Нафтилсодержащие соединения, фармацевтическая композиция, способ смягчения симптомов постклимактерического синдрома и других связанных с эстрогеном физиологических состояний, способы получения нафтилсодержащих соединений - RU2165924C2
Код документа: RU2165924C2
Чертежи







Описание
Изобретение относится к области фармацевтической и органической химии и касается новых нафтилсодержащих соединений, которые могут использоваться при лечении различных медицинских показаний, связанных с постклимактерическим синдромом, и лечения фибромы матки, эндометриоза и пролиферации аортальных гладкомышечных клеток. Настоящее изобретение также относится к промежуточным соединениям и способам, пригодным для получения фармацевтически активных соединений по настоящему изобретению, и к фармацевтическим композициям.
"Постклимактирический синдром" - термин, используемый для описания различных патологических состояний, от которых часто страдают женщины, которые вошли или завершают физиологический метаморфоз, известный как менопауза. Хотя множество патологий подразумевается при использовании этого термина, три основных следствия постклимактерического синдрома являются первопричиной самого пристального длительного внимания медиков: остеопороз, сердечно-сосудистые проявления, такие, как гиперлипидемия и эстрогензависимый рак, в частности рак молочной железы и рак матки.
Остеопороз включает группу болезней, которые возникают из- за отклонений этиологии, но которые характеризуются общей потерей костной массы на единицу объема. Последствием такой потери костной массы и, в результате, перелома является декомпенсация скелета для обеспечения адекватной структурной опоры для тела. Одним из наиболее распространенных типов остеопороза является остеопороз, ассоциированный с менопаузой. Большинство женщин теряет от 20 до 60% костной массы в трабекулярном пространстве кости в течение 3- 6 лет после прекращения менструаций. Такая быстрая потеря связана, как правило, с усилением резорбции и образования кости. Однако резорбционный цикл является преобладающим, и в результате происходит общая потеря костной массы. Остеопороз является распространенным и серьезным заболеванием у женщин в постклимактерический период.
По оценке, 25 миллионов женщин только в Соединенных Штатах поражены этим заболеванием. Последствия остеопороза не только вредны для человека, но также вызывают большие экономические потери вследствие своего хронического характера и необходимости во всесторонней и длительной помощи (госпитализация и лечение на дому) из-за остаточных явлений заболевания. Это особенно справедливо для пациентов преклонного возраста. Кроме того, хотя остеопороз, вообще, не считается состоянием, угрожающим жизни, уровень смертности 20-30% связан с переломом костей тазобедренного сустава у пожилых женщин. Большая часть этого уровень смертности может напрямую ассоциироваться с постклимактерическим остеопорозом.
Тканью в кости, наиболее чувствительной к воздействию постклимактерического остеопороза, является губчатая кость. Эту ткань частно называют губчатым слоем кости или губчатым веществом кости, и она концентрируется, особенно, вблизи концов кости (около суставов) и в позвонках позвоночного столба. Губчатая ткань характеризуется мелкими остеоидными структурами, которые взаимосвязаны друг с другом, так же, как и с более твердой и плотной кортикальной тканью, которая составляет наружную поверхность и основной стержень кости. Такая взаимосвязанная сеть трабекул дает латеральную опору наружной, кортикальной структуре и определяет механическую прочность структуры в целом. При постклимактерическом остеопорозе происходит, в первую очередь, общая резорбция и потеря трабекул, что ведет к повреждению и перелому кости. В свете потери трабекул у женщин в постклимактерический период, не удивительно, что наиболее распространенными переломами являются переломы, связанные с костями, зависящими от трабекулярной поддержки, например, с позвонками, с шейками костей, которые несут массу тела, таких как бедренная кость и предплечье. Действительно, перелом костей тазобедренного сустава, переломы шеек и осколочные переломы позвонков являются признаками постклимактерического остеопороза.
В настоящее время общепризнанным способом лечения постклимактерического остеопороза является эстрогензаместительная терапия. Хотя лечение, как правило, является успешным, податливость пациента лечению сначала низка, поскольку эстрогенное лечение часто дает нежелательные побочные эффекты.
Большинство женщин в предклимактерический период реже болеют сердечно-сосудистыми заболеваниями, чем мужчины соответствующего возраста. После менопаузы, однако, коэффициент сердечно-сосудистых заболеваний у женщин постепенно возрастает до соответствующего коэффициента, наблюдаемого для мужчин. Такая потеря защитных свойств связана с потерей эстрогена и, в частности, с потерей способности эстрогена регулировать уровень липидов в сыворотке. Природа эстрогенной способности регулировать содержание жиров в сыворотке во многом не понятна, но на сегодня данные показывают, что эстроген может положительно регулировать рецепторы липидов с низкой плотностью (LDL) в печени для удаления избытка холестерина. Кроме того, оказывается, что эстроген имеет некоторое влияние на биосинтез холестерина, и оказывает другое благоприятное воздействие на здоровье сердечно-сосудистой системы.
В литературе имеются сообщения, что у женщин в постклимактерическом состоянии, получающих эстрогензаместительную терапию, происходит возврат уровня липидов в сыворотке до концентраций, которые у них имелись в предклимактерическом состоянии. Таким образом, казалось бы, эстроген является обоснованным лечением для такого случая. Однако побочные явления при эстрогензаместительной терапии неприемлемы для многих женщин, что ограничивает, таким образом, применение такого лечения. Идеальной терапией при таком состоянии было бы средство, которое могло бы регулировать уровень липидов в сыворотке, как это делает эстроген, но было бы свободно от побочных действий и риска, связанных с эстрогенной терапией.
Третьей основной патологией, связанной с постклимактерическим синдромом, является эстрогензависимый рак молочной железы и, в меньшей степени, эстрогензависимый рак других органов, в частности, рак матки. Хотя такие неоплазмы присущи не только женщинам в постклимактерическом состоянии, они в большей степени превалируют в более пожилой группе в постклимактерическом состоянии. Современная химиотерапия этих раковых заболеваний зависит, во многом, от применения антиэстрогенных соединений, таких как, например, тамоксифен. Хотя такие смешанные агонист-антагонисты оказывают благоприятное действие при лечении таких раковых заболеваний, а побочные действия эстрогена являются допустимыми при острых, угрожающих жизни ситуациях, они не идеальны. Например, такие средства могут обладать стимулирующим действием на некоторые раковые клеточные популяции в матке вследствие их эстрогенных (агонисты) свойств, и они могут, следовательно, быть в некоторых случаях контрапродуктивными. Лучшей терапией для лечения таких раков было бы средство, которое является антиэстрогенным соединением, обладающим чуть заметными, или не обладающим, свойствами эстрогенного агониста для репродуктивных тканей.
В соответствии с имеющейся потребностью в новых фармацевтических средствах, которые способны смягчать симптомы, среди прочего, посклимактерического синдрома, настоящее изобретение относится к новым нафталиновым соединениям, содержащим эти соединения фармацевтическим композициям и способам применения таких соединений для лечения постклимактерического синдрома и других связанных с эстрогеном физиологических состояний, таких, как указано далее.
Фиброма матки (фиброзное заболевание матки) является давнишней и всегда существующей клинической проблемой, которая имеет хождение под множеством названий, включая фиброзное заболевание матки, гипертрофию матки, лейомиому матки, миометриальную гипертрофию, фиброзную матку и фиброзный метрит. По существу, фиброма матки является состоянием, при котором существует несвойственное отложение фиброзной ткани на стенке матки.
Такое состояние является причиной дисменореи и бесплодия у женщин. Точная причина такого состояния недостаточно понята, но есть основания предполагать, что имеет место несоответствующая реакция фиброзной ткани на эстроген. Такое состояние воспроизведено у кроликов посредством ежесуточного введения эстрогена в течение 3 месяцев. У морских свинок такое состояние получено при ежесуточном введении эстрогена в течение четырех месяцев. Эстроген также вызывает подобную гипертрофию у крыс.
Наиболее распространенное лечение фибромы матки включает хирургические процедуры, которые являются как дорогостоящими, так и вызывающими иногда осложнения, такие как образование абдоминальных спаек и инфекции. Для некоторых пациенток первичная операция является только временным лечением, и фибромы снова растут. В таких случаях осуществляют гистерэктомию, что эффективно убивает фибромы, но также прекращает репродуктивный период пациентки. Также могут вводиться антагонисты гонадотропин-рилизинговых гормонов, но их применение сдерживается тем фактом, что они могут привести к остеопорозу. Таким образом, существует потребность в новых способах лечения фибромы матки, и способы настоящего изобретения удовлетворяют эту потребность.
Эндометриоз является состоянием тяжелой дисменореи, которая сопровождается сильной болью, внутриматочным кровотечением или кровотечением в брюшинной полости, и часто приводит к бесплодию. Оказывается, что причиной симптомов такого состояния являются эктопические эндометриальные новообразования, которые несоответственно откликаются на нормальное гормональное управление и располагаются в несоответствующих тканях. Видимо, вследствие несоответствующего расположения эндометриального новообразования, ткань инициирует локальные реакции, подобные воспалительным, вызывающие макрофаговую инфильтрацию и каскад событий, ведущих к инициации болезненной реакции. Точная этиология этого заболевания далеко не выяснена, и его лечение с помощью гормональной терапии является несходным, недостаточно установленным и отмечаемым многочисленными нежелательными и, вероятно, опасными побочными эффектами.
Одним из способов лечения этого заболевания является применение малых доз эстрогена для подавления эндометриальных новообразований через негативное ответное действие на основную секрецию гонадотропина; однако иногда необходимо постоянно использовать эстроген, чтобы бороться с симптомами. Такое применение эстрогена часто может приводить к нежелательным побочным действиям и даже к опасности возникновения эндометриального рака.
Другой способ лечения состоит в непрерывном введении прогестинов, которые вызывают аменорею и, подавляя овариальное продуцирование эстрогена, могут вызвать рецидивы эндометриальных новообразований. Применение постоянной прогестиновой терапии часто сопровождается неприятными побочными действиями прогестинов на ЦНС, и зачастую приводит к бесплодию вследствие подавления функции яичников.
Третий способ лечения заключается во введении слабых андрогенов, которые являются эффективными при борьбе с эндометриозом: однако они индуцируют серьезные явления маскулинизации. Некоторые из этих способов лечения эндометриоза также связаны с появлением умеренной степени потери костной массы при продолжительной терапии. Поэтому, желательны новые методы лечения эндометриоза.
Пролиферация аортальных гладкомышечных клеток играет важную роль при таких заболеваниях, как атеросклероз и рестеноз. Показано, что сосудистый рестеноз после чрезкожной транслюминальной коронарной ангиопластики (РТСА) является реакцией тканей, характеризующейся ранней и поздней фазами. Ранняя фаза, имеющая место на протяжении часов-суток после РТСА, является следствием тромбоза с некоторыми вазоспазмами, в то время как представляется, что поздняя фаза всецело обязана избыточной пролиферации и миграции аортальных гладкомышечных клеток. При этом заболевании возросшая подвижность клеток и колонизация такими мышечными клетками и макрофагами существенно способствуют патогенезу заболевания. Избыточная пролиферация и миграция васкулярных аортальных гладкомышечных клеток могут быть первоначальным механизмом реокклюзии коронарных артерий после РТСА, атерэктомии, лазерной ангиопластики и операции по обходному сосудистому шунтированию аорты. См. "Intimal Proliferation of Smooth Muscle Cells as an Explanation for Reccurent Coronary Artery Stenosis after Percutaneous Transluminal Coronary Angioplasty", Austin et al., Journal of the American College of Cardiology, 8: 369-375 (Aug. 1985).
Васкулярный рестеноз остается основным длительным осложнением после операции на закупоренных артериях посредством чрезкожной транслюминальной коронарной ангиопластики (РТСА), артерэктомии, лазерной ангиопластики и обходного сосудистого шунтирования аорты. Примерно у 35% пациентов, перенесших РТСА, в течение трех-шести месяцев после процедуры происходит реокклюзия. Сегодняшняя стратегия лечения васкулярного рестеноза включает механическое вмешательство с помощью таких устройств, как стенты, или фармакологическую терапию, включающую гепарин, низкомолекулярный гепарин, кумарин, аспирин, рыбий жир, антагонисты кальция, стероиды и простациклин. Такая стратегия не оправдала ожиданий по сдерживанию степени реокклюзии и неэффективна для лечения и предупреждения васкулярного рестеноза. См. "Prevention of Restenosis after Percutaneous Transluminal Coronary Angioplasty: The Search for a 'Magic Bullet'," Hermans et al., American Heart Journal, 122: 171-187 (July 1991).
В патогенезе рестеноза избыточная клеточная пролиферация и миграция являются результатом факторов роста, продуцированных клеточными составляющими в крови и поврежденной стенке артерии, которая опосредует пролиферацию гладкомышечных клеток при васкулярном рестенозе.
Средства, которые ингибируют пролиферацию и/или миграцию аортальных гладкомышечных клеток, пригодны для лечения и предупреждения рестеноза. Настоящее изобретение относится к применению соединений в качестве ингибиторов пролиферации аортальных гладкомышечных клеток и, таким образом, ингибиторов рестеноза.
Настоящее изобретение относится к соединениям формулы I

где
R1 представляет собой -H, -ОН, -О(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
R2 представляет собой -H, -ОН, -O(C1-C4 алкил),
-OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
n равно 2 или 3; и
R3 представляет собой 1-пиперидинил, 1-пирролидинил, метил-1-пиppoлидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино или 1- гексаметиленимино; или к их фармацевтически приемлемым солям.
Настоящее изобретение также относится к промежуточным соединениям формулы IV, приведенной ниже, которые пригодны для получения фармацевтически активных соединений настоящего изобретения.

где R1a представляет собой -H, -ОН или -O(C1-C4 алкил);
R2a представляет собой -H, -ОН или -O(C1-C4 алкил);
R3 и n имеют указанные выше значения;
или к их фармацевтически приемлемым солям.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединения формулы I, содержащим, необязательно, эстроген или прогестин, и к применению таких соединений, одних или в сочетании с эстрогеном или прогестином, для ослабления симптомов постклимактерического синдрома, в частности остеопороза, родственных сердечно-сосудистых паталогических состояний и эстрогензависимого рака. Используемый здесь термин "эстроген" включает стероидные соединения, обладающие эстрогенной активностью, такие как, например, 17 β -эстрадиол, эстрон, конъюгированный эстроген (Premarin®), конский эстроген, 17 β -этинил-эстрадиол и подобные соединения. Используемый здесь термин "прогестин" включает соединения, обладающие активностью способствовать наступлению или сохранению беременности, такие как, например, прогестерон, норэтилнодрел, нонгестрел, мегестролацетат, норэфиндрон и подобные соединения.
Соединения по настоящему изобретению также пригодны для ингибирования фибромы матки и эндометриоза у женщин и пролиферации аортальных гладкомышечных клеток, особенно, рестеноза, у людей.
Настоящее изобретение также относится к способу получения соединения формулы Ia

где R1a представляет собой -H, -ОН или -O(C1-C4 алкил);
R2a представляет собой -H, -ОН или -O(C1-C4 алкил);
R3 представляет собой 1-пиперидинил, 1-пирролидинил, диметиламино, диэтиламино или 1-гексаметиленимино; и
n равно 2 или 3;
или его фармацевтически приемлемой соли, который включает
а) взаимодействие соединения формулы V

где
R1a, R2a, R3 и n имеют указанные выше значения, с восстановителем в присутствии растворителя, имеющего температуру кипения в интервале от 150 до 200oC, и кипячение смеси с обратным холодильником;
б) когда R1a и/или R2a представляют собой -O(C1-C4 алкил), необязательное удаление защищающих гидроксильную группу групп R1a и/или R2a; и
в) необязательное образование соли продукта реакции со стадии а) или б).
Одним из аспектов настоящего изобретения являются соединения формулы
I

где R1 представляет собой -H, -ОН, -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
R2 представляет собой - H, -ОН, -O(C1 -C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
n равно 2 или 3; и
R3 представляет собой 1-пиперидинил, 1-пирролидинил, метил-1- пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино или 1-гексаметиленимино; или их фармацевтически приемлемые соли.
Общие термины, использованные здесь при описании соединений, имеют свои обычные значения. Например, термин "C1-C6- алкил" относится к линейным или разветвленным алифатическим цепям с 1-6 атомами углерода, включая метил, этил, пропил, изопропил, бутил, н-бутил, пентил, изопентил, гексил, изогексил и подобные группы. Подобным образом, термин "C1-C4-алкоксигруппа" относится к C1-C4-алкильной группе, присоединенной через кислород, такой как, например, метокси, этокси, н-пропокси, изопропокси и подобные группы. Из таких C1-C4-алкоксигрупп особенно предпочтительной является метокси.
Исходные вещества для одного из вариантов получения соединений по настоящему изобретению, соединения формулы II, приведенной ниже, получают, по существу, так, как описано в патенте США N 4230862, выданном 28 октября 1980, который включен в настоящее в качестве ссылки.

