8-фторантрациклингликозиды или их фармацевтически приемлемые соли присоединения кислот, способ их получения, фармацевтическая композиция, промежуточные соединения и способ их получения - RU2095365C1
Код документа: RU2095365C1
Чертежи




Описание
Изобретение относится к гликозидным производным
8-фторантрациклина формулы I:
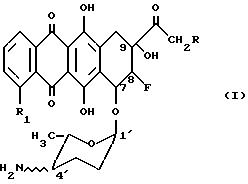
где: R H, OH, OR";
R1 H, OH, OCH3 ;
R" CHO-COCH3 или ацильный остаток, полученный из карбоновой кислоты, содержащей вплоть до 6 атомов углерода; и
≈NH2 показывает, что аминовый заместитель может быть в аксиальной конфигурации (естественная конфигурация) или экваториальной конфигурации (эпи- конфигурация);
и к их фармацевтическим приемлемым солям.
Даунорубицин (дауномицин), 4-деметоксидаунорубицин (гидарубицин) и их производные, содержащие гидроксилированную боковую цепь (доксорубицины), являются гликозидами, обладающими известными противоопухолевыми свойствами, получение и использование которых уже описано. (F.Arcamone "Doxorubicin Anticancer Antibiotics". Medicinal Chemistry Series, Vol.17. Academie Press, 1981).
В настоящее время поразительным образом было найдено, что замена атома H атомом F в положении C-8 не-гликозидной части молекулы повышает активность и селективность этих соединений, которые таким образом неожиданно обладают более высокой способностью (силой) по сравнению с известными антрациклинами, особенно в случае опухолевых клеток, устойчивых к известным соединениям.
Настоящее изобретение относится поэтому к гликозидным производным 8-фторантрациклина формулы (I):

где: R H, OH, OR";
R1 H, OH, OCH3;
R" CHO-COCH3 или ацильный остаток, полученный из карбоновой кислоты, содержащей вплоть до 6 атомов углерода; и
≈NH2 показывает, что аминовый заместитель может быть в аксиальной конфигурации (естественная конфигурация) или экваториальной конфигурации (эпи конфигурация);
и к их фармацевтически приемлемым солям.
Одной из предпочтительных солей настоящего изобретения является хлоргидрат соединений формулы (I).
Более определенно настоящее
изобретение относится к следующим соединениям:
4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-аминодаунорубицин (R R1 H);
4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодаунорубицин (R R1 H);
8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодаунорубицин (R H, R1 OCH3);
8-фтор-3'-дезамино-4'-дезокси-4'-аминодаунорубицин (R H, R1 OCH3);
8-фтор-3'-дезамино-4'-дезокси-4'-аминокарминомицин (R H, R1 OH);
8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминокарминомицин (R H, R1 OH);
4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-аминодоксорубицин (R OH, R1 H) и его сложные эфиры в
положении C-14;
8-фтор-3'-дезамино-4'-дезокси-4'-аминодоксорубицин (R H, R1 OCH3) и его сложные эфиры в положении C-14;
8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодоксорубицин (R H, R1 OCH3) и его сложные эфиры в положении C-14.
Соединения формулы (I) и их фармацевтически приемлемые
соли получаются конденсацией 8-фторантрациклинона формулы (II):
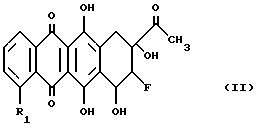
где R1 является таким, как определено выше, с соединением формулы IIIa или IIIe:

в которой X является уходящей группой, способной образовать при условиях конденсации стабильный карбокатион, который может присоединяться гидроксильной группой в положении C-7, а R2 является защищенной амино-группой, чтобы получить гликозид формулы (IV)

