1-//2-(динизший алкиламино)алкил/амино/-4-замещенные тиоксантен-9-оны и фармацевтическая композиция, обладающая противоопухолевой активностью - RU2075477C1
Код документа: RU2075477C1
Чертежи



Описание
Представленное изобретение относится к новым 1-[[(диалкиламино)- алкил] амино] -4-замещенным-тиоксантен-9-онам, к фармацевтическим композициям, содержащим тиоксантеноны, к способам лечения опухолей тиоксантенонами и к способам лечения рака у млекопитающих композициями, содержащими тиоксантеноны.
Nabin и Elsheikh [J. Pharm. Sci 54 1672 1673 /1965/] описывают 1-[[2-(диэтиламино)этил] амино] -4-[(диэтиламино)-метил] тиоксантен-9-он. Однако в публикации не раскрыта полезность соединения.
В пат. США N 3745172 Collins и
Rosi описаны в качестве промежуточного соединения при синтезе противогрибковых и антибактериальных агентов

и в качестве противоглистного и антибактериального агента
Rosi и Peruzotti в пат. США N 3312598 описывают 1-[[2- (диэтиламино)этил] амино]-9-оксо-9Н-тиоксантен-4-карбоксильную кислоту в качестве побочного продукта ферментации, не раскрывая полезности соединения.
Blanz и French [J=med. Chem. 6, 185 191 /1963/] описывают синтез серии тиоксантенонов, относящихся к
лукантону, и результаты испытания соединений в лечении лейкемии и двух видов твердых опухолей.
Среди опубликованных соединений имеются такие

в которых R является метилом, метоксилом и этоксилом.
Jarinsky и Freele [Journal of Tropical Medicine and Hygine, 73, 23 29
/1970/] публикуют

в качестве антишистосомального агента.
Palmer и др. [J. Med. Chem. 31, 707 712 /1988/] публикуют N-[2-(диметиламино)этил] -9-оксо-9Н-тиоксантен-4-карбоксамида моногидрохлорид, который был испытан in vitro при лечении муринового лейкоза /L1210/ и in vivo в борьбе с клеточным лейкозом Р388, при этом было установлено, что "вряд ли имеет смысл рассматривать это соединение" в качестве вероятного противоопухолевого агента.
Данное изобретение
относится к соединениям формулы I

или к кислотно-аддитивной соли или сольвату этих соединений, в которой n равно двум или трем, преимущественно двум, R1 и R2 являются независимо один от другого низшим алкилом, преимущественно оба являются этилом; Q является остатком, выбираемым из групп, состоящих из -CH2NHR3, -CH2N(R4)SO2R7 - -CH2NHCHO; -CH=N-Ar; -C(O)NR5R6, CH2N(R4)C(O)R7; CH2N(C2H5)CHO и CH2N(R4)P(O) (O-низший алкил)2; R3 является водородом или низшим алкилом, R4 является водородом или низшим алкилом, R5 является водородом, низшим алкилом или арилом, преимущественно водородом; R6 является водородом или низшим алкилом, преимущественно водородом; R7 является низшим алкилом, преимущественно метилом или Ar; R8 является водородом, низшим алкилом, низшим алкоксилом или галогеном, а Ar представляет собой фенил или фенил, замещенный метилом, метоксилом, галогеном или нитро-группой. Ar преимущественно представляет собой фенил. Соединения являются полезными для лечения опухолей у млекопитающих.
Использованный здесь термин низший алкил описывает линейные, разветвленные или циклические углеводороды, содержащие четыре или несколько углеродных атомов. Галоген обозначает бром, хлор или фтор.
Далее изобретение относится к композициям для лечения опухолей и рака у млекопитающих, которые содержат соединения формулы I вместе с фармацевтически приемлемыми наполнителями или разбавителями.
Изобретение относится далее к способу использования соединения формулы I для лечения опухолей у млекопитающих животных, который включает прием млекопитающим соединения формулы I.
Кроме того, изобретение относится к способу использования соединения формулы I для лечения рака у млекопитающих, который состоит в приеме млекопитающим композиции соединения формулы I вместе с фармацевтически приемлемыми наполнителями или разбавителями.
