Фосфонооксиметиловые эфиры производных таксана, фармацевтическая композиция, способы ингибирования - RU2128661C1
Код документа: RU2128661C1
Чертежи





























Описание
Настоящая заявка является частичным продолжением нашей одновременно рассматриваемой заявки рег. N США 08/154840, поданной 24 ноября 1993 г., являющейся частичным продолжением заявки рег. N США 08/108015, поданной 17 августа 1993, и в свою очередь, являющейся частичным продолжением заявки с рег. N США 07/996455, поданной 24 декабря 1992, и ныне абандонированной. Заявка с рег. N 08/154840, во всей своей полноте, вводится в настоящее описание посредством ссылки.
Настоящее изобретение относится к противоопухолевым соединениям. В частности, настоящее изобретение относится к новым производным таксана; к фармацевтическим композициям, содержащим указанные производные; а также к их использованию в качестве противоопухолевых средств.
Таксоль® (паклитаксел) представляет собой натуральный продукт, экстрагированный из коры деревьев тихоокеанского тиса (Taxus brevifolia). Было установлено, что этот продукт обладает прекрасной противоопухолевой активностью в in vivo - моделях, а в более поздних исследованиях было выявлено, что соединение обладает уникальным механизмом воздействия, который заключается в аномальной полимеризации тубулина и прекращения митоза. Недавно, это средство было апробировано для лечения рака яичника; а его испытания на активность против рака молочной железы, толстой кишки и легких дали обещающие результаты. Результаты клинических исследований паклитаксела представлены в работе Rowinsky и Donehower "The Clinical Pharmacology and Use of Antimicrotubule Agents in Cancer Chemotherapentics", Pharmac, Ther., 52: 35-84, 1991.
Недавно, было также обнаружено, что полусинтетический аналог паклитаксела, названный Таксотером®, обладает хорошей противоопухолевой активностью в in vivo-моделях. В настоящее время, Таксотер® также проходит испытания в клиниках Европы и США. Ниже представлены структуры паклитаксела и Таксотера, в которых дана стандартная система нумерации в молекуле паклитаксела.

Одним из недостатков паклитаксела является его очень ограниченная водорастворимость, а потому для изготовления лекарственных средств, содержащих паклитаксел, необходимо использовать безводные фармацевтические носители. Таким носителем является часто используемый в таких случаях Cremophor Eh., который сам по себе обладает нежелательными побочными эффектами при введении человеку. В соответствии с этим, был проведен ряд исследований в целях получения водорастворимых производных паклитаксела, которые раскрываются в следующих работах.
(a)
Haugwitz и др., патент США N
4942184;
(b) Kingston и др., патент США N 5059699;
(c) Stella и др., патент США N 4960790;
(d) Заявка на Европатент 0558959 AI, опубликованная
8 сентября 1993 г.
(e) Vyas и др. , Bioorganic 2 Medicinal Chemistry Letters, 1993, 3: 1357-1360; и
(f) Nicolaou и др., Nature, 1993, 364: 464-466.
Соединения настоящего изобретения представляют собой фосфонооксиметиловые простые эфиры производных таксана и их фармацевтически приемлемые соли. Водорастворимость этих солей облегчает изготовление фармацевтических композиций.
Краткое описание изобретения
Настоящее изобретение относится к производным таксана, имеющим формулу (A):
T - [OCH2(OCH2)mOP(O)(OH)2]n (А),
где T представляет собой таксановую часть, несущую на C13-атоме углерода замещенную 3-амино-2-гидроксипропаноилокси-группу;
n = 1,2 или 3;
m = 0
или целому числу от 1 до 6, включительно;
или их фармацевтически приемлемой соли.
Другой вариант настоящего изобретения относится к
производным таксана, имеющим формулу
(B):
T' - [OCH2(OCH2)mSCH3]n (В),
где T представляет собой T, в котором нереактивные
гидроксигруппы были блокированы;
m и n являются такими, как они были определены в формуле (A).
В другом своем варианте, настоящее изобретение относится к промежуточным
соединениям, имеющим формулу (C):
T'-[OCH2(OCH2)mOP(O)(ORy)2]n
где T', m и n являются такими, как они были
определены в формуле (A);
Ry
является фосфонозащитной группой.
В следующем варианте своего осуществления, настоящее изобретение относится к соединениям формулы
(D):
13-OH-txn-[OCH2(OCH2)mSCH3]n,
где m и n определены выше;
txn является таксановой частью;
либо к их
C13-алкоксидам металла.
В еще одном своем варианте, настоящее изобретение относится к способу ингибирования опухолевого роста у млекопитающих, заключающемуся в том, что указанному млекопитающему-хозяину вводят эффективное опухоль-ингибирующее количество соединения формулы (A).
В другом своем варианте, настоящее изобретение относится к способу
ингибирования опухолевого роста у млекопитающих,
заключающийся в том, что указанному млекопитающему вводят эффективное опухоль-ингибирующее количество соединения формулы (B'):

где R1b' является гидроксигруппой, -OCO(O)Rx или -ОC(O)ORx;
R3b' является водородом, гидроксигруппой, -OC(O)ORx, C1-6-алкилоксигруппой, или -OC(O)Rx;
один из R6b' или R7b' является водородом, а другой гидроксигруппой или C1-6 -алканоилоксигруппой; либо R6b' и R7b' вместе образуют оксогруппу;
R4 и R5 независимо представляют собой C1-6-алкил, C2-6 -алкенил; C2-6-алкинил, или -Z-R6;
Z - является прямой связью, C1-6-алкилом, или C2-6 -алкенилом;
R6 является арилом, замещенным арилом, C3-6-циклоалкилом, или гетероарилом;
p = 0 или 1;
Px является C1-6-алкилом необязательно замещенным 1-6 атомами галогена, которые могут быть одинаковыми или различными, C3-6-циклоалкилом, C2-6-алкенилом, или гидроксигруппой; либо является радикалом формулы:

где D является связью или C1-6-алкилом;
Ra, Rb и Rc независимо представляют собой водород, амино, C1-6-алкиламино, ди-C1-6-алкиламино, галоген, C1-6-алкил, или C1-6-алкокси.
И наконец, в еще одном своем варианте, настоящее изобретение относится к фармацевтической композиции, которая включает в себя эффективное опухоль-ингибирующее количество соединения формулы (B) или (A), и фармацевтически приемлемый носитель.
Если это не оговорено особо, то термины, используемые в настоящей заявке имеют следующие значения. "Алкил" означает прямую или разветвленную насыщенную углеродную цепь, имеющую от 1 до 6 атомов углерода, например, такую, как метил, этил, н-пропил, изопропил, н-бутил, втор.-бутил, изобутил, т.-бутил, н-пентил, втор. -пентил, изопентил, и н-гексил. "Алкенил" означает прямую или разветвленную углеродную цепь, имеющую по крайней мере одну "углерод-углеродную", двойную связь, и имеющую от 2 до 6 атомов углерода; например, этенил, пропенил, изопропенил, бутенил, изобутенил, пентенил, и гексенил. "Алкинил" означает прямую или разветвленную углеродную цепь, имеющую по крайней мере одну углерод-углеродную тройную связь, и от двух до шести атомов углерода, например, этинил, пропинил, бутинил, и гексинил.
"Арил" означает ароматический углеводород, имеющий от 6 до 10 атомов углерода, например, такой, как фенил или нафтил. "Замещенный арил" означает арил, замещенный по крайней мере одной группой, выбранной из C1-6-алканоилокси, гидрокси, галогена, C1-6-алкила, трифторометила, C1-6-алкокси, арила, C2-6 -алкенила, C1-6-алканоила, нитро, амино и амидо. "Галоген" означает фтор, хлор, бром и йод.
"Фосфоно-" означает группу -P(O)(OH)2, а "фосфонооксиметокси" или "фосфонооксиметиловый эфир", в общих чертах, означают группу -OCH2(OCH2)mOP(O)(OH)2. "(Метилтио)-тиокарбонил" означает группу -C(S)SCH3. "Метилтиометил" (также сокращенно обозначаемый МТМ), в основном, относится к группе -CH2SCH3.
"Таксановая часть" (также сокращенно обозначаемая txn) означает
части, содержащие основной каркас молекулы таксана, имеющий 20 атомов углерода, и нижеприведенную структурную формулу с абсолютной
конфигурацией

Система нумерации, представленная выше, является стандартной системой, которая применяется в тривиальной номенклатуре таксанов, и используется в настоящем описании. Например, символ CI относится к атому углерода, обозначенному "1"; C5-C20-оксетан означает оксетановое кольцо, образованное атомами углерода, обозначенными 4, 5 и 20, и атомом кислорода; а C9-окси относится к атому кислорода, связанному с атомом углерода, обозначенные "9", где указанным атомом кислорода может быть оксогруппа, α- или β-гидрокси группа, либо α- или β-ацилоксигруппа.
"Замещенная
3-амино-2-гидроксипропаноилокси-группа" означает остаток, имеющий формулу:

где X является группой, не содержащей водород;
X' может быть водородом, или группой, не содержащей водород. Стереохимия этого остатка аналогична боковой цепи паклитаксела. В настоящем описании, эту группу иногда обозначали "C13-боковой цепью".
"Производное таксана" (сокращено обозначаемое "T") относится к соединению, имеющему таксановую часть, несущую C13-боковую цепь.
"Гетероарил" означает 5- или 6-членное ароматическое кольцо, содержащее по крайней мере от 1 до 4 атомов, не являющихся углеродом, и выбранных из атомов кислорода, серы и азота. Примерами гетероарила могут служить тиенил, фурил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, тиазолил, тиадиазолил, оксадиазолил, тетразолил, тиатриазолил, оксатриазолил, пиридил, пиримидил, пиразинил, пиридазинил, триазинил, тетразинил и аналогичные кольца.
"Фосфонозащитные группы" означают группы, которые могут быть использованы для блокирования или защиты функциональной фосфоногруппы; причем, предпочтительными являются такие защитные группы, которые могут быть удалены методами, не оказывающими какого-либо неблагоприятного воздействия на остальную часть молекулы. Подходящие фосфонооксизащитные группы хорошо известны специалистам, и в качестве примера таких групп могут служить бензильная и аллильная группы.
"Гидроксизащитными группами" являются (но не ограничиваются ими) простые эфиры, такие, как т-бутиловый, бензиловый п-метоксибензиловый, п-нитробензиловый, алилловый, тритиловый, метоксиметиловый, метоксиэтоксиметиловый, этоксиэтиловый, тетрагидропираниловый, тетрагидротиопираниловый и триалкилсилиловые эфиры (например, триметилсилиловый, триэтилсилиловый и т-бутилдиметилсилиловый эфир), сложные эфиры, такие, как бензоил, ацетил, фенилацетил, формил, моно-, ди-, и тригалогеноацетил (например, хлороацетил, дихлороацетил, трихлороацетил, трифторацетил); и карбонаты, такие, как метил, этил, 2,2,2-трихлорэтил, аллил, бензил и п-нитрофенил.
Другие примеры гидрокси- и фосфонозащитных групп можно найти в таких известных работах, как Grecne & Wuts, Protective Groups in Organic Synthesis, 2d Ed. , 1991, John Wiley & Jous; и Mc Omie, Protective Groups in Organic chemistry, 1975, plenum Press. В указанных работах также описаны методы введения и удаления защитных групп.
Термин "фармацевтически приемлемая соль" означает металлическую или аминовую соль кислотной фосфоногруппы, где катионы не оказывают неблагоприятного влияния на токсичность или биологическую активность активного соединения. Подходящими металлическими солями являются соли лития, натрия, калия, кальция, бария, магния, цинка, и алюминия. Предпочтительными являются соли натрия и калия. Подходящими аминовыми солями являются, например, соли аммиака, трометамина (TPI), триэтиламина, прокаина, бензатина, дибензиламина, хлоропрокаина, холина, диэтаноламина, триэтаноламина, этилендиамина, глюкамина, N-метилглюкамина, лизина, аргинина, этаноламина, и т.п. Предпочтительными аминовыми солями являются соли лизина, аргинина, триэтаноламина, и N-метилглюкамина. Наиболее предпочтительной является N-метилглюкаминовая соль или триэтаноламиновая соль.
В настоящем описании, термин "-OCH2(OCH2) OP(O)(OH)2" относится как к свободной кислоте, так и к ее фармацевтически приемлемым солям, если только конкретно не указывается, что данное выражение означает свободную кислоту.
В одном из своих вариантов, настоящее изобретения
относится к производным таксана формулы (A):
T-[OCH2(OCH2)mOP(O)(OH)2]n (А),
где T представляет собой таксановую часть,
несущую на C13-атоме углерода замещенную
3-амино-2-гидроксипропаноилоксигруппу; n = 1, 2 или 3; m = 0 или целое число от 1 до 6 включительно;
или к их фармацевтически приемлемым солям.
В другом своем варианте,
настоящее изобретение относится к производным таксана формулы (B):
T'-[OCH2(OCH2)mSCH3] (В),
которые
могут быть использованы для получения
производных таксана формулы (A).
В одном варианте настоящего изобретения, таксановая часть содержит по крайней мере следующие функциональные группы: C1-гидрокси, C2-бензилокси, C4-ацетилокси, C5-C20-оксетан, C9-окси, и C11-C12-двойную связь.
В предпочтительном варианте
осуществления настоящего изобретения, таксановая часть происходит от остатка формулы:

где R2е' является водородом;
R2е является водородом, гидрокси, OC(O)Rx, или -OC(O)OPx; R3е является водородом, гидрокси, OC(O)Rx, -OC(O)OPx, или C1-6-алкилокси; один из R6е или R7е является водородом, а другой гидрокси или -OC(O)Rx; либо R6е и R7е, вместе взятые образуют оксогруппу; а Rx определен ниже.
В другом варианте настоящего изобретения, C13-боковая цепь происходит
от остатка формулы:

где R1е' является водородом, -C(O)Rx или -C(O)ORx; R4 и R5 независимо представляют собой C1-6-алкил, C2-6-алкенил, C2-6-алкинил, или -Z-R6;
Z представляет собой прямую связь, C1-6-алкил, или C2-6-алкенил;
R6 представляет собой арил, замещенный арил, C3-6-циклоалкил, или гетероарил;
Rx представляет собой C1-6 -алкил, необязательно замещенный 1-6 атомами галогена, которые могут быть одинаковыми или различными, C3-6-циклоалкил, C2-6-алкенил, или гидрокси; либо представляет собой радикал формулы:

где D представляет собой связь или C1-6-алкил;
Ra, Rb и Rc независимо представляют собой водород, амино, C1-6-алкиламино, ди-C1-6-алкиламино, галоген, C1-6-алкил, или C1-6-алкокси;
p равно 0 или 1.
В предпочтительном варианте, R4 является C1-6-алкилом, а p равно 1; либо R4 является - Z - R6, а p равно 0. Более предпочтительно, если R4(O)p представляет собой т-бутокси, фенил, изопропилокси, н-пропилокси, или н-бутокси.
В другом предпочтительном варианте, R5 представляет собой C2-6-алкенил или -Z - R6, где Z и R6 определены выше. А более предпочтительно, если R5 представляет собой фенил, 2-фурил, 2-тиенил, изобутенил, 2-пропенил, или C3-6-циклоалкил.
В другом варианте настоящего изобретения, соединение формулы (A) может быть, в
частности, представлено формулой (1):
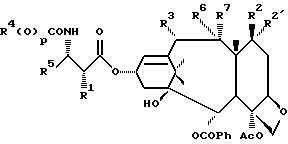
где R1 является гидрокси, OCH2 (OCH2)m OP(O)(OH)2, -OC(O)Rx, или -OC(O)ORx;
R2 является водородом;
R2' является водородом, гидрокси, -OCH2(OCH2)mOP(O) (OH)2, -OC(O)Rx, или -OC(O)ORx;
R3 является водородом, гидрокси, C1-6-алкилокси, -OC(O)Rx, -OCH2(OCH2 )mOP(O)(OH)2, или -OC(O)ORx;
один из R6 или R7 является водородом, а другой гидрокси, C1-6-алканоилокси, или -OCH2 (OCH2)mOP(O)(OH2); либо R6 и R7, взятые вместе, образуют оксогруппу; при условии, что по крайней мере один из R1, R2, R3, R6 и R7 является -OCH2(OCH2)mOP(O)(OH)2);
R4, R5, Rx, m и p являются такими, как они были определены выше;
или его фармацевтически приемлемой солью.
В соединениях формулы (I), примерами Rx могут служить метил, гидроксиметил, этил, н-пропил, изопропил, н-бутил, изобутил, хлорометил, 2,2,2-трихлороэтил, циклопропил, циклобутил, циклопентил, циклогексил, этенил, 2-пропенил, фенил, бензил, бромофенил, 4-аминофенил, 4-метиламинофенил, 4-метилфенил, 4-метоксифенил, и т.п. Примерами R4 и R5 являются 2-пропенил, изобутенил, 3-фуранил(3-фурил), 3-тиенил, фенил, нафтил, 4-гидроксифенил, 4-метоксифенил, 4-фторфенил, 4-трифторметилфенил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, т-бутил, этенил, 2-пропенил, 2-пропинил, бензил, фенетил, фенилэтенил, 3,4-диметоксифенил, 2-фуранил(2-фурил), 2-тиенил, 2-(2-фуранил)этенил, 2-метилпропил, циклопропил, циклобутил, циклопентил, циклогексил, циклогексилметил, циклогексилэтил, и т.п.
В одном из вариантов своего осуществления, настоящее изобретение относится к предпочтительной группе соединений формулы (I), в которых R5 является C2-6-алкенилом или -X-R6, где Z и R6 определены выше. Более предпочтительно, если R5 представляет собой 3-фурил, 3-тиенил, 2-пропенил, изобутенил, 2-фурил, 2-тиенил, или C3-6 -циклоалкил.
В другом предпочтительном варианте осуществления настоящего изобретения. R4 в соединениях формулы (I) представляет собой C1-6-алкил, в случае, если p = 1; либо R4 представляет собой -Z-R6 (где Z и R6 определены выше), в случае, если p = 0. Более предпочтительно, если R4(O)p представляет собой т-бутокси, фенил, изопропилокси, н-пропилокси, н-бутокси.
В другом предпочтительном варианте своего осуществления настоящего изобретения относится к соединению формулы (I), в которых R1 представляет собой -OCH2(OCH2)mOP(O)(OH)2. В более предпочтительном варианте, P2 является гидрокси, -OCH2(OCH2 )mOP(O)(OH)2, -OC(O)OPx, или -OC(O)Rx, а Rx является предпочтительно C1-6-алкилом. В другом более предпочтительном варианте, R3 является гидрокси или ацетокси.
В другом предпочтительном варианте своего осуществления настоящего изобретения относится к соединению формулы (I), в которых R2 представляет собой -OCH2(OCH2)m OP(O)(OH)2; R1 представляет собой гидрокси, -OC(O)Rx или -OC(O)ORx; R3 представляет собой водород, гидроксигруппу, ацетоксигруппу, -OCH2(OCH2)mOP(O)(OH)2 или -OC(O)ORx, а Rx определен выше. В более предпочтительном варианте, R1 является гидрокси, или -OC(O)Rx, Rx предпочтительно является C1-6-алкилом; а R3 является гидрокси или ацетокси.
В другом предпочтительном варианте своего осуществления настоящее изобретение относится к соединению формулы (I), в котором R3 является -OСH2(OCH2)mOP(O)(OH)2; R1 является гидрокси или -OC(O)OPx; R2 является водородом, R2 является водородом, гидрокси, или -OC(O)ORx; а Rx определен выше. В более предпочтительном варианте, R1 является гидрокси или -OC(O)ORx, а Rx является предпочтительно C1-6-алкилом. В другом более предпочтительном варианте, R2 является гидроксигруппой.
В другом предпочтительном варианте, m = 0, 1 или 2, если фосфонооксиметоксигруппа присутствует на C7 таксановой части.
Предпочтительными фармацевтически приемлемыми солями соединения формулы (A) являются соли щелочных металлов, например, соли триэтиламина, триэтаноламина, этаноламина, аргинина, лизина, и N-метилглюкамина. Более предпочтительными являются соли натрия, триэтаноламина и N-метилглюкамина.
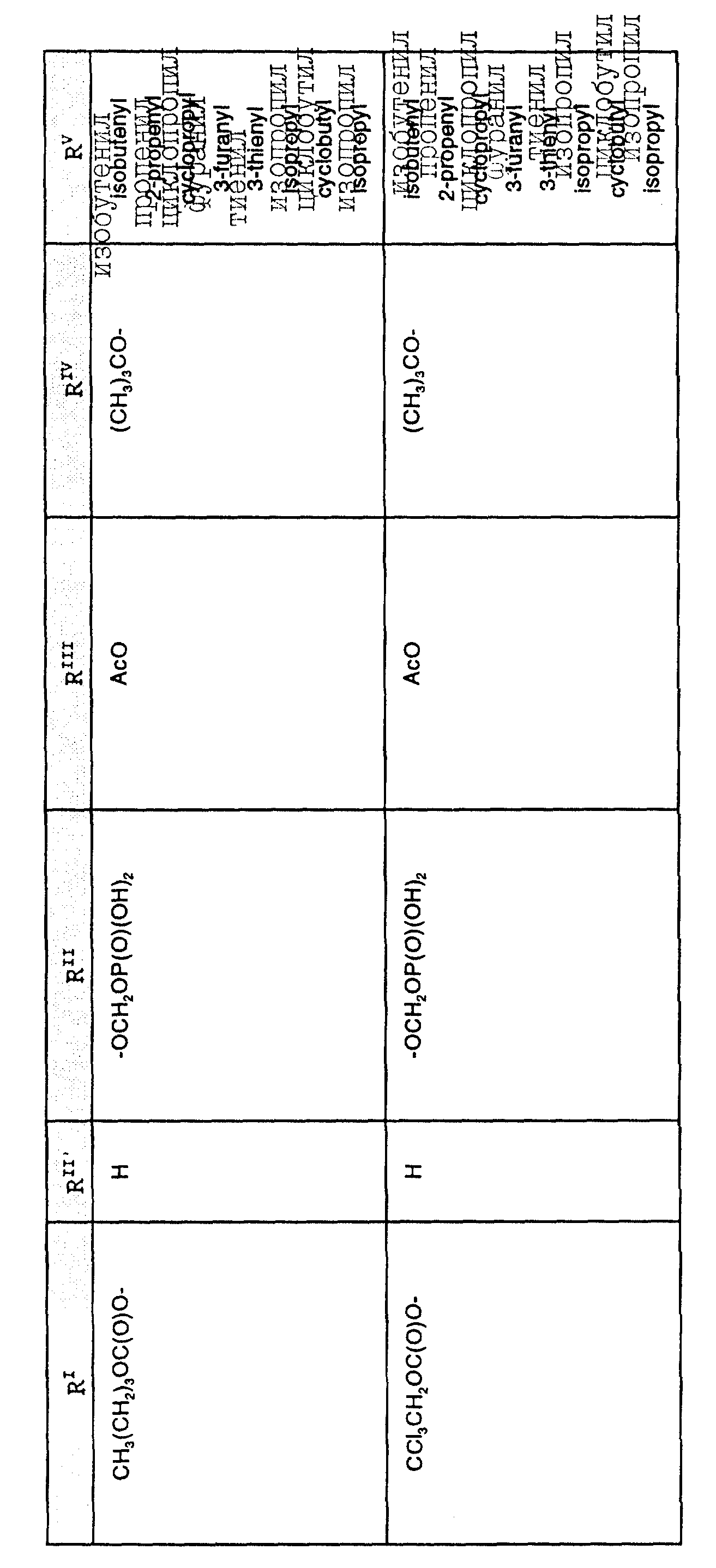
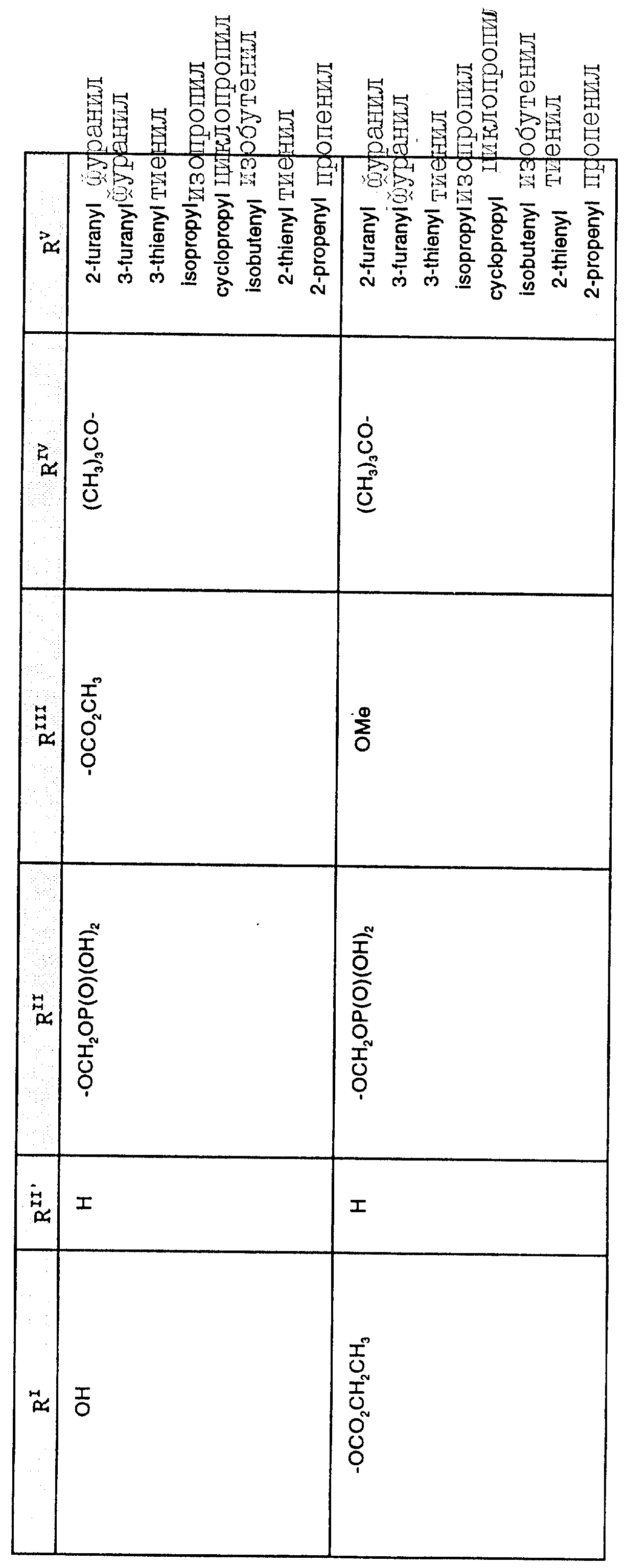
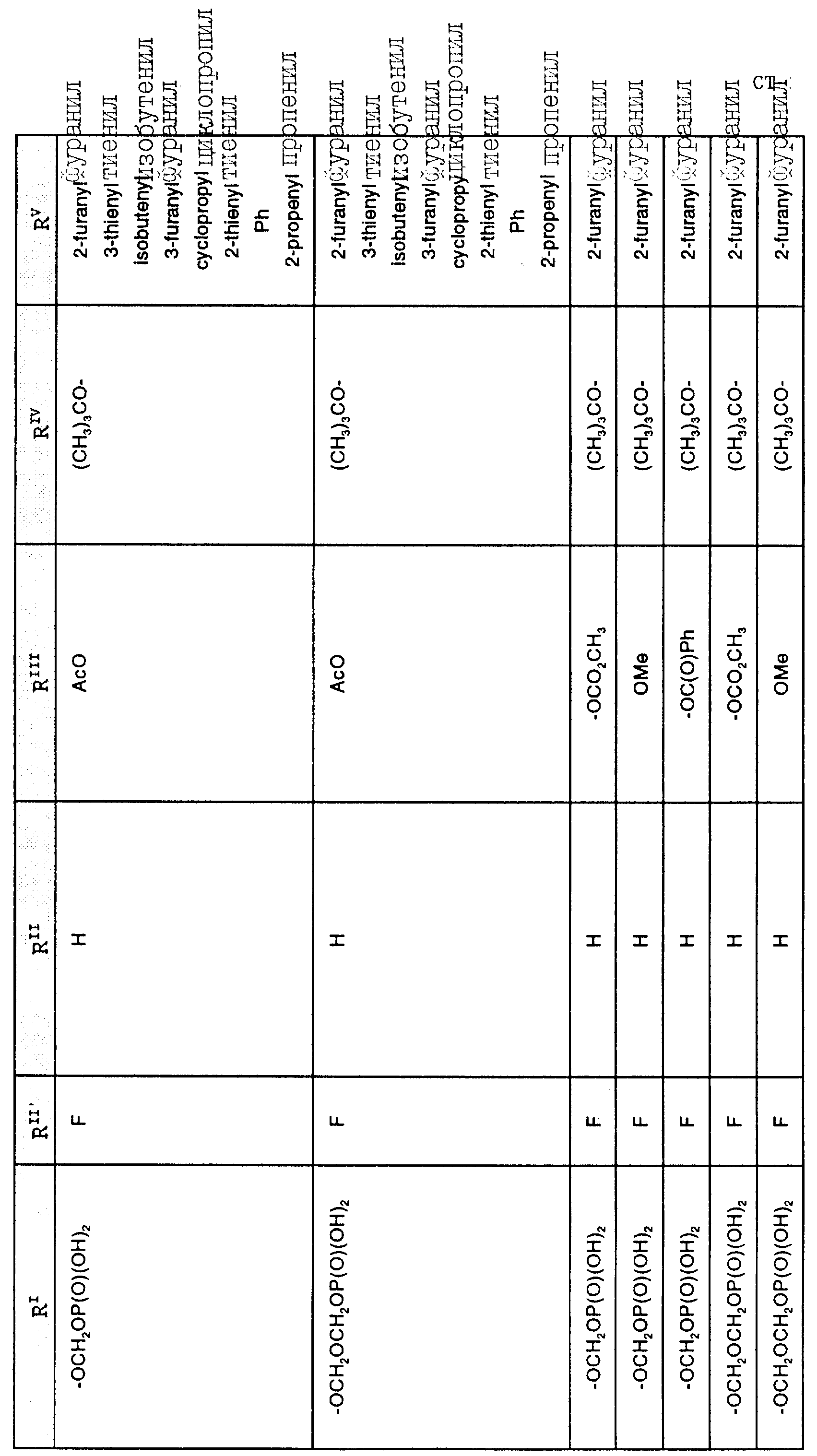
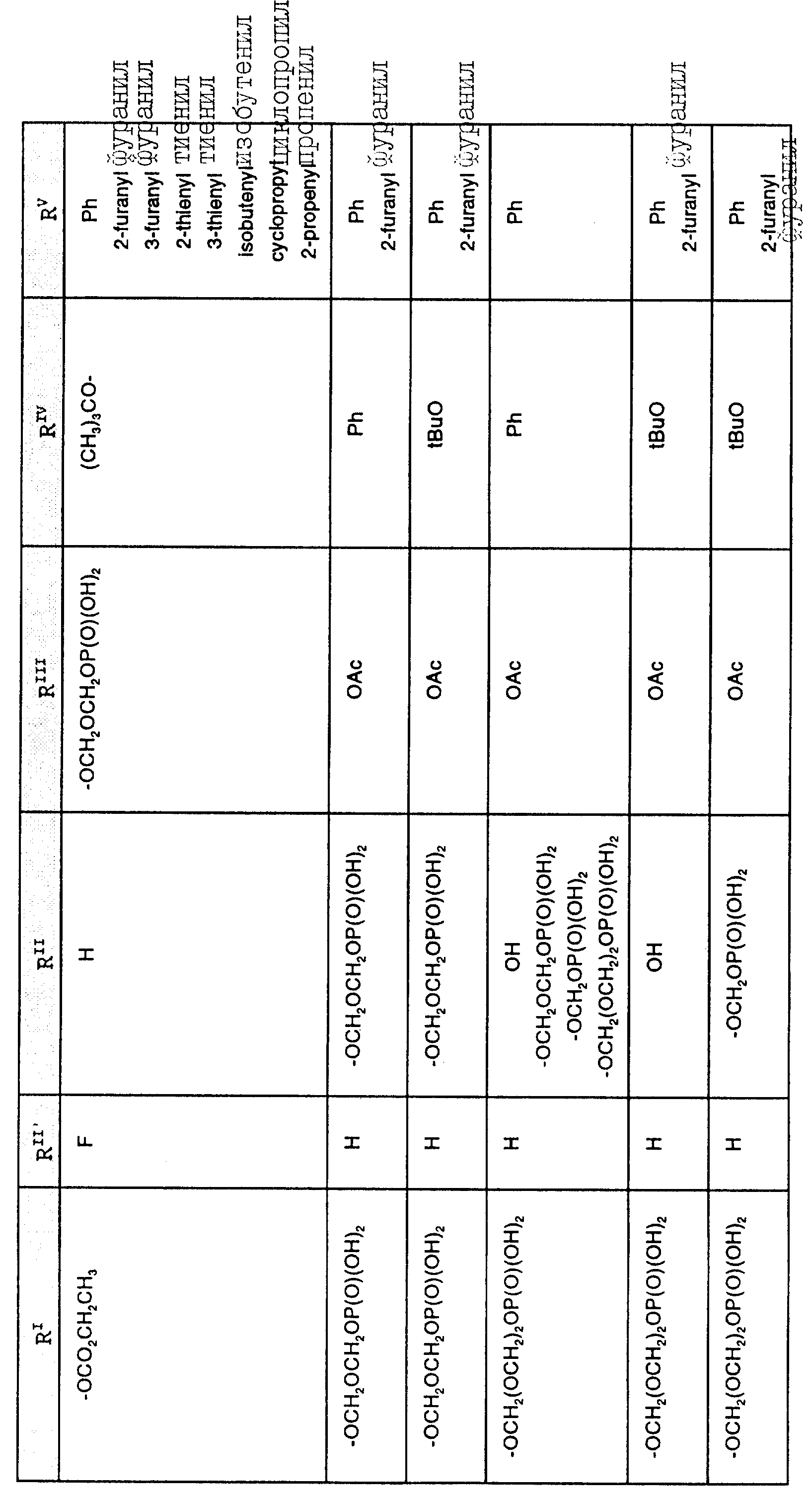
Наиболее предпочтительными производными таксана, имеющими формулу (A), являются следующие соединения: (I)

(2)


(3)

(4)

(5) 3'-N-дебензоил-3'-десфенил-3'-N-(т-бутилоксикарбонил)-3'-(2-фурил)

(6) 3'-N-дебензоил-3'-десфенил-3'-N-(т-бутилоксикарбонил)-3'-(2-тиенил)

(7) 10-дезацетил-3'-десбензоил-3'-N-(т-бутилоксикарбонил)-10-O-(фосфонооксиметил)паклитаксел;
(8)

(9)

(10)

(11)

(12)

и их фармацевтически приемлемые соли, в частности, соли натрия, калия, аргинина, лизина, N-метилглюкамина, этаноламина, триэтиламина и триэтаноламина.
Соединения формулы (A) могут быть получены из таксанового производного T-[OH]n, используемого в
качестве исходного материала, где T' и n определены выше.
Идентичность T'-[OH]n не является строго ограниченной, поскольку имеется по крайней мере одна реактивная гидроксигруппа,
присутствующая либо на таксановой части, либо на C13-боковой цепи, и
способствующая образованию фосфонооксиметилэфирной связи. Следует отметить, что реактивная гидроксигруппа может быть
непосредственно связана с C13-пропаноилокси-каркасом (например, 2-гидроксигруппа
паклитаксела), либо с центральным каркасом таксана (например, 7-гидроксигруппа паклитаксела); или она может
присутствовать на заместителе в C13-боковой цепи, или на заместителе в таксановом ядре. Для
получения соединений формулы (A) может быть использована реакционная схема, показанная в Схеме I:
Схема 1.

В Схеме I, T' представляет собой таксановое производное, в котором нереактивные гидроксигруппы являются блокированными; Ry представляет собой фосфонозащитную группу; а n и m определены выше. Таким образом, соответствующим образом защищенное T', имеющее одну или несколько реактивных гидроксигрупп, сначала превращают в соответствующий метилтиометиловый простой эфир формулы (B). Используя в качестве примера паклитаксел, T' можно определить следующим образом: T' может быть самим паклитакселом (для осуществления 2', 7-бисметилтиометилирования),


ТМТ-эфир, имеющий одну промежуточную метиленоксигруппу (т.е., соединения формулы (B), где m = 1), может быть получен несколькими способами. В одном из них, соединение формулы (B), где m = 0, подвергают реакции с N-иодосукцинимидом (NIS) и метиолтиометанолом, в результате чего цепь удлиняется на одну метиленоксигруппу.

Аналогичная реакция спирта с метилтиометилокси-группой в присутствии NIS описана Veeneman и др., в Tetrahedron, 1991, т. 47, стр. 1547-1562; соответствующие части этой работы вводятся в настоящее описание посредством ссылки. В качестве катализатора предпочтительно используют трифлат серебра. Соединение метилтиометанола и его получение описано в Syn. Comm, 1986, 16 (13): 1607-1610.
В альтернативном способе, T-алкоксид (Ad), полученный путем обработки соединения формулы (Aa) основанием, таким, как н-бутиллитий, диизопропиламид лития, или гексаметилдисилазид лития, подвергают реакции с простым хлорометилметилтиометиловым эфиром, в результате чего получают соединение формулы (B), в котором m = 1.

Соединение (Ae) получают с помощью реакции метилтиометоксида (полученного из метилтиометанола путем обработки основанием таким, как н-бутиллитий, диизопропиламид лития, гексаметилдизилазид лития) с хлороиодометаном. Соединение (Ae) может быть также получено путем обработки 1, 1'-дихлородиметилэфира (ClCH2OCH2 Cl) стехиометрическим количеством или меньшим количеством (например, около 0,8 эквивалентов) иодида натрия, а затем тиометоксидом натрия. 1, 1'-Дихлородиметиловый простой эфир описан в Ind. J. Chem. , 1989, 28B, стр. 454-456.
В другом способе, соединение формулы (Aa) подвергают реакции с бис(МТМ)эфиром, CH3 SCH2OCH2SCH3 и N1S в результате чего получают соединение формулы (B), где m = 1.

Бис-(МТМ)эфир получают путем реакции 1,1'-дихлородиметилового эфира с иодидом натрия, а затем с триметоксидом натрия.
Описанная выше процедура с использованием метилтиометанола и NIS может быть применена к любому реагенту, имеющему МТМ-группу, в целях одновременного удлинения цепи на одно метиленоксизвено. Например, соединение формулы (B), где m = 1, может быть подвергнуто реакции с метилтиометанолом и 1 для получения соединения формулы (B), где m = 2. Эту процедуру можно повторить в целях получения соединений формулы (В), в которых m = 3, 4, 5 или 6.
Во второй стадии схемы 1, метилтиометиловый эфир превращают в соответствующий защищенный фосфонооксиметиловый эфир.
Для этого, МТМ-эфир обрабатывают NIS и защищенным фосфатом HOP(O)(ORy)2. В третьей стадии, фосфонозащитную группу и любую гидроксизащитную группу (или группы) удаляют и получают в результате соединение формулы (A). Например, подходящей фосфонозащитной группой является бензил, который может быть удален путем каталитического галогенолиза; а подходящими гидроксизащитными группами, является триалкилсилил, который может быть удален с помощью фторидного иона, и трихлорэтоксикарбонил, который может быть удален с помощью цинка. Удаление защитных групп описано в справочных пособиях Green и Wuts, hotective Groups in Organic Synthesis John Wiley & Sons, 1991; McOnie, Protective in Organic Chemistry, Plenum Press, 1973. Обе эти стадии более подробно описаны в последующем разделе настоящего описания.
В нижеприведенной схеме II показан другой вариант реакционной последовательности, проиллюстрированной в схеме I.

В схеме II, соединение формулы (Aa) подвергают реакции с соединением формулы (Ca) и N1S, в результате чего получают соединение формулы (C), которое затем подвергают разблокированию и получают соединение формулы (A). Соединение формулы (Ca), в котором m = 0, может быть получено сначала путем обработки метилтиометанола основанием, таким, как гексаметилдисилазид натрия, лития или калия, с получением метилтиометоксида; а затем нужное соединение может быть получено посредством реакции указанного метоксида с защищенным хлорофосфатом, таким, как дибензилхлорофосфат. Соединения формулы (Ca), в которых m = 1, могут быть получены путем обработки CH3SCH2OCH2Cl дизащищенной фосфатной солью, например, дибензилфосфатными солями натрия, калия, или тетра(н-бутил)-аммония; либо CH3SCH2 OCH2Cl может быть сначала превращен в соответствующее иодосоединение с использованием иодида натрия, и это иодосоединение может быть затем подвергнуто реакции с фосфатной солью. Альтернативно, соединения формулы (Ca), в которых m = 1, могут быть получены путем обработки ClCH2OCH2Cl иодидом натрия, а затем тиометоксидом натрия, с образованием соединения CH3CH2OCH2CH3, которое затем обрабатывают NIS и дизащищенным фосфатом, таким, как дибензилфосфат, в результате чего получают нужный продукт. Любой из вышеупомянутых реагентов, имеющих МТМ-группу, может быть удлинен на одно метиленоксизвено при взаимодействии этого реагента с метилтиометанолом и NIS.
В другом способе получения соединения (A), T-алкоксид (Ad) подвергают реакции с иодофосфатом, как показано в схеме III.
Схема III

В схеме III, иодофосфатное соединение получают путем реакции ClCH2(OCH2)mCl с дизащищенной фосфатной солью, в результате которой образуется соединение ClCH2(OCH2)mOP(O)(ORу)2, которое затем обрабатывают иодидом натрия и получают нужный продукт.
В нижеприведенной схеме IV показан еще один метод, который может быть использован для получения подкласса соединений формулы (A), где по крайней мере одна из фосфонооксиметоксигрупп связана с таксоновой частью.
Схема IV.

