Способ получения производных амидоксима о-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты и их солей (варианты), чистое кристаллическое основание амидоксима о-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты - RU2074854C1
Код документа: RU2074854C1
Чертежи



Описание
Изобретение относится к органическому синтезу и касается способа (вариантов) получения производных амидоксима O-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты и их солей. Изобретение также касается чистого кристаллического основания амидоксима O-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты.
Известно, что некоторые производные амидоксима O-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты применяются для лечения диабетической ангиопатии, что практически до сих пор является уникальным. Производные амидоксима проявляют также свойство снижения кровяного давления; с ними также можно наблюдать альфа-блокирующий эффект. Использование этих соединений в качестве лекарственных средств требует экономически выгодного приготовления, которое можно осуществить в промышленном масштабе.
Известно, что O-замещенные производные оксимов обычно
получают путем применения алкилирующих агентов [Houben Weyl том X/4, страницы
217 220 (1968)] -O-замещенные амидоксимы в соответствии с настоящим изобретением получают путем взаимодействия
амидоксимов с эпоксидами (или их функциональными эквивалентами, например с
эпоксисоединениями общей формулы (IV)

или с 1-галоид-2-гидрокси-3-пропанаминами общей формулы (III)

или с 3-гидроксиазетидиновыми солями общей формулы (V)

В этих реакциях в качестве растворителей использовали протонные растворители
такие, как вода, метанол, этанол или смеси воды с водонесмешивающимися растворителями, например бензолом.
Конечные продукты выделяют экстракцией (после упаривания), экстракт промывают несколько раз
концентрированным щелочным раствором и после замены растворителя его подкисляют спиртовой соляной кислотой
и осторожно кристаллизуют. Гидрохлориды продуктов получают с выходами от 6 до 50%
При воспроизведении и увеличении масштаба этих реакций было неожиданно найдено, что амидоксимы общей формулы
(II)

не могут быть полностью превращены. После достижения степени превращения от 60 до 70% реакция прекращается и как увеличение температуры реакции, так и использование избытка реактанта увеличивает количество смоляных побочных продуктов, что снижает выделение продукта.
Таким образом, целью изобретения является промышленный процесс, который не имеет указанных недостатков.
Из литературы известно (J.Am.Chem.Soc. 80, 1257 (1958); Appl.Polym.Sci. II,
145-8 (1967); и J.Chem.Soc. 1950, 2257 2272), что амины общей формулы
(III) нестабильны: при хранении они обратимо превращаются в циклическую соль общей формулы (V) или димеризуются в производное
диоксана общей формулы (VI)

Относительно синтеза предшествующее превращение не является недостатком, так как амидоксимы могут взаимодействовать с солями общей формулы (V) в присутствии основания. Однако последнее превращение необратимо, что может привести к значительной потере вещества во время синтеза.
Было установлено, что соединения
общих формул (III), (IV) или (V), соответственно, реагируют с водой, спиртами и кислотами с получением побочных продуктов общей
формулы (IX)

где R4 и R5 означают гидроксил, С1-C4 алкокси, C2-C8алканоил или кислотный остаток.
В зависимости от размеров R4 и R5 групп в частности в случае гидроксильной или низшей алкокси
группы, может также произойти полимеризация значительной
продолжительности, что приведет, например, к соединениям общей формулы (XI)

