Производные 4-фениламинотиазола, способы их получения и фармацевтическая композиция - RU2146253C1
Код документа: RU2146253C1
Чертежи

Описание
Изобретение касается новых разветвленных аминопроизводных тиазола и способа их получения. Новые производные тиазола обладают антагонистической активностью по отношению к фактору высвобождения кортикотропина (CRF) и, следовательно, могут использоваться как действующие начала в фармацевтических композициях.
Фактор высвобождения кортикотропного гормона (CRF) представляет собой пептид, последовательность которого из сорока одной аминокислоты охарактеризована Vale W. и др. в 1981 г. (Science, 213, 1394-1397 (1981)). CRF является основным эндогенным фактором, принимающим участие в регуляции гипоталамо-гипофизо-надпочечниковой оси (высвобождение адренокортикотропного гормона: ACTH) и ее патологиях, а также в проистекающих от этого депрессивных синдромах. CRF также провоцирует выделение β- эндорфина, β- липотропина и корикостерона.
Следовательно, CRF является физиологическим регулятором выделения адренокортикотропного гормона (ACTH) и более конкретно пептидов, происходящих от проопиомеланокортина (POMC). Хотя он локализован в гипоталамусе, CRF также широко распространен в центральной нервной системе (относящаяся к лимбу зона), в которой он играет роль нейротрансмиттера и/или нейромодулятора, независимо от его воздействий на гипоталамо-гипофизо-надпочечниковую ось.
Многочисленные эксперименты на животных показывают, что центральное введение CRF провоцирует разные анксиогенные эффекты, такие, как изменение поведения в целом: например неофобия, снижение сексуальной рецептивности, уменьшение потребления пищи и продолжительности сна у крыс. Интрацеребровентрикулярная инъекция CRF повышает возбуждение норадренергических нейронов голубого пятна, которое часто у животного ассоциируется с состоянием страха. В случае крысы центральное или периферическое введение CRF вызывает изменения опорожнения желудка, танзита через кишечник, экскреции фекалиев, выделения кислоты, а также оказывает эффект повышения давления. Специфическое участие CRF в этих эффектах выявляют путем использования пептидного антагониста, альфа-геликаля CRF (9-41) (ah-CRF), или специфических антител (Rivier J. и др. Science, 224, 889-891 (1984)); это позволяет подтвердить роль этого пептида в возникновении эндокринных расстройств и нарушений поведения, связанных со стрессом.
В самом деле, эти эксперименты показывают, что CRF играет
значительную роль у человека в интеграции комплексных ответных реакций, наблюдаемых во время физиологического, психологического или иммунологического стресса, как в нейроэндокринном, висцеральном
плане, так и в плане поведения (Morley J.E. и др., Endocrine Rewiew, 8, 3, 256-287 (1987); Smith M.A. и др., Horm.Res., 31, 66-71 (1989)). Кроме того, клинические данные свидетельствуют в пользу
эффективного участия CRF в многочисленных нарушениях, проистекающих от состояния стресса (Gulley L.R. и др., J.Clin. Psychiatry, 54, 1 (дополнение), 16-19 (1993)), таких как:
- существование
теста с участием CRF (введение внутривенно) у человека позволяет показать изменение ответной реакции на уровне ACTH у пациентов, находящихся в состоянии депрессии (Breier A. и др., Am. J. Psychiatry,
144, 1419-1425 (1987));
- обнаружение гиперсекреции эндогенного CRF в случае некоторых патологий, например повышенное количество CRF в спинномозговой жидкости у пациентов неизлечимых,
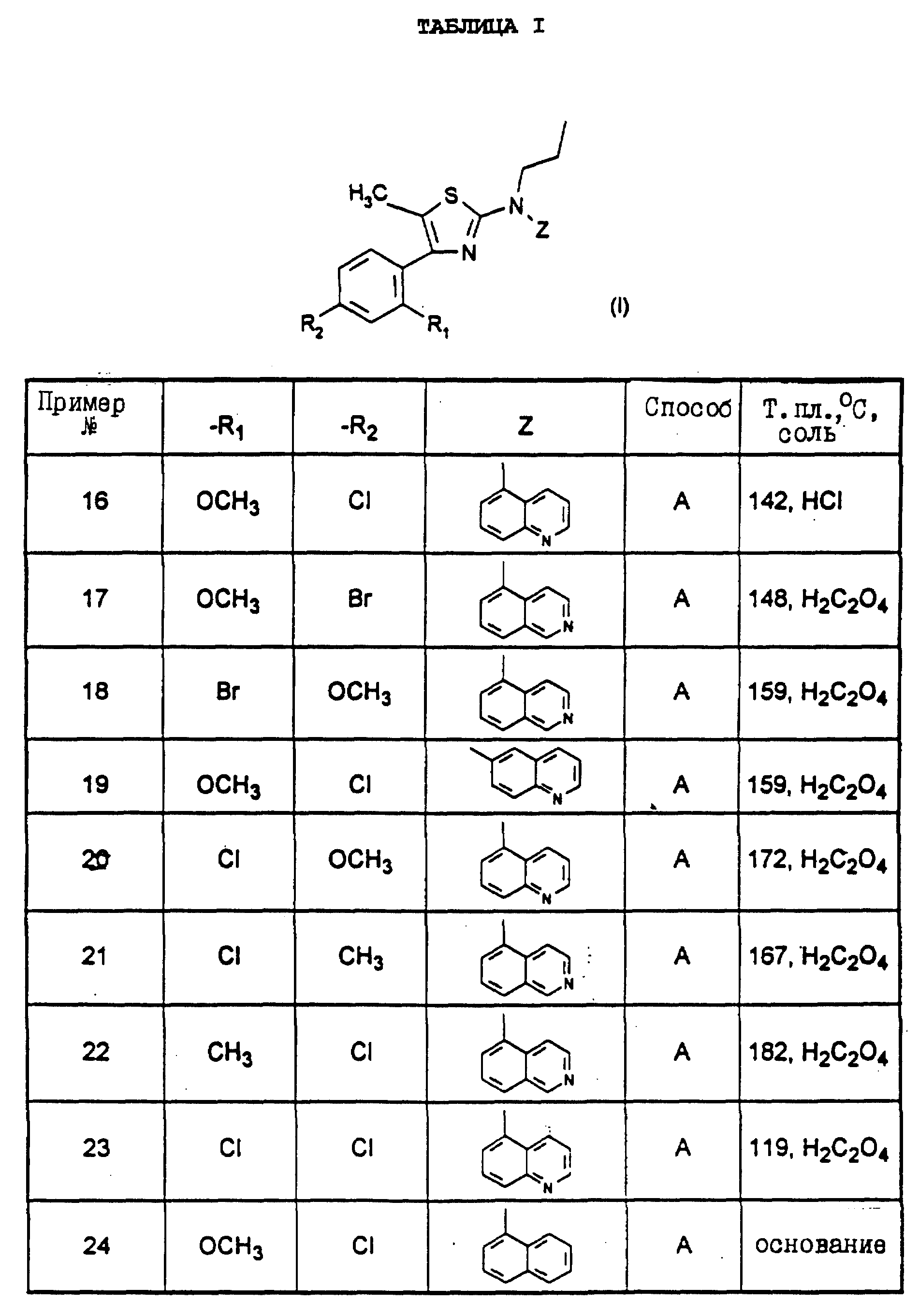
угнетенных или пораженных деменцией типа болезни Альцгеймера (Nemeroff C. B. и др., Science, 226, 4680, 1342-1343 (1984); Regul. Pept. , 25, 123-130 (1989)), или плотность рецепторов CRF, пониженная в
коре головного мозга у самоубийц (Nemeroff C.B. и др., Arch.Gen.Psychiatry, 45, 577-579 (1988));
- дисфункция CRF-зависимых нейронов в случае тяжелых патологий также наводит на мысль, что
имеют место болезни Альцгеймера, Паркинсона, хорея Гентингтона и боковой амиотрофический склероз (De Souza Е.В., Hospital Practice, 23, 59 (1988)).
Центральное введение CRF многочисленным видам животных создает поведенческие эффекты, подобные тем, которые появляются у человека в стрессовых ситуациях. При неоднократном воздействии такими эффектами могут быть различные патологии, такие как: усталость, гипертензия, нарушение работы сердца, изменение опорожнения желудка, экскреции фекалиев (колит, раздражительная ободочная кишка), изменение выделения кислоты, гипергликемия, запоздалое развитие, анорексия, неофобия, нарушения в сфере половой деятельности, иммуноподавляющее действие (воспалительные процессы, множественные инфекции и раковые заболевания) и различные психические расстройства (депрессия, нервная анорексия и беспокойство).
Интрацеребровентрикулярная инъекция стандартного пептидного антагониста ah-CRF предотвращает эффекты, создаваемые либо введением экзогенного CRF, либо при использовании агентов, вызывающих стресс (простой эфир, принуждение, шум, электрический шок, лишение алкоголика этилового спирта, хирургия), которые способны сами по себе индуктировать повышение содержания эндогенного CRF. Эти результаты подтверждаются изучением многочисленных молекул-антагонистов, структурно родственных CRF, и которые обладают более продолжительным периодом действия по отношению к ah-CRF (Rivier J. и др., J. Med.Chem., 36, 2851-2859 (1993); Menzaghi F. и др., Pharmacol.Exp.Ther., 269, 2, 564-572 (1994); Hernandez J.F. и др., J.Med.Chem., 36, 2860-2867 (1993)). Кроме того, предпочтительные исследования показывают, что трициклические антидепрессоры могут модулировать содержание CRF, а также число рецепторов CRF в коре головного мозга (Grigoriadis D. E. и др., Neuropsychopharmacology, 2, 53-60 (1989)). Также было показано, что бензодиазепиновые анксиолитические агенты способны инвертировать воздействие CRF (Britton К.Т. и др., Psychopharmacology, 94, 306, (1988)), причем полностью не разъясняется механизм действия этих веществ. Эти результаты говорят о возрастающей потребности в непептидных молекулах-антагонистах рецепторов CRF.
Также важно отметить три возможных последствия при состоянии хронического стресса, которые представляют собой иммунодепрессию, нарушения деторождения, а также появление диабета.
Уже известно большое количество производных 2-аминотиазола. В европейской заявке на патент 462264 описываются производные 2-аминотиазола, третичная аминогруппа которых в положении 2 содержит два заместителя, каждый из которых включает, по крайней мере, один гетероатом, образуя аминопроизводное. Эти соединения являются антагонистами фактора активации тромбоцитов (PAF-acether) и находят применение при лечении астмы, некоторых аллергических состояний или некоторых воспалительных состояний, сердечно-сосудистых заболеваний, гипертензии и различных почечных патологий, или же в качестве контрацептивных агентов. В заявке на патент Великобритании 2022285 описываются соединения, обладающие регулирующей активностью в отношении иммунного ответа и противовоспалительными свойствами. Речь идет о производных тиазола, замещенных в положении 2 вторичными аминогруппами.
Некоторые гетероциклические производные 2-ациламинотиазола описываются в европейской заявке на патент 432040. Эти соединения являются антагонистами холецистокинина и гастрина. Также известны производные 2-амино-4,5-дифенилтиазола, обладающие противовоспалительными свойствами (заявка на патент Японии 0175475). Также известны производные 2-амино-4-(4-гидроксифенил)тиазола в качестве промежуточных продуктов синтеза производных 2, 2-диарилхроменотиазола (европейская заявка на патент 205069). В J.Chem.Soc.Perkin, Trans. I, 2, 147-153 (1984) и в J.Chem.Soc.Perkin, Trans.I, 2, 341-347 (1983) также описываются производные 2-(N-метил-N-бензиламино)тиазола.
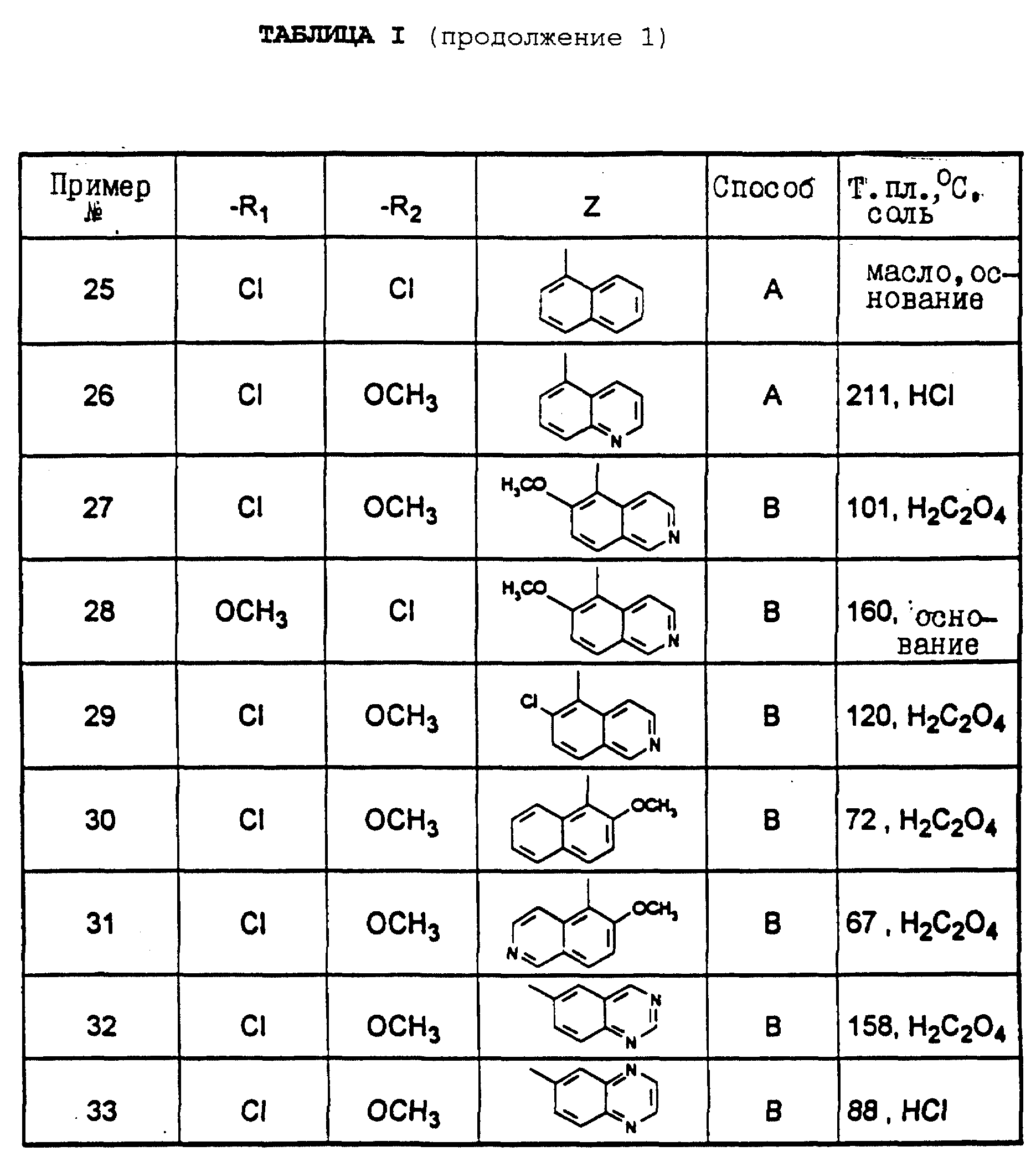
В европейской заявке на патент 283390 в числе других производных тиазола описываются производные 2-(N-алкил-N-пиридилалкиламино) тиазола формулы:
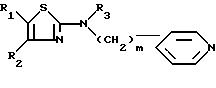
Эти производные, аминогруппа которых в положении 2 замещена неразветвленным пиридилалкильным радикалом, обладают, в частности, активностью, стимулирующей центральную холинергическую трансмиссию. Следовательно, их можно использовать в качестве агонистов мускариновых рецепторов и их можно применять при лечении расстройств памяти и старческого слабоумия.
Производные 2-аминотиазола, аминогруппа которых в положении 2 представляет собой третичную аминогруппу, содержащую разветвленный алкильный или аралкильный заместитель, описываются в европейском патенте 576350, как обладающие сродством к рецепторам CRF.
В патенте США 5063245 описывается антагонист CRF, обладающий микромолярной способностью in vitro. Из многочисленных заявок на патент, касающихся непептидных молекул, опубликованы, например, международные заявки NN 94/13643, 94/13644, 94/13661, 94/13676, 94/13677, 94/10333, 95/00640, 95/10506, 95/13372, 95/33727, 95/33750, 95/34563 или европейский патент 691128.
В настоящее время найдено, что некоторые разветвленные аминопроизводные тиазола, составляющие объект настоящего изобретения, обладают превосходным сродством по отношению к специфическим рецепторам CRF. Более того, учитывая их структуру, эти молекулы обладают хорошей диспергируемостью и/или растворимостью в растворителях или растворах, широко используемых в терапии, которая придает им улучшенную фармакологическую активность, а также позволяет легко получать пероральные и парентеральные галеновые формы.
