Гетероциклические производные замещенных 2-ациламино-5-тиазолов, способы их получения, производное замещенного 2-аминотиазола, производные 2-амино-4-фенилтиазола - RU2059637C1
Код документа: RU2059637C1
Чертежи
Описание
Изобретение относится к новым гетероциклическим производным замещенных 2-ациламино-5-тиазолов, проявляющим сродство к рецепторам холецистокинина и гастрина к способу получения указанных соединений и к фармацевтическим композициям на их основе.
Холецистокинин (ХЦК) является полипептидным гормоном, находящимся in vivo в нескольких формах, содержащих от 8 до 39 аминокислот. Он обладает многочисленными физиологическими действиями на желчные пути, на желудочно-кишечный тракт, здесь можно сослаться на статью J.Е.Morley, Life Sciences, 1982, 30, р. 479-493, которая дает подробный обзор его свойств. Две различных популяции рецепторов ХЦК были выявлены при помощи специфических антагонистов; популяции типа А присутствуют, в частности, в поджелудочной железе, в желчном пузыре и в некоторых зонах центральной нервной системы, тогда как популяции типа В находятся особенно в центральной нервной системе.
Гастрин является полипептидным гормоном, который воздействует, в частности, на кислотную секрецию желудка; его 5 С-концевых аминокислот являются идентичными аминокислотами ХЦК.
Уже были описаны соединения, являющиеся антагонистами по отношению к гастрину и/или ХЦК, в частности, проглумид, п-хлор-бензоил-L-триптофан, или совсем недавно, производные бензо-диазепинов, являющиеся специфичными антагонистами либо рецепторов ХЦК А, такие как 3S[-(N-2-) 1-метил-2-оксо-5-фенил- 2, 3-дигидро-1Н-1, 4-бензодиазепин-3-ил] -2-индолкарбокса- мид ( J.Med. Chem. 1988, 31, 2235-46), либо рецепторов ХЦК В, такие как 3R(+)-N-(1-метил-2-оксо-5-фенил-2,3-дигидро-1Н-1, 4-бен- зодиазепин-3- ил)-N'-(3-метилфенил)-карбамид (Eur, J. Pharmacology, 1989, 162, 273-280).
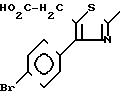
С другой стороны, производные тиазола с формулой А.
H
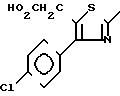
Другие производные тиазола с формулой В
Другие производные тиазола с формулой С
Производные 4-хинолинкарбоксамидов с формулой D

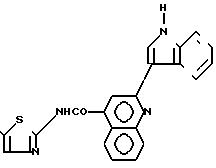
Другие производные тиазола, обладающие свойствами анти-ХЦК, описаны в патенте ЕП-А-0 432 040.
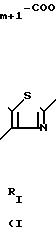
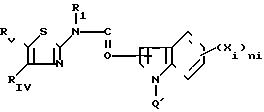
Соединениями по изобретению
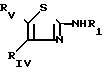
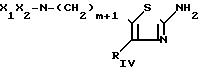
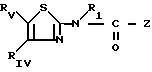
являются гетероциклические производные 2аминотиазолов с формулой I
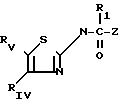
RIV представляет собой циклоалкильную группу С3-С7, незамещенную или замещенную одной или несколькими алкильными группами С1 -С4; ароматическую группу, такую как фенил, незамещенный или имеющий один или несколько заместителей, выбираемых среди атомов галогена, в частности, хлора или фтора, (С1 -С6) алкильных и алкокси- и тиоалкоксигрупп С1-С3нитро- и трифторметильных групп, или такую как гетероцикл, содержащий, по меньшей мере, один гетероатом, выбираемый среди О, S и N, в частности, фурил, тиенил, пирролил, пиразолил, имидазолил, пиридил, пиразинил, оксазолил и тиазолил, замещенные, в случае необходимости, алкильной группой С1-С3 или атомом галогена или алкоксигруппой С1 -С3, или RIV и RV, рассматриваемые вместе, представляет собой группу:
RV представляет собой группу -(СН2)m-X, в которой m составляет от 0 до 5, а Х представляет собой;
атом галогена, предпочтительно атом брома, гидроксил, циклоалкил С3-С7, фенил, который может быть замещен одной из групп, выбираемых среди атомов галогена, алкильных или алкоксигрупп С1-С3, нитро-, амино-, гидрокси- или трифторметильных групп;
группу, выбираемую среди -СООН; -СООХ1; -О-СОХ1-S-COX1; (O)q -S-X1, где q 0, 1 или 2; -О-СООХ1 -
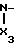




группу, выбираемую среди -CONX1X2; -NX1X2, в которой Х1представляет собой водород, алкил С1-С3 или фенил, незамещенный или замещенный одной или несколькими группами, выбираемыми среди атомов галогена, алкильных или алкоксигрупп С1-С3, нитро-, амино-, гирокси-, трифторметильных групп, а Х2 представляет собой атом водорода, алкил С1-С3, или Х1 и Х2 вместе с атомом азота, c которым они связаны, образуют гетероцикл, выбираемый среди пирролидина или пепиридина, незамещенного или замещенного оксогруппой или гидроксильной группой, причем последний является незамещенным или замещенным ацилом, группой -СООХ1 или -СОNX1X2;
или еще RV представляет собой алкоксигруппу С1-С5; гидроксильную группу; 5- или 6-членный циклический амин, незамещенный или замещенный оксогруппой или гидроксильной группой; пиперазинильную группу, незамещенную или N-замещенную группой -СООAlк, в которой Alк представляет собой алкил С1-С5; группу карбоновой кислоты; группу -NX2X4, где Х4 Н или Х4 -(CH2)t-X5, где t равняется 2, 3 или 4, а Х5 представляет собой -ОН, -O-CO-R2 , -NHCOR2, -NH-

а также соли присоединения этих соединений с минеральными или органическими кислотами и основаниями; причем получаются нетоксичные фармацевтически приемлемые соли, но и другие соли, применяемые для выделения или для очистки соединений с формулой I, также являются объектом изобретения.
Алкильные, алкиленовые, алкокси- и триоалкоксигруппы могут быть линейными или разветвленными.
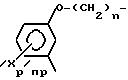
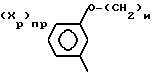
Z представляет собой, в частности, бензотиенильную, бензофуранильную, бензоксазолильную, бензимидазолильную, бензотиазолильную, индолильную, изоиндолильную, индолинильную, изоиндолинильную, хинолильную, изохинолильную хиноксалинильную, хиназолинильную, циннолинильную и (2,3-с)-пиридильную группы.
Когда Z представляет собой индолильную группу с формулой:
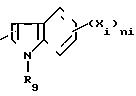
Среди соединений с формулой (I) предпочитают те, в которых R1представляет собой водород, а среди последних, в частности, те, в которых Z представляет собой индолильную группу, незамещенную или замещенную на азоте; среди групп RIV предпочитают фенил.
Объектом изобретения является также
получение
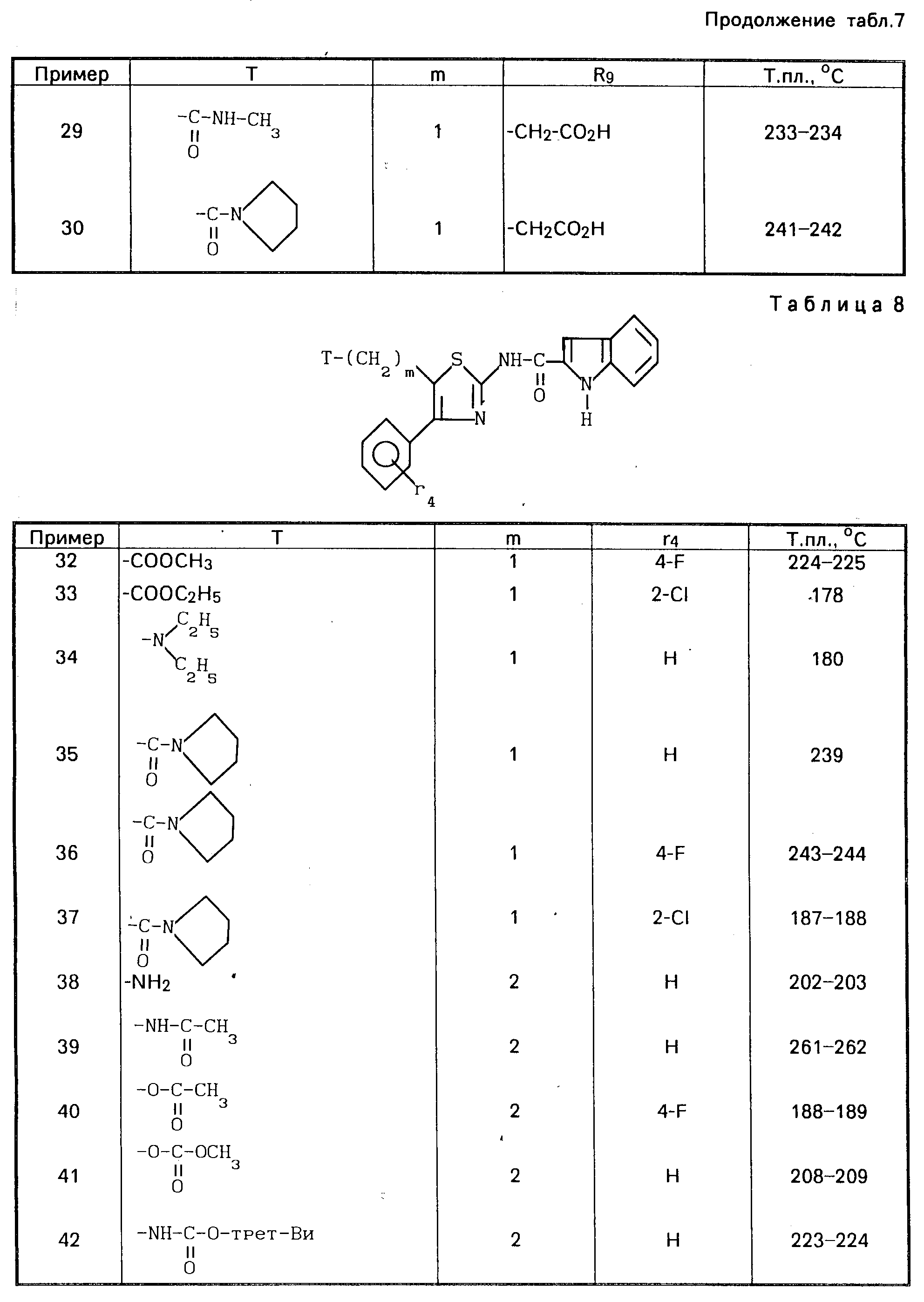
соединений с формулой (I),
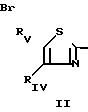
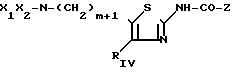
которые образуются в результате реакции конденсации аминотиазола с формулой II
Соединения II могут быть защищены; R в таком случае представляет собой те же самые заместители, что и R1, в котором имеющаяся аминогруппа является N-защищенной, а RIVa и RVa представляют собой те же самые заместители, что и RIV и RV, в которых гидрокси- или аминогруппы являются О- или N-защищенными.
Если функции были защищены, то после конденсации осуществляют, при необходимости, соответствующую реакцию снятия защиты.
Многочисленные аминотиазы с формулой II являются известными.
Новые аминотиазолы могут быть получены по одному из способов, описанных, в частности, в статье Bull. Soc. Chim. (С), 1963, 2498-2503.
