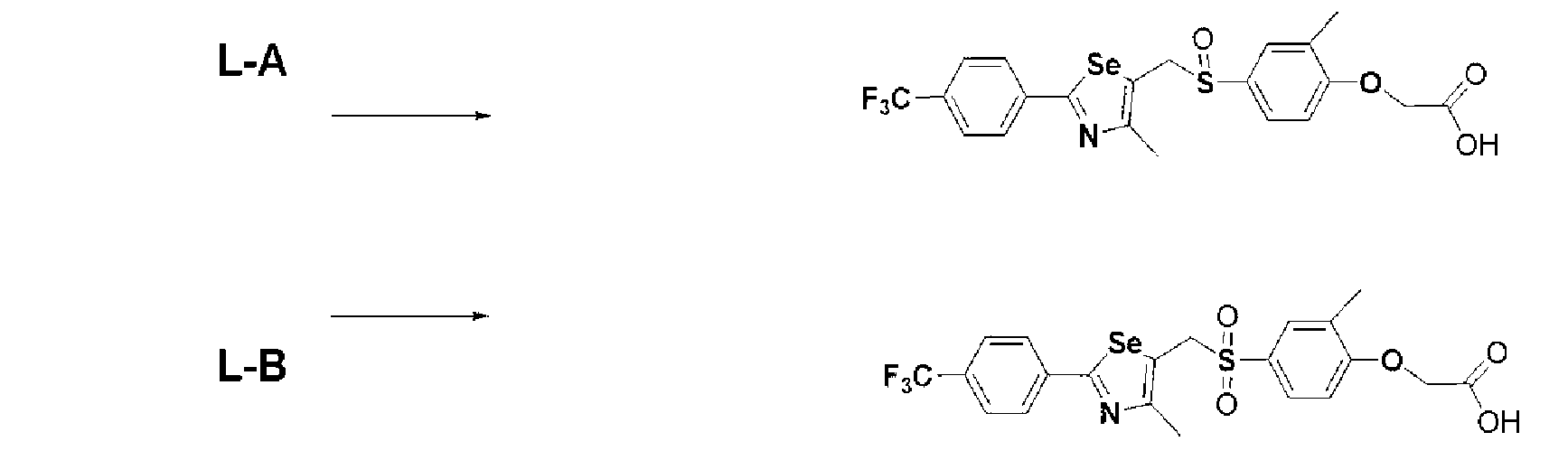
Производное селеназола, имеющее лиганд, который активирует рецептор, активируемый пролифератором пероксисом ( ppar ), способ его получения и применение химических соединений - RU2510394C1
Код документа: RU2510394C1
Чертежи

Описание
Область техники
Настоящее изобретение относится к производному селеназола - соединению, представленному химической формулой I, используемому в качестве лиганда, активирующего рецептор, активируемый пролифератором пероксисом (PPAR), которое может применяться для лечения ожирения, гиперлипемии, жировой инфильтрации печени, атеросклероза и диабета, его гидрату, сольвату, стереоизомеру и фармацевтически приемлемой соли, и фармацевтической композиции, косметической композиции, функциональной пищевой композиции, композиции функционального напитка и кормовой композиции для животных, содержащей указанные соединения:
[Химическая формула I]
Уровень техники
Рецепторы, активируемые пролифератором пероксисом (PPAR), являются ядерными рецепторами. Были идентифицированы три подтипа: PPARα, PPARγ и PPARδ (Nature, 1990, 347, p. 645-650, Proc. Natl. Acad. Sci. USA 1994, 91, p. 7335-7359). PPARα, PPARγ и PPARδ имеют различные функции и экспрессированы в различных тканях. PPARα экспрессируется в основном в сердце, почках, скелетных мышцах и тканях толстого кишечника человека (Mol. Pharmacol. 1998, 53, p. 14-22, Toxicol. Lett. 1999, 110, p. 119-127, J. Biol. Chem. 1998, 273, p. 16710-16714) и вовлечен в β-окисление в пероксисомах и митохондриях (Biol. Cell. 1993, 77, p. 67-76, J. Biol. Chem. 1997, 272, p. 27307-27312). PPARγ слабо экспрессируется в тканях скелетных мышц, но экспрессируется в основном в жировой ткани. Как известно, вовлечен в дифференцировку жировых клеток, аккумулирование энергии в виде жира и регуляцию гомеостаза инсулина и глюкозы (Moll. Cell. 1999, 4, p. 585-594, p. 597-609, p. 611-617). PPARδ эволюционно сохраняется у млекопитающих, включая человека, грызунов и асцидий. Он был идентифицирован как PPARβ у Xenopus laevis (Cell 1992, 68, p. 879-887) и у человека как NUCI (Mol. Endocrinol. 1992, 6, p. 1634-1641), PPARδ (Proc. Natl. Acad. Sci. USA 1994, 91, p. 7355-7359), NUCI (Biochem. Biophys. Res. Commun. 1993, 196, p. 671-677) или FAAR (J. Bio. Chem. 1995, 270, p. 2367-2371). В последнее время он получил унифицированное название - PPARδ. У человека, PPARδ, как известно, присутствует в хромосоме 6p21,1-p21,2. У мышей, мРНК PPARδ обнаружена в различных областях, но в меньшем количестве, чем мРНК PPARα или PPARγ (Endocrinology 1996, 137, p. 354-366, J. Bio. Chem. 1995, 270, p. 2367-2371, Endocrinology 1996, 137, p. 354-366). Согласно исследованиям, проведенным до настоящего времени, PPARδ играет очень важную роль в экспрессии гамет (Genes Dev. 1999, 13, p. 1561-1574). Кроме того, он, как известно, вовлечен в дифференцировку нервных клеток в центральной нервной системе (ЦНС) (J. Chem. Neuroanat. 2000, 19, p. 225-232), заживление ран посредством противовоспалительного действия (Genes Dev. 2001, 15, p. 3263-3277, Proc. Natl. Acad. Sci. USA 2003, 100, p. 6295-6296) или тому подобное. Проведенное в последнее время исследование показало, что PPARδ вовлечен в дифференцировку жировых клеток и метаболизм жировых клеток (Proc. Natl. Acad. Sci. USA 2002, 99, p. 303-308, Mol. Cell. Biol. 2000, 20, p. 5119-5128). Было обнаружено, что PPARδ активирует экспрессию критических генов, связанных с β-окислением и расщеплением белков (UCP), связанных с энергетическим метаболизмом, в процессе расщепления жирных кислот, и таким образом улучшает состояние при ожирении и повышает выносливость (Nature 2000, 406, p. 415-418, Cell 2003, 113, p. 159-170, PLoS Biology 2004, 2, e294, Cell, 2008, 134, 405415). Кроме того, активация PPARδ делает возможным повышение уровня ЛВП (липопротеины высокой плотности) и улучшение состояние при диабете 2 типа без изменения веса тела (Proc. Natl. Acad. Sci. USA 2001, 98, p. 5306-5311, 2003, 100, p. 15924-15929, 2006, 103, p. 3444-3449) и способствует лечению атеросклероза, путем ингибирования связанных с атеросклерозом генов (Science, 2003, 302, p. 453-457, PNAS, 2008, 105, 42714276). Соответственно, регулирование обмена жиров посредством PPARδ предоставляет важный инструмент для лечения ожирения, диабета, гиперлипемии и атеросклероза.
Описание
Техническая задача
Задача настоящего изобретения состоит в получении нового соединения, которое селективно активирует PPARδ. Другой задачей настоящего изобретения является получение фармацевтической композиции, косметической композиции, функциональной пищевой композиции, композиции функционального напитка и кормовой композиции для животных, содержащей новое соединение в соответствии с настоящим изобретением.
Решение технической задачи
Настоящее изобретение относится к соединению - производному селеназола, представленного химической формулой I, которое активирует рецептор, активируемый пролифератором пероксисом (PPAR), его сольвату, стереоизомеру и его фармацевтически приемлемой соли, способу получения указанного соединения, и к фармацевтической композиции, косметической композиции, функциональной пищевой композиции, композиции функционального напитка и кормовой композиции для животных, содержащих указанные соединения:
[Химическая формула I]
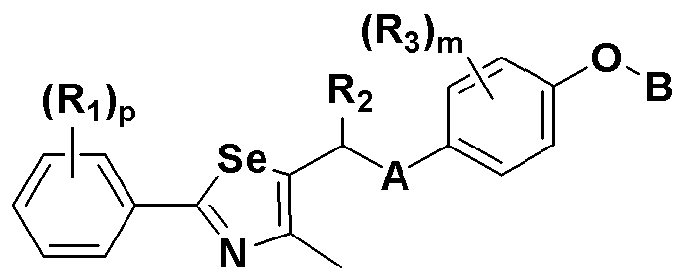
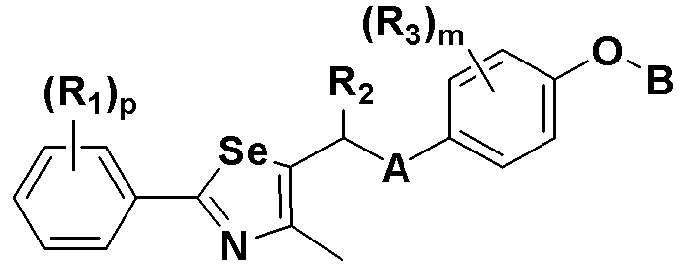
где A представляет собой O, NR, S, S(=O), S(=O)2 или Se; B представляет собой водород или
Особенно предпочтительным производным селеназола, активирующим PPAR, представленным химической формулой I, является производное, в котором: R1 представляет собой водород, C1-C5 алкил, замещенный одним или несколькими фтором, или фтор; R2 представляет собой водород, C1-C8 алкил,
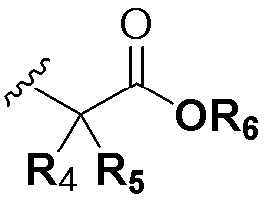
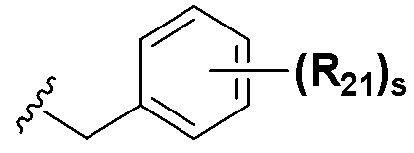
В химической формуле I, R1 может представлять собой водород, метил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 2-этилгексил, фторметил, дифторметил, трифторметил, 2-фторэтил, пентафторэтил, фтор, бром, йод или хлор; R2 может представлять собой водород или замещенный или незамещенный бензил, фенилбензил или пиридилбензил, где фенил, пиридил или бензил в R2 может быть дополнительно замещен фтором, хлором, метилом, этилом, н-пропилом, изо-пропилом, трет-бутилом, фторметилом, дифторметилом, трифторметилом, 2-фторэтил, пентафторэтил, метокси, этокси, пропилокси, н-бутокси, трет-бутокси, фторметокси, дифторметокси, трифторметокси, 2-фторэтокси, пентафторэтокси, CN, NO2, C6-C12 арилом или C3-C12 гетероарилом, содержащим один или несколько гетероатом(ов), выбранных из N, O и S; R3 может представлять собой водород, метил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 2-этилгексил, фторметил, дифторметил, трифторметил, 2-фторэтил, пентафторэтил, фтор, хлор; R4 и R5 могут независимо представлять собой водород, галоген, метил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 2-этилгексил, фторметил, дифторметил, трифторметил, 2-фторэтил или пентафторэтил; и R6 может представлять собой водород, метил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 2-этилгексил, фторметил, дифторметил, трифторметил, 2-фторэтил, пентафторэтил, аллил, этенил, 2-пропенил, 2-бутенил, 3-бутенил, фармацевтически приемлемую органическую соль, Li+, Na+, K+, Ca2+ или Mg2+.
Новые соединения по настоящему изобретению могут быть получены способами, представленными на схемах 1-5. На схеме 1, A представляет собой O, NR, S или Se. На схеме 2, А представляет собой NR. На схеме 3, А представляет собой O.
[Схема 1]
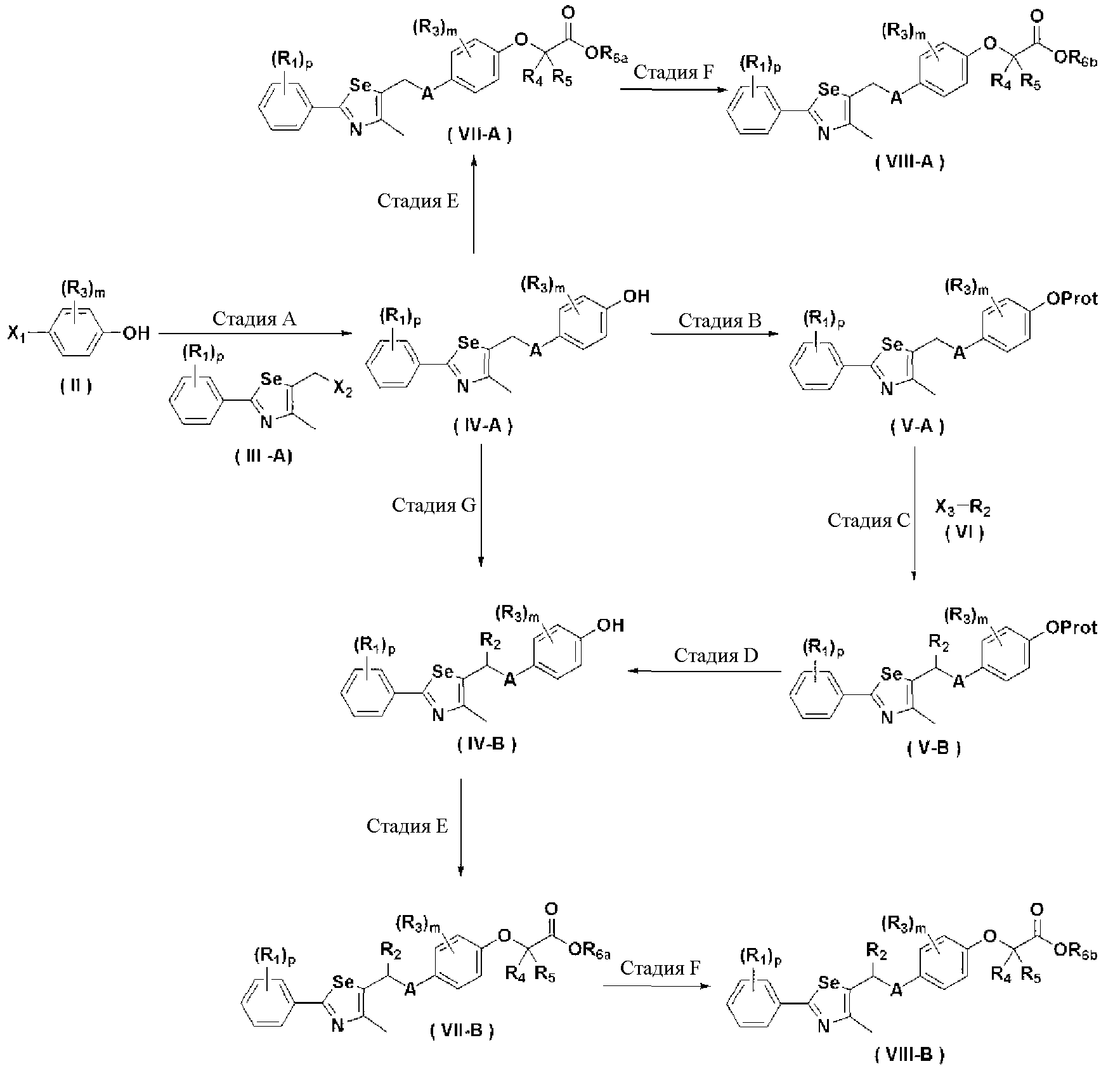
[Схема 2]
[Схема 3]
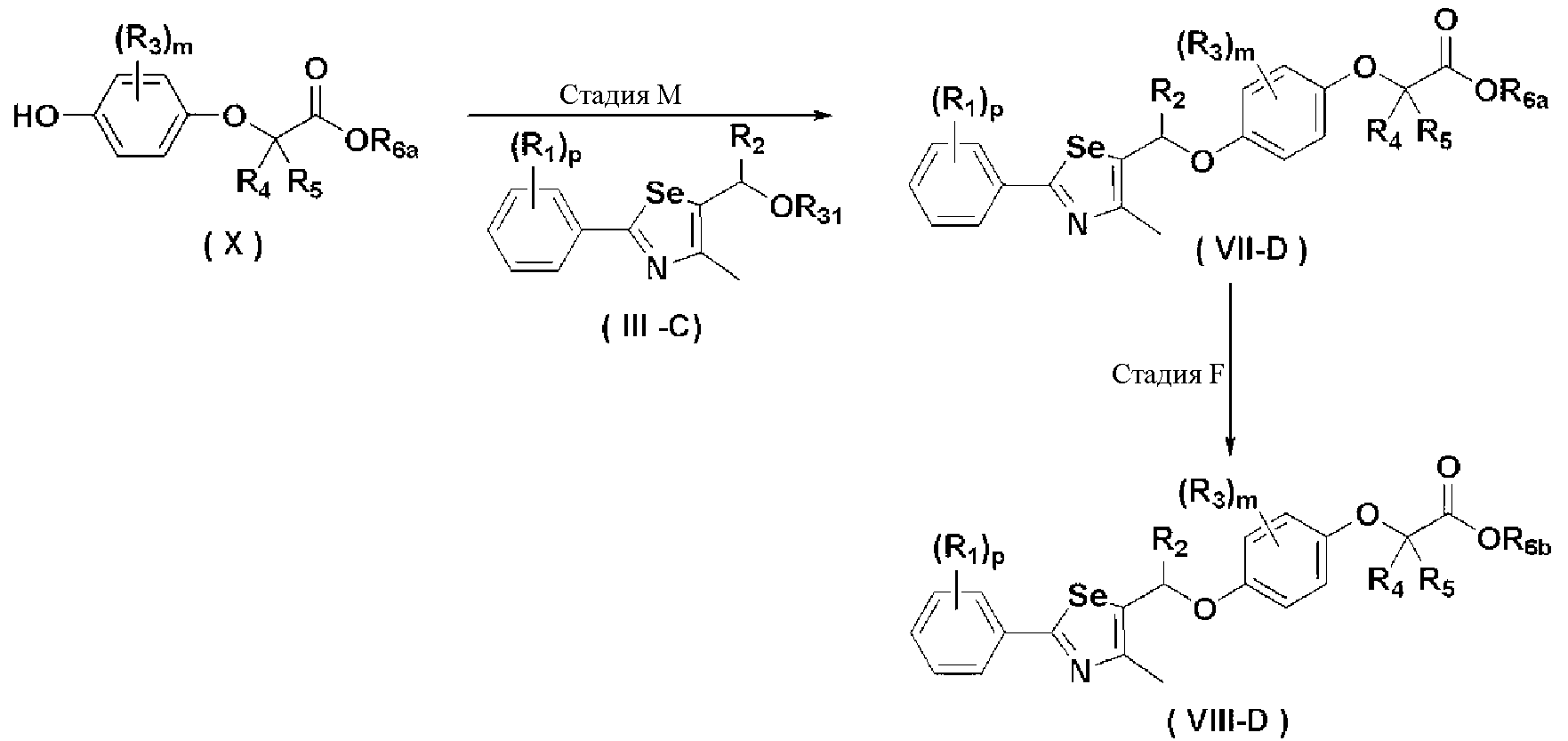
На схемах 1-3, A представляет собой O, NR, S или Se; R1, R2, R3, m, p и s являются такими, как определено для химической формулы I; R6a представляет собой C1-C8 алкил или аллил; R6b представляет собой водород, щелочной металл (Li+, Na+, K+), щелочно-земельный металл (Ca2+, Mg2+) или фармацевтически приемлемую органическую соль; Prot представляет собой защитную группу фенола, выбранную из C1-C4 алкила, аллила, алкилсилила, алкиларилсилила или тетрагидропиранила; X1 представляет собой бром или йод; X2 и X3 независимо представляют собой хлор, бром, йод или другую удаляемую группу, подходящую для нуклеофильного замещения.
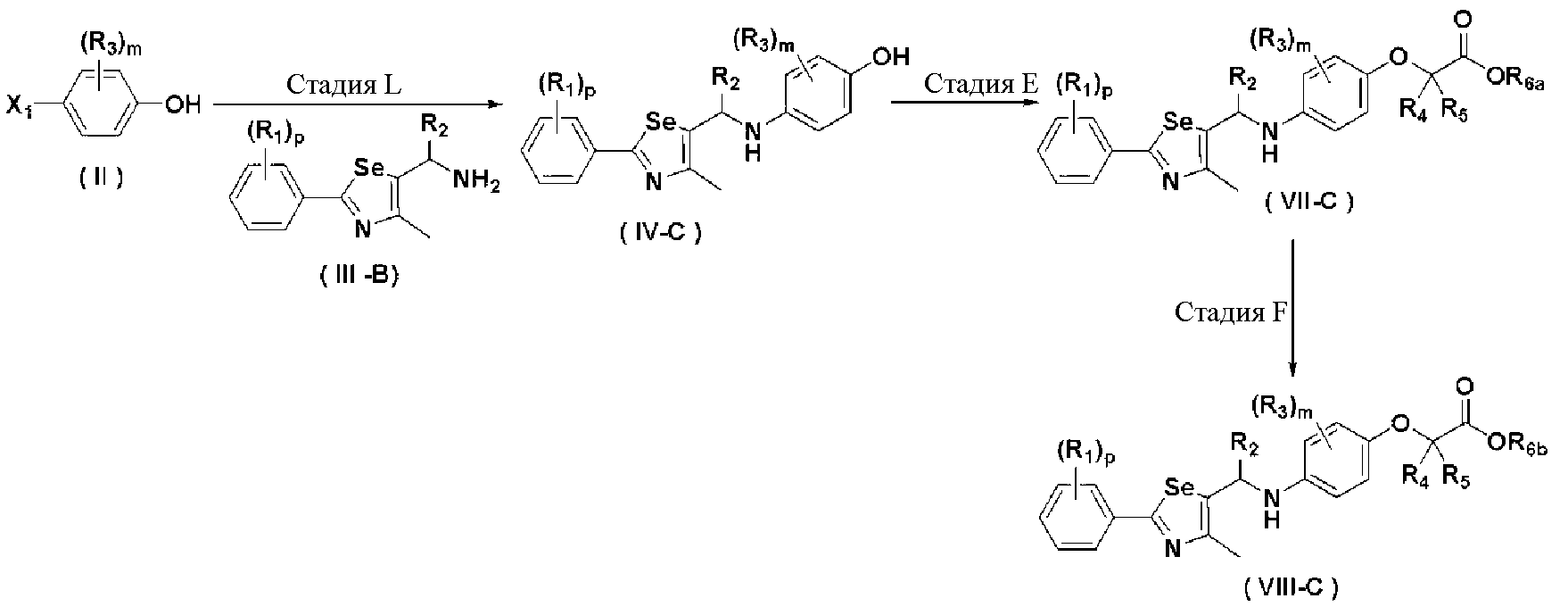
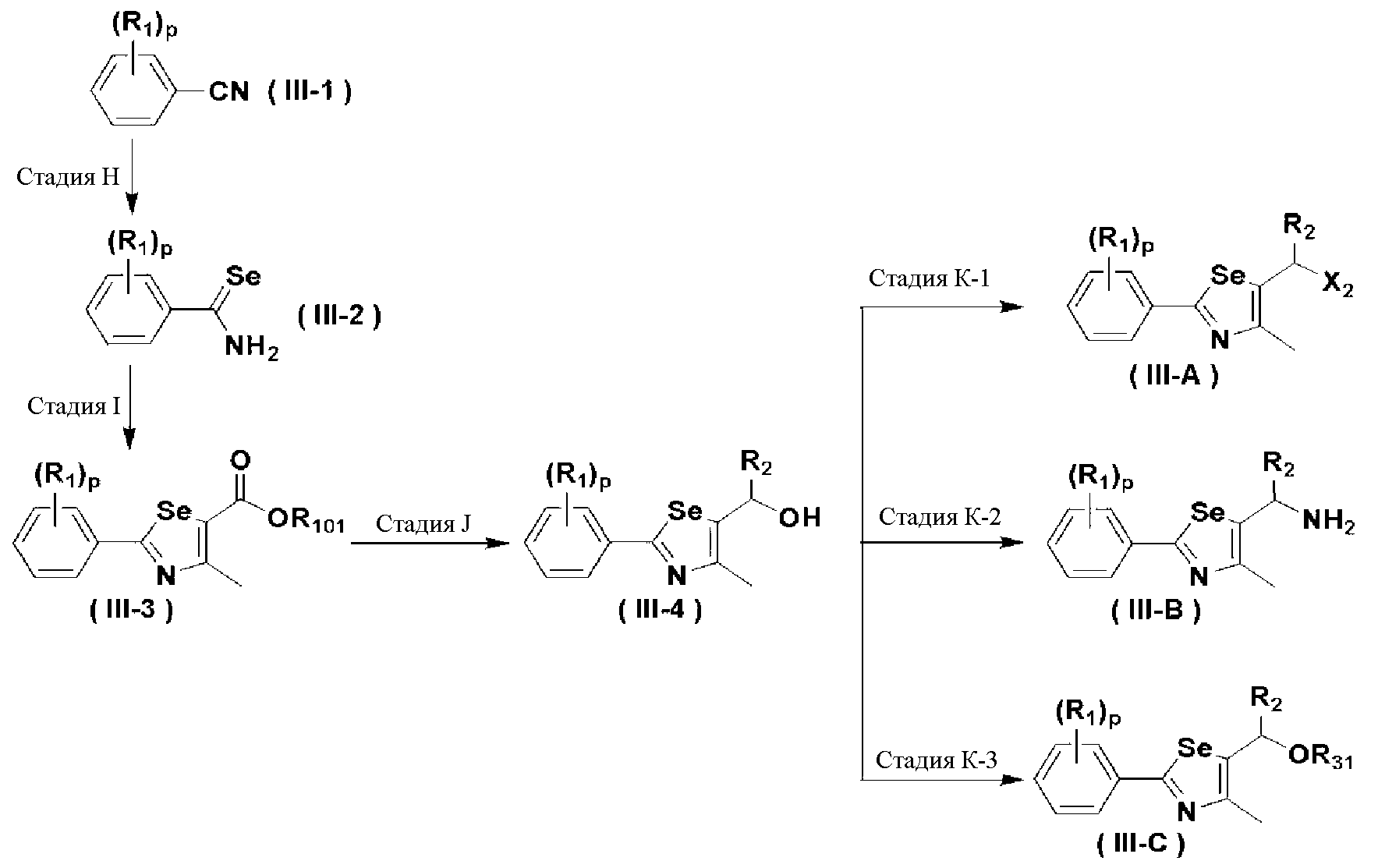
Соединения химической формулы III-A, III-B и III-C могут быть получены в соответствии со схемой 4.
[Схема4]
На схеме 4, R1, R2 и p являются такими, как определено в химической формуле I; R31 представляет собой C1-C4 алкилсульфонил или C6-C12 арилсульфонил, замещенный или незамещенный C1-C4 алкилом; R101 представляет собой C1-C4 алкил; и X2 представляет собой хлор, бром, йод или другую удаляемую группу, подходящую для нуклеофильного замещения.
[Схема 5]
Далее способ получения в соответствии с настоящим изобретением описан более подробно.
[Стадия A] Получение соединения, представленного химической формулой (IV-A)
Для получения соединения, представленного химической формулой (IV-A), в фенольную группу соединения, представленного химической формулой (II), вводят защитную группу с помощью реактива Гриньяра без процесса разделения. Затем полученное соединение подвергают взаимодействию с металлоорганическим реагентом и серой (S) или селеном (Se), и далее с соединением, представленным химической формулой (III-A). Эта стадия включает четырехстадийные взаимодействия, которые выполняются сразу.
Подробное описание представлено далее.
[Защита фенольной группы с помощью реактива Гриньяра]
В качестве безводного растворителя используют диэтиловый эфир, тетрагидрофуран, гексан, гептан или смесь двух или более этих веществ. Из них диэтиловый эфир, тетрагидрофуран или смешанный растворитель, состоящий из диэтилового эфира и тетрагидрофурана, являются предпочтительными. Особенно предпочтительным является полярный растворитель. Наиболее предпочтительным является тетрагидрофуран.
Реактивом Гриньяра может быть хлорид метилмагния, хлорид этилмагния, хлорид н-пропилмагния, хлорид изопропилмагния, хлорид н-бутилмагния, хлорид втор-бутилмагния или бромид алкилмагния. Из них наиболее предпочтительным является хлорид изопропилмагния ((CH3)2CHMgCl).
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно реакцию проводят при температуре в пределах от -20 до 40ºC, предпочтительно, при температуре в пределах от 0ºC до комнатной температуры (25ºC). Время реакции может изменяться в зависимости от температуры реакции и используемого растворителя. Обычно реакцию проводят в течение 10-60 минут, предпочтительно в течение 10-30 минут.
[Галоген-литий замещение и введение серы (S) или селена (Se)]
При галоген-литий замещении может быть использован металлоорганический реагент, такой как н-бутиллитий, втор-бутиллитий, трет-бутиллитий и так далее. Из них, трет-бутиллитий является предпочтительным.
Предпочтительно, сера (S) или селен (Se) представлены в виде тонкодисперсного порошка и добавлялись непосредственно или в виде раствора в безводном тетрагидрофуране.
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно, взаимодействие осуществляют при температуре от -78 до 25ºC. Предпочтительно, галоген-металл замещение осуществляют при -75ºC, и введение серы (S) или селена (Se) начинают при температуре -75ºC и проводят при комнатной температуре (25ºC). Галоген-металл замещение осуществляют в течение от 10 до 30 минут, и введение серы (S) или селена (Se) осуществляют в течение от 30 до 120 минут.
[Добавление соединения, представленного химической формулой (III-A)]
Соединение, представленное химической формулой (III), синтезируют посредством стадий H и K. Галогеном у соединения, представленного химической формулой (III-A), может быть хлор, бром или йод. Из них хлор является предпочтительным.
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно, реакцию проводят при температуре от -78 до 25ºC, предпочтительно, при температуре от 0 до 10ºC. Время реакции обычно составляет от 10 до 120 минут, предпочтительно, от 10 до 60 минут.
[Стадия B] Получение соединения, представленного химической формулой (V-A)
Для получения соединения, представленного химической формулой (V-A), соединение, представленное химической формулой (IV-A), может быть подвергнуто взаимодействию с соединением, обычно используемым для введения защитной группы фенола в присутствии основания.
Защитной группой фенола может быть C1-C4 алкил, аллил, алкилсилил, такой как триметилсилил, трет-бутилдифенилсилил, триизопропилсилил, трет-бутилдиметилсилил и так далее, алкиларилсилил, тетрагидропиранил или тому подобное. Из них, трет-бутил, тетрагидропиранил и силил являются предпочтительными.
На этой стадии может быть использован апротонный полярный растворитель, такой как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ацетонитрил, ацетон, этилацетат, тетрахлорид углерода, хлороформ, дихлорметан или тому подобное. Также может быть использован простой эфир, такой как тетрагидрофуран, диоксан, диметоксиэтан, диметиловый эфир диэтиленгликоля, диметиловый эфир триэтиленгликоля или тому подобное. Также может быть использован ароматический углеводород, такой как бензол, толуол, ксилол или тому подобное. Из них, апротонный полярный растворитель является предпочтительным. Наиболее предпочтительными являются N,N-диметилформамид, хлороформ и дихлорметан.
Основанием может быть основание на основе амина, такое как пиридин, триэтиламин, имидазол, N,N-диметиламинопиридин или тому подобное. Реакцию образования алкильной или аллильной эфирной защитной группы осуществляют с использованием в качестве основания гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия или тому подобное. Из них, имидазол и карбонат калия являются предпочтительными.
Тетрагидропиранильную защитную группу получают путем взаимодействия 3,4-дигидро-2H-пирана с алкил или аллил трифенилфосфонийбромидом в присутствии катализатора.
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно, реакцию проводят при температуре от -10 до 80ºC, предпочтительно, при температуре от 0ºC до комнатной температуры (25ºC). Время реакции может изменяться в зависимости от температуры реакции и используемого растворителя. Обычно, реакцию проводят в течение от 1 часа до 1 дня, предпочтительно, в течение 4 часов или менее.
[Стадия C] Получение соединения, представленного химической формулой (V-B)
Соединение, представленное химической формулой (V-B), получают путем обработки α-протона тио- или селеноэфирного соединения, представленного химической формулой (V-A), с сильным основанием с получением нуклеофила, и далее взаимодействием с различными электрофилами.
На этой стадии, в качестве безводного растворителя используют диэтиловый эфир, тетрагидрофуран, гексан, гептан или смесь двух или более из них. Из них, диэтиловый эфир, тетрагидрофуран или смесь растворителей диэтилового эфира и тетрагидрофурана являются предпочтительными.
Для экстрагирования α-протона может быть использовано сильное основание, такое как трет-бутоксид калия (трет-BuOK), диизопропиламид лития (LDA), н-бутиллитий, втор-бутиллитий, трет-бутиллитий или тому подобное. Из них, LDA является наиболее предпочтительным.
Электрофил, который взаимодействует с нуклеофилом, может быть известным соединением, которое является легкодоступным, или может быть легко получен в соответствии с известным способом. Он может содержать высоко реакционноспособную группу галогена, альдегида или кетона, и его добавляли напрямую или растворенным в безводном растворителе.
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно, реакцию проводят при температуре от -78 до 25ºC. Предпочтительно, экстрагирование α-протона с использованием сильного основания осуществляют при температуре от -75ºC. Электрофил добавляли при температуре -75ºC и потом температуру медленно повышают до комнатной температуры (25ºC). Время реакции может изменяться в зависимости от стадий. Экстрагирование α-протона с использованием сильного основания осуществляют в течение от 10 до 30 минут, и взаимодействие с электрофилом осуществляют в течение от 30 до 90 минут.
[Стадия D] Получение соединения, представленного химической формулой (IV-B)
Соединение, представленное химической формулой (IV-B), получают путем удаления защитной группы фенола у соединения, представленного химической формулой (V-B).
На этой стадии может быть использован полярный растворитель, такой как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ацетонитрил, ацетон, этилацетат, тетрахлорид углерода, хлороформ, дихлорметан или тому подобное. В качестве эфира может быть использован тетрагидрофуран, диоксан, диметоксиэтан, диэтиленгликоль диметиловый эфир или тому подобное. В качестве спирта может быть использован метанол, этанол или тому подобное. В качестве ароматического углеводорода может быть использован бензол, толуол, ксилол или тому подобное. Из них полярный растворитель является предпочтительным. Наиболее предпочтительным является тетрагидрофуран.