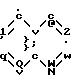
Объектом настоящего изобретения являются соединения формулы (I)
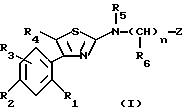
в которой R1 и R2, одинаковые или разные, каждый, независимо друг от друга, означают атом галогена; гидроксиалкильный радикал с 1-5 атомами углерода; алкил с 1-5 атомами углерода; аралкил с 7 - 10 атомами углерода; алкоксил с 1-5 атомами углерода; трифторметил; нитрогруппу; цианогруппу; группу -SR, в которой R означает водород, алкил с 1-5 атомами углерода или аралкил с 7-10 атомами углерода; группу S-CO-R, в которой R означает алкил с 1-5 атомами углерода, или аралкил, в котором арильная часть содержит 6-8 атомов углерода и алкильная часть содержит 1-4 атома углерода; группу -COOR', в которой R' означает водород или алкил с 1-5 атомами углерода; группу -CONR'R'', где R' и R'' имеют указанное выше значение для R'; группу -NR'R'', где R' и R'' имеют указанное выше значение для R'; группу -CONRaRb или -NRaRb, в которых Ra и Rb вместе с атомом азота, с которым они связаны, образуют 5-7-членный гетероцикл; или группу -NHCO-NR'R'', где R' и R'' имеют указанное выше значение для R';
R3 означает водород или имеет указанное выше для R1 и R2 значение;
R4 означает атом водорода; алкил с 1-5 атомами углерода; галоген; гидроксиметил; или формил;
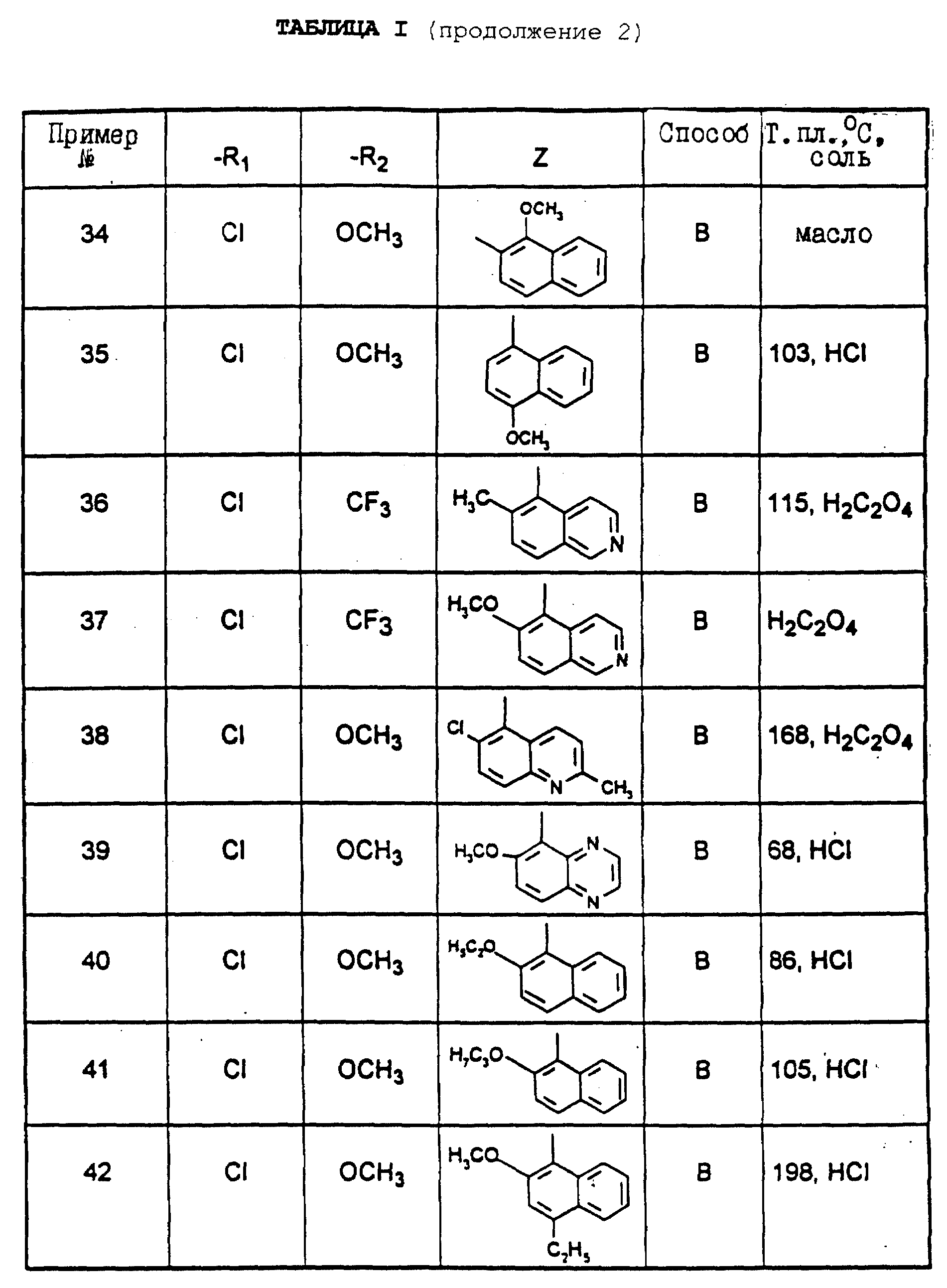
R5 означает алкил с 1-5 атомами углерода; циклоалкил с 3-7 атомами углерода; циклоалкилалкил, в котором циклоалкильная часть содержит 3-7 атомов углерода, а алкильная часть содержит 1-5 атомов углерода; или алкенил с 5-6 атомами углерода;
n означает нуль или единицу;
R6 означает алкил с 1-5 атомами углерода; алкоксиалкил, в котором алкильные части содержат 1-5 атомов углерода; циклоалкил с 3-7 атомами углерода; циклоалкилалкил, в котором циклоалкильная часть содержит 3-7 атомов углерода, а алкильная часть содержит 1-5 атомов углерода; циклоалкилоксиалкил, в котором циклоалкильная часть содержит 3-7 атомов углерода, а алкильная содержит 1-4 атома углерода; гидроксиалкилоксиалкил, в котором алкильные части содержат 2-10 атомов углерода; алкоксиалкилоксиалкил, в котором алкильные части содержат 3-12 атомов углерода;
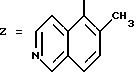
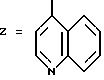
Z означает возможно замещенную би- или трициклическую ароматическую или гетероароматическую группу;
их стереоизомеры и/или их соли присоединения.
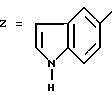
Под би- или трициклической ароматической или гетероароматической группой понимают, в частности, би- или трициклическую арильную группу с 10-14 атомами углерода, или би- или трициклическую гетероарильную группу с 5-13 атомами углерода, включающую 1-5 гетероатомов, выбираемых среди азота, серы и кислорода; причем вышеуказанные группы предпочтительно выбирают из нафталина, хинолина, изохинолина, хиноксалина, хиназолина, циннолина, фталазина, 1,5-нафтиридина, 1,7-нафтиридина, индола, изоиндола, бензотиофена, бензофурана, бензимидазола, индана, индазола, хинолизина, пиридопиримидина, пирролопиримидина, пиразолопиримидина, причем вышеуказанные группы могут быть замещены.
Заместители группы Z выбирают предпочтительно из галогена, алкила с 1-3 атомами углерода, алкоксила с 1-3 атомами углерода, трифторметила, нитрогруппы; -NRdRe, где Rd и Re, независимо друг от друга, означают водород или алкил с 1-3 атомами углерода; алкоксикарбонилалкила, карбоксиалкила, морфолинкарбонилалкила, алкилкарбонилалкила, диалкиламинокарбонилалкила или алкоксиалкоксила, в которых алкильные части содержат 1-3 атома углерода.
В настоящем описании алкильные или алкоксильные группы являются линейными или разветвленными.
Предпочтительными соединениями согласно изобретению являются такие, в которых Z означает нафтильную или гетероароматическую группу, выбираемую из хинолила, изохинолила, хиназолила, хиноксалила, индолила, индазолила, причем указанные группы возможно замещены; a R1, R2, R3, R4, R5 "n" и R6 имеют вышеуказанное в формуле (I) значение; один из их стереоизомеров и/или одна из их солей.
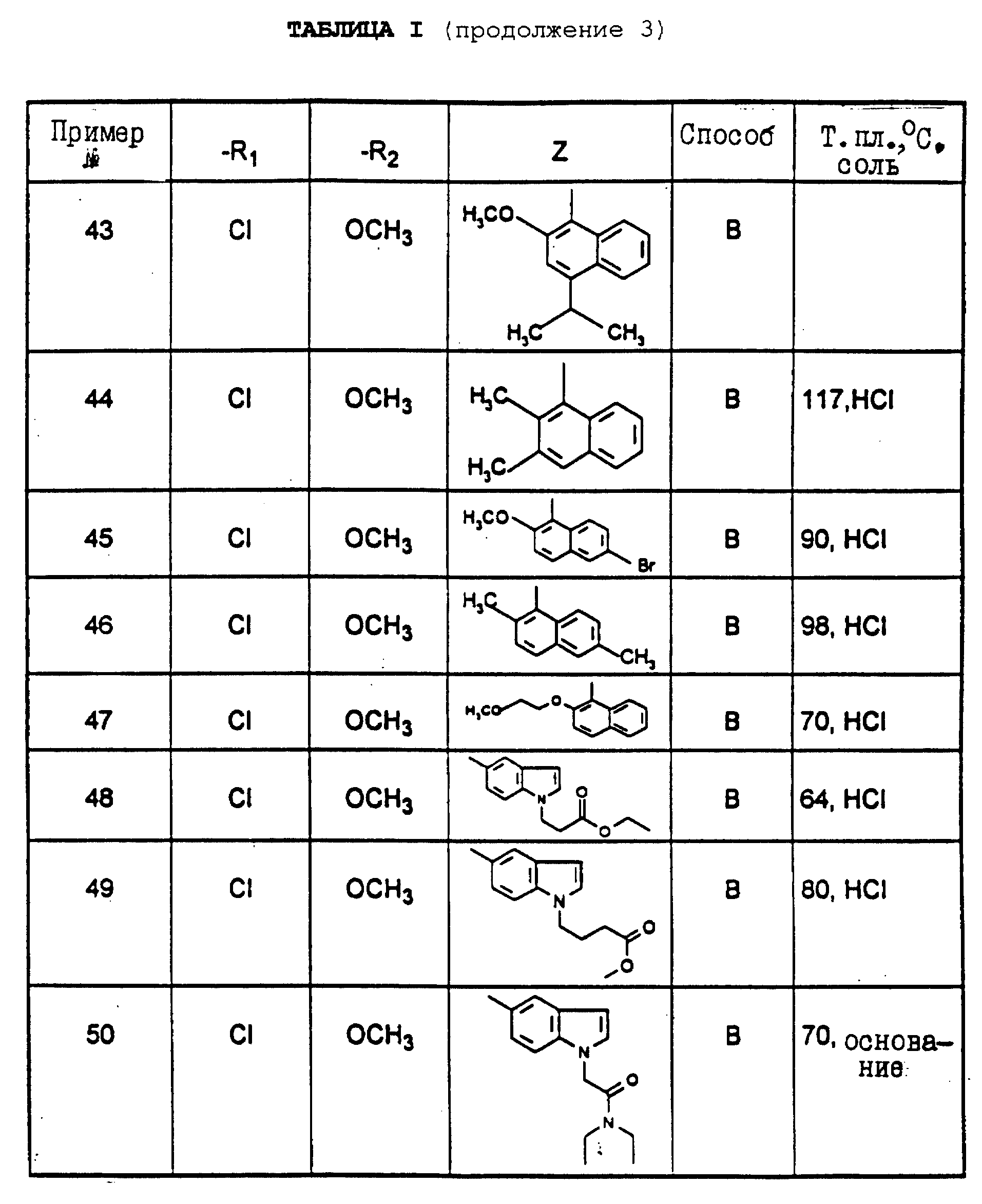
Предпочтительны такие соединения, в которых R3 означает водород, R4 означает метил, R5 означает пропил, "n" означает нуль и R1, R2 и Z имеют указанное для формулы (I) значение, один из их стереоизомеров и/или одна из их солей.
Особенно предпочтительными являются соединения, в которых R3 означает водород, R4 означает метил, R5 означает пропил, "n" означает 1, R6 означает циклопропил и R1, R2 и Z имеют указанное для формулы (I) значение; один из их стереоизомеров и/или одна из их солей.
Также особенно предпочтительными являются соединения, в которых R3 означает водород, R4 означает метил, R5 означает пропил, "n" означает 1, R6 означает метоксиметил и R1, R2 и Z имеют указанное для формулы (I) значение; один из их стереоизомеров и/или одна из их солей.
В особенности предпочтительными являются соединения формулы (I), в которых R3 означает водород, R4 означает метил, R5 означает пропил, R1 или R2 означает галоген, алкил или алкоксил с 1-5 атомами углерода; "n", R6 и Z имеют указанное для формулы (I) значение; один из их стереоизомеров и/или одна из их солей.
Также особенно предпочтительны следующие соединения:
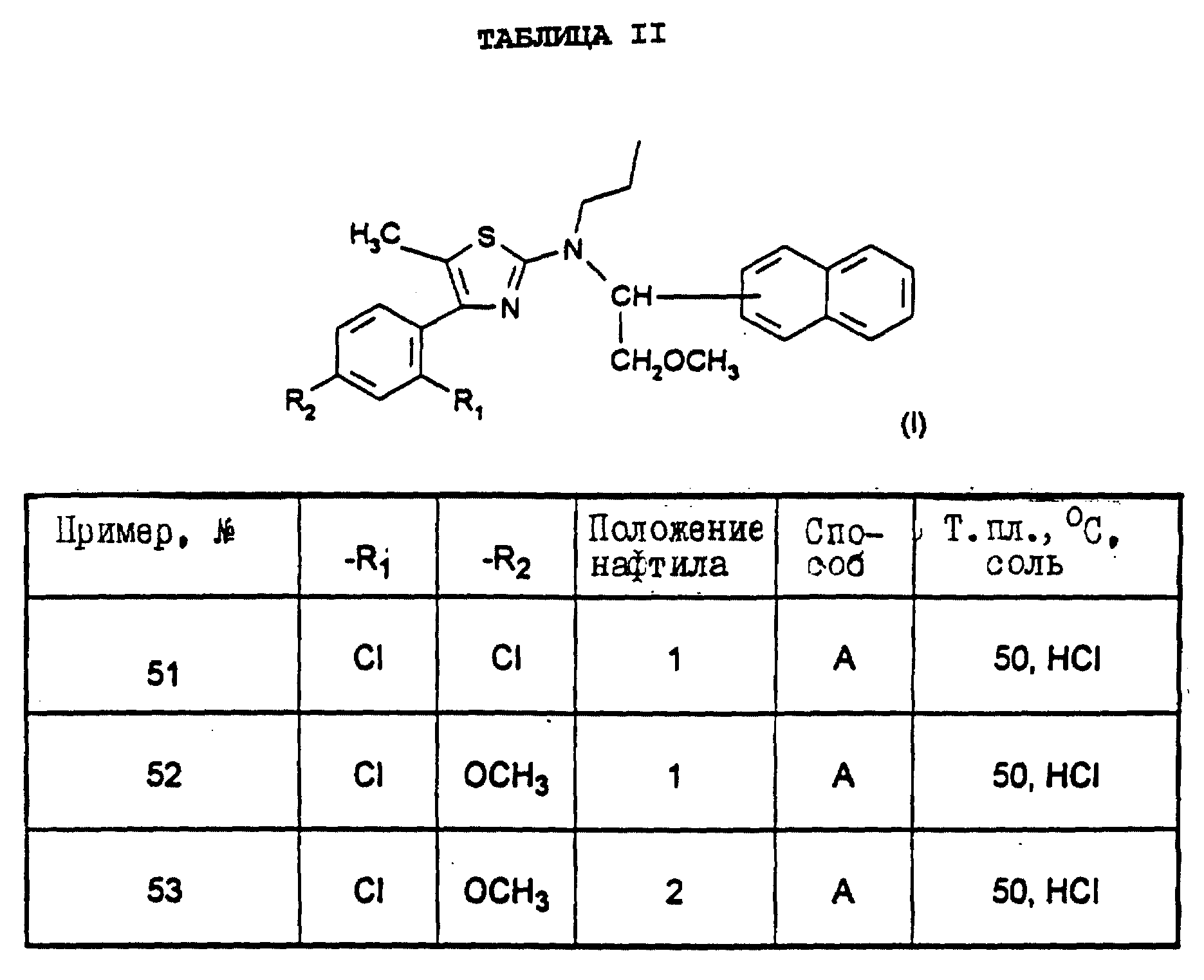
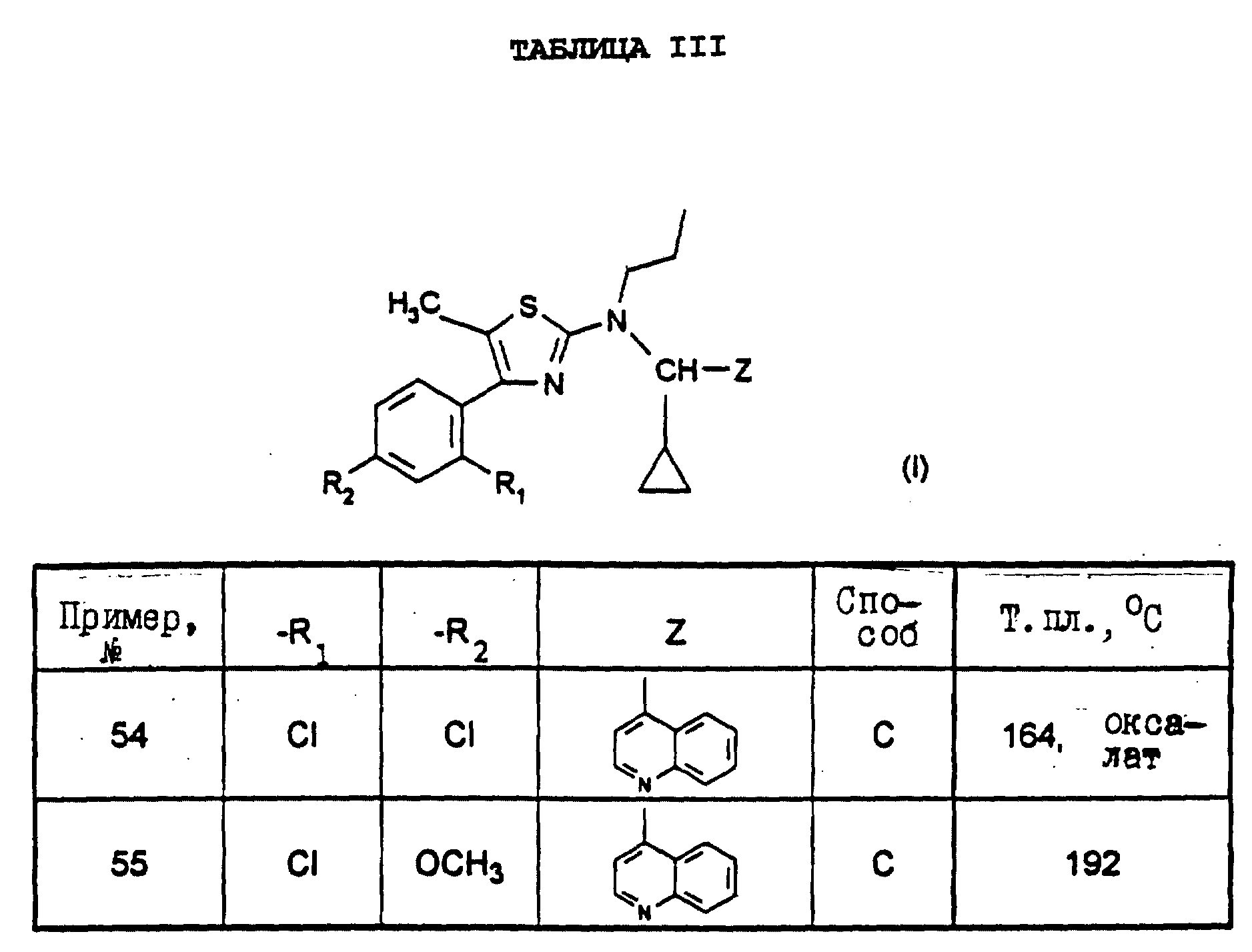
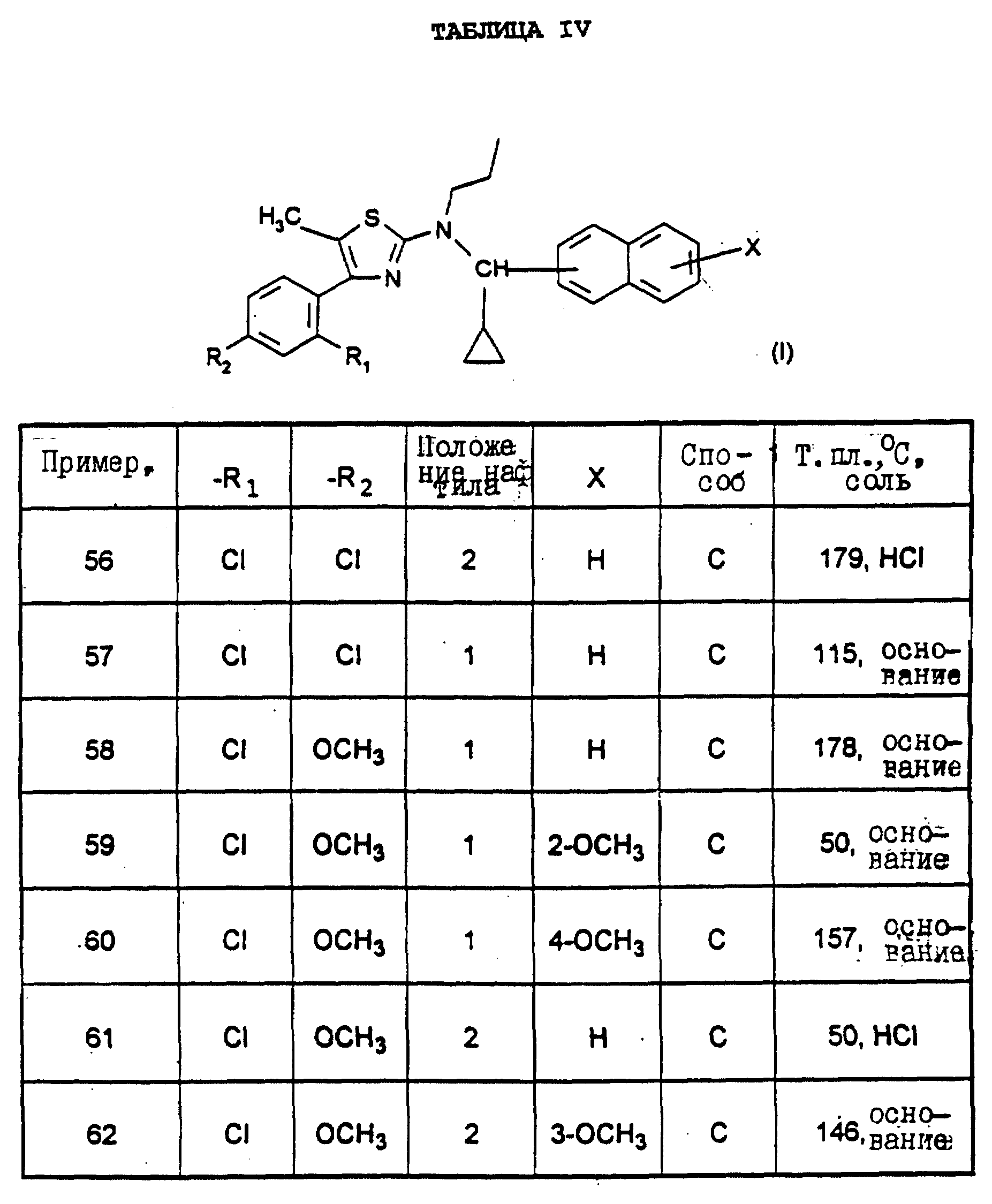
4-(2,
4-дихлорфенил)-5-метил-2-[N-(1-(метоксиметил) -1-(нафт-2-ил)метил)-N-пропиламино]тиазол (пример 3),
оксалат 4-(2,4-дихлорфенил)-5-метил-2-[N-(6-метоксиизо-хинол- 5-ил)-N-пропиламино]тиазола
(пример 4),
оксалат 4-(2-хлор-4- метоксифенил)-5-метил-2-[N-(6-метилизохинол-5-ил)-N- пропиламино]тиазола (пример 5),
4-(2-хлор-4-метоксифенил)-5-метил-2-[N- (1-метоксикарбонилметилиндол-5-ил)-N-пропиламино]тиазол (пример 9),
оксалат
4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6- метоксиизохинол-5-ил)-N-пропиламино]тиазола (пример 27),
оксалат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6-хлоризохинол-5-ил)-N- пропиламино]тиазола
(пример 29),
оксалат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6-метоксиизохинол-5-ил)-N- пропиламино]тиазола (пример 31),
4-(2-хлор-4-метоксифенил)-5-метил-2-[N- (6-метоксинафт-2-ил)-N-пропиламино]тиазол (пример 34),
оксалат
4-(2-хлор-4-трифторметилфенил)-5-метил-2-[N-(6- метоксиизохинол-5-ил)-N-пропиламино]тиазола (пример 37),
хлоргидрат
4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(2- этоксинафт-1-ил)-N-пропиламино]тиазола (пример 40),
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(2,3-диметилнафт-1-ил) -N-пропиламино]тиазола
(пример 44),
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6-бром-2-метоксинафт- 1-ил)-N-пропиламино]тиазола (пример 45),
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(2,
6- диметилнафт-1-ил)-N-пропиламино]тиазола (пример 46),
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(1- (метоксиметил)-1-(нафт-2-ил)-N-пропиламино]тиазола (пример 53),
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(1- (циклопропил)-1-(нафт-2-ил)метил)-N-пропиламино]тиазола (пример 61);
один из их стереоизомеров и/или одна из их солей.