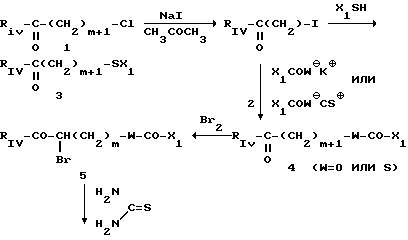
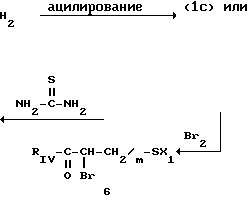
В общем случае будет проводиться реакция тиомочевины с альфа-галогенированным кетоном,
предпочтительно, с альфа-бромированным кетоном,
в соответствии со
следующей реакционной схемой
CXЕМА 1
S C

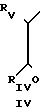
Получение различных соединений II, в которых R1 представляет собой аминоалкильную группу, описано в патенте ЕП-А-О 283390.
Альфа-галогенированные кетоны и тиомочевины могут быть получены при помощи способов, принципы которых описаны в общих руководствах; так альфа-бромированные кетоны IV могут быть получены при действии на RVCH2CORIV брома в уксуснокислой среде или бромида двухвалентной меди в органическом растворителе, таком как этилацетат, хлоросодержащий растворитель или их смеси, Исходные ароматические кетоны получаются в общем случае по реакции Фриделя-Крафтса, тогда как алифатические метилкетоны могут быть получены при действии диазометана на соответствующие хлорангидриды карбоновых кислот с последующим гидролизом полученного диазокетона.
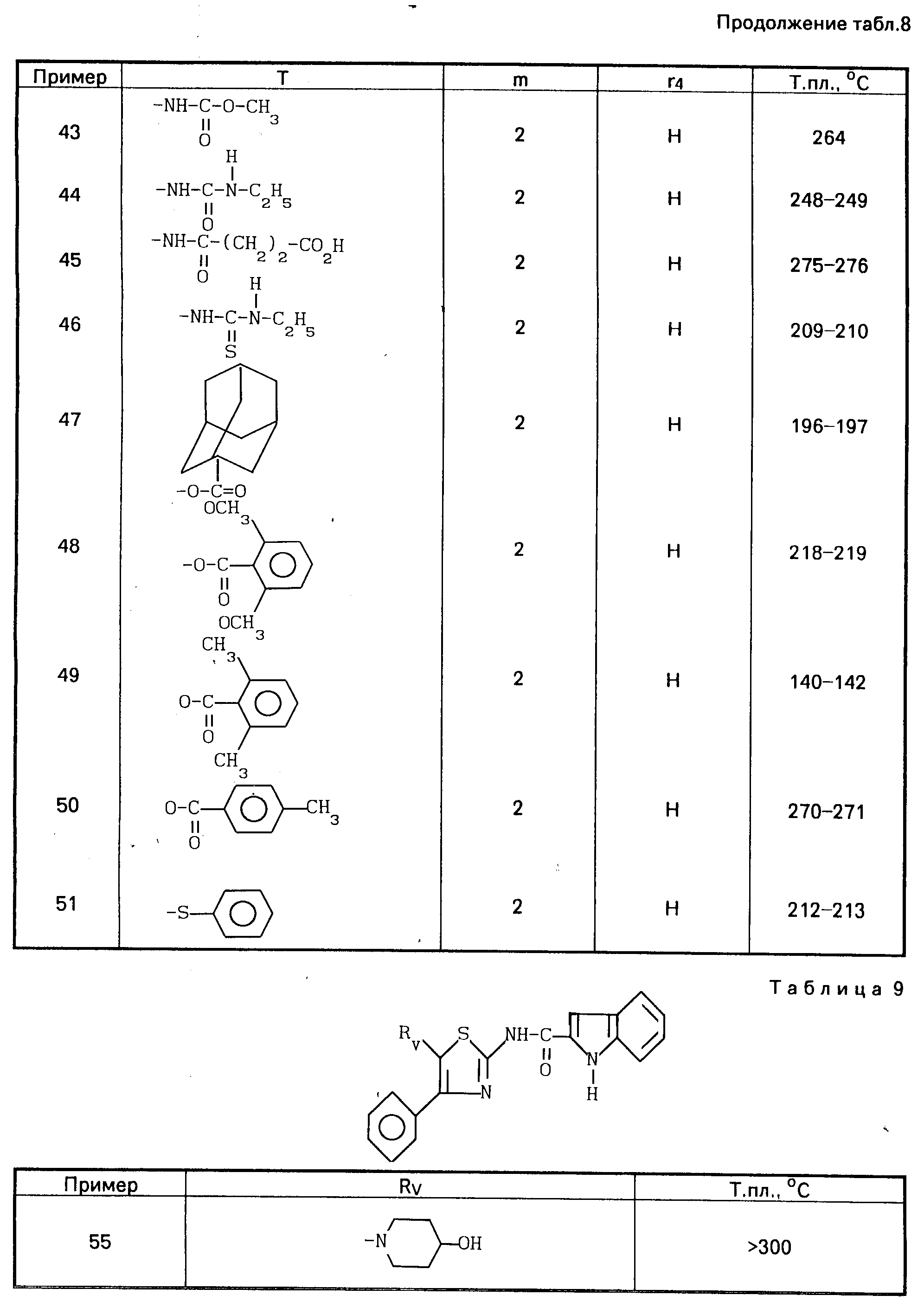
Альфа-хлорированные ароматические кетоны могут быть получены по реакции Фриделя-Крафтса с подходящим хлорангидридом альфа-хлорированной кислоты.
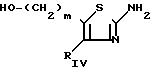
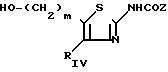
Когда
RV представляет собой сложноэфирную группу (CH2)m СООХ1, то
производные тиазола, соответствующие приведенной ниже формуле (V), для которых RIV,
X1 и m являются такими, как определено для соединений (I), являются известными
или получаются
по известным способам в результате воздействия альфа-бромацетокислоты или альфа-бромкетоэфира
на тиомочевину в соответствии со схемой 2
CXЕМА 2
R
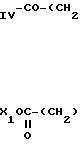
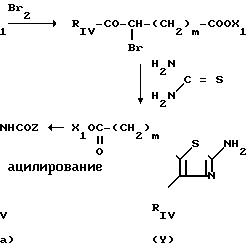
В зависимости от вида заместителя RV используются следующие методы получения:
а) когда RV представляет собой группу -(CH2)m-ОН, то соответствующее производное 2 аминотиазола с приведенной ниже формулой (VI), в которой m является таким, как определено для соединения (I), может быть получено, исходя из предыдущих сложных эфиров (V) в результате восстановления при помощи гидрида щелочного металла, такого как, например, алюмогидрид лития, в апротонном растворителе, таком как, например, тетрагидрофуран, что приводит к аминоспирту с формулой VI
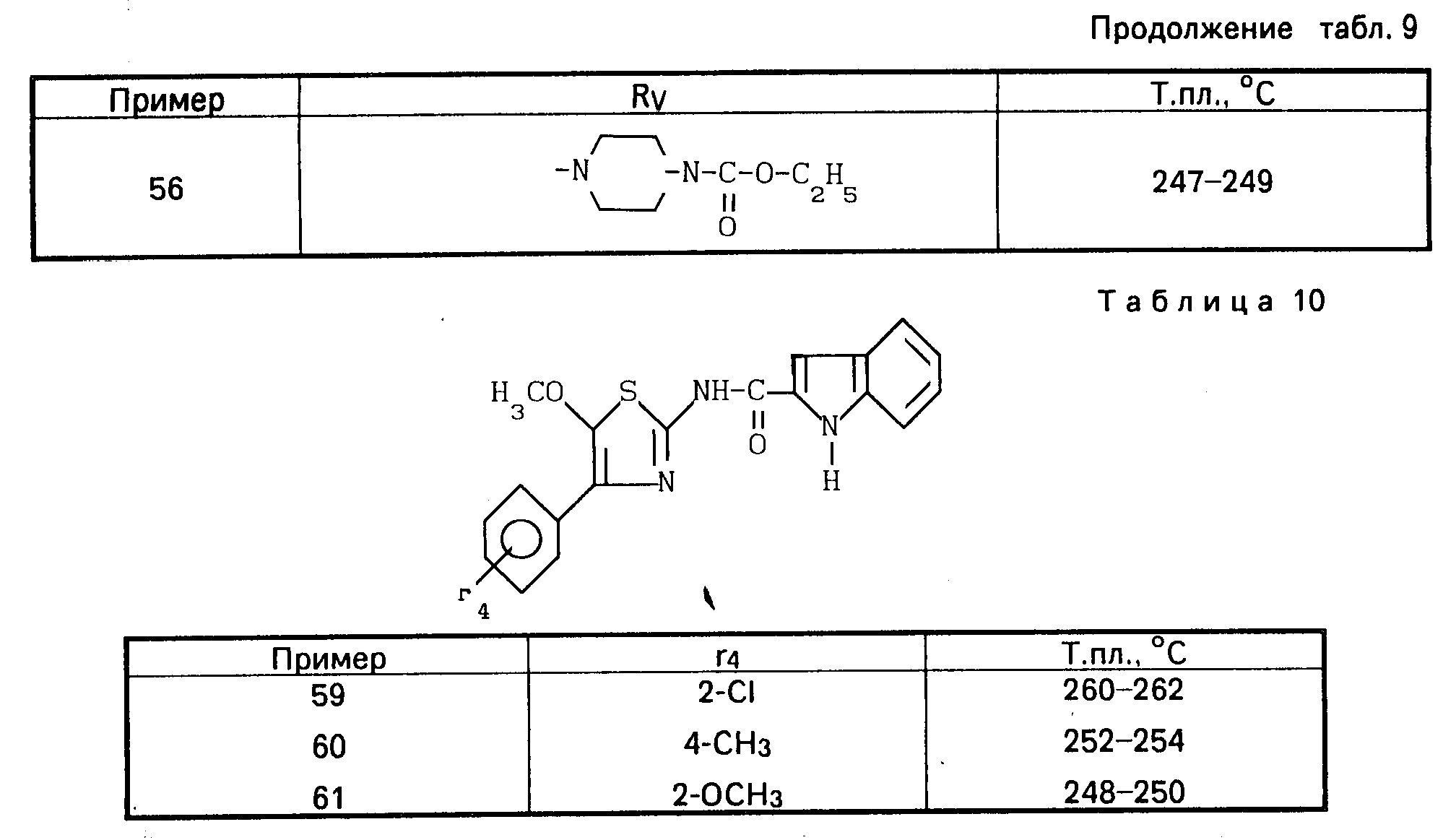
в) когда Ry представляет собой группу сложного эфира -(СH2)m-О-СО-Х1 или сложного тиоэфира -(СН2 )m-S-СОХ1 или группу (О)q-S-X1, в которых m, Х1 и q являются такими, как определено для формулы (I), то производные 2-аминотиазола (VII), (VIIc) или (VIId), в которых RIV q, m, W и Х1 являются такими, как определено для формулы (I), могут быть получены либо в соответствии со следующей схемой 3:
CXЕМА 3
X
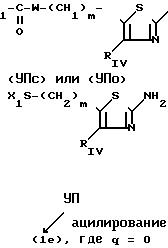
либо исходя из N-замещенных спиртов (VI), таких как определено выше, на которые воздействуют хлорангидридом кислоты, таким как, например, ацетилхлорид, в растворителе, таком как, например, пиридин, с целью получения сложных эфиров с формулой Ic
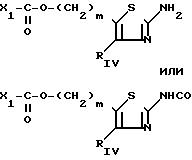
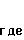



для которых Х1; m, RIV или Z являются такими, как определено выше для соединения (I):
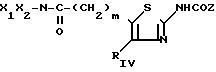
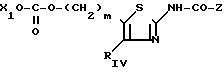
с) когда Rу представляет собой карбамат -(СН2 )m-О-СО-NHX1, в котором m и Х1 являются такими, как определено для соединения (I), то производные тиазола по изобретению получают, исходя из соответствующих гидроксилированных соединений (Iв) при действии изоцианата с формулой X1-N С= О в апротонном растворителе, таком, как например, тетрагидрофуран или дихлорметан, при температуре, заключенной между 20 и 100оС, что приводит к соединению (If) с формулой:
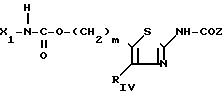
d) когда RV представляет собой амид- (СН2)m-CONX1X2в котором m, Х1 и Х2 являются такими, как определено для соединения (I), то тиазолы по изобретению получают в результате реакции между амином HNX1X2 и соответствующим сложным эфиром с формулой (V) или (Ia) в присутствии без растворителя, такого как алканол, при температуре, заключенной между 20 и 120оС; реакция может также осуществляться в герметически закрытом сосуде в зависимости от того, является ли амин летучим, что приводит к соединению (VIII) или (Id) с формулами:
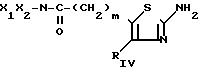
е) когда RV представляет собой аминогруппу -(СН2)m-N-X1 X2, то производные тиазола по изобретению получают, например, в результате восстановления описанных амидов с формулой (VIII), восстанавливая при помощи гидрида щелочного металла такого, как, например, алюмогидрид лития, в растворителе, таком как тетрагидрофуран, при температуре, заключенной между 20оС и температурой кипения растворителя, что приводит к соединению с формулой IX
f) когда RV представляет собой карбонат -(CH2)m-O-COOX1, в котором m и Х1 являются такими, как определено для соединения (I), то тиазолы по изобретению получают, исходя из спиртов (Ib), вводя их в реакцию с хлорформиатом Cl-

g) когда RIV и RV, рассматриваемые вместе, представляют собой группу:
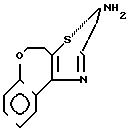
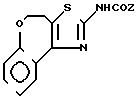
h) когда RV представляет собой аминогруппу -NX2X4, в которой Х2 и Х4 являются такими, как определено для соединения (I), то производное тиазола по изобретению может быть получено, исходя из 2-амино-5-бромтиазола с формулой XI
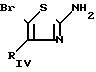
либо замещением амином HNX2X4, а затем ацилируется в положении 2 тиазола, причем обе реакции проводятся в условиях, идентичных тем, которые описаны выше
i) когда RV представляет собой группу -(СН2)m-X, в которой m=0, а Х представляет собой алкоксигруппу С1-С5, то соответствующий 2-аминотиазол получается, исходя из 2-бром-2-алкокси-1-фенилэтанона, замещенного, в случае необходимости, на фениле, что приводит к продукту с формулой XI'
Соединения с формулой XI' являются новыми промежуточными продуктами, составляющими часть изобретения.
