1,3,4-оксадиазол-2-оны в качестве модуляторов ppar-дельта, фармацевтические композиции на их основе и способ лечения - RU2365589C2
Код документа: RU2365589C2
Описание
Область изобретения
Данное изобретение относится к новым соединениям и фармацевтическим композициям, которые действуют как селективные связывающие агенты лигандов PPAR-дельта рецепторов, которые могут использоваться для модуляции PPAR-дельта рецепторов при лечении заболеваний, опосредованных ядерными рецепторами гормонов. Связывающие агенты лигандов PPAR-дельта рецепторов, представленные в данном изобретении, могут использоваться как агонисты или антагонисты PPAR-дельта рецепторов.
Предпосылки изобретения
Рецепторы, активируемые пролифератором пероксисомы, (PPAR), представляют собой подсемейство в семействе ядерных рецепторов. Были обнаружены и клонированы четыре родственные изоформы, которые известны как PPAR-альфа, PPAR-гамма-1, РРАR-гамма-2 и PPAR-дельта. Каждый подтип рецептора имеет характерный ДНК-связывающий домен (DBD) и лигандсвязывающий домен (LBD), каждый из которых необходим для активируемой лигандами экспрессии генов. PPAR связываются как гетеродимеры с ретиноидным X-рецептором. См. J. Berger and D.E. Miller, Annu. Rev. Med., 2002, 53, 409-435.
PPAR-дельта (известный также как PPAR-бета) экспрессируется в широком спектре тканей млекопитающих, но пока очень мало известно о его биологических функциях и полном наборе генов, которые он регулирует. Тем не менее, недавно было обнаружено, что агонисты могут использоваться для лечения таких заболеваний, как дислипидемия, и некоторых дерматологических заболеваниях, а антагонисты - для лечения остеопороза и колоректального рака (D. Sternbach, in Annual Reports in Medicinal Chemistry, Volume 38, A.M. Doherty, ed., Elsevier Academic Press, 2003 pp. 71-80).
PPAR-дельта, по-видимому, в значительной мере экспрессируется в ЦНС, однако его функции по-прежнему остаются неясными. Исключительный интерес представляет то, что PPAR-дельта обнаружили в олигодендроцитах грызунов, основных клетках ЦНС, вырабатывающих липиды (J. Granneman, et al., J. Neurosci. Res., 1998, 51, 563-573). Более того, было также установлено, что селективный агонист PPAR-дельта значительно повышает экспрессию генов олигодендроглиального миелина и диаметр миелиновой оболочки в культурах клеток мышей (I. Saluja et al., Glia, 2001, 33, 194-204). Таким образом, активаторы PPAR-дельта могут использоваться для лечения демиелинизирующих и дисмиелинизирующих заболеваний.
Демиелинизирующие заболевания проявляются в потере миелиновых и нескольких плотных слоев липидов и белков, которые покрывают многие нервные волокна. Такие слои формируются олигодендроглиями в центральной нервной системе (ЦНС) и шванновскими клетками в периферической нервной системе (ПНС). У пациентов с демиелинизирующей патологией демиелинизация может быть необратимой, и она обычно сопровождается или приводит к аксональному вырождению, а часто и клеточному вырождению. Демиелинизация может происходить в результате повреждения нейронов или повреждения самого миелина - в результате аберрантных иммунных реакций, локальных травм, ишемии, нарушений метаболизма, токсических веществ или вирусных инфекций (Prineas and McDonald, Demyelinating Diseases. In Greenfield'sNeuropathology, 6.sup.th ed. (Edward Arnold: New York, 1997) 813-811, Beers and Berkow, eds., The Merck Manual of Diagnosisand Therapy, 17.sup.th ed. (Whitehouse Station, N.J.: Merck Research Laboratories, 1999) 1299, 1437, 1473-76, 1483).
Центральная демиелинизация (демиелинизация ЦНС) происходит при некоторых заболеваниях часто неопределенной этиологии, которые получили название демиелинизирующих заболеваний. Среди них наиболее распространенным является рассеянный склероз (РС). Другими первичными демиелинизирующими заболеваниями являются адренолейкодистрофия (АЛД), адреномиелоневропатия, вакуолярная ВИЧ-миелопатия, миелопатия, связанная с Т-лимфотропным вирусом человека, семейная леберовская атрофия зрительного нерва, прогрессирующая мультифокальная лейкоэнцефалопатия (ПМЛ), подострый склерозирующий панэнцефалит, синдром Гийена-Барре и тропический спастический парапарез. Кроме того, существуют острые состояния, при которых может происходить демиелинизация в ЦНС, например острый рассеянный энцефаломиелит и острый вирусный энцефалит. Более того, острый поперечный миелит, синдром, при котором острая перерезка спинного мозга неизвестного происхождения повреждает как серое, так и белое вещество мозга в одном или нескольких соседних торакальных сегментах, может также приводить к демиелинизации. Кроме того, нарушения, при которых повреждаются образующие миелин глиальные клетки, также включают травмы спинного мозга, нервные заболевания и повреждение нервов.
Селективные модуляторы PPAR-дельта могут быть также пригодны для лечения или профилактики других заболеваний, см., например, Joel Berger et al., Annu. Rev. Med. 2002, 53, 409-435; Timothy Wilson et al. J. Med. Chem., 2000, Vol. 43, No. 4, 527-550; Steven Kliewer et al., Recent Prog Horm Res. 2001; 56: 239-63; Jean-Charles Fruchart, Bart Staels and Patrick Duriez: PPARS, Metabolic Disease and Arteriosclerosis, Pharmacological Research, Vol. 44, No. 5, 345-52; 2001; Sander Kersten, Beatrice Desvergne & Walter Wahli: Roles of PPARs in health and disease, Nature, vol. 405, 25 may 2000; 421-4; Ines Pineda Torra, Giulia Chinetti, Caroline Duval, Jean-Charles Fruchart and Bart Staels: Peroxisome proliferator-actived receptors: from transcriptional control to clinical practice, Curr Opin Lipidol 12: 2001, 245-254).
Соединения, действующие как модуляторы PPAR-дельта, могут быть особенно подходящими для лечения и/или профилактики нарушений метаболизма жирных кислот и нарушений утилизации глюкозы, которые связаны с резистентностью к инсулину.
Сахарный диабет, особенно диабет 2 типа, в том числе профилактика связанных с ним последствий. Частными аспектами этой связи являются гипергликемия, понижение резистентности к инсулину, повышение толерантности к глюкозе, защита бета-клеток поджелудочной железы, предотвращение макро- и микрососудистых нарушений.
Дислипидемии и их осложнения, такие как, например, атеросклероз, коронарная болезнь сердца, нарушения мозгового кровообращения, особенно (среди прочих) характеризующиеся одним или несколькими из следующих факторов: высокие концентрации триглицеридов плазмы, возникающие после приема пищи, пониженные концентрации холестерина альфа-липопротеинов высокой плотности, пониженные концентрации липопротеина ApoA, повышенные концентрации холестерина альфа-липопротеинов низкой плотности, малые размеры частиц плотного холестерина липопротеинов низкой плотности, повышенные концентрации липопротеина ApoB.
С этим метаболическим синдромом могут быть связаны различные другие состояния, например ожирение (избыточный вес), в том числе центральное ожирение, тромбозы, гиперкоагулируемые и протромботические состояния (артериальные и венозные), повышенное кровяное давление, сердечная недостаточность, в том числе (среди прочих) наступающая в результате инфаркта миокарда, гипертонической болезни сердца или кардиомиопатии.
В числе других нарушений или состояний, при которых могут возникать воспалительные реакции или клеточные дифференцировки, могут быть, например, атеросклероз, такой как, например (среди прочих), коронарный склероз, в том числе стенокардия или инфаркт миокарда, инсульт, сосудистый рестеноз или реокклюзия, хронические воспалительные болезни кишечника, такие как, например, болезнь Крона и язвенный колит, панкреатит, другие воспалительные состояния, ретинопатия, опухоли липоцитов, липоматозные карциномы, такие как, например, липосаркомы, солидные опухоли и неоплазмы, такие как, например (среди прочих), карциномы пищеварительного тракта, печени, желчных протоков и поджелудочной железы, эндокринные опухоли, карциномы легких, почек и мочевыводящих путей, половых путей, карциномы простаты и т.п., острые и хронические миелопролиферативные нарушения и ангиогенез лимфом, нейродегенеративные нарушения, болезнь Альцгеймера, болезнь Паркинсона, эритемато-сквамозные дерматозы, такие как, например, псориаз, обыкновенные угри.
Другие кожные нарушения и дерматологические состояния, модулируемые PPAR-дельта: экземы и нейродермиты, дерматиты, такие как, например, себорейная экзема или фотодерматит, кератит и кератозы, такие как, например, себорейные кератозы, старческие кератозы, актиничный кератоз, фотоиндуцируемые кератозы или кератозные фолликулярные келоиды и их профилактика, бородавки, в том числе кондиломы и остроконечные кондиломы, вирусные инфекции вируса папилломы человека (ВПЧ), такие заболевания, как, например, венерическая папиллома, вирусные бородавки, такие как, например, контагиозный моллюск, лейкоплакия-папулезные дерматозы, такие как, например, плоский лишай, рак кожи, такой как, например, базально-клеточная карцинома, меланомы и кожные лимфомы Т-клеток, локализованные доброкачественные эпидермические опухоли, такие как, например, кератодермия, эпидермические родимые пятна и отморожения.
Различные прочие состояния, потенциально модулируемые PPAR-дельта, в том числе синдром X, синдром поликистоза яичников, астматический остеоартрит, красная волчанка (КВ) или воспалительные ревматические заболевания, такие как, например, ревматоидный артрит, васкулит, истощение (кахексия), подагрическая ишемия/синдром реперфузии и синдром острой дыхательной недостаточности (СОДН).
Краткое описание изобретения
Настоящее изобретение относится к соединению формулы I.
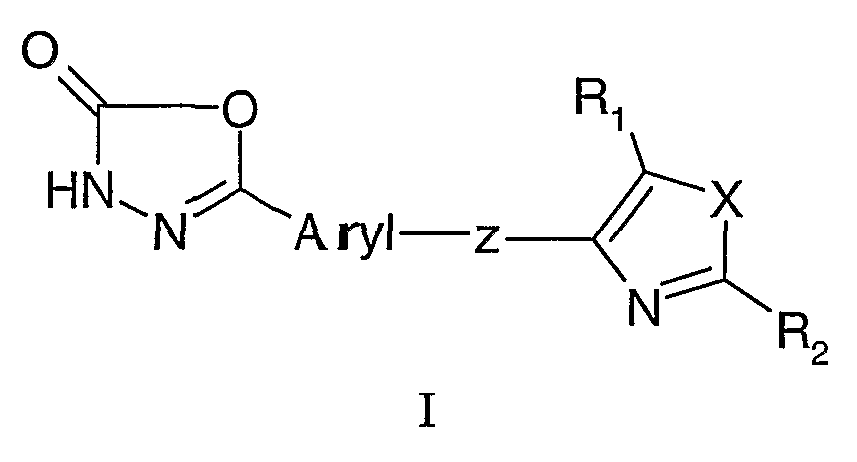
где
ARYL представляет собой фенил или пиридинил, где указанный фенил или пиридинил является необязательно замещенным одним или несколькими заместителями, выбранными из группы, включающей галоген, С1-6алкил, С2-6алкенил, С1-6алкокси, С1-6перфторалкил; С1-6алкилтио, гидрокси, гидроксиС1-6алкил, С1-4ацилокси, нитро, циано, С1-6алкилсульфонил, амино, С1-6алкиламино и С1-6алкоксикарбонил;
Z представляет собой -О(СН2)n-, -SO2(CH2)n-, -(CH2)n-Y-(CH2)n-, -(СH2)n-CO-, -O(CH2)n-CO- или -(CH2)n-Y-(CH2)n-CO-, где Y представляет собой NR3, O или S, и R3 выбирают из группы, включающей H, С1-6алкил, С3-8циклоалкил, С1-6алкилС3-8циклоалкил или бензил, и n представляет собой независимо целое число от 1 до 5;
X представляет собой NR3, O или S, где R3 определен выше;
R1 представляет собой H, галоген, С1-6алкил, С1-6алкокси, С1-6перфторалкил; гидроксиС1-6алкил, нитро, циано и С1-6алкиламино; и
R2 представляет собой замещенный или незамещенный фенил, пиридинил или тиенил, где заместители выбирают из группы, включающей галоген, С1-6алкил, С2-6алкенил, С1-6алкокси, С1-6перфторалкил; С1-6алкилтио, гидрокси, гидроксиС1-6алкил, С1-4ацилокси, нитро, циано, С1-6алкилсульфонил, амино, С1-6алкиламино и С1-6алкоксикарбонил;
при условии, что если Z представляет собой -О(СН2)n- или -SO2(CH2)n-, и ARYL представляет собой фенил, то R2 не является фенилом;
или его стереоизомеру, таутомеру или сольвату, или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к фармацевтическим композициям и способам применения указанных соединений и композиций для модулирования PPAR-дельта у пациентов, нуждающихся в таком модулировании, путем введения соединения, предпочтительно модулирующего активность PPAR-дельта.
