Гидроксиарилфункционализованные полимеры - RU2637124C2
Код документа: RU2637124C2
Описание
Уровень техники
Характеристики сцепления с дорожным покрытием представляют собой один из основных критериев оценки протекторов покрышек, и при данной оценке важным фактором являются эксплуатационные характеристики на мокрых поверхностях, таких как в случае снега и льда.
Некоторыми из сложных взаимосвязанных факторов, которые усложняют тип количественного механистического понимания, необходимого для составления рецептуры протекторных смесей, являются деформация протекторного каучука, вызываемая неровностями поверхности дороги, скорость водоотвода между протектором и поверхностью дороги и возможные адгезионные взаимодействия на межфазной поверхности между протектором и дорогой. Для дополнительного улучшения эксплуатационных характеристик покрышки специалисты, занятые в сфере разработки и изготовления покрышек, продолжают исследовать многочисленные факторы, которые оказывают воздействие на сцепление с мокрым дорожным покрытием.
Резиновые изделия, такие как протекторы покрышек, изготавливают из эластомерных композиций, которые содержат один или несколько армирующих материалов; смотрите, например, публикацию The Vanderbilt Rubber Handbook, 13th ed. (1990), pp.603-04. Первым материалом, обычно использовавшимся в качестве наполнителя, являлся технический углерод, который придает каучуковым композициям хорошие армирующие свойства и превосходную износостойкость. Однако рецептуры, содержащие технический углерод, зачастую характеризуются неблагоприятным повышенным сопротивлением качению, что коррелирует с увеличенными гистерезисом и разогреванием при деформировании во время эксплуатации покрышки - свойствами, которые требуется свести к минимуму для увеличения топливной эффективности автотранспортного средства.
Увеличенный гистерезис, являющийся результатом использования технического углерода, может быть несколько скомпенсирован посредством уменьшения количества (то есть объема) и/или увеличения размера частиц технического углерода, но риски ухудшения армирующих свойств и износостойкости ограничивают степень возможного продвижения по данным маршрутам.
В течение последних нескольких десятилетий в значительной степени возросло использование аморфного диоксида кремния и его обработанных вариантов, как индивидуально, так и в комбинации с техническим углеродом. Использование наполнителей на основе диоксида кремния может в результате привести к получению покрышек, характеризующихся пониженным сопротивлением качению, повышенным сцеплением на мокрых поверхностях и другими улучшенными свойствами.
Несмотря на выдающиеся эксплуатационные характеристики протекторов, использующих в качестве армирующих наполнителей диоксид кремния и технический углерод, все более взыскательные требования регламентации и потребности в эксплуатационных характеристиках привели к продолжению исследований в сфере альтернативных наполнителей. Примеры таких необычных наполнителей включают гидроксид алюминия (смотрите, например, патенты США №№6242522 и 6489389, а также публикацию Н. Mouri et al., «Improved Tire Wet Traction Through the Use of Mineral Fillers», Rubber Chem. and Tech., vol. 72, pp.960-68 (1999)); оксиды металлов, имеющие очень высокие плотности, (смотрите, патент США №6734235); намагничиваемые частицы, такие как оксид железа или стронциевый феррит, использующиеся при изготовлении боковин покрышек, (смотрите патент США №6476110); макроскопические (например, при среднем диаметре 10-5000 мкм) частицы твердых минералов, таких как оксид алюминия, CaCO3 и кварц, (смотрите патент США №5066702); пемзу, содержащую SiO2, (публикация США №2004/0242750 A1); субмикронные частицы ZnO (смотрите патент США №6972307); и частицы ZnSO4, BaSO4 и/или TiO2 микронного диапазона (смотрите патент США №6852785). Более часто потенциально подходящие наполнители просто приводят совместно друг с другом в формате перечня; смотрите, например, патенты США №№4255296 и 4468496. Другие материалы необычных наполнителей включают глины и сложные оксиды.
Как недавно было продемонстрировано, замещение некоторых или всех более часто использующихся типов дисперсных наполнителей неорганическими оксидами, такими как оксид трехвалентного железа, оксид двухвалентного железа, оксид алюминия и тому подобное, обеспечивает получение вулканизатов, демонстрирующих превосходные характеристики сцепления с мокрым дорожным покрытием; смотрите, например, публикацию США №2008/0161467.
Улучшение диспергирования армирующего наполнителя (наполнителей) по всему объему эластомера (эластомеров) может улучшить перерабатываемость и определенные физические свойства. Усилия, приложенные в данной сфере, были направлены на высокотемпературное перемешивание в присутствии селективно реакционно-способных промоторов, поверхностное окисление материалов, составляющих смесь, поверхностную прививку и химическое модифицирование полимера (полимеров).
Химическое модифицирование полимеров зачастую проводят в концевом положении. Концевое химическое модифицирование может быть осуществлено в результате проведения реакции между активным в концевом положении, то есть живым (то есть анионно-инициированным) или псевдоживым, полимером и функциональным агентом, обрывающим цепь. Концевое модифицирование также может быть осуществлено и при использовании функционального инициатора, изолированно или в комбинации с функциональным обрывом цепи. Функциональные инициаторы обычно представляют собой литийорганические соединения, которые дополнительно включают другую функциональность, обычно функциональность, которая включает атом азота. К сожалению, функциональные инициаторы в общем случае характеризуются относительно плохой растворимостью в углеводородных растворителях, относящихся к типу, обычно использующемуся при анионных полимеризациях, и не могут поддерживать рост цепи для живых концевых групп настолько же хорошо, как и более часто использующиеся алкиллитиевые инициаторы, такие как бутиллитий; обе характеристики оказывают негативное воздействие на скорость и эффективность полимеризации.
В течение некоторого времени, зачастую для областей применения клея, синтезировали полимеры, включающие 3,4-дигидроксифенилаланин (ДОФА); смотрите, например, патент США №4908404. Вследствие возможных дороговизны и трудности получения данных полимеров стремились получать так называемые блочные полимеры, приблизительно копирующие их эксплуатационные характеристики; смотрите публикацию Westwood et al., «Simplified Polymer Mimics of Cross-Linking Adhesive Proteins», Macromolecules 2007, 40, 3960-64. Однако стадия снятия защиты, использующаяся в вышеизложенном подходе, не может быть применена в случае содержания в полимере этиленовой ненасыщенности.
Краткое раскрытие изобретения
Вулканизаты, обладающие желательными свойствами, могут быть получены из смесей, использующих полимеры, которые включают арильные функциональности, имеющие гидроксильные группы. Такие полимеры улучшают степень взаимодействия как с обычными, так и с необычными наполнителями.
В одном аспекте предлагается способ получения функционального полимера, который включает один или несколько типов полиеновых мономерных фрагментов и по меньшей мере одно функционализующее звено, которое включает арильную группу, имеющую по меньшей мере один непосредственно связанный заместитель OR, где R представляет собой гидролизуемую защитную группу. В растворе, который включает инициирующее соединение и один или несколько типов этиленненасыщенных мономеров, которые включают по меньшей мере один тип полиена, инициирующему соединению дают возможность анионно инициировать полимеризацию мономеров для получения карбанионного полимера. Карбанионный полимер необязательно может быть введен в реакцию с соединением, обрывающим цепь. Функционализующее звено (звенья) возникает в результате присутствия (то есть представляет/представляют собой радикал (радикалы)) по меньшей мере одного соединения, выбираемого из инициирующего соединения, мономера (мономеров) и необязательного соединения, обрывающего цепь.
Способ может включать дополнительную стадию реакции, на которой защитную группу R гидролизуют для получения арильной группы, имеющей по меньшей мере одну непосредственно связанную гидроксильную группу. Данная дополнительная стадия может представлять собой реакцию между карбанионным полимером и соединением (соединениями), обрывающим цепь.
Арильная группа функционализующего звена может включать по меньшей мере две непосредственно связанные группы OR. В дополнительном или альтернативном варианте, Функционализующее звено может включать вторую арильную группу, в частности, в случае получения звена из соединения, обрывающего цепь.
Инициирующие соединения, которые могут обеспечивать получение функционализующего звена, включают те, которые описываются общей формулой

где М представляет собой атом щелочного металла; R1 представляет собой замещенную или незамещенную арильную группу, имеющую по меньшей мере одну группу заместителя OR2, где каждый R2 представляет собой группу R, которая также является нереакционно-способной по отношению к М; Z представляет собой одинарную связь или замещенную или незамещенную алкиленовую (ациклическую или циклическую) или ариленовую группу; a Q представляет собой группу, связанную с М через атом С, N или Sn. Арильная группа R1 может включать одно ароматическое кольцо (фенильная группа) или два и более конденсированных ароматических кольца. Инициирование данным типом функционального инициатора может в результате привести к получению макромолекулы, которая включает по меньшей мере одну цепь полимера, имеющую концевую функциональность, описывающуюся общей формулой

или функционализованного полимера, который описывается общей формулой

где R3 представляет собой замещенную или незамещенную арильную группу, которая включает по меньшей мере одну группу заместителя OR4 (при этом R4 представляет собой Н или R); Z определяют так же, как и ранее; Q’ представляет собой радикал Q, то есть остаток инициирующего фрагмента, связанного с цепью полимера через атом С, N или Sn; π представляет собой цепь полимера; а κ представляет собой атом водорода или имеющий функциональную группу радикал, образованный в результате прохождения реакции между полимером и соединением, обрывающим цепь. В случае наличия более чем одной группы OR4, каждая может иметься у одних и тех же или различных колец, а в определенных вариантах реализации по меньшей мере два заместителя OR4 могут быть соседними.
В случае результирующего получения функционализующего звена из мономера, мономер может включать арильную группу, предпочтительно фенильную группу, которая имеет по меньшей мере одну непосредственно связанную группу OR. Получающийся в результате полимер может включать несколько мономерных фрагментов, представляющих собой результат включения алкенов, (звенья А) и по меньшей мере три мономерных фрагмента, относящихся к только что описывавшемуся типу, (звенья В), которые могут быть несоседними или могут составлять в пределах полимера блок. В случае наличия блока из звеньев В он может находиться относительно близко к концевому положению полимера, то есть на удалении не более чем в шесть, четыре или два атома цепи полимера от концевого звена. В других вариантах реализации в полимер могут быть введены одно или несколько звеньев В, обычно после проведения полимеризации других мономеров, необязательно с последующим проведением реакции с соединением, которое необязательно может обеспечить получение в полимере дополнительной концевой функциональности. (Данное соединение необязательно должно относиться к типу, способному обеспечить получение конкретной функциональности, продемонстрированной далее в формуле (IV), а вместо этого может обеспечить получение любой из широкого ассортимента функциональностей, включающих, помимо прочего, те, которые содержат один или несколько гетероатомов).
В случае результирующего получения функционализующего звена по реакции между карбанионным полимером и соединением, обрывающим цепь, данная функциональность может быть описана общей формулой

где Z' представляет собой одинарную связь или алкиленовую группу; R3 определяют так же, как и ранее; R6 представляет собой Н, замещенную или незамещенную арильную группу, которая необязательно может включать одну или несколько групп заместителей OR4, R’ или JR’, где J представляет собой О, S или -NR' (при этом каждый R' независимо представляет собой замещенную или незамещенную алкильную группу); a Q” представляет собой остаток функциональности, которая является реакционно-способной по отношению к карбанионным полимерам, но который сам по себе является нереакционно-способным по отношению к таким полимерам. Арильная группа R3 может включать одно ароматическое кольцо (фенильная группа) или два или несколько конденсированных ароматических кольца, а группы OR4 могут иметься у одних и тех же или различных колец арильной группы, хотя в определенных вариантах реализации заместители OR4 в выгодном случае могут быть соседними. В дополнение к этому, R6 и часть R3 могут быть соединены таким образом, чтобы совместно с одним или несколькими атомами группы Q", с которой они связаны, (и необязательно Z') они образовывали бы кольцо, которое является связанным или конденсированным с арильной группой R3; примеры включают любую из широкого ассортимента структур флавонового и антронового типов, которая имеет одну или несколько групп заместителей OR4 по меньшей мере у одной из арильных групп. (Более подробно это описывается далее в связи с формулой (IVb)).
В одной вариации вышеизложенного способа подобное функционализующее звено может представлять собой результат проведения реакции между обрывающим цепь соединением, относящимся к только что описывавшемуся типу, и другими типами реакционно-способных в концевом положении полимеров, например, псевдоживым полимером.
В определенных вариантах реализации полиен (полиены) может представлять собой сопряженные диены. В данных и других вариантах реализации полимер также может включать винилароматические мономерные фрагменты, которые по длине цепи полимера предпочтительно включаются по существу статистически с мономерными фрагментами сопряженного диена.
В каждом из описывавшихся ранее случаев полимер может быть по существу линейным. В определенных вариантах реализации по существу линейный полимер в качестве концевого фрагмента может включать радикал соединения, которое включает по меньшей мере одну арильную группу, имеющую одну или несколько групп заместителей, которые могут быть гидролизованы до гидроксильных групп.
Также предлагаются и композиции, которые включают дисперсные наполнители и полимеры, относящиеся к описывавшемуся ранее типу, как и способы получения и использования таких композиций. Кроме того, предлагаются вулканизаты, полученные из таких наполненных композиций. В любых или во всех случаях полимер может взаимодействовать с дисперсными наполнителями, включающими технический углерод и диоксид кремния, настолько же хорошо, как в выгодном варианте и с необычными наполнителями, такими как неорганические оксиды и гидроксиды, глины и тому подобное.
