Производное дикетен-имина в качестве промежуточного продукта для синтеза ранитидина и способ его получения - RU2042671C1
Код документа: RU2042671C1
Описание
Изобретение касается нового производного дикетен-имина формулы (I)






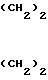





Новое дикетен-иминовое соединение формулы (I) до сих пор не было известно в литературе. Соединение формулы (I) является ценным промежуточным продуктом синтеза 1-{2-[(5-диметиламинометил-2-фурил)метил-тио]этил}-амино-1- метиламино-2-нитроэтилена (родовое название: ранитидин), известного антагониста Н-2 рецептора.
Целью
изобретения является
создание нового промежуточного соединения для синтеза вышеупомянутого Н-2 рецептор-блокирующего ранитидина, который может быть получен одностадийным технологическим процессом, что
особенно удобно в
промышленном масштабе, и который, с другой стороны, может привести к целевому антагонисту Н-2 рецептора в одну простую стадию, а именно взаимодействием производного фурфурилового
спирта формулы
(II)



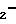
HS-CH2-CH2-NH2˙HCl (III) а затем с 1-нитро-2,2-ди(метилтио)этеном формулы (IV):

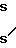

Новое соединение формулы (I) может быть легко превращенo в ранитидин в одну стадию с выходом около 100%
Способ получения соединения формулы (I), предусматривающего реакцию производного фурфурилового спирта формулы (II) с гидрохлоридом цистеамина формулы (III), затем с неорганическим основанием и в
последующем с нитросоединением формулы (IV) и после удаления непрореагировавшего нитросоединения формулы (IV) из реакционной смеси, добавление соли цинка к остатку в С1-4 алканольной среде
в присутствии основания.
Согласно предпочтительному варианту способа реакцию с солью цинка проводят в присутствии органического основания, например, третичного амина; в качестве третичного амина используют триэтиламин.
Преимущество способа по изобретению по сравнению с известным аналогом [1] заключается в том, что ценное новое промежуточное соединение формулы (I) может быть получено с использованием простой технологической процедуры, которая прекрасно подходит для ее применения в промышленном масштабе.
Структура нового соединения формулы (I) была определена посредством УФ, ИР и ЯМР-спектроскопии.
П р и м е р 1. 87 г (0,56 моль) 4-диметиламинометилфурфуриловый спирт формулы (II) и 51,8 г (0,46 моль) гидрохлорида цистеамина формулы (III) порциями добавляют к 36 г концентрированной водной хлористоводородной кислоты, и затем реакционную смесь перемешивают при комнатной температуре в течение 48 ч. После добавления 92 мл этанола смесь нагревают до температуры кипения, а затем снова охлаждают до комнатной температуры (25оС) и при этой же температуре добавляют 132 г 20%-ного водного раствора гидроксида натрия.
Полученный маслянистый коричневый продукт экстрагируют в 270 мл толуола. После разделения водную фазу промывают 80 мл толуола, комбинированный толуольный раствор сушат над 15 г безводного сульфата натрия и затем очищают 9 г смеси, содержащей целит и активированный уголь в соотношении 1:1.
После фильтрования добавляют 61,8 г (0,374 моль) 1-нитро-2, 2(метилтио)этена формулы (IV) (примерно половина которого может быть выделена из реакционной смеси в готовом для применения виде), реакционную смесь нагревают c обратным холодильником в течение 1 ч, а затем охлаждают до комнатной температуры и экстрагируют дважды с 150 мл и один раз 160 мл 0,1 Н хлористоводородной кислоты. Полное удаление непрореагировавшего нитросоединения экстракцией проверяют тонкослойной хроматографией (ТЛС). Когда реакция не завершена, то осуществляют дальнейшую экстракцию с дополнительными 20 мл 0,1 Н раствора хлористоводородной кислоты. После установления уровня рН раствора хлористоводородной кислоты в пределах между 7,5 и 8 посредством добавления 0,2 Н водного раствора гидроксида натрия может быть выделено 23,5 г 1-нитро-2, 2-ди(метилтио)этена формулы (IV). Толуольный раствор экстрагируют 400 мл метанола. Затем 200 г хлорида цинка растворяют в 2000 мл метанола и вышеуказанный метанольный экстракт в количестве 400 мл добавляют по каплям к этому раствору при комнатной температуре. После добавления 100 г триэтиламина реакционную смесь перемешивают при 40оС в течение 18 ч.
В последующем смесь очищают 25 г активированного угля и 25 г целита в течение 30 мин, затем фильтруют и полученный раствор выпаривают при 20оС до одной третьей его начального объема в атмосфере азота при пониженном давлении величиной 10-4 МПа и затем охлаждают до -15оС. После отфильтровывания твердого осадка маслянистый остаток выдерживают при пониженном давлении 100 Па до тех пор, пока его вес не перестанет изменяться в течение 2 ч. Таким путем получают 50 г продукта (выход в расчете на нитросоединение составляет 79,5%) с температурой плавления от -39 до -35оС (свыше 176оС наступает декомпозиция).
УФ ( >макс) 331,6 нм;
ИР 1370 и 1530 см-1 (NO2 группа) 1630 см-1 ( >C