Другой аспект настоящего изобретения раскрывает способ лечения заболевания у млекопитающего, где заболевание может модулироваться активностью связывания PPAR-дельта лигандов, путем введения млекопитающему, страдающему данным заболеванием, терапевтически эффективного количества соединения формулы I
где
ARYL представляет собой фенил или пиридинил, где указанный фенил или пиридинил является необязательно замещенным одним или несколькими заместителями, выбранными из группы, включающей галоген, С1-6алкил, С2-6алкенил, С1-6алкокси, С1-6перфторалкил; С1-6алкилтио, гидрокси, гидроксиС1-6алкил, С1-4ацилокси, нитро, циано, С1-6алкилсульфонил, амино, С1-6алкиламино и С1-6алкоксикарбонил;
Z представляет собой -О(СН2)n-, -SO2(CH2)n-, -(CH2)n-Y-(CH2)n-, -(СH2)n-CO-, -O(CH2)n-CO- или -(CH2)n-Y-(CH2)n-CO-, где Y представляет собой NR3, O или S, и R3 выбирают из группы, включающей H, С1-6алкил, С3-8циклоалкил, С1-6алкилС3-8циклоалкил или бензил, и n представляет собой независимо целое число от 1 до 5;
X представляет собой NR3, O или S, где R3 определен выше;
R1 представляет собой H, галоген, С1-6алкил, С1-6алкокси, С1-6перфторалкил; гидроксиС1-6алкил, нитро, циано и С1-6алкиламино; и
R2 представляет собой замещенный или незамещенный фенил, пиридинил или тиенил, где заместители выбирают из группы, включающей галоген, С1-6алкил, С2-6алкенил, С1-6алкокси, С1-6перфторалкил; С1-6алкилтио, гидрокси, гидроксиС1-6алкил, С1-4ацилокси, нитро, циано, С1-6алкилсульфонил, амино, С1-6алкиламино и С1-6алкоксикарбонил; или его стереоизомера, таутомера или сольвата, или его фармацевтически приемлемой соли.
Подробное описание изобретения
Используемые в данной заявке термины имеют следующие значения:
Выражение «C1-6алкил» включает метильную и этильную группы, а также линейные или разветвленные пропильную, бутильную, пентильную и гексильную группы. Конкретными алкильными группами являются метил, этил, н-пропил, изопропил и трет-бутил. Производные этих обозначений, такие как «С1-6алкокси», «С1-6алкоксиС1-6алкил», «гидроксиС1-6алкил», «С1-6алкилкарбонил», «С1-6алкоксикарбонилС1-6алкил», «С1-6алкоксикарбонил», «аминоС1-6алкил», «С1-6алкилкарбамоилС1-6алкил», «С1-6диалкилкарбамоилС1-6алкил», «моно- или ди-С1-6алкиламиноС1-6алкил», «аминоС1-6алкилкарбонил», «дифенилС1-6алкил», «арилС1-6алкил», «арилкарбонилС1-6алкил» и «арилоксиС1-6алкил», следует толковать соответствующим образом.
Выражение «С2-6алкенил» включает этенильную, а также линейные или разветвленные пропенильную, бутенильную, пентенильную и гексенильную группы. Аналогично, выражение «С2-6алкинил» включает этинильную и пропинильную, а также линейные и разветвленные бутинильную, пентинильную и гексинильную группы.
Термин «С1-4ацилокси» обозначает ацильный радикал, присоединенный к атому кислорода, некоторые примеры включают, но не ограничиваются ими, ацетилокси, пропионилокси, бутаноилокси, изобутаноилокси, втор-бутаноилокси, трет-бутаноилокси и тому подобное.
Термин «арил» обозначает карбоциклическую ароматическую кольцевую систему, такую как фенил, бифенил, нафтил, антраценил, фенантренил, флуоренил, инденил, пенталенил, азуленил, бифениленил и тому подобное. К арилам также относятся частично гидрогенизированные производные карбоциклических ароматических систем, перечисленных выше. Неограничивающими примерами таких частично гидрогенизированных производных являются 1,2,3,4-тетрагидронафтил, 1,4-дигидронафтил и тому подобное.
Термин «арилокси» обозначает группу -О-арил, где арил определен выше.
Термин «гетероарил» (сам по себе или в составе любого значения, например, «гетероарилокси» или «гетероарилалкил») представляет собой 5-10-членную ароматическую кольцевую систему, в которой одно или несколько колец содержат один или несколько гетероатомов, выбранных из группы, включающей N, O или S; таких как, но не ограничивающихся ими, пиррол, пиразол, фуран, тиофен, хинолин, изохинолин, хиназолинил, пиридин, пиримидин, оксазол, тиазол, тиадиазол, тетразол, триазол, имидазол или бензимидазол.
Термин «гетероциклический или гетероциклил» (сам по себе или в составе любого другого значения, например, «гетероциклоалкил») обозначает насыщенную или частично ненасыщенную 4-10-членную кольцевую систему, в которой одно или несколько колец содержат один или несколько гетероатомов, выбранных из группы, включающей N, O или S; таких как, но не ограничивающихся ими, пирролидин, пиперидин, пиперазин, морфолин, тетрагидропиран или имидазолидин.
Термин «С1-6перфторалкил» означает, что все атомы водорода в указанной алкильной группе замещены атомами фтора. Наглядными примерами являются трифторметильная и пентафторэтильная и линейные или разветвленные гептафторпропильная, нонафторбутильная, ундекафторпентильная и тридекафторгексильная группы. Производное выражение «С1-6перфторалкокси» следует толковать соответствующим образом.
Выражение «С3-8циклоалкил» обозначает циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Выражение «С3-8циклоалкилС1-6алкил» означает, что С3-8циклоалкил, определяемый в настоящей заявке, присоединен к С1-6алкилу, как определено в данном описании. Характерными примерами являются циклопропилметил, 1-циклобутилэтил, 2-циклопентилпропил, циклогексилметил, 2-циклогептилэтил и 2-циклооктилбутил и им подобные.
Термин «галоген» или «гало» означает хлор, фтор, бром или йод.
Термин «С1-6алкилсульфонил» в данном контексте обозначает группу -S(=O)2C1-6алкил, где С1-6алкил определен выше. Характерные примеры включают, но не ограничиваются ими, метилсульфонил, этилсульфонил, н-пропилсульфонил, изопропилсульфонил, бутилсульфонил, изобутилсульфонил, втор-бутилсульфонил, трет-бутилсульфонил, н-пентилсульфонил, изопентилсульфонил, неопентилсульфонил, трет-пентилсульфонил, н-гексилсульфонил, изогексилсульфонил и им подобные.
Термин «арилсульфонил» обозначает группу -S(=O)2арил, где арил определен выше.
Термин «гетероарилсульфонил» обозначает группу -S(=O)2гетероарил, где гетероарил определен выше.
Выражение «стереоизомеры» является общим термином, используемым для всех изомеров индивидуальных молекул, которые отличаются только пространственной ориентацией своих атомов. Они, как правило, включают зеркальные изомеры, которые обычно существуют при наличии по меньшей мере одного центра асимметрии (энантиомеры). Если соединения настоящего изобретения обладают двумя или более центрами асимметрии, они могут также существовать в форме диастереоизомеров, кроме того, некоторые индивидуальные молекулы могут существовать в форме геометрических изомеров (цис/транс). Следует понимать, что все такие изомеры и их смеси в любой пропорции также входят в объем настоящего изобретения.
«Замещенный» означает замещенный одним или двумя заместителями, независимо выбранными из группы, включающей С1-6алкил, С1-6перфторалкил, гидрокси, -СО2Н, сложный эфир, амид, С1-6алкокси, С1-6перфторалкокси, -NH2, Cl, Br, I, F, -NH-низший алкил и -N(низший алкил)2.
Соединения и соли настоящего изобретения могут существовать в нескольких таутомерных формах, включая форму енолов и иминов, а также в форме кетонов, енаминов, геометрических изомеров и их смесей. Все такие таутомерные формы также включены в объем настоящего изобретения. Таутомеры существуют в виде смеси набора таутомеров в растворе. В твердом состоянии, как правило, преобладает один из таутомеров. Даже если описан один таутомер, настоящее изобретение включает все таутомеры представленных соединений.
В данной заявке термин «модулятор» обозначает химическое соединение, обладающее способностью усиливать (то есть обладающее «агонистическим» действием) или подавлять (то есть обладающее «антагонистическим» действием) какое-либо функциональное свойство биологической активности или процесс (например, активность фермента или связывание рецептора); такое усиление или подавление может проявляться при выполнении определенных условий, таких как активация или подавление канала передачи сигнала, и/или может проявляться только в клетках определенных типов и приводить к измеряемым биологическим изменениям.
Термин «пациент» означает теплокровное животное, такое как, например, крысы, мыши, собаки, кошки, морские свинки и приматы, такие как человек.
Выражение «фармацевтически приемлемый носитель» обозначает нетоксичный растворитель, диспергирующий агент, эксципиент, вспомогательное или другое вещество, которое смешивают с соединением настоящего изобретения для образования фармацевтической композиции, т.е. лекарственной формы, которую можно вводить пациенту. Одним из примеров такого носителя является фармацевтически приемлемое масло, обычно используемое для парентерального введения.
Термин «фармацевтически приемлемые соли» означает, что соли соединений настоящего изобретения могут использоваться в лекарственных препаратах. Вместе с тем и другие соли могут быть полезны при получении соединений в соответствии с настоящим изобретением или их фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли соединений настоящего изобретения включают кислотно-аддитивные соли, которые могут быть получены, например, смешиванием раствора соединения настоящего изобретения с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, бромистоводородная кислота, серная кислота, метансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, п-толуолсульфоновая кислота, фумаровая кислота, малеиновая кислота, гидроксималеиновая кислота, яблочная кислота, аскорбиновая кислота, янтарная кислота, глутаровая кислота, уксусная кислота, салициловая кислота, коричная кислота, 2-феноксибензойная кислота, гидроксибензойная кислота, фенилуксусная кислота, бензойная кислота, щавелевая кислота, лимонная кислота, винная кислота, гликолевая кислота, молочная кислота, пировиноградная кислота, малоновая кислота, угольная кислота или фосфорная кислота. Также могут быть получены кислые соли металлов, такие как моногидроортофосфат натрия и гидросульфат калия. Кроме того, полученные таким образом соли могут представлять собой моно- или дизамещенные кислые соли и могут существовать в форме гидратов или быть в значительной степени обезвоженными. Более того, если соединения настоящего изобретения включают кислотную группу, то подходящие фармацевтически приемлемые их соли могут включать соли щелочных металлов, например соли натрия или калия, соли щелочноземельных металлов, например соли кальция или магния, и соли, образованные подходящими органическими лигандами, например четвертичные аммониевые соли.
Термин «терапевтически эффективное количество», используемый в данной заявке, означает количество соединения, которое эффективно для лечении данного заболевания или состояния.
В настоящем изобретении предлагаются также фармацевтические композиции, содержащие одно или несколько соединений настоящего изобретения и фармацевтически приемлемый носитель. Предпочтительно такие композиции представлены в стандартных лекарственных формах, таких как таблетки, пилюли, капсулы, порошки, гранулы, стерильные парентеральные растворы или суспензии, дозируемые аэрозоли и жидкие распыляемые растворы, капли, ампулы, автоинжекторные устройства или суппозитории, предназначенные для перорального, парентерального, интраназального, сублингвального или ректального введения, или введения путем ингаляции или инсуффляции. Альтернативно, эти композиции могут быть представлены в форме, подходящей для применения один раз в неделю или один раз в месяц; например, нерастворимые соли активного соединения, такие как деканоатные соли, могут быть приспособлены для приготовления депо-препарата для внутримышечных инъекций. Возможно использование разрушающегося полимера, содержащего активный ингредиент. Для приготовления твердых композиций, таких как таблетки, основное действующее вещество смешивают с фармацевтическим носителем, например с обычными для таблеток ингредиентами, такими как кукурузный крахмал, лактоза, сахароза, сорбит, тальк, стеариновая кислота, стеарат магния, фосфат дикальция или смолы, а также другие фармацевтические разбавители, например вода, чтобы образовать твердый предварительный состав, содержащий гомогенную смесь соединения данного изобретения или его фармацевтически приемлемой соли. Когда такие предварительные составы называют гомогенными, подразумевается, что активный ингредиент перемешан равномерно во всем составе, и состав можно разделить на обладающие равной эффективностью стандартные лекарственные формы, такие как таблетки, пилюли и капсулы. Затем этот твердый предварительный состав делят на стандартные лекарственные формы описанного выше типа, содержащие от 0,1 до приблизительно 500 мг активного ингредиента настоящего изобретения. Ароматизированные лекарственные формы содержат от 1 до 100 мг, например, 1, 2, 5, 10, 25, 50 или 100 мг активного ингредиента. Таблетки или пилюли этих новых составов могут иметь оболочку или иное покрытие, чтобы обеспечить пролонгированное действие лекарственной формы. Например, таблетка или пилюля может содержать внутренний и внешний дозированные компоненты, когда первый находится внутри последнего. Два компонента могут быть разделены энтеральным слоем, который предотвращает разрушение в желудке и позволяет внутреннему компоненту в неразрушенном состоянии попасть в двенадцатиперстную кишку или чтобы его высвобождение произошло с задержкой. В качестве таких энтеральных слоев или покрытий могут быть использованы разнообразные вещества, включая ряд полимерных кислот и смесей полимерных кислот с такими веществами, как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы, в которых новые композиции настоящего изобретения могут быть введены перорально или путем инъекции, включают водные растворы, сиропы с подходящим ароматизатором, водные или масляные суспензии и ароматизированные эмульсии со съедобным маслом, таким как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и аналогичные фармацевтические носители. Подходящие диспергирующие или суспендирующие вещества для водных суспензий включают синтетические и натуральные смолы, такие как трагакант, камедь, альгинат, декстран, натрийкарбоксиметилцеллюлоза, метилцеллюлоза, поливинилпирролидон или желатин.
