Новые производные n-(иминометил)аминов, их получение, их использование в качестве лекарственных средств и содержащие их фармацевтические композиции - RU2230742C2
Код документа: RU2230742C2
Описание
Предметом настоящего изобретения являются новые производные N-(иминометил)аминов, включающие в свой скелет звено аминодифениламина, оксодифениламина, карбазола, феназина, фенотиазина, феноксазина или оксодифенила. Данные производные обладают ингибирующей активностью в отношении ферментов NO-синтаз, продуцирующих монооксид азота NO, и/или активностью, которая состоит в улавливании реакционноспособных кислородных частиц (ROS). Данное изобретение относится к производным, соответствующим общей формуле (I), определенной ниже, способам их получения, содержащим их фармацевтическим препаратам и их использованию для терапевтических целей, в особенности их использованию в качестве ингибиторов NO-синтаэы и селективных или неселективных ловушек для реакционноспособных кислородных частиц.
Учитывая потенциальную роль NO и ROS в физиопатологии, описанные новые производные, соответствующие общей формуле (I), могут давать полезные или благоприятные эффекты при лечении патологий, в которых принимают участие эти химические частицы. В частности, таких потологий, как:
- Пролиферативные и воспалительные заболевания, такие как, например, атеросклероз, легочная гипертензия, респираторный дистресс-синдром, гломерулонефрит, портальная гипертензия, псориаз, артроз и ревматоидный артрит, фиброзы, ангиногенез, амилоидозы, воспаления желудочно-кишечной системы (неспецифический язвенный или неязвенный колит, болезнь Крона), диарея.
- Заболевания, поражающие легочную систему и дыхательные пути (астма, синусит, ринит).
- Сердечно-сосудистые и церебро-васкулярные нарушения, включающие, например, мигрень, артериальную гипертензию, септический шок, ишемические или геморрагические сердечные или церебральные инфаркты, ишемию и тромбозы.
- Нарушения центральной или периферической нервной системы, такие как, например, нейродегенеративные заболевания, где можно в особенности упомянуть церебральные инфаркты, субарахноидальное кровоизлияние, старение, сенильную деменцию, включая болезнь Альцгеймера, хорею Хантингтона, болезнь Паркинсона, болезнь Крейцфельда-Якоба и заболевания, вызванные прионами, боковой амиотрофический склероз; глазные невропатии, такие как глаукома, а также боль, церебральные травмы и травмы костного мозга, аддикцию по отношению к опиатам, алкоголю и аддиктивным веществам, расстройство познавательной способности, энцефалопатии и энцефалопатии вирусного или токсичного происхождения.
Нарушения скелетных мышц и нервно-мышечных суставов (миопатия, миоз), а также кожные заболевания.
- Катаракты.
- Органы-трансплантаты.
- Аутоиммунные и вирусные заболевания, такие как, например, волчанка, СПИД, инфекции, вызванные паразитами и вирусами, диабеты и их осложнения, рассеянный склероз.
- Раковые заболевания.
- Неврологические заболевания, ассоциированные с интоксикацией (отравление кадмием, вдыхание н-гексана, пестицидов, гербицидов), ассоциированные с лечениями (радиотерапия) или нарушениями генетического происхождения (болезнь Вильсона).
- Все патологии, характеризующиеся избыточным продуцированием или дисфункцией NO и/или ROS.
Во всех указанных патологиях имеется экспериментальное доказательство, демонстрирующее вовлеченность NO или ROS в патологический процесс (J. Med. Chem. (1995) 38, 4343-4362; Free Radio. Biol. Med. (1996) 20, 675-705; The Neuroscientist (1997) 3, 327-333).
Более того, в более ранних патентах авторы данного изобретения уже описали ингибиторы NO-синтазы и их использование (патент США 5081148; патент США 5360925) и совсем недавно описали сочетание этих ингибиторов с продуктами, обладающими антиоксидантными или антирадикулярными свойствами (заявка на патент РСТ WO 98/09653). В еще не опубликованных заявках они описали также другие производные амидинов или, совсем недавно, производные аминопиридинов.
Эти производные амидинов или аминопиридинов имеют характерную особенность, заключающуюся в том, что они являются как ингибиторами NO-синтаз, так и ингибиторами ROS.
Предметом настоящего изобретения являются новые производные амидинов, их получение и их использование в терапии.
Соединения данного изобретения соответствуют общей формуле (I):

в которой Ф представляет связь или фениленовый радикал, который может включать помимо двух цепей, уже представленных в общей формуле (I), вплоть до двух заместителей, выбранных из атома водорода, галогена, ОН-группы и неразветвленного или разветвленного алкильного или алкоксирадикала, имеющего от 1 до 6 атомов углерода;
А представляет радикал

в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, галоген, ОН-группу или неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или циано, нитро или радикал NR6R7,
причем R6 и R7 представляют независимо атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или, кроме того, группу –COR8,
причем R8 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или NR9R10,
причем R9 и R10 представляют, независимо, атом водорода, ОН-группу или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
R11 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или радикал –COR12,
причем R12 представляет атом водорода, ОН-группу или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
или радикал

в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, галоген, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или циано, нитро или радикал NR6R7,
причем R6 и R7 представляют независимо атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или, кроме того, группу –COR8,
причем R8 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или NR9R10,
причем R9 и R10 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
В представляет –CH2-NO2, неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода, карбоциклический или гетероциклический арил с 5 или 6 членами, содержащий от 1 до 4 гетероатомов, выбранных из О, S, N, и, в частности, радикал тиофена, фурана, пиррола или тиазола, причем арильный радикал необязательно замещен одной или несколькими группами, выбранными из неразветвленных или разветвленных алкильных, алкенильных и алкоксирадикалов, имеющих от 1 до 6 атомов углерода, или В представляет радикал NR13R14, в котором R13 и R14 представляют независимо атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода, или циано- или нитрорадикал, или R13 и R14 вместе с атомом азота образуют неароматический гетероцикл с пятью или шестью членами, причем элементы цепи выбраны из группы, состоящей из –CH2-, -NH-, -О- или -S-;
W отсутствует или представляет связь или О, S или NR15, где R15 представляет атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода;
X представляет связь или радикал -(СН2)k-NR16-, -О-, -S-, -СО-, -NR16-CO-, -CO-NR16-, -O-СО-, -СО-O-, -NR16-CO-O-, -NR16-CO-NR17-,
причем k равно 0 или 1;
Y представляет связь или радикал, выбранный из -(СН2)m-, -(СН2)m-O-(СН2)n-, -(СН2)m-S-(СН2)n-, -(CH2)m-NR18-(СН2 )n-, -(CH2)m-NR18-CO-(CH2)n-, -(CH2)m-CO-NR18-(CH2)n-, -(СН2)m -Q-(СН2)n-,
причем Q представляет пиперазиновый, гомопиперазиновый, 2-метилпиперазиновый, 2,5-диметилпиперазиновый, 4-оксипиперидиновый или 4-аминопиперидиновый радикал,
m и n равны целым числам от 0 до 6;
R16, R17 и R18 представляют независимо атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
или являются солями продуктов, указанных ранее.
Под термином "разветвленный или неразветвленный алкил, имеющий от 1 до 6 атомов углерода" подразумевается, в частности, метильный, этильный, пропильный, изопропильный, бутильный, изобутильный, втор-бутильный и трет-бутильный, пентильный, неопентильный, изопентильный, гексильный, изогексильный радикалы. Под термином "неразветвленный или разветвленный алкокси, имеющий от 1 до 6 атомов углерода" подразумевается -О-алкильный радикал, где значения алкильного радикала указано ранее. Наконец, под термином "галоген" подразумеваются атомы фтора, хлора, брома и йода.
Предпочтительными соединениями по данному изобретению являются соединения общей формулы (I), у которых:
А представляет радикал

в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода,
R11 представляет атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
или радикал

в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода;
В представляет карбоциклический или гетероциклический арил с 5 или 6 членами, содержащий от 1 до 4 гетероатомов, выбранных из О, S, N, и, в частности, радикалы тиофена, фурана, пиррола или тиазола, причем арильный радикал необязательно замещен одной или несколькими группами, выбранными из неразветвленных или разветвленных алкильных, алкенильных и алкоксирадикалов, имеющих от 1 до 6 атомов углерода;
W отсутствует или представляет связь, S или NR15, где R15 представляет атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода;
Х представляет связь или радикал (CH2)k-NR16-, -О-, -S-, -СО-, -NR16-CO-, -CO-NR16-, -O-СО-, -СО-O-, -NR16-CO-O-, -NR16-CO-NR17-,
причем k равно 0 или 1;
Y представляет связь или радикал, выбранный из -(СН2)m-, -(СН2)m -O-(СН2)n-, -(СН2)m-S-(СН2)n-, -(CH2)m-NR18-(СН2)n-, -(CH2)m-NR18 -CO-(CH2)n-, -(CH2)m-CO-NR18-(CH2)n-, -(СН2)m-Q-(СН2)n-,
причем Q представляет пиперазиновый, гомопиперазиновый, 2-метилпиперазиновый, 2,5-диметилпиперазиновый, 4-оксипиперидиновый или 4-аминопиперидиновый радикал,
m и n равны целым числам от 0 до 6;
или являются солями продуктов, указанных ранее.
Более предпочтительно соединениями по данному изобретению являются соединения общей формулы (I), в которой
А представляет радикал

в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода,
R11 представляет атом водорода или метильный радикал,
или радикал
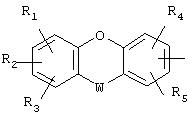
в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода;
В представляет один из фенильных, тиофеновых, фурановых, пиррольных или тиазольных радикалов, необязательно замещенных одной или несколькими группами, выбранными из неразветвленного или разветвленного алкильного, алкенильного или алкоксирадикала, имеющего от 1 до 6 атомов углерода;
W отсутствует или представляет связь, S или NR15, где R15 представляет атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода;
Х представляет связь или радикал -(CH2)k-NR16-, -О-, -S-, -СО-, -NR16-CO-, -CO-NR16-, -O-СО-, -СО-O-, -NR16-CO-O-, -NR16-CO-NR17 -,
причем k равно 0 или 1;
Y представляет связь или радикал, выбранный из -(СН2)m-, -(СН2)m-O-(СН2)n-, -(СН2)m-S-(СН2)n-, (СН2)m-NR18-(CH2)n-, -(CH2)m-NR18-CO-(CH2)n-, -(CH2)m-CO-NR18-(CH2)n-, -(СН2)m-Q-(СН2)n-,
причем Q представляет пиперазиновый, гомопиперазиновый, 2-метилпиперазиновый, 2,5-диметилпиперазиновый, 4-оксипиперидиновый или 4-аминопиперидиновый радикал,
m и n равны целым числам от 0 до 6;
или являются солями продуктов, указанных ранее.
Еще более предпочтительными соединениями по данному изобретению являются соединения общей формулы (I), в которой
А представляет радикал

в котором R1, R2, R3, R4, R5 представляют независимо атом водорода или метильный радикал,
R11 представляет атом водорода или метильный радикал;
В представляет тиофеновый радикал;
W отсутствует или представляет простую связь или S;
Х представляет связь или радикал -(СН2)k-NR16-, -О-, -S-, -СО-, -NR16-CO-, -CO-NR16-, -O-СО-, -СО-O-, -NR16-CO-O-, -NR16-CO-NR17-;
причем k равно 0 или 1;
Y представляет связь или радикал, выбранный из -(СН2)m-, -(СН2)m, -O-(СН2)n-, -(СН2)m-S-(СН2)n-, (СН2)m-NR18-(CH2)n-, -(CH2)m-NR18-CO-(CH2)n-, (СН2)m -CO-NR18-(CH2)n-, -(CH2)m-Q-(CH2)n-,
причем Q представляет пиперазин,
m и n равны целым числам от 0 до 6;
R16, R17 и R18 представляют атом водорода;
или являются солями продуктов, указанных ранее.
Очень предпочтительными являются в особенности следующие соединения, описанные в примерах:
-N-[4-(фениламино)фенил]-2-тиофенкарбоксимидамид;
-4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]-бензолацетамид;
-{4-{[2-тиенил(имино)метил]амино}фенокси}-N-[4-(фениламино)фенил]ацетамид;
-4-{[2-тиенил(имино)метил]амино}-N-[2(фениламино)фенил]-бензолбутанамид;
-4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]-бензолбутанамид;
-4-{[2-тиенил(имино)метил]амино}-N-[4-(4-метоксифениламино)фенил]бензолбутанамид;
-2-{4-{[2-тиенил(имино)метил]амино}фенил}этил-[4-(фениламино)фенил]карбамат;
-N-{2-{4-{[2-тиенил(имино)метил]амино}фенил}этил}-N’-[4-(фениламино)фенил]мочевина;
-4-{4-{[2-тиенил(имино)метил]амино}фенил}-N-[4-(фениламино)фенил]-1-пиперазинацетамид;
-1-{[(4-фениламино)фениламино]карбонил}-4-{4-{[2-тиенил-(имино)метил]амино}фенил}пиперазин;
-4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]-бензолбутанамин;
-3-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]-бензолпропанамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-[2-(4-то-луидино)фенил]бутанамид;
-4-анилинофенил-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)бутаноат;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил-N-[2-(4-то-луидино)фенил]бутанамид;
-N’-{4-[4-(3-анилинофенокси)бутил]фенил}-2-тиофенкарбоксимидамид;
-N’-(9Н-карбазол-3-ил)-2-тиофенкарбоксимидамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(9Н-карбазол-3-ил)бутанамид;
-N’-[4-(10Н-фенотиазин-2-илокси)фенил]-2-тиофенкарбоксимидамид;
-N’-{4-[(10-метил-10Н-фенотиазин-2-ил)окси]фенил)-2-тиофенкарбоксимидамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(10Н-фенотиазин-3-ил)бутанамид;
-N’-(4-{4-[2-(10Н-фенотиазин-2-илокси)этил]-1-пиперазинил}фенил)-2-тиофенкарбоксимидамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-[4-(4-то-луидино)фенил]бутанамид;
-3-анилинофенил-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)бутаноат;
-2-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-[2-(9Н-карбазол-4-илокси)этил]ацетамид;
-N-(4-{[амино(2-тиенил)метилиден]амино}фенетил)-2-анилинобензамид;
-N-(4-{[амино(2-тиенил)метилиден]амино}фенетил)-2-(2,3-ди-метиланилино)бензамид;
-N’-{4-[4-(2-анилинобензоил)-1-пиперазинил]фенил}-2-тио-фенкарбоксимидамид;
-N’-(4-{4-[2-(2,3-диметиланилино)бензоил]-1-пиперазинил}-фенил)-2-тиофенкарбоксимидамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(4-фено-ксифенил)бутанамид;
-N-(4-{[амино(2-тиенил)метилиден]амино}фенетил)-4-(4-гидроксифенокси)бензамид;
-N-[2-(9Н-карбазол-4-илокси)этил]-2-тиофенкарбоксимидамид;
-N-[3-(9Н-карбазол-4-илокси)пропил]-2-тиофенкарбоксимидамид;
-N-{4[4-(10Н-фенотиазин-2-илокси)бутил]фенил}-2-тиофенкарбоксимидамид;
-3-[(3-{[амино(2-тиенил)метилиден]амино}бензил)амино]-N-(4-анилинофенил)пропанамид;
-N’-(4-{2-[(10Н-фенотиазин-3-илметил)амино]этил}фенил)-2-тиофенкарбоксимидамид;
-N-(4-{[амино(2-тиенил)метилиден]амино}фенетил)-2-метокси-10Н-фенотиазин-1-карбоксамид;
-N-[4-(2-{[(2-метокси-10Н-фенотиазин-1-ил)метил]амино}-этил)фенил]-2-тиофенкарбоксимидамид;
-N’-{4-[(10Н-фенотиазин-2-илокси)метил]фенил}-2-тиофенкар-боксимидамид;
или их соли.
Среди указанных в качестве примеров соединений следующие соединения особенно предпочтительны:
-{4-{[2-тиенил(имино)метил]амино}фенокси}-N-[4-(фениламино)фенил]ацетамид;
-4-{[2-тиенил(имино)метил]амино}-N-[2-(фениламино)фенил]-бензолбутанамид;
-4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]-бензолбутанамид;
-2-{4-{[2-тиенил(имино)метил]амино}фенил}этил-[4-(фениламино)фенил] карбамат;
-4-{4-{[2-тиенил(имино)метил]амино}фенил}-N-[4-(фениламино)фенил]-1-пиперазинацетамид;
-3-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]-бензолпропанамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-[2-(4-то-луидино)фенил]бутанамид;
-N’-{4-[4-(3-анилинофенокси)бутил]фенил}-2-тиофенкарбоксимидамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(9Н-карбазол-3-ил)бутанамид;
-N’-[4-(10Н-фенотиазин-2-илокси)фенил]-2-тиофенкарбоксимидамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(10Н-фенотиазин-3-ил)бутанамид;
-N’-(4-{4-[2-(10Н-фенотиазин-2-илокси)этил]-1-пиперазинил}фенил)-2-тиофенкарбоксимидамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(4-фено-ксифенил)бутанамид;
-3-[(3-{[амино(2-тиенил)метилиден]амино}бензил)амино]-N-(4-анилинофенил)пропанамид;
-N’-(4-{2-(10Н-фенотиазин-3-илметил)амино]этил)фенил)-2-тиофенкарбоксимидамид;
-N-(4-{[амино(2-тиенил)метилиден]амино}фенетил)-2-метокси-10Н-фенотиазин-1-карбоксамид;
или их соли.
Особенно более предпочтительны также следующие соединения:
-4-{[2-тиенил(имино)метил]амино}-N-[2-(фениламино)фенил]-бензолбутанамид;
-4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]-бензолбутанамид;
-N’-[4-(10Н-фенотиазин-2-илокси)фенил]-2-тиофенкарбоксимидамид;
-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(10Н-фенотиазин-3-ил)бутанамид;
-3-[(3-{[амино(2-тиенил)метилиден]амино}бензил)амино]-N-(4-анилинофенил)пропанамид;
-N’-(4-{2-[(10Н-фенотиазин-3-илметил)амино]этил}фенил)-2-тиофенкарбоксимидамид;
или их соли.
В общем смысле будут предпочтительны соединения общей формулы (I), в которой Х представляет связь или один из радикалов -О-, -CH2-NR16-, -NR16-CO- или –NR16-CO-O- и Y представляет один из радикалов -(СН2)m- или -(CH2)m-NR18-(СН2)n-.
В некоторых случаях соединения по настоящему изобретению могут содержать асимметричные атомы углерода. В результате этого соединения по настоящему изобретению имеют две возможные энантиомерные формы, то есть конфигурации "R" и "S". Настоящее изобретение включает две энантиомерные формы и все сочетания этих форм, включая рацемические смеси "RS". Чтобы упростить материал, когда в структурных формулах не указывается конкретная конфигурация, следует понимать, что представлены две энантиомерные формы и их смеси.
Данное изобретение, кроме того, относится как к новым промышленным продуктам к промежуточным продуктам синтеза общей формулы (IS), которые пригодны для получения продуктов общей формулы (I), определенных выше
A-X-Y-Ф-Т
(IS)
В общей формуле (IS)
А представляет радикал