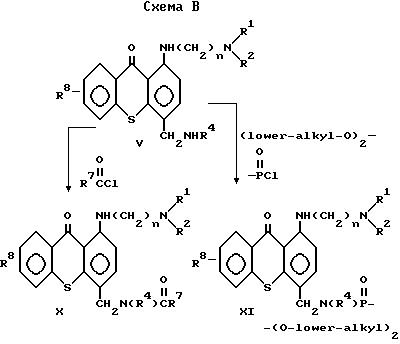
Синтез соединений изобретения в общих чертах может быть описан, как показано на схемах A и B.

Соединения формулы III /формула I, в которой Q представляет собой -CH= N-Ar/ может быть синтезировано нагреванием альдегида формулы II с приблизительно одним эквивалентом соответствующего анилина в инертном, азеотропном растворителе, преимущественно ксилоле или толуоле кипячением с обратным холодильником.
Соединения формула IV могут быть получены из альдегида формулы II нагреванием в интервале от 150o до 185o в присутствии 10 эквивалентов муравьиной кислоты в формамиде или N-алкилформамиде в качестве растворителя. Условия реакции хорошо известны под названием реакции Лейкарта. Затем соединения формулы V получаются кислотным гидролизом формамида.
Соединения формулы VI /формула I, в которой Q представляет собой -CH2N(R4)SO2R7/ могут быть синтезированы сульфонилированием амина V с небольшим избытком низшего алкилсульфонила или арилсульфонилхлорида в подходящем растворителе, преимущественно пиридине, в интервале температур от 0 до 50oC.
Или же соединения формулы VI, в которой R4 является низшим алкилом, могут быть синтезированы сульфонилированием амина V, в котором R4 является водородом, как описано выше, за чем следует действие на образовавшийся сульфонамид VI, в котором R4 является водородом, избытком основания, преимущественно гидридом натрия, в подходящем растворителе, таком как тетрагидрофуран, за которым следует действие избытка соответствующего низшего галлоидного алкила в интервале температуры от 0oC до температуры кипения используемого растворителя.
Карбоксамиды формулы IX могут быть синтезированы взаимодействием альдегида II с 5 6-кратным избытком гидрохлорида гидроксиламина в пиридине, не обязательно содержащем сорастворитель, за чем следует дегидратация оксима (VII) действием избытка уксусного ангидрида и нагревание в инертном, высококипящем растворителе, таком как ксилол и, наконец, частичный гидролиз нитрила (VIII) в концентрированной серной кислоте. В тех случаях, когда желательно, чтобы R5 и R6 имели бы отличное от водорода значение, амид IX может быть далее гидролизован в соответствующую кислоту в 16% спиртовом растворе КОН, и кислота может быть конденсирована с соответствующим амином с использованием хорошо известных способов.
Соединения формулы X /формула I, в которой Q представляет собой CH2N(R4)C(O)R7/ могут быть синтезированы ацилированием амина V избытком хлорангидрида низшей алкильной кислоты или хлорангидридом арильной кислоты в подходящем растворителе, преимущественно пиридине, необязательно в присутствии сорастворителя, преимущественно дихлорметане, при температуре в интервале от 0oC приблизительно до 50oC.
Соединения формулы XI /формула I, в которой Q представляет собой CH2N(R4)P(O) (O низший алкил)2/ могут быть получены действием на амин V избытком соответствующего ди-низшего алкил фосфорхлоридата в подходящем растворителе, таком как ди-хлорметан, в присутствии избытка основания, предпочтительно триэтиламина, в интервале температур приблизительно от 0 до 50oC.
Альдегид II получают по методу, опубликованному в пат. США N 3294803 и окислением спирта MnO2, полученного по способу, описанному в пат. США N 3711512.
Соединения формулы I являются полезными как в виде свободного основания, так и в виде кислотно-аддитивных солей, и обе эти формы охватываются изобретением.