После ознакомления с описанием различных вариантов реализации, которое следует далее, специалистам в соответствующей области техники станут очевидными и другие аспекты настоящего изобретения. В данном описании изобретения по всему ходу изложения используются следующие далее определения, если только контекст не будет однозначно указывать на противоположное намерение:
«полимер» обозначает продукт полимеризации одного или нескольких мономеров и включает гомо-, со-, тер-, тетраполимеры и тому подобное;
«мономерный фрагмент» или «мономерное звено» обозначает ту часть полимера, которая произведена из молекулы одного реагента, (например, этиленовый мономерный фрагмент описывается общей формулой -CH2CH2-);
«сополимер» обозначает полимер, который включает мономерные звенья, произведенные из двух реагентов, обычно мономеров, и включает статистические, блочные, сегментированные, привитые и тому подобные сополимеры;
«интерполимер» обозначает полимер, который включает мономерные звенья, произведенные по меньшей мере из двух реагентов, обычно мономеров, и включает сополимеры, терполимеры, тетраполимеры и тому подобное;
«статистический интерполимер» обозначает интерполимер, содержащий мономерные звенья, произведенные из каждого типа мономерного ингредиента, введенного по существу неповторяющимся образом и по существу в отсутствии блоков, то есть сегментов из трех и более идентичных мономерных фрагментов;
«реакционно-способный полимер» обозначает полимер, содержащий по меньшей мере один центр, который вследствие присутствия ассоциированного катализатора или инициатора, легко вступает в реакцию с другими молекулами, при этом данный термин, помимо прочего, включает псевдоживые и карбанионные полимеры;
«композиция катализатора» представляет собой общий термин, включающий простую смесь ингредиентов, комплекс различных ингредиентов, образование которого вызывают физические или химические силы притяжения, продукт химической реакции между некоторыми или всеми ингредиентами или комбинацию вышеизложенных вариантов, результатом чего является композиция, демонстрирующая каталитическую активность по отношению к одному или нескольким мономерам надлежащего типа;
«вязкость по Муни ненаполненного каучука» является вязкостью по Муни неотвержденного полимера перед добавлением какого-либо наполнителя (наполнителей);
«вязкость по Муни составленной смеси» является вязкостью по Муни композиции, которая, помимо прочего, включает неотвержденный или частично отвержденный полимер и дисперсный наполнитель (наполнители);
«замещенный» обозначает содержащий гетероатом или функциональность (например, гидрокарбильную группу), которые не препятствуют реализации предполагаемого назначения рассматриваемой группы;
«непосредственно связанный» обозначает ковалентно присоединенный без каких-либо промежуточных атомов или групп;
«полиен» обозначает молекулу, содержащую по меньшей мере две двойные связи, расположенные в наиболее длинной ее части или цепи, и, говоря конкретно, включает диены, триены и тому подобное;
«полидиен» обозначает полимер, который включает мономерные звенья одного или нескольких диенов;
«ч./сто ч. каучука» обозначает массовые части (м. ч.) на 100 м. ч. каучука;
«некоординирующий анион» обозначает стерически объемистый анион, который не образует координационных связей с активным центром системы катализатора вследствие стерических затруднений;
«предшественник некоординирующего аниона» обозначает соединение, которое способно образовывать некоординирующий анион в условиях проведения реакции;
«радикал» обозначает ту часть молекулы, которая остается после вступления в реакцию с другой молекулой, вне зависимости от приобретения или утраты каких-либо атомов в результате прохождения реакции;
«арильная группа» обозначает фенильную группу или полициклический ароматический радикал;
«концевое положение» обозначает конец цепи полимерной цепи; а
«концевой фрагмент» обозначает группу или функциональность, расположенные в концевом положении.
По всему ходу изложения данного документа все значения, приведенные в форме процентных величин, представляют собой массовые процентные величины, если только контекст не будет однозначно указывать на противоположное намерение. Соответствующие части любого упомянутого патента или патентной публикации посредством ссылки включаются в настоящий документ.
Детальное раскрытие изобретения
Как с очевидностью следует из приведенного ранее краткого изложения изобретения, способ может включать любые из широкого ассортимента его возможных сочетаний или комбинаций, а получающийся в результате полимер может быть охарактеризован по широкому ассортименту методов. В общем случае полимер включает мономерный фрагмент, произведенный из одного или нескольких полиенов, в частности, диенов, и концевую функциональность, описывающуюся любой или обеими из формул (II) и (IV), и/или одно или несколько описывавшихся ранее мономерных звеньев В. По меньшей мере, в определенных вариантах реализации полимер также может включать и непосредственно связанные боковые ароматические группы.
В последующем изложении описываются получение и использование полимера, который включает несколько мономерных фрагментов А, то есть алкеновых звеньев; необязательно несколько мономерных фрагментов С, то есть звеньев, которые включают боковую арильную группу, в частности фенильную группу, а в случае необходимости получения по меньшей мере некоторой части желательной функционализации из функциональных мономеров, по меньшей мере один мономерный фрагмент В, то есть звено, которое включает боковую арильную, предпочтительно фенильную, группу, имеющую по меньшей мере одну непосредственно связанную группу OR. Каждый из мономерных фрагментов А, В и С может представлять собой результат включения этиленненасыщенных мономеров.
Мономерный фрагмент А обычно представляет собой результат включения полиенов, в частности, триенов (например, мирцен) и диенов, в частности, C4-C12 диенов, а, говоря еще более конкретно, сопряженных диенов, таких как 1,3-бутадиен, 1,3-пентадиен, 1,3-гексадиен, 2,3-диметил-1,3-бутадиен, 2-этил-1,3-бутадиен, 2-метил-1,3-пентадиен, 3-метил-1,3-пентадиен, изопрен, 4-метил-1,3-пентадиен, 2,4-гексадиен и тому подобное. Некоторые или все мономерные фрагменты А могут быть произведены из одного или нескольких типов диенов, в частности, одного или нескольких типов сопряженных диенов, например, 1,3-бутадиена. В некоторых вариантах реализации по существу все (то есть по меньшей мере 95%) из полиенов могут представлять собой диены, в частности, сопряженные диены.
Полиены можно включать в цепи полимеров по более чем одному способу. Регулирование данного способа включения может оказаться желательным, в особенности в случае областей применения протекторов покрышек. Для определенных областей применения вариантов конечного использования желательной может оказаться цепь полимера, демонстрирующая наличие совокупной 1,2-микроструктуры в диапазоне от ~10 до ~80%, возможно от ~25 до ~65%, для выражения через численную процентную величину при расчете на совокупное количество полиеновых звеньев. Полимер, который демонстрирует наличие совокупной 1,2-микроструктуры не более чем на ~50%, предпочтительно не более чем на ~45%, более предпочтительно не более чем на ~40%, еще более предпочтительно не более чем на ~35%, а наиболее предпочтительно не более чем на ~30%, при расчете на совокупный уровень содержания полиена, считается по существу линейным. Для определенных областей применения вариантов конечного использования желательным может оказаться выдерживание еще более низкого уровня содержания 1,2-соединительных звеньев, например меньшего чем приблизительно 7%, меньшего чем 5%, меньшего чем 2% или меньшего чем 1%.
В зависимости от предполагаемого варианта конечного использования одна или несколько цепей полимера могут включать боковые ароматические группы, которые могут быть получены благодаря мономерному фрагменту С, то есть мономерному фрагменту, произведенному из винилароматики, в частности, C8-C20 винилароматики, такой как, например, стирол, α-метилстирол, п-метилстирол, винилтолуолы и винилнафталины. В случае использования в сочетании с одним или несколькими полиенами мономерный фрагмент С может составлять от ~1 до ~50%, от ~10 до ~45% или от ~20 до ~35% от цепи полимера; статистическая микроструктура может обеспечить достижение особенных преимуществ в некоторых областях применения вариантов конечного использования, таких как, например, в случае каучуковых композиций, использующихся при изготовлении протекторов покрышек. В случае желательности получения блочного интерполимера звенья С могут составлять от ~1 до ~90%, в общем случае от ~2 до ~80%, обычно от ~3 до ~75%, а типично от ~5 до ~70%, от цепи полимера. (В данном абзаце все процентные величины являются молярными процентными величинами).
Примеры интерполимеров включают те, у которых для получения звеньев А используют один или несколько сопряженных диенов, то есть полидиены; в их числе, одним из нескольких использующихся полидиенов или единственным использующимся полидиеном может являться 1,3-бутадиен. В случае желательности наличия звеньев С для получения, например, СБК они могут быть получены из стирола. В каждый из вышеупомянутых типов примеров интерполимеров также могут быть включены одно или несколько звеньев В.
Звенья В включают боковую арильную группу, которая включает одну или несколько непосредственно связанных гидроксильных групп. Вследствие активности атомов Н гидроксильных групп и возможного оказания ими помех проведению определенных процессов полимеризации, одно или несколько звеньев В обычно получают из соединений, которые включают группы R, то есть группы, которые являются нереакционно-способными в условиях, использующихся при полимеризации этиленненасыщенных мономеров, но которые впоследствии могут быть удалены, обычно в результате проведения гидролиза или подобной реакции, для получения желательных гидроксильных групп. Конкретный тип (типы) использующейся защитной группы (групп) не должен оказывать помех проведению процесса полимеризации, и способ снятия защиты, использующийся для получения гидроксильных групп, не должен разрушать этиленовую ненасыщенность в полимере, являющуюся результатом присутствия звеньев А, или другим образом вступать с ней в реакцию. Один неограничивающий класс подходящих защитных групп представляет собой триалкилсилоксигруппы, которые могут быть получены в результате проведения реакции между гидроксильными группами и триалкилсилилгалогенидом. Несмотря на использование в следующих далее примерах трет-бутилдиметилсилоксильных групп также могут быть использованы и другие группы, такие как группы ацеталя, простого трет-бутилового эфира, простого 2-метоксиэтоксиэфира и тому подобное.
Количество групп OR в арильной, обычно фенильной, группе каждого звена В необязательно должно быть одним и тем же, в случае, когда число групп одно и то же, группы OR необязательно должны иметься у данных колец в одном и том же положении (положениях). При использовании фенильной группы в качестве представительной арильной группы, можно сказать, что единственная группа OR по отношению к точке присоединения фенильной группы к цепи полимера может иметься у фенильного кольца в положениях орто, мета или пара, в то время как несколько групп OR могут быть получены в фенильном кольце в положениях 2,3, 2,4, 2,5, 2,6, 3,4, 3,5, 3,6, 2,3,4, 2,3,5 и тому подобное.
Звенья В обычно получают из винилароматических соединений, которые включают одну или несколько гидроксилобразующих групп, непосредственно присоединенных к их арильным, обычно фенильным, кольцам. Такие соединения могут быть описаны общей формулой

где R1 определяют так же, как и ранее, и в данном случае он может включать от 1 до 5 групп OR, включительно, при этом каждый R независимо относится к типу описывавшейся ранее защитной группы. (Несмотря на необязательную идентичность каждых R с учетом легкости и простоты результатом обычно является использование в заданном соединении одного типа фрагмента R). Группы OR могут представлять собой заместителей у одного и того же кольца R1 или могут представлять собой заместителей у различных колец, а в случае наличия в R1 трех и более групп OR две из них могут представлять собой заместителей у одного кольца, в то время как другая (другие) будут представлять собой заместителя (заместителей) у другого кольца (колец). В одном варианте реализации две группы OR могут иметься в положениях 3 и 4 одного и того же кольца в пределах арильной группы, предпочтительно фенильной группы. В случае отличия R1 от фенильной группы и включения в него более чем одной группы OR, и в случае наличия групп OR у более чем одного кольца по меньшей мере две из групп OR предпочтительно будут по меньшей мере несколько приближенными, то есть непосредственно связанными с атомами С кольца, которые разделены не более чем 4, предпочтительно 3, а еще более предпочтительно 2, другими атомами кольца. Многие из данных соединений в выгодном случае являются растворимыми в приведенных ранее типах органических растворителей.
В случае полимеризации одного или нескольких соединений, относящихся к типу, описывающемуся формулой (V), оно/они приводит/приводят к получению звена (звеньев) В, после чего каждый из фрагментов R может быть гидролизован для получения фенольных гидроксильных групп.
Количество звеньев В обычно невелико в сопоставлении с количеством звеньев А, а в случае наличия таковых, то и звеньев С; как было установлено, относительно небольшое количество звеньев В приводит к достижению удовлетворительного уровня желательных свойств, при этом дополнительное улучшение данных свойств необязательно будет пропорциональным количеству имеющихся звеньев В. Данное относительно небольшое количество может быть выражено несколькими способами. Например, уровень массового процентного содержания конечного полимера, приписываемого звеньям В, обычно является меньшим чем 2%, более часто находится в диапазоне от ~0,1 до ~1,5%, а обычно от ~0,2 до ~1,0%. Уровень процентного содержания мономерного фрагмента В в сопоставлении с совокупным количеством мономерного фрагмента в полимере обычно является меньшим чем 1%, более часто находящимся в диапазоне от ~0,01 до ~0,75%, а обычно от ~0,05 до ~0,5%. Совокупное количество звеньев В в заданном полимере в общем случае находится в диапазоне от 1 до нескольких дюжин, обычно от 1 до 12, более часто от 1 до 10, а наиболее часто от 1 до 5.
Звенья В могут быть отделены друг от друга, или два и более звена В могут быть смежными по длине цепи полимера. (Несмотря на понимание специалистами в соответствующей области техники того, как синтезировать статистические и блочные интерполимеры, каждый из данных вариантов с определенной детализацией будет обсуждаться далее). Кроме того, звенья В могут быть включены вблизи от начала полимеризации, вблизи от завершения полимеризации или в любых одной или нескольких промежуточных точках; в первых двух из вышеупомянутых возможностей звено В может быть получено в пределах 6 атомов углерода, в пределах 2 звеньев от концевого положения полимера, по соседству с ним или в качестве концевого звена, либо индивидуально, либо в виде части блока.
Вышеупомянутые типы полимеров могут быть получены в результате проведения эмульсионной полимеризации или растворной полимеризации, при этом последняя обеспечивает более значительную степень регулирования в отношении таких свойств, как неупорядоченность, микроструктура и тому подобное. Растворные полимеризации проводили в течение многих десятилетий, так что общие ее аспекты специалистам в соответствующей области техники известны; так что для удобства пользования в настоящем документе приводятся только определенные общие аспекты.
В растворных полимеризациях могут быть использованы как полярные растворители, такие как ТГФ, так и неполярные растворители, при этом последний тип является более часто использующимся в промышленной практике. Примеры неполярных растворителей включают C5-C12 циклические и ациклические алканы, а также их алкилированные производные, определенные жидкие ароматические соединения и их смеси. Специалист в соответствующей области техники имеет представление и о других подходящих возможностях и комбинациях растворителей.
В зависимости от природы желательного полимера конкретные условия проведения растворной полимеризации могут значительно варьироваться. В обсуждении, которое следует далее, сначала будут описываться живые полимеризации с последующим описанием псевдоживых. После данных описаний будут обсуждаться необязательные функционализация и переработка таким образом полученных полимеров.
Растворная полимеризация обычно включает инициатор. Примеры инициаторов включают литийорганические соединения, в особенности, производные алкиллития. Примеры литийорганических инициаторов включают N-литиогексаметиленимин; н-бутиллитий; трибутилолово-литий; производные диалкиламинолития, такие как диметиламинолитий, диэтиламинолитий, дипропиламинолитий, дибутиламинолитий и тому подобное; производные диалкиламиноалкиллития, такие как диэтиламинопропиллитий; и те производные триалкилстанниллития, которые имеют C1-C12, предпочтительно C1-C4, алкильные группы.
Также могут быть использованы и многофункциональные инициаторы, то есть инициаторы, способные приводить к получению полимеров, имеющих более чем одну «живую» концевую группу. Примеры многофункциональных инициаторов включают нижеследующие, но не ограничиваются только этими: 1,4-дилитиобутан, 1,10-дилитиодекан, 1,20-дилитиоэйкозан, 1,4-дилитиобензол, 1,4-дилитионафталин, 1,10-дилитиоантрацен, 1,2-дилитио-1,2-дифенилэтан, 1,3,5-трилитиопентан, 1,5,15-трилитиоэйкозан, 1,3,5-трилитиоциклогексан, 1,3,5,8-тетралитиодекан, 1,5,10,20-тетралитиоэйкозан, 1,2,4,6-тетралитиоциклогексан и 4,4'-дилитиобифенил.
