Способ получения предшественника тетратиафульвалена и способ получения производного тетратиафульвалена - RU2122001C1
Код документа: RU2122001C1
Описание
Изобретение относится к производным тетратиафульвалена, имеющим определенную структуру, а также к предшественникам этих производных, имеющим определенную структуру, которые пригодны в качестве исходных веществ для получения органических комплексов с переносом заряда, которые, как ожидается, будут использоваться для применения, как органические проводники, органические сверхпроводники, органические магнетики, органические электрохромные, органические электролюминесцентные материалы и им подобные. Настоящее изобретение также относится к способам пригодным для получения различных типов производных тетратиафульвалена, их исходных соединений, включая вышеуказанные определенные структуры производных тетратиафульвалена и их соединений предшественников.
Уровень техники
Были предприняты попытки
использования производных тетратиафульвалена в качестве исходных соединений для синтеза органических комплексов с переносом заряда, которые, как ожидалось, будут использованы в таких применениях как
органические проводники, органические сверхпроводники, органические магнетики, органические электрохромные материалы и им подобные. Большое внимание было направлено на развитие новых типов производных
тетратиафульвалена из-за ограничения практически доступных производных тетратиафульфалена и потребности для развития новых органических комплексов с переносом заряда.
Как суммировано
ниже, в предшествующей литературе известно несколько способов получения производных тетратиафульфалена:
i) Получают производное 1,3-дитиол-2-тиона начатая с восстановления дисульфида углерода
щелочным металлом. Полученное таким образом производное превращают в производное 1,3-дитиол-2-она (дитиолон), и двое молекулы превращенного производного подвергают реакции сочетания с получением
производного тетратиафульвалена (A. Mizoe et al, J. Chem. Soc. Chem. Commun. 1978, pp 18; G.Steimecke et al, Phosphorus and Sulfur, vol. 7, pp. 49 - 55 (1979); K. Hartke et al, Chem. Ber., vol 113,
pp. 1898 - 1906 (1980).
ii) Производное тетратиафульвалена получают из 1, 3, 4, 6-тетратиапентален-2,5-диона в качестве исходного соединения, используя катализатор межфазного переноса. (R.R. Schumaker et al, J. Org. Chem. vol 49, pp. 564 - 566 (1984).
iii) Производное тетратиафульфалена получают из 1,2-этандитиола и хлорацетилхлорида в качестве исходных соединений (J. Larsen et al, Synthesis, pp. 134 (1989)).
Вышеприведенный способ (i) имеет ряд недостатков в том, что реакция восстановления дисульфида углерода щелочным металлом имеет тенденцию вызывать взрыв, в способе генерируются побочные продукты реакции, которые трудно удаляются, требует ряд стадий, включая превращение производного 1,3-дитиол-2-тиона в производное 1,3-дитиол-2-она и выход интересующего продукта производного тетратиафульвалена низкий.
Вышеприведенный способ (ii) также имеет проблемы вызванные низким выходом продукта, потому что требует трудоемкого удаления катализатора межфазного переноса. Особенно потому, что используют хроматографическую колонку для удаления катализатора межфазного переноса, требуется продолжительное время для работы и продуктивность становится низкой, вызывая определенные затруднения.
Вышеприведенный способ (iii) также имеет недостатки в том, что требует 5 стадий для завершения синтеза, вызывая таким образом снижение выхода и сужая область применения, так как он может быть применен для получения бис/этилендитио/тетратиафульвалена/ BEDT-TTF) как одного из производных тетратиафульвалена, но труден для получения производных тетратиафульвалена.
Краткое содержание изобретения
В свете вышеизложенного объектом настоящего изобретения становится, таким образом, получение новых
производных тетратиафульвалена и исходных соединений, которые пригодны для получения новых органических комплексов с переносом заряда.
Другой объект настоящего изобретения заключается в обеспечении способа получения производных тетратиафульвалена с высоким выходом и высокой чистотой и их исходных соединений, которые могут применяться не только для вышеприведенных новых производных тетратиафульвалена и их исходных соединений, но также и для широкого ряда других производных тетратиафульвалена и их исходных соединений, и которые свободны от опасности вызывать взрывы, не генерируют достаточно трудно-удалимые побочные продукты реакции и имеют высокую продуктивность благодаря отсутствию трудоемкого удаления катализатора.
Другие объекты и преимущества настоящего изобретения будут очевидны из следующего описания.
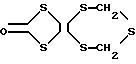
Настоящее изобретение относится к исходным соединениям производного тетратиафульвалена формулы I

где
R1 и R2 могут быть одинаковыми или разными и представляют органические группы, которые могут быть связаны вместе с образованием кольца.
Примеры исходного соединения производных тетратиафульвалена, представленых формулой I, включают те, которые представлены любой из
формул II - V:




Настоящее изобретение также относится к исходному соединению производного тетратиафульфалена, представленному формулой VI

где
R1 и R2 могут быть одинаковыми или разными и представляют органические группы, которые могут связываться вместе с образованием кольца.
Примеры производного тетратиафульфалена, представленного формулой VI, включают те, которые представляются любой из формул VII - X.