Для удаления защитной группы фенола используют кислоту Льюиса, такую как триметилсилил йодид, этантиоалкоголят натрия, йодид лития, галогенид алюминия, галогенид бора, трифторуксусную кислоту и так далее, для метильной, этильной, трет-бутильной, бензильной и аллильной эфирной защитных групп, и используют фторид, такой как тетрабутиламмоний фторид (Bu4N+F-), галогенкислоту (например, фтористоводородную кислоту, хлористоводородную кислоту, бромноватую кислоту или йодноватую кислоту), фторид калия и так далее, для силильных защитных групп, таких как триметилсилил, трет-бутилдифенилсилил, триизопропилсилил, трет-бутилдиметилсилил и так далее. Из них, фторид является предпочтительным для удаления силильной защитной группы. Более предпочтительно, может быть использован тетрабутиламмоний фторид.
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно, реакцию проводят при температуре от 0-120ºC, предпочтительно, при температуре от 10ºC-25ºC. Время реакции может изменяться в зависимости от температуры реакции. Обычно, реакцию проводят в течение от 30 минут до 1 дня, предпочтительно, в течение 2 часов или меньше.
[Стадия E] Получение соединения, представленного химической формулой (VII)
Для получения соединения, представленного химической формулой (VII), соединение, представленное химической формулой (IV), подвергают взаимодействию с алкиловым эфиром галогенуксусной кислоты или алкиловым эфиром алкилгалогенуксусной кислоты в присутствии основания.
Алкиловый эфир галогенуксусной кислоты или алкиловый эфир алкилгалогенуксусной кислоты может быть легко доступным известным соединением. Недоступный алкиловый эфир алкилгалогенуксусной кислоты может быть получен бромированием алкилового эфира алкилуксусной кислоты. Галогеном может быть хлор, бром, йод или тому подобное.
На этой стадии в качестве растворителя может быть использован водорастворимый растворитель, такой как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ацетонитрил, ацетон, этанол и метанол или смесь, содержащая 1-10% воды. Из них ацетон или диметилсульфоксид, содержащий 1-5% воды, является наиболее предпочтительным.
Основанием может быть либо слабое основание, либо сильное основание, без особых ограничений, в той степени, насколько отсутствует отрицательное влияние на реакцию. Сильным основанием может быть гидрид щелочного металла, такой как гидрид натрия, гидрид лития и так далее, гидрид щелочноземельного металла, такой как гидрид калия и так далее, или гидроксид щелочного металла, такой как гидроксид натрия, гидроксид калия и так далее. Кроме того, может быть использован карбонат щелочного металла, такой как карбонат лития, карбонат калия, бикарбонат калия, карбонат цезия и так далее. Предпочтительно, основанием является карбонат щелочного металла, более предпочтительно, карбонат калия.
Температуру реакции особо не ограничивают в той степени, насколько она ниже точки кипения растворителя. Однако реакция при высокой температуре не является предпочтительной, поскольку могут протекать побочные реакции. Обычно, реакцию проводят при температуре от 0 до 90ºC. Время реакции может изменяться в зависимости от температуры реакции. Обычно, реакцию проводят в течение от 30 минут до 1 дня, предпочтительно, в течение 30-120 минут.
[Стадия F-1] Получение соединения, представленного химической формулой (VIII).
Соединение, представленное химической формулой (VIII), получают гидролизом сложного эфира карбоновой кислоты соединения, представленного химической формулой (VII), в растворе водорастворимой неорганической соли и спирта, или гидролизом сложного эфира соединения, представленного химической формулой (VII), в растворе 2,0 M гидроксида лития в ТГФ и воде.
На этой стадии используют смешивающийся с водой спиртовой растворитель, такой как метанол или этанол.
В зависимости от конкретной используемой щелочной соли карбоновой кислоты, в качестве основания используют от 0,1 до 3н водный раствор гидроксида щелочного металла, такого как гидроксид лития, гидроксид натрия, гидроксид калия и так далее. Предпочтительно, кислота, используемая для получения соединения, представленного химической формулой (VIII), в качестве карбоновой кислоты, может быть уксусной кислотой, бисульфатом натрия (NaHSO4) или от 0,1 до 3н HCl. Обычно, для получения соединения, представленного химической формулой (VIII), в качестве карбоновой кислоты, может быть использован 0,5M NaHSO4.
Низкая температура реакции является предпочтительной для предотвращения побочных реакций. Обычно, реакцию проводят при температуре от 0ºC до комнатной температуры. Время реакции может изменяться в зависимости от температуры реакции. Обычно, реакцию проводят в течение от 10 минут до 3 часов, предпочтительно, в течение от 30 минут до 1 часа. В том случае, когда реакцию проводят в растворе 2,0 M гидроксида лития в ТГФ и воде, температура реакции обычно 0ºC, и время реакции, предпочтительно, составляет 1 до 2 часов.
[Стадия F-2] Получение соединения, представленного химической формулой (VIII).
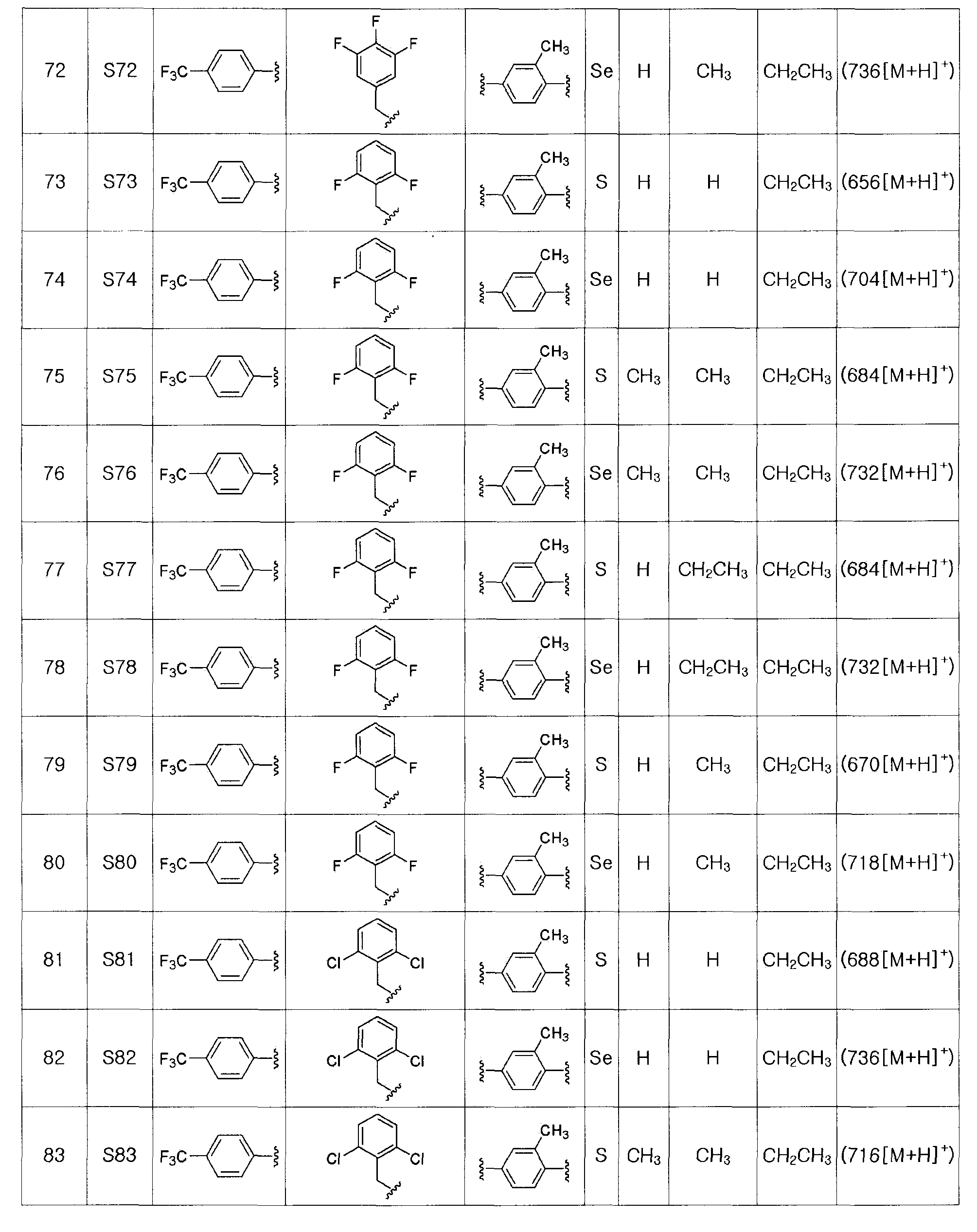
Соединение, представленное химической формулой (VIII), получают замещением соли аллилового эфира соединения, представленного химической формулой (VII), в органическом растворителе с использованием катализатора на основе металла и соли щелочного металла или соли щелочноземельного металла 2-этилгексаноата.
На этой стадии используют безводный органический растворитель, такой как хлороформ, дихлорметан, этилацетат и так далее.
Катализатором на основе металла является тетракис(трифенилфосфин)палладий (Pd(PPh3)4), и катализатор на основе металла может быть использован в количестве от 0,01 до 0,1 эквивалента.
Низкая температура реакции является предпочтительной для предотвращения побочных реакций. Обычно реакцию проводят при температуре от 0ºC до комнатной температуры. Время реакции может изменяться в зависимости от температуры реакции. Обычно, реакцию проводят в течение от 10 минут до 3 часов, предпочтительно, в течение от 30 минут до 1 часа.
Полученную соль соединения отделяют центрифугированием или с использованием ионнообменной смолы. Полученная соль металла соединения, представленного химической формулой (VIII), легче отделяется, чем соль соединения, полученного на стадии F-1 (гидролиз).
[Стадия G] Получение соединения, представленного химической формулой (IV-B)
Соединение, представленное химической формулой (IV-B), получают путем защиты фенольной группы соединения, представленного химической формулой (IV-A), с помощью реактива Гриньяра без процесса разделения, обработкой α-протона полученного тио- или селеноэфира сильным основанием с получением нуклеофила, и последующим взаимодействием с различными электрофилами. Эта стадия включает двух стадийные взаимодействия, которые выполняются сразу.
Далее следует подробное описание.
[Защита фенольной группы с помощью реактива Гриньяра]
На этой стадии в качестве безводного растворителя используют диэтиловый эфир, тетрагидрофуран, гексан, гептан или смесь двух или более этих веществ. Из них наиболее предпочтительным является диэтиловый эфир, тетрагидрофуран или смесь диэтилового эфира и тетрагидрофурана. Особенно предпочтительным является полярный растворитель. Наиболее предпочтительным является тетрагидрофуран.
Реактивом Гриньяра может быть метилмагний хлорид, этилмагний хлорид, н-пропилмагний хлорид, изопропилмагний хлорид, н-бутилмагний хлорид, втор-бутилмагний хлорид или алкилмагний бромид. Из них наиболее предпочтительным является изопропилмагний хлорид ((CH3)2CHMgCl).
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно реакцию проводят при температуре от -20 до 40ºC, предпочтительно, в пределах от 0ºC до комнатной температуры (25ºC). Время реакции может изменяться в зависимости от температуры реакции и используемого растворителя. Обычно, реакцию проводят в течение 10-60 минут, предпочтительно, в течение 10-30 минут.
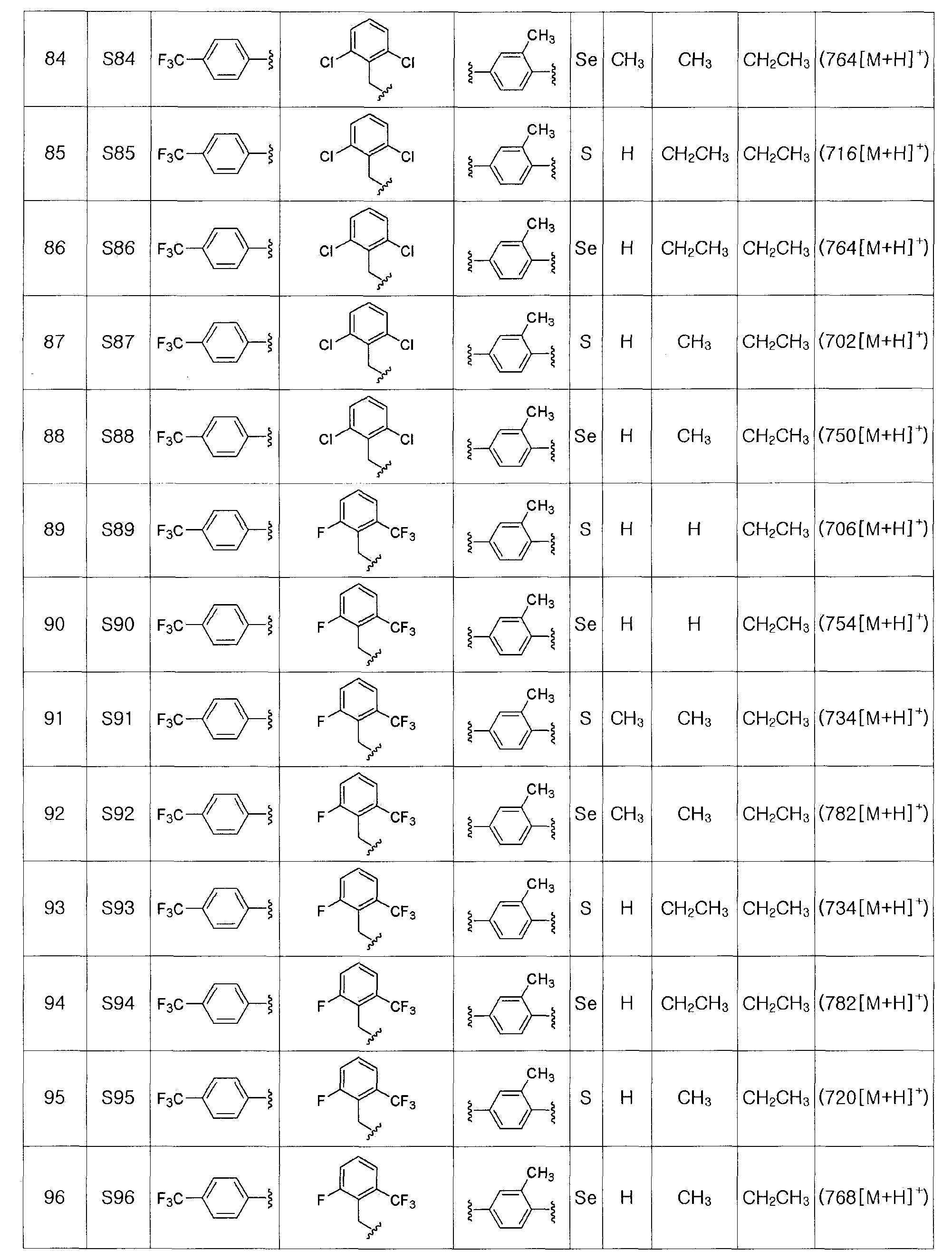
[Экстракция α-протона и электрофильное присоединение]
α-Протон тио- или селеноэфира обрабатывали сильным основанием с получением нуклеофила, который затем взаимодействует с различными электрофилами.
На этой стадии в качестве безводного растворителя используют диэтиловый эфир, тетрагидрофуран, гексан, гептан или смесь двух или более этих веществ. Из них, диэтиловый эфир, тетрагидрофуран или смесь диэтилового эфира и тетрагидрофурана являются наиболее предпочтительными.
Реагентом сильного основания, используемым для экстрагирования α-протона, может быть трет-бутоксид калия (трет-BuOK), диизопропиламид лития (LDA), н-бутиллитий, втор-бутиллитий, трет-бутиллитий или тому подобное. Из них LDA является наиболее предпочтительным.
Электрофил, который подвергают взаимодейставию с нуклеофилом тио- или селеноэфиром, может быть легко доступным известным соединением или может быть легко получен известным способом. Он может содержать высоко реакционноспособную группу галогена, альдегида или кетона, и его добавляли напрямую или растворенным в безводном растворителе.
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно реакцию проводят при температуре в пределах от -78 до 25ºC. Предпочтительно, экстрагирование α-протона с использованием сильного основания выполняют при -75ºC. Электрофил добавляли при -75ºC и потом температуру медленно повышают до комнатной температуры (25ºC). Время реакции может изменяться в зависимости от стадии. Экстрагирование α-протона с использованием сильного основания проводят в течение 10-30 минут, и взаимодействие с электрофилом проводят в течение 30-90 минут.
[Стадия H] Получение соединения, представленного химической формулой (III-2)
Соединение, представленное химической формулой (III-2), может быть получено восстановлением металлического селена сильным восстанавливающим агентом - боргидридом натрия в спиртовом растворителе с получением гидроселенида натрия, взаимодействия его с арилнитрильным соединением, представленным химической формулой (III-1), в среде сильной кислоты, такой как HCl, при нагревании с обратным холодильником с получением селенокарбамата. На этой стадии в качестве растворителя используют спирт, такой как метанол и этанол, а также большое количество пиридина. Предпочтительно, используют боргидрид натрия и порошок металлического селена в эквивалентных количествах и используют 2-3 M HCl кислоту.
[Стадия I] Получение соединения, представленного химической формулой (III-3)
Соединение, представленное химической формулой (III-3), получают взаимодействием соединения, представленного химической формулой (III-2), с C1-C4 алкил 2-хлорацетоацетатом.
На этой стадии в качестве растворителя могут быть использованы спирт, такой как метанол, этанол, пропанол, бутанол и так далее, или эфир, такой как этиловый эфир, тетрагидрофуран, 1,4-диоксан и так далее. Из них этанол и тетрагидрофуран являются предпочтительными.
Температура реакции может изменяться в зависимости от используемого растворителя. Обычно, реакцию проводят при температуре от 25 до 150ºC, предпочтительно, при 60-120ºC. Время реакции может изменяться в зависимости от температуры реакции и используемого растворителя. Обычно, реакцию проводят в течение 6 час - 1 день, предпочтительно, в течение 16 часов или меньше.
[Стадия J] Получение соединения, представленного химической формулой (III-4)
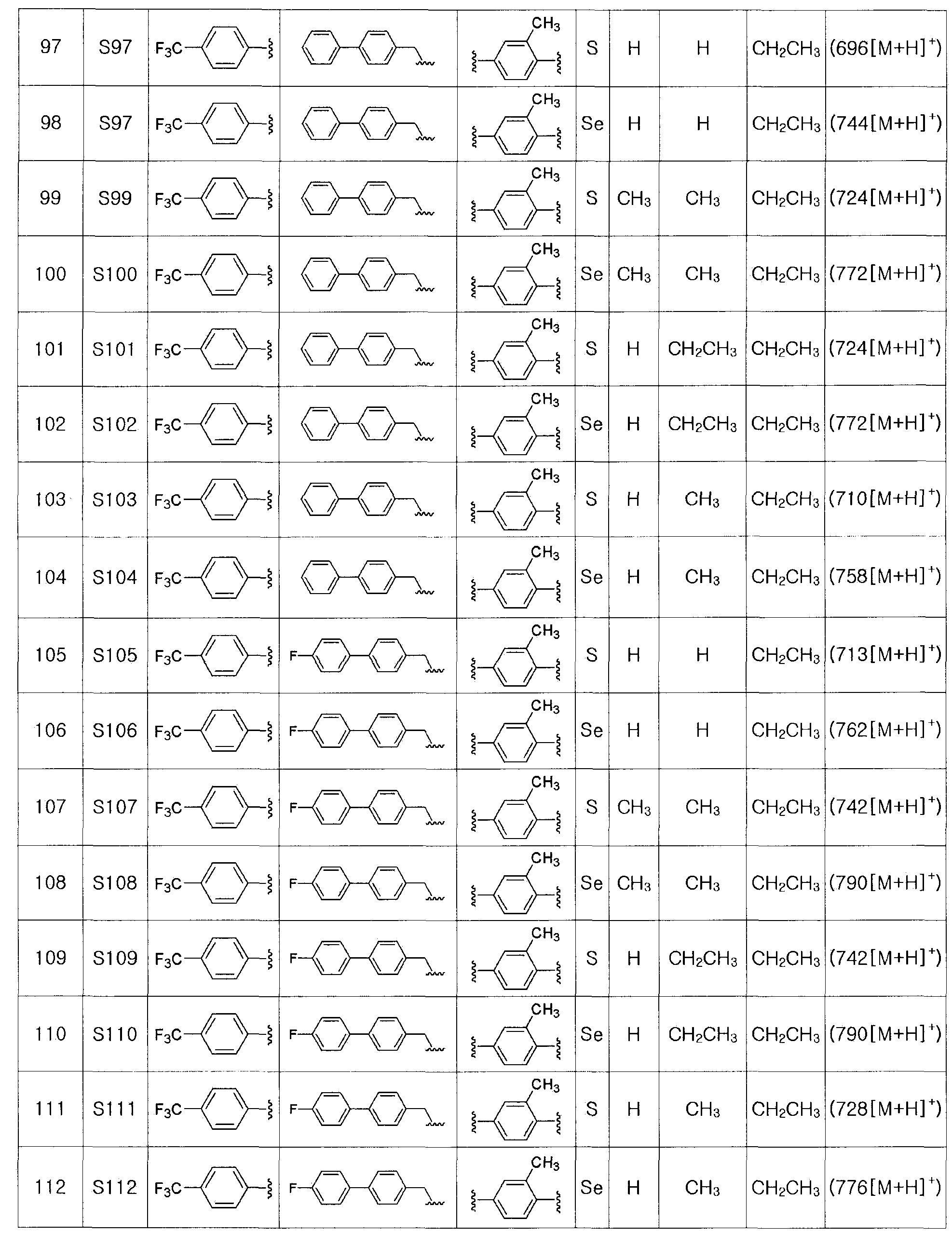
Спиртовое соединение, представленное химической формулой (III-4), получают восстановлением сложного эфира соединения, представленного химической формулой (III-3), с использованием восстанавливающего агента.
Восстанавливающий агент, используемый для восстановления эфира, может представлять собой восстанавливающий агент - гидрид алюминия, такой как алюмогидрид лития (LiAlH4), гидрид диизобутилалюминия (DIBAL-H) и так далее, или восстанавливающий агент - боргидрид, такой как боргидрид натрия, боргидрид лития и так далее. Из них восстанавливающий агент - гидрид алюминия является предпочтительным. Наиболее предпочтительными являются LiAlH4 и DIBAL-H.
На этой стадии в качестве безводного растворителя могут быть использованы диэтиловый эфир, тетрагидрофуран, дихлорметан или тому подобное. Дихлорметан является наиболее предпочтительным.
Время реакции может изменяться в зависимости от растворителя и используемого восстанавливающего агента. Обычно, реакцию проводят при температуре от -100 до 60ºC, предпочтительно, при -78ºC - 25ºC. Время реакции может изменяться в зависимости от температуры реакции и используемого растворителя. Обычно, реакцию проводят в течение от 30 минут до 6 часов, предпочтительно, в течение 2 часов или меньше.
[Стадия K] Получение соединения, представленного химической формулой (III)
Соединение, представленное химической формулой (III-A), может быть получено путем галогенирования спиртовой группы соединения, представленного химической формулой (III-4). Соединение, представленное химической формулой (III-B), может быть получено из соединения, представленного химической формулой (III-4), с использованием NaN3. И, соединение, представленное химической формулой (III-C), может быть получено введением алкил- или арил-замещенного сульфонилхлорида, предпочтительно, метансульфонилхлорида или п-толуолсульфонилхлорида, по гидроксильной группе соединения, представленного химической формулой (III-4).
При галогенировании и введении метансульфонилокси или п-толуолсульфонилокси группы в качестве растворителя может быть использован N,N-диметилформамид, диэтиловый эфир, тетрагидрофуран, тетрахлорид углерода, хлороформ, дихлорметан, пиридин или тому подобное. Из них, дихлорметан является предпочтительным в большинстве случаев при галогенировании, и пиридин является предпочтительным в большинстве случаев при введении метансульфонилокси или п-толуолсульфонилокси группы.
Галогенирование спирта может быть осуществлено с использованием трифенилфосфина (TPP) и N-хлорсукцинимида (NCS), трифенилфосфина и газообразного хлора (Cl2), трифенилфосфина и тетрахлорида углерода (CCl4), пентахлорида фосфора (PCl5), тионилхлорида (SOCl2) или метансульфонил хлорида (MeSO2Cl) или тому подобное для введения хлора, с использованием трифенилфосфина и N-бромсукцинимида (NBS), трифенилфосфина и газообразного брома (BR3), трифенилфосфина и тетрабромида углерода (CBr4), пентабромида фосфора (PBr5) или тионилбромида (SOBr2) или тому подобное для введения брома, и с использованием трифенилфосфина и N-йодсукцинимида, трифенилфосфина и твердого йода, трифенилфосфина и тетрайодида углерода (CI4) или тому подобное для введения йода или путем галоген-йод замещения хлор или бром соединения, представленного химической формулой (IV-A), в ацетоне. Введение метансульфонилокси или п-толуолсульфонилокси группы может быть осуществлено путем взаимодействия с метансульфонилхлоридом или п-толуолсульфонилхлоридом в растворителе пиридине. Наиболее предпочтительной удаляемой группой является хлор или бром, и наиболее предпочтительным способом получения является способ с использованием трифенилфосфина и N-хлорсукцинимида или N-бромсукцинимида.
На этой стадии температура реакции может изменяться в зависимости от способа получения и используемого растворителя. Обычно, реакцию проводят при температуре в пределах от -10 до 40ºC, предпочтительно, при 10-25ºC. Время реакции может изменяться в зависимости от температуры реакции и используемого растворителя. Обычно реакцию проводят в течение от 30 минут до 1 дня, предпочтительно в течение 2 часов или меньше.
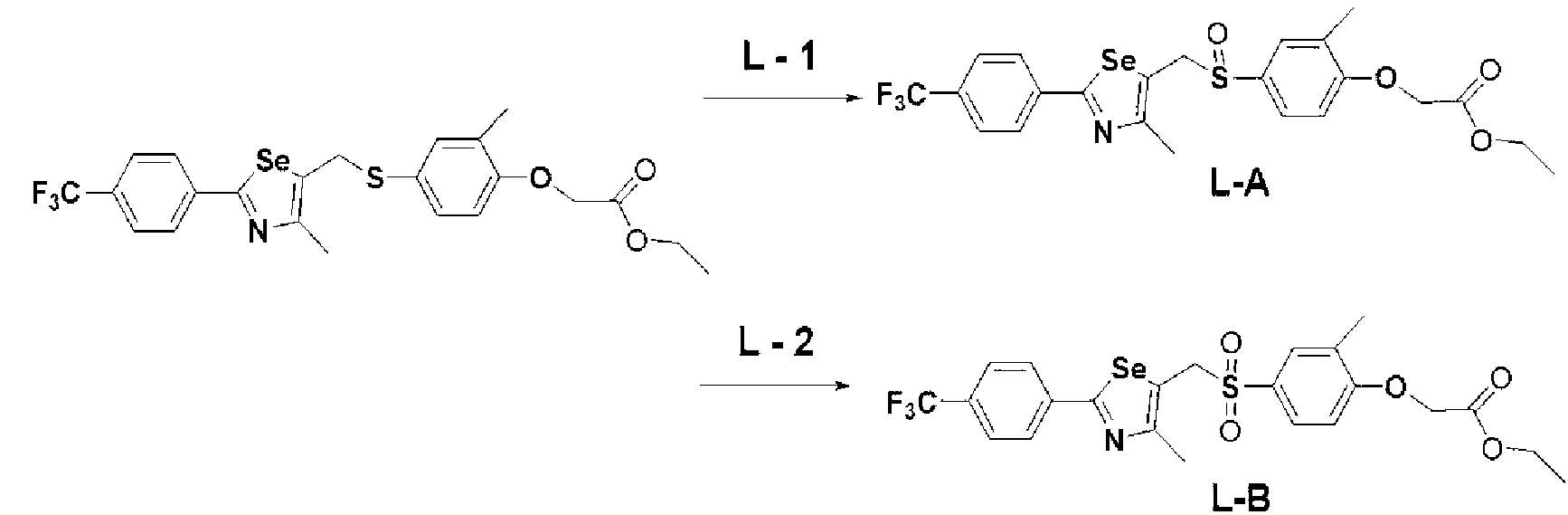
[Стадия L] Получение соединения, представленного химической формулой (L-1) или (L-2)
Соединение, представленное химической формулой (L-1), может быть получено путем растворения соединения, представленного химической формулой (VII), полученного на стадии E, в метиленхлориде (CH2Cl2) и добавления 1 эквивалента м-хлорпербензойной кислоты (м-CPBA), поддерживая температуру реакции при от 0 до 5ºC. И, соединение, представленное химической формулой (L-2), может быть получено добавлением 2 эквивалентов м-CPBA.
Полученное таким образом соединение, представленное химической формулой I, является важным лигандом белка PPAR. Поскольку соединение имеет хиральный углерод, существуют его стереоизомеры. Настоящее изобретение включает соединение производное селеназола химической формулы I, его стереоизомер, его сольват и его соль.
Производное селеназола - соединение, представленное химической формулой I, по настоящему изобретению или фармацевтически приемлемую соль соединения используют в качестве активатора PPAR. Кроме того, производное селеназола, представленное химической формулой I, по настоящему изобретению, его гидрат, сольват, стереоизомер и его фармацевтически приемлемую соль используют для фармацевтической композиции, композиции функциональной пищевой добавки, композиции функционального напитка, композиции пищевой добавки, функциональной косметической композиции или композиции корма для животных для профилактики или лечения атеросклероза, жировой инфильтрации печени или гиперлипемии, профилактики или лечения гиперхолестеримении, профилактики или лечения диабета, профилактики или лечения ожирения, укрепления мышц, улучшения выносливости, улучшения памяти, или профилактики или лечения деменции или болезни Паркинсона, поскольку они активируют PPAR. Производное селеназола, представленное химической формулой I, в соответствии с настоящим изобретением, его гидрат, сольват, стереоизомер и его фармацевтически приемлемую соль используют для функциональной косметической композиции для профилактики или лечения ожирения, профилактики или лечения жировой инфильтрации печени, укрепления мышц или улучшения выносливости. Функциональная косметическая композиция может быть получена в виде мази, лосьона или крема, и может быть нанесена местно на желаемый участок тела до и/или после физических упражнений для укрепления мышц и повышения выносливости. Кроме того, производное селеназола, представленное химической формулой I, в соответствии с настоящим изобретением, его гидрат, его сольват, его стереоизомер и его фармацевтически приемлемая соль могут быть получены в виде мази и нанесено местно с целью профилактики или лечения диабета или диабетической язвы ног.
Фармацевтически приемлемая соль может быть солью карбоновой кислоты производного селеназола, представленного химической формулой I, или любая другая фармацевтически приемлемая органическая соль (например, дициклогексиламин или N-метил-d-глюкамин). Предпочтительная неорганическая соль включает соль щелочного металла и соль щелочноземельного металла Li+, Na+, K+, Ca2+, Mg2+ или тому подобное.
Конечно, количество производного селеназола, представленного химической формулой I, его гидрата, его сольвата, его стереоизомера или его фармацевтически приемлемой соли, требуемое для достижения терапевтического эффекта, зависит от конкретного соединения, способа введения, субъекта, нуждающегося в лечении, и заболевания, требующего лечения, и может быть определено так же, как и для других лекарств. Более предпочтительно, эффективная вводимая доза соединения, представленного химической формулой I, находится в интервале от 1 до 100 мг/кг (вес тела)/день. В объеме дневной эффективной вводимой дозы, оно может быть введено один или несколько раз в день. Также, в зависимости от вида препаратов, оно может быть введено перорально или местно. Фармацевтическая композиция для перорального введения может быть в виде любой существующей формы, включая, например, таблетки, порошки, сухой сироп, жевательные таблетки, гранулы, капсулы, мягкие капсулы, пилюли, питье, сублигвальные таблетки или тому подобное. Таблетки в соответствии с настоящим изобретением могут быть введены пациенту любым биодоступным путем или способом, то есть, посредством перорального пути. Адекватный путь или способ введения может быть легко выбран в зависимости от степени заболевания, которое нужно предотвратить или лечить, развития болезни, или других связанных ситуаций. Когда композицией по настоящему изобретению являются таблетки, они могут содержать один или более фармацевтически приемлемых наполнителей. Состав и свойства наполнителя могут быть определены на основе растворимости и химических свойств выбранных таблеток, выбранного пути введения и обычной фармацевтической практики.
Описание рисунков
Вышеуказанные и другие объекты, признаки и преимущества настоящего изобретения будут ясны из следующего описания предпочтительных вариантов осуществления, приведены в сочетании с прилагаемыми чертежами, в которых:
На фиг. 1 показаны результаты исследования лечебного эффекта при жировой инфильтрация печени.
Методы осуществления изобретения
Далее описаны примеры и эксперименты. Следующие примеры и эксперименты представлены только в целях иллюстрации и не предназначены для ограничения объема настоящего изобретения.
[Примеры]
[Препаративный пример 1] Получение соединения III-2a
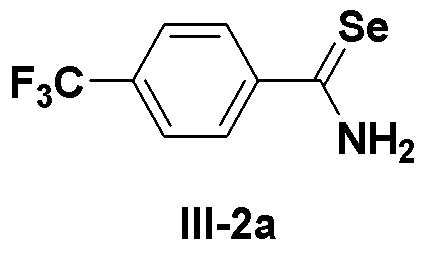
[Стадия H]
К этанолу (50 мл) в атмосфере азота добавляли порошок селена (3,95 г, 50 ммоль). Затем, осторожно добавляли боргидрид натрия (2,02 г, 53 ммоль), медленно, в течение 30 минут (образовывался газообразный водород). К полученному этанольному раствору гидроселенида натрия добавляли 4-(трифторметил)бензонитрил (11,9 г, 70 ммоль) и пиридин (8 мл). Затем медленно, по каплям добавляли в течение 1,5 часа 2M соляную кислоту (25 мл) при кипячении с обратным холодильником при температуре 80ºC. После последующего перемешивания в течение около 30 минут выпавшее в осадок целевое соединение фильтровали и промывали гексаном и водой. Перекристаллизация с использованием растворителя бензола давала соединение III-2a (15,1 г, выход: 91%) в виде твердого продукта желтого цвета.
1H ЯМР (300 МГц, CDCl3) δ 11,07 (шир., 1H), 10,43 (шир., 1H), 7,99 (д, 2H, J=8,5 Гц), 7,77 (д, 2H, J=8,3 Гц).