Некоторые из кислот ZCOOH или Z'COOH являются известными и даже являются коммерческими; другие получают, используя способы, известные для аналогичных молекул.
Так, индолкарбоновые кислоты, обозначаемые ниже Z"COOH, с
формулой:
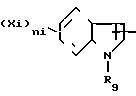
СХЕМА 4
(Xi)
Бензиловые сложные эфиры по схеме 4 получаются при действии соответствующей кислоты на
бензиловый спирт в
присутствии соответствующей кислоты на бензиловый спирт в присутствии
агентов, активирующих кислотную функцию, которые обычно используются в пептидном синтезе, таких как:
1,
1'-карбонилдиимидазол, для которого можно будет сослаться на статью Synthesis, 1982, р.
833;
N, N-дициклогексилкарбодиимид в присутствии 4-диметиламинопиридина, для которого можно будет
сослаться на статью J.Org. Chem, 1990, 54 (4), р. 1390;
гексафторфосфат
бензотриазолилокси-трис-(диметиламино-фосфония) (БОФ), для которого можно
будет сослаться на статью Synthesis, 1977,
р. 413.
Основание, используемое при фиксировании R9 на азоте бензилового сложного эфира, является предпочтительно сильным безводным основанием, таким, как гидрид щелочного металла; в таком случае реакционная смесь является полярным апротонным растворителем, стабильным в присутствии сильного основания, таким как диметилформамид или диметоксиэтан; реакция осуществляется при температуре, заключенной приблизительно между 15 и 80о С.
Отщепление бензильной группы после N-алкилирования осуществляется классическим способом под действием по меньшей мере одного эквивалента водорода в присутствии катализатора, такого как палладий на активированном угле, на сложный эфир, растворенный в спирте или в диметилформамиде, в случае необходимости, под небольшим давлением.
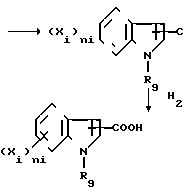
Вместе с тем, некоторые из кислот ZCOOH являются малостабильными или имеют функцию, которая сможет реагировать при конденсации с аминотиазолом, поэтому является предпочтительным использовать их в защищенной форме Z'COOH.
Так, производные (I), в которых Z представляет собой:
(Xi)
(Xi)
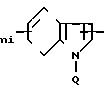
-COO/трет-C4H9/; C6H5 -CH2-O-


БОФ может отщепляться при пиролизе в отсутствие растворителя при температуре, заключенной между 180 и 200оС.
Индолкарбоновые кислоты Z"COOH, в которых R9 является группой COOC(CH3)3 или COOCH2 C6H5, могут быть получены при действии дикарбоната трет-бутила, хлорформиата бензила на Z"COOH, в которой R9 Н, в присутствии основания, такого как триэтиламин или 4-диметиламинопиридин, в растворителе, таком как ацетонитрил или метиленхлорид.
Кислоты Z"COOH, в которых R9 является ацильной группой, могут быть получены при действии хлорангидрида или ангидрида, кислоты Z"COOH, в которой R9 H, в присутствии одного эквивалента триэтиламина и 4-диметиламинопиридина, например, в дихлорметане.
Хлорангидрид кислоты с формулой ZCOCl может быть получен, в частности, при действии SOCl2 или смеси POCl3 и Р2О5 на соответствующую кислоту обычно в отсутствие растворителя и при температуре образования флегмы реакционной смеси.
Среди активированных сложных
эфиров с формулой ZCOOY", Z'COOY" или Z"COOY" можно получить такие, в которых Y"
представляет собой:
Реакция конденсации аминотиазола II с кислотой в форме активированного
сложного эфира может проводиться в растворителе, природа
которого
выбирается в зависимости от растворимости соединений и от типа активации кислотной функции,
предпочтительно в присутствии
основания, например, третичного амина, такого как
триэтиламин;
реакция обычно осуществляется при температуре, заключенной между 0 и 30оС.
Когда соединения с формулой (I) содержат в Z группу карбоновой кислоты, то они могут быть получены результате гидролиза, сложного эфира предпочтительно в основной среде, например, при действии минерального основания, такого как гидроксид щелочного металла, в водно-спиртовой среде или в результате кислотного гидролиза в случае трет-бутилового сложного эфира.
Соли присоединения соединений с формулой (I) с кислотами или основаниями получают обычным способом путем введения кислоты или основания в раствор соединения с формулой (I). Соль выделяется в зависимости от ее растворимостных характеристик после выпаривания растворителя или после прибавления нерастворителя.
Соединения с формулой (I) и их соли ингибируют связывание холецистокинина со своими рецепторами. Они являются более или менее селективными по отношению к рецепторам типа А или В и более или менее мощными антагонистами по отношению к гастрину.
Их сродство к рецептору ХЦК А было определено in vitro при использовании метода, описанного ниже, принцип которого является таким же, как упомянутый в статье Life Sciences, 1985, 37, (26), 2483-2490; он заключается в определении вытеснения иодированного ХЦК 8 S со своих центров связывания на гомогенизате поджелудочной железы крыс: аликвотные количеcтва суспензии из оболочки поджелудочной железы (100 мкг протеинов на мл) в буферном растворе Триса, HCl (50 мМ), рН 7,4, содержащем MgCl2 (5 мМ), бацитрацин (9,1 мг/мл), фторангидрид метилфенилментансульфокислоты (0,1 мг/мл) подвергаются инкубации в течение 40 минут при температуре 25о С в присутствии иодированного ХЦК 8 S (200 Ки/ммоль, т.е. 50 мМ конечной концентрации) и возрастающих концентраций изучаемого вещества; реакция прекращается по истечении 40 мин в результате центрифугирования. После удаления всплывшей фазы измеряется радиоактивность остатка. С другой стороны, неспецифическое связывание определяется в присутствии ХЦK 8 S с концентрацией 1 мкМ.
При этих условиях концентрация, ингибирующая на 50% связывание (Cl50), составляет для продуктов по изобретению менее 10-7 М, а для большого их числа она составляет порядка 10-10 М.
Их сродство к рецепторам ХЦК В было определено при изучении вытеснения иодированного ХЦК 8 S со своих специфических центров связывания, находящихся в гомогенизатах из коры головного мозга морских свинок, путем применения той же самой методики, что и для рецепторов ХЦК А, но для суспензии из оболочек с концентрацией 600 мкг протеинов/мл и с буферным раствором Гепеса (10 мМ) при значении рН 6,5, содержащим NaCl (130 мМ), MgCl2 (5 мМ), ЭДТА (1 мМ) и бацитрацин (250 мг/мл), причем инкубация проводится в течение 2 ч.
При концентрации 10-5 М все продукты вытесняют более 25% меченного ХЦК 8 S из рецепторов В; некоторые имеют Cl50 порядка 10-9 М.
Средство к рецептору гастрина для соединений, наиболее специфичных по отношению к рецептору ХЦК В, было изучено в соответствии со способом, описанным ниже, принцип которого является таким же, как упомянутый в J. Receptor, Res. 1983, 3 (5), 647-655; аликвотные количества желудочных желез морских свинок в буферном растворе Гепеса при значении рН 7,4 (24,5 мМ); содержащем NaCl (98 мМ), HCl (6 мМ); NaH2PO4 (2,5 мМ). пируват (5 мМ), CaCl2 (0,5 мМ), MgCl2 (1 мМ), глюкозу (11,5 мМ), глутамин (1 мМ), бычий альбумин (0,4 г/100 мл), подвергались инкубации в течение 90 мин при 37оС в солевом растворе в присутствии гастрина (2-17), меченного иодом (2000 Ки/ммоль; 70 рМ) и с возрастающими концентрациями изучаемых продуктов. Реакция прекращалась в результате центрифугирования, затем измерялась радиоактивность остатка после центрифугирования; неспецифическое связывание определялось в присутствии гастрина (2-17) при концентрации 1 мкМ. Соединения по изобретению имеют Cl50 в интервале между 10-5 и 10-9 М.
Было также показано, что соединения по изобретению оказывают ингибирующее действие на активность ХЦК. Это было выявлено in vitro путем измерения под действием испытываемых продуктов ингибирования секреции амилазы ацинусами крыс, стимулированной посредством ХЦК 8 S, в соответствии с методом, аналогичным тому, который описан в статье J. Biol. Chem. 1979, 254, (12) 5321-5327, но с панкреатическими тканями морских свинок. Соединения имеют значения Cl50 в интервале от 10-6 до 10-9 М.