В дополнение к литийорганическим инициаторам подходящими для использования также могут являться и так называемые функционализованные инициаторы. Они становятся включенными в полимерную цепь, таким образом, обеспечивая наличие функциональной группы в образованной из инициатора концевой группе цепи. Примеры таких материалов включают литиированные арилтиоацетали (смотрите, например, патент США №7153919) и продукт реакции между литийорганическими соединениями и, например, N-содержащими органическими соединениями, такими как замещенные альдимины, кетимины, вторичные амины и тому подобное, необязательно подвергнутыми предварительной реакции с соединением, таким как диизопропенилбензол, (смотрите, например, патенты США №№5153159 и 5567815). Степень взаимодействия между цепями полимера и частицами технического углерода может быть дополнительно улучшена в результате использования инициатора, содержащего атом N, такого как, например, литиированный ГМИ. Многие из данных функциональных инициаторов являются плохо растворимыми во многих приведенных ранее растворителях, в частности, в тех растворителях, которые являются относительно неполярными.
В противоположность этому, многие соединения, включенные в формулу (I), характеризуются приемлемой растворимостью в тех типах органических жидкостей, которые обычно используются в качестве растворителей при растворных полимеризациях. Соединения, включенные в данную формулу, здесь и далее в настоящем документе называются R1-содержащими инициаторами.
Арильной группой R1-содержащего инициатора могут являться фенильная группа или два и более конденсированных ароматических кольца. В случае включения в арильную группу R1 более чем одной группы OR2 (при этом каждый R2 представляет собой группу R, которая является нереакционно-способной по отношению к М), группы OR2 могут представлять собой заместителей у одного и того же кольца или различных колец в пределах арильной группы; в случае наличия у арильной группы трех и более групп OR2 две из них могут представлять собой заместителей у одного кольца, в то время как другая (другие) будет представлять собой заместителя (заместителей) у другого кольца (колец). В одном варианте реализации две группы OR2 могут иметься в положениях 3 и 4 одного и того же кольца в пределах арильной группы, предпочтительно фенильной группы. В случае отличия R1 от фенильной группы и включения в него более чем одной группы OR2, и в случае наличия групп OR2 у более чем одного кольца по меньшей мере две из групп OR2 предпочтительно будут по меньшей мере несколько приближенными, то есть непосредственно связанными с атомами С кольца, которые разделены не более чем 4, предпочтительно 3, а еще более предпочтительно 2, другими атомами кольца. В случае наличия у фенильной группы одной группы OR2 она может иметься в любом положении кольца, хотя для определенных областей применения предпочтительным может оказаться пара-положение по отношению к Z.
Фрагменты R2 R1-содержащего инициатора обеспечивают отсутствие в арильной группе R1 атомов активного водорода. Такие атомы активного водорода будут оказывать помехи реализации способности R1-содержащего инициатора анионно инициировать полимеризации. Если только конкретный фрагмент R2 не будет составлять группу, которая способна обеспечивать взаимодействие с дисперсным наполнителем, он предпочтительно также будет способен подвергаться гидролизу до атома водорода. Одним неограничивающим примером типа группы, которая может иметь данное двойное назначение, являются триалкилсилоксигруппы; такие группы могут быть получены в результате проведения реакции между гидроксильными группами, присоединенными к арильной группе R1, и триалкилсилилгалогенидом. Несмотря на необязательную идентичность каждых R2 с учетом легкости и простоты результатом обычно является наличие у заданного R1-содержащего инициатора одного типа фрагмента R2.
В случае инициирования R1-содержащим инициатором полимеризации его радикал будет образовывать одну концевую группу цепи полимера (смотрите формулы (II) и (III)). Фрагменты R2 данного радикала обычно подвергаются гидролизу с образованием гидроксильных заместителей у группы R3, описывающейся формулами (II) и (III). Как было установлено, данный тип группы R3 обеспечивает достижение превосходной степени взаимодействия с широким ассортиментом дисперсных наполнителей, включающих технический углерод и диоксид кремния, а также необычные наполнители, такие как неорганические оксиды и гидроксиды, глины и тому подобное.
В R1-содержащем инициаторе М представляет собой атом щелочного металла (предпочтительно атом К, Na или Li, наиболее предпочтительно атом Li), a Q представляет собой группу, связанную с М через атом С, N или Sn. В общем случае Q не содержит каких-либо атомов активного водорода, которые, как должен понимать специалист в соответствующей области техники, оказывают помехи обеспечению эффективности R1-содержащего инициатора. Потенциально подходящие группы Q слишком многочисленны для исчерпывающего перечисления, но несколько неограничивающих примеров может быть приведено; исходя из этого специалист в соответствующей области техники может представить себе и многочисленные другие альтернативы.
Одним типом потенциально подходящей группы Q являются тиоацетали. Данные функциональности описываются общей формулой

где R15 представляет собой С2-С10 алкиленовую группу, предпочтительно C2-C8 алкиленовую группу, более предпочтительно С3-С6 группу; Х выбирают из S, О и NR16, где R16 может представлять собой C1-С6 триалкилсилильную группу, C1-C20 алкильную группу, С4-С20 циклоалкильную группу, С6-С20 арильную группу, при том условии, что может быть присоединено любое из нижеследующего: C1-С10 алкильные группы, С6-С20 арильные группы, С2-С10 алкенильные группы, С3-С10 неконцевые алкинильные группы, группы простых эфиров, третичных аминов, фосфинов, сульфидов, силилов и их смеси. Один предпочтительный тип включает в качестве Х атом S, а в качестве R15 С3 алкилен, то есть 1,3-дитиан. В определенных аспектах Q может представлять собой группу, которая включает гетероатомзамещенный циклический фрагмент, адаптированный для связывания с атомом щелочного металла, таким как Li. Для получения дополнительной информации заинтересованный читатель отсылается к патенту США №7153919.
Другие потенциально подходящие группы Q включают , где каждые R7 независимо представляют собой гидрокарбильную (hydrocarbyl) (например, алкильную, циклоалкильную, арильную, аралкильную, алкарильную и тому подобную) группу или совместно образуют циклоалкильную группу, и NR8, где R8 представляет собой гидрокарбильную группу, в частности, арильную, С3-C8 циклоалкильную или C1-C20 алкильную группу; в число последних включают производные циклоалкилениминоалкиллития, такие как те, которые описываются, например, в патенте США №5574109. Кроме того, потенциально подходящими в качестве групп Q являются любые из широкого ассортимента линейных или разветвленных алкильных групп, неограничивающие примеры которых включают бутил, пентил, гексил, гептил, октил и тому подобное. Все вышеупомянутые инициаторы могут быть получены из гидроксилзамещенных бензальдегидов по методикам синтеза, описывающимся более подробно в примерах, которые следуют далее.
Соединения, описывающиеся формулой (I), могут быть получены по широкому ассортименту способов, при этом выбор маршрута синтеза в большой степени зависит от конкретной природы Q. Например, соединение, включающее несколько гидроксильных групп, присоединенных к арильной группе, и по меньшей мере одну другую функциональность, можно через другую функциональность вводить в реакцию с соединением для получения группы Q; после этого атом (атомы) Н гидроксильной группы (групп) могут быть введены в реакцию с соединением, которое может обеспечить получение вышеупомянутых групп R2, а получающийся в результате материал может быть введен в реакцию с материалом, содержащим щелочной металл, например, литийорганикой. Данный тип подхода к синтезу далее используют в примерах для получения примера инициатора дитианового типа.
R1-содержащий инициатор может быть получен вне полимеризационной емкости, в которой он должен исполнять функцию инициатора. В данном случае в реакционную емкость может быть загружена смесь мономера (мономеров) и растворителя с последующим добавлением инициатора, который зачастую добавляют в виде части раствора или смеси (то есть в растворителе-носителе). По причинам удобства R1-содержващий инициатор обычно синтезируют «по месту».
Хотя специалисты в соответствующей области техники и имеют представление об условиях, обычно используемых при растворной полимеризации, для удобства пользования предлагается представительное описание. Нижеследующее базируется на периодическом способе, хотя специалист в соответствующей области техники сможет адаптировать данное описание и на полупериодический, непрерывный или другой способы.
Растворную полимеризацию обычно начинают с загрузки смеси мономера (мономеров) и растворителя в подходящую реакционную емкость с последующим добавлением координатора (в случае использования такового) и инициатора, которые зачастую добавляют в виде части раствора или смеси; в альтернативном варианте мономер (мономеры) и координатор могут быть добавлены к инициатору. Как неупорядоченность, так и уровень содержания винила (то есть 1,2-микроструктуры) могут быть увеличены благодаря включению координатора - обычно полярного соединения. На один эквивалент инициатора может быть использовано вплоть до 90 и более эквивалентов координатора, при этом данное количество зависит, например, от величины желательного уровня содержания винила, уровня содержания использованного неполиенового мономера, температуры реакции и природы конкретного использованного координатора. Соединения, подходящие для использования в качестве координаторов, включают органические соединения, которые включают гетероатом, имеющий несвязанную пару электронов, (например, О или N). Примеры включают диалкиловые простые эфиры моно- и олигоалкиленгликолей; краун-эфиры; третичные амины, такие как тетраметилэтилендиамин; ТГФ; олигомеры ТГФ; линейные и циклические олигомерные оксоланилалканы (смотрите, например, патент США №4429091), такие как 2,2'-ди(тетрагидрофурил)пропан, дипиперидилэтан, гексаметилфосфорамид, N,N'-диметилпиперазин, диазабициклооктан, диэтиловый эфир, трибутиламин и тому подобное.
Обычно раствор полимеризационного растворителя (растворителей) и мономера (мономеров) получают при температуре в диапазоне от приблизительно -80° до +100°C, более часто от приблизительно -40° до +50°C, а обычно от ~0° до +30°C; к данному раствору добавляют инициирующее соединение или, в случае необходимости получения функционализующего звена из инициатора, и R1-содержащий инициатор (или его предшественник совместно с литийорганикой, обычно алкиллитием). Раствор может иметь температуру в диапазоне от приблизительно -70° до ~150°C, более часто от приблизительно -20° до ~120°C, а обычно от ~10° до ~100°C. Полимеризации дают возможность протекать в безводных анаэробных условиях в течение периода времени, достаточного для получения в результате желательного полимера, и находящегося в диапазоне обычно от ~0,01 до ~100 часов, более часто от ~0,08 до ~48 часов, а типично от ~0,15 до ~2 часов. После достижения желательной степени превращения источник тепла (в случае использования такового) может быть удален, а в случае необходимости резервирования реакционной емкости исключительно для полимеризаций, реакционную смесь удаляют в постполимеризационную емкость для проведения функционализации и/или гашения реакции.
В общем случае полимеры, полученные в соответствии с анионными методиками, имеют среднечисленную молекулярную массу (Mn), доходящую вплоть до ~500000 Да. В определенных вариантах реализации значение Mn может составлять всего лишь ~2000 Да; в данных и/или других вариантах реализации значение Mn в выгодном случае может составлять по меньшей мере ~10000 Да или может находиться в диапазоне от ~50000 до ~250000 Да или от ~75000 до ~150000 Да. Зачастую значение Мn таково, что подвергнутый гашению реакции образец демонстрирует вязкость по Муни ненаполненного каучука (ML4/100°C) в диапазоне от ~2 до ~150, более часто от ~2,5 до ~125, еще более часто от ~5 до ~100, а наиболее часто от ~10 до ~75.
В определенных областях применения вариантов конечного использования требуются полимеры, которые обладают свойствами, получение которых может оказаться трудным или неэффективным в случае анионных (живых) полимеризаций. Например, в некоторых областях применения желательными могут оказаться сопряженные диеновые полимеры, характеризующиеся высокими уровнями содержания цис-1,4-соединительных звеньев. Полидиены могут быть получены по способам, использующим катализаторы (в противоположность инициаторам, использующимся в живых полимеризациях), и могут демонстрировать псевдоживые характеристики.
Как известно, определенные типы систем катализаторов являются подходящими для использования при получении из сопряженных диеновых мономеров очень стереоспецифических 1,4-полидиенов. Некоторые системы катализаторов предпочтительно в результате приводят к получению цис-1,4-полидиенов, в то время как другие предпочтительно приводят к получению транс-1,4-полидиенов, и специалисты в соответствующей области техники знакомы с примерами каждого типа. Последующее описание изобретения базируется на конкретной системе цис-специфического катализатора, хотя она представлена просто в порядке приведения примера и не считается ограничением способа функционализации и соединений.
Примеры систем катализаторов могут использовать металлы лантаноидов, которые, как известно, являются подходящими для использования при полимеризации сопряженных диеновых мономеров. Говоря конкретно, системы катализаторов, которые включают соединение лантаноида, могут быть использованы для получения цис-1,4-полидиенов из одного или нескольких типов сопряженных диенов. Предпочтительные композиции катализаторов на основе лантаноидов включают те, которые описываются в патенте США №6699813 и патентных документах, процитированных в нем. Для удобства и легкости пользования в настоящем документе приводится сжатое описание.
Примеры композиций лантаноидных катализаторов включают (а) соединение лантаноида, алкилирующий агент и галогенсодержащее соединение (хотя в случае содержания атома галогена в соединении лантаноида и/или алкилирующем агенте использование галогенсодержащего соединения является необязательным); (b) соединение лантаноида и алюмоксан; или (с) соединение лантаноида, алкилирующий агент и некоординирующий анион или его предшественник.
Могут быть использованы различные соединения лантаноидов или их смеси, при этом предпочтение отдается тем, которые являются растворимыми в ароматических, алифатических и/или циклоалифатических жидкостях, хотя соединения лантаноидов, нерастворимые в углеводородах, в полимеризационной среде могут быть суспендированы. Предпочтительные соединения лантаноидов включают те, которые включают по меньшей мере один атом Nd, La или Sm, или те, которые включают дидим (смесь оксидов редкоземельных элементов). Атом (атомы) лантаноида в соединениях лантаноидов может находиться в любой из нескольких степеней окисления, хотя более частой является степень окисления +3. Примеры соединений лантаноидов включают карбоксилаты, органофосфаты, органофосфонаты, органофосфинаты, ксантогенаты, карбаматы, дитиокарбаматы, β-дикетонаты, алкоксиды, арилоксиды, галогениды, псевдогалогениды, оксигалогениды и тому подобное.
Обычно соединение лантаноида используют в сочетании с одним или несколькими алкилирующими агентами, то есть металлоорганическими соединениями, которые могут переносить гидрокарбильные группы на другой металл. Обычно данные агенты представляют собой металлоорганические соединения электроположительных металлов, таких как металлы из групп 1, 2 и 3. Примеры алкилирующих агентов включают алюминийорганические соединения и магнийорганические соединения. Первые включают (1) соединения, описывающиеся общей формулой , где n представляет собой целое число в диапазоне от 1 до 3, включительно, каждый R9 независимо представляет собой одновалентную органическую группу (которая может содержать гетероатомы, такие как N, О, В, Si, S, Р и тому подобное), соединенную с атомом Al через атом С, и каждый X' независимо представляет собой атом водорода, атом галогена, карбоксилатную группу, алкоксидную группу или арилоксидную группу; и (2) олигомерные линейные или циклические алюмоксаны, которые могут быть получены в результате проведения реакции между производными тригидрокарбилалюминия и водой. Вторые включают соединения, описывающиеся общей формулой , где X' определяют так же, как и ранее, у представляет собой целое число в диапазоне от 1 до 2, включительно, а R10 представляет собой то же самое, что и R9, за исключением того, что каждая одновалентная органическая группа соединена через атом С с атомом Mg.
