Способы получения (8↔8) димера катехина, и/или эпикатехина, и/или эпикатехингаллата - RU2293081C2
Код документа: RU2293081C2
Описание
Область изобретения
Данное изобретение относится к синтетическим димерам катехина и эпикатехина, их производным и способам их получения и применения.
Предшествующий уровень техники
Полифенолы представляют собой чрезвычайно разнообразную группу соединений (Ferreira, D., Steynberg, J.P., Roux, D.G. and Brandt, E.V., Tetrahedron, 48, (10), 1743-1803 (1992)), которые широко распространены среди растений, некоторые из которых являются звеньями пищевой цепи. Во многих случаях они представляют собой важный класс соединений, входящих в рацион человека. Хотя считается, что некоторые полифенолы не имеют питательной ценности, к данным соединениям возник интерес в силу их возможных положительных эффектов на здоровье.
Например, в экспериментах на животных было показано, что кверцитин (флавоноид) обладает антиканцерогенной активностью (Deschner, E.E., Ruperto, J., Wong, G. and Newmark, H.L., Carcinogenesis, 7, 1193-1196 (1991) and Kato, R., Nakadate, Т., Yamamoto, S. and Sugimura, Т., Carcinogenesis, 4, 1301-1305 (1983)). Было показано, что (+)-катехин и (-)-эпикатехин (флаван-3-олы) ингибируют обратно-транскриптазную активность вируса лейкоза (Chu S.C., Hsieh, Y.S. and Lim, J.Y, J. Nat. Prod., 55, (2), 179-183 (1992)). Также было продемонстрировано, что ноботанин (олигомерный гидролизуемый таннин) обладает противоопухолевой активностью (Okuda Т., Yoshida, Т., and Hatano, Т., Molecular Structures and Pharmacological Activities of Polyphenols - Oligomeric Hydrolyzable Tannins and Others - Presented at the XVIth International Conference of Groupe Polyphenols, Lisbon, Portugal, July 13-16, 1992). Статистические исследования также показали, что смертность от злокачественных опухолей значительно ниже в районах Японии с развитым производством чая. Об эпигаллокатехингаллате сообщалось как о фармакологически активном веществе зеленого чая, которое ингибирует опухоли кожи у мышей (Okuda et al., ibid.). Также было продемонстрировано, что эллаговая кислота обладает антиканцерогенной активностью в различных животных моделях опухолей (Boukharta M., Jalbert, G. and Castonguay, A., Efficacy of Ellagitannins and Ellagic Acid as Cancer Chemopreventive Agents - Presented at the XVIth International Conference of the Groupe Polyphenols, Lisbon, Portugal, July 13-16, 1992).
Kikkoman Corporation были описаны олигомеры проантоцианидина (JP 4-190774), используемые в качестве антимутагенов. Употребление фенольных соединений в пищу и модуляция ими развития опухолей в экспериментальных животных моделях недавно обсуждались в ходе 202nd National Meeting of The American Chemical Society (Phenolic Compounds in Foods and Their Effects on Health I, Analysis, Occurrence 5c Chemistry, Ho, C.T., Lee, C.Y., and Huang, M.T. editors, ACS Symposium Series 506, American Chemical Society, Washington D.C. (1992); Phenolic Compounds in Foods and Their Effects on Health П. Antioxidants & Cancer Prevention, Huang, M.T., Ho, C.T., and Lee, C.Y. editors, ACS Symposium Series 507, American Chemical Society, Washington, D.C. (1992)). Недавно было обнаружено, что процианидины и особенно олигомеры высших процианидинов обладают широким спектром биологической активности.
Были разработаны методики синтеза для определения взаимосвязей структура-активность среди множества возможных регио- и стереоизомеров, содержащих данный олигомер. Данные методики фокусируются на типичных (4↔6), (4↔8), (6↔4) и (8↔4) связях, образующих неразветвленные и разветвленные олигомеры процианидина. Помимо данных связей, стереохимия связей в положении С-4 зависит от мономера, содержащего данные положения связывания.
Например, когда (+)-катехин, обозначенный здесь как С, связан с другим С или с (-)-эпикатехином, обозначаемым здесь как ЕС, связи преимущественно представляют собой (4↔6) или (4↔8). Когда ЕС связан с С или другим ЕС, связи преимущественно представляют собой (4β↔6) или (4β↔8). Что касается связей разветвленного олигомера, стереохимические связи представляют собой (6↔4α), (6↔4β), (8↔4α), (8↔4β).
Однако среди мономеров, составляющих олигомер, возможны и другие положения связывания. Они представляют собой (8↔8), (6↔6) и (6↔8) связи, показательная структура которых приведена ниже. Поскольку олигомеры, образованные такими необычными связями, либо редки, либо не встречаются в природе, путем их биологического исследования могут быть выявлены сходные или новые применения данных соединений или их производных.
Следовательно, существует интерес к синтезу данных олигомеров, содержащих данные связи.
Сущность изобретения
Данное изобретение относится к новым 8↔8, 6↔6, 8↔6 димерам катехина и эпикатехина и галлатированным димерам и к способам их получения. Соединения, полученные в соответствии со способами по данному изобретению, могут быть очищены, например, с помощью ВЭЖХ. Соединения по данному изобретению могут быть применены в качестве противораковых средств.