в которой R1 и R2 являются такими, как определено выше, а (≈) показывает, что заместитель R2 может располагаться в аксиальной или экваториальной конфигурации.
При удалении амино-зацитной группы из соединения формулы IV получается 8-фторантрациклингликозид формулы I, в которой R и R1 являются такими, как определено выше.
Антрациклингликозид формулы I может превращаться в одну из его фармацевтически приемлемых солей, или же соединение формулы I или его фармацевтически приемлемая соль может бромироваться и полученное 14-бром-производное может быть гидролизовано, чтобы получить антрациклингликозид формулы I, в которой R OH, и если это необходимо, указанный антрациклингликозид может быть превращен в одну из его фармацевтически приемлемых солей.
Согласно изобретению предпочтительной уходящей группой X для соединений формулы III является галоген, такой как бром или хлор, предпочтительно хлор, или пара-нитробензоилокси-группа. Амино-зацищенная группа R2 является предпочтительно трифторацетамидной или аллилоксикарбоксиамидной.
Условия реакции для реакции конденсации между соединением формулы II и соединением формулы IIIa или IIIe, чтобы получить соединение формулы IV, могут варьировать в соответствии с типом замещения соединений формулы IIIa или IIIe.
Реакция гликозидации проводится в инертном органическом растворителе в присутствии конденсирующего реагента.
Углеводородные растворители, такие как бензол и толуол, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан, хлорированные растворители, такие как хлороформ, хлористый метилен или дихлорэтан и их смеси могут быть использованы. Хлористый метилен является предпочтительным растворителем.
Конденсирующими агентами могут быть соли, такие как трифторметансульфонат серебра, перхлорат серебра, смеси окиси ртути (2) и бромида ртути, трифторметансульфонат триметилсилила; кислоты Льюиса, такие как галогениды бора, тетрахлорид олова или титана; или кислотные ионнообменные смолы, такие как Амберлиты.
Температура реакции может варьировать от -40oC до 40oC, предпочтительно от -20oC до 20oC, а реакция может продолжаться от 15 мин до 3 ч.
Дегидратирующее средство, такое как активированные молекулярные сита, предпочтительно присутствуют в реакционной смеси.
Во время реакции или в конце ее к реакционной смеси может быть добавлено органическое основание, такое как пиридин, коллидин, N,N-диметиламинопиридин, триэтиламин или акцептор протона.
Согласно изобретению условия для удаления амино-защитной группы соединения формулы IV, в которой R1 и R2 являются такими, как определено выше, для того, чтобы получить соединение формулы I, в которой R H, а R1 является таким, как определено выше, могут варьировать в соответствии с механизмом замещения соединения формулы IV.
Когда амино-защитная группа является трифторацетамидной, реакция депротонирования проводится в полярном растворителе, таком как вода, метанол, этанол, пиридин, диметилформамид или их смеси, в присутствии стехиометрического или более чем стехиометрического количества неорганического основания, такого как NaOH, KOH, LiOH, Ba(OH)2 или их карбонатов. Температура реакции может варьироваться от 0oC до 50oC, а реакция может продолжаться от 3 ч до 3 дней.
Когда амино-защитная группа является аллилкарбоксиамидной, реакция депротонирования проводится в инертном растворителе в присутствии металлического комплекса, такого как (тетракистрифенилфосфин)-палладия, как это описано, например, в Tetrahedron Letters, 30, 3773 (1089), или (тетракарбонил)-никеля, как это описано, например, в J. Org. Chem. 38, 3233 (1973).
Если это необходимо, соединение формулы I, в которой R является водородом, а R1 является таким, как определено выше, может быть превращено, соответственно, в соединение формулы I, в которой R является OH, а R1 является таким, как определено выше, путем бромирования в положение C-14, за которым следует гидролиз 14-бром-производного, полученного по этому способу. Бромирование и последующий гидролиз описываются в пат. США N 4 122 076. Бромирование соединения формулы I, в которой R H, а R1 является таким, как определено выше, проводится с помощью брома в хлороформе, чтобы получить соответствующее 14-бром-производное, из которого после гидролиза при температуре окружающей среды в течение 48 ч с помощью водного раствора формиата натрия получается соединение формулы I в виде свободного основания, в котором R OH, а R1 является таким, как определено выше, и которое путем обработки с хлористоводородной кислотой в метиловом спирте выделяется в виде хлоргидрата.
Изобретение относится также к 8-фторантрациклинонам общей формулы II и к способу их получения.
Этот способ иллюстрируется схемой реакции (A).
Первая стадия способа включает окисление эпоксиспирта общей формулы V, в которой R1 является таким, как определено выше, чтобы получить эпоксикетон общей формулы VI, в которой R1 является таким, как определено выше. Эпоксиспирт V может быть получен так, как это описано в пат. Англии N 2 125 030.
Схема А(см. приложение).
Реакция окисления может быть проведена по методам, обычно известным экспертам в данной области техники. Методы, включающие использование диметилсульфоксида, такие как окисление по методу Моффата и тому подобному, или использование пиридин-хромовых комплексов, таких как хлорхроматы пиридиния, являются предпочтительными.
Вторая стадия способа, представленная в схеме А, включает реакцию эпоксикетона VI с генератором нуклеофильного фтора, чтобы получить 8-фторированное соединение формулы VII, в которой R1 является таким, как определено выше.
Генератором нуклеофильного фтора может быть, например, комплекс пиридина с фтористоводородной кислотой, а предпочтительным методом для осуществления этой стадии
является перемешивание раствора или
суспензии соединения формулы VI с реагентом при температуре окружающей среды в течение ночи. Последняя стадия включает превращение фторогидрокетона формулы VII в
соединение формулы II. Это может быть
осуществлено по известным методам, таким как бромирование и сольволиз, если необходимо, защищая кето-группу [Can. J. Chem. 49, 2712 (1973); J. Amer. Chem. Soc.
98, 1969 (1976); J. Amer. Chem. Soc. 98,
1967 (1976)]
Изобретение относится также к амино-сахарам формулы III, в которой X и R2 являются такими, как определено выше, и к способу
их получения, включающему:
а)
реакцию метил-2,3,6-тридезокси-альфа-L глицеро-гексоксипиранозид-4-улозы формулы VIII:

с гидроксиламином или с солью его при присоединении кислоты, чтобы образовать смесь син и анти оксимов формулы IX:
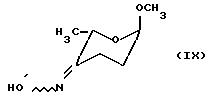
б) восстановление указанной смеси, после этого защиту образующейся амино-группы с помощью трифторацетильной группы и разделение полученных таким образом 4-N-трифторацетилированных эпимеров формулы Xa и Xe:

или, если это предпочитается, защиту образующейся амино-группы с помощью аллилоксикарбонильной группы и разделение 4-N-аллилокси-карбонилированных эпимеров формулы XIa и XIe:
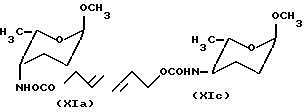
в) если это предпочитается, в виде альтернативы к вышеуказанным стадиям реакцию метил-2,3,6-тридезокси- α -L- глицеро-гексоксипиранозид-4-улозы формулы VIII с восстановителем в присутствии аммонийных солей, защиту образующейся таким образом амино-группы с помощью трифторацетильной группы или, если это предпочитается, с помощью аллилоксикарбонильной группы, и разделение эпимеров формулы Xa и Xe или XIa и XIe;
г) превращение каждого эпимера Xa и Xe в соответствующее 1-гидрокси-производное формулы XIIa и XIIe:

или, если это предпочитается, превращение каждого эпимера XIa и XIe в соответствующее 1-гидрокси-производное формулы XIIIa и XIIIe:

д) превращение указанных 1-гидрокси-производных XIIa и XIIe или XIIIa и XIIIe в соответствующие соединения формулы IIIa и IIIe, в которой X является хлором, а R2 является таким, как определено выше, или, если это желательно, превращение каждого указанного 1-гидрокси-производного XIIa и XIIe или XIIIa и XIIIe в соответствующие соединения формулы IIIa и IIIe, в которой X является пара-нитробензоилокси-группой, а R2 является таким, как определено выше.
В стадии а) метил-2,3,6-тридезокси- α -L- глицеро-гексоксипиранозид-4-улоза VIII может быть обработана солянокислым гидроксиламином в триэтиламине, чтобы получить син и анти оксимы IX.
В стадии б) оксимы IX могут быть восстановлены бораном в тетрагидрофуране или бис-диметоксиэтокси натрий-алюминий-гидридом (Витридом) в тетрагидрофуране, толуоле или диоксане. Температура реакции может находиться в пределах между -20oC и 20oC, а продолжительность реакции между 3 и 12 ч. Восстановленная смесь может быть подвергнута реакции с ангидридом трифторуксусной кислоты в четыреххлористом углероде или диэтиловом эфире при температуре окружающей среды, предпочтительно в присутствии органического основания, такого как триэтиламин или пиридин, чтобы получить смесь N-трифторацетилированных производных, из которых отдельно путем хроматографического разделения получаются соответствующие вещества формулы Xa и Xe. Если это предпочитается, восстановленная смесь может быть подвергнута реакции с аллилхлоркарбонатом в тетрагидрофуране в присутствии органического основания, такого как пиридин, триэтиламин и тому подобные, при температуре между -20oC и 20oC в течение 3-12 ч, чтобы получить смесь N-аллилоксикарбонилатов, из которых соответствующие соединения формулы XIa и XIe получаются путем хроматографического разделения.
Альтернативно, в стадии в) восстановление улозы формулы VIII может быть осуществлено с помощью цианоборогидрида натрия и аммоний-ацетата или аммоний-хлорида в тетрагидрофуране, диэтиловом эфире, метаноле или этаноле при температуре между -20oC и 20o C. Восстановленная смесь может быть обработана так, как описано в стадии б).
В стадии г) превращение каждого эпимера Xa и Xe в соответствующие 1-гидрокси-производные XIIa и
XIIe или,
если предпочитается, превращение каждого эпимера XIa и XIe в соответствующие 1-гидрокси-производные XIIIa и XIIIe может быть осуществлено путем нагревания в присутствии кислоты. Нагревание
может быть
до температуры между -70oC и 100oC в течение времени между 30 мин и 3 ч, используя водный раствор уксусной, трифторуксусной или хлористоводородной кислоты.
Концентрация водного
кислотного раствора может быть между 5 и 30
В стадии д) каждый 1-гидрокси-эпимер формулы XIIa, XIIe, XIIIa и XIIIe может быть сначала обработан в течение ночи при 0oC ангидридом
трифторуксусной кислоты, а затем растворен в диэтиловом эфире и подвергнут реакции в течение ночи газообразным хлористым водородом, чтобы получить каждый индивидуальный эпимер
формулы IIIa и IIIe, в
которой X является хлором, а R2 является таким, как определено выше.
Если предпочитается, каждый из указанных 1-гидрокси-эпимер формулы XIIa, XIIe, XIIIa и XIIIe может быть растворен в пиридине или диметилформамиде или диметилсульфоксиде в присутствии органического основания и обработан пара-нитробензоилхлоридом при температуре между -20oC и 20oC в течение 1-6 ч, чтобы получить каждый индивидуальный эпимер формулы IIIa и IIIe, в которой X является пара-нитробензоилокси-группой, а R2 является таким, как определено выше.
Изобретение относится также к фармацевтическим композициям, содержащим в качестве действующего начала антрациклингликозид формулы I или фармацевтически активную соль его вместе с фармацевтически приемлемым носителем или разбавителем.
Терапевтически эффективное количество соединения изобретения смешивается с инертным носителем. Могут быть использованы обычные носители, а композиция может быть составлена обычным путем.
Соединения изобретения являются эффективными при терапевтическом лечении людей или животных. В частности, соединения изобретения являются полезными в качестве противоопухолевых средств путем назначения терапевтически эффективных количеств соединения вылечиваемому пациенту.
Пример 1. 9-Ацетил-9, 8-(8H)-эпокси-7,10-дигидро-6,11-дигидрокси-5,12- нафтацен-дион (VI, R1 H). 9-(1'-гидроксиэтил)-9,8-(8H)-эпокси-7,10-дигидро-6,11- дигидрокси-5,12-нафтацен-дион (VI, R1 H), полученный так, как описано в пат. Англии N 2 125 030 (1,9 г, 5,6 ммолей) и растворенный в хлористом метилене (150 мл), добавляют при температуре окружающей среды и перемешивании к суспензии хлорхромата пиридиния (1,8 г, 8,5 ммолей). Реакционную смесь перемешивают в течение ночи, разбавляют добавлением хлористого метилена и промывают водой (2 • 100 мл). Органическую фазу высушивают над сульфатом натрия, фильтруют и выпаривают под вакуумом, чтобы получить остаток, который кристаллизуют из этилацетата. Получают 1,78 г (выход 90) соединения с температурой плавления 219-221oC.
ИК (в нуйоле, см-1) 1713.
ЯМР (CDCl3, d ) 2,17 (синглет, 3H); 3,01 (двойной дублет, J 20 Гц, 1H), 3,4 4,2 (мультиплет, 4H); 7,80 (мультиплет, 2H), 8,30 (мультиплет, 2H); 13,39 (синглет, 1H); 13,42 (синглет, 1H).
Действуя подобным образом, получают следующие соединения:
4-метокси-9-ацетил-9,
8-(8H)-эпокси-7,10-дигидро-6,11-дигидрокси- 5,12- нафтацен-дион (VI, R1 OCH3) и
9-ацетил-9,8-(8H)-эпокси-7,10-дигидро-4,6,11-тригидрокси-5,12- нафтацен-дион (VI, R1 OH).