[Препаративный пример 2] Получение соединения III-3a
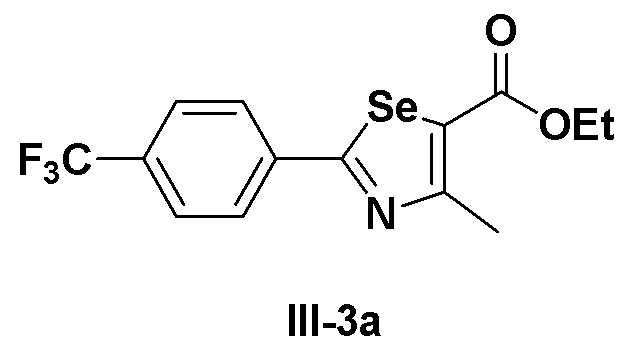
[Стадия I]
Соединение III-2a (2,52 г, 10,0 ммоль) растворяли при комнатной температуре в тетрагидрофуране (35 мл) и медленно в течение 20 минут добавляли этил 2-хлорацетоацетат (1,22 мл, 10,0 ммоль, 1,0 эквивалент). После завершения добавления смесь далее перемешивали при комнатной температуре в течение 30 минут и кипятили с обратным холодильником в течение 12 часов при температуре от 75 до 80ºC. После завершения реакции температуру понижали до комнатной температуры и добавляли 50%-ный водный раствор гидроксида натрия (20 мл). После перемешивания в течение 20 минут органический слой экстрагировали этилацетатом и насыщенным солевым раствором и сушили с помощью сульфата магния. После фильтрования перегонка при пониженном давлении давала соединение III-3a (3,33 г, выход: 95%).
1H ЯМР (300 МГц, CDCl3) δ 8,02 (д, 2H, J=8,1 Гц), 7,69 (д, 2H, J=8,2 Гц), 3,88 (с, 3H), 2,79 (с, 3H).
[Препаративный пример 3] Получение соединения III-4a
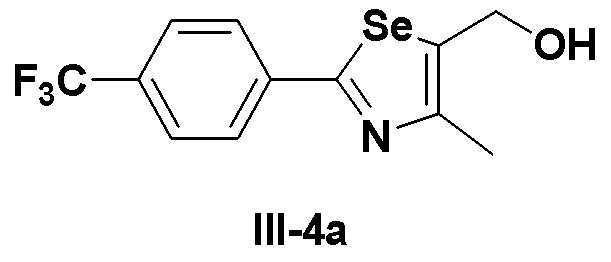
[Стадия J]
Этиловый эфир (соединение III-3a, 2,1 г, 6,0 ммоль), полученный в препаративном примере 2, полностью растворяли в безводном дихлорметане (100 мл) в атмосфере азота и достаточно охлаждали до температуры -78ºC. Медленно, в течение 30 минут добавляли диизобутилалюминий гидрид (DIBAL-H, 16,6 мл, 1,0 M раствор в гексане, 2,5 эквивалента). После проведения взаимодействия при указанной температуре в течение 30 минут, реакцию потом проводили при температуре -10ºC в течение 30 минут. После завершения реакции, реакцию останавливали с использованием этилацетата. После экстракции с помощью 10%-ной серной кислоты и этилацетата продукт сушили с использованием сульфата магния. Фильтрация с использованием короткой колонки с силикагелем и последующей отгонки растворителя при пониженном давлении давала соединение III-4a (1,74 г, выход: 94%).
1H ЯМР (300 МГц, CDCl3) δ 7,96 (д, 2H, J=8,1 Гц), 7,65 (д, 2H, J=8,2 Гц), 4,88 (д, 2H, J=5,3 Гц), 2,45 (с, 3H).
[Препаративный пример 4] Получение соединения III-A-1
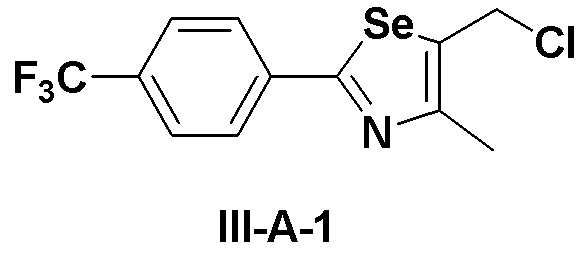
[Стадия K-1]
Соединение III-4a (1,13 г, 3,66 ммоль), полученное в препаративном примере 3, растворяли в безводном дихлорметане (30 мл). Затем добавляли трифенилфосфин (TPP, 1,06 г, 4,03 ммоль, 1,1 эквивалента) и полностью растворяли. При комнатной температуре медленно добавляли N-хлорсукцинимид (717 мг, 4,03 ммоль, 1,1 эквивалента). После дополнительного перемешивания в течение 1 часа растворитель удаляли путем перегонки при пониженном давлении. После высаживания трифенилфосфин оксида с использованием гексана и этилацетата (5:1), фильтрация с последующей перегонкой при пониженном давлении давала соединение III-A-1 (1,07 г, выход: 90%).
1H ЯМР (300 МГц, CDCl3) δ 7,95 (д, 2H, J=8,2 Гц), 7,66 (д, 2H, J=8,3 Гц), 4,84 (с, 2H), 2,48 (с, 3H).
[Препаративный пример 5] Получение соединения III-B-1
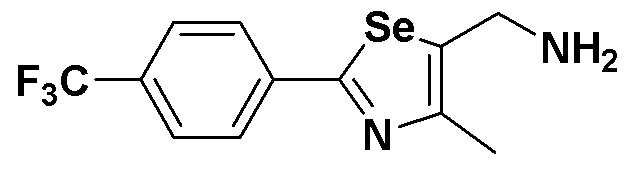
[Стадия K-2]
Соединение III-4a (352 мг, 1,1 эквивалента), полученное в препаративном примере 3, медленно растворяли в CCl4-ДМФ (1:4, 5 мл). После добавления по каплям PPh3 (508 мг, 2,2 эквивалента) и NaN3 (78 мг, 1,2 эквивалента), температуру медленно поднимали до 90ºC. Когда с помощью ТСХ было установлено, что исходный реагент израсходован, температуру понижали до 25ºC. После дальнейшего перемешивания в течение около 10 минут, реакцию останавливали с помощью дистиллированной воды (4 мл). После экстракции этиловым эфиром температуру понижали до 0ºC. Образовавшиеся кристаллы трифенилфосфиноксида удаляли путем фильтрации. Флеш хроматография на силикагеле оставшегося продукта давала соединение III-B-1 (271 мг, выход: 85%) (FABMS: 321[M+H]+).
[Препаративный пример 6] Получение соединения III-C-1
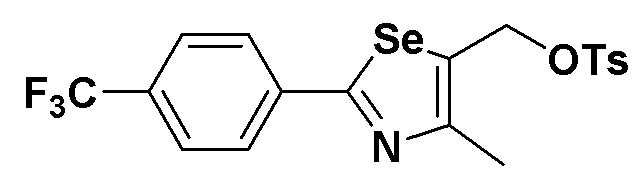
[Стадия K-3]
Соединение III-4a (352 мг, 1,1 эквивалента), полученное в препаративном примере 3, растворяли в метиленхлориде (MC, 5 мл) и охлаждали до температуры 0ºC. Затем, после осторожного добавления п-толуолсульфонилхлорида (п-TsCl, 190 мг, 1,0 эквивалент) и Et3N (1,5 эквивалента), смесь дополнительно перемешивали. После завершения реакции концентрирование органического слоя (MC слой растворителя) с последующей флеш хроматографией на колонке с силикагелем давало соединение III-C-1 (431 мг, выход: 91%) (FABMS: 476[M+H]+).
[Пример 1] Получение соединения S1
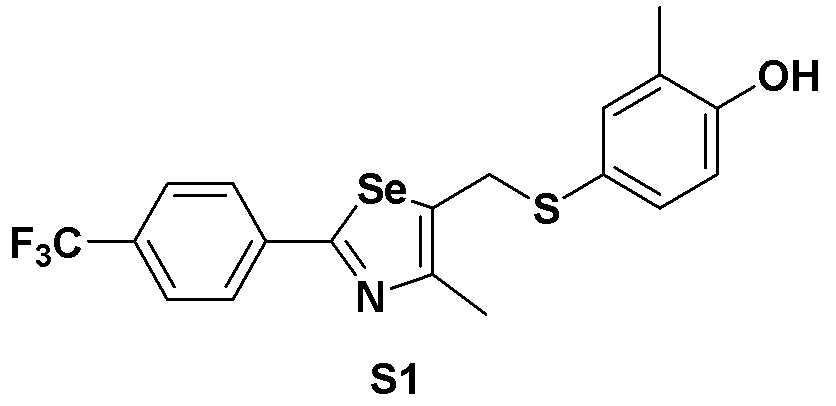
[Стадия A]
4-Йод-2-метилфенол (468 мг, 2 ммоль) растворяли в безводном тетрагидрофуране (20 мл) в атмосфере азота и температуру поддерживали при 0ºC. После медленного добавления изопропилмагнийхлорида (2 M, 1,5 мл) реакцию проводили в течение 10 минут. Смесь достаточно охлаждали до температуры -78ºC и медленно добавляли трет-бутиллитий (2,00 мл, 1,7 M раствор в гексане, 1,0 эквивалент). После перемешивания еще в течение 10 минут, одной порцией добавляли твердую серу (S, 64 мг, 2 ммоль, 1,0 эквивалент) при той же температуре. После проведения реакции в течение 40 минут до повышения температуры до 15ºC, медленно при той же температуре добавляли соединение III-A-1 (652 мг, 2 ммоль, 1,0 эквивалент), полученное в препаративном примере 4, растворенное в безводном ТГФ (10 мл). После дальнейшего взаимодействия в течение около 1 часа, реакцию останавливали водным раствором хлорида аммония. После экстракции органического растворителя этилацетатом и насыщенным солевым раствором, органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (705 мг, выход: 82%).
1H ЯМР (300 МГц, CDCl3) δ 7,91 (д, 2H, J=8,1 Гц), 7,63 (д, 2H, J=8,0 Гц), 7,21 (м, 1H), 7,12 (м, 1H), 6,67 (д, 2H, J=8,2 Гц), 4,15 (с, 2H), 2,19 (с, 3H), 2,17 (с, 3H).
[Пример 2] Получение соединения S2
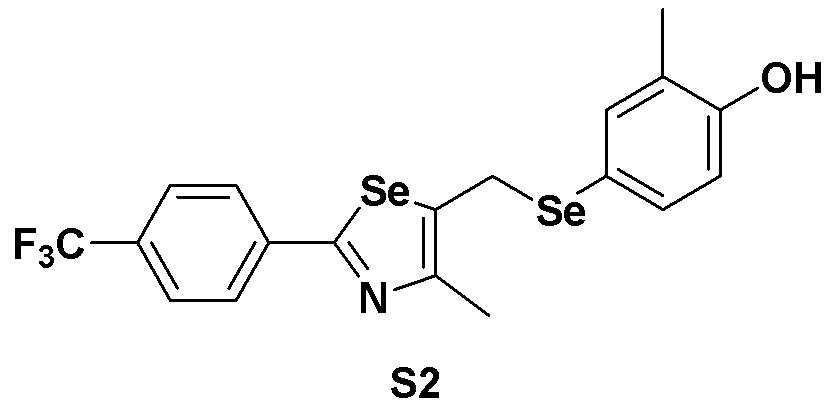
[Стадия A]
4-Йод-2-метилфенол (468 мг, 2 ммоль) растворяли в безводном тетрагидрофуране (20 мл) в атмосфере азота, и температуру поддерживали при 0ºC. После медленного добавления изопропилмагнийхлорида (2 M, 1,5 мл) реакцию проводили в течение 10 минут. Смесь достаточно охлаждали до температуры -78ºC и медленно добавляли трет-бутиллитий (2,00 мл, 1,7 M раствор в гексане, 1,0 эквивалент). После перемешивания еще в течение 10 минут, добавляли одной порцией при той же температуре твердый селен (Se, 158 мг, 2 ммоль, 1,0 эквивалент). После протекания реакции в течение 40 минут до повышения температуры до 15ºC, медленно при той же температуре добавляли соединение III-A-1 (652 мг, 2 ммоль, 1,0 эквивалент), полученное в препаративном примере 4, растворенное в безводном ТГФ (10 мл). После дальнейшего взаимодействия в течение около 1 часа, реакцию останавливали водным раствором хлорида аммония. После экстракции органического растворителя этилацетатом и насыщенным солевым раствором органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (773 мг, выход: 81%).
1H ЯМР (300 МГц, CDCl3) δ 7,89 (д, 2H, J=8,1 Гц), 7,63 (д, 2H, J=8,2 Гц), 7,24 (м, 1H), 7,14 (м, 1H), 6,67 (д, 2H, J=8,2 Гц), 4,17 (с, 2H), 2,18 (с, 3H), 2,13 (с, 3H).
[Пример 3] Получение соединения S3
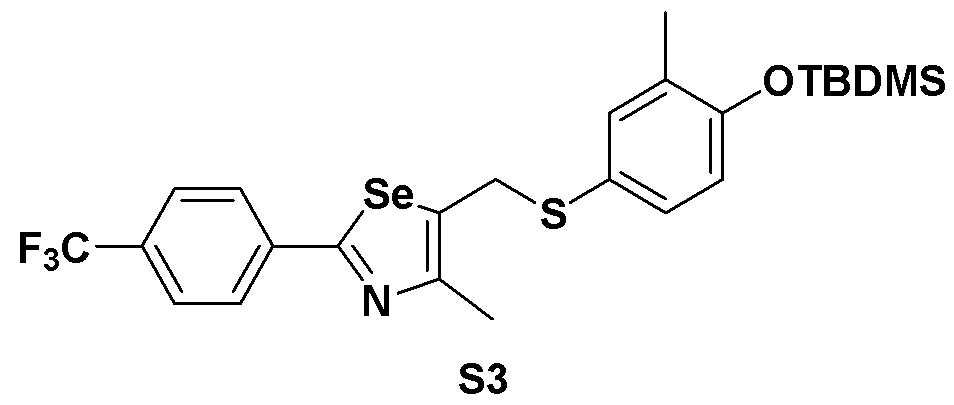
[Стадия B]
Соединение S1 (860 мг, 2 ммоль) и имидазол (290 мг, 2,0 эквивалента) полностью растворяли в диметилформамиде (20 мл). После медленного добавления трет-бутилдиметилсилил хлорида (165 мг, 1,1 эквивалента), реакцию проводили при комнатной температуре в течение 4 часов. После завершения реакции, органический растворитель экстрагировали водным раствором хлорида аммония и этилацетатом, и органический слой сушили с помощью сульфата магния. Очистка с использованием колонки с силикагелем с последующей отгонкой при пониженном давлении давала целевое соединение (1053 мг, выход: 95%). (FABMS: 558 [M+H]+).
[Пример 4] Получение соединения S4
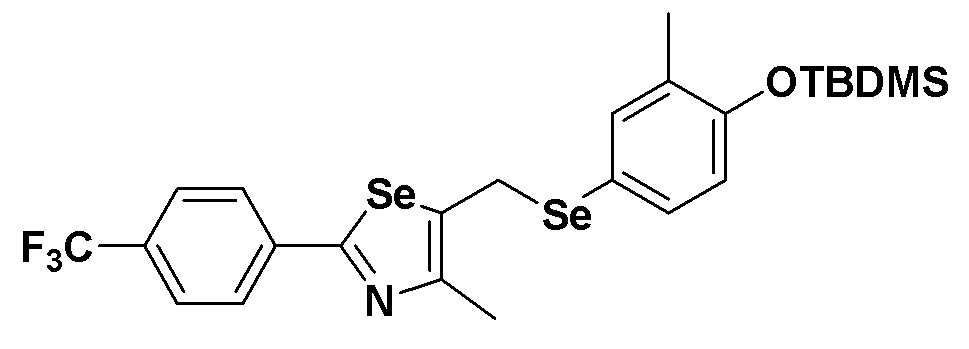
[Стадия B]
Соединение S2 (954 мг, 2 ммоль) и имидазол (290 мг, 2,0 эквивалента) полностью растворяли в диметилформамиде (20 мл). После медленного добавления трет-бутилдиметилсилил хлорида (165 мг, 1,1 эквивалента), реакцию проводили при комнатной температуре в течение 4 часов. После завершения реакции, органический растворитель экстрагировали водным раствором хлорида аммония и этилацетатом, и органический слой сушили с помощью сульфата магния. Очистка на колонке с силикагелем с последующей отгонкой при пониженном давлении давала целевое соединение (1099 мг, выход: 93%). (FABMS: 606[M+H]+).
[Пример 5] Получение соединения S5
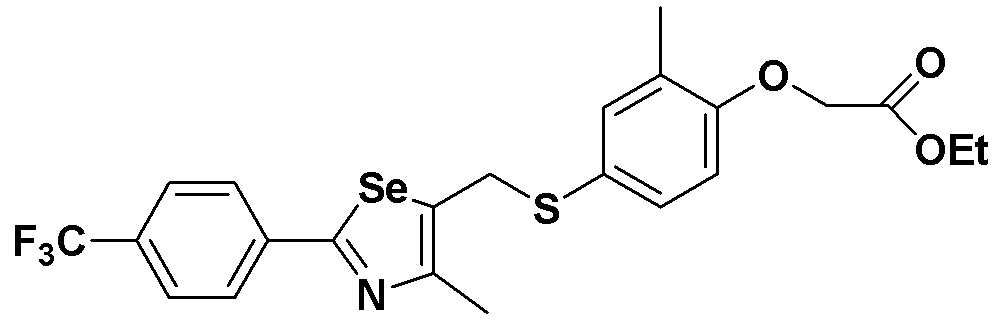
[Стадия E]
Соединение S1 (430 мг, 1 ммоль), ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этилового эфира бромуксусной кислоты (134 мкл, 1,2 ммоль, 1,2 эквивалента) смесь энергично перемешивали в течение 4 часов. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (480 мг, выход: 93%).
1H ЯМР (300 МГц, CDCl3) δ 7,90 (д, 2H, J=8,1 Гц), 7,64 (д, 2H, J=8,2 Гц), 7,25 (м, 1H), 7,14 (м, 1H), 6,67 (д, 2H, J=8,2 Гц), 4,61 (с, 2H), 4,23 (м, 2H), 4,16 (с, 2H), 2,24 (с, 3H), 2,21 (с, 3H), 1,28 (т, 3H, J=3,7 Гц).
[Пример 6] Получение соединения S6
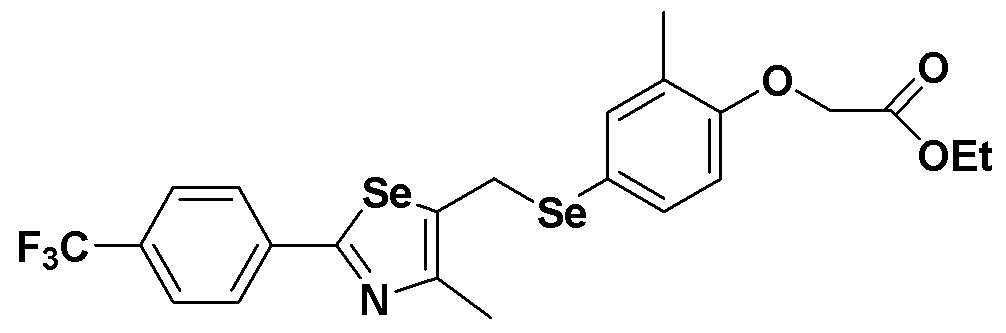
[Стадия E]
Соединение S2 (477 мг, 1 ммоль), ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этилового эфира бромуксусной кислоты (134 мкл, 1,2 ммоль, 1,2 эквивалента) смесь энергично перемешивали в течение 4 часов. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (523 мг, выход: 93%).
1H ЯМР (300 МГц, CDCl3) δ 7,90 (д, 2H, J=8,1 Гц), 7,64 (д, 2H, J=8,2 Гц), 7,27 (м, 1H), 7,20 (м, 1H), 6,68 (д, 2H, J=8,2 Гц), 4,61 (с, 2H), 4,23 (м, 2H), 4,19 (с, 2H), 2,23 (с, 3H), 2,15 (с, 3H), 1,27 (т, 3H, J=3,7 Гц).
[Пример 7] Получение соединения S7
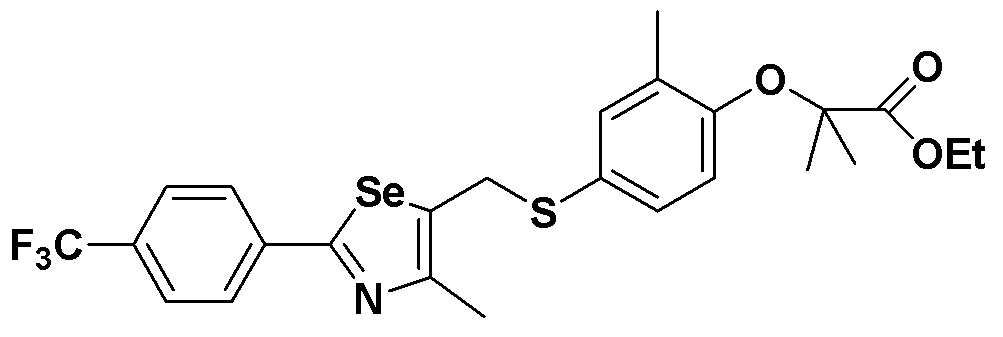
[Стадия E]
Соединение S1 (430 мг, 1 ммоль), ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этил-2-бром-2-метилпропаноата (210 мкл, 1,2 ммоль, 1,2 эквивалента) смесь энергично перемешивали в течение 4 часов, добавляя ацетон, и нагревали при температуре 60-90ºC. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (326 мг, выход: 60%). (FABMS: 558 [M+H]+).
[Пример 8] Получение соединения S8
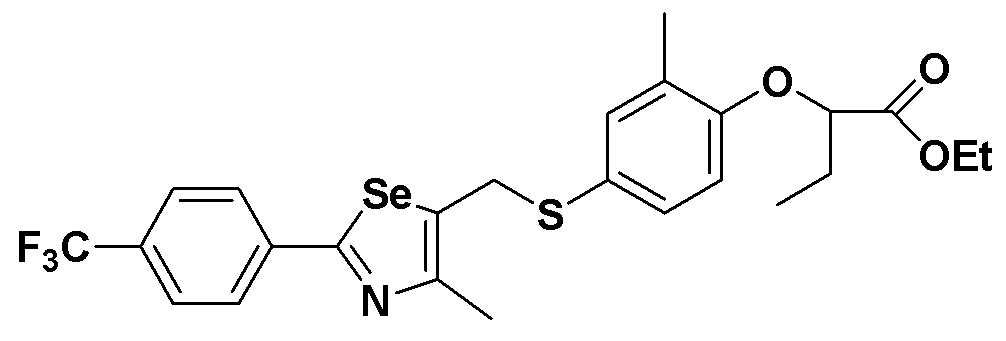
[Стадия E]
Соединение S1 (430 мг, 1 ммоль), ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этил-2-бромбутилата (146 мкл, 1,2 ммоль, 1,2 эквивалента) смесь энергично перемешивали в течение 4 часов, добавляя ацетон, и нагревали при температуре 60-90ºC. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (451 мг, выход: 83%). (FABMS: 558 [M+H]+).
[Пример 9] Получение соединения S9
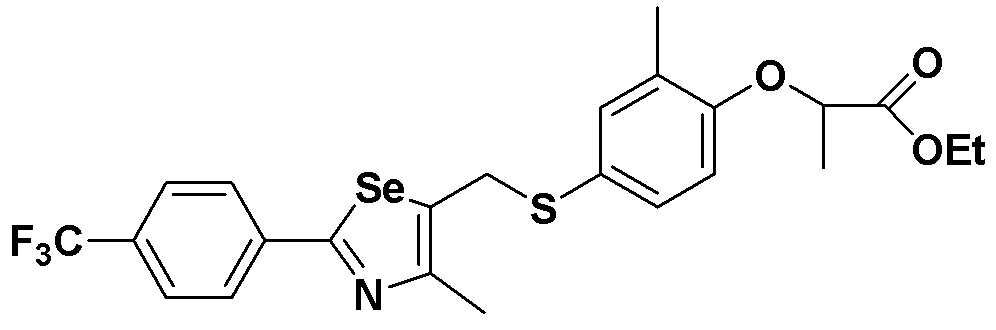
[Стадия E]
Соединение S1 (430 мг, 1 ммоль), ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этил-2-бромпропионата (155 мкл, 1,2 ммоль, 1,2 эквивалента), смесь энергично перемешивали в течение 4 часов, добавляя ацетон, и нагревали при температуре 60-90ºC. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (429 мг, выход: 81%). (FABMS: 544 [M+H]+).
[Пример 10] Получение соединения S10
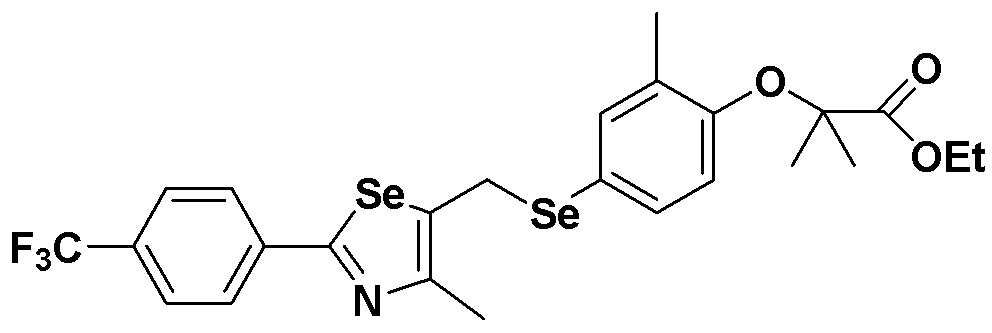
[Стадия E]
Соединение S2 (478 мг, 1 ммоль), ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этил-2-бром-2-метилпропаноата (210 мкл, 1,2 ммоль, 1,2 эквивалента) смесь энергично перемешивали в течение 4 часов, добавляя ацетон, и нагревали при температуре 60-90ºC. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (349 мг, выход: 59%). (FABMS: 606 [M+H]+).
[Пример 11] Получение соединения S11
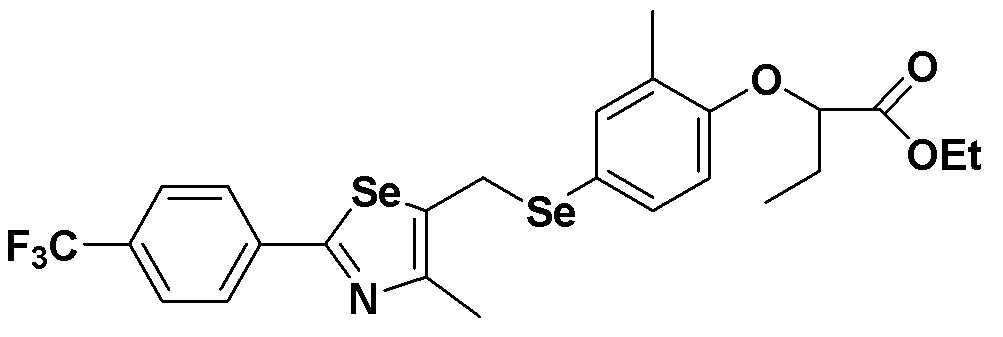
[Стадия E]
Соединение S2 (478 мг, 1 ммоль), ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этил-2-бромбутилата (146 мкл, 1,2 ммоль, 1,2 эквивалента) смесь энергично перемешивали в течение 4 часов, добавляя ацетон, и нагревали при температуре 60-90ºC. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (490 мг, выход: 83%). (FABMS: 606 [M+H]+).
[Пример 12] Получение соединения S12
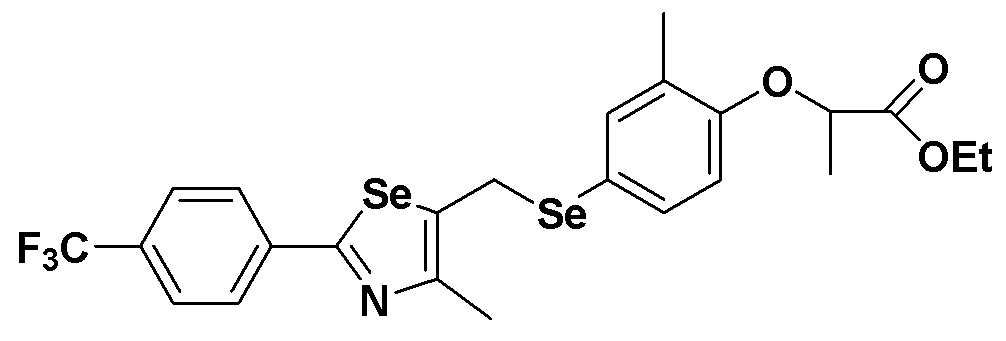
[Стадия E]
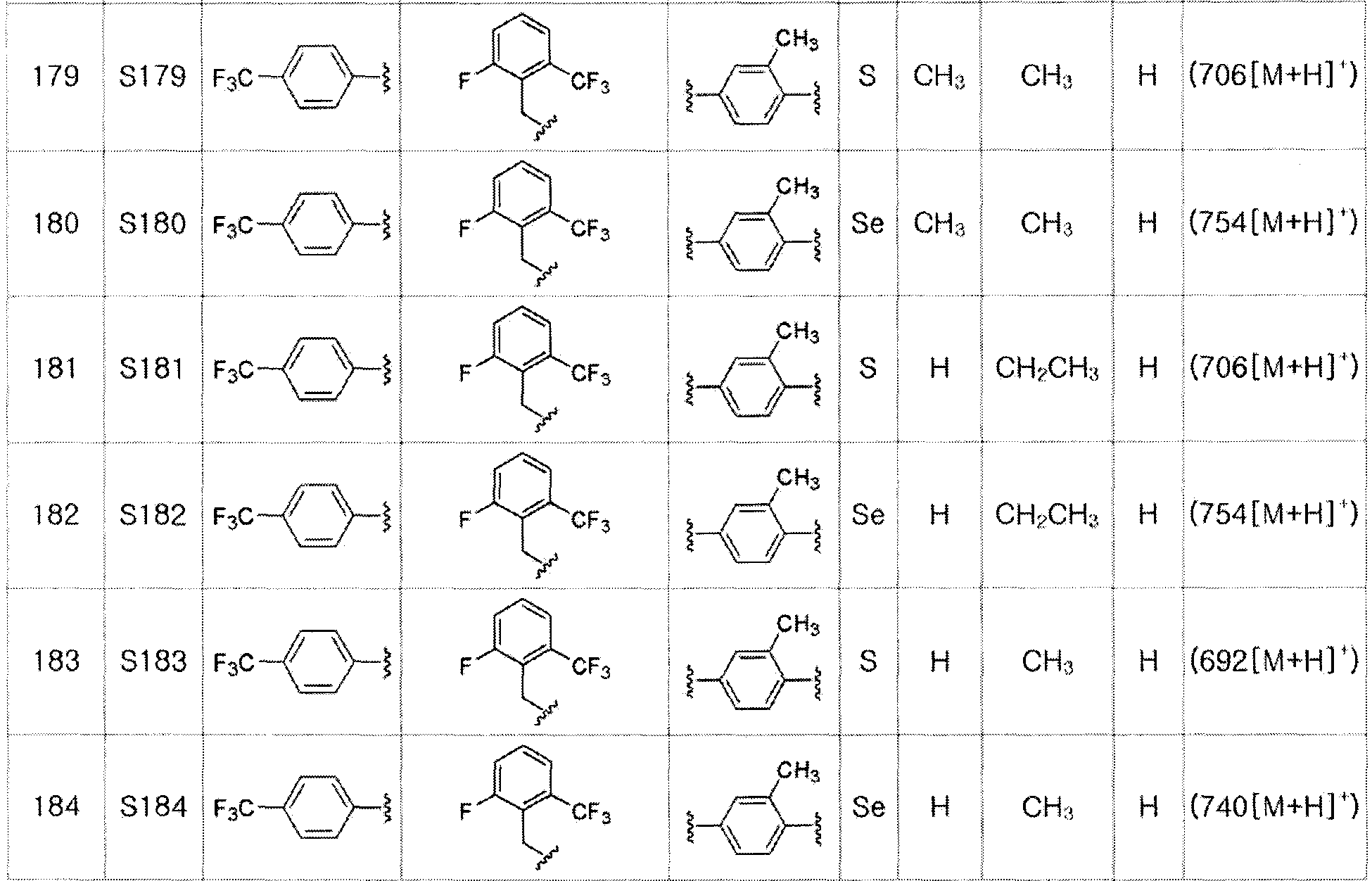
Соединение S2 (478 мг, 1 ммоль), ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этил-2-бромпропионата (155 мкл, 1,2 ммоль, 1,2 эквивалента) смесь энергично перемешивали в течение 4 часов, добавляя ацетон, и нагревали при температуре 60-90ºC. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (462 мг, выход: 80%). (FABMS: 592 [M+H]+).
[Пример 13] Получение соединения S13
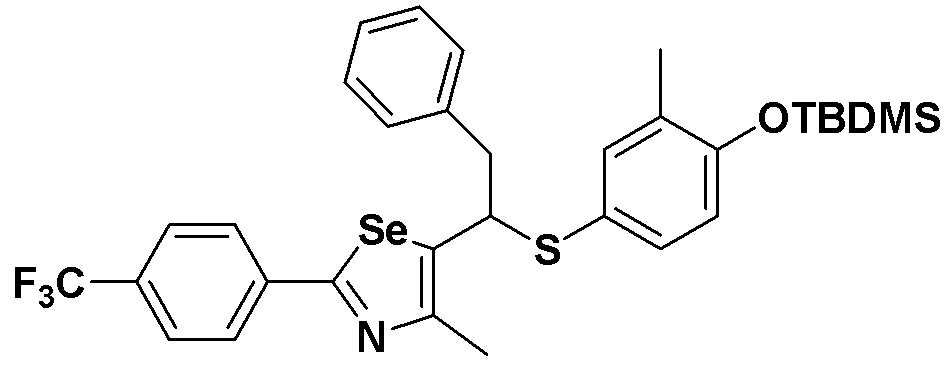
[Стадия C]
Соединение S3 (544 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (10 мл) и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления бензилбромида (137 мкл, 1,0 ммоль) температуру медленно поднимали до комнатной температуры. После дополнительного взаимодействия в течение 30 минут, реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (526 мг, выход: 83%). (FABMS: 648 [M+H]+).