При лечении различных патологических состояний, описанных в настоящей заявке, подходящие дозы составляют примерно от 0,01 до 250 мг/кг в день, предпочтительно примерно от 0,05 до 100 мг/кг в день, а в особенности, примерно от 0,05 до 20 мг/кг в день. Соединения могут быть введены по схеме 1-4 раза в день.
В приведенных ниже примерах и получениях используются термины, имеющие указанные ниже значения: «кг» означает килограммы, «г» означает граммы, «мг» означает миллиграммы, «мкг» означает микрограммы, «пг» означает пикограммы, «моль» означает моли, «ммоль» означает миллимоли, «нмоль» означает наномоли, «л» означает литры, «мл» означает миллилитры, «мкл» означает микролитры, «°C» означает градусы Цельсия, «Rf» означает время удерживания, «тпл» или «т.пл.» означает температуру плавления, «разл.» означает разложение, «ткип» или «т.кип.» означает температуру кипения, «мм рт.ст.» означает миллиметры ртутного столба, «см» означает сантиметры, «нм» означает нанометры, «[α]20D» означает удельное вращение для D-линии натрия при 20°C в 1-дециметровой ячейке, «c» означает концентрацию в г/мл, «ТГФ» означает тетрагидрофуран, «ДМФА» означает диметилформамид, «NMP» означает 1-метил-2-пирролидинон, «насыщенный раствор соли» означает насыщенный водный раствор хлорида натрия, «М» означает концентрацию в молях, «мМ» означает концентрацию в миллимолях, «мкМ» означает концентрацию в микромолях, «нМ» означает концентрацию в наномолях, «ТСХ» означает тонкослойную хроматографию, «ВЭЖХ» означает высокоэффективную жидкостную хроматографию, «HRMS» означает масс-спектрометрию высокого разрешения, «CIMS» означает масс-спектрометрию с химической ионизацией, «ESI» означает масс-спектрометрию с ионизацией электрораспылением, «tR» означает время удерживания, «фт» означает фунты, «гал.» означает галлоны, «П.П.В.» означает потери при высушивании, «мкКи» означает микрокюри, «в/б» означает внутрибрюшинно, «в/в» означает внутривенно.
В одном из аспектов настоящего изобретения описываются новые соединения общей структуры, представленной формулой I:
где
ARYL представляет собой фенил или пиридинил, где указанный фенил или пиридинил является необязательно замещенным одним или несколькими заместителями, выбранными из группы, включающей галоген, С1-6алкил, С2-6алкенил, С1-6алкокси, С1-6перфторалкил; С1-6алкилтио, гидрокси, гидроксиС1-6алкил, С1-4ацилокси, нитро, циано, С1-6алкилсульфонил, амино, С1-6алкиламино и С1-6алкоксикарбонил;
Z представляет собой -О(СН2)n-, -SO2(CH2)n-, -(CH2)n-Y-(CH2)n-, -(СH2)n-CO-, -O(CH2)n-CO- или -(CH2)n-Y-(CH2)n-CO-, где Y представляет собой NR3, O или S, и R3 выбирают из группы, включающей H, С1-6алкил, С3-8циклоалкил, С1-6алкилС3-8циклоалкил или бензил, и n представляет собой независимо целое число от 1 до 5;
X представляет собой NR3, O или S, где R3 определен выше;
R1 представляет собой H, галоген, С1-6алкил, С1-6алкокси, С1-6перфторалкил; гидроксиС1-6алкил, нитро, циано и С1-6алкиламино; и
R2 представляет собой замещенный или незамещенный фенил, пиридинил или тиенил, где заместители выбирают из группы, включающей галоген, С1-6алкил, С2-6алкенил, С1-6алкокси, С1-6перфторалкил; С1-6алкилтио, гидрокси, гидроксиС1-6алкил, С1-4ацилокси, нитро, циано, С1-6алкилсульфонил, амино, С1-6алкиламино и С1-6алкоксикарбонил;
при условии, что если Z представляет собой -О(СН2)n- или -SO2(CH2)n- и ARYL представляет собой фенил, то R2 не является фенилом;
или их стереоизомеры, таутомеры или сольваты, или их фармацевтически приемлемые соли.
В одном из дополнительных аспектов данного осуществления изобретения описано соединение, в котором ARYL представляет собой фенил и X представляет собой O или S.
В другом аспекте данного осуществления описано соединение, в котором X представляет собой O.
Примером соединения данного осуществления является 5-(4-{2-[5-метил-2-(4-трифторметилфенил)тиазол-4-ил]этокси}фенил)-3Н-[1,3,4]оксадиазол-2-он.
В еще одном осуществлении настоящего изобретения описывается фармацевтическая композиция, содержащая эффективное количество соединения формулы I и фармацевтически приемлемый носитель.
В другом аспекте настоящего изобретения раскрывается способ лечения заболевания у млекопитающего, где заболевание может модулироваться активностью связывания PPAR-дельта лигандов, который включает введение млекопитающему, страдающему данным заболеванием, терапевтически эффективного количества соединения формулы I
где
ARYL представляет собой фенил или пиридинил, где указанный фенил или пиридинил является необязательно замещенным одним или несколькими заместителями, выбранными из группы, включающей галоген, С1-6алкил, С2-6алкенил, С1-6алкокси, С1-6перфторалкил; С1-6алкилтио, гидрокси, гидроксиС1-6алкил, С1-4ацилокси, нитро, циано, С1-6алкилсульфонил, амино, С1-6алкиламино и С1-6алкоксикарбонил;
Z представляет собой -О(СН2)n-, -SO2(CH2)n-, -(CH2)n-Y-(CH2)n-, -(СH2)n-CO-, -O(CH2)n-CO- или -(CH2)n-Y-(CH2)n-CO-, где Y представляет собой NR3, O или S, и R3 выбирают из группы, включающей H, С1-6алкил, С3-8циклоалкил, С1-6алкилС3-8циклоалкил или бензил, и n представляет собой независимо целое число от 1 до 5;
X представляет собой NR3, O или S, где R3 определен выше;
R1 представляет собой H, галоген, С1-6алкил, С1-6алкокси, С1-6перфторалкил; гидроксиС1-6алкил, нитро, циано и С1-6алкиламино; и
R2 представляет собой замещенный или незамещенный фенил, пиридинил или тиенил, где заместители выбирают из группы, включающей галоген, С1-6алкил, С2-6алкенил, С1-6алкокси, С1-6перфторалкил; С1-6алкилтио, гидрокси, гидроксиС1-6алкил, С1-4ацилокси, нитро, циано, С1-6алкилсульфонил, амино, С1-6алкиламино и С1-6алкоксикарбонил; или его стереоизомера, таутомера или сольвата, или его фармацевтически приемлемой соли.
В одном из последующих аспектов данного осуществления способа изобретения описано соединение, в котором ARYL представляет собой фенил.
В другом аспекте данного осуществления способа изобретения описано соединение, в котором ARYL представляет собой фенил и R2 представляет собой фенил.
Еще в одном аспекте данного осуществления способа изобретения описано соединение, в котором ARYL представляет собой фенил, Z представляет собой
-О(СН2)n- и R2 представляет собой фенил.
Еще в одном аспекте данного осуществления способа изобретения описано соединение, в котором ARYL представляет собой фенил, Z представляет собой
-О(СН2)n-, X представляет собой O или S и R2 представляет собой фенил.
В другом аспекте данного осуществления способа изобретения описано соединение, в котором ARYL представляет собой фенил, Z представляет собой -О(СН2)n-, X представляет собой O или S, R1 представляет собой С1-6алкил и R2 представляет собой фенил.
В одном из дополнительных аспектов данного осуществления способа изобретения описано соединение, в котором X представляет собой O.
Еще в одном из аспектов настоящего изобретения, данного способа изобретения описано соединение, в котором X представляет собой S.
В одном из последующих аспектов осуществления описан способ, где названным заболеванием является демиелинизирующее заболевание, выбранное из группы, включающей рассеянный склероз, болезнь Шарко-Мари-Тута, болезнь Пелицеуса-Мерцбахера, энцефаломиелит, миелоневрит зрительного нерва, адренолейкодистрофию, синдром Гийена-Барре и нарушения, при которых повреждаются производящие миелин глиальные клетки, включая травмы спинного мозга, невропатии и повреждения нервов.
В еще одном аспекте данного осуществления изобретения описан способ, где демиелинизирующим заболеванием является рассеянный склероз.
В еще одном аспекте данного изобретения описан способ, где указанное заболевание выбрано из группы, включающей ожирение, гипертриглицеридемию, гиперлипидемию, гипоальфалипопротеинемию, гиперхолестеринемию, дислипидемию, синдром X, сахарный диабет II типа и их осложнения, выбранные из группы, включающей невропатию, нефропатию, ретинопатию и катаракты, гиперинсулинемию, нарушение толерантности к глюкозе, резистентность к инсулину, атеросклероз, повышенное артериальное давление, коронарную болезнь сердца, болезнь периферических сосудов или застойную сердечную недостаточность.
Описанные здесь соединения могут быть синтезированы в соответствии со следующими способами по схемам, где заместители ARYL, X, Z и R являются такими, как определено для формулы (I) выше, если не указано иное. Если необходимо, в приведенных ниже схемах синтеза химически активные функциональные группы, присутствующие в соединениях, описанных в данном изобретении, могут быть защищены подходящими защитными группами. Защитную группу можно удалить на одной из последующих стадий синтеза. Методику проведения защиты химически активных функциональных групп и их последующего удаления можно найти в T.W. Greene and P.G.M. Wuts, Protective Groups un Organic Synthesis, Wiley and Sons, 1991.
На схеме А показан синтез соответствующих имидазольных, оксазольных или тиазольных промежуточных соединений для соединений формулы I, где X представляет собой O, S или NR3. Гетероциклические соединения могут быть получены с использованием способов, известных из химической литературы (в качестве обзоров см. Katritzky, A.R.; Rees, C.W. Eds. Comprehensive Heterocyclic Chemistry, Vol. 5; Pergamon Press (1984); Katritzky, A.R.; Rees, C.W.; Scriven, E.F.V. Eds. Comprehensive Heterocyclic Chemistry II; Vols 3 & 4, Pergamon Press (1996)). В частности, указанные оксазолы, имидазолы и тиазолы можно получить конденсацией подходящего α-галогенкетона 1 соответственно с амидом, амидином или тиоамидом (общей формулы 2), при температуре примерно от 40 до 150°С с получением промежуточных гетероциклов 3.
Схема А

На схеме В показан общий синтез соединений формулы I, где Z представляет собой -О(СН2)n-. Соответственно, на стадии В1 соответствующий замещенный сложный эфир карбоновой кислоты 4, который может быть синтезирован, как показано на схеме А, восстанавливают до спирта 5 способами, хорошо известными в данной области. Например, процесс восстановления можно проводить гидридами алюминия, такими как алюмогидрид лития или гидрид диизобутилалюминия в инертном растворителе. На стадии В2 функциональную группу спирта в соединении 5 преобразуют в уходящую группу с образованием соединения 6, где Lg является уходящей группой, такой как галоген или сложный эфир сульфокислоты, например мезилат или тозилат. Преобразование в уходящую группу можно провести путем взаимодействия спирта с такими реагентами, как N-бромсукцинимид, в присутствии трифенилфосфина с образованием соединения, в котором уходящая группа представляет собой бромид, или путем взаимодействия с тионилхлоридом с получением соединения, в котором уходящая группа представляет собой хлорид. Если требуется сложный эфир сульфокислоты, его можно получить путем взаимодействия соединения 5 с соответствующим сульфонилхлоридом в присутствии подходящего основания. Например, взаимодействие соединения 5 с метансульфонилхлоридом в присутствии органического основания, такого как триэтиламин или пиридин, в инертном растворителе приводит к соединению 6, в котором уходящей группой является OSO2CH3.
На стадии В3 соответствующий замещенный гидроксиариловый сложный эфир 7 взаимодействует с гетероциклом 6, замещая уходящую группу, с образованием связанного сложного эфира 8. Реакцию замещения проводят в условиях, хорошо известных специалистам. Как правило, эту реакцию проводят в присутствии основания, такого как гидрид натрия, или другого неорганического основания, такого как карбонат или гидроксид щелочного металла, в инертном растворителе. Температура реакции, хоть она и не является критическим фактором, должна составлять от 0°С до температуры флегмы инертного растворителя.
Затем соединение 8 на стадии В4 обрабатывают гидразином, либо чистым, либо в подходящем органическом растворителе при повышенной температуре с образованием гидразида кислоты 9. Как правило, эту реакцию проводят при температуре от 50°С до температуры флегмы органического растворителя.
Циклизацию гидразида кислоты 9 на стадии В5 до целевых 1,3,4-оксадиазол-2-онов 10 выполняют путем обработки соединения 9 хлорформиатом в присутствии органического основания, такого как пиридин, с последующей обработкой сильным основанием со стерически затрудненной аминогруппой, таким как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), в подходящем органическом растворителе, таком как ацетонитрил, в запаянной колбе при повышенной температуре. Как правило, реакцию проводят при температуре от 100 до 200°С. 1,3,4-Оксадиазол-2-оны можно также синтезировать путем взаимодействия соединения 9 с фосгеном. См. Stempel, A., et al., J. Org. Chem. 1995, 20, 412.