в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, галоген, ОН-группу или неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или циано, нитро или радикал NR6R7,
причем R6 и R7 представляют независимо атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или, кроме того, группу –COR8, причем R8 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или NR9R10,
причем R9 и R10 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
R11 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или радикал –COR12, причем R12 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
или радикал
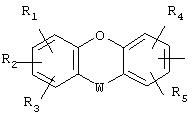
в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, галоген, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или циано, нитро или радикал NR6R7,
причем R6 и R7 представляют независимо атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или группу –COR8,
причем R8 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или NR9R10,
причем R9 и R10 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
W отсутствует или представляет связь или О, S или NR15, где R15 представляет атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода;
Х представляет связь или радикал -(СН2)k -NR16-, -О-, -S-, -СО-, -NR16-CO-, -CO-NR16-, -O-СО-, -СО-O-, -NR16-CO-O-, или –NR16-CO-NR17-,
причем k равно 0 или 1;
Y представляет связь или радикал, выбранный из радикалов -(СН2)m-, -(СН2)m-O-(СН2)n-, -(CH2)m -S-(CH2)n-, -(СН2)m-NR18-(CH2)n-, -(CH2)m-NR18-CO-(CH2)n-, -(CH2)m-CO-NR18-(CH2)n-, -(CH2)n-Q-(CH2)n-.
причем Q представляет пиперазин, гомопиперазин, 2-метилпиперазин, 2,5-диметилпиперазин, 4-оксипиперидин или 4-аминопиперидин,
m и n равны целому числу от 0 до 6;
Ф представляет связь или фениленовый радикал, который может включать в дополнение к двум цепям, уже представленным в общей формуле (I), вплоть до двух заместителей, выбранных из атома водорода, галогена, ОН-группы и неразветвленного или разветвленного алкильного радикала, имеющего от 1 до 6 атомов углерода;
Т представляет NO2 или NH2;
R16, R17 и R18 представляют независимо атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода;
Изобретение далее относится как к новым промышленным продуктам к промежуточным продуктам общей формулы (IS’), которые пригодны для получения продуктов общей формулы (I), в которой Х представляет радикал –NR16-CO- и Y представляет радикал (CH2 )m-NR18-(CH2)n-.

В общей формуле (IS’)
А представляет радикал

в котором R1, R2, R3, R4, и R5 представляют независимо атом водорода, галоген, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или циано, нитро или радикал NR6R7,
причем R6 и R7 представляют, независимо, атом водорода ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или группу –COR8,
причем R8 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или NR9R10,
причем R9 и R10 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
R11 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или группу –COR12,
причем R12 представляет атом водорода, ОН-группу или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода;
или радикал

в котором R1, R2, R3, R4, R5 представляют независимо атом водорода, галоген, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или циано, нитро или радикал NR6R7,
причем R6 и R7 представляют независимо атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или также группу –COR8,
причем R8 представляет атом водорода, ОН-группу, неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода, или NR9R10,
причем R9 и R10 представляют независимо атом водорода, ОН-группу, неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода;
W не присутствует или представляет связь или О, S или NR15, где R15 представляет атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
π представляет атом водорода или защитную группу карбаматного типа;
R16, R17 и R18 представляют независимо атом водорода или неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода,
и m равно целому числу от 0 до 6.
Предметом данного изобретения являются также используемые в качестве лекарственных средств соединения общей формулы (I), описанные ранее, или их фармацевтически приемлемые соли. Оно относится также к фармацевтическим композициям, содержащим указанные соединения или их фармацевтически приемлемые соли, и использованию указанных соединений или их фармацевтически приемлемых солей для получения лекарственных средств, предназначенных для ингибирования нейронной NO-синтазы или индуцируемой NO-синтазы, для ингибирования липидного перокисления или для обеспечения двойной функции ингибирования NO-синтазы и функции ингибирования липидного перокисления;
Под термином "фармацевтически приемлемая соль" подразумеваются, в частности, аддитивные соли неорганических кислот, такие как гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат, дифосфат и нитрат, или органических кислот, таких как ацетат, малеат, фумарат, тартрат, сукцинат, цитрат, лактат, метансульфонат, п-толуолсульфонат, памоат, оксалат и стеарат. Соли, образованные из оснований, таких как гидроксид натрия или калия, также находятся в пределах объема настоящего изобретения, когда их можно использовать. Для других примеров фармацевтически приемлемых солей можно дать ссылку на "Pharmaceutical salts", J. Pharm. Sci. 66:1 (1977).
Фармацевтическая композиция может быть в твердой форме, например в форме порошков, гранул, таблеток, желатиновых капсул, липосом или суппозиторий. Подходящими твердыми носителями могут быть, например, фосфат кальция, стеарат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, по-ливинилпирролидон и воск.
Фармацевтические композиции, содержащие соединение данного изобретения? могут быть представлены также в жидкой форме, например в форме растворов, эмульсий, суспензий или сиропов. Подходящими жидкими носителями могут быть, например, вода, органические растворители, такие как глицерин или гликоли, а также их смеси, в различных пропорциях, с водой.
Лекарственное средство по данному изобретению можно вводить местным, пероральным, парентеральным путем, внутримышечной инъекцией и т.п.
Предусмотренная доза введения для лекарственного средства по данному изобретению находится между 0,1 мг и 10 г в соответствии с типом используемого активного соединения.
В соответствии с данным изобретением соединения общей формулы (I) можно получить способом, описанным ниже.
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ОБЩЕЙ ФОРМУЛЫ (I)
Соединения общей формулы (I) можно получить из промежуточных продуктов общей формулы (II) в соответствии со схемой 1, где А, В, X, Y и Ф имеют указанные выше значения, a Gp представляет защитную группу карбаматного типа, такую как, например, трет-бутоксикарбонильная группа.
Схема 1

Производные анилина общей формулы (II) можно конденсировать с соединениями общей формулы (III), в которой L представляет уходящую группу (например, радикал алкокси, алкилтио, аралкилтио, сульфоновой кислоты, галогенида, арилового спирта или тозильный), чтобы получить конечные соединения общей формулы (I) типа замещенного амидина (ср. схему 1). Например, для случая, когда В = тиофен, производные общей формулы (II) можно конденсировать с гидроиодидом S-метилтиофентиокарбоксамида, полученным по способу, описанному в литературе (Ann. Chim. (1962), 7, 303-337). Конденсацию можно проводить нагреванием в спирте (например, в метаноле или изопропаноле), необязательно в присутствии ДМФА и/или пиридина при температуре, предпочтительно находящейся между 20 и 100°С, в течение времени, обычно находящемся между несколькими часами и всей ночью.
В случае когда В представляет амин, конечными соединениями общей формулы (I) являются гуанидины. Их можно получить, например, конденсацией аминов общей формулы (II) с производными общей формулы (IV) или (IV'). Реагенты общей формулы (IV), в которой L представляет, например, кольцо пиразола, конденсируют с аминами общей формулы (II) в соответствии с условиями, описанными в литературе (J. Оrg. Спет. (1992), 57, 2497-2502), аналогично и для реагентов общей формулы (IV), в которой L представляет, например, кольцо пиразола и Gp представляет трет-ВuОСО-группу (Tetrahedron Lett. (1993) 34 (21), 3389-3392), или, когда L представляет группу -N-SO2-CF3 и Gp представляет трет-BuOCO-группу (J. Org. Chem. (1998) 63, 3804-3805). Во время конечной стадии синтеза снятие защитной группы гуанидиновой функциональной группы проводят в присутствии сильной кислоты, такой как, например, трифторуксусная кислота.
Следовательно, данное изобретение относится также к способу получения продукта общей формулы (I), как определено выше, отличающемуся тем, что промежуточный продукт общей формулы (II)
A-X-Y-Ф-NH2
(II)
в которой А, В, X, Y и Ф имеют указанные выше значения, подвергают взаимодействию с промежуточным продуктом общей формулы (III)

в которой В имеет указанные выше значения и L представляет уходящую группу, например, радикал алкокси, алкилтио, аралкилтио, сульфоновой кислоты, галогенида, арилового спирта или тозильный.
Кроме того, данное изобретение относится к способу получения продукта общей формулы (I), в которой В представляет амин, отличающемуся тем, что промежуточный продукт общей формулы (II)
А-Х-Y-Ф-NН2
(II)
в которой А, В, X, Y и Ф имеют указанные выше значения, подвергают взаимодействию
а) либо с промежуточным продуктом общей формулы (IV)

в которой L представляет уходящую группу, например радикал алкокси, алкилтио, аралкилтио, сульфоновой кислоты, галогенида, арилового спирта или тозильный,
б) либо с промежуточным продуктом общей формулы (IV)

в которой L представляет уходящую группу, например радикал алкокси, алкилтио, аралкилтио, сульфоновой кислоты, галогенида, арилового спирта или тозильный, и Gp представляет защитную группу карбаматного типа, например трет-бутоксикарбонильную группу,
причем за данным взаимодействием следует в случае, когда выбирают взаимодействие с соединением общей формулы (IV), гидролиз в присутствии сильной кислоты, например, трифторуксусной кислоты.
Когда -X-Y-Ф- представляет простую связь:
Промежуточные продукты общей формулы (II) в конкретном случае, когда -Х-Y-Ф- представляет простую связь, сравнимы с соединениями общей формулы (X), A-NH2, описанными в разделе "Синтез промежуточных продуктов". В этом случае указанные амины A-NH2 можно непосредственно конденсировать с производными общей формулы (III) или (IV), как описано в предыдущем разделе.
Получение соединений общей формулы (II):
Некоммерческие промежуточные продукты общей формулы
(II) получают либо удалением защитной группы либо восстановлением предшественника нитридного или нитротипа, как показано в приведенных ниже схемах синтеза.
Снятие защиты у аминогруппы:
Промежуточные продукты общей формулы (II), в которой А, X, Y и Ф имеют указанные выше значения, можно получить из промежуточных продуктов общей формулы (V) (схема 2), которые являются соединениями, включающими защитный амин (N=Gp’) в форме, например, фталимида или 2,5-диметилпиррола. В случае фталимидов защиту снимают стандартным способом с использованием гидразин-гидрата при кипячении этанола с обратным холодильником, а в случае пирролов снятие защиты проводят нагреванием в присутствии гидрохлорида гидроксиламина для получения в конечном счете первичных аминов общей формулы (II).
Схема 2

Восстановление предшественников азидотипа:
Синтетические промежуточные продукты общей формулы (VI) (схема 3, в которой А, X, Y и Ф имеют значения, указанные выше) являются производными азидов, которые превращают в первичный амин общей формулы (II), например, с использованием водорода в присутствии Pd/C в подходящем растворителе, таком как этанол.
Схема 3
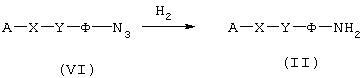
Восстановление предшественников нитротипа:
Восстановление функциональной нитрогруппы промежуточных продуктов общей формулы (VII) (схема 4, в которой А, X, Y и Ф имеют указанные выше значения) обычно проводят каталитическим гидрированием в этаноле в присутствии Pd/C за исключением случая молекул, чувствительных к этим условиям, когда нитрогруппу селективно восстанавливают, например, нагреванием продукта в подходящем растворителе, таком как этилацетат с небольшим количеством этанола, в присутствии SnCl2 (J. Heterocyclic Chem. (1987), 24, 927-930; Tetrahedron Letters (1984), 25 (8), 839-842), с использованием также SnCl2 в присутствии Zn (Synthesis (1996), (9), 1076-1078) или с использованием NaBH4-BiCl3 (Synth. Com. (1995), 25 (23), 3799-3803} в растворителе, таком как этанол, или затем с использованием никеля Ренея с добавленным гидразин-гидратом (Monatshefte fur Chemie, (1995), 126, 725-732; Pharmazie (1993), 48 (11), 817-820)) в случае, например, нитрокарбазолов.
Схема 4