Соединения согласно изобретению в свободной форме обычно обладают основными свойствами. Однако в зависимости от природы некоторых заместителей они могут обладать кислотными свойствами.
Соли соединений формулы (I) с фармацевтически приемлемыми кислотами или основаниями (когда это возможно) являются предпочтительными солями, но также те соли, которые могут обеспечить выделение соединений формулы (I), в частности их очистку или получение чистых изомеров, также входят в объем изобретения.
Из фармацевтически приемлемых кислот для получения аддитивных солей соединений формулы (I) можно назвать соляную, фосфорную, фумаровую, лимонную, щавелевую, серную, аскорбиновую, винную, малеиновую, миндальную, метансульфоновую, лактобионовую, глюконовую, глюкаровую, сукцинилсульфоновую, гидроксипропансульфоновую и т.д. кислоты.
Из фармацевтически приемлемых оснований для получения аддитивных солей соединений формулы (I), когда они обладают кислотными свойствами, можно назвать гидроксид натрия, калия, аммония и т.д.
Соединения согласно изобретению, а также промежуточные продукты получают хорошо известными специалисту способами, например согласно европейскому патенту 576350.
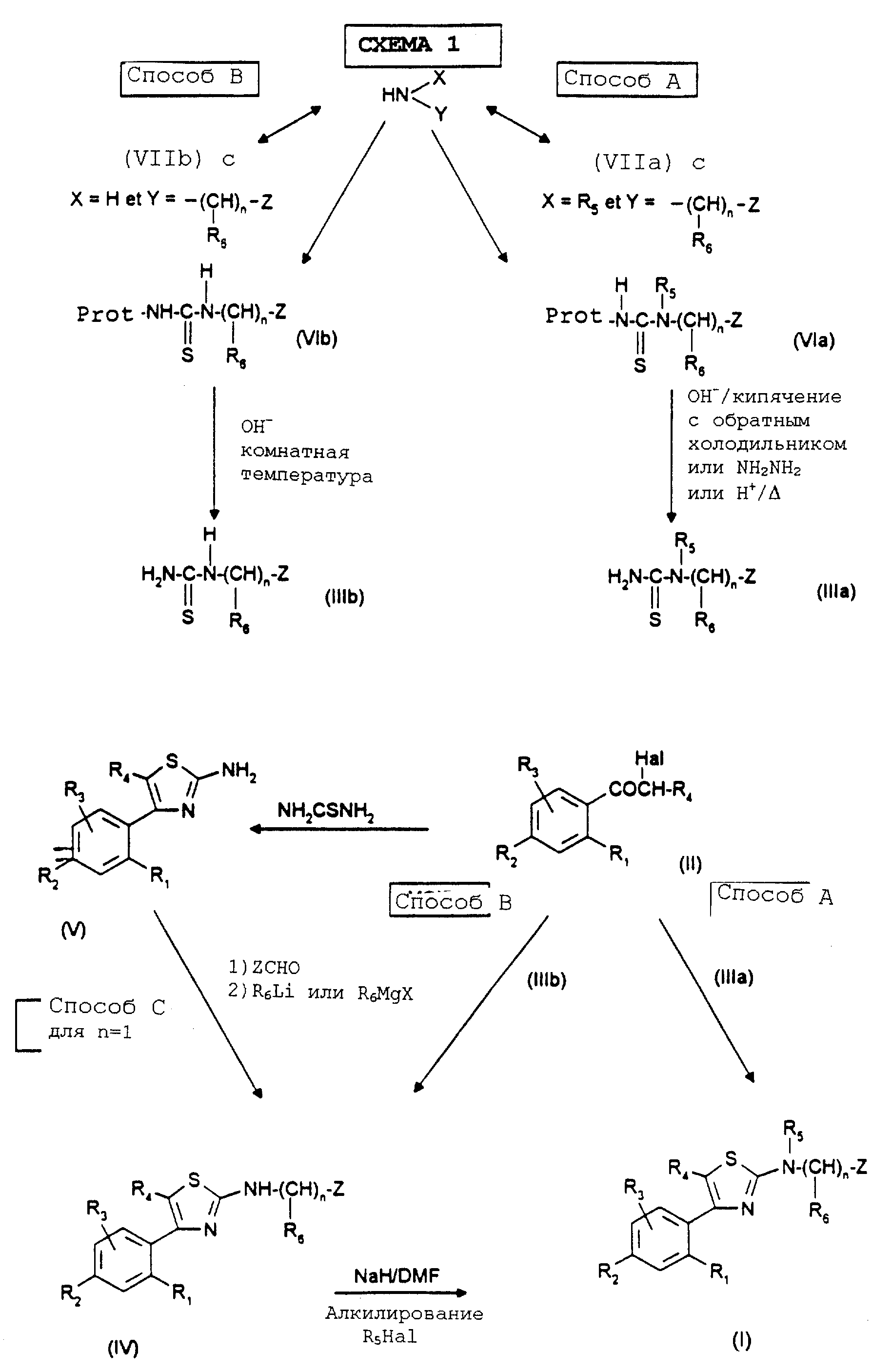
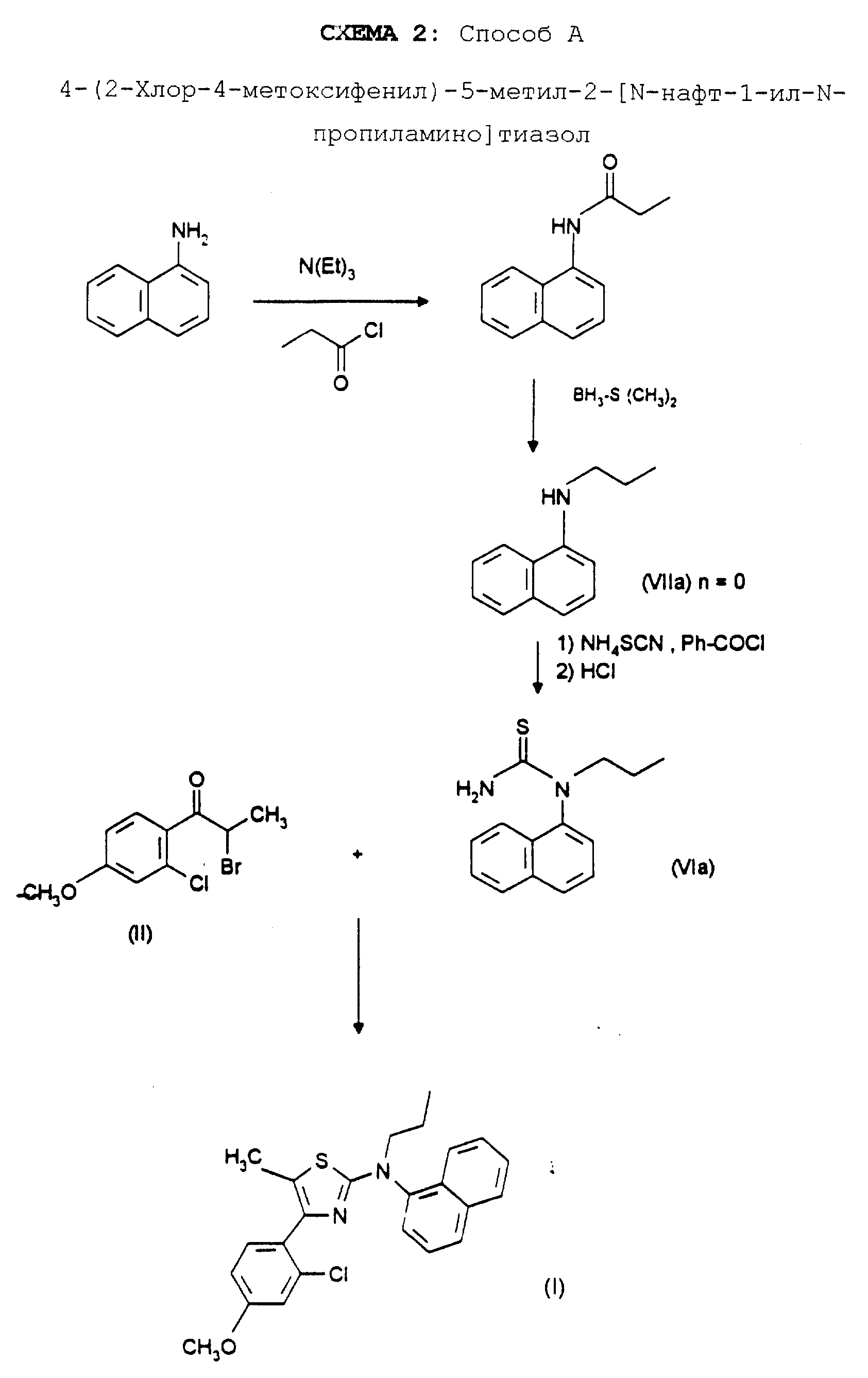
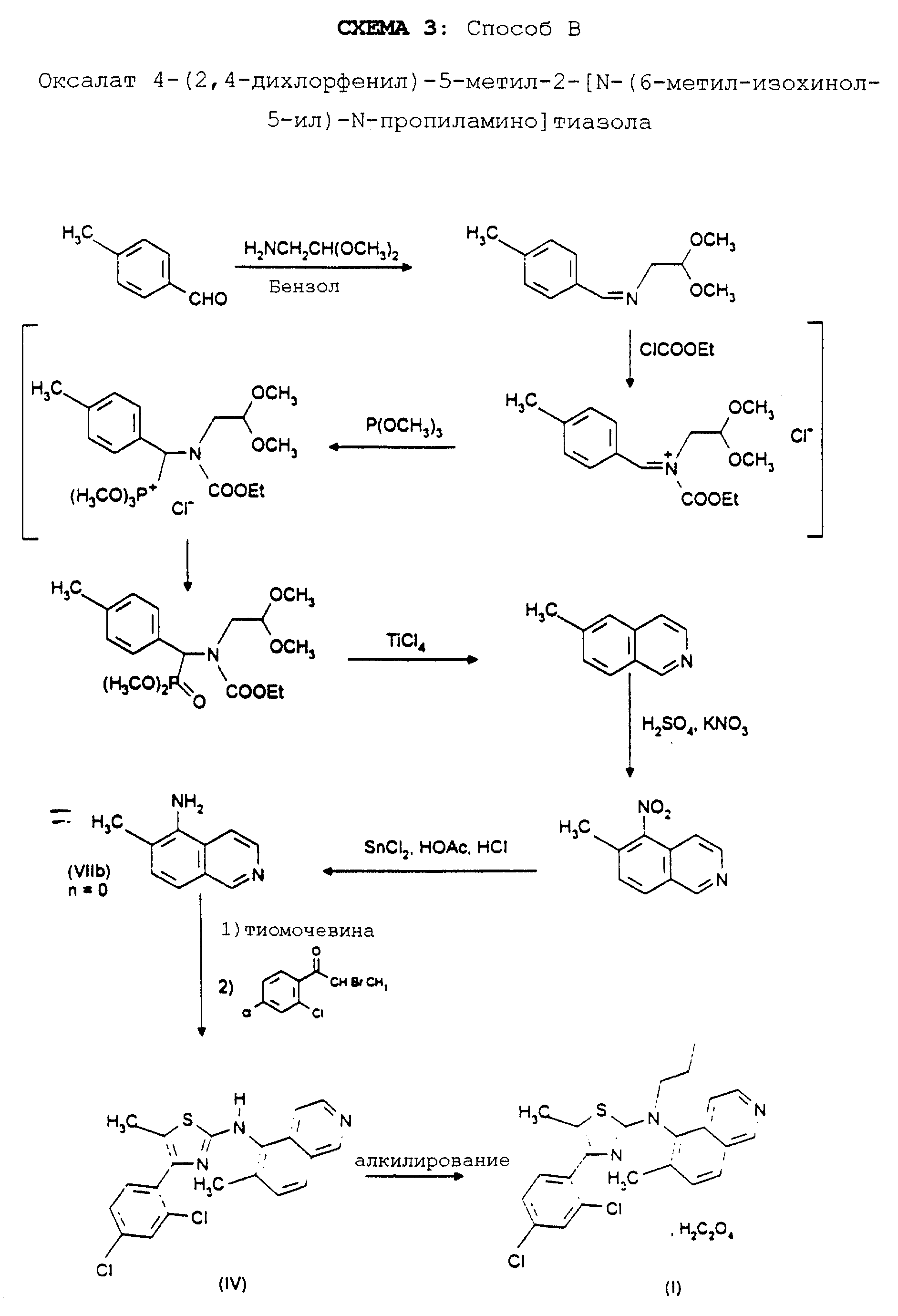
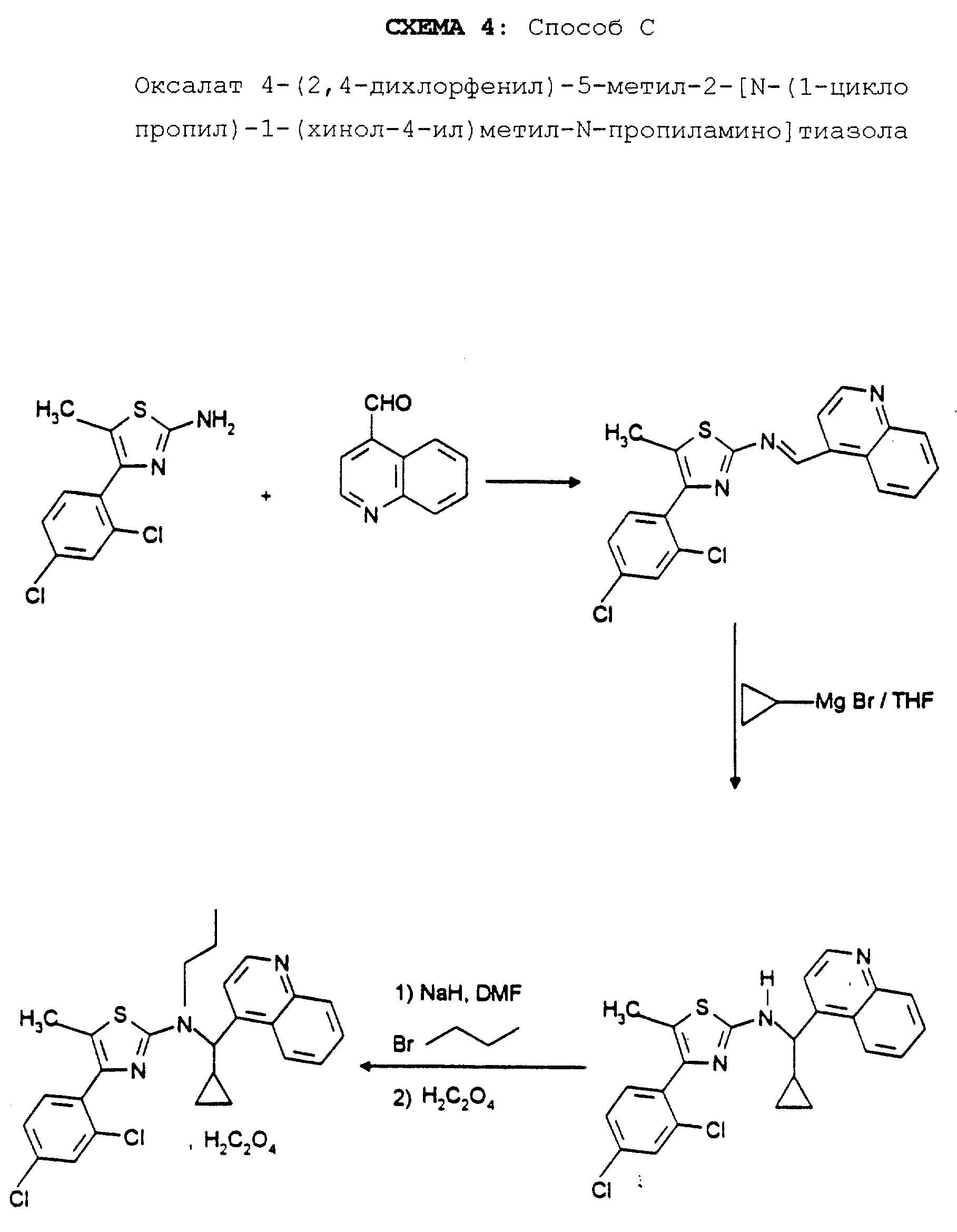
Реакционная схема 1 иллюстрирует способ получения, используемый для синтеза соединений формулы (I); приводимые в качестве примеров реакционные схемы 2, 3 и 4 иллюстрируют синтез отдельных соединений формулы (I) способами A, B и C.