Наконец, у мышей in vivo соединения, имеющие хорошее сродство к рецепторам ХЦК А, оказывали антагонистическое действие на ингибирование опорожнения желудка, вызываемое подкожным введением ХЦК 8 S в соответствии с протоколом опыта, описанным в статье Life Sciences, 1986, 39, 1631-1638; определенная таким образом ДЕ50 (доза с эффективностью 50%) строго меньше аналогичной величины для проглумида, известного антагониста гастрина.
Поскольку эти соединения являются малотоксичными, то они могут использоваться в качестве лекарственных средств для лечения физиологических нарушений, обусловленных гиперсекрецией этих пептидов или расстройством биологических гормональных систем, в которых они задействованы на уровне кишечной сферы или в центральной нервной системе в зависимости от их специфичности. Можно сослаться на обзор терапевтических применений антагонистов ХЦК и гастрина, опубликованный в "Рrоceedings of International Symposium on Gastrin and cholecyctokinin 7-11 сентября 1987 г. Ed. J. P. Bali, J. Martines Elsevier Science Pub. B. BV.
В частности, антагонисты ХЦК будут полезными при лечении кишечных дискинезий, таких как синдром раздраженной ободочной кишки, при лечении острых или хронических панкреатитов или при лечении панкреатических карцином (раковых опухолей), а также для регуляции аппетита или в комбинации с опиумсодержащими болеутоляющими средствами при лечении боли.
Что касается более селективных антагонистов гастринов, то они будут полезными при лечении или для предупреждения желудочных язв, при лечении синдрома Золлингера-Эллисона, при лечении гиперплазий клеток Gr пазухи (полости) или для больных, имеющих раковые опухоли пищевода, желудка или кишечника.
Среди антагонистов
холецистокинина на уровне рецепторов А предпочитают соединения:
2-(2-карбоксиметил-2-индолил)-карбо-ниламино-4-фенил-5-ацетоксиэтилтиазол;
2-(2-индолил)-карбониламино-4-фенил-5-ацетоксиэтилазол.
Лекарственные средства по изобретению содержат по меньшей мере одно из соединений с формулой I или одну из его солей с фармацевтически приемлемыми кислотой или основанием, в случае необходимости, в комбинации с обычными индифферентными веществами для создания фармацевтической формы, вводимой классическим способом оральным, парентеральным ректальным путем или через слизистую оболочку. Вводимые дозы зависят от природы и степени болезни, от соединения и от пути введения. Они будут обычно заключаться между 20 и 100 мг в день для взрослого человека оральным путем и от 3 до 10 мг при инъекции.
Фармацевтические композиции по изобретению для орального введения могут находиться в виде таблеток, пилюль, желатоновых капсул или гранул или еще в виде раствора, суспензии или геля. Для парентерального введения композиции по изобретению будут находиться в виде раствора, суспензии или эмульсии в жидком месте или в любом растворителе для инъекции, в случае необходимости, на водной основе, содержащем классические добавки для этого типа рецептуры.
Для местного введения на кожу или на слизистые оболочки композиции по изобретению будут находиться через кожу, тогда как для ректального введения они будут в виде свечей или ректальных капсул.
Ниже описываются примеры осуществления изобретения, а также способы получения некоторых промежуточных продуктов синтеза с формулами II и IV. Указанные температуры плавления (Тпл) определялись в капилляре. Спектры ядерного магнитного резонанса (ЯМР) были зарегистрированы относительно тетраметилсилана.
Синтез А.
2-Аминотиазол, замещенный в положении 5 группой -(СН2)mX1.
2-Амино-4-(2,4-диметоксифенил)-5-бен- зилтиазол.
R1= H; RIV=
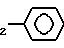
А) 1-(2, 4-Диметоксифенил)-3-фенилпропан-1-он получается в соответствии со статьей Е. Thomas et. al. J. Med. Chem. 1985, 28, 442-446 по реакции Фриделя-Крафтса.
В) 1-(2, 4-Диметоксифенил)-2-бром-3-фенилпропан-1-он получается в соответствии с классическими методами в результате бромирования бромом в растворителе, таком как дихлорметан или четыреххлористый углерод.
С) 2-Амино-4-(2,4-диметоксифенил)-5-бензилтиазол.
К 10 г полученного перед этим бромсодержащего производного, растворенного в 100 мл 95%-ного этанола, прибавляют 4,35 г тиомочевины и нагревают реакционную смесь при температуре образования флегмы в течение 3 ч. Концентрируют под вакуумом и извлекают остаток в дихлорметане, затем промывают насыщенным раствором Na2CO3. Органическая фаза отделяется путем декантации, сушится на MgSO4 и концентрируется под вакуумом. Остаток кристаллизуется в 50 мл дихлорметана.
m 7, 10 г.
Т.пл. 202-203оС.
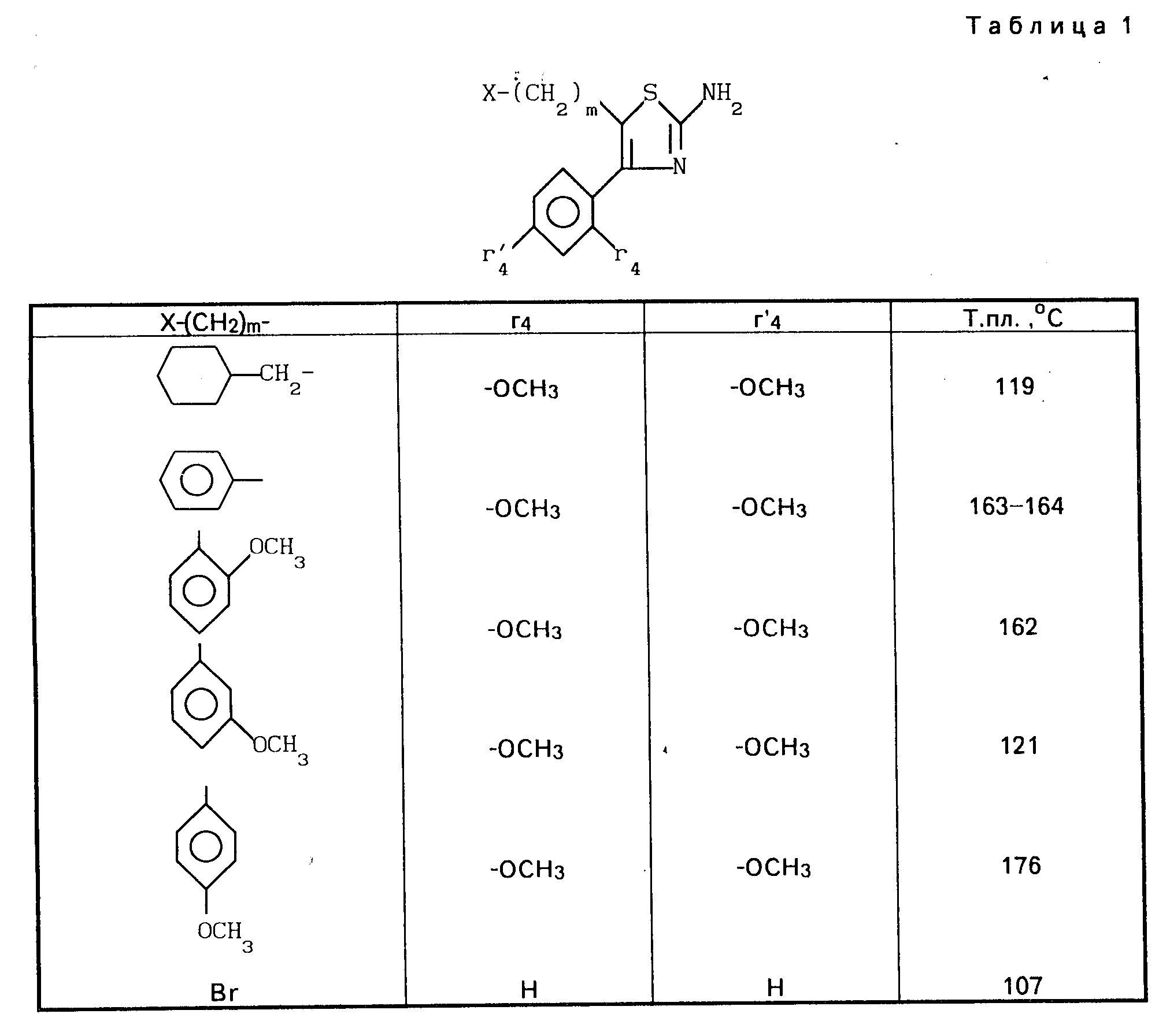
Проводя синтезы, как указано выше, получают 2-аминотиазолы, описанные в таблице 1, приведенной ниже.
Синтез В.
2-Аминотиазол, замещенный в положении 5 группой -(CH2)m-CO2X1 или -(СН2 )m-CH2OH.
А) 2-Амино-4-фенил-5-метоксикарбонилметилтиазол.
(II): R1 H; RIV -C5H6; RV
-CH2-CO2CH3
Получается в соответствии со статьей E. Knott, J. Chem. Soc. 1945, 455.
В) 2-Амино-4-фенил-5-гидроксиэтилтиазол.
К суспензии, содержащей 2 г алюмогидрида лития в 100 мл тетрагидрофурана, охлажденного до 0оС, прибавляют 5 г аминосодержащего сложного эфира, полученного перед этим, и нагревают реакционную смесь при температуре образования флегмы в течение 2 ч. Затем после охлаждения в ледяной бане прибавляют последовательно 2 мл воды, 1 мл концентрированного NaOH и 6 мл воды, потом перемешивают реакционную смесь в течение ночи. Отделяют минеральный осадок путем фильтрования и концентрируют маточные растворы под вакуумом. Остаток извлекается в дихлорметане, промывается водой, затем органическая фаза последовательно декантируется, сушится на MgSO4 и концентрируется под вакуумом. Остаток хроматографируется на силикагеле, элюент: дихлорметан/метанол 100/3 (V:V).
Концентрирование чистых фракций дает 4 г ожидаемого спирта.
Т.пл. 121оС.