На стадии В6 проводится альтернативный синтез связанного сложного эфира 8. Соответственно, спирт 5 может взаимодействовать с гидроксиариловым сложным эфиром 8 в присутствии триарил- или триалкилфосфина, такого как трифенилфосфин или три-н-бутилфосфин, и диэтилазодикарбоксилатом в инертном растворителе, например, ТГФ или дихлорметане, с образованием связанного сложного эфира 8. Как правило, эту реакцию проводят при температуре от комнатной до температуры флегмы инертного растворителя.
Схема В

Схема С иллюстрирует синтез соединения формулы I, в котором Z представляет собой -(CH2)n-Y-(CH2)n-. Эта схема наиболее удобна для синтеза соединений, в которых n в алкиленовой цепи, присоединенной к ARIL, равно 1 или 2. На стадии С1 соединение 5 (Y = O) преобразуют в соединение 6 (в котором Lg представляет собой хлор или бром), как описано на схеме В, стадия В2. Затем соединение 6 взаимодействует с тиомочевиной, соединение 11, в условиях, аналогичных описанным в Treau, M. et al. Heterocycles, 2001, 55 (9), 1727-1735, с образованием тиола 5а.
При взаимодействии соединения 6 с первичным амином 12 образуется аминоалкильный гетероцикл 5b. Такое замещение уходящей группы амином хорошо известно специалистам в данной области. Как правило, реакцию замещения проводят в полярном органическом растворителе в присутствии органического основания, которое используют для нейтрализации кислоты. Хотя это и не является совершенно необходимым условием, реакцию замещения проводят при температуре от комнатной до температуры флегмы растворителя.
На стадии С3 соединения 5, 5а и 5b могут взаимодействовать с соединением 13 с образованием связанного арилового сложного эфира 14, в котором Y представляет собой O, S или NR3. Таким образом, при взаимодействии соединений 5 (Y = O) и 5а (Y = S) с соединением 13 для замещения уходящей группы, такое взаимодействие, как правило, проводят в присутствии сильного основания, например гидрида натрия, в полярном апротонном растворителе, таком как ДМФА или ДМСО, при температурах примерно от 0 до 150°С. Если соединение 5b (Y = NR3) взаимодействует с соединением 14, используют условия, идентичные описанным выше на стадии С2 для первичного амина.
Синтез требуемых 1,3,4-оксадиазол-2-онов 16 из соединения 14 проводят в две стадии (С4 и С5) точно так, как описано на схеме В, стадии В4 и В5.
Схема С

На схеме D показан альтернативный подход к получению соединений формулы I, в которых Z представляет собой -(СН2)n-Y-(CH2)n-. Такая схема наиболее удобна для синтеза соединений, в которых n в алкиленовой цепи, присоединенной к ARIL, равно от 3 до 5.
На стадии D1 соединение с концевым альдегидом 17, которое может быть синтезировано способом, показанным на схеме А, преобразуют в процессе двух стадий в коневой ацетилен 19. Таким образом, взаимодействие соединения 17 с бромметилентрифенилфосфораном (первая стадия) с калий-t-BuOK дает промежуточный бромолефин (не показан), который затем обрабатывают двумя эквивалентами t-BuOK (вторая стадия) с образованием ацетилена 19. Данная последовательность реакций преобразования описана в Pianetti, P., Tet. Letters, 1986, 48, 5853-5856. См. также Corey, E.J., et al. J. Am. Chem. Soc., 1969, 91, 4318-4320. Альтернативно, как показано на стадии D2, промежуточные соединения типа 19 могут быть получены замещением уходящей группы из такого промежуточного соединения, как соединение 6 (см. схему С) с помощью нуклеофила, такого как соединение 18, в который включен концевой ацетилен.
На стадии D3 сочетание Соногаширы ацетиленового промежуточного соединения 19 с арилйодидом 20 проводят в присутствии тетракистрифенилфосфинпалладия(0), йодида меди(I) и подходящего органического основания в инертном растворителе с образованием связанного концевого ацетилена 21. Восстановление ацетилена 21 можно затем проводить на стадии D4 каталитическим гидрированием соединения 21 с образованием насыщенного сложного эфира 14. Как правило, восстановление может проходить под действием катализаторов, таких как палладий на углероде или хлортрис(трифенилфосфин)родий(I), в инертном органическом растворителе с водородом при давлении от 30 до 300 фунтов на кв. дюйм. Восстановление можно проводить при температуре от комнатной до 175°С.
Синтез требуемых 1,3,4-оксадиазол-2-онов 16 из соединения 14 проводят в две стадии (D5 и D6) точно так, как описано на схеме В, стадии В4 и В5.
Схема D
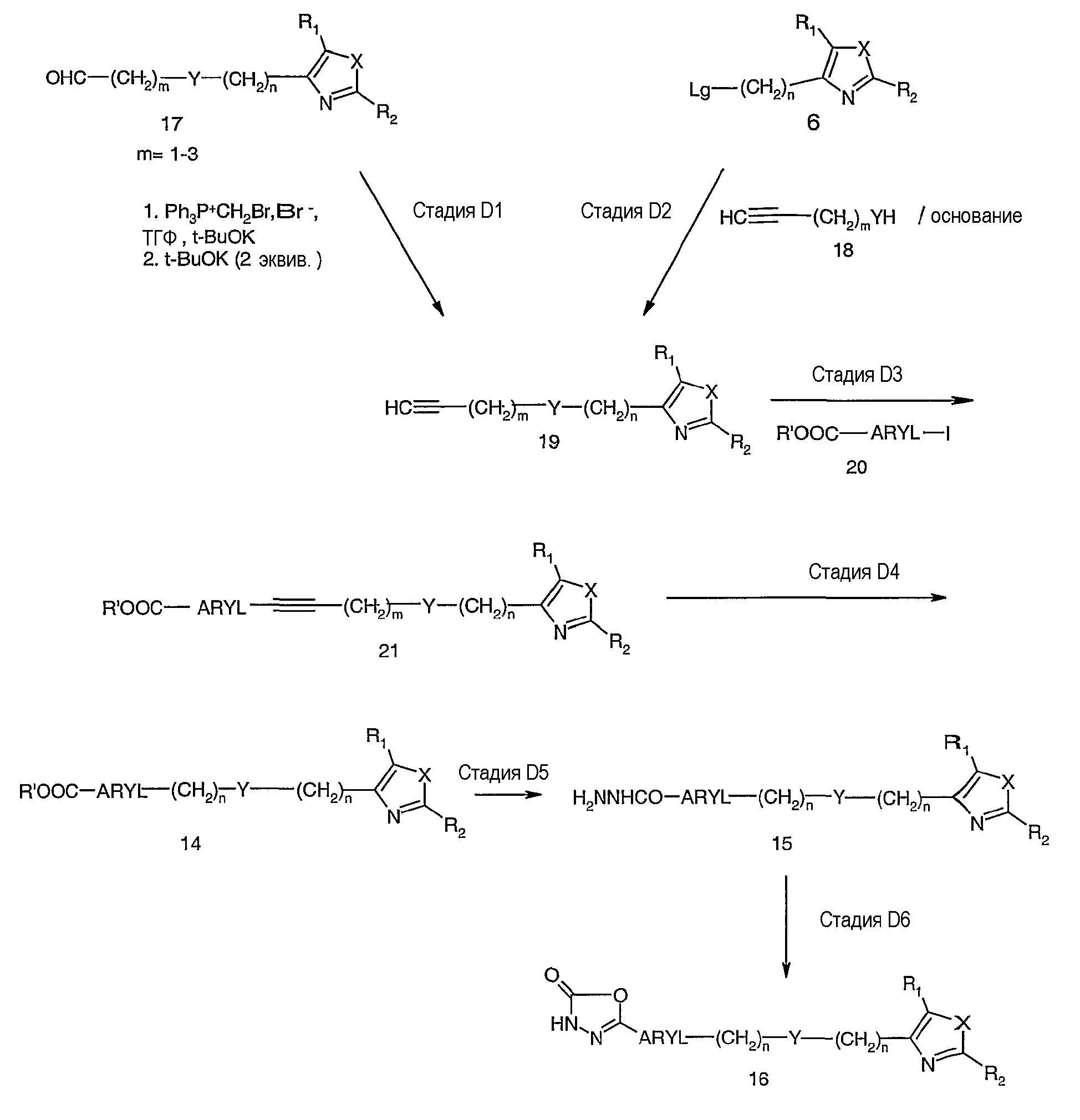
Схема Е иллюстрирует частный случай синтеза соединений формулы I, где Z представляет собой -(СН2)nNR3(CH2)n-. В этом подходе линкер Z конструируют восстановительным аминированием альдегида амином. Например, на стадии Е1 обрабатывают соединение 5b (где Y = NR3) альдегидом, таким как метиловый эфир 4-формилбензойной кислоты (n=1), соединение 22, в полярном растворителе, как правило, спирте или смеси спирта и ТГФ, с последующей обработкой восстановителем, таким как триацетоксиборгидрид натрия, получая требуемое промежуточное соединение 14а (n=1).
Аналогично, на стадии Е2 обработка альдегида, например соединения 17а, амином, таким как метиловый эфир 4-аминоалкилбензойной кислоты (n=1), соединение 23, приводит к соединению 14а, где n равно 1, и R3 в -(СН2)nNR3 представляет собой H. Соединение 14а на стадиях Е3 и Е4 преобразуют в 1,3,4-оксадиазол-2-оны 16а, как показано на схеме В, стадии В4 и В5.
В более общем случае соответствующие амины (R'OOC-ARYL-(CH2)nNHR3) получают из соответствующих нитрилов или нитросоединений каталитическим гидрированием, или из ацетиленаминов и арилйодидов или бромидов сочетанием Соногаширы с последующим каталитическим гидрированием, как описано для схемы D.
Схема E
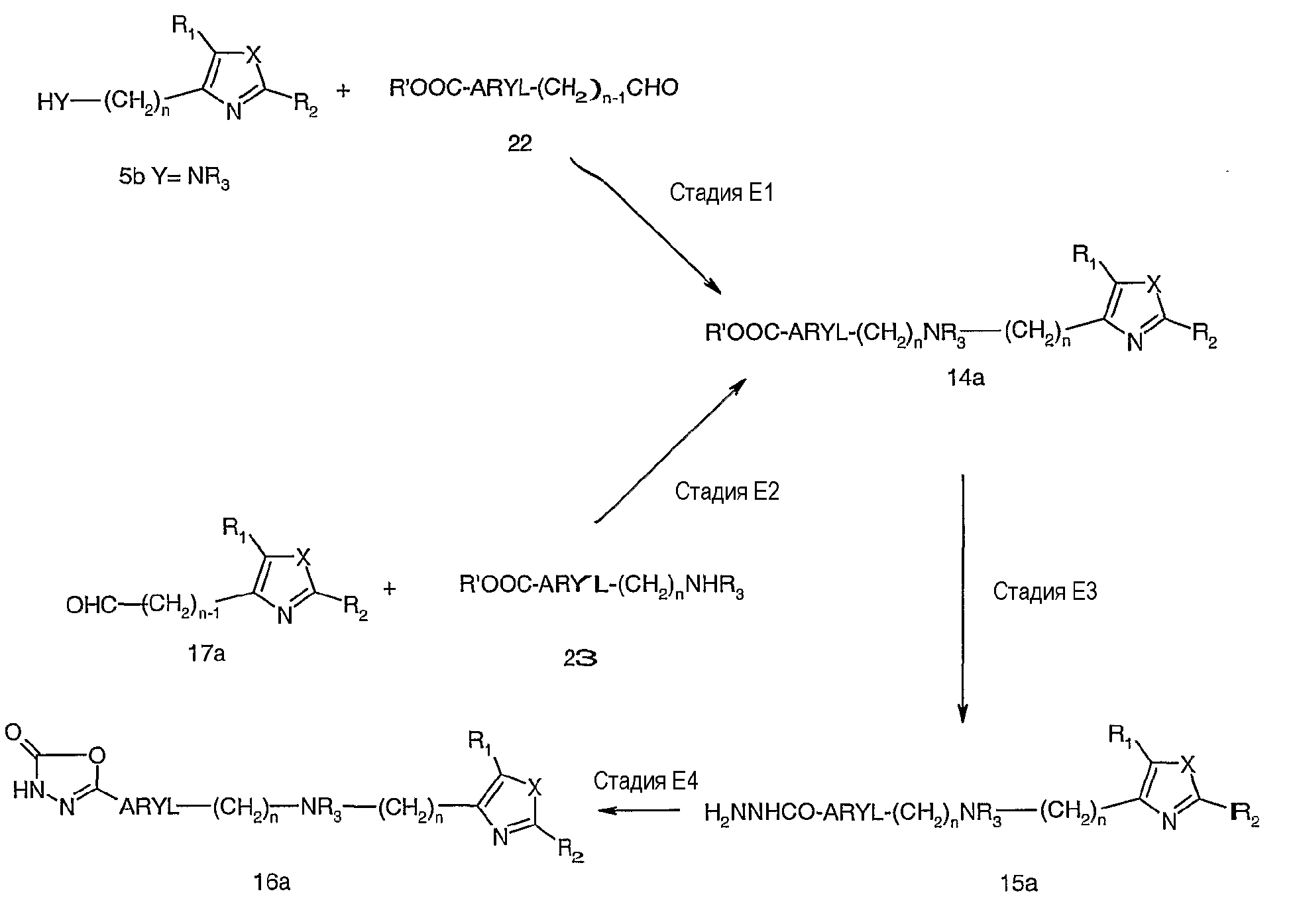
Схема F иллюстрирует синтез соединений формулы I, в которых Z представляет собой -SO2(CH2)n-. На стадии F1 обработка арилсульфонилхлорида 24 водным сульфитом натрия дает сульфиновую кислоту 25. Взаимодействие соединения 25, как показано на стадии F2, с таким промежуточным соединением, как соединение 6, в полярном растворителе, таком как ДМФА, ацетонитрил или этанол, в присутствии основания, такого как DBU, пиридин, метоксид натрия или гидроксид натрия, дает промежуточное соединение 26. Промежуточное соединение 26 преобразуют в соответствующий 1,3,4-оксадиазол-2-он 28 на стадиях F3 и F4, как показано на схеме В, стадии В4 и В5.