Некоторые композиции катализаторов содержат соединения, имеющие один или несколько подвижных атомов галогена. Подходящие галогенсодержащие соединения включают элементарные галогены, смешанные галогены, галогенводороды, органические галогениды, неорганические галогениды, галогениды металлов, металлоорганические галогениды и их смеси. Предпочтительно галогенсодержащие соединения являются растворимыми в растворителях, таких как те, которые были описаны ранее в связи с соединениями лантаноидов, хотя нерастворимые в углеводородах соединения в полимеризационной среде могут быть суспендированы.
Другие композиции катализаторов содержат некоординирующий анион или предшественника некоординирующего аниона. Примеры некоординирующих анионов включают анионы тетраарилборной кислоты, в частности, анионы фторированной тетраарилборной кислоты, и ионные соединения, содержащие некоординирующие анионы и противокатион (например, тетракис(пентафторфенил)борат трифенилкарбония). Примеры предшественников некоординирующих анионов включают соединения бора, которые включают сильные электроноакцепторные группы.
Композиции катализаторов данного типа обладают очень высокой каталитической активностью в отношении полимеризации сопряженных диенов до получения стереоспецифических полидиенов в широком диапазоне концентраций и соотношений, хотя полимеры, обладающие наиболее желательными свойствами, обычно получают из систем, которые используют относительно узкий диапазон концентраций и соотношений ингредиентов. Кроме того, ингредиенты катализатора, как представляется, вступают во взаимодействие с образованием активных частиц катализатора, так что оптимальная концентрация любого одного ингредиента может зависеть от концентраций других ингредиентов. Следующие далее молярные соотношения считаются относительно типичными для широкого ассортимента различных систем на основе вышеупомянутых ингредиентов:
алкилирующий агент к соединению лантаноида (алкилирующий агент/Ln): от ~1:1 до ~200:1, предпочтительно от ~2:1 до ~100:1, более предпочтительно от ~5:1 до ~50:1;
галогенсодержащее соединение к соединению лантаноида (атом галогена/Ln): от ~1:2 до ~20:1, предпочтительно от ~1:1 до ~10:1, более предпочтительно от ~2:1 до ~6:1;
алюмоксан к соединению лантаноида, говоря конкретно, эквиваленты атомов алюминия в алюмоксане к эквивалентам атомов лантаноида в соединении лантаноида (Al/Ln): от ~10:1 до ~50000:1, предпочтительно от ~75:1 до ~30000:1, более предпочтительно от ~100:1 до ~1000:1; и
некоординирующий анион или его предшественник к соединению лантаноида (An/Ln): от ~1:2 до ~20:1, предпочтительно от ~3:4 до ~10:1, более предпочтительно от ~1:1 до ~6:1.
Молекулярная масса полидиенов, полученных при использовании катализаторов на основе лантаноидов, может быть отрегулирована в результате подстраивания количества использующегося катализатора и/или величин концентраций сокатализаторов в системе катализатора. В общем случае увеличение концентраций катализатора и сокатализатора уменьшает молекулярную массу получающихся в результате полидиенов, хотя очень низкомолекулярные полидиены (например, жидкие полидиены) требуют использования чрезвычайно высоких концентраций катализатора, что делает необходимым удаление из полимера остатков катализатора во избежание возникновения неблагоприятных эффектов, таких как замедление скорости серной вулканизации. Включение одного или нескольких Ni-содержащих соединений в композиции катализаторов на основе лантаноидов позволяет выгодным образом проводить простое регулирование молекулярной массы получающегося в результате полидиена без возникновения значительных негативных последствий для активности катализатора и микроструктуры полимера. Могут быть использованы различные Ni-содержащие соединения или их смеси, при этом предпочтение отдается тем, которые являются растворимыми в углеводородных растворителях, таких как те, которые были представлены ранее.
Атом Ni в Ni-содержащих соединениях может находиться в любой из нескольких степеней окисления, хотя в общем случае предпочтительными являются двухвалентные соединения Ni, в которых атом Ni находится в степени окисления +2. Примеры соединений Ni включают карбоксилаты, органофосфаты, органофосфонаты, органофосфинаты, ксантогенаты, карбаматы, дитиокарбаматы, β-дикетонаты, алкоксиды, арилоксиды, галогениды, псевдогалогениды, оксигалогениды, никельорганические соединения (то есть соединения, содержащие по меньшей мере одну связь C-Ni, такие как, например, никелоцен, декаметилникелоцен и тому подобное) и тому подобное.
Молярное соотношение между Ni-содержащим соединением и соединением лантаноида (Ni/Ln) в общем случае находится в диапазоне от ~1:1000 до ~1:1, предпочтительно от ~1:200 до ~1:2, а более предпочтительно от ~1:100 до ~1:5.
Данные типы композиций катализаторов могут быть получены при использовании любого из следующих далее способов:
(1) Получение «по месту». Ингредиенты катализатора добавляют к раствору, содержащему мономер и растворитель (или просто мономер в массе). Добавление может быть проведено постадийно или одновременно. В последнем случае предпочтительно сначала добавляют алкилирующий агент с последующим добавлением в указанном порядке соединения лантаноида, никельсодержащего соединения (в случае использования такового) и (в случае использования такового) галогенсодержащего соединения или некоординирующего аниона или предшественника некоординирующего аниона.
(2) Предварительное смешивание. Ингредиенты перед введением к сопряженному диеновому мономеру (мономерам) могут быть смешаны вне полимеризационной системы, в общем случае при температуре в диапазоне от приблизительно -20° до приблизительно 80°С.
(3) Предварительное получение в присутствии мономера (мономеров). Ингредиенты катализатора смешивают в присутствии небольшого количества сопряженного диенового мономера (мономеров) при температуре в диапазоне от приблизительно -20° до ~80°C. Количество сопряженного диенового мономера может находиться в диапазоне от ~1 до ~500 молей, предпочтительно от ~5 до ~250 молей, а более предпочтительно от ~10 до ~100 молей, на один моль соединения лантаноида. Получающуюся в результате композицию катализатора добавляют к остатку полимеризуемого сопряженного диенового мономера (мономеров).
(4) Двухстадийная методика.
(a) Алкилирующий агент объединяют с соединением лантаноида в отсутствие сопряженного диенового мономера или в присутствии небольшого количества сопряженного диенового мономера при температуре в диапазоне от приблизительно -20° до ~80°C.
(b) Вышеупомянутую смесь и остальные компоненты загружают к остатку полимеризуемого сопряженного диенового мономера (мономеров) либо постадийно, либо одновременно.
(Ni-содержащее соединение в случае использования такового может быть включено на любой стадии).
В случае получения раствора одного или нескольких ингредиентов катализатора в вышеупомянутых способах вне системы полимеризации предпочтительно будут использовать органический растворитель или носитель. Подходящие органические растворители включают те, которые упоминались ранее.
Получение цис-1,4-полидиена осуществляют в результате проведения полимеризации сопряженного диенового мономера в присутствии каталитически эффективного количества композиции катализатора. Совокупная концентрация катализатора, используемая в массе полимеризации, зависит от взаимодействия различных факторов, таких как степень чистоты ингредиентов, температура полимеризации, желательные скорость полимеризации и степень превращения, желательная молекулярная масса и множество других факторов; в соответствии с этим, конкретная совокупная концентрация катализатора не может быть приведена определенно за исключением, скажем, того требования, что должны быть использованы каталитически эффективные количества соответствующих ингредиентов катализатора. Количество использующегося соединения лантаноида в общем случае находится в диапазоне от ~0,01 до ~2 ммоль, предпочтительно от ~0,02 до ~1 ммоль, а более предпочтительно от ~0,03 до ~0,5 ммоль, при расчете на 100 г сопряженного диенового мономера. Все другие ингредиенты в общем случае добавляют в количествах, которые базируются на количестве соединения лантаноида (смотрите различные соотношения, приведенные ранее).
Полимеризацию предпочтительно проводят в органическом растворителе, то есть в виде растворной или осадительной полимеризации, когда мономер находится в конденсированной фазе. Ингредиенты катализатора предпочтительно солюбилизируют или суспендируют в органической жидкости. Количество мономера (% (масс.)), присутствующего в полимеризационной среде в начале полимеризации, в общем случае находится в диапазоне от ~3 до ~80%, предпочтительно от ~5 до ~50%, а более предпочтительно от ~10% до ~30%. (Полимеризация также может быть проведена при использовании полимеризации в массе, проводимой либо в конденсированной жидкой фазе, либо в газовой фазе).
Вне зависимости от использования периодического, непрерывного или полупериодического способа полимеризацию предпочтительно проводят при перемешивании в диапазоне от умеренного до интенсивного в анаэробных условиях при наличии инертного защитного газа. Температура полимеризации может варьироваться в широких пределах, хотя обычно используют температуру в диапазоне от ~20° до ~90°C; тепло может быть отведено при использовании внешнего охлаждения и/или охлаждения в результате испарения мономера или растворителя. Использующееся давление полимеризации может изменяться в широких пределах, хотя обычно используют давление в диапазоне от приблизительно 0,1 до приблизительно 1 МПа.
В случае полимеризации 1,3-бутадиена, цис-1,4-полибутадиен в общем случае характеризуется значением Mn, согласно определению по методу ГПХ при использовании полистирольных стандартов, находящимся в диапазоне от ~5000 до ~200000 Да, от ~25000 до ~150000 Да или от ~50000 до ~125000 Да. Полидисперсность полимеров в общем случае находится в диапазоне от ~1,5 до ~5,0, обычно от ~2,0 до ~4,0.
Получающиеся в результате полидиены в выгодном случае могут характеризоваться уровнем содержания цис-1,4-соединительных звеньев, равным по меньшей мере ~60%, по меньшей мере приблизительно ~75%, по меньшей мере приблизительно ~90% и даже по меньшей мере приблизительно ~95%, и уровнем содержания 1,2-соединительных звеньев меньшим чем ~7%, меньшим чем ~5%, меньшим чем ~2% и даже меньшим чем ~1%.
Вне зависимости от типа использующегося способа полимеризации в данный момент реакционная смесь обычно называется «полимерным клеем» вследствие относительно высокой концентрации полимера в нем.
Получение концевой функциональности, относящейся к типу, приведенному ранее в формуле (IV), может быть проведено в результате функционализации полимера перед гашением реакции, в выгодном варианте при нахождении его в описывавшемся ранее состоянии полимерного клея. Один способ осуществления данной функционализации включает введение в полимерный клей одного или нескольких ароматических соединений, которые включают группу, способную вступать в реакцию с активными в концевом положении полимерами, а также одну или несколько гидроксильных групп или гидролизуемых групп (то есть один или несколько заместителей OR4), и обеспечение прохождения реакции с таким соединением (соединениями) в концевом положении цепи реакционно-способного полимера. Данный тип соединения здесь и далее в настоящем документе называется соединением, обрывающим цепь.
В случае включения в соединение, обрывающее цепь, более чем одного заместителя OR4, каждый из них может иметься у одного и того же кольца арильной группы, или два и более из них могут иметься у различных колец в пределах арильной группы. В случае наличия у арильной группы трех и более заместителей OR4 все из них могут иметься у одного и того же кольца, два из них могут иметься у одного кольца, тогда как другой (другие) будут иметься у другого кольца (колец), или каждый из них может иметься у различных колец.
Одна предпочтительная группа соединений, обрывающих цепь, включает те, которые имеют арильную группу, имеющую по меньшей мере два заместителя OR4, а в их числе предпочтительными являются те, у которых по меньшей мере два заместителя OR4 имеются у одного и того же кольца арильной группы. В числе последних в особенности предпочтительными являются те соединения, у которых заместители OR4 имеются в положениях 3 и 4 у одного и того же кольца в пределах арильной группы, предпочтительно фенильной группы.
Примеры соединений, которые могут быть использованы для получения функциональности, такой как та, которая продемонстрирована в формуле (IV), включают соединения, описывающиеся следующими общими формулами:
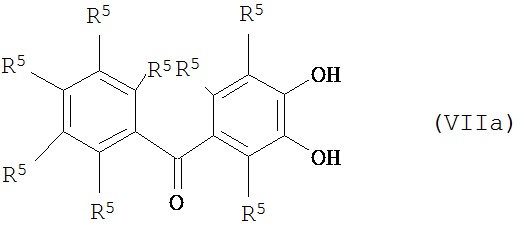




где каждый R5 независимо представляет собой атом водорода, гидроксильную группу, алкоксигруппу или гидрокарбильную группу, предпочтительно алкильную группу, а более предпочтительно C1-С3 алкильную группу; в определенных вариантах реализации каждый R5 может представлять собой Н. В дополнение к вышеизложенному, две и более группы R5 совместно могут образовывать еще одно кольцо, такое как, например, в случае антроны и флавоны:


Как можно видеть в результате сопоставления формул (VIIf) и (VIIg) с приведенной ранее формулой (IV), в концевой функциональности, описывающейся формулой (IV), R6 и часть R3 могут быть соединены таким образом, чтобы совместно с атомом (атомами), к которому каждый присоединен (непосредственно или опосредованно), они образовывали бы кольцо, которое связано или конденсировано с арильной группой R3; иллюстративно это может быть описано общей формулой

где каждую из переменных определяют так же, как и ранее.
Вышеизложенное необходимо рассматривать в качестве примера, а не ограничения. Например, каждое из вышеупомянутых представительных соединений включает соседние гидроксильные группы заместителей (хотя формула (VIIf) не включает одно кольцо, имеющее несоседние гидроксильные заместители), но, как уже описывалось, гидроксильные заместители необязательно должны быть соседними. Конкретно непоказанными в приведенных ранее формулах (VIIa)-(VIIg), но включенными в объем подходящих соединений являются те соединения, которые имеют арильные группы, отличные от фенильных групп, те соединения, которые имеют арильные группы, несвязанные непосредственно с карбонильным атомом С, те соединения, у которых карбонильный атом С связан с атомом S, а не с атомом О, (то есть тиокетоаналоги), те соединения, у которых Z' отличен от одинарной связи, и тому подобное. В случае отличия R3 от фенильной группы гидроксильные группы заместителей могут иметься у одного и того же или различных колец; в случае их наличия у более чем одного кольца, предпочитается, чтобы они были бы по меньшей мере несколько приближенными, то есть чтобы они были бы непосредственно связанными с атомами С кольца, которые разделены не более чем 4, предпочтительно 3, а еще более предпочтительно 2, другими атомами кольца.
Кроме того, как предполагалось ранее, само соединение необязательно должно включать гидроксильные группы, а вместо этого может включать группы, которые являются легко гидролизуемыми до получения гидроксильных групп по завершении реакции. Защищенные соединения в общем случае обладают структурами, подобными тем, которые были приведены ранее в отношении формул (VIIa)-(VIIg), при наличии групп OR вместо некоторых или всех групп ОН. В порядке неограничивающего примера можно сказать то, что защищенное соединение, в общем случае аналогичное соединению, описывающемуся формулой (Vila), при этом каждый R5 представляет собой Н, может быть описано формулой

где каждый R11 независимо представляет собой гидрокарбильную группу, например, линейную или разветвленную алкильную группу. Для защищенных соединений предусматриваются вариации, подобные тем, которые были описаны ранее в отношении гидроксилсодержащих соединений.
