Производные гетероциклических бензоциклоалкенов и способ их получения - RU2197484C2
Код документа: RU2197484C2
Чертежи

Описание
Настоящее изобретение относится к замещенным гетероциклическим бензоциклоалкенам общей формулы I,

где
R1 обозначает ОН, С1-С6алкокси, -O-(С3-С7)циклоалкил;
R2 обозначает С1-С6алкил;
R3 обозначает С1-С6алкил, -(СН2)1-2-арил, -(СН2)1-2 -гетероциклил, -СН2CH=C(R6)2,-СН2-(С3-С7)циклоалкил;
R4 и R5 обозначают одновременно либо в отличие друг от друга Н, Cl, F, CF3, С1-С6алкил, ОН, С1-С6алкокси, O-(С3-С7)циклоалкил, -(СН2)0-2-арил, -O-(CH2)0-2-aрил, α,β-или β,γ-O(СН2)1-2-О-, гетероциклил, α,β-или β,γ-бензогруппу, незамещенные или одно- либо двузамещенные Cl, F, CF3, ОН, С1-С6алкилом, С1-С6алкоксигруппой, -CON(R6R7);
R6 обозначает Н, С1-С6алкил;
R7 обозначает Н, С1-С6алкил, -(CH2)0-2-арил -(СН2)-(С3-С7 )циклоалкил или
R6 и R7 оба вместе обозначают (-СН2-)5-7 или (-СН2)2-O-(СН2)2-;
Х обозначает О, S, SO, SO2 и
Y обозначает -(СН2)1-2-, -СН2-С(СН3)2- или -С(СH3)2-,
или к их применимым в фармацевтике солям, а также к способу получения и к применению указанных соединений в качестве лекарственных средств.
Классические опиоиды, такие как морфин, высокоэффективны при лечении сильных и очень сильных болей. Однако возможности их применения ограничены из-за обусловленных ими известных побочных эффектов, как, например, депрессия дыхания, рвота, необходимость успокоения седативными средствами, запоры и развитие толерантности. Кроме того, их эффективность снижается при невропатических и инцидентальных болях, от которых в первую очередь страдают онкологические больные.
Опиоиды проявляют свое анальгезирующее действие благодаря связыванию с присутствующими на мембране рецепторами, относящимися к семейству так называемых связанных
с G-протеином рецепторов.
Биохимические и фармакологические характеристики подтипов этих рецепторов позволяют надеяться, что специфические по отношению к этим подтипам опиоиды обладают другим
механизмом действия, включая
побочные эффекты, по сравнению, например, с морфином. Если морфин связывается избирательно с так называемыми μ-рецепторами, то эндогенные энкефалины
характеризуются как δ-селективные
пептиды. Дальнейшие фармакологические исследования дают основание предположить, что существует целый ряд подтипов этих опиоидорецепторов (μ1,
μ2, κ1, κ2, κ3, δ1 и δ2).
Сведения о физиологическом значении субстанций,
селективных по отношению к δ
-рецепторам, были существенно расширены благодаря открытию непептидного антагониста налтриндола. На сегодняшний день очевидно, что δ-агонисты обладают
собственным независимым антиноцицептивным
потенциалом. Наряду с данными, полученными при проведении многочисленных экспериментов на животных, имеются также результаты исследований, проводившихся с
пептидным агонистом D-аланин2
-D-лейцин5-энкефалин (DADL) на онкологических больных, у которых морфин более не проявлял анальгетической эффективности. При интратекальном введении
DADL обеспечивал продолжительный
анальгетический эффект. Ярко выраженное отличие δ-агонистов от μ-агонистов проявляется в их взаимодействии с "эндогенным антагонистом опиоидов"
холецистокинином (ХЦК). Наряду с этим
различием в механизме действия δ-агонисты, как полагают, могут отличаться от μ-агонистов также и в той его части, которая касается побочных
эффектов, что проявляется, в частности, в
снижении депрессии дыхания.
Вполне допустима возможность применения этих соединений в терапии в качестве анальгетических средств или, в принципе, при всех патологических состояниях, для лечения которых обычно используют агонисты δ-опиаторецепторов.
Исходя из вышеизложенного в основу настоящего изобретения была положена задача получить субстанции с анальгезирующим действием, биологическая эффективность которых частично либо преимущественно опосредуется агонистами δ-опиаторецепторов.
Благодаря замещенным гетероциклическим бензоциклоалкенам согласно настоящему изобретению удалось получить соединения, которые отвечают указанным требованиям.
Объектом изобретения
являются замещенные гетероциклические
бензоциклоалкены общей формулы I,

где
R1 обозначает ОН, С1 -С6алкокси, -O-(С3-С7)циклоалкил;
R2 обозначает С1-С6алкил;
R3 обозначает С1-С6 алкил, -(СН2)1-2-арил, -(СН2)1-2-гетероциклил, -СН2CH=C(R6)2, -СН2-(С3-С7)циклоалкил;
R4 и R5 обозначают одновременно либо в отличие друг от друга Н, Cl, F, СF3, С1-С6алкил, ОН, C1-С6алкокси, O-(С3-С7)циклоалкил, -(CH2)0-2-арил, -O-(СН2)0-2-арил, α,β- или β,γ -O(CH2)1-2-O-, гетероциклил, α,β- или β,γ-бензогруппу, незамещенные или одно- либо двузамещенные Cl, F, СF3, ОН, С1-С6алкилом, С1-С6 алкоксигруппой, -CON(R6R7);
R6 обозначает Н, С1-С6алкил;
R7 обозначает Н, С1-С6алкил, -(CH2)0-2-aрил, -(СН2)-(С3-С7)циклоалкил
или
R6 и R7 оба вместе обозначают (-СН2-)5-7 или (-СH2)2-O-(CH2)2-;
Х обозначает О, S, SO, SO2 и
Y обозначает -(СН2 )1-2-, -СН2-С(СН3 )2- или -С(СН3)2-,
или их применимые в фармацевтике соли.