где
R1b представляет собой -H или -O(C1-C4 алкил); и
Y представляет собой метокси или R3-(CH2)n-O-, где R3 и n имеют указанные выше значения. Предпочтительно, R1b представляет собой, метокси, Y представляет собой R3-(CH2)n-O-, R3 представляет собой 1-пиперидинил, и n равно 2.
Обычно легко доступный
тетралон или его соль формулы

где R1a имеет указанные выше значения, подвергают взаимодействию с ацилирующим агентом, таким как фенилбензоат формулы

где Y имеет указанные выше значения. Реакцию обычно осуществляют в присутствии основания средней силы, такого как амид натрия, и проводят ее при температуре окружающей среды или при более низкой температуре.
Для следующей стадии, по выбору, выбранное соединение формулы II можно ввести во взаимодействие, после преобразования в енольное фосфатное производное in situ, в условиях реакции Гриньяра, с
реактивом Гриньяра формулы
R2b-MgBr,
где R2b представляет собой -H или -O(C1-C4 алкил), с получением соединений приведенной ниже формулы
III, которые также известны в данной области техники (см., например, патент США N 4230862, цит. выше),

где R1b, R2b и Y имеют указанные выше значения, или их фармацевтически приемлемых солей.
Когда Y в соединении формулы IIIIa представляет собой R3-(CH2)n-O-, такие соединения могут быть восстановлены или подвергнуты снятию защитных групп, как описано ниже. Когда Y в соединениях формулы III представляет собой метоксигруппу, сначала используют один из путей синтеза, показанных ниже на схеме I. На схеме I R1b, R2b, R3 и n имеют указанные выше значения.
Каждая стадия путей синтеза А и Б на схеме I осуществляется через процедуры, хорошо известные специалисту в этой области техники.
Например, соединения формулы IIIc получают путем обработки соединений формулы IIIb гидрохлоридом пиридина при кипячении с обратным холодильником. В этих условиях, если R1b и/или R2b являются алкокси, эти группы будут деалкилироваться до гидроксильных групп. Использование такой методики устранит стадию отщепления защитной группы от такой (таких) алкоксигруппы (алкоксигрупп) на более позднем этапе, если это желательно.
С другой стороны, метоксигруппа Y формулы IIIb может быть селективно деметилирована путем обработки соединения эквивалентным количеством тиоэтоксида натрия в инертном растворителе, таком как N, N-диметилформамид (ДМФА), при умеренно повышенной температуре от 80 до 100oC. Процесс на этой стадии можно контролировать обычными хроматографическими методами, например, тонкослойной хроматографией (ТСХ).
Как только соединение формулы IIIc получено, его можно ввести во взаимодействие с соединением формулы
R3-(CH2)n-Q,
где R3 имеет указанные выше значения, и Q представляет собой бром или, предпочтительно, хлор, с получением соединения формулы IIId. Эта реакция показана в виде последней стадии пути А
на схеме I.
В нормальных условиях алкилирования такая реакция будет осуществляться по каждой гидроксильной группе, которая может присутствовать в молекуле соединения формулы IIIc. Однако селективное алкилирование в 4-гидроксибензоильной группе может быть достигнуто посредством осуществления реакции в присутствии избытка тонко измельченного карбоната калия и использования эквивалентного, или с небольшим избытком, количества реагента Q-(CH2)n-R3.
Чтобы получить соединения формулы IIIe, как показано на пути Б на схеме I,
соединение формулы IIIc подвергают взаимодействию с избытком алкилирующего агента формулы
Z-(CH2)n-Z',
где Z и Z' являются одинаковыми или различными уходящими
группами, в щелочном растворе.
Подходящие уходящие группы включают, например, сульфонаты, такие как метансульфонат, 4-бромбензолсульфонат, толуолсульфонат, этансульфонат, изопропансульфонат, 4-метоксибензолсульфонат, 4-нитробензолсульфонат, 2-хлорбензолсульфонат и подобные группы, галогены, такие как бром, хлор, иод и т.п., и другие родственные группы. Предпочтительным алкилирующим агентом является 1,2-дибромэтан, и на эквивалент основного вещества используют, по крайней мере, 2 эквивалента, предпочтительно более двух эквивалентов, 1,2-дибромэтана.
Предпочтительный щелочной раствор для такой реакции алкилирования содержит карбонат калия в инертном растворителе, например в метилэтилкетоне (МЭК) или ДМФА. В таком растворе 4-гидроксигруппа бензоильного радикала соединения формулы IIId представлена в виде феноксид-иона, который замещает одну из отщепляющихся групп алкилирующего агента.
Эта реакция лучше всего протекает тогда, когда содержащий реагенты и реактивы щелочной раствор кипятят с обратным холодильником и продолжают процесс до его завершения. При использовании МЭК в качестве растворителя время реакции составляет 6 - 20 часов.
Продукт реакции с этой стадии, соединение формулы IIIe, затем подвергают взаимодействию с 1-пиперидином, 1-пирролидином, метил-1-пирролидином, диметил-1-пирролидином, 4-морфолином, диметиламином, диэтиламином или 1-гексаметиленимином, посредством стандартных технических приемов, с образованием соединений формулы IIId. Предпочтительно, гидрохлорид пиперидина подвергают взаимодействию с соединением формулы IIIe в инертном растворителе, таком как безводный ДМФА, и нагревают до 60-110oC. Когда смесь нагревают до предпочтительной температуры порядка 90oC, реакция занимает от 30 минут до 1 часа. Однако изменения условий реакции будут влиять на время, которое требуется для этой реакции, чтобы довести ее до завершения. Конечно, протекание этой реакционной стадии реакции может быть проконтролировано с помощью обычных методов хроматографии.
Соединения формулы IIId представляют собой исходные вещества для получения фармацевтически активных соединений формулы Ia, как показано ниже на схеме II.
На схеме II R1a, R2b, R3 и n имеют указанные выше значения. По схеме II, соединение формулы IIId или его соль добавляют к соответствующему растворителю и подвергают взаимодействию с восстановителем, таким как, например, алюмогидрид лития (ЛАГ). Хотя в этой реакции может быть использовано свободное основание соединения формулы IIId, часто более удобна соль присоединения кислоты, предпочтительно, гидрохлоридная соль.
Количество восстановителя, используемого в этой реакции, является количеством, достаточным для восстановления карбонильной группы соединения формулы IIId с образованием новых карбинольных соединений формулы IV. Как правило, используют произвольный избыток восстановителя на эквивалент основного вещества.
Подходящими растворителями являются любой растворитель или смесь растворителей, которые будут оставаться инертными в условиях восстановления. Подходящими растворителями являются диэтиловый эфир, диоксан и тетрагидрофуран (ТГФ). Предпочтительна безводная форма этих растворителей, и особенно предпочтительным является безводный ТГФ.
Температура, используемая на этой стадии, является температурой, которая достаточна для полного осуществления реакции восстановления. Обычно адекватной является температура окружающей среды в интервале от 17 до 25oC.
Протяженность этой стадии такова, какая необходима для осуществления реакции. Как правило, эта реакция занимает от 1 часа до 20 часов. Оптимальное время можно определить, контролируя развитие реакции с помощью методов хроматографии.
Карбинольные продукты с этой реакционной стадии (соединения формулы IV) экстрагируют, главным образом, по способу, описанному ниже в примере 7, они являются новыми и могут быть использованы в описанных здесь способах.
Как только получат карбинол по настоящему изобретению, такое соединение добавляют к инертному растворителю, например, к этилацетату, затем добавляют сильную протонную кислоту, такую как хлористоводородная кислота, и получают новые соединения формулы Ia. Эту реакцию, как правило, проводят при температуре окружающей среды от 17 до 25oC, и до завершения она, как правило, занимает от нескольких минут до 1 часа. Кристаллизацию конечного продукта осуществляют обычными приемами, по существу, так, как описано ниже в примере 1.
Деалкилирование и отщепление защитных групп у защищенных по концам гидроксильных групп можно осуществлять до получения соединений формулы IV, до получения соединений формулы Ia или после получения защищенных соединений формулы Ia, с помощью метолов, известных специалисту в данной области. Предпочтительно, однако, деалкилировать защищенное соединение формулы Ia после его образования.
Реакция, показанная на схеме II, приводит к новым фармацевтическим активным соединениям формулы Ia, где каждый из R1а и R2а является водородом, гидрокси или C1-C4 -алкокси. Предпочтительные соединения формулы Ia являются соединениями, в которых каждый из R1a и R2a представляет собой метокси, или каждый из R1a и R2a представляет собой гидрокси, R3 представляет собой пиперидинил, и n равно 2. Эти предпочтительные соединения, причем последние являются особенно предпочтительными, как и другие соединения формулы Ia, могут использоваться в качестве фармацевтических средств, или можно также получить их производные, что даст другие соединения формулы I, которые также пригодны для осуществления способов настоящего изобретения.
Как альтернативу реакциям, приведенным на схеме II, можно использовать новый одностадийный процесс для получения соединений настоящего изобретения формулы Ia путем восстановления кетона приведенной ниже формулы V. Точнее, когда R1a и/или R2a представляют собой -O(C1-C4 алкил), эти защищающие гидроксилы группы могут быть удалены перед осуществлением нового способа по настоящему изобретению, или, необязательно, могут быть удалены in situ, после одностадийного процесса восстановления. Кроме того, продукт по этому способу, который может содержать 1 или 2 незащищенных или защищенных гидроксильных радикала, необязательно, можно превратить в соль известными методами, или как описано здесь.
В
этом способе соединение формулы V
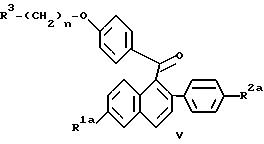
где R1a, R2a, R3 и n имеют указанные выше значения, или его соль, подвергают взаимодействию с восстановителем, таким как алюмогидрид лития или Red-Al® [бис(2-метоксиэтоксиалюмогидрид) натрия] в присутсотвии растворителя, имеющего температуру кипения 150 - 200oC.
Соединение формулы V получают взаимодействием соединения формулы IIIb (описанного выше) с 2 эквивалентами 2,
3-дихлор-5,6-дициано- 1,4-бензохинона (ДДХ) в присутствии инертного растворителя или смеси растворителей, таких как, например, диоксан, дихлорметан, толуол, дихлорэтан или бензол. Реакционную смесь
обычно кипятят с обратным холодильником в течение 1-2 часов и затем перемешивают при температуре окружающей среды в течение 36-72 часов. Полученное соединение формулы VI

где R1b и R2b имеют указанные выше значения, затем деметилируют, как описано выше, и алкилируют соединением формулы
R3-(CH2)n-Q,
где R3 имеет указанные выше значения, с помощью описанных выше процедур.
В случае реакции восстановления по настоящему изобретению количество восстановителя, используемого в этой реакции, является количеством, достаточным для восстановления карбонильной группы соединения формулы V с образованием соединения формулы Ia. Как правило, используют свободный избыток восстановителя на эквивалент основного вещества.
Для использования в этом процессе требуется такой растворитель, у которого имеется относительно высокая температура кипения - (150 - 200oC) как у таких растворителей, как, например, н-пропилбензол, диглим (1, 1'-оксибис[2- метоксиэтан] ) и анизол. Когда R1a и/или R2a представляет собой - OCH3 и -C6H4-4'-O(C1-C4 алкил), из этих растворителей предпочтительным является н-пропилбензол. Когда R1a представляет собой -ОН, и/или R2a представляет собой -C6H4-4'-ОН, предпочтителен Red-AL, используемый как в качестве растворителя, так и в качестве восстановителя.
Температура, используемая при этой реакции, является температурой, которая достаточна для завершения реакции восстановления. Предпочтительно, реакционную смесь кипятят с обратным холодильником от 15 минут до 6 часов, охлаждают ее до температуры окружающей среды, и действуют в соответствии со стандартными методами [см. , например, Fieser and Fieser, Reagents for Organic Synthesis, v. 1, p. 584 (1968)], а также как описано здесь в примерах. Оптимальное время этой реакции, составляющее, как правило, от 10 минут до 1 часа, может быть определено посредством контроля за развитием реакции с помощью стандартных методов.
Продукты одностадийной реакции формулы Ia экстрагируют, по существу, так, как описано ниже в примере 2. Предпочтительные соединения формулы Ia, полученные по этой реакции, являются теми же соединениями, которые являются предпочтительными соединениями формулы Ia, описанными выше, и могут использоваться в качестве фармацевтически активных средств для описанных здесь способов, или можно получить их производные, что даст другие новые соединения формулы I, которые также пригодны для способов настоящего изобретения.
Например, когда R1a и/или R2a в соединении формулы Ia представляют собой защищающие гидроксил C1 -C4- алкильные группы (т.е., не деалкилированы, как предлагает один из вариантов на схеме I), такие группы могут быть удалены с помощью стандартных методов деалкилирования, как описано ниже в примере 2, чтобы получить особенно предпочтительное соединение формулы Ia.
Другие предпочтительные соединения формулы Ia получают путем замещения вновь образовавшихся R1a и/или R2a гидроксильных групп соединения формулы Ia группой формулы -O-CO- (C1-C6 алкил) или -O-SO2-(C1-C6 алкил) через хорошо известные процедуры. См., например, патент США N 4358593.
Например, когда нужна группа -O-CO(C1-C6 алкил), дигидроксисоединение формулы Ia подвергают взаимодействию с таким агентом, как хлорангидрид, бромангидрид, нитрил или азид, или с соответствующим ангидридом или смешанным ангидридом. Реакции удобно осуществлять в основном растворителе, таком как пиридин, лутидин, хинолин или изохинолин, или в третичном амине, таком как триэтиламин, трибутиламин, метилпиперидин и в подобном растворителе. Реакцию также можно осуществлять в таком инертном растворителе, как этилацетат, диметилформамид, диметилсульфоксид, диоксан, диметоксиэтан, ацетонитрил, ацетон, метилэтилкетон и в подобном растворителе, к которому добавлен, по крайней мере, один эквивалент акцептора кислоты (за исключением отмеченных ниже случаев), такого как третичный амин. Если желательно, можно использовать катализатор ацилирования, такой как 4-диметиламинопиридин или 4- пирролидинопиридин. см. , например, Haslam, et al. Tetrahedron, 36: 2409-2433 (1980).
Реакции ацилирования, которые дают вышеуказанные концевые группы R1 и R2 соединений формулы I, осуществляют при умеренных температурах в интервале от -25 до 100oC, часто в инертной атмосфере, такой как азот. Однако для протекания реакции, как правило, адекватной является температура окружающей среды.
Такое ацилирование упомянутых гидроксильных групп также можно осуществить с помощью кислотно-катализированных реакций соответствующих карбоновых кислот в инертных органических растворителях или при нагревании. Используют такие кислотные катализаторы, как серная кислота, полифосфорная кислота, метансульфоновая кислота и подобные кислоты.
Вышеуказанные группы R1 и/или R2соединений формулы I также могут быть введены путем получения активного сложного эфира соответствующей кислоты, такого как эфиры, полученные с помощью таких известных реагентов, как дициклогексилкарбодиимид, ацилимидазолы, нитрофенолы, пентахлорфенол, N-гидроксисукцинимид и 1-гидроксибензотриазол. См., например, Bull. Chem. Soc. Japan, 38: 1979 (1965), и Chem. Ber., 788 и 2024 (1970).
Каждый из вышеуказанных способов, которые дают группы -O-CO- (C1-C6 алкил), осуществляют в растворителях, какие упоминались выше. Те способы, которые не приводят к образованию кислотного продукта в ходе реакции, конечно, не указаны в числе способов, использующих акцептор кислоты в реакционной смеси.
Когда требуется соединение формулы I, где группа R1a и/или R2b соединения формулы I превращается в группу формулы -O-SO2-(C4-C6 алкил), дигидроксисоединение формулы Ia подвергают взаимодействию, например, с ангидридом сульфокислоты или производным соответствующей сульфокислоты, таким как сульфонилхлорид, сульфонилбромид или аммониевая соль сульфокислоты, как описано в King and Monior, J. Am. Chem. Soc., 97:2566-2567 (1975). Дигидроксисоединение также можно подвергнуть взаимодействию с соответствующим ангидридом сульфокислоты или смешанными ангидридами сульфокислот. Такие реакции осуществляют в условиях, которые пояснены выше при описании реакции с галоидангидридами и подобными соединениями.
Все вместе, соединения формулы Ia со своими различно определяемыми заместителями, и их производные, описанные выше, представляют собой соединения по настоящему изобретению формулы I.
Хотя в способах по настоящему изобретению могут использоваться соединения формулы I в форме свободного основания, предпочтительно получать и использовать форму фармацевтически приемлемой соли. Таким образом, соединения, используемые в "способах настоящего изобретения, образуют, преимущественно, фармацевтически приемлемые соли присоединения кислот со многими органическими и неорганическими кислотами и включают физиологически приемлемые соли, которые часто применяются в фармацевтической химии. Такие соли также являются частью настоящего изобретения. Типичными не органическими кислотами, используемыми для образования таких солей, являются хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, азотная, серная, фосфорная, гипофосфорная кислота и подобные кислоты. Также могут использоваться соли, полученные с органическими кислотами, такими как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, оксиалкановые и оксиалкандиовые кислоты, ароматические кислоты, алифатические и ароматические сульфокислоты. Такие фармацевтически приемлемые соли включают, таким образом, ацетаты, фенилацетаты, трифторацетаты, акрилаты, аскорбаты, бензоаты, хлорбензоаты, динитробензоаты, оксибензоаты, метоксибензоаты, метилбензоаты, о- ацетоксибензоаты, нафталин-2-бензоаты, бромиды, изобутираты, фенилбутираты, β - оксибутираты, бутин-1,4-диоаты, гексин-1,4-диоаты, капраты, каприлаты, хлориды, циннаматы, цитраты, формиаты, фумараты, гликоляты, гептаноаты, гиппураты, лактаты, малаты, малеаты, оксималеаты, малонады, манделаты, мезилаты, никотинаты, изоникотинаты, нитраты, оксалаты, фталаты, терефталаты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, прориолаты, пропионаты, фенилпропионаты, салицилаты, себацаты, сукцинаты, субераты, сульфаты, бисульфаты, пиросульфаты, сульфиты, бисульфиты, сульфонаты, бензолсульфонаты, п-бромфенилсульфонаты, хлорбензолсульфонаты, этансульфонаты, 2-оксиэтансульфонаты, метансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты, п- толуолсульфонаты, ксилолсульфонаты, тартраты и подобные соли. Предпочтительной солью является гидрохлоридная соль.
Фармацевтически приемлемые соли присоединения кислот образуются, типично, при взаимодействии соединения формулы I с эквимолярным или избыточным количеством кислоты. Реагенты обычно соединяют в общем растворителе, таком как диэтиловый эфир или этилацетат. Соль обычно выпадает в осадок из раствора в течение периода от 1 часа до 10 суток, и может быть отделена фильтрацией, или растворитель можно отогнать обычными способами.
Фармацевтически приемлемые соли, как правило, имеют улучшенные характеристики растворимости по сравнению с соединением, от которого их производят, и, таким образом, часто более удобны при составлении жидких композиций или эмульсий.
Приведенные далее примеры даются для дополнительной иллюстрации получения соединений настоящего изобретения. Не следует предполагать, что изобретение ограничено по объему на основании какого-либо из нижеследующих примеров.
Данные ЯМР в приведенных ниже примерах получены на приборе ЯМР GE 300 МГц, и в качестве растворителя, если нет других указаний, применяют безводный d-6 ДМСО.
Препаративный пример 1
[3,
4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] (4-метоксифенил)метанон