Каждое из соединений, описывающихся формулами (VIIa)-(VIIg) и (VIII), включает карбонильную группу. Карбонильные группы обеспечивают наличие удобных точек для реакции с цепями карбанионных полимеров и присоединения к ним. Неограничивающие примеры других потенциально подходящих реакционно-способных групп включают группы альдегида, (тио)кетона, сложного (тио)эфира, сложного ди(тио)эфира, амида, эпоксида, галогенсилана и тому подобного.
Реакция данных типов соединения с предварительно полученным реакционно-способным полимером может быть проведена относительно быстро (в течение периода времени продолжительностью от нескольких минут до нескольких часов) при умеренных температурах (например, в диапазоне от 0° до 75°C).
Количество таких соединений, вводимых в реакцию с предварительно полученными реакционно-способными полимерами, может варьироваться в широких пределах, в значительной степени в зависимости от уровня желательного эффекта, количества использующегося необычного наполнителя (наполнителей), соотношения между количествами частиц обычного и необычного наполнителей и тому подобного. При расчете на количество цепей реакционно-способного полимера (в общем случае определяемое, исходя из эквивалентов инициатора или катализатора) количество соединений, в общем случае соответствующих формулам (VIIa)-(VIIg) и (VIII), может находиться в диапазоне от приблизительно 1:10 до приблизительно 5:4, в общем случае от приблизительно 1:5 до приблизительно 9:8, а обычно от приблизительно 1:2 до приблизительно 1:1.
В определенных вариантах реализации для резервирования некоторых концевых положений реакционно-способных полимеров для реакции с другими функционализующими агентами, которые могут быть добавлены до, после только что обсуждавшихся соединений или совместно с ними, могут быть использованы меньшие количества обрывающих цепь соединений, относящихся к только что описывавшемуся типу; данный тип множественной функционализации можно избежать по меньшей мере в некоторой степени в результате использования обсуждавшихся ранее функциональных инициаторов. Кроме того, по меньшей мере некоторые варианты реализации полимеров, содержащих функциональности, описывающиеся формулами (IV) и (IVb), а также защищенные аналоги, могут демонстрировать превосходную степень взаимодействия с техническим углеродом и диоксидом кремния, что, таким образом, устраняет потребность в реакциях множественной функционализации.
В случае неиспользования вышеупомянутого типа соединения, обрывающего цепь, но включения в макромолекулу по меньшей мере одного функционализующего звена, произведенного из одного или обоих соединений, выбираемых из инициатора и мономера, относящегося к типу, описывающемуся формулой (V), дополнительная функционализация может представлять собой результат обрыва цепи гетероатомсодержащим соединением, включающим нижеследующее, но не ограничивающимся только этим: Sn, Si и N. Конкретные примеры альтернативных или дополнительных соединений, обрывающих цепь, включают 1,3-диметил-2-имидазолидинон (ДМИ), 3-бис(триметилсилил)аминопропилметилдиэтоксисилан (АПМДЭОС), а также те соединения, которые описываются в патентах США №№3109871, 4647625, 4677153, 5109907 и 6977281 и ссылках, процитированных в данных патентах, и позднейших публикациях, их цитирующих. Данный тип функционализации описывается далее в примерах 73-75.
В данный момент получающийся в результате полимер включает один или несколько типов полиеновых мономерных фрагментов и по меньшей мере одно функционализующее звено, которое включает арильную группу, имеющую по меньшей мере один непосредственно связанный заместитель OR4. Функционализующее звено (звенья) может быть произведено из инициирующего соединения, мономера (мономеров) или соединения, обрывающего цепь. В определенных аспектах может быть введено более чем одно функционализующее звено, и они могут представлять собой результат включения нескольких мономерных фрагментов, инициатора плюс один или несколько мономерных фрагментов, группы, обрывающей цепь, плюс один или несколько мономерных фрагментов или всех трех вариантов.
Характер фрагмента R4 заместителя (то есть то, будет ли это атом Н или защитная группа) зависит от происхождения звена, частью которого он является. Звенья, произведенные из инициатора и/или мономеров, будут иметь группы OR, в то время как звенья, произведенные из соединения, обрывающего цепь, могут иметь группы OR или ОН. Для промотирования максимальной степени взаимодействия с частицами наполнителя (в случае использования полимера в качестве части каучуковой композиции) желательным обычно является обеспечение превращения в атомы Н большинства, предпочтительно всех, фрагментов R. Описывающиеся далее стадии переработки (включающие гашение реакции) могут оказаться достаточными для гидролиза по меньшей мере некоторых из фрагментов R, что, таким образом, приводит к получению одного или нескольких гидроксильных заместителей у одной или нескольких арильных групп в пределах полимера. В альтернативном варианте, может быть использована отдельная стадия реакции, предназначенная для промотирования обширного, предпочтительно полного, гидролиза; исходя из примера методики, использующейся в нескольких из приведенных далее примеров, специалист в соответствующей области техники может представить себе и другие потенциально эффективные реакции. Кроме того, специалист в соответствующей области техники должен понимать, что группы OR или ОН, присутствующие либо в группе R1, либо в группе R3, либо в группе R6, либо где-либо еще, могут претерпевать дополнительную реакцию во время данной переработки и/или составления смеси с одним или несколькими типами дисперсных наполнителей (описывающихся далее).
Гашение реакции может быть проведено в результате перемешивания полимера и соединения, содержащего активный водород, такого как спирт или кислота, в течение вплоть до приблизительно 120 минут при температурах в диапазоне от приблизительно 25° до приблизительно 150°C.
Растворитель из подвергнутого гашению реакции полимерного клея может быть удален по обычным методикам, таким как высушивание в барабанной сушилке, высушивание в экструдере, вакуумное высушивание и тому подобное, что может быть объединено с коагулированном водой, спиртом или водяным паром, термическим удалением растворителя и тому подобным; в случае проведения коагулирования желательным может оказаться высушивание в печи.
Получающийся в результате полимер может быть использован в наполненной смеси протектора или может быть перемешан с любым обычно использующимся каучуком протекторной смеси, включающим натуральный каучук и/или нефункционализованные синтетические каучуки, такие как, например, один или несколько гомо- и интерполимеров, которые включают только произведенные из полиенов мономерные звенья, (например, поли(бутадиен), поли(изопрен) и сополимеры, включающие бутадиен, изопрен и тому подобное), СБК, бутилкаучук, неопрен, ЭПК, ЭПДК, НБК, силиконовый каучук, фторэластомеры, этилен/акриловый каучук, ЭВА, эпихлоргидриновые каучуки, хлорированные полиэтиленовые каучуки, хлорсульфированные полиэтиленовые каучуки, гидрированный нитрильный каучук, тетрафторэтилен/пропиленовый каучук и тому подобное. В случае перемешивания функционализованного полимера (полимеров) с обычным каучуком (каучуками) количества могут варьироваться в диапазоне от приблизительно 5 до приблизительно 99% от совокупного каучука, при этом обычный каучук (каучуки) составит остаток совокупного каучука. Минимальное количество в значительной степени зависит от желательного уровня уменьшения гистерезиса.
Эластомерные смеси обычно наполняют до объемной доли, которая представляет собой совокупный объем добавленного наполнителя (наполнителей), поделенный на совокупный объем эластомерной смеси, зачастую равной приблизительно 25%; соответственно, типичные (объединенные) количества армирующих наполнителей находятся в диапазоне приблизительно от 30 до 100 ч./сто ч. каучука.
Потенциально подходящие материалы технического углерода включают нижеследующие, но не ограничиваются только этими: разновидности печной сажи, разновидности канальной газовой сажи и разновидности ламповой сажи. Говоря более конкретно, примеры разновидностей технического углерода включают разновидности сверхизносостойкой печной сажи, разновидности износостойкой печной сажи, разновидности быстрошприцуемой печной сажи, разновидности высокодисперсной печной сажи, разновидности высокоизносостойкой печной сажи, разновидности полуусиливающей печной сажи, разновидности среднеобрабатываемой канальной газовой сажи, разновидности труднообрабатываемой канальной газовой сажи, разновидности проводящей канальной газовой сажи и разновидности ацетиленовой сажи; могут быть использованы смеси двух и более из них. Предпочтительными являются разновидности технического углерода (EMSA), характеризующиеся площадью удельной поверхности, равной по меньшей мере 20 м2/г, предпочтительно по меньшей мере приблизительно 35 м2/г; значения площади удельной поверхности могут быть определены по методу ASTM D-1765. Разновидности технического углерода могут иметь гранулированную форму или представлять собой негранулированную хлопьевидную массу, хотя негранулированный технический углерод может оказаться предпочтительным для использования в определенных смесителях.
Исторически использовавшееся количество технического углерода доходило вплоть до ~50 массовых частей (м.ч.) на 100 частей полимера (ч./сто ч. каучука), при этом обычное количество находится в диапазоне от ~5 до ~40 ч./сто ч. каучука. В случае определенных маслонаполненных рецептур количество технического углерода будет еще большим, например, имеющим порядок ~80 ч./сто ч. каучука.
В качестве наполнителя также широко используют аморфный диоксид кремния (SiO2). Разновидности диоксида кремния обычно получают в результате проведения химической реакции в воде, из которой их осаждают в виде ультратонкодисперсных сферических частиц, которые ассоциируются в прочные агрегаты, которые, в свою очередь, объединяются в менее прочные агломераты. Площадь удельной поверхности представляет собой надежную меру армирующего характера различных разновидностей диоксида кремния; при этом в общем случае подходящими считаются определенные по методу БЭТ (смотрите: Brunauer et al., J. Am. Chem. Soc., vol. 60, p.309 et seq.) площади удельной поверхности, меньшие чем 450 м2/г, обычно находящиеся в диапазоне от ~32 до ~400 м2/г и типично от ~100 до ~250 м2/г. Коммерческие поставщики диоксида кремния включают PPG Industries, Inc. (Питтсбург, Пенсильвания), Grace Davison (Балтимор, Мэриленд), Degussa Corp. (Парсиппани, Нью-Джерси), Rhodia Silica Systems (Крэнбери, Нью-Джерси) и J.М. Huber Corp. (Эдисон, Нью-Джерси).
В случае использования в качестве армирующего наполнителя диоксида кремния обычно будут добавлять аппрет, такой как силан, что обеспечит хорошие перемешивание и взаимодействие с эластомером (эластомерами). В общем случае количество силана, которое добавляют, находится в диапазоне приблизительно от 4 до 20% при расчете на массу наполнителя диоксида кремния, присутствующего в смеси. Аппреты могут описываться общей формулой A-T-G, в которой А представляет собой функциональную группу, способную физически и/или химически связываться с группой на поверхности наполнителя диоксида кремния (например, с поверхностными силанольными группами); Т представляет собой соединительное звено в виде углеводородной группы; a G представляет собой функциональную группу, способную связываться с эластомером (например, через серосодержащее соединительное звено). Такие аппреты включают органосиланы, в частности, полисульфированные алкоксисиланы (смотрите, например, патенты США №№3873489, 3978103, 3997581, 4002594, 5580919, 5583245, 5663396, 5684171, 5684172, 5696197 и тому подобное) или полиорганосилоксаны, имеющие вышеупомянутые функциональности G и А. Для уменьшения количества использующегося силана может быть использовано добавление технологической добавки; смотрите, например, патент США №6525118 в отношении описания жирнокислотных сложных эфиров Сахаров, использующихся в качестве технологических добавок. Дополнительные наполнители, использующиеся в качестве технологических добавок, включают минеральные наполнители, такие как глина (водный силикат алюминия), тальк (водный силикат магния) и слюда, а также неминеральные наполнители, такие как мочевина и сульфат натрия. Предпочтительные разновидности слюды в принципе содержат оксид алюминия, диоксид кремния и поташ несмотря на возможность пригодности также и других вариантов. Дополнительные наполнители могут быть использованы в количестве, доходящем вплоть до приблизительно 40 ч./сто ч. каучука, обычно вплоть до приблизительно 20 ч./сто ч. каучука.
Диоксид кремния обычно используют в количествах, доходящих вплоть до ~100 ч./сто ч. каучука, обычно находящихся в диапазоне от ~5 до ~80 ч./сто ч. каучука. Подходящий верхний диапазон ограничивается высокой вязкостью, которую такие наполнители могут придавать. В случае использования также и технического углерода количество диоксида кремния может быть уменьшено до всего лишь ~1 ч./сто ч. каучука; по мере уменьшения количества диоксида кремния могут быть использованы и меньшие количества технологических добавок плюс силана в случае использования таковых.
В сочетании с техническим углеродом и/или диоксидом кремния или вместо них предпочтительно используют один или несколько необычных наполнителей, характеризующихся относительно высокими межфазными свободными энергиями, то есть значениями поверхностной свободной энергии в воде (γpl). Термин «относительно высокий» может быть определен или охарактеризован по различными способами, например, больший, чем значение на межфазной поверхности вода-воздух, предпочтительно в несколько раз (например, по меньшей мере 2х, по меньшей мере 3х или даже по меньшей мере 4х) больший, чем данное значение; по меньшей мере в несколько раз (например, по меньшей мере 2х, по меньшей мере 3х, по меньшей мере 4х, по меньшей мере 5х, по меньшей мере 6х, по меньшей мере 7х, по меньшей мере 8х, по меньшей мере 9х или даже по меньшей мере 10х) больший, чем значение γpl для аморфного диоксида кремния; в абсолютных величинах, например, по меньшей мере ~300, по меньшей мере ~400, по меньшей мере ~500, по меньшей мере ~600, по меньшей мере ~700, по меньшей мере ~750, по меньшей мере ~1000, по меньшей мере ~1500 и по меньшей мере ~2000, мДж/м2; в диапазонах, например, от ~300 до ~5000 мДж/м2, от ~350 до ~4000 мДж/м2, от ~400 до ~5000 мДж/м2, от ~450 до ~4000 мДж/м2, от ~500 до ~5000 мДж/м2 и различные поддиапазоны в пределах вышеупомянутых и/или другие комбинации верхних и нижних значений; и тому подобное.
Неограничивающие примеры встречающихся в природе материалов, характеризующихся относительно высокими межфазными свободными энергиями, включают F-апатит, гетит, гематит, цинкит, тенорит, гиббсит, кварц, каолинит, все формы пирита и тому подобное. Данным типом высокой межфазной свободной энергии также могут характеризоваться и определенные синтетические сложные оксиды.
Вышеупомянутые типы материалов обычно являются более плотными в сопоставлении либо с техническим углеродом, либо с аморфным диоксидом кремния; таким образом, замещение конкретной массы технического углерода или диоксида кремния равной массой необычного наполнителя в результате обычно будет приводить к получению намного меньшего объема совокупного наполнителя, присутствующего в заданной смеси. В соответствии с этим, замещение обычно проводят при расчете на равный объем в противоположность расчету на равную массу.
В общем случае от ~5 до ~60% материала (материалов) обычного дисперсного наполнителя могут быть замещены приблизительно эквивалентным (от ~0,8х до ~1,2х) объемом частиц необычного наполнителя. В определенных вариантах реализации достаточным является замещение от ~10 до ~58% материала (материалов) обычного дисперсного наполнителя приблизительно эквивалентным (от ~0,85х до ~1,15х) объемом частиц другого наполнителя; в других вариантах реализации надлежащим является замещение от ~15 до ~55% материала (материалов) обычного дисперсного наполнителя приблизительно эквивалентным (от ~0,9х до ~1,1х) объемом частиц другого наполнителя; в других еще вариантах реализации предпочтительным может оказаться замещение от ~18 до ~53% материала (материалов) обычного дисперсного наполнителя приблизительно эквивалентным (от ~0,95х до ~1,05х) объемом частиц другого наполнителя.