Каждое из исходных соединений производного тетратиафульвалена и производные тетратиафульвалена настоящего изобретения имеют уникальную и новую структуру, которая не может быть найдена в предшествующей литературе, и представляются полезными для разработки новых органических комплексов с переносом заряда.
Настоящее изобретение относится также к способу получения
исходного соединения производного тетратиафульвалена, представленного формулой I

где
R1 и R2 могут быть одинаковыми или разными и представляют органические группы, которые могут связываться вместе с образованием кольца.
способ, включающий стадии, обработки 1,3,4,6-тетратиапентален-2,5-диона при температуре 30oC или ниже в спиртовом растворе, содержащем метоксид щелочного металла в инертной атмосфере, тем самым вызывая на селективное расщепление одного из его колец с образованием 1,3-дитиол-2-она-4,5-дитиолат дианиона, и проведение взаимодействия 1,3-дитиол-2-он-4,5-дитиолат дианиона с соединением, имеющим моновалентную или бивалентную органическую группу, которая соответствует органическим группам, представленным R1 и R2 в формуле I.
Настоящее изобретение далее также относится к
способу получения производного тетратиафульфалена, представленного формулой VI

где
R1 и R2 могут быть одинаковыми или разными и представляют органические группы, которые могут связываться вместе с образованием кольца,
способ включающий стадии обработки 1,3,4,6-тетратиапентален-2,5-диона при температуре 30o или ниже в спиртовом растворе, содержащем метоксид щелочного металла в инертной атмосфере, тем самым осуществляется селективное расщепление одного из колец с образованием 1,3-дитиол-2-он-4,5-дитиолатдианиона, проведение взаимодействия 1,3-дитиол-2-он-4,5-дитиолат дианиона с соединением, имеющим моновалентную и бивалентную органическую группу, которая соответствует органическим группам, представленным R1 и R2 в формуле VI, с образованием исходного соединения - предшественника тетратиафульфалена, и нагревание и перемешивание соединения - предшественника производного тетратиафульвалена в присутствии триалкилфосфита, тем самым осуществляя реакцию сочетания двух молекул исходного соединения.
Примеры органических групп, представленных R1 и R2 в формуле I и VI, включают алкильную группу (например, метильную, этильную, или им подобные), арильную группу (например, бензильную или ей подобные), гидроксиалкильную группу, триметилсилилэтоксиметильную группу и им подобные. В случае, где R1 и R2 связываются вместе с образованием кольца, примеры групп включают алкиленовую группу (например, этилен, пропилен или им подобные), диметилентиогруппу, диметиленэфирную группу и им подобные.
С целью решения
вышеупомянутых задач, авторы настоящего изобретения провели интенсивные исследования по исходным соединениям и стадиям реакции для получения производных тетратиафульвалена. В результате было найдено,
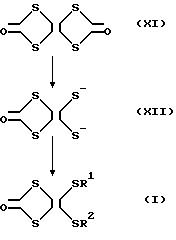
что дитиолон как исходное соединение производных тетратиафульвалена может быть получен с меньшим количеством стадий, чем в предыдущей литературе, если одно из двух колец 1,3,4,6-тетратиапентален-2,
5-диона, представленного формулой XI, может быть расщеплено селективно:

Однако, если 1,3,4, 6-тетратиапентален-2,5-дион подвергать реакции с раскрытием кольца в присутствии сильного основания, такого как алкоксид натрия, оба из его двух колец разрываются как общеизвестно, тогда как ничего неизвестно о селективном разрыве одного из этих колец.
В результате, авторы настоящего изобретения предложили дальнейшие исследования для нахождения способа селективного расщепления одного из двух колец в 1,3,4,6-тетратиапентален-2,5-дионе и нашли, что одно из двух колец может селективно расщепляться без генерирования трудноудаляемых побочных продуктов реакции и без опасности вызвать взрыв, способом в котором алкоксид щелочного металла, такой как метоксид натрия или ему подобный получают в растворе с 1 молярной концентрацией растворением его в спирте, таком как метанол или ему подобный, в инертной атмосфере, свободной от кислорода, влаги и им подобных примесей, раствор алкоксида добавляют к 1,3,4,6-тетратиапентален-2,5-диону и позволяют протекать реакции с раскрытием кольца в течение около 10 минут при относительно низкой температуре 30oC или ниже, особенно при комнатной температуре. Таким образом, способ настоящего изобретения получения производных тетратиафульвалена и их соединений - предшественников, которые являются предметом настоящего изобретения, основываются на указанных исследованиях.
Согласно способу получения рассмотренному в настоящем изобретении, как показано в следующей реакционной схеме, одно из двух колец 1,3,4,6-тетратиапентален-2,5-диона (XI) селективно расщепляется с получением 1,3-дитиол-2-он-4, 5-дитиолатдианиона (XII), которому впоследствии позволяют взаимодействовать с соединением, имеющим моновалентную или двухвалентную органическую группу, которая соответствует R1 и R2 в формуле I. Таким образом, предшественник производного тетратиафульвалена, представленного формулой I, может быть получен однореакторным и одностадийным способом без генерирования трудноудаляемых побочных продуктов реакции. Вследствие этого исходное соединение производного тетратиафульвалена может быть получено с высоким выходом, высокой чистой и за короткий промежуток времени.
Производное тетратиафульвалена может быть получено с использованием вышеуказанной стадии получения предшественника и стадией реакции сочетания двух молекул предшественника, тем самым обеспечивая получение производного тетратиафульвалена с высоким выходом и за короткий промежуток времени.
Кроме того, вышеуказанный способ получения может быть выполнен с высокой безопасностью, потому что не включает взрывоопасные реакции.
Вышеуказанный способ получения имеет также преимущество широкой области применения, так как могут быть легко получены не только различные типы производных тетратиафульвалена и их предшественников, известных из предыдущей литературы, но также и новые производные тетратиафульвалена и их исходные соединения, которые не могут быть получены способами, описанными в предыдущей литературе, такие предшественники производного тетратиафульвалена настоящего изобретения, представленные формулой II - V, как производные тетратиафульвалена настоящего изобретения, представленные формулой VII - X, и им подобные, которые могут быть получены способом настоящего изобретения с выбором соответствующих моновалентных или двухвалентных органических групп для взаимодействия с 1,3-дитиол-2-он-4,5-дитиолатдианином XII.
Настоящее изобретение описывается в порядке следования стадий получения.
Во-первых, согласно способу получения предшественника производного тетратиафульвалена согласно настоящему изобретению одно из двух колец 1,3,4,6-тетратиапентален-2,5-диодна, представленного формулой XII в качестве исходного соединения, селективно расщепляется с получением 1, 3-дитиол-2-он-4,5-дитиолатдианиона, представленного формулой XII.
Как описано выше, реакция селективного раскрытия кольца может осуществляться способом, в котором алкоксид щелочного металла получают в растворе растворения его в соответствующем спирте (например, NaOCH3 в CH3OH, NaOC2H5 в C2H5OH и т. д.) и в инертной атмосфере свободной от кислорода, влаги и подобных примесей, 1,3,4,6-тетратиапентален-2,5-дион (XI) добавляют к раствору алкоксида и позволяют протекать реакции с раскрытием кольца в течение около 5-30 мин, предпочтительно 8-12 мин, при относительно низкой температуре 30oC или ниже, предпочтительно от 18 до 25oC, особенно предпочтительно от 20 до 23oC, особенно при комнатной температуре. Если появляется вероятность интенсивного выделения тепла реакции, как это может быть в случае крупномасштабной реакции, рекомендуется водяной баней (около 20o C).
Концентрация раствора алкоксида щелочного металла обычно представляет около 1 моль в литре, предпочтительно от 0,8 до 1,2 моль/л, и более предпочтительно от 0,95 до 1,05 моль/л. Количество алкоксида щелочного металла в реакции представляет обычно около 2 молей, предпочтительно 1,8-2,2 моль, более предпочтительно от 1,95 до 2,05 моль на моль 1,3,4,6-тетратиапентален-2,5-диона (XI).
Термин "комнатная температура" использованный здесь относится к таким условиям реакции, когда применяют внешнее нагревание или охлаждение, вообще температура лежит в области между 20-25oC.
Могут быть использованы как алкоксид щелочного металла, так и алкоксид натрия и низших спиртов, такие как метоксид натрия, этоксид натрия и им подобные, а также и другие алкоксиды различных щелочных металлов и спиртов. Алкоксид обычно используют в количестве 2 моль на моль 1,3,4,6-тетратиапентален-2,5-диодна (XI).
Продукт, полученный после окончания реакции с раскрытием кольца, затем взаимодействует с соединением, содержащим моновалентную или двухвалентную органическую группу, которая соответствует R1 и R2 в формуле I, при комнатной температуре в течение приблизительно 1 - 30 ч, предпочтительно 2 - 10 ч, более предпочтительно 2 - 4 ч.
После окончания реакции образовавшейся таким образом продукт добавляют к воде и экстрагируют органическим растворителем, таким как метиленхлорид и растворитель удаляют при пониженном давлении с получением сырого продукта. Сырой продукт впоследствии очищают обычным способом таким как перекристаллизация, переосаждение и им подобным с получением исходного соединения производного тетратиафульвалена, представленного формулой I.
В качестве соединения, содержащего органическую группу, соответствующую R1 и R2, может быть использован галоид, такой как хлорид, бромид, иодид и им подобные, предпочтительно, принимая во внимание их реакционную способность с 1,3-дитиол-2-он-4,5-дитиолатдианщионом (XII). Моногалоиды используют если органическая группа моновалентная и дигалоиды если органическая группа двухвалентная.
Точнее, если органическая группа введенная в виде R1 и R2 представляет моновалентную группу, такую как алкильная группа (например, метильная, этильная или им подобные), аралкильную группу (например, бензильную или ей подобные), гидроксиалкильную группу или триметилсилилэтоксиметильную группу, используют соответствующие моногалоиды, которые включают: алкилгалоиды, такие как алкилхлорид, алкилбромид, алкилиодид или им подобные, аралкилгалоиды, такие как аралкилхлорид, аралкилбромид или им подобные и триметилсилилэтоксиметил галоид, такой как триметилсилиэтоксиметилхлорид, триметилсилилэтоксиметилбромид, триметилсилилэтоксиметилиодид или им подобные. Моногалоиды обычно используют в количествах около 2 моделей на моль 1,3-дитиол-2-он-4, 5-дитиолатдианина.
Если органическая группа введенная как R1 и R2 представляет двухвалентную группу, которая образует кольцо взаимным связыванием, такую как алкиленовая группа (например, этиленовая, пропиленовая или им подобная), диметилентио группа, или диметиленэфирная группа, используют соответствующие дигалоиды, которые включают алкилендииодид или им подобные (например, XCH2 -(CH2)n-CH2X, где X представляет атом галоида, и представляет целое число 0 или более); и деметилентиодигалоид, такой как диметилентиодихлорид, диметилентиодибромид, диметиленлентиодииодид или им подобные. Дигалоид обычно используют в количестве около 1 моль на 1 моль 1,3-дитиол-2-он-4,5-дитиолатдианиона.
Когда появляется возможность возникновения слишком быстрого протекания реакции, обусловленная высокой реакционной способность использования галоида, или планируется проведение реакции в большом масштабе, предпочтительно с точки зрения безопасности подавлять реакционную активность разбавлением реакционного раствора после окончания расщепления кольца, спиртом или подобным ему растворителями и растворением галоида в том же само растворителе до его добавления к разбавленному реакционному раствору.
Среди предшественников производного тетратиафульвалена, представленных формулой
I, которые могут быть получены указанным выше способом получения исходного соединения производного тетратиафульвалена настоящего изобретения, предшественники производного тетратиафульвалена настоящего
изобретения, представленные формулами II - X, являются особенно полезными в качестве предшественников производного тетратиафульвалена настоящего изобретения и для разработки новых органических
комплексов с переносом заряда:


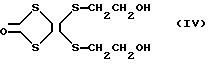

Согласно способу получения производных тетратиафульвалена настоящего изобретения, заранее определенное количество предшественника (исходного соединения) производного тетратиафульвалена, полученного вышеупомянутым способом получения исходного соединения, сначала растворяют или диспергируют в очищенном триалкилфосфита, который выбирают из различных соединений, включая триэтилфосфит. Количество триалкилфосфита, которое должно быть использовано, особенно не ограничивается, но в случае триэтилфосфита, например, его обычно используют в количестве около 5 мл на 1 миллимоль исходного соединения производного тетратиафульвалена.
Нагревание раствора или суспензии происходит при приблизительно 100-120oC с перемешиванием, протекает реакция сочетания двух молекул исходного соединения производного тетратиафульвалена и образуются продукты реакции, содержащие производное тетратиафульвалена, представленное формулой VI, как интересующий продукт в реакционном растворе в виде от красновато-желтого до красновато-коричневого осадка.
После этого полученный таким образом осадок извлекают фильтрованием, промывают растворителем, таким как метанол, и затем подвергают очистке обычным способом, таким как перекристаллизация, переосаждение, хроматографирование на колонке, сублимация и им подобными способами с получением производного тетратиафульвалена, представленного формулой VI.
среди производных тетратиафульвалена, представленных формулой VI, которые могут быть получены
указанным выше способом получения производного тетратиафульвалена настоящего изобретения, производные тетратиафульвалена настоящего изобретения, представленные формулами VII - X, представляют новые
соединения, которые делают возможным разработку новых органических комплексов с переносом заряда:




Как описано выше, согласно настоящему изобретению могут быть получены не только исходные соединения производного тетратиафульвалена простой однореактивной и одностадийной реакционной системой, которая не генерирует трудно удаляемых побочных продуктов и представляется свободной от опасности взрыва и подобных реакций, но также могут быть получены производные тетратиафульвалена из исходных соединений производного тетратиафульвалена только одностадийной реакцией.
В результате, в соответствии со способом настоящего изобретения, исходные соединения производного тетратиафульвалена, имеющие более высокую чистоту, чем те, которые получены способом ранее известным в литературе, могут быть получены безопасной реакционной системой и с более высоким выходом, по сравнению с известным ранее способом, таким образом, делая возможным получение производных тетратиафульвалена в крупном масштабе с низкой стоимостью, которая не может быть достигнута известным ранее способом.
Кроме того, согласно настоящему изобретению могут быть легко получены не только различные типы известных производных тетратиафульвалена и их предшественников, но также новые производные тетратиафульалена и их предшественники, делая таким образом важным дальнейшее развитие новых органических комплексов с переносом заряда, используя производные тетратиафульвалена в виде исходного сырья.
Следующие примеры обеспечивают дальнейшую иллюстрацию настоящего изобретения. Должно быть понятно, однако, что примеры служит только для целей иллюстрации и не являются ограничением настоящего изобретения.
Пример 1.
(1) Получение исходного соединения производного тетратиафульвалена.
9,6 мл стандартного раствора метоксида натрия в метаноле (концентрация:1 моль/л метанола) добавляют сразу к 1 г (4,8 ммоль) 1, 3, 4, 6-тетратиапентален-2,5-диона, и полученный темно-зеленый раствор перемешивают при комнатной температуре в течение 10 мин. К полученному реакционному раствору добавляют 1,36 г (9,6 ммоль) метилиодида сразу, с последующим перемешиванием в течение 2 ч при комнатной температуре.
Полученный таким образом реакционный раствор добавляют к 150 мл воды, трижды экстрагируют 50 мол метиленхлорида (CH2Cl) и сушат над сульфатом магния (MgSO4) с последующим удалением растворителя при пониженном давлении с получением сырого продукта в твердом виде. Затем сырой продукт перекристаллизовывают из этанола с получением 0,7 г очищенного продукта с выходом 70%.
Таким образом, очищенный продукт имел температуру плавления 53-56oC и
был подтвержден, что является 4,5-диметилтио-1,3-дитиол -2-оном(молекулярный вес: 210,3), представленным формулой XIII