К суспензии гидрида натрия (12,75 г, 60% дисперсия в масле, предварительно промытая гексаном, 0,32 моль) в тетрагидрофуране (ТГФ) (650 мл) при перемешивании при 0oС добавляют раствор (3, 4- дигидро-2-гидрокси-6-метокси-1-нафтиленил)(4-метоксифенил)метанона (90,0 г, 0,29 ммоль, см., например, патент США N 4230862) и дифенилхлорфосфата (77,8 г, 0,29 моль) в ТГФ (750 мл). Скорость добавления такова, что температура реакции остается ниже 8oC. После перемешивания в течение 3 часов при 0oC добавляют по каплям 4-MeOC6H4MgBr (1,5 эквивалента, 0,064 г/мл раствор в ТГФ) и полученной смеси дают возможность постепенно нагреваться до комнатной температуры. Через 12 часов раствор гасят, добавляя холодный водный раствор хлорида аммония. Органическую часть отделяют от смеси и водную часть экстрагируют этилацетатом. Объединенные органические экстракты сушат (сульфат натрия), фильтруют и концентрируют. К полученному маслу добавляют ацетонитрил (1 л), после чего образуется осадок. Твердое вещество удаляют фильтрацией, фильтрат концентрируют и получают масло, которое очищают флэш-хроматографией (силикагель, метиленхлорид). Нужный продукт затем очищают кристаллизацией из метанола и получают 96,7 г (83%) названного в заголовке соединения в виде твердого кристаллического вещества желтого цвета. Т.пл. 172-173oC.
1H-ЯМР (ДМСО-d6) δ 7,75 (д, J= 8,7 Гц, 2H), 7,16 (д, J=8,6 Гц, 2H), 6,60-6,90 (комплекс, 7H), 3,74 (с, 3H), 3,71 (с, 3H), 3,64 (с, 3H), 2,96 (м, 2H), 2,69 (м, 2H). М.-с. (FD) (с полевой десорбцией) m/e 400 (М+).
Препаративный пример 2
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил]
(4-гидроксифенил)метанон
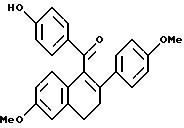
К раствору литийэтантиола [полученного добавлением н-BuLi (87,8 мл, 1,6 М раствор в гексане, 140 ммоль) к раствору этантиола (12,1 мл, 164 ммоль) при перемешивании при 0oC в этиловом эфире (400 мл) с последующим кратковременным перемешиванием и концентрированием] в диметилформамиде (400 мл) при перемешивании добавляют продукт препаративного примера 1 (46,7 г, 117 ммоль). Смесь затем нагревают до 100oC. Через 1 час реакционную смесь концентрируют и полученное коричневое масло растворяют в хлороформе. Этот раствор экстрагируют водным раствором хлорида аммония. Водную часть обрабатывают 1н. соляной кислотой до достижения pH 5 и затем экстрагируют хлороформом. Объединенные органические экстракты промывают солевым раствором, сушат (сульфат натрия), фильтруют и концентрируют. Полученное коричневое масло очищают флэш-хроматографией (силикагель, с градиентом этилацетат/гексан) и получают 30,0 г (66%) названного в заголовке соединения в виде желтого масла.
1H-ЯМР (300 МГц, CDCl3) δ 7,74 (м, 2H), 7,16 (м, 2H), 6,85 (д, J=8,0 Гц, 1H), 6,77 (с, 1H), 6,65 (м, 5H), 6,11 (с, 1H), 3,78 (с, 3H), 3,69 (с, 3H), 3,00 (м, 2H), 2,77 (м, 2H);
13C-ЯМР (75
МГц, CDCl3) δ 201,1, 162,4, 159,7, 159,6, 137,5, 137,2, 134,6, 134,2, 133,3, 130,6, 129,6, 127,6, 127,2, 116,5, 114,7, 114,5, 112,3, 56,2, 56,0, 30,7, 29,6.
Элем. анализ. Вычисл.: C 77,70; H 5,00; найдено: C 77,46; H 5,91.
М.-с. (FD) m/e 386 (M+); ИК (хлороформ) 3400,94, 1641,63, 1601,12 см-1.
Препаративный
пример 3
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-пиперидинил)этокси]фенил]метанон

К раствору продукта препаративного примера 2 (36 г, 93 ммоль) в диметилформамиде (ДМФА; 1 л) при перемешивании добавляют иодид калия (30 мг, 0,18 ммоль), а затем добавляют карбонат калия (64,2 г, 465 ммоль) и моногидрохлорид 1-(2-хлорэтил) пиперидина (18,9 г, 102 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение ночи, затем концентрируют и полученное масло растворяют в хлороформе. Этот раствор тщательно промывают водой и солевым раствором, сушат (сульфат натрия), фильтруют и концентрируют. Полученное масло очищают флэш- хроматографией (силикагель, градиент метанол/хлороформ) и получают 43 г (93%) названного в заголовке соединения в виде желтой пены.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7,72 (д, J=8,0 Гц, 1H), 7,15 (д, J=10 Гц, 3H), 6,87 (д, J=11 Гц, 3H), 6,72 (д, J=8,0 Гц, 2H), 6,62 (с, 2H), 4,05 (м, 2H), 3,69 (с, 3H), 3,63 (с, 3H), 2,95 (м, 2H), 2,62 (м, 4H), 2,38 (м, 4H), 1,44 (м, 4H), 1,33 (м, 2H);13C-ЯМР (75 МГц, ДМСО-d6) δ 197,2, 168,22, 168,18, 162,5, 162,3, 158,4, 158,3, 136,4, 134,9, 133,0, 133,0, 131,3, 129,6, 128,6, 125,9, 114,4, 113,7, 113,6, 113,4, 111,5, 65,7, 62,5, 57,0, 55,0, 55,0, 54,9, 54,1, 29,1, 28,0, 25,4, 23,7.
Элем. анализ. Вычисл. : C 77,24; H 7,09; N 2,81; найдено: C 77,44; H, 7,13; N 2,75.
М.-с. (FD) m/e 497 (M+); ИК (хлороформ) 1672,12 см-1.
Пример 1
Гидрохлорид [2-(4-метоксифенил)-6-метоксинафталин-1-ил]
[4-[2-(1-пиперидинил)этокси]фенил]метана
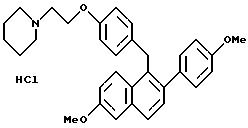
К суспензии алюмогидрида лития (3,80 г, 94,8 ммоль) в сухом ТГФ (100 мл) при перемешивании при 0oC постепенно, в течение 45 минут, добавляют раствор продукта препаративного примера 3 (23,6 г, 47,4 ммоль) в ТГФ (50 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 14 часов, охлаждают ее до 0oC и осторожно гасят водой (5 мл). К этому раствору добавляют по каплям гидроксид натрия (15 мл, 15% (м/м) водный раствор), а затем воду (5 мл). Смесь перемешивают в течение 0,5 часа, фильтруют и твердое вещество тщательно промывают этилацетатом. Фильтрат концентрируют и получают 21 г (89%) промежуточного продукта (карбинол) в виде белой пены, который используют без дополнительной очистки. К промежуточному продукту (23,6 г, 47,2 ммоль) в этилацетате (100 мл) при перемешивании при температуре окружающей среды добавляют хлористоводородную кислоту [100 мл, насыщенный раствор хлористого водорода в этилацетате]. При концентрировании смеси сразу же образуется осадок. Образовавшееся твердое вещество перекристаллизовывают из метанола и получают 19,4 г (79%). названного в заголовке продукта в виде твердого белого кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 10,54 (шс, 1H), 7,72-7,80 (комплекс, 2H), 7,34-7,38 (комплекс, 2H), 7,23 (д, J=8,5 Гц, 2H), 7,08 (дд, J=8,4, 2,3 Гц, 1H), 6,80-6,96 (комплекс, 6H), 4,30 (шс, 4H), 3,85 (с, 3H), 3,76 (с, 3H), 3,37-3,45 (комплекс, 4H), 2,90- 2,99 (м, 2H), 1,61-1,82 (комплекс, 5H), 1,32-1,39 (м, 1H). М.-с. (FD) m/e 481 (M+-хлористоводородная кислота).
Элем. анализ. Вычисл. : C 74,19; H 7,00; N 2,70; найдено: C 74,28; H 7,10; N 2,66.
Пример 2
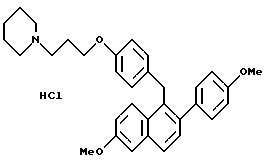
Гидрохлорид [2-(4-гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1-пиперидинил)этокси]фенил]метана

К раствору продукта, полученного в примере 1 (5,0 г, 9,6 ммоль), в 1,2-дихлорэтане (50 мл) при перемешивании при комнатной температуре добавляют трихлорид бора (20 мл, 234 ммоль). Полученную темно-фиолетовую реакционную смесь перемешивают при температуре окружающей среды в течение ночи и затем охлаждают до 0oC. Затем осторожно, по каплям, в течение 2 часов добавляют метанол (50 мл) (осторожно, выделяется газ), и в это время образуется осадок. Твердое вещество отфильтровывают, промывают холодным метанолом, а затем диэтиловым эфиром. Перекристаллизацией из метанола получают названный в заголовке продукт в виде белого порошка.
1H-ЯМР (300 МГц, ДМСО-d6)
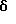
Элем. анализ. Вычисл. : C 73,53; H 6,58; N 2,86; найдено: C 73,48; H 6,57; N 3,01.
Пример 3
[2-(4-Бензоилоксифенил)-6-бензоилоксинафталин-1-ил] [4-[2-(1- пиперидинил)этокси]фенил]метан

К ТГФ суспензии (200 мл) продукта примера 2 (4,1 г, 8,4 ммоль) при перемешивании добавляют N,N-диметиламинопиридин (10 мг, катализатор). Охлаждают до 0oC, добавляют триэтиламин (8,5 г, 83,7 ммоль). Через 10 мин добавляют по каплям бензоилхлорид (4,7 г, 33,5 ммоль) и раствор перемешивают в течение 60 часов. Затем выпавший осадок фильтруют и фильтрат упаривают. Очистка полученного вещества препаративной ВЭЖХ (градиент от хлороформа до хлороформа с 25% этилацетата) с последующей перекристаллизацией из метанола дает 3,78 г названного в заголовке соединения в виде белого порошка.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,18 (прибл. т, J=9,1 Гц, 4H), 7,91-8,05 (компл. , 3H), 7,75 (м, 1H), 7,61-7,69 (м, компл., Н), 7,58 (д, J=8,9 Гц, 1H), 7,42-7,50 (компл., 3H), 7,38 (д, J=8,3 Гц, 2H), 6,91 (д, J=8,5 Гц, 2H), 6,80 (д, J=8,5 Гц, 2H), 4,40 (с, 2H), 3,97 (т, J=3,5 Гц, 2H), 2,60 (т, J=3,3 Гц, 2H), 2,39 (ком., 4H), 1,31-1,52 (компл., 6H). М.-с. (FD) m/e 661 (М+).
Элем. анализ. Вычисл. : C 79,86; H 5,94; N 2,12; Найдено: C 79,59; Н 6,05; N 1,96.
Пример 4
[2-(4-Пивалоилоксифенил)-6-пивалоилоксинафталин-1-ил] [4-[2- (1-пиперидинил)этокси]фенил]метан
К суспензии продукта примера 2 (0,250 г, 0,510 ммоль) в ТГФ (25 мл) при перемешивании добавляют N,N-диметиламинопиридин (2 мг), а затем триэтиламин (0,78 мл, 5,6 ммоль) и триметилацетилхлорид (0,25 мл, 2,0 ммоль). Полученную смесь перемешивают при температуре окружающей среды в течение 2 часов, затем выливают в этилацетат с водой (100 мл, 1:1, о/о). Органический слой отделяют, а водную часть экстрагируют этилацетатом (50 мл). Объединенные органические экстракты промывают насыщенным водным раствором хлорида аммония (1х25 мл), насыщенным водным раствором бикарбоната натрия (2х25 мл) и солевым раствором (1х25 мл). Очистка радиальной хроматографией (силикагель, 2 мм, этилацетат:гексан:триэтиламин:метанол= 10:8:1:1) дает 0,268 г названного в заголовке соединения (85%) в виде густого масла. ИК (хлороформ) 2977, 2939, 1746, 1510, 1167, 1146, 1122 см-1.
1H-ЯМР (300 МГц, CDCl3) δ 7,87-7,90 (д, 1H, J=9,3 Гц), 7,75-7,78 (д, 1H, J= 8,6 Гц), 7,56-7,57 (д, 1H, J=2,4 Гц), 7,43-7,46 (д, 1H, J=8,4 Гц), 7,28-7,31 (м, 3H), 7,10-7,14 (дд, 1H, J=9,2 Гц, 2,4 Гц), 7,03-7,06 (м, 2H), 6,86-6,88 (д, 2H, J=8,5 Гц), 6,71-6,74 (м, 2H), 4,34 (с, 2H), 4,10-4,15 (м, 2H), 2,79-2,83 (м, 2H), 2,52-2,57 (м, 4H), 1,65-1,68 (м, 4H), 1,45-1,51 (м, 2H), 1,39 (с, 9H), 1,36 (с, 9H).
М.-с. (FD) m/e 621 (M+).
Пример 5
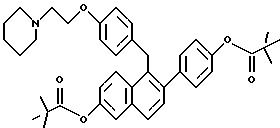
[2-(4-н-Бутилсульфонилоксифенил)-6-н- бутилсульфонилоксинафталин-1-ил] [4-[2-(1-пиперидинил)этокси] фенил]метан

К суспензии продукта примера 2 (0,250 г, 0,510 ммоль) в ТГФ (25 мл) при перемешивании последовательно добавляют N, N- диметиламинопиридин (2 мг), триэтиламин (0, 78 мл, 5,6 ммоль) и бутансульфонилхлорид (0,26 мл, 2,04 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов, затем выливают в этилацетат с водой (100 мл, 1:1, о/о) и органический слой отделяют. Водную часть экстрагируют этилацетатом (50 мл) и объединенные органические слои промывают насыщенным водным раствором хлорида аммония (1х25 мл) и затем насыщенным водным раствором бикарбоната натрия (2х25 мл) и солевым раствором (1х25 мл). Очистка радиальной хроматографией (силикагель, 2 мм, этилацетат: гексан:триэтиламин:метанол=10:8:1:1) дает 0,289 г названного в заголовке соединения (85%) в виде густого сиропа.
ИК (хлороформ) 3032, 2966, 2940, 2879, 1609, 1510, 1375, 1245, 1171, 1149, 1129, 870, 839 см-1.
1H-ЯМР (300 МГц, CDCl3) δ 7,92-7,95 (д, 1H, J=9,3 Гц), 7,81-7,84 (д, 1H, J= 8,6 Гц), 7,77-7,78 (д, 1H, J=2,5 Гц), 7,46-7,49 (д, 1H, J=8,4 Гц), 7,24-7,34 (м, 5H), 6,84-6,87 (д, 2H, J=8,6 Гц), 6,74-6,77 (д, 2H, J=8,6 Гц), 4,33 (с, 2H), 4,05-4,09 (м, 2H), 3,25-3,32 (м, 4H), 2,76-2,81 (м, 2H), 2,48-2,52 (м, 4H), 1,93-2,06 (м, 4H), 1,44-1,61 (м, 10H), 0,96-1,01 (м, 3H). М.-с. (FD) m/e 694 (M+).
Пример 6
[2-(4-н-Гексилсульфонилоксифенил)-6-н- гексилсульфонилоксинафталин-1-ил] [4-[2-(1-пиперидинил)этокси]фенил]метан
К раствору продукта примера 2 (0,49 г, 1,00 ммоль) в ТГФ (200 мл) при перемешивании при температуре окружающей среды последовательно добавляют N, N-диметилформамид (10 мг), триэтиламин (0,50 мл, 5 ммоль) и гексилсульфонилхлорид (0,46 мл, 2,5 ммоль). Через 18 часов реакционную смесь концентрируют и полученное темное масло обрабатывают этилацетатом и насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяют, сушат (сульфат натрия) и концентрируют. Сырое вещество растворяют в этилацетате и добавляют раствор хлористого водорода в эфире (10 мл насыщенного раствора). Образующийся осадок обрабатывают Et2O и сушат, получают 1,2 г целевого продукта в виде густого, смолистого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) соответствует строению. М.-с. (FD) m/e 938 (M+-хлористоводородная кислота).
Препаративный пример 4
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил](4-гидроксифенил) метанон