8↔8, 6↔6, 8↔6 димеры катехина и/или эпикатехина, обладающие следующими структурами:
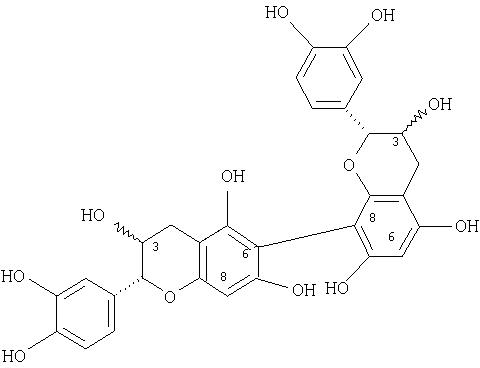

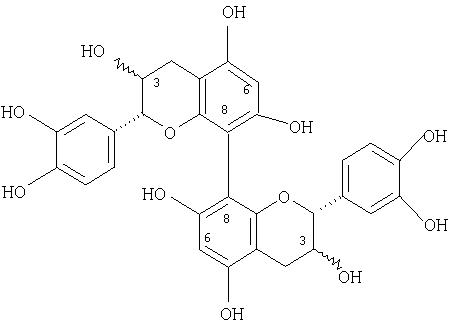
8↔8, 6↔6, 8↔6 димеры катехин- и/или эпикатехиндигаллата, обладающие следующими структурами:


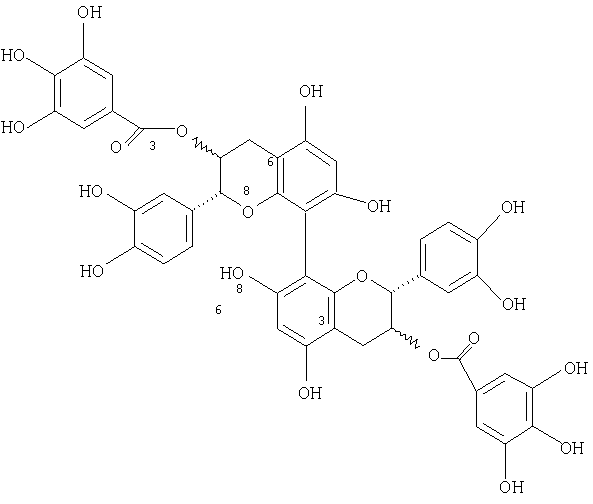
(8↔8) Димеры получали способом, предусматривающим следующие стадии: (а) защита фенольных гидроксильных групп эпикатехиновых и/или катехиновых мономеров первой защитной группой; (b) защита 3-гидроксильных групп мономеров второй защитной группой; (с) галогенирование положения С-8, например, N-бромсукцинимидом, с получением 8-бромпроизводных; (d) взаимодействие 8-бромпроизводных с алкиллитием, например с трет-бутиллитием или бутиллитием, с получением ариллитиевых производных; (е) проведение окислительного или восстановительного связывания ариллитиевых производных путем обмена галогена на металл, где добавляют ариллитий с получением 8-литиевого производного с последующим добавлением хлорида железа (III) для осуществления (8↔8) связывания; и (f) снятие защиты с (8↔8) соединений.
Альтернативный способ предусматривает выполнение описанных выше стадий (а)-(с) включительно с последующим проведением восстановительного связывания галогенированных соединений, например, путем использования реактива на основе никеля с нулевой валентностью в сочетании с порошкообразным соответствующим металлом с последующим снятием защиты.
Если для защиты фенольных гидроксигрупп или 3-гидроксигрупп используют бензильные группы, снятие защиты осуществляют гидрогенолизом. Если для защиты 3-гидроксигрупп используют тетрагидропиранил, то сначала удаляют тетрагидропиранильные защитные группы, а затем бензильные защитные группы.
Способ получения (6↔6) димера катехин- и/или эпикатехиндигаллата предусматривает следующие стадии:
(а) защита фенольных гидроксильных групп эпикатехиновых или катехиновых мономеров первой защитной группой; (b) галогенирование положений С-6 и С-8; (с) защита 3-гидроксильных групп второй защитной группой, например трет-бутилдиметилсилилом; (d) селективное удаление 8-галогеновых групп, например брома; (е) проведение окислительного или восстановительного связывания 6-галогенпроизводных; (f) снятие защиты с 3-гидроксильных групп; (д) этерификация 3-положений три-o-бензилгаллоилхлоридом; и (h) в положении С-3; и (i) снятие защиты с фенольных гидроксильных групп с получением свободного дигаллата (6↔ 6) димера.
(6↔8) Димеры получают из катехина и/или эпикатехина способом, который предусматривает следующие стадии: (а) осуществление окислительного связывания смеси 6-бром- и 8-бром-катехиновых и/или эпикатехиновых мономеров с получением смеси (8↔8), (6↔8) и (6↔6) димеров; и (b) разделение смеси с помощью ВЭЖХ. Альтернативно (6↔8) димеры получают способом, который предусматривает следующие стадии: (а) получение арилбороновой кислоты либо из 6-бром-, либо из 8-бром-катехиновых и/или эпикатехиновых мономеров с помощью реакции обмена галогена на металл, которую гасят триметилборатом с последющей водно-кислотной обработкой с получением свободной кислоты; и (b) взаимодействие смеси с палладиевым катализатором для осуществления связывания.
Подробное описание изобретения
Подходящие защитные группы для фенольных гидроксильных групп мономеров, применимые здесь, включают такие защитные группы, которые могут быть введены в мономеры и удалены без рацемизации или разрушения мономеров и которые стабильны в условиях, применяемых для проведения реакции окислительного или восстановительного связывания. Способы защиты и снятия защиты с гидроксильных групп хорошо известны специалистам в данной области и описаны в "Protective Groups in Organic Synthesis," T.W.Greene, John Wiley & Sons. Предпочтительно защитные группы представляют собой бензильные группы, каждая из которых легко удаляется в одну стадию.
Подходящие защитные группы для 3-гидроксильных групп включают бензил, тетрагидропиранил и тому подобное. Ссылки, в которых описано применение хлорида железа (FeCl3) в окислительном связывании 2 арила с арилом 2: С.A.Broka, Tetrahedron Lett. 32, 859 (1991).
Ссылки, в которых описано восстановительное связывание 2 арила с арилом 2 с помощью реактивов на основе никеля с нулевой валентностью (в большинстве случаев никель с нулевой валентностью получают in situ из солей/комплексов Ni(II) и восстановителя), включают следующие:
R.H.Mitchell et al., J. Am. Chem. Soc. 106, 7776 (1984); H.Matsumoto et al., J. Org. Chem. 48, 840 (1983); S. Inaba et al. Tetrahedron Lett. 23, 4215 (1982); S.Knapp et al., J. Org. Chem. 58, 997 (1993); К.Takagi et al., Bull. Chem. Soc. Jpn. 57, 1887 (1984); M.Iyoda et al., Bull. Chem. Soc. Jpn. 63, 80 (1990) (наиболее важная ссылка) К. Takagi et al., Chem. Lett., 917 (1979); M.A.Fox et al., J. Org. Chem. 56, 3246 (1991); Y.Rollin et al., J.Organomet. Chem. 303, 131 (1986); R.Vanderesse et al., J.Organomet. Chem. 264, 263 (1984); В.Loubinoux et al., Tetrahedron Lett., 3951 (1977).