В схеме IV, m и n являются такими, как они были определены ранее, X является группой, не содержащей водород, P является гидроксизащитной группой; а txn является таксоновой частью. Соединения формулы (I) представляют собой имеющие 13-альфа-гидроксигруппу и одну или несколько метилтиометилоэфирных групп, непосредственно или опосредованно связанных с таксановым ядром; а также C13-алкоксиды металлов формулы (D). Примером соединения формулы (D) является


Реакция взаимодействия таксана (D) с азетидиноном является аналогичной реакции, показанной в схеме VI (см. ниже); поэтому процедура, описанная для получения соединения формулы (1d), может быть также использована для получения соединения формулы (Ba)(т.е., соединения формулы (B), в котором, по крайней мере, одна из МТМ-групп непосредственно или опосредованно связана с таксановой частью), в том случае, если соединение (II) в схеме IV заменить соединением формулы (D). Сначала, таксан предпочтительно превращают в C13-алкоксид металла, например, алкоксид натрия, калия, или лития, а более предпочтительно, в алкоксид лития. Азетидинон служит в качестве предшественника C13-боковой цепи. После реакции присоединения с таксаном, гидроксизащитную группу P удаляют, и если это необходимо, то свободная гидроксигруппа на боковой цепи может быть превращена в МТМ-эфир, либо дериватизирована с образованием сложного эфира или карбоната, как описано ниже.
Азетидинон может быть получен описанными ниже способами, которые хорошо известны любому специалисту. Соединения формулы (D) могут быть получены при помощи общей процедуры, описанной выше для получения соединений формулы (B) с использованием соответствующим образом защищенного таксана. Однако предпочтительно, эти соединения могут получены из соединения формулы (Ba) путем расщепления 13-боковой цепи с использованием борогидрида, например, борогидрида натрия или тетрабутиламмония. Например,
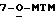
Общая процедура, показанная в схеме 1 для получения соединения формулы (A), более наглядно представлены в схеме V, где проиллюстрировано получение соединения формулы (I') (т. е., соединения формулы (I), в котором m = 0). Процедура, используемая в этой последовательности реакций, может быть, в основном, применена к другим производным таксана, которые конкретно не относятся к соединениям формулы (I). Кроме того, процедура, используемая в схеме (V), может быть модифицирована в соответствии с указаниями, приведенными в данном описании в отношении получения производных таксана формулы (A), в которых m=1,2 или 3.
При этом, следует отметить, что в схеме V, а также в других разделах настоящего описания, термин
"гидроксизащитные группы" может включать в себя соответствующие карбонаты (например,
-OC(O)ORx, где Rx не содержит гидроксигруппу); поэтому, если карбонат используется в
качестве гидроксизащитной
Схема V




группы, то в более поздней стадии, эта группа должна быть удалена с образованием свободной гидроксигруппы; либо, в противном случае, эта карбонатная группа останется как часть конечного продукта.
В схему V, R1a является гидроксигруппой, защищенной гидроксигруппой, -OC(O)Rx, или -OC(O)ORx ; P2' является водородом, R2a является водородом, гидроксигруппой, защищенной гидроксигруппой, -OC(O)Rx или -OC(O)OR

В первой стадии, свободную гидроксигруппу соединения формулы (1a) превращают в соответствующую метилтиометилоэфирную (-OCH2CH3) группу. Для этого осуществляют одну из двух процедур: 1a (диметилсульфидный метод) и 1b (диметилсульфоксидный метод). Диметилсульфидный метод превращения спиртов в метилтиометиловые эфиры описан Medina и др. (Jet. Lett 1988, стр. 3773-3776; соответствующие части этой работы вводятся в настоящее описание посредством ссылки). Диметилсульфоксидный метод хорошо известен специалистам как реакция Пуммерера.
При этом следует отметить, что реакционная способность гидроксигруппы варьируется в зависимости от ее расположения на исходном соединении таксанового производного формулы (1a). Хотя, обычно, 2-гидроксигруппа является более реактивной, в реакциях ацилирования, чем 7-гидроксигруппа, которая, в свою очередь, является более реактивной, чем 10-гидроксигруппа, однако, используя диметилсульфидный метод, было неожиданно обнаружено, что 7-гидроксигруппа легче превращается в метилтиометилоэфирную группу, чем 2-гидроксигруппа. В основном, наименее реактивной является третичная гидроксигруппа в C-1. Такое различие в реакционной способности гидроксигрупп может быть использовано для регулирования места и степени метилтиометилирования.
Так, например, в случае использования соединения формулы (1a), где R1a и R2a являются гидроксигруппами, преимущественным продуктом метилтиометилирования в диметилсульфидном методе будет соответствующий

Кроме того, реакционные условия могут быть подобраны таким образом, чтобы они благоприятствовали образованию бис- или трис-метилтиометилоэфирных производных таксана. Например, в случае паклитаксела, увеличение реакционного времени или использование избыточного количества метилтиометилирующих реагентов может привести к более высокому содержанию 2,7-бис(метилтиометил)-эфир-паклитаксела в смеси конечного продукта.
Возвращаясь к схеме V, следует напомнить, что в процедуре (1a), соединение формулы (1a) обрабатывают диметилсульфидом и органическим пероксидом, таким, как бензоилпероксид. Эту реакцию осуществляют в инертном органическом растворителе, таком, как ацетонитрил, метиленхлорид, и т.д., при температуре, способствующей образованию нужного продукта. В основном, указанную реакцию проводят при температуре от около - 40oC до приблизительно комнатной температуры. Диметилсульфид и бензоилпероксид используются в избыточном количестве по отношению к исходному таксановому производному (1a) а диметилсульфид используется в избыточном количестве по отношению к бензоилпероксиду.
Относительные количества используемых исходных материалов зависят от желаемой степени метилтиометилирования. Так, например, если свободную гидроксигруппу исходного таксанового производного (1a) превращают в метилтиометилоэфирную группу, то диметилсульфид и бензоилпероксид могут быть использованы в количестве, которое в 10 раз превышает количество таксанового производного (Ia); а предпочтительно, если количество диметилсульфида примерно в 2 - 3 раза превышает количество бензоилпероксида. В случае, если исходное соединение (1a) имеет 2'-7-гидроксигруппы, то количество полученного 2',7-бис(метилтиометил)эфира возрастает в соответствии с относительными количествами диметилсульфида и бензоилпероксида. Если целевым продуктом является 2', 7-бис(метилтиометил)эфир, то предпочтительно, чтобы количество диметилсульфида примерно в 15 - 20 раз превышало количество исходного соединения таксанового производного, а количество используемого бензоилпероксида примерно в 5 - 10 раз превышало количество исходного таксанового производного.
Альтернативно, соединение формулы (Ib) может быть получено с помощью реакции соединения формулы (Ia) с диметилсульфоксидом и уксусным ангидридом (процедура (Ib). Эта процедура может быть использована для реиватизации группы, не являющейся 2-гидроксигруппой, в ее метилтиометиловый эфир. В процедуре (Ib), соединение формулы (Ia) растворяют в диметилсульфоксиде, и к полученному раствору добавляют уксусный ангидрид. Эту реакцию, в основном, осуществляют при комнатной температуре, в течение 18 - 24 часов, с получением монометилтиометилового эфира.
Во второй стадии этой реакционной последовательности, метилтиометиловый эфир превращают в соответствующий защищенный фосфонооксиметиловый эфир. Эта реакция превращения может быть осуществлена с использованием общего метода, описанного Veeneman и др. , Tetrahedron, 1991, т. 47, стр. 1547 - 1562 (соответствующие разделы этой работы вводятся в настоящее описание посредством ссылки. Так, например, соединение формулы (Ib), имеющее по крайней мере одну метилтиометилоэфирную группу, обрабатывают N-иодосукцинимидом и защищенной фосфорной кислотой, такой, как дибензилфосфат. Эту реакцию проводят в инертном органическом растворителе, таком, как тетрагидрофуран или галогенированный углеводород, такой, как 1, 2-дихлороэтан или метиленхлорид, и необязательно в присутствии дегидратирующего реагента, такого как молекулярное сито. Для ускорения реакции может быть также добавлен катализатор, такой, как трифторометансульфонат серебра. Эту реакцию осуществляют при температуре от около 0oC и примерно до комнатной температуры, а предпочтительно, при комнатной температуре. N-Иодосукцинамид и защищенную фосфорную кислоту используют примерно в тех же молярных эквивалентах, что и метилтиометиловый эфир (Io), однако, предпочтительно, чтобы количества этих реагентов слегка превышали (например, приблизительно от 1,3 до 1,5 эквивалентов) количество соединения формулы (Io).
В третьей стадии реакционной последовательности, удаляют фосфонозащитную и гидроксизащитную группы (если таковые имеются). Эту реакцию разблокирования осуществляют с использованием стандартной техники, хорошо известной специалистам, например, такой, как кислотный или основный гидролиз, гидрогенолиз, восстановление, и т.п. Например, каталитический гидрогенолиз может быть использован для удаления бензильной фосфонозащитной группы, а также бензилоксикарбонильной гидроксизащитной группы. Методы разблокирования подробно описаны в известных руководствах Greene и Wutz, или McOmie (см., выше). Само собой разумеется, что если соединение формулы (Ia) содержит гидроксигруппы в радикале Rx , то предпочтительно, чтобы эти гидроксигруппы были защищены соответствующими защитными группами вплоть до описанной стадии разблокирования.
Как указывалось выше, метод, проиллюстрированный в схеме V, может быть модифицирован в соответствии с нижеприведенными указаниями, таким образом, чтобы этот метод можно было использовать для получения производных таксана формулы A, в которых m = 1, 2 или 3.
Ниже приводятся схемы Va и Vb, в соответствии с которыми каждый специалист может модифицировать вышеописанный метод для получения конкретных соединений формулы A, в которых по крайней мере один заместитель представляет собой -OCH2(OCH2)2OP(O)(OH)2. Аналогично, могут быть легко получены и другие соединения формулы (A), в которых m = 3.
Схема Va





Схема Vb





Основные соли соединения формулы (I) могут быть получены стандартными способами, например, путем реакции взаимодействия соединения формулы (I) в виде свободной кислоты с металлическим или аминовым основаниями. Подходящими для этой цели металлическими основаниями являются гидроксиды, карбонаты, и бикарбонаты натрия, калия, лития, кальция, бария, магния, цинка и алюминия, а подходящими аминами являются триэтиламин, аммиак, лизин, аргинин, N-метилглюкамин, этаноламин, прокаин, бензатин, дибензиламин, трометамин (TRIS), хлоропрокаин, холин, диэтаноамин, триэтаноламин, и т.п. Указанные основные соли могут быть затем очищены путем хроматографии с последующей лиофилизацией или кристаллизацией.
Производные таксана, используемые в качестве исходных материалов
Для получения соединений (A) в вышеописанных способах могут быть использованы производные
таксана формулы T-[OH]n. В литературе приводится множество примеров соединений формулы T-[OH]n, и некоторые из них перечислены ниже, а именно: (a) паклитаксел; (b) Таксотер®; (c) 10-дезацетилпаклитаксел; (d) таксановые производные, раскрытые в PCT-заявке 93/06079 (опубликованной 1 апреля 1993 г.), и имеющие формулу:
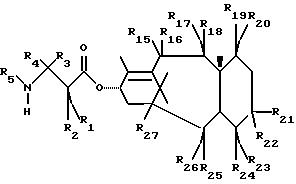
где R1 представляет собой -OR6, -SR7, или -NR8R9;
R2 представляет собой алкил, алкенил, алкинил, арил, или гетероарил;
R3 и R4 независимо представляют собой водород, алкил, алкенил, алкинил, арил, гетероарил, или ацил, при условии, однако, что R3 и R4 не являются оба ацилом;
R5 представляет собой -COR10, -COOR10, -COSR10, -CONR8 R10, -SO2R11, или -POR12R13;
R6 представляет собой водород, алкил, алкенил, алкинил, арил, гетероарил, гидроксизащитную группу, или функциональную группу, способствующую увеличению водорастворимости таксанового производного;
R7 представляет собой алкил, алкенил, алкинил, арил, гетероарил, или сульфидрильную защитную группу;
R8 представляет собой водород, алкил, алкенил, алкинил, арил, гетероарил;
R9 представляет собой аминозащитную группу;
R10 представляет собой алкил, алкенил, алкинил, арил, гетероарил, -OR10, или -NR8R14;
R12 и R13 независимо представляют собой алкил, акенил, алкинил, арил, гетероарил, -OR10, или -NR8R14;
R14 представляет собой водород, алкил, алкенил, алкинил, арил, гетероарил;
R15 и R16 независимо представляют собой водород, гидрокси, низшую алканоилоксигруппу, алкеноилокси, алкиноилокси, арилоилоксигруппу, или R15 и R16, вместе взятые, образуют оксогруппу;
R17 и R18 независимо представляют собой водород, гидроксигруппу, низшую алканоилоксигруппу, алкеноилоксигруппу, алкиноилоксигруппу, ариноилоксигруппу; либо R17 и R18, взятые вместе, образуют оксогруппу;
R19 и R20 независимо представляют собой водород, гидроксигруппу, низшую алканоилоксигруппу, алкеноилоксигруппу, алкиноилоксигруппу, или арилоилоксигруппу;
R19 и R20 независимо представляют собой водород, или низшую алканоилокси-, алкеноилокси, алкиноилокси- или арилоилоксигруппу;
либо R21 и R22, вместе взятые, образуют оксогруппу;
R23 представляет собой водород, гидрокси, или низшую алканоилокси, алкеноилокси, алкиноилокси, или арилоилоксигруппу; либо R23 и R24, взятые вместе, образуют оксогруппу или метилен; либо R23 и R24, взятые вместе с атомом углерода, с которым они связаны, образуют оксирановое кольцо; либо R23 и R24, взятые вместе с атомом углерода, с которым они связаны, образуют оксетановое кольцо;
R25 представляет собой водород, гидроксигруппу, или низшую алканоилокси-, алкеноилокси-, алкиноилокси-, или арилоилокси-группу;
R26 представляет собой водород, гидроксигруппу, или низшую алканоилокси, алкеноилокси, алкиноилокси, или ариноилоксигруппу; либо R26 и R25, взятые вместе, образуют оксогруппу;
R27 представляет собой водород, гидроксигруппу, или низшую алкокси-, алканоилокси-, алкеноилокси-, алкиноилокси-, или арилоилоксигруппу; (e) таксановые производные, раскрытые в патенте США 5227400, 3'-десфенил-3'-(2-фурил)- или 3'-(2-тиенил)-производные паклитаксела, Таксотер®, (f) таксановые производные, раскрытые в EP 534709, опубл. 31 марта 1993 (производные паклитаксела, где фенильные группы боковой цепи являются независимо замещенными нафтилом, стиролом, или замещенным фенилом). См. также PCT 92/09589, опублик. 11 июня 1992; (g) производные таксана, раскрытые в EP 534707, опубл. 31.3.1993 (производные паклитаксела, в которых 3'-N-бензоильная группа замещена этоксикарбонилом или метоксикарбонилом); (h) PCT-заявка 93/06093, опубликованная 1 апреля 1993 (10-дезацетокси-производные паклитаксела и Таксотер®; (i) EP 524093, опубл. 20 января 1993 (10-, 7-, или 7,10-бис-

Свободная гидроксигруппа или группы производных таксона могут быть стандартными методами превращены в соответствующий сложный эфир или карбонат; например, в соединениях формулы (Ia), один из R1a, R2a или R3a является -OC(O)Rx или -OC(O)ORx, где Rx определен выше. Таким образом, таксановое производное T-OH может быть подвергнуто реакции с соединением формулы L-C(O)ORx (где L - уходящая группа), таким, как хлороформат, в присутствии основания, такого, как третичный амин, с образованием соответствующего карбоната. Например, при реакции паклитаксела с этилхлороформатом в присутствии диизопропилэтиламина образуется 2'-

Кроме того, производные таксана Т-[OH]n могут быть получены путем ацилирования таксановой части, имеющей C13-гидроксигруппу, с использованием соответствующим образом замещенной 3-амино-2-гидроксипропановой кислоты, либо ее ацилирующего эквивалента, или его предшественника. Подходящими предшественниками замещенной 3-амино-2-гидроксипропановой кислоты являются, например, азетидиноны формулы (III). Примером реакция ацилирования может служить реакция присоединения гидроксизащищенного баккатина III или гидроксизащищенного 10-деацетилбаккатина III с производным фенилизосерина с образованием производных паклитаксела, раскрытых, например, в патентах США 4924011 и 4924012 (Denis и др.); и реакция присоединения защищенного баккатина III и азетидинона с образованием паклитаксела и его производных, раскрытых в EP-заявке 400971, опубликованных 5 декабря 1990 (ныне патенты США N 5175315 и 5229526).
Способ, раскрываемый в EP 400971 (способ Холтона), заключается в том, что 1-бензоил-3-(1-этокси)этокси-4-фенил-2-азетидинон подвергают реакции с 7-


Способ с использованием баккатина/азетидинона, адаптированный для получения соединений формулы (Ia), проиллюстрирован в схеме VI. При этом, указанным способом с использованием соответствующих исходных материалов могут быть получены также и другие производные таксана, которые конкретно не входят в объем формулы (Ia).

В схеме VI, R2' является водородом, R2d является водородом защищенной гидроксигруппой, -OC(O)Rx, или -OC(O)ORx; R3d является водородом, -OC(O)Rx, C1-6-алкилоксигруппой, защищенной гидроксигруппой, или OC(O)ORx, один из P6d и P7d является водородом, а другой является гидроксигруппой, защищенной гидроксигруппой, или C1-6-алканоилоксигруппой; либо R6d и R7d, взятые вместе, образуют оксогруппу; P является гидроксизащитной группой; M является водородом или металлом группы IA, таким, как литий, натрий, или калий; а p, R4, R5 и Rx являются такими, как они определены выше. Указанная реакция может быть проведена в соответствии с процедурой, описанной в EP 400971, где производное баккатина III формулы (II) (где M является водородом) подвергают реакции с азетидиноном формулы (III) в присутствии органического основания, такого, как N, N-диметиламинопиридин. Однако предпочтительно, если производное баккатина III сначала превращают в 13-алкоксид путем обработки указанного производного сильным основанием, таким, как гидриды, алкиламиды, и бис(триалкилсилил)амиды металлов группы IA, как описано в патенте США 5229526 и в работе Ojima (см. выше). Более предпочтительно, если 13-алкоксидом является алкоксид лития. Литиевая соль может быть получена посредством реакции соединения формулы (II), где M является водородом, с сильным металлическим основанием, таким, как диизопропиламид лития, C1-6-алкиллитий, бис(триметилсилил)амид лития, фениллитий, гидрид лития, и т.п. Само собой разумеется, что если соединение формулы (II) содержит гидроксигруппы в радикале Rx, то указанные гидроксигруппы должны быть предпочтительно защищенными соответствующими гидроксизащитными группами.
Реакцию присоединения между таксаном формулы (II) и азетидиноном формулы (III) осуществляют в инертном растворителе, таком, как тетрагидрофуран при приведенной температуре в диапазоне от около 0oC до около -78oC. Азетидиноны формулы (III) могут быть использованы в виде рацемической смеси для проведения реакции сочетания с металлоалкоксидами таксана формулы (II) (где M является металлом группы IA); причем, в этом случае, предпочтительно, чтобы количество используемого азетидинона составляло по крайней мере 2 эквивалента по отношению таксановому реагенту, а более предпочтительно, чтобы это количество составляло от около 3 до около 6 эквивалентов. Могут быть также использованы и хиральные азетидиноны, и в этом случае, достаточным количеством азетидинона по отношению к таксану может быть 1 эквивалент, однако, предпочтительно, чтобы используемое количество азетидинона слегка превышало (например, до 1,5 экв.) количество таксана.
Гидроксизащитные группы могут быть аналогичными, либо они могут быть выбраны таким образом, чтобы их можно было избирательно удалить, не оказывая, при этом, какого-либо неблагоприятного воздействия на остальные группы. Например, в соединении формулы (Id), где R2b и PO оба могут быть триэтилсилилоксигруппой, а R3b может быть бензилоксикарбонилом, указанная бензилоксикарбонильная защитная группа может быть легко удалена, при этом, с сохранением триэтилсилильной группы, посредством каталитического гидрогенолиза в присутствии палладированного угля. Таким образом, гидроксизащитные группы соединения формулы (Id) могут быть селективно удалены с получением соединения формулы (Ia).
Соединения формулы (II) являются либо известными в литературе (например, баккатин III, 10-деацетилбаккатин III, и их гидроксизащищенные производные), либо они могут быть получены из известных соединений стандартными методами, например, путем превращения гидрокси-группы в карбонат. Другие соединения формулы (II) могут быть получены в соответствии с процедурами, описанными ниже в разделе "Получение исходных материалов".
Соединения формулы (III) могут быть получены из соединений (IIIa) в соответствии с общим методом, описанным а EP 400971 и в работе Ojima и др. Tetrahedion 48: 6985-7012, 1992.

Так, например, соединение формулы (IIIa) сначала обрабатывают основанием, таким, как н-бутиллитий или триэтиламин, а затем соединением формулы R4(O)pCO-L, где L является уходящей группой, в результате чего получают соединение формулы (III).
Соединения формулы (IIIa) могут быть получены в соответствии с общим методом, описанным в EP 400971, с использованием промежуточного соединения 3-ацетокси-4-замещенного-2-азетидинона (IIIb); либо в соответствии с методом, описанным в пат. США N 5229526, с использованием промежуточного соединения 3-триэтилсилилокси-4-замещенного-2-азетидинона. В предпочтительном варианте, соединения (IIIb) могут быть получены путем конденсирования ацетоксиацетилхлорида с бисимином с последующим проведением гидрогенолиза или кислотного расщепления для удаления N-иминовой группы. Этот способ показан в нижеприведенной схеме, где R5 является необязательно замещенным арилом, или гетероарильной группой, такой, как фурил и тиенил. Указанный способ описан в одновременно рассматриваемой заявке рег. N 08/165610, поданной 13 декабря 1993, и вводимой в настоящее описание посредством ссылки.

Продукты (IIIb), полученные в результате описанных реакций циклоприсоединения, обычно представляют собой рацемическую смесь двух цис-азетидинонов. Эта рацемическая смесь может быть разделена стандартными методами, например, такими, как превращение в диастереомеры, дифференциальная адсорбция на колонке, упакованной хиральными адсорбентами, или ферментное разделение. Например, рацемическая смесь соединений формулы (IIIb) может быть подвергнута взаимодействию с ферментом, который катализирует гидролиз сложного эфира (например, такой фермент, как эстераза или липаза), и способствует селективному отщеплению 3-ацильной группы одного энантиомера, не оказывая, при этом, неблагоприятного воздействия на другие группы (см., например, Brilva и др., J. Org. Chem. 1993, 58: 1068-1075, а также одновременно рассматриваемую заявку рег. N 092170, поданную 14 июля 1993, и EP - заявку N 552041, опубликованную 21 июля 1993). Альтернативно, рацемическая смесь может быть сначала подвергнута основному гидролизу в целях удаления 3-ациальной группы, и получения рацемической и смеси соответствующего 3-гидрокси-β-лактама, а затем, эту рацемическую смесь 3-гидрокси-β-лактама подвергали взаимодействию с ферментом, способным катализировать ацилирование гидроксигруппы, в результате чего осуществляют селективное ацилирование гидроксигруппы одного энантиомера, не оказывая какого-либо воздействия на другой энантиомер. Либо, рацемическая смесь 3-гидрокси-β-лактами может быть подвергнута ацилированию с использованием хиральной карбоновой кислоты, а полученная в результате диастереомерная смесь может быть затем разделена стандартными методами, и после удаления хирального вспомогательного соединения, может быть получен нужный энантиомер.
Ojima
и др., в "J. Org. Chem. 56: 1681 - 1683, 1991; Tet. hetl, 33: 5737 - 5440, 1992" описывают синтез ряда хиральных азетидинонов формулы (IIIa) и/или их соответствующие -(p-метоксифенил)-аналог, где P
является триизопропилсилильной гидроксизащитной группой; а R5 представляет собой 4-метоксифенил; 3,4-диметилоксифенил; фенил; 4-фторфенил, 4-трифторометилфенил, 2-фурил, 2-фенилэтенил,
2-(2-фурил)этенил, 2-метилпропил, циклогексилметил, изопропил, фенетил, 2-циклогексилэтил, или н-пропил. Другие примеры получения азетидинонов формулы (IIIa) и/или (III) могут быть найдены в
следующих
работах: EP-заявки 0534709 AI, 0534708 AI и 0534707 AI (все три опубликованы 31 марта 1993); PCT-заявка 0 93/06079, опубликованная 1 апреля 1993;
- Bioorgaanic and Medicinal
Chemistu hetters,
3 N 11, pp 2475 - 2478 (1993); и Bioerganic and Medicinal chetistry Zetters, 3, N 11, pp 2479 - 2482 (1993); J. Org. Chem., 58, pp. 1068 - 1075; Tetrahedron hetteis 31, N 44, pp.
6429 - 6432 (1990);
Bioorganic and Medicinal Chenustrg hetters, 3, N 11, pp. 2467 - 2470 (1993); Еврозаявка 552041, опубликованная 21 июля 1993; и наша одновременно рассматриваемая заявка на пат. США
рег. N 092170,
поданная 14 июля 1993. Соответствующие разделы вышеуказанных работ вводятся в настоящее описание посредством ссылки. Другие азетидиноны, подпадающие под определение формулы (III), но
конкретно не
рассматриваемые в вышеуказанных работах, могут быть получены любым специалистом известными способами.
Биологические испытания
Соединения формулы (B) настоящего
изобретения
представляют собой промежуточные соединения, которые могут быть использованы для получения противоопухолевых соединений формулы (A). Кроме того, некоторые соединения, входящие в объем
формулы (B), а
именно, соединения формулы (B), сами по себе могут быть использованы в качестве противоопухолевых агентов. В "Биологической части I", представленной ниже, иллюстрируется
противоопухолевая активность
соединений формулы (A), а в "Биологической части II" (см. ниже) иллюстрируется противоопухолевая активность соединений формулы (B).
Биологическая часть
I
Данные in vitro
- цитотоксичности
Соединения формулы (A) показали цитотоксичную in vitro активность против клеток HCT-116 и HCT-116/M46 карциномы толстой кишки человека. Клетки
HCT-116/M46 были
предварительно подвергнуты селекции на устойчивость против teniposide и на экспрессию фенотипа устойчивости против нескольких лекарственных средств, включая устойчивость против
паклитаксела.
Цитотоксичность в клетках HCT-116 карциномы толстой кишки человека оценивали посредством анализа с использованием XTT 2,3-бис(2-метокси-4-нитро-5-сульфенил)-5-(фениламино)карбонил
2H-тетразолия
гидроксид, как описано в работе D.A.Scudiero и др., "Evaluation of soluble tetrazolium/formazan assay for cell grokth and drug sensitvitu in culture using human ahd other tumor cell
lines", Cancer
Res. -48: 4827 - 4833, 1988. Эти клетки засевали при плотности 4000 клеток на лунку в 96-луночные планшеты для микротитрования, а через 24 часа в лунки добавляли лекарственные средства
и осуществляли
серийные разведения. Клетки инкубировали при 37oC в течение 72 часов, а затем добавляли тетразолиевый краситель, XTT. Дегидрогеназа (фермент), присутствующая в живых клетках,
способствует
восстановлению XTT до его формы, способной абсорбировать световое излучение при 450 нм, которое может быть количественно оценено путем спектрофотометрии. Причем, чем больше оптическая
плотность, тем
больше число живых клеток. Полученные результаты выражали в IC50-значениях, которые представляют собой концентрацию лекарственного средства, необходимую для ингибирования
пролиферации
клеток (т.е., оптич. пл. при 450 нм) на 50% по сравнению с необработанными контрольными клетками. IC50-величины для характерных соединений, оцененные в данном анализе,
представлены в
таблице 1.
Соединение 7-

Противоопухолевая -in vivo-активность
Гибридным мышам (Balb/CxDBA2F1(CDF1)) подкожно (SC) имплантировали 0,1 мл 2% (мас./об.)-ного состава клеток карциномы легких M109 (как описано W. ROSE ""Evaluation of Madison 109 Lung Carcinoma as Model for Screening Antirumor Drugs", Caneer Treatment Reports" 65, N 3 - 4, pp. 299 - 312 (1981). Группам мышей вводили испытуемые соединения и стандартное лекарственное средство, паклитаксел (внутривенно); причем, каждая группа получала различные дозы испытуемых соединений, и каждое соединение оценивали при трех или четырех различных дозах. Мышей ежедневно наблюдали на выживаемость вплоть до их гибели, либо примерно 75 дней после имплантации опухоли независимо от исхода эксперимента. Одна группа мышей в данном эксперименте оставалась необработанной и служила в качестве контрольной. Один или два раза в неделю также измеряли размер опухоли (в мм), и эти измерения использовали для оценки массы опухоли в соответствии с процедурой, описанной в вышеприведенной работе.
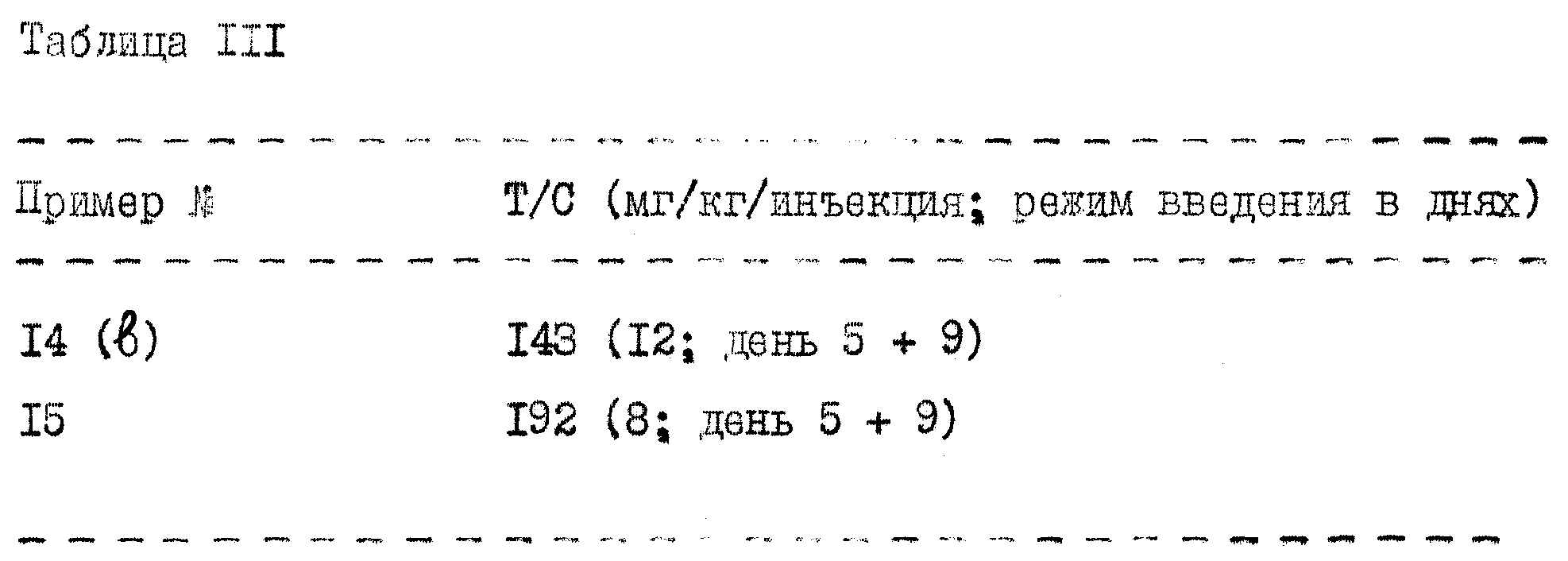
Среднее время выживания мышей, обработанных испытуемым соединение (T), сравнивали со средним временем выживания контрольных мышей (C). Отношение этих двух величин для каждой группы T-группы мышей (т.е., обработанных мышей) умножали на 100 и в виде процентного соотношения (т.е., % T/C) систематизировали в таблице II для наиболее типичных соединений. Кроме того, в таблице II также приводится разница между средним временем выживания для обработанных групп и средним временем выживания для контрольной группы (выраженная в днях, T-C) в случае увеличения опухоли до 1 грамма. Чем больше величины T-C, тем больше замедление первичного роста опухоли. Соединения, показавшие значения %T/C ≥ 125% и/или T-C ≥ 4,0 дня, рассматривались как активные соединения в исследуемой модели M109SC.
Кроме того, соединение примера 3 (в виде триэтаноламиновой соли) испытывали на моделях опухолевого ксенотрансплантата мыши и человека (M109, A2780/cDDP - карцинома яичника человека, обладающая резистентностью к цисплатину; и HTC-116 - карцинома толстой кишки человека) по сравнению с паклитаксела, используемого в качестве позитивного контроля. A2780/сDDP - модель описана в работе Rose и Basler, in vivo 1990, 4: 391-396; HCT-116-модель описана Rose и Basler in vivo, 1989, 3:249-254. M109 подкожно вводили Balb /C-мышам, и подкожно имплантировали CDF1 - мышам для анализа на противоопухолевую активность. A2780/cDDP и HCT-116 культивировали у "голых" мышей для проведения экспериментов по пассированию (каждые 2-3 недели) и экспериментов по терапии. Соединение примера 3 вводили i.v. в воде, либо перорально в воде, содержащей несколько капель Твина 80, а паклитаксел либо суспендировали в воде, содержащей Твин 80, либо растворяли в Кремофоре/этаноле (50%/50%), и разводили солевым раствором. В испытаниях с подкожным введением M109-опухоли, обработку проводили на 4-й день после имитации опухоли и ежедневно в течение 5-ти последующих дней. В испытаниях с использованием опухолевого ксенотрансплантата человека, соединения вводили 5 раз: - один раз в день на каждый второй день (т.е., через день), начиная с того момента, когда размер опухоли составлял от 50 до 100 мг.
В одном M109-эксперименте, соединения примера 3, введенные внутривенно достигали максимальных значений % T/C = 155 (T-C=19 дней) при 36 мг/кг/инъекц. (для паклитаксела % T/C= 132 (T-C = 13 дней) при 36 или 18 мг/кг/инъекц. В том же самом эксперименте, соединения примера 3, вводимые перорально достигали максимальных значений % T/C = 158 (T-C = 22,8 дней) при дозе 160 мг/кг, тогда как паклитаксел при той же дозе (наиболее высокая исп. доза) (суспендированный в воде и Твине 80) не показал какой-либо активности. В другом M109-эксперименте, соединение примера 3, введенное внутривенно, продуцировало макс. значение % T/C = 170 (T-C = 17 дней) при дозе 48 мг/кг/инъекц. (для паклитаксела, макс.% T/C = 167 (T-C = 14 дн.) при 48 или 36 мг/кг/инъекц.). В том же самом эксперименте, пероральное введение содениения примера 3 давало макс. % T/C = 172 (T-C = 17 дней) при дозе введения 200 мг/кг, тогда как паклитаксел, растворенный в Кремофоре/этаноле/солевом растворе, не показывал активности при дозе 60 мг/кг/инъекц. В этом эксперименте, паклитаксел, растворенный в Кремофоре/этаноле/солевом растворе, не может быть введен в дозе более, чем 60 мг/кг/инъекц., из-за плохой растворимости и токсичности.
В A2780/cDDP-эксперименте, соединения примера 3, вводимые внутривенно, показали макс. величину T-C 29,8 дней при дозе 36 мг/кг/инъекц. (для паклитаксела, макс. T-C = 26,3 дн. при 36 мг/кг/инъекц.) Пероральное введение соединения примера 3 давало макс. T-C = 20 дней при дозе введения 160 мг/кг. В HCT-116-эксперименте, внутривенное введение дозы 24 или 36 мг/кг/инъекц. паклитаксела приводило к 6 извлечениям из 7 или 6 извлечениям из 8 отработанных мышей, соответственно; а пероральное введение соединения примера 3 при дозе введения 160 или 240 мг/кг давало 6 или 7 извлечений из 8 обработанных мышей, соответственно. "Извлечение" означает отсутствие опухоли на 80 день после имплантации опухоли.
Триэтаноламиновая соль соединения примера 1 также показала пероральную активность в экспериментах с M109- и HCT-116-моделями.
Из литературы хорошо известно, что иногда наблюдаются небольшие изменения в противоопухолевой активности в зависимости от конкретно используемой солевой формы.
Фармацевтически приемлемая соль фосфонооксиметиловых эфиров таксоновых производных формулы (A) обладает более высокой водорастворимостью по сравнению с паклитакселом, что позволяет использовать вышеуказанные соли для изготовления более удобных для употребления лекарственных композиций. Не претендуя на какую-либо конкретную теорию, можно лишь отметить, что фосфонооксиметиловые эфиры настоящего изобретения являются, очевидно, предшественниками паклитаксела или его производных, где фосфонооксиметильная часть этих предшественников отщепляется под действием in vivo-фосфотазы, что приводит к последующему продуцированию исходного соединения.
Биологическая часть II
Мышиная M109-модель
Гибридным мышам (Balb/cxDBA12 F1)вводили
внутрибрюшинно (как описано William Roseb'' Evaluation of Madison 109 Lung Carеinoma as a Model for Screening Antitumor Drugs, Cancer Treatment Reports, 65, N 3-4 (1981)) 0,5 мл 2% (мас./об.)-состава
карциномы легких M109.
Мышей обрабатывали испытуемым соединение путем внутрибрюшинных инъекций данного соединения в различных дозах, вводимых либо через 1, 5 или 9 дней после имплантации опухоли, либо на 5-й и 8-й день после имплантации. Мышей ежедневно наблюдали на выживаемость в течение приблизительно 75-90 дней после имплантации опухоли. Одна группа мышей в данном эксперименте оставалась неотработанной и служила в качестве контрольной группы. Среднее время выживания мышей, обработанных испытуемым соединением (T), сравнивали со средним временем выживания контрольных мышей (C). Отношение этих двух величин для каждой обрабатываемой группы мышей умножали на 100 и в виде процентного соотношения (т.е., % T/C) систематизировали в таблице III для наиболее типичных соединений формулы (B').
Как показано выше, соединения формул (A) и (B') настоящего изобретения являются эффективными опухоль-ингибирующими средствами, а поэтому они могут быть использованы в медицине и/или ветеринарии. Таким образом, в другом варианте своего осуществления, настоящее изобретение относится к способу ингибирования роста опухолей у человека и/или других млекопитающих, заключающемуся в том, что указанному человеку или млекопитающему, имеющему опухоль, вводят эффективное опухоль-ингибирующее количество соединения формулы (A) или (B).
Соединения настоящего изобретения, имеющие формулы (A) и (B), могут быть использованы в соответствии с той же лечебной схемой, которая обычно используется для паклитаксела, а поэтому специалист-онколог может самостоятельно, без излишнего экспериментирования, определить нужную дозу соединения настоящего изобретения при соответствующей схеме приема. Доза, способ и схема введения соединений настоящего изобретения не имеют каких-либо конкретных ограничений, и могут варьироваться в зависимости от типа используемого соединения. Так, например, соединения настоящего изобретения могут быть введены любым подходящим способом (предпочтительно парентерально) в дозе, составляющей, например, от около 1 до около 10 мг/кг веса тела, или от около 20 до около 500 мг/м2. Соединения настоящего изобретения могут быть также введены перорально в дозе, составляющей от около 5 до около 500 мг/кг веса тела. Конкретно используемая доза может варьироваться в зависимости от конкретно используемой композиции, способа ее введения, конкретного места введения, от самого индивидуума-хозяина, и от типа обрабатываемой опухоли. При отделении нужной дозы введения, необходимо учитывать и многие другие факторы, которые могут оказывать влияние на действие лекарственного средства, например, такие факторы, как возраст, вес тела, пол, пищевой рацион, и физическое состояние пациента.
Настоящее изобретение также относится к фармацевтическим композициям (лекарственным препаратам), содержащим эффективное опухоль-ингибирующее количество соединения формулы (A) или (B) в сочетании с одним или несколькими фармацевтически приемлемыми носителями, наполнителями, разбавителями или адъювантами. В качестве примеров изготовления композиций настоящего изобретения могут быть взяты примеры изготовления препаратов паклитаксела или его производных, описанные в патентах США NN 4960790 и 4814470. Например, соединения настоящего изобретения могут быть изготовлены в виде таблеток, пилюль, порошковых смесей, капсул, инъекцируемых растворов, суппозиториев, эмульсий, дисперсий, пищевых добавок, и других форм. Они могут быть также изготовлены в виде стерильных твердых композиций, например, лиофилизованных препаратов, в сочетании, если это необходимо, с другими фармацевтически приемлемыми наполнителями. Такие твердые композиции могут быть затем разведены стерильной водой, физиологическим раствором, или смесью воды и органического растворителя, такого, как пропиленгликоль, этанол, и т.п., или какой-либо другой стерильной инъецируемой средой непосредственно перед парентеральным введением.
Типичными примерами фармацевтически приемлемых носителей являются манит, мочевина, декстраны, лактоза, картофельный и кукурузный крахмалы, стеарат магния, тальк, растительные масла, полиалкиленгликоли, этилцеллюлоза, поли(винилпиролидон), карбонат кальция, этилолеат, изопропилмиристат, бензилбензоат, карбонат натрия, желатин, карбонат калия, салициловая кислота. Фармацевтическая композиция может также содержать нетоксичные добавки, такие как, эмульгаторы, консерванты, смачивающие агенты, и т.п., как например, сорбитан-монолаурат, олеат триэтаноламина, моностеарат полиоксиэтилена, трипальмитат глицерина, сульфосукцинат диоктилнатрия, и т.п.
В нижеприведенных экспериментальных процедурах, все температуры даны в градусах Цельсия, если это не оговорено особо. Спектральные данные ядерного магнитного резонанса (ЯМР) относятся к химическим сдвигам (о), выраженных в миллионных долях (м.д.) по отношению к тетраметилсилану (TMS), используемому в качестве стандарта. Относительные площади, указанные для различных сдвигов в данных протонного ЯМР, соответствуют числу атомов водорода конкретного функционального типа в молекуле. Природа сдвигов, а также их мультиплетность обозначены следующим образом: (шир.с) - широкий синглет, (шир.д.) - широкий дублет, (шир.кв.) - широкий квартет, (с) - синглет, (м) - мультиплет, (д) - дублет, (кв.) - квартет, (т) - триплет, (дд) - дублет дублетов, (дт) - дублет триплетов и (дкв) - дублет квартетов. Для получения ЯМР-спектров использовались следующие растворители: ацетон-d6 (дейтерированный ацетон), ДМСО-d6 (пердейтеродиметилсульфоксид), D2O - тяжелая вода, CDCl3 - (дейтерохлороформ) и другие стандартные дейтерированные растворители. В ИК-спектрах указываются лишь поглощение в области определенных волновых чисел (см-1), которое позволяет идентифицировать функциональные группы соединения.
"Целит" является торговым знаком для диатомовой земли, поставляемой корпорацией.
В нижеприведенных примерах используются следующие сокращения: МС (MS) - масс-спектрометрия; ВРМС (HRMS) - масс-спектрометрия высокого разрешения; Ac - ацетил; Ph - фенил, об./об. - объем/объем; FAB - спектрометрия путем бомбардировки быстрыми атомами; NOBA - m - нитробензиловый спирт, мин - минуты, час - часы (h, hr), NIS N - йодосукцинимид; BOC - т-бутоксикарбонил; CBZ или Cbz - бензилоксикарбонил; Bn - бензил; Bz - бензоил; TES - триэтилсилил; ДМСО (DMCO) - диметилсульфоксид; ТГФ (THF) - тетрагидрофуран; HMDS - гексаметилдисилазан.
Получение исходных материалов
Получение различных
специфических исходных материалов,
используемых в последующем получении соединений формулы (A), проиллюстрированы ниже.
Получение 1:
10-Дезацетоксипаклитаксел