Схемы 1-4 приведены в конце описания.
Синтез промежуточных соединений подробно описывается в разделе ПРИМЕРЫ ПРИГОТОВЛЕНИЯ; синтез соединений формулы (I) согласно изобретению описывается в разделе ПРИМЕРЫ, а также в ТАБЛИЦАХ I-IY,приведенных в конце описания.
Объектом настоящего изобретения также является способ получения соединений формулы (I), отличающийся тем, что альфа-галогенированное, предпочтительно альфа-бромированное или
альфа-хлорированное производное формулы (II):
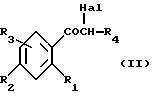
в которой R1, R2, R3, Hal и R4 имеют указанное для формулы (I) значение,
вводят во взаимодействие либо с тиомочевиной (Способ B) формулы
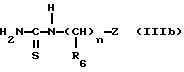
в которой R6 и Z имеют указанное для формулы (I) значение,
для получения соединения формулы (IV):
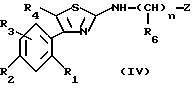
в которой R1, R2, R3, R4, "n", R6 и Z имеют указанное для формулы (I) значение, которое затем подвергают реакции алкилирования с получением соединения формулы (I), в частности с получением, когда Z означает азотсодержащий гетероцикл, такой как индол или индазол, либо моноалкилированных соединений, замещая предварительно реакционноспособный азот цикла защитной группой, предпочтительно типа тетрагидропиранила, либо диалкилированных соединений, осуществляя после удаления защитной группы из цикла полученного моноалкилированного соединения, алкилирование высвобожденного реакционноспособного азота, причем эти диалкилированные соединения в зависимости от природы второй алкильной группы могут приводить к диалкилированным продуктам, содержащим различные или идентичные алкильные группы, и причем в этом последнем случае эти соединения могут быть также получены прямо путем диалкилирования из соединения формулы (IV), в котором реакционноспособный азот гетероцикла не защищен;
либо с тиомочевиной (Способ A) формулы:
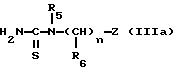
в которой R5, "n", R6 и Z имеют указанные для формулы (I) значения,
для получения непосредственно соединения формулы (I);
либо с тиомочевиной (Способ C) для получения аминотиазола формулы (V):
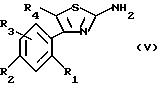
в которой R1, R2, R3 и R4 имеют указанное для формулы (I) значение, который затем при необходимости вводят во взаимодействие с альдегидом формулы HCO-Z для получения имина, который в свою очередь под действием магнийорганического соединения формулы R6MgX (где X означает галоген) или литийорганического соединения формулы R6Li приводит к получению соединения формулы (IV), которое подвергают алкилированию, например, путем обработки соединением формулы R5X (где X означает удаляемую группу, такую как галоген) с целью получения соединения формулы (I), и при желании таким образом полученные соединения формулы (I) затем разделяют на их возможные стереоизомеры и/или подвергают солеобразованию для получения соответствующих солей.
Осуществляемые в вышеуказанном способе реакции алкилирования проводят в обычных условиях, известных специалисту, путем воздействия соответствующего алкилирующего агента, такого как, например, алкилгалогенид.
Производные формулы (II) могут быть получены из соответствующих негалогенированных
кетонов формулы:
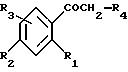
либо путем воздействия брома в соответствующем органическом растворителе, таком как уксусная кислота, тетрахлорид углерода или диэтиловый эфир, либо путем воздействия четвертичных трибромидов аммония согласно способу, описанному в Bull. Chem.Soc. Japan, 60, 1159-1160 и 2667-2668 (1987), либо путем воздействия бромида двухвалентной меди в органическом растворителе, таком как смесь хлороформа с этилацетатом, согласно J.Org.Chem., 29, 3451-3461 (1964).
В качестве варианта соединения формулы (II) могут быть получены путем воздействия 2-бромпропионилбромида на замещенный бензол формулы:
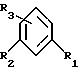
по реакции Фриделя-Крафтса.
Кетоны обычно являются известными или имеющимися в продаже продуктами. Эти соединения могут быть получены путем реакции Фриделя-Крафтса в присутствии кислоты Льюиса, согласно хорошо известным специалисту способам.
Производные тиомочевины формулы (IIIa) и (IIIb) получают из
соединений формулы (VIa) и формулы (VIb):
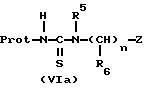
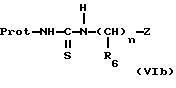
в которых Prot означает защитную группу, например бензоил, пивалоил или тетрагидропиранил, R5, R6, "n" и Z имеют указанные выше для формулы (I) значения,
либо путем обработки основанием, используя предпочтительно гидроксид аммония, гидроксид натрия или гидразин, при температуре от комнатной до температуры кипения с обратным холодильником реакционной смеси,
либо путем обработки кислотой, используя предпочтительно соляную кислоту.
Соединения формул (VIa) и (VIb)
получают путем введения во взаимодействие согласно известным способам изотиоцианата, например бензоилизотиоцианата или пивалоилизотиоцианата, с соответствующими аминами HNXY формулы (VIIa) и формулы
(VIIb):
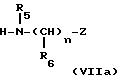
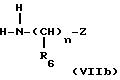
в которых Y означает
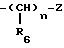
где "n", R6 и Z имеют указанное для формулы (I) значение, а X означает водород или R5, имеющий указанное для формулы (I) значение.
Когда Z означает азотсодержащую гетероциклическую группу типа индола или индазола, используют Способ B, в котором блокируют реакционноспособный циклический азот, замещая его защитной группой типа тетрагидропиранила. После алкилирования экзо-азота 2-аминотиазольного производного можно осуществлять удаление защитной группы с защищенного азота гетероцикла путем обработки кислотой, предпочтительно соляной кислотой. Полученное соединение можно затем подвергнуть реакции нуклеофильного замещения при использовании галогенированных производных, таких как алкилбромиды или алкилиодиды, для получения соединения формулы (I). Hекоторые производные можно затем подвергнуть классическим реакциям, как например гидролиз сложноэфирной или нитрильной функции для получения кислот, реакция магнийорганического соединения с нитрильной группой для получения соответствующих кетонов. Активация кислотной функции либо в форме хлорангидрида кислоты, либо в форме активированного сложного эфира путем воздействия азотистого основания, такого как морфолин, позволяет получать соответствующие амиды.
Вторичные амины формулы (VIIa) получают из первичных аминов
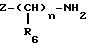
либо путем введения во взаимодействие с альдегидом
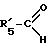
где R'5-CH2 - означает R5,
затем восстановления имина, например, с помощью NaBH4, предпочтительно в этаноле или метаноле при комнатной температуре;
либо путем введения во взаимодействие с галоидангидридом или ангидридом кислоты в органическом растворителе, выбираемом среди галогенированных углеводородов, таких как дихлорметан, в присутствии акцептора протонов, предпочтительно триэтиламина. Получаемый путем этой реакции амид затем восстанавливают с помощью гидрида, такого как литийалюминийгидрид, в органических растворителях типа диэтилового эфира.
Оба вышеописанных способа используют предпочтительно для получения соединений формулы (VIIa) в виде чистых энантиомеров из оптически чистых первичных аминов.
Другой способ
получения соединений формулы (VIIa) состоит в конденсации кетона
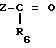
в котором Z и R6 имеют указанное для формулы (I) значение,
с амином R5NH2, где R5 имеет указанное для формулы (I) значение, в обезвоживающей среде для получения соответствующего имина, который затем восстанавливают классическим способом с помощью гидрида металла, предпочтительно боргидрида натрия, или с помощью водорода в присутствии соответствующего катализатора. Во время реакции первичного амина с кетоном в обезвоживающей среде предпочтительно используют либо тетра-хлорид титана (TiCl4), либо катализ с помощью п-толуолсульфокислоты.
Вышеуказанные соединения формулы (I) также включают соединения, в которых один или несколько атомов водорода или углерода, например те, которые находятся в R4, в частности, когда этот радикал означает метил, заменены на их радиоактивный изотоп, например тритий или углерод-14. Такие меченые соединения пригодны для исследований в области метаболизма или фармакокинетики или же в биохимических опытах в качестве лигандов рецептора.
Соединения настоящего изобретения были объектом биохимических и фармакологических исследований. Они обладают очень ценными фармакологическими свойствами. Соединения согласно изобретению, в частности, в концентрациях ниже 10 мкмоль (0,01-10 мкмоль), смещают связь125I-CRF с его специфическими рецепторами, присутствующими в мембранах головного мозга человека (или трансфецированных клеток яичника китайского хомячка, экспрессирующих клонированный рецептор головного мозга человека) и/или головного мозга животных (крыса, мышь), по методу, описанному De Souza Е.В. (J.Neurosci., 7 (1), 88-100 (1987)).
Этот факт является удивительным и неожиданным, так как соединения со структурой, близкой к таковой соединений согласно изобретению, не смещают заметным образом связь125I-CRF.
CRF представляет собой нейропептид, который контролирует активность гипоталамо-гипофизо-надпочечниковой оси. Этот фактор является ответственным за ответные реакции эндокринной системы и в отношении поведения, связанные со стрессом.
Так, было показано, что CRF может модулировать поведение так же, как некоторые функции автономной нервной системы (G.F. Koob, F.E. Bloom, Fed. Proc. , 44, 259 (1985); M.R. Brown, L.A.Fisher, Fed.Proc., 44, 243 (1985)). Более конкретно CRF индуктирует выделение кортикотропина (ACTH), β- эндорфинов и других пептидов, происходящих от проопиомеланокортина (A. Tazi и др., Regul. Peptides, 18, 37 (1987); M.R. Brown и др., Regul.Peptides, 16, 321 (1986); C.L. Williams и др., Am.J.Physiol., G 582, 253 (1987)).
Соединения согласно изобретению, следовательно, могут быть пригодны для регуляции секреции этих эндогенных веществ. Они находят свое применение, в частности, в качестве действующих начал лекарственных средств для уменьшения ответной реакции на стресс (поведение, эмоциональные состояния, желудочно-кишечные расстройства и сердечно-сосудистые нарушения, расстройства иммунной системы) и в более широком случае при патологиях, в которых принимает участие CRF, например психические расстройства, беспокойство, депрессия, нервная анорексия, нарушения половой активности и фертильности, болезнь Альцгеймера, или другие.
Полученные во время различных фармакокинетических исследований, осуществляемых при использовании соединений согласно изобретению, результаты показывают, что все эти соединения очень хорошо абсорбируются.
Эти исследования также показывают, что фармацевтические композиции, приготовленные при использовании продуктов формулы (I), могут абсорбироваться пищеварительным трактом, причем не требуется вводить слишком большие количества для использования в терапии человека. Соединения согласно изобретению, следовательно, пригодны для получения фармацевтических композиций, вводимых одинаково хорошо как парентерально, так и перорально.
Соединения согласно изобретению являются очень стабильными и, следовательно, особенно пригодны в качестве действующего начала лекарственных средств.
Изобретение также относится к фармацевтическим композициям, содержащим в качестве действующего начала эффективное количество соединения формулы (I) или одной из его фармацевтически приемлемых солей, возможно в сочетании с одним или несколькими соответствующими инертными эксципиентами.
В каждой разовой дозе действующее начало формулы (I) находится в количествах, соответствующих предусматриваемым суточным дозам. Обычно каждую разовую дозу подбирают в зависимости от дозировки и предусматриваемого пути введения, как например таблетки, желатиновые капсулы и тому подобное, пакетики с лекарством, ампулы, сиропы и аналогичные формы, капли, пластырь для введения через кожу или через слизистую оболочку таким образом, чтобы такая разовая доза содержала 0,5-200 мг действующего начала, предпочтительно 0,5-800 мг для введения ежедневно.
Соединения согласно изобретению также можно использовать в сочетании с другим действующим началом, необходимым для проведения желаемой терапии, таким как, например, анксиолитические средства, антидепрессанты или лекарственные средства, снижающие аппетит.
Соединения формулы (I) являются малотоксичными; их токсичность совместима с их использованием в качестве лекарственного средства для лечения расстройств и заболеваний, указанных выше.
Соединения формулы (I) можно вводить в фармацевтические композиции, которые вводят млекопитающим, в том числе и человеку, для лечения вышеуказанных заболеваний.
Таким образом, полученные фармацевтические композиции предпочтительно находятся в различных формах, таких как, например, растворы для инъекции или питья, драже, таблетки или желатиновые капсулы. Фармацевтические композиции, содержащие в качестве действующего начала, по крайней мере, одно соединение формулы (I) или одну из его солей, особенно пригодны для профилактики или лечения заболеваний, связанных со стрессом, и более конкретно для лечения любых патологий, в которых принимает участие CRF, таких как, например, нейропсихиатрические расстройства как беспокойство, паника, фобии, ухудшения настроения, нарушения поведения, анорексия, булимия, гипергликемия, запоздалое развитие, нарушения сна и депрессии всех типов; болезнь Альцгеймера, Паркинсона; хорея Гентингтона; боковой амиотрофический склероз; сердечно-сосудистые нарушения; расстройства половой активности и фертильности; иммунодепрессия, иммуносупрессия и связанные с ними заболевания, такие как воспалительные процессы, множественные инфекции, раковые заболевания, ревматоидный артрит, остеоартрит, псориаз, так же как диабет; желудочно-кишечные расстройства и проистекающие от этого воспаления (раздражимость ободочной кишки; диареи); нарушения болевого восприятия, фибромиалгии, связанные или нет с нарушениями сна, усталость, мигрень; симптомы, связанные с лекарственной зависимостью или лишением приема лекарств.
Дозировку можно изменять в широких пределах в зависимости от возраста, массы и состояния здоровья пациента, природы и тяжести заболевания, а также от пути введения. Эта дозировка включает введение одной или нескольких доз около 0,5 - 200 мг в день, предпочтительно около 0,5-800 мг в день.
В фармацевтических композициях согласно настоящему изобретению для введения перорально, подъязычно, подкожно, внутримышечно, внутривенно, через кожу, через слизистую оболочку, локально или ректально действующее начало может вводиться в виде лекарственных форм в смеси с классическими фармацевтическими носителями, животным и в том числе людям. Соответствующие лекарственные формы включают пероральные формы, такие как таблетки, желатиновыне капсулы с лекарством, порошки, гранулы и пероральные растворы или суспензии; подъязычные формы введения и формы введения через рот, формы введения покожно, внутримышечно, внутривенно, через нос или внутриглазные формы введения и ректальные формы введения.
Когда получают твердую композицию в виде таблеток, основное действующее начало смешивают с фармацевтическим эксципиентом, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные продукты. Таблетки можно покрывать сахарозой или другими соответствующими веществами, или их можно обрабатывать таким образом, что они приобретают пролонгированную или замедленную активность и непрерывно высвобождают заданное количество действующего начала.
Препарат в виде желатиновых капсул с лекарством получают путем смешения действующего начала с разбавителем и внесения полученной смеси в мягкие или твердые желатиновые капсулы.
Препарат в виде сиропа или эликсира может содержать действующее начало вместе с подслащивающим, предпочтительно некалорийным средством, метилпарабеном или пропилпарабеном в качестве антисептика, а также с придающим вкус агентом и соответствующим красителем.
Диспергируемые в воде порошки или гранулы могут содержать действующее начало в смеси с диспергаторами или смачивателями или суспендирующими агентами, такими как поливинилпирролидон, а также с подслащивающими средствами или улучшающими вкус веществами.
Для ректального введения используют суппозитории, которые получают с помощью связующих, плавящихся при ректальной температуре, например масло какао или полиэтиленгликоли.
Для парентерального, внутриносового или внутриглазного введения используют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекции, которые содержат фармакологически приемлемые диспергаторы и/или смачиватели, например пропиленгликоль или бутиленгликоль.
Для введения через слизистую оболочку действующее начало может быть переведено в лекарственную форму в присутствии промотора, такого как соль желчной кислоты, гидрофильного полимера, такого как, например, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза, этилцеллюлоза, карбоксиметилцеллюлоза, декстран, поливинилпирролидон, пектины, крахмалы, желатина, казеин, акриловые кислоты, эфиры акриловых кислот и их сополимеры, виниловые полимеры или сополимеры, виниловые спирты, алкоксиполимеры, полиэтиленоксиды, простые полиэфиры или их смеси.
Действующее начало также может выпускаться в форме микрокапсул, возможно с одним или несколькими носителями или добавками.
Действующее начало также может находиться в виде комплекса с циклодекстрином, например, в виде α-,β- или γ- циклодекстрина, 2-гидроксипропил -β- циклодекстрина или метил- β- циклодекстрина.
Нижеприводимые ПРИМЕРЫ, не ограничивающие объема охраны изобретения, иллюстрируют изобретение.
В различных ПРИМЕРАХ ПРИГОТОВЛЕНИЯ описываются способы синтеза различных промежуточных соединений, позволяющих получать соединения согласно изобретению. Эти промежуточные соединения получают хорошо известными специалисту способами.
Температуры плавления определяют по микроспособу Кофлера и выражают в градусах Цельсия.
Спектры протонного ядерного магнитного резонанса (1H-ЯМР) соединений формулы (I) регистрируют, в зависимости от случая при 200 мГц или 100 мГц. Химические сдвиги даются в м. д. (миллионные доли), а константы связывания приводятся в герцах. Для соединений согласно изобретению указывается выход в процентах в расчете на теоретически рассчитанный.
Соединения согласно изобретению, описанные в ТАБЛИЦАХ I-IV, также характеризуются ЯМР-спектрами в соответствии с их структурой.
ПРИМЕРЫ ПРИГОТОВЛЕНИЯ
Получение кетонов формулы
(II)
ПРИГОТОВЛЕНИЕ I 2-Бром-1-(2,4-дихлорфенил)пропан-1-он (соединение 1)
К раствору 7 г 1-(2,4-дихлорфенил)пропан-1-она в смеси из 420 см дихлорметана и 140 мл метанола при
комнатной температуре добавляют 17,4 г тетрабутиламмонийтрибромида. Спустя 24 часа реакционную смесь концентрируют в вакууме. Остаток обрабатывают водой, экстрагируют этилацетатом, органическую фазу
сушат над сульфатом натрия и выпаривают в вакууме, затем очищают на колонке с силикагелем, используя в качестве элюирующего средства смесь циклогексана с этилацетатом в соотношении 20:1 по объему, с
получением масла.
Таким образом могут быть также получены при использовании соответствующих кетонов следующие соединения:
2-бром-1-(4-хлор-2-метоксифенил)пропан-1-он
(соединение 2);
2-бром-1-(4-бром-2-метоксифенил)пропан-1-он (соединение 3);
2-бром-1-(2-бром-4-метоксифенил)пропан-1-он (соединение 4);
2-бром-1-(2-хлор-4-метоксифенил)пропан-1-он (соединение 5);
2-бром-1-(2-хлор-4-метилфенил)пропан-1-он (соединение 6);
2-бром-1-(4-хлор-2-метилфенил)пропан-1-он (соединение 7).