Ссылки, в которых описано связывание Suzuki для синтеза несимметричных биарилов (арил Br + арил' В(ОН)2 с ArAr' в присутствии соединения палладия (Pd) в качестве катализатора), включают следующие:
R.В.Miller and S.Dugar, Organometallics 3, 1261 (1984); M.A.F.Brandao et al., Tetrahedron Lett. 34, 2437 (1993); M. Sato et al., Chem. Lett., 1405 (1989); S.P.Maddaford and В.А.Кеау, J. Org. Chem. 59, 6501-3 (1994) (ключевая ссылка); M.J.Burk et al., J. Am. Chem. Soc. 116, 10847-8 (1994); S.W.Wright et al., J. Org. Chem. 59, 6095-7 (1994) (ключевая ссылка); G.В.Smith et al., J. Org. Chem. 59, 8151-6 (1994); Т.I.Wallow and B.M.Novak, J. Org. Chem. 59, 5034-7 (1994) (ключевая ссылка); X.Yue et al., Tetrahedron Lett. 37, 8213-6 (1996); J.W.Benbow and B.L.Martinez, Tetrahedron Lett. 37, 8829-32 (1996); M.Beller et al., Angew. Chem. 107, 1992-3 (1995), (иногда свободным бороновым кислотам предпочитают сложные эфиры боронатов).
Стереоизомеры димеров подпадают под объем настоящего изобретения. Стереохимия заместителей полифенольного мономерного звена димера может быть охарактеризована с учетом их взаимной стереохимии, "альфа/бета" или "цис/транс", или абсолютной стереохимии, "R/S". Термин "альфа" (α) свидетельствует о том, что заместитель ориентирован под плоскостью флаванового кольца, тогда как "бета" (β) свидетельствует о том, что заместитель ориентирован над плоскостью кольца. Термин "цис" свидетельствует о том, что два заместителя ориентированы на одной стороне кольца, тогда как "транс" свидетельствует о том, что два заместителя ориентированы на противоположных сторонах кольца. Термины R и S применяют для обозначения расположения заместителей относительно хирального центра, основываясь на определении старшинства групп, непосредственно связанных с указанным центром. Межфлавановая связь между замещенными ароматическими кольцами образует хиральную ось, на которой может возникать два атропоизомера.
Пример 1
Получение (2R,3S,транс)-5,7, 3′,4′-тетра-O-бензилкатехина
Раствор (+)-катехина (65,8 г, 226,7 ммоль, безводный), растворенный в безводном диметилформамиде (ДМФ, 720 мл), добавляли капельно при комнатной температуре в течение 80 мин к перемешиваемой суспензии гидрида натрия, 60% в масле, (39 г, 975 ммоль, 4,3 экв.) в ДМФ (180 мл) (S. Miura et al., Radioisotopes, 32, 225-230 1993). После перемешивания в течение 50 мин колбу помещали на NaCl/ледяную баню с температурой -10°С. Бензилбромид (121 мл, 1,02 моль, 4,5 экв.) добавляли капельно в течение 80 мин и коричневую реакционную смесь нагревали до комнатной температуры при перемешивании в течение ночи. Полученную реакционную смесь выпаривали и полученное леденцеобразное твердое вещество растворяли с нагреванием и перемешиванием в двух порциях растворителя, каждая из которых состояла из 200 мл хлороформа (CHCl3) и 100 мл воды. Фазы разделяли и водную фазу экстрагировали добавлением хлороформа (20 мл) и объединенные органические фазы промывали водой (100 мл), сушили над сульфатом магния (MgSO4) и выпаривали. Остаток очищали с помощью хроматографии на силикагеле (42×10 см; этилацетат/хлороформ/гексан 1:12:7) с получением после выпаривания и вакуумной сушки 85 г сырого продукта, который перекристаллизовывают из трихлорэтилена (1/3 л) с получением 35,1 г (24%) не совсем белого порошка.1Н ЯМР (CDCl3) δ 7,47-7,25 (м, 20Н), 7,03 (с, 1Н), 6,95 (с, 2Н), 6,27, 6,21 (АВкв, 2Н, J=2 Гц), 5,18 (с, 2Н), 5,17 (узкий АВкв, 2Н), 5,03 (с, 2Н), 4,99 (с, 2Н), 4,63 (д, 1Н, J=8,5 Гц), 4,00 (м, 1Н), 3,11, 2,65 (АВкв, 2Н, J=16,5 Гц, обе части д с J=5,5 и 9 Гц соотв.), 1,59 (д, 1Н, J=3,5 Гц); ИК (пленка) 3440 (ушир.), 1618, 1593, 1513, 1499, 1144, 1116, 733, 696 см-1; МС m/z 650 (М+, 0,5%), 319, 181, 91.
Альтернативно тетра-О-бензил-(+)-катехин может быть получен в соответствии с методикой, описанной в Н.Kawamoto et al., Mokazai Gakkaishi, 37, (5) 488-493 (1991), с использованием карбоната калия и бензилбромида в ДМФ. О частичной рацемизации катехина и в положении 2, и в положении 3 сообщалось в М.-С.Pierre et al., Tetrahedron Letters, 38, (32) 5639-5642 (1997).