(a) 2',7-O-бис(2,2,2-трихлороэтоксикарбонил)-10-деацетилпаклитаксел
10-Деацетилпаклитаксел (140 мг, 0,173 мМ) в безводном дихлорметане (3,5 мл), при 0oC, обрабатывали пиридином (0,028 мл, 0,346 мМ) и трихлороэтилхлороформатом (0,0724 мл, 0,260 мМ). После выдерживания смеси в течение 1 часа при той же температуре, холодную баню удаляли и смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали, а полученный остаток хроматографировали на силикагеле (элюент: 30-50% этилацетат в гексане), и получали целевое соединение в виде пены (92,3 мг, 45%). После дальнейшего элюирования получали непрореагировавший исходный материал (35 мг, 25%) и 2, 10-О-бис(2,2, 2-трихлороэтоксикарбонил)- 10-деацетилпаклитаксел (выход - 16%).
(b) 2',7-O-бис(2,2,2-трихлорэтоксикарбонил)-10-дезацетокси-11,12- дигидропаклитаксел-10,12(18)-диен
Продукт,
полученный в стадии (a) (92,3 мг, 0,079 мМ) в безводном дихлорметане (2 мл) обрабатывали при комнатной температуре 1,1,2-трифторо-2-хлоротриэтиламином (0,0384 мл, 0,238 мМ). Полученный
раствор
перемешивали в течение ночи. Растворитель выпаривали, и остаток очищали с помощью колоночной хроматографии (элюент: 25% этилацетат в гексане), в результате чего получали целевое соединение в
виде
белого порошка (42,8 мг, 47,3%).
(c) 10-Дезацетокси-11,12-дигидропаклитаксел-10,12(18)-диен
Продукт стадии (b) (39 мг, 0,034 мМ) растворяли в метаноле (0,5 мл) и
уксусной
кислоте (0,5 мл), а затем обрабатывали промытой кислотой цинковой пылью (66,4 мг, 1,020 мМ). Суспензию нагревали при 40oC в течение часа, фильтровали, а фильтрат выпаривали.
Остаток
хроматографировали (элюент: 60% этилацетат/гексан), и получали целевое соединение в виде пены (22 мг, 81%).
(d) 10-Дезацетоксипаклитаксел
Продукт стадии (c) (22 мг,
0,028 мМ)
в этилацетате (0,7 мл) гидрировали при атмосферном давлении в присутствии палладированного угля (10%, 14,7 мг, 0,014 мМ Pd). После выдерживания смеси в течение 5,5 часа при комнатной
температуре, ее
фильтровали (промывая этилацетатом), выпаривали и подвергали хроматографии (60% этилацетат в гексане). В результате этой процедуры получали целевой продукт (15,0 мг, 688) в виде белой
пены.
Получение 2:
7-Дезокси-7α-фторопаклитаксел

(a) 2'-O-Бензилоксикарбонил-7-дезокси-7α-фторопаклитаксел
Трифторид диэтиламиносеры (DAST, 18,7 мкл, 0,141 мМ) растворяли в безводном дихлорметане (0,5 мл), и этот раствор охлаждали до 0oC. Затем добавляли раствор 2'-


(b)
7-Дезокси-7-фторопаклитаксел
Смесь продукта стадии (a) (89 мг) растворяли в этилацетате (3 мл), а затем слегка перемешивали при давлении водорода в 1 атм, в присутствии палладированного угля
(10% Pd, 29 мг, 0,027 мМ). Через 12
часов растворитель удаляли, а остаток очищали с помощью хроматографии на силикагеле (элюент: 40% этилацетат в гексане), в результате чего получали 67,7 мг целевого
соединения - 8-дезметил-7,
8-циклопропапаклитаксела.
Для выделения 7-дезокси-7α-фторопаклитаксела и 8-дезметил-7,8-циклопропапаклитаксела использовали следующий ВЭЖХ-метод.
Оборудование:
Насос: Серия 4 РЕ
Колонка: Shandon Hypercarb (графитированный углерод), 7 мкм, 100 х 4,6 мм, # 59864750 (данные о размерах препаративных колонок могут
быть получены от
фирмы Keystone Scietific, Bellefonte, PA)
Инжектор: PEISS - 100
Детектор: HP-104ОM
Условия
Подвижная фаза: метиленхлорид: гексан = 85:15.
Выделение не дает потерь
при отношении: метиленхлорид: гексан: изопропиловый спирт = 80:19:1
Скорость потока: 2,5 мл/мин
Детектор: 254 нм
Разбавитель: Образец, растворенный
в метиленхлориде.
Получение 3:
7-Дезокси-7-фторобаккатин III
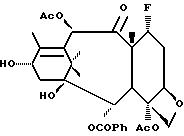
В сухую колбу, в атмосфере инертного газа, добавляли 2'-O-(бензилоксикарбонил)паклитаксел (4 г, 4 мМ) и безводный толуол (80 мл). Полученную суспензию перемешивали при комнатной температуре, добавляли при этом по капле безводный тетрагидрофуран (16 мл) до образования бесцветного раствора. Вышеуказанный раствор охлаждали до -78oC в бане из сухого льда и ацетона, а затем обрабатывали трифторидом диэтиламиносеры (DAST, 1,2 мл, 2,5 экв.). Реакционную смесь оставляли для перемешивания в течение 16 часов, постепенно нагревая до комнатной температуры. Полученную суспензию фильтровали, и фильтрат (разбавленный 30 миллилитрами этилацетата) промывали насыщенным водным бикарбонатом натрия, а затем солевым раствором. Органическую фракцию осушали сульфатом магния и концентрировали, в результате чего получали неочищенный продукт в виде белой пены. Этот неочищенный материал частично очищали с помощью колоночной хроматографии на силикагеле (элюент: 10% CH3CN в CH2Cl2) и получали 1,45 г смеси


Вышеописанную смесь (1,45 мг) растворяли в этилацетате (60 мл) и обрабатывали 300 миллиграммами палладированного угля. После 4-часового встряхивания в атмосфере водорода при 50 фунт/кв.дюйм (3,515 кг/см2), реакционную смесь продували, фильтровали через узкую пробку из силикагеля и концентрировали. Таким образом, получали нужный продукт в виде смеси 7-дезокси-7α -фторопаклитаксела и 8-дезметил-7,8-циклопропапаклитаксела в качестве белой пены (1,24 г, выход - 99%, 90:10, определенной с помощью1H ЯМР-анализа). Полученную смесь растворяли в сухом метиленхлориде (30 мл) и обрабатывали борогидридом тетрабутиламмония (745 мг, 2,9 мМ, 2 экв), а затем оставляли для перемешивания в течение 6 часов. Реакцию гасили путем добавления уксусной кислоты (1 мл), разбавляли еще 30 миллилитрами метиленхлорида, и после этого промывали насыщенным водным раствором бикарбонатом натрия. Органическую фракцию осушали сульфатом магния и концентрировали. Неочищенную маточную смесь замещенного таксана частично очищали с помощью колоночной хроматографии на силикагеле (элюент: 10% CH3CN в CH2Cl2) и получали смесь (90: 10, определенную с помощью1H-ЯМР-анализа) 7-дезокси-7α-фторобаккатина III и 8-дезметил-7,8-циклопропабаккатина III (510 мг, 60%) в виде белой пены. Полученное пенообразное вещество кристаллизовали из горячего изопропанола и получали 7-дезокси-7α-фторобаккатин III в виде небольших белых игольчатых кристаллов (выход - 410 мг) с т.пл. 234-236oC (с разлож.).
Получение 4:
10-Дезацетокси-7-дезокси-7α-фторопаклитаксел

(a) 2'-O-Бензилоксикарбонил-10-дезацетоксипаклитаксел
10-Дезацетоксипаклитаксел (27 мл, 0,034 мМ) в дихлорметане (1 мл) обрабатывали бензилхлороформатом (0,0146 мл, 0,102 мМ), а затем диизопропилэтиламином (0,0177 мл, 0,102 мМ). Реакционную смесь перемешивали при 0oC в течение 45 минут и при комнатной температуре в течение 12 часов. После выпаривания растворителя и хроматографирования на силикагеле (элюент: 40% этилацетат в гексане), получали 25,5 мг (выход - 81%) целевого соединения в виде пены.
(b) 10-Дезакцетокси-7-дезокси-7α
-фторопаклитаксел
Продукт стадии (a) (25,5 мг, 0,028 мМ) в дихлорметане (0,8 мл), при 0oC, обрабатывали трифторидом
диэтиламиносеры (0,0071 мл, 0,055 мМ). После выдерживания в
течение 45 минут при 0oC, реакционную смесь оставляли для реакции на 5 часов при комнатной температуре. Растворитель выпаривали,
а смесь подвергали хроматографии и получали
Получение 5:
10-Деацетил-7-дезокси-7α-фторопаклитаксел

Раствор 2', 10

Получение 6:
7-Дезоксибаккатин III

(a)

Баккатин III (750 мг, 1,278 мМ) растворяли в сухом тетрагидрофуране (20 мл) и добавляли одну порцию имидазола (8,7 мг, 0,128 мМ). После этого добавляли (при комнатной температуре) гидрид натрия (50%) в минеральном масле, 77 мг, 1,597 мМ). После прекращения выделения газа (10 мин), добавляли (одной порцией) 4,6 мл сероуглерода. После отстаивания в течение 3 часов при комнатной температуре, желтый раствор обрабатывали метилиодидом (0,238 мл, 3,835 мМ) и перемешивали в течение ночи. После обработки этилацетатом и водой, получали целевое соединение в виде неочищенного маслообразного вещества.
Альтернативная процедура
Баккатин III (394 мг, 0,672 мМ) растворяли в
тетрагидрофуране (5 мл) и сероуглероде (1 мл). Затем к этому раствору добавляли гидрид натрия
(40,3 мг, 60%, 1,009 мМ). После этого также добавляли каталитическое количество имидазола. Полученную
реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа, а затем добавляли
метилиодид (122,8 мкл, 2,016 мМ). Через 40 минут, растворитель удаляли в вакууме, и остаток
хроматографировали на силикагеле (элюент: 20%-50%-60% этилацетата в гексанах), в результате чего получали
целевой продукт (260 мг, выход - 57,2%) вместе с 7-епибаккатином (98,5 мг, 25%).
(b)

Продукт, полученный в стадии (a) (в виде неочищенного маслообразного вещества) растворяли в сухом диметилформамиде (5 мл) и обрабатывали имидазолом (870 мг, 12,78 мМ) и триэтилсилилхлоридом (2,10 мл, 12,78 мМ) при комнатной температуре в течение 15 часов. После этого добавляли воду, и раствор экстрагировали в этилацетате. Органический слой промывали тщательно водой, а затем осушали. После флеш-хроматографии на силикагеле (элюент: 20% этилацетат в гексане) получали целевое соединение в виде стеклообразного твердого вещества (выход - 20% из двух стадий; 209 мг).
Альтернативная процедура
Продукт,
полученный в стадии (a) (193,4 мг, 0,286 мМ) растворяли в сухом диметилформамиде (2,86 мл). К этому раствору
добавляли имидазол (77,9 мг, 1,14 мМ), а затем триэтилсилилхлорид (192 мкл, 1,14 мМ).
Полученную реакционную смесь перемешивали в течение ночи при комнатной температуре. Через 12 часов реакционную
смесь разбавляли 150 миллилитрами этилацетата. Органический слой промывали водой (3 х 10
мл), солевым раствором (1 х 10 мл), осушали и концентрировали в вакууме. Полученный остаток хроматографировали
на силикагеле (элюент: 20% этилацетат в гексане) и получали целевой продукт (163 мг,
выход - 72,0%).
(c) 7-Дезокси

Продукт, полученный в стадии (о) (182 мг, 0,230 мМ) в сухом бензоле (5 мл) нагревали до 80oC в присутствии гидрида трибутилолова (0,310 мл, 1,150 мМ) и 2', 2'-азобисизобутиронитрила (AIBN 10 мг). Через 3 часа раствор оставляли для охлаждения, а растворитель выпаривали в вакууме. Полученный остаток хроматографировали на силикагеле (элюент: 20% этилацетат в гексане) и получали целевое соединение в виде маслообразного продукта.
(d) 7-Дезоксибаккатин III
Продукт,
полученный в стадии (c) растворяли в тетрагидрофуране (5 мл) и обрабатывали фторидом тетрабутиламмония (1M в
тетрагидрофуране, 0,50 мл, 0,50 мМ) в течение 2 часов при комнатной температуре. Этот
раствор разбавляли этилацетатом, промывали водой и солевым раствором, а затем подвергали хроматографии на
силикагеле (элюент: этилатетат/гексан, 1:1), в результате чего получали целевое соединение в
виде белого стеклообразного твердого вещества (63 мг, выход - 58% от двух стадий).
Получение 7:
10-Дезацетоксибаккатин III

(a) 10-Деацетил-10-


(b) 10-Дезацетокси

Продукт, полученный в стадии (a) (1119 мг, 0,135 мМ) растворяли в сухом толуоле (3 мл) и обрабатывали 2 миллиграммами AIBN. Полученный раствор обезгаживали безводным азотом, после чего добавляли гидрид трибутилолова (0,055 мл, 0,202 мМ). Затем раствор нагревали в течение часа при 90oC. После этого растворитель выпаривали, а остаток хроматографировали на силикагеле (элюент: 40% этилацетат в гексане) и получали 87 мг (выход - 99%) целевого соединения в виде бесцветной пены.
(c)
10-Дезацетоксибаккатин III
Продукт, полученный в стадии (b) (120 мг, 0,187 мМ) растворяли в ацетонитриле (3,5 мл), и раствор охлаждали
до -10oC. К раствору добавляли
концентрированную соляную кислоту (36%, 0,060 мл), и этот раствор перемешивали в течение 30 минут. Полученную смесь разбавляли 75 миллилитрами этилацетата,
промывали насыщенным водным бикарбонатом
натрия и солевым раствором, а затем осушали и концентрировали. Остаток очищали с помощью флеш-хроматографии на двуокиси кремния (элюент: 70% этилацетат в
гексане) и получали 10-деацетилоксибаккатин
III в виде пены (75 мг, выход - 76%).
Получение 8:
10-Деацетокси-7-дезоксибаккатин III

(a)

10-Дезацетоксибаккатин III (75 мг, 0,142 мМ) растворяли в сухом тетрагидрофуране (2 мл) и сероуглероде (0,5 мд). Затем к раствору добавляли гидрид натрия (60% в минеральном масле, 8,5 мг, 0,213 мМ), и смесь перемешивали при комнатной температуре в течение 2 часов. После добавляли иодометана (0,026 мл, 0,426 мМ), реакционную смесь оставляли в течение ночи для продолжения реакции. Растворитель удаляли, и остаток очищали с помощью хроматографии на силикагеле (элюент: 50 - 70% этилацетата в гексане), в результате чего получали целевое соединение в виде пены (46,4 мг, выход - 53%).
(b) 10-Дезацетокси-7-дезоксибаккатин III
Продукт, полученный в стадии (a) (36 мг, 0,058 мМ) в течение 3 часов нагревали
с обратным холодильником в бензоле (1 мл) в присутствии
AIBN (2 мг) и гидрида трибутилолова (0,079 мл, 0,290 мМ), и в атмосфере аргона. Реакционную смест концентрировали, а остаток подвергали
флеш-хроматографии на силикагеле (элюент: 40% этилацетат в
гексане) с последующим отделением от других компонентов с помощью ЖХВД (жидкостная хроматография высокого давления), в результате чего
получали целевое соединение в виде пены (выход - 56%; 16,8
мг).
Альтернативная процедура
К раствору 7-0-[(метилтио)карбонотиоил]-13-


К раствору продукта вышеописанной стадии (16,0 мг, 0,255 мМ) в сухом тетрагидрофуране (2 мл) при комнатной температуре, добавляли фторид тетрабутиламмония (766 мкл, 1М, 0,766 мМ). Реакционную смесь перемешивали 1 час при комнатной температуре. Растворитель удаляли, а остаток хроматографировали на силикагеле (элюент: 50 - 70% этилацетат в гексане), в результате чего получали нужный целевой продукт (115 мг, выход - 87,9%).
Получение 9:
(3R,
4S)-1-т-Бутоксикарбонил-4-фенил-3-триэтилсилилокси-2-азетидинон

К перемешенному раствору (3R, 4S)-4-фенил-3-триэтилсилилокси-2-азетидинона (2,200 г, 7,92 мМ) в сухом тетрагидрофуране, добавляли (при 0oC и в атмосфере аргона) N, N-диизопропилэтиламин (1,65 мл, 9,510 мМ, 1,2 экв). Полученный раствор перемешивали 5 минут с последующим добавлением ди-т-бутилдикарбоната (2,080 г, 9,510 мМ, 1,2 экв) и 4-диметиламинопиридина (193,6 мг, 1,581 мМ, 0,20 экв). Реакционную смесь перемешивали 60 минут при 0oC, а затем разбавляли этилацетатом (25 мл). После этого раствор промывали солевым раствором, 10%-ным NaHCO3, 10% раствором соляной кислоты, осушали сульфатом магния и концентрировали с получением неочищенного продукта (маслообразного вещества). Это соединение очищали с помощью флэш-хроматографии на силикагеле (элюент: 15% этилацетат в гексане) и получали целевое соединение в виде белого твердого вещества (2,4 г, выход - 83%).
Получение 10:
(±)-цис-3-Ацетилокси-4-фенилазетидин-2-он

(a) В литровую 3-горлую круглодонную колбу, снабженную термометром, магнитной мешалкой и капельной воронкой, добавляли гидробензамид (30,00 г, 100,5 мМ) и этилацетат (150 мл). После перемешивания в атмосфере аргона, реакционную смесь охлаждали до 5oC и добавляли триэтиламин (16,8 мл, 121 мМ). После этого по капле, в течение 90 минут, добавляли раствор ацетоксиацетилхлорида (12,4 мл, 116 мМ) в этилацетате (300 мл). После выдерживания в течение 16 часов при той же температуре, реакционную смесь оставляли для нагревания до комнатной температуры (1,5 часа) и переносили в делительную воронку. Органический слой последовательно промывали насыщенным водным NH4Cl (150 мл, 100 мл), насыщенным водным NaHCO3 (120 мл) и солевым раствором (120 мл). В целях характеризации, целевое соединение может быть выделено на этом этапе путем осушки органической фазы сульфатом магния, фильтрации и удаления растворителя в вакууме. Таким образом, получали неочищенный (±)-цис-3-ацетилокси-1-[(фенил)(бензилиденимино)метил] -4-фенил-азетидин-2-он (с количественным выходом) в виде красного стеклообразного продукта.
(b) Раствор соединения, полученного в части (a) в этилацетата (500 мл), в потоке аргона, осторожно переносили в 2, 0-литровую колбу Парра, содержащую 6,00 г 10% палладия на активированном угле. Эту смесь обрабатывали водородом (4 атм) в течение 20 часов, после чего катализатор удаляли путем фильтрации через слой Целита. Осадок на фильтре суспендировали в этилацетате (200 мл), перемешивали (10 минут) и фильтровали. После этого осадок на фильтре промывали этилацетатом (100 мл), а фильтраты объединяли. Органический слой промывали 10% соляной кислотой (300 мл), и оба слоя фильтровали через воронку из спеченного стекла для удаления белого осадка (дибензиламин HCl), который промывали 100 миллилитрами этилацетата. Затем фазы отделяли, и органический слой промывали еще одной порцией 10% соляной кислоты (200 мл). Объединенные 10% HCl-промывки снова экстрагировали этилацетатом (200 мл), а объединенные органические слои промывали насыщенным водным NaHCO3 (300 мл) и солевым раствором (250 мл). Органический слой осушали сульфатом магния, фильтровали и концентрировали в вакууме до конечного объема 75 мл. Эту смесь охлаждали до 4oC и осажденный продукт выделяли путем фильтрации. Осадок на фильтре промывали гексаном (200 мл) и получали 16,12 г (полный выход из гидробензамида - 78,1%) целевое осаждение в виде белых игольчатых кристаллов, т. пл. 150 - 151oC.
Получение 11:
(±
)-цис-3-Триэтилсилилокси-4-(2-фурил)-N-т-бутоксикарбонилазетидин-2-он

(a) Осуществляли процедуру, описанную в получении 10 (часть (a), за исключением того, что вместо гидробензамида использовали гидрофурамид [(т.е. , 2-фурил-CH-(N= CH-2фурил)2] и реакцию проводили на шкале 18, 6 мМ (вместо 100 миллимоль). Таким образом, в результате реакции гидрофурамида (5,00 г, 18,6 мМ), триэтиламина (3,11 мл, 22,3 мМ) и ацетоксиацетилхлорида (2,30 мл, 21,4 мМ), получали 6,192 г (выход -90,4% (±)-цис-3-ацетилокси-1- [(2-фурил)-2-фурилметиленимино)метил] -4-(2-фурил)азетидина-2-она в виде бледного красного сиропообразного вещества.
(b) Осуществляли процедуру, описанную в получении 10 (части b), за исключением того, что, указанный продукт выделяли с помощью препаративной ТСХ и реакцию осуществляли в масштабе 2,7 мМ исходя из первоначального количества гидрофурамида. Таким образом, неочищенный продукт, полученный в части (a), описанной выше, снова растворяли в этилацетате (50 мл) и добавляли 150 мг 10% палладия на активированном угле. Неочищенное твердое вещество очищали с помощью препаративной ТСХ (2 мм силикагеля, элюент: этилацетет/гексан, 1 : 1) и получали 386 мг (полный выход из гидрофурамида составлял 65,8%) (± )-цис-3-(ацетилокси)-4-(2-фурил)азетидин-2-она в виде желтого твердого вещества, которое затем перекристаллизовывали из этилацетата/гексана, т.пл. 118 - 119oC.
(c) Соединение, полученное в вышеописанной части (b) (3,78 г, 19,4 мМ) в 60% метаноле, перемешивали с карбонатом калия в течение 90 минут (20 мг, 0,014 мМ), и раствор нейтрализовали смолой DOWEX 50W-X8 и фильтровали. Фильтрат концентрировали, а остаток растворяли в 80 миллилитрах безводного ТГФ и перемешивали (при 0oC) с имидазолом (1,44 г, 21,2 мМ) и TESCT (3,4 мл, 20,2 мМ) в течение 30 минут. Раствор разбавляли этилацетатом, промывали солевым раствором, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 3 : 1) и получали 4,47 г (выход - 86%) (±)-цис-3-триэтилсилилокси-4-(2-фурил)-азетидин-2-она в виде бесцветного маслообразного вещества.
(d) Продукт части (c) (2,05 г, 7,7 мМ) в 30 мл дихлорметана перемешивали при 0oC с диизопропилэтиламином (1,5 мл, 8,6 мМ) и ди-т-бутилдикарбонатом (2,0 г, 9,2 мМ) в дополнение к каталитическому количеству диметиламинопиридина (DMAP). Полученный раствор разбавляли дихлорметаном, промывали солевым раствором, осушали сульфатом магния и концентрировали. Полученный остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 8 : 1) и получали 2,0 г (выход - 70%) целевого соединения в виде воскообразного твердого продукта.
Рацемическая смесь, полученная в части (b) может быть использована в качестве субстрата для ферментного гидролиза, осуществляемого с помощью липазы, такой как PS-30, поставляемой фирмой Pseudomonas sp. (Amano International Co. ) с получением (3R, 4R)-3-гидрокси-4-(2-фурил)-азетидин-2-она. Метод ферментного разделения с использованием липазы и других ферментов раскрывается в нашей одновременно рассматриваемой заявке на патент США, рег. N 092170, поданной 14 июля 1993 года, которая целиком вводится в настоящее описание посредством ссылки.
Процедуры в части (c) и части (d) осуществляли с использованием (3R, 4R)-3-гидрокси-4-(2-фурил)-азетидин-2-она, в результате чего получали (3R, 4R)-N-(т-бутоксикарбонил)-3-триэтилсилилокси-4-(2-фурил)азетидин-2-он.
Получение 12:
(±
)-цис-3-Триэтилсилилокси-4-(2-тиенил)-N-т-бутоксикарбонилазетидин-2-он
(a) Осуществляли процедуру, описанную в получении 10 (стадия (a)), за исключением того, что вместо гидробензамида
использовали гидротиенамид [(т. е. , 2-тиенил-CH-(N=CH-2-тиенил)2]. Таким образом, гидротиенамид (30 г, 94,7 мМ), триэтиламин (15,84 мл, 114 мМ) и ацетоксиацетилхлорид (11,6 мл, 108 мМ)
подвергали реакции, и получали (±)-цис-3-ацетилокси-1-[(2-тиенил)(2-триенилметиленимино)метил] -4-(2-тиенил)азетидин-2-он в виде вязкого маслообразного вещества.
(b) К перемешанному раствору продукта, полученного в части (a) (0,431 г, 1,03 мМ) в дихлорметане (2,93 мл) (при 25oC) добавляли одной порцией 70% водный раствор уксусной кислоты (0,35 мл ледяной кислоты и 0,15 мл воды). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 2,5 часа. После этого реакционную смесь разбавляли 50 миллилитрами дихлорметана, а затем промывали двумя 75-миллилитровыми порциями насыщенного водного бикарбоната натрия и одной 50-миллилитровой порции насыщенного солевого раствора. Органический экстракт концентрировали в вакууме с получением коричневого маслообразного продукта, который растворяли в минимальном количестве дихлорметана, а затем помещали на колонку с силикагелем, имеющую размер 4 x 0,5 (101,6 x 12,7 мм). В результате элюирования градиентом 10% - 60% EtOAc в гексане, получали менее полярные побочные продукты, а затем (±)-цис-3-ацетилокси-4- (2-тиенил)-азетидин-2-он (0,154 г, выход - 75%) в виде белого твердого вещества.
(c) Раствор продукта, полученного в части (b) (2,5 г, 11,8 мМ) растворяли в метаноле (10 мл) и полученную суспензию оставляли для перемешивания при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли этилацетатом (20 мл) и промывали водой (15 мл). Затем водную фракцию снова несколько раз экстрагировали этилацетатом, и объединенные органические фракции осушали сульфатом магния и концентрировали с получением желтого твердого продукта (выход - 1,7 г). Неочищенный материал растворяли в безводном тетрагидрофуране (20 мл), и раствор охлаждали до 5oC в ледяно/водяной бане. После этого добавляли имидазол (752 мг, 1,1 экв). После перемешивания в течение 5 минут, к раствору по капле добавляли триэтилхлоросилан (1,85 мл, 1, 1 экв. ). Полученную суспензию перемешивали в течение 3 часов при той же температуре, а затем твердые вещества удаляли путем фильтрации. Органическую фракцию промывали водой (2 x 20 мл), осушали сульфатом магния и концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (элюент : гексан/этилацетат, 7 : 3) и получали (± )-цис-3-триэтилсилилокси-4-(2-тиенил)-азетидин-2-он в виде бесцветного твердого вещества (1,5 г, выход - 45%) с т.пл. 70 - 71oC.
Альтернативная процедура
Продукт,
полученный в части (b) (2,0 г, 9,37 мМ) в 40 миллилитрах метанола, перемешивали с карбонатом калия (60 мг, 0,43 мМ) в течение 30 минут, и раствор нейтрализовали смолой DOWEX 50 W-X8 и фильтровали.
Полученный фильтрат концентрировали, а остаток растворяли в 50 мл безводного ТГФ, а затем перемешивали при 0oC с имидазолом (0,85 г, 11,3, мМ) и TESCE (1,9 мл, 12,5 мМ) в течение 30 минут.
Раствор разбавляли этилацетатом, промывали солевым раствором, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 3:1) и получали 2,13 г
(выход - 86%) целевого продукта в виде бесцветного маслообразного вещества.
(d) Раствор продукта, полученного в части (c) (427,7 мг, 1,48 мМ) растворяли в дихлорметане (10 мл) и охлаждали до 5oC в бане из льда и воды. Полученную реакционную смесь обрабатывали каталитическим количеством DMAP, диизопропилэтиламином (TESCI, 0,25 мл, 1,0 экв.), а затем ди-т-бутил-дикарбонатом (388,4 мг, 1,2 экв.). После перемешивания в течение 2 часов при той же температуре, реакцию гасили насыщенным водным бикарбонатом натрия (5 мл), органическую фракцию промывали водой (5 мл), затем осушали сульфатом магния, пропускали через короткую пробку с силикагелем, и концентрировали, в результате чего получали нужный продукт в виде бесцветного маслообразного вещества (523,3 мг, выход - 93%).
Получение 13:
(3R, 4R)-3-Триэтилсилилокси-4-(2-фурил)-N-n-бутилоксикарбонилазетидин-2-он

(3R, 4R)-3-Триэтилсилилокси-4-(2-фурил)азетидин-2-он (0,51 г, 1,91 мМ) в 25 мл дихлорметана, перемешивали с диизопропилэтиламином (0,78 мл, 4, 4 мМ) и N-пропилхлороформатом (4,0 мл, 1,0 М в толуоле), 4,0 мМ) в дополнение к каталитическому количеству DMAP. Полученный раствор перемешивали 1 час, разбавляли дихлорметаном, промывали солевым раствором, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 5:1) и получали 649 мг целевого соединения (выход - 96%).
ИК (KBr): 1822, 1812, 1716, 1374, 1314, 1186, 1018, 1004, 746 см-1.
1H-ЯМР (CDCl3, 300 МГц) δ: 7,39 (м, 1H), 6,35 (м., 2H), 5,08 (ABкв., J = 15,
6, 5,6 Гц, 2H), 4,96 (d, J = 10,0 Гц, 1H), 1,25 (д, J = 7,3 Гц, 3H), 1,17 (д, J = 6,3 Гц, 3H), 0,83 (т, J = 7,8 Гц, 9H), 0,50 (м, 6H);
13C-ЯМР (CDCl3, 75,5 Гц) δ:
165,5, 148,6, 147,8, 142,9, 110,5, 109,9, 77,6, 71,1, 55,9, 21,7, 21,6, 6,3, 4,4;
DCI-MC (M+H) для C17H18NO5Si:
Вычислено: 354,
Найдено:
354.
Получение 15:
±-цис-3-Триэтилсилилокси-4-изобутенил-N-т-бутоксикарбонил-азетидин-2-он
(a) N-4-метокси-N-(3-метил-2-бутенил)-бензоламин

Раствор p-анизидина (5,7 г, 46,3 мМ) растворяли в диэтиловом эфире (100 мл) и обрабатывали каталитическим количеством p-толуолсульфоновой кислоты (10 мг). К этой смеси добавляли одной порцией 3-метил-2-бутеналь (2,67 мл, 50,9 мМ) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем растворитель выпаривали на роторном испарителе при 0,5 Торр. , и получали нужный имин (8,7 г, 100%) в виде коричневого маслообразного вещества.
1H-ЯМР (300 МГц, CDCl3) δ: 8,38 (д., 1H, J = 9,5 Гц), 7,11 (дд., 2H, J = 2,2, 6,7 Гц), 6,88 (дд, 2H, J = 2,2, 6,7 Гц); 6,22 - 6,18 (м, 1H); 3,81 (с, 3H); 2,01 (с. 3H); 1,95 (с, 3H).
(b) ±-цис-N-(4-метоксифенил)-3-ацетилокси-4-изобутенилазетидин-2-он
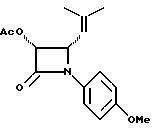
Раствор ацетоксиацетилхлорида (6,9 г, 50,5 мМ) растворяли в этилацетате (100 мл) и охлаждали до -30oC в атмосфере инертного газа. К этому раствору добавляли триэтиламин (7,0 мл, 50,5 мМ) в течение 5 минут. Полученную белую суспензию обрабатывали по капле в течение 20 минут этилацетатом и раствором N-4-метокси-N-(3-метил-2-бутенил)-бензоламина (8,7 г, 40 мл). Затем полученную таким образом зеленовато-коричневую суспензию постепенно нагревали до комнатной температуры в течение 4 часов. После этого суспензию фильтровали через слой целита, и фильтрат промывали водой, а затем солевым раствором. Органическую фракцию осушали сульфатом магния и концентрировали с получением коричневого маслообразного вещества. Неочищенный продукт тщательно очищали с помощью хроматографии на силикагеле (элюент: гексан/этилацетат, 8:2) и получали оранжевое маслообразное вещество, которое отверждали при отстаивании. Затем это вещество перекристаллизовывали из дихлорметана/гексана, и получали нужный продукт в виде бледно-желтого твердого продукта (4,4 г, 32%).
1H-ЯМР (300 МГц, CDCl3) δ: 7,32 (д, 2H, J = 9,1 Гц), 6,86 (д, 2H, J = 9,1 Гц), 5,59 (дд, 1H, J = 3,0, 7,8 Гц), 5,14 - 5,10 (м, 1H), 4,96 (дд, 1H, J = 4,8, 9,3 Гц), 3,77 (с, 3H), 2,11 (с, 3H), 1,81 (с, 3H), 1,78 (с, 3H).
(c) (±
)-цис-3-Ацетилокси-4-изобутенилазетидин-2-он

Раствор (± )-цис-N-(4-метоксифенил)-3-ацетилокси-4-изобутенилазетидин-2-она (4,88 г, 16,2 мМ) растворяли в ацетонитриле (50 мл) и охлаждали до 0-5oC в ледяной бане. К этому раствору добавляли холодный раствор аммониевого нитрата церия (26,6 г, 48,6 мМ, 50 мл) одной порцией). Темно-красную реакционную смесь оставляли для перемешивания в течение 10 минут, и в течение этого времени цвет смеси постепенно приобретал оранжевый оттенок. Холодный раствор переносили в делительную воронку, разбавляли водой, экстрагировали этилацетатом. Органическую фракцию промывали несколькими порциями 10% водного сульфита натрия, а затем насыщенным водным бикарбонатом натрия. После этого органическую фракцию осушали сульфатом магния и концентрировали с получением нужного продукта (2,71 г, 91%) желто-оранжевого цвета, который использовали непосредственно в последующей стадии.
1H-ЯМР (300 МГц, CDCl3) δ: 6,11 (шир.с, 1H); 5,73 (дд, 1H, J = 2,2, 4,7 Гц), 5,12 - 5,08 (м, 1H), 4,63 (дд, 1H, 4,7, 9,1 Гц), 2,09 (с, 3H), 1,75 (с, 3H), 1,67 (с, 3H).
(d) (±)-цис-3-Триэтилсилилокси-4-изобутенилазетидин-2-он
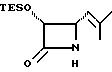
(±)-цис-3-ацетилокси-4-изобутенилазетидин-2-он (1,47 г, 8,0 мМ) растворяли в метаноле (15 мл) и перемешивали с карбонатом калия (110,5 мг, 0,8 мМ) в течение 3 часов при комнатной температуре. Полученный раствор нейтрализовали с силой DOWEX 50W-X8, а затем фильтровали. Фильтрат концентрировали, а неочищенный твердый продукт растворяли в ТГФ (25 мл) и охлаждали до 5oC в ледяной бане. Затем добавляли имидазол (544,0 мг, 8,0 мМ) и после его растворения, по капле, с помощью шприца, добавляли триэтилсилилхлорид (1,34 мл, 8,0 мМ). Полученную суспензию нагревали до комнатной температуры и перемешивали в течение ночи. Раствор фильтровали, а фильтрат промывали водой, а затем солевым раствором. Органическую фракцию осушали сульфатом магния и концентрировали. Полученное таким образом неочищенное твердое вещество очищали с помощью хроматографии на силикагеле (элюент: гексан/этилацетат, 3: 1) и получали нужный продукт (612 мг, 30%) в виде бледно-желтого твердого вещества.
1H- ЯМР (300 МГц, CDCl3) δ: 5,87 (шир.с, 1H), 5, 31 - 5,26 (м, 1H), 4,90 (дд, J = 2,2, 4,7 Гц), 4,42 (дд, 1H, J = 4,7, 9,3 Гц), 1,74 (с, 3H), 1,28 (с, 3H), 0,98 - 0,91 (м, 9H), 0,71 - 0,55 (м, 6H).
(e) (±
)-цис-3-Триэтилсилилокси-4-изобутенил-N-т- бутоксикарбонилазетидин-2-он

(± )-цис-3-Триэтилсилилокси-4-изобутенилазетидин-2-он (1,01 г, 3,95 мМ) растворяли в дихлорметане (20 мл) и обрабатывали диизопропилэтиламином (0,68 мл, 3,95 мМ) и каталитическим количеством диметиламинопиридина. К этому раствору добавляли ди-т-бутилдикарбонат (1,02 г, 4,68 мМ), и раствор оставляли для перемешивания при комнатной температуре на 24 часа. Полученный раствор разбавляли еще раз дихлорметаном, промывали водой, а затем солевым раствором. Органическую фракцию осушали сульфатом магния и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (элюент: гексан/этилацетат, 8:2) и получали целевое соединение (1,26 г, 90%) в виде бесцветного маслообразного вещества.
1H-ЯМР (300 МГц, CDCl3) δ: 5,24 (д, 1H, J = 9,6 Гц); 4,86 (д, 1H, J = 5,7 Гц); 4,72 (дд, 1H, J = 6,0, 9,9 Гц); 1,78 (д, 3H, J = 1,1 Гц); 1,75 (д, 3H, J = 1,1 Гц); 1,47 (с, 9H), 0,96 - 0,91 (м, 9H), 0,64 - 0,55 (м, 6H).
Процедура, описанная в получениях 9, 11(d), 12(d), 13, 14, и 15(е) может быть применена для получения других N-замещенных азетидинонов, которые используются в получении соединений настоящего изобретения.
Примеры таких азетидинонов представлены в следующей таблице 4. P представляет собой гидрокси-защитную группу, такую как триэтилсилил, триизопропилсилил и этоксиэтил.

