CaCl2 1,8.
Кривые зависимости ответа от концентрации агонистов рецептора 5-НТ получают в условиях, в которых активность циклооксигеназы инактивируется 3 мкМ индометацина, активность моноаминоксидазы инактивируется 0,1 мМ паргилина, а механизмы накопления инактивируются 30 мкМ кокаина и 30 мкМ кортикостерона.
Действие лекарств отслеживают по преобразователям напряжения и записывают на полиграф. Ответ ткани определяют как изменение изометрического напряжения (g). Среднюю эффективность (ЕС50) и максимальный ответ определяют стандартными итерационными методами подбора кривой.
Действие антагонистов определяют измерением смещения направо по сравнению с кривой ответа на концентрацию агониста после уравновешивания антагониста в течение, по крайней мере, 1 часа. Отношения концентраций измеряют при половине максимальных уровней ответа, а аффинность антагониста одной концентрации определяют по уравнению:
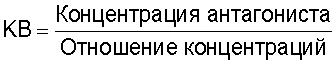
В случае, если для соединения обнаружено конкурентное поведение, для множества концентраций антагониста применяют регрессионный анализ Шильда (Schild).
В результате описанных выше исследований было обнаружено, что соединения настоящего изобретения являются селективными антагонистами рецептора 5-НТ2B.
Пример 6:
Исследование связывания с клонированными рецепторами H1, H2 и Н3 человека
Клетки COS-7 транзиторно трансфицируют экспрессионной плазмидой pCineohH1, pCineohH2, pCineohH3 и pCineohH4, кодирующей рецептор гистамина H1, Н2, Н3 или Н4 человека соответственно. Трансфицированные клетки собирают через 48 часов, гомогенизируют в ледяном 50 мМ Na2/калий фосфатном буфере (рН 7,4) и используют в исследованиях связывания с радиолигандом. Гомогенаты клеток (40-50 мкг белка) инкубируют в течение 30 минут при 25°С в 50 мМ Na2/калий фосфатном буфере (рН 7, 4) в 400 мкл с различными концентрациями [3Н]-мепирамина, [3H]-тиотидина, [3H]-R-α-метилгистамина или [3H]-пирамиламина для клеток, экспрессирующих рекомбинантные рецепторы H1, Н2, Н3 и Н4 человека соответственно. Неспецифическое связывание определяют в присутствии 1 мкМ миансерина. В исследованиях вытеснения гомогенаты клеток инкубируют с 1 нМ [3H]-мепирамина, 15 нМ [3H]-тиотидина, 0,5 нМ [3H]-R-α-метилгистамина или 15 нМ [3H]-пирамиламина и возрастающими концентрациями конкурирующих лигандов. Инкубацию останавливают быстрым разбавлением 3 мл ледяного 50 мМ Na2/калий фосфатного буфера (рН 7,4). Связанную радиоактивность отделяют фильтрованием через фильтры Whatman GF/C, обработанные 0,3% полиэтиленимином. Фильтры дважды промывают в 3 мл буфера и оставшуюся на фильтрах радиоактивность измеряют методом жидкой сцинтилляции.
При использовании итерационного метода подбора кривой определяют концентрацию 1-метил-4-(3-этокси-9Н-тиоксантен-9-илиден)-пиперидина, дающую 50% ингибирование связывания (IC50).
В результате описанных выше исследований было обнаружено, что каждое из соединений настоящего изобретения обладает низкой аффинностью к рецептору H1 человека.
Пример 7:
Функциональный анализ с рецептором гистамина H1 человека
Для измерения образования [3H]-инозитол фосфата транзиторно трансфицированные клетки НЕК-293 сеют в 24-луночные планшеты и метят до установления равновесия мио-[2-3H]-инозитолом (3 мкКю/мл) в течение 24 часов в ростовой среде. Отбирают среду, а клетки промывают один раз 500 мкл буфера HBS (130 мМ NaCl, 900 мкМ NaH2PO4, 800 мкМ MgSO4, 5,4 мМ KCl, 1,8 мМ CaCl2, 25 мМ глюкозы в 20 мМ HEPES рН 7,4). Через две минуты после внесения 20 мМ Li+ клетки стимулируют добавлением агониста в буфере HBS. Инкубацию останавливают отбором культуральной среды и добавлением холодной 10 мМ муравьиной кислоты. [3Н]-инозитол фосфаты выделяют методом анионообменной хроматографии (см. Seuwen et al., 1988, EMBO J., 7, 161-168). Величину рКB вычисляют по формуле: рКB=log(A'/A-1)-log[В], где А'/А - это соотношение концентраций агониста (ЕС50 в присутствии / ЕС50 в отсутствии антагониста), а [В] - концентрация антагониста.
По результатам исследований, описанных в вышеприведенном примере, было обнаружено, что соединения настоящего изобретения являются антагонистами более низкой эффективности к рецептору H1 человека. Результаты приведены в Таблице.
По результатам, приведенным в Таблице 1, видно, что соединения настоящего изобретения обладают селективной аффинностью к рецептору 5НТ2B, особенно они обладают гораздо большей активностью к рецептору 5НТ2В, чем к рецепторам H1 и 5НТ2C.
Реферат
Настоящее изобретение относится к производным 4-(тио- или селеноксантен-9-илиден)-пиперидина или акридина общей формулы I
в которой R1 - алкил, алкокси; R2 и R3 - водород; R4 - гидрокси-, алкокси, алкил, водород, R5 - водород, R6 - метил или этил; Х-S, N или Se, или их фармацевтически приемлемые соли. Соединения могут быть использованы для производства лекарственного средства для лечения заболевания, которое может быть облегчено лечением антагонистом 5-НТ2В. Описан также способ получения соединений I, фармацевтическая композиция и применение соединений. 6 н. и 10 з.п. ф-лы, 1 табл.
Формула
Комментарии