Образование этих продуктов разложения вызывают "остановку" реакции сочетания. Допускают, что появление продуктов разложения положительно влияет на реакции SNI и поэтому приводит к протонной системе, способствующей дальнейшему разложению и замедляющей превращение амидоксима. Используя модели реакций, было установлено, что реагенты общих формул (III), (IV) и (V) очень нестабильны при условиях, описанных до сих пор и побочные реакции ускоряются или за счет основного или кислотного катализа. Таким образом, в случае эпоксида общей формулы (IV) основной катализ способствует замещению конечных групп, в то время как кислотный катализ способствует замещению у С-2 атома углерода. В реакции сочетания с амидоксимом эпоксид общей формулы (IV) полностью разлагается в течение 2 4 ч.
На основании вышесказанного прежде всего следовало найти реакционную среду или условия реакции, соответственно, где реагенты не разлагаются и селективное сочетание амидоксима общей формулы (II) и образование конечного продукта ускоряются или путем подавления образования загрязнений или путем выделения конечного продукта из этих загрязнений, которые в результате образуются в небольших количествах.
Объектом данного изобретения является
Способ получения производных амидоксима O-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой
кислоты общей формулы I:

и их солей, путем (А) взаимодействия амидоксима формулы II

с гидроокисью щелочного металла или алкоксидом щелочного металла в присутствии органического растворителя с последующим (Б) взаимодействием полученного на стадии (А) комплекса амидоксима
(Б1) с галогенидом общей формулы III

где Х атом галогена,
в условиях реакции нуклеофильного замещения с образованием из двух реагирующих молекул одного переходного комплекса в среде диполярного апротонного растворителя при температуре от 0oC до 100oC и (В) выделением полученного производного амидоксима формулы I, и, при необходимости, (Г) превращением производного амидоксима формулы I с одной или несколькими кислотами в соответствующие соль или смесь солей, соответственно, и, при необходимости, превращением солей в свободные основания, и, при необходимости, превращением свободного основания в соответствующую соль, отличающийся тем, что на стадии (А) в качестве органического растворителя используют диметилформамид или 1,3-диметил-2-имидазолидинон.
Другим объектом изобретения является
Способ получения производных амидоксима
O-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты формулы I:

и их солей путем (А) взаимодействия амидоксима формулы II

с гидроокисью щелочного металла или алкоксидом щелочного металла в присутствии органического растворителя с последующим (Б) взаимодействием полученного на стадии (А) комплекса амидоксима.
(Б2) c амином формулы IV

в условиях реакции нуклеофильного замещения с образованием из двух реагирующих молекул одного переходного комплекса в среде диполярного апротонного растворителя при температуре от 0oC до 100oC и выделяют производное амидоксима формулы I, которое при необходимости подвергают взаимодействию с одной или несколькими кислотами с получением соответствующей соли или смеси солей и при необходимости, соли превращают в свободное основание и при необходимости, свободное основание превращают в соль, отличающийся тем, что в качестве органического основания используют диметилформамид или 1, 3-диметил-2-имидазолидинон.
Еще одним объектом изобретения является
Способ получения производных амидоксима O-(2-гидрокси-3-пипеиридино-1-пропил)-никотиновой кислоты формулы
I:

и их солей (А) взаимодействием амидоксима формулы II:

с гидроксидом щелочного металла или алкоксидом щелочного металла в присутствии органического растворителя с получением комплекса амидоксима, который подвергают взаимодействию с аминосоединением формулы V
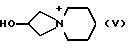
в условиях реакции нуклеофильного замещения с образованием из двух реагирующих молекул одного переходного комплекса в среде диполярного апротонного растворителя при температуре от 0oC до 100oC и (В) выделением полученного производного амидоксима формулы I, и, при необходимости, (Г) превращением производного амидоксима формулы I с одной или несколькими кислотами в соответствующие соль или смесь солей, соответственно, и, при необходимости, превращением солей в свободные основания и при необходимости превращением свободного основания в соответствующую соль, отличающийся тем, что на стадии (А) в качестве органического растворителя используют диметилформамид или 1,3-диметил-2-имидазолидинон.
Согласно изобретению предпочтительно взаимодействие на стадии (А) осуществляют в присутствии соединения донора протонов.
Предпочтительно также в качестве донора протонов на стадии (А) используют третичный бутанол.
При необходимости согласно изобретению полученный на стадии (А) комплекс амидоксима используют на стадии (Б) без выделения. Взаимодействие комплекса амидоксима с амином на стадии (Б) осуществляют при температуре от 30 до 75oC.
Предпочтительно также взаимодействие комплекса амидоксима с амином на стадии (Б) осуществляют в присутствии катализатора, содержащего соль металла. В частности, в качестве катализатора, содержащего соль металла, используют окись алюминия, окись олова, окись дибутилолова или их органические комплексы.
При необходимости согласно изобретению производное амидоксима формулы I селективно отделяют от побочных продуктов.
Для селективного выделения производного амидоксима формулы I
нейтрализуют реакционную смесь кислотой при температуре от 40 до 70oC, при
необходимости, после разбавления реакционной смеси растворителем и/или после фильтрации,
подкисляют
полученную смесь,
устанавливают значение рН 4 5 при 50oC или рН 2-3
при 70oC и выделяют осажденные соли с примесями из смеси, и
кристаллизуют соль производного
амидоксима формулы I, образованную с кислотой при установлении в смеси значения рН
1-3.
Амин формулы IV получают путем реакции эпихлоргидрина формулы VI