(2) Получение производного тетратиафульвалена
2,1 г (10 ммоль) порцию 4,5-диметилтио-1,3-дитиол-2-она, полученного способом приведенным выше, растворяют в 50 мл триэтилфосфита (P)OC2H5)3), который был свежеперегнанным и очищенным. Таким образом, полученный раствор перемешивали в течение 3 ч при 100-120oC. Затем осадок, образовавшийся в реакционном растворе, удаляли фильтрованием, трижды промывали 10 мл метанола, сушили и затем перекристаллизовывали из этанола, тем самым получая 0,37 г очищенного продукта с выходом 50%.
По данным элементного анализа (Е.А.) подтверждено, что очищенный таким образом продукт представляет производное тетратиафульфалена, представленное
формулой XIV

Пример 2.
(1) Получение исходного соединения производного
тетратиафульвалена
Сырой продукт исходного соединения был получен в твердом виде повторением способа получения исходного соединения производного тетратиафульвалена (1) как в примере 1,
приведенном выше, за исключением того, что был использован 1,22 г (9,6 ммоль) бензилхлорид вместо метилиодида. Затем сырой продукт растворяют в метиленхлориде и переосаждают в пентан с получением 1,44
г очищенного продукта с выходом 85-99%.
Очищенный таким образом продукт подвергают измерению температуры плавления ЭА, анализу с помощью инфракрасной спектроскопии (ИК) анализу с
использованием таблетки КВr, масс-спектральному (МС) анализу и ядерному магнитному резонансу (1H-ЯМР), получая тем самым следующие результаты:
Температура плавления: 59-59,5oC.
ЭА:
вычислено,%: C 56,32; H 3,88;
найдено,%: C 56,04; H 3,78;
ИК (KBr) ν (см-1): 1679 (см. C=O), 1454, 1240, 898, 762, 692.
МС(ЕИ) м/ч: 362 (M+), 271, 243, 211, 91.
1H-ЯМР д/м.д стандарт ТМС (:3,87 (с. 4H, CH2), 7,27 (мс, 10Hаром).
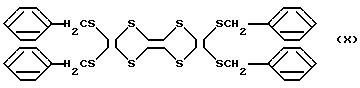
На основании этих данных, очищенный таким образом продукт подтверждает образование 4,5-дибензилтио-1,3-дитиол-2-она) (молекулярный вес: 362,5), представленного формулой V

(2) Получение производного тетратиафульвалена
Способу получения производного тетративфульвалена повторяют как в примере 1, за исключением того, что используют 3,6 г (10 ммоль) 4,5дибензилтио-1,3-дитиол-2-она, полученного в стадии I или II с получением 0,38 г очищенного продукта с выходом 80%.
Очищенный таким образом продукт подвергают анализу по данным температуры плавления ЭА, ИК, МС и1H-ЯМР-спектров, получая тем самым следующие результаты:
Температура
плавления: 166,5-168,5oC.
ЭА:
вычислено,%: C 58,92; H 4,07;
найдено,%: C 58,26; H 3,83.
ИК (KBr) ν (см-1): 1493, 1451, 893, 768 (срав), 701 (срав), 660.
МС (ЕИ) м/ч: 692 (M+), 567, 536, 490, 444, 380, 357, 324, 212.
1H-ЯМР д/м.д стандарт ТМС) :3,85 (с. 8H, CH2), 7,28 (мс, 20Hаромат).
На основании этих данных, очищенный таким образом продукт является производным тетратиафульвалена, представленным формулой X
Пример 3.
(1) Получение исходного соединения производного тетратиафульвалена
Сырой продукт исходного соединения в твердой форме получают повторением способа получения исходного соединения производного тетратиафульвалена 1, как в примере 1, приведенном выше, за
исключением того, что используют 1,6 г (9,6 ммоль) триметилсилилэтоксиметилхлорида вместо метилиодида. Затем сырой продукт
полученный таким образом, подвергают очистке на хроматографической колонке с
использованием силикагеля в качестве носителя и смеси гексан/этилацетат (80/20) в качестве растворителя с получением 1,36 г (выход 60%) 4,5-бис/триметилсилилэтоксиметил/тио-1,3-дитиол-2-она
(молекулярный вес: 442,81), представленного формулой XV