ПРИГОТОВЛЕНИЕ II. 2-Бром-1-(2-хлор-4-трифторметил-фенил)- пропан-1-он (соединение 8)
Стадия 1:
Суспензию 10 г 2-хлор-4-трифторметиланилина в 18 г 95%-ной серной кислоты и
65 мл воды медленно при температуре 15oC добавляют к раствору 3,57 г нитрата натрия в 7 мл воды. Реакционную смесь перемешивают при температуре 40-45oC в течение двух часов,
затем осторожно выливают в доведенную до температуры 95oC следующую смесь: 10,77 г цианида натрия, 0,51 г цианида меди, 25,8 г гидрокарбоната натрия и 0,46 г гидратированного сульфата
никеля в 30 мл воды. Реакционную смесь перемешивают при температуре 100oC в течение 1 часа, затем после охлаждения добавляют 30 мл водного насыщенного раствора гидрокарбоната натрия и
экстрагируют дихлорметаном. Экстракт фильтруют через целит, затем промывают последовательно водой, соленой водой, сушат над сульфатом натрия и выпаривают досуха. Остаток очищают на колонке с
силикагелем, элюируя смесью циклогексана с этилацетатом в объемном соотношении 20:1, и получают 3,55 г 2-хлор-4-трифторметилбензонитрила в виде каштанового цвета масла.
1 H-ЯМР (дейтерохлороформ): 7.4-7.8 (м, 3H).
Стадия 2:
Раствор 3,6 г полученного выше продукта в 50 мл бензола перемешивают при температуре 20oC и добавляют 11,7 мл 3
М раствора этилмагнийбромида в диэтиловом эфире. Реакционную смесь перемешивают при кипячении с обратным холодильником в течение двух часов, затем охлаждают до 0oC и медленно добавляют 17,5
мл 6 н. соляной кислоты. После перемешивания при кипячении с обратным холодильником в течение трех часов, затем охлаждения, реакционную смесь экстрагируют диэтиловым эфиром. Экстракт промывают
насыщенным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают досуха. Остаток после выпаривания очищают путем хроматографии на колонке с силикагелем, элюируя смесью циклогексана с
этилацетатом в объемном соотношении 20:1, и получают 83,2 т 1-(2-хлор-4-трифторметилфенил) пропан-1-она.
1H-ЯМР (дейтерохлороформ): 1.2 (м, 3H), 2.9 (м, 2H), 7.45-7.62-(м, 3H).
Стадия 3:
К раствору 3,5 г полученного выше продукта в 150 мл дихлорметана добавляют 7,65 г тетрабутиламмонийтрибромида. Реакционную смесь перемешивают при температуре
35oC в течение 4,5 часов, затем после охлаждения промывают 3 раза водой до нейтральной реакции. Органическую фазу выпаривают и остаток обрабатывают диэтиловым эфиром. Эфирную фазу промывают
последовательно водой, насыщенным раствором хлорида натрия, затем сушат над сульфатом натрия и выпаривают досуха, получая 4,6 г 2-бром-1-(2-хлор-4-трифторметилфенил)пропан-1-она.
1H-ЯМР (дейтерохлороформ): 1.9 (д, 3H), 5.2 (к, 1H), 7.5-7.7 (м, 3H).
Получение аминов
ПРИГОТОВЛЕНИЕ III. N-нафт-1-ил-N-пропиламин (соединение 9)
Стадия 1:
4,0 г 1-нафт-ил-амина растворяют в 40 мл тетрагидрофурана, затем прикапывают 2,6 г пропионилхлорида, реакционную смесь перемешивают в течение двух часов, затем выпаривают досуха. Полученный
остаток обрабатывают дихлорметаном и полученный раствор промывают водным раствором хлорида натрия. Сушат над сульфатом натрия и выпаривают досуха, получая 5,5 г кристаллов белого цвета
N-пропионил-1-нафтил-амина с т.пл. 127oC.
1H-ЯМР (дейтерохлороформ): 1.28 (т, J=7.3, 3H, -CH2-CH3), 2.51 (к, J= 7.3, 2H, -CH2 -CH3), 7.30-7.50 (м, 3H, H2, H3 и H8), 7.55-7.85 (м, 4H, H6, H4 и H5).
Стадия 2:
В трехгорлой
колбе, снабженной капельной воронкой и с поддерживаемой атмосферой аргона, 5,5 г полученного выше амида растворяют в 50 мл безводного тетрагидрофурана, затем нагревают до 50oC и прикапывают
42 мл 2 М диметилсульфида борана и реакционную смесь кипятят с обратным холодильником в течение трех часов и выдерживают в течение ночи при комнатной температуре. Смесь охлаждают на бане со льдом;
после этого добавляют 100 мл 6 н. соляной кислоты, затем кипятят с обратным холодильником в течение трех часов. Тетрагидрофуран выпаривают, затем остаток последовательно подщелачивают и экстрагируют
этилацетатом, экстракт сушат над сульфатом натрия и выпаривают досуха, получая 3,9 г бесцветного масла.
1H-ЯМР (дейтерохлороформ): 1.10 (т, J=7.3, 3H, -CH2-CH3), 2.51 (секстет, J= 7.3, 2H, -CH2-CH3), 3.26 (т, J=7.3, 2H, -CH2-CH2- CH3), 4.33 (м, 1H, NH), 6.61 (д, J= 7.3, 1H, H2), 7.23 (д, J=8.4, 1H, H3), 7.34 (д, J=1H, H8), 7.40-7.50 (м, 2H, H6 и H7), 7.75-7.90 (м, 2H, H4 и H5).
ПРИГОТОВЛЕНИЕ IV.
N-Пропил-N-хинол-5-ил-амин (соединение 10)
В колбе емкостью 250 мл 5 г 5-аминохинолина, 3 мл пропионового альдегида и 4,7 г п-тиокрезола растворяют в 100 мл этанола. Реакционную смесь кипятят
с обратным холодильником в течение двух часов, затем выпаривают досуха. Остаток растворяют в 100 мл этанола, охлаждают на бане со льдом, после чего маленькими порциями добавляют 6,5 г боргидрида
натрия. По окончании добавления реакционную смесь кипятят с обратным холодильником в течение двух часов, затем последовательно добавляют 30 мл воды, подщелачивают с помощью 20 мл концентрированного
раствора гидроксида натрия, перемешивают в течение 15 минут и органические растворители выпаривают. Экстрагируют дихлорметаном, экстракт промывают водой, затем сушат над сульфатом натрия. Выпаривают
досуха и остаток очищают на колонке с силикагелем, элюируя этилацетатом, с получением 4 г масла, которое кристаллизуется.
1H-ЯМР (дейтерохлороформ): 1.05 (т, J=3H, -CH3), 1.82-1.71 (м, 2H, -CH2-CH3), 3.15-3.26 (м, 2H, -NH-CH2-), 4.37 (уш. с, 1H, -NH-) 6.61 (дд, J= 1,0 и J= 8.6, 1H, H4), 7.26 (дд, J=4.2 и J=8.6, 1H, H6), 7.44-7.59 (м, 2H, H3, H2), 8.12 (дд, J=0.95 и J=8.6, 1H, H8), 8.84 (дд, J=1.5 и J=4.2, H2).
Следуя той же самой методике, получают N-пропил-N-хинол-6-ил-амин (соединение 11).
ПРИГОТОВЛЕНИЕ V. H-(1-Hафт-1-ил-2-метоксиэтил)-N-пропиламин (соединение 12)
Стадия 1:
Получают
нафталин-1-магнийбромид из 25 г 1-бромнафталина и 3,5 г магния в 50 мл диэтилового эфира, затем раствор охлаждают на льду и прикапывают 9 мл метоксиацетонитрила в виде раствора в 20 мл диэтилового
эфира. Реакционную смесь затем перемешивают в течение двух часов при комнатной температуре, затем охлаждают до 0oC. После этого добавляют 100 мл насыщенного раствора хлорида аммония и
экстрагируют диэтиловым эфиром. Органическую фазу последовательно промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают досуха, получая 29 г маслянистого остатка
метоксиметилнафт-1-ил-кетона.
Стадия 2:
Полученный выше кетон растворяют в 350 мл дихлорметана, затем добавляют 50 мл пропиламина и после этого при температуре 5oC
прикапывают 120 мл 1 М раствора тетрахлорида титана в дихлорметане. Реакционную смесь перемешивают при комнатной температуре в течение 20 часов, затем добавляют 200 мл метанола. Раствор охлаждают на
бане со льдом, затем маленькими порциями добавляют 4,6 г NaBH4 и оставляют стоять для постепенного повышения температуры до комнатной. После перемешивания в течение трех часов реакционную
смесь фильтруют через целит и фильтрат выпаривают досуха. Остаток обрабатывают дихлорметаном, полученный раствор промывают 1н. соляной кислотой, объединенные водные фазы подщелачивают и продукт
экстрагируют 4 раза по 200 мл дихлорметаном, получая затем 5,1 г целевого амина.
1H-ЯМР (диметилсульфоксид): 0.77 (м, 3H), 1.27-1.44 (м, 2H), 2.00-2.47 (м, 2H), 3.25 (с, 3H), 3.28-3.49 (м, 2H), 4.64-4.70 (м, 1H), 7.19- 8.29 (м, 8H).
Следуя вышеуказанной методике ПРИГОТОВЛЕНИЯ V, получают N-(1-нафт-2-ил-2-метоксиэтил)-N-пропиламин (соединение 13).
ПРИГОТОВЛЕНИЕ VI. 5-Амино-6-метоксихинолин (соединение 14)
Стадия 1:
4,0 г 6-Метоксихинолина растворяют в 70 мл уксусной кислоты, затем охлаждают до 0oC и
добавляют 5,5 г нитрата калия. Реакционную смесь перемешивают при температуре 0oC в течение 1 часа, после чего подщелачивают с помощью 10 н. раствора гидроксида натрия. Полученный осадок
желтого цвета отфильтровывают и промывают его обильно водой, получая 4,9 г 6-метокси-5-нитрохинолина в виде порошка желтого цвета.
1H-ЯМР (дейтерохлороформ): 4.07 (с, 3H, -OCH3), 7.48-7.54 (м, 1H, H3), 7.58 (д, J=9.5, 1H, H7), 8.04 (д, J=8.8, 1H, H4), 8.25 (д, J=9.5, 1H, H8), 8.86 (дд, J=1.5, J=4.2, 1H, H2).
Стадия 2:
4,9 г 6-Метокси-5-нитрохинолина растворяют в 100 мл уксусной кислоты и 60 мл 37%-ной соляной кислоты, затем добавляют 51 г SnCl2 и реакционную
смесь кипятят с обратным холодильником в течение трех часов, затем оставляют стоять при комнатной температуре в течение 12 часов. Потом выпаривают досуха, остаток обрабатывают водой и подщелачивают
насыщенным раствором гидрокарбоната натрия. Экстрагируют этилацетатом, экстракт сушат над сульфатом натрия, затем выпаривают досуха, получая 2,9 г 6- метокси-5-аминохинолина в виде порошка желтого
цвета.
1H-ЯМР (дейтерохлороформ): 4.00 (с, 3H, -OCH3), 4.28 (м, 2H, -NH2), 7.28-7.34 (м, 1H, H3), 7.44 (д, J=9.1, 1H, H7), 7.60 (д, J=9.1, 1H, H4), 8.14 (дд, J=0.7 и J=9.5, 1H, H8), 8.78 (дд, J=1.8 и J-4.2, 1H, H2).
Следуя вышеуказанной в ПРИГОТОВЛЕНИИ VI методике, получают 5-амино-6-хлор-2-метилхинолин (соединение 15).
ПРИГОТОВЛЕНИЕ VII. 2-Амино-1-метоксинафталин (соединение 16)
Стадия 1:
10 г 1-Метоксинафталина растворяют в 100 мл
уксусного ангидрида, затем прикапывают 2,6 мл концентрированной азотной кислоты в виде раствора в 15 мл уксусного ангидрида. Реакционную смесь перемешивают в течение 30 минут при комнатной температуре,
затем подщелачивают с помощью насыщенного раствора гидрокарбоната натрия. Выпавший осадок коричневого цвета отфильтровывают, обрабатывают его этилацетатом, затем последовательно обильно промывают
насыщенным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают досуха. Полученный остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью этилацетата с гексаном в
объемном соотношении 5:95 и получая первый изомер; затем элюируя смесью этилацетата с гексаном в объемном соотношении 10: 90 и получая второй изомер. Таким образом получают 2.45 г целевого продукта
(выход=19%)
1H-ЯМР (дейтерохлороформ): 4.15 (с, 3H, -OCH3), 7.64-7.71 (м, 5H, H3, H4, H6, H7, H8), 8.30-8.34 (м, 1,
H5).
Стадия 2:
2,45 г вышеполученного продукта растворяют в 50 мл уксусной кислоты и 25 мл концентрированной соляной кислоты. Добавляют 8,2 г SnCl2
•H2O, кипятят с обратным холодильником в течение трех часов, после чего реакционную смесь перемешивают в течение 12 часов при комнатной температуре. Осадок отфильтровывают, затем
обрабатывают его насыщенным раствором гидрокарбоната натрия и экстрагируют этилацетатом. Фильтрат выпаривают, подщелачивают с помощью насыщенного раствора гидрокарбоната натрия, после чего
экстрагируют этилацетатом. Оба этилацетатных раствора объединяют, затем высушивают и выпаривают досуха. Полученный остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью
этилацетата с гексаном в объемном соотношении 50:50. Таким образом получают 1,75 г целевого продукта в виде масла (выход=84%).
1H-ЯМР (дейтерохлороформ): 3.91 (с, 3H, -OCH3), 3.98 (м, 2H, -NH2), 7.03 (д, J=8.8, 1H, H3), 7.27 (т, J=7.7, 1H, H7), 7.47-7.52 (м, 2H, H4, H6), 7.73 (д, J=8.0, 1H, H8), 7.94 (д, J=8.4, 1H, H5).
Следуя указанной выше в ПРИГОТОВЛЕНИИ VII методике, получают следующие соединения:
1-амино-4-метоксинафталин (соединение 17),
5-амино-6-метоксихиноксалин (соединение 18), заменяя SnCl2 на TiCl3,
1-амино-2-этоксинафталин (соединение 19),
1-амино-2-пропоксинафталин (соединение 20),
1-амино-2,3-диметилнафталин (соединение 21),
1-амино-2-метокси-6-бромнафталин (соединение 22),
1-амино-2,6-диметилнафталин (соединение 23),
1-амино-2-(этокси-2-метокси)нафталин (соединение 24).
ПРИГОТОВЛЕНИЕ VIII. 1-Амино-2-метокси-4-этилнафталин (соединение 25)
Стадия 1:
7,5 г 2-Метоксинафталина
растворяют в 80 мл уксусной кислоты, охлаждают до 0oC, затем добавляют 2,2 мл концентрированной азотной кислоты и перемешивают в течение 1 часа при 0oC. Реакционную смесь
выдерживают в течение 12 часов при комнатной температуре. Выпавший осадок желтого цвета отфильтровывают, затем промывают водой. Фильтрат обрабатывают водой, затем экстрагируют этилацетатом и в
результате получают 4 г целевого продукта в виде порошка желтого цвета, (выход = 42%).
1H-ЯМР (дейтерохлороформ): 4.02 (с, 3H, -OCH3), 7.33 (д, J=9.1, 1H, H3), 7.45 (т, J=7.3, 1H, H7), 7.70-7.55 (м, 2H, H6, H8), 7.83 (д, J=7.7, 1H, H5), 7.95 (д, J=9.1, 1H, H4).
Стадия 2:
4 г вышеполученного продукта растворяют в 80 мл безводного тетрагидрофурана при температуре 0oC, затем добавляют раствор магнийорганического соединения (0,96 г магния в 50 мл
безводного тетрагидрофурана, к которому добавляют 2,9 мл этилбромида и несколько кристаллов иода). Реакционную смесь перемешивают в течение 1 минуты при 0oC, после чего добавляют 50 мл
насыщенного раствора хлорида аммония. Экстрагируют этилацетатом, сушат экстракт над сульфатом натрия, затем выпаривают досуха. Остаток обрабатывают в 50 мл безводного тетрагидрофурана, затем добавляют
5,3 г 2,3-дициано-5,6-дихлор-1,4-бензохинона. Кипятят с обратным холодильником в течение 4 часов, после чего выпаривают досуха. Полученный остаток очищают путем хроматографии на колонке с силикагелем,
элюируя смесью этилацетата с гексаном в объемном соотношении 25: 75. Получают 1,8 г целевого продукта в виде порошка белого цвета (выход=40%). Т.пл. = 80oC.
1 H-ЯМР (дейтерохлороформ): 1.41 (т, J=7.5, 3H, -CH2-CH3), (к, J=7.5, 2H, -CH2-CH3), 4.04 (с, 3H, -OCH3), 7.20 (с, 1H, H3), 7.46-8.01 (м, 4H, H5, H6, H8).
Стадия 3:
1,8 г вышеполученного продукта растворяют в 40 мл уксусной кислоты и 20 мл 37%-ной соляной кислоты. Добавляют 5,3
г SnCl2•H2O и кипятят с обратным холодильником в течение 12 часов. Полученный осадок отфильтровывают, затем обрабатывают водой и подщелачивают насыщенным раствором
гидрокарбоната натрия. Экстрагируют этилацетатом, сушат над сульфатом натрия, затем выпаривают досуха и получают 1,4 г целевого продукта в виде порошка желтого цвета (выход=90%).
1H-ЯМР (дейтерохлороформ): 1.37 (т, J-7.5, 3H, -CH2-CH3), 3.06 (к, J= 7.5, 2H, -CH2-CH3), 3.98 (с, 3H, OCH3), 7.13 (с, 1H, H3), 7.34-7.50 (н, 2H, H6, H7), 7.82 (д, J=8.2, 1H, H5), 7.88 (д, J=8.2, 1H, H8).
Следуя методике, указанной в ПРИГОТОВЛЕНИИ VIII, получают 1-амино-2-метокси-4-изопропилнафталин (соединение 26).
ПРИГОТОВЛЕНИЕ IX. 5-Амино-6-метоксиизохинолин (соединение 27)
Стадия 1:
В установку, снабженную насадкой
Дина-Старка, вводят 9,5 г 4-метоксибензальдегида и 7,8 г диметилацеталь-амино- ацетальдегида, разбавленные в 50 мл бензола. Реакционную смесь кипятят с обратным холодильником в течение 12 часов.
Раствор выпаривают досуха, затем остаток обрабатывают два раза бензолом перед выпариванием досуха. Полученное масло растворяют в безводном тетрагидрофуране и выдерживают при температуре -10o
C, затем добавляют один эквивалент этилхлорформиата при интенсивном перемешивании и реакционную смесь перемешивают еще в течение 5 минут, после чего баню со льдом удаляют (появление желтого осадка).