Кислотно-аддитивные соли в некоторых случаях являются более удобной формой для использования, и на практике использование солевой формы, по существу, приближается к использованию основной формы. Кислоты, которые могут быть использованы для получения кислотно-аддитивных солей, включают предпочтительно такие, которые образуют в сочетании со свободным основанием медицински приемлемые соли, то есть соли, анионы которых являются относительно безвредными для живого организма в лечебных дозах так, чтобы полезные свойства, присущие свободному основанию, не были бы испорчены побочными эффектами, обусловленными анионами. При реализации настоящего изобретения удобно создавать солянокислые, фумаратные, полиолсульфонатные, метансульфонатные или малеатные соли. Однако данное изобретение охватывает и другие соответствующие медицински приемлемые соли производные других минеральных кислот и органических кислот. Кислотно-аддитивные соли основных соединений получаются либо растворением свободных оснований в водном спиртовом растворе, содержащем соответствующую кислоту, и выделением соли выпариванием раствора, либо взаимодействием свободного основания и кислоты в органическом растворителе, и в этом случае соль выделяется непосредственно, осаждается вторым органическим растворителем или может быть получена концентрированием раствора. Несмотря на то, что предпочтительными являются медицински приемлемые соли основных соединений, кислотно-аддитивные соли охватываются действием данного изобретения. Все кислотно-аддитивные соли являются благоприятными в качестве источника образования свободного основания, даже если конкретная соль per se является нужной только как промежуточный продукт, как, например, когда соль образуется только с целью очистки или идентификации, или когда она используется как промежуточный продукт при получении медицински приемлемой соли по способу ионного обмена.
Структуры соединений изобретения были установлены методом синтеза, элементарным анализом и инфракрасной, ультрафиолетовой и ядерной магнитной резонансной спектроскопией. Течение реакций и идентичность и однородность продуктов анализировались методом тонкослойной хроматографии /ТСХ/ или газожидкостной хроматографии /ГЖХ/. Точки плавления даны в градусах C и не исправлены. Исходные материалы являются коммерчески доступными или могут быть получены с использованием хорошо известных методов.
Итак, изобретение может быть описано более конкретно со ссылкой на следующие примеры, которые никоим образом не ограничивают объем изобретения.
Пример 1.
1-[[2-(Диэтиламино)-этил] амино] -4-(N-фенилформимидоил)-тиоксантен-9-он /I:
R1=R2=Et; Q=CH=N-C6H5;
R8=H, n=2/ /III; Ar=Ph/
Смесь 17,7 г /50 ммоль /1-[[2-(диэтиламино)-этил]амино]тиоксантен-4-карбоксиальдегида и
15,1 г /150 ммоль/ анилина в 100 мл толуола в течение 8 часов
нагревалась с обратным холодильником с ловушкой Дина-Старка. ТСХ на окиси алюминия с хлороформ /гексан/ изопропиламином 10 10 2 показала
неполноту реакции. Толуол отгонялся, было добавлено 25 мл
анилина и смесь в течение 4 часов кипятилась с обратным холодильником. Добавлялось 50 мл ксилола и реакционная смесь снова кипятилась с
обратным холодильником в течение 3 часов. Растворитель и
избыток анилина удалялись в вакууме, а остаток перекристаллизовывался из бензола с выходом 19,9 г неочищенного продукта. Этот продукт был
повторно перекристаллизован из приблизительно 1,5 л гексана с
выходом 15,8 г /86%/ продукта, т. пл 125 126oC.
Пример 2.
N-[[1-[[2-(Диэтиламино)-этил] амино]
-9-оксотиоксантен-4-ил]метил]форма- мид /I/: R1
=R2=Et; Q=CH2NHCHO; R8 H, n 2/ /IV, R4 H/
Раствор из 35,4 г /0,1
моль/ 1-[[2- (диэтиламино)-этил]амино]тиоксантен-4-карбоксиальдегида, 420 мл
формамида и 50 мл /1 моль/ муравьиной кислоты нагревался в течение 1 часа при 160oC. Реакционная смесь
охлаждалась, выливалась в 2 л воды и подщелачивалась приблизительно 50 мл 35%-ного
раствора гидроокиси натрия. Клейкий осадок отфильтровывался и сушился в вакууме. Высушенный осадок растворялся
приблизительно в 1,5 л горячего этилацетата, обрабатывался активированным углем и
кристаллизовался с использованием охлаждения. Продукт отфильтровывался, промывался этилацетатом и сушился, давая 29,0
г /75%/ продукта, т. пл. 154 155oC.