1 H-ЯМР ( δ ррм): 2,25 (сингль, 12Н, две группы

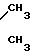
2, 75 (триплет, 4Н, две группы -S-СН2-СН
3,53 (сингль, 4Н, две группы -СН2-S-)
3,60 (м, 4Н, две группы -S-СН2 -СН2-N
3,80 (сингль, 4Н, две группы -N-СH2-)
6,20 (сингль, 4Н, два олефина)
6,80 (сингль, 4Н, >С-СН-NO2)
П р и м е р 2. 8,5 г (0,015 моль) дикетен-иминового производного формулы (П) растворяют в 30 мл воды и в течение 15 мин при комнатной температуре добавляют 41 г (0,5 моль) 40%-ного водного раствора метиламина. После перемешивания в течение 1 ч смеси осветляют 0,5 г целита и 0,5 г активированного угля при комнатной температуре в течение 15 мин, а затем фильтруют. Фильтрат экстрагируют с 40 мл и затем дважды с 20 мл хлороформа. После сушки комбинированного экстракта над безводным сульфатом натрия высушивающий агент отфильтровывается, растворитель выпаривается и маслянистый остаток перекристаллизуют из 35 мл этилацетата с получением 9 г (94%) 1-{2-[(5-диметиламинометил-2-фурил)метилтио]-этил}-амино-1-метиламино -2-нитроэтилена c температурой плавления 71-73оС. Этот продукт не содержит никаких загрязнений, которые могли бы быть идентифицированы тонкослойной хроматографией (ТДС).
Реферат
Использование: в производстве ранитидина известного антагониста H рецептора в качестве промежуточного продукта. Сущность изобретения: продукт производное дикетон-имина: 1-{ 2-[ (5-N, N-диметиламинометал-2-фурил) -метилтио] -этил} -2-{ 1-[ (5-N, N-диметиламинометал-2-фурил) -метилтио]-1-этиламино} -3-нитро-4-нитрометилен-2-азетина. Tпл (-39) (-35)°С. Реагент 1: 5-(N, N-диметиламинометил)-фурфуриловый спирт. Реагент 2: гидрохлорид цистеамина. Условия процесса: в присутствии кислоты при температуре от комнатной до температуры кипения реакционной среды. Реагент 3: неорганическое основание с последующей обработкой реакционной смеси избытком 1-нитро-2,2-ди(метилтио)этеном при кипячении с удалением непрореагированного 1-нитро-2,2-ди(метилтио)-этена и добавлением соли цинка к остатку в среде C1-C4 -алканола в присутствии основания. Предлагаемое соединение позволяет получить ранитидин в одну стадию с выходом 100% 2 с. и 3 з.п. ф-лы.
Формула

в качестве промежуточного продукта для синтеза ранитидина.
Комментарии