Получение 16:
10-Дезокситаксотер

10-Дезацетокси-7-


Общая процедура, использованная в получении 16 может быть применена к получению других соединений формулы (1a), исходя из соответствующего баккатина III и азетидинона. Примеры других соединений формулы (1a) проиллюстрированы в нижеследующей таблице 5. При этом, следует отметить, что хотя представленные ниже соединения имеют свободные гидроксильные группы, однако, при целесообразном выборе различных гидроксильных групп, любая из этих защитных групп в 2'-, 7-, или 10-положении, может быть селективно удалена без какого-либо воздействия на другие присутствующие защитные группы.
Получение 17:
Бис(метилтиометил)эфир
CH3SCH2OCH2SCH3
К раствору 1,1'-дихлородиметилового эфира (3,0 г, 26,3 мМ) в ацетоне (100 мл), при 0oC, добавляли иодид натрия (8,23 г,
55,23 мМ), и смесь перемешивали при той же температуре в течение 20 минут. Затем к смеси четырьмя порциями добавляли тиометоксид натрия (1,84 г, 5,23 мМ), и полученный раствор перемешивали еще 1
час.
После этого гетерогенный раствор фильтровали через слой целита, а фильтрат концентрировали в вакууме. Остаток в виде маслообразного вещества распределяли между этилацетатом и насыщенным водным
раствором бикарбоната натрия. Водный слой удаляли, а затем экстрагировали этилацетатом. Объединенные органические слои обрабатывали смесью (1:1, об/об) насыщенного водного бикарбоната натрия и 5%
водного раствора тиосульфата натрия. Затем органические слои промывали солевым раствором, осушали сульфатом натрия, и концентрировали в вакууме. Остаточный маслообразный продукт очищали с помощью
флеш-хроматографии (30:1, гексан/этилацетат) и получали 1,9 г желтого маслообразного вещества, которое затем подвергали Kugelrohr дистилляции (120-130oC, 20 мм рт.ст.), в результате чего
получали 1,5 г (45%) целевого соединения в виде бесцветного маслообразного вещества.
1H-ЯМР (300 МГц, CDCl13) δ: 4,73 (4H, с), 2,15 (6H, с).
Получение 18:
Дибензилметилтиометилфосфат
CH3SCH2OP(O)(OBu)2
К раствору бис(метилтиометил)эфира (30 мг, 2,34 мМ) и молекулярных
сит (300 мг) в ТГФ (100 мл), при комнатной температуре, добавляли дибензилфосфат (2,74 г, 9,85 мМ), а затем N-иодосукцинимид (608 мг, 2,71 мМ), и раствор перемешивали в течение 4 часов. Затем
реакционную смесь разбавляли этилацетатом и фильтровали через слой целита. Фильтрат обрабатывали смесью (1:1, об/об) раствора насыщенного водного бикарбоната натрия и 5% водного тиосульфата натрия.
Бесцветный органический экстракт промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме, в результате чего получали 600 мг (69%) целевого соединения
1
H-ЯМР (300 МГц, CDCl3) δ: 7,35 (10H, с); 5,29 (2H, д, J = 12,2 Гц), 5,08 (4H, дд, J = 8,0, 1,0 Гц), 4,68 (2H, с), 2,10 (3H, с),
Примеры.
Нижеприведенные примеры иллюстрируют синтез характерных соединений настоящего изобретения и не должны рассматриваться как некое ограничение объема настоящего изобретения. Каждый специалист может использовать описанные методы, не прибегая при этом к излишнему экспериментированию, для синтеза соединений, входящих в объем настоящего изобретения, но конкретно не раскрываемых в настоящем описании.
Пример 1:
7-O-фосфонооксиметилпаклитаксел и его мононатриевая соль
(a) Получение 7-O-метилтиометилпаклитаксела

При 0oC, к энергично перемешанной смеси паклитаксела (0,85 г, 1 мМ) и диметилсульфида (0,72 мл, 8 мМ) в безводном ацетонитриле (10 мл) добавляли бензоилпероксид (0,98 г, 4 мМ). Перемешивание продолжали в течение 2,5 часа при 0oC. За ходом реакции наблюдали с помощью ТСХ на силикагеле (в системе растворителей: толуол/ацетон = 2:1, по объему, Rfтакс = 0,38, Rfпрод. = 0,64), и если наблюдалось образование более мобильных продуктов, реакцию гасили путем выпаривания растворителей на роторном испарителе при 30oC. ТСХ-анализ реакционной смеси указывал на присутствие некоторых количеств непрореагировавших паклитаксела и 2', 7-O-бис(метилтиометил)паклитаксела. Выделение целевого соединения из реакционной смеси осуществляли с помощью флеш-хроматографии на колонках с силикагелем 60(40-63 мкм) EM Science (100 мл), (диаметр колонки-2 дюйма) с использованием системы растворителей: этилацетат/гексан (1:1, по объему) (Rfпр. = 0,34 ). Полученный продукт (552 мг, выход - 60%) выделяли из фракций 12-18 (каждая фракция составляла приблизительно 20 мл).
MC (FAB/матрикс NOBA, NaI, KI): [M+H]+, m/z : 914, [M+Na]+, m/z : 936; [M+K]+, m/z 952.
Элементный анализ:
Вычислено: C 64,28
Найдено: 5,85
Элементный анализ для 64,39:
Вычислено: 6,07
Найдено: H 5,85
Вычислено: C 1,53; N 1,
46
УФ (MeOH): λмакс= 226 нм, E(1%/1 см) = 150, A = 0,2653.
ИК (KBr): 3432, 3066, 2940, 1726, 1668, 1602, 1582, 1514, 1484, 1452, 1372, 1242, 1178, 1142, 1108, 1068, 1026, 990, 916, 884, 852, 802, 774, 710, 608, 570, 538, 482 см-1.
1H-ЯМР (CDCl3) δ: 1,15 (3H, с); 1,19 (3H, с), 1,73 (3H, с), 1,79 (H, с), 1,90 (3H, д), 2,09 (3H, с), 2,16 (3H, с), 2,29 (2H, д), 2,35 (3H, с), 2,77 (H, м), 3,70 (H, д), 3,83 (H, д), 4,17 (H, д), 4,26 (H, м, перекрывающийся с H, д); 4,63 (2H, т), 4,77 (H, дд);4,91 (H, д), 5,65 (H, д), 5,77 (H, дд), 6,16 (H, дд), 6,48 (H, с), 7,07 (H, д), 7,29-7,50 (10H, м), 7,57 (H, м), 7,73 (2H, д), 8,08 (2H, д).
(b) Получение
7-O-дибензилфосфонооксиметилпаклитаксела

Раствор N-йодосукцинимида (45 мг, 0,2 мМ) и дибензилфосфата (55 мг, 0,2 мМ) в безводном тетрагидрофуране (4 мл) добавляли к смеси 7-O-метилтиометилпаклитаксела (119 мг, 0,13 мМ) и порошкообразных молекулярных сит 4A (приблизительно 120 мг) в безводном 1,2-дихлороэтане (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. За ходом реакции наблюдали с помощью ТСХ (в системе растворителей: толуол/ацетон, 2: 1, по объему) (Pfпр. = 0,48). Молекулярные сита удаляли путем фильтрации через слой целита (Целит 545), и фильтрат экстрагировали метиленхлоридом (100 мл). Органический слой промывали 1% раствором тиосульфата натрия (прибл. 100 мл) и 0,5 молями бикарбоната натрия (100 мл), а затем солевым раствором. Экстракт фильтровали через Whatman Phase Separator, а растворители выпаривали. После очистки с помощью флеш-хроматографии на силикагеле 60 (в метиленхлориде/этилацетате, 2: 1, по объему), получали 7-O-дибензилфосфонооксиметилпаклитаксел (41,5 мг).
(c) Получение 7-O-фосфонооксиметилпаклитаксела и его мононатриевой соли

7-O-Дибензилфосфонооксиметилпаклитаксел (41,5 мг) растворяли в этилацетате (5 мл) и добавляли 10% палладированный уголь (20 мл). Гидрогенизацию осуществляли при 40 фунт/кв.дюйм (275 кПа) в течение часа и при комнатной температуре. За ходом реакции наблюдали с помощью ТСХ в смеси хлороформа, метанола и воды (120:45:8, объему). После очистки с помощью препаративной ТСХ (20х20х0,05-см-пластина с силикагелем в аналитической системе), получали 7-O-фосфонооксиметилпаклитаксел (26 мг, выход - 75%).
Поскольку в процессе силикагелевой очистки наблюдалось разложение 7-O-дибензилфосфонооксиметилпаклитаксела, то процедура гидрогенизации была модифицирована. Таким образом, неочищенный экстракт 7-O-дибензилфосфонооксиметилпаклитаксела гидрировали без дополнительной очистки. Гидрогенизацию неочищенного экстракта 7-O-дибензилфосфонооксиметилпаклитаксела осуществляли при 60 фунт/кв.дюйм (400 кПа) в течение 24 часов.
7-O-Фосфонооксиметилпаклитаксел (70 мг) растворяли в 5 мл раствора ацетона и воды (1:1), и разбавляли водой до объема 50 мл. После этого добавляли безводный бикарбонат натрия (18 мг, 1,2 экв.). Ацетон выпаривали при комнатной температуре с помощью роторного испарителя, а оставшийся водный раствор лиофилизовали. Неочищенную мононатриевую соль 7-O-фосфонооксиметилпаклитаксела очищали с помощью обращеннофазовой колоночной (C18) хроматография в смеси воды/ацетонитрила (70:30, по объему). Элюат контролировали с помощью аналитической ЖХВД (15 см; Jones C18-колонки, 1 мл/мин, 1=230-270 нм) в смеси: ацетонитрил/0,05 М буфера ацетата аммония (45:55, по объему), pH 7, Rt = 2,09 мин. Фракции, содержащие целевой продукт, объединяли, ацетонитрил выпаривали, и остаточный водный раствор лиофилизовали, в результате чего получали 7-O-фосфонооксиметилпаклитаксела мононатриевую соль (112 мг).
MC (FAB): [M+H]+ m/z 986; [M+Na]+ m/z: 1008.
УФ (MeOH) λмакс=: 230 нм, E (1%/1 см) = 248.
ИК (KBr): 3430, 3066, 2948, 1724, 1652, 1602, 1580, 1518, 1486, 1452, 1372, 1316, 1246, 1178, 1154, 1108, 1070, 1000, 982, 946, 856, 802, 776, 710, 628, 538 см-1.
1H-ЯМР (ацетон-d6/D2O) δ: 8,05 (2H, d); 7,92 (2H, д) ; 7,65 (1H, дд), 7,58-7,35 (9H, м, перекрывающийся), 7,23 (1H, дд), 6,38 (1H, с), 6,08 (1H, т), 5,65 (1H, д), 5,60 (1H, д), 5,10 (1H, шир. с), 4,99 (1H, д); 4,97 (1H, шир. с), 4,80 (1H, д), 4,28 (1H, дд), 4,11 (2H, с), 3,79 (1H, д), 2, 94 (1H, м), 2,35 (3H, с), 2,35-2,10 (1H, м), 2,13 (3H, с), 1,95 (3H, с), 1,84 (1H, м), 16,7 (3H, с), 1,13 (6H, с, перекрывающийся).
Пример 2
Альтернативный способ получения
7-O-фосфонооксиметилпаклитаксела
(a) Получение 2'-O-(бензилоксикарбонил)паклитаксела

К перемешанному раствору паклитаксела (150 мг, 0,176 мМ) и N,N-диизопропилэтиламина (93 мкл, 0,534 мМ, 3 экв.) в безводном метиленхлориде (4 мл) (при комнатной температуре), добавляли бензилхлороформат (75 мкл, 0,525 мМ, 3 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, концентрировали до объема 2 мл, и очищали на колонке с силикагелем (элюент: этилацетат/гексан) (1:1), и получали целевое соединение в виде белого порошка (150 мг, выход - 86%), т.пл. 140-150oC (с разлож.).
(b) Получение
2'-O-(бензилоксикарбонил)-7-O-метилтиометилпаклитаксела
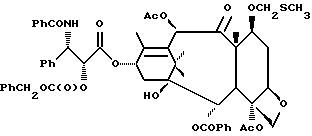
К охлажденному (в бане из сухого льда/CCI4, при температуре -30oC) раствору 2'-O-(бензилоксикарбонил)паклитаксела (4,935 г, 5,0 мМ) в сухом ацетонитриле (80 мл) добавляли последовательно диметилсульфид (3,6 мл, 40 мМ) и бензоилхлорид (4,9 г, 20,247 мМ). После выдерживания в течение 10 минут при -30oC, холодную баню удаляли, а реакционную смесь энергично перемешивали в течение 2 часов при комнатной температуре. Затем реакционную смесь разбавляли этилацетатом до объема 200 мл, и промывали водой и солевым раствором. Органический слой осушали сульфатом магния, а растворитель выпаривали, в результате чего образовывался остаток, который выдерживали в вакууме в течение 18 часов для удаления диметилсульфоксида, который присутствовал в качестве реакционного побочного продукта. После этого остаток очищали на колонках с силикагелем, используя в качестве элюента для удаления менее полярных примесей, этилацетат/гексан (1:2), а затем этилацетат/гексан (1: 1), в результате чего получали целевое соединение в виде пены. Затем это соединение перетирали с сухим эфиром и фильтровали, в результате чего получали целевое соединение в виде хлопьеобразного твердого вещества (5,0 г 95%) с т.пл. 120-122oC).
MC (FAB): [MH]+ : m/z: 1048; [M+Na]+ m/z; 1070; [M+K]+ m/z : 108.
ИК (KBr): 3440, 3066, 1750, 1722, 1664, 1602, 1583, 1538 см-1.
ЯМР (CDCl3) δ: 1,177 (3H, с), 1,236 (3H, с), 1,745 (3H, с); 2,023 (3H, с), 2,121 (3H, с), 2,162 (3H, с), 2,436 (3H, с), 3,887 (H, d), 4, 134 (H, о), 4,197 (H, о), 4,295 (H, м), 4,964 (H, д), 5,161 (2H, д), 5,450 (H, д), 5,703 (H, д), 5,981 (H, дд), 6,257 (H, т), 6,541 (H, с), 6,920 (H, д), NH), 7,332-8,22 (15H, м).
Целевое соединение также получали альтернативным способом, описанным ниже.
К раствору 2'-O-(бензилоксикарбонил)паклитаксела (2,0 г, 2,0263 мМ) в безводном диметилсульфоксиде (10 мл) по капле добавляли уксусный ангидрид (10 мл). Полученную смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере азота, разбавляли этилацетатом (100 мл), и промывали холодным 6% раствором бикарбоната натрия (6х30 мл), холодной водой (6х30 мл), и солевым раствором. Органический слой осушали сульфатом магния, а растворитель выпаривали, в результате чего образовывался остаток. Этот остаток очищали на колонках с силикагелем (элюент: метиленхлорид, метиленхлорид/5% ацетонитрил, и метиленхлорид/10% ацетонитрил) и получали нужное соединение (1,86 г, 87,7%). Это соединение является идентичным соединению, полученному ранее описанным методом с использованием диметилсульфида/бензоилпероксида.
(c) Получение
2'-O-(бензилоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксела

К раствору 2'-O-(бензилоксикарбонил)-7-O-метилтиометилпаклитаксела (5,0 г, 5,5369 мМ) в сухом 1,2-дихлорэтане (120 мл) добавляли активированные порошкообразные молекулярные сита 4A (5,0 г). К этой смеси по капле при комнатной температуре добавляли раствор смеси N-йодосукцинимида (1,61 г, 7,1632 мМ) и дибензилфосфата (1,97 г, 7,1632 мМ) в сухом тетрагидрофуране (90 мл). После энергичного перемешивания при комнатной температуре в течение 30 минут, реакционную смесь фильтровали через слой целита, а фильтрат выпаривали досуха, в результате чего образовывался красный остаток. Этот остаток растворяли в этилацетате (100 мл), промывали холодным 6% раствором NaHSO3 (2x50 мл), холодным 6% раствором NaHCO3 (2x50 мл), и силевым раствором (1x50 мл). Органический слой осушали сульфатом магния, а растворитель выпаривали, и получали твердую массу, которую перетирали с безводным эфиром и фильтровали, в результате чего получали целевое соединение в виде твердого вещества, имеющего цвет слоновой кости (5,9 г, 97%), т.пл. 124-127oC
MC (FAB): [MH]+ m/z: 1278, [M+Na]+ m/z: 1301; [M+K]+ m/z 1316.
ИК
(KBr): 3430, 3066,
3032, 1750, 1726, 1664, 1582, 1532 см-1
ЯМР (CDCl3) δ: 1,160 (3H, с), 1,703 (3H, с), 1,985 (3H, с), 2,164 (3H, с), 2,420 (3H, с), 3,854 (H, д),
4,151 (H, д), 4,
216 (H, м), 4,298 (H, д), 4,873 (H, д), 5,043 (6H, м), 5,140 (2H, д), 5,417 (H, д), 5,670 (H, д), 5,971 (H, дд), 6,241 (H, т), 6,317 (H, с), 6,912 (H, д, NH), 7,280-8,115 (25H,
м).
(d) Получение 7-O-фосфонооксиметилпаклитаксела
К раствору 2'-O-(бензилоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксела (6,0 г, 4,7095 мМ) в этилацетате (120 мл)
добавляли 10% Pd/C (6,0
г) и полученную смесь гидрогенизировали при 60 фунт/кв. дюйм (400 кПа) в течение 24 часов. Реакционную смесь фильтровали через слой целита, и растворитель выпаривали, в
результате чего получали 4,07 г
неочищенного остатка. Этот остаток очищали с помощью узкой колонки с силикагелем, последовательно элюируя хлороформом/10%, 20% и 40% метанолом, и получали целевое
соединение в виде твердого белого
вещества (3,2 г, 71%) с т.пл. 155-158oC.
Этот продукт имел такое же значение Rf (ТСХ) и такое же время удержания (ЖХВД), как и аутентичный образец.
МС (FAB): [MH]+ m/z: 964; [M+Na]+ m/z: 986; [M+K]+ m/z 1002. [M+K++Na+-H]+ m/z: 1024, [M+2K-H]+, m/z : 1040.
УФ (MeOH): λмакс= 230 нм, E (1%/1 см) = 252,5
ИК (KBr): 3432, 3066, 2992, 1722, 1648, 1602, 1580, 1522, 488, 1452, 1372,
1316, 1246, 1178, 1154, 1110, 1070,
1000, 980, 946, 854, 802, 776, 710, 628, 538 см-1.
ЯМР (ацетон-d6/D2O) δ: 1,08 (3H, с), 1,10 (3H, с), 1,63 (3H, с), 1,88 (3H, с), 1,96 (H, м), 2,13 (3H, с), 2,32 (3H, с), 2,89 (H, м), 3,76 (H, д), 4,19 (H, м), 4,89 (H, дд), 5,09 (H, дд), 5,55-5,60 (2H, перекрывающийся с с), 6,04 (H, т), 6,32 (H, с), 720 (H, т), 7,34-7,67 (10H, перекрывающийся с с), 7,87 (2H, дд), 8,02 (2H, дд).
Пример 3.
2'-O-(этоксикарбонил)-7-O-фосфонооксиметилпаклитаксел
(a) Получение
2'-O-(этоксикарбонил)паклитаксела

К раствору паклитаксела (4,35 г, 5,1 мМ) в безводном метиленхлориде (51 мл) добавляли N,N-диизопропилэтиламин (2,67 мл, 15,3 мМ), а затем этилхлороформат (1,46 мл, 15,3 мМ). Полученную реакционную смесь перемешивали при 0oC в течение 2 часов, а затем при комнатной температуре еще 1 час. Реакционную смесь разбавляли этилацетатом (400 мл), а органическую фазу промывали насыщенным раствором NaHCO3 (2x30 мл) и солевым раствором (30 мл). Полученную органическую фазу осушали сульфатом магния и получали неочищенный целевой продукт (93%), который использовали в последующей стадии без дополнительной очистки.
MC (FAB/NOBA, NaI, KI): [M+H]+ m/z: 926; [M+Na]+ m/z: 948; [M+K]+ m/z: 964.
HPMC (FAB/NOBA, внешний стандарт CSI/GIy): [M+H]+ m/z C26, 3588
вычислено: 926, 3588,
Анализ для C50H56NO16:
Вычислено: 926,3599 (отклонение Δ = 1,2 ppm).
1H-ЯМР (CDCl3
) δ: 1,13 (3H, с), 1,23 (3H, с), 1,30 (3H, т), 1,67 (3H, с), 1,92 (3H, с), 2,21 (3H, с), 2,37 (H, д), 2,45 (3H, с), 2,54 (H, м), 3,80 (H, д), 4,15-4,32 (4H, м, перекрывающийся), 4,43 (H, дд),
4,
93 (H, д), 5,42 (H, д), 5,68 (H, д), 5,98 (H, дд), 6,28 (2H, м, перекрывающийся),7,00 (H, д), 7,34-7,59 (11H, перекрывающийся с м); 7,74 (2H, д), 8,12 (2H, д),
***Альтернативная
процедура
Паклитаксел (5,40 г, 6,324 мМ) в безводном дихлорметане (63 мл) охлаждали до 0oC и обрабатывали чистым N,N-диизопропилэтиламином (3,30 мл, 3 экв. ), а затем в течение 5
минут по капле
добавляли чистый этилхлороформат (1,81 мл, 3 экв.). За ходом реакции наблюдали с помощью ТСХ (50% этилацетат в гексане). После выдерживания в течение 2 часов при 0oC и 16
часов при
комнатной температуре, реакцию завершали, и образовавшийся желто-оранжевый раствор разбавляли этилацетатом (300 мл) и промывали насыщенным бикарбонатом натрия (3x75 мл) и солевым раствором
(75 мл).
После осушки сульфатом магния и выпаривания, получали неочищенное целевое соединение, которое очищали путем осаждения: то есть, сначала добавляли дихлорметан (прибл. 100 мл), а затем
охлаждали и
добавляли гексан (прибл. 60 мл) до точки помутнения. После охлаждения во льду в течение нескольких часов, полученное твердое вещество собирали путем фильтрации (выход 5,17 г, 88%).
Альтернативная процедура
В осушенной пламенем, одногорлой трехлитровой колбе растворяли паклитаксел (99,0 г, 115,9 мМ) в 3,150 мл сухого метиленхлорида, в атмосфере азота.
Полученный раствор
охлаждали до - 10oC. После этого медленно добавляли N, N-диизопропилэтиламин (5+4 г, 405,7 мМ) (дополнительное время 3 мин), а затем ClCO2Et (31,45 г, 289,8
мМ, дополн. время 15
минут). Полученную смесь перемешивали в течение ночи (16 часов) при -4oC. ТСХ указывала на то, что реакция не была завершена. Поэтому добавляли другую партию N,
N-диизопропилэтиламина (2,62
г, 20,28 мМ), затем ClCO2Et (2,20 г, 20,28 мМ), и перемешивание продолжали в течение 3 часов при -4oC. При этом ТСХ не обнаруживала присутствия
исходного материала. Холодную
смесь разбавляли этилацетатом (1,5 л) и переносили в делительную воронку. Затем эту смесь промывали 5% KHSO4 (2 x 500 мл), водой (1 х 500 мл), 5% KHSO4 (1 х 500 мл), снова водой
(1 х 500 мл), затем насыщенным NaHCO3 (2 x 500 мл) и солевым раствором (2 х 500 мл). После осушки сульфатом магния и выпаривания растворителей в вакууме,
получали 147 г неочищенного
продукта. Остаток растворяли в горячем метиленхлориде (8000 мл, температура бани -42oC) и при перемешивании, по капле добавляли гексан (530 мл), поддерживая при
этом температуру 42oC. Полученную кристаллическую смесь отстаивали в течение 3 часов при комнатной температуре, а затем при 0oC в течение ночи в холодной комнате. Образовавшиеся
тяжелые белые кристаллы
собирали путем фильтрации и промывали гексано-/метиленхлоридом (1: 1, по объему, 2 х 200 мл). После осушки на всасывающем фильтре в течение 1 часа, кристаллы осушали в вакууме
(≈ 1,0 мм рт. ст)
в течение ночи, и получали 95,7 г (выход - 89%) целевое соединение (показатель гомогенности, измеренный с помощью ЖХВД, составлял 98,5%).
(b) Получение
2'-О-(этоксикарбонил)-7-О-метилтиометилпаклитексела

К раствору 2-О-(этоксикарбонил)паклитаксела (4,38 г, 4,7 мМ) в сухом диметилсульфоксиде (12,5 мл) добавляли уксусный ангидрид (12,5 мл). Реакционную смесь перемешивали 24 часа при комнатной температуре, а затем разбавляли этилацетатом (500 мл), промывали насыщенным раствором NaHCO3 (3 x 40 мол) и водой (2 х 40 мл). Полученный органический слой осушали сульфатом магния, а растворители выпаривали в вакууме досуха. Остаток очищали с помощью хроматографии на силикагеле (элюент: 40% этилацетат в гексане) и получали целевое соединение (4,39 г, 94%). МС (FAB/NOBA, NaI, KI):[M+H]+ m/z: 986; [M+Na]+ m/z: 1008, [M+K]+ m/z: 1024.
HPMC (FAВ/NOBA, внешний стандарт CSI/GLy): [M+H]+m/z: 9863646 (Вычислено: 9863633, отклонение Δ = 1,3
ppm).
1H-ЯМР (CDCl3): δ 1,18 (3H, с); 1,20 (3H, с), 1,30 (3H, с), 1,75 (3H, с), 1,84 (H, м), 2,09 (3H. с), 2,11 (3H, с), 2,16 (3H, с), 2,24 (H, д), 2,37 (H, д), 2,
45 (3H, с), 2,80 (H, м), 3,68 (H, д), 4,08 -4,33 (5H, м, перекрывающийся), 4,65 (2H, с), 4,96 (H, д), 5,43 (H, д), 5,69 (HH, д), 5,98 (H, дд), 6,26 (H, т), 6,55 (H, с), 7,00 (H, д), 7,32-7,61 (11H, м,
перекрывающийся), 7,73 (2H, дд), 8,11 (2H, дд).
Альтернативная процедура
2'-О-(Этоксикарбонил)паклитаксел (2,260 г, 2,4406 мМ) растворяли в безводном диметилсульфоксиде (6
мл), и одной порцией (при комнатной температуре) добавляли уксусный ангидрид (6 мл). За ходом реакции наблюдали с помощью ЖХВД (аналитическая C18-колонка, элюент: 60% ацетонитрил/40% 10 мМ
аммонийфосфатный буфер, pH 6). Через 30 часов раствор разбавляли этилацетатом (250 мл) и промывали насыщенным водным бикарбонатом (3 раза), а затем водой и солевым раствором. После осушки сульфатом
магния и фильтрации, получали неочищенный продукт. Этот продукт хроматографировали на двуокиси кремния (элюент: 40% этилацетат в гексане) и получали целевое соединение в виде белой пены (2,030 г,
91%),
которое имело чистоту 90% (ЖХВД). Кроме того, часть этого соединения очищали на второй колонке (элюент: 5% ацетонитрил в дихлорметане), в результате чего получали продукт, который имел чистоту
примерно 97% (ЖХВД).
Альтернативный способ получения 2'-О-(этоксикарбонил)-7-О-метилтиометилпаклитаксела
2-О-(Этоксикарбонил)паклитаксел (4,170 г, 4,503 мМ) растворяли в
безводном ацетонитриле (68 мл) при -40oC, и добавляли диметилсульфид (3,2 мл, 44,10 мМ), а затем бензоилпероксид (4,400 г, 18,24 мМ). Смесь помещали в ледяную баню и перемешивали при 0oC, после чего за ходом реакции наблюдали с помощью ТСХ (40% этилацетат в гексане). Через 3 часа исходный материал не был обнаружен, и поэтому раствор обрабатывали путем добавления этилацетата
(250 мл) и насыщенного водного бикарбоната натрия (100 мл). Кроме того, органическую фазу промывали бикарбонатом, водой, и солевым раствором, а затем осушали сульфатом магния и фильтровали.
Полученный
остаток очищали с помощью флеш-хроматографии на силикагеле (элюент: 4% ацетонитрил в дихлорметане), и получали целевое соединение в виде белой пены (2,571 г, выход - 58%). Чистота этого
образца
составляла ≈ 97% (ЖХВД).
ЯМР-спектр был идентичен спектру первого соединения, описанного выше.
Альтернативная процедура получения
2-О-(этоксикарбонил)-7-О-метилтиометилпаклитаксела
2'О-(Этоксикарбонил)паклитаксел (49,3 г, 53,2 мМ) помещали в осушенную пламенем, одногорлую литровую колбу и растворяли в сухом
ацетонитриле
(500 мл) при комнатной температуре. Затем к смеси медленно, посредством шприца, добавляли метилсульфид (39,1 мл, 0,532 М). Перемешанную реакционную смесь охлаждали до -16oC в
ледяной/солевой бане, и к смеси одной порцией добавляли твердый бензоилпероксид (51,6 г, 0,213 М). (Для протекания реакции, вплоть до ее завершения, требуется не менее четырех эквивалентов).
Перемешивание продолжали в течение 30 минут, а температуру понижали до ≈ -10oC. Реакционная среда оставалась гетерогенной на протяжении всего периода (бензоилпероксид не был
полностью растворен). Охлаждающую баню заменяли на ледяную/водяную, температуру, повышали до 0oC, а оставшийся бензоил-пероксид растворяли в течение приблизительно 5 минут, после чего
нагревали. После перемешивания при 0oC в течение 2,5 часа, реакцию прекращали с помощью ТСХ. Объем этого раствора снижали приблизительно до 20 мл путем удаления растворителя на роторном
испарителе, после чего раствор переносили в делительную воронку, где его промывали гептаном (5 х 500 мл). Ацетонитриловый слой разбавляли этилацетатом (1,5 л) и промывали смесью насыщенного NaHCO3/5% K2CO3 (по объему) (2 х 500 мл), насыщенным NaHCO3 (2 x 500 мл), полунасыщенным солевым раствором (1 х 500 мл) и солевым раствором (1 х 500 мл), а затем
осушали сульфатом магния, и растворители удаляли в вакууме, в результате чего получали 67,0 г неочищенного продукта. Этот продукт растворяли в ацетоне (200 мл), нагревали до 40oC в водяной
бане, и по капле, при перемешивании, добавляли гексан до тех пор, пока раствор не становился мутным (400 мл). После этого кристаллическую смесь отстаивали в течение 3 часов при комнатной температуре,
а затем переносили в холодную комнату (0oC), где хранили в течение ночи (16 часов). Таким образом, образовывался толстый осадок. Этот твердый продукт собирали путем фильтрации и промывали
гексаном/ацетоном (3:1 по объему) (2 х 50 мл). Полученные белые кристаллы осушали на всасывающем фильтре в течение часа, а затем осушали в вакууме (≈ 0,5 мм рт. ст.) в течение ночи, в
результате чего получали 47,5 г (выход - 91%) целевого соединения (показатель гомогенности составлял 94,8%, измеренный с помощью ЖХВД).
(с) Получение
2'-О-(этоксикарбонил)-7-О-дибензилфосфоно-оксиметилпаклитаксела
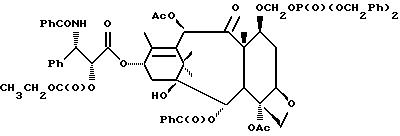
Раствор N-иодосукцинимида (1, 953 г, 8,65 мМ) и дибензилфосфата (2,41 г, 8,65 мМ) в тетрагидрофуране, добавляли к смеси 2-О-(этоксикарбонил)-7-О-метилтиометилпаклитаксела (5,677 г, 5,76 мМ) и молекулярных сит 4А (5,7 г) в метиленхлориде (100 мл), при комнатной температуре. Полученную реакционную смесь перемешивали в течение 40 минут при комнатной температуре. После этого реакция считалась завершенной, о чем свидетельствовали ТСХ. Реакционную смесь фильтровали через слой целита, и фильтрат концентрировали в вакууме, и получали коричневый остаток, который разбавляли этилацетатом (800 мл). Органическую фазу промывали 1% Na2SO3 (2 x 80 мл), а затем 5% солевым раствором (2х50 мл). После этого органическую фазу концентрировали в вакууме и осушали. Полученный остаток хроматографировали (элюент: 50-60% этилацетат в гексане), и получали целевое соединение (6,23 г, 89%).
МС (FAB/NOBA, Nal, Kl): [M+Na]+ m/z: 1238; [M+K]+ m/z: 1254.
НРМС (FAB/NOBA, внешний стандарт CSl/Gly): [M+Na]+ m/z: 12154291 (Вычислено для C65H71NO20P: 12164307; отклонение Δ = 1,3
м.д.).
1
H-ЯМР (CDCl3): δ 1,18 (3H, с), 1,21 (3H, с), 1,30 (3H, т), 1,67 (6H, с), 1,80 (H, с), 1,93 (H, м), 1,99 (3H, д), 2,18 (3H, с), 2,23 (H, м), 2,38 (H, м), 2,
45 (3H, с), 2,80 (H, м), 3,
86 (H, д), 4,14-4,32 (5H, перекрывающийся с с), 4,88 (H, д), 5,00-5,07 (4H, перекрывающийся с с), 5,42 (H, д), 5,68 (H, д), 5,96 (H, дд), 6,26 (H, т), 6,33 (H, с), 6,95 (H,
д), 7,30-7,61 (11H,
перекрывающийся с с), 7,75 (2H, дд), 8,12 (2H, дд).
Альтернативная процедура
К раствору 2'-O-(этоксикарбонил)-7-O-метилтиометилпаклитаксела (350 мг, 0,
355 мМ) в безводном
тетрагидрофуране (8 мл), добавляли раствор N-иодосукцинимида (120 мг, 0,532 мМ) и дибензилфосфата (148 мг, 0,532 мМ) в тетрагидрофуране (5 мл). Ход реакции прослеживали с помощью
ТСХ (C18-колонка;
смесь: 70% ацетонитрила/30% 10 мМ фосфата аммония, pH 6). Через 2 часа обнаруживалось содержание менее 5% исходного материала, после чего реакцию завершали. Раствор разбавляли
этилацетатом (75 мл) и
промывали 1%-ным водным бисульфитом
натрия (2х50 мл) и солевым раствором (50 мл). После быстрой осушки сульфатом магния и фильтрации, растворитель выпаривали. Остаток
подвергали флеш-хроматографии,
на силикагеле (элюент: 45% этилацетат в гексане), и получали целевое соединение в виде белой пены (281 мг, 65%). ЖХВД-анализ указывал на чистоту примерно 95%.
Альтернативная
процедура
Измельченные молекулярные сита 4a помещали в осушенную пламенем одногорловую литровую колбу, которую затем подсоединяли к вакуумному трубопроводу (≈
0,5 мм рт. ст). Сита
нагревали с использованием бытового фена в течение примерно 10 минут, встряхивая при этом вручную. После охлаждения в вакууме, в колбу вводили аргон и добавляли
2'-O-(этоксикарбонил)-7-O-метилтиометилпаклитаксел (37,5 г, 38,03 мМ), а затем дибензилфосфат (14,8 г, 53,24 мМ) и ТГФ (400 мл). В течение 15 минут при комнатной температуре, гетерогенную смесь
энергично перемешивали в магнитной мешалке. После этого, в отдельную, осушенную пламенем, колбу загружали N-иодосукцинимид (10,7 г, 47,54 мМ) в атмосфере аргона), растворенный в ТГФ (50 мл). (В
процессе получения NIS-раствора, переноса жидкости, и во время протекания реакции, сосуды покрывали алюминиевой фольгой для защиты от воздействия света). Затем этот раствор медленно, посредством
шприца, в течение 10 минут добавляли к реакционной смеси. Колбу, содержащую NIS-раствор, промывали 5 миллилитрами тетрагидрофурана и переносили в реакционную смесь, после чего перемешивали 2 часа при
комнатной температуре. ТСХ-анализ указывал при этом на отсутствие исходного материала. Темно-красный раствор фильтровали через слой Целита®, непосредственно в энергично перемешанную
смесь (двухфазную), содержащую этилацетат (500 мл), 10% водный тиосульфат натрия (300 мл) и насыщенный бикарбонат натрия (200 мл). В течение двух секунд красный цвет исчезал, и образовывался
бесцветный раствор. Слой Целита® промывали этилацетатом (прибл. 100 мл) и оба жидкостных слоя переносили в делительную воронку. Органический слой разбавляли этилацетатом (1 л), слои
разделяли, а органический слой промывали смесью насыщенного NaHCO3 (2х500 мл) и 5% K2CO3 (3:1, по объему, 2х500 мл), а затем насыщенным NaHCO3 (2х500 мл),
полунасыщенным солевым раствором (1х500 мл), и солевым раствором (1х500 мл). Полученный экстракт осушали безводным сульфатом магния и фильтровали. Затем этот экстракт обрабатывали 5,0 граммами
нейтрального Norit (уголь), перемешивая при этом в течение 15 минут при комнатной температуре. После этого экстракт снова фильтровали через слой Целита®, растворитель удаляли при
пониженном давлении, и получали 52 г неочищенного продукта. Этот продукт растворяли в толуоле/метиленхлориде (280 мл/25 мл), и по капле добавляли гексан (20 мл). После отстаивания в течение 3 часов
при комнатной температуре, кристаллическую смесь оставляли в течение ночи при температуре 0oC. Таким образом, на стенках колбы образовывался бледно-желтый твердый осадок. После
декантирования меточного раствора, остаток растирали с толуолом (50 мл), фильтровали, промывали толуолом, и осушали на всасывающем фильтре в течение 30 минут. Затем остаток переносили в эксикатор
Drierite®, и осушали в вакууме (≈ 0,5 мм рт.ст.) в течение 4 часов, в результате чего получали 24,4 г (выход - 53%) целевого соединения (показатель гомогенности, измеренный с
помощью ЖХВД, составлял 95,9%). Маточный раствор выпаривали досуха, растирали с толуолом (100 мл), фильтровали, промывали толуолом и осушали на всасывающем фильтре в течение 30 минут. После осушки в
эксикаторе, описанном выше, получали 12,5 г (выход - 27%) того же продукта (показатель гомогенности, измеренный с помощью ЖХВД, составлял 97,1%).
(d) Получение
2'-O-(этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела и его мононатриевой, монокалиевой, триэтиламиновой, аргининовой, лизиновой, этаноламиновой, -метилглюкаминовой и триэтаноламиновой соли