К раствору литийэтантиола [полученного добавлением н-BuLi (63,7 мл, 1,6 М раствор в гексане, 101,4 ммоль) к раствору этантиола (101,4 ммоль) при перемешивании при 0oC в Et2O (400 мл) с последующим концентрированием] в диметилформамиде (400 мл) при перемешивании добавляют (3,4-дигидро-6-метокси-2-фенил-1- нафталинил) (4-метоксифенил) метанон (30,0 г, 78,0 ммоль), полученный так, как описано в Jones, et al., J. Med. Chem., 53: 931-938 (1992), см. выше. Затем смесь нагревают до 85oC. Через 0,5 часа смесь концентрируют и полученное твердое коричневое вещество растворяют в хлороформе. Этот раствор экстрагируют насыщенным водным раствором хлорида аммония, водную часть обрабатывают 1н. соляной кислотой до получения pH 5, а затем экстрагируют хлороформом. Объединенные органические экстракты промывают солевым раствором, сушат (сульфат натрия), фильтруют и концентрируют. Полученное коричневое масло очищают флэш- хроматографией (силикагель, градиент этилацет/гексан) и получают 24,7 г (87%) целевого продукта в виде желтой пены.
1H-ЯМР (300 МГц, CDCl3) δ 7,74 (д, J=8,6 Гц, 2H), 7,15-7,18 (м, 2H), 7,05-7,18 (м, 3H), 6,86 (д, J=8,6 Гц, 1H), 6,78 (д, J=2,7 Гц 1H), 6,60-6,70 (м, 3H), 6,23 (шс, 1H), 3,78 (с, 3H), 2,95-3,05 (м, 2H), 2,75-2,85 (м, 2H).
Элем. анализ. Вычисл.: C 80,87; H 5,66; найдено: C 80,66; H 5,48. М.-с. (FD) m/e 354 (M+).
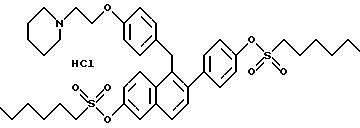
Препаративный пример 5
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил]
[4-[2- (1-пиперидинил)этокси]фенил]метанон
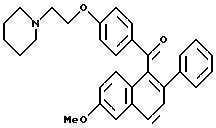
К раствору продукта препаративного примера 4 (20,4 г, 57,0 ммоль) в диметилформамиде (400 мл) при перемешивании при температуре окружающей среды добавляют иодид калия (30 мг, 0,18 ммоль), а затем карбонат калия (39,3 г, 285 ммоль) и моногидрохлорид 1-(2-хлорэтил) пиперидина (11,6 г, 62,7 ммоль). Через 16 часов реакционную смесь концентрируют и полученное масло растворяют в хлороформе. Этот раствор тщательно промывают водой и солевым раствором, сушат (сульфат натрия), фильтруют и концентрируют. Полученное масло очищают флэш-хроматографией (силикагель, градиент метанол/хлороформ) и получают 25,1 г (94%) целевого продукта в виде коричневого масла.
1H-ЯМР (300 МГц, CDCl3), δ 7,79 (д, J=8,7 Гц, 2H), 7,20-7,33 (м, 2H), 7,04-7,20 (м, 3H), 6,88 (д, J=8,5 Гц, 1H), 6,70-6,82 (м, 3H), 6,62 (м, 1H), 4,
08 (т, J=6,0 Гц, 2H), 3,70 (с, 3H), 3,03 (т, J=7,5 Гц, 2H), 2,70-2,90 (м, 4H), 2,40-2,60 (м, 4H), 1,55-1,65 (м, 4H), 1,40-1,52 (м, 2H);
13C-ЯМР (75 МГц, CDCl3) δ
198,33, 162,84, 158,97, 141,21, 136,71, 135,97, 137,78, 131,79, 130,44, 128,08, 127,48, 127,24, 126,59, 126,49, 114,17, 113,80, 111,37, 66,15, 57,68, 55,23, 55,05, 29,73, 28,80, 25,89, 24,12.
Элем. анализ. Вычисл. : C 79,63; H 7,11; N 2,99; найдено: C 79,92; H 7,15; N 3,07. M.-c. (FD) m/e 467 (M+).
Препаративный пример 6
[3,
4-Дигидро-2-фенил-6-метоксинафталин-1-ил] [4-[2- (1-пирролидинил)этокси]фенил]метанон

Взаимодействие продукта препаративного примера 4 (1,9 г, 5,3 ммоль), моногидрохлорида 1-(2-хлорэтил)пирролидина (0,99 г, 5,8 ммоль) и карбоната калия (3,65 г, 29,1 ммоль) в диметилформамиде (50 мл) в соответствии с методикой препаративного примера 5 дает названное в заголовке соединение в виде густого масла с выходом 81%.
1H-ЯМР (300 МГц, CDCl3) δ 7,79 (д, J=7,8 Гц, 2H), 7,20-7,30 (м, 2H), 7,05-7,20 (м, 3H), 6,87 (д, J=8,6 Гц, 1H), 6,73-6,84 (м, 3H), 6,60 (д, J=8,6 Гц, 1H), 4,08 (т, J= 5,8 Гц, 2H), 3,78 (с, 3H), 3,00 (т, J=8,0 Гц, 2H), 2,76-2,96 (м, 4H), 2,50-2,70 (м, 4H), 1,75-1,85 (м, 4H). М.-с. (FD) m/e 453 (M+).
Пример 7
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил]
[4-[2-(1-пиперидинил)этокси]фенил]метанол
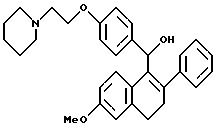
К суспензии алюмогидрида лития (1,60 г, 42,8 ммоль) в сухом ТГФ (200 мл) при перемешивании при 0oC добавляют по каплям в течение 5 минут раствор продукта препаративного примера 5 (10,0 г, 21,4 ммоль) в ТГФ (125 мл). Реакционной смеси позволяют нагреваться до температуры окружающей среды и затем перемешивают в течение 1 часа. Затем раствор охлаждают до 0oC и осторожно гасят водой (1,6 мл). К этому раствору добавляют по каплям гидроксид натрия (4,8 мл, 15% (м/м) водный раствор) и затем добавляют воду (1,6 мл). После перемешивания в течение 30 минут смесь фильтруют и твердое вещество тщательно промывают ТГФ. Затем концентрируют фильтрат и получают 8,7 г (87%) целевого продукта в виде желтого масла, которое используют без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3) δ 7,20-7,45 (м, 7H), 6,82 (д, J=8,3 Гц, 2H), 6,71 (с, 1H), 6,53 (м, 1H), 5,83 (шс, 1H), 4,07 (т, J=6,1 Гц, 2H), 3,75 (с, 3H), 2,91 (т, J=6,1 Гц, 2H), 2,60-2,80 (м, 4H), 2,40-2,60 (м, 4H), 1, 80-1,95 (м, 2H), 1,52-1,70 (м, 4H), 1,43 (с, 1H). M.-c. (FD) m/e 469 (M+).
Пример 8
[3,4-Дигидро-2-фенил-6-метоксинафталин-1-ил]
[4-[2- (1-пирролидинил)этокси]фенил]метанол

Взаимодействие продукта препаративного примера 4 (1,8 г, 4,0 ммоль), алюмогидрида лития (0,31 г, 8,0 ммоль) в ТГФ (65 мл) в соответствии с методом получения продукта примера 7 дает названное в заголовке соединение в виде белой пены с выходом 87%.
1H-ЯМР (300 МГц, CDCl3) δ 7,20-7,40 (м, 7H), 6,84 (д, J=8,6 Гц, 2H), 6,71 (с, 1H), 6,51 (м, 1H), 5,83 (д, J=4,9 Гц, 1H), 4,07 (т, J=6,3 Гц, 2H), 3,75 (с, 3H), 2,82-2,95 (м, 4H), 2,55-2,73 (м, 6H), 2,27 (д, J=3,8 Гц, 1H), 1,70-1,90 (м, 4H), 1,67 (с, 1H). М.-с. (FD) m/e 455 (M+). М.-с.ВР FAB+ (м. -с. высокого разрешения при бомбардировке быстрыми атомами): для C30H33NO3 вычислено 456,2539, найдено 456,2531.
Пример 9
Гидрохлорид [2-фенил-6-метоксинафталин-1-ил]
[4-[2-(1-пиперидинил)этокси]фенил]метана
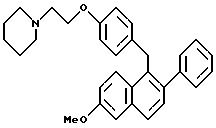
К раствору продукта примера 7 (8,7 г, 18,5 ммоль) в этилацетате (100 мл) при перемешивании добавляют насыщенный раствор хлористого водорода в этилацетате (250 мл). Через 0,5 минуты полученный раствор концентрируют и получают 8,0 г (89%) целевого продукта в виде белой пены, который используют без дополнительной очистки.
1H-ЯМР (300 МГц, ДМСО δ 7,70-7,85 (м, 4H), 7,30-7,50 (м, 7H), 7,10 (с, 1H), 6,80-7,00 (м, 2H), 4,25-4,40 (м, 4H), 4,00-4,20 (шс, 3H), 3,35-3,55 (м, 4H), 2,85-3,55 (м, 2H), 1,70- 1,90 (м, 4H), 1,30-1,45 (м, 2H).
Элем. анализ. Вычислено: C 76,29; H 7,02; N 2,87; найдено: C 76,56; H 7,18; H 2,91. М.-с. (FD) m/e 452 (M+ - хлористоводородная кислота).
Пример 10
Гидрохлорид [2-фенил-6-метоксинафталин-1-ил]
[4-[2-(1-пирролидинил)этокси]фенил]метана
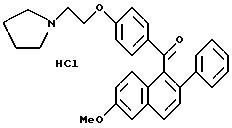
Взаимодействие (продукта препаративного примера 8) (1,57 г, 3,4 ммоль) с раствором хлористого водорода в этилацетате в соответствии с методикой примера 9 дает названное в заголовке соединение с количественным выходом.
1H-ЯМР (300 МГц, ДМСО)

М. -с. (FD) m/e 437 (M+ -хлористоводородная кислота). Элем. анализ. Вычислено: C 76,01; H 6, 80; N 2,95; найдено: C 75,71; H 6,35; N 2,82.
Пример 11
[2-Фенил-6-гидроксинафталин-1-ил] [4-[2-(1-пиперидинил) этокси]фенил]метан(ол)

К раствору продукта примера 9 (4,0 г, 8,0 ммоль) в 1,2- дихлорэтане (50 мл) при перемешивании при 0oC добавляют трихлорид бора (10 мл, 117,0 ммоль). Полученный темно-фиолетовый раствор перемешивают при комнатной температуре в течение ночи в плотно закрытой пробирке, и затем охлаждают до 0oC. Осторожно, по каплям, в течение 30 минут, добавляют метанол (50 мл) (осторожно, выделяется газ). Полученный раствор концентрируют и остаток растворяют в этилацетате. Органический экстракт промывают насыщенным водным раствором бикарбоната натрия и солевым раствором, сушат (сульфат натрия), фильтруют и концентрируют. Полученную коричневую пену очищают флэш-хроматографией (силикагель, градиент метанол/хлороформ) и получают 2,7 г (63%) целевого продукта в виде белой пены.
1H-ЯМР (300 МГц, ДМСО) δ 9,72 (шс, 1H), 7,62-7,80 (м, 2H), 7,22-7,50 (м, 6H), 7,10-7,22 (м, 2H), 7,00 (м, 1H), 6,80-6,90 (м, 2H), 6,78 (м, 1H), 4,23 (с, 2H), 3,85-4,10 (м, 2H), 2,50-2,75 (м, 2H), 2,25-2,50 (м, 4H), 1,25-1,56 (м, 6H).
Элем. анализ. Вычислено: C 82,35; H 7, 14; N 3,20; найдено: C 82,17; H 7,11; N 3,35. М.-с. (FD) m/e 437 (M+).
ИК (KBr) 2935,07, 2855,01, 1621,38, 1597,26 см-1.
Пример 12
[2-Фенил-6-гидроксинафталин-1-ил] [4-[2-(1-пирролидинил) этокси] фенил] метанол

Взаимодействие продукта примера 10 (1,27 г, 2,7 ммоль) с трихлоридом бора (10 мл, 117 ммоль) в 1,2-дихлорэтане (30 мл) в соответствии с методикой примера II дает нужный продукт в виде белого твердого вещества с выходом 32%.
ИК (KBr) 2932,17, 2876,23, 2815,47, 1620,41, 1597,26 см-1.
1H-ЯМР (300 МГц, CDCl3) δ 7,74 (д, J=8,5 Гц, 1H), 7,61 (д, J=8,5 Гц, 1H), 7,20-7,40 (м, 7H), 7,13 (с, 1H), 7,00 (м, 1H), 6,85 (д, J= 8,3 Гц, 2H), 6,66 (д, J=8,3 Гц, 2H), 4,31 (с, 2H), 4,06 (т, J=5,8 Гц, 2H), 2,95 (т, J=5,8 Гц, 2H), 2,65-2,80 (м, 4H), 1,77-1,90 (м, 4H).
М.-с. (FD) m/e 424 (M+).
Элем. анализ. Вычислено: C 82,24; H 6,90; N 3,31; найдено: C 82,01; H 6,84; N 3,37.
Пример 13
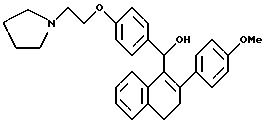
[3,4-Дигидро-2-(4-метоксифенил)нафталин-2-ил] [4-[2-(1-пиперидинил)этокси]фенил]метанол
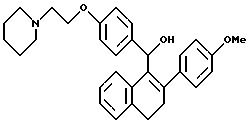
К суспензии мезилата [2-(4-метокcифенил)-3,4-дигидронафт 1-ил][4-[2-(1-пиперидинил)этокси] фенил] метанона [Jones et al., J. Med. Chem., 35:931 (1992), см. выше] (2,00 г, 3,35 ммоль) в ТГФ (100 мл) при перемешивании при температуре окружающей среды постепенно, в течение 20 минут, добавляют алюмогидрид лития (1,0 г, 26 ммоль). Через 18 часов раствор концентрируют почти досуха и затем осторожно добавляют воду (50 мл). Получающуюся в результате смесь экстрагируют этилацетатом (3х100 мл). Объединенные органические экстракты промывают водой, сушат (сульфат натрия) и концентрируют. Очистка жидкостной хроматографией (Waters Prep 500, силикагель, градиент от хлороформа до 25% метанола в хлороформе) дает 1,0 г целевого продукта в виде желтовато-коричневого аморфного порошка.
1H-ЯМР (300 МГц, CDCl3) соответствует строению.
М.-с. (FD) m/e 469 (M+).
Пример 14
[3,
4-Дигидро-2-(4-метоксифенил)нафталин-2-ил] [4-[2-(1-пирролидинил)этокси]фенил]метанол
Взаимодействие мезилата [2-(4-метоксифенил)-3,4-дигидронафт- 1-ил][4-[2-(1-пирролидинил)этокси] фенил]-метанона [Jones et al., J. Med. Chem., 15: 931 (1992), см. выше] (0,85 г, 1,9 ммоль) и алюмогидрида лития (0,16 г, 4,0 ммоль) в ТГФ (150 мл) в соответствии с методикой эксперимента в примере 13 дает 670 мг целевого соединения в виде желтовато-коричневого твердого вещества.
1H-ЯМР (CDCl3, 300 МГц) соответствует строению.
М.-с. (FD) m/e 455(M+).
Элем. анализ. Вычислено: C 79,20; H 7,26; N 3,08; найдено: C 79,11; H 7,47; N 2,93.
Пример 15
Гидрохлорид [2-(4-метоксифенил)нафталин-1-ил] [4-[2-(1-пиперидинил)этокси]фенил]метана

К раствору продукта примера 13 (1,90 г, 4,21 ммоль) в метаноле (40 мл) при перемешивании при температуре окружающей среды добавляют раствор хлористого водорода в метаноле (10 мл насыщенного раствора). Через 48 часов реакционную смесь концентрируют и сушат. Обработка эфиром с последующей фильтрацией и высушиванием дает 580 мг целевого соединения в виде белого порошка.
1H-ЯМР (CDCl3, 300 МГц) соответствует строению.
М.-с. (FD) m/e 451 (M+-хлористоводородная кислота).
Пример 16
Гидрохлорид [2-(4-метоксифенил)нафталин-1-ил] [4- [2-(1-пирролидинил)этокси]фенил]метана

К раствору продукта примера 14 (2,0 г, 4,58 ммоль) в метаноле (50 мл) при перемешивании при температуре окружающей среды добавляют раствор хлористого водорода в метаноле (10 мл насыщенного раствора). Реакционную смесь затем концентрируют до 20 мл и охлаждают при -20oC в течение нескольких часов. Фильтрация дает 0,62 г целевого продукта в виде белого порошка.
1H-ЯМР (CDCl3, 300 МГц) соответствует строению.
М.-с. (FD) m/e 437 (M+-хлористоводородная кислота).
Элем. анализ. Вычислено: C 76,01; H 6,80; N 2,96; найдено: C 75,95; H 6,76; N 2,98.
Препаративный пример 7
[3,
4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-пирролидинил)этокси]фенил]метанон