Пример 2. 9-Ацетил-8-(8H)-фтор-7,10-дигидро-6,9,11-тригидрокси-5,12- нафтацен-дион (VII, R1 H). Смесь 9-ацетил-9,8-(8H)-эпокси-7,10-дигидро-6,11-дигидрокси-5, 12- нафтацен-диона (0,6 г, 1,7 ммолей), полученного, как в примере 1, и комплекса фтористоводородная кислота/пиридин (70-ный раствор, 21 мл) перемешивают в течение 24 ч в токе азоте при температуре окружающей среды. Реакционную смесь выливают затем на лед и воду и перемешивают в течение 30 мин. Полученный осадок фильтруют, промывают водой до тех пор, пока не станет нейтральной и высушивают при 40oC под вакуумом. После кристаллизации получают 0,380 г (выход 60) соединения с температурой плавления 248-252oC.
ЯМР (CDCl3, d ): 2,50 (дублет, 3H, Jnf 2,6 Гц); 3,0-3,4 (мультиплет, 4H); 4,25 (синглет, 1H); 4,78 (три дублета, 1H, Jnf 48,4 Гц); 7,80 (мультиплет, 2H); 8,30 (мультиплет, 2H), 13,0 (синглет, 1H); 13,05 (синглет, 1H).
Следуя подобным путем, получают следующие соединения:
4-метокси-9-ацетил-8(8H)-фтор-7,10-дигидро-6,9,11-тригидрокси- 5,12-нафтацен-дион (VII, R1 OCH3
) и
9-ацетил-8(8H)-фтор-7,10-дигидро-4,6,9,11-тетрагидрокси-5,12- нафтацен-дион (VII, R1 OH).
Пример 3. 9-Ацетил-8(8H)-фтор-10-гидро-6,7(7H),9, 11-тетрагидрокси-5, 12- нафтацен-дион (II, R1 H). Смесь 9-ацетил-8(8H)-фтор-7,10-дигидро-6,9,11-тригидрокси-5,12- нафтацен-диона (0,1 г, 0,26 ммолей), полученного, как в примере 2, и брома (0,4 ммолей) в четыреххлористом углероде (30 мл) облучают в течение одного часа 500-ваттной солнечной лампой (кварцевой). Реакционную смесь промывают насыщенным водным раствором бикарбоната натрия, а затем водой, высушивают над сульфатом натрия, фильтруют и выпаривают под вакуумом, чтобы получить остаток, который очищают флеш-хроматографией. Получают 0,028 г (выход 30) продукта реакции.
ЯМР (CDCl3, d ): 2,54 (дублет, 3H, Jnf 2,2 Гц); 3,18 (два дублета, 1H, J 18 Гц); 3,30 (два дублета, 1H, J 18 Гц); 3,65 (дублет, 1H, Jnf 3 Гц); 4,60 (уширенный синглет, 1H); 4,98 (два дублета, 1H, Jnf 46,2 Гц); 5,18 (уширенный дублет, 1H, Jnf 13 Гц); 7,80-7,90 (мультиплет, 2H); 8,20-8,43 (мультиплет, 2H); 13,29 (синглет, 1H); 13,53 (синглет, 1H).
Следуя подобным путем, получают следующие соединения:
4-метокси-9-ацетил-8(8H)-фтор-10-гидро-6,7(7H), 9,11- тетрагидрокси-5,12-нафтацен-дион (II, R1 OCH3)
и
9-ацетил-8(8H)-фтор-10-гидро-4,6,7(7H), 9,11-пентагидрокси-5,12- нафтацен-дион (II, R1 OH).
Пример 4. Метил-2,3,4, 6-тетрадезокси-4-трифторацетамидо- a -L-трео-гексоксипиранозид (Xa) и метил-2,3,4,6-тетрадезокси-4- трифторацетамидо- a -L-эритро-гексоксипиранозид (Xe). Смесь 2,5 г метил-2,3,6-тридезокси- a -L- глицерогексоксипиранозид-4-улозы (VIII), 2,4 г солянокислого гидроксиламина и 3,33 мл метанола кипятят с обратным стеканием флегмы в течение 2 ч. Растворитель выпаривают под вакуумом и получающийся маслянистый остаток смешивают со 150 мл воды и экстрагируют с помощью 200 мл хлористого метилена (трижды). Экстракты высушивают над сульфатом натрия, фильтруют и выпаривают под вакуумом, чтобы получить остаток, который очищают флеш-хроматографией, элюируя 6/4 смесью гексана и диэтилового эфира. Получают 2,42 син/анти-смеси оксимов (IX) в вязкой жидкой форме. Эту вязкую жидкость (0, 830, 5,21 ммолей) растворяют в толуоле (15 мл) и к этому раствору в токе азота добавляют по каплям при -40oC бис-метоксиэтокси-натрий-алюминий-гидрид (24,5 ммолей). Реакционную смесь перемешивают при -40oC в течение 1/2 ч и при температуре окружающей среды в течение 4 ч, после чего добавляют воду (0,5 мл), водный раствор гидроокиси натрия (0,5 мл) и воду (1,5 мл). Две фазы отделяют, органическую фазу высушивают над сульфитом натрия, фильтруют через целит и выпаривают под вакуумом, чтобы получить сырое соединение, которое суспендируют в четыреххлористом углероде. По каплям добавляют триэтиламин (2,27 мл, 16,3 ммолей) и ангидрид трифторуксусной кислоты (1,15 мл, 6,24 ммолей) в таком порядке к суспензии при 0oC с перемешиванием. После 3 ч дальнейшего перемешивания при 0oC реакционную смесь разбавляют хлористым метиленом и промывают водой, 10-ным водным раствором бисульфата натрия и 8-ным водным раствором бикарбоната натрия. Органическую фазу высушивают над сульфатом натрия, фильтруют и выпаривают над вакуумом, чтобы получить сырой продукт реакции, который очищают флеш-хроматографией. Элюирование смесью 15/1 хлористого метилена/диэтилового эфира дает соединение Xa (0,480 г, выход 38).
ЯМР (CDCl3, d ): 1,10 (дублет, 3H, J 8 Гц); 1,5-2,0 (мультиплет, 4H); 3,31 (синглет, 3H); 3, 8-4,3 (мультиплет, 2H); 4,6-4,8 (мультиплет, 1H); 6,8 (синглет, 1H).
Элюирование 10/1 смесью хлористого метилена/диэтилового эфира дает соединение Xe (0,480 г, выход 38).
ЯМР (CDCl3, d ): 1,15 (дублет, 3H, J 8 Гц); 1,7-1, 9 (мультиплет, 4H); 3,32 (синглет, 3H); 3,6-3,8 (мультиплет, 2H); 4,6-4,7 (мультиплет, 1H); 6,8 (синглет, 1H).
Пример 5. Метил-2,3,4,6-тетрадезокси-4-трифторацетамидо- a -L-трео-гексоксипиранозид (Xa) и метил-2,3,4,6-тетрадезокси-4- трифторацетамидо- a -L-эритро-гексоксипиранозид (Xe). Смесь метил-2,3, 6-тридезокси- a -L-трео-гексоксипиранозид-4- улозы (VIII) (2 г, 13, 8 ммолей), ацетата аммония (10,7 г, 139 ммолей) и цианоборогидрида натрия (2,84 г, 45 ммолей) в метаноле (480 мл) перемешивают при температуре окружающей среды в течение 48 ч, а затем обрабатывают 5-ным водным раствором уксусной кислоты (240 мл). После 10 мин дополнительного перемешивания полученный раствор промывают хлороформом (400 мл). Водную фазу отделяют, доводят до pH 8 добавлением бикарбоната натрия и экстрагируют 7 раз с 300 мл хлороформа. Экстракты высушивают над сульфитом натрия, фильтруют и выпаривают под вакуумом, чтобы получить сырой продукт реакции, который после обработки ангидридом трифторуксусной кислоты (5,8 мл), как это описано в примере 4, дает возможность получить два соединения Xa (0,28 г) и Xe (1,5 г).