Пример 3.
N-[[1-[[2-(Диэтиламино)этил] амино] -9-оксотиоксантен-4-ил]метил]-N-мет- илформамид. /IV: R1=R2=Et; R4=Me; R8=H, n=2/
По методу,
аналогичному описанному в примере 2, было получено 24,6 г N-метилформамида из 35,4 г /0,1
моль/ 1-[[2-(диэтиламино)этил]амино]-тиоксантен-4-карбоксиальдегида, 394 г N-метилформамида и 50 мл
муравьиной кислоты. Продукт был перекристаллизован из 150 мл ацетона до т. пл. 127 130o
C.
Пример 4.
4-(Аминометил)-1-[[2-(диэтиламино)этил] амино]
-тиоксантен-9-он. /I: R1= R2=Et; Q=CH2NH2; R8=H,
n=2/ /V, R4=H/
Раствор из 24,4 г /64 ммоль/ формамида примера 2 в 240 мл 2
н. соляной кислоты нагревался в течение часа на паровой бане. Реакционная смесь охлаждалась до комнатной
температуры, подщелачивалась 35%-ным водным раствором гидроокиси натрия и образующийся желтый
осадок собирался фильтрацией. Продукт растворялся в бензоле, обрабатывался активированным углем, сушился
сульфатом магния, фильтровался и подвергался азеотропной перегонке для удаления следов воды.
Высушенный остаток перекристаллизовывался из метанола и изопропанола добавлением эфирного хлористого
водорода. Образующееся твердое вещество перекристаллизовывалось с несколькими сборами из метанола
с выходом 10,6 г продукта, т.пл. 270 272oC, в виде дигидрохлоридной соли.
Пример 5.
1-[[2-(Диэтиламино)этил] амино]-4[(метиламино-метил]тиоксантен-9-он. /I:
R1=R2=Et; Q=CH2NHCH3; R8=H, n=2/ /V, R4=Me/
По методу, в точности аналогичному тому, что описан в примере 4, было получено
10,5 г метиламина в виде полугидрата дигидрохлорида из 14,6 г /37 ммоль/ N-метилформамида из примера
3 и 150 мл 2 н. хлористоводородной кислоты. Продукт плавился при 241 243oC.
Пример 6.
N-[[1-(2-Диэтиламино)этил] амино]
-9-оксотиоксантен-4-ил]метил]метансул- ьфонамид. /I: R1=R2=Et; Q=CH2NHSO2CH3; R8=H, n=2/ /VI, R4=H; R7=Me/
Раствор 10,65 г /30 ммоль/ свободного основания амина из примера 4 в 100 мл пиридина был охлажден на ледяной бане и к
нему был добавлен в виде одной порции 4 г /35 ммоль/ метансульфонилхлорид.
Смесь перемешивалась в течение 2 часов при комнатной температуре и выливалась в 750 мл воды, содержащей 2 г гидроокиси
натрия. Темно-желтый осадок собирался, промывался водой и в течение ночи сушился
в вакууме. Второе осаждение было получено добавлением избытка гидроокиси натрия к фильтрату и отфильтровыванием
образующегося твердого вещества. Соединенные осадки после высушивания
перекристаллизовывались из бензола с выходом 6,4 г метансульфонамида, т. пл. 169 170oC.
Пример 7.
1-[[2'-(Диэтиламино)этил]
амино]-9-оксотиоксантен-4-карбоксамид. /I: R1= R2=Et; Q=CONH2; R8=H, n=2/ /IX/
Суспензия из 74 г /0,23
моль/ 1-[[2-(диэтиламино)-этил]амино]тиоксантен-4-карбоксальдегида и 74 г /1,06 моль/ гидрохлорида гидроксиламина в 400 мл пиридина и 400 мл этанола нагревалась с обратным холодильником в течение 0,5
часа и для образования гомогенного раствора добавлялось 70 мл воды. В течение следующих двух часов раствор нагревали и оставляли стоять при комнатной температуре в течение 14 часов. Образующийся
кристаллический оксим /VII/ отфильтровывался с количественным выходом, т. пл. 215 218oC.