Способ получения производного тетратиафульвалена повторяют как в примере 1, приведенном выше, за исключением того, что используют 4,4 г (10 ммоль) 4,5-бис-/триметилсилилэтокси/тио-1,3-дитиол-2-она, полученного выше в стадии I или II, с получением 0,32 г (выход 30%) сырого производного тетратиафульвалена, представленного формулой XVI

Пример 4.
(1) Получение исходного соединения производного тетратриафульвалена
Сырой продукт исходного соединения в твердой форме получают повторением способа
получения исходного соединения производного тетратриафульвалена 1 как в примере 1, приведенном выше, за исключением того, что используют 0,9 г (4,8 ммоль) этилендибромид (1,2-диброметана), вместо
метилиодида. Затем сырой продукт перекристаллизовывают из этанола с получением 0,5 г очищенного продукта с выходом 45 - 50%.
Очищенный таким образом продукт имеет температуру плавления
127 - 128oC и свидетельствует об образовании 4,5-этилендитио-1,3-дитио-2-она, представленного формулой XVII

(2) Получение производного тетратиафульвалена
Способ получения производного тетратиафульвалена повторяют как в примере 1, приведенном выше, за исключением того, что используют 2,1 г (10 ммоль) 4,5-этилендитио-1,3-дитиол-2-она, полученного в стадиях I или II, приведенных выше, с получением 1,6 г очищенного продукта с выходом 85%.
На основании результатов элементного анализа подтверждается образование производного тетратиафульвалена (бисэтилендитиотетратиафульвалена, ВЕДТ-TTF), представленного формулой XVIII

Пример 5.
(1) Получение исходного соединения производного тетратиафульвалена
Сырой продукт исходного соединения получают в твердой форме повторением способа получения исходного соединения производного тетратиафульвалена 1, как в примере 1, приведенном выше, за исключением того,
что используют 0,49 г (4,8 ммоль) пропилендибромид (1,3-дибромпропана) вместо метилиодида. Затем сырой продукт перекристаллизовывают из этанола с получением 0,58 г очищенного продукта с выходом
55%.
Очищенный таким образом продукт имеет температуру плавления 103 - 104oC при измерении и подтверждает образование 4,5-пропиленлитио-1,3-дитиол-2-она (молекулярный вес
222,3), представленного формулой XIX

(2) Получение производного тетратиафульвалена
Способ получения производного тетратриафульвалена повторяют как в примере 1, приведенном выше, за исключением того, что используют 2,2 г (10 ммоль) 4,5-пропилендитио-1,3-дитиол-2-она, полученного в приведенной выше стадии I или II, с получением 0,47 г очищенного продукта с выходом 65%.
На основании результатов элементного анализа подтверждено, что очищенный таким образом продукт
был производным тетратиафульвалена (бис-(пропилендитио(тетратиафульвален, ВРДТ-TTF), представленным формулой XX

Пример 6.
(1) Получение исходного соединения производного тетратиафульвалена
Сырой продукт исходного соединения получают в твердой
форме повторением способа получения исходного соединения производного тетратиафульвалена 1 как в примере 1, приведенном выше, за исключением того, что используют 0,315 г (4,8 ммоль)
диметилентиодихлорида (бисхлорметилсульфида) вместо метилиодида. Затем сырой продукт перекристаллизовывают из изопропилового спирта с получением 0,29 г очищенного продукта с выходом 20 - 25%.
Очищенный таким образом продукт подвергают измерению температуры плавления, ЭА, ИК, МС и1H-ЯМР спектров, получая тем самым следующие результаты:
Температура плавления:
197 - 198oC.
ЭА:
вычислено, %: C 24,98; H 1,68;
найдено, %: C 25,13; H 1,66.
ИК (KBr) ν (см-1): 1682 (с), 1651 (СТ), 1612 (с), 1357, 1223, 1162, 1128, 885, 856, 720.
МС (ЕИ) м/ч: 240 /M+/, 180, 166, 88.
1H-ЯМР д/ м.д. ст. ТМС//: 4.00 /с, 4H/.
На
основании этих результатов, очищенный таким образом продукт представляет 4,5-/2-тиапропилен/-дитио-1,3-дитио-2-он (молекулярный вес, 240.3), представленный формулой II

(2) Получение производного тетратиафульвалена
Способ получения производного тетратиафульвалена как в примере 1, приведенном выше, повторяют за исключением того, что используют 2,4 г (10 ммоль) 4,5-/2-тиапропилен/-дитио-1,3-дитиол-2-она, полученного в приведенной выше стадии I или II, с получением 1,79 г очищенного продукта с выходом 80%.
Очищенный таким образом продукт анализируют с помощью ЭА, ИК и МС, получая тем самым следующие результаты:
ЭА:
вычислено, %: C 26,
76; H 1,79;
найдено, %: C 26,94; H 1,69.
ИК (KBr) ν (см-1): 2958, 1364, 1218, 1164, 1126, 878, 852, 770, 722.
МС (ЕИ) м/ч: 448 /M+ /, 370, 268, 222, 180, 148, 88.
На основании этих результатов, очищенный таким образом продукт, представляет производное тетратиафульвалена, представленное формулой VII