[Пример 14] Получение соединения S14
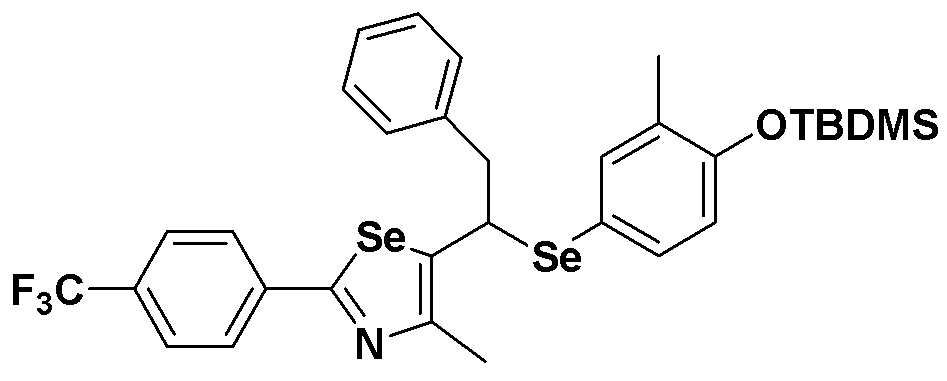
[Стадия C]
Соединение S4 (591 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (10 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления бензилбромида (137 мкл, 1,0 ммоль) температуру медленно поднимали до комнатной температуры. После дальнейшего взаимодействия в течение 30 минут, реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (538 мг, выход: 79%). (FABMS: 694 [M+H]+).
[Пример 15] Получение соединения S15
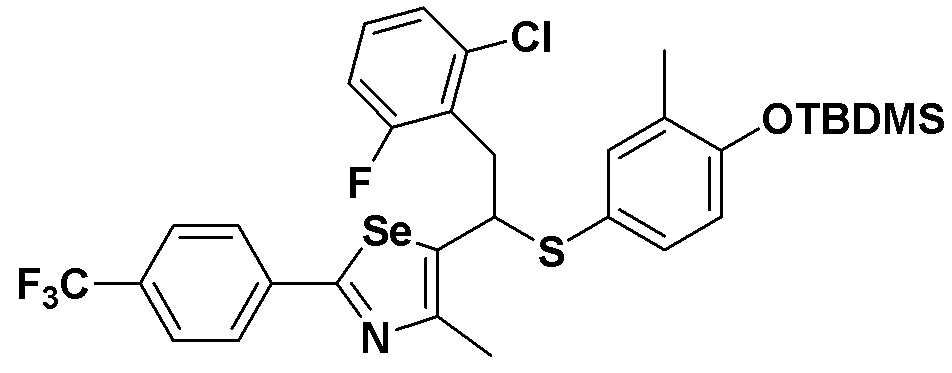
[Стадия C]
Соединение S3 (699 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 2-хлор-5-фторбензилбромида (270 мкл, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дополнительного взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (531 мг, выход: 76%). (FABMS: 700 [M+H]+).
[Пример 16] Получение соединения S16
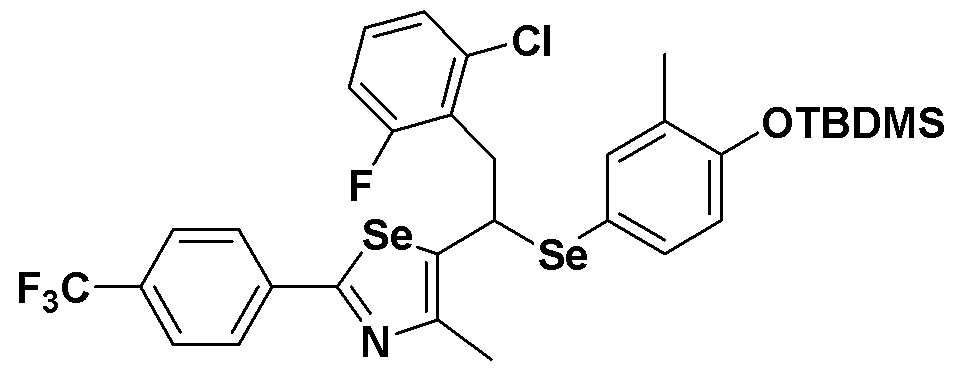
[Стадия C]
Соединение S4 (746 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл, и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 2-хлор-5-фторбензилбромида (270 мкл, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дополнительного взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (552 мг, выход: 74%). (FABMS: 746 [M+H]+).
[Пример 17] Получение соединения S17
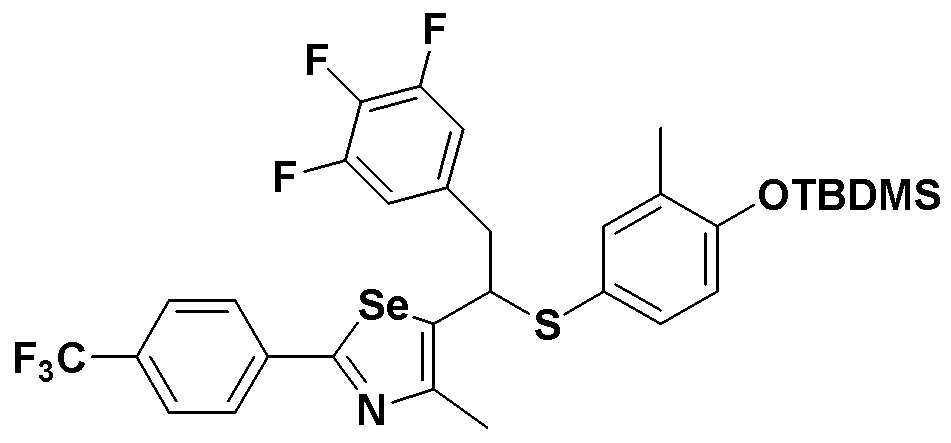
[Стадия C]
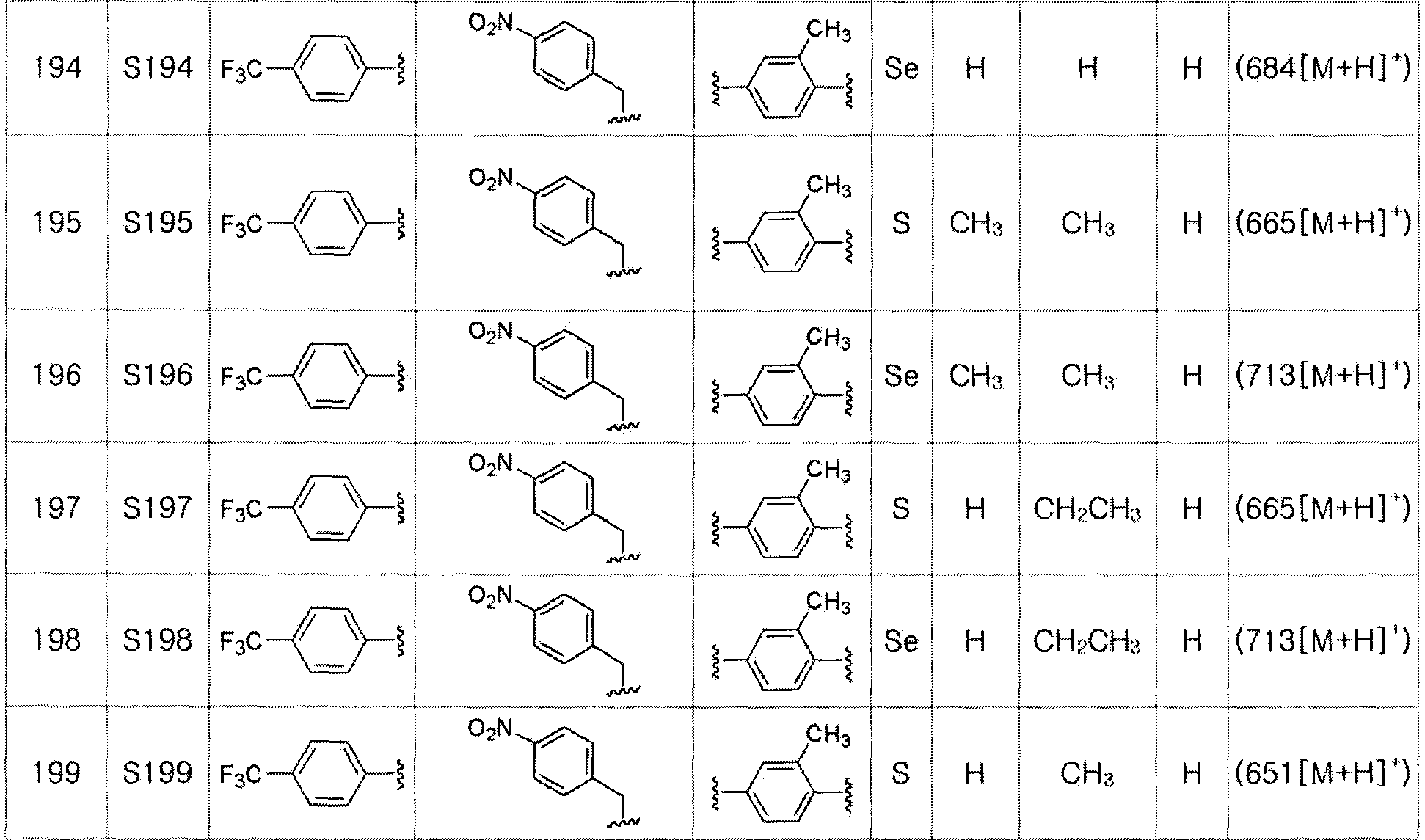
Соединение S3 (700 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 3,4,5-трифторбензилбромида (282 мкл, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дополнительного взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (525 мг, выход: 75%). (FABMS: 702 [M+H]+).
[Пример 18] Получение соединения S18
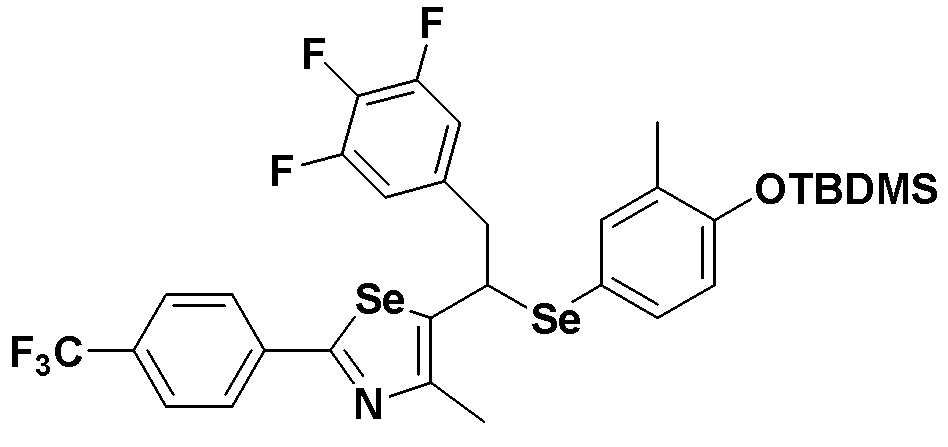
[Стадия C]
Соединение S4 (747 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 3,4,5-трифторбензилбромида (282 мкл, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дальнейшего взаимодействия в течение 30 минут, реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (523 мг, выход: 70%). (FABMS: 750 [M+H]+).
[Пример 19] Получение соединения S19
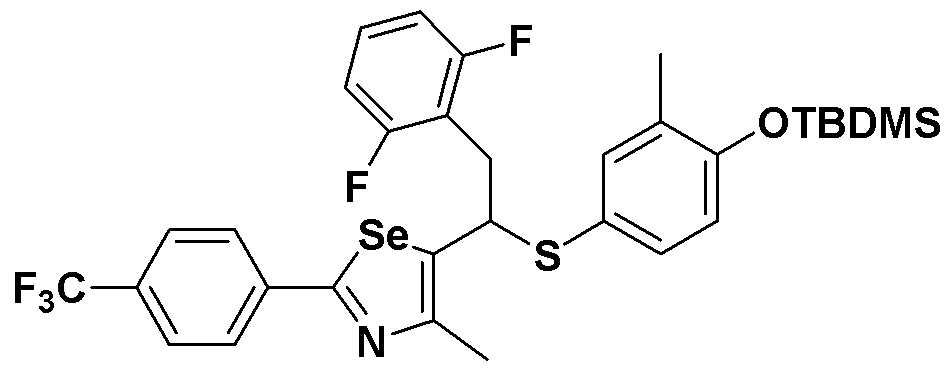
[Стадия C]
Соединение S3 (682 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 2,5-дифторбензилбромида (259 мкл, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дополнительного взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (518 мг, выход: 76%). (FABMS: 684 [M+H]+).
[Пример 20] Получение соединения S20
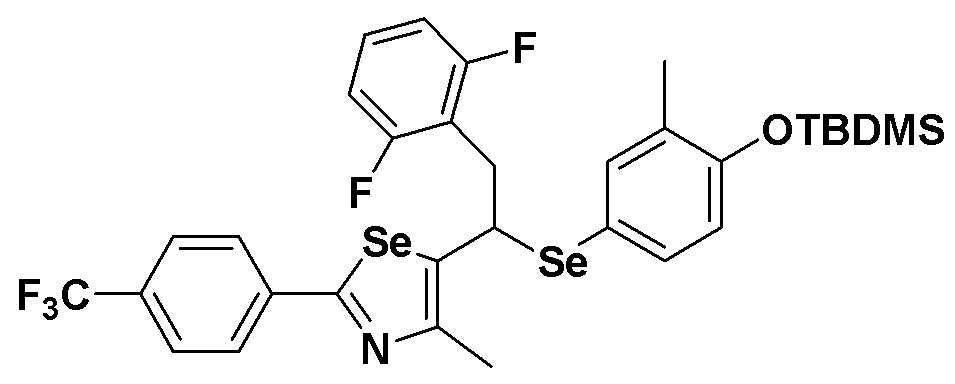
[Стадия C]
Соединение S4 (724 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 2,5-дифторбензилбромида (259 мкл, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дополнительного взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (514 мг, выход: 71%). (FABMS: 732 [M+H]+).
[Пример 21] Получение соединения S21
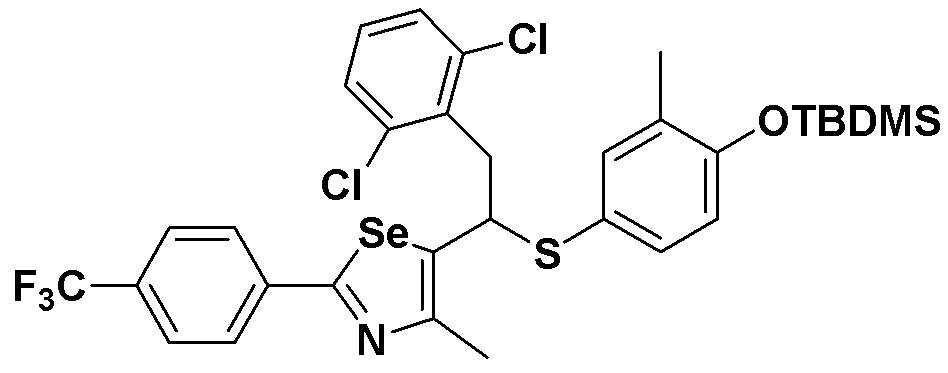
[Стадия C]
Соединение S3 (715 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 2,5-дихлорбензилбромида (300 мкл, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дальнейшего взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (529 мг, выход: 74%). (FABMS: 716 [M+H]+).
[Пример 22] Получение соединения S22
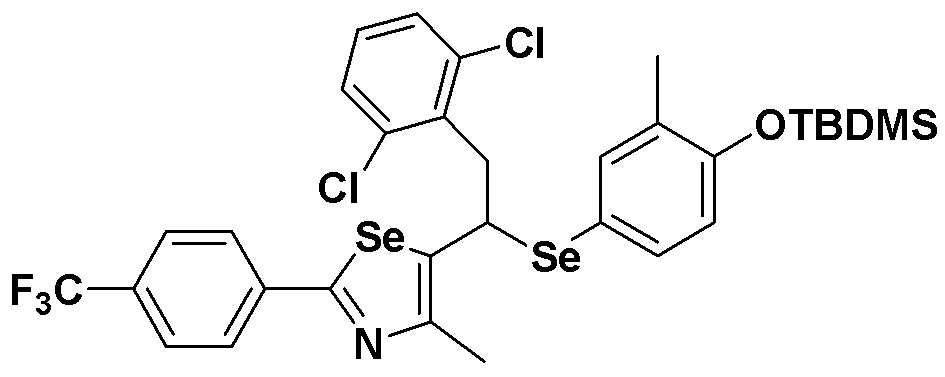
[Стадия C]
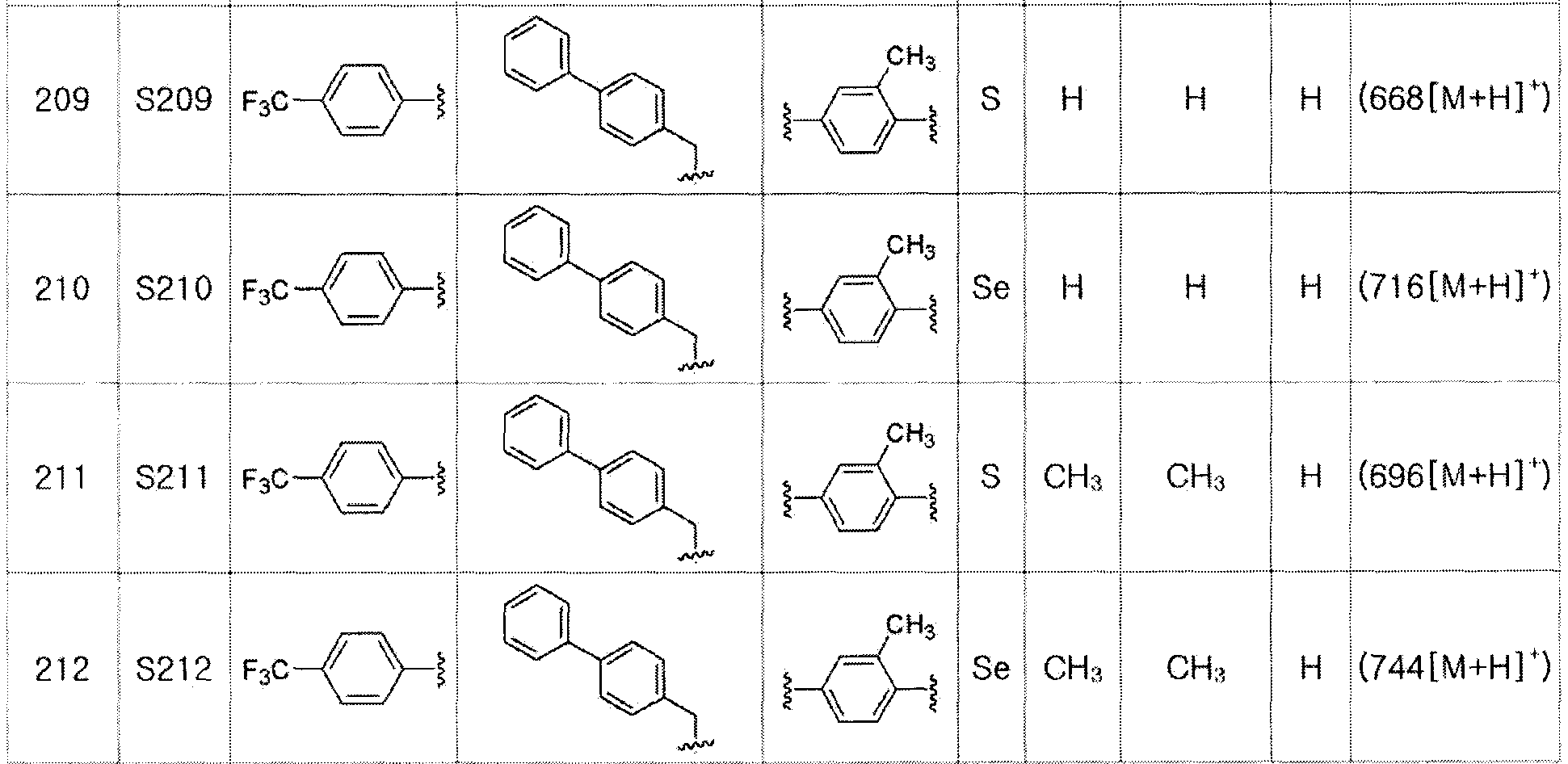
Соединение S4 (762 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 2,5-дихлорбензилбромида (300 мкл, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дальнейшего взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (541 мг, выход: 71%). (FABMS: 762 [M+H]+).
[Пример 23] Получение соединения S23
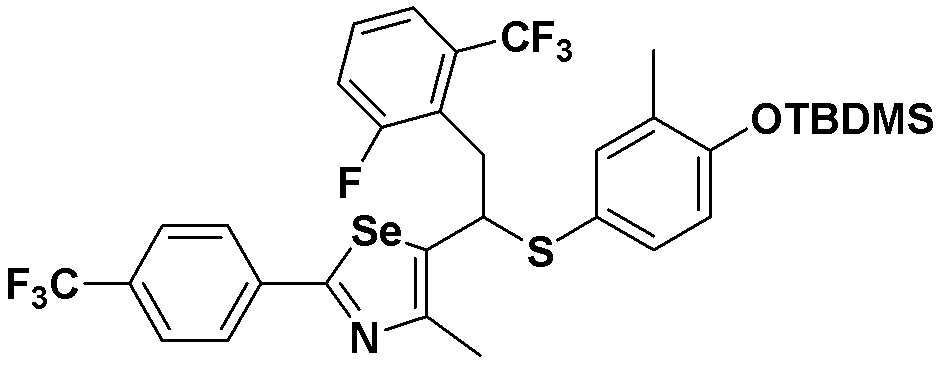
[Стадия C]
Соединение S3 (732 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 2-фтор-5-трифторметилбензилбромида (514 мг, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дополнительного взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (534 мг, выход: 73%). (FABMS: 734 [M+H]+).
[Пример 24] Получение соединения S24
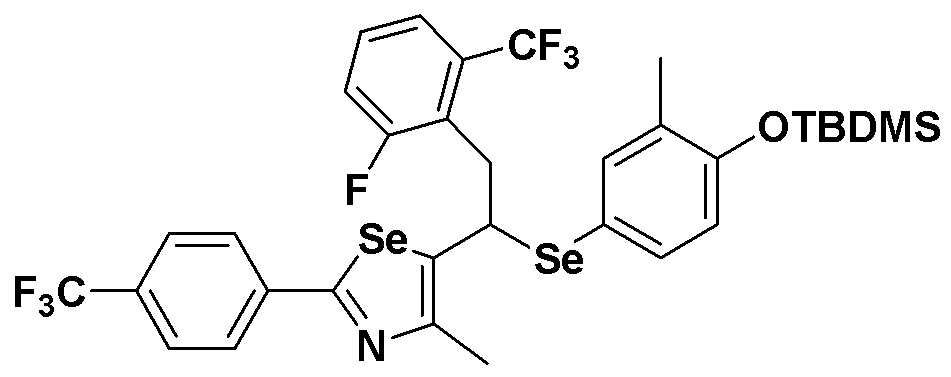
[Стадия C]
Соединение S4 (779 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали до -78ºC. Затем медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления 2-фтор-5-трифторметилбензилбромида (514 мг, 2,0 ммоль) температуру медленно поднимали до комнатной температуры. После дальнейшего взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (545 мг, выход: 70%). (FABMS: 782 [M+H]+).
[Пример 25] Получение соединения S25
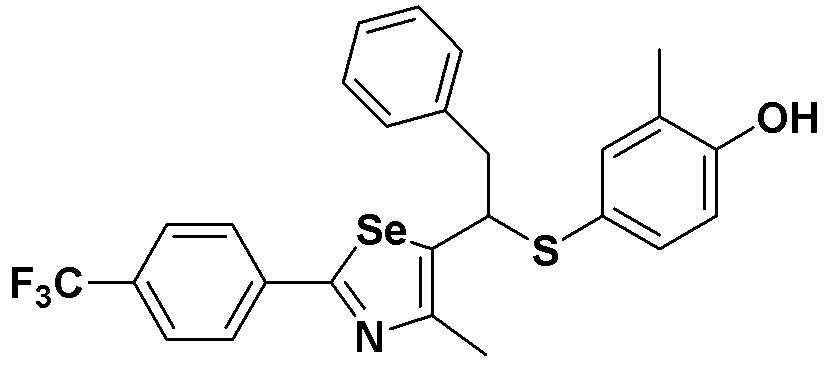
[Стадия G]
Соединение S1 (430 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (10 мл) в атмосфере азота, и температуру поддерживали при 0ºC. После медленного добавления изопропилмагнийхлорида (2M, 1 мл) реакцию проводили в течение 10 минут. После достаточного охлаждения до температуры -78ºC медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления бензилбромида (137 мкл, 1,0 ммоль), температуру медленно поднимали до комнатной температуры. После дополнительного взаимодействия в течение 30 минут реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (426 мг, выход: 80%). (FABMS: 534 [M+H]+).
[Пример 26] Получение соединения S26
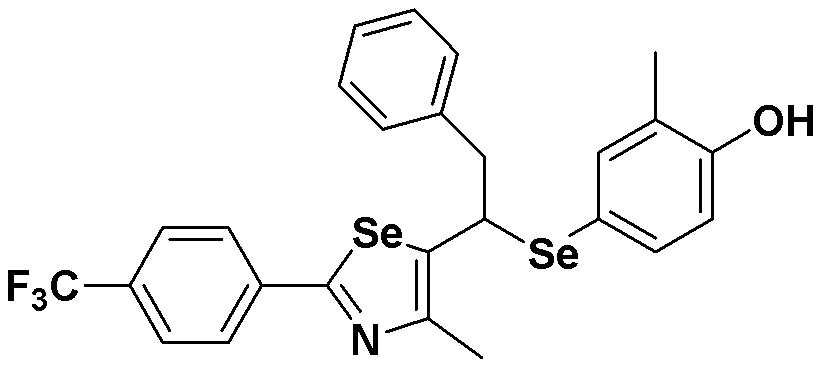
[Стадия G]
Соединение S2 (478 мг, 1 ммоль) растворяли в безводном тетрагидрофуране (10 мл) в атмосфере азота, и температуру поддерживали при 0ºC. После медленного добавления изопропилмагнийхлорида (2 M, 1 мл) реакцию проводили в течение 10 минут. После достаточного охлаждения до температуры -78ºC медленно добавляли диизопропиламид лития (LDA, 1,8 мл, 1,8 M, 2,0 эквивалента). После добавления бензилбромида (137 мкл, 1,0 ммоль) температуру медленно поднимали до комнатной температуры. После дальнейшего взаимодействия в течение 30 минут, реакцию останавливали водным раствором хлорида аммония. Органический растворитель экстрагировали этилацетатом и насыщенным солевым раствором, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (457 мг, выход: 78%). (FABMS: 582 [M+H]+).
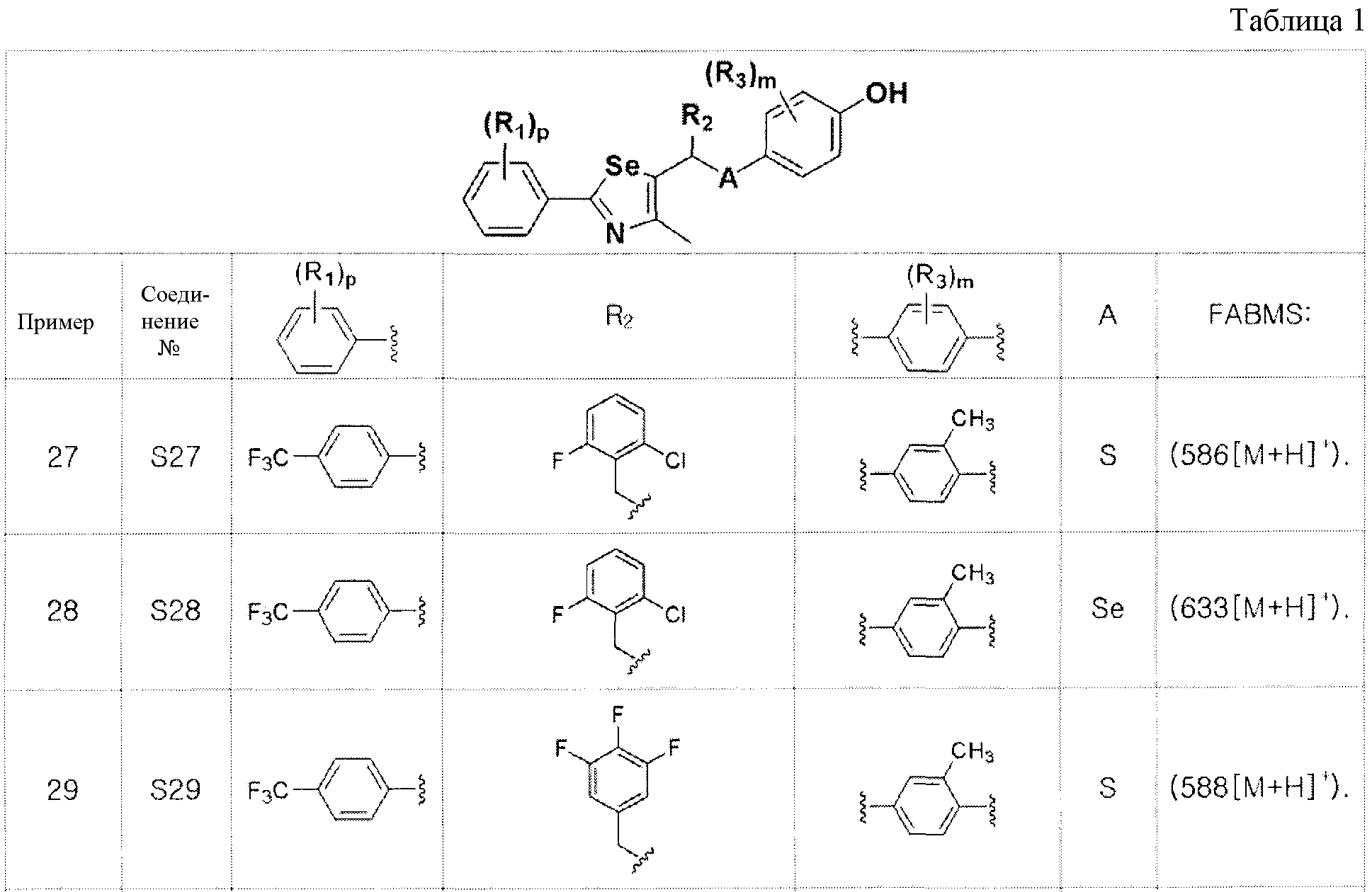
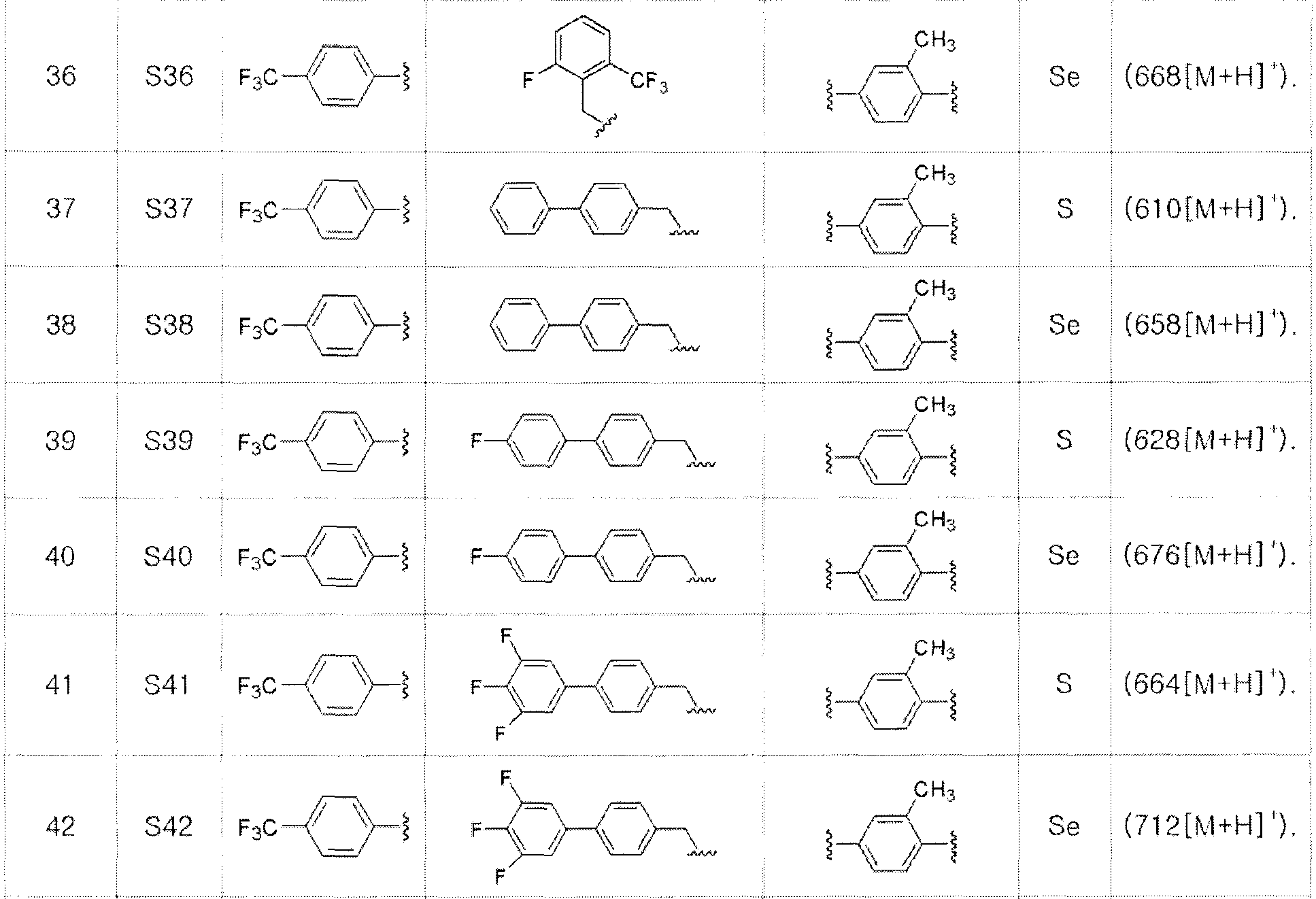
[Примеры 27-36]
Соединения S27-S46, представленные в таблице 1, были получены в соответствии со способом по примерам 25 и 26. В таблице приведены данные МС анализа.