Пример 6. Метил-2,3,4, 6-тетрадезокси-4-аллилоксикарбоксиамид- a -L-трео- гексоксипиранозид (XIa) и метил-2,3,4,6-тетрадезокси-4- аллилоксикарбоксиамид- a -L-эритро-гексоксипиранозид (XIe). Восстановленную смесь (0,58 г, 4 ммоля), полученную так, как описано в примере 4 и растворенную в тетрагидрофуране (6 мл), обрабатывают при 0oC пиридином (0,55 мл) и аллилхлоркарбонатом (0,82 г, 6,8 ммолей), растворенным в тетрагидрофуране (2 мл), в указанном порядке. Реакционную смесь перемешивают в течение 6 ч при температуре окружающей среды, после чего добавляют воду и смесь экстрагируют хлористым метиленом (5 раз, каждый раз с 10 мл). Экстракты высушивают над сульфатом натрия, фильтруют и выпаривают под вакуумом, чтобы получить остаток, который очищают флеш-хроматографией. Элюирование 9/1 смесью петролейного эфира/ ацетона дает последовательно названное выше соединение XIa (0,366 г, выход 40).
ЯМР (200 МГц, CDCl3, d ): 1,08 (дублет, J 65 Гц, 3H); 1,2-2,1 (мультиплет, 4H, ); 3,31 (синглет, 3H); 3,4-3,5 (мультиплет, 1H); 3,63 (дублет, 1H, J 10,73); 4,02 (триплет, дублет, 1H, Jn 6,5 Гц, Jn 1,6 Гц); 4,54 (дублет, 2H, J 5,6 Гц); 5,18 (два дублета, 1H); 5,27 (два дублета, 1H); 5,9 (мультиплет, 1H).
Соединение XIe (0,395 г выход 43).
ЯМР (200 МГц, CDCl3, d ): 1,08 (дублет, J 6,5 Гц, 3H); 1,4-1,9 (мультиплет, 4H); 3,31 (синглет, 3H); 3,4-3,5 (мультиплет, 1H); 3,52 (квартет, дублет, 1H, J H-Me 6,5 Гц, JH-H 9,9 Гц); 4,46 (дублет, 1H, J 10,7 Гц); 4,54 (дублет, 2H, J 5,6 гц); 4,63 (дублет, 1H, J < 1 Гц); 5,18 (два дублета, 1H); 5,27 (два дублета, 1H); 5,9 (мультиплет, 1H).
Пример 7. 2,3,4, 6-тетрадезокси-4-трифторацетамидо- a -L-эритро- гексокси-пиранозилпара-нитробензоат (IIIe, R2 NHCOCF3, X пара NO2 С6H4 COO). Смесь 150 мг метил-2,3,4,6-тетрадезокси-4-трифтор-ацетамидо- a L - эритро-гексоксипиранозида (Xe), полученного, как это описано в примере 5, и 20-ного водного раствора уксусной кислоты (15 мл) нагревают до 80oC в течение 3 ч. Реакционную смесь выпаривают под вакуумом, чтобы получить 2,3,4, 6-тетрадезокси-4-трифторацетамидо- L-эритро-гексоксипиранозид (XIIe) в форме белого твердого вещества, которое растворяют в 2,5 мл пиридина и обрабатывают при 0oC со 177 мг пара-нитробензоилхлорида. Реакционную смесь перемешивают в течение 2 ч при температуре 0oC, добавляют воду и смесь экстрагируют хлороформом (5 раз, каждый раз с 3 мл). Экстракты промывают 3 N серной кислотой, водой и 8-ным раствором бикарбоната натрия. Органическую фазу высушивают над сульфатом натрия, фильтруют и выпаривают под вакуумом, чтобы получить соединение IIIe (200 мг, выход 85) в виде вязкой желтой жидкости.
ЯМР (60 МГц, CDCl3, d ): 1,25 (дублет, J 6 Гц, 3H); 1,9-2,2 (мультиплет, 4H, ); 3,6-4,0 (мультиплет, 2H); 6,0 (синглет, 1H); 6,35 (мультиплет, 1H); 8,2 (синглет, 4H).
Пример 8. 2,3,4,6-тетрадезокси-4-трифторацетамидо- a L -трео-гексоксипиразонил-пара-нитробензоат (IIIa, R2 NHCOCF3, X пара NO2-С6H4-COO). Исходя из метил-2,3,4,6-тетрадезокси-4-трифторацетамидо- a -L треогексоксипиранозида (Xa), полученного, как это описано в примере 4, получают указанное соединение по методике, описанной в примере 7. TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей: гексан/этилацетат (3:1), дает Rf 0,43.
Пример 9. 2,3,4,6-тетрадезокси-4-аллилоксикарбоксиамидо- a -L-трео-гексоксипиразонил-пара-нитробензоат (IIIa, R2 NHCOOCH2CH=CH2, X пара NO2-С6 H4-COO). Смесь 200 мг метил-2,3,4,6-тетрадезокси-4-аллилоксикарбоксиамид- a - L-трео-гексоксипиранозида (XIa), полученного, как это описано в примере 6, и 20 мл 20-ного водного раствора уксусной кислоты нагревают до 80oC в течение 2 ч. Реакционную смесь выпаривают под вакуумом, чтобы получить 2,3,4,6-тетрадезокси-4- аллилокси-карбоксиамидо- a -L-трео-гексоксипиранозу (XIIIa) в виде вязкой жидкости, которую растворяют в 4 мл пиридина и обрабатывают со 120 мг пара-нитробензоилхлорида. Реакционную смесь перемешивают в течение 2 ч при 0oC, добавляют воду и смесь экстрагируют хлороформом (5 раз, каждый раз с 4 мл). Экстракты промывают 3 N серной кислотой, водой и 8-ным раствором бикарбоната натрия. Органическую фазу высушивают над сульфатом натрия, фильтруют и выпаривают под вакуумом, чтобы получить соединение IIIa (260 мг, выход 81). TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей: хлористый метилен/ацетон (95:5), дает Rf 0,40.
Пример 10. 2,3,4, 6-тетрадезокси-4-аллилоксикарбоксиамидо- a - L-эритро-гексоксипиранозил-пара-нитробензоат (IIIe, R2 NHCOOCH2 CH=CH2, X пара- NO2-С6H4 -COO). Исходя из метил-2,3,4,6-тетрадезокси-4-аллилокси-карбоксиамид- a -L-эритро-гексоксипиранозида (XIe), полученного, как это описано в примере 6, получают указанное соединение по методике, описанной в примере 9. TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей: гексан/этилацетат (95:5), дает Rf 0,31.
Пример 11. 2,3,4, 6-тетрадезокси-4-трифторацетамидо- a -L- эритро-гексокси-пиранозилхлорид (IIIe, R2 NHCOCF3, X Cl). Раствор 210 мг 2,3,4, 6-тетрадезокси-4-трифторацетамидо- a -L-эритро-гексокси-пиранозил-пара-нитробензоата (IIIe, R2 NHCOCF3, X пара-NO2-С6H4-COO), полученного в соответствии с примером 7, в 5 мл безводного хлористого метилена насыщают при 0oC безводным хлористым водородом. После стояния в течение ночи при 0oC выпадающую в осадок п-нитробензойную кислоту отфильтровывают и раствор выпаривают под вакуумом, чтобы получить соединение, которое используют для реакции сочетания без дополнительной очистки.