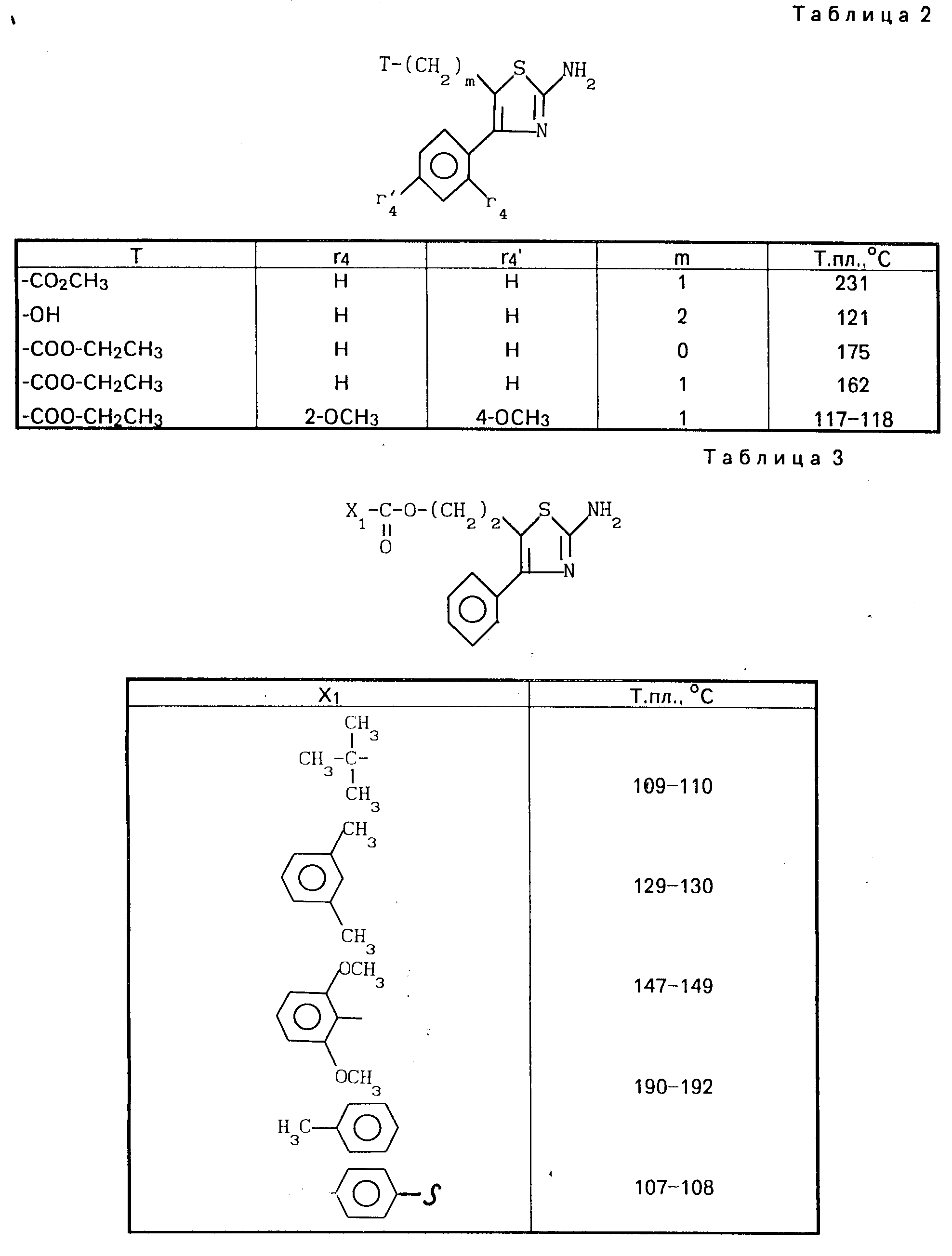
Проводя синтезы, как указано выше, получают 2-аминотиазолы, описанные в табл.2, приведенной ниже.
Синтез С.
2-Аминотиазолы, замещенные в положении 5 группой -(СН2)m-O-C-X1 или (СН2)m
-S-X1
2-Амино-5-(1-адамантил-1-карбонило-ксиэтил)-4-фенилтиазол.
RI= H; RIV= -C6H5; RV= -(CH2)2-O-
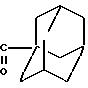
А) 4-(1-Адамантилкарбонилокси)-1-фенил-1-бутан.
Получают в соответствии со статьей J.Org. Chem. 1977, 42, 8, 1286 цезиевую соль 1-адамантилкарбоновой кислоты, исходя из 12 г 1-адамантилкарбоновой кислоты, и 10,96 г карбоната цезия. Полученная смесь растворяется в 70 мл ДМФА, затем прибавляют 18 г 4-иод-1-фенилбутан-1-она и нагревают реакционную смесь при температуре образования флегмы в течение ночи. Выпаривают ДМФА под вакуумом, извлекают, остаток в 5%-ном растворе Na2CO3 и экстрагируют в CH2Cl2. Органическая фаза промывается водой, а затем сушится на Na2SO4. Концентрируют под вакуумом и хроматографируют остаток на силикагеле, элюент: CH2Cl2.
Концентрирование чистых фракций дает 10 г ожидаемого соединения.
В) 2-Амино-5-(1-адамантил-1-карбонилоксиэтил)-4-фенил-тиазол.
Растворяют 10 г полученного перед этим соединения в 100 мл CCl4. Прибавляют 4,9 г брома, растворенного в 50 мл CCl4 и оставляют реакционную смесь при перемешивании в течение 30 мин. Промывают водой, декантируют органическую фазу, сушат ее на MgSO4, фильтруют и концентрируют под вакуумом.
Остаток извлекается в 50 мл 95%-ного этанола. К раствору прибавляют 3,9 г тиомочевины и оставляют реакционную смесь на ночь при температуре окружающей среды. Концентрируют смесь под вакуумом, извлекают остаток в CH2Cl2, промывают при помощи 5%-ного раствора NaHCO3, декантируют органическую фазу, сушат на MgSO4, фильтруют и концентрируют под вакуумом. Остаток извлекается в эфире и сушится.
m 6,8 г.
Т.пл. 167оС.
Проводя синтезы, как указано выше, получают 2-аминотиазолы, описанные в табл.3, приведенной ниже.
Синтез D.
2-Аминотиазолы, замещенные в положении 5 группой -(СН2)mХ,
в которой Х
представляет собой группу NX1X2, где X1 X2 ± H.
Получение 2-амино-5-аминоэтил-4-фенилтиазола.
(II): R1 H;
RIV -C6H5; RV -CH2CH2NH2
А) 4-фталимидо-1-фенилбутан-1-он.
Нагревают 27,4 г 4-иод-1-фенилбутан-1-она и 27 г калиевого фталимида при температуре 120оС в течение 24 часов в 100 мл ДМФА. Концентрируют ДМФА под вакуумом, а остаток последовательно промывается водой, 1 н.раствором NaOH и экстрагируется этилацетатом. Органические фазы декантируются, сушатся на MgSO4, фильтруются и концентрируются под вакуумом.
m 11 г.
В) 2-Амино-5-фталимидоэтил-4-фенилтиазол.
Растворяют 9,6 г полученного перед этим соединения в 50 мл CCl4 80 мл CH2Cl2. К раствору прибавляют по капле раствор, содержащий 5,6 г брома в 30 мл CCl4. Реакционная смесь промывается водой, органические фазы сушатся на MgSO4, фильтруются и концентрируются под вакуумом. Остаток извлекается в 70 мл этанола, затем прибавляют 4,5 г тиомочевины и оставляют реакционную смесь на ночь при температуре окружающей среды.
Смесь охлаждается, бромгидрат отделяется путем фильтрования, промывается этанолом, затем интенсивно перемешивается в смеси 5%-ный Na2CO3/этиловый эфир. Отфильтровывают кристаллы.
m 8 г.
Т.пл. 209оС.
С) 2-Амино-5-аминоэтил-4-фенилтиазол.
Обрабатывают 8 г полученного перед продукта посредством 1,5 г гидрата гидразина, растворенного в 100 мл абсолютного этанола. Реакционная смесь нагревается при температуре образования флегмы в течение ночи, затем последовательно концентрируют этанол под вакуумом, извлекают остаток в воде, подкисляют путем добавления концентрированной HCl до значения рН 1, отделяют фталазиндион путем фильтрования, подщелачивают охлажденную в ледяной бане водную фазу путем прибавления концентрированного NaOH до значения рН 9, отфильтровывают осадок, промывают водой и сушат в сушильном шкафу.
m 3,7 г.
Т.пл. 136-137оС.
Синтез Е.
2-Аминотиазолы, замещенные в положении 5 группой -(СН2)mХ, в которой Х представляет собой группу -NX1X2,
где Х1 Н и Х2
-СО-СН3.
2-Амино-5-(2-ацетиламино-1-этил)-4-фе- нилтиазол.
(II): R1 H; RIV -C6H5;
Rv
CH3CONH(CH2)2
Обрабатывают 1 г полученного согласно синтезу Д 2-аминотиазола, растворенного в 60 мл ТГФ в присутствии 0,7 мл триэтиламина, посредством 0,44
мл
уксусного ангидрида, растворенного в 20 мл ТГФ. Реакционная смесь оставляется при температуре окружающей среды в течение 2 ч и концентрируется под вакуумом. Остаток промывается при помощи 5%-ного
раствора NaHCO3, осадок отделяют путем фильтрования, промывают водой и сушат.
m 1,12 г
Т.пл. 208-209оС.
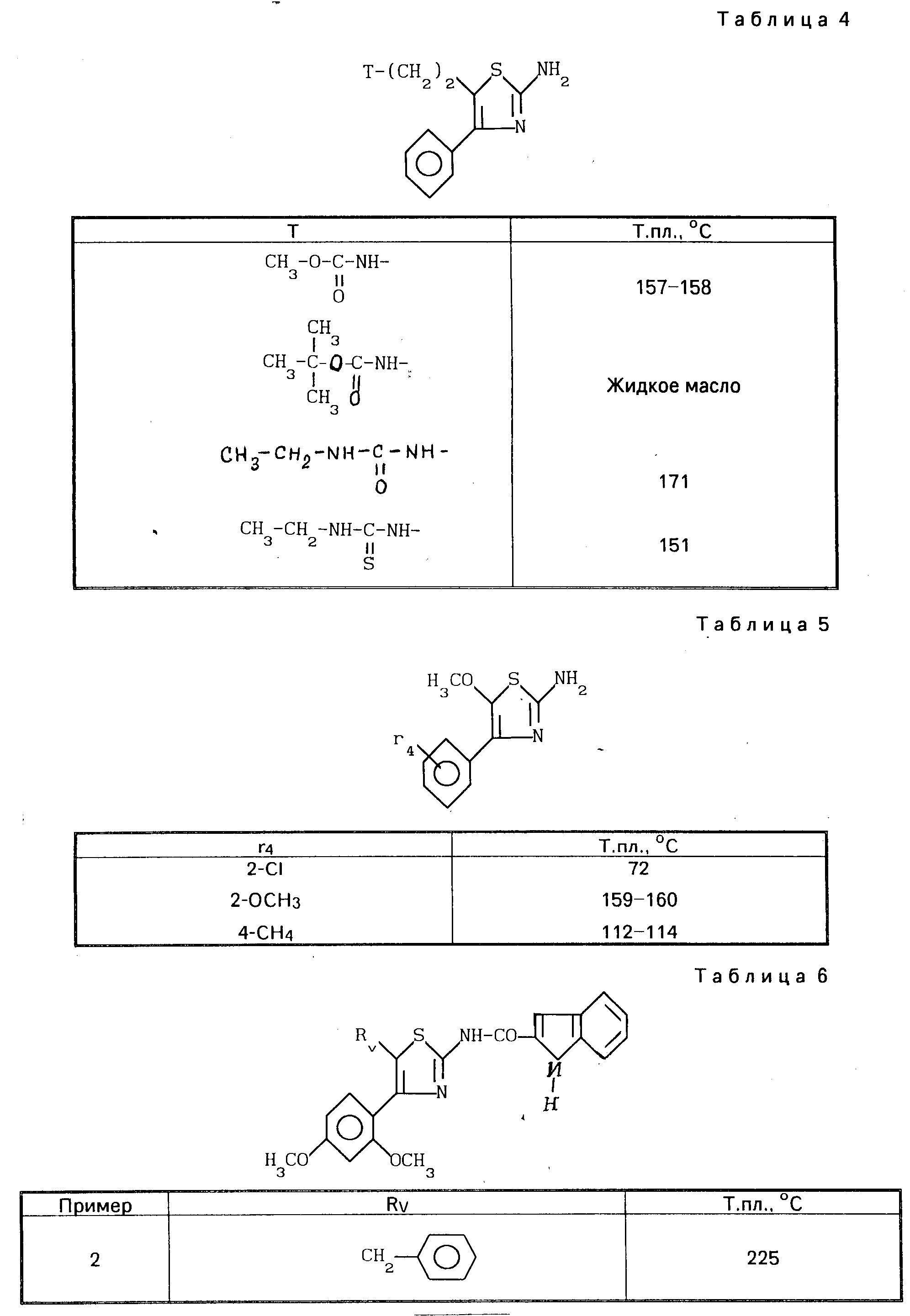
Исходя из полученного согласно синтезу Д 2-аминотиазола и проводя синтезы согласно синтезу Е, получают промежуточные соединения, описанные в табл. 4, приведенной ниже.