Предпочтительными являются
соединения общей формулы I, в которых Х обозначает О, S
либо SO и Y обозначает -(СН2)1-2-, a R1-R7 имеют значение согласно расшифровке общей формулы I,
или
Х обозначает О, Y обозначает -(СН2
)1-2-, а R1 -R7 имеют значение согласно расшифровке общей формулы I, или R и R одновременно либо в отличие
друг от друга обозначают -O-(СН2)0-2
-арил, -(CH2)0-2-арил либо гетероциклил, a R1-R3, R6, R7, Х и Y имеют
указанное выше значение, или R1 обозначает С1-С6алкокси либо -O-(С3-С7)циклоалкил, R4 и R5 одновременно либо в отличие
друг от друга обозначают -(CH2)0-2-арил
либо гетероциклил, а R2, R3, R6, R7, Х и Y имеют указанное выше значение, или R1
обозначает ОН либо С1-С6алкокси, a R2-R7, Х и Y имеют указанное выше значение, или
Y обозначает -(CH2)1-2-, R2
обозначает C1-С6алкил, а X, R1 и
R3-R7 имеют значение согласно расшифровке общей формулы I, или
Y обозначает -(СН2)1-2-, R1 обозначает ОН либо С2-С6
алкокси, R2 и R3 обозначают С1-С6алкил, а Х и R4-R7 имеют
значение согласно расшифровке общей формулы I, или
Y обозначает
-(СН2)1-2-, R1 обозначает ОН, R2 и R3 обозначают С1-С6алкил, Х обозначает О, а R4 -R7 имеют значение
согласно расшифровке общей формулы I.
Понятие "C1-С6алкил" в контексте настоящего изобретения обозначает линейные либо разветвленные углеводороды с 1-6 атомами углерода. В качестве примеров можно назвать метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, неопентил и н-гексил.
Понятие "С1-С6 алкоксигруппа" в рамках настоящего изобретения обозначает линейные либо разветвленные углеводороды с 1-6 атомами углерода, как указано выше, связанные через атом кислорода.
Понятие "арил" в рамках настоящего изобретения обозначает незамещенные либо одно- или многократно замещенные ОН, F, Cl, CF3 , C1-С6алкилом, С1-С6 алкоксигруппой, С3-С7циклоалкилом, С2-С6алкиленом, гетероциклилом либо фенилом фенилы. Под этим понятием при определенных условиях может иметься в виду также нафтил.
Под понятием "гетероциклил" в рамках настоящего изобретения имеются в виду 5- или 6-членные насыщенные либо ненасыщенные, при необходимости с присоединенной путем конденсации арильной системой, гетероциклические соединения, содержащие 1 или 2 гетероатома из группы, включающей азот, кислород и/или серу.
В качестве примеров насыщенных гетероциклилов можно привести 1,4-диоксан, тетрагидрофуран, 1,2-оксатиолан, пирролидин и пеперазин.
В качестве примеров группы ненасыщенных гетероциклилов в рамках настоящего изобретения можно назвать фуран, тиофен, пиррол, пиридин, пиримидин, 1,3-тиазол, оксазол, изоксазол, имидазол, пиразол, γ-пиран, γ-тиапиран, пирадизин, пиразин, 1,4-тиазин, хинолин, изохинолин и хиназолин.
Под понятием "силанильное соединение" в рамках настоящего изобретения имеются в виду триалкил- либо триарилсилилы, диалкиларилсилилы либо диарилалкилсилилы, которые используют в качестве защитной группы для гидроксильной функции. Примерами таких соединений могут служить триэтилсилил, трипропилсилил, диметилфенилсилил, ди-трет-бутилфенилсилил, триизопропилсилил, диметилизопропилсилил, диэтилизопропилсилил, диметилтексилсилил, трет-бутилдиметилсилил, трет-бутилдифенилсилил, трибензилсилил, три-п-ксилилсилил, трифенилсилил, дифенилметилсилил или пропилдифенилсилил.
Объектом изобретения является также способ получения замещенных гетероциклических бензоциклоалкенов общей
формулы (I), который отличается тем, что осуществляют
взаимодействие между третичным спиртом общей формулы II,

в которой R1-R7 , Х и Y имеют то же значение, что и в формуле I, и полуконцентрированными либо концентрированными органическими либо неорганическими кислотами, особенно предпочтительно с муравьиной кислотой или соляной кислотой, при температурах в интервале от 0 до 100o С, предпочтительно при 50oС, причем третичные спирты общей формулы II получают благодаря тому, что основания Манниха общей формулы III,

где R2, R3, X и Y имеют то же значение, что и в формуле I, a R8 идентичен R4 и R9 идентичен R5, за исключением тех случаев, когда необязательно имеющаяся в наличии гидроксильная функция представлена в защищенной форме в виде бензилокси- или силанилоксигруппы, подвергают взаимодействию с металлорганическим соединением формулы IV,

в которой Z представляет собой MgCl, MgBr, Mgl или Li, a R10 имеет значение, идентичное R1 за исключением тех случаев, когда необязательно имеющаяся в наличии гидроксильная функция представлена в защищенной форме в виде бензилокси- или силанилоксисоединения, например в виде трет-бутилдифенилсилилоксигруппы, с получением соединения формулы IIа,

которое затем переводят в соединения формулы II.
Реакцию с соединениями формул III и IV проводят в простом алифатическом эфире, например в диэтиловом эфире и/или тетрагидрофуране, при температурах в интервале от -70 до +60o С. Соединения формулы IV, в которых Z представляет собой атом лития, получают при этом из соединений формулы IV, в которых Z обозначает Вr или I, за счет обмена галоген-литий, осуществляемого с помощью, например, раствора н-бутиллития/н-гексана.
Для получения из соединения формулы IIа соединения формулы II имеется в зависимости от значений R8, R9, соответственно R10 целый ряд методов.
Так, если R8, R9 и/или R10 представляют собой бензилоксигруппу, то целесообразно применять метод восстановительного дебензилирования с использованием каталитически активированного водорода, причем в качестве катализатора служит платина либо палладий, абсорбированные на носителе, таком как активированный уголь. Реакцию проводят в растворителе, таком как уксусная кислота, или в С1-С4 алкиловом спирте при давлении в диапазоне от 1 до 100 бар и при температурах в интервале от 20 до 100oС, причем соединение IIа применяют предпочтительно в виде одной из его солей.
Если R и/или R представляют собой силанилоксигруппу, то отщепление защитной группы осуществляют благодаря тому, что соответствующее соединение формулы IIа при температуре 20oС подвергают в инертном растворителе, таком как тетрагидрофуран, диоксан либо диэтиловый эфир, взаимодействию с фторидом тетра-н-бутиламмония или же обрабатывают метанольным раствором хлористого водорода.