Добавляют 10,5 мл триметилфосфита при комнатной температуре. Перемешивают в течение 15 минут и затем реакционную смесь выпаривают досуха. Для удаления всех следов триметилфосфита масло обрабатывают
толуолом и выпаривают досуха, повторяя эту операцию два раза. Масло растворяют в безводном дихлорметане, добавляют 6 эквивалентов тетрахлорида титана и раствор кипятят с обратным холодильником в
течение 36 часов в условиях отсутствия влаги. Раствор охлаждают и при перемешивании добавляют один эквивалент водного раствора гидроксида натрия до нейтрализации. Осаждается TiO2 в виде
твердого вещества белого цвета. Его отфильтровывают и раствор экстрагируют с помощью 3 н. раствора соляной кислоты, водную фазу промывают дихлорметаном, подщелачивают с помощью сильного основания и
экстрагируют дихлорметаном, экстракт сушат над сульфатом натрия и выпаривают досуха, получая 6,2 г 6-метоксиизохинолина в виде масла светло-оранжевого цвета.
1H-ЯМР (дейтерохлороформ): 3.95 (с, 3H, -OCH3), 7.06 (д, J=2.2, 1H, H5), 7.21 (дд, J= 8.7, J=2.2, 1H, H7), 7.55 (д, J=5.8, 1H, H4), 7.85 (д, J=8.7, 1H, H8), 8.44 (д, J=5.8, 1H, H3), 9.11 (с, 1H, H1).
Стадия 2:
1,2 г нитрата калия добавляют к раствору 1,0 г вышеполученного 6-метоксиизохинолина в 20
мл концентрированной серной кислоты, причем все выдерживают на бане со льдом. После перемешивания в течение 1 часа добавляют дистиллированную воду, затем последовательно подщелачивают, отфильтровывают
осадок и высушивают его, получая 1,2 г 6-метокси-5-нитроизохинолина в виде кристаллов желтого цвета.
1H-ЯМР (дейтерохлороформ): 4.08 (с, 3H, -OCH3), 7.44 (д, J=9.1, 1H, H7), 7.52 (д, J=6.2, 1H, H4), 8.12 (д, J=9.1, 1H, H8), 8.57 (д, J=6.2, 1H, H3), 9.20 (д, J=0.7, 1H, H1).
Стадия 3:
0,9 г вышеполученного нитропроизводного растворяют в смеси из 40 мл уксусной кислоты и 22 мл концентрированной соляной кислоты, затем добавляют 10,2 г хлорида олова и реакционную смесь кипятят с
обратным холодильником в течение трех часов, затем в течение 12 часов выдерживают при комнатной температуре. Подщелачивают и экстрагируют дихлорметаном, органическую фазу сушат и выпаривают досуха,
получая 0,7 г кристаллов желтого цвета целевого амина.
1H-ЯМР (дейтерохлороформ): 3.96 (с, 3H, -OCH3), 4.26 (уш.с, обмениваемый на D2O, 2H, -NH2), 7.25 (д, J=9.1, 1H, H8), 7.40 (д, J=9.1, 1H, H8), 7.48 (д, J=5.8, 1H, H4), 8.35 (д, J=6.2, 1H, H3), 9.07 (с, 1H, H1).
ПРИГОТОВЛЕНИЕ X. 5-Амино-6-метилизохинолин (соединение 28)
Следуя методикам стадий 1 и 2 ПРИГОТОВЛЕНИЯ IX и используя 4-метилбензальдегид в качестве исходного реагента, получают
6-метил-5-нитроизохинолин. 4,0 г 6-Метил-5-нитроизохинолина растворяют в смеси из 80 мл уксусной кислоты и 40 мл концентрированной соляной кислоты, затем добавляют 40,0 г хлорида олова и реакционную
смесь кипятят с обратным холодильником в течение трех часов, затем выдерживают ее в течение 12 часов при комнатной температуре. Отфильтровывают образовавшиеся кристаллы, обрабатывают их водой и
подщелачивают полученный раствор с помощью 10H раствора гидроксида натрия. Экстрагируют дихлорметаном, органическую фазу сушат и выпаривают досуха, получая 0,52 г кристаллов желтого цвета.
1H-ЯМР (дейтерохлороформ): 2.37 (с, 3H, -CH3), 4,18 (уш.с, обмениваемый на D2O, 2H, -NH2), 7.29 (д, J=8.8, 1H, H7), 7.44 (д, J=8.8, 1H, H4, 7.53 (д, J=6.2, 1H, H8), 8.49 (д, J=5.8, 1H, H3), 9.13 (с, 1H, H1).
ПРИГОТОВЛЕНИЕ XI. 5-Амино-1-тетрагидропиран-2-ил-индол (соединение
29)
Стадия 1:
К раствору 20 г 5-нитроиндола в 200 мл диметилформамида при температуре 0oC и в атмосфере аргона добавляют 6,3 г 55%-ного гидрида натрия. После перемешивания
в течение 15 минут при температуре 0oC к реакционной смеси добавляют 27,1 г тетрагидропиранилхлорида. После перемешивания в течение 24 часов при комнатной температуре реакционную смесь
выливают в 1200 мл воды со льдом и экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия и выпаривают досуха. Остаток очищают путем хроматографии на колонке с
силикагелем, элюируя смесью циклогексана с этилацетатом в объемном соотношении 4:1, с получением 19 г 5-нитро-1-тетрагидропиран-2-ил-индола.
1H-ЯМР (диметилсульфоксид): 1.34- 2.12 (м, 6H), 3. 67-4.48 (м, 2H), 5.70 (дд, J=2.0 и J=10.2, 1H), 6.75 (д, J=3.4, 1H), 7.72-7.76 (м, 2H), 8.02 (дд, J=2.2 и 9.2, 1H), 8.54 (д, J=2.2, 1H).
Стадия 2:
17 г
5-Hитро-1-тетрагидропиран-2-ил-индола растворяют в 170 мл метанола, затем добавляют 3 г 10%-ного палладия-на-угле с последующим добавлением маленькими порциями и при температуре 0oC 20,6 г
формиата аммония. После перемешивания в течение полутора часов при комнатной температуре реакционную смесь отфильтровывают и фильтрат выпаривают досуха. Остаток обрабатывают 300 мл этилацетата.
Полученный раствор промывают с помощью 800 мл воды, сушат над сульфатом натрия и выпаривают досуха. Получают 14,2 г 5-амино-1-тетрагидропиран-2-ил-индола в виде твердого вещества. Образец очищают
путем хроматографии на колонке с силикагелем, элюируя смесью циклогексана с этилацетатом в объемном соотношении 3:1, и получают соединение 29.
1H-ЯМР (диметилсульфоксид): 1.30-2.20 (м, 6H), 3.59-3.93 (м, 2H), 4.72 (м, 2H), 5.41 (дд, J=1,8 и 1.20, 1H), 6.16 (д., J=3.2, 1H), 6.52 (дд, J=2.0 и 8.6, 1H), 6.66 (д, J=2.0, 1H), 7.20 (д, J=8.6, 1H), 7.27(д, J=3.2, 1H).
Получение тиомочевин
ПРИГОТОВЛЕНИЕ XII. N-Hафт-1-ил-N-пропилтиомочевина (соединение 30)
В трехгорлую колбу с атмосферой аргона и поддерживаемую при температуре 0-5oC вводят 3,9 г бензоилхлорида и 2,1 г тиоцианата аммония в безводном ацетоне, и реакционную смесь перемешивают в течение 15 минут, затем прикапывают раствор 3,9 г N-нафт-1-ил-N- пропиламина в
ацетоне. После этого реакционную смесь кипятят с обратным холодильником в течение 1 часа, выпаривают досуха и остаток обрабатывают концентрированной соляной кислотой, кипятят с обратным холодильником
в течение трех часов и оставляют стоять до возврата температуры к комнатной. Органические продукты экстрагируют диэтиловым эфиром, затем последовательно подщелачивают с помощью 33%-ного раствора
гидроксида натрия, экстрагируют этилацетатом, выпаривают досуха и очищают на колонке с силикагелем, элюируя смесью гексана с этилацетатом в объемном соотношении 75:25, получая 3,2 г кристаллов белого
цвета. Т.пл. = 170 -171oC.
1H-ЯМР (дейтерохлороформ): 0.86 (т, J=7.3, 3H, -CH2-CH3), 1.68 (м, 2H, -CH2-CH3), 3.72 (м, 2H, -CH2-CH2-CH3), 5.58 (м,-2H, -NH2), 7.37 (д, J=7.3, 1H, H2), 7.23 (м, 2H, H3 и H8), 7.75-7.80 (м, 2H, H6 и H7), 7.85-7.95 (м, 2H, H4 и H5).
ПРИГОТОВЛЕНИЕ XIII. N-Пропил-N-хинол-5-ил-тиомочевина (соединение 31)
В трехгорлой колбе, снабженной
капельной воронкой и в атмосфере аргона, растворяют 1,54 г тиоцианата аммония в 50 мл ацетона, затем прикапывают 2,5 мл бензоилхлорида и наблюдают выпадение осадка белого цвета. Сразу по окончании
добавления нагревают в течение 15 минут при 60oC (слегка кипение с образованием флегмы), затем реакционную смесь оставляют стоять до возврата температуры к комнатной и прикапывают раствор 3,
6 г N-пропил-N-хинол-5-ил-амина (соединение 10) в 10 мл ацетона. Реакционную смесь кипятят с обратным холодильником в течение трех часов, затем последовательно выпаривают досуха, добавляют воду,
экстрагируют дихлорметаном и выпаривают досуха.
Удаление защитной группы в полученном соединении осуществляют путем обработки с помощью 30 мл 15%-ного раствора гидроксида аммония в этаноле при кипячении с обратным холодильником в течение одной ночи, после чего последовательно выпаривают растворитель, экстрагируют дихлорметаном, сушат над сульфатом натрия и выпаривают досуха. Остаток очищают на колонке с силикагелем, элюируя смесью этилацетата с гексаном в объемном соотношении 6:4. Выделяют 1,5 г целевой тиомочевины в виде порошка желтого цвета.
1 H-ЯМР (дейтерохлороформ): 0.91 (т, J=, 3H, -CH3), 1.64-1.79 (м, 2H, -CH2-CH3), 4.19-4.24 (м, 2H, > N-CH2-), 5.73 (уш.с, 2H, -NH2), 7.47 (дд, J= 4.2 и 8.3, 1H, H4), 7.55 (дд, J=2.3 и 8.9, 1H, H7), 7.71 (д, J=2.3, 1H, H5), 8.14-8.22 (м, 2H, H3, H8), 8.97 (дд, J=1.7 и 4.2, H2).
Следуя вышеописанной методике ПРИГОТОВЛЕНИЯ XIII, получают N-пропил-N-хинол-6-ил-тиомочевину (соединение 32).
ПРИГОТОВЛЕНИЕ XIV.
N-(1-нафт-1-ил-2-метоксиэтил)-N- пропилтиомочевина (соединение 33)
Стадия 1:
1,68 г тиоцианата аммония суспендируют в 65 мл ацетона. Реакционную смесь охлаждают на льду и добавляют
раствор 2,5 мл бензоилхлорида в 5 мл ацетона. После перемешивания в течение 15 минут при температуре 5oC прикапывают раствор 5,1 г N-(1-нафт-1-ил-2-метоксиэтил)-N-пропиламина в 60 мл
ацетона. После перемешивания в течение трех часов при температуре в пределах от 5 до 25oC ацетон выпаривают досуха, остаток обрабатывают дихлорметаном и полученный раствор промывают водой.
Органическую фазу затем сушат над сульфатом натрия и выпаривают досуха. Остаток очищают путем хроматографии на силикагеле, элюируя смесью циклогексана с этилацетатом в объемном соотношении 9:1 с
получением 5,50 г N'-бензоил-N-(1-нафт-1-ил-2-метоксиэтил)-N- пропиламина.
Стадия 2:
5,50 г вышеполученого соединения растворяют в 100 мл метанола, затем добавляют 0,60 мл
гидразингидрата. Реакционную смесь перемешивают при комнатной температуре в течение 20 часов. Метанол выпаривают и остаток очищают путем хроматографии на силикагеле, элюируя смесью циклогексана с
этилацетатом в объемном соотношении 3:1, с получением 2,19 г тиомочевины в виде твердого вещества желтого цвета.
ПРИГОТОВЛЕНИЕ XV. N-(6-Метоксихинол-5-ил)тиомочевина (соединение
34)
1,7 г тиоцианата аммония растворяют в 50 мл ацетона, затем добавляют 2,5 мл бензоилхлорида и реакционную смесь кипятят с обратным холодильником в течение 15 минут. Добавляют 2,9 г амина
(соединение 14), растворенных в 20 мл ацетона, затем реакционную смесь кипятят с обратным холодильником в течение 30 минут. Выпаривают досуха, остаток обрабатывают водой, затем экстрагируют
этилацетатом. Удаление защитной группы у остатка осуществляют путем обработки с помощью 5 мл 33%-ного раствора гидроксида аммония в 10 мл этанола при кипячении с обратным холодильником в течение двух
часов. Выпавший осадок отфильтровывают, фильтрат выпаривают и остаток порошкуют с помощью смеси этилацетата с гексаном в объемном соотношении 75:25, получая 3,5 г порошка белого цвета.
1H-ЯМР (дейтерохлороформ): 3.93 (с, 3H, -OCH3), 7.50-7.53 (м, 1H, H3), 7.70 (д, J=9.5, 1H, H7), 8.00 (д, J=9.5, 1H, H4), 8.07 (д, 1H, H8), 8.75 (д, J=2.5, 1H, H2).
ПРИГОТОВЛЕНИЕ XVI. N-(6-метоксиизохинол-5-ил)тиомочевины (соединение 35)
Тиомочевину получают в обычных условиях, используя 3,8
г вышеполученного амина (соединение 27), 1,8 г тиоцианата аммония и 4,8 мл бензоилхлорида в безводном ацетоне. Удаление защитной группы осуществляют в основной среде с помощью 33%-ного аммиака и
получают после очистки путем хроматографии на колонке с силикагелем, элюируя этилацетатом, 3,24 г кристаллов белого цвета с т.пл. 186oC.
1H-ЯМР (диметилсульфоксид): 3.96 (с, 3H, -OCH3), 7.55 (м, 4H, где считают при 7.50 (д, J=6.6, 1H, H4) и при 7.61 (д, J=9.1, 1H, H7) 2H, -NH2), 8.12 (д, J= 9.1, 1H, H8), 8.42 (д, J=5.8, 1H, H3), 9.20-(м,2H, где считают при 9.19 (с, 1H, H7), 1H, -NH).
ПРИГОТОВЛЕНИЕ XVII. N-(6-метилизохинол-5-ил)тиомочевина
(соединение 36)
1,7 г тиоцианата аммония растворяют в 30 мл ацетона, затем добавляют 2,6 мл бензоилхлорида и реакционную смесь кипятят с обратным холодильником в течение 15 минут. Добавляют 2,
7 г 6-метил-5-изохинолиламина (соединение 28), растворенные в 20 мл ацетона. Реакционную смесь нагревают в течение 30 минут, затем выпаривают досуха. Остаток обрабатывают водой и отфильтровывают
остающийся осадок. У полученного соединения удаляют защитную группу с помощью 10 мл 30%-ного раствора гидроксида аммония в 20 мл этанола при кипячении с обратным холодильником в течение двух часов.
Выпаривают досуха и остаток порошкуют в смеси этилацетата с гексаном в объемном соотношении 75:25, получая 3,5 г порошка белого цвета.
1H-ЯМР (дейтерохлороформ): 2.38 (с, 3H, -CH3), 3,36 (с, 2H, -NH2), 6.58 (с, 1H, -NH-), 7.47-7.60 (м, 2H, H7, H4), 7.98 (м, 1H, H8), 8.50 (м, 1H, H3), 9.26 (с, 1H, H1).
Следуя вышеописанным методикам ПРИГОТОВЛЕНИЙ I-XVII и используя соответствующие исходные продукты, получают промежуточные соединения, применяемые в синтезе соединений формулы (I) согласно изобретению.
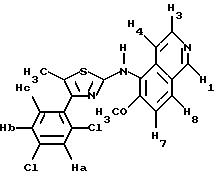
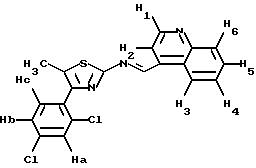
ПРИМЕР 1. 4-(2-Хлор-4-метоксифенил)-5-метил-2-(N-нафт-1- ил-N-пропиламино)тиазол
формула (I); R1= хлор; R2= OCH3; R3=H; R4=CH3; R5=-CH2CH2CH3; n=0; Z=
Раствор, содержащий 0,5 г N-нафт-1-ил-N-пропил-тиомочевины и 0,5 г 2-бром-1-(2-хлор-4-метоксифенил)пропан-1-она в 15 мл метанола, кипятят с обратным холодильником в течение 6 часов. Затем реакционную смесь выпаривают досуха, после чего последовательно остаток обрабатывают водой, полученный раствор подщелачивают с помощью 33%-ного раствора гидроксида натрия, экстрагируют этилацетатом, экстракт сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток затем очищают на колонке с силикагелем, элюируя этилацетатом, с получением 0,56 г масла желтого цвета.
1H-ЯМР (дейтерохлороформ): 0.97 (т, J=7.7, 3H, -CH2-CH3), 1.73-(секстет, 2H, -CH2-CH3), 2.04 (с, 3H, -CH3), 3.82 (с, 3H, -OCH3), 3.90 (м, 2H, -CH2-CH2-CH3), 6.89 (дд, J= 1.8 и 8.0, 1H, H5аром), 7.02 (д, J=1.8, 1H, H3аром), 7.50-7.70 (м, 4H, H2, H3, H8 и H6аром), 7.85-8.10 (м, H, H6, H7, H4 и H5).
ПРИМЕР
2. Хлоргидрат 4-(4-хлор-2-метоксифенил)-5-метил-2- (N-пропил-N-хинол-5-иламино)тиазола
формула (I); R1= OCH3; R2= Cl; R3= H; R4=CH;
R5=-CH2CH2CH3; n=0; Z =
Раствор, содержащий 0,4 г N-пропил-N-хинол-5-ил-тиомочевины (соединение 31) и 0,5 г 2-бром-1-(4-хлор-2-метоксифенил)пропан-1- она в 15 мл этанола, кипятят с обратным холодильником в течение 6 часов. Реакционную смесь выпаривают досуха, затем последовательно обрабатывают остаток водой, полученный раствор подщелачивают с помощью 33%-ного раствора гидроксида натрия и экстрагируют этилацетатом. Остаток очищают на колонке с силикагелем, элюируя смесью этилацетата с гексаном в объемном соотношении 1:1, с получением 0,46 г масла желтого цвета.
1H-ЯМР (дейтерохлороформ): 0.91 (т, J=7.3, 3H, -CH3), 1.61 -1.78 (м, 2H, -CH2-CH3), 2.02 (с, 3H, -CH3 гетероциклического остатка), 3.83 (с, 3H, -OCH3), 3.97 (т, J=7.5, 2H, -CH2 -N <), 6.88-(дд, J=2.5 и 8.4, 1H, H5), 7.0 (д, J= 2.5, 1H, H3), 7.36 (д, J=8.5, 1H, H6), 7.46 (дд, J=4.2 и 8.6, 1H, H3'), 7.63 (д, J= 7.2, 1H, H4'), 7.80 (т, J=7.9, 1H, H7'), 8.17 (д, J=8.4, 1H, H6'), 8.33 (д, J=8.4, 1H, H8'), 8.96-8.99 (м, 1H, H2').
Получение хлоргидрата:
К эфирному раствору 0,46 г вышеполученного соединения добавляют насыщенный раствор газообразного хлороводорода в диэтиловом эфире. Отфильтровывают порошок оранжевого цвета, затем
перекристаллизуют из изопропанола, получая целевой хлоргидрат с т.пл. 142oC.