Схема F

Схема G иллюстрирует синтез соединения формулы I, в котором Z представляет собой -O(CH2)nCO-. В показанном на этой схеме случае n равно 1. Исходный 2-ацильный гетероцикл 29 может быть синтезирован из соответствующей карбоновой кислоты (полученной способом, показанным на схеме А) добавлением соответствующего реактива Гриньяра к промежуточному N-метокси-N-метилкарбоксамиду (Khlestkin, V.K. et al.; Current Organic Chemistry, 2003, 7(10), 967-993; and Singh, J. et al., Journal für Praktische Chemie, 2000, 342, 340-347). Получение промежуточного N-метокси-N-метилкарбоксамида чаще всего проводят путем взаимодействия кислоты с гидрохлоридом N-метокси-N-метилгидроксиламина в присутствии пептидных реагентов сочетания, таких как EDC, DCC, DMPU, и третичного аминного основания, такого как диизопропилэтиламин или триэтиламин.
Полученное соединение 29 бромируют с получением бромкетона 30, как показано на стадии G1. Бромирование можно провести хорошо известными способами, например, взаимодействием соединения 29 с бромидом пиридиния в уксусной кислоте или взаимодействием соединения 29 с Br2 в инертном органическом растворителе, таком как дихлорметан. Полученный бромкетон 30 взаимодействует на стадии G2 с арилгидроксиэфиром 7 в условиях, описанных на схеме В (стадия B3) с образованием связанного сложного эфира 31. Кетоновую группу в соединении 31 защищают в виде кеталя 32, как показано на стадии G3, способами, известными специалистам в данной области. Соединение 32 затем преобразуют в кеталь 1,3,4-оксадиазол-2-она 34 на стадиях G4 и G5 по стандартной последовательности, описанной на схеме В (В4 и В5). Наконец, на стадии G6 кетальную группу в соединении 34 расщепляют, например, минеральной кислотой в смеси ТГФ-метанол-вода или другими известными способами, с получением требуемого соединения 35.
Для специалиста очевидно, что описанную выше методику для схемы G можно использовать для синтеза аналогичных соединений, где n в соединении 35 равно 2-5, если в качестве исходного соединения использовать бромкетон, соединение 30, с более крупным бромалканоильным заместителем (Br(CH2)nCO-, где n равно 2-5.
Схема G

Схема H иллюстрирует процесс получения соединений формулы I, в которых Z представляет собой -(СН2)nСО-. На стадии H1 соответствующий метоксикарбонилзамещенный гетероцикл 36 обрабатывают 2 эквивалентами литиевого енолята трет-бутилацетата в растворителе, таком как ТГФ или ДМЭ, при температуре от -78°С до комнатной с образованием кетоацетатного промежуточного соединения 37. На стадии Н2 обработка соединения 37 основанием, таким как гидрид натрия, в инертном растворителе при температуре от -10°С до комнатной с последующим алкилированием полученного аниона электрофилом, таким как соединение 13, дает разветвленный промежуточный кетодиэфир 38. Декарбоксилирование, показанное на стадии Н3, можно проводить обработкой соединения 38 ТФУК в инертном растворителе, таком как дихлорметан, с последующим термолизом при температуре от 70 до 150°С, получая промежуточный кетоэфир 39. Кетоновую группу в соединении 39 защищают в виде кеталя 40, как показано на стадии Н4, способами, хорошо известными специалистам. Соединение 40 затем преобразуют в кеталь 1,3,4-оксадиазол-2-она 42 на стадиях H5 и H6 по стандартной последовательности, как описано на схеме В (В4 и В5). Наконец, на стадии Н7 кетальную группу в соединении 42 расщепляют, как описано выше на схеме G, стадия G6, с получением требуемого 1,3,4-оксадиазол-2-она, соединение 43.
Схема H

Биологические примеры
Приведенные ниже протоколы анализов используются для подтверждения биологических свойств соединений настоящего изобретения. Последующие примеры приводятся для более подробной иллюстрации изобретения. Тем не менее, их не следует понимать как ограничивающие каким-либо образом настоящее изобретение.
Определение значений EC50 в клеточном анализе PPAR-дельта-GAL4:
Принцип анализа
Активность веществ, которые связывают PPAR-дельта человека и активируют его агонистическим образом, анализируют, используя стабильно трансфицированную линию клеток HEK (НЕК - почка эмбриона человека), которая здесь называется репортерной линией клеток PPAR-дельта. Репортерная линия клеток PPAR-дельта содержит два генетических элемента - репортерный элемент люциферазы (pdeltaM-GAL4-Luc-Zeo) и PPAR-дельта слитый белок (GR-GAL4-human PPAR delta-LBD), который регулирует экспрессию репортерного элемента люциферазы в зависимости от PPAR-дельта лиганда. Стабильно и конститутивно экспрессируемый слитый белок GR-GAL4-human PPAR delta-LBD связывается в ядре клетки репортерной линии клеток PPAR-дельта через часть белка GAL4 со связывающими мотивами GAL4 ДНК в 5'-направлении от репортерного элемента люциферазы, который стабильно интегрируется в геном линии клеток. Наблюдается лишь слабая экспрессия репортерного гена люциферазы в отсутствие PPAR-дельта лиганда, если в анализе используется обедненная жирными кислотами фетальная телячья сыворотка (cs-FCS). Лиганды PPAR-дельта связывают и активируют слитый белок PPAR-дельта и, таким образом, стимулируют экспрессию репортерного гена люциферазы. Образующуюся люциферазу можно регистрировать с помощью хемилюминесценции при участии подходящего субстрата.
Конструкция репортерной линии клеток PPAR-дельта
Получение стабильной репортерной линии клеток PPAR-дельта основано на стабильном HEK-клеточном клоне, который стабильно трансфицирован репортерным элементом люциферазы. Данная стадия уже описана выше в разделе «конструкция репортерной линии клеток PPAR-альфа». На второй стадии слитый белок PPAR-дельта (GR-GAL4-human PPAR delta-LBD) стабильно вводили в этот клеточный клон. Для этого кодирование кДНК для N-концевых 76 аминокислот глюкокортикоидного рецептора (№ по каталогу P04150) связывали с областью кДНК, кодирующей аминокислоты 1-147 транскрипционного фактора дрожжей GAL4 (№ по каталогу P04386). кДНК лиганд-связывающей области PPAR-дельта рецептора человека (аминокислоты S139-Y441; № по каталогу L07592) клонировали на 3'-конце этой конструкции GR-GAL4. Полученная, таким образом, слитая конструкция (GR-GAL4-human PPAR delta-LBD) была повторно клонирована в плазмиду pcDNA3 (Invitrogen), чтобы сделать возможной конститутивную экспрессию промотора цитомегаловируса. Данная плазмида была линеаризована рестриктазой и стабильно трансфицирована в ранее описанный клеточный клон, содержащий элемент репортера люциферазы. Полученная репортерная линия клеток PPAR-дельта, которая содержит элемент репортера люциферазы и конститутивно экспрессирует слитый белок PPAR-дельта (GR-GAL4-human PPAR delta-LBD), была изолирована селекцией зеоцином (0,5 мг/мл) и G418 (0,5 мг/мл).
Порядок проведения анализа и оценка
Активность агонистов PPAR-дельта определяют в ходе 3-дневного анализа, который описан ниже.
День 1
Репортерную линию клеток PPAR-дельта культивируют до конфлюентности 80% в DMEM (#41965-039, Invitrogen), которую смешивают со следующими добавками: 10% сs-FCS (фетальная телячья сыворотка; #SH-30068.03, Hyclone), 0,5 мг/мл зеоцина (#R250-01, Invitrogen), 0,5 мг/мл G418 (#10131-027, Invitrogen), 1% раствора пенициллин-стрептомицин (#15140-122, Invitrogen) и 2 мМ L-глутамина (#25030-024, Invitrogen). Культивация происходила в стандартных флаконах для выращивания клеток (#353112, Becton Dickinson) в инкубаторе для выращивания клеток при 37°С в присутствии 5% СО2. Клетки с конфлюентностью 80% один раз промывают 15 мл PBS (забуференный фосфатом физиологический раствор) (#14190-094, Invitrogen), обрабатывают 3 мл раствора трипсина (#25300-054, Invitrogen) при 37°С в течение 2 минут, помещают в 5 мл описанной DMEM и считают в цитометре. После разбавления до 500000 клеток/мл, в каждую ячейку в объеме 180 мкл высевают 35000 клеток в 96-ячеечные планшеты для микротитрования с основанием из прозрачной пластмассы (#3610, Corning Costar). Планшеты инкубируют в инкубаторе для клеточных культур при 37°С и 5% СО2 в течение 24 часов.
День 2
PPAR-дельта агонисты, которые необходимо исследовать, растворяют в ДМСО в концентрации 10 мМ. Полученный маточный раствор разбавляют в DMEM (#41965-039, Invitrogen), которую смешивают с 5% сs-FCS (#SH-30068.03, Hyclone), 2 мМ L-глутамина (#25030-024, Invitrogen) и описанными ранее антибиотиками (зеоцин, G418, пенициллин и стрептомицин).
Анализируемые вещества тестируют в 11 разных концентрациях в диапазоне от 10 мкМ до 100 пМ. Наиболее активные соединения тестируют в диапазонах концентраций от 1 мкМ до 10 пМ или от 100 нМ до 1 пМ.
Среду репортерной линии клеток PPAR-дельта, посеянную в день 1, либо полностью удаляют отсасыванием, либо оставляют, и разбавленные в среде испытуемые вещества сразу же добавляют к клеткам. Разбавление и добавление веществ выполняет лабораторный робот (Beckman FX). Конечный объем анализируемых веществ, разбавленный в среде, составляет 100 мкл на ячейку 96-ячеечного планшета для микротитрования. Концентрация ДМСО в аналитической смеси составляет менее 0,1% по объему во избежание цитотоксических эффектов растворителя.
В каждый планшет вводили стандартный агонист PPAR-дельта, который аналогичным образом разбавляли в 11 разных концентрациях, чтобы продемонстрировать процесс анализа в каждом отдельном планшете. Аналитические планшеты инкубируют в инкубаторе при 37°С и 5% СО2 в течение 24 часов.
В качестве альтернативы, 20 мкл 10-кратной конечной концентрации анализируемого вещества добавляют непосредственно к 180 мкл клеток в ячейках планшета. Анализируемые вещества трижды тестируют при 8 разных концентрациях в таком аналитическом планшете.
День 3
Репортерные клетки PPAR-дельта обрабатывают анализируемыми веществами и убирают из инкубатора, а затем среду отсасывают. Клетки лизируют, добавляя пипеткой 50 мкл реагента Bright Glo (производства Promega) в каждую ячейку 96-ячеечного планшета для микротитрования. После инкубации при комнатной температуре в темноте в течение 10 минут планшеты для микротитрования анализируют в люминометре (Trilux производства Wallac). Время измерения каждой ячейки планшета для микротитрования составляет 1 сек.
Оценка
Необработанные данные люминометра переносят в файл Microsoft Excel. Графики эффекта дозы и значения EC50 PPAR-агонистов вычисляют с помощью программы XL.Fit согласно описанию производителя (IDBS).
Измеряют значения EC50 PPAR-дельта в диапазоне от 1 нМ до 10 мкМ для модуляторов PPAR, приведенных в примерах данной заявки. Соединения формулы I данного изобретения могут выполнять функцию агонистов или антагонистов. Ниже описан анализ для определения парциальной агонистической или антагонистической активности.
Определение эффективности парциальных агонистов или антагонистов на рецепторе PPAR-дельта
Данный анализ позволяет определить, действуют ли соединения в качестве парциальных агонистов или антагонистов на рецепторе PPAR-дельта.
Культивирование и сбор клеток в аналитических планшетах описаны в разделах «День 1» и «День 3» выше.
День 2
Парциальный агонист или антагонист и известный селективный агонист разбавляют в DMEM (#41965-039, Invitrogen), которую смешивают с 10% сs-FCS (#SH-30068.03, Hyclone), 2 мМ L-глутамина (#25030-024, Invitrogen) и описанными ранее антибиотиками (зеоцин, G418, пенициллин и стрептомицин) до 20-кратной желаемой конечной концентрации. В аналитический планшет с клетками добавляют десять микролитров парциального агониста или антагониста. Аналитические планшеты инкубируют в инкубаторе при 37°С и 5% СО2 в течение 30 минут. Затем после предварительной инкубации парциального агониста или антагониста добавляют десять микролитров известных селективных агонистов в 20-кратной концентрации. Аналитические планшеты инкубируют в инкубаторе при 37°С и 5% СО2 в течение 24 часов. Определяют эффект известных селективных агонистов EC50 для каждой концентрации парциального агониста или антагониста.