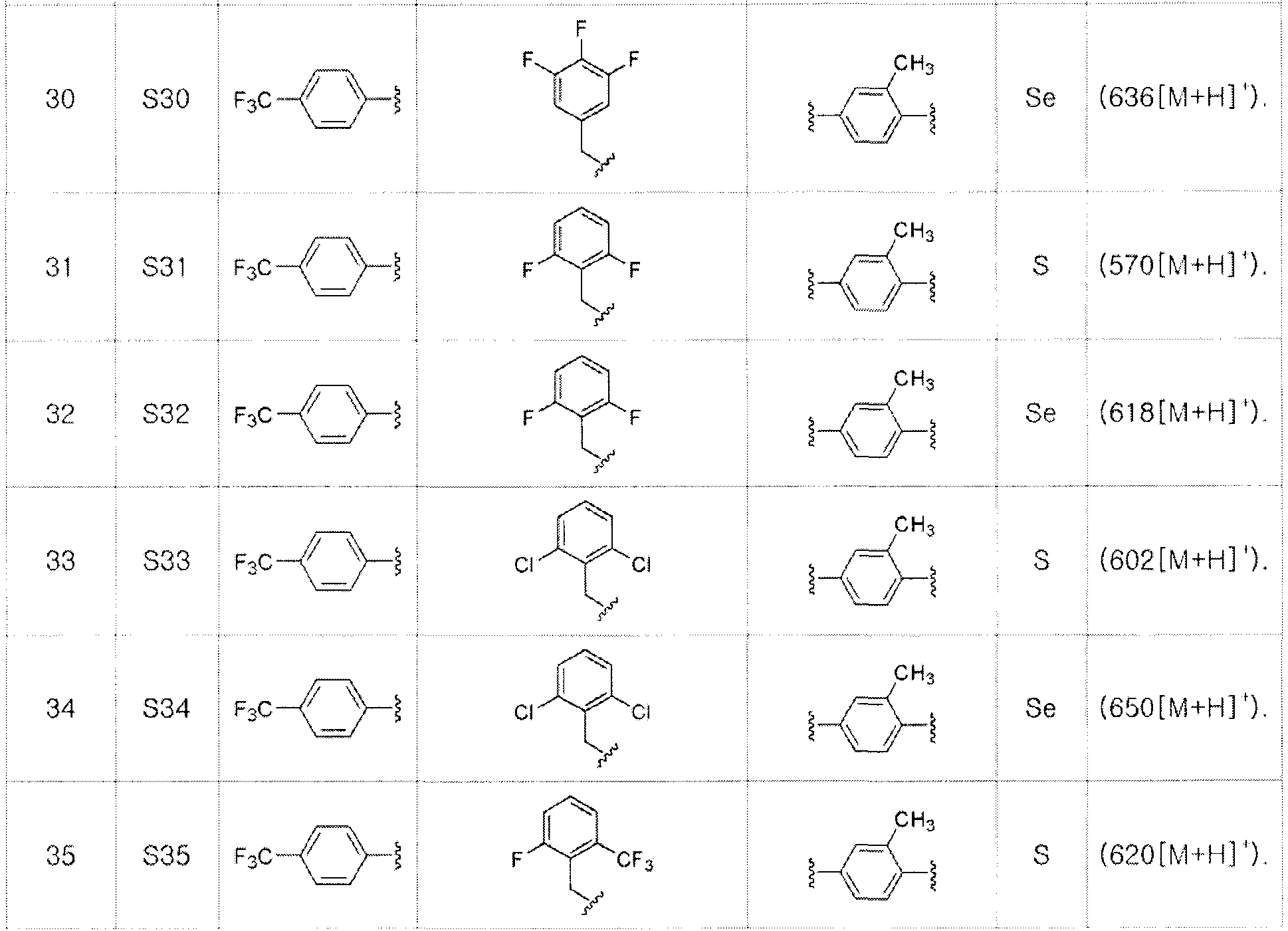
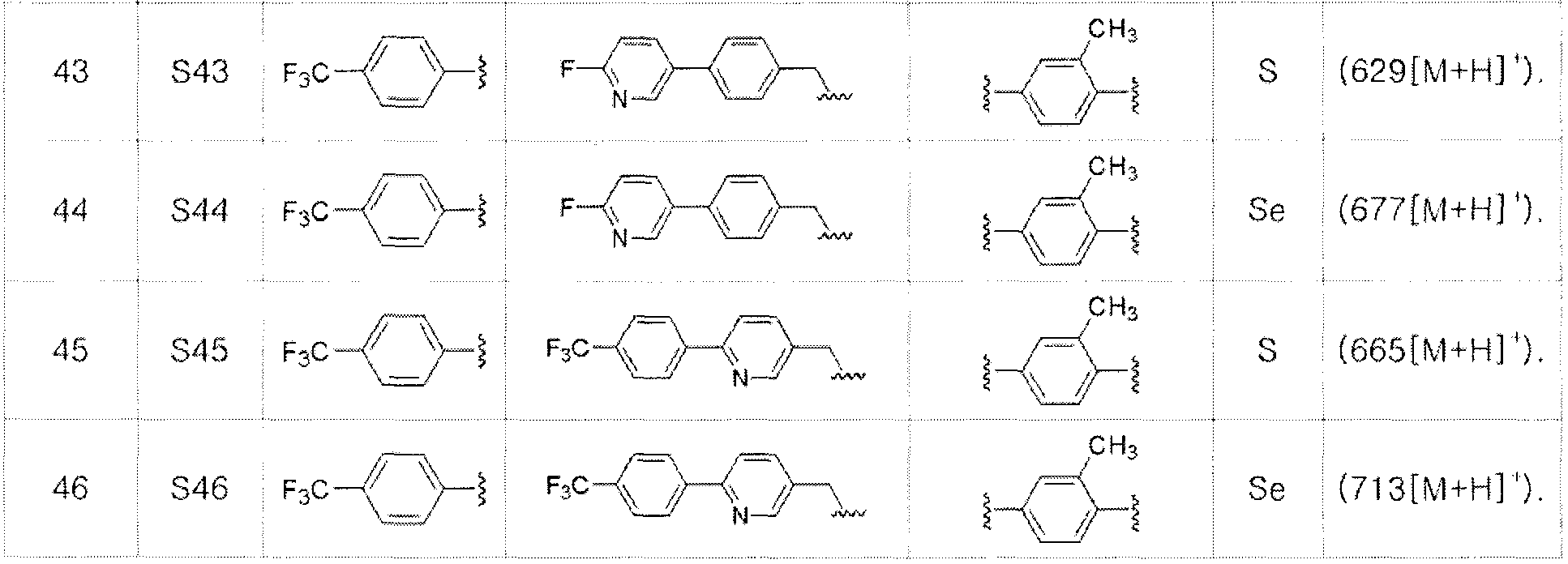
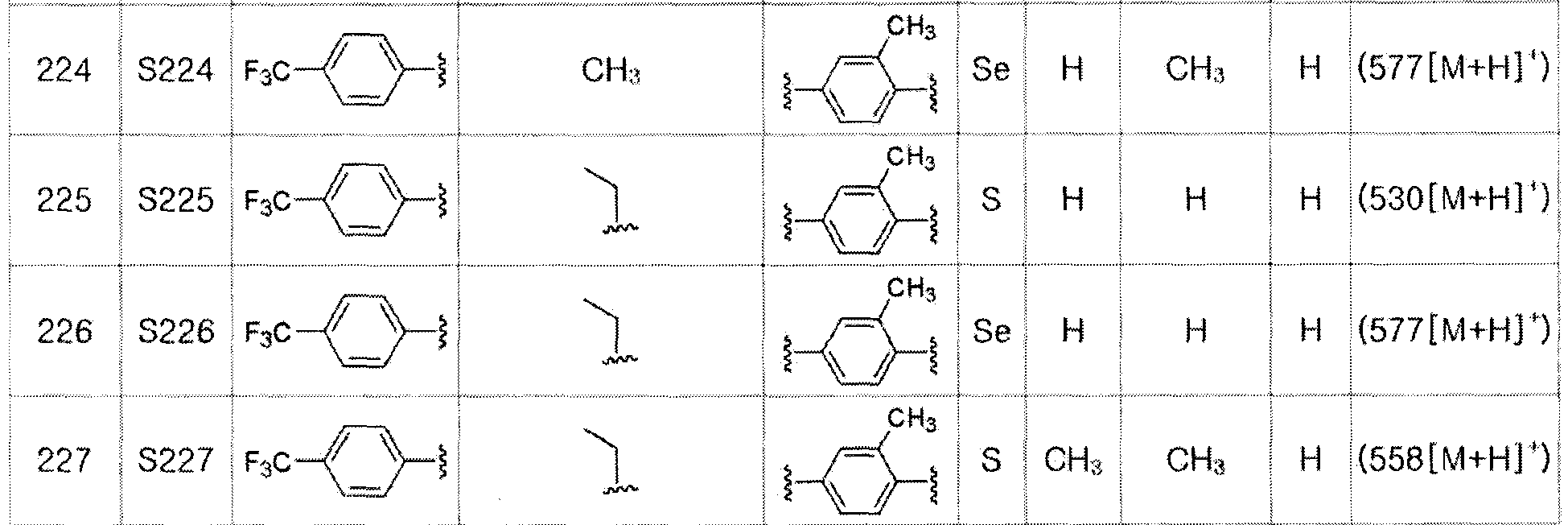
[Пример 47] Получение соединения S47
[Стадия D]
Соединение S13 (646 мг, 1 ммоль) полностью растворяли в тетрагидрофуране (10 мл). Затем при комнатной температуре медленно добавляли тетрабутиламмоний фторид (TBAF, 2,5 мл, 1M раствор в тетрагидрофуране, 2,5 эквивалента). После взаимодействия в течение 30 минут, органический растворитель экстрагировали водным раствором хлорида аммония и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (479 мг, выход: 92%). (FABMS: 534 [M+H]+).
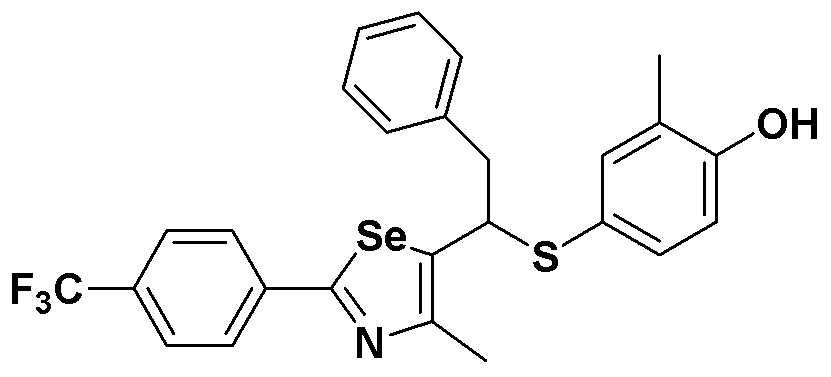
[Пример 48] Получение соединения S48
[Стадия D]
Соединение S14 (693 мг, 1 ммоль) полностью растворяли в тетрагидрофуране (10 мл). Затем при комнатной температуре медленно добавляли тетрабутиламмоний фторид (TBAF, 2,5 мл, 1 M раствор в тетрагидрофуране, 2,5 эквивалента). После взаимодействия в течение 30 минут органический растворитель экстрагировали водным раствором хлорида аммония и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем давала целевое соединение (521 мг, выход: 90%). (FABMS: 582 [M+H]+).
Соединения S27 - S46 могут быть получены в соответствии со способом по примерам 47 и 48.
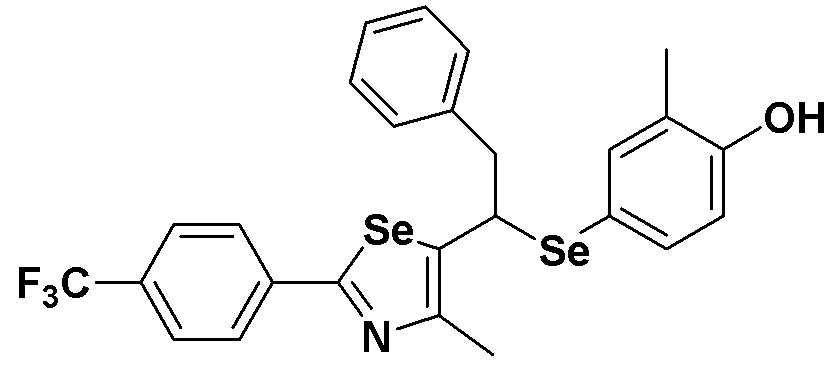
[Пример 49] Получение соединения S49
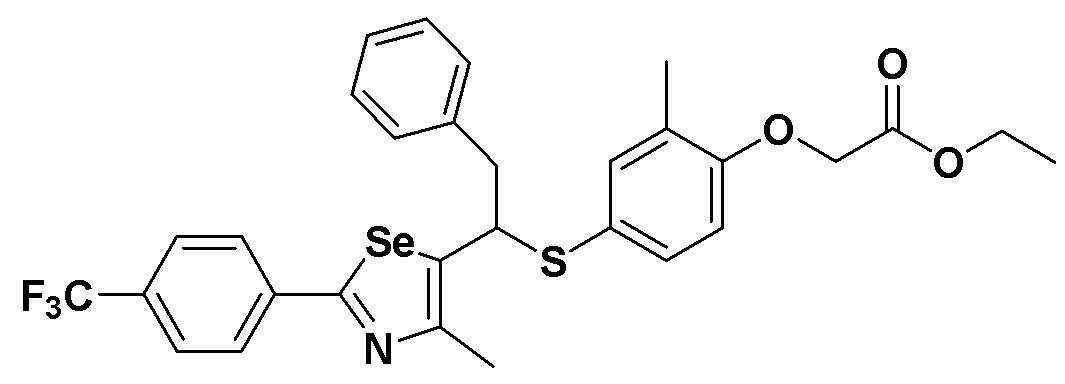
[Стадия E]
Соединение S25 (532 мг, 1 ммоль) и ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этилового эфира бромуксусной кислоты (134 мкл, 1,2 ммоль, 1,2 эквивалента) смесь энергично перемешивали в течение 4 часов. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (575 мг, выход: 93%). (FABMS: 620 [M+H]+).
[Пример 50] Получение соединения S50
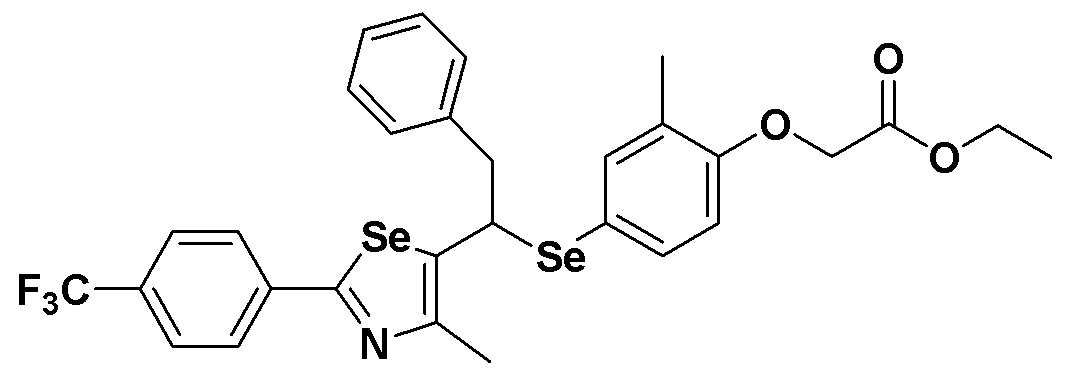
[Стадия E]
Соединение S26 (579 мг, 1 ммоль) и ацетон (10 мл), содержащий 5% воды, и карбонат калия (346 мг, 2,5 ммоль, 2,5 эквивалента) хорошо смешивали при комнатной температуре. После добавления этилового эфира бромуксусной кислоты (134 мкл, 1,2 ммоль, 1,2 эквивалента), смесь энергично перемешивали в течение 4 часов. После завершения реакции органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, и органический слой сушили с помощью сульфата магния. После фильтрования с последующей перегонкой при пониженном давлении, очистка остатка путем хроматографии на колонке с силикагелем с использованием смеси гексан/этилацетат (об./об.=5:1) давала целевое соединение (605 мг, выход: 91%). (FABMS: 668 [M+H]+).
[Примеры 51 - 136]
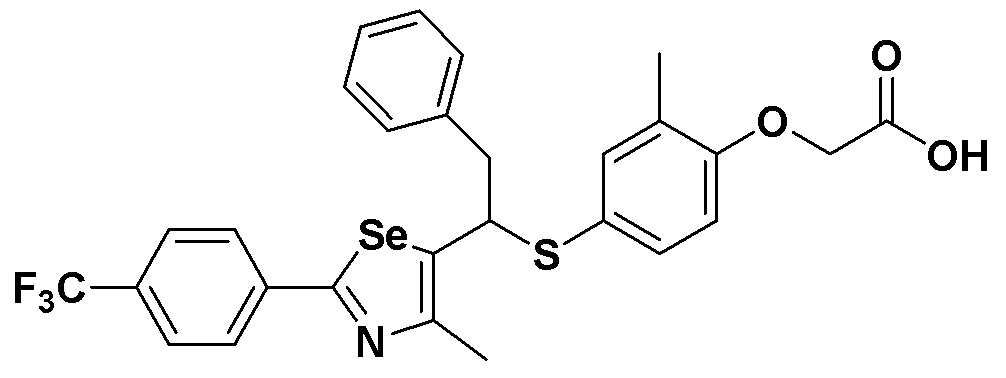
Соединения S51-S136, представленные в таблице 2, были получены в соответствии со способом по примерам 49 и 50. В таблице приведены данные МС анализа.
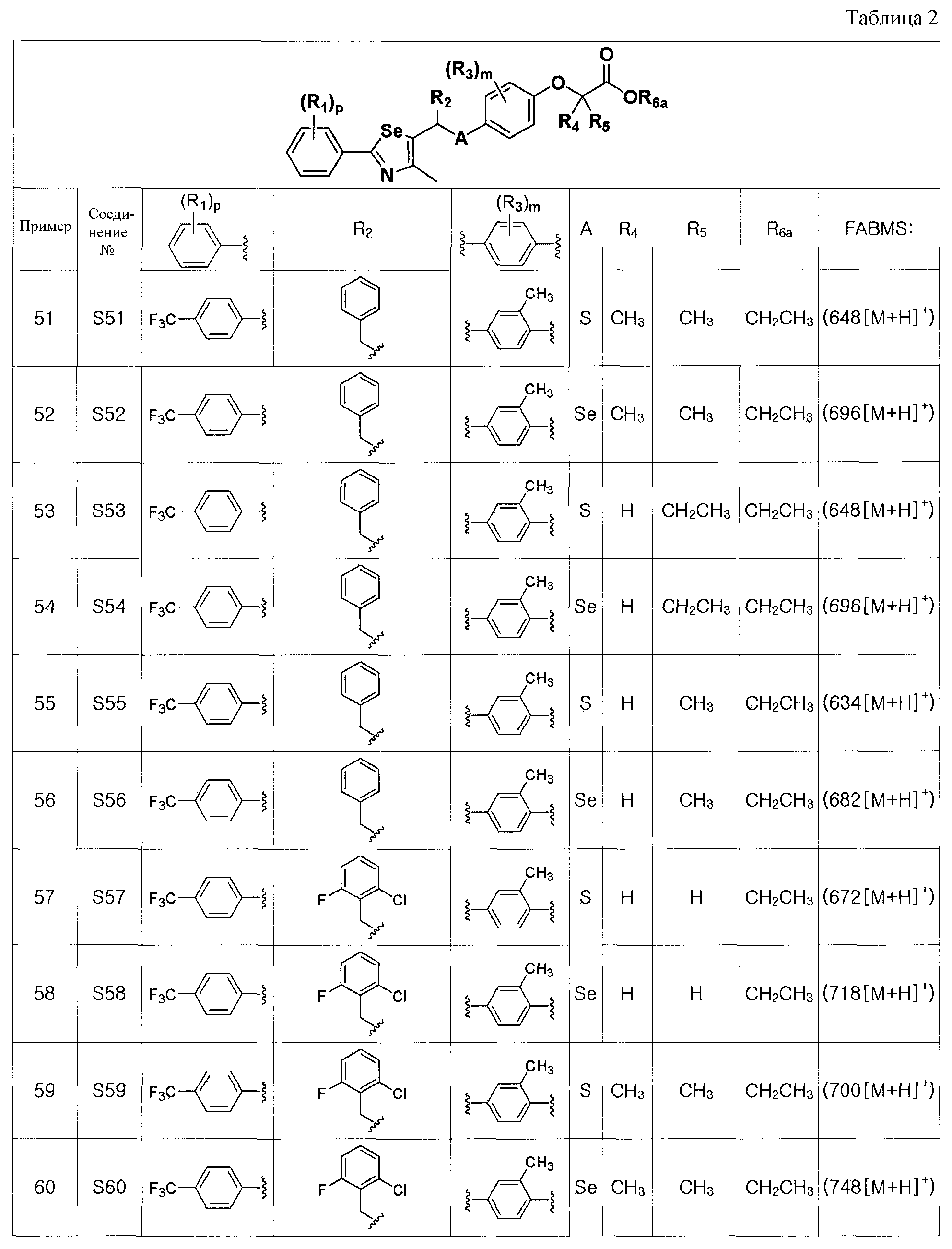
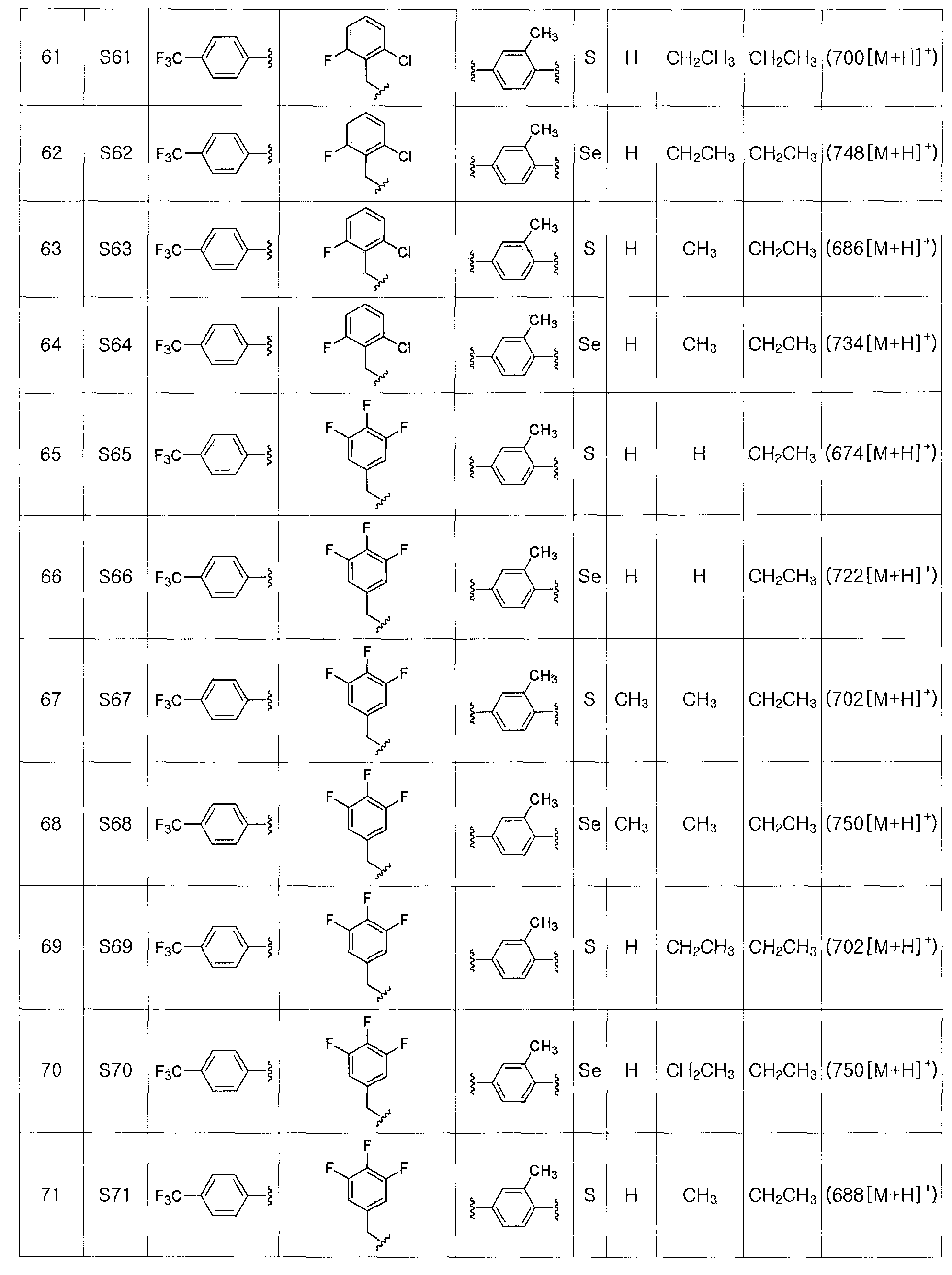

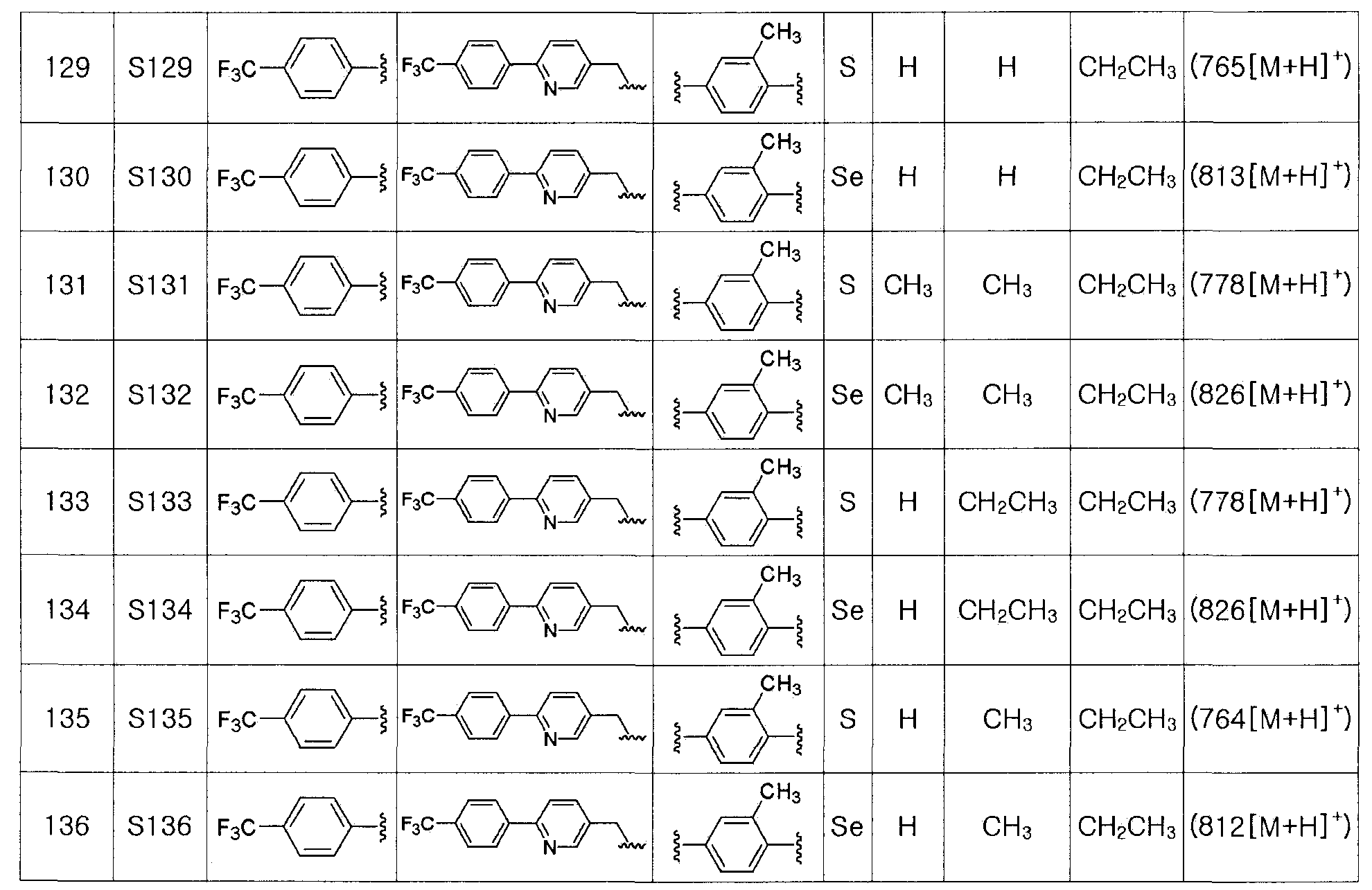
[Пример 137] Получение соединения S137
[Стадия F]
Соединение S49 (618 мг, 1 ммоль) хорошо смешивали с ТГФ (15 мл) и водой (10 мл) и при 0ºC медленно добавляли 2,0 M водный раствор гидроксида лития (0,6 мл). После дополнительного перемешивания в течение 60 минут при 0ºC, после завершения реакции добавляли 0,5 M NaHSO4 (2,5 мл). Затем органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, фильтровали и перегоняли при пониженном давлении. Очистка остатка с помощью хроматографии на колонке LH-20 давала целевое соединение (566 мг, выход: 96%). (FABMS: 592 [M+H]+).
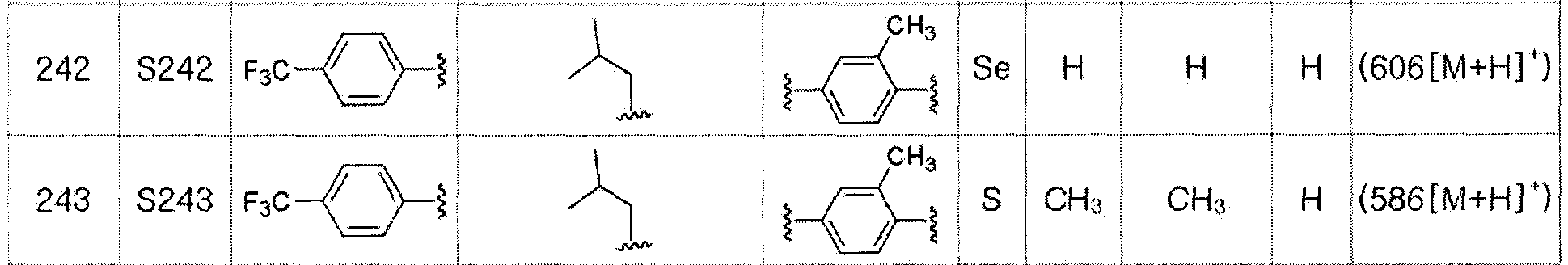
[Пример 138] Получение соединения S138
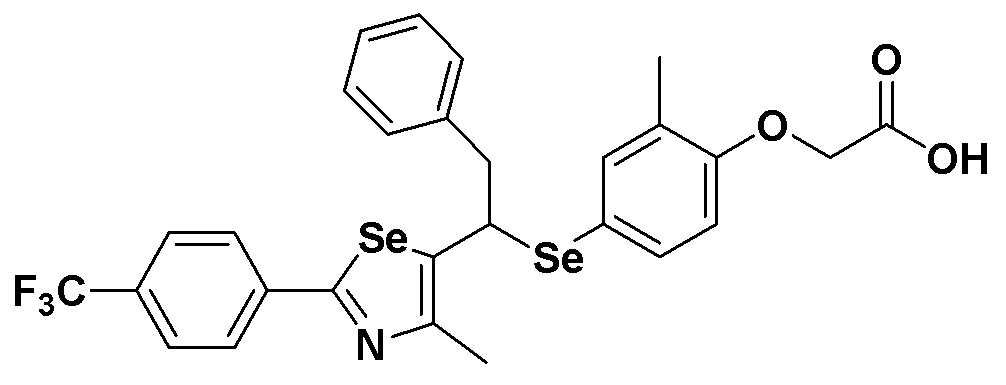
Соединение S50 (665 мг, 1 ммоль) хорошо смешивали с ТГФ (15 мл) и водой (10 мл) и при 0ºC медленно добавляли 2,0 M водный раствор гидроксида лития (0,6 мл). После дополнительного перемешивания в течение 60 минут при 0ºC, после завершения реакции добавляли 0,5 M NaHSO4 (2,5 мл). Затем органический растворитель экстрагировали насыщенным солевым раствором и этилацетатом, фильтровали и перегоняли при пониженном давлении. Очистка остатка с помощью хроматографии на колонке LH-20 давала целевое соединение (605 мг, выход: 95%). (FABMS: 640 [M+H]+).