Пример 7.
(1) Получение исходного соединения производного тетратиафульвалена
Сырой продукт исходного соединения получают в твердой форме повторением способа получения исходного соединения производного тетратиафульвалена 1 как в примере 1, приведенном выше, за исключением того,
что используют 0,55 г (4,8 ммоль) диметиленоксидихлорида (бисхлорметилового эфира) вместо метилиодида. Затем, сырой продукт перекристаллизовывают из изопропилового спирта с получением 0,5 г очищенного
продукта с выходом 50%.
Очищенный таким образом продукт подвергают измерению температуры плавления, ЭА, ИК и1H-ЯМР, получая тем самым следующие результаты:
Температура плавления: 159 - 161oC.
ЭА:
вычислено, %: C 26,77; H 1,79;
найдено, %: C 26,785; H 1,69.
ИК (KBr) ν (см-1): 1682 (с), 1670 (ст, C=O), 1421, 1299, 1226, 1051 (с, C=O), 911 (с).
1H-ЯМР д (м.д. ст. ТМС): 4.89 (с, 4H).
На основании этих результатов, очищенный таким
образом продукт представляет 4,5//2-оксапропилен/-дитио-1,3-дитио-2-он/ (молекулярный вес, 224.32), представленный формулой III

(2) Получение производного тетратиафульвалена
Способ получения производного тетратиафульвалена как в примере 1, приведенном выше, повторяют за исключением того, что используют 2,2 г (10 ммоль) 4,5-/2-оксапропилен/дитио-1,3-дитиол-2-она, полученного в приведенной выше стадии I или II, с получением 1,45 г очищенного продукта с выходом 80%.
Очищенный таким образом продукт анализируют с помощью ЭА, ИК и МС, получая тем самым следующие результаты.
ЭА:
вычислено, %: C 28,82; H 1,93;
найдено, %:
C 28,86; H 1,82.
ИК (KBr) ν (см-1): 2921, 1423, 1288, 1225, 1040, 974, 908, 773, 696, 664.
МС (ЕИ) м/ч: 416 /M+/, 386, 355, 222, 178, 88.
1H-ЯМР д/ м.д. ст. ТМС//: 4.00 /с, 4H/.
На основании этих результатов очищенный таким образом продукт представляет производное тетратиафульвалена,
представленное формулой VIII

Пример 8.
(1) Получение исходного соединения
производного тетратиафульвалена
Сырой продукт исходного соединения получают в твердой форме повторением способа получения исходного соединения производного тетратиафульвалена 1 как в примере
1, приведенном выше, за исключением того, что используют 0,56 г (4,8 ммоль) бромэтанола вместо метилиодида. Затем сырой продукт перекристаллизовывают из изопропилового спирта с получением 0,71 г
очищенного продукта с выходом 55%.
Очищенный таким образом продукт подвергают измерению температуры плавления, ЭА, ИК, МС и1H-ЯМР, получая тем самым следующие
результаты:
Температура плавления: 90 - 92oC.
ЭА:
вычислено, %: C 31,09; H 3,73;
найдено, %: C 31,09; H 3,43.
ИК (KBr) ν (см-1): 1682 (с), 3278 (с, бр, OH), 1675.5 (ст C=O), 1407, 1076, 1055, 883, 793.
МС (ЕИ) м/ч: 270 /M+/, 242, 199, 149, 121, 45.
1H-ЯМР д (м.д. ст. ТМС): 3.05 (мс, 4H, - SCH2), 3.8 (м, 4H, HOCH2).
На основании этих результатов, очищенный таким образом продукт представляет 4,
5-бис/гидроксиэтил/-дитио-1,3-дитиол-2-он (молекулярный вес, 270.39), представленный формулой IV

(2) Получение производного тетратиафульвалена
Способ получения производного тетратиафульвалена как в примере 1, приведенном выше, повторяют за исключением того, что используют 2,7 г (10 ммоль) 4,5-бис/гидроксиэтил/-дитио-1,3-дитиол-2-она, полученного в стадиях I или II, приведенных выше, с получением производного тетратиафульвалена, представленного формулой IX