Пример 2
Получение (2R)-5,7,3′,4′-тетракис(бензилокси)флаван-3-она
Свежеприготовленный перйодинан Dess-Martin (39,0 г, 92 ммоль, полученный методом D.B.Dess and J.C.Martin, J. Am. Chem. Soc. 113, 7277-7287 (1991) и R.E.Ireland and L. Liu, J. Org. Chem. 58, 2899 (1993)) добавляли при комнатной температуре весь сразу к перемешиваемому раствору тетра-O-бензилкатехина, полученному, как описано в предыдущем примере, (54,4 г, 83,8 ммоль) в метиленхлориде (420 мл). В течение 1,5 часов в реакционную смесь по каплям добавляли приблизительно 30 мл насыщенного водой метиленхлорида с образованием мутного раствора янтарного цвета. (S.D.Meyer and S.L.Schreiber, J. Org. Chem., 59, 7549-7552 (1994)). Двадцать минут спустя реакционную смесь разбавляли насыщенным раствором карбоната натрия (NaHCO3, 500 мл) и 10% водным раствором Na2S2O3· 5Н2О (200 мл). Фазы разделяли и водную фазу экстрагировали добавлением 50 мл метиленхлорида. Объединенные органические фазы фильтровали через силикагель (24×9 см, хлороформ/этилацетат/9:1). Элюат упаривали и сушили в вакууме с получением 50,1 г (92%) кетона, который очищали путем перекристаллизации из хлороформа/эфира: т.пл. 144-144,5°С; [α]D+38,5, [α]546+48,7° (хлороформ, с 20,8 г/л);1Н ЯМР (CDCl3) δ 7,45-7,26 (м, 2OН), 6,96 (с, 1Н), 6,88, 6,86 (АВкв, 2Н, J=8 Гц обе части д с J=1,5 Гц), 6,35 (узкий АВкв, 2Н), 5,24 (с, 1Н), 5,14 (с, 2Н), 5,10 (узкий АВкв, 2Н), 5,02 (с, 2Н), 5,01 (с, 2Н), 3,61, 3,45 (АВкв, 2Н, J=21,5 Гц).
Пример 3
Получение 8-бром-5,7,3′,4′-тетра-O-бензилэпикатехина
Способ А: К раствору 116 мг (178 мкмоль) тетра-O-бензилэпикатехина в 4 мл безводного CH2Cl2 добавляли при охлаждении на льду и перемешивании 32 мг (180 мкмоль) N-бромсукцинимида (NBS). Перемешивание при 0°С продолжали в течение 100 мин, раствор концентрировали и остаток очищали путем хроматографии на силикагеле (15×1,8 см) хлороформом/этилацетатом (CHC3/EtOAc) (25:1). Кристаллизация из CHCl3/этанола позволяла получить 110 мг (85%) бесцветного хлопкоподобного твердого вещества. Т.пл. 137,5°С; αD - 50,4°, α546 - 60,7° (с 17,3 г/л, EtOAc);1Н ЯМР (300 МГц, CDCl3, TMC) δ 7, 5-7,25 (м, 20 Н), 7,23 (д, 1Н, J=1,5 Гц), 7,03, 6,98 (АВкв, 2H, J=8,5 Гц, часть А д с J=1 Гц), 6,25 (с, 1Н), 5,22 (с, 2Н), 5,19 (с, 2Н), 5,11 (с, 2Н), 5,02, 4,96 (АВкв, 2Н, J=9 Гц), 4,98 (с, 1Н) Н, J=9 Гц), 4,27 (ушир. с, 1Н), 3,04, 2,90 (АВкв, 2Н, J=17,5 Гц, обе части д с J=1,5 и 4 Гц соотв.), 1,58 (д, 1Н, J=4,5 Гц);13С ЯМР (75 МГц, CDCl3) δ 156,86, 154,79, 151,65, 149,09, 148,73, 137,31, 137,15, 136,77, 136,72, 130,82, 128,67, 128,65, 128,58, 128,56, 128,09, 127,98, 127,87, 127,50, 127,31, 127,25, 127,13, 118,91, 115,17, 113,07, 102,85, 93,07, 78,62, 71,35, 71, 20, 70,31, 65,92, 28,00; ИК (суспензия в минеральном масле) 3571, 1606, 1581, 1518, 1184, 1129, 771, 732, 694 см-1; МС m/z 399/397 (1/1%), 332 (1%), 181 (8%), 91 (100%). Анал. рассч. для С43Н37O6Br: С 70,78; Н 5,11. Найдено С 70,47; Н 5,10.
Способ В: К 563 мг (771 мкмоль) 5,7,3′,4-тетра-О-бензил-8-бромкатехина, полученного в соответствии с методикой, описанной в примере 1, в 5 мл СН2Cl2 при комнатной температуре добавляли сразу 425 мг (1,00 ммоль) перйодинана Dess-Martin. Насыщенный водой СН2Cl2 добавляли по каплям в течение 40 минут с получением легкой мутности. Спустя еще 20 минут добавляли по 20 мл насыщенного раствора NaHCO3 и 10% водного раствора Na2S2O3·5Н2О. Фазы разделяли и водную фазу экстрагировали добавлением 3×15 мл эфира. Объединенные органические фазы концентрировали и остаток фильтровали через силикагель (20×2,5 см, эфир/гексан 1:1). Элюат выпаривали и сушили в вакууме с получением 522 мг (93%) кетона в виде бесцветной пены:1Н ЯМР (CDCl3) 7, 47-7,25 (м, 20 Н), 7,04 (д, 1Н, J=1 Гц), 6,85, 6,81 (АВкв, 2Н, J=8,5 Гц, часть В д с J=8,5 Гц), 3,52, 3,48 (АВкв, 2Н, J-21,5 Гц);13С ЯМР (CDCl3) 203,99, 155,55, 155,40, 150,68, 148,98, 137,06, 136,90, 136,28, 136,04, 128,64, 128,62, 128,46, 128,41, 128,22, 128,05, 127,78, 127,76, 127,35, 127,17, 127,13, 127,08, 126,99, 118,86, 114,59, 112,43, 103,54, 93,96, 93,87, 82,91, 71, 25, 71,04, 70,98, 70,38, 33,30; ИК (пленка) 1734, 1605, 1513, 1099, 737 696 см-1.