Проблему неравенства массы может оказаться возможным устранить или сгладить в результате использования нестандартных частиц. Например, можно предусмотреть по существу пустотелые частицы одного или нескольких типов необычных наполнителей, а также относительно легкие частицы, имеющие покрытие, обеспечивающее наличие поверхности, которая включает один или несколько типов соединений необычных наполнителей.
Частицы необычных наполнителей в общем случае могут иметь приблизительно тот же самый размер, что и обычные наполнители, использующиеся в смесях. Другими словами, не требуются ни чрезмерно крупные частицы, такие как те, которые используются в вышеупомянутом патенте США №5066702, ни чрезмерно мелкие частицы, такие как те, которые используются в вышеупомянутом патенте США №6972307. В общем случае как в целях армирования, так и для обеспечения доступности большого количества частиц на поверхности протектора предпочтительными являются относительно небольшие частицы.
Также могут быть добавлены и другие обычные добавки к каучукам. Они включают, например, технологические масла, пластификаторы, противостарители, такие как антиоксиданты и противоозоностарители, отвердители и тому подобное.
Все ингредиенты могут быть перемешаны при использовании стандартного оборудования, такого как, например, смесители Banbury или Brabender. Обычно перемешивание проводят в две или более стадии. Во время первой стадии (также известной под наименованием стадии маточной смеси) перемешивание обычно начинают при температурах в диапазоне от ~120° до ~130°C, и температуру увеличивают вплоть до достижения так называемой температуры каплепадения, обычно равной ~165°C.
В случае включения в рецептуру наполнителей, отличных от технического углерода, для отдельного добавления силанового компонента (компонентов) зачастую используют отдельную стадию перевальцевания. Данную стадию зачастую проводят при температурах, подобных тем, которые используют на стадии маточной смеси, хотя зачастую и несколько меньших их, то есть линейно изменяющихся от ~90°C до температуры каплепадения ~150°C.
Армированные каучуковые смеси обычно отверждают при использовании от приблизительно 0,2 до приблизительно 5 ч./сто ч. каучука одного или нескольких известных вулканизаторов, таких как, например, системы отверждения на серной или пероксидной основе. Для получения общего описания подходящих вулканизаторов заинтересованный читатель отсылается к обзору, такому как тот, который представлен в публикации Kirk-Othmer, Encyclopedia of Chem. Tech., 3d ed., (Wiley Interscience, New York, 1982), vol. 20, pp.365-468. Вулканизаторы, ускорители и тому подобное добавляют на стадии конечного перемешивания. Во избежание возникновения нежелательной подвулканизации и/или преждевременного начала вулканизации данный этап перемешивания зачастую проводят при более низких температурах, например, начиная со значений в диапазоне от ~60°C до ~65°C и не выходя за значения в диапазоне от ~105°C до ~110°C.
После этого составленную смесь перерабатывают (например, вальцуют), получая листы, затем профилируют в форме любого из широкого ассортимента компонентов, а после этого вулканизуют, что обычно осуществляют при температуре, превышающей наивысшие температуры, использующиеся во время стадий перемешивания, на величину в диапазоне от ~5°C до ~15°C, наиболее часто равной приблизительно 170°C.
Следующие далее неограничивающие иллюстративные примеры представляют подробное описание условий и материалов, которые могут оказаться подходящими для использования в практике только что описывавшегося изобретения.
ПРИМЕРЫ
Во всех примерах при всех получениях использовали просушенные стеклянные емкости, предварительно запечатанные извлекаемыми мембранными прокладками и перфорированными крончатыми крышками, с продувкой при положительном давлении N2.
Все испытания по методу ядерного магнитного резонанса (ЯМР) проводили на спектрометре Varian™ 300 MHz (Varian, Inc.; Пало-Альто, Калифорния).
Данные, соответствующие «связанному каучуку», определяли при использовании методики, описывавшейся в публикации J.J. Brennan et al., Rubber Chem. and Tech., 40, 817 (1967).
Испытание на хладотекучесть проводили при использовании испытательного прибора Scott™. Образцы получали в результате прессования из расплава в течение 20 минут 2,5 г полимера в форме при 100°C с использованием предварительно нагретого пресса. Получающимся в результате цилиндрическим образцам, которые имели однородную толщину ~12 мм, перед удалением из формы давали возможность охладиться до комнатной температуры. Образцы по отдельности помещали под груз гири разновеса в 5 кг. Испытания проводили в течение ~30 мин для образцов СБК и ~8 мин для полибутадиеновых образцов (согласно измерению от момента времени устранения нагрузки), при этом толщины образца регистрировали в зависимости от времени. Толщину образца по истечении надлежащего времени (~30 мин или ~8 мин) в общем случае рассматривают в качестве приемлемого показателя стойкости к хладотекучести.
Значения вязкости по Муни (ML1+4) определяли при использовании вискозиметра Муни Alpha Technologies™ (большой ротор) с использованием времени прогревания в одну минуту и времени работы в четыре минуты; механические свойства при растяжении определяли при использовании стандартной методики, описывавшейся в документе ASTM-D412; данные по эффекту Пейна (ΔG', то есть разница между G’ при деформации 0,25% и при деформации 14%) и гистерезису (tan δ) получали в динамических экспериментах, проведенных при 60°C и 10 Гц (развертка по деформации) и при деформации 2% и 10 Гц (развертка по температуре). Что касается механических свойств при растяжении, то MY представляет собой модуль упругости при относительном удлинении Y %, Tb представляет собой прочность при растяжении в момент разрыва, а Eb представляет собой процент относительного удлинения в момент разрыва.
А. Примеры 1-33 (агенты, обрывающие цепь)
В данных примерах использовали стирол (33% в гексане), гексан, н-бутиллитий (1,60 моль/л в гексане), 2,2-бис(2'-тетрагидрофурил)пропан (раствор в гексане с концентрацией 1,6 моль/л, хранившийся над СаН2) и раствор в гексане 2,6-ди-трет-бутил-4-метилфенола (БГТ).
Коммерчески доступные реагенты и исходные материалы включали нижеследующие, все из которых приобретали в компании Sigma-Aldrich Co. (Сент-Луис, Миссури) и использовали без дополнительной очистки, если только в конкретном примере не будет указано другого: 3,4-дигидроксибензальдегид, 3,4-дигидроксибензофенон, имидазол, трет-бутил(хлор)диметилсилан, диэтиловый эфир, NH4Cl, MgSO4 (безводный), ТГФ, этилацетат, метилалюмоксан (МАО), диизобутилалюминийгидрид, ГМИ, диэтилалюминийхлорид, 4,4'-бис(диэтиламино)бензофенон (ДЭАБ) и фторид тетрабутиламмония (ФТБА).
Данные по испытаниям в большинстве примеров получали для наполненных композиций, полученных в соответствии с рецептурами, продемонстрированные далее в
Таблице 1а (диоксид титана, рутил),
Таблице 1b (технический углерод и гидроксид алюминия),
Таблице 1с (технический углерод и диоксид титана) и
Таблице 1d (технический углерод).
Диоксид титана, использующийся в данных рецептурах, представлял собой TiO2, стабилизированный оксидом алюминия, Tronox™ CR-834, характеризующийся размером частиц ~0,17 мкм и относительной плотностью ~4,2, (Tronox Inc.; Оклахома-Сити, Оклахома), а использовавшийся гидроксид алюминия представлял собой частицы Аl(ОН)3 Hydral™ PGA-HD, характеризующиеся срединным диаметром частиц ~1 мкм и плотностью 2,42 г/см3, (Almatis, Inc.; Литсдейл, Пенсильвания). В данных (и подобных последующих таблицах) в качестве антиоксиданта используют N-фенил-N'-(1,3-диметилбутил)-п-фенилендиамин (6PPD), а в качестве ускорителей используют 2,2'-дитиобис(бензотиазол) (MBTS), N-трет-бутилбензотиазол-2-сульфенамид (ТББС) и N,N'-дифенилгуанидин (ДФГ). Маслом для наполнения являлось темное масло, которое содержит относительно небольшое количество полициклических ароматических соединений (ПЦА).
Пример 1: Синтез 3,4-бис(трет-бутилдиметилсилилокси)бензофенона
В сухую колбу в атмосфере азота загружали ~6,0 г 3,4-дигидроксибензофенона, ~6,3 г триэтиламина, ~0,14 г 4-(диметиламино)пиридина и 30 мл ДМФА. После этого в покапельном режиме добавляли раствор ~9,3 г трет-бутил(хлор)диметилсилана в 30 мл ДМФА.
Реакционную смесь перед выливанием в ~100 мл гексана и ~30 мл насыщенного раствора NH4Cl в течение ~4 часов перемешивали при комнатной температуре. Органическую фазу три раза промывали порциями воды в 50 мл и высушивали при использовании безводного MgSO4.
После удаления растворителя остаток разделяли по методу силикагелевой колоночной флэш-хроматографии при использовании в качестве элюента смеси гексан/этилацетат (85:15, (об./об.)). Получали приблизительно 11,5 г (93%-ный выход) белого твердого вещества. Спектроскопический анализ по методам протонного и13С ЯМР подтвердил, что данным продуктом является 3,4-бис(трет-бутилдиметилсилилокси)бензофенон(БТБДМСБФ).
Пример 2: Синтез 3,4-бис(трет-бутилдиметилсилилокси)бензальдегида
В сухую колбу в атмосфере азота загружали ~10,0 г 3,4-дигидроксибензальдегида, ~16,1 г триэтиламина, ~0,35 г 4-(диметиламино)пиридина и 60 мл ДМФА. После этого в покапельном режиме добавляли раствор ~24,0 г трет-бутил(хлор)диметилсилана в 60 мл ДМФА.
Реакционную смесь перед выливанием в ~200 мл гексана и ~100 мл насыщенного раствора NH4Cl в течение ~4 часов перемешивали при комнатной температуре. Органическую фазу три раза промывали порциями воды в 100 мл и высушивали при использовании безводного MgSO4.
После удаления растворителя остаток разделяли по методу флэш-хроматографии на силикагелевой колонке при использовании в качестве элюента смеси гексан/этилацетат (95:5, (об./об.)). Получали приблизительно 25,5 г (96%-ный выход) белого твердого вещества. Спектроскопический анализ по методам протонного и13С ЯМР подтвердил то, что данным продуктом является 3,4-бис(трет-бутилдиметилсилилокси)бензальдегид (БТБДМСБА).
Примеры 3-5: Стирол/бутадиеновые сополимеры
В продуваемый при использовании N2 реактор, снабженный перемешивающим устройством, добавляли 1,39 кг гексана, 0,37 кг раствора стирола и 2,27 кг раствора бутадиена (21,6% (масс.) в гексане). В реактор загружали 3,19 мл раствора н-бутиллития с последующим добавлением 1,13 мл раствора 2,2-бис(2'-тетрагидрофурил)пропана. Рубашку реактора нагревали до 50°C и полимеризации давали возможность продолжаться в течение ~75 минут. Перед гашением реакции в изопропаноле, содержащем 2,6-ди-трет-бутил-4-метилфенол, а после этого высушиванием в барабанной сушилке полимерный клей охлаждали до комнатной температуры. Далее это обозначают как пример 3.
Вышеупомянутую полимеризацию по существу повторяли; однако перед коагулированном в реактор добавляли 6,0 мл раствора БТБДМСБФ (из примера 1) в гексане с концентрацией 0,83 моль/л и перед обеспечением охлаждения полимерного клея до комнатной температуры его перемешивали в течение еще ~30 минут при 50°C. Полимерный клей переводили в колбу, продуваемую при использовании N2, в которую добавляли ~130 мл раствора ФТБА (0,23 моль/л в ТГФ). Колбу в течение ~4 часов вращали в водяной бане при 25°C с последующими еще ~30 минутами в водяной бане при 50°C. После этого содержимое колбы коагулировали и высушивали в барабанной сушилке так, как это описывалось ранее. Далее это обозначают как пример 4.
Полимеризацию еще раз по существу повторяли за исключением использования ~2,24 кг раствора бутадиена с концентрацией 21,9% (масс.). Перед коагулированном в реактор добавляли 4,9 мл раствора БТБДМСБА (из примера 2) в гексане с концентрацией 1,0 моль/л и полимерный клей перерабатывали подобно полимерному клею из примера 4. Далее это обозначают как пример 5.
Свойства данных стирол/бутадиеновых сополимеров суммарно представлены в следующей далее таблице. Молекулярные массы определяли по методу ГПХ при использовании стандартов СБК, а уровни содержания 1,2-соединительных звеньев определяли по методу спектроскопии1Н ЯМР.
Примеры 6-11: Наполненные композиции
Полимеры из примеров 3-5 использовали совместно с рецептурой, продемонстрированной ранее в таблице 1а, для получения композиций, из которых получали вулканизаты, обозначенные далее как примеры 6-8.
Те же самые полимеры использовали совместно с рецептурой, продемонстрированной ранее в таблице 1b, для получения композиций, из которых получали вулканизаты, обозначенные далее как примеры 9-11.
Перемешивание каждой смеси (то есть наполненной композиции) проводили при использовании закрытого резиносмесителя Brabender™ на 65 г. После вулканизации при высоком давлении и высокой температуре определяли физические свойства смесей, а результаты суммарно представлены далее в таблице 3.
В сопоставлении с примерами 6 и 9 (нефункционализованные контрольные образцы) соответственно примеры 7-8 и 10-11 демонстрируют значительно повышенные значения твердости по Шору А при комнатной температуре, существенно повышенные значения эластичности по отскоку при 50°C (что в общем случае соответствует уменьшению гистерезиса), улучшенные пределы прочности при растяжении при комнатной температуре, пониженные значения модуля упругости при -20°C (что в общем случае соответствует лучшему сцеплению со льдом), повышенный модуль упругости при 60°C и высокой деформации (что в общем случае соответствует лучшим характеристикам управляемости) и значительно уменьшенный тангенс потерь при 60°C (что в общем случае соответствует уменьшению гистерезиса).
Примеры 12-14: Функционально инициированные стирол/бутадиеновые сополимеры
В продуваемый при использовании N2 реактор, снабженный перемешивающим устройством, добавляли 1,39 кг гексана, 0,37 кг раствора стирола и 2,27 кг раствора бутадиена (21,6% (масс.) в гексане). В реактор загружали 1,62 мл раствора ГМИ в толуоле с концентрацией 3,0 моль/л и 3,19 мл раствора н-бутиллития с последующим добавлением 1,13 мл раствора 2,2-бис(2'-тетрагидрофурил)пропана. Рубашку реактора нагревали до 50°C и полимеризации давали возможность продолжаться в течение ~60 минут. Перед гашением реакции и высушиванием в барабанной сушилке так, как это описывалось ранее, полимерный клей охлаждали до комнатной температуры. Далее это обозначают как пример 12.