К раствору продукта препаративного примера 2 (2,0 г, 5,2 ммоль) в диметилформамиде (50 мл) при перемешивании добавляют карбонат калия (3,6 г, 26 ммоль) и моногидрохлорид 1-(2- хлорэтил)пирролидина (0,8 г, 5,7 ммоль). Реакционную смесь перемешивают в течение ночи при температуре окружающей среды и концентрируют. Полученное масло растворяют в хлороформе и полученный раствор тщательно промывают водой и солевым раствором, сушат (сульфат натрия), фильтруют и концентрируют. Полученное масло очищают флэш-хроматографией (силикагель, градиент метанол/хлороформ) и получают 2,25 г (90%) целевого продукта в виде коричневого масла.
1H-ЯМР (300 МГц, CDCl3) δ 7,80 (д, J=9,4 Гц, 2H), 7,18 (д, J= 6,8 Гц, 2H), 6,87 (д, J=8,6 Гц, 2H), 6,65-6,85 (м, 4H), 6,60 (м, 1H), 4,09 (т, J=5,8 Гц, 2H), 3,78 (с, 3H), 3,71 (с, 3H), 3,01 (т, J=7,5 Гц, 2H), 2,88 (т, J=5,8 Гц, 2H), 2,65-2,85 (м, 2H), 2,60-2,75 (м, 4H), 1,80-1,90 (м, 4H). М.-с. (FD) m/e 483 (M+).
Пример 17
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-пирролидинил)этокси]фенил]метанол

К суспензии алюмогидрида лития (0,34 г, 8,80 ммоль) в ТГФ (40 мл) при перемешивании при 0oC постепенно, в течение 5 минут, добавляют раствор продукта препаративного примера 7 (2,14 г, 4,4 ммоль) в ТГФ (25 мл). Реакционную смесь нагревают до температуры окружающей среды. Через 1 час смесь охлаждают до 0oC и осторожно добавляют воду (0,4 мл). К этому раствору добавляют по каплям гидроксид натрия (1,2 мл, 155 (м/м) водный раствор), а затем воду (0,4 мл). После перемешивания в течение 30 минут смесь фильтруют и твердое вещество тщательно промывают ТГФ. Фильтрат концентрируют и получают 1,60 г (75%) целевого продукта в виде белой пены, который используют без дополнительной очистки.
1H-ЯМР (300 МГц, ДМСО) δ 7,40 (д, J=8,2 Гц, 2H), 7,33 (д, J= 7,6 Гц, 1H), 7,16 (д, J=8,l Гц, 2H), 6,90 (д, J=7,7 Гц, 2H), 6,75 (д, J=7,8 Гц, 2H), 6,66 (с, 1H), 6,45 (д, J=7,6 Гц, 1H), 5,69 (с, 1H), 5,64 (с, 1H), 3,95 (т, J= 5,5 Гц, 2Н), 3,72 (с, 3Н), 3,64 (с, 3Н), 2,65-2,85 (м, 4Н), 2,40-2,65 (м, 6Н), 1,60-1,80 (м, 4Н).
М.-с. (FD) m/e 485 (M+).
Пример 18
Гидрохлорид [2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-пирролидинил)этокси]фенил]метана

К раствору продукта примера 17 (1,61 г, 3,30 ммоль) в этилацетате (50 мл) при перемешивании при температуре окружающей среды добавляют насыщенный раствор хлористого водорода в этилацетате (50 мл). Полученную смесь концентрируют и получают 1,66 г (100%) целевого продукта в виде белой пены, который используют без дополнительной очистки.
1H-ЯРМ (300 МГц, ДМСО) δ 7,70-7,80 (м, 2Н), 7,30-7,40 (м, 2Н), 7,20-7,30 (м, 2Н), 7,05 (м, 1Н), 6,80-7,00 (м, 6Н), 4,29 (с, 2Н), 4,20-4,25 (м, 2Н), 3,84 (с, 3Н), 3,75 (с, 3Н), 3,42-3,75 (м, 4Н), 3,00-3,15 (м, 2Н), 1,80-2,00 (м, 4Н).
М.-с. (FD) m/e 467 (M+-хлористоводородная кислота).
Пример 19
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1-пирролидинил)этокси]фенил]метан

К раствору продукта примера 18 (1,61 г, 2,60 ммоль) в 1,2- дихлорэтане (30 мл) при перемешивании при 0oC добавляют трихлорид бора (10 мл, 117 ммоль). Полученный темно-фиолетовый раствор перемешивают в течение ночи при температуре окружающей среды в плотно закрытой пробирке. После охлаждения раствора до 0oC осторожно, в течение 30 минут, добавляют метанол (25 мл) (осторожно, выделяется газ). Затем раствор концентрируют и полученное вещество растворяют в 30% изопропанола в хлороформе, затем промывают насыщенным раствором бикарбоната натрия, солевым раствором, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Сырое вещество очищают радиальной хроматографией (градиент метанол/хлороформ) и получают 0,34 г (27%) целевого продукта в виде белой пены.
1H-ЯМР (300 МГц, ДМСО-d6) δ 9,45 (с, 1H), 9,36 (с, 1H), 7,72 (д, J=8,8 Гц, 1H), 7,62 (д, J=9,2 Гц, 1H), 7,28 (д, J=8,7 Гц, 1H), 7,00-7,10 (м, 2H), 6,80-6,90 (м, 2H), 6,70-6,80 (м, 4H), 5,45 (с, 1H), 4,84 (с, 1H), 4,25 (с, 2H), 3,90-4,05 (м, 2H), 2,75- 2,90 (м, 2H), 2,50-2,65 (м, 4H), 1,60-1,80 (м, 4H);13C-ЯМР (75 МГц, ДМСО-d6) δ 203,32 191,97, 188,16, 186,14, 135,95, 177,43, 173,46, 169,60, 167,74, 163,48, 162,30, 159,87, 158,14, 154,98, 152,43, 60,50, 56,25, 54,00, 45,05, 41,00, 37,50, 35,00, 30,05, 27,50, 26,00, 22,50, 20, 00.
Элем. анализ. Вычисл. : C 79,24; H 6,65; N 3,19; найдено: C 78,99; H 6,51; N 2,92.
М.-с. (FD) m/e 440 (M+).
ИК (KBr) 3382,61, 2964,00, 1610,77, 1509,49 см-1.
Препаративный пример 8
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(N,N-диметилaминo)этокси]фенил]метанон
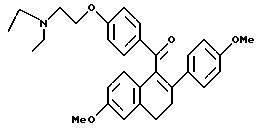
Взаимодействие продукта препаративного примера 2 (1,6 г, 4,1 ммоль), гидрохлорида 2-диэтиламиноэтилхлорида (0,8 г, 4,5 ммоль) и карбоната калия (2,3 г, 16,4 ммоль) в диметилформамиде (50 мл) в соответствии с методикой получения соединения в препаративном примере 3 дает нужный продукт с выходом 95%.
1H-ЯМР (300 МГц, CDCl3)

Элем. анализ. Вычисл. : C 76,67; H 7,26; N 2,88; найдено: C 76,97; H 7,43; N 2,91.
Препаративный пример 9
[3,
4-Дигидро-2-(4-метоксифенил)-2,4-дигидро-6- метоксинафталин-1-ил] [4-[3-(1-пиперидинил)пропокси]-фенил]метанон

Взаимодействие продукта препаративного примера 2 (1,6 г, 4,1 ммоль), гидрохлорида 1-(3-хлорпропил) пиперидина (0,9 г, 4,5 ммоль) и карбоната калия (2,3 г, 16,4 ммоль) в ДМФА (50 мл) в соответствии с методикой получения соединения в препаративном примере 7 дает целевой продукт с выходом 95%.
1H-ЯМР (300 МГц, CDCl3) δ 7,80 (д, J=8,7 Гц, 2H), 7,19 (д, J= 5,0 Гц, 2H), 6,86 (д, J=8,4 Гц, 1H), 6,63-6,80 (м, 5H), 6,60 (м, 1H), 3,98 (т, J=6,4 Гц, 2H), 3,78 (с, 3H), 3,70 (с, 3H), 3,00 (т, J=7,7 Гц, 2H), 2,75-2,85 (м, 2H), 2,30-2,50 (м, 6H), 1,90-2,00 (м, 2H), 1,50-1,65 (м, 2H), 1,40-1,50 (м, 2H). М.-с. (FD) m/e 511 (M+).
Элем. анализ. Вычисл. : C 77,47; H 7,29; N 2,74; найдено: C 77, 42; H 7,36; N 2,72.
Пример 20
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(N, N-диэтиламино)этокси]фенил]метанол
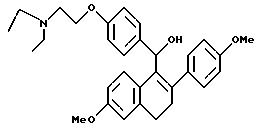
Взаимодействие продукта препаративного примера 8 (1,7 г, 3,4 ммоль) с алюмогидридом лития (0,3 г, 6,8 ммоль) в ТГФ (80 мл) в соответствии с методикой примера 17 дает целевой продукт с количественным выходом.
1H-ЯМР (300 МГц, CDCl3) δ 7,33 (д, J=8,5 Гц, 2H), 7,20-7,30 (м, 3H), 6, 80-6,90 (м, 4H), 6,71 (с, 1H), 6,50 (м, 1H), 5,85 (д, J=3,9 Гц, 1H), 4,01 (т, J=6,4 Гц, 2H), 3,78 (с, 3H), 3,74 (с, 3H), 2,86 (ABk, J=8,2 Гц, Dn=14,7 Гц, 4H), 2,60-2,70 (м, 6H), 1,85 (м, 1H), 1,05 (т, J=7,2 Гц, 6H). М.-с. (FD) m/e 487 (M+).
Пример 21
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[3-(1-пиперидинил)пропокси]фенил]метанол

Взаимодействие продукта препаративного примера 9 (1,77 г, 3,50 ммоль) с алюмогидридом лития (0,27 г, 7,00 ммоль) в ТГФ (50 мл) в соответствии с методикой примера 17 дает целевой продукт с выходом 97%.
1H-ЯМР (300 МГц, CDCl3) δ 7,32 (д, J=8,4 Гц, 2H), 7,20- 7,30 (м, 4H), 6,80-6,90 (м, 3H), 6,70 (с, 1H), 6,50 (м, 1H), 5,85 (с, 1H), 3,96 (т, J=6,3 Гц, 2H), 3,78 (с, 3H), 3,74 (с, 3H), 2,85- 2,95 (м, 2H), 2,60-2,70 (м, 2H), 2,25-2,50 (м, 6H), 1,90-2,00 (м, 1H), 1, 54-1,60 (м, 4H), 1,43 (с, 2H). М.-с. (FD) m/e 514 (М+).
Пример 22
Гидрохлорид [2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(N,
N-диэтиламино)этокси]фенил]метана
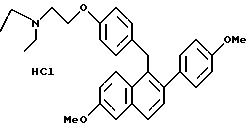
Взаимодействие продукта примера 20 (1,6 г, 3,3 ммоль) с хлористоводородной кислотой (100 мл насыщенного раствора хлористого водорода в этилацетате) в этилацетате (100 мл) в соответствии с методикой примера 18 дает целевой продукт с выходом 90%.
ИК (KBr) 3416,37, 2935,07, 2835,72, 2575,30, 2437,37, 1624,27, 1608,84, 1510,45 см-1.
1H-ЯМР (300 МГц, CDCl3) δ 7,72 (т, J=8,6 Гц, 2H), 7,15-7,30 (м, 4H), 7,05 (м, 1H), 6,85-6,95 (м, 3H), 6,72 (д, J=8,6 Гц, 2H), 4,40-4,50 (м, 2H), 4,35 (с, 3H), 3,92 (с, 3H), 3,83 (с, 3H), 3,35-3,45 (м, 2H), 3,20-3,35 (м, 4H), 1,43 (т, J=7,2 Гц, 6H).
М.-с. (FD) m/e 470 (M+-хлористоводородная кислота).
Элем. анализ. Вычисл. : C 73,57; H 7,17; N 2,77; найдено: C 73,80; H 7,35; N 2,77.
Пример 23
Гидрохлорид [2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[3-(1-пиперидинил)пропокси]фенил]метана
Взаимодействие продукта примера 21 (1,5 г, 2,9 ммоль) с хлористоводородной кислотой (50 мл насыщенного раствора хлористого водорода в этилацетате) в этилацетате (50 мл) в соответствии с методикой примера 18 дает целевой продукт с выходом 97%.
1H-ЯМР (300 МГц, CDCl3) δ 7,70-7,80 (м, 2H), 7,42 (д, J=8,4 Гц, 1H), 7, 15-7,30 (м, 3H), 7,05 (м, 1H), 6,85-6,95 (м, 4H), 6,69 (д, J=8,6 Гц, 2H), 4,34 (с, 2H), 3,97-4,03 (м, 2H), 3,92 (с, 3H), 3,82 (с, 3H), 3,50-3,60 (м, 2H), 3,05-3,20 (м, 2H), 2,57-2,70 (м, 2H), 2,20-2, 50 (м, 4H), 1,80-2,00 (м, 4H).
М.-с. (FD) m/e 495 (М+-хлористоводородная кислота).
Элем. анализ. Вычисл. : C 74,49; H 7,20; N 2,63; найдено: C 74,74; H 7, 36; N 2,75.
Пример 24
[2- (4-Гидpoкcифенил)-6-гидроксинафталин-1-ил][4-[2-(1-N, N-диэтиламино)этокси]фенил]метан
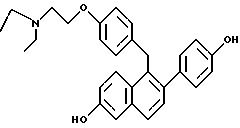
Взаимодействие продукта примера 22 (1,32 г, 2,60 ммоль) с трихлоридом бора (10,0 мл, 117,0 ммоль) в 1,2-дихлорэтане (30 мл) в соответствии с методикой примера 19 дает целевой продукт в виде белого порошка с выходом 76%.
ИК (KBr) 3356,57, 2973,65, 1734,23, 1704,33, 1610,77, 1509,49 см-1.
1H-ЯМР (300 МГц, ДМСО-d6) δ 9,62 (с, 1H), 9,43 (с, 1H), 7,56-7,70 (м, 2H), 7,24 (д, J=8,4 Гц, 1H), 7,00-7,15 (м, 3H), 6,95 (м, 1H), 6,82 (д, J=8,6 Гц, 2H), 6,65-6,78 (м, 4H), 4, 23 (c, 2H), 4,00 (т, J=6,4 Гц, 2H), 2,65-2,75 (М, 2H), 2,40-2,60 (м, 4H), 0,90 (т, J=7,1 Гц, 6H);13C-ЯМР (75 МГц, ДМСО-d6) δ 156,53, 156,45, 154,87, 136,65, 134,44, 133, 49, 132,66, 132,28, 130,14, 128,90, 128,73, 126,93, 126,57, 125,18, 118,73, 115,01, 114,32, 109,43, 66,22, 51,43, 47,00, 39,00, 33,81, 11,87.
M. -c. (FD) m/e 442 (M+). М. -с. ВР (FAB+) для C29H31NO3 вычислено 442,2382, найдено 442,2381.
Препаративный пример 10
[3,
4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-бром] этокси]фенил]метанон

К раствору продукта препаративного примера 2 (4,00 г, 10,0 ммоль) в 2-бутаноне (100 мл) при перемешивании при температуре окружающей среды добавляют карбонат калия (2,76 г, 20,0 ммоль) и 1,2-дибромэтан (17,2 мл, 100 ммоль). Этот раствор кипятят с обратным холодильником в течение ночи, затем фильтруют и концентрируют. Полученное коричневое масло очищают флэш- хроматографией (силикагель, 20% этилацетата в гексане) и получают 4,40 г (89%) целевого продукта в виде коричневого масла.
1H-ЯМР (300 МГц, CDCl3) δ 7,81 (д, J=8,7 Гц, 2H), 7,18 (д, J=8,7 Гц, 2H), 6,86 (д, J=8,6 Гц, 1H), 6,76 (д, J=8,7 Гц, 3H), 6,78 (д, J=8,6 Гц, 2H), 6,60 (м, 1H), 4,26 (т, J=6,1 Гц, 2H), 3,78 (с, 3H), 3,70 (с, 3H), 3,60 (т, J=6,4 Гц, 2H), 3,01 (т, J=7,7 Гц, 2H), 2, 75-2,85 (м, 2H).
Элем. анализ. Вычислено: C 65,73; H 5,11; найдено: C 65,96; H 5,28.
Препаративный пример 11
[3,
4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-гексаметилениминил)этокси]фенил]метанон

К раствору продукта препаративного примера 10 (2,1 г, 4,3 ммоль) в диметилформамиде (50 мл) при перемешивании при температуре окружающей среды добавляют карбонат калия (1,8 г, 13 ммоль) и гексаметиленимин (0,9 мл, 13 ммоль). Затем раствор нагревают до 100oC. После перемешивания в течение ночи смесь концентрируют и полученное коричневое масло обрабатывают хлороформом и водой. Органический экстракт промывают солевым раствором, сушат (сульфат натрия), фильтруют и концентрируют. Полученное желтое масло очищают радиальной хроматографией (градиент, этилацет/гексан/метанол) и получают 0,95 г (43%) целевого продукта в виде желтого масла.
1H-ЯМР (300 МГц, CDCl3)
Элем. анализ. Вычислено: C 77,47; H 7,29; N 2,74 найдено; С 77,25; H 7,16; N 2,71. М.-с. (FD) m/e 511 (M+).
Пример 25
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1- гексаметиленимин)этокси]фенил]метанол