К 598 мг (822 мкмоль) указанного выше неочищенного кетона в 8,2 мл безводного ТГФ по каплям в течение 10 минут добавляли 1,23 мл 1 М раствора три-втор-бутилборгидрида лития (L-Selectride®). После перемешивания при -78°С в течение 3 часов исходное вещество еще определялось в реакционной смеси путем тонкослойной хроматографии (ТСХ), (SiO2, EtOAc/гексан 1:3), и добавляли еще 1,23 мл восстановителя. Перемешивание продолжали еще в течение 4 часов, в то время как температура постепенно поднялась до -4°С. Водный гидроксид натрия (NaOH) (2,5 М, 6 мл) и 4 мл 35% водного пероксида водорода (Н2O2) добавляли с охлаждением; протекающая экзотермическая реакция поднимала температуру бани до +12°С. Перемешивание на водяной бане продолжали в течение ночи, затем смесь частично упаривали и добавляли 20 мл эфира и 10 мл этилацетата (EtOAc). Фазы разделяли и водную фазу экстрагировали добавлением 50 мл EtOAc. Объединенные органические фазы упаривали и остаток очищали путем хроматографии на силикагеле (23×2,5 см) EtOAc/гексаном 1:3 с получением 327 мг (55%) продукта в виде светло-желтой пены.
Пример 4
5,7,3′,4′ -Тетра-О-бензил-8-бром-3-O-(тетрагидропиран-2-ил)катехин
К раствору 297 мг (407 мкмоль) 5,7,3′,4′-тетра-О-бензил-8-бромэпикатехина в 2 мл безводного метиленхлорида (СН2Cl2) при комнатной температуре добавляли 56 мкл (0,61 мкмоль) дигидропирана с последующим добавлением 2,6 мкл (40 мкмоль) метансульфоновой кислоты. Постепенно темнеющий раствор перемешивали при комнатной температуре в течение 25 минут, после чего добавляли 0,15 мл насыщенного водного раствора карбоната натрия (Na2CO3). После упаривания остаток хроматографировали через силикагель (SiO2) этилацетатом/гексаном. Первая фракция элюировалась с отношением концентраций компонентов 1:4, продукт (215 мг, 65%) с отношением 1:3 и непрореагировавшее исходное вещество (97 мг, 33%) с отношением 1:2. Продукт:1H ЯМР (CDCl3) δ 7,50-7,25 (м, 20Н), 7,10(с) и 7,08 (д, J=1 Гц) (1Н два эпимера), 6,94, 6,91 (АВкв, 2Н, J=8,5 Гц), 6,22 (с, 1Н), 5,20-4,97 (м, 8Н), 4,88 (с) и 4,86 (с) (1Н два эпимера), 4,13-3,80 (м, 3Н), 3,42-2,87 (м, 3Н), 2,78 (дд, J=16,5 8,5 Гц), и 2,61 (дд, J=16,5, 7 Гц) (1Н два эпимера), 1,77-1,18 (м, 5Н); ИК (пленка) 1605, 1121, 1031, 735, 696 см-1.
Пример 5
5,7,3′,4′,5′′,7′′,3'′′, 4'′′-Окта-O-бензил-8,8′′-бикатехин
К раствору 527 мг (648 мкмоль) тетрагидропиранилового эфира из примера 4 в 6,5 мл безводного ТГФ при -78°С в течение 5 мин по каплям добавляли 0,91 мл (1,55 ммоль) трет-бутиллития (1,7 М в пентане). Полученный раствор перемешивали при -78°С в течение 5 минут, в то время как к 147 мг (0,91 ммоль) безводного хлорида железа (FeCl3) добавляли 1,5 мл безводного тетрагидрофурана (ТГФ) (бурная экзотермическая реакция). Полученный раствор/суспензию добавляли в течение 2 минут к органолитиевому реагенту с получением черно-коричневого раствора. Реакционную смесь выдерживали в течение 5 минут при -78°С, затем оттаивали до 0°С в течение 1 часа. После добавления 1 мл 5% HCl и частичного упаривания продукт экстрагировали добавлением 15 мл хлороформа (CHCl3) и органическую фазу промывали 2×5 мл 5% соляной кислоты (HCl) и сушили над сульфатом магния (MgSO4). Растворитель выпаривали и заменяли 4 мл ТТФ, к которому было добавлено 0,4 мл 5% HCl. Через 65 минут при комнатной температуре реакционную смесь упаривали и остаток хроматографировали на силикагеле (SiO2) смесями этилацетата/гексана. Элюирование головной фракции с отношением 2:5 позволяло получать 64 мг (15%) 5,7,3′,4′ -тетра-О-бензилкатехина. Последующее элюирование с отношением 1:2 позволяло получить два неидентифицированных побочных продукта, и в итоге элюировали 94 мг (22%) желаемого димера с отношением 2:3. Образец массой 92 мг далее очищали путем препаративной ВЭЖХ (Waters μPorasil 125 Å, размер частиц 10 мкм, 30×5 см, EtOAc/гексан 2:3, 80 мл/мин, УФ определение при 280 нм) с получением 65 мг (16%) чистого продукта в виде бесцветной пленки: αD-75,2°, α546-91,4° (EtAc, с 18,3 gL-1);1H ЯМР (CDCl3) δ 7,42-7,20 (м, 40Н), 6,90 (д, 2Н, J=1 Гц), 6,75, 6,67 (АВкв, 4Н, J=8H), 6,28 (с, 2Н), 5,03 (с, 4Н), 5,00-4,85 (м, 12Н), 4,59 (д, 2Н, J=8,5 Гц), 3,84 (м, 2Н), 2,95, 2,66 (АВкв, 4Н, J=16,5 Гц, обе части д с J=5,5 и 8 Гц соотв.), 1,67 (ушир., 2Н);13С ЯМР (CDCl3) δ 156,64, 156,45, 153,02, 148,78, 148,68, 137,85, 137,22, 137,05, 131,80, 128,38, 128,31, 128,22, 127,71, 127,67, 127,58, 127,26, 127,17, 127,09, 126,58, 119,88, 114,44, 113,19, 105,46, 102,61, 92,51, 80,64, 71,14, 71,10, 69,78, 68,11, 27,20; ИК (пленка) 3563, 3440 (ушир.), 1602, 1264, 1120, 736, 697 см-1; МС (Электровпрыск, 0,1% НСООН в СН3CN) m/z 1323,1/1322,0 (M+Na)+рассч. для13C12C85H74Ol2 Na/12C86H74O12Na: 1322,5/1321,5), 968,8/967,8 (М+H)+, затем ретро-реакция Дильса-Альдера; рассч. дляl3C12C63 H55О9/12C64H55O9: 968,4/967,4).