Если же R8, R9 и/или R10 в соединении формулы IIа представляют собой метоксильный радикал, то взаимодействием с гидридом диизобутилалюминия, осуществляемом в ароматическом углеводороде, таком, как толуол, при температуре в интервале от 60 до 130oС, можно получить соединение формулы II, в котором R1 представляет собой гидроксильную группу. Существует также возможность непосредственного получения аналогичного соединения формулы I, состоящая в том, что соединение IIа нагревают с обратным холодильником либо с раствором бромистого водорода в ледяном уксусе, либо с концентрированной бромистоводородной кислотой.
Также из соединений формулы I, в которых R1 и/или R4, соответственно R5 представляют собой метоксильную группу, по описанной выше технологии взаимодействием с гидридом диизобутилалюминия можно получить соединения формулы I, где R1 и/или R2, соответственно R5 обозначают ОН. Аналогичный путь получения возможен также осуществлением взаимодействия с метансульфокислотой/метионином при температурах в интервале от 20 до 50oС.
Из соединений формулы I, в которых Х обозначает S-атом, могут быть получены соединения формулы I, в которых Х представляет собой SO- или SO2 -группу; эту возможность реализуют окислением пероксидом водорода (30 мас. % в воде) и с использованием уксусной кислоты в качестве растворителя при температурах в интервале от 20 до 60o С.
Соединения формулы I с помощью физиологически приемлемых кислот, таких как соляная кислота, бромистоводородная кислота, серная кислота, метансульфоновая кислота, муравьиная кислота, уксусная кислота, щавелевая кислота, янтарная кислота, винная кислота, миндальная кислота, фумаровая кислота, молочная кислота, лимонная кислота, глутаминовая кислота и/или аспарагиновая кислота, могут переводиться по известной методике в их соли. Предпочтительно процесс солеобразования проводить в растворителе, таком как диэтиловый эфир, диизопропиловый эфир, алкиловый эфир уксусной кислоты, ацетон и/или 2-бутанон. Для получения гидрохлоридов особенно предпочтительным является триметилхлорсилан в водосодержащем растворе.
Примеры
Представленные ниже примеры
предназначены для более подробного пояснения настоящего изобретения.
В качестве неподвижной фазы для колоночной хроматографии использовали силикагель 60 (0,040-0,063 мм) фирмы E.Merck, Дармштадт.
Исследования посредством тонкослойной хроматографии проводили с помощью готовых ТСХВР-пластин, силикагель 60 F 254, фирмы Е. Merck, Дармштадт.
Соотношения компонентов в смесях растворителей для хроматографического анализа во всех случаях указаны в отношении объем/объем.
Понятие "трис-НСl" обозначает трис(гидроксиметил)аминометангидрохлорид.
"(мас./об.)" означает соотношение масса/объем.
Пример 1
3-(4-диметиламинометил-2,3-дигидробензо[b] тиепин-5-ил)
фенол, гидрохлорид
Стадия 1
(RS)-4-диметиламинометил-3,4-дигидро-2Н-бензо[b]тиепин-5-он, гидрохлорид
Раствор из 32,1
г 3,4-дигидро-2Н-бензо[b]тиепин-5-она в 320 мл
ацетонитрила смешивали с 16,9 г хлорида N,N-диметилметилениммония и тремя каплями ацетилхлорида и смесь перемешивали в течение 72 часов при 20o
С. Затем разбавляли 100 мл диэтилового эфира,
кристаллический продукт выделяли, промывали диэтиловым эфиром и сушили под вакуумом при 40o С. Таким путем получили 44,1 г (90% от теории)
указанного в заголовке соединения в виде белых
кристаллов.
Температура плавления: 183-185oС.
Стадия 2
(4RS,
5RS)-5-[3-трет-бутилдифенилсиланилокси)фенил] -4-диметиламинометил-2,3,4,
5-тетрагидробензо[b]тиепин-5-ол
Раствор из 32,9 г (3-бромфенокси)трет-бутилдифенилсилана в 250 мл сухого
тетрагидрофурана при -40oС, перемешивая и подавая сухой азот, смешивали по
каплям с 50 мл 1,6-молярного раствора н-бутиллития в н-гексане. По завершении процесса добавления продолжали
перемешивание еще в течение 30 минут при температуре в интервале от -40 до -30o
С, после чего при той же температуре по каплям добавляли раствор из 15,1 г свободного основания продукта из
стадии 1 в 40 мл сухого тетрагидрофурана. Далее продолжали перемешивать еще в течение 4
часов при указанной температуре, после чего разлагали добавлением 50 мл насыщенного раствора хлорида аммония.
Затем органическую фазу отделяли, а водную фазу еще дважды экстрагировали этиловым эфиром
уксусной кислоты. Объединенные органические фазы промывали насыщенным раствором хлорида натрия и сушили над
сульфатом натрия. После отфильтровывания и выпаривания под вакуумом растворителей получили
11,4 г (31,3% от теории) указанного в заголовке соединения в виде бесцветного вязкого масла.
Стадия 3
(4RS, 5RS)-4-диметиламинометил-5-(3-гидроксифенил)-2,3,4,
5-тетрагидробензо[b]тиепин-5-ол
Раствор из 11,4 г продукта из стадии 2 в 200 мл сухого тетрагидрофурана при 5oС и при перемешивании по каплям смешивали с 22 мл 1 М раствора
фторида тетра-н-бутиламмония в тетрагидрофуране. По завершении процесса добавления перемешивали в течение 3 часов при 20oС, смешивали с 50 мл насыщенного раствора хлорида аммония и трижды
экстрагировали порциями по 50 мл этилового эфира уксусной кислоты соответственно. Затем экстракты промывали насыщенным
раствором хлорида натрия, сушили над сульфатом натрия и упаривали под вакуумом.
Остаток очищали посредством колоночной хроматографии с использованием в качестве элюирующих агентов этиловый эфир
уксусной кислоты/метанол в соотношении 9:1, получив таким путем 6 г (90,8% от теории)
указанного в заголовке соединения в виде белых кристаллов с температурой плавления 188-190oС.
Стадия 4
3-(4-диметиламинометил-2,3-дигидробензо[b]тиепин-5-ил)фенол,
гидрохлорид
Раствор из 4,95 г соединения из стадии 3 в 50 мл тетрагидрофурана смешивали со 150 мл 6н.
соляной кислоты и смесь перемешивали сначала в течение 48 часов при 20oС, а
затем в течение 24 часов при 60oС. Далее подщелачивали едким натром и трижды экстрагировали порциями
по 100 мл этилового эфира уксусной кислоты соответственно. Экстракты промывали
насыщенным раствором хлорида натрия, сушили над сульфатом натрия и упаривали под вакуумом. В результате получили 4,44 г
(95,0% от теории) свободного основания указанного в заголовке соединения в виде
твердого вещества белого цвета (температура плавления: 217-219oС), которое с помощью триметилхлорсилана/воды
переводили в смеси растворителей, состоявшей из 2-бутанона и тетрагидрофурана
(1:2), в гидрохлорид.