Получение соединений общей формулы (V):
Промежуточные продукты общей формулы (V) (схема 5) содержат амин, защищенный в форме фталимида, в которой Х представляет -О-, Y представляет -(СН2)m- и A, R1, R2, R3, R4, R5, W, m и Ф имеют значения, указанные выше, можно получить из гидроксилированных ароматических колец общей формулы (VIII). В частном случае гидроксикарбазолов соединения общей формулы (VIII) получают а соответствии с экспериментальным протоколом в литературе (J. Chem. Soc. (1955), 3475-3477; J. Med. Сhem. (1964), 7, 158-161), и в случае гидроксифенотиазинов протокол описывается в J. Med. Сhem. (1992), 35, 716. Соединения общей формулы (VIII) конденсируют с коммерческими галогеналкилфталимидами в присутствии основания, например NaH, в растворителе, таком как ДМФА, для получения промежуточных продуктов общей формулы (V).
Схема 5

Получение соединений общей формулы (VI):
Промежуточные продукты общей формулы (VI) (схема 6) в которой А, X, Y, R1, R2, R3, R4, R5, W, m и Ф имеют значения, указанные выше, являются производными азидотипа. Их получают в две стадии из промежуточных продуктов общей формулы (VIII) (схема 5). Радикал ОН соединений общей формулы (VIII) можно алкилировать дигалогенированными производными типа дибромалкана в присутствии основания, например NaH или NaOH, для получения соединений общей формулы (IX), которые затем замещают с использованием азида натрия в ДМФА для получения промежуточных продуктов общей формулы (VI).
Схема 6

Синтезы соединений общей формулы (VII), которые содержат концевую нитрогруппу и у которых А, X, Y и Ф имеют значения, указанные выше, иллюстрируются в следующих схемах синтезов.
Синтез карбоксамидов общей формулы (VII):
Карбоксамиды общей формулы (VII) (схема 7), в которой Х представляет –NR16-CO- и А, Y, Ф и R16 имеют указанные выше значения, получают конденсацией коммерческих аминов общей формулы (X) с коммерческими кислотами общей формулы (XI). Карбоксамидные связи образуются в стандартных условиях синтеза пептидов (М. Bodanszky and A. Bodanszky, The Practice of Peptide Synthesis, 145 (Springer-Verlag, 1984)) в ТГФ, дихлорметане или ДМФА в присутствии конденсирующего реагента, такого как дициклогексилкарбодиимид (ДЦК), 1,1’-карбонилдиимидазол (КДИ) (J. Med. Chem. (1992), 35 (23), 4464-4472) или гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC или WSCI) (John Jones, The chemical synthesis of peptides, 54 (Clarendon Press, Oxford, 1991)). Синтез некоммерческих аминов общей формулы (X) и синтез некоммерческих карбоновых кислот общей формулы (XI) описывается в разделе "Получение промежуточных продуктов".
Схема 7

Карбоксамиды общей формулы (VII) (схема 8), в которой Х представляет -CO-NR16- и A, Y, Q, Ф и R16 имеют указанные выше значения, получают конденсацией коммерческих кислот общей формулы (XII) с коммерческими аминами общей формулы (XIII) или аминами общей формулы (XIV) в стандартных условиях для синтеза пептидов, описанного ранее. Синтезы некоммерческих кислот общей формулы (XII) и аминов общей формулы (XIV) описываются в разделе "Получение промежуточных продуктов".
Схема 8

Карбоксамиды общей формулы (VII) (схема 9), в которой Х представляет -О-, Y представляет (CH2)m-NR18-CO(CH2)n- и A, R18, m, n и Ф имеют значения, указанные выше, получают стандартной пептидной конденсацией кислот общей формулы (XI) (схема 7) с аминами общей формулы (II), синтез которых был описан в схемах 2 и 3.
Схема 9

Синтез аминов общей формулы (VII):
Амины общей формулы (VII), в которой Х представляет –NR16- и Y представляет -(СН2 )m- и A, R16, m и Ф имеют значения, указанные выше, получают (схема 10) из карбоксамидов общей формулы (VII). Восстановление карбоксамидной функциональной группы проводят в присутствии избытка (5 экв.) диборана в ТГФ нагреванием смеси до кипения растворетеля с обратным холодильником с получением аминов общей формулы (VII).
Схема 10

Синтез карбаматов общей формулы (VII):
Производные карбаматов общей формулы (VII), в которой Х представляет –NR16-CO-O- и Y представляет -(СН2)m- и A, m и Ф имеют значения, указанные выше, получают (схема 11) конденсацией амина общей формулы (X) (схема 7) с коммерческим спиртом общей формулы (XV) в присутствии трифосгена и основания, такого как, например, N,N-диметиланилин, в инертном растворителе, таком как, например, дихлорметан, в соответствии с протоколом, описанным в Tetrahedron Lett. (1993), 34 (44), 7129-7132.
Схема 11

Синтез мочевин общей формулы (VII):
Мочевины общей формулы (VII), в которой Х представляет –NR16-CO-NR17- и Y представляет -(СН2)m- или X-Y представляет –NR16-CO-Q- (в случае азотистого гетероцикла) и А, R17, m, Q и Ф имеют значения, указанные выше, получают (схема 12) из первичных аминов общей формулы (X) (схема 7) и аминов общей формулы (XIII) или (XIV) (схема 8) в присутствии трифосгена и третичного амина, такого как, например, диизопропилэтиламин, в нейтральном растворителе, таком как дихлорметан (J. Org. Chem. (1994), 59 (7), 1937-1938).
Схема 12

Синтез сложных эфиров общей формулы (VII):
Эфиры карбоновых кислот общей формулы (VII), в которой Х представляет -O-СО- или -СО-O- и Y представляет -(CH2)m-, а А, m и Ф имеют значения, указанные выше, получают в одну стадию из спиртов общей формулы (VIII) (схема 5) и карбоновых кислот общей формулы (XI) (схема 7) или кислот общей формулы (XII) (схема 8) и спиртов общей формулы (XV) (схема 11) в присутствии конденсирующего агента, такого как, например, карбонилдиимидазол или дициклогексилкарбодиимид, в подходящем растворителе, таком как, например, дихлорметан.
Схема 13

Синтез простых эфиров общей формулы (VII)
Простые эфиры общей формулы (VII), в которой Х представляет –О- и Y представляет -(СН2)m-, a A, m и Ф имеют указанные выше значения, (схема 14), получают в одну стадию конденсацией ароматических спиртов общей формулы (VIII) (схема 5) и спиртов общей формулы (XV) (схема 11) в стандартных условиях реакции Мицунобу (Synthesis (1981), 1) в присутствии, например, диэтилазодикарбоксилата и трибутилфосфина, в растворителе, таком как, например, ТГФ.
Схема 14

Когда Х представляет -О-, Y представляет связь и Ф представляет фенилен, а А и n имеют указанные выше значения, простые эфиры общей формулы (VII) (схема 15) можно также получить в одну стадию конденсацией ароматических спиртов общей формулы (VIII) (схема 5) с галогенированными производными общей формулы (XVI), в которой Hal представляет атом галогена, в присутствии основания, такого как, например, К2СО3, в полярном растворителе, таком как, например, ТГФ или ДМФА при температуре реакции, находящейся между 20 и 140°С. Схема 15

Когда X представляет –О- и Y представляет -(CH2)m-Q-(СН2)n-, а А, Ф, Q и m имеют значения, указанные выше, простые эфиры общей формулы (VII) (схема 16) можно также получить конденсацией ароматических спиртов общей формулы (VIII) (схема 5) с галогенированными производными общей формулы (XVII), в которой Hal представляет атом галогена, в присутствии основания, такого как, например, К2СО3, в инертном растворителе, таком как, например, СН2Сl2, при температуре, находящейся между 40°С и температурой дефлегмации реакционной смеси. Синтез соединений общей формулы (XVII) описывается в разделе "Получение промежуточных продуктов".

Синтез аминов общей формулы (VII) восстановительным аминированием
Амины общей формулы (VII), в которой Х представляет –NR16 -CO- и Y представляет (CH2)m-NR18-(СН2)n-, а А, Ф, R16, R18, m и n имеют указанные выше значения, получают (схема 17) конденсацией альдегида общей формулы (XIX) с амином общей формулы (XVIII) в восстановительной среде. Реакцию проводят в спиртовом растворителе, таком как, например, метанол, в присутствии порошкообразного молекулярного сита 4

Схема 17

Аналогичным способом амины общей формулы (VII), в которой Х представляет –CH2-NR16-, a A, Y, Ф и R16 имеют значения, указанные выше, получают (схема 18) конденсацией альдегидов общей формулы (XX) с аминами общей формулы (XIII) (схема 8) в восстановительной среде в условиях, описанных ранее. Получение некоммерческих альдегидов общей формулы (XX) описывается в разделе "Получение промежуточных продуктов".
Схема 18
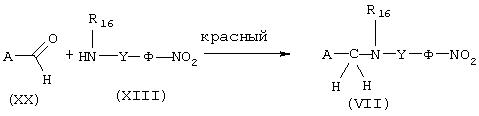
Модификация радикала А в соединениях общей формулы (VII):
Промежуточные продукты общей формулы (VII), в которой А, X, Y, Ф, R1, R2, R3, R4 и R5 имеют значения, указанные выше, можно подвергнуть химической модификации на уровне радикала А (схема 19), в особенности на уровне атома азота, который может быть алкилирован, с использованием реагента R11-Hal, как определено выше, и в особенности с использованием метилиодида в присутствии основания, такого как, например, NaH, в инертном растворителе, таком как, например, ТГФ.
Схема 19

Получение различных промежуточных продуктов синтеза:
Синтез промежуточных продуктов (X);
Промежуточные продукты общей формулы (X), в которой А представляет дифениламин (W отсутствует) доступны с использованием способов, описанных в литературе (Synthesis (1990), 430; Indian J. Chem. (1981), 20B, 611-613; J. Med. Chem. (1975, 18(4), 386-391), которые проводят посредством восстановления промежуточного нитродифениламина. Восстановление функциональной нитрогруппы проводят стандартным способом гидрированием в присутствии каталитического количества Pd/C для получения аминодифениламинов общей формулы (X).
Когда А представляет производное карбазола (W тогда представляет простую связь), способы получения аминокарбазолов общей формулы (X) проводят посредством синтеза промежуточного нитрокарбазола. Эти способы описываются в Pharmazie (1993), 48(11), 817-820; Synth. Commun. (1994), 24(1), 1-10; J. Org. Chem. (1980), 45, 1493-1496; J. Org. Chem. (1964), 29(8), 2474-2476; Org. Prep. Proced. Int. (1981), 13(6), 419-421 или J. Org. Chem. (1963), 28, 884. Восстановление функциональной нитрогруппы промежуточных нитрокарбазолов в этом случае предпочтительно проводят с использованием гидразин-гидрата в присутствии никеля Ренея.
Промежуточные продукты общей формулы (X), в которой А представляет производное фенотиазина (W представляет атом серы) достижимы посредством способов, описанных в литературе, которые проводят посредством синтеза производного нитрофенотиазина. В частности, 3-нитрофенотиазин описывается в J. Org. Chem. (1972), 37, 2691. Восстановление функциональной нитрогруппы для получения аминофенотиазинов общей формулы (X) проводят стандартным способом посредством гидрирования в присутствии каталитического количества Pd/C в растворителе, таком как этанол.
Синтез промежуточных продуктов (XI):
Синтез некоммерческих кислот общей формулы (XI) описывается в схемах 7.1 и 7.2
В конкретном случае, когда Y представляет -(CH2 )m-Q-(СН2)n- и Ф представляет фениленовый радикал, a Q, m и n имеют значения, указанные выше, карбоновые кислоты общей формулы (XI) (схема 7.1) получают в 2 стадии из гетероциклического амина общей формулы (XIV) (схема 8), например 4-нитрофенилпиперазина, и сложного галогенэфира общей формулы (XI.1), такого как, например, этилбромацетат. Конденсацию проводят при 20°С в присутствии основания, такого как, например, триэтиламин, в инертном растворителе, таком как, например, дихлорметан, для получения промежуточных продуктов общей формулы (XI.2). Омыление при помощи LiOH при 20°С дает карбоновые кислоты общей формулы (XI).
В случае когда Y представляет -(CH2)m-O-(CH2)n- и Ф представляет фениленовый радикал, a m и n имеют значения, указанные выше, синтез карбоновых кислот общей формулы (XI) (схема 7.1) проводят посредством конденсации галогенированных производных общей формулы (XI.1) со спиртами общей формулы (XI.3) в присутствии основания, такого как, например, триэтиламин или карбонат калия, при кипячении с обратным холодильником полярного растворителя, такого как, например, ТГФ или ДМФА. Снятие защиты сложной эфирной функции промежуточного продукта общей формулы (XI.4) затем проводят стандартным способом в присутствии основания или сильной кислоты в случае сложных трет-бутиловых эфиров.
Схема 7.1

Карбоновые кислоты общей формулы (XI), в которой Y представляет -(СН2)m- и Ф представляет замещенную фениленовую группу, а m имеет значения, указанные выше, получают в 3 стадии из коммерческих спиртов общей формулы (XI.3), (схема 7.2). Активацию спирта проводят стандартным способом с использованием метансульфонилхлорида (MsCl) в присутствии основания, такого как триэтиламин, в инертном растворителе, таком как дихлорметан, с получением промежуточных продуктов общей формулы (XI.4). Мезилат затем замещают цианидом натрия в ДМФА для получения промежуточных продуктов общей формулы (XI.5). Нитрильную функциональную группу затем гидролизуют нагреванием в смеси этанола и концентрированной НСl с получением кислот общей формулы (XI).
Схема 7.2

Синтез промежуточных продуктов (XII):
Синтез являющихся производными карбоновых кислот фенотиазинов общей формулы (XII) описывается в литературе (J. Med. Chem. (1992), 35(4), 716-724).
Синтез промежуточных продуктов (XIV):
Некоммерческие амины общей формулы (XIV), определенной ранее, в которой Q представляет гомопиперазин, 2-метилпиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, синтезируют в три стадии из соответствующих коммерческих диаминов. Диамины селективно монозащищают в форме карбамата (Synthesis (1984), (12), 1032-1033; Synth. Commun. (1990), 20, (16), 2559-2564) до реакции нуклеофильного замещения с галогеннитробензолом, в особенности с 4-фторнитробензолом. Амины, которые были предварительно защищены, выделяют в свободном состоянии на последней стадии в соответствии со способами, описанными в литературе (T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, Second edition (Wiley-Interscience, 1991)) с получением промежуточных продуктов общей формулы (XIV).
Синтез промежуточных продуктов (XVII):
Галогенированные производные общей формулы (XVII), определенной ранее (схема 16.1) доступны в две стадии из аминов общей формулы (XIII) или (XIV) (схема 8) и коммерческих галогенированных производных общей формулы (XVII.1). Конденсацию для получения промежуточных продуктов общей формулы (XVII.2) или (XVII.3) проводят стандартным способом в присутствии основания, такого как, например, К2СО3, в подходящем инертном растворителе, таком как, например, дихлорметан. Затем спиртовую функциональную группу активируют в форме галогенированного производного с использованием, например, тетрабромида углерода в присутствии трифенилфосфина с получением промежуточных продуктов общей формулы (XVII).
Схема 16.1