Пример 6
5,7,3′,4′,5′′,7′′, 3'′′,4'′′-Окта-O-бензил-3,3′′-ди-O-(три-O-бензилгаллоил)-8,8′′-бикатехин
К раствору 63,5 мг (144 мкмоль, 5 экв.) три-O-бензилгалловой кислоты и 1,5 мкл диметилформамида (ДМФ) в 1 мл метиленхлорида (СН2Cl2) добавляли 25 мкл (0,29 мкмоль, 10 экв.) оксалилхлорида. После перемешивания при комнатной температуре в посуде, снабженной хлоркальциевой трубкой (CaCl2), в течение 35 мин смесь упаривали и сушили в вакууме. К неочищенному хлорангидриду добавляли раствор 37,5 мг (28,9 мкмоль) 8,8′′-димера из примера 7 в 0,8 мл безводного пиридина и 17,6 мг (144 мкмоль, 5 экв.) 4-ди(метиламино)пиридина (ДМАП). Смесь перемешивали при комнатной температуре в закрытой посуде в течение 24,5 часов. После добавления 50 мкл воды перемешивание при комнатной температуре продолжали в течение 4 часов. Затем добавляли 15 мл 5% соляной кислоты (HCl) и продукт экстрагировали добавлением 3×5 мл метиленхлорида (CH2Cl2). Органические фазы сушили над сульфатом магния (MgSO4) и упаривали и неочищенный продукт очищали путем фильтрации через силикагель (SiO2) (15×1,8 см) этилацетатом (EtOAc)/CHCl3/гексаном 1:9:10. Упаривание и сушка в вакууме позволяли получить 58,2 мг бесцветной пленки, которую далее очищали путем препаративной ТСХ (SiO2, 200×200×2 мм, EtOAc/гексан 1:2) с получением 55,0 мг (89%) продукта: δD- 31,4, δ546- 36,9 (EtOAc, с 15,4 gL-1);1Н ЯМР (CDCl3) δ 7,40-7,15 (т, 70 Н), 6,85 (с, 2 Н), 6,68, 6,36 (АВкв, 4Н, J=8,5 Гц), 6,34 (с, 2Н), 5,25 (м, 2Н), 5,05 (с, 4Н), 5,03-4,92 (м, 10 Н), 4,84 (с, 8Н), 4,83 (с, 4Н), 4,77, 4,71 (АВкв, 4Н, J=11,5 Гц), 2,87, 2,78 (АВкв, 4Н, J=16,5 Гц, обе части д с J=5,5 и 4,5 Гц соотв.);13С ЯМР (CDCl3) δ 164,84, 156,63, 156,46, 153,16, 152,24, 148,65, 148,41, 142,52, 137,82, 137,64, 137,30, 137,06, 137,02, 136,69, 131,90, 128,46, 128,39, 128,31, 128,20, 128,11, 127,80, 127,74, 127,60, 127,53, 127,31, 127,13, 127,06, 126,47, 124,99, 119,19, 114,34, 112,39, 109,08, 105,40, 102,00, 91,93, 75,06, 70,98, 70,89, 70,02, 69,94, 23,02; ИК (пленка) 1714, 1596, 1428, 1125, 735, 696 см-1. Анал. рассч. для С142Н118O20: С 79,42; Н 5,81. Найдено: С 79,53; Н 5,55.
Пример 7
3,3"-Ди-O-галлоил-8,8"-бикатехин
Раствор 29,2 мг (13,6 мкмоль) описанного выше соединения в 2 мл ТГФ и 2 мл МеОН гидрировали при атмосферном давлении (баллон) над 34,5 мг коммерческого (влажного) 20% Pd(OH)2,/C в течение 105 мин. Катализатор отфильтровывали через вату и промывали 2 мл МеОН. После упаривания неочищенный продукт очищали путем препаративной ВЭЖХ (Waters Bondapak С18, 300×19 мм, скорость элюции 9 мл/мин, УФ определение при 280 нм), используя следующий градиент растворителя В (0,5% АсОН в денатурированном EtOH) в растворителе А (0,5% АсОН в Н2O): 0-1 мин, 15% В; 1-15 мин, 15-26% В; 15-16 мин, 26-80% В; 16-20 мин, 80% В. Объединенные элюаты, содержащие главный компонент, упаривали и сушили в вакууме с получением 6,7 мг (56%) продукта в виде багрянистой пленки:1H ЯМР (ацетон - d6/D2O 3: lv/v) 7,06 (с, 4Н), 7,03 (д, 2Н, J=2 Гц), 6,86, 6,76 (АВкв, 4Н, J=8 Гц, часть А д c J=1,5 Гц), 6,19 (с, 2Н), 5,23 (м, 2Н), 4,99 (д, 2Н, J=8 Гц), 3,05, 2,64 (АВкв, 4Н, J=16 Гц, обе части д с J=5,5 и 8 Гц соотв.);13С ЯМР (ацетон-d6/D2O 3: lv/v) 166,59, 155,92, 155,59, 154,26, 145,81, 145,27, 145,19, 139,00, 131,30, 121,08, 119,58, 115,74, 114,76, 109,95, 100,89, 99,41, 96,08, 78,89, 71,36, 25,97; МС (Электровпрыск, МсОН/СН3CN) m/z 906,4/905,4 (М+Na+рассч. для13С12С43Н34О20Na/12С44Н34O20Na: 906, 2/905,2), 735,6 (М+Na+-галловая кислота; рассч. для12С37Н28О15Na: 735,1) 601,7 (М+Na+- затем ретро-реакция Дильса-Альдера; рассч. для12С29Н22O13Na: 601,1).
Пример 8
Получение 5,7,3′,4′-тетра-O-бензилкатехина
Раствор (+)-катехина (65,8 г, 226,7 ммоль, безводный) растворяли в безводном диметилформамиде (ДМФ, 720 мл), добавляли по каплям при комнатной температуре в течение 80 минут к перемешиваемой суспензии гидрида натрия (60% в масле, 39 г, 975 ммоль, 4,3 экв.) в диметилформамиде (ДМФ) (180 мл).