Синтез F.
2-Амино-4,
5-дигидро-[5,4-d]-тиазоло(-)1-бензоксэпин II
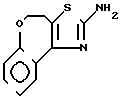
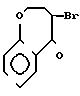
А) 4-Бром-4-2Н-3, 4-дигидро-(1)-бензоксэпин-5-он получают в соответствии со статьей G.Fontaine, P.Maite, C.R.Acad, Sci, 1964, 258, 4583.
В) 2-Амино-4,5-дигидро-[5,4-d]-тиазоло-(1)-бензоксэпин
К 0,027 моль бромсодержащего
производного, растворенного в 100 мл этанола, прибавляют 2,05 г тиомочевины.
Смесь нагревается при температуре образования флегмы в течение 3 ч. Этанол выпаривается, остаток извлекается водным раствором карбоната натрия. Экстрагируют этилацетатом, сушат органическую фазу на Na2SO4 и выпаривают досуха. Получают 2,4 г кристаллов белого цвета.
Т.пл. 216оС.
Синтез G.
2-Аминотиазолы, замещенные в положении 5 группой -(СН2)m-Х, в которой m 0, а Х
представляет собой
алкоксигруппу С1-С5, галоген
Реакционная схема
Стадия 1:
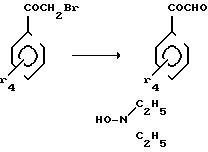
Стадия 2:
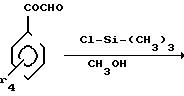
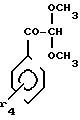
Стадия 3:
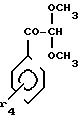
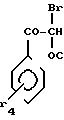
2-Амино-5-метокси-4-фенилтиазол.
(II):
R1 H; RIV -C6H5; RV -OCH3
Растворяют 15,65 г
2-бром-2-метокси-1-фенилэтанола и 5,52 г тиомочевины в 70 мл метанола.
Реакционная смесь нагревается при температуре образования флегмы в течение ночи, затем концентрируется под вакуумом. Остаток
извлекается в 10% -ном водном растворе Na2CO3,
экстрагируют при помощи CH2Cl2, отделяют органическую фазу, которая последовательно сушится на Na2SO4, фильтруется и концентрируется под вакуумом. Остаток
перекристаллизовывается из изопропилового эфира.
m 7,5 г
Т.пл. 96оС.
Синтез Н.
Получение индолкарбоновых кислот:
А)
1-Трет-бутилоксикарбонилметилиндол-2-карбоновая кислота
а) Индол-2-карбоксилат бензила.
Вводят 5 г N,N'-карбонилдиимидазола в раствор, содержащий 5 г индол-2 карбоновой кислоты в 50 мл безводного тетрагидрофурана; после перемешивания в течение 12 ч при температуре окружающей среды прибавляют 3,7 г бензилового спирта и нагревают реакционную смесь до температуры образования ее флегмы; последняя поддерживается в течение 8 ч перед тем, как удалить растворитель путем перегонки при пониженном давлении. Остаток растворяется в этилацетате, а органическая фаза промывается 1 н. водным раствором NaOH, затем сушится перед выпариванием растворителя.
Остаток желтого цвета перекристаллизовывается в изопропаноле.
Т.пл. 136оС.
Выход 85%
в) 1-Трет-бутилоксикарбонилметилиндол-2-карбоксилат-бензила.
К раствору, содержащему индол-2-карбоксилат бензила (0,072 моль; 18,18 г) в 200 мл диметилформамида, прибавляют в атмосфере азота порциями гидрид натрия (0,075 моль; 2,25 г), находящийся в жидком масле с концентрацией 80% при температуре, заключенной между 0 и 5о С. Дают смеси вернуться к температуре окружающей среды и перемешивают смесь в течение 1 ч. Затем прибавляют по капле при 10оС 14 г (0,072 моль) бромацетата трет-бутила. Реакционная смесь оставляется на 3 ч при температуре окружающей среды. Выпаривают диметилформамид, затем последовательно извлекают в воде, экстрагируют метиленхлоридом, сушат органическую фазу на сульфате натрия и выпаривают досуха. В результате кристаллизации остатка в диизопропиловом эфире получают 23,8 г кристаллов белого цвета.
Т.пл. 95оС.
с) 2-Трет-бутоксикарбонилметилиндол-2-карбоновая кислота.
Полученный перед этим сложный эфир (0,065 моль; 23,8 г) растворяется в смеси, состоящей из 400 мл этанола и 100 мл диметилформамида. Прибавляют 1 г палладия, нанесенного на активированный уголь с содержанием 5% и гидрируют под атмосферным давлением при температуре окружающей среды. После перемешивания в течение 30 мин поглощается теоретически рассчитанный объем водорода. Отфильтровывают на тальке катализатор, выпаривают досуха растворители. Получают кристаллический остаток, который промывают диизопропиловым эфиром. Получают 15,3 г кристаллов белого цвета.
Т.пл. 177оС.
В) 1-Ацетилиндол-2-карбоновая кислота.
Смесь, состоящая из индол-2-карбоновой кислоты (0,06 моль; 10 г), триэтиламина (0,15 моль; 21,25 г), уксусного ангидрида (0,075 моль; 7,5 г) и 4-диметиламинопиридина (0,006 моль; 0,8 г) в метиленхлориде, перемешивается при температуре окружающей среды в течение 18 ч. Затем реакционная смесь приливается к водному буферному раствору со значением рН 2.
Образовавшийся осадок отфильтровывается, потом сушится в сушильном шкафу под вакуумом. Фаза на основе метиленхлорида декантируется, сушится на сульфате натрия и выпаривается на 3/4. Осаждается вторая партия 1-ацетилиндол-2-карбоновой кислоты. Обе партии объединяются, что дает 9,4 г кристаллов бежевого цвета.
Т.пл.168оС.
С) 1-Бензилоксикарбониламиноиндол-2-карбоновая кислота.
Растворяют 8 г индола-2-карбоновой кислоты в 120 мл дихлорметана, затем прибавляют 10 г триэтиламина и 1 г диметиламинопиридина.
Реакционная смесь перемешивается, затем охлаждается до 0-5оС. Прибавляют по капле 8,5 г бензилоксикарбонилхлорида при температуре менее 5о С. Смесь оставляется при перемешивании на ночь, затем концентрируется под вакуумом. Остаток извлекается в 500 мл этилацетата, затем фильтруется. Маточные растворы концентрируются под вакуумом и извлекаются в 50 мл дихлорметана. Фильтруют и концентрируют маточные растворы под вакуумом.
m 2,4 г жидкого масла.
ЯМР(ДМО): 2Н при 5,38 (с, СН2-С6Н5); 10Н между 7,0 и 8,0 (м, Н ароматич. ).
Д) 1-Трет-бутилоксикарбонилиндол-2-карбоновая кислота.
Вводят по капле 30 мл раствора, содержащего 6 г дикарбоната дитрет-бутила в 30 мл смеси, состоящей из 4 г индол-2-карбоновой кислоты, 4 мл триэтиламина и 0,4 г 4-диметиламинопиридина в ацетонитриле. После перемешивания в течение 2 ч при температуре окружающей среды и удаления образовавшегося осадка ацетонитрил удаляется путем перегонки, а остаток растворяется в метиленхлориде. Органическая фаза промывается водой, сушится и концентрируется досуха.
Т.пл. 117оС
Выход 66%
П
р
и м е р 1. 2-(Индолил-2-карбониламино)-4-(2,4-диметоксифенил)-5-бензилтиа- зол

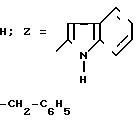
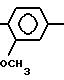
Растворяют 1,96 г 2-амино-4-(2,4-диметоксифенил)-5-бензилтиазола, полученного перед этим согласно синтезу А, 1,22 г 1-ацетилиндол-2-карбоновой кислоты, 0,85 г триэтиламина и 2,95 г БОФ в 20 мл ди метилформамида.
Pеакционная смесь перемешивается в течение 24 ч при температуре окружающей среды, затем приливается к буферному раствору со значением рН 2. Осадок желтого цвета отделяется путем фильтрования, промывается водой и растворяется в этилацетате. Раствор промывается последовательно буферным раствором со значением рН 2, водой, 5%-ным раствором NaHCO3 и водой, затем сушится на MgSO4 и концентрируется под вакуумом. Остаток очищается методом хроматографии на силикагеле, элюент: дихлорметан/этилацетат 98/2 (V/V).
Концентрирование фракций чистого продукта дает остаток, который обрабатывается при перемешивании в течение 24 ч при помощи 3 г Na2CO3 в 80 мл метанола. Метанол концентрируется под вакуумом, а остаток извлекается в смеси вода/эфир. Осадок белого цвета отделяется путем фильтрования и промывается в эфире.
m 0,97 г
Т.пл. 201-202оС.
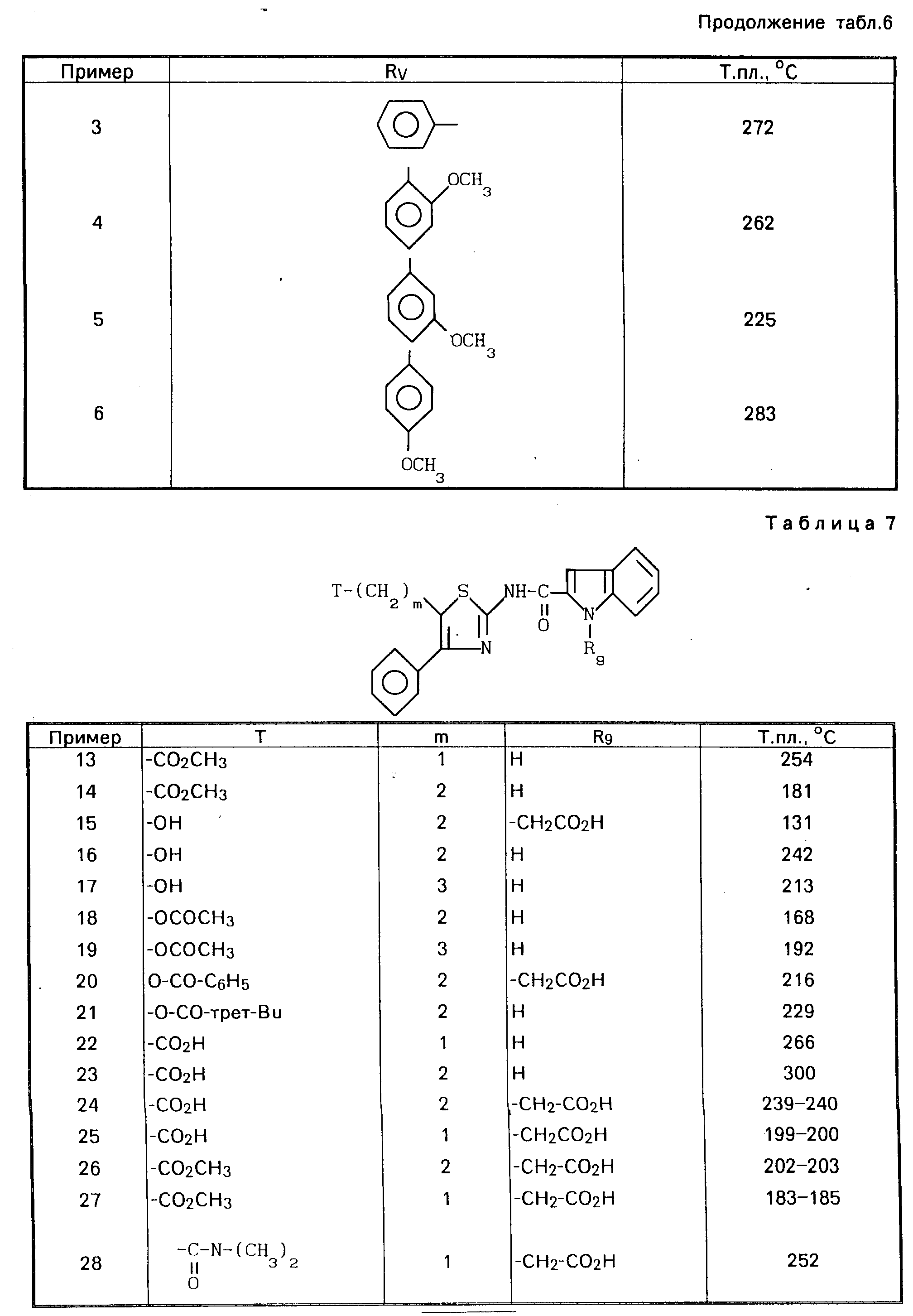
Тем же самым способом получают соединения по изобретению, описанные в табл.6, приведенной ниже.