К раствору 2'-O-(этоксикарбонил)-7-O- дибензилфосфонооксиметилпаклитаксела (1,23 г, 1,01 мМ) в безводном этилацетате (40 мл) добавляли 10% палладированный уголь (428 мг, 0,404 мМ). Реакционную смесь гидрировали (60 фунт/кв. дюйм - 400 кПа) при встряхивании, в течение 24 часов. Твердый продукт отфильтровывали через слой Целита®, после чего Целит® промывали несколько раз этилацетатом. Фильтрат концентрировали и получали целевое соединение в форме свободной кислоты (1,01 г, чистота, определенная с помощью ЖХВД, составляла 80%). В последующей стадии примеси удаляли путем препаративной хроматографии на C18-колонках.
МС (FAB/NOBA, NaI, KI): [M+Na]+ m/z: 1058, [M+K]+ m/z: 1074, [M+Na-H]+ m/z: 1080; [M+Na+K-H]+ m/z: 1096; [M+2K-Н]+ m/z: 1112.
МС-анализ
(FAB/NOBA,
внешний стандарт CSl/Gly): [M+Na]+ m/z: 10583163 (вычислено для C51H58NO20PNa: 10583188; отклонение Δ = 2,3 м.д.).
1
H-ЯМР
(ацетон-d6/D2O): δ 1,13 (3H, с), 1,21 (3H, с), 1,66 (3H, с), 1,87 (H, м), 1,93 (3H, с), 1,21 (3H, с), 2,18 (H, м), 2,44 (3H, с), 2,95 (H, м), 3,81 (H, д), 4,12 (2H,
с), 4,
15-4,27 (3H, перекрывающийся с с), 4,92-4,99 (2H, шир. перекрывающийся с), 5,15 (H, шир. с), 5,48 (H, д), 5,61 (H, д), 5,84 (H, дд), 6,07 (H, т), 6,36 (H, с), 7,25 (H, т), 7,28-7,69 (10H,
перекрывающийся с), 7,89 (2H, дд), 8,08 (2H, дд), 8,86 (H, д).
Альтернативная процедура
2'-O-(Этоксикарбонил)-7-O-(дибензилфосфонооксиметил)паклитаксел (490 мг, 0,402 мМ) в
этилацетате (20 мл) гидрогенизировали в шейкера Парра при 60 фунт/кв. дюйм (400 кПа) в присутствии палладированного угля (10% об/об, 150 мг). Ход реакции прослеживали с помощью ТСХ и ЖХВД. Если
больше
не обнаруживалось ни исходного материала, ни промежуточного соединения (предположительно, монобензилфосфата) (26 ч.), то суспензию фильтровали через Целит® и выпаривали
досуха.
ЖХВД-анализ показывал чистоту 88-92%.
Альтернативная процедура
Триэтиламиновая соль 2'-O-(Этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела, описанную ниже (5,4 г, 4,
75 мМ)
энергично распределяли между EtOAc (100 мл) и 5% NaHSO4 (45 мл) и перемешивали при 0oC в течение 30 минут. Водный слой отделяли и экстрагировали этилацетатом (20 мл).
Объединенный EtOAc-слой промывали наполовину солевым раствором (25 мл), солевым раствором (25 мл х 2), осушали сульфатом натрия и фильтровали, в результате чего получали раствор кислоты (≈ 4,
75
мМ) в EtOAc (≈ 150 мл). Затем этот EtOAc-раствор концентрировали досуха на роторном испарителе, и получали 3,75 г целевого соединения в виде свободной кислоты с выходом 95%. ЖХВД-анализ
показал,
что показатель гомогенности составлял 96,1%.
Мононатриевую соль получали следующим образом:
Образец 2'-O-(этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела (1,6 г, 1,
55 мМ)
растворяли в ацетонитриле (30 мл) путем обработки ультразвуком. Этот раствор разбавляли водой (30 мл) и добавляли 1,1 М раствора NaHCO3 (2,11 мл, 2,32 мМ), временами встряхивая, и
обрабатывали ультразвуком (5-20 минут) до получения раствора. Раствор несколько беловатого цвета наносили на C18-колонку и промывали двумя колоночными объемами воды, а затем мононатриевую соль
элюировали 25% ацетонитрилом/водой. Затем соответствующие фракции объединяли, ацетонитрил выпаривали, водную фазу лиофилизовывали, и получали мононатриевую соль целевого соединения (850 мг, прибл.
50%), имеющую чистоту 97% (ЖХВД).
MC (FAB/NOBA, NaI, KI): [M +Na]+ m/z: 1180.
HP-MC (FAB/NOBA, CSI/Gly-внешний стандарт): [M + Na]+ m/z: 10802968 (Анализ для C51H57NO20PNa2: вычислено: 10803007; отклонение D = 3,6 м.д.).
Элементный анализ: C 52,65 (выч. 56,72); H 5,06 (выч. 5, 23); N 1,20 (выч. 1,30); Na 2,74 (выч. 2,12).
ИК (KBr): 3430, 3066, 2988, 1646, 1722, 1660, 1602, 1582, 1526, 1488, 1452, 1374, 1246, 1178, 1150, 1108, 1070, 1052, 1026, 1002, 966, 912, 834, 792, 776, 710, 628, 538 cм-1.
1H-ЯМР (ДМСО-d6, D2O, ацетон-d6): δ 1,10 (6H, с), 1,23 (3H, т), 1,64 (3H< с), 1,70 (H, м), 1,90 (3H, с), 1,99 (H, м), 2,14 (3H, с), 2,37 (3H, с), 2,98 (H, м), 3,74 (H, д), 4,07 (2H, д), 4,13 - 4,26 (3H, м, перекрывающийся), 4,80 (H, шир. дд), 4,97 (H, д), 5,09 (H, шир. т), 5, 44 (H, д), 5,55 (H, д), 5,99 (H, т), 6,34 (H, с), 7,22 (H, т), 7,43 - 7,69 (10H, м, перекрывающийся); 7,92 (2H, дд), 8,06 (2H, дд).
Натриевая соль была также получена следующим
образом:
Неочищенный 2'-O-(этоксикарбонил)-7-O-фосфонооксиметилпаклитаксел (89%, 70 мг, 0,060 мМ) в EtOAc (2 мл), при комнатной температуре, обрабатывали раствором этилгексаноата натрия (8,
75
мМ в EtOAc, 1,0 мл, 0,0875 мМ), перемешивая при этом. После перемешивания при комнатной температуре в течение часа, к раствору добавляли гексан (1,2 мл) до точки помутнения. После хранения смеси
при
-20oC в течение 2 часов, тонкодисперсный аморфный порошок фильтровали с некоторыми трудностями очень медленно) через тонкую фильтровальную бумагу, и получали 45 мг (70%) натриевой
соли. Эта
соль имела чистоту 95,2% (ЖХВД) и содержала небольшое количество этилгексановой кислоты (ЯМР-анализ).
Триэтаноламиновую соль получали следующим образом:
2'-O-(Этоксикарбонил)-7-O-фосфонооксиметилпаклитаксел (неочищенный), полученный в результате гидрирования (чистота составляла 89% (ЖХВД)) (0,69 г, 0,593 мМ после внесения поправок на примеси)
растворяли в этилацетате (10 мл), и медленно перемешивали, добавляя при этом по капле раствор триэтаноламина (0,11 М в EtOAc, 5,1 мл, 0,95 экв.). Полученный в результате этой процедуры, раствор
беловатого цвета автоклавировали при 0oC в течение 2 часов, а затем фильтровали через тонкую фильтровальную бумагу, и промывали холодным этилацетатом. В результате этой процедуры получали
499 мг (80%) аморфного, тонкодисперсного, неэлектростатического порошка, который осушали в вакууме в течение ночи. Анализ, проведенный с помощью ЖХВД показал чистоту 96,6% (C-18, 45% 5 мМ Q12 + 10 мМ фосфата аммония; pH 6; 55% - ацетонитрил). ЯМР-спектр (D2O/ацетон/ДМСО) показал наличие следовых количеств этилацетата и никаких других явных примесей. Также проводили
анализы на 2-3 х гидрат.
Кроме того, триэтаноламиновую соль, полученную в результате другого эксперимента, очищали с помощью следующей процедуры. Триэтаноламиновую соль (приблизительно 2 г) растворяли в смеси примерно 30% ацетонитрила и воды. Затем этот раствор элюировали при низком давлении азота на C18-колонке (Bakerbond) градиентом 20 - 40% ацетонитрила в воде. Фракции, содержащие нужную триэтаноламиновую соль, собирали, а ацетонитрил удаляли с помощью роторного испарителя при пониженном давлении. После этого водные растворы замораживали и лиофилизовали в течение ночи, в результате чего получали 1,4 г триэтаноламиновой соли с чистотой 97,5%.
Триэтаноламиновая соль может быть также получена следующим образом:
Триэтиламиновую
соль 2'-O-(этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела (3,0 г, 2,64 мМ) распределяли между EtOAc (60 мл) и 5% NaHSO4 (30 мл), энергично перемешивая при этом в течение 15 минут (0oC). Водный слой отделяли и экстрагировали этилацетатом (10 мл). Объединенный этилацетатный слой промывали солевым раствором (15 мл), осушали сульфатом натрия, фильтровали и получали раствор
кислоты (прибл. 2,64 мМ) в EtOAc (прибл. 70 мл). К этому этилацетатному раствору при комнатной температуре в течение 5 минут по капле добавляли N(CH2CH2OH)3 (0,35 мл,
2,64 мМ), энергично перемешивая при этом. Полученную суспензию перемешивали еще 1 час, а затем фильтровали, промывали этилацетатом (15 мл х 2) и осушали в вакууме, в результате чего получали 2,8 г
триэтаноламиновой соли с выходом 89% и т. пл. > 157oC (при разлож. ). Показатель гомогенности, измеренный с помощью ЖХВД, составлял 98,7%.
Элементный анализ для
C56H73N2O23P • 2,0 • H2O 0,3 EtOAc:
Вычислено: C 55,60; H 6,48; N 2,27; KF (H2O) : 2,92.
Найдено: C 55,94; H 6,59; N 2,43; KF (H2O) : 3,50.
Триэтиламиновую соль получали следующим образом:
К раствору
2'-O-(этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксела (10 г, 8,23 мМ) в EtOAc (350 мл), при комнатной температуре, добавляли 10% палладированный уголь (2 г, 20%-ная загрузка). Полученную
суспензию дегазировали путем откачки воздуха, а затем продували аргоном. Эту процедуру повторяли еще 2 раза. После этого аргон заменяли на водород, и проводили аналогичную процедуру дегазирования.
Полученную суспензию перемешивали под давлением балонного водорода 2 - 3 фунт/кв.дюйм (0,141 - 0,211 кг/см2) в течение 16 часов при комнатной температуре, энергично перемешивая при этом.
Водород откачивали и загружали аргон (3 раза), а затем проводили аналогичную процедуру дегазирования. Полученную суспензию фильтровали через слой целита. Затем к этому гомогенному фильтрату медленно
добавляли Et3N (8,23 мМ, 1,14 мл) (5 минут), энергично перемешивая при этом. Полученную тонкодисперсную белую суспензию перемешивали еще 30 минут, после чего фильтровали через воронку из
спеченного материала. Осадок на фильтре осушали в вакууме (1 мм рт. ст. ) в течение 16 часов, и получали 8,22 г целевого соединения, а именно, триэтиламиновой соли с выходом 88% и т. пл. >
178oC (с разлож.). Показатель гомогенности, измеренный с помощью ЖХВД, составлял 97,4%.
Элементный анализ для C57H73N2O20P
• 4,5 H2O:
Вычислено: C 56,19; H 6,79; N 2,30; KF (H2): 6,65.
Найдено: C 56,33; H 6,87; N 2,32; KF (H2O): 7,96.
Альтернативный способ получения триэтиламиновой соли:
2'-O-(Этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксел (5,67 г, 4,66 мМ) загружали в 250-миллилитровую колбу и растворяли в
этилацетате (150 мл). Колба была снабжена трехпозиционным клапаном и подсоединена к низковакуумному трубопроводу и трубопроводу, через который подавали аргон. С использованием этого клапана,
содержимое колбы частично откачивали, а затем колбу продували аргоном. Эту процедуру повторяли еще 2 раза. Затем в колбу добавляли 10% палладий на активированном угле (0,85 г). Трубопровод, через
который подавали аргон и связанный с трехпозиционным клапаном, заменяли на баллон, заполненный водородом. Используя клапан, содержимое колбы частично удаляли, и продували аргоном. Эту процедуру
повторяли еще 4 раза. Полученную смесь перемешивали при комнатной температуре в атмосфере баллонного водорода в течение ночи. ТСХ-анализ, проведенный после первоначального воздействия водородом,
показал, на отсутствие исходных материалов. После этого баллон с водородом, связанный с трехпозиционным клапаном, заменяли на трубопровод, через который подавали аргон. Используя трехпозиционный
клапан, колбу частично откачивали, а затем продували аргоном. Эту процедуру также повторяли еще 2 раза. Содержимое колбы подвергали вакуумной фильтрации через слой Целита. Целит промывали
этилацетатом
(2 х 10 мл), и к перемешиваемому фильтрату добавляли Et3N (0,650 мл, 4,66 мМ). Полученную суспензию перемешивали в течение 2 часов при комнатной температуре, а объем снижали
до ≈ 150
мл с использованием роторного испарителя. Твердый продукт фильтровали, промывали этилацетатом (2 х 10 мл) и осушали в вакууме, в результате чего получали 4,76 г (выход - 90%) целевого
соединения в
качестве триэтиламиновой соли и в виде белого порошка (показатель гомогенности, измеренный с помощью ЖХВД, составлял 96,6%).
Альтернативный способ получения
триэтиламиновой соли:
2'-O-(Этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксел (5,17 г, 4,25 мМ) добавляли в 250-миллилитровую колбу и растворяли в этилацетате (150 мл). Колба была
снабжена трехпозиционным
клапаном, и подсоединена к низковакуумному трубопроводу и трубопроводу, через который подавали аргон. С использованием этого клапана, содержимое колбы частично откачивали, а
затем продували аргоном.
Эту процедуру повторяли еще 2 раза. Затем в колбу добавляли 10% палладий на активированном угле (0,86 г). Трубопровод, через который подавали аргон, связанный с
трехпозиционным клапаном, заменяли на
баллон, заполненный водородом. С использованием клапана, содержимое колбы частично откачивали, а затем продували аргоном. Эту процедуру повторяли еще 5 раз.
Полученную смесь перемешивали при комнатной
температуре в атмосфере баллонного водорода. 16-часовой ТСХ-анализ, проведенный после первоначального воздействия водородом, показал на отсутствие исходных
материалов. Баллон с водородом, связанный с
трехпозиционным клапаном, заменяли на трубопровод, через который подавали аргон. Используя трехпозиционный клапан, содержимое колбы частично откачивали, а
затем продували аргоном. Эту процедуру
повторяли еще 2 раза. Содержимое колбы, подвергали вакуумной фильтрации через слой целита. Целит промывали этилацетатом (4 х 10 мл). К перемешиваемому фильтрату
добавляли Et3N (0,590 мл, 4,
25 мМ). Полученную суспензию перемешивали при комнатной температуре в течение часа, а затем объем этой суспензии снижали приблизительно до 140 мл посредством
роторного испарителя. Твердый продукт
фильтровали, промывали этилацетатом (10 мл) и осушали в вакууме, в результате чего получали 4,46 г (выход - 92%) целевой триэтиламиновой соли в виде белого
порошка (показатель гомогенности,
определенный с помощью ЖХВД, составлял 96,7%).
Лизиновую соль получали следующим образом:
2'-O-(Этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксел (15,0 г, 12,34 мМ)
порциями добавляли к суспензии 10% палладированного угля (20%-ная загрузка, 3 г) в EtOH (600 мл, испыт. 200) при 0oC. Полученную суспензию дегазировали путем откачки воздуха и продували
аргоном. Эту процедуру повторяли еще 2 часа. После этого аргон заменяли на водород, и при энергичном перемешивании,
продолжали аналогичную процедуру дегазирования. Полученную смесь перемешивали при
0oC в течение 2 часов. Охлаждающую баню удаляли, а реакционный раствор перемешивали при комнатной
температуре еще в течение 4,5 часа. Реакционную смесь дегазировали путем откачки водорода,
а затем 3 раза продували аргоном. Затем эту смесь фильтровали (в атмосфере аргона) через слой целита. К
полученному фильтрату медленно, при энергичном перемешивании, добавляли раствор лизина (1,63 г,
0,94 экв.) в смеси H2O : EtOH (1 : 1, испыт. 200) (20 мл) в течение 5 минут. После этого к
полученной белой суспензии добавляли дистиллированную воду (110 мл) и перемешивали 30 минут. Эту
суспензию также нагревали до около 55oC. Полученный гомогенный раствор поддерживали в
масляной бане при 50oC, и медленно охлаждали до комнатной температуры в течение 16 часов, а
затем при 4oC в течение 3 часов. После фильтрации и осушки на всасывающем фильтре,
получали 11,8 г (выход ≈ 80%) лизиновой соли с т. пл. > 170oC ( с разлож.).
Показатель гомогенности, определенный с помощью ЖХВД, составлял 99,0%.
Элементный
анализ для C57H72N3O22P • 8,0 H2O:
Вычислено: C 51,62; H 6,69; N 3,17; KF (H2O): 10,87.
Найдено: C 51, 76; H 6,57; N 3,48; KF (H2O): 11,42.
Этаноламиновую соль получали
следующим образом:
2'-O-(Этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела триэтиламиновую соль
(3,0 г, 2,64 мМ) распределяли между EtOAc (60 мл) и 5% NaHSO4 (30 мл) при энергичном
перемешивании при 0oC в течение 15 минут. Водный слой отделяли и экстрагировали этилацетатом
(15 мл). Объединенный этилацетатный слой промывали солевым раствором (15 мл), осушали сульфатом
натрия, фильтровали, и получали раствор свободной кислоты (≈ 2,64 мМ) в EtOAc (≈ 70 мл).
К этому этилацетатному раствору при комнатной температуре, по капле и в течение 5 минут,
добавляли раствор H2NCH2CH2OH (0,15 мл, 2,64 мМ) в EtOAc (5 мл), энергично
перемешивая при этом. Полученную суспензию перемешивали еще 1 час, а затем фильтровали,
промывали этилацетатом (15 мл х 2), и осушали в вакууме, в результате чего получали 2,6 г целевой
этаноламиновой соли с выходом 89%, т. пл. > 130oC (с разлож.). Показатель
гомогенности, определенный с помощью ЖХВД-анализа, составлял 97,8%.
Элементный анализ для
C53H65N2O21P • 2,5 H2O:
Вычислено: C 55,73; H 6,18; N 2,45; KF (H2O) 3,94;
Найдено: C 55,76; H 6,39; N 2,45;
KF (H2O) 6,00.
Аргининовую соль получали следующим образом:

Этот материал (12,95 г) растворяли в смеси 15% H2O в EtOH (прибл. 700 мл) при 55oC. Полученный раствор охлаждали и поддерживали при 30oC в течение 3,5 часа, затем при комнатной температуре в течение 16 часов, и при 4oC в течение 3 часов. Полученные кристаллы фильтровали, промывали холодной 2% H2O в EtOH (50 мл х 2), осушали путем отсасывания в течение 4 часов, а затем осушали в вакууме (1 мм. рт. ст.) в течение 16 часов, и получали 10,2 г (выход примерно 80%) целевой аргининовой соли (показатель гомогенности - 98,5%) с т. пл. > 176oC (при разлож.).
Элементный анализ для C57
H72N5O22P • 6,4 H2O:
Вычислено: C 51,65; H 6,45; N 5,28; KF (H2O) 8,7;
Найдено: C 51,86; H 6,65; N 5,53; KF (H2O) 8,72.
N-Метилглюкаминовую соль получали
следующим образом:
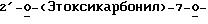
Этот продукт (9,65 г) растворяли в смеси 15% H2O в EtOH (≈ 450 мл) при 52o C. Затем раствор охлаждали и выдерживали при 28oC в течение 3,5 часа, при комнатной температуре в течение 16 часов, и при 4oC в течение 3 часов. Полученные кристаллы фильтровали, промывали холодной 2% H2O в EtOH (50 мл х 2), осушали путем отсасывания в течение 4 часов, а затем осушали в вакууме (1 мм рт. ст.) в течение 16 часов, и получали целевую N-метилглюкаминовую соль (7,5 г, выход приблизительно 80%) с т. пл. > 154oC (при разлож.). Показатель гомогенности, определенный с помощью ЖХВД-анализа, составлял 98,6%.
Элементный анализ для C58H75N2O25P • 5,0 H2O):
Вычислено: C 52,72; H 6,48; N 2,12; KF (H2O) 6,82.
Найдено: C 53,09; H 6,50; N 2,08; KF (H2O) 7,12.
Пример 4:
2'-O-(фосфонооксиметил)паклитаксел
(a) Получение
2'-O-метилтиометил)-7-O-(триэтилсилил)паклитаксела

К холодному (0 - 5oC) раствору 7'-O-(триэтилсилил)паклитаксела (2,46 г, 2,5439 мМ) в безводном ацетонитриле (100 мл) добавляли диметилсульфид (1,348 г, 1,59 мл, 21,6976 мМ), а затем бензоилпероксид (2,628 г, 10,8488 мМ). Полученную гетерогенную смесь перемешивали при 0oC в течение часа, и выдерживали при 5oC в течение 18 часов. В результате этой процедуры образовывался желтый раствор. Этот раствор выпаривали досуха и очищали на колонках с силикагелем (элюент: этилацетат/гексан, 1 : 4, 1 : 3, и 1 : 2), в результате чего получали 1,0 г целевого соединения (выход - 38%). Это соединение использовали также в последующей стадии.
MC: [M + H]+: 1028; [M + Na]+: 1050; [M + K]+: 1066.
(b) Получение
2'-O-(метилтиометил)паклитаксела

К холодному (-15oC) раствору продукта части (a) (1,0 г, 0,9737 мМ) в безводном ацетонитриле (30 мл) по капле добавляли 0,5 н. соляную кислоту (3 мл). Полученный раствор перемешивали при -15oC в течение часа и при 5oC в течение 18 часов. Этот раствор разбавляли этилацетатом (20 мл) и промывали холодным 6% NaHCO3-раствором, и солевым раствором. После этого раствор осушали сульфатом магния и выпаривали досуха. Полученный раствор очищали на пластинах с силикагелем (элюент: метиленхлорид/15% ацетонитрил), и получали чистое целевое соединение (280 мг, 31,4%).
ИК (KBr): 3446, 3064, 2940, 1726, 1666, 1582, 1516, 1486.
ЯМР (CDCl3): δ 1,118 (с, 3H); 1,229 (с, 3H); 1,662 (с, 3H); 1,689 (с, 3H); 1,871 (с, 3H); 2,209 (с, 3H); 2,450 (с, 3H); 3, 800 (д, H); 4,119 (д, H); 4,305 (д, H); 4,413 (м, H); 4,563 (д, H); 4,703 (д, H); 4,940 (д, H); 4,958 (дд, H); 5,667 (д, H); 5,882 (дд, H); 6,263 (м, 2H); 7,019 (д, H); 7,293 - 8,127 (м, 15H).
MC: [M + H]+ : 914; [M + Na]+ : 936; [M + K]+ 952.
BPMC: MH+ : 9143394 (вычислено: 9143422)
(c) Получение
2-O-дибензилфосфонооксиметил)паклитаксела

К перемешанному раствору продукта стадии (b) (0,89 г, 0,9748 мМ) в безводном 1,2-дихлорэтане (12 мл) добавляли порошкообразные молекулярные сита 4A (1,0 г), а затем по капле добавляли раствор смеси N-иодосукцинимида (0,33 г, 1,4622 мМ) и дибензилфосфата (0,41 г, 1,4622 мМ) в безводном тетрагидрофуране (8 мл). Полученную смесь перемешивали при комнатной температуре в течение часа, а затем фильтровали через слой целита. Фильтрат выпаривали досуха, и образовавшийся красный остаток растворяли в этилацетате (50 мл) и промывали холодным 6% NaHSO3, холодным 6% гидрокарбонатом натрия, и солевым раствором. После осушки сульфатом магния и выпаривания, получали пенообразный продукт. Этот продукт очищали на пластинах с силикагелем (элюент: метиленхлорид/20% ацетонитрил), и получали чистый продукт (0,77 г, 69%).
ИК (KBr): 3854, 3744, 3362, 3066, 1960, 1722, 1602, 1580.
ЯМР (CDCl3): δ 1,075 (с, 3H); 1,167 (с, 3H); 1,651 (с, 3H); 1,799 (с, 3H); 2,209 (с, 3H); 2,296 (с, 3H); 2,464 (м, H); 3,686 (д, H); 4,121 (д, H); 4,240(д, H); 4,293 (м, H); 4,408 - 4,957 (м, 6H); 5,006 (м, H); 5,565 - 5,649 (м, 2H); 6,034 (т, H); 6,194 (3H, с); 7,100 - 8,132 (м, 26H).
MC: [M + H]+ : 1144; [M + Na]+ : 1166; [M + K]+ : 1182.
(d) Получение 2'-O-(фосфонооксиметил)паклитаксела

Смесь продукта стадии (c) (0,9 г, 0,8874 мМ) и 10% палладированного угля (1,0 г) в этилацетате (20 мл) гидрировали при 60 фунт/кв.дюйм (400 кПа) в течение 24 часов. Реакционную смесь фильтровали через целит, а фильтрат выпаривали досуха. Остаток очищали на пластинах с силикагелем (элюент : метиленхлорид/40% метанол), и получали целевой продукт (0,254 г, 33,4%) с т. пл. 202 - 205oC (с разлож.).
ИК (KBr): 3438, 3066, 2942, 1722, 1652, 1602 см-1.
ЯМР (ацетон-d6/D2O): δ 1,081 (с, 6H), 1,571 (с, 3H), 1,847 (с, 3H), 2,115 (с, 3H), 2,357 (с, 3H), 3,707 (д, 1H), 4,08 (м, 2H), 4,275 (м, H), 4,941 - 5,085 (м, 4H), 5,231 (т, H), 5,430 (д, H), 5,544 (д, H), 5,970 (т, H), 6,376 (с, H), 6,961 - 8,017 (м, 16H).
MC: [M + Na]+ : 986, [M + K]+ : 1002, [M + 2Na - H]+; 1008, [M + Na - K + H]+: 10,24, [M + 2K - H]+ : 1040.
BPMC: MNa+, 9862955 (вычислено: 9862976).
Пример 5:
2'-7-O-бис-(фосфонооксиметил)паклитаксела натриевая
соль
(a) Получение 2',7-O-бис(метилтиометил)паклитаксела

К перемешанному раствору паклитаксела (0,853 г, 1 мМ) и диметилсульфида (1,465 г, 20 мМ) в ацетонитриле (20 мл) (при 0oC) добавляли твердый бензоилпероксид (1,995 г, 8 мМ). Полученную реакционную смесь энергично перемешивали при 0oC в течение 3 часов. За ходом реакции наблюдали с помощью ТСХ (в гексане : этилацетате; 1 : 1, по объему, Pfпаклитаксел = 0,24, Pfпродукта = 0,60). После исчезновения исходного материала (приблизительно через 3 часа), реакцию гасили путем выпаривания растворителей досуха в низком вакууме при 25oC. Безводный остаток выделяли с использованием колонок с силикагелем (EM Science, 40 - 63 мкм; 100 мл безводного силикагеля; размер колонки: ⌀ = 3/4 дюйм; система растворителей: гексан/этилацетат (3 : 2, по объему); объем каждой фракции составлял приблизительно 25 мл). Полученное целевое соединение (0,515 г, выход - 53%) выделяли из фракций 15 - 19.
MC (FAB /матрик. NOBA, NaI, KI): [M + H]+ m/z: 974; [M + Na]+ : m/z : 996; [M + K]+ m/z 1012.
УФ (MeOH): λмакс= 204 нм, E (1% /1см) = 243,45; λмакс= 228 нм, E (1%/1 см) = 313,99.
ИК (KBr): 3440, 3064, 2926, 1724, 1668, 1602, 1582, 1514, 1484, 1452, 1372, 1314, 1266, 1242, 1178, 1142, 1068, 1026, 990, 916, 886, 848, 800, 774, 710, 646, 606, 570, 540, 480 см-1.
1H-ЯМР (CDCl3): δ 1,17 (3H, с); 1,20 (3H, с); 1,68 (3H, с); 1,74 (3H, с); 1,84 (H, дд); 2,04 (3H, д); 2,09 (3H, с); 2,15 (3H, с, перекрывающийся с (H, м); 2,37 (H, дд); 2,51 (3H, с); 2,79 (H, ддд); 3,78 (H, д); 4,18 (H, д); 4,28 (H, м); 4,31 (H, д); 4,53 - 4,74 (4H, два перекрывающихся AB, м); 4,93 (H, д); 4,95 (H, д); 5,68 (H, д); 5,82 (H, дд); 6,24 (H, дд); 6,54 (H, с); 7,05 (H, д); 7,28 - 7,59 (10H, перекрывающийся); 7,57 (H, м); 7,76 (2H, д); 8,09 (2H, д).
(b) Получение 2',
7'-O-бис(дибензилфосфонооксиметил)паклитаксела

Раствор N-йодосукцинимида (135 мг, 0,5 мМ) и дибензилфосфата (167 мг, 0,5 мМ) в безводном тетрагидрофуране (8 мл), при комнатной температуре, добавляли к смеси 2', 7-бис(метилтиометил)паклитаксела (198 мг, 0,2 мМ) и молекулярных сит 5A (приблизительно 200 мг) в метиленхлориде (12 мл). Реакционную смесь перемешивали в течение 1,5 часа, а затем молекулярные сита фильтровали через целит, промывали метиленхлоридом (10 мл), и растворители выпаривали досуха при комнатной температуре в низком вакууме. Остаток растворяли в этилацетате (100 мл) и промывали в делительной воронке 50 миллилитрами 1% тиосульфата натрия, 0,5 молями бикарбоната натрия (50 мл), а затем дважды промывали водой (2 х 50 мл). Органическую фазу осушали сульфатом магния, выпаривали досуха и снова растворяли в этилацетате (1 мл). Продукт осаждали путем добавления смеси 50 мл этилового эфира/гексана, (1 : 1) и дважды промывали, используя ту же самую систему растворителей (2 х 50 мл). Таким образом был получен неочищенный продукт (218 мг) с выходом 74%. Этот продукт очищали путем загрузки его метиленхлоридного раствора (3 мл) на пластины с силикагелем (⌀ = 3/4 дюйм х L = 1 дюйм) и элюировали системой растворителей (метиленхлорид/этилацетат), (3 : 1) (50 мл). Таким образом получали 172,7 мг целевого соединения (выход - 59,3%).
MC (FAB, матрич. NOBA / NaI, KI): [M + Na]+ m/z : 1456; [M + K]+ m/z : 1472.
УФ (MeCN): λмакс= 194 нм, E (1%/1 см) = 1078,36; λмакс= 228 нм, E (1%/1 см) = 311,95.
ИК (KBr): 3430, 3066, 3032, 2958, 1744, 1726, 1664, 1602, 1582, 1532, 1488, 1456, 1372, 1270, 1244, 1158, 1108, 1068, 1016, 1000, 952, 886, 800, 776, 738, 698, 604, 498 см-1.
1 H-ЯМР (CDCl3): δ 1,12 (3H, с); 1,14 (3H, с); 1,56 (H, м); 1,67 (3H, с); 1,84 (3H, д); 1,90 (H, м); 2,17 (3H, с); 2,29 (3H, с); 2,73 (H, м); 3,73 (H, д); 4,08 (H, д); 4,15 (H, м); 4, 20 (H, д); 4,77 (H, м); 4,79 (H, д); 4,91 - 5,04 (10H, перекрывающийся м); 5,25 (H, дд); 5,38 (H, дд); 5,54 - 5,56 (2H, перекрывающийся м); 5,99 (H, шир. дд); 6,25 (H, с); 7,11 - 7,14 (2H, м); 7,24 - 7,64 (28H, перекрывающийся м); 7,94 (2H, дд); 8,04 (2H, дд); 8,30 (H, д).
(c) Получение натриевой соли 2',
7-О-бис(фосфонооксиметил)паклитаксела

Образец 2'-7-О-бис(дибензилфосфонооксиметил)паклитаксела (112 мг, 0,078 мМ) растворяли в этилацетате (7 мл) и гидрировали, при комнатной температуре, в присутствии 10% палладированного угля (50 мг) при 60 фунт/кв. дюйм (400 кПа) в течение 2 часов. Катализатор удаляли путем фильтрации через слой целита. Целит промывали этилацетатом (10 мл). Фильтрат обрабатывали твердым бикарбонатом натрия (20 мл, 3 экв.), а затем растворитель выпаривали досуха. Полученный сухой остаток снова растворяли в 5 мл воды:ацетоне (4:1, по объему) и очищали на C-18-колонках с использованием обращенно-фазовой хроматографии (55 - 105 мкм C-18, Jones, 50 мл сухого C-18, ⌀ = 3/4 дюйм в системе: вода/ацетон, 4:1, по объему). Элюент контролировали с помощью аналитической ЖХВД на C18-колонках Jonex (15 см, 1 мл/мин, λ = 230 мн) в ацетонитриле:фосфатном буфере (pH 6, 50/50, по объему) с добавлением смеси Q12-ионных пар (Regis), при Rt = 4,7 мин. Фракции, содержащие целевой продукт, объединяли, ацетон выпаривали в условиях невысокого вакуума при 20oC, и раствор лиофилизовали. Таким образом получали целевой продукт (44,2 мг, 58,8% - выход).
МС FAB, матрич. NOBA /NaI, KI): [M + H]+ m/z : 1118; [M + Na]+: m/z 1140.
УФ (MeCN): λмакс= 192 нм, E (1%/1 см) = 129,73; λмакс= 230 нм, E (1%/1 см) = 26,43.
ИК (KBr): 3430, 3066, 2956, 1724, 1658, 1604, 1582, 1520, 1486, 1452, 1374, 1316, 1256, 1152, 1110, 1070, 1026, 966, 914, 802, 772, 710, 538 см-1.
1H-ЯМР (ацетон-d6/D2O): δ 0,97 (3H, с); 1,02 (3H, с); 1,47 (H, м); 1,54 (3H, с); 1,70 (H, м); 1,75 (3H, с); 1,85 (H, м); 2,11 (3H, с); 2,30 (3H, с); 2,88 (H, м); 3,64 (H, д); 4,03 (H, м); 4,06 (H, д); 4,16 (H, д); 4,74 (H, м), 4,86 (H, м); 5,11 (H, шир. т); 5,22 (H, д); 5,42 (H, д); 5, 90 (H, шир. т); 6,21 (H, с); 7,06 (H, шир. т); 7,32 - 7,69 (10H, перекрывающийся m); 7,80 (2H, д); 7,93 (2H, д).
Пример 6. 7'-О-Метилтиометилбаккатин III (7-МТМ-баккатин III)

К раствору 2'-О-этилоксикарбонил-7-О-метилтиометилпаклитаксела (соединение примера 3 (b), 27 г, 27,4 мМ) в 100 мл ТГФ и 500 мл метанола, добавляли свежеразмолотый карбонат калия (2,7 г, 19 мМ). Полученный раствор перемешивали 30 минут, нейтрализовали смолой IP-120 (H+), фильтровали и концентрировали. Неочищенный фильтрат растворяли в 200 мл дихлорметана и перемешивали в течение 24 часов с борогидридом тетрабутиламмония (10 г). Затем раствор разбавляли дихлорметаном и промывали водой, насыщенным бикарбонатом и солевым раствором. После этого органическую фракцию осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 1:1) и получали 9,4 г целевого соединения (53%) с т. пл. 269oC.
FAB-MC (NOBA): [M + H]+ для C33H43 NO11: вычислено: 647. Найдено: 647.
ИК (KBr): 3474, 1746, 1724, 1712, 1270, 1240, 1070 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,08 (д, J = 7,1 Гц, 2H); 7, 58 (т, J = 7,5 Гц, 1H); 7,45 (т, J = 7,8 Гц, 2H); 6,55 (с, 1H); 4,94 (д, J = 8,1 Гц, 1H); 4,83 (кв, J = 5,1 Гц, 1H); 4,66 (AB кв, J = 14,7, 12,3 Гц, 2H); 4,30 (м, 2H): 4,13 (д, J = 8,4 Гц, 1H); 3,91 (д, J = 6,6 Гц, 1H); 2,79 (м, 1H); 2,27 (c, 3H); 2,25 (м, 2H); 2,19 (с, 3H); 2,16 (c, 3H); 2,10 (с, 4H); 1,81 (м, 1H); 1,72 (с, 3H); 1,61 (м, 2H); 1,16 (с, 3H); 1,03 (с, 3H).
13C-ЯМР (CDCL3, 75,5 Гц): δ 202,3, 170,8, 169,3, 167,0, 144,2, 132,6, 132,1, 130,1, 129,4, 128,6, 83,9, 80,9, 78,7, 75,7, 74,5, 73,9, 57,9, 57,6, 47,6, 42,7, 38,3, 26,7, 22,6, 21,0, 20,1, 15,2, 15,0, 10,8.
Пример 7.
Триэтаноламиновая соль
3'-N-дебензоил-3'-дезфенил-3'-N-(т-бутоксикарбонил)-3'-(2-фурил)-2'-О- этилоксикарбонил-7-О-фосфонооксиметилпаклитаксела
(a) Получение
3'-дебензоил-3'-дезфенил-3'-N-(т- бутилоксикарбонил)-3'-(2-фурил)-7-О-метилтиометилпаклитаксела

К раствору HMDS (0,40 мл, 1,90 мМ) в 15 мл ТГФ добавляли раствор n-бутиллития (0,75 мл, 2,5 M в гексане, 1,88 мМ) и перемешивали в течение 5 минут при -55oC. К этому раствору добавляют баккатин III 7-МТМ (соединение примера 6, 1,03 г, 1,59 мМ) в 10 мл ТГФ и перемешивали в течение 10 минут, после чего добавляли 10 мл раствора (3R, 4R)-1-(т-бутилоксикарбонил)-4-(2-фурил)-3-(триэтилсилилокси)-2- азетидинона (883 мг, 2,40 мМ). Холодную баню удаляли и заменяли на баню с т. 0oC, и реакционную смесь перемешивали в течение 30 минут. Полученный раствор разбавляли этилацетатом и промывали насыщенным раствором NH4Cl, осушали сульфатом магния, а затем концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 2: 5: 1), и получали 1,5 г продукта присоединения 3'-N-дебензоил-3'-дезфенил-3'-N-(т-бутилоксикарбонил)-3'-(2-фурил)-7-О-метилтиометил-2'-О-триэтилсилилпаклитаксела (93%).
FAB-MC (NOBA) [M+Na] для C50H71NSSiO16:
Вычислено: 1036; Найдено: 1036.
ИК (пленка): 3446 (с), 1720, 1368, 1242, 1166, 1144, 1124, 1066 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,07 (д, J = 7,2 Гц, 2H); 7,56 (м, 1H); 7,46 (т, J = 7,5 Гц, 2H); 7,36 (м, 1H); 6,56 (с, 1H); 6,33 (м, 1H); 6,20 (м, 2H); 5,67 (д, J = 6,9 Гц, 1H); 5,29 (шир. с, 2H); 4,94 (д, J = 7,8 Гц, 1H); 4,75 (с, 1H), 4,65 (с, 2H); 4,28 (м, 2H); 4,16 (д, J = 8,1 Гц, 1H); 3,89 (д, J = 6,9 Гц, 1H); 2,80 (м, 1H); 2,46 (с, 3H); 2,37 (м, 1H); 2,22 (м, 1H); 2,16 (с, 3H); 2,10 (с, 3H); 2,04 (c, 3H); 1,84 (м, 1H); 1,74 (с, 3H); 1,65 (м, 1H); 1,33 (с, 9H); 1,20 (с, 3H); 1,19 (с, 3H); 0,81 (т, J = 7,8 Гц, 9H); 0,47 (м, 6H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 202,0, 171,2, 170,3, 169,3, 167,1, 155,3, 152,0, 141,9, 141,0, 133,6, 132,9, 130,2, 129,2, 128,7, 110,7, 107,3, 84,0, 81,1, 80,2, 78,7, 76,1, 75,7, 74,1, 72,4, 71,1, 57,4, 52,8, 47,1, 43,3, 35,2, 33,0, 28,1, 26,3, 22,9, 21,2, 21,0, 15,0, 14,5, 10,9, 6,5, 4,3.
К раствору 2-триэтилсилилового эфира, полученного выше (330 мг, 0,32 мМ) в 7 мл ТГФ, добавляли фторид тетрабутиламмония (0,35 мл, 1,0 М в ТГФ, 0,35 мМ) и перемешивали 10 минут. Раствор разбавляли этилацетатом, промывали солевым раствором, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 2:1), и получали 301 мг целевого соединения (95%).
FAB-МС (NOBA) [M + H] для C45H58NO16S: Вычислено: 900; Найдено: 900.
ИК (пленка): 3442, 1720, 1242, 1066, 1026 см-1.
1 H-ЯМР (CDCl3, 300 МГц): δ 8,07 (д, J = 7,3 Гц, 2H); 7,57 (т, J = 7,3 Гц, 1H); 7,45 (т, J = 7,8 Гц, 2H); 7,38 (с, 1H); 6,53 (с, 1H); 6,34 (д, J = 3,2 Гц, 1H); 6, 29 (д, J = 3,2 Гц, 1H); 6,17 (т, J = 8,1 Гц, 1H); 5,65 (д, J = 6,9 Гц, 1H); 5,29 (м, 2H); 4,92 (д, J = 8,0 Гц, 1H); 4,70 (м, 1H); 4,64 (д, J = 4,6 Гц, 2H); 4,29 (м, 2H); 4,14 (д, J = 8,3 Гц, 1H); 3, 86 (д, J = 6,8 Гц, 1H); 3, 37 (д, J = 5,8 Гц, 1H); 2,77 (м, 1H); 2,38 (с, 3H); 2,32 (м, 2H); 2,16 (с, 3H); 2,10 (с, 3H); 2,02 (c, 3H); 1,77 (м, 3H); 1,73 (с, 3H); 1,33 (с, 9H); 1,17 (с, 3H); 1,12 (с, 3H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 202,0, 172,6, 170,3, 169,2, 167,0, 155,2, 151,3, 142,4, 140,4, 133,7, 133,2, 130,2, 129,1, 128,7, 110,7, 107,4, 83,9, 81, 2, 80,5, 78,6, 76,5, 76,1, 75,4, 74,6, 74,0, 72,5, 71,8, 57,4, 51,7, 47,2, 43,2, 35,2, 32,8, 28,1, 26,4, 22,6, 20,9, 15,2, 14,6, 10,9, 8,3.
(b) Получение
3'-N-дебензоил-3'-дезфенил-3'-N-(т- бутоксикарбонил)-3'-(2-фурил)-2'-О-этилоксикарбонил-7-О- метилтиометилпаклитаксела

К раствору продукта стадии (a) (864 мг, 0,96 мМ) в 50 мл дихлорметана при 0oC, добавляли диизопропилэтиламин (2,0 мл, 11,5 мМ) и этилхлороформат (0, 50 мл, 5,25 мМ), а затем полученный раствор перемешивали в течение 4 часов. После этого раствор разбавляли дихлорметаном и промывали насышенным бикарбонатом, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 1:1) и получали 884 мг целевого соединения (2-этилкарбоната) с выходом 95%.
FAB-MC (NOBA) [M + H] для C48H62NO18S: Вычислено: 9723668; Найдено: 9723654.
ИК (пленка): 1752, 1720, 1370, 1244, 1196, 1176, 1064 см-1.
1H-ЯМР (CDCl3, 300 Мгц): δ 8,09 (д, J = 7,8 Гц, 2H); 7,57 (т, J = 7,5 Гц, 1H); 7,46 (т, J = 7,8 Гц, 2H); 7,38 (с, 1H); 6,55 (с, 1H); 6,35 (м, 1H); 6,27 (м, 1H); 6,22 (т, J = 7, 8 Гц, 1H); 5,67 (д, J = 7,2 Гц, 1H); 5,51 (д, J = 9,9 Гц, 1H); 5,34 (д, J = 2,4 Гц, 1H); 5,25 (д, J = 10,2, Гц, 1H); 4,95 (д, J = 8,1 Гц); 4,65 (с, 2H); 4,30 (м, 2H); 4,22 (м, 2H); 3,88 (д, J = 7,2 Гц, 1H); 2,81 (м, 1H); 2,41 (с, 3H); 2,36 - 2,21 (м, 2H); 2,16 (с, 3H); 2,11 (с, 3H); 2,09 (с, 3H); 1,83 (м, 1H); 1,74 (с, 3H); 1,67 (c, 3H); 1,59 (с, 1H); 1,34 (с, 9H); 1,29 (т, J = 7,2 Гц, 3H); 1, 20 (с, 3H); 1,18 (с, 3H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 202,1, 169,9, 169,1, 167,6, 167,0, 154,0, 150,1, 142,6, 141,0, 133,6, 132,9, 130,2, 129,2, 128,7, 110,7, 107,5, 83,9, 81,1, 80,7, 78,7, 76,0, 75,7, 75,1, 74,7, 74,2, 71,8, 65,1, 57,4, 49,7, 46,1, 43,2, 35,0, 33,0, 28,1, 26,3, 22,6, 21,1, 20,9, 15,1, 14,5, 14,1, 10,9.
(c)
Получение
3'-дебензоил-3'-дезфенил-3'- (т-бутилоксикарбонил)-3'-(2-фурил)-2'-О-этилоксикарбонил-7-О-дибензил-фосфонооксиметилпаклитаксела