Синтез промежуточных продуктов (XVIII):
Амины общей формулы (XVIII), определенной ранее (схема 17.1) в которой A, R16, R18 и m имеют значения, указанные выше, получают конденсацией аминов общей формулы (X) (схема 7) с защищенными аминокислотами (Gp: защитная группа) общей формулы (XVIII.1) в стандартных условиях синтеза пептидов (см. раздел "синтез карбоксамидов"). Снятие защиты у амина соединений общей формулы (XVIII.2) проводят стандартным способом в соответствии с условиями, описанными в литературе (Т.W. Greene et P.G.M. Wuts, Protective Groups in Organic Synthesis, Second edition (Wiley-Interscience, 1991)).
Схема 17.1

Синтез промежуточных продуктов (XX):
Синтез альдегидов фенотиазинов общей формулы (XX), определенной ранее, описывается в литературе (J. Сhеm. Sоc. (1951), 1834; Bull. Soc. Chim. Fr. (1969), 1769).
Если все технические и научные термины, использованные здесь, не определяются по-другому, они имеют такие же значения, как значения, обычно подразумеваемые средним специалистом в области, к которой принадлежит данное изобретение. Аналогично этому все публикации, заявки на патенты, патенты и другие ссылки, указанные здесь, включаются сюда для сведения.
Следующие примеры представлены для иллюстрации вышеуказанных процедур и не должны никоим образом рассматриваться в качестве ограничения объема данного изобретения.
Пример 1.
Гидроиодид N-[4-(фениламино)фенил]-2-тио-фенкарбоксимидамид: 1
4-Аминодифениламин в количестве 0,92 г (5 ммоль) и 2,85 г (10 ммоль) гидроиодида S-метил-2-тиофентиокарбоксимида в 15 мл изопропанола смешивают вместе в 50 мл колбе в атмосфере аргона. Реакционную смесь нагревают при 70°С в течение 48 часов. Растворитель частично выпаривают в вакууме и полученное твердое вещество фильтруют и промывают несколько раз последовательно изопропанолом и этиловым эфиром. Получают желтый порошок с выходом 98%. Точка плавления: 216,3-216,8°С.
1H ЯМР (400 МГц, ДМСО-d6, δ): 6,90 (м, 1Н, аром.); 7,10-7,30 (м, 8Н, аром.); 7,40 (м, 1Н, тиофен); 8,10-8,20 (м, 2Н, тиофен); 8,50 (с, 1Н, NH); 8,75 (с, 1Н, NH+); 9,70 (С, 1Н, NH+); 11,15 (С, 1Н, NH+ ).
ИК: νC=N (амидин): 1590 см-1.
Пример 2.
Гидрохлорид 4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]бензолацетамида: 2
2.1) 4-Hитрo-N-[4-(фениламино)фенил]бензолацетамид:
4-Аминодифениламин в количестве 1,84 г (10 ммоль), 1,81 г (10 ммоль) 4-нитрофенилуксусной кислоты и 1,48 г (11 ммоль) гидроксибензотриазола в 40 мл ТГФ растворяют последовательно в 100 мл колбе. Затем добавляют 2,27 г (11 ммоль) 1, 3-дициклогексилкарбодиимида (ДЦК) и реакционную смесь перемешивают в течение 15 часов. Образуется осадок дициклогексилмочевины (ДЦМ), который фильтруют и промывают 100 мл этилацетата. Фильтрат затем промывают последовательно 50 мл насыщенного раствора Na2CO3, 50 мл воды, 50 мл молярного раствора НСl и, наконец, 2х50 мл соленой воды. Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток быстро очищают на колонке с силикагелем (элюент: гептан/этилацетат, 1/1). Самые чистые фракции собирают и упаривают в вакууме с получением коричневого порошка. Продукт непосредственно используют в следующей стадии.
1H ЯМР (100 МГц, CDCl3, δ): 1,61 (шир. с, 1Н, NH); 3,82 (с, 2Н, СН2); 5,70 (шир. с, 1Н, NH); 6,85-7,50 (м, 10Н, аром., NH-CO); 7,90 (АВ, 4Н, Ph-NO2).
2.2) 4-Aмино-N-[4-(фениламино)фенил]бензолацетамид:
Раствор промежуточного продукта 2.1 (0,54 г, 1,54 ммоль) в 40 мл смеси этилацетат/этанол (1/1), а также 0,1 г Pd/C при 10% вводят в автоклав из нержавеющей стали, снабженной магнитной мешалкой. Реакционную смесь перемешивают при давлении водорода 1,5 бара (1,48 атм) в течение 1 часа и 30 минут при температуре 20°С. Pd/C затем удаляют фильтрованием и фильтрат концентрируют в вакууме. Остаток после упаривания очищают на колонке с силикагелем (элюент: гептан/этилацетат, 4/6), чистые фракции собирают и концентрируют в вакууме. Получают белый порошок с выходом 90%. Точка плавления: 162-163° С.
1H ЯМР (100 МГц, CDCl3, δ): 1,61 (шир. с, 1Н, NH); 3,61 (с, 2Н, СН2); 3,70 (шир. с, 2Н, NH2); 5,62 (шир. с, 1H, NH-CO); 6, 68-7,40 (м, 13H, аром.).
2.3) Гидрохлорид 4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]бензолацетамида: 2
Промежуточный продукт 2.2 в количестве 0,44 г (1,39 ммоль) и 0,47 г (1,67 ммоль) гидроиодида S-метил-2-тиофен-тиокарбоксимида в 15 мл изопропанола растворяют в 50 мл колбе. Реакционную смесь перемешивают в течение 20 часов при температуре 60°С. После выпаривания растворителя в вакууме остаток растворяют в 100 мл смеси 1н. соды и этилацетата (1/1). После декантации органическую фазу промывают 50 мл воды, затем 50 мл соленой воды. Органический раствор сушат над сульфатом магния, фильтруют, концентрируют в вакууме и остаток очищают на колонке с силикагелем (элюент: этилацетат). Чистые фракции собирают и концентрируют в вакууме. Получают белый порошок с выходом 25%. Соединение затем растворяют в метаноле и превращают в соль добавлением 1н. раствора НСl в этиловом эфире (1 мл). После перемешивания в течение одного часа при 20°С реакционную смесь концентрируют в вакууме с получением бледно-желтого порошка. Точка плавления: продукт превращается в пену.
1H ЯМР (400 МГц, ДМСО-d6, δ): 3,71 (с, 2Н, СН2); 4,60 (шир. с, 1Н, NH); 6,75 (м, 1Н, тиофен); 7,00 (м, 4Н, аром.); 7,19 (м, 2Н, аром.); 7,40 (м, 3Н, аром.); 7,55 (м, 4Н, аром.); 8,14 (м, 2Н, тиофен); 8,95 (шир. с, 1Н, NH+); 9,86 (шир. с, 1Н, NH+); 10,41 (с, 1Н, NH-CO); 11,60 (шир. с, 1Н, NH+).
ИК: νC=O (амид): 1649 см-1; νC=N (амидин): 1597 см-1.
Пример 3.
Гидроиодид {4-{[2-тиенил(имино)метил]-амино}фенокси}-N-[4-(фениламино)фенил]ацетамида: 3
3.1) трет-Бутил-4-нитрофеноксиацетат:
Паранитрофенол в количестве 3 г (21,6 ммоль), 8,94 г (64,8 ммоль) карбоната калия и 8,42 г (43,2 ммоль) трет-бутилбромацетата вводят в атмосфере азота в колбу на 250 мл, содержащую 100 мл ТГФ. Реакционную смесь перемешивают при кипячении с обратным холодильником в течение 2 часов. Твердую часть отделяют фильтрованием и фильтрат концентрируют при пониженном давлении. Остаток растворяют в 50 мл этилацетата и промывают последовательно 50 мл воды и 50 мл соленой воды. Органическую фазу сушат над сульфатом натрия, фильтруют и упаривают в вакууме. После очистки чистых фракций на колонке с силикагелем (элюент: этилацетат/гептан, 1:8) и концентрирования в вакууме получают белый порошок с выходом 50%. Точка плавления: 81-83°С.
1H ЯМР (100 МГц, CDCl3, δ): 1,50 (с, 9Н, 3хСН3); 4,60 (с, 2Н, СН2); 7,57 (АВ, 4Н, Ph-NO2).
3.2) 4-Нитрофеноксиуксусная кислота:
Промежуточный продукт 3.1 в количестве 2,58 г (10,2 ммоль) растворяют в 45 мл дихлорметана в 100 мл колбе в атмосфере азота. Смесь охлаждают до 0°С и по каплям добавляют 7,85 мл (102 ммоль) трифторуксусной кислоты. Реакционную смесь перемешивают в течение 3 с половиной часов при комнатной температуре. Раствор затем концентрируют при пониженном давлении. Остаток после упаривания растворяют в 30 мл этилацетата и промывают 20 мл воды. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Получают желтое твердое вещество с выходом 89%. Полученный продукт является достаточно чистым, чтобы его можно было использовать непосредственно в следующей стадии. Точка плавления: 190-192°С.
1H ЯМР (100 МГц, CDCl3, δ): 2,00 (шир. с, 1Н, СООН); 4,80 (с, 2Н, СН2); 7,60 (АВ, 4Н, Ph-NO2).
3.3) (4-Нитрофенокси)-N-[(4-фениламино)фенил]ацетамид:
4-Аминодифениламин в количестве 1,65 г (8,98 ммоль), 1,77 г (8,98 ммоль) промежуточного продукта 3.2 и 1,27 г (9,42 ммоль) гидроксибензотриазола растворяют в 40 мл ТГФ в 100 мл колбе в атмосфере азота. Когда все растворится, добавляют 1,94 г (9,42 ммоль) 1,3-дициклогексилкарбодиимида и реакционную среду оставляют при перемешивании в течение 15 часов. Осадок образовавшейся дициклогексилмочевины фильтруют и промывают этилацетатом. Фильтрат упаривают в вакууме и остаток после упаривания растворяют в этилацетате, он затем образует осадок, который отфильтровывают и промывают с использованием того же самого растворителя. Получают зеленоватое твердое вещество с выходом 65%. Полученный продукт является достаточно чистым, чтобы его можно было использовать непосредственно в следующей стадии. Точка плавления: 192-195°С.
1H ЯМР (100 МГц, CDCl3, δ): 4,75 (с, 2Н, СН2-O); 5,70 (шир. с 1Н, NH); 7,10 (м, 9Н, аром.); 7,85 (АВ, 4Н, Рh-NO2); 8,05 (шир. с, 1Н, NH-CO).
3.4) (4-Аминофенокси)-N-[(4-фениламино)фенил]ацетамид:
Промежуточный продукт 3.3 в количестве 1 г (2,75 ммоль), растворенный в 200 мл смеси растворителей (этанол/дихлорметан/ТГФ, 1:1:1), и 0,1 г палладия на угле при 10% вводят в 300 мл автоклав. Смесь помещают под давление водорода 1,5 бара (1,48 атм) и перемешивают при комнатной температуре в течение 15 минут. Катализатор отфильтровывают и растворители упаривают при пониженном давлении с получением розовато-бежевого твердого вещества с выходом 71%. Точка плавления: 146-148°С.
1 H ЯМР (100 МГц, CDCl3, δ): 3,50 (шир. с, 2Н, NH2); 4,50 (с, 2Н, CH2-O); 5,70 (шир. с, 1Н, NH); 6,70 (м, 4Н, аром.); 7,10 (м, 4Н, аром.); 7,25 (м, 5Н, аром.); 8, 20 (шир. с, 1Н, NH-CO).
3.5) Гидроиодид [4-{[имино(2-тиенил)метил]амино}фенокси]-N-[(4-фениламино)фенил]ацетамида: 3
Смесь 0,3 г (0,9 ммоль) промежуточного продукта 3.4 в присутствии 0,25 г (0,9 ммоль) гидроиодида S-метил-2-тиофентиокарбоксимида в растворе в 20 мл изопропанола нагревают при 50°С в течение 15 часов. Реакционную смесь фильтруют и полученное твердое вещество промывают этиловым эфиром. Желтый порошок получают с выходом 78%. Точка плавления: 163-166°С.
1H ЯМР (400 МГц, ДМСО-d6, δ): 4,75 (с, 2Н, СН2О); 6,77 (м, 1Н, тиофен); 7,04 (м, 4Н, аром.); 7,19 (м, 4Н, аром.); 7,40 (м, 3Н, аром.); 7,50 (м, 2Н, аром.); 8,12 (м, 2Н, тиофен); 8,81 (шир. с, 1Н, NH+); 9,70 (шир. с, 1Н, NH+); 10,01 (с, 1Н, CO-NH); 11,20 (шир. с, 1Н, NH+).
ИК: νC=O (амид): 1647 см-1; νC=N (амидин): 1598 см-1.
Пример 4.
4-{[2-Тиенил(имино)метил]амино)-N-[2(фениламино)фенил]бензолбутанамид: 4
Используемый экспериментальный протокол является таким же, как протокол, описанный для примера 2. Продукт получают в форме свободного основания (белое твердое вещество). Точка плавления 164-167°С.
1H ЯМР (400 МГц, ДМСО-d6, δ): 1,86 (м, 2Н, CH2); 2,35 (м, 2Н, CH2); 2,55 (м, 2Н, CH2); 6,37 (шир. с, 2Н, NH2); 6,76 (м, 3Н, аром.); 6,87 (м, 2Н, аром.); 6,96 (м, 1Н, тиофен); 7,10 (м, 3Н, тиофен); 7,18 (м, 2Н, аром.); 7,25 (м, 1Н, аром.); 7,33 (с, 1Н, NH); 7,52 (м, 1Н, тиофен); 7,73 (м, 1Н, тиофен); 9,36 (с, 1Н, NH-CO).
ИК: νC=O (амид): 1627 см-1; νC=N (амидин): 1591 см-1.
Пример 5.
Гидрохлорид 4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]бензолбутанамида: 5
Используемый экспериментальный протокол является таким же, как протокол, описанный в примере 2. Продукт получают в форме розовато-оранжевого порошка. Точка плавления 167-170°С.
1H ЯМР (400 МГц, ДМСО-d6, δ): 1,90 (м, 2Н, CH2); 2,35 (м, 2Н, СН2); 2,70 (м, 2Н, СН2); 6,70 (м, 1Н, тиофен); 7,00 (м, 4Н, аром.); 7,20 (м, 2Н, аром.); 7,40 (м, 5Н, аром.); 7,50 (м, 2Н, аром.); 8,20 (м, 2Н, тиофен); 8,90 (с, 1Н, NH+); 9,85 (с, 1Н, NH+); 9,90 (с, 1Н, NHCO); 11,55 (с, 1Н, NH+).
ИК: νC=O (амид): 1654 см-1; νC=N (амидин): 1597 см-1.
Пример 6.
Гидрохлорид 4-{[2-тиенил(имино)метил]амино}-N-[4-{4-метоксифениламино)фенил]бензолбутанамида: 6
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.3, причем промежуточный продукт 6.2 заменяет 4-амино-N-[4-(фениламино)фенил]бензолацетамид. Получают бежевый продукт с выходом 65%. Точка плавления 200-202°С.
1H ЯМР (400 МГц, ДМСО-d6, δ); 1,91 (м, 2Н, СН2); 2,33 (м, 2Н, СН2); 2,67 (м, 2Н, СН2); 3,69 (с, 3Н, O-СН3); 4,71 (шир. с, 1Н, NH); 6,81-7,00 (м, 6Н, аром.); 7,37-7,45 (м, 7Н, аром.); 8,20 (м, 2Н, тиофен); 8,90 (шир. с, 1Н, NH+ ); 9,87 (шир. с, 1Н, NH+); 9,92 (с, 1Н, NH-CO); 11,67 (шир. с, 1Н, NH+).
ИК: νC=O (амид): 1664 см-1; νC=N (амидин): 1603 см-1.
Пример 7.
Гидрохлорид 2-{4-{[2-тиенил(имино)метил]-амино}фенил}этил-[4-(фениламино)фенил]карбамата: 7
7.1) 2-(4-Нитрофенил)этил-[4-(фениламино)фенил]карбамат:
Трифосген в количестве 1,18 г (3,9 ммоль) растворяют в 15 мл дихлорметана в 250 мл колбе в атмосфере аргона. С использованием шприца с мотором раствор 2 г (12 ммоль) 4-нитрофенилэтанола и 1,7 мл (13 ммоль) N,N-диметиланилина в 40 мл дихлорметана добавляют в течение 1 часа. Реакционную смесь перемешивают в течение нескольких минут при 20°С до добавления в виде одной порции раствора 2,2 г (12 ммоль) 4-аминодифениламина и 1,7 мл (13 ммоль) N,N-диметиланилина в 40 мл дихлорметана. После перемешивания в течение одного часа при 20°С содержимое колбы выливают в 100 мл воды. Смесь разбавляют 100 мл дихлорметана и перемешивают. Органическую фазу декантируют, сушат над сульфатом магния, фильтруют и упаривают в вакууме. Полученное твердое вещество растворяют в этиловом эфире, растирают и фильтруют. После сушки получают зеленоватый порошок с выходом 22%. Точка плавления: 146,4-148°С.
1H ЯМР (400 МГц, CDCl3, δ): 3,10 (м, 2Н, CH2); 4,40 (м, 2Н, СН2); 5,65 (с, 1Н, NH); 6,50 (с, 1Н, NH); 6,80-7,60 (м, 11Н, аром.); 8,20 (м, 2Н, аром.).
7.2) 2(4-Аминофенил)этил-[4-(фениламино)фенил]карбамат:
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.2, причем промежуточный продукт 7.1 заменяет 4-нитро-N-[4-(фениламино)фенил]бензолацетамид. Получают белое твердое вещество с выходом 48%. Точка плавления 140-140,5°С.
1H ЯМР (400 МГц, ДМСО-d6, δ): 2,75 (м, 2Н, СН2); 4,15 (м, 2Н, СН2); 5,20 (с, 2Н, NH2); 6,50 (м, 2Н, аром.); 6,70 (м, 1Н, аром.); 7,00 (м, 6Н, аром.); 7,15 (м, 2Н, аром.); 7,30 (м, 2Н, аром.); 8,00 (с, 1Н, NH); 9,40 (с, 1Н, NH).
7.3) Гидрохлорид 2-{4-{[2-тиенил(имино)метил]амино}-фенил этил-[4-(фениламино)фенил]карбамата: 7
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.3, причем промежуточный продукт 7.2 заменяет 4-амино-N-[4-(фениламино)фенил]бензолацетамид. Получают белое твердое вещество с выходом 34%. Точка плавления 153-159°С.
1H ЯМР (400 МГц, ДМСО-d6, δ): 3,00 (м, 2Н, СН2); 4,30 (м, 2Н, CH2); 6,60-7,70 (м, 14Н, аром.); 8,20 (м, 2Н, тиофен); 8,90 (с, 1Н, NH+); 9,50 (с, 1Н, NH-CO); 9,90 (с, 1Н, NH+), 11,70 (с, 1Н, NH+).
ИК: νC=O (карбамат): 1719 см-1; νC=N (амидин): 1598 см-1.
Пример 8.
Гидрохлорид N-{2-{4-{[3-тиенил(имино)метил]амино}фенил}этил}-N’-[4-(фениламино)фенил]мочевины: 8
8.1) N-[2-(4-Нитрофенил)этил]-N’-[4-(фениламино)фенил] мочевина
Трифосген в количестве 0,5 г (1,7 ммоль) растворяют в 8 мл дихлорметана в 100 мл колбе в атмосфере аргона. С использованием шприца с мотором в течение одного часа добавляют 0, 92 г (5 ммоль) 4-аминодифениламина и 1,44 мл (8,2 ммоль) диизопропилэтиламина в 15 мл дихлорметана. Через пять минут после окончания добавления добавляют 1,01 г (5 ммоль) гидрохлорида 4-нитрофенетиламина, и затем раствор 1,44 мл (8,2 ммоль) диизопропилэтиламина в 10 мл дихлорметана в виде одной порции. После перемешивания в течение двух часов при 20°С реакционную смесь разбавляют 50 мл дихлорметана и 20 мл воды. Органическую фазу декантируют и снова промывают 20 мл воды. После сушки над MgSO4 и фильтрования органический раствор частично концентрируют в вакууме. Образованный осадок собирают фильтрованием и промывают дихлорметаном. Получают желтое твердое вещество с выходом 40%. Точка плавления: 204-205°С.
1H ЯМР (100 МГц, ДМСО-d6, δ): 2,96 (м, 2Н, СН2); 3,50 (м, 2Н, CH2-NH); 5,78 (м, 1Н, NH-CH2); 6,45 (шир. с, 1Н, Ph-NH-CO); 6,72-7,49 (м, 11Н, аром.}; 7, 81 (шир. с, 1Н, NH); 8,15 (м, 2Н, аром.).
8.2) N-[2-(4-Аминофенил)этил]-N’-[4-(фениламино)фенил]мочевина