с амином формулы VII
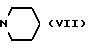
в присутствии третичного бутанола при массовом соотношении третичного бутанола к амину формулы VII от 1:0,8 до 1:1,2.
В соответствии с предпочтительным осуществлением процесса по изобретению реагенты общих формул (III), (IV) и (V) используют в избытке, равном 0,05 до 0,15 эквивалентов.
Удобно использовать диметилформамид (ДМФ) или N,N-диметил-2-имидазолидинон (ДМИ) с содержанием воды 1% или менее, которые свободны от загрязнений, например, от разложения, так как в противном случае выход может быть снижен, или может произойти нежелательная трансформация ДМФ или ДМИ.
Образование и целевое получение, соответственно, комплекса являются существом настоящего изобретения. Наблюдают очень значительное количество осадка в реакции, осуществляемой в диметилформамиде при 50oC, используя гидроокись щелочи в качестве основания. Этот осадок постепенно исчезает по мере продолжения реакции. Для получения комплекса не обязательно отделять его от смеси. Для улучшения условий растворимости и обеспечения передачи протонов предпочтительно добавлять источник протонов, предпочтительно третичный бутанол, к смеси, в результате чего выход увеличивается на 3-5% и время реакции снижается. Далее комплекс реагирует предпочтительно при 30 до 75oC.
Было установлено, что чистота исходных реагентов общих формул (III), (IV) или (V) соответственно важна для хода реакции. Все эти три соединения могут отлично реагировать в чистом состоянии, в то время как очистка и хранение этих реагентов может быть решена с трудом в промышленном масштабе из-за их высокой реактивности. Поэтому был разработан простой метод получения этих продуктов для того, чтобы достичь облегчения решения проблем настоящего изобретения.
Было найдено, что удобно подвергать
взаимодействию комплекс амидоксима, полученного в соответствии с настоящим изобретением,
с реагентом общей формулы (III), (IV) или (V), соответственно, который получают путем взаимодействия
эпихлоргидрина с амином общей формулы (VII)