К суспензии алюмогидрида лития (0,3 г, 7,2 ммоль) в ТГФ (40 мл) при перемешивании при 0oC постепенно, в течение 5 минут, добавляют раствор продукта препаративного примера 11 (1,8 г, 3,6 ммоль) в ТГФ (25 мл). Реакционной смеси дают нагреться до температуры окружающей среды. Через 1 час смесь охлаждают до 0oC и осторожно добавляют воду (0,4 мл). К этому раствору постепенно добавляют гидроксид натрия (1,2 мл, 15% водный раствор), и затем добавляют воду (0,4 мл). После перемешивания в течение 30 минут смесь фильтруют и твердое вещество тщательно промывают ТГФ. Фильтрат концентрируют и получают 1,71 г (93%) целевого продукта в виде белой пены, который используют без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3) δ 7,34 (д, J=8,5 Гц, 2H), 7,20- 7,30 (м, 3H), 6,80-6,90 (м, 4H), 6,73 (с, 1H), 6,55 (м, 1H), 5,88 (с, 1H), 4,06 (т, J=6,3 Гц, 2H), 3,81 (с, 3H), 3,76 (с, 3H), 2,85-3,00 (м, 4H), 2,75-2,85 (м, 4H), 2, 63-2,75 (м, 2H), 2,95 (м, 1H), 1,60-1,75 (м, 8H).
Элем. анализ. Вычислено: C 77,16; H 7,65; N 2,73 найдено: C 77,33; H 7,79; N 2,71. М.-с. (FD) m/e 513 (M+).
Пример 26
Хлористоводородная соль [2-(4-метoкcифенил)-6-метокcи- нафталин-1-ил] [4-[2-(1-гексаметилениминил)этокси]-фенил]метана

К раствору продукта примера 25 (1,7 г, 3,3 ммоль) в этилацетате (100 мл) при перемешивании при температуре окружающей среды добавляют хлористоводородную кислоту (100 мл насыщенного раствора хлористого водорода в этилацетате). Полученную смесь концентрируют и получают 1,66 г (94%) целевого продукта, который используют без очистки.
1H-ЯМР (300 МГц, CDCl3) δ 7,48 (т, J=8,9 Гц, 2H), 7,43 (д, J= 8,6 Гц, 1H), 7,20-7,35 (м, 3H), 7,10 (м, 1H), 6,85-7,00 (м, 4H), 6,75 (д, J=8,6 Гц, 2H), 4,45-4, 60 (м, 2H), 4,37 (с, 2H), 3,94 (с, 3H), 3,85 (с, 3H), 3,55-3,70 (м, 2H), 3,40-3,50 (м, 2H), 3,00-3,20 (м, 2H), 2,10-2,25 (м, 2H), 1,80-2,00 (м, 4H), 1,60-1,80 (М, 2H);13C-ЯМР (75 МГц, ДМСО) δ 155,6, 137,15, 134,29, 134,19, 134,08, 132,29, 130,15, 129,01, 128,79, 127,28, 126,91, 125,95, 124,94, 118,63, 114,61, 113,70, 106,79, 62,42, 55,20, 55,13, 55,10, 54,85, 54,10, 33,77, 30,44, 26,05, 22,72.
Элем. анализ. Вычислено: C 74,49; H 7,20; N 2,63; найдено: C 74,73; H 7,16; N 2,62. M.-c. (FD) m/e 495 (M+ -хлористоводородная кислота). ИК (KBr) 2934,
10, 2862,73, 2835,72, 2448,94, 1624,27, 1608,84, 1511,42 см-1
Пример 27
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1- гексаметилениминил)этокси]фенил]метан

К раствору продукта примера 26 (1,3 г, 2,4 ммоль) в 1,2- дихлорэтане (30 мл) при перемешивании при 0oC добавляют трихлорид бора (10 мл, 117 ммоль). Полученный темно-фиолетовый раствор перемешивают в течение ночи при температуре окружающей среды в плотно закрытой пробирке и затем охлаждают до 0oC. Постепенно, в течение 30 минут, добавляют метанол (25 мл) (осторожно, выделяется газ) и полученный раствор концентрируют. Сырое вещество растворяют в 20%-ном растворе метанола в хлороформе и затем раствор промывают насыщенным раствором бикарбоната натрия и солевым раствором. Органический экстракт сушат (сульфат натрия), фильтруют и концентрируют. Полученную коричневую пену очищают радиальной хроматографией (градиент, этилацетат/триэтиламин/метанол/гексан) и получают желтовато-коричневое твердое вещество. Это вещество растворяют в этилацетате и затем промывают раствор насыщенным раствором бикарбоната натрия. Органический экстракт концентрируют и получают 0,60 г (54%) целевого продукта в виде белой пены.
1H-ЯМР (300 МГц, ДМСО-d6) δ 9,64 (с, 1H), 9,41 (с, 1H), 7,55-7,70 (м, 2H), 7,24 (д, J=8,5 Гц, 1H), 7,00-7,10 (м, 3H), 6,95 (м, 1H), 6,81 (д, J=8,6 Гц, 2H), 6,70-6,78 (м, 4H), 4,23 (с, 2H), 3,91 (т, J=6,0 Гц, 2H), 2,70-2,80 (м, 2H), 2,55-2,70 (м, 4H), 1,40-1,60 (м, 8H).
Элем. анализ. Вычислено: C 79,63; H 7,11; N 2,99; найдено: C 79,35; H 6,87; N 2,75. М.-с. (FD) m/e 468 (M+). ИК (KBr) 3362,35, 2926,39, 2855,98, 1734,23, 1704,33, 1610,77, 1509,49 см-1.
Препаративный пример 12
[3,4-Дигидро-2-(4-метоксифенил)-3,
4-дигидро-6-метокси-нафталин-1-ил] [4-[2-(1-морфолинил)этокси]фенил]метанон

Взаимодействие продукта препаративного примера 10 (2,1 г, 4,3 ммоль), морфолина (1,13 мл, 12,9 ммоль) и карбоната калия (1,78 г, 12,9 ммоль) в ДМФА (50 мл) в соответствии с процедурой препаративного примера 11 дает целевой продукт в виде густого масла с выходом 80%.
1H-ЯМР (300 МГц, CDCl3) δ 7,83 (д, J=8,7 Гц, 2H), 7,60 (м, 1H), 7,20 (д, J=8,8 Гц, 2H), 6,88 (д, J=8,7 Гц, 1H), 6,65-6,80 (м, 5H), 4,05-4,20 (м, 2H), 3,80 (с, 3H), 3,73 (с, 3H), 3,70-3,80 (м, 4H), 2,90 (т, J=7,9 Гц, 2H), 2,75-2,85 (м, 4H), 2,50-2,60 (м, 4H).
М.-с. (FD) m/e 499 (M+).
Элем. анализ. Вычислено: C 74,53; H 6,66; N 2,80; найдено: C 74,75; H 6,58; N 2,83.
Препаративный пример 13
[3,
4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-(3,3-диметил)пирролидинил)этокси]фенил]метанон
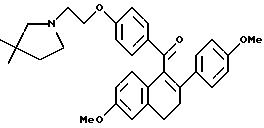
Взаимодействие продукта препаративного примера 10 (2,1 г, 4,3 ммоль), 3,3-диметилпирролидина (1,2 г, 12 ммоль) и карбоната калия (1,8 г, 13 ммоль) в ДМФА (100 мл) в соответствии с процедурой препаративного примера 11 дает целевой продукт в виде густого масла с выходом 60%.
1H-ЯМР (300 МГц, CDCl3) δ 7,80 (д, J=8,7 Гц, 2H), 7,18 (д, J= 8,7 Гц, 2H), 6,87 (д, J= 8,6 Гц, 1H), 6,73-6,80 (м, 3H), 6,67 (д, J= 8,6 Гц, 2H), 6,60 (м, 1H), 4,05 (м, 2H), 3,78 (с, 3H), 3,71 (с, 3H), 2,89-3,05 (м, 2H), 2,73-2,86 (м, 4H), 2,64-2,75 (м, 2H), 2,04 (с, 2H), 1,60 (т, J=6,9 Гц, 2H), 1,07 (с, 6H). М.-с. (FD) m/e 511 (M+).
Пример 28
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил]
[4-[2-(1-(морфолинил)пирролидинил)этокси]фенил]метанол

Взаимодействие продукта препаративного примера 12 (1,6 г, 3,2 ммоль) с алюмогидридом лития (0,3 г, 7,2 ммоль) в ТГФ (65 мл) в соответствии с процедурой примера 25 дает целевой продукт в виде белой пены с выходом 98%.
1H-ЯМР (300 МГц, CDCl3) δ 7,39 (д, J=8,7 Гц, 2H), 7,20-7,30 (м, 4H), 6,80-7,00 (м, 3H), 6,73 (м, 1H), 6,55 (м, 1H), 5,86 (д, J=4,2 Гц, 1H), 4,09 (т, J=5,6 Гц, 2H), 3,80 (с, 3H), 3,70-3,80 (м, 4H), 3,76 (с, 3H), 2,85-3,00 (м, 2H), 2,75-2,85 (м, 2H), 2,65 (м, 1H), 2,55-2,65 (м, 4H), 1,05-1,10 (м, 2H). М.-с. (FD) m/e 501 (M+ ).
Элем. анализ. Вычислено: C 74,23; H 7,03; N 2,79; найдено: C 74,51; H 7,18; N 2,79.
Пример 29
[3,4-Дигидро-2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-(3,
3-диметил)пирролидинил)этокси]фенил]метанол

Взаимодействие продукта препаративного примера 13 (1,3 г, 2,5 ммоль) с алюмогидридом лития (0,2 г, 5,0 ммоль) в ТГФ (65 мл) в соответствии с процедурой примера 25 дает целевой продукт в виде белой пены с выходом 98%.
1H-ЯМР (300 МГц, CDCl3) δ 3,33 (д, J=8,6 Гц, 2H), 7,20-7,30 (м, 3H), 6,80-6,90 (м, 4H), 6,70 (м, 1H), 6,52 (м, 1H), 5,85 (с, 1H), 4,04 (т, J=6,1 Гц, 2H), 3,79 (с, 3H), 3,74 (с, 3H), 2,80-2, 95 (м, 4H), 2,60-2,75 (м, 4H), 2,42 (с, 2H), 2,20 (м, 1H), 1,85 (м, 1H), 1,61 (м, 1H), 1,08 (с, 6H). М.-с. (FD) m/e 513 (M+). Элем. анализ. Вычислено: C 77,16; H 7,65; N 2,73; найдено: C 77, 33; H 7,51; N 2,69.
Пример 30
Гидрохлорид [2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-морфолинил)этокси]фенил]метана
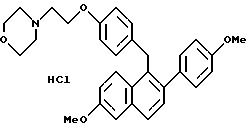
Взаимодействие продукта примера 28 (1,58 г, 3,1 ммоль) с хлористоводородной кислотой (100 мл насыщенного раствора хлористого водорода в этилацетате) в этилацетате (100 мл) в соответствии с процедурой примера 26 дает целевой продукт в виде белой пены с выходом 94%.
1H-ЯМР (300 МГц, CDCl3) δ 7,70-7,85 (м, 2H), 7,44 (д, J=8,4 Гц, 1H), 7,20-7,40 (м, 4H), 6,86-7,15 (м, 4H), 6,70-6,86 (м, 2H), 4,50-4,65 (м, 2H), 4,25-4,50 (м, 4H), 3,83-4,10 (м, 2H), 3,94 (с, 3H), 3,85 (с, 3H), 3,50-3,70 (м, 2H), 3,40-3,50 (м, 2H), 3,00-3,20 (м, 2H).
М.-с. (FD) m/e 483 (M+-хлористоводородная кислота).
Пример 31
Гидрохлорид
[2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-(3,3-диметил)пирролидинил)этокси]фенил]метана
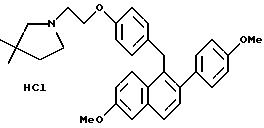
Взаимодействие продукта примера 29 (1,2 г, 2,4 ммоль) с хлористоводородной кислотой (100 мл насыщенного раствора хлористого водорода в этилацетате) в этилацетате (100 мл) в соответствии с процедурой примера 26 дает целевой продукт в виде белой пены с выходом 92%.
1H-ЯМР (300 МГц, CDCl3) δ 7,29 (т, J=9,3 Гц, 2H), 7,41 (д, J= 8,2 Гц, 1H), 7, 15-7,30 (м, 3H), 7,19 (д, J=6,8 Гц, 1H), 6,85-7,00 (м, 4H), 6,73 (д, J=7,52 Гц, 2H), 4,48 (с, 2H), 4,35 (с, 2H), 3,93 (с, 3H), 3,83 (с, 3H), 3,60 (м, 1H), 3,15- 3,50 (м, 1H), 3,15 (м, 1H), 2,76 (м, 1H), 2,05 (м, 1H), 1,85 (м, 1H), 1,75 (м, 1H), 1,33 (с, 3H), 1,22 (с, 3H). М.-с. (FD) m/e 495 (M+ -хлористоводородная кислота).
Элем. анализ. Вычислено: C 74,49; H 7,20; N 2,63; найдено: C 74,70; H 7,18; N 2,47.
Пример 32
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1- морфолинил)этокси]фенил]метан

Взаимодействие продукта примера 30 (1,28 г, 2,40 ммоль) с трихлоридом бора (10 мл, 117 ммоль) в 1,2-дихлорэтане (30 мл) в соответствии с процедурой примера 27 дает целевой продукт в виде белого твердого вещества с выходом 28%.
ИК (KBr) 3317,99, 2927,35, 2868,51, 1610,77, 1509,49 см-1
1H-ЯМР (300 МГц, CDCl3) δ 7,75 д, J=9,3 Гц, 1H), 7,55 (м, 1H), 7,37 д, J= 8,5 Гц, 1H), 7,10-7,20 (м, 2H), 6,65- 7,05 (м, 8H), 5,55 (шс, 2H), 4,32 (с, 2H), 4,00-4,20 (м,
2H), 3,70-3,30 (м, 4H), 2,70-2,85 (м, 2H), 2,50-2,70 (м, 24). М.-с. (FD) m/e 456 (M+).
Элем. анализ. Вычислено: C 76,46; H 6,42; N 3,07; найдено: C 76,75; H 6,44; N 3, 02.
Пример 33
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2-(1-(3,3-диметил)пирролидинил)этокси]фенил]метан

Взаимодействие продукта примера 31 (1,2 г, 2,3 ммоль) с трихлоридом бора (10 мл, 117 ммоль) в 1,2-дихлорэтане (30 мл) в соответствии с процедурой примера 27 дает целевой продукт в виде белой пены с выходом 58%.
ИК (KBr) 3370,07, 2955,32, 2869,48, 1711,08, 1610,77, 1510,46 см-1
1H-ЯМР
(300 МГц, CDCl3)
М.-с. (FD) m/e 468 (M+).
Элем. анализ. Вычислено: C 79,63; H 7,11; N 3,00; найдено: C 79,65; H 7,24; N 2,72.
Препаративный
пример 14
[2-(4-Метоксифенил)-6-метоксинафталин-1-ил] (4-метэтоксифенил)метанон

К 50 мл диоксана добавляют 6,0 г (15 ммоль) [3,4-дигидро-2- (4-метоксифенил)-6-метоксинафталин-1-ил] (4-метоксифенил)метанона и 7,3 г (32 ммоль) 2,3-дихлор-5,6-дициано-1,4-бензохинона. Смесь кипятят с обратным холодильником в течение 2 часов и затем перемешивают при температуре окружающей среды в течение 60 часов. Затем смесь концентрируют досуха и остаток растворяют в 500 мл метиленхлорида и раствор 3 раза промывают 400 мл 2н. раствора гидроксида натрия и затем один раз промывают 500 мл деионизованной воды. Полученный органический слой отделяют, сушат над сульфатом натрия и удаляют растворитель в вакууме. Полученное вещество затем очищают флэш-хроматографией (силикагель, 20% этилацетата в гексане, градиент) и получают 4,75 г (80%) названного в заголовке соединения в виде белой пены.
ЯМР QE 300 МГц в CDCl3: (3,80 м.д., с, 3H), (4,00 м.д., с, 3H), (6,75 м. д. , д, 2H), (6,85 м.д., д, 2H), (7,20 м.д., дд, 1H), (7,30 м.д., дс, 1H), (7,40 м. д., д, 2H), (7,60 м.д., д, 1H), (7,75 м.д., д, 2H), (7,95 м.д., д, 1H). М.-с. (FD) m/e 398 (M+).
Элем. анализ. Вычислено: C 78,37; H 5,57; найдено: C 78,55; H 5,78.
Пример 34
Гидрохлорид [2-(4-метоксифенил)-6-метоксинафталин-1-ил] [4-[2-(1-пиперидинил)этокси]фенил]метана
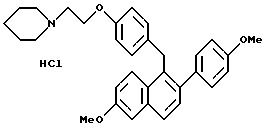
К 20 мл пропилбензола добавляют 240 мг (6,01 ммоль) 95% алюмогидрида лития и 240 мг (0,484 ммоль) соединения, полученного в препаративном примере 14. Смесь кипятят с обратным холодильником в течение 35 минут и охлаждают ее до температуры окружающей среды. К смеси осторожно добавляют 1 мл деионизованной воды, затем добавляют 3 мл 15% раствора гидроксида натрия в деионизованной воде (м/м), а затем добавляют еще 1 мл деионизованной воды. Смесь перемешивают в течение 15 минут при температуре окружающей среды и удаляют осадок на вакуум-фильтре. Маточную жидкость затем разбавляют метиленхлоридом (100 мл), промывают один раз солевым раствором, сушат над сульфатом натрия и упаривают досуха на роторном испарителе. Коричневую смолу очищают радиальной хроматографией на 4 мм пластине, со смесью метиленхлорида с метанолом в качестве элюента и получают названное в заголовке соединение.
ЯМР QE 300 МГц в CDCl3: (1,55 м.л., м, 2H), (1,75 м.д., комплекс, 4H), (2,60 м.д., комплекс, 4H), (2,85 м.д., т, 2H), (3,95 м.д., с, 3H), (4,05 м. д. , с, 3H), (4,20 м.д., т, 2H), (4,45 м.л., с, 2H), (6,85 м.д., д, 2H), (7,00 м. д., комплекс, 4H), (7,15 м.л., дд, 1H), (7,25 м.л., дс, 1H), (7,35 м.д., д, 2H), (7,50 м.л., д, 1H), (7,80 м.л., д, 2H), (7,90 м.д., д, 1H). М. -с. (FD) me/e 481 (M+).
Пример
35
[2-(4-Гидроксифенил)-6-гидроксинафталин-1-ил] [4-[2- (1-пиперидинил)этокси]фенил]метан