(S.Miura, et al., Radioisotopes, 32, 225-230 (1983)). После перемешивания в течение 50 минут колбу помещали в NaCl/ледяную баню с температурой -10°С. Бензилбромид (121 мл, 1,02 моль, 4,5 экв.) добавляли по каплям в течение 80 мин и коричневую реакционную смесь нагревали до комнатной температуры при перемешивании в течение ночи. Полученную реакционную смесь упаривали и полученное леденцеобразное твердое вещество растворяли с нагреванием и перемешиванием в двух порциях растворителя, каждая из которых состояла из 200 мл хлороформа (CHCl3) и 100 мл воды. Фазы разделяли, водную фазу экстрагировали добавлением CHCl3 (20 мл) и объединенные органические фазы промывали водой (100 мл), сушили над сульфатом магния (MgSO4) и упаривали. Остаток очищали путем хроматографии на силикагеле (42×10 см; этилацетат/хлороформ/гексан 1:12:7) с получением после упаривания и сушки в вакууме 85 г неочищенного продукта, который перекристаллизовывали из трихлорэтилена (1,3 л) с получением 35,1 г (24%) не совсем белого порошка.1Н ЯМР (CDCl3) 7,47-7,25 (м, 20 Н), 7,03 (с, 1Н), 6,95 (с, 2Н), 6,27, 6,21 (АВкв, 2Н, J=2 Гц), 5,18 (с, 2Н), 5,17 (узкий АВкв, 2Н), 5,03 (s 2Н), 4,99 (s 2Н), 4,63 (д, 1Н, J=8,5 Гц), 4,00 (м, 1Н), 3,11, 2,65 (АВкв, 2Н, J=16,5 Гц, обе части д с J=5,5 и 9 Гц соотв.), 1,59 (д, 1Н, J=3,5 Гц); ИК (пленка) 3440 (ушир.), 1618, 1593, 1513, 1499, 1144, 1116, 733, 696 см1; МС m/z 650 (M+, 0,5%), 319, 181, 91.
Альтернативно тетра-O-бензил-(+)-катехин может быть получен в соответствии с методикой, описанной в Н. Kawamoto et al, Mokazai Gakkaishi, 37, (5) 488-493 (1991), с использованием карбоната калия и бензилбромида в диметилформамиде (ДМФ). О частичной рацемизации катехина и в положении 2, и в положении 3 сообщалось в М.-С.Pierre et al., Tetrahedron Letters, 38, (32) 5639-5642 (1997).
Пример 9
Получение 5,7,3′,4′ -тетра-O-бензилэпикатехина
1 М раствор три-втор-бутилборгидрида лития в тетрагидрофуране (ТГФ) (100 мл, L-Selectride®, продается Aldrich Chemical Co, Inc., Milwaukee, WI) добавляли в атмосфере аргона к перемешиваемому раствору безводного бромида лития (LiBr) с температурой 0°С (34,9 г, 402 ммоль) в 100 мл безводного ТГФ. Полученную смесь охлаждали до -78° С, применяя ацетоновую/СО2баню с последующим капельным добавлением раствора защищенного эпикатехина (50,1 г, 77,2 ммоль) в 400 мл безводного ТГФ в течение 50 мин. Перемешивание продолжали при -78°С в течение 135 минут. Охлаждающую баню удаляли и 360 мл 2,5 М водного гидроксида натрия (NaOH) добавляли в реакционную смесь. Реакционную колбу помещали в водяную баню комнатной температуры и смесь 35% водного пероксида водорода H2O2 (90 мл) и этанола (270 мл) добавляли в течение 130 мин. Перемешивание продолжали в течение ночи. Для растворения выкристаллизованного продукта добавляли хлороформ (700 мл), фазы разделяли, водную фазу экстрагировали добавлением хлороформа (CHCl3) (50 мл), объединенные органические фазы сушили над сульфатом магния (MgSO4, упаривали и сушили в вакууме с получением 56,6 г неочищенного продукта. Данный продукт растворяли в 600 мл кипящей смеси этилацетата (EtOAc) и этанола (EtOH) (2:3) и выкристаллизовывали при комнатной температуре, а затем в холодильнике. Продукт выделяли путем вакуумного фильтрования, промывали 2×50 мл холодного (-20°С) EtOAc/EtOH (1:3) и сушили в вакууме сначала при комнатной температуре, а затем при 80°С с получением 35,4 г (70%) светло-желтого твердого вещества. Упаренный маточный раствор фильтровали через силикагель (SiO2), (14×6,5 см, хлороформ (CHCl3), а затем CHCl3/EtOAc 12:1), элюат концентрировали до 40 мл и остаток разбавляли 60 мл этанола с получением дополнительных 5,5 г (11%) O-бензилэпикатехина в виде желтого твердого вещества: т.пл. 129,5-130°С (из EtOAc/EtOH); αD- 27,7°, α546- 33,4° (EtOAc, с 21,6 г/л);1H ЯМР (CDCl3) 7,48-7,25 (м, 20 Н), 7,14 (с, 1Н), 7,00, 6,97 (АВкв, 2Н, J=8,5 Гц, часть А д c J=1,5 Гц), 6,27 (с, 2Н), 5,19 (с, 2Н), 5,18 (с, 2Н), 5,02 (с, 2Н), 5,01 (s2H, 4,91 (s 1H), 4,21 (ушир.с, 1 Н), 3,00 2,92 (АВкв, 2Н, J=17,5 Гц, обе части д с J=1,5 и 4 Гц соотв.), 1,66 (д, 1H, J=5,5 Гц); Анал. рассч. для С43Н48O6: С 79,36; Н 5,89, Найдено: С 79,12; Н 5,99.