Тепература плавления: 251:253oС.
Пример 2
Используя соответствующие циклические кетоны вместо 3,4-дигидро-2Н-бензо[b]
тиепин-5-она из стадии 1, по технологии, аналогично описанной в примере 1, получили следующие соединения:
2а:
3-(8-хлор-3-диметиламинометил-2Н-хромен-4-ил)фенол, гидрохлорид,
2б:
3-(3-диметиламинометил-2Н-бензо[g] хромен-4-ил)фенол, гидрохлорид, температура плавления: 232-235oС,
2в: 3-(2-диметиламинометил-3Н-4-тиафенантрен-1-ил)фенол, гидрохлорид,
температура плавления: 246-248,5oС,
2г: 3-(7-диметиламинометил-2,3-дигидро-6Н-1,4,
5-триоксафенантрен-8-ил)фенол, гидрохлорид, температура плавления: 229-231oС,
2д: 3-(4-диметиламинометил-2,3-дигидробензо[b]оксепин-5-ил)фенол, гидрохлорид, температура плавления:
235-237oС,
2е: 3-(3-диметиламинометил-6-метокси-2,
2-диметил-2Н-хромен-4-ил)фенол, гидрохлорид, температура плавления: 217-219oС,
2ж:
3-(4-диметиламинометил-8-метокси-2,3-дигидробензо[b] оксепин-5-ил)фенол, гидрохлорид,
температура плавления: 206-208oС,
2з: 3-(4-диметиламинометил-8-метокси-2,3-дигидробензо[b]
тиепин-5-ил)фенол, гидрохлорид, температура плавления: 232-235oС,
2и: 3-(4-диметиламинометил-8-фтор-2,3-дигидробензо[b]тиепин-5-ил)фенол, гидрохлорид, температура плавления:
разложение выше 130oС,
2к: 3-(4-диметиламинометил-8-фтор-2,
3-дигидробензо[b]оксепин-5-ил)фенол, гидрохлорид, температура плавления: 245-247oС,
2л:
3-(7-трет-бутил-4-диметиламинометил-2,3-дигидробензо[b]оксепин-5-ил)фенол, гидрохлорид,
температура плавления: 264-266oС,
2м: 3-(7,8-дихлор-4-диметиламинометил-2,3-дигидробензо[b]
оксепин-5-ил)фенол, гидрохлорид, температура плавления: 219-220oС,
2н: 3-(4-диметиламинометил-9-метокси-2,3-дигидробензо[b] оксепин-5-ил)фенол, гидрохлорид, температура плавления:
194-196oС,
2о: 3-(8-бензилокси-4-диметиламинометил-2,
3-дигидробензо[b]оксепин-5-ил)фенол, гидрохлорид, температура плавления: 234-236oС.
Пример 3
3-(4-диметиламинометил-1-оксо-2,3-дигидро-1Н-1λ4
-бензо[b] тиепин-5-ил)фенол, гидрохлорид (рацемат и энантиомеры)
Смесь из 1,74 г продукта из примера 1, 17 мл ледяного
уксуса и 1,6 мл водного раствора пероксида водорода (30 мас.% Н2О2) перемешивали в течение 2 часов при 20oС. Затем смесь разбавляли 50 мл воды и подщелачивали едким
натром до значения рН 9. После этого экстрагировали трижды порциями
по 30 мл этилового эфира уксусной кислоты соответственно. Объединенные экстракты промывали насыщенным раствором хлорида натрия,
сушили над сульфатом натрия и упаривали под вакуумом. Остаток,
аналогично тому, как это описано в примере 1, стадия 4, переводили в гидрохлорид. Таким путем получили 1,68 г (92,4% от теории)
рацемического соединения, указанного в заголовке, в виде белых
кристаллов с температурой плавления 208-210o С. Посредством высокоэффективной жидкостной хроматографии с неподвижной фазой,
включавшей н-гексан/изопропанол/диэтиламин в соотношении
950:50:1, из вышеуказанного соединения получили энантиомеры в чистом виде.
Пример 4
3-(4-диметиламинометил-1,
1-диоксо-2,3 -дигидро-1H-1λ6-бензо[b]
тиепин-5-ил)фенол, гидрохлорид
0,91 г продукта из примера 3 в 4,5 мл ледяного уксуса перемешивали с 0,5 мл водного раствора пероксида
водорода (30 мас.% Н2О2) в течение
24 часов при 45oС. После переработки, аналогично описанной в примере 2, очистки сырого продукта посредством колоночной хроматографии
с использованием элюирующих агентов этиловый эфир
уксусной кислоты/метанол в соотношении 5:1 и перевода очищенной субстанции в гидрохлорид получили 0,67 г (70,3% от теории) указанного в заголовке
соединения в виде белых кристаллов с температурой
плавления 263-266oС.
Пример 5
3-(3-диметиламинометил-2Н-тиохромен-4-ил)фенол, гидрохлорид
Стадия 1
(3RS,
4RS)-3-диметиламинометил-4-(3-метоксифенил)тиохроман-4-ол
Из 0,73 г магниевой стружки и 5,61 г 1-бром-3-метоксибензола в 20 мл сухого тетрагидрофурана получали при слабом кипении
соответствующий реагент Гриньяра. К этому реагенту при температуре в интервале от 5 до 10oС по каплям добавляли раствор из 4,43 г (RS)-3-диметиламинометилтиохроман-4-она в 10 мл сухого
тетрагидрофурана. Далее в течение 6 часов перемешивали при 20oС, после чего разлагали 10 мл насыщенного раствора хлорида аммония. Затем трижды экстрагировали диэтиловым эфиром,
объединенные
экстракты промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. Образовавшийся после выпаривания летучих компонентов сырой продукт очищали посредством колоночной
хроматографии с
использованием в качестве элюирующего агента этилового эфира уксусной кислоты, получив в результате 3,68 г (55,8% от теории) указанного в заголовке соединения.