К раствору продукта стадии (b) (230 мг, 0,236 мМ) в 10 мл безводного ТГФ, добавляли 300 мг молекулярных сит 4A 270 мг (0,98 мМ) дибензилфосфата и 62 мг (0,28 мМ) перекристаллизованного NIS. К этому раствору добавляли 45 мг (0,17 мМ) трифторометансульфоната серебра, и раствор перемешивали в течение 3 часов. Затем раствор фильтровали через целит и разбавляли этилацетатом, промывали 10%-ным NaS2O8, насыщенным бикарбонатом, и солевым раствором, после чего осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: 15% ацетонитрил/хлороформ), в результате чего получали 219 мг целевого дибензилфосфата (77 %).
FAB-MC (NOBA) [M + Na] : для C61H72NPO22Na:
Вычислено: 1224; Найдено: 1224.
ИК (пленка): 3422 (шир. ), 1750, 1722, 1370, 1244, 1160, 1036, 1016, 1000, 976, 944 см-1 .
1 H-ЯМР (CDCl3, 300 МГц): δ 8,08 (д, J = 6,9 Гц, 2H); 7,58 (т, J = 7,2 Гц, 1H); 7,46 (т, J = 7,8 Гц, 2H); 7,39 (с, 1H); 7,31 (м, 10H); 6,35 (м, 2H); 6,28 (с, 1H); 6,21 (т, J = 7,8 Гц, 1H); 5,64 (д, J = 6,9 Гц, 1H); 5,50 (д, J = 10,5 Гц, 1H); 5,39 (д, J = 6,6 Гц, 1H); 5,32 (д, J = 2,4 Гц, 1H); 5,25 (д, J = 9,9 Гц, 1H); 5,01 (дд, J = 8,1, 6,3 Гц, 5H); 4, 86 (д, J = 8,4 Гц, 1H); 4,29 - 4,09 (м, 4H); 3,85 (д, J = 8,4 Гц, 1H); 2,77 (м, 1H); 2,40 (с, 3H); 2,30 (м, 2H); 2,16 (с, 3H); 1,99 (с, 3H); 1,94 (м, 1H); 1,70 (с, 3H); 1,67 (с, 1H); 1,54 (с, 1H); 1, 34 (с, 9H); 1,28 (т, J = 7,2 Гц, 3H); 1,20 (с, 3H); 1,17 (с, 3H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 201,8, 169,9, 169,2, 167,7, 167,0, 155,1, 154,0, 150,0, 142, 74, 141,1, 133,7, 132,9, 130,2, 129,1, 128,7, 128,5, 128,4, 128,0, 110,7, 107,6, 93,8, 84,1, 81,6, 80,8, 80,7, 78,8, 76,3, 75,1, 74,6, 71,8, 69,3, 69,2, 65,1, 57,0, 49,7, 46,7, 43,2, 35,0, 28,1, 26,4, 22,6, 21,2, 20,8, 14,6, 14,1, 10,5.
(d) Получение триэтаноламиновой соли
3'-N-дебензоил-3'-N-дезфенил-3'-N-(т-бутоксикарбонил)-3'-(2-фурил)-2'-О- этилоксикарбонил-7-О-фосфонооксиметилпаклитаксела

К раствору продукта стадии (c) (311 мг, 0,259 мМ) в 25 мл этилацетата добавляли 60 мг 10% палладированного угля и полученный раствор перемешивали в атмосфере водорода в течение 30 минут. Катализатор удаляли путем фильтрации через целит, и фильтрат концентрировали в вакууме. Остаток растворяли в 3 мл этилацетата и добавляли триэтаноламин (2,3 мл, 0,1 М в этилацетате, 0,23 мМ). Полученный раствор концентрировали, а остаток хроматографировали на C18-колонках (элюент: 40% ацетонитрил/вода) и лиофилизовали, в результате чего получали 205 мг фосфаттриэтаноламиновой соли (67%).
FAB-MC (NOBA) [M+Na] для C47H60HPO22Na: вычислено: 1044, найдено: 1044.
ИК (пленка): 3432 (шир. ), 1752, 1722, 1372, 1246, 1158, 1108, 1096, 1070, 1002 см-1.
1H-ЯМР (d6-ацетон/D2O, 300 МГц): δ 8,09 (д, J = 7,2 Гц, 2H); 7,62 (м, 2H); 7,52 (т, J = 7,5 Гц, 2H); 6,48 (д, J = 3,3 Гц, 1H); 6,42 (м, 2H); 6,16 (т, J = 8,7 Гц, 1H); 5,65 (д, J = 6,9 Гц, 1H); 5,46 (д, J = 3,6 Гц, 1H); 5, 30 (д, J = 3,6 Гц, 1H); 5,17 (шир.с, 1H); 5,01 (шир.д, J = 9,0 Гц, 1H); 4,19 (шир.с, 1H); 4,18 (м, 5H); 3,95 (м, 4H); 3,87 (д, J = 6,9 Гц, 1H); 3,68 (с, 10H); 3,50 (шир.триплет, J = 4,8 Гц, 4H); 2,95 (м, 1H); 2,44 (с, 3H); 2,41 (м, 2H); 2,16 (с, 3H); 1,99 (с, 3H); 1,94 (м, 1H); 1,68 (с, 3H); 1,34 (с, 9H); 1,24 (т, J = 6,9 Гц, 3H); 1,17 (с, 6H).
Пример 8. Триэтаноламиновая соль
3'-N-дебензоил-3'-дезфенил-3'-N-(бутилоксикарбонил)-3'-(2-тиенил-2'-O-этилоксикарбонил-7-O-фосфонооксиметилпаклитаксела
(а) Получение
3'-N-дебензоил-3'-дезфенил-3'-N-(т-бутилоксикарбонил)-3'-(2-тиенил)-7-O-метилтиометилпаклитаксела

К раствору HDMS (0,5 мл, 2,4 мМ) в 18 миллилитрах ТГФ, при -55oC, добавляли n-бутиллитий (0,85 мл, 2,5 М в гексане, 2,1 мМ). Через 10 минут к раствору по капле добавляли баккатин III 7-МТМ (1,15 г, 1,78 мМ) в 18 миллилитрах ТГФ, и полученный раствор перемешивали в холоде в течение 10 минут. После этого добавляли (± )-цис-1-(т-бутилоксикарбонил)-4-(2-тиенил)-3- (триэтилсилилокси)-2-азетидинон (2,80 г, 7,3 мМ) в 18 мл ТГФ, и холодную баню медленно нагревали до 0oC в течение 30 минут. Раствор разбавляли этилацетатом и промывали насыщенным раствором NH4Cl, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 3: 1) и получали 1,87 г выделенного лактама (элюент: гексан/этилацетат, 3: 1) и 1,44 г продукта присоединения - 3'-N-дебензоил-3'-дезфенил-3'-N-(т-бутилоксикарбонил)-3'-(2-тиенил)- 7-O-метилтиометил-2'-O-триэтилсилилпаклитаксела (выход - 78%).
FAB-MC (NOBA) [M + Na] для C51H71NO15S2SiNa:
Вычислено: 1052; Найдено: 1052.
ИК (пленка): 3442 (шир. ), 1720, 1490, 1368, 1270, 1242, 1162, 1110, 1064, 1024, 984, 754 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,09 (д, J = 7,2 Гц, 2H); 7,57 (т, J = 7,6 Гц, 1H); 7,47 (т, J = 7,8 Гц, 2H); 7,22 (м, 1H), 6,95 (м, 2H); 6,55 (с, 1H); 6,21 (т, J = 9,3 Гц, 1H); 5,68 (д, J = 6,9 Гц, 1H); 5,49 (шир.д, 1H); 5,39 (шир. д, J = 9,6 Гц, 1H); 4,94 (д, J = 7,8 Гц, 1H); 4,65 (с, 2H); 4,57 (с, 1H); 4,28 (м, 2H); 4,17 (д, J = 8,4 Гц, 1H); 3,88 (д, J = 6,9 Гц, 1H); 2,80 (м, 1H); 2,46 (с, 3H); 2,37 (м, 1H); 2,20 (м, 1H); 2,17 (с, 3H); 2,10 (с, 3H); 2,03 (с, 3H); 1,84 (м, 1H); 1,74 (с, 3H); 1,68 (с, 1H); 1,62 (с, 1H); 1,31 (с, 9H); 1,20 (с, 6H); 0,84 (т, J = 7,8 Гц, 9H); 0,50 (м, 6H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 201,9, 171,1, 170,7, 170,1, 169,3, 167,0, 155,1, 142,8, 140,9, 133,6, 132,9, 130,2, 129,2, 128,7, 126,9, 124,6, 83,9, 81,2, 80,1, 78,8, 77,4, 76,0, 75,7, 75,2, 74,8, 74,1, 71,3, 57,4, 53,8, 47,0, 43,3, 35,3, 33,3, 28,1, 26,3, 23,0, 21,3, 20,9, 14,9, 14,4, 10,9, 6,6, 4,5.
К раствору 2'-триэтилсилилового эфира, полученного выше (1,41 г, 1,37 мМ) в 14 мл ТГФ, добавляли фторид тетрабутиламмония (1,4 мл, 1,0 М в ТГФ, 1,40 мМ). Раствор перемешивали 30 минут, разбавляли этилацетатом, промывали солевым раствором, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 1:1) и получали 1,16 г целевого соединения (92%).
FAB-MC (NOBA) [M + Na] для C45H57NO15S2Na:
Вычислено: 938; Найдено: 938.
ИК (пленка): 3440 (шир.), 1720, 1368, 1242, 1168, 1106, 1066, 710 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,08 (д, J = 7,2 Гц, 2H); 7,59 (м, 1H); 7,47 (т, J = 7,8 Гц, 2H); 7,24 (м, 1H), 7,07 (м, 1H); 6,99 (м, 1H); 6,53 (с, 1H); 6,18 (т, J = 8,1 Гц, 1H); 5,66 (д, J = 6,9 Гц, 1H); 5,49 (д, J = 9,6 Гц, 1H); 5,32 (д, J = 9,6 Гц, 1H); 4,92 (д, J = 7,8 Гц, 1H); 4,63 (м, 3H); 4,28 (м, 2H); 4,15 (д, J = 8,4 Гц, 1H); 3,86 (д, J = 6,9 Гц, 1H); 3,47 (д, J = 5,4 Гц, 1H); 2,78 (м, 1H); 2,36 (с, 3H); 2,34 (с, 2H); 2,17 (с, 3H); 2,10 (с, 3H); 2,00 (с, 3H); 1,83 (м, 1H); 1,74 (с, 3H); 1,72 (с, 1H); 1,61 (с, 1H); 1,33 (с, 9H); 1,21 (с, 3H); 1,18 (с, 3H).
13C-ЯМР (CDCl3, 75,5 Гц) δ 201,9, 172,3, 170,3, 169,2, 167,0, 154,0, 141,5, 140,2, 133,7, 133,3, 130,2, 129,1, 128,7, 127,0, 125,4, 83,9, 81,3, 80,4, 78,6, 76,1, 75,4, 74,5, 74,0, 73,4, 72,5, 57,5, 52,8, 47,2, 43,2, 35,3, 32,9, 28,2, 26,4, 22,6, 20,9, 15,1, 14,7, 10,8.
(b) Получение 3'-дебензоил-3'-дезфенил-3'-N-(т-бутилоксикарбонил)-3'-(2-тиенил)-2'-O-этилоксикарбонил-7-O-метилтиометилпаклитаксела

К раствору продукта стадии (a) (621 мг, 0,677 мМ) в 35 мл дихлорметана, при 0oC, добавляли диизопропилэтиламин (1,20 мл, 6,89 мМ) и этилхлороформат (0,35 мл, 3,7 мМ). Полученную смесь перемешивали в течение 1 часа. Холодную баню удаляли, а раствор перемешивали в течение 2 часов и разбавляли дихлорметаном, промывали насыщенным бикарбонатом, осушали сульфатом магния, и концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат, 1:1), и получали 528 мг целевого соединения (79%).
FAB-MC
(NOBA) [M + Na] для C48H61NO17S2Na:
Вычислено: 1010; Найдено: 1010.
ИК (пленка): 3510, 3440, 1752, 1720, 1370, 1244, 1198, 1170, 1026, 988, 756 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,09 (д, J = 7,2 Гц, 2H); 7,58 (м, 1H); 7,48 (т, J = 7,8 Гц, 2H); 7,26 (м, 1H), 6,99 (м, 2H); 6, 55 (с, 1H); 6,23 (т, J = 8,0 Гц, 1H); 5,68 (д, J = 6,9 Гц, 1H); 5,33 (д, J = 9,9 Гц, 1H); 5,25 (д, J = 2,4 Гц, 1H); 4,94 (д, J = 7,8 Гц, 1H); 4,65 (c, 2H); 4,33 - 4,08 (м, 5H); 3,88 (д, J = 6,9 Гц, 1H); 2,80 (м, 1H); 2,40 (с, 3H); 2,40 - 2,20 (м, 2H); 2,16 (с, 3H); 2,11 (с, 3H); 2,07 (с, 3H); 1,83 (м, 1H); 1,74 (с, 3H); 1,69 (с, 1H); 1,60 (с, 1H); 1,33 (с, 9H); 1,31 (т, J = 7,2 Гц, 9H); 1,20 (с, 3H); 1,19 (с, 3H).
13C-ЯМР (CDCl3, 75,5 Гц): δ: 202,0, 169,7, 169,1, 167,5, 167,1, 154,0, 140,9, 133,6, 132,9, 130,2, 129,2, 128,7, 127,2, 125,4, 125,3, 83, 9, 81,2, 80,6, 78,8, 76,9, 76,0, 75,7, 74,7, 74,2, 72,8, 72,0, 65,2, 57,4, 50,9, 47,1, 43,3, 35,1, 33,0, 28,1, 26,4, 22,7, 21,2, 20,9, 15,1, 14,5, 14,1, 10,9.
(c) Получение
3'-N-дебензоил-3'-дезфенил-3'-N-(т-бутилоксикарбонил)-3-(2-тиенил)-2'-O-этилоксикарбонил-7-O- дибензилфосфонооксиметилпаклитаксела

К раствору продукта стадии (b) (516 мг, 0,522 мМ) в 15 мл безводного тетрагидрофурана добавляли 530 мг молекулярных сит 4A, 576 мг (2,09 мМ) дибензилфосфата и 136 мг перекристаллизованного NIS (0,604 мМ). К этому раствору добавляли трифторометансульфонат серебра (50 мг, 0,194 мМ), и полученный раствор перемешивали 1 час. После этого раствор фильтровали через целит, разбавляли этилацетатом, промывали 10% NaS2O8, насыщенным бикарбонатом и солевым раствором, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (15% ацетонитрил/-хлороформ) и получали 535 мг целевого соединения (84%).
FAB-MC (NOBA) [M + Na] для C61H72NO21PSNa:
Вычислено:
1240; Найдено: 1240.
ИК (пленка): 3424 (шир.), 1750, 1722, 1370, 1244, 1016, 1000, 944 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,08 (д, J = 7,0 Гц, 2H); 7,58 (м, 1H); 7,47 (т, J = 7,5 Гц, 2H); 7,28 (м, 11H), 6,99 (м, 2H); 6,33 (с, 1H); 6,22 (т, J = 7,8 Гц, 1H); 5,66 (м, 2H); 5,39 (т, J = 6,6 Гц, 1H); 5,34 (д, J = 12 Гц, 1H); 5,22 (д, J = 2,4 Гц, 1H); 5,01 (д, J = 8,1, 6,0 Гц, 5H); 4,86 (д, J = 7,8 Гц, 1H); 4,29 - 4,08 (м, 5H); 3,85 (д, J = 6,6 Гц, 1H); 2,76 (м, 1H); 2,39 (с, 3H); 2,35 - 2,18 (м, 2H); 2,16 (с, 3H); 1,97 (с, 4H); 1, 69 (с, 4H); 1,33 (с, 9H); 1,30 (т, J = 7,2 Гц, 3H); 1,20 (с, 3H); 1,17 (с, 3H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 197,4, 165,4, 164,9, 163, 3, 162,7, 150,6, 149,7, 136,7, 136,0, 129,4, 128,6, 125,9, 124,7, 124,3, 124,2, 124,1, 123,6, 122,9, 121,1, 121,0, 89,4, 79,8, 77,3, 76,5, 76,3, 74,4, 72,0, 70,7, 70,3, 67,7, 64,9, 60,9, 52,7, 46,5, 42,3, 38,9, 30,7, 23,8, 22,0, 13,3, 17,0, 16,4, 10,3, 9,8, 6,2.
(d) Получение триэтаноламиновой соли
3'-N-дебензоил-3-дезфенил-3'-N-(т-бутилоксикарбонил)-3'-(2-тиенил)-2-O-этилоксикарбонил-7-O-фосфонооксиметилпаклитаксела

К раствору продукта стадии (c) (512 мг, 0,42 мМ) в 30 мл этилацетата, добавляли 53 мг 10% палладированного угля и раствор перемешивали в атмосфере водорода в течение 3 часов. Катализатор удаляли путем фильтрации через целит, а фильтрат концентрировали в вакууме. Остаток растворяли в 2 мл этилацетата и добавляли триэтаноламин (4,0 мл, 0,1 М в этилацетате, 0, 40 мМ). Полученный раствор концентрировали, и остаток хроматографировали на C18-колонках (элюент: 40% ацетонитрил/вода), а затем лиофилизовали, и получали 280 мг фосфаттриэтаноламиновой соли (56%). Чистота соли, определенная с помощью ЖХВД-анализа, составляла 96%.
FAB-MC (NOBA) [M + Na) для C47H60NO21PS:
Вычислено: 1060; Найдено:
1060.
ИК (KBr): 3422 (шир.), 1750, 1720, 1372, 1246, 1162, 1096, 1068, 1000 см-1.
1H-ЯМР (d6-ацетон/D2O, 300 МГц): δ 8,06 (д, J = 7,2 Гц, 2H); 7,63 (т, J = 7,2 Гц, 1H); 7,52 (т, J = 7,8 Гц, 2H); 7,38 (д, J = 4,2 Гц, 1H); 7,16 (д, J = 3,5 Гц, 1H); 7,01 (дд, J = 5,1, 3,6 Гц, 1H); 6,37 (с, 1H); 6,11 (т, J = 8,7 Гц, 1H); 5,61 (д, J = 6,9 Гц, 1H); 5,60 (с, 1H); 5,26 (д, J = 4,5 Гц, 1H); 5,14 (т, J = 6,6 Гц, 1H); 5,00 (д, J = 8,4 Гц, 1H); 4,86 (дд, J = 12,0, 6,3 Гц, 1H); 4,17 (м, 5H); 4,00 (с, 7H); 3,92 (т, J = 4, 8 Гц, 6H); 3,84 (д, J = 6,9 Гц, 1H); 3,48 (т, J = 4,8 Гц, 6H); 2,94 (м, 1H); 2,42 (с, 3H); 2,36 (м, 1H); 2,27 (м, 1H); 2,15 (с, 3H); 1,95 (с, 4H); 1,66 (с, 3H); 1,30 (с, 9H); 1,23 (т, J = 7,2 Гц, 3H); 1,14 (с, 6H).
Пример 9. 10-Дезацетил-3'-N-дезбензоил-3'-N-(т-бутилоксикарбонил)-10-O-(фосфонооксиметил)паклитаксел
(a) Получение
10-дезацетил-10-O-бензилоксикарбонил-7-O-триэтилсилилбаккатина III

В сухую колбу (в атмосфере аргона), содержащую 7-O-триэтилсилил-10-дезацетилбаккатин III (2,093 г, 3,177 мМ), добавляли безводный тетрагидрофуран (30 мл), и раствор охлаждали до -70oC. К этому раствору по капле добавляли 2,38 мМ (3,81 мМ) 1,6 М n-бутиллития. Через 15 минут к этому раствору по капле добавляли бензилхлороформат (0,91 мл, 6,35 мМ). Полученную смесь перемешивали 3 часа при постепенном нагревании до комнатной температуры. Реакцию гасили путем добавления 25 мл насыщенного NH4Cl, промывали солевым раствором, и осушали в вакууме сульфатом магния. После флеш-хроматографии (силикагель, 30 - 45%: этилацетат/гексан) получали 2,24 г (89%) целевого соединения в виде белой пены.
1H-ЯМР (300 МГц, CDCl3): δ 8,10 (д, J = 8,0 Гц, 2H); 7,63 - 7,58 (м, 1H); 7,47 (т, J = 8,0 Гц, 2H); 7,41 - 7,26 (м, 5H); 6,29 (с, 1H); 5,61 (д, J = 7,0 Гц, 1H); 5,20 (кв., J = 12,2 Гц, 2H); 4,96 (д, J = 9,0 Гц, 1H); 4,87 - 4,84 (м, 1H); 4,48 (дд, J = 6,7, 10,4 Гц, 1H); 4,30 (д, J = 8,5 Гц, 1H); 4,14 (д, J = 8,5 Гц, 1H); 3,84 (д, J = 7,0 Гц, 1H); 2,58 - 2,48 (м, 1H); 2,29 (м, 4H); 2,20 (с, 3H); 2,03 (д, J = 5,0 Гц, 1H); 1,92 - 1,83 (м, 1H); 1,68 (с, 3H); 1,17 (с, 3H); 1,04 (с, 3H); 0,91 (т, J = 7,5 Гц, 9H); 0,57 (кв., J = 7,4 Гц, 6H).
(b) Получение
10-дезацетил-10-O-бензилоксикарбонил-3'-N-дебензоил-3'-N-(т-бутилоксикарбонил)-2',
7-бис-O- триэтилсилилпаклитаксела

В сухую колбу, содержащую продукт стадии (a) (3,50 г, 4,42 мМ), добавляли небольшое количество толуола, и раствор концентрировали в вакууме. Затем эту колбу помещали в атмосферу аргона, и добавляли 100 мл сухого ТГФ. Колбу охлаждали до -70oC и по капле добавляли 1,0 М гексаметилдизилазида лития (6,19 мл, 6,19 мМ). После перемешивания в течение 20 минут, к раствору по капле добавляли раствор (3R, 4S)-1-(т-бутилоксикарбонил)-4-фенил-3-триэтилсилилокси-2-азетидинона (2,58 г, 7,07 мМ) в 10 мл сухого ТГФ. Реакционную смесь перемешивали в течение 3,5 часа, постепенно нагревая до комнатной температуры. После этого реакцию гасили путем добавления 70 мл насыщенного NH4Cl, промывали солевым раствором, осушали сульфатом магния и концентрировали. После флеш-хроматографии (силикагель, 5 - 15% этилацетат/гексан), получали 5,12 г (99%) целевого соединения в виде белой пены.
1H-ЯМР (300 МГц, CDCl3): δ 8,11 (д, J = 8,0 Гц, 2H); 7,60 - 7,58 (м, 1H); 7,48 (т, J = 8,0 Гц, 2H); 7,24 - 7,26 (м, 10H); 6,32 - 6,26 (м, 2H); 5,69 (д, J = 7,0 Гц, 1H); 5,47 (шир. д, J = 9,7 Гц, 1H); 5,31 - 5,10 (м, 3H); 4,94 (д, J = 8,5 Гц, 1H); 4,56 (с, 1H); 4,46 (дд, J = 6,9 J = 10,6 Гц, 1H); 4,31 (д, J = 8,3 Гц, 1H); 4,17 (д, J = 8,3 Гц, 1H); 3,81 (д, J = 7,0 Гц, 1H); 2,53 (с, 3H); 2,48 - 2,33 (м, 1H); 2,22 - 2,17 (м, 1H); 2,09 (с, 3H); 1,95 - 1,86 (м, 1H); 1,70 (с, 3H); 1,95 - 1,86 (м, 1H); 1,65 (с, 1H); 1,52 (с, 1H); 1,30 (с, 9H); 1,26 - 1,19 (м, 6H); 0,94 - 0,87 (м, 9H); 0,80 - 0,75 (м, 9H); 0,61 - 0,53 (м, 6H); 0,48 - 0,30 (м, 6H).
(c) Получение 10-дезацетил-3'-N-дебензоил-3'-N-(т-бутилоксикарбонил)-7-O- триэтилсилилпаклитаксела

Продукт стадии (b) (5,12 г, 4,40 мМ) растворяли в 100 миллилитрах этилацетата, переносили в сосуд Парра и помещали в атмосферу аргона. Затем добавляли 2,4 г 10% палладированного угля, и реакционную смесь помещали в аппарат для гидрогенизации Парра (55 фунт/кв.дюйм - 3,867 кг/см2) в течение 8 часов. Реакционную смесь фильтровали через слой целита и концентрировали. После флеш-хроматографии (силикагель; 15 - 20% этилацетат/гексан), получали 3,24 г (79%) целевого соединения в виде белой пены. В результате гидролиза 2'-триэтилсилиловой группы продукта стадии (b) в аппарате Парра, получали кислотные остатки в следовых количествах.
1H-ЯМР (300 МГц, CDCl3): δ 8,10 (д, J = 8,0 2H); 7,63-7,58 (м, 1H); 7,49 (д, J = 8,0, 2H); 7,39 - 7,26 (м, 5H); 6,27 - 6,17 (м, 1H); 5,64 (д, J = 7,2); 5,42 (д, J = 9,4, 1H); 5,28 - 5,25 (м, 1H); 5,12 (с, 1H); 4,92 (д, J = 8,6, 1H); 4,62 (шир. с, 1H); 4,38 - 4,28 (м, 3H); 4,17 (д, J = 8,5, 1H); 3,85 (д, J = 6,7, 1H); 3,36 (д, J = 5,3, 1H); 2,49 - 2,40 (м, 1H); 2,36 (с, 3H); 2,25 (шир. дублет. J = 8,7, 2H); 1,99 - 1,91 (м, 1H); 1,85 (с, 3H); 1,74 (с, 3H); 1,69 (с, 1H); 1,67 (с, 1H); 1,35 (с, 9H); 1,22 (с, 3H); 1,11 (с, 3H); 0,93 (т, J = 7,5 Гц, 9H), 0,61 - 0,49 (м, 6H).
(d) Получение
10-дезацетил-2'-O-бензилоксикарбонил-3'-N-дебензоил-3'-N-(т-бутилоксикарбонил)-7-O-триэтилсилилпаклитаксела

В колбу, содержащую продукт стадии (с) (3,24 г, 3,51 мМ) добавляли 30 мл сухого дихлорметана. После этого колбу помещали в атмосферу аргона и охлаждали до 0oC. К реакционной смеси добавляли N,N-диизопропилэтиламин (1,22 мл, 7,02 мМ), а затем по капле добавляли бензилхлороформат (1,00 мл, 7,02 мМ). Через 15 минут охлаждающую баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 7 часов. Полученную смесь делали нереакционной путем добавления 30 мл насыщенного NH4Cl, промывали солевым раствором и осушали сульфатом магния. После флеш-хроматографии (силикагель, 7-20% этилацетат/гексан) получали 3,24 г (89%) целевого соединения в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 8,10 (д, J = 8,0, 2H); 7,62-7,57 (м, 1H); 7,48 (т, J = 8,0, 2H); 7,40 - 7,26 (м, 10H); 6,33 - 6,27 (м, 1H); 5,66 (д, J = 7,0, 1H); 5,49 - 5,42 (м, 5H); 5,31 (с, 1H); 5,22 - 5,13 (м, 3H); 4, 93 (д, J = 9,4 Гц, 1H); 4,38 (дд, J = 6,5, J = 10,7, 1H); 4,34 - 4,28 (м, 2H); 4,18 (д, J = 8,3, 1H); 3,90 (д, J = 6,7, 1H); 2,52 - 2,30 (м, 4H); 2,24 - 2,20 (м, 1H); 1,97 - 1,87 (м, 3H); 1,74 (с, 3H); 1, 59 (с, 3H); 1,32 (с, 9H); 1,26 (с, 3H); 1,11 (с, 3H); 0,96 - 0,88 (м, 9H); 0,61 - 0,48 (м, 6H).
(e) Получение
10-дезацетил-2'-O-бензилоксикарбонил-3'-N-дебензоил-3'-N-(т-бутилоксикарбонил) 10-O-(дебензоил-фосфонооксиметил)-7-O-триэтилсилилпаклитаксела

Продукт стадии (d) растворяли в 13,5 мл (54%) ДМСО, 8,75 мл (35%) уксусного ангидрида и 2,75 мл (11%) ледяной уксусной кислоты, а затем помещали в атмосферу аргона. Реакционную смесь перемешивали 56 часов, после чего ее разбавляли этилацетатом до объема 60 мл. Раствор промывали насыщенным гидрокарбонатом натрия до тех пор, пока он не становился нейтральным, на что указывала pH-фильтровальная бумага, а затем промывали солевым раствором. Органическую фракцию осушали сульфатом магния и концентрировали. После флеш-хроматографии (элюент: 15-20% EtOAc /гексан) получали 3,12 г неочищенного белого пенистого вещества, содержащего нужный тиометилацеталевый продукт (т. е. , 10-дезацетил-2'-N-O-бензилоксикарбонил-3'-N-дебензоил-3'-N- (т-бутилоксикарбонил)-10-O-(метилтиометил)-7-O- триэтилсилилпаклитаксел, из расчета на 70% материала (ЯМР-анализ).
Вышеописанную неочищенную смесь (3,12) растворяли в 1,2-дихлорэтане (61 мл) и помещали в атмосферу аргона. После этого добавляли порошкообразные молекулярные сита 4А (3,12 г) и полученную гетерогенную смесь энергично перемешивали. К этой перемешанной смеси добавляли раствор перекристаллизованного N-йодсукцинимида (0,830 г, 3,69 мМ), а затем посредством каннюлирования добавляли дибензилфосфат (1,027 г, 3,69 мМ) в безводном ТГФ (46 мл). Полученную смесь перемешивали 5 часов, фильтровали через слой целита, и разбавляли этилацетатом до объема 250 мл. После промывки холодным 2% NaHSO3 (2х125 мл), холодным 6%-ным NaHCO3 (2х125 мл) и солевым раствором, органическую фазу осушали сульфатом магния, и концентрировали. После флеш-хроматографии (силикагель; элюент: 25-35% этилацетат/гексан), получали 1,52 г (40%) целевого соединения в виде белого твердого вещества.
1H-ЯМР (CDCl3), 300 МГц): δ 8,08 (д, J = 7,0, 2H); 7,59 - 7, 55 (м, 1H); 7,46 (т, J = 7,2, 2H); 7,38 - 7,25 (м, 20H); 6,30 (т, J = 8,5, 1H); 5,65 (д, J = 6,8, 1H); 5,49 - 5,39 (м, 4H); 5,32 (с, 1H); 5,18 - 4,19 (м, 4H); 4,93 (д, J = 9,2, 1H), 4, 44 (дд, J = 6,6, J = 10,2, 1H); 4,31 (д, J = 8,4, 1H); 4,16 (д, J = 8,5, 1H); 3,80 (д, J = 6,9, 1H); 2,69 - 2,39 (м, 4H); 2,33 - 2,23 (м, 3H); 2,03 (с, 3H); 1,90 (т, J = 12,6, 1H); 1,68 - 1,63 (м, 6H); 1,28 (с, 9Н); 1, 16 - 1,10 (м, 6H); 0,93 (т, J = 7,4, 9H), 0,55 (кв., J = 7,8, 6H).
13С-ЯМР (CDCl3, 75,5 МГц): δ 204,1, 169,7, 157,9, 167,1, 151,1, 140,7, 135,7, 133,6, 130, 2, 129,2, 128,9, 128,8, 128,7, 128,6, 128,5, 128,4, 128,3, 128,2, 128,0, 127,8, 126,4, 90,4, 84,2, 81,1, 80,4, 79,3, 78,8, 74,9, 72,8, 72,0, 70,5, 69,2, 69,1, 69,0, 58,1, 46, 8, 43,2, 37,1, 35,0, 28,1, 26,5, 22,8, 21,0, 14,1, 10,0, 6,9, 5,5.
MC (FAB) m/z + : 1345
(f) Получение
10-дезацетил-2-O-бензилоксикарбонил-3'-N-дебензоил-3'-N-(т-бутилоксикарбонил)-10-O-(дибензил-фосфонооксиметил)паклитаксела

В атмосфере аргона, раствор продукта стадии (e) (50,8 мг, 0,038 мМ) в безводном ТГФ (2,5 мл) охлаждали до -40oC. К этому раствору по капле добавляли фторид тетрабутиламмония (0,057 мл, 0,057 мМ) в ТГФ (1,0 М). Реакционную смесь перемешивали в течение 1,5 часа, постепенно нагревали до -20oC. Затем реакционную смесь гасили 15 миллилитрами насыщенного NH4Cl и разбавляли 30 миллилитрами EtOAc. Органическую фазу промывали гидрокарбонатом натрия (2х15 мл) и солевым раствором. После этого смесь осушали сульфатом магния и концентрировали. После препаративной хроматографии (силикагель; 50% этилацетат/гексан), получали 36 мг (77%) целевого соединения в виде белого порошка.
1H-ЯМР (CDCl3, 300 МГц): δ 8,10 (д, J = 8,5, 2H); 7,60 - 7,55 (м, 1H); 7,49 - 7,44 (м, 2H); 7,36 - 7,18 (м, 20H); 6,27 - 6,22 (м, 1H); 5,78 (с, 1H); 5,67 (д, J = 7,0, 1H); 5,44 - 5,34 (м, 3H); 5,27 (д, J = 2,2, 1H); 5,24 - 5,05 (м, 4H); 5,01 - 4,91 (м, 4H); 4,39 - 4,28 (м, 2H); 4,17 (д, J = 8,2, 1H); 3,87 (д, J = 7,0, 1H); 2,58 - 2,51 (м, 1H); 2,41 (с, 3H); 2,40 - 2,18 (м, 2H); 2,00 - 1,87 (м, 5H); 1,73 - 1,69 (м, 4H); 1,30 (с, 9H); 1,22 - 1,15 (м, 6H).
MC (FAB) m/z: 1231.
(g) Получение
10-дезацетил-3'-N-дезбензоил-3'-N-(т-бутилоксикарбонил)-10-O- (фосфонооксиметил)паклитаксела триэтаноламиновой соли

500-Миллилитровую колбу Парра загружали 10-дезацетил-2-O-бензилоксикарбонил-3'-N-дебензоил-3'-N- (т-бутилоксикарбонил)-10-O-(дибензилфосфонооксиметил)паклитакселом (264,9 мг, 0,215 мМ) и этилацетатом (20 мл). Колбу продували аргоном и добавляли 10% палладированный уголь (318 мг). Полученную смесь помещали в аппарат Парра под давлением водорода 55 фунт/кв. дюйм (3,586 кг/см2). За ходом реакции прослеживали с помощью ЖХВД (70: 30, CH3CN/Q8-буфер; pH 6,0, 1,00 мл/мин, C-18 колонка Zorbax, 25,0 см, λ = 230 нм) до исчезновения исходного материала (12,5 часа). Полученную смесь фильтровали через целит и промывали этилацетатом и небольшим количеством дихлорметана. Полученный фильтрат концентрировали, и остаток растворяли в дихлорметане (5 мл). В результате добавления гексана, образовывался белый осадок, в виде свободной кислоты (80% чистоты по ЖХВД), из которой выделяли 140,3 мг белого твердого вещества. Этот материал непосредственно использовали в следующей стадии.
В колбу, содержащую вышеописанную свободную кислоту (140 мг, 0,153 мМ) добавляли дихлорметан (10 мл). Полученный раствор обрабатывали 0,100 молями раствора триэтаноламина в этилацетате (1,16 мл, 0,116 мМ), в результате чего этот раствор приобретал мутный цвет. Затем добавляли приблизительно 2 мл гексана и смесь оставляли в течение ночи при -20oC. Полученный осадок фильтровали через воронку из спеченного стекла (4,0-5,5 мкм). Образовавшийся твердый продукт удаляли и помещали в вакуум на 4 часа. Таким образом, получали 69,9 мг (42%) целевой триэтаноламиновой соли в виде серого порошка (ЖХВД-чистота: 95 - 96%; TR = 2,05 мин, элюент: CH3CN/ Q8-буфер; pH 6,0, 1,00 мл/мин, C-18-колонки Zorbax - 25,0 см, λ = 230 нм).
1H-ЯМР (d6-ацетон /D2O, 300 МГц): δ 8,03 (д, J = 7,4, 2H); 7,65 (т, J = 7,3, 1H); 7,54 (т, J = 7,6, 2H); 7,42 - 7,33 (м, 5H); 7,21 (т, J = 7,0, 1H); 6,09 (т, J = 9,0, 1H); 5,88 (с, 1H); 5,59 (д, J = 7,0, 1H); 5,12 (шир. с, 2H); 4,93 (д, J = 8,4, 2H); 4,56 (д, J = 4,9, 1H); 4,31 - 4,26 (м, 1H); 4,11 (с, 2H); 3,41- 3,37 (м, 6H); 2,42 - 2,32 (м, 5H); 2,15 (шир. с, 1H); 1,97 (с, 3H); 1, 77 - 1,64 (м, 2H); 1,58 (с, 3H); 1,13 (с, 9H); 1,15 - 1,07 (м, 6H).
13С-ЯМР (d6-ацетон, D2O, 75,6 МГц): δ 171,6, 166,9, 156,6, 141,8, 135,1, 134, 2, 131,0, 130,7, 129,4, 129,3, 128,4, 128,1, 88,3, 85,4, 81,9, 79,7, 78,6, 78,1, 76,8, 76,0, 74,8, 71,9, 71,2, 47,4, 44,0, 37,1, 36,3, 28,5, 27,0, 23,1, 22,0, 14,7, 10,4.
BPMC: MNA+ для C44H56NO18PNa: вычислено: 9403142; найдено: 9403133.
Пример 10. 2'-O-Фосфонооксиметоксиметилпаклитаксел
(a) Получение
2'-O-(метилтиометоксиметил)-7-O-триэтил-силилпаклитаксела

К раствору 7-O-триэтилсилилпаклитаксела (70,0 мг, 72,2 мМ), бис(метилтиометил)эфира (90 мг, 72,2 мМ), молекулярных сит (70 мг), и N-йодосукцинимида (160 мг, 72,2 мМ) в ТГФ (2,0 мл), при комнатной температуре, добавляли трифлат серебра (5,0 мг, 19,5 мМ), и полученный раствор перемешивали в течение 2 часов. Затем реакционную смесь разбавляли этилацетатом и фильтровали через слой целита. Фильтрат промывали насыщенным водным раствором бикарбоната натрия, смесью (1:1, по объему) насыщенного водного бикарбоната натрия и 5% водного раствора тиосульфата натрия, и наконец, солевым раствором. Органические слои осушали сульфатом натрия и концентрировали в вакууме. Остаточный маслообразный продукт с помощью флеш-хроматографии (гексан/этилацетат, 3:1), и получали 22,0 г (29%) целевого соединения в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 8,12 - 7,20 (15H, м); 7,04 (1H, д, J = 8,9 Гц); 6,41 (1H, с); 6,25 (1H, м); 5,81 (1H, дд, J = 8,9, 2,
4 Гц); 5,
68 (1H, д, J = 7,0 Гц); 4,93 (1H, д, J = 7,0 Гц); 4,79 (2H, м); 4,71 (1H, д, J = 2,4 Гц); 4,45 (1H, дд, J = 10,5, 6,6 Гц); 4,30 (1H, д, J = 8,3 Гц); 4,28 (1H, д, J = 11,7 Гц); 4,17 (1H, д, J
= 8,3 Гц);
4,04 (1H, д, J = 11,7 Гц); 3,80 (1H, д, J = 6,9 Гц); 2,48 - 1,13 (25H, м, включая синглеты при 2,51, 2,13, 2,05, 2,01, 1,69, 1,19, 1,16); 0,98 - 0,85 (9H, м); 0,65 - 0,50 (6H, м);
(b)
Получение 2'-O-(дибензилфосфонооксиметоксиметил)-7- триэтилсилилпаклитаксела

К раствору продукта, полученного в стадии (a) (15 мг, 0,0141 мМ) и молекулярный сит (15 мг) в ТГФ (0,5 мл), при комнатной температуре, добавляя дибензилфосфат (20,0 мг, 0,089 мМ), а затем N-йодосукцинимид (4,2 мг, 0,0187 мМ), и раствор перемешивали 1 час. В этот период времени ТСХ-анализ реакционной смеси указывал на присутствие только исходного материала. Затем к раствору тремя порциями добавляли трифлат серебра (5,0 мг, 0,019 мМ) (в течение 2 часов), и реакционную смесь перемешивали еще 1 час. Затем реакционную смесь разбавляли этилацетатом, и полученный раствор фильтровали через слой целита. После этого фильтрат обрабатывали смесью (1:1, по объему) раствора насыщенного водного бикарбоната натрия и 5% водного раствора тиосульфата натрия. Органический экстракт промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Остаточный маслообразный продукт очищали с помощью флеш-хроматографии (гексан/этилацетат, 1:1), и получали 5,0 мг (33%) целевого соединения.
1H-ЯМР (300 МГц, CDCl3): δ 8,08 - 7,16 (25H, м); 7,18 (1H, д, J = 8,8 Гц); 6.41 (1H, с); 6,21 (1H, м); 5,82 (1H, дд, J = 9,0, 3,1 Гц); 5,66 (1H, д, J = 7,0 Гц); 5,01 - 4,65 (10H, м); 4,56 (1H, дд, J = 14,7, 5,6 Гц); 4,43 (1H, дд, J = 10,4, 6,7 Гц); 4,29 (1H, д, J = 8,3 Гц); 4,16 (1H, д, J = 8,3 Гц); 3,78 (1H, д, J = 8,3 Гц); 2,60 - 1,13 (22H, м, включая синглеты при 2,49, 2,15, 1,93, 1,66, 1,15, 1,13, и 3H-каждый), 0,95 - 0,84 (9H, м); 0,63 - 0,45 (6H, м).
(с) Получение 2'-O-фосфонооксиметоксиметилпаклитаксела