Таким образом, как описано детально в приведенных выше примерах, каждое из производных тетратиафульвалена и их исходные соединения (предшественника) настоящего изобретения имеют новые структуры, которые не могут быть найдены в предыдущей литературе и используются для разработки новых органических комплексов с переносом заряда.
Согласно способу получения исходных соединений производного тетратиафульвалена настоящего изобретения, исходные соединения производного тетратиафульвалена могут быть получены преимущественно однореакторной и одностадийной реакционной системой, которая не генерирует трудноудаляемым побочным продуктом реакции и свободна от опасности взрыва и подобных процессов. Кроме того, согласно способу получения производных тетратиафульвалена настоящего изобретения, производные тетратиафульвалена могут быть получены из исходных соединений производного тетратиофульвалена только одностадийной реакцией.
В результате, в соответствии с настоящим изобретением, исходные соединения производного тетратиафульвалена и производные тетратиафульвалена, имеющие более высокую чистоту, чем те, которые получены способом, известным из предыдущей литературы, могут быть получены с помощью реакционной системы и с более высокими выходами, по сравнению с предыдущими способами, таким образом делая возможным получение производных тетратиафульвалена в большом масштабе с низкой стоимостью, что не может быть достигнуто предыдущими способами.
Поэтому способ получения настоящего изобретения имеет большое промышленное значение, так как позволяет получать производные тетратиафульвалена в большом масштабе и с низкой стоимостью, которые как ожидается используются в таких применениях как органические проводники, органические сверхпроводники, органические магнетики, органические электрохромные материалы, органические электролюминесцентные материалы и им подобные.
Хотя изобретение описано в деталях и со ссылками на конкретные примеры, для специалистов будет очевидным, что различные изменения и модификации могут быть осуществлены без отклонения от сути и объема изобретения.
Реферат
Производные тетратиафульвалена (6) могут использоваться как органические проводники, органические сверхпроводники, органические магнетики и им подобные материалы. Объектом изобретения является способ получения предшественника тетратиафульвалена формулы I, где R1 и R2 представляют органические группы: алкил, аралкил, алкен, алкоксил, арил, гидроксиалкил, триметилсилил и триметилсилилэтоксиметил, в случае, когда R1 и R2 связаны с образованием кольца, они представляют алкилен, диметилентиогруппу, диметиленэфирную группу. Соединение I получают однореакторным и одностадийным способом без генерирования трудноудаляемых побочных продуктов, следовательно, с высоким выходом и высокой чистотой за короткий промежуток времени: одно из двух колец 1,3,4,6-тетратиапентален-2,5-диона селективно расщепляют в спиртовом растворе, содержащем метоксид щелочного металла, в инертной атмосфере при 30oC или ниже. Полученный 1,3-диол-2-он-4,5-дитиолатдианион подвергают взаимодействию с соединением, имеющим моновалентную или двухвалентную группу, которая соответствует R1 и R2 в формуле I. Производные формулы VI получают с использованием вышеуказанной стадии получения предшественника и стадией реакции сочетания двух молекул предшественника, обеспечивая получение производного тетратиафульвалена с высоким выходом и за коротки период. 2 с. и 8 з.п. ф-лы.


Формула

где R1 и R2 могут быть одинаковыми или разными и представляют органические группы, которые могут быть связаны вместе с образованием кольца, выбранные из группы, включающей алкил, аралкил, алкен, алкоксил, арил, гидроксиалкил, триметилсилил и триметилсилилэтоксиметил, и в случае, когда R1 и R2 связаны с образованием кольца, они представляют алкилен, диметилентиогруппу, диметиленэфирную группу,
отличающийся тем, что 1,3,4,6-тетратиапентален-2,5-дион обрабатывают спиртовым раствором, содержащим метоксид щелочного металла, в инертной атмосфере при температуре 30oC или ниже с получением 1,3-дитиол-2-он-4,5-дитиолатдианиона, который затем подвергают взаимодействию с соединением, имеющим одновалентную или двухвалентную органическую группу, которая соответствует вышеприведенным органическим группам, представленным R1 и R2 в формуле I, выбранным из моногалоидного соединения, имеющего органическую группу, соответствующую R1 и R2 или дигалоидного соединения, имеющего органическую группу, которая соответствует кольцевой группе, образованной R1 и R2 как указано выше.
3. Способ по п. 1, отличающийся тем, что предшественник производного тетратиафульвалена представлен формулой III

4. Способ по п. 1, отличающийся тем, что предшественник производного тетрафульвалена по п.1, представлен формулой IV

5. Способ по п. 1, отличающийся тем, что предшественник производного тетратиафульвалена по п.1, представлен формулой V

6. Способ получения производного тетратиафульвалена, общей формулы VI

где R1 и R2 определены в п.1, взаимодействием предшественника тетратиафульвалена общей формулы I по п.1 с триалкилфосфитом, отличающийся тем, что предшественник тетратиафульвалена общей формулы I по п.1 получают путем обработки 1,3,4,6-тетратиапентален-2,5-диона при температуре 30oC или ниже спиртовым раствором, содержащим метоксид щелочного металла, в инертной атмосфере с получением 1,3-дитиол-2-он-4,5-дитиолат-дианиона с последующим взаимодействием последнего с соединением, имеющим одновалентную или двухвалентную органическую группу, которая соответствует органическим группам, представленным R1 и R2, как определено в п.1.
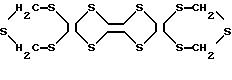
8. Способ по п.6, отличающийся тем, что производное тетратиафульвалена представлено формулой VIII
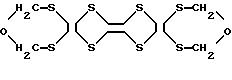
9. Способ по п.6, отличающийся тем, что производное тетратиафульвалена представлено формулой IX
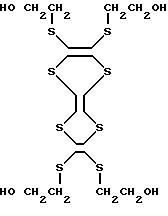
10. Способ по п.6, отличающийся тем, что производное тетратиафульвалена представлено формулой X
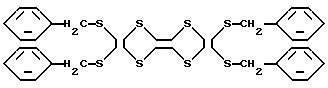
Комментарии