Раствор промежуточного продукта 8.1 (0,68 г, 1,81 ммоль) в 40 мл смеси ТГФ/этанол (3/1), а также 0,1 г Pd/C при 10% вводят в автоклав из нержавеющей стали, снабженный магнитной мешалкой. Реакционную смесь перемешивают под давлением водорода 1,5 бара (1,48 атм) в течение 1 часа при температуре 20°С. Pd/C затем удаляют фильтрованием и фильтрат концентрируют в вакууме. Полученное твердое вещество промывают последовательно этилацетатом и дихлорметаном. Получают бежевый порошок с выходом 61%. Точка плавления >260°С.
1H ЯМР (100 МГц, ДМСО-d6, δ): 2,70 (м, 2Н, СН2); 3,40 (м, 2Н, CH2-NH); 5,18 (шир. с, 2Н, NH2); 6,07 (м, 1Н, NH-CH2); 6,60-7,45 (м, 13Н, аром.); 8,00 (шир. с, 1Н, NH); 8,41 (шир. с, 1Н, Ph-NH-CO).
8.3) Гидрохлорид N-{2-{4-{[2-тиенил(имино)метил]амино}фенил}этил}-N’-[4-(фениламино)фенил]мочевины: 8
Промежуточный продукт 8.2 в количестве 0,38 г (1,10 ммоль) и 0,34 г (1,21 ммоль) гидроиодида S-метил-2-тиофен-тиокарбоксимида растворяют в 20 мл изопропанола в 50 мл колбе. Реакционную смесь перемешивают в течение 20 часов при температуре 60°С. После выпаривания растворителя в вакууме остаток растворяют в 50 мл смеси 1/1 насыщенного раствора Na2CO3 и этилацетата. Реакционную среду энергично перемешивают, и через некоторое время появляется осадок. Осадок собирают, фильтруют и промывают последовательно этилацетатом и водой. После сушки осадок очищают на колонке с силикагелем (элюент ТГФ). Чистые фракции собирают и концентрируют в вакууме. Полученное твердое вещество (300 мг) снова растворяют в 80 мл ТГФ, к которому добавляют 2 мл 1н. раствора НСl в этиловом эфире. Образованный гидрохлорид осаждается, его отделяют фильтрованием и промывают ТГФ, затем этиловым эфиром с получением светло-серого порошка. Точка плавления: продукт становится пеной.
1H ЯМР (400 МГц, ДМСО-d6, δ); 2,80 (м, 2Н, СН2); 3,37 (м, 2Н, CH2); 4,46 (шир. с, 1Н, NH); 6,40 (шир. с, 1Н, NH-CH2); 6,70 (м, 1Н, тиофен); 6,94 (м, 4Н, аром.); 7,15 (м, 2Н, аром.); 7,28 (м, 2Н, аром.); 7,40 (м, 5Н, аром.); 8,17 (м, 2Н, тиофен); 8,78 (шир. с, 1Н, Ph-NH-CO); 8,93 (шир. с, 1Н, NH+); 9,84 (шир. с, 1Н, NH+); 11,52 (шир. с, 1Н, NH+).
ИК: νC=O (мочевина): 1654 см-1; νC=N (амидин): 1598 см-1.
Пример 9.
Гидрохлорид 4-{4-{[2-тиенил(имино)метил]-амино}фенил}-N-[4-(фениламино)фенил]-1-пиперазинацетамида: 9
9.1) Этил-4-(нитрофенил)-1-пиперазинацетат:
1-(4-Нитрофенилпиперазин) в количестве 3 г (14,5 ммоль) и 1,8 мл (15,9 ммоль) бромэтилацетата растворяют в 60 мл дихлорметана в 100 мл колбе. После добавления 2,42 мл (17,4 ммоль) триэтиламина реакционную смесь перемешивают при 20°С в течение одного часа. Раствор затем выливают в 100 мл воды и экстрагируют 100 мл дихлорметана. После декантации органическую фазу сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученное твердое вещество растворяют в этиловом эфире, растирают и фильтруют. Получают желтый порошок с выходом 89%. Точка плавления: 122,1-122,5° С.
1H ЯМР (400 МГц, СDСl3, δ): 1,3 (т, 3Н, СН3, J=7 Гц); 2,75 (м, 4Н, пиперазин); 3,30 (с, 2Н, CO-CH2); 3,50 (м, 4Н, пиперазин); 4,20 (к, 2Н, СН2-СН3, J=7 Гц); 7,45 (АВ, 4Н, Ph-NO2).
9.2) 4-(Нитрофенил)-1-пиперазинуксусная кислота:
1 М водный раствор LiOH в количестве 32,4 мл добавляют по каплям при 20°С в колбу, содержащую раствор 3,8 г (13 ммоль) промежуточного продукта 9.1 в растворе в 80 мл ТГФ. После перемешивания в течение одного часа реакционную смесь подкисляют до рН 5 при помощи 2н. раствора хлористовородной кислоты. Полученный осадок фильтруют и промывают минимальным количеством ТГФ и воды. Продукт используют непосредственно в следующей стадии.
1H ЯМР (100 МГц, D2O, δ): 3,30 (м, 4Н, пиперазин); 3,60 (м, 6Н, пиперазин + СО-СН2); 7,45 (АВ, 4Н, Ph-NO2 ).
9.3) 4-(4-Нитрофенил)-N-[4-(фениламино)фенил]-1-пиперазинацетамид:
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.1, причем промежуточный продукт 9.2 заменяет 4-нитро-фенилуксусную кислоту. Получают желтое твердое вещество с выходом 84%. Точка плавления 212-213°С.
1H ЯМР (400 МГц, CDCl3, δ); 2,80 (м, 4Н, пиперазин); 3,25 (с, 2Н, CO-CH2); 3,50 (м, 4Н, пиперазин}; 5,70 (с, 1Н, NH); 6,90 (м, 3Н, аром.); 7,10 (м, 4Н, аром.); 7,30 (м, 2Н, аром.); 7,85 (АВ, 4Н, Ph-NО2); 8,90 (с, 1Н, NHCO).
9.4) 4-(4-Аминофенил)-N-[4-(фениламино)фенил]-1-пиперазинацетамид:
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.2, причем промежуточный продукт 9.3 заменяет 4-нитро-N-[4-(фениламино)фенил]бензолацетамид. Получают коричневое масло с выходом 71%.
1H ЯМР (400 МГц, CDCl3, δ); 2,80 (м, 4Н, пиперазин); 3,15 (м, 4Н, пиперазин); 3,20 (с, 2Н, CO-CH2); 5,70 (с, 1Н, NH); 6,70 (м, 2Н, аром.); 6,90 (м, 3Н, аром.); 7,10 (м, 4Н, аром.); 7,30 (м, 2Н, аром.); 7,50 (м, 2Н, аром.); 9,10 (с, 1Н, NHCO).
9.5) Гидрохлорид 4-{4-{[2-тиенил(имино)метил]амино}-фенил}-N-[4-(фениламино)фенил]-1-пипераэинацетамида: 9
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.3, причем промежуточный продукт 9.4 заменяет 4-амино-N-[4-(фениламино)фенил]бензолацетамид. Получают желтое твердое вещество с выходом 30%. Точка плавления: 230-240° С.
1H ЯМР (400 МГц, ДМСО-d6, δ); 3,10-3,50 (м, 4Н, пиперазин); 3,65 (м, 2Н, пиперазин); 3,90 (м, 2Н, пиперазин); 4,30 (с, 2Н, СО-СН2); 6, 80 (м, 1Н, тиофен); 6,90-7,40 (м, 11Н, аром.); 7,50 (м, 2Н, аром.); 8,15 (м, 2Н, тиофен); 8,75 (с, 1Н, NH+); 9,80 (с, 1Н, NH+); 10,9 (м, 2Н, NHCO + NH+); 11,40 (с, 1Н, NH+).
ИК: νC=O (амид): 1680 см-1; νC=N (амидин): 1512 см-1.
Пример 10.
Гидрохлорид 1-{[(4-фениламино)фениламино]-карбонил}-4-{4-{[2-тиенил(имино)метил]амино}фенил}пиперазина: 10
Используемый экспериментальный протокол является таким же, как протокол, описанный для примера 8. Продукт превращают в соль в условиях, которые идентичны таковым для соединения 2, за исключением того, что ТГФ заменяет метанол. Получают желтый порошок. Точка плавления: 239-240° С.
1H ЯМР (400 МГц, ДМСО-d6, δ); 3,30 (шир. с, 4Н, пиперазин); 3,70 (шир. с, 4Н, пиперазин); 5,80 (шир. с, 1Н, NH); 6,73 (м, 1Н, тиофен); 6,98 (м, 4Н, аром.); 7,17 (м, 2Н, аром.); 7,28-7,37 (м, 7Н, аром.); 8,16 (м, 2Н, тиофен); 8,65 (шир. с, 1Н, Ph-NH-CO); 8,80 (шир. с, 1Н, NH+); 9,80 (шир. с, 1Н, NH+); 11,52 (шир. с, 1Н, NH+).
ИК: νC=O (мочевина): 1654 см-1; νC=N (амидин): 1597 см-1.
Пример 11.
Гидрохлорид 4-{[2-тиенил(имино)метил]амино}-N-[4 -(фениламино)фенил]бензолбутанамина: 11
11.1) 4-Hитpo-N-[4-(фениламино)фенил]бензолбутанамин:
4-Нитро-N-[4-(фениламино)фенил]бензолбутанамид (получен в тех же условиях, что и промежуточный продукт 2.1) в количестве 1,12 г (3 ммоль) растворяют в 50 мл безводного ТГФ в 250 мл трехгорлой колбе в атмосфере аргона. Раствор охлаждают с использованием ледяной бани, а затем добавляют по каплям 15 мл (15 ммоль) раствора диборан/ТГФ. Реакционную смесь нагревают до кипения с обратным холодильником в течение 5 часов. После охлаждения до 20°С медленно добавляют по каплям 25 мл раствора НСl (6н.) и смесь кипятят с обратным холодильником в течение 2 часов. Раствор затем охлаждают с использованием ледяной бани до добавления 20% раствора карбоната натрия до достижения основного значения рН. Продукт экстрагируют с использованием этилового эфира (2x50 мл), органический раствор промывают соленой водой (2х50 мл) и сушат над сульфатом магния. После фильтрования и концентрирования в вакууме остаток очищают на колонке с силикагелем (элюент: гептан/AcOEt, 1/1). Чистые фракции собирают и упаривают в вакууме с получением коричневого масла с выходом 28%.
1H ЯМР (400 МГц, ДМСО-d6, δ): 1,55 (м, 2Н, СН2); 1,71 (м, 2Н, CH2); 2,75 (м, 2Н, СН2-аром.); 2,98 (м, 2Н, NH-СН2); 5,29 (м, 1Н, NH); 6,51-7,51 (м, 12Н, аром. + NH); 8,15 (м, 2Н, Ph-NO2).
11.2) 4-Амино-N-[4-(фениламино}фенил]бензолбутанамин:
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.2, причем промежуточный продукт 11.1 заменяет 4-нитро-N-[4-(фениламино)фенил]бензолацетамид. Получают коричневое масло с выходом 36%.
1H ЯМР (400 МГц, ДМСО-d6, δ): 1,55 (м, 4Н, 2хСН2); 2,44 (м, 2Н, СН2); 2,97 (м, 2Н, СН2); 4,81 (с, 2Н, NH2); 5,27 (м, 1Н, NH); 6,47-7,10 (м, 13Н, аром.); 7,49 (с, 1Н, NH).
11.3) Гидрохлорид 4-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино}фенил]бензолбутанамина: 11
Промежуточный продукт 11.2 в количестве 0,10 г (0,3 ммоль) и 0,11 г (0,37 ммоль) гидроиодида S-метил-2-тиофен-тиокарбоксимида растворяют в 5 мл изопропанола с добавленным в него 0,05 мл (0,6 ммоль) пиридина, в 50 мл колбе. Реакционную смесь перемешивают в течение 20 часов при 23°С. После выпаривания растворителя в вакууме остаток растворяют в 25 мл смеси (1/1) насыщенного раствора NаНСО3 и дихлорметана. После декантации органическую фазу промывают 2х25 мл соленой воды. Органический раствор сушат над сульфатом магния, фильтруют, концентрируют в вакууме и остаток очищают на колонке с силикагелем (элюент: дихлорметан + 5% этанола). Чистые фракции собирают и концентрируют в вакууме. Получают розоватый порошок, который превращают в соль добавлением 1н. раствора НСl в этиловом эфире (1 мл) к раствору основания в ацетоне. После перемешивания в течение одного часа при 20°С реакционную смесь фильтруют и порошок промывают последовательно 20 мл ацетона и 20 мл этилового эфира. Точка плавления: 165-166°С.
1H ЯМР (400 МГц, ДМСО-d6, δ): 1,71 (м, 4Н, 2хCH2); 2,66 (м, 2Н, СН2); 3,20 (м, 2Н, СН3); 6,85-7,41 (м, 14Н, аром.); 8,16 (м, 2Н, тиофен); 8,53 (шир. с, 1Н, NH); 8,87 (шир. с, 1Н, NH+); 9,83 (шир. с, 1Н, NH+); 11,19 (шир. с, 2Н, 2хNH+); 11,56 (шир. с, 1Н, NH+).
ИК: νC=N (амидин): 1595 см-1.
Пример 12
Гидрохлорид 3-{[3-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]бенэолпропанамида: 12
12.1) Меэилат 3-нитрофенилэтанола:
Раствор 4,63 мл (59,8 ммоль) метансульфонилхлорида, разбавленного 20 мл дихлорметана, добавляют по каплям к раствору 10 г (59,8 ммоль) 3-нитрофенилэтанола и 8,31 мл (59,8 ммоль) триэтиламина в 120 мл дихлорметана, охлаждают с помощью ледяной бани. Перемешивание поддерживают в течение 1 часа при 0°С и в течение 2 часов при 20°С. Реакционную смесь затем концентрируют в вакууме и остаток растворяют в 125 мл этилацетата и 100 мл воды. После перемешивания и декантации органическую фазу промывают последовательно 100 мл воды и 100 мл соленой воды. Органический раствор сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток упаривания очищают на колонке с диоксидом кремния (элюент гептан/этилацетат, 6/4) и чистые фракции собирают и упаривают с получением желтого масла с выходом 71%.
1H ЯМР (100 МГц, СDСl3, δ): 3,00 (с, 3Н, СН3); 3,20 (т, 2Н, CH2 , J=5,8 Гц); 4,50 (т, 2Н, СН2); 7,60 (м, 2Н, аром.); 8,20 (м, 2Н, аром).
12.2) 3-Нитробензолпропаннитрил:
NaCN в количестве 0,49 г (10 ммоль) вводят в виде одной порции в 100 мл колбу с атмосферой аргона, содержащую раствор 1,22 г (5 ммоль) промежуточного продукта 12.1 в 20 мл безводного ДМФА. Реакционную смесь нагревают при 60°С в течение 3 часов и после того, как смеси дают охладиться до 20°С, ее выливают в 100 мл воды. Раствор экстрагируют 5х50 мл этилацетата, органические фазы собирают и промывают последовательно 100 мл воды и 100 мл соленой воды. После сушки над сульфатом магния органический раствор концентрируют в вакууме и остаток очищают на колонке с диоксидом кремния (элюент: гептан/этилацетат, 7/3). Чистые фракции собирают и концентрируют в вакууме с получением светло-желтого порошка с выходом 78%. Точка плавления: 86-88°С.
1H ЯМР (100 МГц, CDCl3, δ): 2,70 (т, 2Н, CH2, J=5,8 Гц); 3,10 (т, 2Н, СН2); 7,60 (м, 2Н, аром.); 8,20 (м, 2Н, аром.)
12.3) 3-Нитробензолпропановая кислота:
Раствор 2,33 г (19,2 ммоль) промежуточного продукта 12.2 в 100 мл 10% водного раствора НСl и 100 мл этанола кипятят с обратным холодильником в течение 72 часов. После того как температуре дают снизиться до 20°С, реакционную смесь концентрируют досуха в вакууме. Остаток растворяют в 100 мл этилацетата и промывают 3х100 мл воды и 50 мл соленой воды. После сушки над сульфатом натрия органический раствор фильтруют и концентрируют в вакууме. Остаток упаривания очищают на колонке с диоксидом кремния (элюент: гептан/этилацетат, от 95/5 до 80/20). Получают светло-желтый порошок с выходом 21%. Точка плавления: 107-109°С.
1H ЯМР (100 МГц, CDCl3, δ): 2,70 (м, 2Н, СН2); 3,10 (м, 2Н, СН2); 5,40 (шир. с, 1Н,); 7,50 (м, 2Н, аром.); 8,10 (м, 2Н, аром.)
12. 4) 3-Нитро-N-[4-(фениламино)фенил]бензолпропанамид:
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.1, причем промежуточный продукт 12.3 заменяет 4-нитрофенилуксусную кислоту. Коричневый порошок получают с выходом 70%. Точка плавления: 130-132°С.
1H ЯМР (CDCl3, 100 МГц, δ): 2,70 (т, 2Н, СН2, J=5,8 Гц); 3,20 (т, 2Н, CH2); 5,70 (шир. с, 1Н, NH); 6,90-7,60 (м, 13Н, аром.).
12.5) 3-Амино-N-[4-(фениламино)фенил]бензолпропанамид:
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.2, причем промежуточный продукт 12.4 заменяет 4-нитро-N-[4-(фениламино)фенил]бензолацетамид. Получают белый порошок с выходом 64%. Точка плавления: 164-166°С.
1H ЯМР (CDCl3, 100 МГц, δ): 2,80 (т, 2Н, СН2); 3,50 (м, 2Н, СН2); 5,10 (шир. с, 2Н, NH2); 6,50 (м, 3Н, аром.); 6,80-7,45 (м, 8Н, аром.); 7,60 (м, 2Н, аром); 8,15 (с, 1Н, NH); 9,88 (с, 1Н, NH-CO).
12.6) Гидрохлорид 3-{[2-тиенил(имино)метил]амино}-N-[4-(фениламино)фенил]бензолпропанамида: 12
Используемый экспериментальный протокол является таким же, как протокол, описанный для промежуточного продукта 2.3, причем промежуточный продукт 12.5 заменяет 4-амино-N-[4-(фениламино)фенил]бензолацетамид. После превращения в соль получают светло-бежевый порошок с выходом 78%. Точка плавления: 228-230°С.
1H ЯМР (400 МГц, ДМСО-d6, δ); 2,70 (м, 2Н, СН2); 2,96 (м, 2Н, СН2); 5,20 (шир. с, 1Н, NH); 6,74 (м, 1Н, тиофен); 7,00 (м, 4Н, аром.); 7,19 (м, 2Н, аром.); 7,29 (м, 1Н, аром.); 7,39 (м, 3Н, аром.); 7,47 (м, 3Н, аром.); 8, 18 (м, 2Н, тиофен); 8,96 (шир. с, 1Н, NH+); 9,90 (шир. с, 1Н, NH+); 10,07 (с, 1Н, NH-CO); 11,60 (шир. с, 1Н, NH+).
ИК: νC=O (амид): 1649 см-1; νC=N (амидин): 1596 см-1.
Пример 13.
4-(4-{[Амино(2-тиенил)метилиден]амино}фенил)-N-[2-(4-толуидино)фенил]бутанамид: 13
13.1) N’-(4-Метилфенил)-1,2-бензолдиамин:
Восстановление функциональной нитрогруппы N-(4-метилфенил)-2-нитроанилина (Synthesis (1990), 430) проводят в присутствии Pd/C в этаноле в условиях, описанных ранее для промежуточного продукта 2.2. Получают фиолетовый продукт в полумасляной-полукристаллической форме с выходом 90%.
13.2) 4-(4-{[Амино(2-тиенил)метилиден]амино}фенил)-N-[2-(4-толуидино)фенил]бутанамид:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный в примере 2, исходящим из 4-нитрофенилмасляной кислоты и промежуточного продукта 13.1. Продукт выделяют в форме свободного основания. Белое твердое вещество. Точка плавления: 66-68°С.
Пример 14.
4-Анилинофенил-4-{4-{[амино(2-тиенил)метилиден]амино}фенил)бутаноат: 14
14.1) 4-Анилинофенил-4-(4-нитрофенил)бутаноат:
1, 1’-Карбонилдиимидазол в количестве 0,98 г (6,02 ммоль) медленно добавляют при 20°С к раствору 1,25 г (5,96 ммоль) 4-нитрофенилмасляной кислоты в 25 мл CH2Cl2. Реакционную смесь перемешивают в течение 30 минут до добавления 1 г (5,42 ммоль) 4-гидроксидифениламина. После перемешивания в течение 3 часов реакцию останавливают добавлением 3 мл МеОН и растворитель выпаривают в вакууме. Остаток выпаривания очищают на колонке с диоксидом кремния (элюент:гептан/AcOEt: от 100/0 до 80/20). Получают желтое твердое вещество с выходом 89%.
14.2) 4-Анилинофенил-4-(4-{[амино(2-тиенил)метилиден]амино}фенил)бутаноат:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточных продуктов 2.2 и 2.3, исходящим из промежуточного продукта 14.1. Продукт выделяют в форме свободного основания. Бледно-желтое твердое вещество. Точка плавления: 147-148°С.
Пример 15.
4-(4-{[Амино(2-тиенил)метилиден]амино}фенил)-N-[2-(4-толуидино)фенил]бутанамид: 15
15.1) N-{4-[4-(4-Нитрофенил)бутокси]фенил}-N-фениламин:
4-Гидроксидифениламин в количестве 2,0 г (10,88 ммоль), 2,0 мл (12 ммоль) 4-(4-нитрофенил)-1-бутанола и 1,66 мл (12 ммоль) трибутилфосфина последовательно вводят в колбу, содержащую 10 мл CH2Cl2. Затем по каплям добавляют 1,90 мл (12 ммоль) диэтилазодикарбоксилата и всю смесь перемешивают при 20°С в течение 16 часов. Растворитель выпаривают в вакууме и остаток очищают на колонке с диоксидом кремния (элюент: гептан/AcOEt: от 100/0 до 80/20). Ожидаемый продукт получают в форме темно-красного масла с выходом 35%.
15.2) N’-{4-[4-(4-Aнилинoфeнoкcи)бутил]фенил}-2-тио-фенкарбоксимидамид:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточных продуктов 2.2 и 2.3, исходящим из промежуточного продукта 14.1. Продукт выделяют в форме свободного основания. Белый твердый продукт. Точка плавления: 120-121°С.
Пример 16.
N’-{4-[4-(3-Анилинофенокси)бутил]фенил)-2-тиофенкарбоксимидамид: 16
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 15, исходящим из 3-гидроксидифениламина. Продукт выделяют в форме свободного основания. Белое твердое вещество. Точка плавления: 73-74°С.
Пример 17. N’-(9Н-Карбазол-3-ил)-2-тиофенкарбоксимидамид: 17
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 1, исходящим из 3-аминокарбазола (Pharmazie (1993) 48 (11), 817-820). Продукт выделяют в форме свободного основания. Светло-бежевое твердое вещество. Точка плавления: 243-244°С.
Пример 18.
Гидрохлорид 4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(9Н-кар6азол-3-ил)бутанамида: 18
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из 3-аминокарбазола (Pharmazie (1993) 48 (11), 817-820) и 4-нитрофенилмасляной кислоты. Светло-бежевое твердое вещество. Точка плавления >250° С.
МС: MH+: 452,2.
Пример 19.
Гидроиодид N’-[4-(10Н-фенотиазин-2-илокси)фенил]-2-тиофенкарбоксимидамида: 19
19.1) 2-(4-Нитрофенокси)-10Н-фенотиазин:
2-Гидрокси-10Н-фенотиазин (J. Med. Chem. (1992), 35, 716) в количестве 1,1 г (5,11 ммоль), 1,34 г (9,71 ммоль) К2СО3 и 0,94 г (6,64 ммоль) 4-фтор-1-нитробензола смешивают в 25 мл безводного ДМФА в колбе с атмосферой аргона. Реакционную смесь нагревают при 70°С в течение 18 часов. Растворитель затем выпаривают в вакууме и остаток растворяют в 50 мл AcOEt и 50 мл воды. После перемешивания и декантации органическую фазу промывают 50 мл соленой воды. Органический раствор сушат над МgSO4, фильтруют и концентрируют в вакууме. Остаток кристаллизуют из диизопропилового эфира. После сушки получают желтое твердое вещество с выходом 83%. Точка плавления: 210-211°С.
19.2) Гидроиодид N’-[4-(10Н-фенотиазин-2-илокси)фенил]-2-тиофенкарбоксимидамида:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточных продуктов 2.2 и 2.3, исходящим из промежуточного продукта 19.1. Ожидаемый конечный продукт осаждается непосредственно из реакционной смеси, его выделяют фильтрованием и промывают с использованием изо-РrОН. Желтое твердое вещество. Точка плавления: 175-180°С.
Пример 20.
Гидрохлорид N’-{4-[(10Н-метил-10Н-фенотиазин-2-ил)окси]фенил}-2-тиофенкарбоксимидамида: 20
20.1) 10-Метил-10Н-фенотиазин-2-ил-4-нитрофениловый эфир:
NaH (60%) в количестве 0,014 г (0,58 ммоль) добавляют в колбу с атмосферой аргона, содержащей раствор 0,1 г (0,29 ммоль) промежуточного продукта 19.1 в 10 мл безводного ДМФА. Перемешивание поддерживают при 20°С в течение 16 часов. Затем к реакционной смеси при перемешивании при 20°С добавляют 0,04 мл (0,58 ммоль) МеI. В конце реакции всю реакционную массу выливают в 50 мл охлажденной льдом воды и продукт экстрагируют с использованием 50 мл AcOEt. Органическую фазу декантируют, промывают 50 мл соленой воды, сушат над МgSO4, фильтруют и концентрируют в вакууме. Остаток очищают на колонке с диоксидом кремния (элюент:гептан/AcOEt: 80/20). Получают оранжевое масло с выходом 50%.
20.2) Гидрохлорид N’-(4-[(10-метил-10Н-фенотиазин-2-ил)окси]фенил}-2-тиофенкарбоксимидамида:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточных продуктов 2.2 и 2.3, исходящим из промежуточного продукта 20.1. Получают белый твердый продукт. Точка плавления: 256-257°С.
Пример 21.
Гидрохлорид 4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(10Н-фенотиазин-3-ил)бутанамида: 21
21.1) 3-Амино-10Н-фенотиазин:
Восстановление функциональной нитрогруппы 3-нитро-10Н-фенотиазина (J. Org. Chem. (1972), 37, 2691) проводят в присутствии Pd/C в смеси EtOH/ТГФ в условиях, описанных для промежуточного продукта 2.2. Получают серое твердое вещество с выходом 97%. Точка плавления: 150-156°С.
21.2) Гидрохлорид N’-(4-{[амино(2-тиeнил)мeтилилeн]-амино}фенил)-N-(10Н-фенотиазин-3-ил)бутанамида:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из промежуточного продукта 21.1 Светло-серое твердое вещество. Точка плавления: 170-176°С.
Пример 22.
Дигидрохлорид N’-(4-{4[2(10H-фенотиазин-2-илокси)этил]-1-пиперазинил}фенил)-2-тиофенкарбоксимида: 22
22.1) 2-[4-(4-Нитрофенил)-1-пиперазинил]-1-этанол:
2-Бромэтанол в количестве 7,5 г (60 ммоль) добавляют в атмосфере аргона к смеси 10,35 г (50 ммоль) 1-(4-нитрофенил)пиперазина, 7,6 г (55 ммоль) К2СО3 и 9 мл (65 ммоль) Et3N в 200 мл СН2 Сl2. Всю реакционную массу затем нагревают при 45°С в течение 18 часов. Реакционную смесь, наконец, разбавляют 50 мл воды, перемешивают и декантируют. Органическую фазу промывают 50 мл соленой воды, сушат над МgSO4, фильтруют и концентрируют в вакууме. Остаток кристаллизуют из диизопропилового эфира. Получают желтое твердое вещество с выходом 89%. Точка плавления: 98-99°С.
22.2) 1-(2-Бромэтил)-4-(4-нитрофенил)пиперазин:
СВr4 в количестве 8,6 г (26 ммоль) добавляют к раствору 5 г (20 ммоль) промежуточного продукта 22.1 в 75 мл CH2Cl2. Всю реакционную смесь охлаждают с использованием ледяной бани до добавления по порциям 6,3 г (24 ммоль) трифенилфосфина. Перемешивание поддерживают в течение 2 часов при 20°С. После добавления 50 мл воды, перемешивания и декантации органическую фазу промывают 50 мл соленой воды, сушат над МgSO4, фильтруют и концентрируют в вакууме. Остаток после выпаривания очищают на колонке с диоксидом кремния (элюент: CH2Cl2/EtOH: 95/5) и, наконец, кристаллизуют из этилового эфира. Получают желто-оранжевое твердое вещество с выходом 40%. Точка плавления: 134-135°С.
22.3) 2-{2-[4(4-Нитрофенил)-1-пиперазинил]этокси}-10Н-фенотиазин:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточного продукта 19.1, исходящим из промежуточного продукта 22.2. Получают желтое твердое вещество с выходом 43%. Точка плавления: 224-225°С.
22.4) Дигидрохлорид N’-(4-{4-[2-(10Н-фенотиазин-2-илокси)этил]-1-пиперазинил}фенил)-2-тиофенкарбоксимидамида:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточных продуктов 2.2 и 2.3, исходящим из промежуточного продукта 22.3. Светло-бежевое твердое вещество. Точка плавления: 198-200°С.
Пример 23.
Гидрохлорид 4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-[4-(4-толуидино)фенил]бутанамида: 23
23.1) N’-(4-Метилфенил)-1,4-бензолдиамин:
Восстановление функциональной нитрогруппы N-(4-метилфенил)-4-нитроанилина (Indian J. Chem. (1981), 20В, 611-613) проводят в присутствии Pd/C в этаноле в условиях, описанных для промежуточного продукта 2.2. Получают серое твердое вещество с выходом 85%.
23.2) Гидрохлорид 4-(4-{[амино(2-тиенил)метилиден]-амино}фенил)-N-[4-(4-толуидино)фенил]бутанамида:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из промежуточного продукта 22.3 и 4-нитрофенилмасляной кислоты. Желтое твердое вещество. Точка плавления: 142-145°С.
Пример 24.
3-Анилинофенил-4(4-{[амино(2-тиенил)метилиден]амино}фенил)бутаноат: 24
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 14, исходящим из 3-гидроксидифениламина и 4-нитрофенилмасляной кислоты. Белое твердое вещество. Точка плавления: 110-112°С.
Пример 25.
Гидрохлорид 2-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-[2-(9Н-карбазол-4-илокси)этил]ацетамида: 25
25.1) 3-(2-Бромэтокси)-9Н-кар6азол:
Смесь 1,83 г (10 ммоль) 4-гидроксикарбазола (J. Chem. Sоc. (1955), 3475-3477; J. Med. Chem (1964), 7, 158-161), 1,08 мл (12,5 ммоль) 1,2-дибромэтана и 2,6 мл (10,5 ммоль) 4 М водного раствора NaOH в 2 мл воды нагревают при кипячении с обратным холодильником в течение 5 часов. После того как температуре дают снизиться до 20°С, продукт экстрагируют с использованием 2 раза по 30 мл CH2Cl2. Собранные органические растворы затем последовательно промывают 20 мл воды и 20 мл соленой воды. После сушки над МgSO4, фильтрования и концентрирования в вакууме остаток очищают на колонке с диоксидом кремния (элюент: гептан/AcOEt: 80/20). Получают бежевый порошок с выходом 32%. Точка плавления: 135-136°С.
25.2) 3-(2-Азидоэтокси)-9Н-карбазол:
Смесь 0,9 г (3,1 ммоль) промежуточного продукта 25.1 и 0,20 г (3,1 ммоль) NaN3 в 10 мл ДМФА нагревают при 70°С в течение 1 часа. Всю реакционную смесь затем выливают в 30 мл смеси воды и льда. После добавления 50 мл AcOEt и перемешивания органическую фазу декантируют и промывают последовательно 20 мл воды и 20 мл соленой воды. Органический раствор затем сушат над Na2SО4, фильтруют и концентрируют в вакууме. После сушки получают бежевый порошок (количественный выход), который используют непосредственно в следующей стадии.
25.3) 2-(9Н-Карбазол-3-илокси)этиламин:
Раствор промежуточного продукта 25.2 в 50 мл EtOH, a также 0,3 г Pd/C (10%) вводят в автоклав из нержавеющей стали, снабженный магнитной мешалкой. Реакционную смесь перемешивают в течение 2 часов под давлением H2 1,5 бара (1,48 атм) при температуре 25°С. Pd/C затем удаляют фильтрованием и фильтрат концентрируют в вакууме досуха. Остаток растворяют в этиловый эфир и образованные кристаллы фильтруют и обильно промывают этиловым эфиром. После сушки получают белый порошок с выходом 82%. Точка плавления: 145-146° С.
25.4) Гидрохлорид 2-(4-{[амино(2-тиенил)метилиден]-амино}фенил)-N-[2-(9Н-карбазол-4-илокси)этил]ацетамида:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из промежуточного продукта 25.3 и 4-нитрофенилмасляной кислоты. Светло-бежевое твердое вещество. Точка плавления: 233-234°С.
Пример 26.
Гидрохлорид N-(4-{[амино{2-тиенил)метилиден]амино}фенетил)-2-анилинобензамида: 26
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из N-фенилантраниловой кислоты и 4-нитрофенетиламина. Светло-желтое твердое вещество. Точка плавления: 163-165°С.
Пример 27.
Гидрохлорид N-(4-{[амино(2-тиенил)метилиден]амино}фенетил)-2-(2,3-диметиланилино)бензамида: 27
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из мефенамовой кислоты и 4-нитрофенетиламина. Светло-желтое твердое вещество. Точка плавления: 168-170°С.
Пример 28.
Дигидрохлорид N’-{4-[4-(2-анилинобензо-ил)-1-пиперазинил]фенил}-2-тиофенкарбоксимидамида: 28
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из N-фенилантраниловой кислоты и 4-нитрофенилпиперазина. Светло-желтое твердое вещество. Точка плавления: 168-170°С.
Пример 29.
Дигидрохлорид N’-{4-{4-(2-(2,3-диметиланилино)бензоил]-1-пиперазинил)фенил)-2-тиофенкарбоксимидамида: 29
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из мефенамовой кислоты и 4-нитрофенилпиперазина. Светло-желтое твердое вещество. Точка плавления: 166-168°С.
Пример 30.
Гидрохлорид 4-(4-{[амино(2-тиенил)метилиден]амино}фенил)-N-(4-феноксифенил)бутанамида: 30
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 14, исходящим из 4-нитрофенилмасляной кислоты и 4-феноксифенола. Светло-желтое твердое вещество. Точка плавления: 119-123°С.
Пример 31.
Гидрохлорид N-(4-{[амино(2-тиенил)метилиден]амино}фенетил)-4-(4-гидроксифенокси)бензамида: 31
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из 4-(4-гидроксифенокси)бензойной кислоты и 4-нитрофенетиламина. Светло-желтое твердое вещество. Точка плавления: 155-157°С.
Пример 32.
N-[2-(9Н-Карбазол-4-илокси)этил]-2-тиофен-карбоксимидамид: 32
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2.3, исходящим из промежуточного продукта 25.3. Ожидаемый продукт выделяют в форме свободного основания. Белое твердое вещество. Точка плавления: 180-181°С.
Пример 33.
N-[3-(9Н-Карбазол-4-илокси)пропил]-2-тио-фенкарбоксимидамид: 33
33.1) 2-[3-(9Н-Карбазол-4-илокси)пропил]-1H-изоиндол-1,3(2Н)-дион:
4-Гидроксикарбазол (J. Chem. Soc. (1955), 3475-3477; J. Med. Chem. (1964), 7, 158-161) в количестве 1 г (5,46 ммоль) добавляют в атмосфере аргона к суспензии 0,23 г (5,73 ммоль) NaH (60%) в 20 мл безводного ДМФА. После перемешивания в течение 30 минут при 20°С к реакционной смеси добавляют по каплям 1,46 г (5,46 ммоль) 3-бромпропилфталимида в растворе в 10 мл безводного ДМФА. Всю реакционную смесь нагревают при 80°С в течение 16 часов. После того как температуре дают понизиться до 20°С, добавляют 5 мл воды, и смесь концентрируют в вакууме. Остаток растворяют в 300 мл CH2Cl2 и органический раствор промывают последовательно 50 мл 1 М NaOH, 100 мл воды и 100 мл соленой воды. После сушки над MgSO4, фильтрования и концентрирования в вакууме получают маслянистый остаток, который медленно кристаллизуется. Кристаллы промывают с использованием этилового эфира. Получают бежевое твердое вещество с выходом 40%. Точка плавления: 171-172°С.
33.2) 3-(9Н-Карбазол-4-илокси)пропиламин:
Раствор 0,13 мл (3,24 ммоль) гидразин-гидрата в 5 мл этанола добавляют по каплям к раствору 0,8 г (2,16 ммоль) промежуточного продукта 33.1 в 30 мл этанола, нагревают для кипячения с обратным холодильником. Реакционную смесь перемешивают и нагревают при кипячении с обратным холодильником в течение 4 часов. После того как температура понизиться до 20°С, продукт распределяют между 100 мл AcOEt и 50 мл 1 М NaOH. После декантации органическую фазу промывают последовательно 50 мл воды и 50 мл соленой воды. Органический раствор сушат над Na2SО4, фильтруют и концентрируют в вакууме. Получают бежевый порошок с выходом 41%. Точка плавления: 146-147°С.
33.3) N-[3-(9Н-Карбазол-4-илокси)пропил]-2-тиофенкарбоксимидамид:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2.3, исходящим из промежуточного продукта 33.2. Ожидаемый продукт выделяют в форме свободного основания. Бледно-бежевое твердое вещество. Точка плавления: 189-190°С.
Пример 34.
Гидроиодид N-{4-[4-(10Н-фенотиазин-2-ил-окси)бутил]фенил}-2-тиофенкарбоксимидамида: 34
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 15, исходящим из 4-(4-нитрофенил)-1-бутанола и 2-гидрокси-10Н-фенотиазина (J. Med. Chem. (1992), 35, 716). Ожидаемый конечный продукт осаждается непосредственно из реакционной смеси, его выделяют фильтрованием и промывают с использованием iPrOH. Желтое твердое вещество. Точка плавления: 262-270°С.
Пример 35.
Тригидрохлорид 3-[(3-{[амино(2-тиенил)метилиден]амино}бензил)амино]-N-(4-анилинофенил) пропанамида: 35
35.1) трет-Бутил-3-(4-анилиноанилино)-3-оксопропилкарбамат:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточного продукта 2.1, исходящим из Вос-β-аланина и 4-аминодифениламина. После быстрого фильтрования на колонке с диоксидом кремния (элюент: гептан/AcOEt: 1/1) ожидаемый продукт получают с количественным выходом.
35.2) 3-Амино-N-(4-анилинофенил)пропанамид:
Промежуточный продукт 35.1 в количестве 15 г (42,2 ммоль) растворяют в 300 мл AcOEt и добавляют 120 мл водного 6н. раствора НСl. Реакционную смесь энергично перемешивают при 20°С в течение 1 часа. После декантации водную фазу выделяют и делают основной (рН>11) добавлением водного 2 М раствора NaOH. Продукт затем экстрагируют с использованием два раза по 50 мл CH2Cl2 и органическую фазу промывают 50 мл соленой воды. После сушки над МgSO4, фильтрования и концентрирования в вакууме остаток очищают на колонке с диоксидом кремния (элюент: CH2Cl2/EtOH/NH4OH (20%): 20/5/0,5). Получают фиолетовый порошок с выходом 73%. Точка плавления: 108-110°С.
35.3) N-(4-Анилинофенил)-3-[(3-нитробензил)амино]пропанамид:
Промежуточный продукт 35.2 в количестве 1,40 г (5,5 ммоль), 0,92 г (6 ммоль) 3-нитробензальдегида и 3 г порошкообразного молекулярного сита 4