в присутствии третичного бутанола, взятого в соотношении 1:0,8 к 1:1,2 в расчете на массу амина.
Предпочтительно реагенты формул (III), (IV) или (V) взаимодействуют с комплексом без выделения из реакционной среды после их получения путем использования диметилформамида как последующего растворителя. Таким образом может быть получено производное амидоксима общей формулы (I) с хорошим выходом в том же оборудовании (реакция в "одном реакторе").
Однако реакция продолжается дольше из-за влияния продуктов разложения, как было описано выше. В этих случаях использование катализаторов, указанных выше, особенно предпочтительно.
Таким образом в соответствии с настоящим изобретением решена задача получения целевого соединения с хорошей эффективностью и возможным подавлением побочных реакций.
Способ в соответствии с изобретением гораздо проще, чем известные до сих пор, и
чистота веществ, полученных как сырые продукты, значительно выше. Что касается приемов способа,
то легко увеличить их до промышленного производства и осуществить. Выходы отличные (75 97%) и способ
можно усовершенствовать путем подбора маточных растворов кристаллизации. После упаривания и
щелочной экстракции маточного раствора, раствор можно опять кристаллизовать путем подкисления, как описано
выше,
Используя способ в соответствии с настоящим изобретением стало возможным
выделять основание амидоксима O-(2-гидрокси-3-пиперидино-1-пропил)никотиновой кислоты в виде чистых кристаллов,
т.пл. 70 73oC, что не описано до сих пор в литературе. Наиболее важные
физико-химические характеристики этого вещества следующие.
ИК-спектр (KBr): ν-O-C=N 1642 см-1
1Н-ЯМР спектр (CDCl3, δмлнд: 1,48
м, (6Н), CH2-пиперидин; 2,42 м, (6Н), 3 х CH2N; 3,36, широкий, (1Н), CH-O; 4,08 м, (3Н), 1-CH2, OH; 5,2, шир. (2Н), NH2; 7,30 м (1Н), пиридин-5'; 7,92 м,
4', 8,62 м (1Н), 6', 8,88 м, (1Н), 2'.
1 г этого вещества, растворенного в 10 мл концентрированной серной кислоты, дает желтоватый гомогенный раствор.
Изобретение относится также к этому новому веществу. Таким же образом удалось получить в очень чистом состоянии гидрохлоридные и гидробромидные соли амидоксима O-(2-гидрокси-3-пиперидино-1-(пропил)никотиновой кислоты. Гидробромид до настоящего времени не был описан вообще; о гидрохлориде упоминалось в литературе только как о веществе с низкой степенью чистоты. Спектральные характеристики этих новых соединений следующие.
УФ спектр: λмакс 274,237 нм
ИК спектр (KBr): ν-O-C=N
1649 см-1
1Н-ЯМР спектр (DMSO-d6, δмлнд): 1,80, шир. (6Н), CH2-пиперидин; 2,65 3,75 м (6Н 3 х CH2-N); 4,00 м, (2H), 1-CH2; 4,40 м (1Н), CH-O; 7,00 шир. (4Н), 2 х NH+, H2
; 8,00 дд, (1Н), пириди-5'; 8,75 м, (1Н), 4'; 8,95 м, (1Н), 6'; 9,13 д, (1Н), 2'; 10,35, шир. (1Н), OH.
1 г этого вещества, растворенного в 10 мл концентрированной серной кислоты дает желтоватый гомогенный раствор. Способ по настоящему изобретению иллюстрируется подробно следующими примерами, не ограничивающими объем притязаний заявителя.
Пример 1. 1,0 (25 ммоль) порошка гидроокиси натрия и 4,7 мл (50 ммол) трет-бутанола добавляют к раствору 6,9 г (50 ммол) амидоксима никотиновой кислоты в 50 мл чистого сухого ДМФ.
К полученной суспензии добавляют 7,8 г (55 ммол) перегнанного -(2,3-эпоксипропил) пиперидина [J.Am.Chem.Soc. 80, 1257-9 (1958)] при 50oC и перемешивают при 70oC.
Ход реакции наблюдают
при помощи тонкослойной хроматографии (ТСХ). После полного завершения реакции рН раствора доводят до 6 и после
его очистки и фильтрации, фильтрат подкисляют до рН 2,5 и кристаллизуют. Бледно желтый