Сто двадцать три грамма оксима в 180 мл уксусного ангидрида быстро нагревалось на паровой бане для получения раствора. Раствор охлаждался, к нему добавлялось 100 мл 1,8 M HCl в эфире и образующаяся суспензия разбавлялась 500 мл эфира. Суспензию оставляли на 14 часов при 0oC и фильтровали. Остаток /123 г, т. пл. 109 112oC/ выливался в 250 мл ксилола и кипятился в течение 20 минут с обратным холодильником. Смесь охлаждалась и отфильтровывалась 71,3 г нитрила /VIII/, т. пл. 265oC.
Десять грамм нитрила в течение 3 дней перемешивались в 200 мл конц. H2SO4 при комнатной температуре. Реакционная смесь нейтрализовалась конц. NH4OH, и остаток отфильтровывался. Остаток вываривался в теплой смеси EtOAc/EtOH, фильтровался и продукт перекристаллизовывался из охлажденного раствора, т. пл. 241 243oC. Он растворялся в этаноле и добавлялся один эквивалент HCl в этаноле. Было получено шесть грамм гидрохлорида амида, т. пл. 271 272oC.
Пример 8.
N-[[1-[[2-(Диэтиламино(этил] амино] -9-оксотиоксантен-4-ил]метил]N-мети- лметансульфонамид. /I: R1=R2=Et; Q=CH2N(CH3)SO2CH3;
R8=H; n=2/ /VI, R4=Me, R7=Me/
Раствор 1,5 г /3,5 ммоль/ метансульфонамида примера 6 в ТГФ /60 мл/ охлаждался до 0oC на ледяной бане и добавлялся
NaH 0,16 г /4,0 ммоль/. Реакционная смесь нагревалась до комнатной температуры, перемешивалась в течение 10 минут, затем добавлялся метилйодид 0,25 мл /4,0 ммоль/. Реакционная смесь перемешивалась в
течение 24 часов при комнатной температуре и растворитель удалялся в вакууме. Остаток очищался на хроматографической колонке с двуокисью кремния с элюированием хлороформом /100% /, затем 1%
изопропиламин/хлороформ, давая 1,15 г /74% / N-метилметансульфонамида в виде желтого порошка, т. пл. 175 177oC.
Пример 9.
N-[[1-[[2-(Диэтиламино)этил]
амино]
-9-оксотиоксантен-4-ил]метил]фенилс- ульфонамид. /I: R1=R2=Et; Q=CH2NHSO2Ph; R8=H, n=2/ /VI, R4=H; R7=Ph/
Следуя
методу, в основном аналогичному описанному в примере 6, было получено 2,4 г /57%/ фенилсульфонамида в виде соли метансульфоновой кислоты из 2,54 г /7,15 ммоль/ свободного основания амина
примера 4,
пиридина /50 мл/ и бензолсульфонилхлорида /1,1 мл, 8,62 ммоль/, затем следовала обработка полученного таким образом остатка метансульфоновой кислотой в метаноле. Продукт
перекристаллизовывался из
этанола.
Пример 10.
N-[[1-[[2-(Диэтиламино)этил] амино] -9-оксотиоксантен-4-ил]метил]ацетам- ид. /I: R1=R2=Et; Q=CH2NHC(O)CH3
; R8=H, n=2/ /X, R4=H, R7=Me/
Следуя методу, в основном аналогичном тому, что описан в примере 6, было получено 2,3 г
/52%/ ацетамида в виде оранжевого
твердого вещества из 4,15 г /11,7 ммоль/ свободного основания амина из примера 4, пиридина /60 мл/ и ацетилхлорида /0,82 мл, 11,53 ммоль/. Продукт
перекристаллизовывался из ацетона и плавился при 182
183oC.
Пример 11.