Продукт стадии (b) обрабатывали фторидом тетрабутиламмония в соответствии с процедурой, описанной в примере 9(-), для удаления 7-O-триэтилсилиловой защитной группы. В соответствии с процедурой, описанной в предыдущих примерах, полученное таким образом соединение подвергали каталитической гидрогенизации, и получали целевое соединение.
Пример 11. 2-O-Фосфонооксиметоксиметилпаклитаксел (Альтернативная процедура)
(a) Получение 2-O-Триэтилсилилпаклитаксела
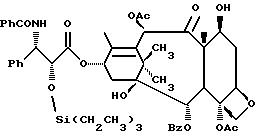
К раствору паклитаксела (20,0 г, 0,0234 М) и имидазола (3,59 г, 0,052 М) в 150 мл ДМФ, при 0oC, добавляли триэтилсилилхлорид (6,0 мл, 0,053 М) 2,0-миллилитровыми порциями в течение 20 минут. Реакционную смесь перемешивали при 0oC в течение часа. Затем эту смесь разбавляли этилацетатом и насыщенным водным хлоридом аммония. Органический слой удаляли, промывали солевым раствором, осушали сульфатом магния и концентрировали в вакууме с получением желтого маслообразного вещества. Этот неочищенный продукт очищали с помощью флеш-хроматографии (гексан/этилацетат, 1:3, а затем гексан/этилацетат, 1:1), и получали 21,07 г (выход 98%) целевого соединения в виде бесцветного твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 8,15 (2H, м); 7,70 (2H, м); 7,65 - 7,30 (11H, м); 7,15 (1H, д, J = 8,9 Гц); 6,30 (1H, с); 6,25 (1H, м); 6,70 - 6,10 (2H, м); 4,94 (1H, д, J = 7,9 Гц); 4,67 (1H, д, J = 2,0 Гц); 4,40 (1H, м); 4,29 (1H, д, J = 8,4 Гц); 4,18 (1H, д , J = 8,4 Гц); 3,81 (1H, д, J = 7,1 Гц); 2,65 - 1,10 (22H, включая синглеты при 2,55, 2,20, 1,88, 1,69, 1,22, 1,13, 3H - каждый).
(b) Получение 2'-O-триэтилсилил-7-O-бензилоксикарбонилпаклитаксела

К раствору 2-O-триэтилсилилпаклитаксела (22,3 г, 24,1 мМ) в ТГФ (250 мл), охлажденном до -50oC, по капле, в течение 10 минут, добавляли бутиллитий (1, 6 М в гексане, 12,9 ил, 8,06 мМ). Полученный раствор перемешивали 20 минут, а температуру поддерживали в пределах от -50oC до -35oC. Затем реакционную смесь охлаждали до -50oC, и по капле в течение 5 минут добавляли бензилхлороформат (5,08 г, 29,8 мМ). После этого реакционную смесь поддерживали при -40oC в течение 30 минут, а затем в течение 30 минут энергично перемешивали до температуры 0oC. Полученную смесь разбавляли этилацетатом и насыщенным водным хлоридом аммония. Полученный органический слой промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме.1H-ЯМР - анализ неочищенной реакционной смеси указал на присутствие нужного 2'-О-триэтилсилил-7-О-бензилоксикарбонилпаклитаксела, а также 2'-О-триэтилсилил-7-эпигидроксипаклитаксела (соответственно, отношение равно 3:1). Этот продукт (смесь) использовали в последующей стадии без дополнительной очистки, а затем изомеры отделяли. Аналитический образец первого продукта-2'-0-триэтилсилил-7-0-бензилоксикарбонилпаклитаксела очищали с помощью флеш-хроматографии.
1H-ЯМР (300 МГц, CDCl3): δ 8,12 (2H, м); 7,72 (1H, м); 7,65 - 7,27 (1H, д, J = 8,8 Гц); 6,41 (1H, м); 6,20 (1H, м); 5,72 - 5,65 (2H, м); 5,52 (1H, м); 5,24 (1H, д, J = 12, 3 Гц); 5,16 (1H, д, J = 12,3 Гц); 4,95 (1H, д, J = 8,7 Гц); 4,69 (1H, с); 4,35 (1H, д, J = 8,3 Гц); 4,25 (1H, д, J = 8,3 Гц); 3,94 (1H, д, J = 6,8 Гц); 2,70 - 1,12 (22H, включая синглеты при 2,54, 2, 14, 2,01, 1,80, 1,20, 1,15, 3H - каждый); 0,81 - 0,73 (9H, м); 0,55 - 0,31 (6H, м).
(c) Получение 7-О-бензилоксикарбонилпаклитаксела

К раствору продукта стадии (b) (24,0 г, 22,6 мМ) в ацетонитриле (250 мл), охлажденном до 0oC, добавляли соляную кислоту (6н 1,0 мл, 6,0 мМ). Через 10 минут, ТСХ-анализ (гексан/этилацетат, 1:1) указывал на завершение реакции. Полученную реакционную смесь разбавляли насыщенным водным бикарбонатом натрия, а затем этилацетатом, и органический слой удаляли, промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Остаточное маслообразное вещество очищали с помощью флеш-хроматографии (гексан/этилацетат, 1:3, а затем гексан/этилацетат, 1:1), и получали 11,4 г (выход по отношению к двум стадиям составлял 48%) целевого соединения и 4,8 г (выход - 20%) 7-эпигидроксипаклитаксела.
1H-ЯМР (300 МГц, CDCl3):
δ 8,09 (2H, м); 7,71 (2H, м); 7,65 - 7,27 (16H, м) 7,10 (1H, д, J = 8,9 Гц); 6,39 (1H, с); 6,16 (1H, м); 5,81 (1H, д, J = 8,9, 2,4 Гц);); 5,65 (1H, д,
J = 6,9 Гц); 5,49 (1H, дд, J = 10,6, 7,2
Гц); 5,20 (1H, д, J = 11,9 Гц); 5,12 (1H, д, J = 11,9 Гц); 4,91 (1H, д, J = 8,4 Гц); 5,78 (1H, м); 4,30 (1H, д, J = 8,4 Гц); 4,15 (1H, д, J = 8,4 Гц); 3,91
(1H, д, J = 6,8 Гц); 3,69 (1H, д, J = 4,9
Гц); 2,65 - 1,10 (22H, включая синглеты при 2,39, 2,18, 1,81, 1,75, 1,21, 1,15, 3H - каждый);
(d) Получение
2'-О-(метилтиометилметокси)-7-О-бензилоксикарбонилпаклитаксела

К раствору 7-0-бензилоксикарбонилпаклитаксела (5,53 г, 5,71 мМ), 1', 1'-дитиометилдиметилового эфира (7,8 г, 57,1 мМ) N-йодосукцинимида (6,35 г, 28,3 мМ) и осушенных в печи, порошкообразных молекулярных сит (5, 0 г) в ТГФ (110 мл), при комнатной температуре, добавляли трифлат серебра (300 мг, 1,17 мМ). ТСХ-анализ (гексан: этилацетат, 1:1) реакционной смеси через 20 минут указывал на превращение приблизительно 40% исходного материала в более высокую фракцию продукта. Затем добавляли трифлат серебра (150 мг, 0,585 мМ) и ход реакции прослеживали с помощью ТСХ, которая через 30 минут свидетельствовала о приблизительно 65%-ном завершении реакции. После этого смесь разбавляли этилацетатом (100 мл), фильтровали через слой целита, и полученный фильтрат выливали в делительную воронку, содержащую 200 мл насыщенного водного раствора бикарбоната натрия и 50 мл 5% водного раствора тиосульфата натрия. Органический слой удаляли, промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Остаток в виде маслообразного продукта очищали с помощью флеш-хроматографии (элюент: градиент этилацетата/гексана, 4: 1 - 3:2), и получали 3,0 г (выход - 54%) целевого продукта в виде светло-желтого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 8,10 (2H, м); 7,74 (2H, м); 7,66 - 7,25 (18H, м) 7,05 (1H, д, J = 8,9 Гц); 6, 40 (1H, с); 6,26 (1H, м); 5,77 (1H, дд, J = 8,8, 2,5 Гц); ); 5,71 (1H, д, J = 6,9 Гц); 5,51 (1H, д, J = 11,9 Гц); 5,21 (1H, д, J = 11,9 Гц); 5,14 (1H, д, J = 11,9 Гц); 4,92 (1H, м); 4,79 (2H, м); 4, 68 (1H, д, J = 2,5 Гц); 4,31 (1H, дд, J = 11,8 Гц); 4,30 (1H, д, J = 11,8 Гц); 4,16 (1H, д, J = 8,5 Гц); 4,10 (1H, д, J = 11,8 Гц); 3,93 (1H, д, J = 6,9 Гц); 2,65 - 1,10 (25H, включая синглеты при каждом 2,50, 2,15, 2,05, 1,74, 1,72, 1,20, 1,15, 3H).
(e) Получение
2-О-(дибензилфосфонооксиметоксиметил)-7-О-бензилоксикарбонилпаклитаксела

К раствору 2'-О-(метилтиометоксиметил)-7-О-бензилоксикарбонилпаклитаксела (1,06 г, 1,07 мМ) и осушенных в печи, порошкообразных молекулярных сит (1,0 г) в ТГФ (20 мл), при комнатной температуре, добавляли дибензилфосфат (1,49 г, 5,30 мМ), а затем сразу добавляли N-йодосукцинимид (2,65 г, 1,18 мМ). ТСХ-анализ (элюент: гексан/этилацетат, 1:1) реакционной смеси через 2,5 часа свидетельствовал о том, что реакция была завершена приблизительно на 60%. После добавления N-йодосукцинимида (175 мг, 0,78 мМ), реакционную смесь перемешивали еще 30 минут, после чего ТСХ-анализ указывал на завершение реакции. Полученную реакционную смесь разбавляли этилацетатом (50 мл) и фильтровали через слой целита. После этого фильтрат выливали в делительную воронку, содержащую 100 мл насыщенного водного раствора бикарбоната натрия и 20 мл 5% водного раствора тиосульфата натрия. Органический слой удаляли, промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Остаток в виде маслообразного продукта очищали с помощью флеш-хроматографии (элюент: градиент смеси гексана/этилацетата, 3:1 - 1:1), в результате чего получали 750 мг (выход - 62%) целевого соединения в виде белого твердого вещества.
1H-ЯМР (360 МГц, CDCl3): δ 8,10 (2H, м); 7,79 (2H, м); 7,65 - 7,24 (26H, м); 7,10 (1H, м); 6,41 (1H, с); 6,20 (1H, м); 5,79 (1H, дд, J = 8,8, 3,6 Гц); 5,65 (1H, д, J = 7,0 Гц); 5,52 (1H, м); 5,20 (1H, д, J = 11,8 Гц); 5,11 (1H, д, J = 11,8 Гц); 5,04 - 4, 85 (6H, м); 4,75 - 4,60 (4H, м); 4,30 (1H, д, J = 8,4 Гц); 4,15 (1H, д, J = 8,4 Гц); 3,92 (1H, д, J = 8,4 Гц); 2,65 - 1,10 (22H, включая синглеты при 2,48, 2,19, 1,95, 1,80, 1,20, 1,10, 3H - каждом).
(f) Получение 2'-О-фосфонооксиметоксиметилпаклитаксела
триэтаноламиновой соли
К раствору 2'-О-(дибензилфосфонооксиметоксиметил)-7-О-бензилоксикарбонилпаклитаксела
(500 мг, 0,382 мМ) в этилацетате (40 мл), помещенном в сосуд Парра, добавляли 10%
палладированный угол. Сосуд прикрепляли к аппарату Парра, и реакционную смесь подвергали взаимодействию с водородом
при давлении 50 фунт/кв. дюйм (3,515 кг/см2), затем реакционную смесь
встряхивали в течение 6,5 часов, и после этого фильтровали через воронку из спеченного стекла. К полученному фильтрату
добавляли триэтаноламин (0,1 н. в этилацетате, 4,0 мл), и полученный раствор
концентрировали в вакууме. Неочищенный твердый продукт суспендировали в этилацетате (приблизительно 5,0 мл), а
растворитель декантировали. Эту процедуру повторяли три раза и полученную целевую
триэтаноламиновую соль (300 мг) подвергали ЖХВД-анализу, который свидетельствовал о чистоте соли 87%. Кроме того, это
соединение очищали с помощью хроматографии на C18-колонках (элюент:
вода/ацетонитрил, 3:1) и получали целевое соединение (120 мг, 34%) с чистотой 95% (ЖХВД).
1H-ЯМР (300 МГц, CD3COCD3, D2O): δ 9,05 (1H, д, J = 8,7 Гц); 8,15 - 7,12 (21H, м); 6,40 (1H, м); 6,05 (1H, м); 5,69 - 5,55 (2H, м); 5,01 - 4,85 (6H, м); 4,35 (1H, м); 4,14 (2H, м); 3,96 - 3,85 (6H, м); 3,25 (1H, д, J = 7,1 Гц); 3,30 - 3,15 (6H, м); 2,50 - 1,04 (22H, включая синглеты при каждом 2,49, 2,15, 2,05, 1,81, 1,60, 3H).
Пример 12.
3'-N-Дебензоил-3'-N-(изопропилоксикарбонил)-7-О-метилтиометилпаклитаксел

К раствору 7-0-метилтиометилбаккатина III (408 мг, 0,630 мМ) в 10 мл ТГФ, при -60oC, добавляли n-бутиллитий (0,30 мл, 2,5 М, 0,75 мМ) и перемешивали 10 минут. Затем к раствору по капле добавляли (3R, 4S)-3-триэтилсилилокси-4-фенил-N- изопропилоксикарбонилазетидин-2-он (320 мг, 0,88 мМ) в 6 мл ТГФ, а затем реакцию доводили до 0oC в течение 30 минут. Реакционный раствор гасили путем добавления насыщенного NH4Cl, экстрагировали этилацетатом, встряхивали с 1,0 миллилитрами (1,0 М, 1,0 мМ) Bu4NF, а затем промывали соленым раствором, осушали сульфатом магния, и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 1, 5:1); и получали 545 мг продукта, который кристаллизовывали из ацетона/гексана. В результате этой процедуры получали 476 мг целевого продукта в виде белого твердого вещества, выход - 84%.
ИК (KBr): 3460, 1720, 1266, 1244, 1230 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,07 (д, J=7,2 Гц, 2H); 7,59 (т, J=7,2 Гц, 1H); 7,47 (т, J=7, 5 Гц, 2H); 7,32 (м, 5H); 6,51 (с, 1H); 6,18 (т, J=8,7 Гц, 1H); 5,65 (д, J=6,6 Гц, 1H); 5,50 (д, J=9,3 Гц, 1H); 5,28 (д, J=8,4 Гц, 1H); 4,91 (д, J= 8,1 Гц, 1H); 4,77 (м, 1H); 4,64 (шир. с, 3H); 4,26 (м, 2H); 4,15 (д, J= 8,4 Гц, 1H); 3,83 (д, J=6,9 Гц, 1H); 3,44 (д, J=5,1 Гц, 1H); 2,78 (м, 1H); 2,34 (с, 3H); 2, 25 (д, J=9,0 Гц, 2H); 2,17 (с, 3H); 2,14 (с, 1H); 2,10 (с, 3H); 1,96 (с, 3H); 1,83 (м, 1H); 1,73 (с, 3H); 1,15 (м, 12H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 201,8, 170,4, 169,2, 167,0, 156,3, 140,1, 138,3, 133,7, 133,3, 130,2, 129,1, 128,8, 128,6, 128,1, 126,8, 83,8, 81,4, 78,7, 76,0, 75,5 74,5, 74,0, 73,6, 72,2, 68,9, 57,5, 56,4, 47,1, 43,2, 35,3, 32,9, 26,6, 22,6, 22,0, 21,9, 20,9, 15,1, 14,6, 10,9.
MC (FAB) (NOBA) [M+Na] для C46H57NSO15:
Вычислено: 918; Найдено: 918.
Элементный анализ для C46H57NO15: Вычислено: C 61,66; H 6,41; N 1,56; Найдено: C 61,63; H 6,36; N 1,68.
Пример 13.
3'-N-Дебензоил-3'-N-(т-бутилоксикарбонил)-7-O-метилтиометилпаклитаксел

К раствору 7-O-метилтиометилбаккатина III (425 мг, 0,66 мМ) в 10 мл ТГФ, при -60oC, добавляли n-бутиллитий (0,30 мл, 2,5 М, 0,75 мМ), и раствор перемешивали в течение 10 минут. Затем к раствору по капле добавляли (3R, 4S)-3-триэтилсилилокси-4-фенил-N-(n-бутилоксикарбонил)азетидин-2-он (350 мг, 0,93 мМ) в 6 мл ТГФ, и реакцию доводили до 0oC в течение 30 минут. Реакционный раствор гасили путем добавления насыщенного NH4Cl и экстрагировали этилацетатом, после чего встряхивали с 1,0 миллилитрами (1,0 М, 1,0 мМ) Bu4NF и промывали солевым раствором, осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 1,5:1) и получали 581 мг целевого продукта, который кристаллизовали из толуола/гексана, в результате чего получали 464 мг белого твердого вещества (77%).
ИК (KBr): 3444, 1722, 1372, 1242, 1108, 1066, 1026, 988 см-1 .
1H-ЯМР (CDCl3, 300 МГц): δ 8,08 (д, J=7,2 Гц, 2H); 7,59 (т, J=7,5 Гц, 1H), 7,47 (т, J=7,2 Гц, 2H); 7,39 - 7,11 (м, 5H); 6,51 (с, 1H); 6,20 (т, J= 8,7 Гц, 1H); 5,65 (д, J=6,9 Гц, 1H); 5,56 (д, J=9,3 Гц, 1H); 5,29 (д, J=8,4 Гц, 1H); 4,91 (д, J=8,1 Гц, 1H); 4,65 (шир. с, 3H); 4,27 (м, 2H); 4,15 (д, J=8,4 Гц, 1H); 3,97 (м, 2H); 3,84 (д, J=6,9 Гц, 1H); 3,45 (д, J=4,8 Гц, 1H); 2,78 (м, 1H); 2,33 (с, 6H); 2,25 (д, J=8,7, 2H); 2,17 (с, 3H); 2,10 (с, 3H); 1,96 (с, 3H); 1,83 (м, 1H); 1,74 (с, 3H); 1,62 (с, 1H); 1,48 (м, 2H); 1,19 (м, 5H); 0,83 (т, J=7,2 Гц, 3H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 201,9, 172,3, 170,5, 169,2, 167,0, 156,3, 140,1, 138,4, 133,8, 133,4, 130,2, 129,2, 129,0, 128,9, 128,7, 128,2, 126,8, 125, 3, 83,9, 81,4, 78,8, 77,3, 76,0, 75,6, 74,1, 73,7, 72,2, 65,4, 57,5, 56,5, 47,2, 43,2, 35,4, 26,6, 22,6, 21,5, 21,0, 18,9, 15,1, 14,7, 13,7, 10,9.
MC (FAB) (NOBA) [M+H] для C47H60NSO15:
Вычислено: 910; Найдено: 910.
Элементный анализ для C47H59NSO15:
Вычислено: C 62,03; H 6,53;
N 1,54;
Найдено: C 62,16; H 6,45;
N 1,57.
Пример 14. 3'-N-Дебензоил-3'-N-(т-бутоксикарбонил)-7-O- метилтиометилпаклитаксел
(a) Получение
3'-N-дебензоил-3'-N-(т-бутоксикарбонил)-2-O- триэтилсилил-7-O-метилтиометилпаклитаксела

К раствору HMDS (0,275 мл, 1,30 мМ) в 8 мл ТГФ добавляли раствор n-бутиллития (0,48 мл, 2,5 М в гексане, 1,20 мМ) и перемешивали 5 минут при -55oC. К этому раствору добавляли 7-O-метилтиометилбаккатин III (639 мг, 0,99 мМ) в 8 миллилитрах ТГФ и перемешивали в течение 10 минут, после чего добавляли 8 мл раствора (3R, 4S)-3-триэтилсилилокси-4-фенил-N-(т-бутоксикарбонил)азетидин-2-она (1,52 мМ). Холодную баню удаляли и заменяли на баню с температурой 0oC, а затем реакционную смесь перемешивали в течение 30 минут. Полученный раствор разбавляли этилацетатом, промывали насыщенным раствором NH4Cl, осушали сульфатом магния, и концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат, 3:1), и получали 1,0 г целевого продукта (выход - 98%).
1H-ЯМР (CDCl3, 300 МГц): δ 8,09 (д, J=6,9 Гц, 2H); 7,57 (м, 1H); 7,46 (т, J=7, 8 Гц, 2H); 7,35 (м, 2H); 7,26 (м, 3H); 6,55 (с, 1H); 6,25 (т, J=9,6 Гц, 1H); 5,68 (д, J=6,9 Гц, 1H); 5,45 (шир. д, J=9,3 Гц, 1H); 5,27 (шир. д, 1H); 4,95 (д, J=7,8, 1H); 4,65 (с, 2H); 4,53 (с, 1H); 4, 29 (м, 2H); 4,17 (д, J= 8,4 Гц); 3,89 (д, J=6,9 Гц, 1H); 2,81 (м, 1H); 2,51 (с, 3H); 2,37 (дд, J= 15,3, 9,6 Гц, 1H); 2,17 (с, 3H); 2,10 (с, 3H); 2,03 (с, 3H); 1,85 (м, 1H); 1,74 (с, 3H); 1,63 (д, J=14,1 Гц, 1H); 1,29 (с, 9H); 1,21 (c, 6H); 0,76 (т, J=7,8 Гц, 9H); 0,36 (м, 6H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 202,0, 171,6, 170,1, 169,3, 167,1, 155,2, 141,0, 139, 0, 133,6, 132,8, 130,2, 129,2, 128,7, 128,5, 127,7, 126,4, 83,9, 81,2, 79,9, 78,9, 76,0, 75,7, 75,2, 74,8, 74,2, 71,3, 57,3, 56,7, 47,0, 43,3, 35,3, 33,0, 28,2, 26,4, 23,0, 21,5, 21,0, 15, 0, 14,4, 10, 9, 6,5, 4,3.
ИК(пленка): 3448 (с.,), 1720, 1242, 1120, 1056 см-1.
МС(FAB) (NOBA) [M+H]: для C53H74NSSiO15:
Вычислено: 10244549. Найдено: 10244583.
(b) Получение 3'-N-дебензоил-3'-N-(т-бутоксикарбонил)-7-O-метилтиометилпаклитаксела

К раствору 3'-N-дебензоил-3'-N-(т-бутоксикарбонил)-2-O-триэтилсилил-7-O- метилтиометилпаклитаксела (269 мг, 0,26 мМ) в 6 мл ТГФ, добавляли фторид тетрабутиламмония (0,3 мл, 1,0 М в ТГФ, 0,3 мМ) и полученную смесь перемешивали 10 минут. Полученный раствор разбавляли этилацетатом, промывали солевым раствором, осушали сульфатом магния, и концентрировали. Остаток подвергали хроматографии на силикагеле (элюент: гексан/этилацетат, 1:1), и получали 240 мг целевого продукта (95%).
ИК (пленка): 3440, 1720, 1370, 1242, 1170, 1108, 1066, 756, см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,06 (д, J = 7,2 Гц, 2H); 7,57 (т, J = 7,2 Гц, 1H); 7,46 (т, J = 7,8 Гц, 2H); 7,35 (м, 5H); 6, 52 (c, 1H); 6,16 (т, J = 7,8 Гц, 1H); 5,64 (д, J = 6,9 Гц, 1H); 5,43 (шир, д, J = 9,3 Гц, 1H); 5,24 (шир, д, J = 8,1 Гц, 1H); 4,91 (д, J = 8,1 Гц, 1H); 4,63 (м, 3H); 4,26 (м, 2H), 4,14 (д, J = 8,4 Гц, 1H), 3,83 (д, J = 6,9 Гц, 1H); 3,46 (д, J = 5,4 Гц, 1H); 2,77 (м, 1H); 2,34 (с, 3H); 2,27 (д, J = 8,7 Гц, 2H); 2,16 (c, 3H); 2,09 (c, 3H); 1,97 (с, 3H); 1,79 (м, 2H); 1,72 (с, 3H); 1,32 (c, 9H); 1,19 (c, 3H); 1,18 (с, 3H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 202,0, 172,7, 170,3, 169,2, 167,0, 155,3, 140,3, 138,4, 133,7, 133,2, 130,2, 129,1, 128,8, 128,7, 128,0, 126,7, 83,9, 81,3, 60,2, 78,6, 76,5, 76,1, 75,4, 74,6, 74,0, 73,6, 72,3, 57,4, 56,1, 47,1, 43,2, 35,3, 32,8, 28,2, 26,5, 22,6, 21,0, 15,1, 14,6, 10,9.
FAB-MC (NOBA) [M + N]
для C47H60NO15S:
Вычислено: 9103684. Найдено:
9103706.
Пример 15.
3'-N-Дебензоил-3'-N-(т-бутоксикарбонил)-2-O-этилоксикарбонил-7-O-метилтиометилпаклитаксел
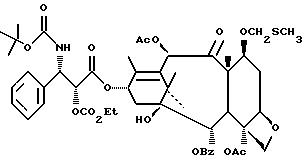
К раствору 3'-N-дебензоил-3'-N-(т-бутоксикарбонил)-7-O-метилтиометилпаклитаксела (428 мг, 0,47 мМ) в 10 мл дихлорметана, добавляли диизопропилэтиламин (0,85 мл, 4,8 мМ) и DMAP (20 мл), и полученный раствор охлаждали до 0oC. После этого добавляли этилхлороформат (0,25 мл, 2,6 мл) и раствор перемешивали в течение 1 часа. Раствор разбавляли этилацетатом, промывали бикарбонатом и солевым раствором, осушали сульфатом магния и концентрировали. Полученный таким образом остаток хроматографировали на силикагеле (гексан/этилацетат, 1:1), и получали 428 мг целевого этилкарбоната (92%).
ИК (пленка): 3448 (м), 1750, 1720, 1370, 1244, 1064 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,09 (д, J = 7,2 Гц, 2H); 7,59 (т, J = 7,2 Гц, 2H); 7,48 (т, J = 7,8 Гц, 2H); 7,39 (м, 2H); 7,31 (м, 3H); 6,55 (с, 1H); 6,25 (т, J = 9,0 Гц, 1H); 5,68 (д, J = 7,2 Гц, 1H); 5,40 (шир, м, 2H); 5,25 (c, 1H); 4,95 (д, J = 8,1 Гц, 1H); 4,65 (д, 2H); 4, 29 (м, 2H); 4,15 (м, 3H); 3,88 (д, J = 6,9 Гц, 1H); 2,81 (м, 1H); 2,43 (с, 3H); 2,32 (м, 1H); 2,21 (м, 1H); 2,16 (с, 3H); 2,11 (с, 3H); 2,08 (с, 3H); 1,84 (м, 1H); 1,74 (с, 3H); 1,62 (с, 1H); 1,32 (c, 3H); 1,28 (т, J = 7,2 Гц, 3H); 1,20 (с, 6H).
13C-ЯМР (CDCl3, 75,5 Гц): δ 202,0 169,7, 169,1, 168,1, 167,0, 155,1, 154,1, 141,0, 137,2, 133,6, 132,9, 130,2, 129,2, 128,9, 128,7, 128,2, 126,4, 83,9, 81,2, 80,4, 78,9, 76,5, 76,0, 75,8, 74,8, 74,2, 72,0, 65,1, 57,4, 47,1, 43,3, 35,1, 33,0, 28,1, 26,4, 22,7, 21,3, 20,9, 15,0, 14,5, 14,1, 10,9.
FAB-MC (NOBA) [M + N]: для C50H64NSO17:
Вычислено: 9823895. Найдено: 9823874.
Пример 16.
3'-N-Дебензоил-3'-N-(т-бутоксикарбонил)-7-O-метилтиометил-10-деацетил-10-гидроксиметилкарбонил (паклитаксел)
(a) Получение 7-O-Триэтилсилил-10-деацетил-10-бензилоксиметилкарбонилбаккатина
III

К раствору 7-O-триэтилсилил-10-дезоксиацетилбаккатина III (3,85 г, 5,85 мМ) в 40 мл ТГФ, при -60oC, добавляли n-бутиллитий (2,6 мл, 2,5 М в гексане, 6,5 мМ) и перемешивали в течение 5 минут, после чего добавляли бензилоксиацетилхлорид (1,0 мл, 6,5 мМ). После перемешивания в течение 30 минут при -60oC и последующего нагревания до комнатной температуры раствор разбавляли этилацетатом и промывали бикарбонатом. После этого раствор осушали сульфатом магния и концентрировали, а полученный остаток хроматографировали на силикагеле (элюент: гексан/этилацетат 2:1, и гексан/этилацетат, 1:1), в результате чего получали 4,36 г продукта (92%).
ИК (пленка): 3478 (шир.), 1724, 1270, 1244, 1136, 1110, 1070 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,08 (д, J = 7,2 Гц, 2H); 7,60 - 7,23 (м, 8H), 6,54 (с, 1H); 5,60 (д, J = 6,9 Гц, 1H); 4,94 (д, J = 7,8 Гц, 1H); 4,79 (шир, кв., 1H); 4,69 (с, 2H); 4,49 (дд, J = 10,5, 6,6 Гц, 1H); 4,26 (м, 2H); 4,12 (м, 1H); 3,85 (д, J = 6,9 Гц, 1H); 2,52 (м, 1H); 2, 26 (с, 3H); 2,23 (м, 2H); 2,18 (с, 3H); 2,10 (м, 1H); 1,86 (м, 1H); 1,66 (с, 3H); 1,14 (с, 3H); 0,99 (с, 3H); 0,91 (т, J = 7,5 Гц, 9H); 0,56 (м, 6H).
Элементный анализ для C44H58SiO12:
Вычислено: C 65,49; H 7,24
Найдено: C 65,33; H 7,27.
FAB-MC (NOBA) M + N: для C44H59SiO12
:
Вычислено: 807; Найдено: 807.
(b) 3'-N-Дебензоил-3'-N-(т-бутоксикарбонил)-10-дезоксиацетил-10-бензилоксиметилкарбонил (паклитаксел)

К раствору 7-O-триэтилсилил-10-дезоксиацетил-10-бензилоксиметилкарбонилбаккатина III (1,21 г, 1,66 мМ) в 50 мл ТГФ, при -60o C, добавляли n-бутиллитий (0,7 мл, 2,5 М в гексане, 1,75 мМ), и раствор перемешивали в течение 5 минут с последующим добавлением (3R, 4S)-3-триэтилсилилокси-4-фенил-N-(т-бутоксикарбонил)азетидин-2-она, (1,2 г, 3,2 мМ). После перемешивания в течение 5 минут при -60oC, а затем в течение 30 минут при 0oC, раствор разбавляли этилацетатом и промывали насыщенным NH4Cl. После этого раствор осушали сульфатом магния и концентрировали. Остаток подвергали хроматографии на силикагеле (гексан/этилацетат, 3:1, а затем гексан/этилацетат, 1:1), и получали 980 мг продукта (53%). Этот продукт растворяли в 6 мл ацетонитрила и охлаждали до 0oC, а затем перемешивали с 0,60 миллилитрами 6 н. соляной кислоты в течение 19 часов. После этого раствор разбавляли этилацетатом, промывали насыщенным бикарбонатом, осушали сульфатом магния и хроматографировали на силикагеле (гексан/этилацетат, 1:1), в результате чего получали 570 мг продукта (35%).
ИК (пленка): 3448 (шир. ), 1716, 1496, 1368, 1316, 1270, 1246, 1176, 1108, 1070, 1026 см-1.
1 H-ЯМР (CDCl3, 300 МГц): δ 8,08 (д, J = 7,5 Гц, 2H); 7,59 (т, J = 7,8 Гц, 1H); 7,47 (т, J = 7,8 Гц, 2H); 7,36 (м, 10H); 6,38 (с, 1H); 6,20 (т, J = 9,0 Гц, 1H); 5,65 (д, J = 6,9 Гц, 1H); 5,39 (шир. д, J = 9,3 Гц, 1H); 4,93 (д, J = 7,8 Гц, 1H); 4,69 (с, 2H); 4,60 (шир. с, 1H); 4,39 (м, 1H); 4,28 (м, 3H); 4,15 (д, J = 8,4 Гц, 1H); 3,78 (д, J = 6,9 Гц, 1H); 3,40 (шир. с, 1H); 2,54 (м, 1H); 2,43 (м, 1H); 2,36 (с, 1H); 2,28 (м, 2H); 1,84 (с, 4H); 1,72 (м, 1H); 1,67 (с, 3H); 1,31 (с, 9H); 1,23 (м, 1H); 1,21 (с, 3H); 1,10 (с, 3H).
Элементный анализ для C52H61NO16:
Вычислено: C 65,33; H 6,43; N 1,
46.
Найдено: C 64,97; H 6,44; N 1,43.
FAB-MC (NOBA) M+Na: для C52H61NO16Na: Вычислено: 978; Найдено: 978.
(c)
Получение
3'-N-дебензоил-3'-N-(т-бутоксикарбонил)-2-O- бензилоксикарбонил-7-O-метилтиометил-10-дезацетил-10- бензилоксиметилкарбонила (паклитаксел)

К раствору 3'-N-дебензоил-3'-N-(т-бутоксикарбонил)-10-дезацетил- 10-бензилоксиметилкарбонила (паклитаксела) (570 мг, 0,59 мМ) в 10 миллилитрах метиленхлорида, при 0oC, добавляли диизопропилэтиламин (0,15 мл, 0,86 мМ) и CbzCl (0,10 мл, 0,70 мМ). Полученный раствор перемешивали в течение часа, медленно нагревая до комнатной температуры. Раствор промывали бикарбонатом, осушали сульфатом магния и концентрировали. Полученный остаток в 10 миллилитрах ацетонитрила (при 0oC) перемешивали с бензоилпероксидом (780 мг, 3,22 мМ) и диметилсульфидом (0,50 мл, 6,8 мМ), медленно нагревая до комнатной температуры в течение 75 часов. После этого раствор разбавляли этилацетатом, промывали насыщенным бикарбонатом, осушали сульфатом магния, и хроматографировали на силикагеле (элюент: гексан/этилацетат, 2:1), в результате чего получали 412 мг целевого продукта (65%).
ИК (пленка): 3438, 1754, 1722, 1368, 1272, 1244, 1176, 1110, 1066, 1028 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,11 (д, J = 7,2 Гц, 2H); 7,61 (т, J = 7,2 Гц, 1H); 7,49 (т, J = 7,8 Гц, 2H); 7,35 (м, 15H); 6,67 (с, 1H); 6,26 (т, J = 8,7 Гц, 1H); 5,69 (д, J = 6,6 Гц, 1H); 5,41 (шир. м, 2H); 5,29 (с, 1H); 5,14 (AB кв., J = 12, 5,7 Гц, 2H); 4,98 (д, J = 8 Гц, 1H); 4,72 (м, 4H); 4,32 (м, 3H); 4,19 (м, 2H); 3,90 (д, J = 6,0 Гц, 1H); 2,85 (м, 1H); 2,45 (м, 1H); 2,44 (с, 3H); 2,34 (м, 1H); 2,24 (м, 1H); 2,15 (с, 3H); 2,12 (с, 3H); 1,87 (м, 1H); 1,77 (с, 3H); 1,33 (с, 9H); 1,19 (с, 6H).
13C-ЯМР (CDCl3, 75,5 МГц): δ 201,6, 169,7, 168,7, 168,0, 167,0, 155,1, 154,1, 141,6, 137,1, 134,4, 133,7, 132,5, 130,2, 129,2, 128,9, 128,8, 128,7, 128,5, 128,4, 128,2, 128,0, 128,0, 126,4, 83,9, 81,2, 80,4, 78,8, 77,2, 76,2, 75,8, 74,7, 74,3, 73,4, 72,0, 70,6, 67,1, 57,4, 54,1, 47,1, 43,2, 35,2, 32,9, 28,1, 26,4, 22,7, 21,3, 15,2, 14,6, 10, 9.
МС (FAB) (NOBA) (M+Na) для C62H71NO18SNa: Вычислено: 1172, Найдено: 1172.
(d) Получение
3'-N-дебензоил-3'-N-(т-бутоксикарбонил)-7-O- метилтиометил-10-деацетил-10-гидроксиметилкарбонила (паклитаксела)

К раствору 3'-N-дебензоил-3'-N-(т-бутоксикарбонил)-2-O- бензилоксиметилкарбонил-7-O-метилтиометил-10-деацетил-10- бензилоксикарбонила (паклитаксела) (377 мг, 0, 35 мМ) в 30 миллилитрах этанола, добавляли 450 мг 10% палладированного угля, и раствор перемешивали в атмосфере водорода в течение 120 часов. Катализатор удаляли путем фильтрации через целит, и раствор концентрировали. Остаток хроматографировали на силикагеле (элюент: 20% CH3CN /79% CH2Cl2/1% MeOH), и получали 190 мг целевого продукта (65%).
ИК (пленка): 3444 (шир.), 1724, 1368, 1246, 1174, 1096, 1070, 1026, 988 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,07 (д, J = 7,2 Гц, 2H); 7,59 (т, J = 7,2 Гц, 1H); 7,47 (т, J = 7,8 Гц, 2H); 7,35 (м, 5H); 6,65 (с, 1H); 6,17 (т, J = 8,7 Гц, 2H); 5,65 (д, J = 6,6 Гц, 1H); 5,39 (шир. д, J = 9,6 Гц, 1H); 5,26 (шир. д, 1H); 4,93 (д, J = 8,4 Гц, 1H); 4,67 (м, 3H); 4,28 (м, 5H); 3,83 (д, J = 6,0 Гц, 1H); 3,44 (д, J = 5,1 Гц, 1H); 2,77 (м, 1H); 2,50 (м, 1H); 2,36 (с, 3H); 2,29 (д, J = 8,4 Гц, 2H); 2,13 (шир. с, 3H); 2,01 (с, 3H); 1,82 (с, 3H); 1,74 (с, 3H); 1,33 (с, 9H); 1,18 (с, 3H); 1,16 (с, 3H).
13C-ЯМР (CDCl3, 75,5 МГц): δ 201,5, 171,5, 170,3, 167,0, 155,4, 141,3, 133,7, 132,7, 130,2, 129,0, 128,8, 128,7, 128,1, 126,8, 83,8, 81,3, 80,2, 78,6, 75,0, 74,4, 74,0, 73,6, 72,3, 60,6, 57,4, 56,2, 47,2, 43,2, 35,3, 32,6, 28,2, 26,5, 22,6, 21,0, 15,5, 14,7 10,8.
FAB-МС (NOBA) (M+Na) для
C47H59NO16SNa:
Вычислено: 948; Найдено: 948.
Пример 17.
3'-N-Дебензоил-3'-N-(т-бутоксикарбонил)-7-O-метилтиометил-3'-дезоксифенил-3'-изобутенилпаклитаксел

К раствору 7-O-метилтиометилбаккатина III (1,5 г, 2,3 мМ) в 30 мл ТГФ, при -60oC, добавляли n-бутиллитий (1,0 мл, 2,5 М в гексане, 2,5 мМ), и полученный раствор перемешивали в течение 10 минут. Затем к этому раствору по капле добавляли раствор (±)-цис-3-триэтилсилилокси-4-изобутенил-N-т-бутоксикарбонилазетидин-2-она (3,3 г, 9,3 мМ) в 10 мл ТГФ. После этого раствор перемешивали при 0oC в течение 30 минут и гасили насыщенным раствором NH4Cl, а затем экстрагировали этилацетатом. Полученный раствор осушали сульфатом магния, концентрировали, и остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 3:1). Полученный продукт растворяли в 100 мл ТГФ, встряхивали с 2,3 миллилитрами (1,0 М в ТГФ, 2,3 мМ) Bu4NF, разбавляли этилацетатом и промывали солевым раствором. Затем этот раствор осушали сульфатом магния и концентрировали, а остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 1,5:1), в результате чего получали 1,6 г целевого продукта (78%).
ИК (пленка): 3452 (шир.), 1724, 1370, 1242, 1096, 1066 см-1.
1H-ЯМР (CDCl3, 300 МГц): δ 8,07 (д, J = 7,2 Гц, 2H); 7,59 (т, J = 7,5 Гц, 1H); 7,45 (т, J = 7,8 Гц, 2H); 6,54 (с, 1H); 6,11 (т, J = 9,3 Гц, 1H); 5,66 (д, J = 6,0 Гц, 1H); 5,29 (д, J = 6,0 Гц, 1H); 4,94 (д, J = 8,1 Гц, 1H); 4,75 (м, 2H); 4,64 (AB кв, J = 12,0, 2,7 Гц, 2H); 4,29 (м, 2H); 4,20 (м, 2H); 3,86 (д, J = 6,0 Гц, 1H); 3,37 (шир.д, 1H); 2,79 (м, 1H); 2,35 (с, 6H); 2,16 (с, 3H); 2,10 (с, 3H); 2,04 (с, 3H); 1, 82 (м, 1H); 1,74 (с, 9H); 1,34 (с, 9H); 1,23 (с, 3H); 1,20 (с, 3H).
13C-ЯМР (CDCl3, 75,5 МГц): δ 202,0, 170,2, 169,2, 166,9, 155,4, 140,6, 138, 0, 133,7, 133,1, 130,1, 129,2, 128,6, 120,6, 83,8, 81,2, 79,9, 78,7, 77,2, 76,1, 75,5, 74,6, 74,0, 73,7, 72,2, 57,7, 57,4, 51,5, 47,1, 43,2, 35,4, 32,9, 28,2, 26,4, 25,8, 22,4, 21,0, 18,6, 15,1, 14,8, 10,9.
FAB-MC (NOBA) M + H для C45H62NSO15:
Вычислено: 888; Найдено: 888.
Пример 18.
7-O-Метилтиометил-3'-дезоксифенил-3'-изобутенилпаклитаксел
В соответствии с процедурой, описанной в примере 17, целевое соединение получалииз7-O-метилтиометилбаккатинаIIIи(±
)-цис-3-триэтилсилилокси-4-изобутенил-N-бензоилазетидина-2-она.
Пример 19. 3'-Дезоксифенил-3'-(2-фурил)-2'-O- этилоксикарбонил-7-O-метилтиометилпаклитаксел
В соответствии с
процедурами, описанными в примерах 7(a) и 7(b), целевое соединение может быть получено из (3R, 4R)-1-бензоил-4-(2-фурил)-3-триэтилсилилокси-2-азетидинона и 7-O-метилтиометилбаккатина III.
Пример 20. 2-O-n-Пропилкарбонил-7-O-фосфонооксиметилпаклитаксел
(a) Получение 2-O-n-пропилкарбонилпаклитаксела