Стадия
2
3-(3-диметиламинометил-2Н-тиохромен-4-ил)фенол, гидрохлорид
3,3 г продукта из стадии 1 перемешивали с 90 мл раствора бромистого водорода в ледяном уксусе (33% НВr) в течение 8
часов при
температуре в интервале от 100 до 110oС. Затем упаривали под вакуумом и остаток растворяли с помощью 100 мл воды. Далее подщелачивали карбонатом натрия и трижды экстрагировали
порциями по
30 мл дихлорметана соответственно. Затем экстракты сушили над сульфатом натрия, упаривали и остаток очищали посредством колоночной хроматографии с использованием в качестве элюирующего
агента
этилового эфира уксусной кислоты. Полученное таким путем основание указанного в заголовке соединения с помощью триметилхлорсилана/воды в 2-бутаноне переводили в гидрохлорид.
Выход: 1, 40 г (41,7% от теории).
Температура плавления: 203-208oС.
Пример 6
[4-(3-метоксифенил)-2Н-хромен-3-илметил]диметиламин
Стадия
1
(3RS,4RS)-3-диметиламинометил-4-(3-метоксифенил)хроман-4-ол
Взаимодействием 4,11 г (RS)-3-диметиламинометилхроман-4-она с реагентом Гриньяра из 0,73 г магниевой стружки и 5,61 г
1-бром-3-метоксибензола по технологии, описанной в примере 5, стадия 1, и после аналогичной очистки получили 3,91 г (62,4% от теории) указанного в заголовке соединения.
Стадия 2
[4-(3-метоксифенил)-2Н-хромен-3-илметил]диметиламин
Раствор из 3,9 г продукта из стадии 1 в 20 мл этанола аналогично тому, как описано в примере 1, стадия 4, смешивали с 14,5 мл 6н.
соляной кислоты в течение 2 часов при 20oС. После аналогичной переработки и очистки посредством колоночной хроматографии с использованием в качестве элюирующего агента этилового эфира
уксусной кислоты получили 2,1 г (57,1% от теории) указанного в заголовке соединения в виде почти бесцветного масла, отверждающегося при 4oС (температура плавления: 68-71oС).
Пример 7
3-(3-диметиламинометил-2Н-хромен-4-ил)фенол, гидрохлорид
Раствор из 0,59 г продукта из примера 6 в 10 мл метансульфоновой кислоты в атмосфере N2
смешивали с 0,59 г метионина, в результате чего образовывался раствор коричневого цвета. Этот раствор перемешивали в течение одного часа при 20oС и затем сливали на лед/воду. Далее
добавляли 50 мл этилового эфира уксусной кислоты и подщелачивали водным раствором карбоната натрия. Органическую фазу отделяли, а водную фазу еще дважды экстрагировали порциями по 20 мл этилового
эфира уксусной кислоты соответственно. Органические фазы промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия и под вакуумом удаляли летучие компоненты. Остаток очищали
посредством колоночной хроматографии с использованием в качестве элюирующих агентов этиловый эфир уксусной кислоты/метанол в соотношении 5:1. Полученный таким путем продукт по описанной в примере 1,
стадия 4, технологии переводили в гидрохлорид.
Выход: 0,26 г (48,1% от теории).
Температура плавления: 213-215oС.
Пример 8
3-диметиламинометил-4-(3-гидроксифенил)-2,2-диметил-2Н-хромен-6-ол, гидрохлорид
Стадия 1
(3RS, 4RS)-6-бензилокси-3-диметиламинометил-4-(3-гидроксифенил) -2,2-диметилхроман-4-ол,
гидрохлорид
Осуществляя реакцию в последовательности, описанной в примере 1, стадии 1-3, и применяя использовавшиеся в ней реагенты, с тем, однако, отличием, что вместо 3,
4-дигидро-2Н-бензо[b]тиепин-5-она использовали 6-бензилокси-2,2-диметилхроман-4-он, получили свободное основание указанного в заголовке соединения, которое по описанной в примере 1, стадия 4,
технологии переводили в гидрохлорид.
Температура плавления: 142-143oС.
Стадия 2
(3RS, 4RS)-3-диметиламинометил-4-(3-гидроксифенил)-2,
2-диметилхроман-4,6-диол, гидрохлорид
1,08 г продукта из стадии 1, растворенного в 15 мл безводного метанола, подвергали каталитическому гидрированию в присутствии 0,11 г палладия на
активированном угле (10% Pd). После отфильтровывания катализатора и выпаривания растворителя под вакуумом получили 0,83 г указанного в заголовке соединения со степенью чистоты, достаточной для
проведения последующей реакции.
Стадия 3
3-диметиламинометил-4-(3-гидроксифенил)-2,2-диметил-2Н-хромен-6-ол, гидрохлорид
0,83 г продукта из стадии 2 смешивали с 20
мл
6н. соляной кислоты и полученный раствор в течение 2 часов перемешивали при 20oС. Затем подщелачивали разбавленным едким натром (рН 8-9) и трижды экстрагировали порциями по 20 мл
дихлорметана соответственно. Объединенные экстракты сушили над сульфатом натрия и концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии с использованием в качестве
элюирующих агентов этиловый эфир уксусной кислоты/метанол в соотношении 5:1. После перевода в гидрохлорид с помощью хлортриметилсилана/воды в 2-бутаноне получили 0,57 г (72,2% от теории) указанного в
заголовке соединения в виде белых кристаллов с температурой плавления 195-198oС.
Пример 9
Применяя вместо продукта из примера 1 продукты из примеров 2з и 2и по
описанной в примере 3 технологии аналогичным путем получили:
9а: 3-(4-диметиламинометил-8-метокси-1-оксо-2,3-дигидро-1Н-1λ4-бензо[b] тиепин-5-ил)фенол, гидрохлорид, температура
плавления: выше 222oС разложение,
9б: 3-(4-диметиламинометил-8-фтор-1-оксо-2,3-дигидро-1Н-1λ4-бензо[b]тиепин-5-ил)фенол, гидрохлорид, температура плавления: выше 198oС разложение.
Пример 10
Применяя продукт из примера 9а вместо продукта из примера 3, по описанной в примере 4 технологии аналогичным путем получили:
3-(4-диметиламинометил-8-метокси-1,1-диоксо-2,3-дигидро-1Н-1λ6-бензо[b] тиепин-5-ил)фенол, гидрохлорид
Температура плавления: 253-256oС
Пример 11
По
описанной в примере 7 технологии с тем, однако, отличием, что вместо продукта из примера 6 изменяли продукты из примера 2ж, соответственно 2з, аналогичным путем получили:
11а:
4-диметиламинометил-5-(3-гидроксифенил)-2,3-дигидробензо[b]оксепин-8-ол, гидрохлорид, температура плавления: выше 103oС разложение,
11б: 4-диметиламинометил-5-(3-гидроксифенил)-2,
3-дигидробензо[b] тиепин-8-ол, гидрохлорид, температура плавления: выше 117oС разложение.