35.4) 3-[(3-Аминобензил)амино]-N-(4-анилинофенил)пропанамид:
Восстановление функциональной нитрогруппы промежуточного продукта 35.3 проводят в присутствии Pd/C в этаноле в условиях, описанных для промежуточного продукта 2.2. После фильтрования Pd/C и концентрирования в вакууме продукт непосредственно используют в следующей стадии.
35.5) Тригидрохлорид 3-[(3-{[амино(2-тиенил)метилиден]амино}бензил)амино]-N-(4-анилинофенил)пропанамида:
Промежуточный продукт 35.4 в количестве 0,50 г (1,40 ммоль) и 0,50 г (1,75 ммоль) гидроиодида S-метил-2-тиофентиокарбоксимида растворяют в 15 мл изопропанола и 15 мл ДМФА в 50 мл колбе в присутствии 0,11 мл (1,40 ммоль) пиридина. Реакционную смесь перемешивают в течение 20 часов при 23°С. После выпаривания растворителя в вакууме остаток растворяют в 100 мл смеси 1н. NaOH и этилацетата (1/1). После декантации органическую фазу промывают 50 мл воды, затем 50 мл соленой воды. Органический раствор сушат над сульфатом магния, фильтруют, концентрируют в вакууме и остаток очищают на колонке с диоксидом кремния (элюент: CH2Cl2/EtOH/NH4OH (20%): 20/5/0,5). Чистые фракции собирают и концентрируют в вакууме. Соединение затем растворяют в метаноле и превращают в соль добавлением 1н. раствора НСl в этиловом эфире (10 мл). После перемешивания в течение одного часа при 20°С реакционную смесь концентрируют в вакууме с получением бледно-желтого порошка. Точка плавления: 184-186°С.
Пример 36.
N’-(4-{2-[(10Н-Фенотиазин-3-илметил)амино]этил}фенил)-2-тиофенкарбоксимидамид: 36
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточных продуктов 35.3, 35.4 и 35.5, исходящим из 10Н-фенотиазин-3-карбальдегида (J. Chem. Soc. (1951), 1834; Bull. Soc. Chim. Fr. (1969), 1769) и 4-нитрофенетиламина. Бежевая пена. МС: МН+: 457,1.
Пример 37.
Гидрохлорид N-(4-{[амино(2-тиенил)метилиден]амино}фенетил)-2-метокси-10Н-фенотиазин-1-карбоксамида: 37
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 2, исходящим из 2-метокси-10Н-фенотиазин-1-карбоновой кислоты (J. Med. Chem. (1992) 35(4), 716-724) и 4-нитрофенетиламина. Бледно-желтое твердое вещество. Точка плавления > 200°С (разложение).
Пример 38.
N’-[4-(2-{[(2-Метокси-10Н-фенотиазин-1-ил)метил]амино}этил)фенил]-2-тиофенкарбоксимидамид: 38
38.1) 3-Метокси-10Н-фенотиазин-1-карбальдегид:
2-Метокси-10Н-фенотиазин в количестве 4,6 г (20 ммоль) растворяют в атмосфере аргона в трехгорлой колбе, содержащей 140 мл безводного этилового эфира. Затем при 20°С по каплям добавляют 20 мл (50 ммоль) раствора н-BuLi (2,5 М) в гексане. Реакционную смесь перемешивают в течение 3 часов при 20°С до добавления по каплям 6,2 мл (80 ммоль) безводного ДМФА. Перемешивание поддерживают в течение дополнительных 15 часов при 20°С. Всю смесь затем выливают в 150 мл охлажденной льдом воды и продукт экстрагируют два раза с использованием 200 мл этилацетата. Органический раствор промывают 100 мл соленой воды, сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток выпаривания растворяют в диизопропиловом эфире, фильтруют и сушат с получением красного твердого вещества с выходом 30%. Точка плавления: 155-160°С.
38.2) N’-[4-{2-{[(2-Метокси-10Н-фенотиазин-1-ил)метил]амино}этил)фенил]-2-тиофенкарбоксимидамид:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для примера 36, исходящим из промежуточного продукта 38.1 и 4-нитрофенетиламина. Серое твердое вещество.
МС: МН+: 487,2.
Пример 39.
N’-{4-[(10Н-Фенотиазин-2-илокси)метил]фенил}-2-тиофенкарбоксимидамид: 39
39.1) 2-[(4-Нитробензил)окси]-10Н-фенотиазин:
2-Гидрокси-10Н-фенотиазин (J. Med. Chem. (1992), 35, 716) в количестве 1,08 г (5 ммоль) растворяют в атмосфере аргона в 20 мл безводного ТГФ в колбе. Раствор затем охлаждают до 0°С и по порциям добавляют 0,22 г (5,5 ммоль) NaH (60%). После перемешивания в течение 15 минут по частям добавляют 1,2 г (5,5 ммоль) 4-нитробензилбромида и реакционную смесь перемешивают в течение 15 часов при 20°С до выливания в 50 мл охлажденной льдом воды. Продукт экстрагируют два раза по 25 мл СН2Сl2 и органический раствор промывают последовательно 25 мл воды и 25 мл соленой воды. После сушки над МgSO4, фильтрования и концентрирования в вакууме остаток выпаривания очищают на колонке с диоксидом кремния (элюент: CH2Cl2/EtOH: от 99/1 до 98/2). После концентрирования самых чистых фракций получают твердое вещество каштанового цвета, которое перекристаллизовывают с использованием изопропилацетата. Наконец, получают твердое вещество каштанового цвета с выходом 37%.
39.2) 4-[(10Н-Фенотиазин-2-илокси)метил]анилин:
SnСl2·2Н2О в количестве 1,02 г (4,52 ммоль) и 0,29 г (4,52 ммоль) Zn добавляют последовательно к раствору 0,65 г (1,86 ммоль) промежуточного продукта 39.1 в смеси 9,3 мл уксусной кислоты и 1,2 мл НСl (12н.). Всю реакционную смесь перемешивают в течение 18 часов при 20°С. Реакционную смесь затем подщелачивают добавлением 30% водного раствора NaOH. Продукт затем экстрагируют два раза с использованием по 50 мл CH2Cl2. Органический раствор промывают 50 мл соленой воды, сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают на колонке с диоксидом кремния (элюент: гептан/AcOEt: 1/1). Получают светло-желтое твердое вещество с выходом 20%. Точка плавления: >175°С (разложение).
39.3) N’-{4-[(10Н-Фенотиазин-2-илокси)метил]фенил}-2-тиофенкарбоксимидамид:
Использованный экспериментальный протокол является таким же протоколом, как протокол, описанный для промежуточного продукта 2.3, исходящим из промежуточного продукта 39.2. Оранжево-розовое твердое вещество. Точка плавления: 105-116°С.
Фармакологическое изучение продуктов данного изобретения
Изучение влияния на нейронную конститутивную NO-синтазу мозжечка крыс
Ингибирующую активность продуктов данного изобретения определяют посредством измерения их действия на превращение NO-синтазой [3H]L-аргинина в [3Н]L-цитруллин по модифицированному способу Bredt и Snyder (Proc. Natl. Acad. Sci, USA, (1990), 87: 682-685). Мозжечок крыс Sprague-Dawley (300 г - Charles River) быстро удаляют, иссекают при 4°С и гомогенизируют в объеме буфера для экстракции (50 мМ HEPES, 1 мМ ЭДТК, рН 7,4, 10 мг/мл пепстатина А, 10 мг/мл лейпептина). Гомогенаты затем центрифугируют при 21000 g в течение 15 мин при 4°С. Дозирование проводят в стеклянных трубках для испытания, в которых распределяют 100 мкл буфера для инкубации, содержащего 100 мМ HEPES (рН 7,4), 2 мМ ЭДТК, 2,5 мМ CaCl2, 2 мМ дитиотреита, 2 мМ восстановленного НАДФ и 10 мкг/мл калмодулина. Добавляют 25 мкл раствора, содержащего 100 нМ [3H]L-аргинин (удельная активность: 56,4 Ки/ммоль, Amercham) и 40 мкМ нерадиоактивный L-аргинин. Реакцию инициируют добавлением 50 мкл гомогената, причем конечный объем составляет 200 мкл (недостающими 25 мкл являются либо вода либо испытуемый продукт). Спустя 15 мин реакцию останавливают 2 мл останавливающего буфера (20 мМ HEPES, рН 5,5, 2 мМ ЭДТК). После пропускания образцов через 1 мл колонку со смолой DOWEX радиоактивность количественно определяют жидкостным сцинтилляционным спектрометром. Соединения примеров 3-5, 1, 9-12, 15, 16, 18, 19, 21, 22, 26, 27, 30, 31 и 35-37, описанные выше, проявляют IC50 ниже, чем 3,5 мкМ.
Изучение влияния на липидное перокисление коры головного мозга крыс
Ингибирующую активность продуктов данного изобретения определяют измерением их влияния на степень липидного перокисления, определяемую концентрацией малонового диальдегида (MDA). MDA, продуцированный перокислением ненасыщенных жирных кислот, представляет собой хороший указатель липидного перокисления (Н. Esterbauer and KH Cheeseman, Meth. Enzymol. (1990), 186: 407-421). Самцов крыс Sprague Dawley, весящих от 200 до 250 г (Charles River) умерщвляли декапитацией. Кору головного мозга удаляли, затем гомогенизировали с использованием керамической посуды Thomas в 20 мМ буфере Трис-HCl, рН 7,4. Гомогенат центрифугировали два раза при 50000 g в течение 10 минут при 4°С. Осадок центрифугирования хранили при -80°С. В день эксперимента осадок центрифугирования помещали в суспензию при концентрации 1 г/15 мл и центрифугировали при 515 g в течение 10 минут при 4°С. Супернатант используют сразу для определения липидного перокисления. Гомогенат коры головного мозга (500 мкл) инкубируют при 37°С в течение 15 минут в присутствии испытуемых соединений или растворителя (10 мкл). Реакцию липидного перокисления инициируют добавлением 50 мкл FeCl2 в концентрации 1 мМ, ЭДТК в концентрации 1 мМ и аскорбиновой кислоты в концентрации 4 мМ. После инкубации в течение 30 минут при 37°С реакцию останавливают добавлением 50 мкл раствора гидроксилированного ди-трет-бутилтолуола (ВНТ, 0,2%). MDA количественно определяют с использованием колориметрической пробы реакцией хромогенного реагента (R) а именнно N-метил-2-фенилиндола (650 мкл) с 200 мкл гомогената в течение 1 часа при 45°С. Конденсация молекулы MDA с двумя молекулами реагента R образует стабильный хромофор, максимальная длина волны поглощения которого равна 586 нм. (Caldwell et al. European J. Pharmacol. (1995), 285, 203-206). Соединения примеров 1-9, 12-19, 21-23, 30 и 35-37, описанные выше, проявляют IC50, ниже, чем 30 мкМ.
Методика испытаний на токсичность
Тест на жизнеспособность линии мышиных макрофаговых клеток J774-A1
Линия клеток
Линию мышиных макрофаговых клеток J774 А1 получали из коллекции АТСС, сохраняли при 37°С с 5% СО2 в модифицированной по Дульбекко минимальной эссенциальной среде (DMEM)/10% фетальной коровьей сыворотке (FBS).
Обработка клеток
Клетки собирали после центрифугирования, промывали и суспендировали в DMEM/10% FBS. Эти клетки (5х104 клеток на лунку) культивировали на 96-луночных многолуночных планшетах в течение 24 часов и затем в течение еще 18 часов в присутствии тестируемых соединений в концентрации 10, 30, 50 или 100 мкМ. Тестируемые соединения растворяли в диметилсульфоксиде (ДМСО) или этаноле в виде исходных растворов кратностью 250Х, 500Х или 1000Х. Последующие разбавления выполняли в DMEM. Конечная концентрация ДМСО или этанола не влияла на клеточную морфологию или жизнеспособность клеток.
Оценка жизнеспособности клеток.
Испытание с красителем Alamar Blue выполняли в соответствии с инструкциями фирмы-производителя (Interchim). В кратком изложении, после отсасывания среды добавляли разбавленный в соотношении 1/10 раствор Alamar Blue в DMEM без фенолового красного. 10% FBS добавляли в течение 3 часов при 37°С (5% CO2). Колориметрическое определение проводили на волнах с длиной 570 нм и 620 нм на планшет-ридере. Процент ингибирования метаболической активности в результате реакции на испытуемые соединения в сравнении с необработанными клетками рассчитывали по формуле:
% жизнеспособности клеток =