К суспензии лишенного защитных групп (отщепление защитных групп осуществляют стандартными способами, описанными здесь) продукта препаративного примера 12 (0,51 г, 1,00 мл) в н- пропилбензоле при перемешивании добавляют Red-AiR (0,87 г, 6,00 ммоль) и смесь кипятят с обратным холодильником. Через 3 часа раствор охлаждают до температуры окружающей среды и осторожно гасят избытком 1, 0н. соляной кислоты. Полученную двухфазную смесь экстрагируют этилацетатом и объединенные органические экстракты промывают насыщенным водным раствором бикарбоната и солевым раствором, сушат (MgSO4), фильтруют и концентрируют. Очистка сырого вещества радиальной хроматографией (силикагель, этилацетат: гексан: метанол:триэтиламин=2,5:2,5:0,7:0,3) дает названное в заголовке вещество.
Методы испытаний
Общая подготовительная процедура
В примерах, иллюстрирующих способы изобретения, используют постклимактерическую модель, на которой определяют влияние
различных обработок на циркулирующие липиды. Самок крыс Sprague Dawley в возрасте семидесяти пяти дней (масса от 200 до 225 г) получают от фирмы Charles River Laboratories (Portage, MI). У животных
либо удаляют яичники (OVX), либо их подвергают хирургической процедуре Sham в Charles River Laboratories, и затем, спустя неделю, отправляют. После доставки животных размещают в металлических
подвесных клетках группами по 3 или по 4 особи в клетке, и они имеют доступ к пище (содержание кальция приблизительно 0,5%) и воде по желанию в течение одной недели. Комнатную температуру поддерживают
в интервале 22,2o±1,7oC при минимальной относительной влажности 40%. Световой период в помещении составляют 12 часов освещения и 12 часов темноты.
Режим дозирования и сбор образцов ткани. После однонедельной акклиматизации (т. е. , через две недели после OVX) начинают ежесуточное дозирование исследуемого соединения. Исследуемое соединение или 17a-этинилэстрадиол вводят перорально, если нет других указаний, в виде суспензии в 1% карбоксиметилцеллюлозе или растворенным в 20% циклодекстрине. Животным дают дозы ежесуточно в течение 4 суток. После режима дозирования животных взвешивают и дают наркоз кетамином с ксилазином (2:1, о/о) и берут образцы крови посредством кардиальной пункции. Затем животных умерщвляют посредством асфиксии CO2, удаляют матку через средний разрез и определяют массу влажной матки.
Анализ на холестерин. Образцы крови оставляют коагулировать при комнатной температуре в течение 2 часов и затем при центрифугировании в течение 10 минут при 3000 об/мин получают сыворотку. Определяют содержание холестерина в сыворотке, используя высокоэффективную диагностику Boehringer Mannheim для количественного определения холестерина. Коротко, холестерин окисляют до холес-4-ен-3-она и пероксида водорода. Пероксид водорода затем вводят в реакцию с фенолом и 4-аминофеназоном в присутствии пероксилазы и получают краситель п-хинонимин, который определяют спекрофотометрически при 500 нм. Затем по стандартной кривой вычисляют концентрацию холестерина. Весь анализ автоматизируется при использовании автоматизированного рабочего места Biomek (Biomek Automated Workstation).
Анализ маточной эозинофил-пероксидазы (ЕРО). До начала ферментного анализа ткань матки хранят при 4oC. Затем ткань матки гомогенизируют в 50 объемах 50 мМ трис-буфера (pH 8,0), содержащего 0,005% тритона Х-100. После добавления 0,01% пероксида водорода и 10 мМ о-фенилендиамина (конечные концентрации) в трис-буфере контролируют возрастание поглощения в течение одной минуты при 450 нм. Присутствие эозинофилов в матке указывает на эстрогенную активность соединения. Определяют максимальную скорость в 15-секундном интервале в начальной, линейной части кривой.
Источник соединения. 17α-Этинилэстрадиол получают от фирмы Sigma Chemical Co., Сент-Луис, Миссури.
Влияние соединений формулы I на сывороточной холестерин и определение активности агонист/не-агонист
Данные, приведенные ниже в табл.1, показывают сравнительные
результаты, полученные для крыс с удаленными яичниками, крыс, обработанных 17α-этинилэстрадиолом (EE2; пригодная для перорального приема форма эстрогена), и крыс, обработанных
некоторыми соединениями настоящего изобретения. Хотя EE2 вызывает снижение холестерина в сыворотке, когда вводится перорально при норме 0,1 мг/кг/сутки, он также проявляет стимулирующее
действие на матку, так что масса матки при использовании EE2 значительно больше, чем масса матки у подопытных животных с удаленными яичниками. Такая реакция матки на эстроген общепризнана в
данной области.
При использовании соединения по настоящему изобретению в большинстве случаев не только снижается холестерин в сыворотке по сравнению с контрольными животными с удаленными яичниками, но масса матки только минимально увеличивается или немного уменьшается при применении большинства проверенных соединений формулы I. При сравнении с эстрогенными соединениями, известными в технике, это достижение по снижению содержания холестерина в сыворотке без вредного влияния на массу матки яавляется редким и очень желательным.
Как показывают приведенные ниже данные, эстрогенность также определяется путем оценки неблагоприятной реакции эозинофильной инфильтрации в матку. Соединения по настоящему изобретению не вызывают какого-либо увеличения числа эозинофилов, наблюдаемого в стромальном слое у крыс с удаленными яичниками, в то время как эстрадиол вызывает существенное, ожидаемое возрастание инфильтрации эозинофилов.
Данные, приведенные ниже в табл. 1, отражают реакцию 5-6 крыс в случае каждой обработки.
Кроме показанных преимуществ соединений по настоящему изобретению, особенно, при сравнении с эстрадиолом, приведенные выше данные четко демонстрируют, что соединения формулы I не подобны эстрогену. Более того, не наблюдается токсикологического действия (выживание) ни при какой обработке.
Методика теста на остеопороз
Следуя общей подготовительной процедуре, см. выше, крыс обрабатывают ежесуточно в течение 35 суток (6 крыс на группу для обработки) и
умерщвляют их асфиксией диоксидом углерода на 36-е сутки. Период в 35 суток является достаточным для получения максимального уменьшения плотности костей, измеряемой так, как описано здесь. Во время
умерщвления матку удаляют, отсекают посторонние ткани и перед определением влажной массы жидкое содержимое изгоняют, чтобы подтвердить отсутствие эстрогена, ассоциированное с полной овариэктомией. В
ответ на овариэктомию, масса матки снижается, как правило, примерно на 75%. Затем ткань матки помещают в 10%-ный нейтральный забуференный формалин для последующего гистологического анализа.
Иссекают правые бедренные кости и дигитилизируют рентгеновским излучением и анализируют на дистальный метафиз с помощью программы анализа изображения (NIH image). Далее с помощью количественной компьютерной томографии сканируют проксимальную сторону большеберцовых костей этих животных.
В соответствии с вышеописанными процедурами подопытным животным вводят перорально соединения по настоящему изобретению и этинилэстрадиол (EE2) в 20% гидроксипропил- -β- циклодекстрине. Данные по дистальному бедренному метафизу, приведенные ниже в таблицах 2 и 3, являются результатами обработки соединением формулы I при сравнении с интактными животными и животными с удаленными яичниками. Результаты даются как среднее ± стандартное отклонение от среднего.
Таким образом, овариэктомия подопытных животных вызывает значительное уменьшение плотности бедренной кости при сравнении с плотностью у интактных, обработанных носителем контрольных животных. Пероральное введение этинилэстрадиола (EE2) предотвращает такую потерю, но всегда существует опасность стимуляции матки.
Соединения по настоящему изобретению также предотвращают потерю костной массы в зависимости от дозы. Соответственно, соединения по настоящему изобретению пригодны для лечения постклимактерического синдрома, в частности, остеопороза.
Испытания на пролиферацию MCF-7
Клетки аденокарциномы молочной железы MCF-7 (АТСС НТВ 22) хранят в MEM (минимальная поддерживающая среда, без
фенолового красного, Sigma, Сент-Луис, Миссури) с добавлением 10% фетальной сыворотки теленка (FBS) (о/о), L-глутамина (2 мM), пирувата натрия (1 мМ), ГЕПЕС
{(N-[2-гидроксиэтил]-пипеpaзин-N'- [2-этaнcульфoнoвaя кислота], 10 мМ}, заменимых аминокислот и бычьего инсулина (1 едг(ug)/мл (поддерживающая среда). За десять суток до испытаний клетки MCF-7
переносят в поддерживающую среду с добавлением 10% сыворотки плода коровы с покрытой декстраном угольной пылью (DCC-FBS) (среда для испытаний) вместо 10% FBS, чтобы истощить внутренние запасы
стероидов. Клетки MCF-7 извлекают из флаконов для хранения, используя диссоциирующую среду [свободный от Ca++/Mg++ HBSS (сбалансированный солевой раствор Хэнкса) (без фенолового
красного) с добавлением 10 мМ HEPES и 2 мМ ЭДТК]. Клетки дважды промывают средой для испытаний и доводят до концентрации 80000 клеток/мл. Приблизительно 100 мкл (8000 клеток) добавляют в плоскодонные
лунки для микрокультуры (Costar 3596) и инкубируют при 37oC во влажной камере с 5% CO2 в течение 48 часов, чтобы создать клеткам возможность для слипания и уравновешивания после
переноса. Готовят серийные разведения лекарственных средств или контрольного ДМСО в среде для испытаний и 50 мкл переносят в микрокультуры, повторяя три раза, а затем добавляют 50 мкл среды для
испытаний для конечного объема 200 мкл. После дополнительной выдержки в течение 48 часов при 37oC во влажной камере с 5% CO2 микрокультуры импульсно метят тимидином, меченным
тритием (1 едКи/лунку) в течение 4 часов. Получение культур завершают замораживанием при -70oC в течение 24 часов с последующим оттаиванием и сбором микрокультур с использованием
полуавтоматического харвестера клеток Skatron. Подсчитывают число импульсов образцов в жидкой фазе, используя счетчик β- частиц Wallac BetaPlace. Результаты, приведенные ниже в табл. 4,
показывают IC50 для некоторых соединений настоящего изобретения.
Ингибирование опухоли молочной железы, вызванной DMBA, формируют эстрогензависимые опухоли молочной железы у самок крыс Sprague-Davley, которых закупают у Harlan Industries, Индианполис, Индиана. В возрасте 55 дней крысы получают однократно перорально 20 мг 7,12-диметил-бенз[а] антрацена (DMBA). Через 6 недель после введения DMBA молочные железы еженедельно пальпируют с целью выявления появления опухоли. Всякий раз, когда появляется одна или несколько опухолей, замеряют штангенциркулем наибольший и наименьший диаметры каждой опухоли, измерения записывают и такое животное отбирают для эксперимента. Делают попытку равномерно распределить опухоли по размеру в обрабатываемой и контрольной группах, так, чтобы усредненные по размеру опухоли эквивалентно распределялись между группами. В контрольных и тестовых группах при каждом эксперименте содержится 5-9 животных.
Соединения формулы I вводят либо внутрибрюшинно инъекциями в 2% аравийской камеди, либо перорально. При пероральном введении соединения либо растворяют, либо суспендируют в 0,2 мл кукурузного масла. Каждую обработку, включая контрольные обработки аравийской камедью и кукурузным маслом, осуществляют один раз в сутки для каждого подопытного животного. После начального измерения опухоли и отбора подопытных животных опухоли измеряют каждую неделю вышеописанным способом. Обработку и измерения продолжают в течение 3-5 недель, затем определяют конечную площадь опухолей. Определяют изменение средней площади опухоли при обработке каждым соединением и при контрольной обработке.
Методы испытаний в случае фибромы матки
Тест 1
Соединение по настоящему изобретению вводят 3-20
женщинам с фибромой матки. Количество вводимого соединения составляет от 0,1 до 1000 мг/сутки, и продолжительность введения составляет 3 месяца.
За женщинами наблюдают по поводу явлений фибромы матки в течение всего времени введения и еще в течение 3 месяцев после прекращения введения.
Тест 2
Осуществляют ту же процедуру, что в тесте 1, за
исключением того, что продолжительность введения составляет 6 месяцев.
Тест 3
Осуществляют ту же процедуру, что в тесте 1, за исключением того, что продолжительность введения
составляет 1 год.
Тест 4
А. Индуцирование фибром у морской свинки
Для индуцирования лейомиом у половозрелых морских свинок применяют длительную эстрогенную
стимуляцию. Животным посредством инъекций вводят дозы эстрадиола 3-5 раз в неделю в течение 2-4 месяцев, пока не появятся опухоли. Обработку, включающую соединением изобретения или носителем,
назначают ежесуточно в течение 3-16 недель, а затем животных умерщвляют, извлекают матки и анализируют на регрессию опухолей.
Б. Имплантация ткани фибромы матки человека мыши без
волосяного покрова
Ткань человеческой лейомиомы имплантируют в брюшинную полость и/или в миометрий половозрелой кастрированной самки мыши без волосяного покрова. Добавляют экзогенный эстроген,
чтобы индуцировать рост эксплантата. В некоторых случаях харвестированные опухолевые клетки перед имплантацией культивируют in vitro. Обработка, включающая введение соединение по настоящему
изобретению или носитель, ежедневно дополняется промыванием желудка в течение 3-16 недель, и имплантаты удаляют и измеряют на предмет роста или регрессии. Во время умерщвления матку извлекают и
оценивают состояние органа.
Тест 5
А. Собирают и сохраняют, in vitro, ткань фибромы матки человека как первичные нетрансформированные культуры. Операционные препараты
просеивают через стерильную сетку или сито, или, с другой стороны, препарируют с помощью иглы из окружающей ткани, чтобы получить только клеточную суспензию. Клетки сохраняют в среде, содержащей 10%
сыворотки и антибиотик. Определяют скорость роста в присутствии и в отсутствии эстрогена. Клетки оценивают на их способность продуцировать комплемент компонента C3 и на их реакцию на факторы роста и
гормоны роста. In vitro культуры оценивают на их пролиферативную реакцию после обработки прогестинами, GnRH, соединением по настоящему изобретению и носителем. Еженедельно оценивают уровни стероидных
гормонных рецепторов, чтобы определить, сохраняются ли важные клеточные характеристики in vitro. Используют ткани от 5-25 пациенток.
Активность, по крайней мере, в одном из вышеперечисленных тестов показывает, что соединения по настоящему изобретению относятся к потенциально применимым при лечении фибромы матки.
Методы испытаний в случае эндометриоза
По тестам 1 и 2 можно проверить влияние 14-дневного и 21-дневного введения соединений по настоящему изобретению на рост эксплантата эндометриальной ткани.
Тест 1
В
качестве подопытных животных используют двадцать-тридцать взрослых самок крыс линии CD. Их разделяют на три равные группы. Контролируют эстральный цикл всех животных. На день проэструса осуществляют
операцию на каждом животном. Самкам в каждой группе удаляют левую трубу матки, иссекают на небольшие квадраты и квадраты сшивают редкими швами в различных местах вблизи мезентериального кровотока.
Кроме того, самкам в группе 2 удаляют яичники.
Начиная со следующего дня после операции животные в группах 1 и 2 получают интраперитонеальные инъекции воды в течение 14 суток, в то
время как животные в группе 3 в тот
же период получают интраперитонеальные инъекции 1,0 мг соединения по настоящему изобретению на килограмм массы тела. После обработки в течение 14 дней
каждую самку умерщвляют и удаляют эндометриальные эксплантаты, надпочечники, оставшуюся часть матки и яичники, где это возможно, и препарируют для гистологической проверки. Яичники и надпочечники
взвешивают.
Тест 2
В качестве подопытных животных используют двадцать-тридцать взрослых самок крыс линии CD. Их разделяют на две равные группы. Контролируют эстральный цикл
всех животных. На день проэструса осуществляют операцию на каждой самке. Самкам в каждой группе удаляют левый рог матки, иссекают на небольшие квадраты и квадраты сшивают редкими швами в различных
местах вблизи мезентериального кровотока.
Приблизительно через 50 дней после операции животные, приписанные к группе 1, получают интраперитонеальные инъекции воды в течение 21 дня, в то время как животные в группе 2 в тот же период получают интраперитонеальные инъекции 1,0 мг соединения по настоящему изобретению на килограмм массы тела. Через 21 день после обработки каждую самку умерщвляют и удаляют, взвешивают эндометриальные эксплантаты и надпочечники. Эксплантаты измеряют как указание на рост. Контролируют эстральные циклы.
Тест 3
А. Хирургическая
индукция эндометриоза
Чтобы вызвать эндометриоз у крыс и/или кроликов, используют аутотрансплантаты эндометриальной ткани. Самок репродуктивной зрелости подвергают двусторонней овариэктомии и
экзогенно дают эстроген, обеспечивая, таким образом, специфический и постоянный уровень гормона. Аутологическую эндометриальную ткань имплантируют в брюшину 5-150 животных и дают эстроген, чтобы
индуцировать рост эксплантата. Обработка, включающая введение соединения по настоящему изобретению, дополняется ежедневным промыванием желудка в течение 3-16 недель, и имплантаты удаляют и измеряют на
предмет роста или регрессии. При умерщвлении извлекают интактный рог матки и оценивают состояние эндометрия.
Б. Имплантация человеческой эндометриальной ткани мыши без волосяного
покрова
Ткань эндометриальных повреждений человека имплантируют в брюшину половозрелой кастрированной самки мыши без волосяного покрова. Добавляют экзогенный эстроген, чтобы индуцировать рост
эксплантата. В некоторых случаях собранные эндометриальные клетки культивируют in vitro перед имплантацией. Обработку, включающую введение соединения по настоящему изобретению, ежедневно дополняют
промыванием желудка в течение 3-16 недель, и имплантаты удаляют и измеряют на предмет роста или регрессии. Во время умерщвления извлекают матки и оценивают состояние интактного эндометрия.
Тест 4
А. Ткань человеческого поврежденного эндометрия собирают и хранят in vitro как первичные нетрансформированные культуры. Операционные препараты просеивают через стерильную
сетку или сито, или, с другой стороны, препарируют с помощью иглы из окружающей ткани, чтобы получить только клеточную суспензию. Клетки сохраняют в среде, содержащей 10% сыворотки и антибиотик.
Определяют скорость роста в присутствии и в отсутствии эстрогена. Клетки оценивают на их способность продуцировать комплемент компонента C3 и на их реакцию на факторы роста и гормоны роста. In vitro
культуры оценивают на их пролиферативную реакцию после обработки прогестинами, GnRH, соединением по настоящему изобретению и носителем. Еженедельно оценивают уровни стероидных гормонных рецепторов,
чтобы определить, сохраняются ли важные клеточные характеристики in vitro. Используют ткани от 5-25 пациенток.
Активность, по крайней мере, в одном из вышеперечисленных тестов показывает, что соединения по настоящему изобретению относятся к потенциально применимым при лечении эндометриоза.
Методика испытания на ингибирование пролиферации аортальных
гладкомышечных клеток или рестеноза
Соединения по настоящему изобретению обладают способностью ингибировать пролиферацию аортальных гладкомышечных клеток. Это можно показать, используя
культивированные гладкомышечные клетки, полученные из кроличьей аорты, причем пролиферацию определяют, измеряя синтез ДНК. Клетки получают методом эксплантата, как описано в Ross, J. of Cell Bio.,
50:172 (1971). Клетки культивируют в 96-луночных титрационных микропланшетах в течение пяти суток. Культуры начинают сливаться и рост прекращается. Затем клетки переносят в модифицированную по способу
Дульбекко среду Игла (DMEM), содержащую 0,5-2% обедненной тромбоцитами плазмы, 2 мМ L- глутамина, 100 E/мл пенициллина, 100 мг/мл стрептомицина, 1 мКи/мл3H-тимидина, 20 нг/мл фактора роста
тромбоцитов и различные концентрации соединений по настоящему изобретению. Исходный раствор соединений готовят в диметилсульфоксиде и затем разводят его до соответствующей концентрации (0,01-30 мМ) в
вышеупомянутой среде для испытаний. Затем клетки инкубируют при 37oC в течение 24 часов в атмосфере с 5% CO2 и 95% воздуха. По прошествии 24 часов клетки фиксируют в метаноле.
Затем определяют включение3H-тимидина в ДНК, подсчитывая импульсы, как описано в Bonin, et al., Exp. Cell Res., 181:475-482 (1989).
Затем демонстрируют ингибирование пролиферации аортальных гладкомышечных клеток соединениями по настоящему изобретению, определяя их действие на экспоненциально растущие клетки. Гладкомышечные клетки из кроличьей аорты высевают в 12-луночные тканевые культуральные планшеты в DMEM, содержащую 10% сыворотки плода коровы, 2 мМ L-глутамина, 100 E/мл пенициллина и 100 мг/мл стрептомицина. Через 24 часа клетки сливаются и среду заменяют DMEM, содержащей 10% сыворотки, 2 мМ L-глутамина, 100 E/мл пенициллина, 100 мг/мл стрептомицина и нужные концентрации соединений. Клеткам позволяют расти в течение 4 суток. Клетки обрабатывают трипсином и определяют число клеток в каждой культуре, производя подсчет с помощью цитометра ZM.
Активность в вышеописанных испытаниях указывает, что соединения по настоящему изобретению могут быть полезными при лечении рестеноза.
Настоящее изобретение также относится к способу ослабления постклимактерического синдрома у женщин, который включает вышеупомянутый способ, использующий соединения формулы I, а также включает введение женщине эффективного количества эстрогена или прогестина. Такая обработка особенно пригодна для лечения остеопороза и снижения содержания холестерина в сыворотке, поскольку пациентка будет получать пользу от каждого фармацевтического средства, так как соединения по настоящему изобретению могли бы ингибировать нежелательные побочные действия эстрогена и прогестина. Активность таких комбинированных обработок в любом из упомянутых выше постклимактерических тестов указывает, что комбинированное лечение пригодно для ослабления симптомов постклимактерического синдрома у женщин.
Коммерчески доступны различные формы эстрогена и прогестина. Содержащими эстроген средствами являются, например, этинилэстроген (0,01-0,03 мг/сутки), местранол (0,05-0,15 мг/сутки) и конъюгированные эстрогенные гормоны, такие как Premarin® (Wyet-Ayerst; 0,3-2,5 мг/сутки). Средства на основе прогестинов включают, например, медроксипрогестерон, такой как Provera® ((Upjohn; 2,5-10 мг/сутки), норэтилнодрел (1,0-10,0 мг/сутки) и норэтиндрон (0,5-2,0 мг/сутки). Предпочтительным эстрогенсодержащим соединением является Premarin, а норэтилнодрел и норэтиндрон являются предпочтительными прогестинсодержащими средствами.
Способ введения каждого средства на основе эстрогена и прогестина является способом, который известен в технике. В большинстве способов настоящего изобретения соединения формулы 1 вводят непрерывно от 1 до 3 раз в сутки. Однако при лечении эндометриоза может быть особенно полезна периодическая терапия, или она может быть срочно применена при приступе болезни, сопровождающемся сильной болью. В случае рестеноза лечение может ограничиваться небольшими периодами (1-6 месяцев) после таких медицинских процедур, как ангиопластика.
Используемое здесь выражение "эффективное количество" означает количество соединения по настоящему изобретению, которое способно ослабить симптомы различных патологических состояний, описанных здесь. Специфическая доза соединения, вводимого по настоящему изобретению, будет, конечно, определяться конкретными условиями, сопровождающими данный случай, включающими, например, вводимое соединение, способ введения, состояние пациента и патологическое состояние, которое лечат. Типичная суточная доза будет содержать нетоксичную дозу соединения по настоящему изобретению, составляющую от 5 мг до 600 мг в сутки. Предпочтительные суточные дозы будут составлять, как правило, от 15 мг до 80 мг в сутки.
Соединения по настоящему изобретению могут вводиться различными способами, включая пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный. Эти соединения включают в составы, предпочтительно, перед введением, выбор способа которого остается за лечащим врачом. Таким образом, другим аспектом настоящего изобретения является фармацевтическая композиция, содержащая эффективное количество соединения формулы 1 или его фармацевтически приемлемой соли, содержащая, необязательно, эффективное количество эстрогена или прогестина и фармацевтически приемлемый носитель, разбавитель или эксципиент.
Общее количество активных ингредиентов в таких составах составляет от 0,1 мас. % до 99,9 мас.% от массы состава. "Фармацевтически приемлемый" означает, что носитель, разбавитель, эксципиенты и соль должны быть совместимыми с другими ингредиентами состава и не приносить вреда их реципиенту.
Фармацевтические композиции по настоящему изобретению могут быть получены способами, известными в технике, с использованием известных и легко доступных ингредиентов. Например, соединения формулы I, вместе или без эстрогена или прогестина, можно ввести в состав смеси с обычными эксципиентами, разбавителями или носителями и сформировать в виде таблеток, капсул, суспензий, порошков и так далее. Примеры эксципиентов, разбавителей и носителей, которые являются подходящими для таких составов, включают следующие вещества: наполнители и сухие разбавители, такие как крахмал, сахара, маннит и кремниевые производные; связующие, такие как карбоксиметилцеллюлоза и другие производные целлюлозы, альгинаты, желатин и поливинилпирролидон; увлажнители, такие как глицерин; дезинтегрирующие агенты, такие как карбонат кальция и бикарбонат натрия; вещества, замедляющие растворение, такие как парафин; ускорители резорбции, такие как четвертичные аммониевые основания; поверхностно-активные вещества, такие как цетиловый спирт, моностеарат глицерина; носители-адсорбенты, такие как каолин и бентонит; и смазки, такие как тальк, стеараты магния и кальция и твердые полиэтиленгликоли.
Соединения также могут входить в составы эликсиров или растворов для традиционного перорального приема или растворов, подходящих для парентерального введения, например, внутримышечным, подкожным или внутривенным путем. Кроме того, соединения весьма подходят для приготовления лекарственных форм с длительным выделением лекарственного средства и подобных форм. Составы могут быть составлены таким образом, что они выделяют активный ингредиент только, или предпочтительно, в определенном участке организма, возможно, в течение некоторого времени. Покрытия, оболочки и защитные матрицы могут быть изготовлены, например, из полимерных веществ, или восков.
Соединения формулы I, одни или в сочетании с фармацевтическим средством по настоящему изобретению, как правило, будут вводиться в обычные составы. Далее приводятся примеры составов, которые являются только иллюстративными и не предназначаются для ограничения объема настоящего изобретения.
Составы
В приведенных ниже составах "активный ингредиент" означает соединение формулы 1, или его соль, или его сольват.
Состав 1.
Желатиновые капсулы
Твердые желатиновые капсулы готовят, используя перечисленные далее ингредиенты, (мг, капсулы):
Активный ингредиент - 0,1 - 1000
Крахмал, нф (нетекучий)
- 0 - 650
Крахмал, текучий порошок - 0 - 650
Силиконовая жидкость, 350 сСт - 0 - 15
Приведенный выше состав может быть изменен в требуемых разумных пределах.
Состав 2. Таблетки (м/таблетку):
Активный ингредиент - 2,5 - 1000
Микрокристаллическая целлюлоза - 200 - 650
Диоксид кремния, высокодисперсный - 10 - 650
Стеариновая кислота - 5 - 15
Компоненты смешивают и прессуют в форме таблеток. С другой стороны, таблетки, каждая из которых содержит 2,5-1000 мг активного ингредиента, готовят как описано
ниже.
Состав 3. Таблетки (мг/таблетку):
Активный ингредиент - 25
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде
10%-ного раствора в воде) - 4
Натрийкарбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк - 1
Активный ингредиент, крахмал и целлюлозу просеивают через сито N 45
меш, США, и тщательно смешивают. Раствор поливинилпирролидона смешивают с полученными порошками, которые затем пропускают через сито N 14 меш, США. Полученные таким образом гранулы сушат при 50-60oC и пропускают через сито N 18 меш, США. Затем к гранулам добавляют натрийкарбоксиметилкрахмал, стеарат магния и тальк, предварительно просеянные через сито N 60 меш, США, и после перемешивания
прессуют на таблетировочной машине и получают таблетки.
Суспензии, каждая из которых в 5-мл дозе содержит 1-1000 мг лекарственного средства, готовят следующим образом.
Состав 4. Суспензии, (мг/ 5 мл):
Активный ингредиент - 0,1 - 1000
Натрийкарбоксиметилцеллюлоза - 50
Сироп - 1,25
Раствор бензойной кислоты, мл - 0,10
Корригент - q.v.
Краситель - q.v.
Очищенная вода - До 5
Лекарственное средство просеивают через сито N 45 меш, США, и смешивают с
натрийкарбоксиметилцеллюлозой и сиропом до образования равномерной пасты. Раствор бензойной кислоты, корригент и краситель разбавляют некоторым количеством воды и при перемешивании добавляют к пасте.
Затем добавляют воду в количестве, достаточном для получения требуемого объема.
Готовят аэрозольный раствор, содержащий перечисленные ниже ингредиенты.
Состав 5.
Аэрозоль, мас.%:
Активный ингредиент - 0,25
Этанол - 25,75
Пропеллент 22 (хлордифторметан) - 70,00
Активный ингредиент смешивают с этанолом и смесь добавляют к
части пропеллента 22, охлаждают до 30oC и переносят в заливочное устройство. Затем требуемое количество загружают в емкость из нержавеющей стали и разбавляют оставшимся пропеллентом. Затем
емкость снабжают клапанами.
Суппозитории готовят следующим образом.
Состав 6. Суппозитории, (мг/суппозиторий):
Активный ингредиент - 250
Глицериды
насыщенных жирных кислот - 2000
Активный ингредиент пропускают через сито N 60 меш, США, и суспендируют в глицеридах насыщенных жирных кислот, предварительно расплавленных при минимальном
необходимом для этого нагреве. Затем смесь выливают в форму для суппозиториев с номинальной емкостью 2 г и охлаждают.
Состав для внутривенного введения готовят следующим образом.
Состав 6. Раствор для внутривенного введения
Активный ингредиент, мг - 50
Изотонический физиологический раствор, мл - 1000
Раствор вышеуказанных ингредиентов
вводят пациенту внутривенно со скоростью около 1 мл в минуту.
Состав 8. Комбинированная капсула I, (мг/капсулу):
Активный ингредиент - 50
Премарин - 1
Авицел pH 101 - 50
Крахмал 1500 - 117,50
Силиконовое масло - 2
Твин 80 - 0,50
Cab-O-Sil - 0,25
Состав 9. Комбинированная капсула II, (мг/капсулу):
Активный ингредиент - 50
Норэтилнодрел - 5
Авицел pH 101 - 82,50
Крахмал 1500 - 90
Силиконовое масло - 2
Твин 80 - 0,50,
Состав 10.
Комбинированная таблетка, (мг/капсулу):
Активный ингредиент - 50
Премарин - 1
Кукурузный крахмал, нф - 50
Повидон К29-32 - 6
Авицел pH 101 - 41,50
Авицел pH 102 - 136,50
Кросповидон XL10 - 2,50
Стеарат магния - 0,50
Cab-O-Sil - 0,50
Реферат
Изобретение относится к новым нафтилсодержащим соединениям формулы I, где R1 и R2 - H, -OH, -O(C1-C4алкил), -OCOC6H5, -OCO(C1-C6алкил), -OSO2(C4-C6 алкил); R3 - 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино, 1-гексаметиленимино; промежуточным соединениям, которые пригодны для ослабления симптомов постклимактерического синдрома, включая остеопороз, гиперлипемию и эстрогензависимый рак, и ингибирования фибромы матки, эндометриоза и пролиферации аортальных гладкомышечных клеток. Также описываются способы получения таких соединений. 6 с. и 40 з.п.ф-лы, 4 табл.