Пример 12. 2, 3,4, 6-тетрадезокси-4-трифторацетамидо-альфа-L- трео-гексокси-пиранозилхлорид (IIIa, R2 NHCOOF3, X Cl). Исходя из 2,3,4, 6-тетрадезокси-4-трифторацетамидо- L-трео-гексоксипиранозил-пара-нитробензоата (IIIa, R2 NHCOCF3, X пара-NO2-С6H4-COO), полученного, как это описано в примере 8, получают указанное соединение, следуя методике, описанной в примере 11.
Пример 13. 2,3,4,6-тетрадезокси-4-аллилоксикарбоксиамидо-L-трет-гексокси-пиранозилхлорид (IIIa, R2 NHCOOCH2CH= CH2, X Cl). Раствор 270 мг 2,3,4,6-тетрадезокси-4-аллилоксикарбоксиамидо-L-трео-гексоксипиранозил-пара-нитробензоата (IIIa, R2 NHCOOCH2-CH=CH2, X пара-NO2-С6H4-COO), полученного в соответствии с примером 9, в 6 мл безводного хлористого метилена насыщают безводным хлористым водородом. После стояния в течение ночи при 0oC выпадающую в осадок п-нитробензойную кислоту отфильтровывают и раствор выпаривают под вакуумом, чтобы получить соединение, которое используют для реакции сочетания без дополнительной очистки.
Пример 14. 2,3,4,6-тетрадезокси-4-аллилоксикарбоксиамидо-L -эритро- гексоксипиранозилхлорид (IIIe, R2 NHCOOCH2CH=CH2, X Cl). Исходя из 2,3,4,6-тетрадезокси-4-аллилоксикарбоксиамидо-L-эритрогексоксипиранозил-паранитробензоата (IIIe, R2 NHCOOCH2-CH=CH2, X пара-NO2-С6H4-COO), полученного в соответствии с примером 10, получают указанное соединение, следуя методике, описанной в примере 13.
Пример 15. 4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-эпи-амино-даунорубицина хлоргидрат (I, R H, R1 H). Смесь 120 мг 9-ацетил-8(8H)-фтор-10-гидро-6,7(7H), 9,11-тетрагидрокси-5, 12-нафтацендиона (II, R1 H), полученного, как это описано в примере 3, 240 мг 2,3,4,6-тетрадезокси-4-трифторацетамидо-а-1-эритро-гексопиранозил-пара-нитробензоата (IIIe, R2 NHCOCF3, X NO2-С6H4-COO), полученного, как это описано в примере 7, в 60 мл сухого хлористого метилена и 20 мл диэтилового эфира и в присутствии молекулярных сит (4 ангстрем) при 0oC обрабатывают с 0,225 мл трифторметансульфонатом триметилсилила. Реакционную смесь перемешивают в течение 15 мин при 0oC и гасят с помощью 266 мг 1, 8-бис-(диметиламино)-нафталина, после перемешивая в течение 10 мин. Смесь фильтруют, чтобы удалить получающийся белый осадок, и фильтрат выпаривают под вакуумом, чтобы получить остаток, который очищают флеш-хроматографией. Элюирование смесью хлористого метилена/ацетона (95:5) дает 4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-эпи-трифторацетамидодаунорубицин (IV, R1 H, R2 NHCOCF3) (108 мг, выход 60).
H-ЯМР (200 МГц, CDCl3, d ): 1,15 (дублет, J 8 Гц, 3H); 1,7-1,9 (мультиплет, 4H); 2,45 (синглет, 3H); 3,19 (дублет, J 19 Гц, 1H); 3, 33 (два дублета, JH-F 3,6, J 19 Гц, 1H); 3,6-3,8 (мультиплет, 2H); 3,98 (синглет, 1H); 4,91 (два дублета, JH-F 50 Гц, 1H); 5,17 (два дублета, JH-F 12 Гц, 1H); 5,26 (мультиплет, 1H); 6,21 (уширенный дублет, 1H); 7,86 (мультиплет, 2H); 8,4 (мультиплет, 2H); 13,35 (синглет, 1H); 13,36 (синглет, 1H). Суспензию 78 мг N-трифторацетильного производного в 15, 6 мл 0,2 М раствора гидроокиси бария перемешивают в течение 2 ч в токе азота при комнатной температуре. Реакционную смесь нейтрализуют двуокисью углерода, а затем экстрагируют хлороформом; объединенные экстракты высушивают над безводным сульфатом натрия и концентрируют до незначительного объема. Хлористый водород и диэтиловый эфир добавляют, чтобы получить названное выше соединение (I, R H, R1 H) (56 мг, выход 80). TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей: хлороформ/ метанол/вода (60:30:10), дает Rf 0,45. Действуя аналогичным образом, могут быть получены следующие соединения.
Хлоргидрат 4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-амино-даунорубицина (I, R H, R1 H).
TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей: хлороформ/метанол/вода (65:30:10), дает Rf 0,47.
H-ЯМР соответствующего
трифторацетамидо-производного сообщается:
H-ЯМР (200 МГц, CDCl3, между прочим d ): 3,17 (дублет, J 19 Гц, 1H, H -10 акс); 3,31 (два дублета, JH-F 3,6, J 19 Гц, 1H, H -10
экв); 3,8-4,3 (мультиплет, 2H, H-4', H-5');
4,89 (два дублета, JH-F 50 гц, 1H, H-8); 5,25 (два дублета, JH-F 12 Гц, 1H, H-7); 5,46 (мультиплет, 1H, H-1').
Хлоргидрат 8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодаунорубицина (I, R H, R1 OCH3). TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей хлороформ/метанол/вода (60:30:10), дает Rf 0,41.
H-ЯМР соответствующего трифторацетамидо-производного сообщается: H-ЯМР (200 МГц, CDCl3, между прочим d ): 3,17 (дублет, J 19 Гц, H-10 акс); 3,5 (два дублета, JH-F 3,6 Гц, J 19 Гц, 1H, H-10 экв); 4,10 (синглет, 3H, OCH3).
Хлоргидрат 8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодаунорубицина (I, R H, R1 OCH3). TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей хлороформ/метанол/вода (60:30:10), дает Rf 0,42.
H-ЯМР соответствующего трифторацетамидо-производного сообщается: H-ЯМР (200 МГц, CDCl3, между прочим d ): 3,15 (дублет, J 19 Гц, 1H, H-10 акс); 3,49 (два дублета, JH-F 3,6 Гц, J 19 Гц, 1H, H-10 экв); 4,08 (синглет, 3H, OCH3).
Пример 16. Хлоргидрат 4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-аминодаунорубицина (I, R H, R1 H). Смесь 200 мг 9-ацетил-8(8H)-фтор-10-гидро-6,7(7H), 9,11-тетрагидрокси-5,12-нафтацендиона (II, R1 H), полученного, как это описано в примере 3, 157 мг 2,3,4,6-тетрадезокси-4- аллилоксикарбоксиамидо-L-трео- гексопиранозилхлорида (IIIa, R NHCOOCH2CH=CH2, X Cl), полученного, как это описано в примере 13, в 60 мл сухого хлористого метилена и в присутствии молекулярных сит (4