[Примеры 139-288]
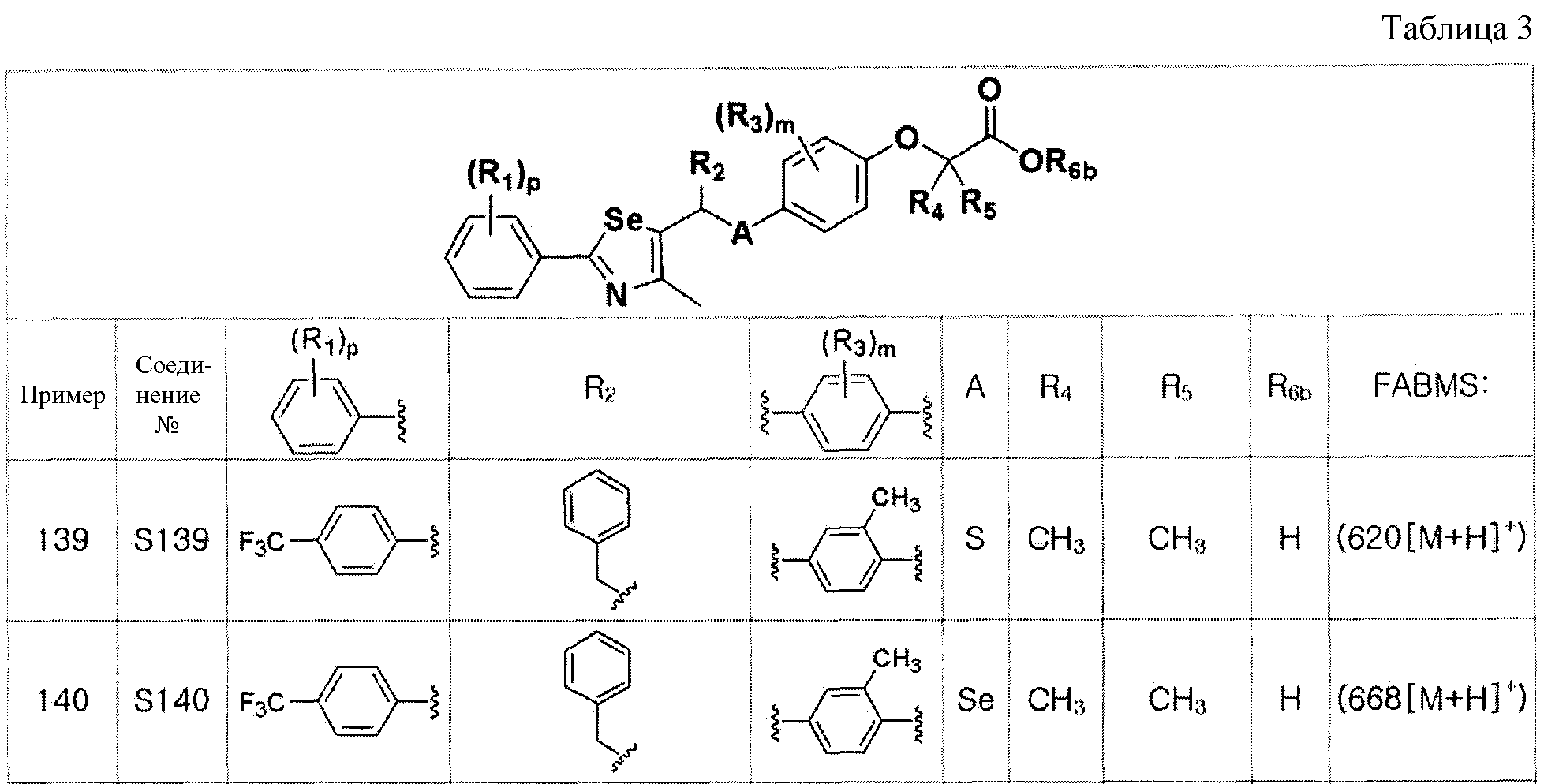
Соединения S139-S288, представленные в таблице 3, были получены в соответствии со способом по примерам 87 и 88. В таблице приведены данные МС анализа.
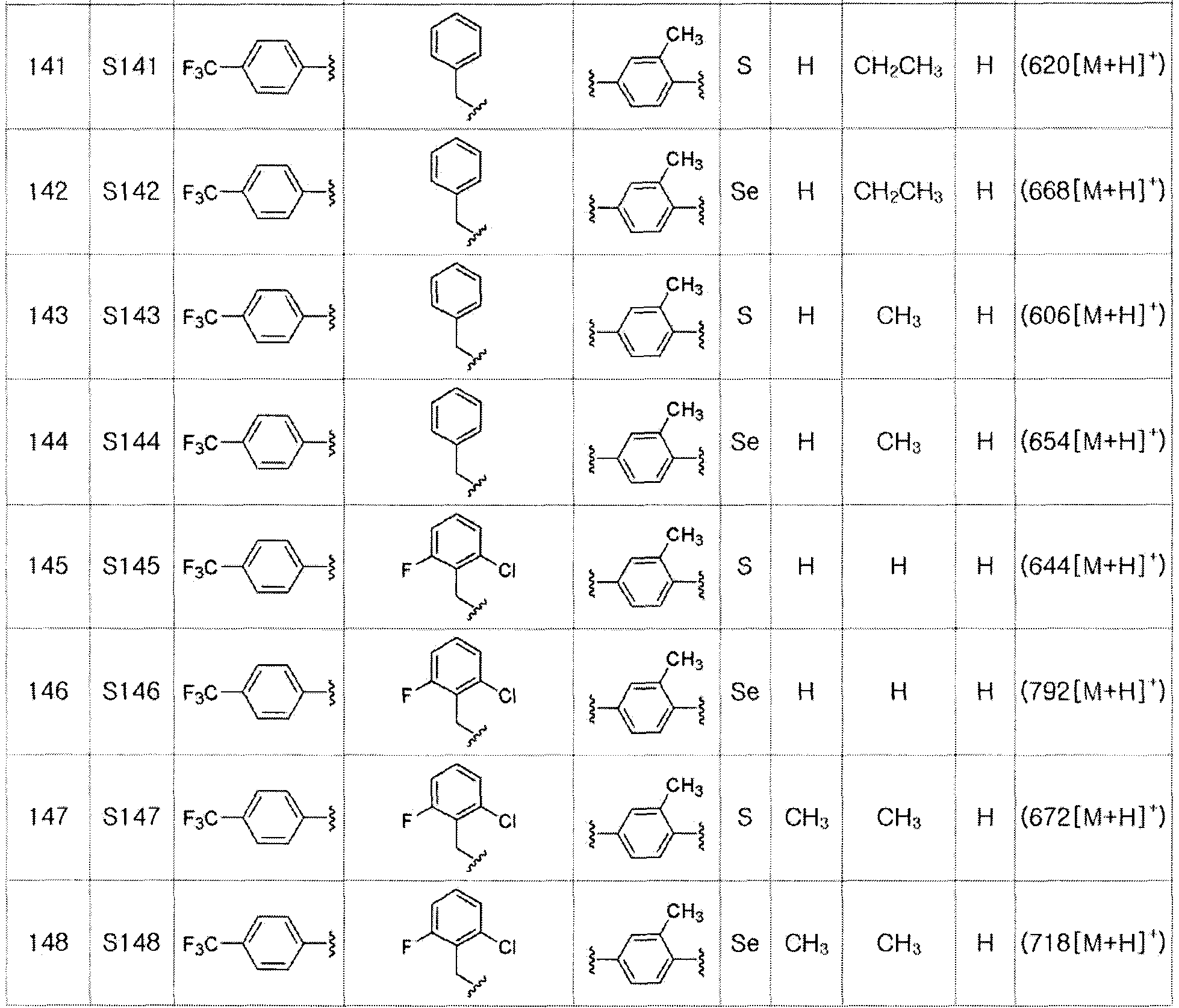
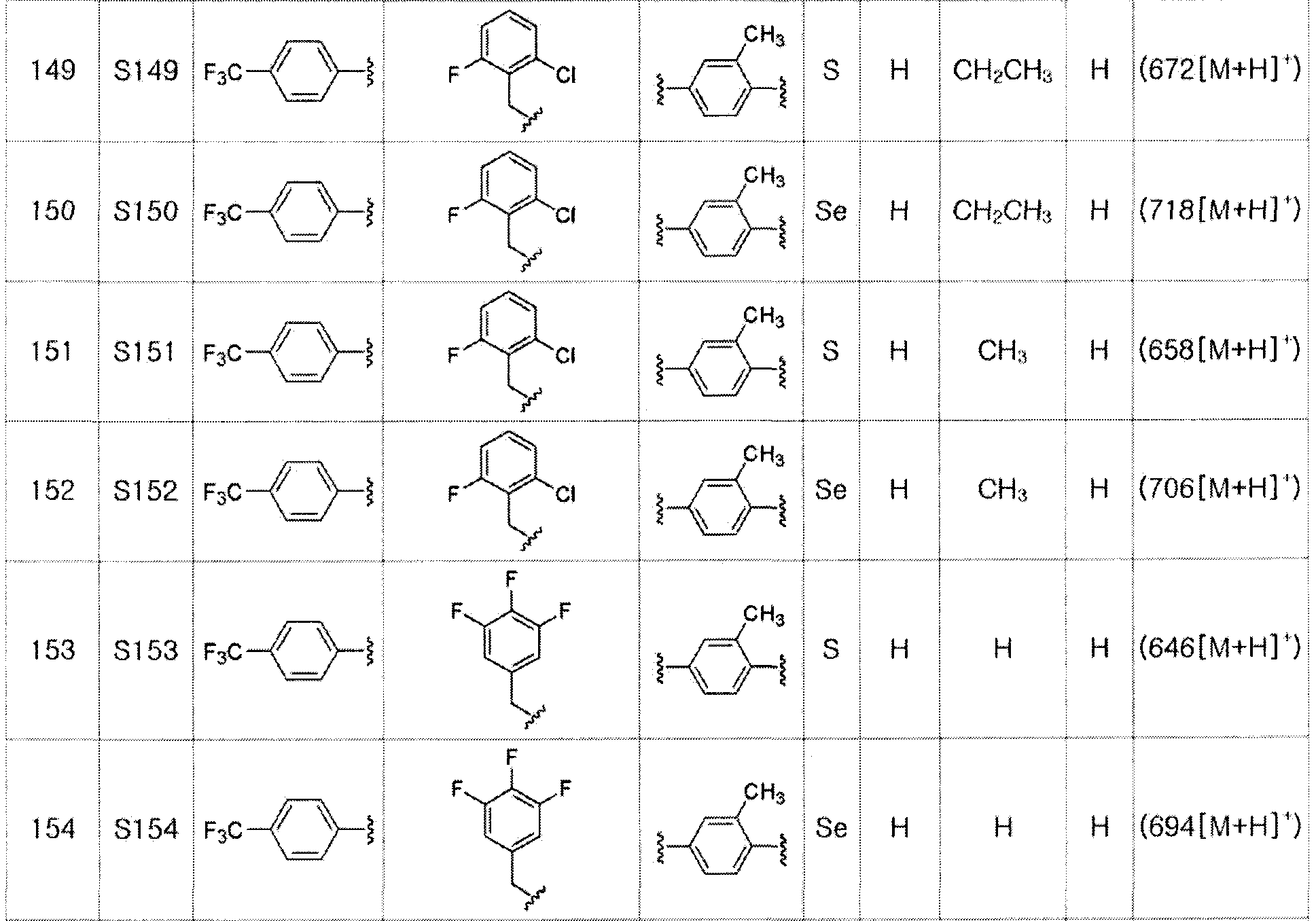
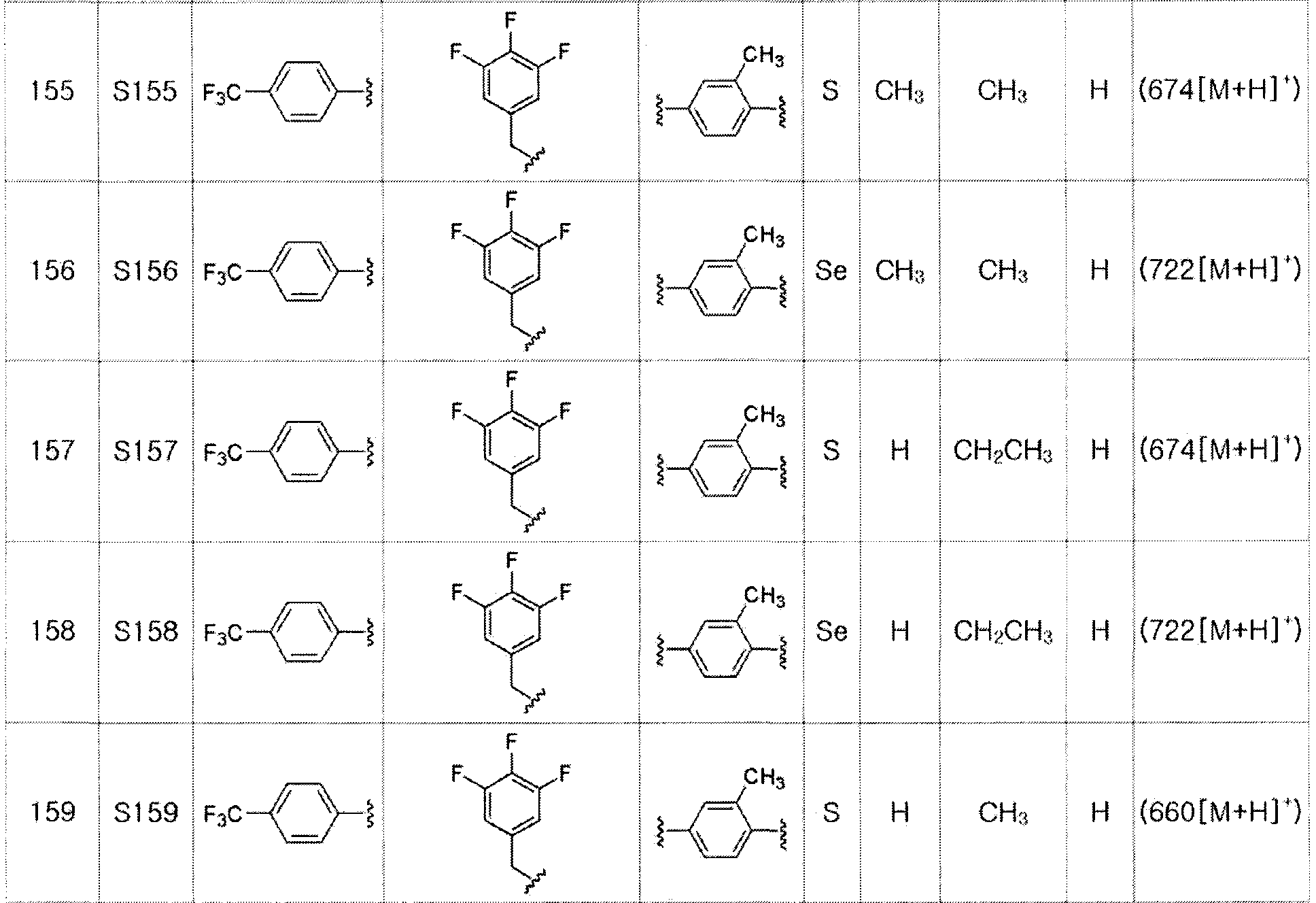
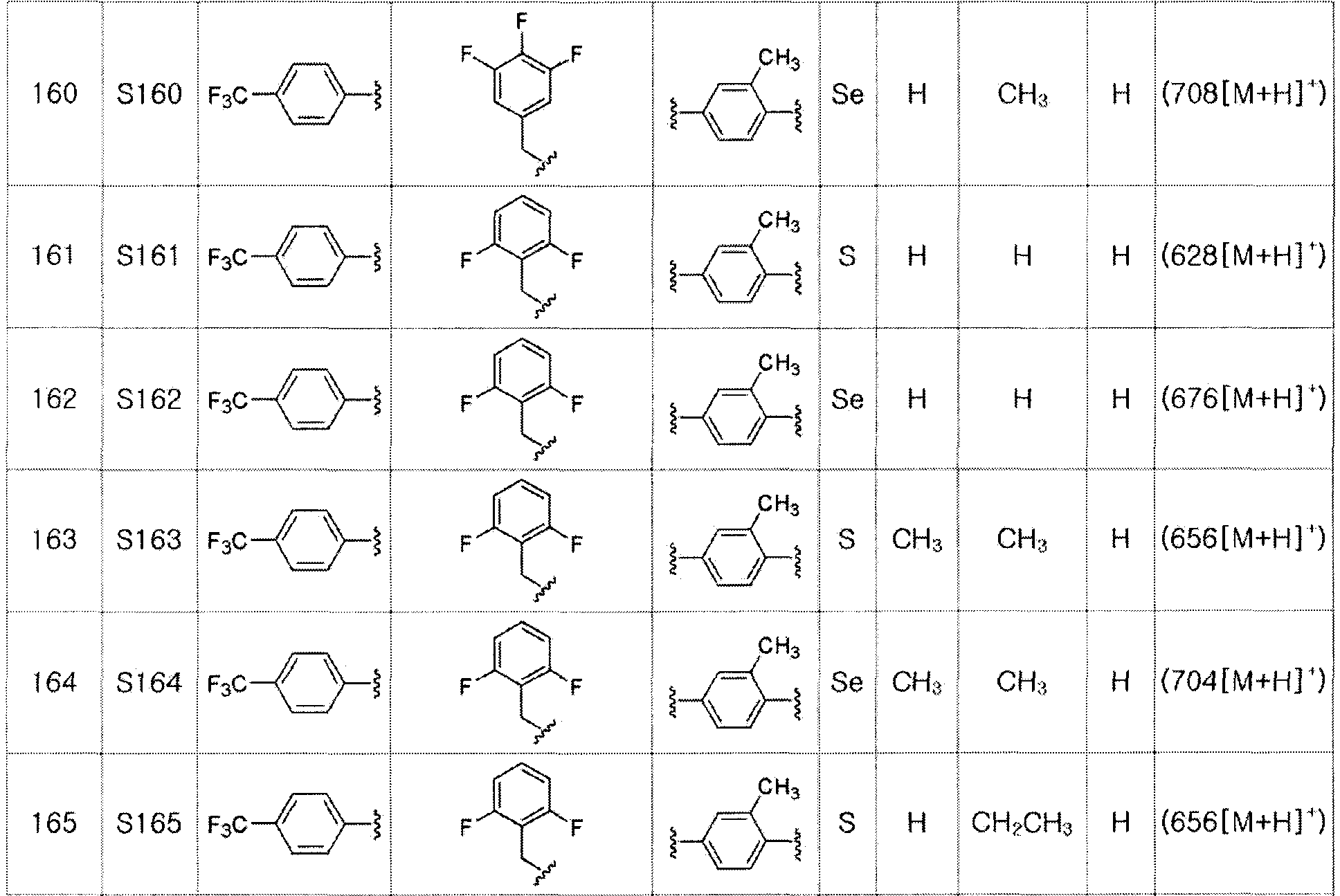
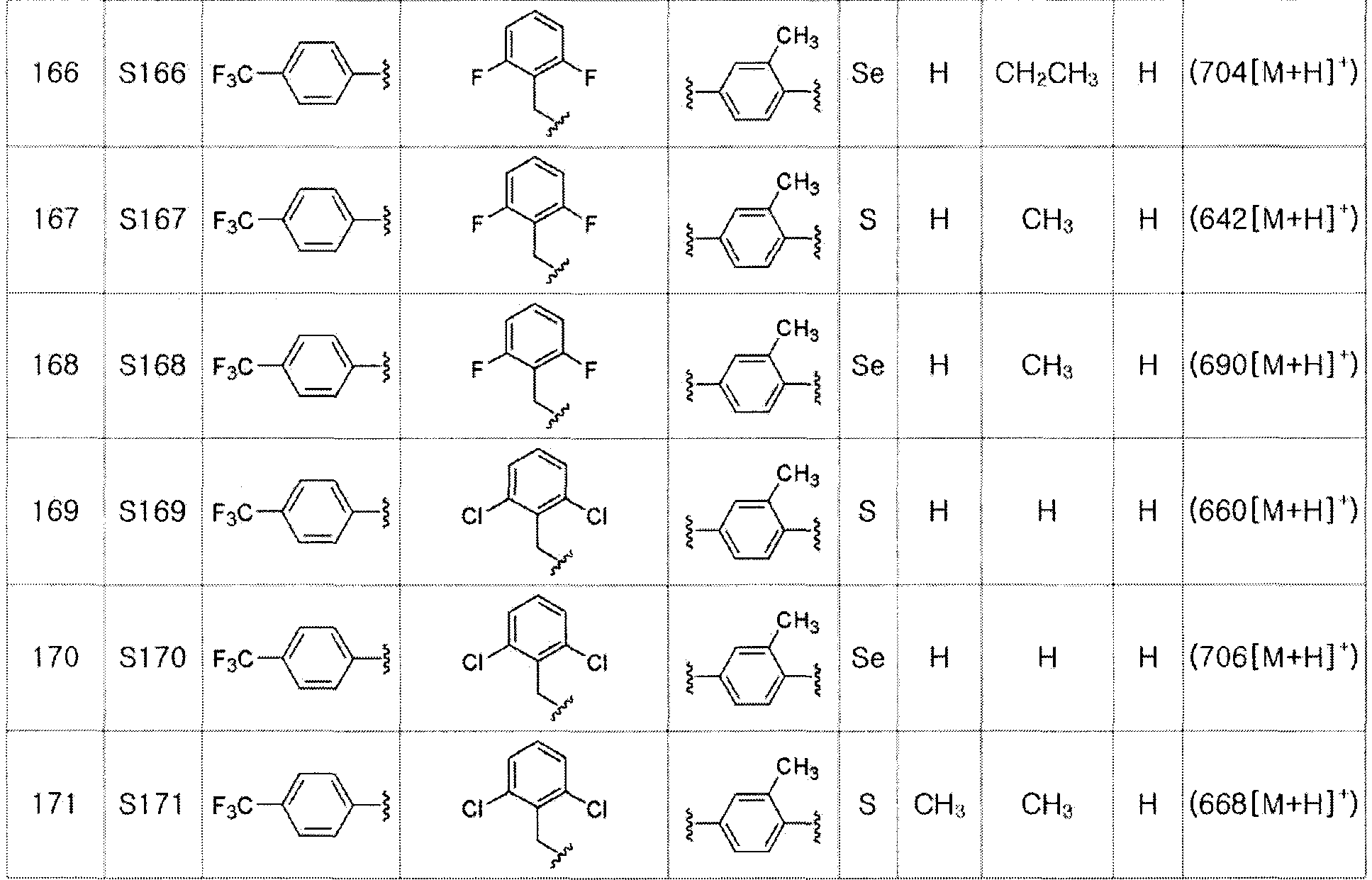
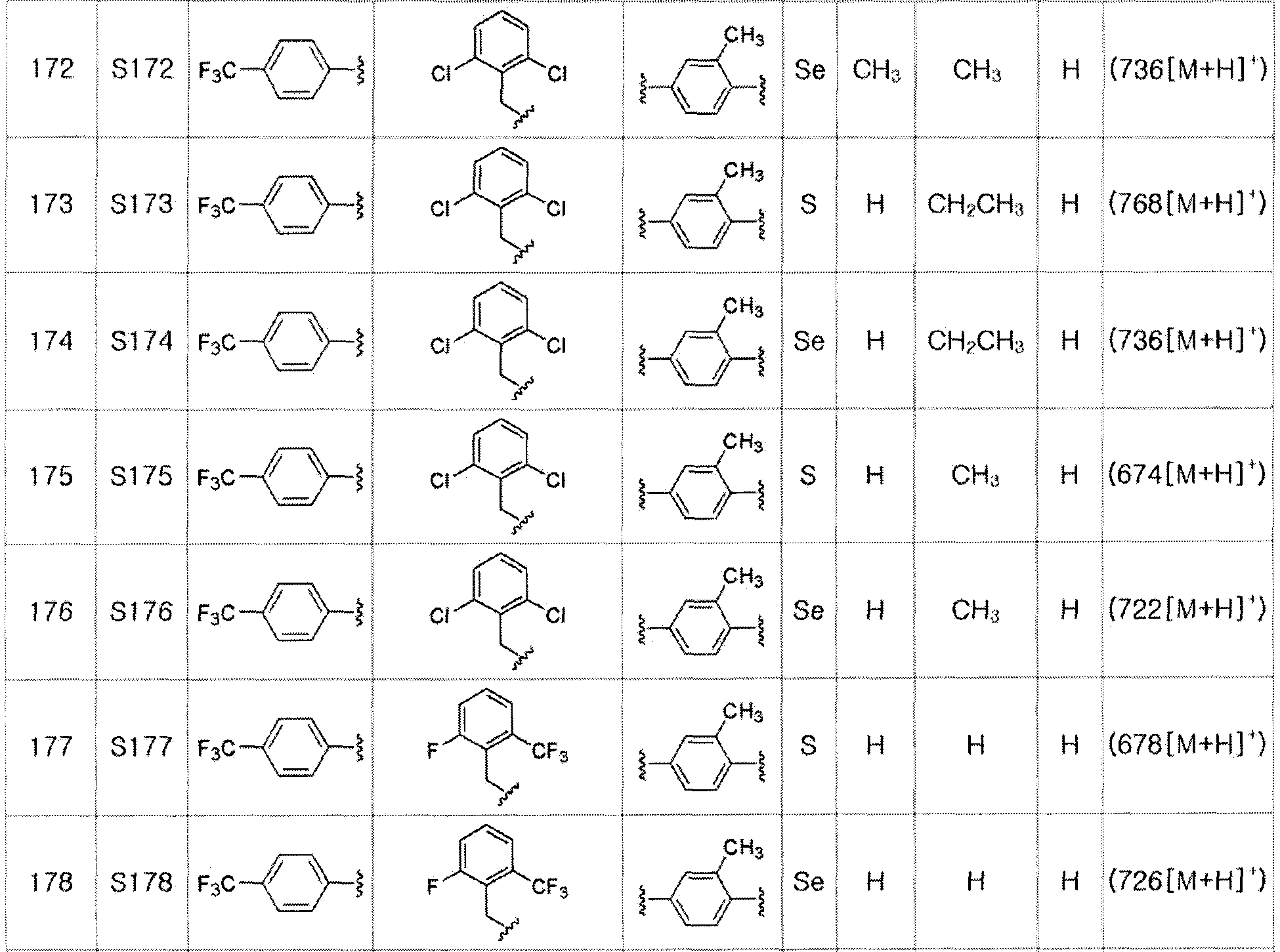
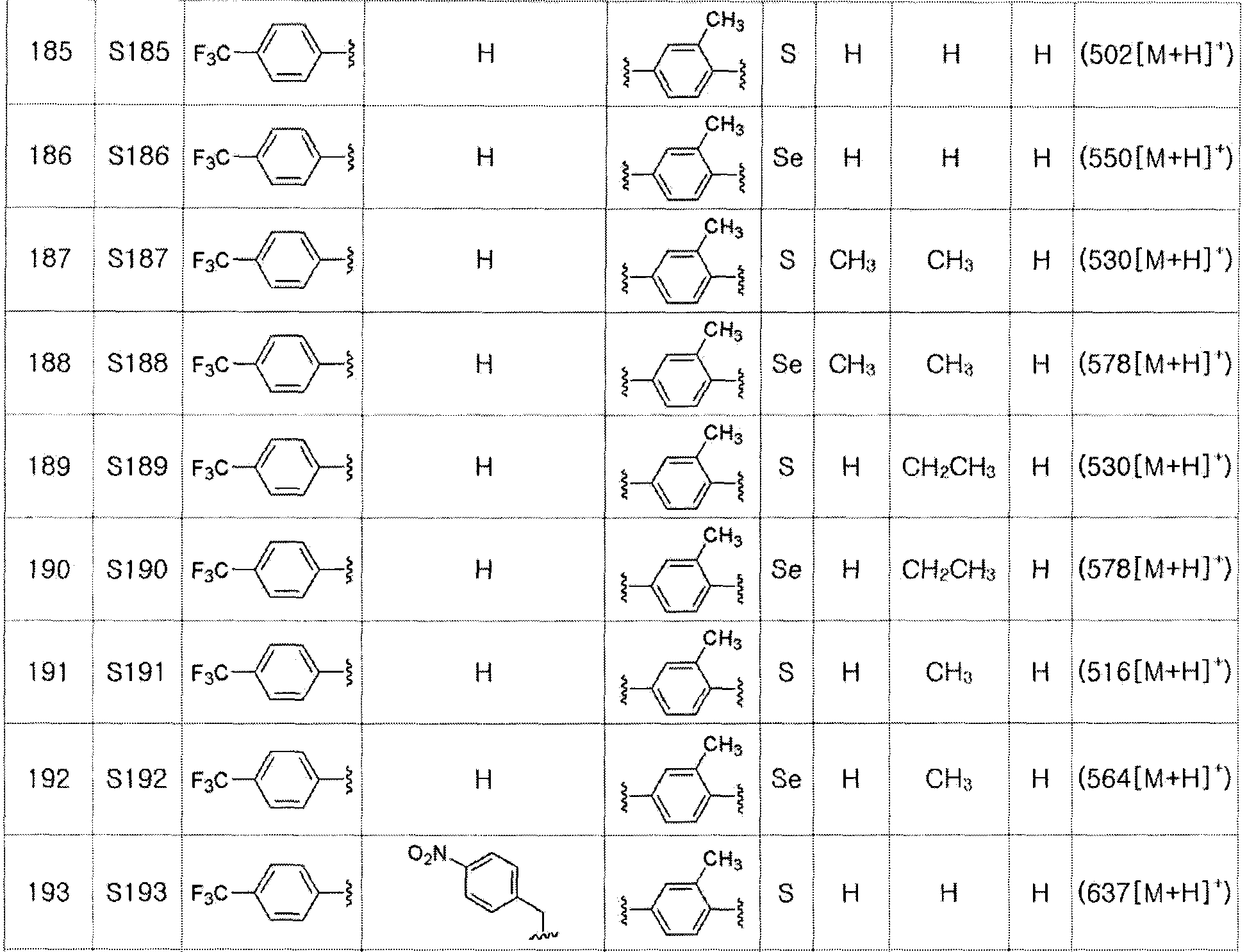

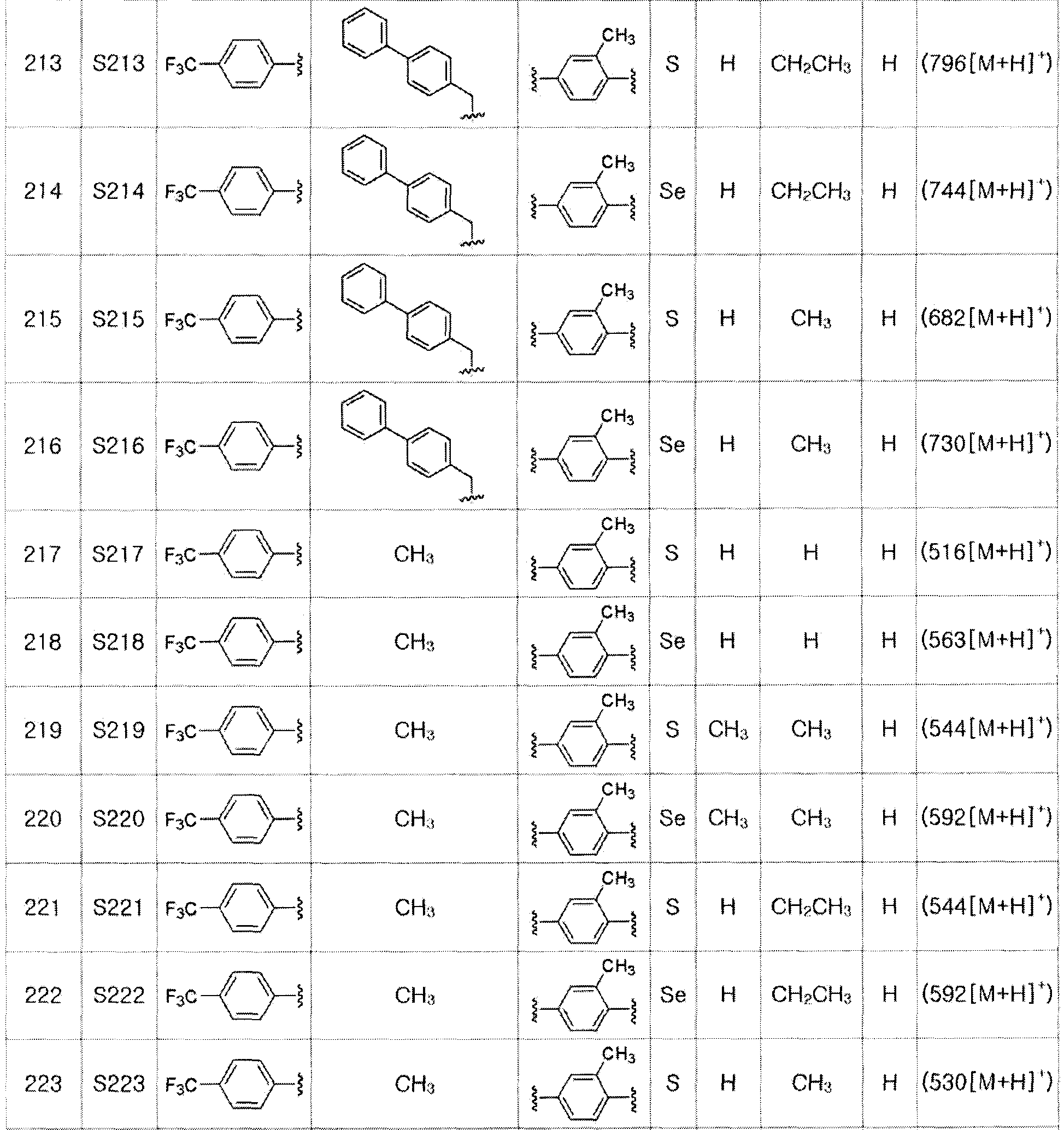
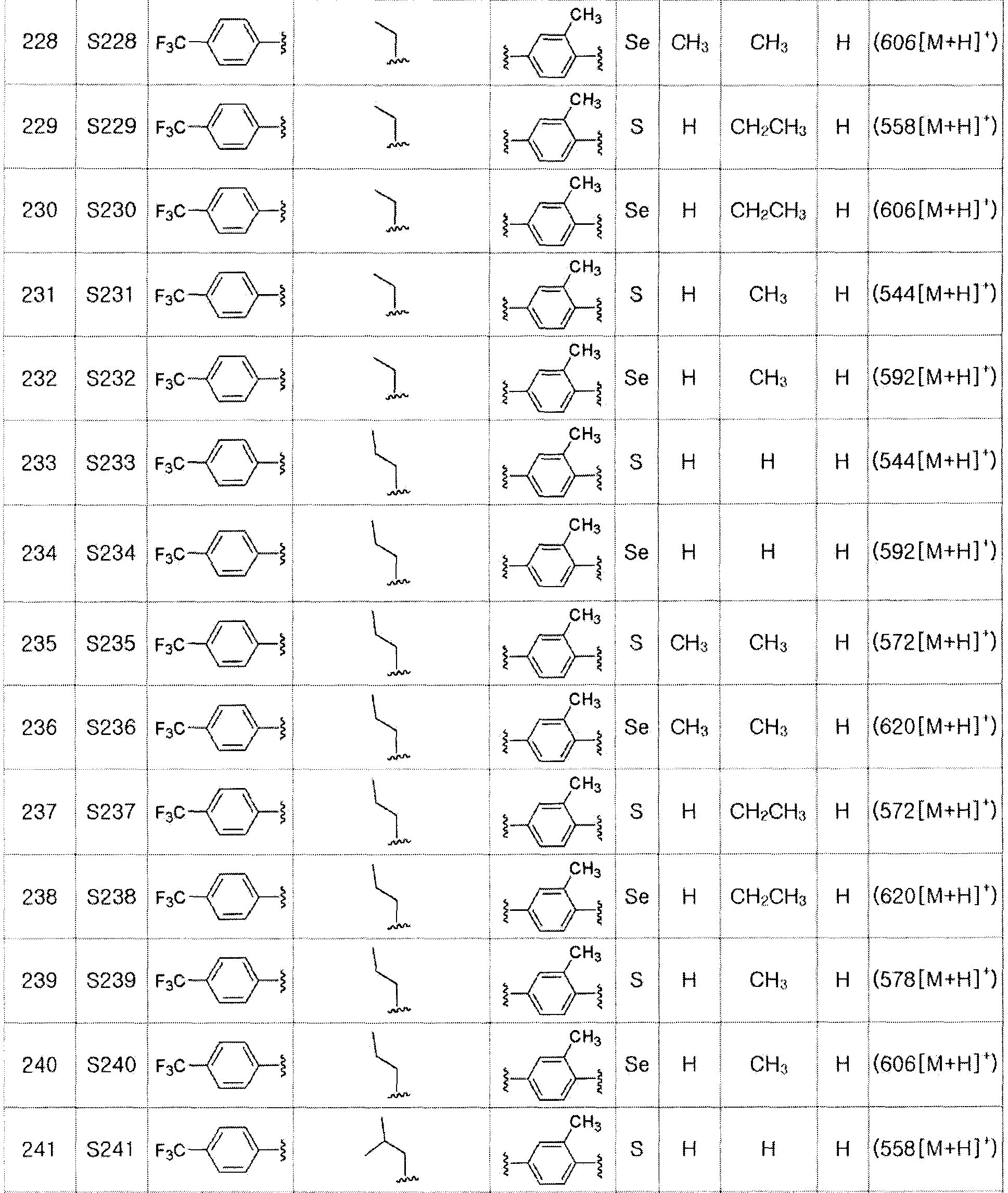
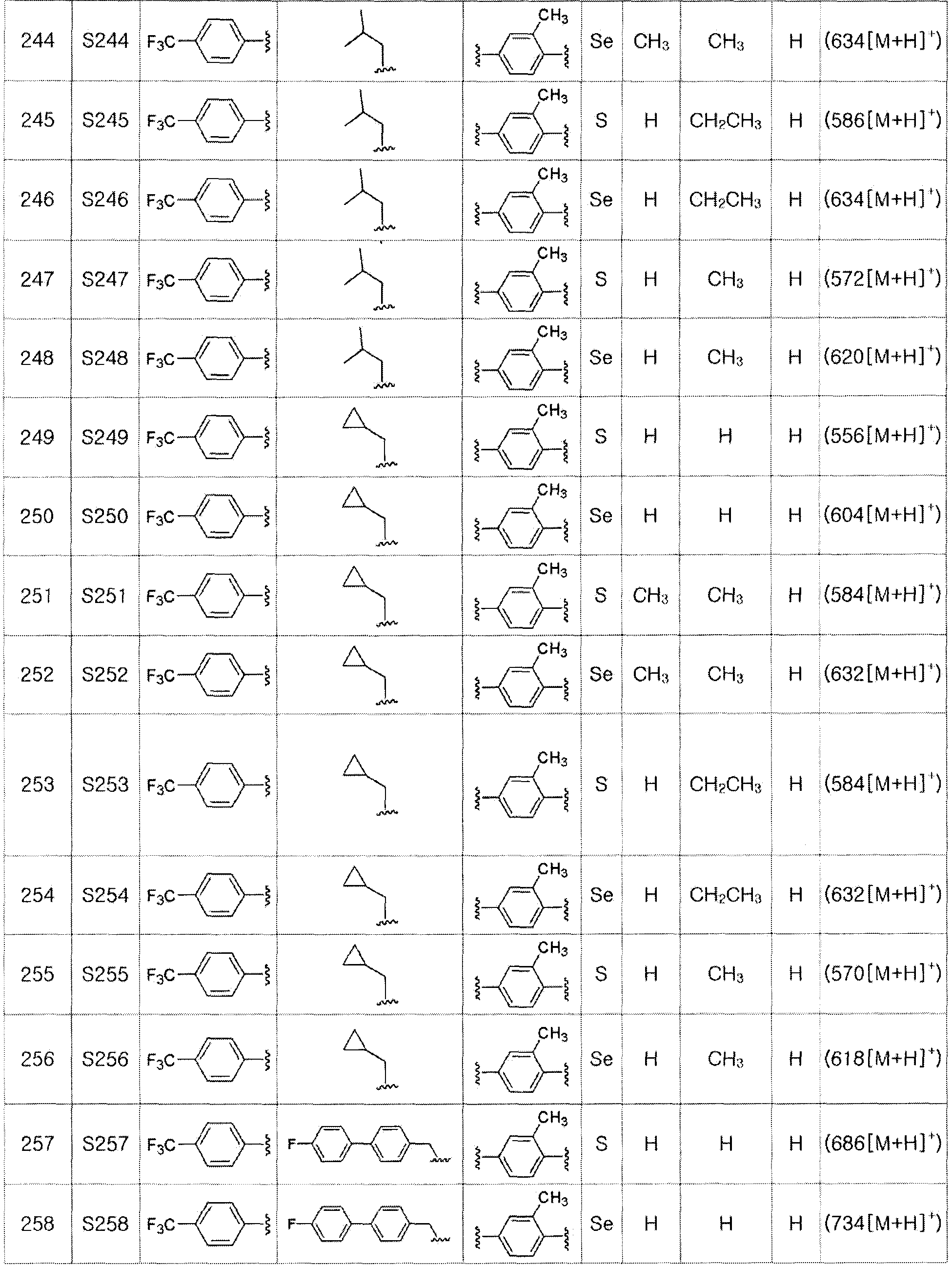
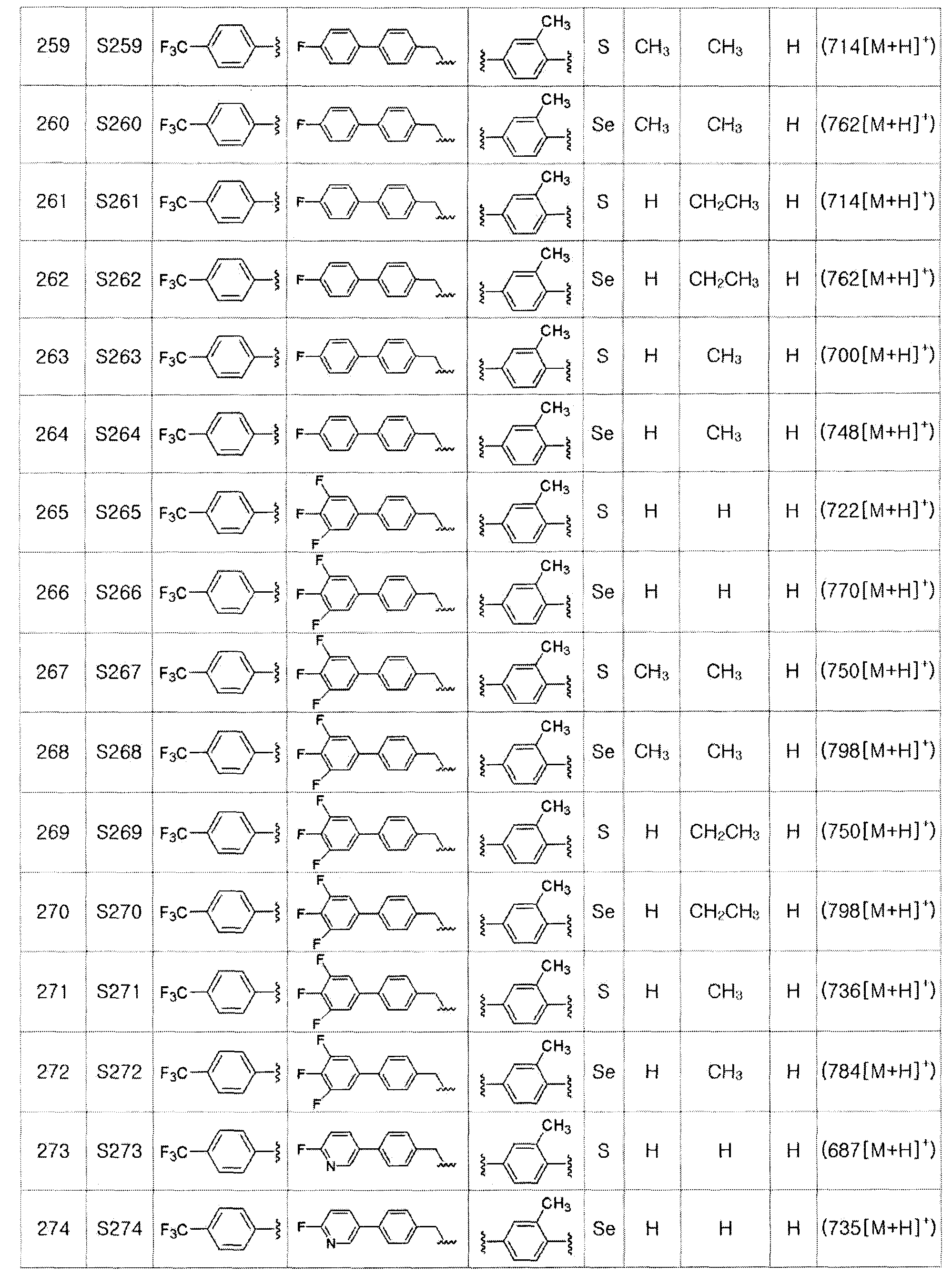
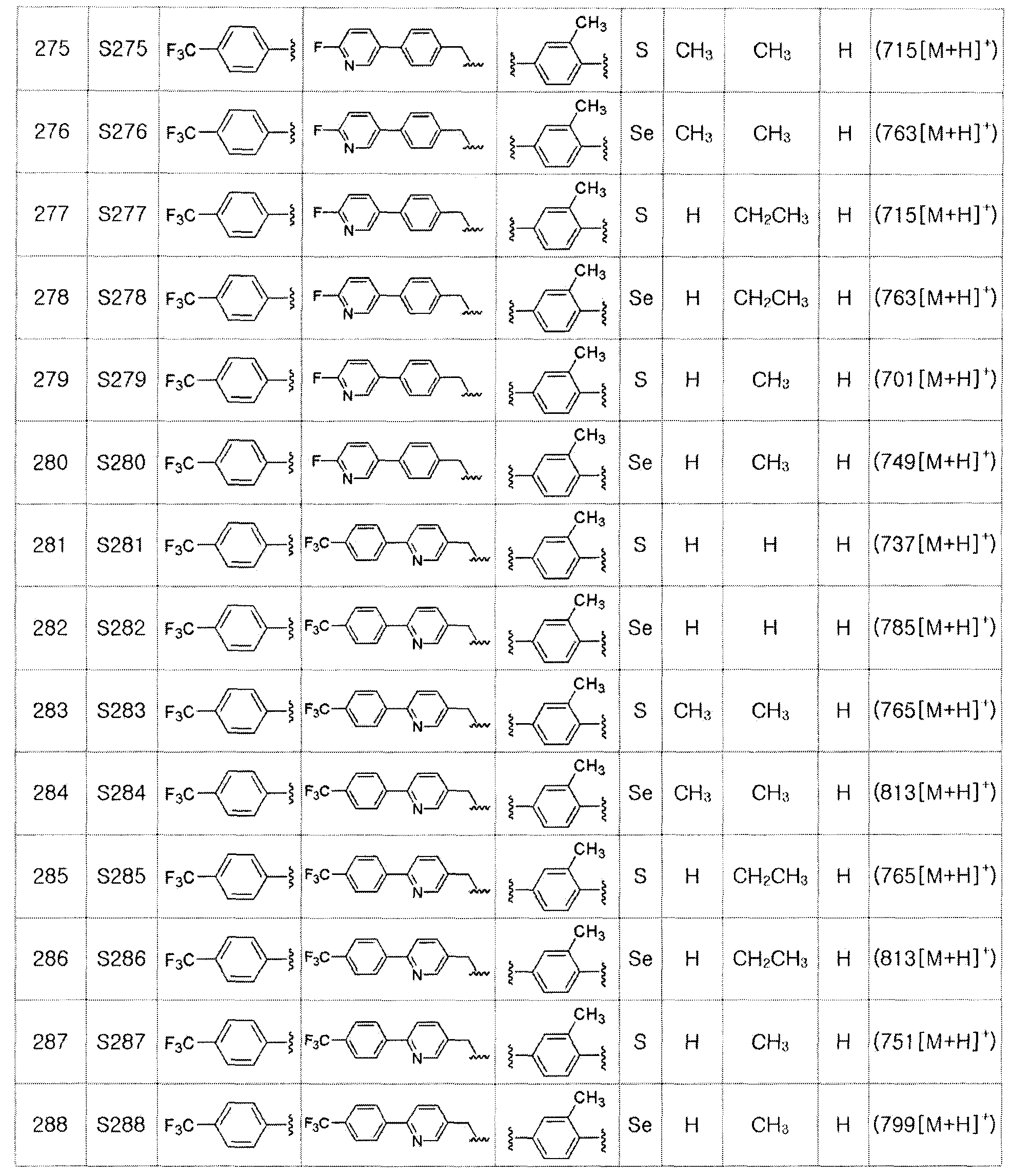
[Пример 289] Получение соединения S289
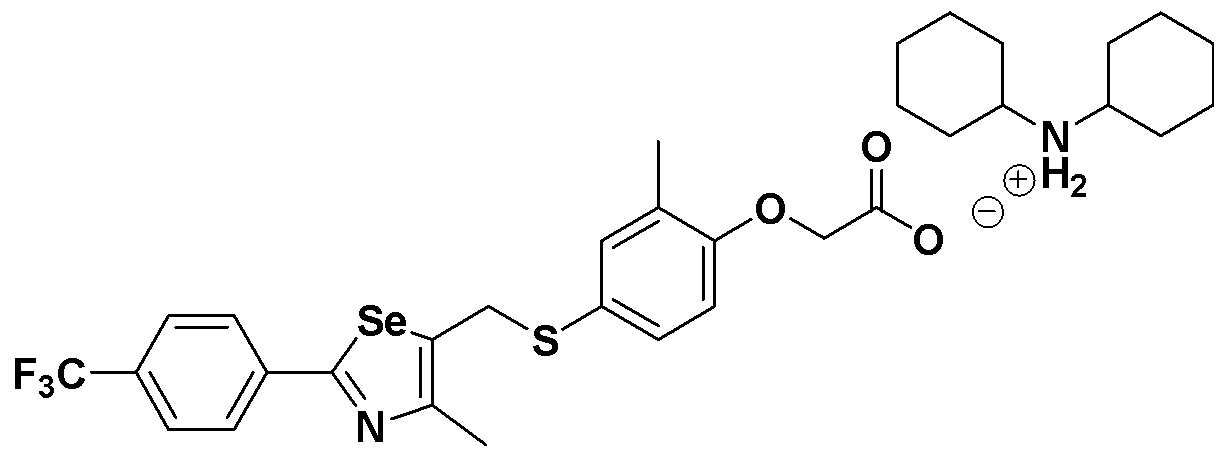
Соединение S185 (500 мг, 1 ммоль) растворяли в ацетонитриле (10 мл). Реакцию проводили в течение около 20 минут, осторожно по каплям добавляя дициклогексиламин (181 мг, 1 ммоль). Затем, после добавления дистиллированной воды (8 мл) и дополнительного взаимодействия в течение около 10 минут, лиофилизация растворителя давала целевое соединение (674 мг, выход: 99%). (FABMS: 683 [M+H]+).
[Пример 290] Получение соединения S290
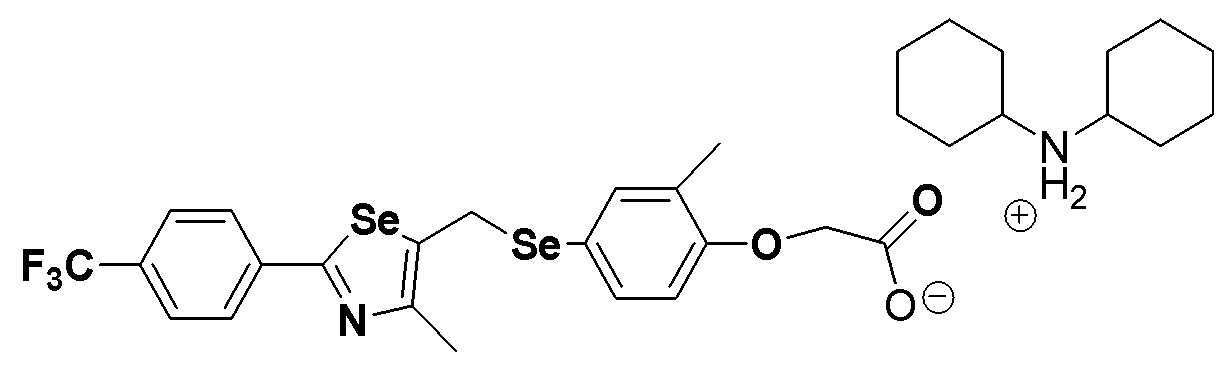
Соединение S186 (590 мг, 1 ммоль) растворяли в ацетонитриле (10 мл). Реакцию проводили в течение около 20 минут, осторожно по каплям добавляя дициклогексиламин (181 мг, 1 ммоль). Затем, после добавления дистиллированной воды (8 мл) и дополнительного взаимодействия в течение около 10 минут, лиофилизация растворителя давала целевое соединение (768 мг, выход: 99%). (FABMS: 731 [M+H]+).
[Пример 291] Получение соединения S291
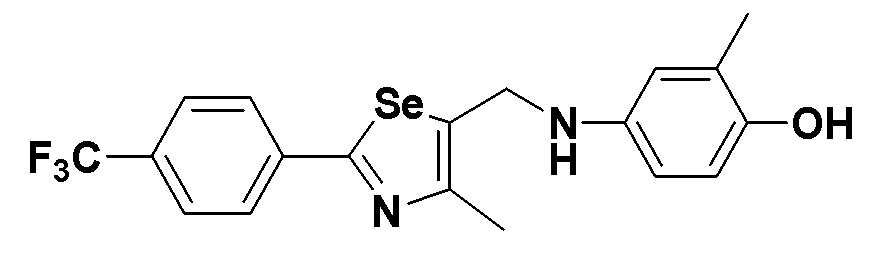
[Стадия L]
CuI (10 мг, 0,05 ммоль, 5 моль%), CsCO3 (845 мг, 2,6 эквивалента) и 4-йод-2-метилфенол (234 мг, 1 ммоль) добавляли к безводному ДМФ (0,7 мл) в атмосфере азота. После герметичного закупоривания тефлоновой лентой, заполняли азотом. Затем, после осторожного добавления 2-изобутирилциклогексанона (34 мг, 0,2 ммоль, 20 моль%) и соединения III-B-1 (320 мг, 1 эквивалент), полученного в препаративном примере 5, смесь перемешивали при комнатной температуре в течение 8 часов. После нейтрализации продукта до pH 4 с использованием 10%-ного раствора HCl, концентрирование органического слоя с последующей хроматографией на колонке с силикагелем давало целевое соединение (355 мг, выход: 79%) (FABMS: 427 [M+H]+).
[Пример 292] Получение соединения S292
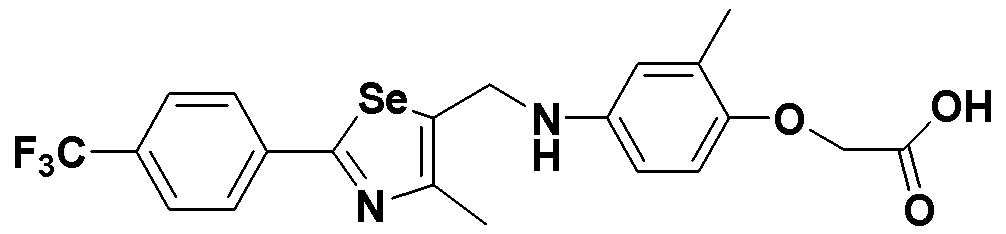
[Стадии E и F]
Целевое соединение (348 мг, выход: 82%) получали исходя из соединения S291 (425 мг, 1 эквивалент) в соответствии со способом по примерам 39 и 87 (FABMS: 485 [M+H]+).
[Пример 293] Получение соединения S293
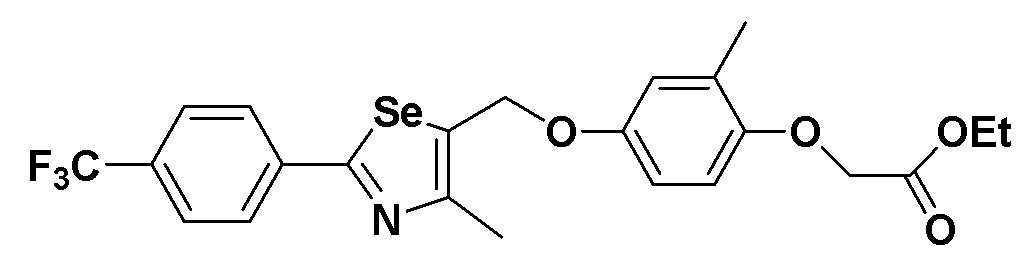
[Стадия M]
Соединение III-C-1 (474 мг, 1 эквивалент), полученное в препаративном примере 6, растворяли в ацетонитриле (5 мл) и ДМФ (0,5 мл). Затем, после медленного добавления CsCO3 (490 мг, 1,5 эквивалента) и
[Пример 294] Получение соединения S294
[Стадия F]
Целевое соединение (454 мг, выход: 94%) получали исходя из соединения S293 (510 мг, 1 эквивалент) в соответствии со способом по примеру 87 (FABMS: 486 [M+H]+).
[Пример 295] Получение соединения S295
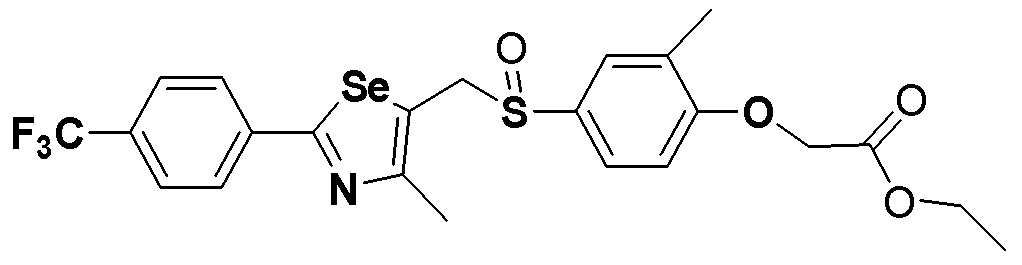
Соединение S5 (530 мг, 1 ммоль) растворяли в CH2Cl2 (10 мл). После добавления м-хлорпербензойной кислоты (м-CPBA, 170 мг, 1 ммоль), температуру реакционной смеси поддерживали при 0-5ºC. Реакцию проводили в течение около 1 часа при этой температуре. После завершения реакции (установлено с помощью ТСХ), разделение полученной смеси путем хроматографии на колонке с силикагелем давало соединение S295 (485 мг, 89%) в виде мутного масла желтого цвета. (FABMS: 546[M+H]+).
[Пример 296] Получение соединения S296
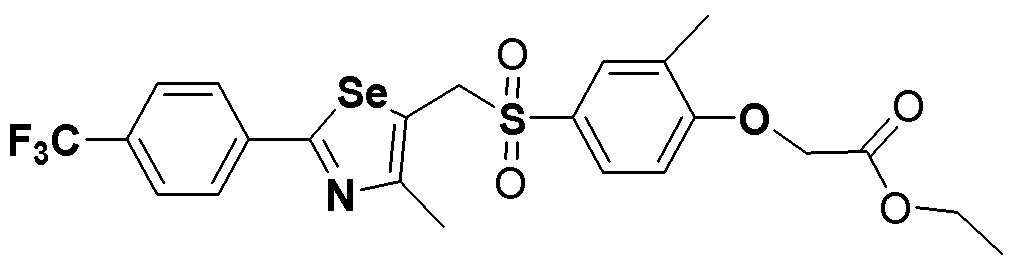
Соединение S5 (530 мг, 1 ммоль) растворяли в CH2Cl2 (10 мл). После добавления м-хлорпербензойной кислоты (м-CPBA, 340 мг, 2 ммоль), температуру реакционной смеси поддерживали при 0-5ºC. Реакцию проводили в течение около 2 часов при этой температуре. После завершения реакции (установлено с помощью ТСХ), разделение полученной смеси путем хроматографии на колонке с силикагелем давало соединение S296 (516 мг, 92%) в виде твердого вещества белого цвета. (FABMS: 562 [M+H]+).
[Пример 297] Получение соединения S297
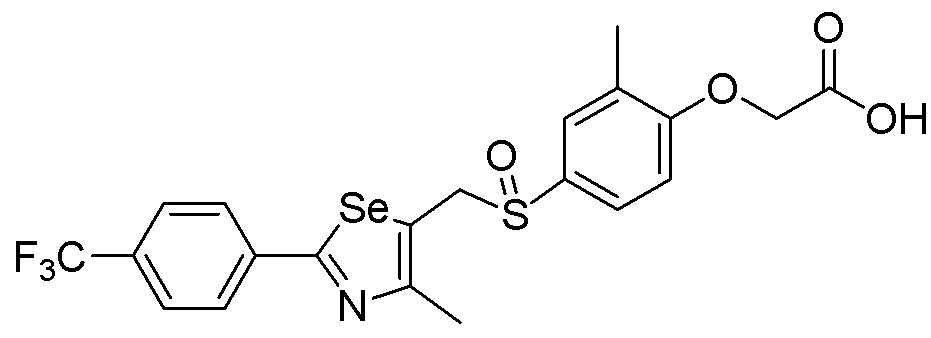
[Стадия F]
Целевое соединение (476 мг, выход: 92%) получали исходя из соединения S295 (545 мг, 1 эквивалент) в соответствии со способом по примеру 87 (FABMS: 518 [M+H]+).
[Пример 298] Получение соединения S298
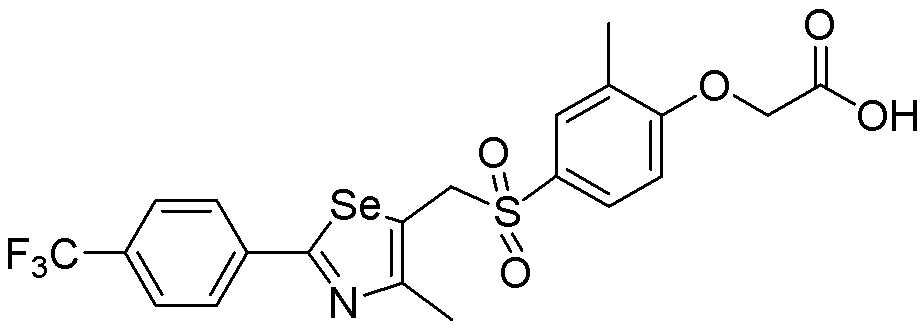
Целевое соединение (490 мг, выход: 92%) получали исходя из соединения S296 (561 мг, 1 эквивалент) в соответствии со способом по примеру 87 (FABMS: 534 [M+H]+).
[Тестовый пример 1] Тест на активность и токсичность
Эффект активации PPARδ соединения, представленного химической формулой I, в соответствии с настоящим изобретением, определяли исследованием трансфекции. Кроме того, выполняли тест на селективность для других PPAR подтипов PPARα и PPARγ, тест на токсичность проводили с помощью MTT анализа, и активность in vivo подтверждали тестом на животном.
[Трансфекционный анализ]
Для трансфекционного анализа использовали клетки CV-1. Клетки культивировали в 5% CO2 инкубаторе при 37ºC, на 96-луночном планшете, используя среду DMEM, содержащую 10% FBS, DBS (делипидизированный) и 1% пенициллина/стрептомицина. Тест выполняли в четыре стадии: посев клеток, трансфекция, обработка соединением по настоящему изобретению и подтверждение результата. Клетки CV-1 высеивали на 96-луночном планшете при концентрации 5000 клеток на лунку и трансфицировали через 24 часа. Исследование трансфекции выполняли с использованием полноразмерной плазмидной ДНК PPAR, репортерной ДНК, которая обладает активностью по люциферазе и таким образом позволяет подтверждать активность в отношении PPAR, и ДНК β-галактозидазы, которая предоставляет информацию об эффективности трансфекции. Соединение по настоящему изобретению растворяли в диметилсульфоксиде (ДМСО), разбавляли, используя среду, и затем обрабатывали им клетки при различных концентрациях. После 24 часов культивирования в инкубаторе клетки лизировали с помощью лизирующего буфера и измеряли активность по люциферазе и активность по β-галактозидазе с использованием люминометра и микропланшетного ридера. Измеренную активность по люциферазе сопоставляли с активностью по β-галактозидазе и результат изображали на графике для определения значения EC50.