N-[[1-[[2-(Диэтиламино)этил] амино] -9-оксотиоксантен-4-ил]метил]бензам- ид. /I:
R1=R2=Et; Q=CH2NHC(O)Ph;
R8=H, n=2/ /X, R4=H, R7=Ph/
Следуя методу, в основном аналогичному тому, что описан в примере 6,
было получено 1,02 г /68%/ бензамида в виде желтого порошка
из 1,17 г /3,29 ммоль/ свободного основания амина из примера 4, пиридина /25 мл/ и бензоилхлорида /0,42 мл, 3,62 ммоль/. Продукт очищался
на хроматографической колонке с двуокисью кремния и
элюированием хлороформом /100%/ и смесью 1% изопропиламин / хлороформ, за чем следовала перекристаллизация из этилацетата. Продукт плавился при 161
163oC.
Пример 12.
N-[[1-[[2-(Диэтиламино)этил] амино] -9-оксотиоксантен-4-ил]диэтилфосфор- амид. /I: R1=R2=Et; Q=CH2
NPHP(O)(OEt)2, R8=H, n=2/ /XI, R4=H/
Раствор 2,28 г /6,41 ммоль/ свободного основания амина примера 4, CH2Cl2 /50 мл/ и триэтиламин /2
мл/ обрабатывались при 0oC диэтилфосфорхлоридатом
/1,0 мл, 6,9 ммоль/. Реакционная смесь в течение 2 часов перемешивалась при 0oC, затем 1 час при комнатной температуре.
Растворитель удалялся в вакууме, а остаток очищался на
хроматографической колонке с двуокисью кремния с элюированием этилацетатом /100%/, затем смесью 5% метанола / этилацета и, наконец, смесью
метанол / изопропиламин / этилацетат /5:5:90/, образуя 2,28
/72% / диэтилфосфорамида в виде желтого твердого вещества, т. пл. 108 110oC после перекристаллизации из этилацетата.
Пример 13.
N-[[1-[[2-(Диэтиламино)этил]
амино] -9-оксотиоксантен-4-ил]метил]-N-эти- лформамид. /I: R1=R2=Et; Q=CH2N Et CHO; R8=H,
n=2/ /IV, R4=Et/
Раствор, содержащий 2,0 г /5,
6 ммоль/ 1-[[2-(диэтиламино)этил]амино] тиоксантен-4-карбоксальдегид-этилформам- ид /24 мл/ и муравьиную кислоту /3,0 мл, 79,5
ммоль/, нагревался в течение 4 часов при 170oC. Реакционная
смесь охлаждалась, выливалась в воду и превращалась с основание действием 10% гидроокиси натрия. Было получено твердое вещество,
которое собиралось фильтрацией и промывалось водой. Твердый остаток
растворялся в смеси хлороформ / вода, и органический слой отделялся и сушился над Na2SO4.
Растворитель удалялся в вакууме, а остаток очищался с использованием радиальной хроматографии с элюированием смесью изопропиламин / метанол / этилацетат /0,5/1/98,5/, давая 1,32 г /57%/ N-этилформамида в виде оранжевого твердого вещества, т.пл. 75 77oC.
Пример 14.
1-[[2-(Диэтиламино)этил]амино]-4-[(этиламино)-метил]тиоксантен-9-он. /I:
R1=R2=Et; Q=CH2NHC2H5, R8=H, n=2/ /V, R4=Et/
При использовании метода, в основном аналогичного тому, что описан в
примере 4, было получено 1,29 г (92%) этиламина в виде дигидрохлорида из 1,3 г /3,2
ммоль/ N-этилформамида из примера 13 и 10,8 мл 2 н. соляной кислоты. Продукт был перекристаллизован из смеси этанол
/ тетрагидрофуран и плавился при 160oC /разл./.
Другие представители соединений вида I могут быть получены по способу, аналогичному изложенным в примерах 1 14 с замещением соответствующего 1-[[2-(диалкиламино)-этил]амино] или 1-[[3-(диалкиламино)-пропил]амино]-тиоксантен-4-карбоксальдегида 1-[[2-(диэтиламино)этил] амино] тиоксантен-4-карбоксальдегидом. Альдегиды и предшествующие им соединения описаны в пат. США N 3294803, который включен здесь в ссылку.
Представленные примеры изобретения были испытаны на антиопухолевую активность у мышей в соответствии со следующей методикой.