кристаллический осадок фильтруют с получением дигидрохлорида амидоксима O-(2-гидрокси-3-пиперидино-1-пропил)
никотиновой кислоты при выходе 16,5 г (94% ), т.пл. 202 204oC после
перекристаллизации из этанола. Первым образованием является 14,9 г бледно-желтого продукта, который имеет 99,1% чистоту
на основе метода вытеснения или определением Cl- и 99,2% на основе
спектрофотометрического и азотного определения, т.пл. 202 204oC. Выход 85%
УФ спектр: λмакс 274,237 нм
ИК спектр: (KBr): -O-C=N 1649 см-1
1Н-ЯМР спектр (DMSO-d6, δмлнд): 1,80, шир. (6Н), CH2-пиперидин; 2,65 375
нм, (6Н, 3 х CH2-N); 4,00 м; (2Н), 1-CH2; 4,40 м, (1Н),
СН-O; 7,00, шир. (4Н), 2 х NH+, NH2; 8,00 дд, (1Н), пиридин-5'; 8,75 м, (1Н), 4'; 8,95 м (1Н), 6'; 9,
13 д, (1Н), 2'; 10,35, шир. (1Н), OH.
При растворении 1 г продукта в 10 мл концентрированной серной кислоты получают желтоватый гомогенный раствор.
Пример 2. 435 мл
сухого трет-бутанола и 676 г (4,785 мол) N-(2,3-эпоксипропил) пиперидина
добавляют к смеси, содержащей 596 г (4,35 мол) амидоксима никотиновой кислоты, 4350 мл чистого сухого ДМФ и 100 г (2,5 мол)
гидроокиси натрия. Бледную коричневую суспензию перемешивают при 65oC в течение 3 ч. Ход реакции наблюдают как описано в примере 1. После подкисления смеси до рН 6, ее очищают, фильтруют и
темно-желтый раствор доводят до рН 2,5. Бледно-желтый кристаллический
осадок фильтруют с получением 1480 г дигидрохлорида амидоксима O-(2-гидрокси-3-пиперидино-1-пропил) никотиновой кислоты при
чистоте 95% Выход равен 92%
После перекристаллизации общий выход
вместе со вторым и третьим продуктом составляет до 88% рассчитанный по исходным соединениям. Чистота кристаллического продукта
равна 99,5% т.пл. 202 204oC. Продукт идентичен продукту
примера 1.
Пример 3. После смешения 41,3 г амидоксима никотиновой кислоты, 750 мл сухого ДМФ и 6 г NaOH, 46,6 г N-(2, 3-эпоксипропил) пиперидина добавляют к полученному комплексу и смесь реагирует при 65oC в течение 4 часов. В это время оранжево-красная густая суспензия переходит в бледно-коричневый раствор, в соответствии с ходом реакции. После перегонки 400 мл ДМФ из раствора в течение 90 минут смесь охлаждают до комнатной температуры и разбавляют 500 мл сухого изопропанола.
После доведения рН раствора до 6 7 и затем фильтруют загрязнения, рН
бледно-желтого раствора доводят до 2,5 путем добавления соляной кислоты и оставляют медленно кристаллизоваться. Получают 81 г
кристаллического гидрохлорида цвета масла, т.пл. 202 205oC.
Этот продукт аналитически идентичен, тем, которые описаны в предшествующих примерах. Из маточного раствора получают 14,5 г
бледно-желтого кристаллического продукта как 2-ой выход, т.пл. 195 200oC. Общий выход составляет до 90,6%
Пример 4. Путем обработки дигидрохлорида амидоксима
O-(2-гидрокси-3-пиперидино-1-пропил)никотиновой кислоты раствором гидроокиси натрия получают
основание и белое кристаллическое вещество амидоксим O-(2-гидрокси-3-пиперидино-1-пропил) никотиновой
кислоты выделяют в чистом состоянии, т.пл. 70 73oC, ИК-спектр (KBr): ν 1642
см-1.
1Н-ЯМР спектр (CDCl3, dмлнд): 1,48 м, (6Н), CH2-пиперидин; 2,42 (6Н), 3 х CH2N 3,36, шир, (1Н), CH-O; 4,08 м, (3Н), 1-CH2, OH; 5,2 шир. (2Н), NH2; 7,30 м, (1Н), пиридин 5'; 7,92 м, (1Н), 4'; 8,62 м, (1Н), 6'; 8,88 м (1Н), 2'.
1 г этого вещества, растворенного в 10 мл концентрированной серной кислоты дает желтоватый гомогенный раствор.
Пример 5. Амидоксим O-(2-гидрокси-3-пиперидино-1-пропил) никотиновой кислоты получают как описано в примере 1, за исключением того, что подкисление проводят при помощи этанольного раствора сухого бромистого водорода с получением 20,5 г дигидробромида амидоксима никотиновой кислоты с выходом 93% (вместе с 2-ым выходом), т.пл. 180 184oC.