Как видно из таблицы 4, соединение по настоящему изобретению обладает высокой селективностью в отношении PPARδ.
Соединение по настоящему изобретению демонстрировало активность от 0,66 до 300 нМ в отношении PPARδ.
[MTT анализ]
Токсичность соединения, представленного химической формулой I, по настоящему изобретению определяли с помощью MTT анализа. MTT является водорастворимым желтым веществом. Однако при введении в живую клетку оно превращается под действием митохондриальной дегидрогеназы в водонерастворимые пурпурные кристаллы. Клеточная токсичность может быть оценена путем растворения этого вещества в диметилсульфоксиде и измерения поглощения света при 550 нм. Тест проводили следующим образом.
Сначала, CV-1 клетки высевали на 96-луночном планшете при концентрации 5000 клеток на лунку. После культивирования в течение 24 часов в увлажненной системе культивирования при 37°C, содержащей 5%, культивированные клетки CV-1 обрабатывали соединением по настоящему изобретению (соединение S185) при различных концентрациях. После 24 часов культивирования добавляли MTT реагент. После культивирования в течение около 15 минут кристаллы пурпурного цвета растворяли в диметилсульфоксиде и измеряли поглощение с помощью микропланшетного ридера.
В результате, соединение, представленное химической формулой I, не показало токсичность даже при концентрациях в 100-1000 раз большей, чем EC50 относительно PPAR.
[Тест на животных]
[Эффект предотвращения ожирения]
Для того, чтобы подтвердить in vivo эффект соединения по настоящему изобретению, тест проводили на мышах. Использовали мышь C57BL/6 (SLC Co.) 8-недельного возраста и давали ей корм, содержащий 35% жира, для вызывания ожирения. При рационе с высоким содержанием жира в течение 60 дней перорально вводили (10 мг/кг/день) носитель, соединение S185 или соединение S186. В результате, группа, получавшая S185, показала только 39% увеличение веса, а группа, получавшая S186, показала только 42% увеличение веса по сравнению с группой, которой вводили носитель.
[Эффект предотвращения атеросклероза]
Для подтверждения эффекта предотвращения атеросклероза соединения по настоящему изобретению, in vivo эксперимент выполняли с использованием модели атеросклероза на мышах линии ApoE-/-, Ldlr-/-. При рационе с высоким содержанием жира и высоким содержанием холестерина (20% жир, 1,25% холестерин; диета AIN-93G) соединение по настоящему изобретению (соединение S185) вводили перорально в дозе 2 мг/кг/день. Через 28 дней артериальные бляшки окрашивали Суданом IV, и эффект предотвращения атеросклероза сравнивали с контрольной группой. В результате, мышь линии ApoE-/-, которой вводили соединение S185, показала улучшенный на 60% эффект предотвращения атеросклероза по сравнению с контрольной группой. А мышь линии Ldlr-/-, которой вводили соединение S185, показала улучшенный на 36% эффект предотвращения атеросклероза.
[Эффект лечения диабета]
Для подтверждения эффекта лечения диабета с помощью соединения по настоящему изобретению, выполняли тест толерантности к глюкозе (GTT). Мышам, которым перорально вводили исследуемое соединение в течение 57 дней, вводили внутрибрюшинно глюкозу (1,5 г/кг) и контролировали изменение уровня глюкозы. Группа, которой вводили соединение S185 или S186 (10 мг/кг/день), показала более низкий уровень глюкозы при голодании, чем контрольная группа. Кроме того, группа, которой вводили соединение по настоящему изобретению, показала быстрое снижение уровня глюкозы в крови между 20 и 40 минутами и полное исчезновение глюкозы через 100 минут. Для сравнения, в группе, в которой вводили носитель, уровень сахара в крови не приходил в норму даже через 120 минут. Этот результат подтверждает, что соединения S185 и S186 являются эффективными в лечении диабета.
[Эффект улучшения мышечной выносливости и функции мышц]
Эксперимент на животных выполняли для подтверждения эффекта улучшения мышечной выносливости и функции мышц с помощью соединения по настоящему изобретению. Так как формирование мышц происходит главным образом на стадии развития, соединение S185 или S186 (10 мг/кг/день) перорально вводили мыши в период беременности, лактации, или в период беременности и лактации. Различий в весе тела или темпе роста зародыша между контрольной группой и группой, которой вводили соединение, не наблюдали. После удаления кожи наблюдали, что мышцы в группе, которой вводили соединение, имели более красную окраску, чем в контрольной группе. Окрашивание АТФазы и иммунное окрашивание также показали увеличение мышечных волокон I типа в группе, которой вводили соединение. Для подтверждения эффекта изменения мышечных волокон относительно улучшения выносливости мышц и функция мышц, анализ выполняли с использованием “беговой дорожки” в виде колеса. В результате, группа, которой вводили соединение, показала значительно большее время пробега по сравнению с контрольной группой.
Кроме того, улучшение выносливости мышц и функции мышц подтверждали в случае, когда соединение по настоящему изобретению вводили взрослой особи. Мышь линии C57BL/6 10-недельного возраста подвергали физической нагрузке при пероральном введении соединения S185 или S186 (10 мг/кг). Мышь подвергали физической нагрузке на “беговой дорожке” в виде колеса в течение 30 дней, один раз в день в течение 30 минут. Режим физической нагрузки был следующим: 5 минут со скоростью 2 м/мин, 5 минут со скоростью 5 м/мин, 5 минут со скоростью 8 м/мин, далее 5 минут со скоростью 20 м/мин. В конце теста эффект улучшения мышечной выносливости и функции мышц анализировали с применением “беговой дорожки” в виде колеса. В результате, группа, которой вводили соединение, показала лучшие показатели как относительно времени физических нагрузок, так и дистанции, по сравнению с контрольной группой.