H-ЯМР (200 МГц, CDCl3, δ ): 1,08 (дублет, J 6,5 Гц, 3H); 1,2-2,1 (мультиплет, 4H); 2,45 (синглет, 3H); 3,18 (два дублета, J 19 Гц, 1H); 3,31 (два дублета, JH-F 3,6 Гц, J 19 Гц, 1H); 3,63 (уширенный дублет, 1H); 3,38-3,52 (мультиплет, 1H); 4,02 (мультиплет, 1H); 4,54 (дублет, 2H); 4,89 (два дублета, JH-F 50 Гц, 1H); 5,15 (два дублета, JH-F 12 Гц, 1H); 5,21-5,27 (мультиплет, 3H); 5,9 (мультиплет, 1H).
Смесь 102 мг N-аллилоксикарбонильного производного, 4,4 мг тетракис-(трифенилфосфин)-палладия, 4,4 мг трифенилфосфина и 44,5 мг 2-этилгексановой кислоты в 15 мл безводного хлористого метилена перемешивают при 25oC в течение 21 ч. Добавляют хлористый метилен, органическую фазу промывают 8-ным водным раствором бикарбоната натрия, высушивают над безводным сульфатом натрия и выпаривают под вакуумом. Остаток смешивают с водной соляной кислотой (pH 3), а затем экстрагируют хлористым метиленом, и объединенные экстракты высушивают над безводным сульфатом натрия и концентрируют до незначительного объема. Добавляют хлористый водород и диэтиловый эфир, чтобы получить названное выше соединение (I, R H, R1 H) (76 мг, выход 81). TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей хлороформ/метанол/вода (65:30:10), дает Rf 0, 48.
Действуя аналогичным образом, могут быть получены следующие соединения.
Хлоргидрат 4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодаунорубицина (I, R H,
R1 H)
H-ЯМР соответствующего аллилоксикарбоксиамидо-производного сообщается: H-ЯМР (200 МГц, CDCl3, между прочим d ): 3,19 (дублет, J 19 Гц, 1H, H -10 акс); 3,33 (два
дублета, JH-F 3,6 Гц, J 19 Гц, 1H, H -10 экв); 4,91 (два дублета, JH-F 50 Гц, 1H, H-8); 5,14 (два дублета, JH-F 12 Гц, 1H, H-7); 5,18-5,27 (мультиплет, 3H, H-1' +
-CH=CH2).
Хлоргидрат 8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодаунорубицина (I, R H, R1 OCH3).
H-ЯМР соответствующего аллилоксикарбоксиамидо-производного сообщается: H-ЯМР (200 МГц, CDCl3, между прочим d ): 3,19 (дублет, J 19 Гц, 1H, H-10 акс); 3,48 (два дублета, JH-F 3,6 Гц, J 19 Гц, 1H, H-10 экв); 4,10 (синглет, 3H, OCH3).
Хлоргидрат 8-фтор-3'-дезамино-4'-дезокси-4'- аминодаунорубицина (I, R H, R1 OCH3).
H-ЯМР соответствующего аллилоксикарбоксиамидо-производного сообщается: H-ЯМР (200 МГц, CDCl3, между прочим d ): 3,17 (дублет, J 19 Гц, 1H, H-10 акс); 3,52 (два дублета, JH-F 3,6 Гц, J 19 Гц, 1H, H-10 экв); 4,08 (синглет, 3H, OCH3).
Пример 17. Хлоргидрат 4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодоксорубицина (I, R OH, R1 H). Согласно методике, описанной в пат. США N 4 122 076, раствор 100 мг хлоргидрата 4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-эци-аминодаунорубицина (IV, R1 H, R2 NH2), полученного, как это описано, в 2,5 мл безводного метанола и 5 мл диоксана обрабатывают при перемешивании раствором 490 мг брома в 5 мл хлористого метилена, чтобы получить сырой 14-бром-4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-эпи- даунорубицин, который выпадает в осадок добавлением диэтилового эфира. Сырой продукт реакции растворяют в смеси 5 мл ацетона и 1 мл воды и обрабатывают со 150 мг формиата натрия. Реакционную смесь перемешивают при комнатной температуре в течение 36 ч, затем добавляют воду и экстрагируют хлористым метиленом. Водную фазу смешивают с 3 мл 8-ного раствора бикарбоната натрия и экстрагируют хлористым метиленом. Объединенные органические экстракты высушивают над безводным сульфатом натрия, концентрируют под вакуумом до незначительного объема и обрабатывают метанольным раствором хлористого водорода (pH 3,5) и диэтиловым эфиром, чтобы получить названное выше соединение (I, R1 H, R OH) (77 мг, выход 75). TCX на пластинах Кизельгеля (Мерк F254), используя систему растворителей хлороформ/метанол/вода (65:30:10), дает Rf 0,31.
Действуя аналогичным образом и исходя из соответствующих производных даунорубицина (I, R H, R1 H или OCH3), могут быть получены следующие соединения.
Хлоргидрат 4-деметокси-8-фтор-3'-дезамино-4'-дезокси-4'-амино-доксорубицина (I, R OH, R1 H). TCX на Кизельгеле (Мерк F254), используя систему растворителей хлороформ/метанол/вода (65:30:10), дает Rf 0,34.
Хлоргидрат 8-фтор-3'-дезамино-4'-дезокси-4'-аминодоксорубицина (I, R OH, R1 OCH3). TCX на Кизельгеле (Мерк F254), используя систему растворителей хлороформ/метанол/вода (65:30:10), дает Rf 0,29.
Хлоргидрат 8-фтор-3'-дезамино-4'-дезокси-4'-эпи-аминодоксорубицина (I, R OH, R1 OCH3). TCX на Кизельгеле (Мерк F254), используя систему растворителей хлороформ/метанол/вода (65:30:10), дает Rf 0,27.
Чтобы получить фармацевтическую композицию, пригодную для терапевтического использования, соответствующее количество действующей основы может быть, например, растворено в бидистиллированной воде для использований при инъекциях до концентрации 1-10 мг/мл. Полученный таким образом раствор может быть затем лиофилизирован после добавления соответствующего носителя (такого как маннит или лактоза), или вместе, или без подходящих консервирующих средств, а затем разделен в стерильные сосуды (ампулы), готовые для последующего использования.
Приложение 2.
Биологическая активность. Биологическая активность соединений.
1a: 4-деметокси-8-фтор-3'-деамино-4'-деокси-4'-эпи-амино-даунорубицина (I, R R1 H),
1b:
8-фтор-3'-деамино-4'-деокси-4'-эпи-аминодаунорубицина
(I, R H, R1 OCH3),
1c: 4-деметокси-8-фтор-3'-деамино-4'-деокси-4'-эпи-амино-доксорубицина (I, R OH, R1
H)
была испытана в сравнении с доксорубицином
по отношению к мышиной P 388 лейкемии и различным опухолям человека.
Активность ин витро (табл. 1).
Соединения 1a и 1c, как оказалось, являются более циотоксичными, чем исходное соединение, против как P388, так и POYD раковых клеток легких, чувствительных к DXR.
Такая повышенная циотоксичность, однако, является значительно более очевидной, если мы рассмотрим активность этих соединений против соответствующих DXR-устойчивых опухолей (P388/DXR и POYD/DXR).
Активность ин виво (табл. 2-4).
Против лейкемии P388 и раковых клеток яичников оба соединения, 1a и 1c, показывают противоопухолевую активность, определенно более высокую, чем противоопухолевая активность, DXR, увеличивая время выживания мышей и степень ингибирования роста опухоли в меньших дозах, чем дозы, в которых применяется исходное соединение.
На аденокарциноме легких H460, особенно устойчивой к действию DXR, 1a и 1c показывают по меньшей мере тот же уровень активности, что исходное соединение, а соединение 1b оказалось неактивным.
Реферат
Сущность изобретения: новые 8-фторпроизводные
антрациклингликозиды общей формулы I:

где R - H, OH; R1 - H, OH, MeO; ≈ NH2 показывает, что аминозаместитель может находиться в аксиальной или экваториальной конфигурации, или их фармацевтически приемлемые соли присоединения кислот. Соединения получают путем конденсации соответствующего 8-фторантрациклинона с соединением формулы IIIa или IIIe

где X-хлор или n-нитробензоилоксигруппа; R2-трифторацетамидная или аллилоксикарбоксиамидная группа с последующим удалением защитных групп и в случае необходимости трансформированием радикала R - водород в R - гидроксигруппа через соответствующие 14-бромпроизводное; промежуточные соединения IIIa и IIIe и способ их получения, заключающийся во взаимодействии метил-2,3,6-тридезокси- α -L-глицерогексоксипиранозид-4-улозы с гидроксиламином, с последующим восстановлением полученной смеси син- и антиоксимов, защиты образующейся аминогруппы и разделением соответствующих эпимеров, с последующим последовательным превращением последних в соответствующие 1-гидроксипроизводные и целевой продукт. Промежуточные соединения формул II и VII

где R - H, OCH3, OH; и фармацевтическая композиция. Соединения обладают противоопухолевыми свойствами. 9 с. и 7 з.п. ф-лы, 4 табл.
Формула

где R водород или гидроксигруппа;
R1 водород, гидрокси или метоксигруппа и ~ NH2 показывает, что аминозаместитель может находиться в аксиальной или экваториальной конфигурации,
или их фармацевтически приемлемые соли присоединения кислот.

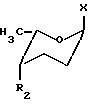
с соединением формулы IIIа
или IIIe

где Х хлор или паранитробензоилоксигруппа;
R2 трифторацетамидная или аллилоксикарбоксиамидная группа,
с получением N-защищенного гликозида общей формулы IV

где R1 и R2 имеют приведенные выше значения, R2~ показывает, что заместитель R2 находится в аксиальной или экваториальной конфигурации, из которого удаляют N-защитную трифторацетильную или предпочтительно аллилоксикарбонильную группу, получая целевой продукт, где R водород, R1 как указано выше, и в случае необходимости превращает целевой продукт в его фармацевтически приемлемую соль, или в случае необходимости бромируют указанный гликозид формулы I, где R водород, или его фармацевтически приемлемую соль с последующим гидролизом соответствующего 14-бромпроизводного с получением целевого продукта формулы I, где R - гидроксигруппа, который в случае необходимости также превращают в фармацевтически приемлемую соль.

где R1 водород, ОСН3, ОН.

где R1 водород, ОСН3, ОН.

где Х хлор или п-нитробензоилоксирадикал;
R2 трифторацетамидо- или аллилоксикарбоксиамидорадикал.

где Х хлор или п-нитробензоилокси радикал;
R2 трифторацетамидо- или аллилоксикарбоксиамидорадикал.
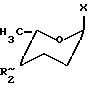
где Х хлор или п-нитробензоилоксирадикал;
R2 трифторацетамидо- или аллилоксикарбоксиамидорадикал,
R2~ означает, что данный заместитель может находиться в аксиальной IIIа или экваториальной конфигурации IIIе, отличающийся тем, что метил-2,3,6-тридезокси-α-L-глицерогексоксипиранозид-4-улозу формулы VIII
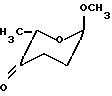
предвергают взаимодействию с гидроксилаином или его солью присоединения кислоты, полученную смесь син- и антиоксимов формулы IX

восстанавливают, затем после защиты образующейся аминогруппы с помощью трифторацетильной группы осуществляют разделение 4-N-трифторацетилированных эпимеров формулы Ха

и Хе
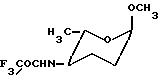
или предпочтительно после защиты образующейся аминогруппы аллилоксикарбонильной группой осуществляют разделение 4-N-аллилоксикарбонилированных эпимеров формулы XIа

и XIе

с последующим последовательным превращением каждого эпимера Ха и Хе или XIа и XIe в соответствующие I-гидроксипроизводные формулы XIIa

и XIIe

или XIIIа

и XIIIe

и целевой продукт, где Х хлор или предпочтительно пара-нитробензоилоксирадикал, а R указанные выше значения.
Комментарии