Животные были объединены, подвергнуты подкожному имплантированию опухолевыми фрагментами от 30 до 60 мг с использованием троакара 12-го размера и снова объединены до проведения неселективного распределения на различные воздействия и контрольные группы. На ранней стадии лечения химиотерапия начиналась в интервале от 1 до 5 дней после имплантации опухоли, когда некоторое количество клеток было относительно малым /107 108 клеток/. На прогрессирующей стадии лечения химиотерапия была замедлена до тех пор, пока опухоль не стала относительно большой /размером от 200 до 300 мг/. Опухоль в 300 мг содержит в общем приблизительно 3 • 108 клеток. Для 90% животных опухоли, относящиеся к данной прогрессирующей стадии, имели приблизительное увеличение в 2,5 раза. Опухоли измерялись штанген-циркулем еженедельно или дважды в неделю при более быстром росте опухолей. /Мыши были умерщвлены, когда их опухоль достигла 1500 мг, то есть до того, как она вызывала недомогание животного/. Веса опухолей определялись на основании двух величин измерений.
Измерения в лечебной и контрольной группе проводились тогда, когда опухоли достигли приблизительно от 700 до 1200 мг размером /средняя группа/. Определялся средний вес опухоли в каждой группе /включая ее отсутствие/. Антиопухолевая эффективность определялась как величина Т/С опухолей, на которые осуществлялось воздействие, по отношению к соответствующему весу контрольной опухоли, в процентах, значение Т/С, равное или меньшее чем 42% рассматривается как значительная антиопухолевая активность Drug Evaluation Branch. Отделения Раковой Терапии /NCJ/. Значение Т/С < 10% рассматривается как высокая антиопухолевая активность. Нижний предел потери веса тела /средняя для группы/ величиною более 20% или более чем 20% гибели от лекарства рассматривается как чрезвычайно токсичная дозировка.
Результаты приведены в таблице 1 для аденокарциномы протоков поджелудочной железы N03 и в таблице 2 для аденокарциномы толстой кишки N38.
Соединение примера 5 испытывалось в условиях внутривенного вливания против ряда других опухолей, как это показано в таблице 3, и выявило активность при 300 мг/кг против аденокарциномы толстой кишки N38.
Соединение примера 6 испытывалось при введении шариковой внутривенной инъекцией против ряда других опухолей, как это показано в таблице 4.
Фармацевтические композиции представленного изобретения включают одно или большее число соединений данного изобретения, объединенные в композиции вместе с одним или большим числом нетоксичных, физиологически приемлемых носителей, вспомогательных веществ или наполнителей, которые объединенно названы здесь как носители для парентеральных инъекций, для орального приема в твердой или жидкой форме, для ректального или местного приема и т.п.
Композиции могут приниматься людьми или животными либо орально, ректально, парентерально /внутривенно, внутримышечно или подкожно/, внутриполостно, внутривагинально, внутриперитонально, локально /порошки, мази или капли/, или как ротовой или носовой аэрозоль.
Композиции, подходящие для парентеральной инъекции, могут содержать физиологически допустимые стерильные водные или неводные растворы, суспензии, дисперсии или эмульсии и стерильные порошки для включения в стерильные, пригодные для инъекций растворы или дисперсии. Примерами подходящих водных и неводных носителей, разбавителей, растворителей или наполнителей являются вода, этанол, полиспирты /пропиленгликоль, полиэтиленгликоль, глицерин и тому подобные/, их подходящие смеси, растительные масла /такие, как оливковое масло/ и пригодные для инъекции органические эфиры, такие как этилолеат. Соответствующая подвижность может поддерживаться, например, использованием покрытия, такого как лецитин, сохранением необходимого размера частиц в случае применения дисперсий и использованием поверхностно-активных веществ.
Композиции могут также содержать вспомогательные вещества, такие как консервирующие, увлажняющие, эмульгирующие и диспергирующие агенты. Предотвращение действия микроорганизмов может быть осуществлено использованием различных антибактериальных и антигрибковых агентов, например парабенов, хлорбутанола, фенола, сорбиновой кислоты и т.п. Желательным может быть также включение в композиции изотонических агентов, например сахаров, хлористого натрия и подобных им. Пролонгирующая абсорбция пригодных для инъекции фармацевтических форм может быть достигнута использованием агентов, задерживающих абсорбцию, например моностеарата алюминия и желатина.