Пример 12
Диэтиламид 4-диметиламинометил-5-(3-гидроксифенил)-2,
3-дигидробензо[b] оксепин-7-карбоновой кислоты, гидрохлорид
Стадия 1
7-бром-3,4-дигидро-2Н-бензо[b]оксепин-5-он, этиленацеталь
Смесь из 24,1 г 7-бром-3,
4-дигидро-2Н-бензо[b]оксепин-5-она, 8,5 мл этиленгликоля и 0,35 г моногидрата п-толуолсульфоновой кислоты нагревали с обратным холодильником в течение 24 часов в водоотделителе. После отделения
реакционной воды в смесь добавляли 4 г порошкообразного карбоната калия и перемешивали в течение одного часа при 20oС. Затем отфильтровывали от соли и фильтрат упаривали под вакуумом.
Таким
путем получили 27 г указанного в заголовке соединения в виде масла коричневого цвета.
Стадия 2
5-оксо-2,3,4,5-тетрагидробензо[b] оксепин-7-карбоновая кислота,
этиленацеталь
Раствор из 27 г продукта из стадии 1 в 280 мл безводного тетрагидрофурана при -50oС при перемешивании и подаче азота по каплям смешивали с 62 мл 1,6-молярного
раствора н-бутиллития в
н-гексане. По завершении процесса добавления перемешивание продолжали еще в течение 30 минут, после чего при температуре в интервале от -40 до -50oС в раствор
подавали до насыщения
последнего диоксид углерода. Затем раствору в течение 3 часов давали нагреться до 20oС, разлагали добавлением 50 мл насыщенного раствора хлорида аммония, органическую
фазу отделяли, а
водную фазу дважды экстрагировали порциями по 50 мл этилового эфира уксусной кислоты соответственно. Объединенные органические фазы промывали насыщенным раствором хлорида натрия и
сушили над сульфатом
натрия. Полученный после упаривания под вакуумом остаток очищали посредством колоночной хроматографии с использованием в качестве элюирующего агента этилового эфира уксусной
кислоты, получив в
результате 16,4 г (69,1% от теории) указанного в заголовке соединения в виде масла.
Стадия 3
Диэтиламид 5-оксо-2,3,4,5-тетрагидробензо[b]
оксепин-7-карбоновой кислоты,
этиленацеталь
Раствор из 16,1 г продукта из стадии 2 в 40 мл циклогексана и 25 мл тионилхлорида в атмосфере N2 перемешивали в течение 2 часов при
20oС. Затем тщательно
выпаривали под вакуумом летучие компоненты. Образовавшийся сырой хлорангидрид карбоновой кислоты растворяли в 70 мл тетрагидрофурана и раствор при перемешивании и
охлаждении ледяной водой добавляли по
каплям к раствору из 8,7 мл диэтиламина в 150 мл тетрагидрофурана, после чего перемешивали еще в течение 2 часов при 20oС. Затем с помощью
вакуум-фильтра отфильтровывали от твердого
продукта, этот последний тщательно промывали тетрагидрофураном и фильтрат концентрировали под вакуумом. Остаток очищали посредством колоночной хроматографии
с использованием в качестве элюирующих
агентов этиловый эфир уксусной кислоты/н-гексан в соотношении 3:1, получив в результате 8,9 г (45,5% от теории) указанного в заголовке соединения в виде
масла.
Стадия 4
Диэтиламид 5-оксо-2,3,4,5-тетрагидробензо[b]оксепин-7-карбоновой кислоты
Смесь из 8,7 г продукта из стадии 3, 15 мл тетрагидрофурана и 30 мл 1н.
соляной кислоты перемешивали в течение 20
часов при 20oС. Затем трижды экстрагировали этиловым эфиром уксусной кислоты. Экстракты промывали насыщенным раствором хлорида натрия, сушили над
сульфатом натрия и упаривали под вакуумом.
В результате получили 7,37 г (98,9% от теории) указанного в заголовке соединения в виде слегка окрашенного в желтый цвет вязкого масла.
Стадия 5
Диэтиламид
(RS)-4-диметиламинометил-5-оксо-2,3,4,5-тетрагидробензо[b]оксепин-7-карбоновой кислоты, гидрохлорид
7,2 г продукта из стадии 4 аналогично тому, как описано в
примере 1, стадия 1, подвергали
взаимодействию с 2,7 г хлорида N,N-диметилметилениммония. Таким путем получили 8,8 г (90,1% от теории) указанного в заголовке соединения в виде белых кристаллов с
температурой плавления 178-180oС.
Стадия 6
Диэтиламид (4RS, 5RS)-5-[3-(трет-бутилдиметил)силанилокси)фенил]-4-диметиламинометил-5-гидрокси-2,3,4,
5-тетрагидробензо[b]оксепин-7-карбоновой кислоты
9,5 г (3-бромфенокси)-третбутилдифенилсилана, 20,2 мл 1,6-молярного раствора н-бутиллития в н-гексане и 7,8 г продукта из стадии 5 в виде
свободного основания подвергали взаимодействию по
описанной в примере 1, стадия 2, технологии. После аналогичной переработки и очистки посредством колоночной хроматографии с использованием в качестве
элюирующих агентов этиловый эфир уксусной
кислоты/метанол в соотношении 5:1 получили 8,29 г (64,3% от теории) указанного в заголовке соединения в виде слегка окрашенного в желтый цвет масла.
Стадия 7
Диэтиламид (4RS,
5RS)-4-диметиламинометил-5-гидрокси-5-(3-гидроксифенил)-2,3,4,5-тетрагидробензо[b]оксепин-7-карбоновой кислоты
8,2 г продукта из стадии 6 и 50 мл
6н. соляной кислоты перемешивали в течение 48
часов при 20oС. После подщелачивания едким натром трижды экстрагировали порциями по 30 мл этилового эфира уксусной кислоты соответственно.
Экстракты промывали насыщенным раствором хлорида
натрия, сушили над сульфатом натрия и упаривали под вакуумом. После очистки посредством колоночной хроматографии с использованием в качестве
элюирующих агентов этиловый эфир уксусной кислоты/метанол в
соотношении 3:1 получили 5,32 г (82,8% от теории) указанного в заголовке соединения.