где OD – оптическая плотность при соответствующей длине волны.
Значение CT50 представляет собой концентрацию, при которой жизнеспособность клеток снижается на 50%, и это значение получают экстраполяцией графиков зависимости степени реакции от дозы соединения.

Реферат
Изобретение относится к соединениям формулы (I)

в которой Ф представляет фениленовый радикал, А представляет радикал

в котором Rl, R2, R3, R4, R5 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода; R11 представляет атом водорода, неразветвленный или разветвленный алкильный радикал, имеющий от 1 до 6 атомов углерода, или радикал

в котором Rl, R2, R3, R4, R5 представляют независимо атом водорода, ОН-группу или неразветвленный или разветвленный алкильный или алкоксирадикал, имеющий от 1 до 6 атомов углерода; В представляет тиофен; W отсутствует или представляет связь или S; Х представляет связь или радикал -(CH2)k-NR16-, -О-, -СО-, -NR16-CO-, и так далее, причем k равно 0 или 1; Y представляет связь или радикал, выбранный из радикалов -(СН2)m-, -(CH2)m-O-(CH2)n, -(CH2)m-NR18-(CH2)n-, -(CH2)m-NR18 -CO-(CH2)n-, -(CH2)m-Q-(CH2)n; причем Q представляет пиперазиновый радикал, m и n равны целым числам от 0 до 6; R16, R17, R18 представляют независимо атом водорода, или соль указанного соединения. Предложены соединения, представленные формулами (IS) и (IS’),
А-Х-У-Ф-Т (IS)

используемые в качестве промежуточных продуктов для получения соединений формулы (I). Также предложена фармацевтическая композиция, обладающая свойством ингибирования NO-синтазы, включающая активное соединение. В качестве активного соединения фармацевтическая композиция содержит соединение формулы (I) или его фармацевтически приемлемую соль. Технический результат - производные N-(иминометил)аминов, обладающие ингибирующей активностью в отношении ферментов NO-синтаз, продуцирующих монооксид азота NO. 4 с. и 9 з.п. ф-лы, 1 табл.
Формула












Комментарии