В случае необходимости и для более эффективного распределения соединения могут быть объединены в медленно выделяющие системы или системы целевой поставки, такие как полимерные матрицы, липосомы и микросферы. Они могут быть стерилизованы, например, фильтрованием через фильтр, задерживающий бактерии, или сочетанием стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены в стерилизованной воде или некоторых других стерильных, пригодных для инъекции средах непосредственно перед использованием.
Твердые дозированные формы для
орального приема включают капсулы, таблетки, пилюли, порошки и гранулы. Во всех твердых
дозированных формах активное соединение смешивается по меньшей мере с одним инертным обычным наполнителем /или
носителем/, таким как цитрат натрия или двукальциевый фосфат, или
/а/ наполнителями или разбавителями, как, например, крахмалы, лактоза, сахароза, глюкоза, маннитол и салициловая кислота,
/b/ связующими,
как, например, карбоксиметилцеллюлоза, альгинаты,
желатин, поливинилпирролидон, сахароза и акация
/с/ увлажняющими агентами, как, например, глицерин, /d/ измельчающими
агентами, как, например, агар агар, карбонат кальция, картофельный
крахмал или крахмал из тапиоки, альгиновая кислота, отдельные комплексные силикатные соединения и карбонат натрия.
/e/ растворами замедлителей, как, например, парафина,
/f/ ускорителями абсорбции, как, например, четвертичные аммониевые соединения,
/g/ увлажняющими агентами, как, например,
цетиловый спирт или моностеарат глицерина,
/h/ адсорбентами,
как, например, каолин и бентонит, и
/i/ смазывающими веществами, как, например, тальк, стеарат кальция, стеарат магния,
твердые полиэтиленгликоли, натрий лаурилсульфат или их смеси. В случает
капсул, таблеток и пилюль дозированные формы могут также включать буферные агенты.
Процентное содержание активного компонента в композиции и способ лечения опухолей или рака может варьироваться таким образом, чтобы была получена подходящая доза. Доза, принимаемая конкретным пациентом, изменяется в зависимости от клинической оценки с использованием в качестве критерия: пути приема, длительности лечения, возраста и условий пациента, эффективности активного компонента и реакции пациента на лекарство. Таким образом, эффективное дозированное количество активного компонента может быть легко определено в клинических условиях с учетом всех критериев и с использованием лучших решений, исходя из интересов пациента.
Реферат
Использование: для лечения опухолей у млекопитающих животных. Продукт: N-[[1-[[2-(диэтиламино)этил] амино]-9-оксотиоксантен-4-ил]метил]-N-эти- лформамид, т.пл. 75 - 77oC. N-[[1-[[2-(диэтиламино)этил]амино]-9-оксотиоксантен-4-ил] метил]-N-мет- илметансульфонамид, т.пл. 175 - 177oC. 2 с. и 13 з. п. ф-лы, 4 табл.
Формула

где n 2 или 3,
заместители R1 и R2 независимо друг от друга низший алкил;
заместитель О остаток, выбираемый из группы, включающей СН2NHR3, CH2N(R4)S O2R7, CH2NHCHO, CH=N-Ph, C(O)NR5R6, CH2N(R4)C(O)R7, CH2 N(C2H5) CHO и CH2N(R4)P(O) (O-низший алкил)2;
заместитель R3 водород или низший алкил;
заместитель R4 водород или низший алкил;
заместитель R5 водород, низший алкил, фенил или фенил, замещенный метилом, метокси или галогеном;
заместитель R6 водород или низший алкил;
заместитель R7 низший алкил, фенил или фенил, замещенный метилом, метоксигруппой или галоидом;
заместитель R8 водород, низший алкил или низшая алкоксигруппа, при условии, что когда n 2, заместители R1 и R2 этил, заместитель R8 атом водорода и Q CH2NHSO2R7, где R7 не может быть 4-монозамещенным метилом или атомом галогена фенилом,
или его фармацевтически приемлемая кислотно-аддитивная соль или сольват.
13.02.92 при Q CH2N(R4)C(O)R7, CH2 N(C2H5)CHO, CH2N(R4)Р(О)(О-низший алкил)2.
Комментарии