Стадия 8
Диэтиламид
4-диметиламинометил-5-(3-гидроксифенил)-2,3-дигидробензо[b]
оксепин-7-карбоновой кислоты, гидрохлорид
5,6 г продукта из стадии 7 в виде гидрохлорида нагревали при перемешивании с 70 мл
муравьиной кислоты в течение 2 часов до температуры ванны 110oС. После охлаждения подщелачивали едким натром и карбонатом калия (рН 9) и трижды экстрагировали дихлорметаном. Экстракты
промывали насыщенным раствором хлорида натрия, сушили над сульфатом
натрия и упаривали под вакуумом. Из остатка с помощью триметилхлорсилана/воды в 2-бутаноне получали гидрохлорид. Таким путем
получили 4,91 г (91,3% от теории) указанного в заголовке соединения в виде
белых кристаллов, которые при температуре плавления выше 234oС разлагались.
Пример 13
3-{ 4-[(метилфенетиламино)метил] -2,3-дигидробензо[b]оксепин-5-ил}фенол,
гидрохлорид
Стадия 1
(RS)-4-[(метилфенетиламино)метил]-3,4-дигидро-2Н-бензо[b]оксепин-5-он
Смесь из
24,4 г 3,4-дигидро-2Н-бензо[b]оксепин-5-она, 17,2 г
N-метил-2-фенилэтиламина, гидрохлорида, и 3,0 г пара-формальдегида в 200 мл ледяного уксуса нагревали в течение 3 часов до 100oС, после
чего под вакуумом выпаривали растворитель, а остаток
растворяли с помощью 200 мл воды. Затем трижды экстрагировали порциями по 100 мл диэтилового эфира соответственно. Водную фазу подщелачивали
карбонатом калия и трижды экстрагировали порциями по 100 мл
дихлорметана соответственно. После промывки экстрактов насыщенным раствором хлорида натрия, сушки над сульфатом натрия и упаривания под
вакуумом получили 23,9 г (77,2% от теории) указанного в
заголовке соединения в виде желтоватого масла.
Конечная стадия
3-{ 4-[(метилфенетиламино)метил]-2,
3-дигидробензо[6]оксепин-5-ил} фенол, гидрохлорид
Продукт из стадии
1 по технологии, описанной в примере 1, стадии 2-4, подвергали дальнейшей переработке. Таким путем получили указанное в
заголовке соединение в виде белых кристаллов, которые при температуре плавления
выше 135oС разлагались.
Пример 14
3-{ 4-[(циклопропилметиламино)метил]-2,
3-дигидробензо[b]оксепин-5-ил}фенол, гидрохлорид
Используя N-(циклопропилметил)
метиламин, гидрохлорид, из примера 13, стадия 1, и подвергнув полученный таким путем продукт дальнейшей
переработке по технологии, аналогично описанной в примере 1, стадии 2-4, получили указанное в
заголовке соединение в виде белых кристаллов с температурой плавления 216-218oС.
Исследования по связыванию δ-опиаторецептора
Исследования по определению
сродства предлагаемых согласно изобретению соединений формулы I к δ-опиаторецептору, которое
является решающим условием для наличия анальгетических свойств, проводили на гомогенатах мембран
головного мозга (гомогенат головного мозга самцов крыс линии Wistar без мозжечка, моста и
продолговатого мозга). Для выявления δ-опиаторецепторных свойств использовали выборочные соединения
формулы I.
С этой целью свежепрепарированный для каждого эксперимента мозг крыс при охлаждении льдом гомогенизировали в 50 ммоль/л трис-НСl (рН 7,4) и в течение 10 минут центрифугировали при 5000 g и 4oС. После декантирования и отбрасывания надосадной жидкости, повторного растворения и гомогенизации мембранного осадка в 50 ммоль/л трис-НСl (рН 7,4) гомогенат в течение 20 минут центрифугировали при 20000 g и 4oС. Эту операцию по промывке повторяли еще раз. Затем надосадную жидкость декантировали, а мембранный осадок гомогенизировали в холодном 50 ммоль/л трис-HCl, 20% глицерина (маc./об.), 0,01% бацитрацина (мас. /об.) (рН 7,4) и в аликвотных количествах замораживали до начала тестирования. Для проведения исследований по связыванию рецептора аликвоты оттаивали и разбавляли соответствующим буфером в соотношении 1:10.
В качестве буфера в исследованиях на связывание применяли 50 ммоль/л трис-HCl, 5 ммоль/л MgCl (рН 7,4), дополненные 0,1% (мас./об.) бычьего сывороточного альбумина, а в качестве радиоактивного лиганда использовали 1 нмоль/л (3H)-2-D-Аla-дельторфин II. Долю неспецифического связывания определяли в присутствии 10 ммоль/л налоксона.
В других смесях соединения по изобретению добавляли в последовательно варьируемых концентрациях и определяли вытеснение радиоактивного лиганда из его специфически связанного состояния. Соответствующие смеси, которые применяли в трех параллельных опытах, инкубировали в течение 90 минут при 37oС, после чего для выявления связанного с гомогенатом мембран радиоактивного лиганда последний собирали путем фильтрации через фильтр из стекловолокна (GF/B). Радиоактивность дисков этого стекловолоконного фильтра измеряли после добавления сцинциллятора с помощью счетчика β-излучения.
Сродство соединений по изобретению к δ-опиаторецептору рассчитывали в качестве IC50 согласно закону действующих масс методом нелинейной регрессии. Кi - значения в таблице 1 указаны в качестве средних значений ± стандартные отклонения, равные 3, в проведенных независимо друг от друга опытах.