Формула

где R1 представляет собой -H, -OH-, -O(C1-C4алкил), OCOC6H5, -OCO(C1-C6алкил) или -OSO2(C4-C6алкил);
R2 представляет собой -H, -OH, -O(C1-C4алкил), OCOC6H5, -OCO(C1-C6алкил) или -OSO2(C4-C6алкил);
n = 2 или 3;
R3 - 1-пипридинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино или 1-гексаметиленимино,
или его фармацевтически приемлемая соль.
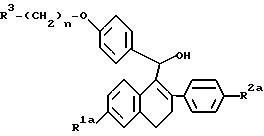
где R1а представляет собой -H, -OH или -O(C1-C4алкил);
R2а представляет собой -H, -OH или -O(C1-C4алкил);
R3 - 1-пиперидинил, 1-пирролидинил, диметиламино, диэтиламино или 1-гексаметиленимино;
n = 2 или 3,
или его фармацевтически приемлемая соль.

где R1а представляет собой -H, -OH или -O(C1-C4алкил);
R2a - -H, -OH или -O(C1-C4алкил);
R3 - 1-пиперидинил, 1-пирролидинил, диметиламино, диэтиламино или 1-гексаметиленимино; и
n = 2 или 3,
или его фармацевтически приемлемой соли, заключающийся в том, что осуществляют а) взаимодействие соединения формулы V
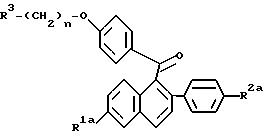
где R1а, R2а, R3 и n имеют указанные выше значения,
с восстановлением в присутствии растворителя, имеющего температуру кипения 150 - 200oC, и кипячение смеси с обратным холодильником; а) когда R1а и/или R2а представляет собой -O(C1-C4 алкил), необязательное удаление гидроксизащитных групп R1а и/или R2а и в) необязательное получение соли продукта реакции со стадии а) или б).

где R1 - -H, -OH или -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
R2 - -H, -OH, или -O(C1 -C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C4-C6 алкил);
R3 - 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино или 1-гексаметиленимино;
M - -CH(OH)- или -CH2-;
n = 2 или 3,
или его фармацевтически приемлемой соли, заключающийся в том, что осуществляют а) взаимодействие соединения формулы IIId

где R1b - -H, или O(C1-C4 алкил);
R2b - -H, или O(C1-C4 алкил);
R3 и n имеют указанные выше значения,
с восстановителем; б) когда R1b и/или R2b - -O(C1-C4 алкил), необязательное удаление гидроксизащитных групп R1b и/или R2b, в) когда R1 и R2 - -OH, необязательную замену указанных гидроксильных групп заместителем формулы -O(C1-C4 алкил), -OCOC6H5, -OCO(C1-C6 алкил) или -OSO2(C1-C4 алкил) и г) необязательное получение соли продукта реакции со стадии а), б) или в).
Комментарии