Пример 6. Повторяют способ, описанный в примере 1, за
исключением того, что соль получают, используя изопропаноловый раствор, насыщенный сухим
хлористым водородом (с концентрацией примерно 8,5 ммол/мл) с получением дигидрохлорида амидоксима
O-(2-гидрокси-3-пиперидино-1-пропил) никотиновой кислоты с выходом 95%
Пример 7. Повторяют
способ, описанный в примере 1, за исключением того, что не используют трет-бутанол. Реакционную
смесь перемешивают в течение 6 часов при 70oC с получением общего выхода 94%
Пример
8. Повторяют способ, описанный в примере 1, за исключением того, что не используют трет-бутанол
и в качестве основания используют 1,12 г (10 ммол) трет-бутоксида калия. После перемешивания
реакционной смеси при 70oC в течение 3 ч получают общий выход 16,67 г (95%).
Пример 9. Повторяют способ, описанный в примере 1 за исключением того, что используют 70 мл ДМФ, 5,0 г (125 ммол) гидроокиси натрия и в качестве реагента добавляют 11,8 г (55 мол) гидрохлорида 1-хлор-2-гидрокси-3-пиперидинопропана (Monatshefte fur chemie, 15, 119).
После перемешивания реакционной смеси при 70oC в течение 6 часов получают продукт с общим выходом 16, 7 г (95%).
Пример 10. Повторяют способ в примере 1 за исключением того, что используют 3,2 г (80 ммол) порошка гидроокиси натрия, 7,5 мл трет-бутанола и в качестве реагента добавляют 9, 8 г (55 ммол) хлорида 1,1-пентаметилен-3-гидроксиазетидина (J.Org.Chem. 33(2) (523).
После перемешивания суспензии при 70oC в течение 3 ч получают дигидрохлорид амидоксима O-(2-гидрокси-3-пиперидин-1-пропил) никотиновой кислоты с общим выходом 15,8 г (90%).
Пример 11. Повторяют способ, описанный в примере 1, за исключением того, что в качестве катализатора используют 1,0 г (10 ммол) окиси алюминия. После перекристаллизации получают продукт с выходом 14,9 г (85%), т.пл. 206 - 209oC.
Пример 12. Повторяют способ, описанный в примере 1, за исключением того, что в качестве катализатора используют 1,5 г (10 ммол) окиси олова. После перекристаллизации продукт получают с выходом 14,7 г (84%), т.пл. 207 - 210oC.
Пример 13. 1 г (3,6 ммоля) амидоксина O-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты, полученного по примеру 3, растворяют в 60 мл этилацетата и добавляют 890 мг (7, 2 ммоля) никотиновой кислоты. Смесь кипятят в течение 30 мин, затем охлаждают. Выпадает белый кристаллический осадок, который отфильтровывают с получением 0,95 г соли никотината, т-ра плавл. 109 111oC.
Пример 14. Повторяют способ, описанный в примере 1, за исключением того, что в качестве компонента амидоксима кислоты используют 9,3 г (50 ммол) 2-амино-5-хлорбензамидоксима и после подкисления реакционную смесь упаривают при пониженном давлении. Вязкий масляный остаток растворяют в 50 мл горячего изопропанола и оставляют кристаллизоваться с получением выхода 15 г, т.пл. 189 190oC.
Пример 15. 4, 7 г (5,5 мл; 55 ммол) пиперидина добавляют в течение 20 минут к раствору 5,1 г (4,3 мл; 55 ммол) эпихлоргидрина в 5,2 мл трет-бутанола при охлаждении водой при сильном перемешивании. Реакционную смесь перемешивают при комнатной температуре в течение 1 ч, затем добавляют 50 мл чистого сухого ДМФ, 6,9 г (50 ммол), амидоксима никотиновой кислоты и 3,2 г (80 ммол) порошка гидроокиси натрия. После перемешивания суспензии при 70oC в течение 12 ч, реакционную смесь обрабатывают, как описано в примере 1 с получением 13,2 г (75%) дигидрохлорида амидоксима O-(3-амино-2-гидроксипропил) никотиновой кислоты.