1H-ЯМР (дейтерохлороформ): 0.85 (т, J=7.2, 3H, -CH2-CH3), 1.62-(секстет, J= 7.2, 2H, -CH2-CH3), 1.99 (с, -CH3), 3.78 (с, 3H, -OCH3), 3.92 (т, J=7.2, 2H, -N-CH2-CH2), 6.95 (дд, J=8.4 и 2.2, 1H, H5фенила), 7.08 (д, J=2.2, 1H, H3фенила), 7.30 (д, J=8.4, 1H, H6фенила), 7.80-8.00 (м, 2H, H3' и H4'), 8.09 (т, J= 7.8, 1H, H7'), 8.36 (д, J=8.1, 1H, H6'), 8.70 (д, J=8.4, 1H, H8'), 9.20 (д, J=4.4, 1H, H2').
ПРИМЕР 3. 4-(2,4-Дихлорфенил)-5-метил-2-[N-(1-(метоксиметил) -1-(нафт-2-ил)
метил)-N-пропиламино]тиазол
формула (I); R1=R2=Cl; R3=H; R4=CH3; R5=-CH2CH2CH3; n=1 R6=-CH2OCH3; Z =
0,93 г 2-Бром-1-(2,4-дихлорфенил)пропан-1-она, 1 г N-(1- нафт-2-ил-2-метоксиэтил)-N-пропилтиомочевины и 0,47 мл триэтиламина растворяют в 20 мл этанола и реакционную смесь нагревают в течение трех часов при температуре 70oC. Этанол удаляют путем выпаривания, затем последовательно добавляют к остатку воду, экстрагируют дихлорметаном, экстракт промывают, органические фазы сушат над сульфатом натрия, отфильтровывают и фильтрат концентрируют в вакууме. Остаток очищают на колонке с силикагелем, используя в качестве элюирующего средства смесь циклогексана с этилацетатом в объемном соотношении 9:1, с получением 0,9 г целевого продукта, из которого получают хлоргидрат. Т.пл. = 50oC.
1H-ЯМР (диметилсульфоксид): 0.73 (м, 3H), 1.12-1.59 (м, 2H), 2.09 (с, 3H), 3.35 (с, 3H), 3.31-3.46 (м, 2H), 3.97-4.23 (м, 2H), 5.48-5.60 (м, 1H), 7.48-7.95 (м, 10H).
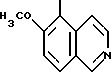
ПРИМЕР 4. Оксалат 4-(2,4-дихлорфенил)-5-метил-2-[N-(6- метоксиизохинол-5-ил)-N-пропиламино]тиазола
формула (I);
R1=R2=Cl; R3=H; R4=CH3; R5=-CH2CH2CH3; n = 0 Z =
Стадия 1:
Раствор, содержащий 0,5 г N-(6-метилизохинол-5-ил)тиомочевины (соединение 35) и 0,6 г 2-бром-1-(2, 4-дихлорфенил)-пропан-1-она в метаноле, кипятят с обратным холодильником. Смесь выпаривают досуха, остаток обрабатывают насыщенным раствором гидрокарбоната калия, затем экстрагируют дихлорметаном, экстракт сушат над сульфатом натрия и выпаривают досуха, получая 0,3 г кристаллов желтого цвета с т.пл. 187-188oC.
1H-ЯМР (дейтерохлороформ): 1.99 (с, 3H, -CH3), 4.02 (с, 3H, -OCH3), 6.52 (дд, J= 8.4, J= 1.8, 1H, Hb), 6.87 (д, J=1.8, 1H, Ha), 7.03 (д, J=8.4, 1H, Hc), 7.39 (д, J=9.1, 1H, H7'), 7.71 (д, J=5.8, 1H, H4'), 7.94 (д, J=9.1, 1H, H8'), 8.40 (д, J=6.2, 1H, H3'), 9.15 (с, 1H, H1).
Стадия 2:
К раствору 0,3 г полученного выше в стадии 1 амина в 50 мл безводного диметилформамида добавляют 0,05 г 55%-ного гидрида натрия и реакционную смесь перемешивают в атмосфере аргона в течение 15 минут перед добавлением 0,3 мл 1-бром-пропана. После этого смесь нагревают при 80oC в течение двух часов, затем добавляют 1 эквивалент гидрида натрия и 1 эквивалент бромпропана и реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Выпаривают досуха, остаток обрабатывают насыщенным раствором гидрокарбоната натрия и экстрагируют дихлорметаном. Экстракт выпаривают досуха и остаток очищают на колонке с силикагелем, элюируя смесью этилацетата с метанолом в объемном соотношении 9:1, с получением 0,3 г вязкого масла.
1H-ЯМР (дейтерохлороформ): 0.86 (т, J=7.3, 3H, -CH2-CH3), 1.61 (секстет, J= 7.3, 2H, -CH2-CH3),
1.99 (с, 3H, -CH3), 3.80 (м, 2H, -N-CH2-), 4.02 (с, 3H, -OCH3), 7.25 (дд, J=8.4, J=2.1, 1H, Hb), 7.40 (д, J=8.4, 1H, Hc), 7.43 (д, J=2.1, 1H,
Ha), 7.44 (д, J=
9.1, 1H, H7), 7.68 (д, J=5.8, 1H, H4, 8.03 (д, J=9.1, 1H, H8), 8.46 (д, J=5.8, 1H, H3), 9.19 (c, 1H, H1).
Получение оксалата:
0,06 г щавелевой кислоты, растворенные в минимальном количестве изопропанола, добавляют к эфирному раствору 0,3 г аминотиазола, полученного выше в стадии 2.
Выпавший осадок перекристаллизуют из изопропанола, получая кристаллы бледно-желтого цвета с т.пл. 162- 163oC.
1H-ЯМР (диметилсульфоксид): 0.82 (т, J= 7.3, 3H, -CH2-CH3), 1.57 (секстет, J-7.3, 2H, -CH2-CH3), 1.96 (с, 3H, -CH3), 3.75 (м, 2H, -N-CH2-), 4.00 (с, 3H, -OCH3), 7.46 (м, 2H, Hb и Hc), 7.57 (д, J=6.2, 1H, H4), 7.66 (д, J= 1.1, 1H, Ha), 7.78 (д, J=8.7, 1H, H7), 8.30 (д, J=9.1, 1H, H8), 8.45 (д, J=5.5, 1H, H3), 9.31 (с, 1H, H1).
ПРИМЕР 5. Оксалат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N- (6-метилизохинол-5-ил)-N-пропиламино]тиазола
формула (I); R1
=Cl; R2=OCH3; R3=H; R4=CH3; R5=-CH2CH2CH3; n = 0
Стадия 1:
1,5 г N-(6-метилизохинол-5-ил)тиомочевины (соединение 36) растворяют в 40 мл метанола, затем добавляют 2,1 г 2-бром-1-(2-хлор-4-метоксифенил) пропан-1-она и реакционную смесь кипятят с обратным холодильником в течение 12 часов. Выпаривают досуха, остаток обрабатывают насыщенным раствором гидрокарбоната натрия, затем последовательно экстрагируют этилацетатом, сушат экстракт над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью этилацетата с гексаном в объемном соотношении 75:25, с получением после концентрирования чистых фракций, 1,3 г 4-(2-хлор-4- метоксифенил)-5-метил-2-[N-(6-метилизохинол-5-ил)амино]тиазола в виде порошка желтого цвета.
1H-ЯМР (дейтерохлороформ): 1.95 (с, 3H, -CH3), 2.52 (с, 3H, -CH3), 3.57 (с, 3H, -OCH3), 5.96 (дд, J=8.4, J=1.8, 1H, H4), 6.23 (д, J=1.8, 1H, H2), 6.86 (д, J=8.4, 1H, H5), 7.45 (д, J=8.4, 1H, H7), 7.79 (м, 2H, H4, H8), 8.44 (д, J=5.8, 1H, H3), 9.16 (с, 1H, H1).
Стадия 2:
К раствору 0,2 г 55%-ного гидрида натрия в 20
мл безводного диметилформамида добавляют 1,3 г полученного выше в стадии 1 амина и реакционную смесь перемешивают в атмосфере аргона в течение 15 минут перед добавлением 0,6 мл 1-бром-пропана.
Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, добавляют 100 мл насыщенного раствора хлорида аммония, затем последовательно экстрагируют этилацетатом, эту фазу промывают
насыщенным раствором хлорида натрия, выпаривают досуха и очищают остаток на колонке с силикагелем, элюируя смесью этилацетата с гексаном в объемном соотношении 25:75, с получением после
концентрирования чистых фракций 0,8 г масла желтого цвета.
1H-ЯМР (дейтерохлороформ): 0.88 (т, J=7.3, 3H, -CH2-CH3), 1.66 -1.78 (м, 2H, -CH2 -CH3), 2.03 (с, 3H, -CH3), 2.52 (с, 3H, -CH3), 3.87-3.90 (м, 2H, -N-CH2-), 4.86 (с, 3H, -OCH3), 6.86 (дд, J=8.4 и 2.5, 1H, H5), 7.00 (д, J= 2.5, 1H, H3), 7.39 (д, J=8.8, 1H, H6), 7.57 (д, J=8.4, 1H, H7), 7.70 (д, J= 5.8, 1H, H4), 7.94 (д, J=8.4, 1H, H8), 8.54 (д, J=5.8, 1H, H3), 9.27 (с, 1H, H1).
Получение оксалата:
0,16 г щавелевой кислоты, растворенные в минимальном количестве изопропанола, добавляют к раствору 0,8 г
аминотиазола, полученного выше в стадии 2, в минимальном количестве эфира и петролейного эфира, получая целевой продукт в виде кристаллов желтого цвета.
1H-ЯМР (диметилсульфоксид): 0.82 (т, J=7.3, 3H, -CH2-CH3), 1.64 (м, 2H, -CH2-CH3), 1.96 (с, 3H, -CH3), 2.44 (с, 3H, - CH3), 3.76-3.79 (м, 2H, -N-CH2), 3.79 (с, 3H, -OCH3), 6.94 (дд, J=2.5, J=8.4, 1H, H5), 7.08 (д, J=2.5, 1H, H3), 7.33 (д, J=8.8, 1H, H6), 7.62 (д, J=8.4, 1H, H7 ), 7.72 (д, J=5.8, 1H, H4), 8.15 (д, J=9.1, 1H, H8), 8.53 (д, J=5.5, 1H, H3), 9.38 (с, 1H, H1).
ПРИМЕР 6. Оксалат 4-(2,
4-дихлорфенил)-5-метил-2-[N-(1- (циклопропил)-1-(хинол-4-ил)метил)-N-пропил-амино]тиазола
формула (I); R1=R2=Cl; R3=H; R4=CH3; R5=-CH2CH2CH3; n = 1
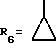
Стадия 1:
В колбе, снабженной насадкой Дина-Старка, растворяют 3,0 г 4-хинолинкарбоксальдегида и 5,0 г 2-амино-4-(2, 4- дихлорфенил)-5-метилтиазола в 50 мл бензола. Реакционную смесь кипятят с обратным холодильником в течение 24 часов. Выпаривают досуха, остаток обрабатывают насыщенным раствором гидрокарбоната натрия и экстрагируют дихлорметаном. Выпаривают досуха и остаток очищают путем хроматографии на колонке с силикагелем, элюируя этилацетатом с 2% триэтиламина. Концентрирование чистых фракций дает 8,2 г кристаллов ярко-желтого цвета с т.пл. 140-141oC 4-(2,4-дихлорфенил)-5-метил-2-(N-(хинол-4-ил- метил)имино)тиазола.
1H-ЯМР (дейтерохлороформ): 2.27 (с, 3H, - CH3), 7.23 (дд, J=7.6, J=1.8, 1H, Hb), 7.29 (д, J=7.6, 1H, Hc), 7.43 (д, J=1.8, 1H, Ha), 7.53 (тд, J-8.4 и 1.4, 1H, H5), 7.65 (тд, J=8.4 и 1.4, 1H, H4), 7.84 (д, J=4.4, 1H, H2), 8.07 (д, J= 8.4, 1H, H3), 8.75 (д, J=8.4, 1H, H6), 8.92 (д, J=4.4, 1H, H1), 9.51 (c, 1H, - N=CH-).
Стадия 2:
В трехгорлой колбе, в которой поддерживают атмосферу аргона, к раствору, содержащему магнийорганическое соединение, полученное из 1,0 г магния и 4,8 г циклопропилбромида, медленно добавляют 8,5 г вышеполученного в стадии 1 имина, разбавленных в тетрагидрофуране. Избыток: магнийорганического соединения разлагают путем добавления насыщенного раствора хлорида аммония и экстрагируют диэтиловым эфиром. Выпаривают досуха и остаток очищают путем хроматографии на колонке с силикагелем, элюируя этилацетатом, с получением после концентрирования чистых фракций 5,1 г 4-(2,4-дихлорфенил)-5-ме-тил-2-[N-(1- (циклопропил)-1-(хинол-4-ил)метил)амино] тиазола в виде очень вязкого желтого масла.
1H-ЯМР (дейтерохлороформ): 0,40-0,65 (м, 4H, -CH2-CH2), 1.15-1.35 (м, 1H, -CH-CH-CH2), 2.03 (с, 3H, -CH3), 4.93 (дд, J=8.1 и 3.9, 1H, -NH-CH-), 6.31 (J=4.0, 1H, -NH-CH-), 7.23 (дд, J=7.6, J=1.8, 1H, Hb), 7.29 (д, J=8.4, 1H, Hc), 7.43 (д, J=1.8, 1H, Ha), 7.53 (J=8.4 и 1.4, 1H, H5), 7.65 (тд, J= 8.4 и 1.4, 1H, H4), 7.84 (д, J=4.4, 1H, H2), 8.07 (д, J=8.4, 1H, H3), 8.75 (д, J=8.4, 1H, H6), 8.92 (д, J=4.4, 1H, H1 ).
Стадия 3:
К раствору 5,46 г полученного выше в стадии 2 амина в 50 мл диметилформамида добавляют 0,6 г 55%-ного гидрида натрия и реакционную смесь перемешивают в атмосфере
аргона в течение 15 минут перед добавлением 1,8 г 1-бромпропана. Реакционную смесь нагревают при 80oC в течение двух часов, добавляют один эквивалент гидрида натрия и один эквивалент
бромпропана, после чего перемешивают при комнатной температуре в течение 12 часов. Выпаривают досуха, остаток обрабатывают насыщенным раствором гидрокарбоната натрия и экстрагируют дихлорметаном.
Экстракт выпаривают досуха и остаток очищают путем хроматографии на колонке с силикагелем, элюируя этилацетатом, с получением 0,79 г целевого продукта в виде бесцветного масла.
1H-ЯМР (дейтерохлороформ): 0,35-0,75 (м, 7H, где считают при 0,71 (т, J= 7.6, 3H, -CH2-CH3), -CH2-CH2), 1.15- 1.35 (м, 1H, CH-CH2-CH2), 1,68 (секстет, J= 7.6, 2H, -CH2-CH3), 2.10 (с, 3H, -CH3), 3.14 (м, J=7.6, 2H, -N-CH2), 5.58 (д, J=9.9, 1H, -N-CH-), 7.20 (дд, J=8.4 и 1.8, 1H, Hb), 7.29 (д, J=8.4, 1H, Hc), 7.35-(тд, J=8.4 и 1.4, 1H, H5), 7.40 (д, J=1.8, 1H, Ha), 7.55 (тд, J=8.4 и 1.4, 1H, H), 7.65 (д, J=4.4, 1H, H), 8.01 (д, J=8.4, 1H, H), 8.15 (д, J=8.4, 1H, H6), 8.84 (д, J=4.4, 1H, H1).
Получение оксалата:
0,1 г щавелевой кислоты, разбавленный в минимальном количестве
изопропанола, добавляют в раствору диэтилового эфира, содержащему 0,8 г вышеполученного в стадии 3 аминотиазола, затем выпавший осадок перекристаллизуют из изопропанола, получая кристаллы желтого
цвета с т.пл. 164-165oC.
1H-ЯМР (диметилсульфоксид): 0,35-0,75 (м,7H, где считают при 0,71 (т, J= 7.6, 3H, -CH2-CH3), -CH2-CH2), 1.15-1.35 (м, 1H, CH-CH-CH2), 1.68 (секстет, J= 7.6, 2H, -CH2-CH3), 2.10 (с, 3H, -CH3), 3.14 (м, J=7.6, 2H, -N-CH2), 5.46 (д, J=10.2, 1H, -N-CH-) 7.30-7.50 (м, 3H, Ha, Hb и Hc), 7.65-7.80 (м, 2H, H5 и H4), 7.92-(д, J=3.6, 1H, H2), 8.02 (д, J=7.7, 1H, H3), 8.21 (д, J=8.4, 1H, H6), 8.95 (д, J=3.6, 1H, H1).
ПРИМЕР 7. 4-(2-Хлор-4-метоксифенил)-5-метил-2-[N-(индол-5-ил)-N-пропиламино]тиазол
формула (I); R1=Cl; R2=OCH3; R3=H; R4=CH3; R5=-CH2CH2CH3; n=0
Стадия 1:
10,9 г 4-(2-Хлор-4-метоксифенил)-5-метил-2-[N-(1-(тетрагидропиран-2-ил)индол-5-ил)амино] тиазола растворяют в 110 мл диметилформамида. При температуре 0oC добавляют 1,06 г 60%-ного гидрида натрия в масле, затем 3,2 мл пропилбромида. После перемешивания в течение 16 часов при комнатной температуре реакционную смесь выливают в 500 мл воды и экстрагируют три раза по 250 мл этилацетатом. Органическую фазу промывают три раза по 200 мл водой, сушат над сульфатом натрия и выпаривают досуха. Остаток очищают путем флеш-хроматографии на силикагеле, элюируя смесью циклогексана с этилацетатом в объемном соотношении 3:1. Получают 10,17 г целевого продукта (выход= 85%).
1 H-ЯМР (дейтерохлороформ): 0,90 (м, 3H), 1.57-1.75 (м, 6H), 2.02 (с, 3H), 1.95-2.25 (м, 2H), 3.70-4.17 (м, 7H), 5.50 (дд, J=2.6 и 10.0, 1H,), 6.54-7.61 (м, 8H).
Стадия 2:
К 6,
6 г 4-(2-Хлор-4-метоксифенил)-5-метил-2-[N-(1-(тетрагидропиран-2-ил) индол-5-ил)-N-пропиламино] тиазола, растворенным в 50 мл метанола, добавляют 8 мл 35%-ной соляной кислоты. Реакционную смесь
перемешивают в течение 24 часов, затем разбавляют водой и нейтрализуют 30%-ным раствором гидроксида натрия. Смесь экстрагируют дихлорметаном. Органическую фазу промывают несколько раз водой и сушат
над сульфатом натрия. После выпаривания растворителя остаток очищают путем хроматографии на колонке с силикагелем, элюируя с помощью ступенчатообразного градиента от 10 до 50% этилацетата в
циклогексане. Выделяют 3,5 г защищенного продукта и 0,85 г целевого продукта в виде твердого белого вещества; т.пл. = 154oC.
1H-ЯМР (диметилсульфоксид): 0,83 (м, 3H), 1.42-1.65 (м, 2H), 1.96 (с, 3H), 3.74 (с, 3H), 3.59- 3.89 (м, 2H), 6.35 (д, J=3.2, 1H), 6.94 (дд, J=2.6 и 8.6, 1H). 7.02- 7.08 (м, 2H), 7.31 (д, J=8.5, 1H), 7.38-7.42 (м, 1H), 7.43 (д, J=8.6, 1H), 7.55 (д, J=2.0, 1H).