[Эффект улучшения памяти]
Эксперимент на животном (мышь линии C57BL/6 8-недельного возраста) выполняли для подтверждения эффекта лечения деменции и болезни Паркинсона с помощью соединения по настоящему изобретению путем улучшения памяти. Перед проведением эксперимента, модель заболевания мозга на животном получали путем инъекции ЛПС в мозг животного с использованием стереотаксиса. Исследуемые группы разделяли в зависимости от введения исследуемого соединения и физической нагрузки. Режим физической нагрузки был следующим: 5 минут со скоростью 2 м/мин, 5 минут со скоростью 5 м/мин, 5 минут со скоростью 8 м/мин, далее 5 минут со скоростью 20 м/мин. Тест водного лабиринта Морриса выполняли в конце эксперимента. Результат представлен в следующей таблице. Эксперимент подтверждает, что соединение в соответствии с настоящим изобретением и физическая нагрузка эффективны в лечении деменции и болезни Паркинсона посредством улучшения памяти.
[Эффект лечения жировой инфильтраци печени]
Эксперимент на животном (мышь C57BL/6 8-недельного возраста) выполняли для подтверждения эффекта лечения жировой инфильтрации печени с помощью соединения по настоящему изобретению. Для вызывания жировой инфильтрации печени использовали корм, содержащий 35% жира. В течение 78 дней при рационе с высоким содержанием жира перорально вводили соединение S185 (10 мг/кг/день). В конце эксперимента получали ткань печени и фиксировали в растворе параформальдегида и затем окрашивали гематоксилином и эозином. Результат показан на фиг. 1. Как показано на фигуре, соединение по настоящему изобретению является эффективным в предотвращении жировой инфильтрации печени.
Промышленная применимость
Как описано, новые соединения по настоящему изобретению являются эффективными в качестве лиганда, который активирует PPAR и используется для фармацевтической композиции, композиции функциональной пищевой добавки, композиции функционального напитка, композиции пищевой добавки, функциональной косметической композиции или композиции корма для животных для профилактики или лечения жировой инфильтрации печени, атеросклероза или гиперлипемии, профилактики или лечения гиперхолистеринемии, профилактики или лечения диабета, профилактики или лечения ожирения, укрепления мышц, профилактики или лечения мышечного заболевания, повышения выносливости, улучшения памяти, или профилактики или лечения деменции или болезни Паркинсона.
Реферат
Изобретение относится к производному селеназола или его фармацевтически приемлемой соли. Соединение представлено формулой I:где А представляет собой О, NR, S, S(=O), S(=O)или Se; В представляет собой водород или; Rпредставляет собой водород, С1-С8 алкил или галоген; Rпредставляет собой водород, С1-С8 алкил,или; Xи Xнезависимо представляют собой CR или N; R представляет собой водород или С1-С8 алкил; Rпредставляет собой водород, С1-С8 алкил; Rи Rнезависимо представляют собой водород, галоген или С1-С8 алкил; Rпредставляет собой водород, С1-С8 алкил, или фармацевтически приемлемую органическую соль; R, Rи Rнезависимо представляют собой водород, галоген, NO, С1-С7 алкил, замещенный или незамещенный галогеном, С3-С12 гетероарил, содержащий один или несколько гетероатом(ов), выбранных из N, О и S; m равно целому числу от 1 до 4; р равно целому числу от 1 до 5; s равно целому числу от 1 до 5; и равно целому числу от 1 до 3; w равно целому числу от 1 до 4; и алкил в R, R, R, Rи Rмогут быть дополнительно замещены одним или несколькими галогеном, С3-С7 циклоалкилом или С1-С5 алкиламином. Также предложены способы получения производных селеназола, фармацевтическая композиция, композиция функциональной пищевой добавки, функционального напитка, пищевой добавки, корма для животных, функциональная косметическая композиция, композиция активатора рецептора, активируемого пролифератором пероксисом (PPAR). Изобретение позволяет получить производное селеназола, которое активирует рецептор, активируемый пролифератором пероксисом (PPAR). 10 н. и 5 з.п. ф-лы, 1 ил., 6 табл., 298 пр.
Формула
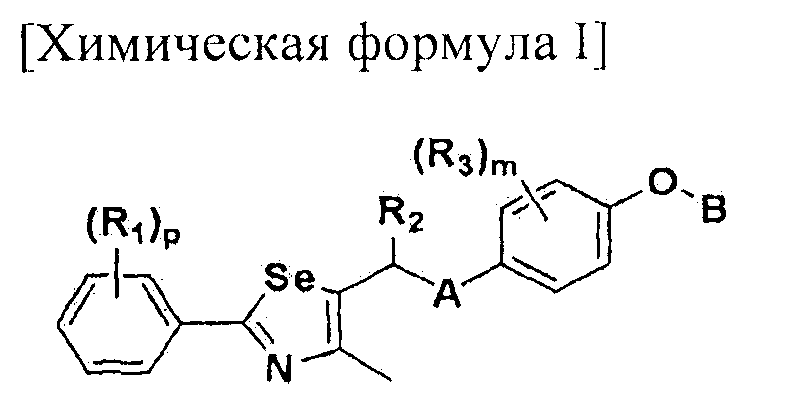
где А представляет собой О, NR, S, S(=O), S(=O)2 или Se;
В представляет собой водород или
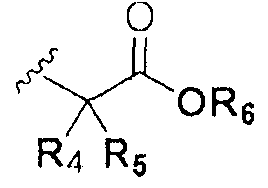
R1 представляет собой водород, С1-С8 алкил или галоген;
R2 представляет собой водород, С1-С8 алкил,
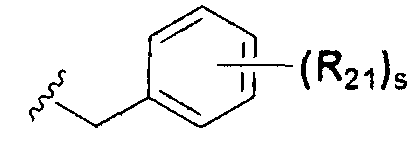
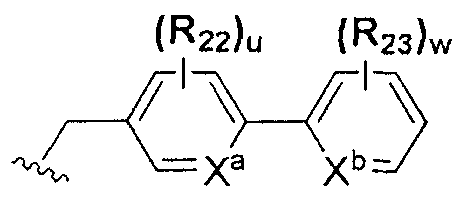
Xa и Xb независимо представляют собой CR или N;
R представляет собой водород или С1-С8 алкил;
R3 представляет собой водород, С1-С8 алкил;
R4 и R5 независимо представляют собой водород, галоген или С1-С8 алкил;
R6 представляет собой водород, С1-С8 алкил, или фармацевтически приемлемую органическую соль;
R21, R22 и R23 независимо представляют собой водород, галоген, NO2, С1-С7 алкил, замещенный или незамещенный галогеном, С3-С12 гетероарил, содержащий один или несколько гетероатом(ов), выбранных из N, О и S;
m равно целому числу от 1 до 4;
p равно целому числу от 1 до 5;
s равно целому числу от 1 до 5;
u равно целому числу от 1 до 3;
w равно целому числу от 1 до 4; и
алкил в R1, R3, R4, R5 и R6 могут быть дополнительно замещены одним или несколькими галогеном, С3-С7 циклоалкилом или С1-С5 алкиламином.
R1 представляет собой водород, С1-С5 алкил, замещенный одним или несколькими атомами фтора, или фтор:
R2 представляет собой водород, С1-С8 алкил,
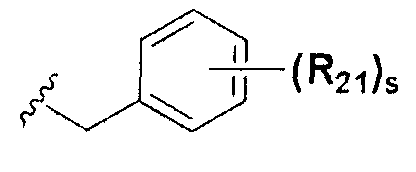
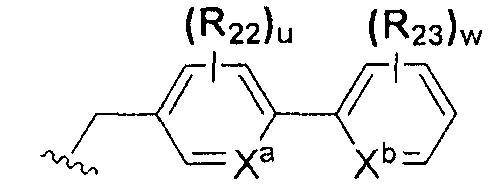
Xa и Xb независимо представляют собой CR или N;
R представляет собой водород или С1-С8 алкил;
R3 представляет собой водород или С1-С5 алкил, замещенный или незамещенный галогеном;
R4 и R5 независимо представляют собой водород, С1-С5 алкил, замещенный или незамещенный галогеном;
R6 представляет собой водород, С1-С8 алкил или фармацевтически приемлемую органическую соль; и
R21, R22 и R23 независимо представляют собой водород, галоген, NO2, С1-С7 алкил, замещенный или незамещенный галогеном, или С3-С12 гетероарил, содержащий один или несколько гетероатом(ов), выбранных из N, О и S.
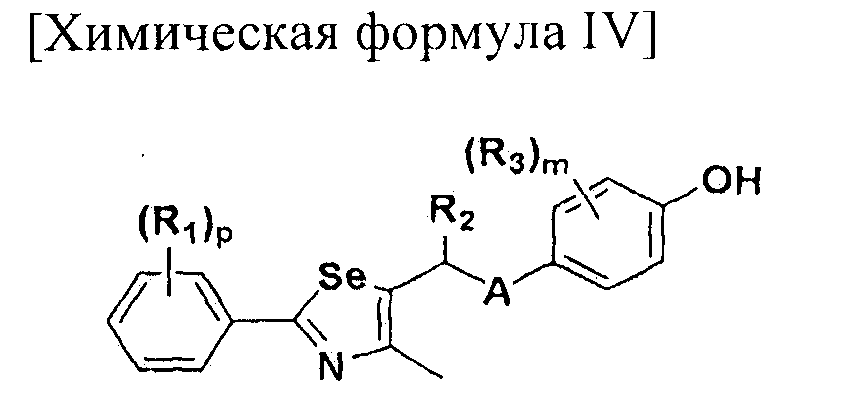
где А представляет собой О, NR, S или Se; и
R1, R2, R3, m и p являются такими, как определено в химической формуле I в п.1.
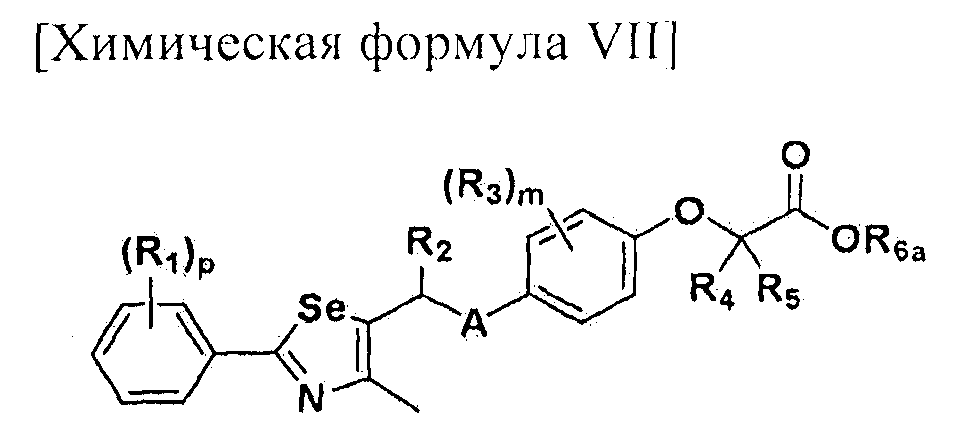
где А, R1, R2, R3, R4, R5, m и p являются такими, как определено в химической формуле I в п.1; и
R6a представляет собой С1-С8 алкил.
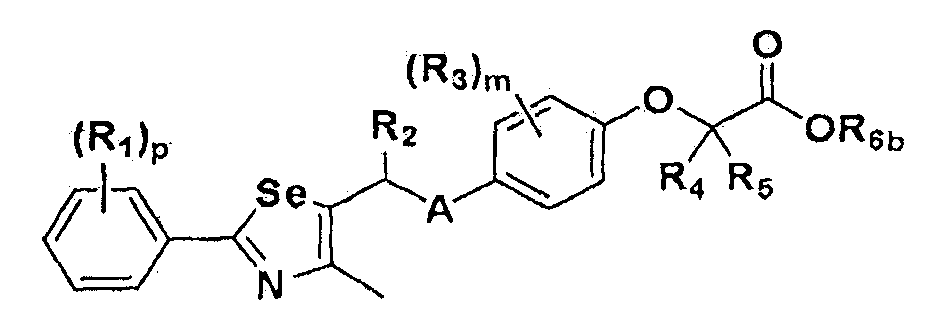
где A, R1, R2, R3, R4, R5, m и p являются такими, как определено в химической формуле I в п.1; и
R6b представляет собой водород или фармацевтически приемлемую органическую соль.
взаимодействие соединения, представленного химической формулой II, с реактивом Гриньяра и далее с соединением органолития;
последующее добавление порошка серы (S) или селена (Se);
последующее взаимодействие с соединением, представленным химической формулой III-A, с получением соединения, представленного химической формулой IV-A; и
защиту фенольной группы соединения, представленного химической формулой IV-A, путем введения алкилсилильной группы, обработку α-протона полученного тио- или селеноэфирного соединения сильным основанием, добавление соединения, представленного химической формулой VI, и последующее удаление защитной группы с получением соединения, представленного химической формулой IV-B:
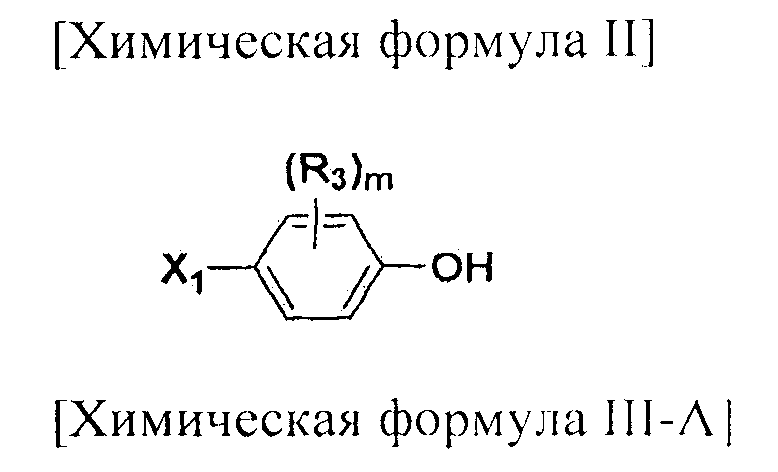
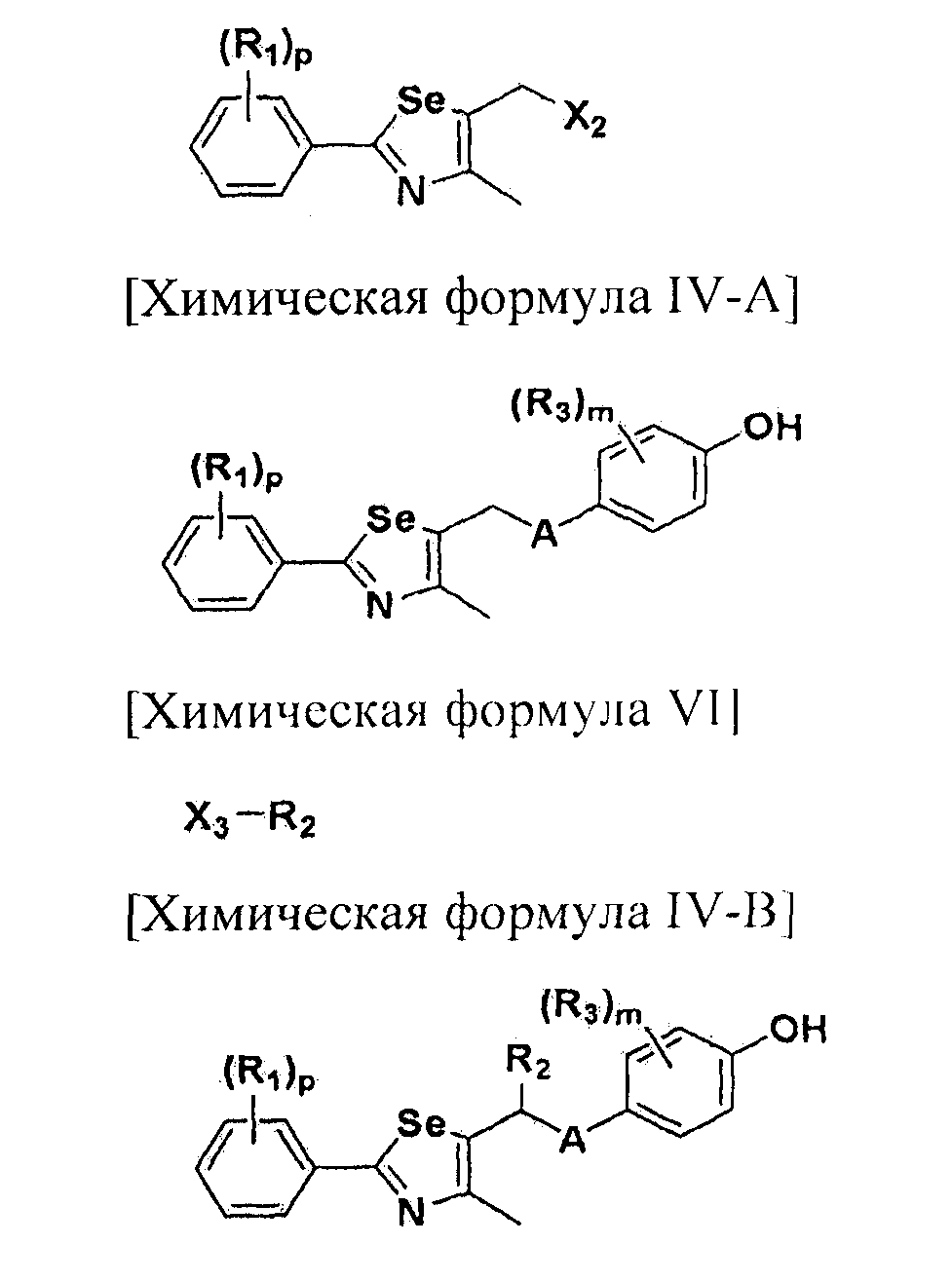
где А представляет собой S или Se; R2 представляет собой
взаимодействие соединения, представленного химической формулой IV-A, с реактивом Гриньяра;
последующую обработку α-протона полученного тио- или селеноэфирного соединения сильным основанием; и
взаимодействие с соединением, представленным химической формулой VI, с получением соединения, представленного химической формулой IV-B:
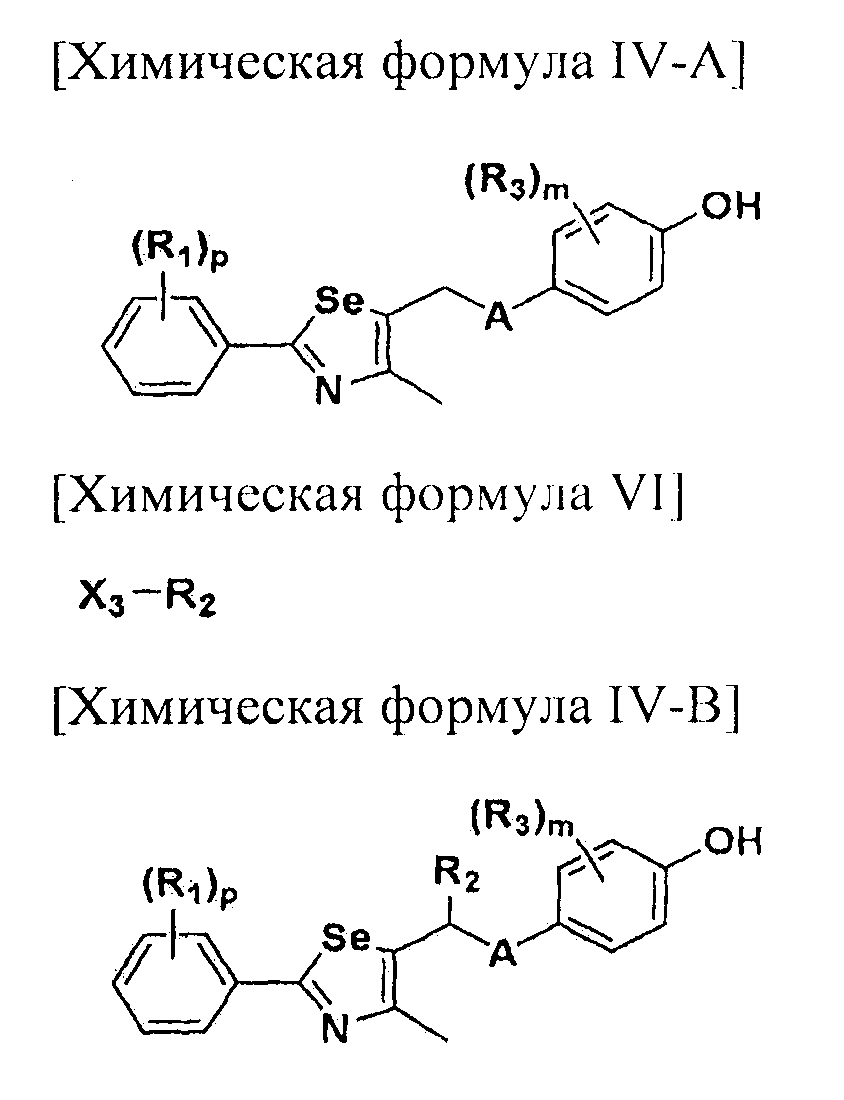
где А представляет собой S или Sc; R2 представляет собой
взаимодействие соединения, представленного химической формулой II, с соединением, представленным химической формулой III-B, в присутствии йодида меди (CuI) и 2-изобутирилциклогексанона с получением соединения, представленного химической формулой IV-C:
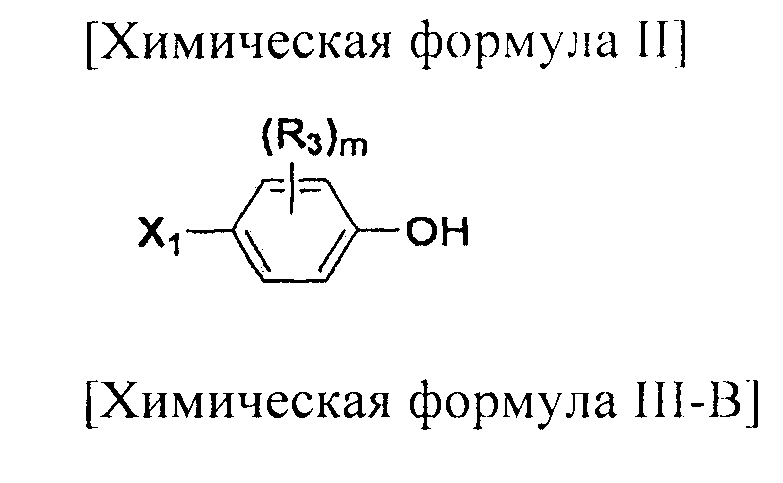
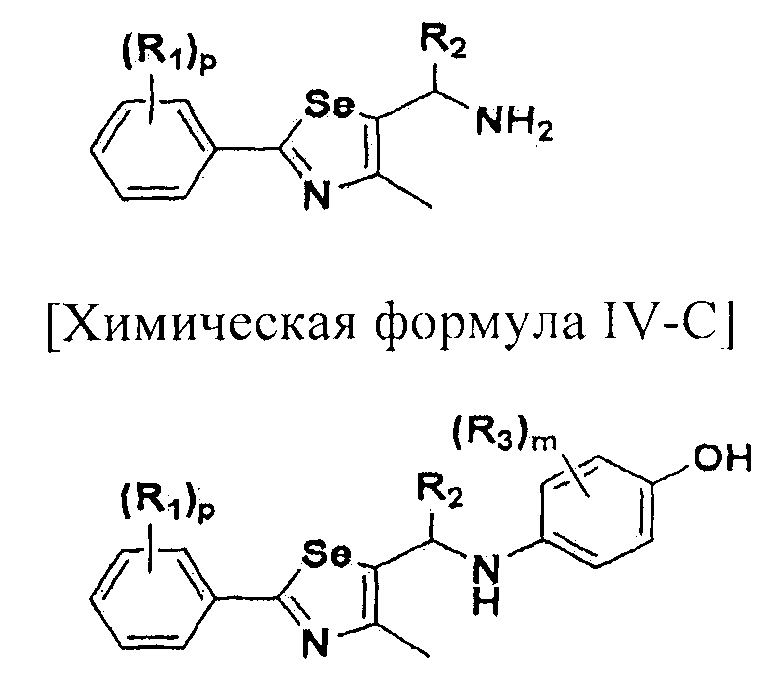
где R1, R2, R3, m и p являются такими, как определено в химической формуле I в п.1; и X1 представляет собой бром или йод.
взаимодействие соединения, представленного химической формулой IV, с алкил галогенацетатом или алкиловым эфиром алкилгалогенуксусной кислоты с получением сложноэфирного соединения, представленного химической формулой VII:
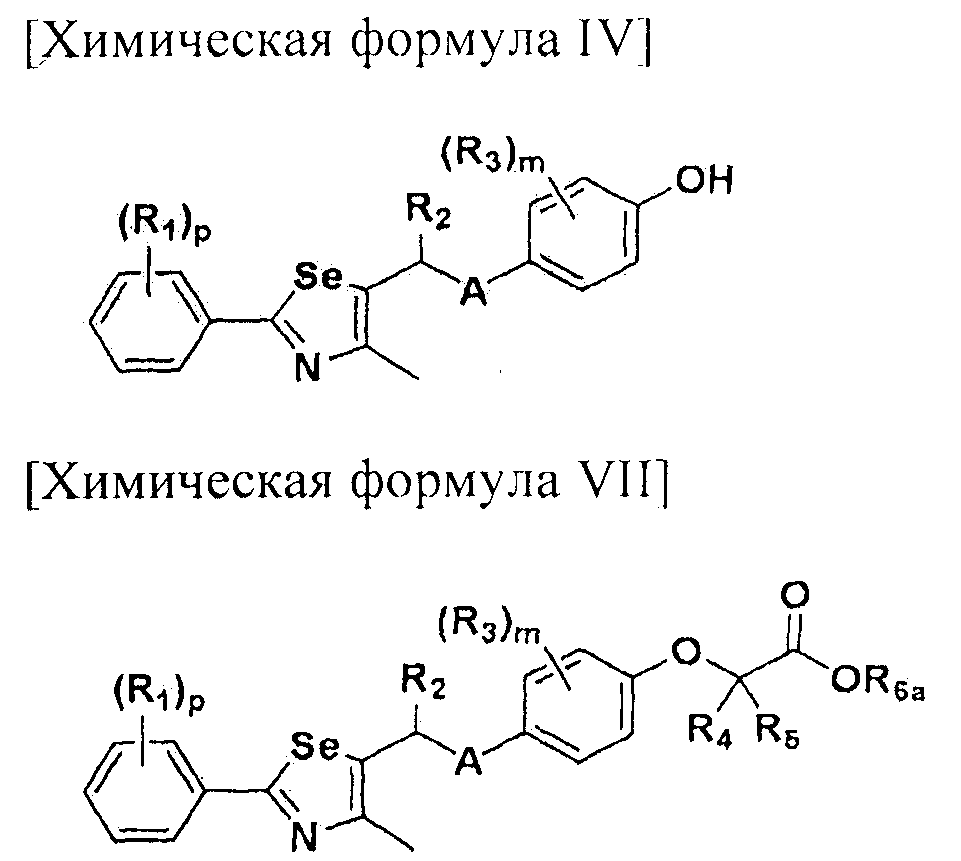
где A, R1, R2, R3, R4, R5, m и p являются такими, как определено в химической формуле I в п.1; и R6a представляет собой C1-C8 алкил.
взаимодействие соединения, представленного химической формулой X, с соединением, представленным химической формулой III-C, с получением соединения, представленного химической формулой VII-D:
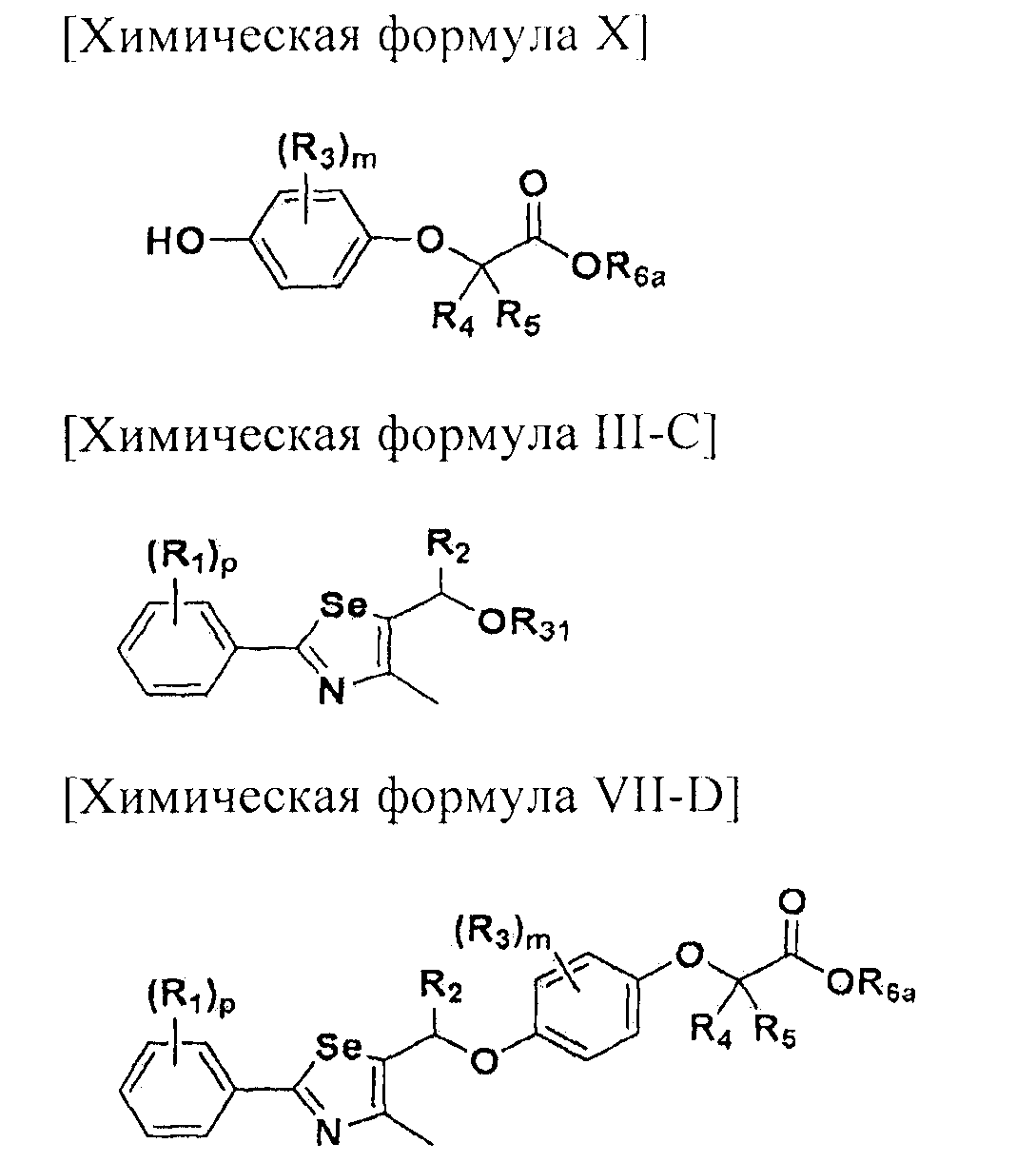
где R1, R2, R3, R4, R5, m и p являются такими, как определено в химической формуле I в п.1; R6a представляет собой С1-С8 алкил; R31 представляет собой C1-С4 алкилсульфонил или C6-С12 арилсульфонил, замещенный или незамещенный C1-С4 алкилом.
гидролиз сложноэфирного соединения, представленного химической формулой VII, с получением соединения, представленного химической формулой VIII:
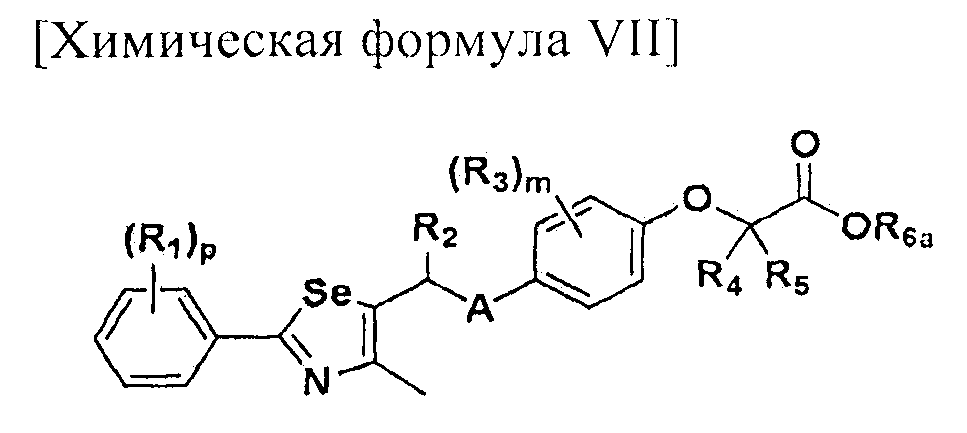
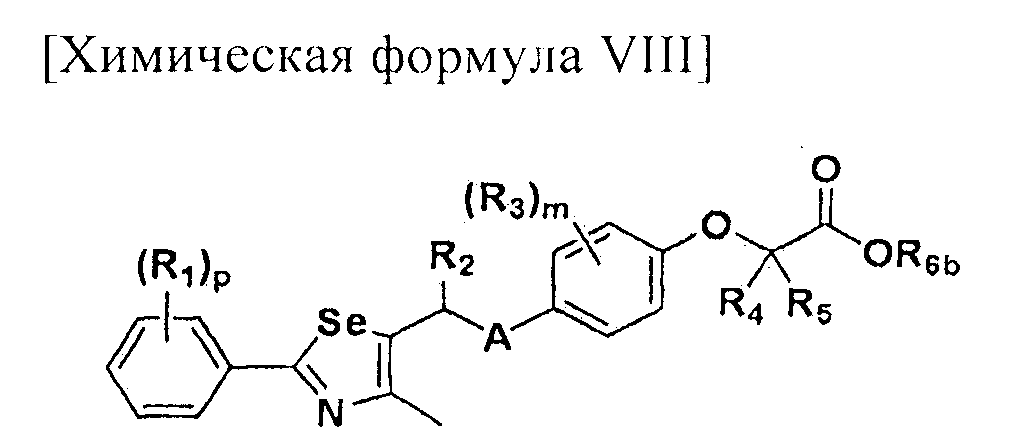
где A, R1, R2, R3, R4, R5, m и p являются такими, как определено в химической формуле I в п.1; R6a представляет собой C1-C8 алкил; R6bпредставляет собой водород или фармацевтически приемлемую органическую соль.
Комментарии