Примеры 16 26. По нижеописанным методикам получают производные амидоксима O-алкилированной кислоты общей формулы (I), приведенные в табл.1. Способ А 50 ммол кислотного амидоксима общей формулы (П) растворяют в 50 мл чистого сухого ДМФ, затем добавляют 25 ммол основного катализатора, 4,7 мл (50 ммол) трет-бутанола и наконец 55 ммол 1-(2,3-эпоксипропил)амина общей формылу (IY). После перемешивания при 70oC до полного завершения реакции смесь подкисляют, как описано в примере 1, и затем продукт общей формулы (1) выделяют. Способ В Повторяют способ, описанный как способ А, за исключением того, что реакционную смесь подкисляют и упаривают, как описано в примере 14 и полученный продукт кристаллизуют из растворителя. Способ С Повторяют способ, описанный как способ А, за исключением того, что выделяют свободное основание как описано в примере 4 и затем кристаллизуют из растворителя или выделяют в виде масла.
Для доказательства преимущества способа по изобретению в таблице ниже приводятся сравнительные данные по синтезу наиболее предпочтительного из соединений по изобретению O-(3-пиперидино-2-окси-1-пропил)никотинамидоксим-дигидрохлорида (NP-51) заявленным способом и способом по патенту США N 4187220.
Как видно из таблицы, предлагаемый способ позволяет получать соединение NP-51 с выходом, в 2 раза превышающим выход при использовании прототипа нового способа, защищенного патентом США N 4187220.
Способ по изобретению имеет и другие преимущества.
Чистота продукта и технические недостатки ранее описанного способа.
Соединение NP-51, получаемое по способу, описанному в патенте США N 4187220, представляет собой смолоподобное вещество, из которого получают кристаллическое вещество, вводят сухой газообразный хлористый водород или сухой этанольный раствор хлористого водорода.
На этой стадии происходят дополнительные потери и конечный выход продукта не превышает 30% при содержании NP-51 90 95% и температуре плавления 180
195oC.
Для получения NP-51 с чистотой 99% и такого же качества, как при использовании способа по данной заявке, необходима трехкратная перекристаллизация NP-51.
Таким образом, получаемый по патенту США N 4187220 продукт не пригоден для применения в промышленном масштабе из-за очень низкого выхода и необходимости многостадийной очистки для получения приемлемого для фармацевтических целей материала. Способ по данной заявке не требует использования сухого газообразного хлористого водорода или сухой смеси хлористого водорода с этанолом, которые характеризуются высокой чувствительностью к увлажнению.
Заявитель указывает, что было проведено сравнение биологических свойств, в частности, токсичности соединения по изобретению (пример 7 данной заявки) с известным соединением близкой химической природы по патенту США 4187220 (пример 5 этого патента).
Определяли величину ЛД50 на мышах при введении внутривенной инъекцией.
Получили следующие результаты:
Известное соединение 125 мг/кг веса тела
Соединение по изобретению 265 мг/кг веса тела самца 250 мг/кг веса
тела самки
Таким
образом, соединение по изобретению является менее токсичным, чем известное; безопасность соединения по изобретению выше более, чем в два раза.
Реферат
Использование: в медицине в качестве лекарственных средств. Сущность изобретения: продукт - производные амидоксима O-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты. Реагент 1 - амидоксин O-(2-гидрокси-3-пиперидино-1-пропил)-никотиновой кислоты. Реагент 2: гидроксид или алкоксид щелочного металла. Условия реакции: растворитель - диметилформамид или 1,3-2-имидазолидинон. 4 с. и 9 з.п.ф-лы. 2 табл.
Формула