Вышеупомянутую полимеризацию по существу повторяли; однако полимеризации давали возможность продолжаться в течение ~75 минут при 50°C, а перед коагулированном в реактор добавляли 6,0 мл раствора БТБДМСБФ (из примера 1) в гексане с концентрацией 0,83 моль/л и перед обеспечением охлаждения полимерного клея до комнатной температуры его перемешивали в течение еще ~30 минут при 50°C. Полимерный клей переводили в колбу, продуваемую при использовании N2, в которую добавляли ~130 мл раствора ФТБА (0,23 моль/л в ТГФ). Колбу в течение ~4 часов вращали в водяной бане при 25°C с последующими еще ~30 минутами в водяной бане при 50°C. После этого содержимое колбы коагулировали и высушивали в барабанной сушилке так, как это описывалось ранее. Далее это обозначают как пример 13.
Полимеризацию и переработку, описывавшуюся в связи с примером 13, еще раз по существу повторяли за исключением использования ~ 2,24 кг раствора бутадиена с концентрацией 21,9% (масс.), а перед коагулированном в реактор добавляли 4,9 мл раствора БТБДМСБА (из примера 2) в гексане с концентрацией 1,0 моль/л. Далее это обозначают как пример 14.
Свойства данных сополимеров суммарно представлены в следующей далее таблице.
Примеры 15-20: Наполненные композиции
Полимеры из примеров 12-14 использовали совместно с рецептурой, продемонстрированной ранее в таблице 1с, для получения композиций, из которых получали вулканизаты, обозначенные далее как примеры 15-17 соответственно.
Те же самые полимеры использовали совместно с рецептурой, продемонстрированной ранее в таблице 1b, для получения других композиций, из которых получали вулканизаты, обозначенные далее как примеры 18-20.
Перемешивание, вулканизацию и испытания для данных наполненных композиций проводили подобно тому, что было описано ранее в связи с примерами 6-11. Физические свойства суммарно представлены далее в таблице 5.
Данные из таблицы 5 демонстрируют тенденции, подобные тем, которые наблюдали ранее в связи с таблицей 3.
Пример 21: Синтез 3,4-бис(триметилсилилокси)бвнзалъдегида
В сухую колбу в атмосфере азота загружали ~4,83 г 3,4-дигидроксибензальдегида, ~4,80 г имидазола и 50 мл ТГФ. Раствор перед добавлением в покапельном режиме ~43,8 мл раствора н-бутиллития с концентрацией 1,6 моль/л охлаждали до - 78°C.
Реакционную смесь перед добавлением в покапельном режиме ~7,61 г хлортриметилсилана нагревали до комнатной температуры, а после этого реакционную смесь в течение ~2 часов перемешивали при комнатной температуре.
После осаждения LiCl на дно емкости в приведенных далее примерах 26-27 сразу же использовали ~0,33 моль/л 3,4-бис(триметилсилилокси)бензальдегида (БТМСБА) в качестве соединения, обрывающего цепь.
Примеры 22-27: Цис-1,4-полибутадиены
Катализаторы получали в результате добавления в просушенные, запечатанные, продутые при использовании N2 колбы следующих далее ингредиентов:
Смеси перед использованием в следующих далее полимеризациях в течение 15 минут подвергали старению при комнатной температуре.
В продуваемый при использовании N2 реактор, снабженный перемешивающим устройством, добавляли 1,22 кг гексана и 2,86 кг раствора бутадиена (21,4% (масс.) в гексане). В реактор загружали смесь катализатора А, а температуру рубашки выставляли на 60°C. Полимеризации давали возможность продолжаться в течение ~60 минут. Перед гашением реакции и высушиванием в барабанной сушилке так, как это описывалось ранее, полимерный клей охлаждали до комнатной температуры. Далее это обозначают как пример 22.
Вышеупомянутую полимеризацию по существу повторяли еще три раза. В первый раз использовали 1,32 кг гексана, 2,76 кг раствора бутадиена (22,2% (масс.) в гексане), а также смесь катализатора В. Далее это обозначают как пример 23.
Во второй раз использовали смесь катализатора С, а перед коагулированном добавляли 0,5 моль/л ДЭАБ в толуоле (1,25 мл на 100 г полимерного клея) и реакции давали возможность протекать в течение ~30 минут в водяной бане при 65°C. Далее это обозначают как пример 24.
В третий раз использовали 1,32 кг гексана, 2,76 кг раствора бутадиена (22,2% (масс.) в гексане), а также смесь катализатора D. Три раздельные порции полимерного клея переводили в колбы и перед гашением реакции и высушиванием в барабанной сушилке так, как это описывалось ранее, подвергали переработке следующим далее образом.
Пример 25: Добавляли 1,0 моль/л БТБДМСБА (из примера 2) в гексане (0,3 мл на 100 г полимерного клея) и перед добавлением раствора ФТБА (1,0 моль/л в ТГФ, 0,7 мл на 100 г полимерного клея) и вращением колбы в течение ~4 часов в водяной бане при 25°C реакции с клеем давали возможность протекать в течение ~30 мин в водяной бане при 65°C.
Пример 26: Добавляли 0,33 моль/л БТМСБА (из примера 21) в гексане (0,91 мл на 100 г полимерного клея) и реакции с клеем давали возможность протекать в течение ~30 мин в водяной бане при 65°C.
Пример 27: Добавляли 0,33 моль/л БТМСБА (из примера 21) в гексане (0,91 мл на 100 г полимерного клея) и перед добавлением раствора HCl в изопропаноле с концентрацией 1,0 моль/л (1,0 мл на 100 г полимерного клея) и вращением колбы в течение ~30 мин в водяной бане при 50°C реакции с клеем давали возможность протекать в течение ~30 мин в водяной бане при 65°C.
Свойства данных полибутадиенов суммарно представлены в следующей далее таблице. Как и прежде, молекулярные массы определяли по методу ГПХ, но микроструктуру полимеров определяли в результате проведения ИК-спектроскопического анализа. Значения стойкости к хладотекучести определяли так же, как и ранее.
Примеры 28-33: Наполненные композиции
Полимеры из примеров 22-27 использовали совместно с рецептурой, продемонстрированной ранее в таблице 1d, для получения композиций, из которых получали вулканизаты, обозначенные далее как примеры 28-33 соответственно.
Перемешивание, вулканизацию и испытания для данных наполненных композиций проводили подобно тому, что было описано ранее в связи с примерами 6-11, при этом физические свойства суммарно представлены в следующей далее таблице.
Данные из таблицы 8, помимо прочего, демонстрируют то, что в сопоставлении с вулканизатами, полученными из нефункционализованных контрольных полимеров (примеры 28-29) или даже сравнительного функционализованного полимера (пример 30), вулканизаты, полученные при использовании цис-1,4-полибутадиенов, функционализованных арильными группами, которые включают гидроксильные заместители, непосредственно связанные с соседними атомами углерода кольца, (примеры 31-33), характеризуются довольно существенным уменьшением значения tan δ при 50°C (что соответствует уменьшению гистерезиса).
В. Примеры 34-50 (инициаторы)
В данных примерах использовали растворы бутадиена (все в гексане), стирол (33% в гексане), гексан, н-бутиллитий (н-BuLi, 1,60 моль/л в гексане), 2,2-бис(2'-тетрагидрофурил)пропан (раствор в гексане с концентрацией 1,6 моль/л, хранившийся над СаН2) и раствор бутилированного гидрокситолуола (БГТ) в гексане.
Коммерчески доступные реагенты и исходные материалы включали нижеследующие, все из которых использовали без дополнительной очистки, если только в конкретном примере не было указано другого:
от компании Sigma-Aldrich Co. - 3,4-дигидроксибензальдегид (97%), 1,3-пропандитиол (99%), моногидрат п-толуолсульфоновой кислоты (98,5%), этилацетат (99,5%) и 4-ди(метиламино)пиридин (ДМАП, 99%) и
от компании ACROS Organics - трет-бутилдиметилсилилхлорид (98%) и ФТБА (1 моль/л в ТГФ, содержащем ~ 5% воды).
Испытания проводили для наполненных композиций, полученных в соответствии с рецептурами, продемонстрированными в таблицах 9а (рецептура, использующая в качестве дисперсного наполнителя только диоксид кремния) и 9b (рецептура, использующая в качестве дисперсного наполнителя только технический углерод).
Пример 34: 3,4-ди(трет-бутилдиметилсилокси)фенил-1,3-дитиан
В просушенную колбу объемом 500 мл, включающую якорь магнитной мешалки и дефлегматор, добавляли 8,2 г 3,4-дигидроксибензальдегида, 1,6 г моногидрата п-толуолсульфоновой кислоты и 100 мл ТГФ с последующим добавлением 6 мл 1,3-пропандитиола в 30 мл ТГФ. Данную смесь выдерживали при кипячении в атмосфере азота в течение ~12 часов. После охлаждения до комнатной температуры смесь отфильтровывали, при этом фильтрат перед высушиванием над безводным MgSO4 промывали два раза насыщенным раствором NaHCO3 (100 мл) и один раз насыщенным раствором NaCl (100 мл). Растворитель выпаривали, а остаток очищали по методу силикагелевой колоночной хроматографии при использовании в качестве элюирующего растворителя 50%-ного этилацетата в гексане. Получали маслянистый продукт (13,3 г, 99 %-ный выход) и по методам1H и13С ЯМР в CDCl3 подтвердили наличие структуры в виде

В просушенную колбу объемом 500 мл, включающую якорь магнитной мешалки, добавляли 13,3 г данного дитиана, 0.5 г ДМАП, 100 мл ТГФ и 30 мл триэтиламина с последующим добавлением шприцом раствора 18,7 г трет-бутилдиметилсилилхлорида в 50 мл ТГФ. Данной смеси в течение приблизительно одного часа давали возможность перемешиваться (в атмосфере азота) при комнатной температуре. Твердое вещество из смеси отфильтровывали, а перед очисткой фильтрата по методу силикагелевой колоночной хроматографии при использовании в качестве элюирующего растворителя 5 %-ного этилацетата в гексане растворитель выпаривали. Получали белое твердое вещество (24,9 г, 92%-ный выход) и по методам1Н и13С ЯМР в CDCl3 подтвердили то, что данный продукт представляет собой 3,4-ди(трет-бутилдиметилсилокси)фенил-1,3-дитиан.
Подобным же образом при использовании соответствующих дигидроксибензальдегидов могут быть получены 3,5-, 2,5-, 2,3- и тому подобные аналоги 3,4-ди(трет-бутилдиметилсилокси)фенил-1,3-дитиана. Кроме того, при использовании в качествен исходного материала 4-гидроксибензальдегида может быть получен 4-(трет-бутилдиметилсилокси)фенил-1,3-дитиан. Все бензальдегиды могут быть получены от коммерческого поставщика, такого как, например, компания Sigma-Aldrich.
Пример 35: БТБДМСБА (альтернатива синтезу из примера 2)
В просушенную колбу объемом 500 мл, включающую якорь магнитной мешалки, добавляли ~8,2 г 3,4-дигидроксибензальдегида, ~0.5 г ДМАП, 100 мл ТГФ и 30 мл триэтиламина с последующим добавлением шприцом раствора ~19,0 г трет-бутилдиметилсилилхлорида в 50 мл ТГФ. Данной смеси в течение приблизительно одного часа давали возможность перемешиваться (в атмосфере азота) при комнатной температуре. Твердое вещество из смеси отфильтровывали, а перед очисткой фильтрата по методу силикагелевой колоночной хроматографии при использовании в качестве элюирующего растворителя 10%-ного этилацетата в гексане растворитель выпаривали. Получали воскообразное маслянистое полутвердое вещество (21,3 г, 97%-ный выход). По методам1Н и13С ЯМР подтвердили то, что данный продукт представляет собой БТБДМСБА.
Пример 36: СБК (контрольный образец)
В продуваемый при использовании N2 реактор, снабженный перемешивающим устройством, добавляли 1,47 кг гексана, 0,41 кг раствора стирола и 2,60 кг раствора бутадиена (20,9% в гексане). В реактор загружали ~3,2 мл раствора н-BuLi с последующим добавлением 1,1 мл раствора 2,2-бис(2'-тетрагидрофурил)пропана. Рубашку реактора нагревали до 50°C, и по истечении ~30 минут температура партии достигала пикового значения ~64°C. По истечении еще ~30 минут полимерный клей прикалывали в изопропанол, содержащий БГТ, и высушивали в барабанной сушилке.
Данный полимер в приведенной далее таблице 10 обозначают как образец 36.
Примеры 37-40: 3,4-ди(трет-бутилдиметилсилокси)фенил-1,3-дитиан в качестве предшественника инициатора
В продуваемый при использовании N2 реактор, подобный тому, который использовался в примере 36, добавляли 1,37 кг гексана, 0,41 кг раствора стирола и 2,71 кг раствора бутадиена (20,1% в гексане). В реактор загружали 5,9 мл раствора дитиана из примера 34 в гексане с концентрацией 1,0 моль/л с последующим добавлением 3,9 мл раствора н-BuLi. По истечении ~5 минут добавляли 1,1 мл раствора 2,2-бис(2'-тетрагидрофурил)пропана. Рубашку реактора нагревали до 50°C, и по истечении ~35 минут температура партии достигала пикового значения ~67°C.
По истечении еще ~30 минут порции полимерного клея переводили в стеклянные колбы и подвергали реакции обрыва при использовании
Каждый образец перемешивали в течение еще ~30 минут в водяной бане при 50°C. Защитные группы образцов 5-7 гидролизовали при комнатной температуре (~2 часа) по реакции с раствором ФТБА (~20%-ный молярный избыток в сопоставлении с рассчитанным количеством защитных групп).
Каждый полимерный клей коагулировали и высушивали так же, как и в примере 36.
Свойства полимеров из примеров 36-40 суммарно представлены далее в таблице 10, где Mp представляет собой пиковую молекулярную массу.
При реализации описанной ранее методики испытаний образец 39 продемонстрировал превосходные результаты по хладотекучести.
Примеры 41-49: Получение и испытания вулканизатов
При использовании рецептур из приведенных ранее таблиц 9а и 9b из образцов 36-40 получали вулканизуемые эластомерные смеси, содержащие армирующие наполнители. Смеси, полученные из рецептуры таблицы 9а, обозначают как примеры 41-45 соответственно, в то время как смеси, полученные из рецептуры таблицы 9b, обозначают как примеры 46-50 соответственно. Смеси в течение ~15 минут отверждали при 171°C.
Результаты испытаний с разверткой по деформации табулированы в таблицах Паи 11b, в то время как результаты испытаний с разверткой по температуре табулированы в таблицах 12а и 12b.
Как демонстрируют таблицы 11а, 11b, 12а и 12b, в сопоставлении со смесями, использующими контрольный СБК, инициированный при использовании н-BuLi, вулканизаты, использующие интерполимеры СБК, инициированные функциональными инициаторами, характеризуются значительным уменьшением гистерезиса, и данный эффект улучшается в случае наличия также и функциональной группы, возникающей в результате концевой функционализации, и/или в случае гидролиза защитных групп до получения гидроксильных групп. Подобные позитивные тенденции можно наблюдать и на примере данных по G' в таблицах 11а и 11b.