П р и м е р 7. 2-(1-Трет-бутоксикарбонилоксиметил-2-индолил)-карбонил-амино-4-фенил- 5-гидроксиэтилтиазол. (I): RV= -(CH2 )2-OH; RIV= -C6H5; RI=H; Z
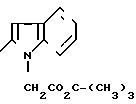
Растворяют 2 г аминоспирта, полученного перед этим (согласно синтезу В, табл. 2), 2,75 г 1-трет-бутоксикарбонилиндол-2-карбоновой кислоты, 1,4 г триэтиламина и 4,9 г БОФ в 15 мл диметилформамида. После выдерживания в течение ночи при температуре окружающей среды реакционная смесь приливается к фосфатному буферному раствору со значением рН 2. Осадок отделяется путем фильтрования, промывается водой и растворяется в этилацетате. Раствор последовательно промывается при помощи 5%-ного водного раствора NaHCO3, отделяется путем декантации, органическая фаза сушится на MgSO4 и концентрируется под вакуумом.
Остаток очищается методом хроматографии на силикагеле, элюент: дихлорметан/метанол 100/0,5 (V/V).
Продукт, элюированный первым, соответствует диацилированному соединению (0- и N-ацилирование, Т.пл. 70оС). Ожидаемый продукт элюируется вторым.
m 1,2 г.
Т.пл. 180-181оС.
П р и м е р 8. 2-(1-Трет-бутоксикарбонилметил-2-индолил)- карбониламино-4-фенил-5-ацетоксиэтилтиазол. (I): RI= H; Z
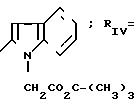
RV CH3-COO-(CH2)2-
Растворяют 0,30 г полученного перед этим продукта в 5 мл пиридина с образованием суспензии.
Прибавляют 1,2 мл уксусного ангидрида и перемешивают реакционную смесь в течение ночи при температуре окружающей среды. Затем смесь приливается к сульфатному буферному раствору со значением рН 2, и осадок отделяется путем фильтрования, промывается водой, затем извлекается в дихлорметане. Раствор последовательно промывается при помощи 5%-ного раствора NaHCO3, отделяется путем декантации, органическая фаза сушится на MgSO4, фильтруется и концентрируется под вакуумом. Остаток очищается методом хроматографии на силикагеле, элюент: дихлорметан/этилацетат-98/2 (V/V).
m 0,16 г.
ЯМР (ДМСО): 9Н при 1,48 (с, трет-

П р и м е р 9. 2-(1-Карбоксилметил-2-индолил)-карбониламино-4-фенил-5-ацеток-сиэтилтиазол. (I): RI= H; Z
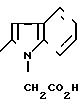
RV СН3-СОО-(СН2)2-
Растворяют 0,15 г полученного перед этим соединения в 2 мл анизола и 10 мл трифторуксусной кислоты.
Смесь оставляется в течение 45 мин при температуре окружающей среды, затем концентрируется под вакуумом. Полученный остаток промывается смесью, состоящей из гексана и диэтилового эфира (50/50), затем сушится.
m 0,14 г.
Т.пл. 117-218оС.
П р и м е р 10. 2-(1-Ацетил-2-индолил)-карбониламино-4-(2,4-диметоксифенил)-5-этоксикарбонил мети (I): RI= H; Z
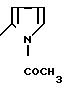
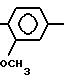
RV CH3CH2-OCO-CH2.
Растворяют 1,5 г 2-амино-4-(2,4-диметоксифенил)-5-этокси-карбонилметилтиазо- ла, 1 г ацетилиндол-2-карбоновой кислоты, 0,7 мл триэтиламина и 2,39 г БОФ в 15 мл дихлорметана. Реакционная смесь перемешивается в течение ночи при температуре окружающей среды, затем концентрируется под вакуумом. Остаток извлекается в этилацетате, а раствор последовательно промывается буферным раствором со значением рН 2, 5%-ным раствором NaHCO3 и водой, затем органическая фаза сушится на MgSO4 и концентрируется под вакуумом. Остаток очищается методом хроматографии на силикагеле, элюент: дихлорметан/этилацетат-100/2,5 (V/V).
m 1,2 г
Т.пл. 130-135оС.
П р и м е р 11. 2-(2-Индолил)-карбониламино-5-этоксикарбонил-4-фенил-тиазол. (I): R

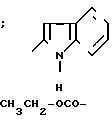
Проводя синтез в соответствии с описанным перед этим примером 10, получают соединение 2-(1-ацетил-2-индолил)-карбонил-амино-4-фенил-5-этоксикарбонилтиа-зол (1,5 г), которое растворяют в 100 мл этанола в присутствии 0,6 г Na2CO3. Смесь перемешивается при температуре окружающей среды в течение 48 ч, затем концентрируется под вакуумом. Остаток превращается в порошок в воде, потом в минимальном количестве дихлорметана, отфильтровывается и сушится.
m 1,1 г
Т.пл. 248оС.
П р и м е р 12. 2-(2-Индолил)-карбониламино-4-(2,4-диметоксифенил)-5-карбокси-метилтиазол. (I): RI= H; Z
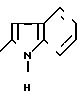
RV -CH2-COOH.
Растворяют 0,5 г 2-(1-ацетил-2-индолил)-5-карбониламино-4-(2, 4-диметоксифе- нил)-5-этоксикарбонилметилтиазола, полученного перед этим в соответствии с примером 10, в 10 мл 95%-ного этанола, затем прибавляют 1,5 мл 2 н. NaOH. Реакционная смесь перемешивается в течение ночи при температуре окружающей среды, затем концентрируется под вакуумом. Остаток извлекается в буферном растворе со значением рН 2, осадок отделяется путем фильтрования, промывается водой, отфильтровывается, затем споласкивается эфиром.
m 0,
28 г
Т.пл. 284оС.
Проводя синтезы в соответствии с приведенными примерами 7-12, получают соединения, описанные в табл.7.
П р и м е р 31. 2-(2-Индолил-)карбониламино-4-фенил-5-[2-(1-пирролидино-кар- бонил)-1- этил]-тиазол. (I):

Прибавляют 0,5 г сложного эфира, описанного в примере 14, 5 мл пирролидина, перемешивают смесь в течение ночи при температуре окружающей среды, затем приливают к буферному раствору со значением рН 2. Осадок отделяется путем фильтрования, затем растворяется в этилацетате. Раствор промывается буферным раствором со значением рН 2, затем водой, органическая фаза отделяется путем декантации, сушится на MgSO4 и концентрируется под вакуумом.
m 0,48 г.
Т.пл. 179оС.
Проводя синтезы в соответствии с приведенным выше примером 10, получают соединения 32-51, описанные в табл.8.
П р и м е р 52. 2-(2-Индолил)-карбониламино-4-фенил-5-фениламинокарбонилокси- этилтиазол. (I): RI= H; Z
А) 2-(1-Ацетил-2-индолил)-карбониламино-5-фенил-5-фениламинокарбонилокси- этилтиазол.
Перемешивают в течение ночи при температуре окружающей среды 0,7 г спирта, полученного в соответствии с примером 16, и 0,2 мл фенилизоцианата в 5 мл дихлорметана. Образовавшийся осадок отделяется путем фильтрования, затем очищается методом хроматографии на силикагеле, элюент: дихлорметан/этилцетат 95/5 (V/V).
m 0,52 г.
Т.пл. 156оС.
В) Соединение 52.
Растворяют 0,5 г
полученного перед этим соединения в 30 мл этанола и 5 мл воды, затем прибавляют 0,21 г Na2CO3
Реакционная смесь
перемешивается в течение ночи при температуре окружающей
среды, затем концентрируется под вакуумом. Остаток извлекается в этилацетате и промывается последовательно раствором Na2CO3и водой. Органическая фаза отделяется путем декантации
и концентрируется под вакуумом. Остаток извлекается в эфире.
m 0,4 г
Т.пл. 249оС.
П р и м е р 53. 2-(2-Индолил)-карбониламино-4, 5-дигидро-[5,4-d] 1Н-тиазолбензоксэпин. (I): RI= H; Z
В 30 мл диметилформамида смешивают 1 г полученного перед этим (синтез ± ) тиазола 2,6 г БОФ, 0,93 г 1-ацетилиндол-2-карбоновой кислоты и 0,46 г триэтиламина. Реакционная смесь перемешивается в течение 30 ч при температуре окружающей среды. Выпаривают диметилформамид, извлекают остаток в этилацетате и промывают водой. Органическая фаза сушится на сульфате натрия и выпаривается. Остаток хроматографируется на силикагеле, элюент: дихлорметан/метанол 100/0,5 (V/V). Получают 0,9 г пенообразного вещества желтого цвета, которое растворяют в дихлорметане с добавкой этанола (100 мл). Прибавляют 10 мл 2 н. NAOH и перемешивают смесь при температуре окружающей среды в течение 1 ч. После выпаривания органических растворителей извлекают остаток этилацетатом и промывают буферным раствором со значением рН 2. Сушат органическую фазу на MgSO4, фильтруют и выпаривают. Получают кристаллы желтого цвета, которые затем промываются дихлорметаном, потом этанолом.
m 0,45 г
Т.пл. > 260оС.
П р и м е р 54. 2-(2-Индолил)-карбониламино-4-фенил-5-(1-пиперидинил)-тиазол. (I): RI= H; Z
А) 2-Амино-4-фенил-5-(1-пиперидинил)-тиазол.
Нагревают при температуре образования флегмы в течение 48 ч смесь, состоящую из 1 г 2-амино-4-фенил-5-бромтиазол и 1,7 г пиперидина в 25 мл абсолютного этанола. Этанол концентрируется под вакуумом, а остаток извлекается в 50 мл воды и 10 мл 30%-ного NaOH. Экстрагируют этилацетатом, сушат органическую фазу на Na2SO4 и фильтруют. Концентрируют под вакуумом и перекристаллизовывают остаток в изопропиловом эфире.
m 0,41 г.
Т.пл. 135-137оС.
В) Соединение 54.