Анализ связывания SPA PPAR-дельта-LBD
Исходные растворы
1 М Tris (pH 8,0 или рН 7,6)(Gene Medicine Stock Room)
2 М КСl (порошок в N2140)
Tween 20
100 мМ DTT
13,9 мкМ GW2331 в EtOH ГОРЯЧИЙ
10 мМ GW2331 в ДМСО ХОЛОДНЫЙ
PPAR-альфа (концентрация варьируется)
EX.: 0,884 мкг/мкл
Промывной буфер: (Хранить при 4°С. Срок годности буфера одна неделя).
Связывающий буфер: (Каждый раз готовить свежий связывающий буфер).
Подготовка реакционных реагентов для одного планшета
Покрытые глутатионом SPA-гранулы
Каждый флакон с SPA-гранулами содержит 500 мг гранул.
Растворить 500 мг SPA-гранул в 5 мл водного буфера, и он сохраняет свою пригодность для использования в течение нескольких недель.
Хранить при 4°С.
Приготовить разбавленные SPA-гранулы в связывающем буфере.
Добавить 1 мл растворенных SPA-гранул в 60 мл связывающего буфера.
Добавить 20 мкл растворенных гранул в каждую ячейку 96-ячеечного планшета.
Использовать 2 мл растворенных таким образом гранул для каждого планшета (без учета мертвого объема).
3Н-GW2331 плюс GST-PPAR-дельта-LBD (для одного 96-ячеечного планшета без мертвого объема) 13,9 мкМ 40 нМ на ячейку.
3,0 мл на планшет (с учетом мертвого объема).
Если удельная активность 3Н-GW2331 составляет 1 мкКи/мл (производство Amersham), разбавить 17 мкл 3Н-GW2331 в 3,0 мл связывающего буфера = 0,08 мкМ.
Если концентрация белка составляет 1 мг/мл, добавить 21 мкл белков в 3,0 мл связывающего буфера.
В заключение: ОДИН 96-ячеечный планшет: 3000 мкл связывающего буфера + 17 мкл 3Н-GW2331 + 21 мкл GST-PPAR-дельта (1 мг/мл).
Контрольные планшеты
96-Ячеечный маточный планшет (для 2 контрольных планшетов)
В колонке № 1
Добавить 5 мкл холодного GW2331 (10 мМ) в ячейки E-H.
Добавить 45 мкл ДМСО в ячейки А-Н.
В колонке № 12 (3-кратное разбавление)
Добавить 10 мкл холодного GW2331 (10 мМ) в ячейку А.
Затем добавить 90 мкл ДМСО в ячейку А и хорошо перемешать раствор.
Добавить 20 мкл ДМСО в лунки В-Н.
Взять 10 мкл раствора из ячеек А и В и хорошо перемешать,
затем взять 10 мкл раствора из ячеек от В до С и хорошо перемешать,
затем взять 10 мкл раствора из ячеек от С до D и хорошо перемешать.
Наконец, взять 10 мкл раствора из ячеек от F до H.
Контрольный планшет (для 8 реакционных планшетов)
В контрольном планшете содержится раствор маточного планшета, разбавленный 1:10. Буфером разбавления является промывочный буфер.
Планшеты с образцами
В свежий планшет библиотеки CPC добавить 90 мкл ДМСО.
Взять 10 мкл раствора, разбавленного ДМСО и добавить его в 90 мкл промывочного буфера в планшет с образцом.
Реакционные планшеты
Добавить 20 мкл гранул полиакрилата натрия и 30 мкл 3Н-GW2331 с GST-PPAR-дельта в каждую лунку реакционного планшета.
Добавить 5 мкл соединений из каждой лунки планшета с образцом в колонки со 2 по 11 реакционного планшета.
Добавить 5 мкл соединений из колонки 1 и колонки 12 контрольного планшета в колонку 1 и колонку 12 реакционного планшета.
Протокол для 96-ячеечного анализа SPA
Дождаться равновесия в реакционных планшетах - это займет от 20 минут до 2 часов.
Запечатать планшеты, прежде чем проводить подсчет в цитометре Microbeta (Wallac).
Вычислить IC50.
В анализе связывания SPA PPAR-дельта-LBD для модуляторов PPAR, приведенных в примерах данной заявки, были измерены величины IC50 в диапазоне от 1 нМ до >10 мкМ. Соединения формулы I данного изобретения могут выполнять функцию агонистов или антагонистов.
Культуры олигодендроцитов КРЫС/МЫШЕЙ
Приготовление клеток
1. Первичные клетки-предшественники олигодендроцитов получают из коры головного мозга новорожденных (2-3 постнатальных дня) крыс или мышей после удаления микроглий путем механического отделения сепарированием от астроцитного монослоя, используя модифицированный способ, впервые описанный в работе McCarthy and de Vellis (1980).
2. Удаляют мягкие мозговые оболочки из мозга новорожденной крысы и механически отделяют ткань. Помещают клетки в колбы T75 и добавляют питательную среду для клеток DMEM/F12 + 10% FBS.
3. Собирают олигодендроциты, растущие на астроцитовой подложке, методом стряхивания через четырнадцать дней после исходного приготовления. Центрифугируют суспензию и снова взвешивают клеточный осадок в не содержащей сыворотки среде (SFM; DMEM, смешанной с 25 мкг/мл переносимого вещества, 30 нМ трийодотиронина, 20 нМ гидрокортизона, 20 нМ прогестерона, 10 нМ биотина, 1× следовых элементов, 30 нМ селена, 1 мкг/мл путресцина, 0,1% бычьего сывороточного альбумина, 5 ед./мл PenStrep, 10 мкг/мл инсулина) c добавлением следующих факторов роста: тромбоцитарный фактор роста-АА (PDGF) и фактор роста фибробластов-2 (FGF).
4. Помещают клетки в чашки, покрытые PDL (поли-D-лизин), и инкубируют при 37°С с 6-7% СО2.
5. Компоненты среды заменяют каждые 48 часов, чтобы клетки оставались в исходном состоянии.
Пассирование клеток-предшественников для увеличения числа клеток для скринингового анализа
1. Когда культура станет конфлюентной, промывают ее физиологическим раствором с PBS буфером, добавляют трипсин и инкубируют в течение ~2-3 минут при 37°C.
2. Нейтрализуют и центрифугируют суспензию клеток при 900 g в течение 5 минут.
3. Ресуспендируют клеточный осадок в SFM + PDGF/FGF.
4. Добавляют питание клеткам со свежими факторами роста каждые 48 часов, чтобы поддерживать их в обогащенном состоянии для быстрого деления клеток-предшественников.
5. Клетки пассируют не более 4-5-раз перед экспериментальным анализом.
6. Все эксперименты с клетками-предшественниками олигодендроцитов выполняли с клетками, которые постоянно поддерживались в этих условиях. Более 95% всех клеток были A2B5-иммунопозитивными и экспрессировали мРНК 2'3'-циклической нуклеотидной 3'-фосфодиэстеразы II.
7. Для генерации зрелых олигодендроцитов, через 24 часа после помещения клеток-предшественников в планшеты, их перемещали в свободную от сыворотки среду с добавлением инсулинового фактора роста IGF-I или без него и выращивали в этих условиях в течение 7 дней перед экспериментальным анализом.
8. В качестве альтернативы могут использоваться обогащенные линии клеток-предшественников Central Glia (CG4) крыс, которые сохраняются в основной среде (DMEM с 2 мМ глутамина, 1 мМ пирувата натрия, биотином (40 нМ), инсулином (1 мкМ) и N1) с добавлением 30% кондиционированной среды из линии клеток нейробластомы B-104. Чтобы индуцировать дифференцировку, клетки CG4 помещают в основную среду с 1% фетальной телячьей сыворотки (удаляемой через 2 дня) и инсулином (500 нМ). Для подтверждения обогащения >95% в зрелых и незрелых культурах используется иммунореактивность A2B5 и ОБМ (основной белок миелина) соответственно.
Обработка соединением культуры крыс/мышей
1. Помещают 10000-15000 клеток на ячейку в 24-ячеечные планшеты, покрытые PDL, и культивируют клетки в присутствии митогена (10 нг/мл) в течение ночи.
2. В присутствии митогена:
a. На следующий день удаляют старую среду и добавляют соединения в свежую среду (с митогеном).
b. Оценки эффекта дозы соединений выполняют при 6 разных концентрациях (10 мкМ, 1 мкМ, 100 нМ, 10 нМ, 1 нМ и 0,1 нМ).
c. Для каждой концентрации соединения оценку выполняют на трех ячейках.
3. В отсутствие митогена:
a. На следующий день удаляют старую среду и добавляют соединения в свежую среду (без митогена).
b. Оценки эффекта дозы соединений выполняют при 6 концентрациях (10 мкМ, 1 мкМ, 100 нМ, 10 нМ, 1 нМ и 0,1 нМ).
c. Для каждой концентрации соединения оценку выполняют на трех ячейках.
4. Культивируют обработанные клетки в течение 7 дней перед их использованием в экспериментальном анализе.
Культуры олигодендроцитов ЧЕЛОВЕКА
Приготовление клеток
1. Нейросферы человека, взятые из кортекса эмбриона человека E19.5-E22, культивируют в течение 2 недель в среде клеток-предшественников: DMEM/F12, содержащей 100 мкг/мл переносимого вещества, 30 нМ трийодотиронина, 30 нМ гидрокортизона, 20 нМ прогестерона, 10 нМ биотина, 1× следовых элементов 30 нМ селена, 60 мкМ путресцина, 0,1% бычьего сывороточного альбумина, 5 ед./мл PenStrep, 25 мкг/мл инсулина, с добавлением PDGF и FGF.
2. Нейросферы диссоциировали 20 ед./мл папаина при 37°C в течение 30-50 минут.
3. Клетки помещали в чашки с PDL-покрытием при плотности 50000-100000 клеток на ячейку в среде клеток-предшественников, содержащей PDGF/FGF и инкубируемой при 37°C с 5-6% CO2.
4. Среду и факторы роста добавляли каждые 48 часов.
Обработка соединением культуры человека
1. Через 24-48 часов после помещения в планшет старую среду удаляют и добавляют соединения в свежую среду (с митогеном).
2. Оценки эффекта дозы соединений выполняют при 3-6 разных концентрациях (10 мкМ, 1 мкМ, 100 нМ, 10 нМ, 1 нМ и 0,1 нМ).
3. Для каждой концентрации соединения оценку выполняют на трех ячейках.
5. Культивируют клетки в течение 7 дней перед их использованием в экспериментальном анализе.
Удельное иммунное окрашивание олигодендроцитов КРЫСЫ/МЫШИ/ЧЕЛОВЕКА
После обработки соединением используют олигодендроцитспецифические антитела, чтобы оценить способность соединения ускорять или способствовать дифференциации олигодендроцитов (например, иммунореактивность O4, O1 или основного миелинового белка через некоторое время после обработки соединением находится между обработанными и необработанными культурами).
1. Клетки помещают на обработанные поли-D-лизином 4-ячеечные камерные планшеты при плотности от 5×103 до 20×103 клеток на ячейку и выращивают, как описано выше. Последующее окрашивание выполняют на популяциях олигодендроцитов с нарастающей скоростью дифференциации клеток, как описано по дням “in vitro” без PDGF и FGF.
2. Для определения стадиеспецифической экспрессии маркера поверхности клеток олигодендроцитов (включая A2B5, O4 и O1) используют окрашивание живых клеток в течение 30 минут при 37°C.
3. Затем клетки фиксируют 4% параформальдегидом в течение 10 минут при комнатной температуре.
4. Процедуру окрашивания фиксированных клеток используют для определения стадиеспецифической экспрессии маркера олигодендроцитов (включая основной миелиновый белок, ОБМ).
5. Промывают буфером PBS.
6. Пермеабилизируют с 0,1% Triton/0,01% NaAz, разбавленного в 1Х буфере PBS в течение 10 минут при комнатной температуре.
7. Блокируют 5-10% козьей сыворотки в буфере разбавления антител (0,1% Triton-X 100 и 1% альбумина бычьей сыворотки, не содержащей IgG; также используется для разбавления антител) в течение 15 минут при комнатной температуре.
8. Добавляют первичное антитело, разбавленное в буфере разбавления антител.
9. Инкубируют в течение ночи при плавном покачивании при 4°C.
10. На следующий день промывают буфером PBS 1X в течение 5 минут, а затем 3Х в течение 15 минут каждым при комнатной температуре.
11. Инкубируют с подходящими вторичными антителами в течение 45 минут при комнатной температуре.
12. Ядра клеток окрашивают 4,6-диамидино-2-фенилиндолом (DAPI) в течение 15 минут при комнатной температуре.
13. Промывают несколько раз буфером PBS и исследуют с помощью флуоресцентной микроскопии.
14. По времени и при разных дозах соединения сопоставляют следующие условия: только PDGF/FGF, только SFM, только SFM-IGF1, PDGF/FGF и соединение, SFM и соединение.
Иммунное окрашивание бромдезоксиуридина (БДУ) КРЫСЫ/МЫШИ/ЧЕЛОВЕКА
Для того, чтобы убедиться, что соединения не способствуют пролиферации клеток
1. Клетки-предшественники олигодендроцитов маркируют 10 мкМ БДУ в течение 20 часов, а затем фиксируют с помощью либо 70% этанола, либо 4% параформальдегида.