и их солей, взаимодействием амидоксима формулы II

с гидроксидом или алкоксидом щелочного металла в присутствии органического растворителя с получением комплекса амидоксима, который подвергают взаимодействию с галогенидом формулы III

где Х галоген,
в условиях реакции нуклеофильного замещения с образованием из двух реагирующих молекул одного переходного комплекса в среде диполярного апротонного растворителя при 0 100oC и выделяют полученное производное амидоксима формулы I, которое при необходимости подвергают взаимодействию с одной или несколькими кислотами с получением соли или смеси солей и при необходимости соль превращают в свободное основание и при необходимости превращают свободное основание в соль, отличающийся тем, что в качестве органического растворителя используют диметилформамид или 1,3-диметил-2-имидазолидинон.

и их солей взаимодействием амидоксима формулы II

с гидроксидом или алкоксидом щелочного металла в присутствии органического растворителя с получением комплекса амидоксима, который подвергают взаимодействию с эпоксидом формулы IV

в условиях реакции нуклеофильного замещения с образованием из двух реагирующих молекул одного переходного комплекса в среде диполярного апротонного растворителя при 0 100oC и выделяют производное амидоксима формулы I, которое при необходимости подвергают взаимодействию с одной или несколькими кислотами с получением соответствующей соли или смеси солей и при необходимости соли превращают в свободное основание и при необходимости свободное основание превращают в соль, отличающийся тем, что в качестве органического растворителя используют диметилформамид или 1,3-диметил-2-имидазолидинон.

и их солей взаимодействием амидоксима формулы II

с гидроксидом щелочного металла или алкоксидом щелочного металла в присутствии органического растворителя (А) с получением комплекса амидоксима, который подвергают взаимодействию с 3-гидроксиазетидиновой солью формулы V

где X галоген,
в условиях реакции нуклеофильного замещения с образованием из двух реагирующих молекул одного переходного комплекса в среде диполярного апротонного растворителя при 0 100o C и выделяют производное амидоксима формулы I, которое при необходимости подвергают взаимодействию с одной или несколькими кислотами с получением соответствующей соли или смеси солей (Б), при необходимости соли превращают в свободное основание и при необходимости свободное основание превращают в соль, отличающийся тем, что в качестве органического растворителя используют диметилформамид или 1,3-диметил-2-имидазолидинон.

с амином формулы VII

в присутствии третичного бутанола при массовом соотношении третичного бутанола и амина формулы VII 1 0,8 1 1,2.

с точкой плавления 70 73oC, при растворении 1 г которого в 10 мл концентрированной серной кислоты образуется желтый гомогенный раствор, имеющее следующие спектральные характеристики: ИК-спектр (KBr): ν -O-C=N 1642 см; Н-ЯМР спектр (СDCl3, δ млн.доли): 1,48, м, (6Н), CH2 пиперидин; 2,42, м, (6Н), 3хCH2-N; 3,36, шир, (1Н), СН-О; 4,08, м, (3Н), 1-СН2, ОН; 5,2, шир, (2Н), NH2; 7,30, м, (1Н), пиридин-5'; 7,92, м, 4'; 8,62, м, (1Н), 6'; 8,88, м, (1Н), 2'.
Комментарии