Пример 10
5,7,3′,4′-Тетра-O-бензил-6,8-дибромэпикатехин
К раствору 334 мг (914 мкмоль) 5,7,3′,4′ -тетра-O-бензилэпикатехина в 10 мл безводного СН2Cl2 добавляли с охлаждением на льду сразу все 192 мг (1,08 ммоль) перекристаллизованного N-бромсукцинимида (NBS). Реакционную смесь перемешивали при 0°С в течение 45 мин и при комнатной температуре в течение 17 ч. Добавляли раствор 200 мг Na2S2O3·5H2O в 5 мл воды. После непродолжительного перемешивания фазы разделяли, водную фазу экстрагировали добавлением 5 мл СН2Cl2 и объединенные органические фазы сушили над MgSO4 и выпаривали. Хроматографию на силикагеле (30×2,6 см) EtOAc/CHCl3/гексаном 1:12:7 (для удаления следов побочного продукта), затем 3:12:7 с последующим упариванием и сушки в вакууме с получением 362 мг (87%) дибромида в виде бесцветной пены: []546 -58,2° (EtOAc, с 13,5 gL-1);1H ЯМР (CDCl3) δ 7,64 (д, 2Н, J=7 Гц), 7,52-7,26 (м, 18Н), 7,17 (с, 1Н), 7,03, 6,97 (с, 2Н), 5,20 (с, 2Н), 5,17 (с, 2Н), 5,03 (с, 2Н), 5,01, 4,97 (АВкв, 2Н, J=11 Гц), 4,99 (с, 1Н), 4,19 (узкий м, 1Н), 3,04, 2,87 (АВкв, J=17,5 Гц, обе части д с J=1,5 и 3,5 Гц соотв.), 1,55 (д, 1H, J=3,5 Гц);13С ЯМР (CDCl3) δ 154,43, 152,57, 151,09, 149,03, 148,82, 137,10, 136,94, 136,50, 136,37, 130,13, 128,52, 128,50, 128,48, 128,47, 128,43, 128,35, 128,32, 128,16, 127,82, 127,81, 127,36, 127,20, 118,81, 115,06, 112,91, 112,30, 105,23, 103,25, 78,80, 74,61, 74,55, 71,24, 71,14, 65,33, 28,75; ИК (пленка) 1734, 1606, 1513, 1369, 1266, 1184, 1113, 1083, 735, 697 см-1, Анал. рассч. для С43H36О6 Br2: С 63,88; Н 4,49, Найдено С 64,17; Н 4,45.
Пример 11
5,7,3′,4′-Тетра-O-бензил-6-бром-3-О-(тетрагидропиран-2-ил)катехин
К раствору 297 мг (407 мкмоль) 5, 7,3′,4′-тетра-О-бензил-6-бромэпикатехина в 2 мл безводного СН2Cl2 при комнатной температуре добавляли 56 мкл (0,61 ммоль) дигидропирана с последующим добавлением 2,6 мкл (40 мкмоль) метансульфоновой кислоты. Раствор перемешивали при комнатной температуре в течение 25 мин, после чего добавляли 0,15 мл насыщенного водного раствора Na2CO3. После упаривания остаток хроматографировали на SiO2 EtOAc/гексаном.
Пример 12
5,7,3′,4′,5′′,7′′,3′′,4'′′-Окта-О-бензил-6,6′′ -бикатехин
К раствору 527 мг (648 мкмоль) соединения тетрагидропиранилового эфира из примера 9 в 6,5 мл безводного ТГФ при -78°С в течение 5 мин по каплям добавляли 0,91 мл (1,55 ммоль) трет-бутиллития (1,7 М в пентане). Полученный раствор перемешивали при -78°С в течение 5 минут, в то время как к 147 мг (0,91 ммоль) безводного хлорида железа (FeCl3) добавляли 1,5 мл безводного тетрагидрофурана (ТГФ) (бурная экзотермическая реакция). Полученный раствор/суспензию добавляли в течение 2 минут к органолитиевому реагенту. Реакционную смесь выдерживали в течение 5 минут при -78°С, затем оттаивали до 0°С в течение 1 часа. После добавления 1 мл 5% HCl и частичного упаривания продукт экстрагировали добавлением 15 мл CHCl3 и органическую фазу промывали 2×5 мл 5% HCl и сушили над MgSO4. Растворитель выпаривали и заменяли 4 мл ТГФ, к которому было добавлено 0,4 мл 5% HCl, и остаток очищали.
Пример 13
5,7,3′,4′,5′′,7′′,3'′′,4'′′-Окта-О-бензил-3,3′′-ди-O-(три-O-бензилгаллоил)-6,6′ ′-бикатехин
К раствору 63,5 мг (144 мкмоль, 5 экв.) три-O-бензилгалловой кислоты и 1,5 мкл ДМФ в 1 мл СН2Cl2 добавляли 25 мкл (0,29 ммоль, 10 экв.) оксалилхлорида. После перемешивания при комнатной температуре в посуде, снабженной хлоркальциевой трубкой (CaCl2), в течение 35 мин смесь упаривали и сушили в вакууме. К неочищенному хлорангидриду добавляли раствор 37,5 мг (28,9 мкмоль) 6,6′′-димера из примера 10 в 0,8 мл безводного пиридина и 17,6 мг (144 мкмоль, 5 экв.) ДМАП. Смесь перемешивали при комнатной температуре в закрытой посуде в течение 24,5 часов. После добавления 50 мкл воды перемешивание при комнатной температуре продолжали в течение 4 часов. Затем добавляли 15 мл 5% HCl и продукт экстрагировали добавлением 3×5 мл СН2Cl2. Органические фазы сушили над MgSO4 и упаривали и неочищенный продукт очищали путем фильтрации через SiO2 (15×1,8 см) EtOAc/CHCl3/гексаном 1:9:10. Упаривание и сушка в вакууме позволяли получить пленку, которую далее очищали путем хроматографии с получением продукта.
Пример 14
3.3′′-Ди-O-галлоил-6,6′′-бикатехин
Раствор 29,2 мг (13,6 мкмоль) соединения из примера 8 в 2 мл ТГФ и 2 мл МеОН гидрировали при атмосферном давлении (баллон) над 34,5 мг коммерческого (влажного) 20% Pd(OH)2/C в течение 105 мин. Катализатор отфильтровывали через вату и промывали 2 мл МеОН. После упаривания неочищенный продукт очищали путем препаративной ВЭЖХ.
Реферат
Изобретение относится к новым способам получения (8↔8) димера катехина, и/или эпикатехина, и/или эпикатехингаллата, которые включают окислительное или восстановительное связывание защищенных ариллитиевых соединений. 2 н. и 7 з.п. ф-лы.
Комментарии