2. Клетки последовательно инкубируют биотинилированным анти-БДУ мышей и стрептавидин-пероксидазой с тремя промежуточными промывками PBS буфером.
3. Колориметрическую визуализацию иммунореактивности БДУ проводят с помощью DAB (диаминобензидин) и оценивают полное количество клеток с использованием контрастирующего гематоксилина.
4. Подсчет БДУ-иммунопозитивных клеток проводят два независимых исследователя.
Анализ изображения культуры КРЫСЫ/МЫШИ/ЧЕЛОВЕКА
Для количественной оценки степени дифференциации олигодендроцитов после их обработки соединением используют флуоресцентную микроскопию. Этот анализ показывает, что селективные агонисты ускоряют или благоприятствуют дифференциации олигодендроцитов.
1. Ручной подсчет клеток: для каждого экспериментального условия выбирают четыре произвольных поля и в каждой области насчитывают 500-600 клеток. Процентное содержание ОБМ (или О4) иммунопозитивных клеток (зрелые клетки, в которых протекает процесс, с миелиновыми листами или без них) в зависимости от DAPI-позитивных клеток (полного количества клеток) сравнивают в контрольных и обработанных лекарственным препаратом группах.
2. Автоматический подсчет клеток: для количественной оценки степени дифференциации олигодендроцитов после их обработки соединением используют флуоресцентную микроскопию. Для оценки количества дифференцирующих олигодендроцитов на всю популяцию (подсчитывается от ~8 до 15×103 клеток на ячейку) произвольно выбирали шесть областей/ячеек. Изображения иммунофлуоресценции получали, используя систему формирования цифровых изображений Zeiss Axio Vision с охлаждаемой ПЗС камерой Zeiss AxioCam HRc, подключенной к тому же самому микроскопу. Все параметры формирования микроскопического изображения были установлены с учетом того, что изображения будут использоваться для анализа интенсивности иммунофлуоресценции клеток. Процентное содержание ОБМ-позитивных (дифференцированных) клеток по отношению к полному количеству клеток (ядрами, окрашенными DAPI) сравнивали в контрольной и обработанной лекарственным препаратом группах. В условиях формирования изображений зарегистрировать автофлуоресценцию было невозможно.
3. Анализ дифференциации олигодендроцитов человека: ручной подсчет полного количества О4 иммунопозитивных клеток на ячейку (биполярных и многополярных).
Количественный анализ полимеразной цепной реакции (ПЦР) КРЫСЫ/МЫШИ/ЧЕЛОВЕКА
Чтобы оценить вызванную соединением активацию пути PPAR-дельта и степень зрелости олигодендроцитов (изменения в уровнях мРНК):
1. Всю РНК экстрагируют из культивированных олигодендроцитов с использованием реагента TriZol.
2. Затем мРНК обрабатывают ДНКазой, не содержащей РНКазы, снова очищают, а затем превращают в матрицу кДНК с помощью реакции обратной транскрипции (набор Clontech Advantage RT for PCR).
3. Экспрессию транскрипта члена пути PPAR-дельта количественно оценивают, используя Sybr Green PCR Master Mix.
4. Смесь праймера и зонда рибосомной РНК 18S RNA (186 пар оснований), взвешенную в Taqman 2Х PCR Master Mix, используют для внутреннего контроля.
5. Количественную ПЦР проводят с использованием технологии реального времени Taqman™ (Gibson, et al., 1996) на модели 7700 Sequence Detector System (Applied Biosystems, Foster City, CA).
6. Результаты анализируют, используя программное обеспечение Sequence Detection Systems версии 1.91.
Твердофазный иммуноферментный анализ культур КРЫС
Чтобы оценить вызванную соединением активацию пути PPAR-дельта и степень зрелости олигодендроцитов (изменения в уровнях белка):
1. Планшеты промывают буфером PBS, а затем хранят на льду. Добавляют 200 мкл ледяного лизирующего буфера (Tris 50 мМ, рН 7,4, MgCl2 2 мМ, EDTA 1 мМ, β-меркаптоэтанол 5 мМ, Nonidet P-40 1%, коктейль ингибиторов протеазы (Roche): 1 таблетка/50 мл) в каждую ячейку.
2. Клетки лизируют, прокачивая пипеткой, и вращают планшеты со скоростью 2000 об/мин при 4°C в течение 5 минут. Супернатант готов к использованию.
3. Пипеткой помещают в ячейки 50 мкл стандарта, контрольного раствора и образцов.
4. В каждую ячейку добавляют 50 мкл аналитического буфера основного миелинового белка.
5. Инкубируют ячейку, встряхивая ее при 500-700 об/мин на ротационном микропланшетном шейкере в течение 2 часов при комнатной температуре.
6. Добавляют в каждую ячейку 100 мкл конъюгата антитела основного миелинового белка ОБМ с биотином.
7. Инкубируют ячейку, встряхивая ее при 500-700 об/мин на ротационном микропланшетном шейкере в течение 1 часа при комнатной температуре.
8. Промывают 5-кратно ячейку промывным раствором. Промокают планшет досуха, переворачивая его на абсорбирующий материал.
9. Разбавляют концентрат конъюгата стрептавидина и фермента 1:50 аналитическим буфером ОБМ ИФА (разбавить необходимо непосредственно перед использованием в анализе).
10. Добавляют 100 мкл раствора конъюгата стрептавидина и фермента в каждую ячейку.
11. Инкубируют ячейку, встряхивая ее при 500-700 об/мин на ротационном микропланшетном шейкере в течение 30 минут при комнатной температуре.
12. Промывают 5-кратно ячейку промывным раствором. Промокают планшет досуха, переворачивая его на абсорбирующий материал.
13. Добавляют 100 мкл раствора хромогена ТМБ в каждую лунку.
14. Инкубируют ячейку, встряхивая ее при 500-700 об/мин на ротационном микропланшетном шейкере в течение 10-20 минут при комнатной температуре. Не следует допускать воздействия прямого солнечного света.
15. Добавляют 100 мкл стоп-раствора в каждую лунку.
Измеряют поглощение раствора в лунках в течение 30 минут, используя микропланшетный аппарат для чтения, установленный на 450 нМ.
Проверка моделей концепции «in vivo»
Местные поражения: (используется для оценки способности соединений защищать миелиновую целостность и ускорять или повышать скорость ремиелинизации)
1. Крысам в возрасте 7 недель дают свободный доступ к пище и воде, и они акклиматизируются в течение минимум 4 дней перед их использованием в эксперименте.
2. Перед операцией каждое животное взвешивают. Затем крысу подвергают анестезии кетамином (100 мг/мл) в сочетании с ксилазином (20 мг/мл) в отношении
1,8:1. Перед операцией крысам вводят в/б 0,15 мл/180 г массы тела анестезирующего раствора. Животных готовят к операции в асептических условиях в соответствии с руководящими принципами IACUC. Все хирургические инструменты автоклавируют. Волосы между ушами состригают, и эту область очищают раствором бетадина, промывают стерильным физиологическим раствором и, наконец, протирают спиртовыми тампонами в стерильной упаковке.
3. Для проведения операции крысу кладут на брюшную поверхность в небольшом стереотаксическом устройстве для животных, предназначенном для устойчивого удержания головы. Резцовую дугу устанавливают на -3,9 мм, так, как было показано, что это помогает установить плоское положение черепа крыс SD.
4. Разрез делают по предварительно обритой коже, покрывающей череп между ушами.
5. Небольшую область кости (0,75 мм в диаметре) просверливают в следующих координатах: АР -1,8, ML -3,1 от лямбда.
6. Кость убирают и крысе инжектируют 2 мкл этидийбромида, лизолецитина или SIN-1 в правую каудальную мозжечковую ножку, DV -7,1 мм, в течение 2 минут микролитровым шприцом и иглой Hamilton. В качестве альтернативы инъекцию можно делать в спинной мозг, мозолистое тело или кору головного мозга.
7. Иглу оставляют в этом положении в течение следующих 2 минут.
8. После вынимания иглы на разрез накладывают шов.
9. Каждая крыса получает в/м инъекцию 0,003 мг бупренорфина в заднюю конечность.
10. Крысу помещают в тепловой шкаф то тех пор, пока она не придет в сознание. Тогда ее возвращают в клетку. Нельзя допускать, чтобы в клетке находилось более двух крыс, поскольку они могут повредить друг другу швы.
11. Аналогичные процедуры выполняют и с мышами.
Модель экспериментального аллергического энцефаломиелита крыс (ЭАЭ крыс)
Экспериментальный аллергический энцефаломиелит (ЭАЭ) представляет собой аутоиммунное заболевание нервной системы, опосредованное Т-клетками, которое развивается у восприимчивых животных после сенсибилизации либо клеточным гомогенатом спинного мозга, либо одним из компонентов (основной миелиновый белок). Модель ЭАЭ грызунов является подходящим средством изучения воспаления головного и спинного мозга, наблюдаемого у пациентов, страдающих рассеянным склерозом. У грызунов инъекция спинного мозга или компонентов спинного мозга, таких как основной миелиновый белок, вызывает аутоиммунную реакцию, основанную на активации Т-лимфоцитов. Клиническое заболевание, как правило, проявляется примерно через 8-10 дней после инокуляции и наблюдается в виде широкого спектра аномалий поведения, от слабых нарушений походки и атонии хвоста до полного паралича и смерти. Как правило, происходит потеря веса. У выживших животных происходит спонтанное выздоровление, сопровождаемое восстановлением большинства двигательных функций. В зависимости от вида, аллергена и используемых методов животные, исследованные по модели ЭАЭ, могут испытывать один (острый ЭАЭ) или несколько (хронический рецидивирующий ЭАЭ) приступов. Можно использовать несколько подходов к лечению. Выбранное лекарство или способ лечения можно применять до иммунизации, во время бессимптомного периода или во время клинического заболевания.
Животные
Самки мышей Льюиса 160-220 г (Charles River)
Антиген
Цельный спинной мозг морской свинки (Harlan Biosciences).
Полный адъювант Фрейнда Н37 Ra [1 мг/мл туберкулезной бациллы Mycobacterium Tuberculosis H37 Ra] (Difco).
Дополнительный антиген
Туберкулезная бацилла (Difco).
Бордетеллы коклюша [убитые нагреванием] (Difco).
Подготовка антигена (примерно на 720 животных):
1. Взвесить 5 граммов замороженного спинного мозга морских свинок.
2. Добавить 5 г спинного мозга в 5 мл 0,9% физиологического раствора (1 г/мл) в круглодонной центрифужной пробирке.
3. Гомогенизировать на льду при помощи Tissue-tech до полного разрушения ткани (примерно 5 минут).
4. Добавить 10 мл полного адъюванта Фрейнда Н37 Ra, дополненного 200 мг туберкулезной бациллы (20 мг/мл полного адъюванта Фрейнда Н37 Ra).
5. Экстрагировать гомогенат/адъювант из пробирки, отсасывая его в 10-мл шприц с эмульгирующей иглой 18 размера.
6. Эмульгировать между двумя 30 мл стеклянными шприцами, пока не возникнут трудности отбора материала через иглу. (Примерно 5 минут {не должно быть никакого разделения между жировой и водной фазой }).
7. Использовать немедленно либо хранить на льду, пока не потребуется (не более 30 минут) (не замораживать).
Протокол
1. Самкам крыс Льюиса (Charles River) дают свободный доступ к пище и воде, и они акклиматизируются не менее 3 дней перед использованием в экспериментах.
2. Крысы весом 160 и 220 граммов сначала индуцируют 5% изофлураном (Aerrane, Fort Dodge), 30% О2, 70% N2O в течение 2-5 минут.
3. Затем крысу помещают на одеяло с обогревом циркулирующей водой (Gaymar) (дорсальной поверхностью вверх) и в носовой конус для самопроизвольного вдыхания анестезирующих газов. Концентрацию изофлурана снижают до 2%.
4. Делают две подкожные инъекции (0,1 мл каждая) антигена или нормального физиологического раствора в вентральную поверхность задних лап.
5. Животных убирают из носового конуса, взвешивают и нумеруют.
6. Крысам дают возможность выйти из наркоза, а затем помещают их в отдельные клетки.
7. Животных ежедневно наблюдают на предмет индукции ЭАЭ (см. критерии ниже).
СТАДИЯ: О НОРМА
СТАДИЯ: 1 Патологическое поведение и атония хвоста
СТАДИЯ: 2 Небольшая, но определенная слабость одной или обеих задних лап.
СТАДИЯ: 3 Ярко выраженная слабость одной или обеих задних лап, или легкая атаксия.
СТАДИЯ: 4 Тяжелый парапарез и минимальная подвижность задних лап.
СТАДИЯ: 5 Отсутствие движений задних лап и паралич нижней части тела.
СТАДИЯ: 6 Предсмертное состояние без спонтанных движений и с нарушенным дыханием.
Также наблюдается растущая степень поражения передних лап и мочевое и фекальное недержание.
СТАДИЯ: 7 СМЕРТЬ
Лечение было начато на 10-й день после иммунизации. Так как симптомы заболевания в этой модели проявляются, как правило, через 10-11 дней после инокуляции, можно считать, что этот момент представляет начальную фазу острого периода рассеянного склероза. Считается, что эта задержка начала лечения более точно моделирует клиническую ситуацию, чем традиционно используемые протоколы, когда лекарство применяют в момент инокуляции или даже до нее (Teitelbaum D. et al., Proc Natl Acad Sci USA 1999; 96: 3842-3847 и Brod S. A., et al., Ann Neurol 2000; 47: 127-131).