ПРИМЕР 8. 4-(2-Хлор-4-метоксифенил)-5-метил-2-[N-(1- метилиндол-5-ил)-N-пропиламино]тиазол
формула (I); R1=Cl; R2=OCH3;
R3=H; R4=CH3; R5=-CH2CH2CH3; n = 0
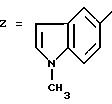
К раствору 0,39 г вышеполученного (Пример 7) 4-(2-хлор-4- метоксифенил)-5-метил-2-[N-(индол-5-ил)-N-пропиламино]тиазола в 5 мл диметилформамида при температуре 4oC добавляют 0,045 г 55%-ного гидрида натрия в масле, затем 0,12 мл метилиодида. После перемешивания в течение 5 часов при комнатной температуре реакционную смесь выливают в воду со льдом. Смесь экстрагируют и органическую фазу промывают несколько раз водой, сушат над сульфатом натрия и выпаривают досуха. Остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью циклогексана с этилацетатом в объемном соотношении 9: 1. Целевой продукт выделяют в виде твердого белого вещества. Т.пл. = 146oC.
1H-ЯМР (дейтерохлороформ): 0,92 (м, 3H), 1.58-1.73 (м, 2H), 2.01 (с, 3H), 3.81-4.0 (м, 8H), 6.51 (д, J=2.8, 1H), 6.85 (дд, J=2.2 и 8.4, 1H,), 6.99 (д, J=2.0, 1H), 7.09-7.20 (м, 2H), 7.35-7.40-(м, 2H), 7.62 (с,1H).
ПРИМЕР 9. 4-(2-Хлор-4-метоксифенил)-5-метил-2-[N-(1- метоксикарбонилметилиндол-5-ил)-N-пропиламино]тиазол
формула (1); R1=Cl; R2=OCH3; R3=H; R4=CH3; R5=-CH2CH2CH3; n = 0;
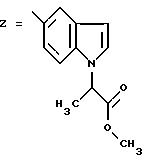
К раствору 0,8 г вышеполученного (Пример 7) 4-(2-хлор-4- метоксифенил)-5-метил-2-[N-(индол-5-ил)-N-пропиламино] тиазола в 20 мл диметилформамида при температуре 0oC добавляют 0,093 г 55%-ного гидрида натрия в масле, затем 0,95 мл метилбромацетата. После перемешивания в течение 12 часов при комнатной температуре реакционную смесь выливают в воду со льдом. Смесь экстрагируют этилацетатом и органическую фазу промывают несколько раз водой, сушат над сульфатом натрия и выпаривают досуха. Остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью циклогексана с этилацетатом в объемном соотношении 9:1. Целевой продукт выделяют в виде твердого белого вещества (выход=95%); т.пл.=80oC.
1H-ЯМР (диметилсульфоксид): 0,84 (т, 3H), 1.51-1.62 (м, 2H), 1.94 (с, 3H), 3.68 (с, 3H), 3.79 (с, 3H), 3.75-3.82 (м, 2H), 5.17 (с, 2H,), 6.50 (д, 1H), 6.91-7.58 (м, 7H).
ПРИМЕР 10. Хлоргидрат
4-(2-хлор-4-метоксифенил)-5-метил-2-[N- (1-(1-(метоксикарбонил)этил)индол-5-ил)-N-пропиламино]тиазола
формула (I); R1=Cl; R2=OCH3; R3=H; R4=CH3; R5=-CH2CH2CH3; n = 0;
Стадия 1:
К раствору 1,2 г вышеполученного (Пример 7) 4-(2-хлор-4-метоксифенил) -5-метил-2-[N-(индол-5-ил)-N-пропиламино] тиазола в 20 мл метанола при температуре 0oC и в атмосфере аргона добавляют 0,14 г 55%-ного гидрида натрия в масле, затем 1,6 мл метил-2-бромпропионата. После перемешивания в течение 24 часов реакционную смесь выливают в воду со льдом и экстрагируют этилацетатом. После высушивания и выпаривания досуха в вакууме остаток обрабатывают с помощью 20 мл метанола. После добавления 1,1 мл 2 н. раствора гидроксида натрия перемешивают в течение 24 часов, затем этанол выпаривают, остаток обрабатывают водой, доводят значение pH до 6 путем добавления 2 н. соляной кислоты и экстрагируют этилацетатом. Органическую фазу промывают несколько раз, сушат над сульфатом натрия и выпаривают. Твердый остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью дихлорметана с метанолом в объемном соотношении 98:2. Получают 0,80 г соответствующей кислоты.
Стадия 2:
К раствору 0,8 г вышеполученного продукта в 15 мл диметилформамида добавляют 0,294 г карбоната цезия, затем 0,31 мл метилиодида. После
перемешивания в течение трех часов реакционную смесь разбавляют этилацетатом, промывают несколько раз насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, затем выпаривают досуха в
вакууме. Остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью циклогексана с этилацетатом в объемном соотношении 5:1. Целевой продукт выделяют в виде бесцветного масла (0,66
г). Моногидрат хлоргидрата получают путем добавления 0,1 н. раствора хлороводорода в изопропаноле в виде твердого белого вещества. Т. пл. = 80oC.
1H-ЯМР (дейтерохлороформ): 0,92 (т, 3H), 1.62-1.74 (м, 2H), 1.83 (д, 3H), 2.02 (с, 3H), 3.74 (с, 3H), 3.81 (с, 3H), 3.88-3.98 (м, 2H), 5.13- 5.20 (м, 1H,), 6.59 (д, 1H), 6.82-7.62 (м, 7H).
ПРИМЕР 11. 4-(2-Хлор-4-метоксифенил)-5-метил-2-[N-(1- карбоксиметилиндол-5-ил)-N-пропиламино]тиазол
формула (I); R1=Cl; R2=OCH3; R3=H; R4
=CH3; R5=-CH2CH2CH3; n = 0;
К раствору 0,52 г вышеполученного продукта (Пример 9) в 10 мл этанола добавляют 1,2 мл 1 М водного раствора гидроксида натрия. После перемешивания в течение 18 часов при комнатной температуре этанол выпаривают, остаток обрабатывают водой, доводят значение pH до 6 путем добавления 2 н. соляной кислоты, затем экстрагируют дихлорметаном. Органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают. Твердый остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью дихлорметана с метанолом в объемном соотношении 92:8. Целевой продукт выделяют в виде твердого белого вещества (полугидрат). Т.пл. = 120oC.
1H-ЯМР (диметилсульфоксид): 0,88 (т, 3H), 1. 51-1.62 (м, 2H), 1.93 (с, 3H), 3.78 (с, 3H), 3.74- 3.82 (м, 2H), 4.48 (м, 2H), 6.38-(д, 1H), 6.91-7.51 (м, 7H).
ПРИМЕР 12. Хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2- [N-(1-морфолинокарбонилметилиндол-5-ил)-N-пропиламино)тиазол
формула (I); R1=Cl; R2=OCH3; R3=H; R4=CH3; R5=-CH2CH2CH3; n = 0;
К раствору 1,1 г вышеполученного продукта (Пример 11) в 20 мл диметилформамида при температуре -10oC и в атмосфере аргона добавляют 0,36 мл триэтиламина, затем 0,34 мл изобутилхлорформиата. После перемешивания в течение 10 минут при температуре -10oC добавляют 0,74 мл свежеперегнанного морфолина. После выдерживания в течение двух часов при температуре -10oC реакционную смесь оставляют стоять до повышения температуры до комнатной, затем органическую фазу разбавляют этилацетатом. Органическую фазу промывают несколько раз водой, сушат над сульфатом натрия и выпаривают. Остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью дихлорметана с метанолом в объемном соотношении 98: 2. Образующийся в форме гидрата (дигидрата) хлоргидрат получают при использовании 0,1 н. раствора хлороводорода в изопропаноле. Т.пл. = 134oC.
1H-ЯМР (дейтерохлороформ): 0,93 (т, 3H), 1.59-1.77 (м, 2H), 1.99 (с, 3H), 2.85-2.93 (м, 4H), 3.43-7.47 (м, 4H), 3.80 (с, 3H), 3.88- 4.15 (м, 2H), 4.93 (с, 2H,), 6.59 (д, 1H), 6.81-7.60 (м, 7H).
ПРИМЕР 13. Хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2- [N-(1-(метилкарбонилметил)индол-3-ил)-N-пропиламино]тиазола
формула (I); R1=Cl; R2=OCH3; R3=H; R4=CH3; R5=-CH2CH2CH3; n = 0;
Стадия 1:
К раствору 1,1 г вышеполученного (Пример 7) 4-(2-хлор-4-метоксифенил)-5- метил-2-[N-(индол-5-ил)-N-пропиламино] тиазола в 10 мл диметилформамида при температуре 0oC и в атмосфере аргона добавляют 0,35 г 55%-ного гидрата натрия в масле, затем 1 мл бромацетонитрила. После перемешивания в течение 18 часов при комнатной температуре реакционную смесь выливают в воду со льдом. Смесь экстрагируют этилацетатом и органическую фазу промывают несколько раз водой, сушат над сульфатом натрия и выпаривают досуха. Остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью циклогексана с этилацетатом в объемном соотношении 9:1.
Стадия 2:
К раствору 0,66 г
полученного в предыдущей стадии продукта в 10 мл безводного диэтилового эфира при температуре 0oC добавляют 1,3 мл 1,4 М раствора метилмагнийбромида. После выдерживания в течение 5 часов
при комнатной температуре реакционную смесь гидролизуют путем добавления насыщенного раствора хлорида аммония. Смесь экстрагируют этилацетатом и органическую фазу промывают несколько раз водой, сушат,
затем выпаривают досуха. Остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью дихлорметана с метанолом в объемном соотношении 98:2. Целевой продукт выделяют в виде масла.
Хлоргидрат получают в моногидратированной форме путем добавления 0,1 М раствора хлороводорода в изопропаноле в виде твердого белого вещества. Т.пл. = 189oC.
1 H-ЯМР (дейтерохлороформ): 0,90 (т, 3H), 1.41 (с, 3H), 1.57-1.68 (м, 2H), 2.01 (с, 3H), 3.81 (с, 3H), 3.85-3.92 (м, 2H), 6.60-7.63-(м, 8H).
ПРИМЕРЫ 14 И 15.
4-(2-Хлор-4-метоксифенил)-5-метил-2-[N- [1-пропилиндазол-6-ил]-N-пропиламино]тиазол
(Пример 14)
4-(2-Хлор-4-метоксифенил)-5-метил-2-[N-[(2-пропил)
индазол-6-ил]-N-пропиламино]тиазол
(Пример 15)
формула (I); R1=Cl; R2=OCH3; R3=H; R4=CH3; R5=-CH2CH2CH3; n = 0;
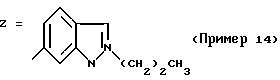
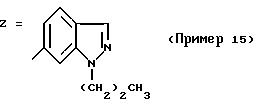
К раствору 0,94 г 4-(2-хлор-4-метоксифенил)-5-метил-2-[N- индазол-6-иламино]тиазола в 20 мл диметилформамида при температуре 0oC добавляют 0,24 г 55%-ного гидрида натрия в виде суспензии в масле, затем после перемешивания в течение 10 минут добавляют 0,55 мл пропилбромида. После перемешивания в течение полутора часов при комнатной температуре реакционную смесь выливают в 100 мл воды со льдом и экстрагируют этилацетатом. Органическую фазу промывают 4 раза по 100 мл водой, сушат над сульфатом натрия и выпаривают в вакууме. Остаток очищают путем хроматографии на колонке с силикагелем, элюируя смесью циклогексана с этилацетатом в объемном соотношении 4:1. Получают 0,34 г 4-(2-хлор-4- метоксифенил)-5-метил-2-[N-(1-пропилиндазол-6-ил)-N- пропиламино] тиазола в виде масла и 0,28 г 4-(2-хлор-4- метоксифенил)-5-метил-2-[N-(2-пропил)индазол-6-ил-N-пропиламино] тиазола в виде масла.
1H-ЯМР (дейтерохлороформ) (соединение Примера 14): 0,88-1.08- (м, 6H), 1.62-1.81 (м, 2H), 1.95-2.14 (м, 5H), 3.81 (c, 3H), 3.90-3.98 (м, 2H), 4.36 (т, J=7.0, 2H, ), 6.84 (дд, J=2.6 и 8.4, 1H), 6.97 (д, J=2.4, 1H), 7.10 (дд, J=1.7 и 8.9, 1H), 7.33 (д, J= 8.6, 1H), 7.69-7.88 (м, 2H), 7.90 (с, 1H).
1H-ЯМР (дейтерохлороформ) (соединение Примера 15): 0.89-0.98-(м, 6H), 1.63-1.82 (м, 2H), 1.83- 2.05 (м, J=7.0, 2H), 2.08 (с, 3H), 3.82 (с, 3H), 3.93-4.00 (м, 2H), 4.32 (т, J=7.0, 2H), 6.85-(дд, J=2.6 и 8.4, 1H), 6.98 (д, J= 2.6, 1H), 7.14 (дд, J=1.7 и 8.6, 1H), 7.34 (д, J=8.6, 1H), 7.48 (м, 1H), 7.75 (д, J=8.6, 1H), 7.98 (с, 1H).
Реферат
Производные 4-фениламинотиазола формулы I, в которой R1, R2 -галоген, С1-5-алкил, С1-5-алкоксил, трифторметил; R3 - Н; R4- С1-5-алкил; R5 - С1-5-алкил, циклоалкил; n = 0 или 1; R6 - С1-5-алкил, алкоксиалкил, циклоалкил; Z - нафтил или гетероароматическая или бензоконденсированная группа, содержащая 1 или 2 атома азота в качестве гетероатома, возможно замещенная 1-2 заместителями, выбранными из галогена, алкила, алкоксила, алкоксикарбонилалкила, карбоксиалкила, морфолинкарбонила, алкилкарбонила, диалкиламинокарбонилалкила или алкоксиалкоксила, их стереоизомеры или их соли обладают антагонистической активностью по отношению к фактору высвобождения кортикотропина. 5 с. и 7 з.п. ф-лы, 4 табл.
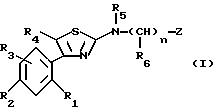
Формула

в которой R1 и R2, одинаковые или разные, каждый, независимо друг от друга, означают атом галогена, алкил с 1 - 5 атомами углерода, алкоксил с 1 - 5 атомами углерода, трифторметил;
R3 означает водород;
R4 означает алкил с 1 - 5 атомами углерода;
R5 означает алкил с 1 - 5 атомами углерода, циклоалкил с 3 - 7 атомами углерода;
n означает нуль или единицу;
R6 означает алкил с 1 - 5 атомами углерода, алкоксиалкил, в котором алкильные части содержат 1 - 5 атомов углерода, циклоалкил с 3 - 7 атомами углерода;
Z означает нафтильную или гетероароматическую бензоконденсированную группу, содержащую 1 или 2 атома азота в качестве гетероатома, возможно замещенную 1 или 2 заместителями, выбранными из галогена, низшего алкила, низшего алкоксила, алкоксикарбонилалкила, карбоксиалкила, морфолинкарбонилалкила, алкилкарбонилалкила, диалкиламинокарбонилалкила или алкоксиалкоксила,
их стереоизомеры и/или их соли присоединения.
4-(2, 4-дихлорфенил)-5-метил-2-[N-(1-(метоксиметил)-1-(нафт-2-ил)метил-N-пропиламино]тиазол;
оксалат 4-(2,4-дихлорфенил)-5-метил-2-[N-(6-метоксиизохинол-5-ил)-N-пропиламино]тиазола;
оксалат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6-метилизохинол-5-ил)-N-пропиламино]тиазола;
4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(1-метоксикарбонилметилиндол-5-ил)-N-пропиламино]тиазол;
оксалат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6-метоксиизохинол-5-ил)-N-пропиламино]тиазола;
оксалат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6-хлоризохинол-5-ил)-N-пропиламино]тиазола;
оксалат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6-метоксиизохинол-5-ил)-N-пропиламино]тиазола;
4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(1-метоксинафт-2-ил)-N-пропиламино]тиазол;
оксалат 4-(2-хлор-4-трифторметилфенил)-5-метил-2-[N-(6-метоксиизохинол-5-ил)-N-пропиламино]тиазола;
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(2-этоксинафт-1-ил)-N-пропиламино]тиазола;
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(2,3-диметилнафт-1-ил)-N-пропиламино]тиазола;
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(6-бром-2-метоксинафт-1-ил)-N-пропиламино]тиазола;
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(2, 6-диметилнафт-1-ил)-N-пропиламино]тиазола;
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(1-(метоксиметил)-1-(нафт-2-ил)метил)-N-пропиламино]тиазола;
хлоргидрат 4-(2-хлор-4-метоксифенил)-5-метил-2-[N-(1-(циклопропил)-1-(нафт-2-ил)метил)-N-пропиламино] тиазола; один из его стереоизомеров и/или возможно одна из его солей.
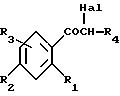
в которой R1, R2, R3 и R4 имеют указанные для формулы I значения, а Hal означает атом галогена, вводят во взаимодействие с тиомочевиной (Способ В) формулы (IIIb)
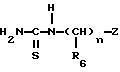
в которой R6 и Z имеют указанные для формулы I значения,
для получения соединения формулы (IV)
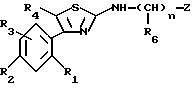
в которой R1, R2, R3, R4, "n", R6 и Z имеют указанные для формулы I значения,
которое затем подвергают реакции алкилирования для получения соединения формулы I и, в частности, для получения в случае, когда Z означает азотсодержащий гетероцикл, такой, как индол или индазол, либо моноалкилированных соединений, замещая предварительно реакционноспособный азот цикла защитной группой, предпочтительно типа тетрагидропиранила, либо диалкилированных соединений, осуществляя, после удаления защитной группы в цикле полученного моноалкилированного соединения, алкилирование высвобожденного реакционноспособного азота, причем эти диалкилированные соединения, в зависимости от природы второй алкильной группы, могут приводить к диалкилированным продуктам, содержащим различные или идентичные алкильные группы, при этом, в последнем случае, диалкилированные соединения могут быть получены непосредственно путем диалкилирования соединения формулы IV, в котором реакционноспособный азот гетероцикла не защищен, и, при желании, таким образом полученные соединения формулы I затем разделяют на их возможные стереоизомеры и/или подвергают солеобразованию для получения соответствующих солей.
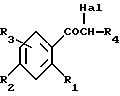
в которой R1, R2, R3 и R4 имеют указанные для формулы I значения, а Hal означает атом галогена,
вводят во взаимодействие с тиомочевиной (Способ А) формулы IIIa

в которой R5, n, R4 и Z имеют указанные для формулы I значения,
для получения непосредственно соединения формулы I, и, при желании, таким образом полученные соединения формулы I затем разделяют на их возможные стереоизомеры и/или подвергают солеобразованию для получения соответствующих солей.

в которой R1, R2, R3 и R4 имеют указанные для формулы I значения, а Hal означает атом галогена,
вводят во взаимодействие с тиомочевиной (Способ С) формулы

для получения аминотиазола формулы V
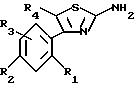
в которой R1, R2, R3 и R4 имеют указанные для формулы I значения,
который затем, при необходимости, вводят во взаимодействие с альдегидом формулы HCO-Z для получения имина, который в свою очередь, под действием магнийорганического соединения формулы R4MgX (где X означает галоген) или литийорганического соединения формулы R4Li приводит к соединению формулы IV, которое подвергают алкилированию, например, путем воздействия соединения формулы R5X (где X означает удаляемую группу, такую, как галоген) с целью получения соединения формулы I, и, при желании, таким образом полученные соединения формулы I затем разделяют на их возможные стереоизомеры и/или подвергают солеобразованию для получения соответствующих солей.
Комментарии