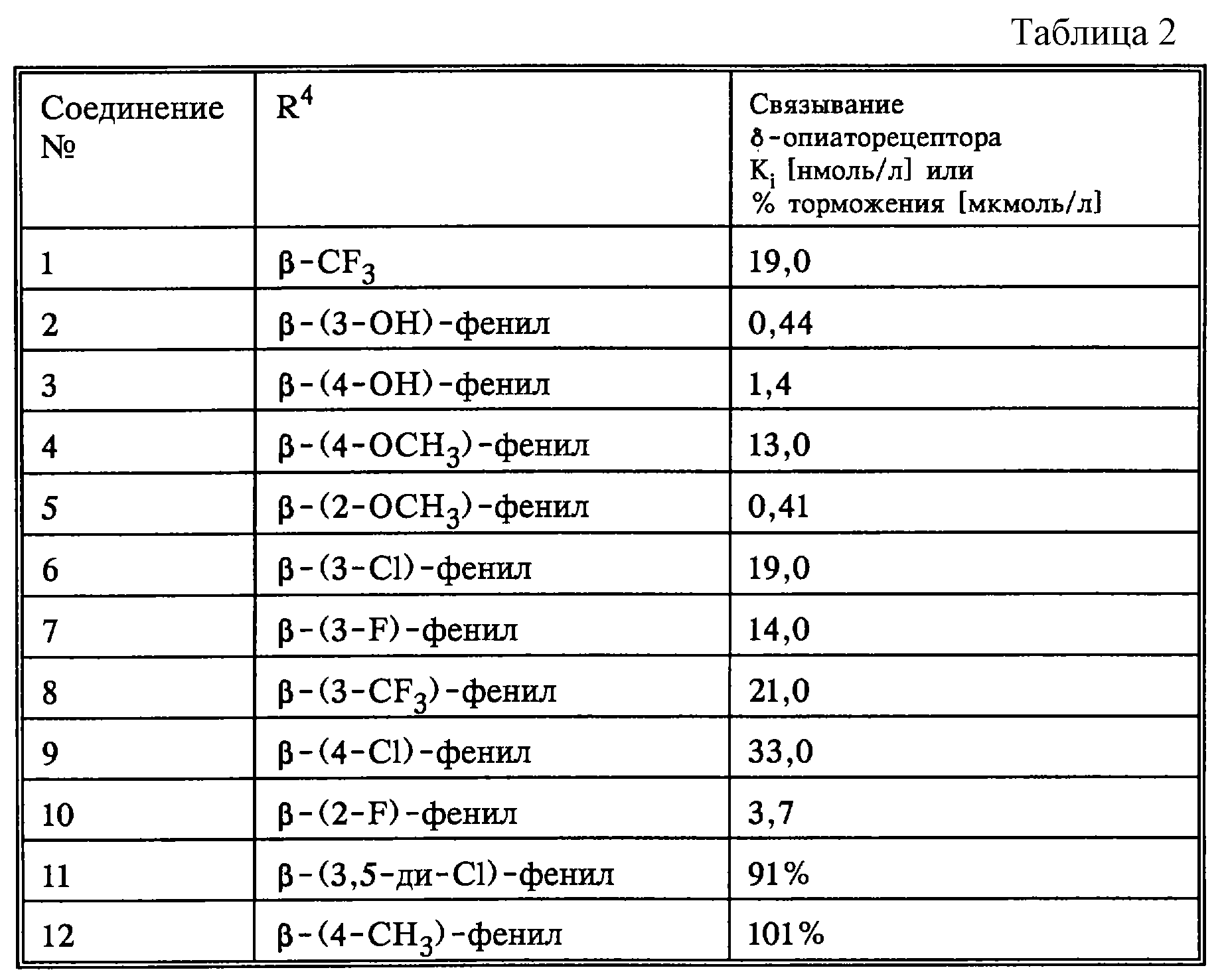
Отчет о фармакологических испытаниях.
Исследования по связыванию δ-опиарецептора.
Исследования по определению сродства заявленных соединений общей формулы I, где заместители имеют значения, указанные в таблице 2, проводили по методике, охарактеризованной в описании. Все соединения были получены в соответствии с методикой, описанной в заявке.
R1 = OH
R2 = R3 = CH3
R5 = H
X = O
Y = (CH2)2н
Реферат
Изобретение относится к производным гетероциклических бензоциклоалкенов общей формулы I, где R1 обозначает ОН, C1-С6 алкокси; R2 обозначает C1-С6 алкил; R3 обозначает C1-С6 алкил, -(СН2)1-2-арил, где арил - это фенил, СН2 -(С3-С7) циклоалкил; R4 и R5 одновременно или в отличие друг от друга обозначают Н, Cl, F, C1-С6 алкил, ОН, C1-С6 алкокси, -(СН2)0-2-арил, -O-(СН2)0-2-арил, цепарил - это фенил, незамещенный или однозамещенный, -СОN(R6R7). Описан также способ получения соединений. Замещенные гетероциклические бензоциклоалкены обладают анальгезирующим действием, биологическая эффективность которых частично либо преимущественно опосредуется агонистами δ-опиаторецепторов. 2 с. и 8 з.п.ф-лы, 2 табл.

Формула

где R1 обозначает ОН, С1-С6алкокси;
R2 обозначает С1-С6алкил;
R3 обозначает С1-С6алкил, -(СН2)1-2 -арил, где арил представляет собой фенил, и СН2-(С3-С7)циклоалкил;
R4 и R5 одновременно или в отличие друг от друга обозначают Н, Cl, F, СF3, С1-С6алкил, ОН, С1-С6алкокси, -(CH1)0-2-арил, -O-(СН2)0-2-арил, где арил представляет собой фенил либо одно- или многократнозамещенный ОН, F, Cl, СF3, С1-С6 алкилом или С1-С6алкоксигруппой фенил, и -CON(R6R7); или
R4 и R5 вместе образуют α, β- либо β, γ -O-(СН2)1-2-O-группу, присоединенную к атомам углерода бензольного кольца, или α, β- либо β, γ-бензогруппу;
R6 обозначает Н, С1-С6алкил;
R7 обозначает Н, С1-С6 алкил;
Х обозначает О, S, SO, SO2 и
Y обозначает -(СН2)1-2 , -СН2-С(СН3)2- или -С(СН3)2-,
и их применимые в фармацевтике соли.

где R1-R7, Х и Y имеют значение по п. 1, отличающийся тем, что третичный спирт общей формулы II

где R1 -R7, Х и Y имеют то же значение, что и в формуле I, подвергают взаимодействию с кислотами в интервале температур от 0 до 100oС, причем третичные спирты общей формулы II получают благодаря тому, что сначала основание Манниха общей формулы III

где R2, R3, Х и Y имеют то же значение, что и в общей формуле I, a R8 идентичен R4 и R9 идентичен R5, за исключением тех случаев, когда имеющаяся в наличии гидроксильная группа представлена в защищенной форме в виде силанилокси- или бензилоксигруппы, подвергают взаимодействию с металлоорганическим соединением общей формулы IV

где Z представляет собой MgCl, MgBr, MgI или Li, a R10 имеет значение, идентичное R1, за исключением тех случаев, когда необязательно имеющаяся в наличии гидроксильная функция представлена в защищенной форме в виде бензилокси- или силанилоксигруппы, с получением соединения общей формулы IIа

где R8, R9 и R10 имеют вышеуказанные значения,
которое затем при наличии одной или более гидроксильных групп, представленных в защищенной форме в виде силанилокси- или бензилоксигрупп, переводят в соединение общей формулы II путем удаления этих защитных групп.
Комментарии