Растворяют 0,4 г полученного перед этим продукта в 50 мл дихлорметана. Прибавляют последовательно 0,33 г 1-ацетил-индол-2-карбоновой кислоты, 0,82 г БOФ и 0,19 г триэтиламина. Реакционная смесь перемешивается в течение 4 дней при температуре окружающей среды. Прибавляют 25 мл воды и отделяют органическую фазу путем декантации, сушат на Na2SO4 и концентрируют под вакуумом. Остаток извлекается в 50 мл абсолютного этанола, прибавляют 10 мл 2,5 н. NaOH и перемешивают смесь в течение 3 ч при температуре окружающей среды. Концентрируют под вакуумом извлекают остаток в 50 мл воды, экстрагируют этилацетатом, отделяют органическую фазу путем декантации, сушат на Na2SO4 и концентрируют под вакуумом. Остаток перекристаллизовывается из этилацетата.
m 0,26 г
Т.пл. > 260оС.
Проводя синтез в соответствии с примером 54, получают соединения 55 и 56, описанные в табл.9.
П р и м е р 57. 2-(2-Индолил)-карбониламино-5-бром-4-фенилтиазол.
(I): RI= H; Z
Растворяют 3,9 г (0,015 моль) 2-амино-5-бромтиазола в 60 мл дихлорметана.
Прибавляют 3,26 г 1-ацетилиндол-2-карбоновой кислоты, 8,1 г БОФ, 1,85 г триэтиламина, перемешивают реакционную смесь при температуре окружающей среды в течение 8 дней, затем прибавляют 100 мл водного буферного раствора со значением рН 2, декантируют, сушат органическую фазу на Na2SO4, концентрируют под вакуумом. Извлекают остаток в 100 мл метанола, затем прибавляют 5 г Na2 CO3. Перемешивают в течение 3 ч при температуре окружающей среды, концентрируют под вакуумом на холоду, извлекают посредством 100 мл водного буферного раствора со значением рН 2, экстрагируют этилацетатом и сушат на Na2SO4 органическую фазу. Фильтруют и концентрируют под вакуумом. Хроматографируют остаток на силикагеле, элюент: CH2Cl2. После удаления головных примесей элюируют ожидаемый продукт, который перекристаллизовывают (после выпаривания растворителя) из смеси СH2Cl2 -/диизопропиловый эфир.
m 2,8 г.
Т.пл. 224-226оС.
П р и м е р 58. 2-(2-Индолил)-карбониламино-5-метокси-4-фенилтиазол. (I): RI= H; Z
Растворяют 3,15 г 2-амино-5-метокси-4-фенилтиазола в 60 мл CH2Cl2. Прибавляют 3,26 г 1-ацетилиндол-2-карбоновой кислоты, 8,12 г БОФ и 1,86 г триэтиламина, затем перемешивают реакционную смесь при температуре окружающей среды в течение одной недели. Прибавляют 50 мл воды, декантируют органическую фазу, сушат на Na2SO4, фильтруют и концентрируют под вакуумом. Остаток извлекается в 100 мл метанола, прибавляют 10 г К2 СO3 и перемешивают смесь в течение ночи при температуре окружающей среды. Концентрируют под вакуумом, извлекают остаток в воде, отделяют образовавшийся осадок путем фильтрования и промывают его в CH2Cl2.
m 1,7 г.
Т.пл. > 260оС.
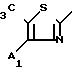
Проводя синтезы в соответствии с приведенным выше примером, получают соединения 59-61, описанные в табл.10.
П р и м е р 62. 2-(2-Индолил)-карбониламино-5-гидрокси-5-фенилтиазол. (I): RI= H; Z
Растворяют 0,58 г тиазола, полученного в соответствии с примером 58, в 100 мл CH2Cl2. Прибавляют при температуре окружающей среды 4,98 мл 2М раствора трибромида бора в CH2Cl2 и оставляют реакционную смесь при перемешивании в течение 24 ч. Снова прибавляют 4,98 мл BBr3, оставляют смесь в течение 5 дней при температуре окружающей среды и прибавляют последний раз 4,98 мл BBr3, оставляя реакционную смесь в течение 48 ч при температуре окружающей среды. Эта смесь затем доводится до значения рH 5-6 путем прибавления 4 н. раствора NaOH, затем экстрагируют органическую фазу 2 раза по 50 мл 4 н. NaOH. Нейтрализуют водную фазу, прибавляя 2 н. раствор HCl. Экстрагируют в CH2Cl2, сушат органические фазы на Na2 SO4, фильтруют и концентрируют под вакуумом, в результате чего получают остаток, который кристаллизуется.
m 0,5 г.
Т.пл. 237-239оС.
Реферат
Использование: в химии гетероциклических соединений, в качестве веществ, ингибирующих связывание холецистокинина с рецепторами. Сущность изобретения: продукт 1: гетероциклические производные замещенных 2-ациламино-5-тиазолов ф-лы I с соответствующими значениями радикалов. Получение соединений I ведут реакцией соединения II с функциональным производным кислоты ф-лы Z COOH, либо ацилированием соединения ф-лы III соединением ф-лы IV. Предложены новые промежуточные соединения ф-лы V и VI. Структура соединений ф-лы I, II, III, IV, V и VI; представлены в тексте описания. 5 с. и 2 з. п. ф-лы.
Формула
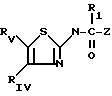
где R1 водород;
RIV фенил, незамещенный или имеющий один или несколько заместителей, выбираемых среди атомов галогена, в частности хлора или фтора, C1 C6-алкильных или C1 C3-алкоксигрупп, или RIV и RV , взятые вместе, представляют собой группу
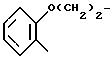
присоединенную через углерод фенила в положение 4 тиазольного кольца,
RV группа -(CH2)m -X, где m- 0 5, X галоген, предпочтительно бром; гидроксил, циклогексил, фенил, который может быть замещен C1 C3-алкоксигруппой, группа, выбираемая среди -COOH, -COOX1, -OCOX1, -SX1, -OCOOX1, -NHCOX1,
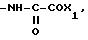
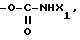
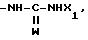
где W кислород или сера,
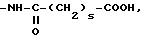
где S 2, 3, 4, где X1 C1 C5-алкил, фенил, который может быть замещен одной или несколькими C1 C3-алкильными или C1 C3 -алкоксигруппами, адамантильная группа;
либо X группа, выбираемая среди -CONX1 X2, - NX1X2, где X1 водород или C1 C3-алкил, X2 водород или C1 C3-алкил или X1 и X2 вместе с атомом азота, с которым они связаны, образуют пирролидин или пиперидин, незамещенный или замещенный оксогруппой или гидроксильной группой;
или RV C1 -С5-алкоксигруппа, гидроксильная группа, пиперазинильная группа, возможно N-замещенная группой -COOAlk, где AIk - C1-C5-алкил, группа карбоновой кислоты;
Z- индолил, незамещенный или замещенный по азоту карбоксиалкиленом - Z4 COOR10, где Z4 C1-C4-алкилен, R10 водород или C1-C6-алкил, ацилом COR13, где R13 C1-C4-алкил, алкоксикарбонилом -COOR14, где R14 трет-бутил.
COOR14, где R14 трет-бутил.
где R1 водород;
RIV фенил, незамещенный или имеющий один или несколько заместителей, выбираемых среди атомов галогена, в частности хлора или фтора, C1-C6-алкильных или C1-C3 -алкоксигрупп, или RIV и RV , взятые вместе, представляют собой группу
присоединенную через углерод фенила в положение 4 тиазольного кольца;
RV группа -(CH2)m -X, где m 0 5, X галоген, предпочтительно бром, гидроксил, циклогексил, фенил, который может быть замещен C1 -C3-алкоксигруппой, группа, выбираемая среди -COOH, -COOX1, -OCOX1, SX1, -OCOOX1, -NHCOX1,
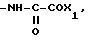
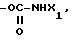
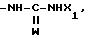
где W кислород или сера;
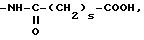
где s-2, 3, 4;
где X1 C1-C5-алкил, фенил, который может быть замещен одной или несколькими C1-C3-алкильными или C1-C3-алкоксигруппами, адамантильная группа, либо X группа, выбираемая среди -CONX1 X2, -NX1X2, где X1 - водород или C1-C3 -алкил, X2 водород или C1-C3-алкил, или X1 и X2 вместе с атомом азота, с которым они связаны, образуют пирродилин или пиперидин, незамещенный или замещенный оксогруппой или гидроксильной группой;
или RV C1 -C5-алкоксигруппа, гидроксильная группа, пиперазинильная группа, N-замещенная группой -COOAlk, где Alk - C1-C5-алкил, группа карбоновой кислоты,
Z индолил, незамещенный или замещенный по азоту карбоксиалкиленом - Z4 -COOR10 , где Z4 C1-C4-алкилен, R10 водород или C1-C6-алкил, ацилом -COR13, где R13 C1-C4-алкил, алкоксикарбонилом -COOR14, где R14 трет-бутил, отличающийся тем, что замещенный 2-амино-5-тиазол общей формулы
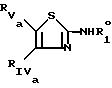
где R водород;
RIVa и RVa имеют значения, указанные для RIV и RV, где гидрокси- или аминогруппы являются O- или N-замещенными,
обрабатывают функциональным производным кислоты общей формулы
Z0 COOH,
где Z0 имеет указанные для Z значения, где группа NH является N-замещенной,
затем, когда RIVa, RVa содержат N-защищенные или O-защищенные группы, осуществляют снятие защиты и подвергают, в случае необходимости, полученный таким образом продукт ацилированию или алкилированию с целью повышения соединений общей формулы I, где RI, RIY и RV имеют указанные значения.
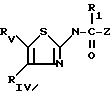
где Z индолил;
R1 водород;
RIV фенил, незамещенный или имеющий один или несколько заместителей, выбираемых среди атомов галогена, в частности хлора или фтора, C1-C6-алкильных или C1-C3 -алкоксильных групп, или RIY и RV, взятые вместе, представляют собой группу
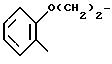
присоединенную через углерод фенила в положение 4 тиазольного кольца;
RV группа -(CH2)m -X, где m 0 5, X галоген, предпочтительно бром, гидроксил, циклогексил, фенил, который может быть замещен C1-C3-алкоксигруппой; группа, выбираемая среди -COOH, -COOX1, -OCOX1, -SCOX1, -OCOOX1, NHCOX1,
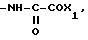
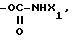
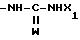
где W кислород или сера;
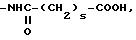
где s 2, 3, 4; X1 C1-C5-алкил, фенил, который может быть замещен одной или несколькими C1-C3-алкильными или C1-C3-алкоксигруппами, адамантильная группа;
или X группа, выбираемая среди -CONX1X2, -NX1X2, где X1 водород или C1-C3-алкил, X2 водород или C1-C3-алкил,
или X1 и X2 вместе с атомом азота, с которым они связаны, образуют пирролидин или пиперидин, незамещенный или замещенный оксогруппой или гидроксильной группой, или RV C1-C5-алкоксигруппа, гидроксильная группа, пиперазинильная группа, N-замещенная группой -COOAlk, где Alk C1-C3-алкил, группа карбоновой кислоты,
отличающийся тем, что иминотиазол общей формулы
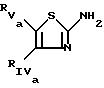
где RIVa и RVa имеют значения, указанные для RIV и RV , где гидрокси- или аминогруппы являются О- или N-зашищенными,
подвергают ацилированию активированной формой кислоты формулы
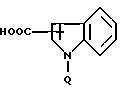
где Q защищающая группа,
и осуществляют снятие защиты с азота.
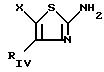
где RIV фенил, незамещенный или замещенный галогеном, C1 -C6-алкилом или C1-C3-алкоксилом;
X C1-C5-алкоксигруппа, пиперидин, незамещенный или замещенный 4-гидроксигруппой.
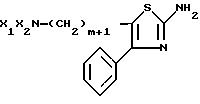
где m=1 или 2;
X1X2N фталимидо- или NH2-группа.
Комментарии