Настоящее изобретение далее иллюстрируется следующими примерами соединений, которые приводятся для целей пояснения и никоим образом не ограничивают объем настоящего изобретения.
Примеры синтеза
Общие сведения
Коммерчески доступные реагенты и растворители использовали без предварительной обработки. Спектры1H-ЯМР записывали на спектрометре Varian MercuryPlus (300 мГц) или Varian Unity Inova (400 мГц), как указано в тексте. Протонные химические сдвиги приведены в δ м.д. относительно внутреннего стандарта тетраметилсилана (0,0 м.д.). Данные масс-спектрометрии (ЖХ/МС) получены с помощью времяпролетного масс-спектрометра Micromass LCT с ионизацией электрораспылением и временем измерения 5 минут для m/z от 100 до 1000. ЖХ (ЖХ/МС) проводят на колонке Hypersil C18 (4,6×50 мм, 3,5 мкм) с подвижной фазой 0,1% ТФУК в H2O (A) и 0,1% ТФУК в ACN (B) и градиентом от 5 до 100% B в течение 3 минут, а затем 2 минут при 100% B. В качестве альтернативы можно использовать Platform LC-MS с электрораспылительным источником с системой HP1100 LC, работающей при 2,0 мл/мин, 200 мкл/мин, состоящей из источника ионизации электрораспылением со встроенным детектором HP1100 DAD и детектором SEDEX ELS. Используют колонку Luna C18(2) (30×4,6 мм 3 мкм) с градиентом от 5 до 95% B в течение 4,5 минут с подвижной фазой 0,1% муравьиной кислоты в H2O и 0,1% муравьиной кислоты в ACN (B). Очистку ВЭЖХ выполняют на системе Varian ProStar, используя колонку для обращенно-фазовой хроматографии C18 с линейным градиентом ACN/H2O, содержащим 0,1% трифторуксусной кислоты. Микроволновой синтез выполняли с использованием микроволновой системы Personal Chemistry Smithcreator с двумя реакторами объемом 2 или 5 мл.
Пример 1
Промежуточное соединение: этиловый эфир [5-метил-2-(4-трифторметилфенил)тиазол-4-ил]уксусной кислоты
К раствору 4-трифторметилбензолтиоамида (1,845 г, 9 ммоль) в этаноле (15 мл, 200%) добавляют этил-4-бром-3-оксопентаноат (2,07 г, 9 ммоль). Раствор плотно закрывают и нагревают до 170°C в микроволновой печи Personal Chemistry™ и перемешивают при этой температуре в течение 20 мин. Полученный раствор охлаждают до комнатной температуры, концентрируют при пониженном давлении и очищают флэш-хроматографией (элюируя смесью 30% этилацетата/10% дихлорметана в гептане) с получением указанного в заголовке соединения в виде белого твердого вещества (1,4 г).
MS (ESI) m/z 330 (M+H);1H-ЯМР (CDCl3) δ 1,87 (шир.с, 1H), 2,49 (с, 3H), 4,86 (с, 2H), 7,67 (д, J=8Гц, 2H), 8,02 (д, J=8Гц, 2H).
Пример 2

Промежуточное соединение: 4-(2-гидроксиэтил)-5-метил-2-(4-трифторметилфенил)тиазол
Охлаждают (0°C) раствор алюмогидрида лития (5,3 мл, 1 М в ТГФ) и добавляют раствор этилового эфира [5-метил-2-(4-трифторметилфенил)тиазол-4-ил]уксусной кислоты (пример 1, 1,4 г, 4,25 ммоль) в ТГФ (15 мл). После добавления удаляют холодную баню и перемешивают в течение 2 часов. Раствор охлаждают до 5°C, а затем по каплям добавляют воду (0,2 мл), после чего добавляют раствор NaOH (0,2 мл, 5 М в воде) и воду (0,2 мл). Полученную смесь разбавляют этилацетатом и затем отфильтровывают через слой целита. Твердые частицы промывают дихлорметаном и затем концентрируют смесь фильтратов при пониженном давлении. Остаток очищают флэш-хроматографией (элюируя смесью 30% этилацетата и 40% дихлорметана в гептане) с получением указанного в заголовке соединения в виде желтого твердого вещества (0,879 г). В качестве исходного материала используют соединение примера 1 для получения указанного в заголовке соединения.
MS (ESI)m/z 288 (M+H);1H-ЯМР (CDCl3) δ 2,44 (с, 3H), 2,91 (т, J=7Гц, 2H), 3,62 (т, J=6Гц, 1H), 4,01 (дт, J=7, 6Гц, 2H), 7,66 (д, J=8Гц, 2H), 7,96 (д, J=8Гц, 2H).
Пример 3
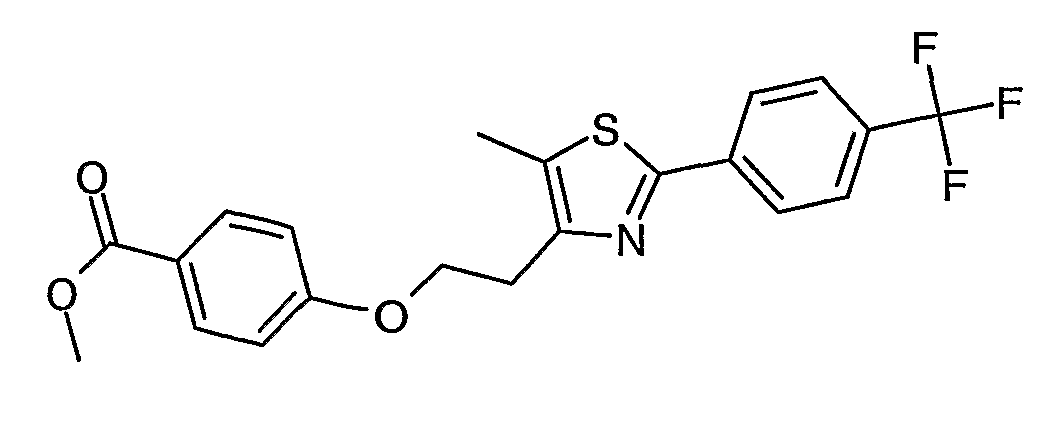
Промежуточное соединение: метиловый эфир 4-[5-метил-2-(4-трифторметилфенил)тиазол-4-илэтокси]бензойной кислоты
К раствору 4-(2-гидроксиэтил)-5-метил-2-(4-трифторметилфенил)тиазола (пример 3, 288 мг, 1,0 ммоль) в ТГФ (3 мл) добавляют метиловый эфир 4-гидроксибензойной кислоты (167 мг, 1,1 ммоль) и потом трифенилфосфин (288 мг, 1,1 ммоль). К полученному раствору по каплям добавляют диэтилазодикарбоксилат (174 мкл, 1,1 ммоль). После добавления полученный красный раствор перемешивают в течение 20 минут. Полученную смесь концентрируют при пониженном давлении и остаток очищают флэш-хроматографией (элюируя смесью 15% этилацетата/15% дихлорметана в гептане) с получением указанного в заголовке соединения в виде белого твердого вещества (410 мг).
MS (ESI)m/z 422 (M+H);1H-ЯМР (ДМСО) δ 2,51 (с, 3H), 3,19 (т, J=7Гц, 2H), 3,80 (с, 3H), 4,40 (т, J=7Гц, 2H), 7,05 (д, J=9Гц, 2H), 7,83 (д, J=8Гц, 2H), 7,88 (д, J=8Гц, 2H) 8,05 (д, J=8Гц, 2H).
Пример 4

Промежуточное соединение: гидразид 4-{2-[5-метил-2-(4-трифторметилфенил)тиазол-4-ил]этокси}бензойной кислоты
К суспензии метилового эфира 4-[5-метил-2-(4-трифторметилфенил)тиазол-4-илэтокси]бензойной кислоты (пример 4, 410 мг, 1 ммоль) в метаноле (3 мл) добавляют безводный гидразин (0,32 мл, 10 ммоль). Полученную смесь нагревают до 60°C и перемешивают при этой температуре в течение 66 часов. Полученный раствор охлаждают до комнатной температуры и добавляют три капли воды. Осадок отделяют фильтрованием, промывают эфиром с получением указанного в заголовке соединения (279 мг).
MS (ESI)m/z 422 (M+H);1H-ЯМР (ДМСО) δ 2,51 (с, 3H), 3,17 (т, J=7Гц, 2H), 4,36 (т, J=7Гц, 2H), 4,38 (шир.с, 2H), 6,98 (д, J=9Гц, 2H), 7,77 (д, J=9Гц, 2H), 7,83 (д, J=8Гц, 2H) 8,06 (д, J=8Гц, 2H) 9,58 (шир.с, 1H).
Пример 5
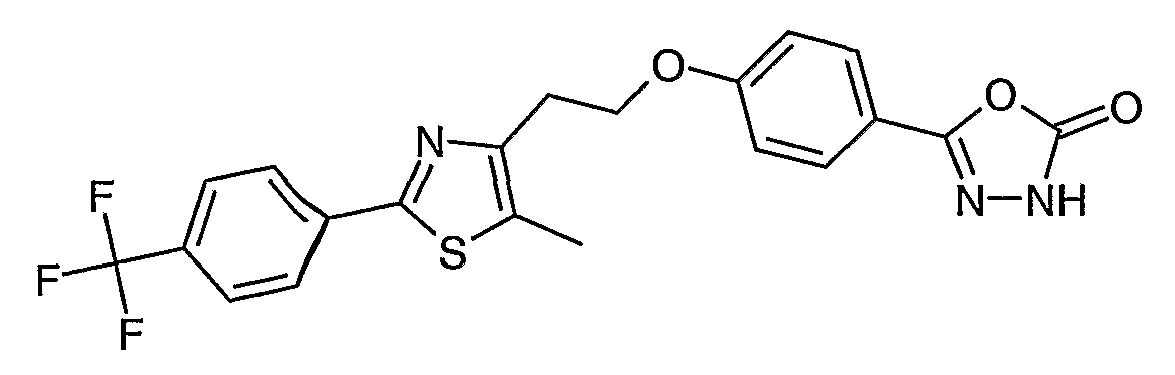
5-(4-{2-[5-метил-2-(4-трифторметилфенил)тиазол-4-ил]этокси}фенил)-3H-[1,3,4]оксадиазол-2-он
К суспензии гидразида 4-{2-[5-метил-2-(4-трифторметилфенил)тиазол-4-ил]этокси}бензойной кислоты (пример 4, 276 мг, 0,65 ммоль) в дихлорметане (4 мл) добавляют пиридин (104 мкл, 1,3 ммоль) и затем фенилхлорформиат (0,88 мкл, 0,71 ммоль). Полученную смесь перемешивают при комнатной температуре, пока не будет израсходовано все исходное вещество (по анализу ТСХ). Смесь разбавляют этилацетатом, промывают водой, затем насыщенным раствором соли, сушат над MgSO4 и концентрируют при пониженном давлении. Остаток помещают в ацетонитрил (5 мл). К полученной смеси добавляют DBU (106 мкл, 0,71 ммоль). Полученный раствор плотно закрывают, нагревают до 170°C в микроволновой печи Personal Chemistry™ и перемешивают при этой температуре в течение 120 минут. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом, промывают раствором 1 М HCl (или насыщенным раствором NaH2PO4), сушат над MgSO4 и концентрируют. Полученный остаток растирают несколько раз в дихлорметане с получением указанного в заголовке соединения в виде коричневого твердого вещества (137 мг) (перекристаллизованного из этилацетата в запаянной трубке при 140°C).
MS (ESI)m/z 448 (M+H);1H-ЯМР (ДМСО) δ 2,51 (с, 3H), 3,19 (т, J=7Гц, 2H), 4,40 (т, J=7Гц, 2H), 7,10 (д, J=8Гц, 2H), 7,70 (д, J=8Гц, 2H), 7,83 (д, J=8Гц, 2H) 8,05 (д, J=8Гц, 2H) 12,41 (шир.с, 1H).
Реферат
Описываются производные 1,3,4-оксадиазол-2-она формулы (I), где Aryl представляет собой фенил; Z представляет собой -O(CH2)n- и n представляет собой независимое целое число от 1 до 5; X представляет собой S; R1 представляет собой C1-6алкил; R2 представляет собой фенил, замещенный C1-6перфторалкилом; или его фармацевтически приемлемая соль; фармацевтическая композиция на его основе и способ лечения заболевания, где заболевание может модулироваться ативностью связывания PPAR-дельта. Используемые соединения обладают агонистической или антагонистической активностью. 4 н. и 3 з.п. ф-лы.
Формула

где Aryl представляет собой фенил;
z представляет собой -O(CH2)n- и n представляет собой независимо целое число от 1 до 5;
Х представляет собой S;
R1 представляет собой С1-6алкил; и
R2 представляет собой фенил, замещенный С1-6перфторалкилом;
или его фармацевтически приемлемая соль.
где Aryl представляет собой фенил;
z представляет собой -O(CH2)n- и n представляет собой независимо целое число от 1 до 5;
Х представляет собой S;
R1 представляет собой С1-6алкил; и
R2 представляет собой фенил, замещенный С1-6перфторалкилом;
или его фармацевтически приемлемой соли.
Документы, цитированные в отчёте о поиске
Модуляторы pparγ
Комментарии