Более полный набор данных по физическим характеристикам получили для примеров 41, 43-44, 46 и 48-49. Эти данные суммарно представлены далее в таблице 13.
С.Примеры 51-87 (мономеры)
Растворы бутадиена (все в гексане), раствор стирола (33% в гексане), гексан, н-бутиллитий (н-BuLi, 1,60 моль/л в гексане), 2,2-бис(2'-тетрагидрофурил)пропан (раствор в гексане с концентрацией 1,6 моль/л, хранившийся над СаH2 и раствор БГТ в гексане, использовавшиеся в данных примерах, получали из запаса складского помещения.
Коммерчески доступные реагенты и исходные материалы включали нижеследующие, все из которых использовали без дополнительной очистки, если только в конкретном примере не было указано другого:
от компании Sigma-Aldrich Co. - 2,3-дигидроксибензальдегид (97%), 3,4-дигидроксибензальдегид (97%), 3,5-дигидроксибензальдегид (98%), 2,5-дигидроксибензальдегид (98%), моногидрат 3,4,5-тригидроксибензальдегида (98 %), бромид метилтрифенилфосфония (МТФ-Br, 98%), этилацетат (99,5%) и
ДМАП (99%) и
от компании ACROS Organics - трет-бутилдиметилсилилхлорид (98%) и ФТБА (1 моль/л в ТГФ, содержащем ~5% воды).
Колоночную хроматографию проводили при использовании силикагелевого сорбента на 200-425 меш (Fisher Scientific; Питтсбург, Пенсильвания). Тонкослойную хроматографию проводили на хроматографических пластинках, полученных в компании Sigma-Aldrich.
Испытания проводили для вулканизатов, полученных из каучуковых смесей в соответствии с рецептурами, продемонстрированными в таблицах 9а и 9b (смотрите выше).
Пример 51: Синтез 3.4-ди(трет-бутилдиметилсилокси)стирола
К перемешиваемому холодному (0°C) раствору 23,2 г МТФ-Br в 100 мл высушенного ТГФ в атмосфере азота покапельно добавляли 40,6 мл раствора н-BuLi. По истечении ~15 минут шприцом покапельно добавляли раствор ~22,3 г БТБДМСБА (из примера 35) в 30 мл ТГФ. Получающуюся в результате желтую суспензию перед обработкой при использовании NH4Cl перемешивали в течение ~4 часов. Данный раствор отфильтровывали и концентрировали в вакууме. Остаток очищали по методу силикагелевой колоночной хроматографии при использовании в качестве элюирующего растворителя 5%-ного этилацетата в гексане, что в результате привело к сбору ~20,6 г (94%-ный выход) бесцветного масла. По методам1Н и13С ЯМР подтвердили то, что данное соединение представляет собой 3,4-ди(трет-бутилдиметилсилокси)стирол (ДТБДМСОС).
Пример 52: СБК (контрольный образец)
В продуваемый при использовании N2 реактор, снабженный перемешивающим устройством, добавляли 0,81 кг гексана, 0,21 кг раствора стирола и 1,20 кг раствора бутадиена (22,6% в гексане). В реактор загружали ~1,9 мл раствора н-BuLi с последующим добавлением 0,55 мл раствора 2,2-бис(2'-тетрагидрофурил)пропана. Рубашку реактора нагревали до 50°C, и по истечении ~30 минут температура партии достигала пикового значения ~59°C.
По истечении еще ~30 минут полимерный клей прикалывали в изопропанол, содержащий БГТ, и высушивали в барабанной сушилке. Данный полимер в приведенной далее таблице 14 обозначают как образец 52.
Примеры 53-55: Интерполимеры, включающие звенья 3,4-ди(трет-бутилдиметилсилокси) стирола
Для смесей, включающих 0,81 кг гексана, 0,21 кг раствора стирола и 1,20 кг раствора бутадиена (22,6% в гексане), проводили серии полимеризаций в продуваемом при использовании N2 реакторе, подобном тому, что и в примере 53. Смеси различались по количествам использующихся ДТБДМСОС (1,0 моль/л в гексане) и раствора н-BuLi, говоря конкретно
К каждой смеси также добавляли 0,55 мл раствора 2,2-бис(2'-тетрагидрофурил)пропана. В каждом случае рубашку реактора нагревали до 50°C, и температура партии достигала пикового значения, соответственно ~56°C (по истечении ~32 минут), ~57°C (по истечении ~30 минут) и ~56°C (по истечении ~30 минут). Добавляли достаточное количество раствора ФТБА таким образом, чтобы соотношение между количествами ФТБА и ДТБДМСОС в каждом случае составляло бы ~6:5, и данные смеси в каждом случае в течение ~2 часов перемешивали при комнатной температуре.
Каждый из полимерных клеев прикалывали в изопропанол, содержащий БГТ, и высушивали в барабанной сушилке. Данные полимеры в приведенной далее таблице 14 обозначают как образцы 53-55.
Примеры 56-58: Интерполимеры, включающие блок 3-4-ди(трет-бутилдиметилсилокси) стирола
В продуваемый при использовании N2 реактор, подобный тому, что и в примере 52, добавляли 1,55 кг гексана, 0,41 кг раствора стирола и 2,52 кг раствора бутадиена (21,6% в гексане). В реактор загружали ~3,3 мл раствора н-BuLi с последующим добавлением 1,1 мл раствора 2,2-бис(2'-тетрагидрофурил)пропана. Рубашку реактора нагревали до 50°C, и по истечении ~30 минут температура партии достигала пикового значения ~63°C.
По истечении еще ~30 минут порции полимерного клея прикалывали в стеклянные колбы. Добавляли переменные количества раствора ДТБДМСОС таким образом, чтобы получить, соответственно, соотношения между количествами ДТБДМСОС и атомов Li 1:1, 3:1 и 5:1. Данные смеси в течение еще ~40 минут перемешивали в водяной бане при 50°C.
Добавляли раствор ФТБА таким образом, чтобы соотношение между количествами ФТБА и ДТБДМСОС в каждом случае составляло бы ~6:5, и данные смеси в каждом случае в течение ~2 часов перемешивали при комнатной температуре.
Каждый из полимерных клеев прикалывали в изопропанол, содержащий БГТ, и высушивали в барабанной сушилке.
Пример 59: Испытания на хладотекучесть
Образцы для испытаний получали и подвергали испытаниям так, как это описывалось ранее.
Как свидетельствуют результаты испытаний, образец, полученный из полимера из примера 53, по характеристикам хладотекучести был почти что идентичен образцу, полученному из полимера из примера 52, в то время как все образцы, полученные из полимеров из примеров 54 и 56-58, были значительно улучшены (с кратностью в диапазоне от ~1,5х до ~3,5х) в сопоставлении с образцом, полученным из полимера из примера 52, при этом в сопоставлении со статистическими интерполимерами лучшими являются блочные интерполимеры.
Примеры 60-67: Получение и испытания вулканизатов
При использовании рецептур из приведенных ранее таблиц 9а и 9b из образцов 52-55 получали вулканизуемые эластомерные смеси, содержащие армирующие наполнители. Смеси, полученные из рецептуры таблицы 9а, обозначают как примеры 60-63 соответственно, в то время как смеси, полученные из рецептуры таблицы 9b, обозначают как примеры 64-67 соответственно. Вулканизаты получали в результате отверждения данных смесей в течение ~15 минут при 171°C.
Данные по испытаниям с разверткой по деформации табулированы в таблицах 15а (как для G', так и для tan δ) и 15b (только для tan δ), в то время как результаты испытаний с разверткой по температуре табулированы в таблицах 16а и 16b.
Как демонстрируют данные из таблиц 15а и 15b (развертка по деформации при 60°C), присутствие звеньев В, помимо прочего, в результате приводит к уменьшению значения tan δ, что свидетельствует об уменьшенном гистерезисе. Как демонстрируют данные из таблиц 16а и 16b, присутствие звеньев В в результате приводит к общему увеличению пикового значения tan δ и значения tan δ при 0°C, что свидетельствует, помимо прочего, об улучшении характеристик хладотекучести и сцепления с мокрым дорожным покрытием.
Пример 68: Синтез 3.4,5-три(трет-бутилдиметилсилокси)бензалъдегида
В просушенную колбу объемом 250 мл, включающую якорь магнитной мешалки, добавляли ~5,0 г 3,4,5-тригидроксибензальдегида, ~0.3 г ДМАП, 60 мл ТГФ и 10 мл триэтиламина с последующим добавлением шприцом раствора ~15,2 г трет-бутилдиметилсилилхлорида в 30 мл ТГФ. Данной смеси в течение приблизительно одного часа давали возможность перемешиваться (в атмосфере азота) при комнатной температуре. Твердое вещество из смеси отфильтровывали, а перед очисткой фильтрата по методу силикагелевой колоночной хроматографии при использовании в качестве элюирующего растворителя 10%-ного этилацетата в гексане растворитель выпаривали. Получали воскообразный продукт (15,3 г, 96%-ный выход). По методам1Н и13С ЯМР подтвердили то, что данное соединение представляет собой 3.4,5-три(трет-бутилдиметилсилокси)бензальдегид.
Пример 69: Синтез 3.4,5-три(трет-бутилдиметилсилокси)стирола
К перемешиваемому холодному (0°C) раствору ~11,5 г МТФ-Br в 100 мл высушенного ТГФ в атмосфере азота покапельно добавляли ~19,5 мл раствора н-BuLi. По истечении ~10 минут шприцом покапельно добавляли раствор 15,0 г продукта из примера 68 в 30 мл ТГФ. Получающуюся в результате желтую суспензию перед обработкой при использовании NH4Cl перемешивали в течение ~4 часов. Данный раствор отфильтровывали и концентрировали в вакууме. Остаток очищали по методу силикагелевой колоночной хроматографии при использовании в качестве элюирующего растворителя 5%-ного этилацетата в гексане, что в результате привело к сбору ~13,4 г (90%-ный выход) бесцветного масла. По методам1Н и13С ЯМР подтвердили то, что данное соединение представляет собой 3,4,5-три(трет-бутилдиметилсилокси)стирол (ТТБДМСОС).
Пример 70: СБК (контрольный образец)
В продуваемый при использовании N2 реактор, снабженный перемешивающим устройством, добавляли ~1,55 кг гексана, ~0,41 кг раствора стирола и ~2,52 кг раствора бутадиена (21,6% в гексане).
В реактор загружали ~3,0 мл раствора н-BuLi (1,7 моль/л) с последующим добавлением 1,10 мл раствора 2,2-бис(2'-тетрагидрофурил)пропана. Рубашку реактора нагревали до 50°C, и по истечении ~34 минут температура партии достигала пикового значения ~63°C.
По истечении еще ~30 минут полимерный клей прикалывали в изопропанол, содержащий БГТ, и высушивали в барабанной сушилке. Данный полимер в приведенной далее таблице 17 обозначают как образец 70.
Примеры 71-74: Интерполимеры, включающие звенья 3,4,5-три(трет-бутилдиметилсилокси) стирола
Проводили полимеризацию в продуваемом при использовании N2 реакторе, подобном тому, что и в примере 70. В отличие от количества раствора инициатора (в данном случае ~2,9 мл) количества добавлявшихся материалов были идентичны тем, что и в примере 70. Рубашку реактора нагревали до 50°C, а по истечении ~35 минут температура партии достигала пикового значения ~64°C.
По истечении еще ~30 минут в реактор загружали 5 мл раствора ТТБДМСОС с концентрацией 1,0 моль/л (1,0 моль/л в гексане); данную смесь перед переводом порций полимерного клея в стеклянные колбы и проведением реакции обрыва при использовании
в течение ~30 минут перемешивали при 50°С. Каждый образец в течение еще ~30 минут перемешивали в водяной бане при 50°C.
Половину образца 71 переводили в другую колбу, и далее это обозначают как образец 71а.
Защитные группы образцов 71а и 73-74 в течение ~60 минут гидролизовали при комнатной температуре по реакции с раствором ФТБА (раствор с концентрацией 1 моль/л в ТГФ, содержащем ~5% воды и использующем такое количество, чтобы в результате получить соотношение между количествами ФТБА и ТТБДМСОС 11:10).
Каждый полимерный клей коагулировали и высушивали так же, как и в примере 70.
Пример 75: Испытания на хладотекучесть
При получении образцов для испытаний по приведенной ранее методике использовали полимеры из примеров 70 и 73-74, а также СБК, подвергнутые реакции обрыва при использовании ДМИ и АПМДЭОС, (то есть в цепь полимера не включали каких-либо звеньев В).
Как свидетельствуют результаты испытаний, оба образца, полученные из полимеров из примеров 73-74, являлись лучшими (имеющими ~ на 2 мм большую толщину при любой заданной температуре) в сопоставлении с образцом, полученным из подобного полимера, который не включал звенья В.
Примеры 76-87: Получение и испытания вулканизатов
При использовании рецептур из приведенных ранее таблиц 9а и 9b из образцов 70-74 получали вулканизуемые эластомерные смеси, содержащие армирующие наполнители. Смеси, полученные из рецептуры таблицы 9а, обозначают как примеры 76-81 соответственно, в то время как смеси, полученные из рецептуры таблицы 9b, обозначают как примеры 82-87 соответственно. Вулканизаты получали в результате отверждения данных смесей в течение ~15 минут при 171°C.
Как продемонстрировали физические испытания, подобные тем, которые представлены ранее, (то есть зависимость значения tan δ как от % деформации (при 60°C), так и от температуры, в обоих случаях при 10 Гц), вулканизаты, использующие интерполимеры СБК, которые разработаны включающими один или несколько мономерных фрагментов В по соседству с концевыми функциональностями, характеризуются значительным уменьшением гистерезиса и другими желательными свойствами в случае вулканизатов, содержащих как технический углерод, так и диоксид кремния.
Получили полный комплект данных по физическим характеристикам, который суммарно представлен далее в таблицах 18 и 19.
Реферат
Изобретение относится к способу получения полимера, включающему по меньшей мере одно функционализующее звено и один или несколько типов полиеновых мономерных фрагментов. Указанный способ включает а) получение раствора, содержащего (1) один или более типов этиленненасыщенных мономеров, включающих по меньшей мере один тип полиена, и (2) инициирующее соединение, и b) обеспечение анионного инициирования для получения карбанионного полимера, имеющего среднечисленную молекулярную массу в диапазоне от 75000 до 150000 Да. Указанное инициирующее соединение описывается общей формулой RZQ-М, где М представляет собой атом щелочного металла, Rпредставляет собой замещенную или незамещенную арильную группу, содержащую по меньшей мере одну группу OR, где каждый Rпредставляет собой группу, которая является нереакционно-способной по отношению к М и способной гидролизоваться, Z представляет собой одинарную связь или замещенную или незамещенную циклическую алкиленовую, ациклическую алкиленовую или ариленовую группу, a Q представляет собой группу, связанную с М через атом С, N или Sn. Полученные полимеры характеризуются превосходной степенью взаимодействия как с обычными, так и с необычными наполнителями и могут быть использованы для получения вулканизатов, обладающих желательными свойствами. 2 н. и 20 з.п. ф-лы, 18 табл., 87 пр.
Формула
Документы, цитированные в отчёте о поиске
Способ получения карбоцепных полимеров
Комментарии