К раствору паклитаксела (15,0 г, 17,5 мМ) и диизопропилэтиламина (18,3 мл, 105 мМ) в дихлорметане (175 мл), охлажденном до 0oC, по капле в течение 2 минут добавляли бутирилхлорид (5,49 мл, 52,4 мМ). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. После этого реакционную смесь распределяли между этилацетатом и насыщенным водным раствором хлорида аммония. Органическую фазу промывали насыщенным раствором бикарбоната натрия, а затем солевым раствором, после чего осушали сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (элюент: гексан/-этилацетат), и получали целевой сложный эфир (15,9 г, 98% - выход) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц): δ 8,13 - 8,05 (2H, м); 7,75 - 7,65 (2H, м); 7,62 - 7,30 (11H, м); 6,88 (1H, д, J = 9,0 Гц); 6,26 (1H, с); 6,23 (1H, дд, J = 8,4 Гц); 5, 92 (1H, дд, J = 9,3, 6,0 Гц); 5,65 (1H, д, J = 7,1 Гц); 5,48 (1H, дд, J = 10,4, 6,5 Гц); 4,94 (1H, д, J = 7,9 Гц); 4,21 (1H, дд, J = 10,4, 6,5 Гц); 4,28 (1H, д, J = 8,4 Гц); 4,17 (1H, д, J = 8,4 Гц); 3,78 (1H, д, J = 7,0 Гц); 2,64 - 1,02 (26H, м, включая синглеты при каждом 2,43, 2,19, 1,91, 1,65 1,65, 1,20, 1,10, 3H); 0,87 (3H, дд, J = 8,2 Гц).
(b) Получение
2'-O-n-пропилкарбонил-7-O- метилтиометилпаклитаксела

К раствору 2'-O-n-пропилкарбонилпаклитаксела (14,4 г, 15,6 мМ) и диметилсульфида (9,23 мл, 124,8 мМ) в ацетонитриле (312 мл), охлажденном до -40oC, добавляли бензоилпероксид (15,1 г, 62,3 мМ), и реакционную смесь нагревали до комнатной температуры в течение часа. В этот период времени ТСХ-анализ (элюент: гексан/этилацетат) указывал на завершение реакции. После этого реакционную смесь разбавляли этилацетатом, а полученный органический раствор промывали три раза насыщенным раствором бикарбоната натрия, а затем солевым раствором. Органическую фазу осушали сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), и получали целевое соединение (14,4 г, 93%) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц): δ 8,21 - 8,19 (2H, м); 7,72 - 7,70 (2H, м); 7,62 - 7,26 (11H, м); 6,92 (3H, с); 6,20 (1H, дд, J = 8,4 Гц); 5,92 (1H, дд, J = 9,0, 3,1 Гц); 5,66 (1H, д, J = 6,9 Гц); 5,51 (1H, д, J = 3,2 Гц); 4,92 (1H, д, J = 8,2 Гц); 4,68 - 4,59 (2H, м); 4,32 - 4,26 (2H, м); 4,15 (1H, д, J = 8,3 Гц); 3,86 (1H, д, J = 6,8 Гц); 2,77 (1H, м); 2,50 - 1, 05 (25H, м); 0,87 (3H, д, J = 7,3 Гц).
(c) Получение 2'-O-n-пропилкарбонил-7-O-(дибензилфосфонооксиметил)паклитаксела

К раствору 2'-O-n-пропилкарбонил-7-O-метилтиометилпаклитаксела (10,7 г, 11,0 мМ), дибензилфосфата (15,3 г, 55,0 мМ) и 5 г осушенных в печи сит Angstrom 3 в ТГФ (200 мл), при комнатной температуре, одной порцией добавляли N-йодосукцинимид (4,9 г, 21,8 мМ), и полученную смесь перемешивали в течение 1 часа. В этот период времени ТСХ-анализ (элюент: гексан/этилацетат) указывал на завершение реакции. Эту реакционную смесь разбавляли этилацетатом до объема, в два раза превышающего исходный объем, и фильтровали через целит. Затем фильтрат выливали в насыщенный раствор бикарбоната натрия, содержащий 1% тиосульфат натрия (по массе). Органический слой промывали четыре раза насыщенным водным раствором бикарбоната натрия, а затем солевым раствором. Водный слой снова экстрагировали этилацетатом, и объединенные органические экстракты осушали сульфатом натрия и концентрировали в вакууме. Остаточное маслообразное вещество очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат) и получали целевой дибензилфосфат (9,9 г, выход - 76%) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3): δ 8,10 - 8,08 (2H, м); 7,74 - 7,71 (2H, м); 7,61 - 7,25 (21H, м); 6,94 (1H, д, J = 9,0 Гц); 6,31 (1H, с); 6,20 (1H, дд, J = 8,7 Гц); 5,91 (1H, дд, J = 9,0, 3,1 Гц); 5,64 (1H, д, J = 6, 9 Гц); 5,49 (1H, д, J = 3,0 Гц); 5,39 (1H, дд, J = 6,6 Гц); 5,05 - 4,98 (5H, м); 4,86 (1H, д, J = 6,6 Гц); 4,26 - 4,12 (3H, м); 3,84 (1H, д, J = 8,4 Гц); 2,82 - 2,71 (1H, м); 2,52 - 1,05 (26H, м, включая синглеты при каждом 2,43, 2,18, 1,97, 1,69, 1,22, 1,20, 3H); 0,90 - 0,85 (3H, дд, J = 7,3 Гц).
(d) Получение 2'-O-n-пропилкарбонил-7-O-фосфонооксиметилпаклитаксела

В продутую азотом, колбу для гидрирования Парра, добавляли 2,5 г 10% палладированного угля, а затем чистый этилацетат (150 мл) и раствор 2'-O-n-пропилкарбонил-7-O- (дибензилфосфонооксиметил)паклитаксела (4,9 г, 4,14 мМ) в этилацетате (40 мл). Затем этот реакционный сосуд подсоединяли к гидрогенизатору Парра, помещали в вакууме и подавали водород при давлении 50 фунт/кв. дюйм (3,515 кг/см2). Полученную гетерогенную смесь перемешивали при встряхивании в течение 5 часов, после чего ТСХ-анализ (элюент: гексан/этилацетат) указывал на полное израсходование исходного материала. Затем реакционную смесь помещали в вакуум, после чего продували азотом. Полученную смесь фильтровали через воронку из спеченного стекла, а фильтрат концентрировали в вакууме, в результате чего получали целевое соединение (3,7 г, выход - 91%), которое очищали с помощью1 H-ЯМР-анализа.
(е) Получение триэтаноламиновой соли 2'-O-n-пропилкарбонил-7- O-фосфонооксиметилпаклитаксела
К раствору 2'-O-n-пропилкарбонил-7-O-фосфонооксиметилпаклитаксела
(1,1 г, 1,09 мМ) в дихлорметане (50 мл), добавляли 0,1 М раствора триэтаноламина (10,9 мл, 1,09 мл) в этилацетате, и полученную смесь перемешивали в течение 5 минут при комнатной температуре.
Реакционную смесь концентрировали в вакууме, и полученное белое твердое вещество очищали сначала путем растворения неочищенного материала в минимальном количестве смеси метиленхлорида и этилацетата,
а
затем к этому раствору добавляли гексан, в результате чего образовывалась нужная аминовая соль в виде белого твердого вещества. Затем эту смесь декантировали, и получали аминовую соль в виде белого
твердого вещества, которая имела чистоту (определенную с помощью ЖХВД-анализа) более 95%.
1H-ЯМР (ацетон-d6, D2O, 300 МГц): δ 8,09 - 8,07 (2H, м); 7,86 - 7,84 (2H, ); 7,69 - 7,24 (11H, м); 7,24 (1H, дд, J = 7,5 Гц); 6,36 (1H, с); 6,05 (1H, дд, J = 8,4 Гц); 5,85 (1H, д, J = 6,7 Гц); 5,61 (1H, д, J = 7,0 Гц); 5,49 (1H, д, J = 6,9 Гц); 5,15 - 5, 13 (1H, м); 4,98 (1H, д, J = 8,2 Гц); 4,87 (1H, дд, J = 12,1 Гц, 6,4 Гц); 4,12 (шир, с, 2H); 3,89 - 3,80 (7H, м); 3,36 - 3,30 (6H, м); 2,95 - 2,93 (1H, м); 2,42 - 1,50 (25H, м, включая синглеты при каждом 2,42, 2,22, 1,93, 1,66, 3H); 1,13 (шир. с, 6H); 0,86 - 0,81 (2H, дд, J = 7,4 Гц).
Пример 21. 2'-O-Метилкарбонил-7-O-фосфонооксиметилпаклитаксел
(a) Получение
2'-O-ацетилпаклитаксела

К раствору паклитаксела (8,0 г, 9,37 мМ) и диизопропилэтиламина (4, 89 мл, 28,1 мМ) в дихлорметане (140 мл), охлажденном до 0oC, по капле в течение 2 минут добавляли ацетилхлорид (1,0 мл, 14,1 мМ). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 10 часов. Полученную реакционную смесь распределяли между этилацетатом и насыщенным водным раствором хлорида аммония. Органическую фазу промывали насыщенным раствором бикарбоната натрия, а затем солевым раствором, после чего осушали сульфатом натрия и концентрировали в вакууме. Остаточный маслообразный продукт очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), и получали 2-O-ацетилпаклитаксел (7,7 г, 92%) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц): δ 8,10 - 8,08 (2H, м); 7,92 - 7, 90 (1H, м); 7,89 - 7,70 (2H, м); 7,60 - 7,29 (11H, м): 6,94 (1H, д, J = 9,2 Гц); 6,26 (1H, с); 6,23 (1H, дд, J = 9,5 Гц); 5,93 (1H, дд, J = 9,2, 3,1 Гц); 5,65 (1H, д, J = 7,8 Гц); 5,48 (1H, д, J = 3,2 Гц); 4,94 (1H, д, J = 7,8 Гц); 4,42 (1H, дд, J = 10,8 Гц, 6,6 Гц); 4,28 (1H, д, J = 8,4 Гц); 4,16 (1H, д, J = 8,4 Гц); 3,78 (1H, д, J = 6,9 Гц); 2,60 - 1,02 (25H, м, включая синглеты при каждом 2,42, 2,19, 2,12, 1,90, 1,65, 1,25, 1,11 и 3H).
(b) Получение 2'-O-ацетил-7-O-метилтиометилпаклитаксела

К раствору 2'-O-ацетилпаклитаксела (7,7 г, 8,60 мМ) и диметилсульфида (5,1 мл, 68,8 мМ) в ацетонитриле (200 мл), охлажденном до -40oC, добавляли бензоилпероксид (8,3 г, 34,4 мМ), и реакционную смесь нагревали до комнатной температуры в течение часа. В это время ТСХ-анализ (элюент: гексан/этилацетат, 1 : 1) указывал на завершение реакции. Полученную реакционную смесь разбавляли этилацетатом, а органический раствор три раза промывали насыщенным раствором бикарбоната натрия, а затем солевым раствором. Органическую фазу осушали сульфатом натрия и концентрировали в вакууме. Остаточное маслообразное вещество очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), и получали целевой метилтиометиловый эфир (7,39 г, 90%) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц): δ 8,10 - 8,08 (2H, м); 7,77-7,73 (2H, м); 7,65 - 7,26 (11H, м); 6,53 (3H, 2); 6,20 (1H, дд, J = 8,3 Гц); 5,92 (1H, дд, J = 12,2, 3,1 Гц); 5,67 (1H, д, J = 7,0 Гц); 5,51 (1H, д, J = 3,2 Гц); 4,94 (1H, д, J = 8,2 Гц); 4,69 - 4,60 (3H, м); 4,33 - 4,28 (2H, м); 4,27 (1H, д, J = 8,4 Гц); 3,86 (1H, д, J = 6, 9 Гц); 2,84 - 2,74 (1H, м); 2,50 - 1,1 (28H, м, включая синглеты при каждом 2,41, 2,15, 2,13, 2,11, 2,06, 1,73, 1,18, 1,15, 3H).
(c) Получение
2'-O-ацетил-7-O-(дибензилфосфонооксиметил)-паклитаксела
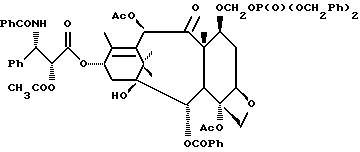
К раствору 2-O-ацетил-7-O-метилтиометилпаклитаксела (5,0 г, 5,23 мМ), дибензилфосфата (5,0 г, 5,23 мМ) и 5 г осушенных в печи сит Angstrom 3 в ТГФ (104 мл), при комнатной температуре, одной порцией добавляли N-йодосукцинимид (1,75 г, 7,85 мМ), и полученную смесь перемешивали в течение 1,5 часа. В этот период времени ТСХ-анализ (элюент: гексан/этилацетат, 1 : 1) указывал на завершение реакции. После этого реакционную смесь разбавляли этилацетатом до объема, в два раза превышающего исходный объем, и фильтровали через слой целита. Затем фильтрат выливали в насыщенный раствор бикарбоната натрия, содержащий 1% тиосульфат натрия (по массе). Органический слой промывали четыре раза насыщенным водным раствором бикарбоната натрия, а затем солевым раствором. После этого водные слои снова экстрагировали этилацетатом, а объединенные органические экстракты осушали сульфатом натрия и концентрировали в вакууме. Остаточное маслообразное вещество очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), в результате чего получали целевой дибензилфосфат (4,9 г, 80%) в виде белого твердого вещества.
(d) Получение
2'-O-ацетил-7-O-фосфонооксиметилпаклитаксела
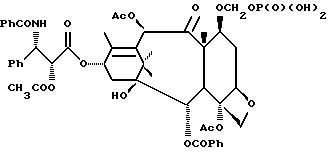
В сосуд Парра для гидрогенизации (продутый азотом) добавляли 700 мг 10% палладированного угля, а затем 130 мл чистого этилацетата, и раствор 2'-O-ацетил-7-O-(дибензилфосфонооксиметил)паклитаксела (1,0 г, 0,84 мМ) в этилацетате (40 мл). Реакционную колбу подсоединяли к гидрогенизатору Парра, помещали в вакуум, и подавали водород при давлении 50 фунт/кв.дюйм (3,515 кг/см2). Затем реакционную смесь перемешивали при встряхивании в течение 6 часов, после чего ТСХ-анализ (элюент: гексан/этилацетат) указывал на полное израсходование исходного материала. Эту реакционную смесь помещали в вакуум, а затем продували азотом. Образовавшийся гетерогенный раствор фильтровали через воронку из спеченного стекла, а фильтрат концентрировали в вакууме, и получали 848 мг белого твердого продукта. Это вещество подвергали1H-ЯМР-анализу, который указывал на присутствие смеси целевого соединения (50%) и 2-О-ацетилпаклитаксела.
(e) Получение 2'-O-ацетил-7-O-фосфонооксиметилпаклитаксела
триэтаноламиновой соли
К раствору
2'-O-ацетил-7-O-фосфонооксиметилпаклитаксела (424 мг, 0,42 мМ) и вышеупомянутого побочного продукта 2-O-ацетилпаклитаксела в дихлорметане (15 мл), добавляли
0,1 М раствора триэтаноламина (3,7 мл, 3,8
мМ) в этилацетате, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. После этого реакционную смесь концентрировали в вакууме и
полученное белое твердое вещество очищали с
помощью хроматографии на C18-колонках (элюент: вода/ацетонитрил, 2,3:1), в результате чего получали нужную аминовую соль (310 мг, 72%), чистота которой
составляла (ЖХВД-анализ) более 96%.
1H-ЯМР (ацетон-d6, D2O, 300 МГц): δ 8,05 - 8,08 (2H, м); 7,86 - 7,83 (2H, м); 7,69 - 7,24 (11H, м); 7,23 (1H, дд, J = 7,4 Гц); 6,35 (1H, с); 6,02 (1H, дд, J = 8,3 Гц); 5,69 (1H, д, J = 6,9 Гц); 5,59 (1H, д, J = 7,1 Гц); 5,45 (1H, д, J = 8,4 Гц); 4,86 (1H, дд, J = 11,5, 6,5 Гц); 4,24 - 4,18 (1H, м); 4,12 (2H, шир.с); 3,92 - 3,89 (6H, м); 3,80 - 3,77 (1H, м); 3,46 - 3,43 (6H, м); 3,00 - 2,89 (6H, м); 2,39 - 1,65 (21H, включая синглеты при каждом 2,39, 2,14, 2,12, 1,92, 1,65, 1,11 и 3H); 5,12 (1H, дд, J = 6,4 Гц); 4,95 (1H, д, J = 8,4 Гц); 1,11 (6H, шир.с).
Пример 22. 2'-O-Метоксикарбонил-7-O-фосфонооксиметилпаклитаксел
(a) Получение 2'-O-метоксикарбонилпаклитаксела

К раствору паклитаксела (8,0 г, 9,60 мМ) и диизопропилэтиламина (5,0 мл, 28,8 мМ) в дихлорметане (96 мл), охлажденном до 0oC, по капле в течение 2 минут добавляли хлорометилкарбонат (1,11 мл, 14,4 мМ). Полученную реакционную смесь нагревали до комнатной температуры и перемешивали в течение 20 часов. После этого реакционную смесь распределяли между этилацетатом и насыщенным раствором хлорида аммония. Органическую фазу промывали насыщенным раствором бикарбоната натрия, а затем солевым раствором, после чего осушали сульфатом натрия и концентрировали в вакууме. Остаточный маслообразный продукт очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), и получали целевое соединение (7,8 г, 91,3%) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц): δ 8,12 - 8,09 (2H, м); 7,72 - 7,70 (2H, м); 7,62 - 7,30 (11H, м); 6,96 (1H, д, J = 9,3 Гц); 6,29 - 6,23 (3H, м); 5,95 (1H, дд, J = 9,3, 2,5 Гц); 5,66 (1H, д, J = 7,1 Гц); 5,38 (1H, д, J = 2,6 Гц); 4,94 (1H, д, J = 7,8 Гц); 4,41 (1H, дд, J = 10,8, 6,6 Гц); 4,28 (1H, д, J = 7,8 Гц); 4,17 (1H, дд, J = 10,8, 6,6 Гц); 3,79 - 3,78 (3H, м); 2,60 - 1,04 (22H, м, включая синглеты при каждом 2,43, 2,19, 1,90, 1,65, 1,22, 1,10, 3H).
(b) Получение
2'-O-метоксикарбонил-7-O-метилтиометилпаклитаксела

К раствору 2'-O-метоксикарбонилпаклитаксела (7,4 г, 8,10 мМ) и диметилсульфида (4,8 мл, 64,8 мМ) в ацетонитриле (162 мл), охлажденном до -40oC, добавляли бензоилпероксид (7,48 г, 32,4 мМ), и реакционную смесь нагревали до комнатной температуры в течение часа. В этот период времени ТСХ-анализ (элюент: гексан/этилацетат) указывал на завершение реакции. Полученную реакционную смесь разбавляли этилацетатом, а полученный органический раствор 3 раза промывали насыщенным раствором бикарбоната натрия, а затем солевым раствором. Органическую фазу осушали сульфатом натрия и концентрировали в вакууме. Остаток в виде маслообразного вещества очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), и получали целевое соединение (7,4 г, 95%) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц); δ 8,25 - 8,23 (2H, м); 7,87 - 7,77 (2H, м); 7,60 - 7,30 (11H, м); 6,93 (1H, д, J = 9,2 ГЦ); 6,53 (1H, с); 6,25 (1H, дд, J = 8,2 Гц); 5,95 (1H, дд, J = 11,7, 2,4 Гц); 5,68 (1H, д, J = 6,9 Гц); 5,40 (1H, д, J = 2,6 Гц); 4,95 (1H, д, J = 8,1 Гц); 4,69 - 4,60 (2H, м); 4,31 - 4,26 (2H, м); 4,16 (1H, д, J = 8,4 Гц); 3,86 (1H, д, J = 6,9 Гц); 3,79 (3H, с); 2,84 - 2,74 (1H, м); 2,43 - 1,10 (25H, включая синглеты при каждом 2,44, 2,15, 2,10, 0,08, 1,73, 1,19, 1,16, 3H).
(c) Получение
2'-O-метоксикарбонил-7-O-(дибензилфосфонооксиметил)паклитаксела

К раствору 2'-O-метоксикарбонилпаклитаксела (5,04 г, 5,18 мМ), дибензилфосфата (7,2 г, 25,8 мМ) и 5 г осушенных в печи молекулярных сит Angstron 3 в ТГФ (100 мл), при комнатной температуре, одной порцией добавляли 1,74 г (7,77 мМ) N-йодосукцинимида, и полученную смесь перемешивали в течение 1,5 часа. В это время ТСХ-анализ (элюент: гексан/этилацетат, 1:1) свидетельствовал о завершении реакции. Полученную реакционную смесь разбавляли этилацетатом до объема, в два раза превышающего исходный объем, и фильтровали через слой целита. Фильтрат выливали в насыщенный раствор бикарбоната натрия, содержащий 1% тиосульфат натрия (по массе). После этого органический слой четыре раза промывали насыщенным водным раствором бикарбоната натрия, а затем солевым раствором. Водный слой снова экстрагировали этилацетатом, и объединенные органические экстракты осушали сульфатом натрия и концентрировали в вакууме. Остаточное маслообразное вещество очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), и получали целевое соединение (5,1 г, 96%) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц): δ 8,12 - 8,08 (2H, м); 7, 73 - 7,70 (2H, м); 7,62 - 7,27 (21H, м); 7,00 (1H, д, J = 9,2 Гц); 6,31 (1H, с); 6,24 - 6,21 (1H, м); 5,96 - 5,92 (1H, м); 5,66 - 5,64 (1H, м); 5,40 - 5,36 (2H, м); 5,05 - 4,93 (5H, м); 4,87 - 4,84 (1H, м); 4, 29 - 4,05 (3H, м); 3,85 - 3,83 (1H, м); 3,77 (3H, с); 2,81 - 2,71 (1H, м); 2,62 - 1,05 (22H, м, включая синглеты при каждом 2,43, 2,19, 2,01, 1,73, 1,22, 1,15, 3H).
(d)
Получение
2'-O-метоксикарбонил-7-O-фосфонооксиметилпаклитаксела

В продутый азотом сосуд Парра для гидрогенизации, добавляли 1,3 г 10% палладированного угля, а затем чистый этилацетат (140 мл) и раствор 2'-O-метоксикарбонил-7-O-(дибензилфосфонооксиметил)паклитаксела (3,4 г, 3,32 мМ) в этилацетате (40 мл). Затем реакционный сосуд подсоединяли к гидрогенизатору Парра, помещяли в вакуум, и подавали водород при давлении 50/фунт/кв. дюйм (3,515 кг/см2). Полученную смесь перемешивали при встряхивании в течение 8,5 часа, после чего ТСХ-анализ (элюент: гексан/этилацетат) указывал на полное израсходование исходного материала. После этого реакционную смесь помещяли в вакуум, а затем продували азотом. Гетерогенный раствор фильтровали через воронку из спеченного стекла, и фильтрат концентрировали в вакууме, в результате чего получали 2,9 г белого твердого вещества. В результате1H-ЯМР-анализа обнаруживалось наличие смеси целевого продукта (67%) и 2-O-метоксикарбонилпаклитаксела (33%).
(e) Получение
2'-O-метоксикарбонил-7-O-фосфонооксиметилпаклитаксела триэтаноламиновой соли
К раствору 2'-О-метоксикарбонил-7-О-фосфонооксиметилпаклитаксела (1,91 г, 1,87 мМ) и вышеупомянутого побочного
продукта 2-О-метоксикарбонилпаклитаксела в дихлорметане (11 мл) добавляли 0,1 М раствора триэтаноламина (18,9 мл, 1,89 мМ) в этилацетате, и полученную смесь перемешивали в течение 5 минут при
комнатной температуре. Затем реакционную смесь концентрировали в вакууме и полученное белое твердое вещество очищали с помощью хроматографии на C18-колонках (элюент: вода/ацетонитрил, 2,3:1), в
результате чего после лиофилизации, получали триэтаноламиновую соль. Эту соль анализировали с помощью ЖХВД, которая указывала на чистоту соли, составляющую 97%.
1H-ЯМР (ацетон-d6, D2О, 300 МГц): δ 8,08- 8,06 (2H, м); 7,88-7,55 (2H, м); 7,69-7,24 (11H, м); 7,24 (1H, дд, J=7,3 Гц); 6,36 (1H, м); 6,05 (1H, дд, J= 8,8 Гц); 5,82 (1H, д, J=6, 8 Гц); 5,60 (1H, д, J=7,1 Гц); 5,46 (1H, д, J= 6,9 Гц); 5,13 (1H, дд, J=6,5 Гц); 5,98 (1H, д, J=8,1 Гц); 4,87 (1H, дд, J= 11,8 Гц, 6,3 Гц); 4,21(1H, дд, J=10,3, 6,9 Гц); 4,13 (шир. с, 6H); 3,92-3,89 (6H, м); 3,81 (1H, д, J=7,02); 3,76 (3H, с); 3,46-3,42 (6H, м); 3,01 - 2,90 (1H, м); 2,42 (3H, с); 2,20-1,80 (10H, включая синглеты при 2,20, 1,93); 1,66 (3H, с); 1,12 (6H, шир. с).
Пример 23. Получение 2'-О-фосфонооксиметоксиметил-7-О-фосфонооксиметилпаклитаксела
(а) Получение 2'-О-метилтиометилметоксипаклитаксела

К раствору 2'-О-метилтиометоксиметил-7-О-бензилоксикарбонилпаклитаксела (1,2 г, 1,11 мМ) в этилацетате (100 мл) и этаноле (70 мл), помещенном в колбу Парра, добавляли 3 г 10% палладированного угля. Этот реакционный сосуд подсоединяли к аппарату Парра, и реакционную смесь помещали в атмосферу водорода под давлением 50 фунт/ кв. дюйм (3,515 кг/см2). После этого реакционную смесь перемешивали при встряхивании в течение 20,5 часа, фильтровали через воронку из спеченного стекла, и фильтрат концентрировали в вакууме. Остаточное маслообразное вещество очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), и получали нужное твердое вещество (0,98 г, 93%).
1H-ЯМР (CDCl3; 300 МГц); δ 8,12-8,10 (2H, м); 7,76 - 7,73 (2H, м); 7,61-7,27 (11H, м); 7,03 (1H, д, J= 8,9 Гц); 6,40 - 6,27 (11H, м); 6,25 (1H, с); 5,80 (1H, дд, J=8,9, 2,4 Гц); 5,66 (1H, д, J=7,1 Гц); 4,98-4,94 (1H, м); 4,86-4,79 (2H, м); 4,75 - 4,68 (1H, м); 4,43 - 4,39 (1H, м); 4,05 (1H, д, J= 11,7 Гц); 3,78 (1H, д, J=7,1 Гц); 2,60 - 1,06 (25H, м, включающий синглеты при каждом 2,45, 2,21, 2,02, 1,85, 1, 66, 1, 22, 1,11, 3H).
(b) Получение 2'-О-метилтиометоксиметил-7-О-метилтиометилпаклитаксела

В раствору 2'-О-метилтиометоксиметилпаклитаксела (0,98 г, 1,03 мМ) и диметилсульфида (0,6 мл, 8,11 мМ) а ацетонитриле (20 мл), охлажденном до -40oC, добавляли бензоилпероксид (1,0 г, 4,13 мМ), и реакционную смесь нагревали до комнатной температуры в течение 30 минут. В этот период времени ТСХ-анализ (элюент: гексан/этилацетат) указывал на завершение реакции. После этого реакционную смесь разбавляли этилацетатом, и полученный органический раствор промывали три раза насыщенным раствором бикарбоната натрия, а затем солевым раствором. Органическую фазу осушали сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (элюент: этилацетат/гексан), и получали целевой продукт (0,945 г, 91%) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц): δ 8,13 - 8,11 (2H, м), 7,79 - 7,77 (2H, м); 7,61 - 7,29 (11H, м); 6,54 (1H, с); 6,30 - 6,26 (1H, м); 5, 83 - 5,80 (1H, м); 5,71 - 5,69 (1H, м); 5,01 - 4,66 (6H, м); 4,34 - 4,04 (5H, м); 3,88 (1H, д, J=6,6 Гц); 2,90 - 2,80 (1H, м); 2,55 - 1,05 (27H, м, включающий синглеты при 2,51, 2,18, 2,11, 1,80, 1, 21, 1,20, 3H-каждом).
(c) Получение 2'-О-дибензилфосфонооксиметилметокси-7-О-(дибензилфосфонооксиметил)паклитаксела

К раствору 2'-0-метилтиометоксиметил-7-0-метилтиометилпаклитаксела (0,92 г, 0,916 мМ), дибензилфосфата (2,03 г, 7,30 мМ) и 1 г осушенных в печи молекулярных сит Angstrom 3 в ТГФ (18 мл), при комнатной температуре, одной порцией добавляли N-йодосукцинимид (0,615 г, 2,74 мМ), и полученную смесь перемешивали в течение 30 минут. В это время ТСХ-анализ (элюент: гексан/этилацетат) указывал на завершение реакции. Затем реакционную смесь разбавляли этилацетатом до объема, в два раза превышающего исходный объем, и фильтровали через слой целита. Фильтрат выливали в насыщенный раствор бикарбоната натрия, содержащий 1% тиосульфат натрия (по массе). Органический слой 4 раза промывали насыщенным водным раствором бикарбоната натрия, а затем солевым раствором. После этого органический слой снова экстрагировали этилацетатом, и объединенные органические экстракты осушали сульфатом натрия и концентрировали в вакууме. Остаточный маслообразный продукт очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат), и получали целевое соединение (0,768 г, 58%) в виде белого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц); δ 8,10 - 8,05 (2H, м); 7,80 - 7,64 (2H, м); 7,65 - 7,27 (11H, м); 6,30 (1H, c); 6,25 - 6,18 (1H, м); 5,82 (1H, дд, J= 9,1, 3,4 Гц); 5,63 (1H, дд, J=6,9 Гц); 5,38 (1H, дд, J=6,6 Гц); 5,10 - 4,60 (15H, м); 4,30 - 4,10 (3H, м); 3,80 (1H, д, J=6,8 Гц); 2,85 - 2,65 (1H, м); 2,50 - 1,60 (22H, м, включающий синглеты при каждом 2,47, 2,16, 1,91, 1,72, 1,88, 1,15, 3H).
(d) Получение
2'-О-фосфонооксиметоксиметил-7-О-фосфонооксиметилпаклитаксела
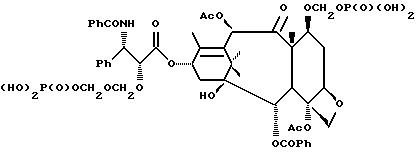
В продутый азотом сосуд Парра для гидрогенизации, добавляли 1,3 г 10% палладированного угля, а затем добавляли чистый этилацетат (110 мл) и раствор 2-О-дибензилфосфонооксиметоксиметил-7-О-(дибензилфосфанооксиметоксиметил)паклитаксела (0,721 г, 0,498 мМ) в этилацетате (40 мл). Этот реакционный сосуд подсоединяли к аппарату Парра, помещяли в вакуум, а затем подавали водород при давлении 50 фунт/кв. дюйм (3,515 кг/см2). Полученную гетерогенную смесь перемешивали при встряхивании в течение 16 часов, после чего ТСХ-анализ (элюент: гексан/этилацетат) указывал на полное израсходование исходного материала. После этого реакционную смесь помещали в вакуум и продували азотом. Затем смесь фильтровали через воронку из спеченного стекла, а фильтрат концентрировали в вакууме, в результате чего получали 0,413 г целевого продукта, который имел чистоту 60% (ЖХВД-анализ).
(e) Получение
бис-триэтаноламиновой соли
2-О-фосфонооксиметоксиметил-7-О-фосфонооксиметилпаклитаксела
К раствору неочищенного 2'-О-фосфонооксиметоксиметил-7-О-фосфонооксиметилпаклитаксела (413 мг) в
дихлорметане (10 мл) добавляли 0,
1 М раствора триэтаноламина (7,6 мл, 0,076 мМ) в этилацетате, и полученную смесь перемешивали в течение 5 минут при комнатной температуре. Затем реакционную смесь
концентрировали в вакууме, и
полученное белое твердое вещество очищали с помощью хроматографии на C18-колонках (элюент: вода/ацетонитрил, 9:1 - 5,6: 1). Фракции элюента, содержащие нужную соль
(ЖХВД-чистота составляла более 96%)
объединяли, а ацетонитрил удаляли на роторном испарителе. Полученный водный раствор аминовой соли лиофилизовали, в результате чего получали нужную соль (0,210 г,
выход по отношению к двум стадиям
составил 30%) в виде белого твердого вещества.
1H-ЯМР (ацетон-d6/D2O, 300 МГц): δ 7,97 - 7,94 (2H, м); 7,79 - 7,76 (2H, м); 7,67 - 7,33 (11H, м); 7,12 - 7,07 (1H, м); 6,26 (1H, с); 5,89 (1H, дд J = 8,6 Гц); 5,48 (1H, д, J=7,9 Гц); 5,00 - 4,79 (8H, м); 4,70 (1H, д, J = 8,1 Гц); 4,15 - 4,03 (3H, м); 3,74 - 3, 66 (7H, м); 3,14 - 2,86 (8H, м), 2,33 - 1,00 (20H, м, включающий синглеты при каждом 2,33, 2,10, 1,88, 1,56, 1,02, 1,00, 3H).
Дополнительные примеры
Общие процедуры,
представленные в предыдущих примерах и
описаниях, были использованы для получения нижеследующих соединений, входящих в объем формулы (А) (таблица 6).
Реферат
Предложены
фосфонооксиметиловые эфиры производных таксана общей формулы А
T-[OCH2(OCH2)mOP(O)(OH)2]n,
где Т представляет собой таксановую
часть, несущую на C13-атоме углерода замещенную 3-амино-2-гидроксипропаноилоксигруппу;
m=0 или 1;
n=1,2,
или его фармацевтически приемлемые соли. Тиопроизводные
баккатина формулы D
13-OH-txn-[OCH2(OCH2)mSCH3]n,
где txn
является таксановой частью; либо его C13-алкоксид металла.
Тиопроизводные таксана формулы В T1-[OCH2(OCH2)mSCH3]n, где
T1 представляет собой Т, в котором переактивные гидроксигруппы
являются блокированными.
Сложноэфирные производные фосфонооксиметиловых эфиров производных таксана формулы С
Т'-[OCH2(OCH2)m OP(O)(OR7)2]n, где T1, m, n определены выше;
R7 является фосфонозащитной группой.
Фармацевтическая композиция, обладающая ингибирующей опухолеактивностью, способы ингибирования роста опухоли у млекопитающего-хозяина. 7 с. и 74 з.п. ф-лы, 6 табл.
Формула
T-[OCH2(OCH2)mOP(o)(OH)2]n,
где Т представляет собой таксановую часть, несущую на С13-атоме углерода замещенную 3-амино-2-гидроксипропаноилоксигруппу,
m = 0 или 1;
n = 1, 2,
или его фармацевтически приемлемые соли.
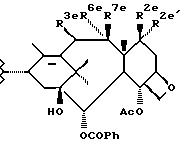
где R2е' является водородом;
R2е является водородом, гидрокси, -ОС(О)ОRх;
R3е является водородом, гидрокси-, -ОС(О)Rх, -ОС(О)ОRх или ацетилокигруппой;
R6е и R7е, взятые вместе, образуют оксогруппу; Rх представляет собой С1-6-алкил, либо Rх представляет собой радикал формулы

где D представляет С1-6-алкил;
Rа, Rв и Rс независимо представляют собой водород.

где R1е представляет собой водород или С(О)Rх; -С(О)ОRх;
R4 и R5 независимо представляют собой С1-6-алкил, С2-6-алкенил или Z-R6;
Z представляет собой прямую связь;
R6 представляет собой арил или гетероарил;
р = 0 или 1,
Rх такой, как определен ранее.
указанная замещенная 3-амино-2-гидроксипропаноилоксигруппа является производной от остатка, имеющего формулу

где R1е, R4 и R5 и р являются такими, как они определены выше.

где R1 является гидрокси, -ОСН2(ОСН2)mОР(О)(ОН)2, -ОС(О)Rх или -ОС(О)ОRх;
R2' является водородом;
R2 является водородом, гидрокси, -ОСН2(ОСН2)mОР(О)(ОН)2; -ОС(О)ОRх;
R3 является водородом, гидрокси, ацетилоксигруппой, -ОС(О)Rх, -ОСН2(ОСН2)mОР(О)(ОН)2 или -ОС(О)ОRх;
R6 и R7, взятые вместе, образуют оксогруппу, при условии, что по крайней мере один из R1, R2, R3, R6 и R7 являются -ОСН2(ОСН2)mОР(О)(ОН)2;
m равно 0 или 1;
R4, R5, Rх и р являются такими, как они определены выше;
и его фармацевтически приемлемая соль.















13-ОН-txn-[OCH2(OCH2)mSCH3]n
где txn является таксановой частью;
m и n являются такими, как они определены выше, либо его С13-алкоксид металла.

где R2е, R2е', R3е, R6е и R7е являются такими, как они определены выше.

или его С13-алкоксид металла.
T'-[OCH2(OCH2)mSCH3]n
где Т' представляет собой Т, в котором нереактивные гидроксигруппы являются блокированными;
m и n определены выше.

где R1в является гидроксигруппой, защищенной гидроксигруппой, -OCH2SCH3, -ОС(О)Rх или -ОС(О)ОRх;
R2' является водородом;
R2в является, водородом, гидроксигруппой, защищенной гидроксигруппой, -OCH2SCH3 или -ОС(О)ОRх;
R3в является водородом, гидроксигруппой, ацетилоксигруппой, -ОС(О)ОRх, -ОС(О)Rх или -OCH2SCH3;
R6в и R7в, взятые вместе, образуют оксогруппу; при условии, что по крайней мере один из R1в, R2в, R3в, R6в или R7в являются -OCH2SCH3,
р, R4, R5 и Rх являются такими, как они определены выше.







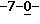




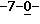





где R2', R2в, R3в, R4, R5, R6в, R7в и р являются такими, как они были определены выше.


T'-[OCH2(OCH2)mОР(о)(ОRy)2]n (с)
где Т', m и n - такие, как они были определены выше;
Ry - фосфонозащитная группа.

где R1с - гидроксигруппа, защищенная гидроксигруппа, -ОСН2ОР(О)(ОСН2Ry)2 , -ОС(О)Rх или -ОС(О)ОRх;
R2' - водород;
R2с - водород, гидроксигруппа, защищенная гидроксигруппа, -ОСН2ОР(О)(ОСН2 Ry)2, -ОС(О)Rх;
R3с - водород, гидроксигруппа, ацетилоксигруппа или -ОСН2ОР(О)(ОСН2Ry)2;
R6с и R7с, взятые вместе, образуют оксогруппу; при условии, что по крайней мере один из R1с, R2с, R3с, R6с или R7с является -ОСН2ОР(О)(ОСН2Ry)2; р, R4, R5, Rх, Ry являются такими, как они определены выше.

где R2', R2с, R3с, R4, R5, R6с, R7с, Ry и р являются такими, как они определены выше.

где R2' - водород;
R2в является ОСН2SCH3;
R1в - гидроксигруппа, -ОС(О)Rх или -ОС(О)ОRх;
R3в является -ОС(О)Rх;
R6в и R7в, взятые вместе, образуют оксогруппу;
R4, R5, р и Rх являются такими, как они были определены выше, при условии, что соединение этой формулы не может быть 3'-N-дебензоил-3'-дезфенил-3'-N-(т-бутоксикарбонил)-3'-(2-фурил)-7-0-метилтиометилпаклитакселом или 3'-N-дебензоил-3'-дезфенил-3'-N-(т-бутоксикарбонил)-3'(2-фурил)
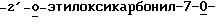
17.08.93 - по пп.1-26, 30-39, 41-43, 46-51, 70, 73-78;
24.11.93 - по пп.40, 44, 45, 52-60, 71, 72;
17.05.94 - по пп.27, 28, 29, 61-69, 79-81.
Комментарии