Производные 1,4,10,13-тетраокса-7,16-диазациклооктадекана, обладающие свойством выводить радиоактивные изотопы из живого организма, и фармацевтическая композиция для выведения из организма радиоактивных изотопов - RU2060256C1
Код документа: RU2060256C1
Чертежи
Описание
Изобретение относится к частично новым производным 1,4,10-13-тетраокса-7,16-диазациклооктадекана и использованию таких соединений для
удаления из живых
организмов металлических ионов, особенно радиоактивных изотопов, оказывающих поражающее действие. В частности, изобретение касается металлических комплексов, солей и двойных солей 1,
4,10,
13-тетраокса-7,16-диазациклооктаденовых производных формулы (1)
M
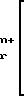
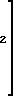
-
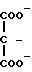
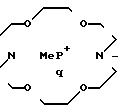
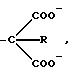
В группе с формулой 2 R может быть водородом, прямой или разветвленной С1-5 аклкильной группировкой, прямой или разветвленной С2-5 алкенильной группировкой, фенильной или фенил С1-5 алкильной группировкой, причем две последние группировки в случае необходимости могут быть замещены по ароматическому компоненту одним или несколькими галогенами, С1-5 алкильной, С1-5 алкокси, циано- или нитрогруппами при условии, что по меньшей мере Q1 или Q2 не является водородом. Ме представляет собой щелочной металл или щелочной редкоземельный металл, или переходный металлический ион; q 0 или 1; М и N независимо друг от друга представляют собой водород или щелочной металл, щелочной редкоземельный металл или в случае необходимости замещенный ион аммония; m, n и р эквиваленты зарядов соответственно N, M или Ме; S и r независимо друг от друга 0, 1, 2, 3 или 4 при условии, что r, s и q одновременно не могут быть 0 и количество водородных атомов в М и N может составлять 0, 1 или 2.
Изобретение относится также к фармацевтическим композициям на основе этих соединений.
Из числа соединений с формулой (1) соединения, содержащие в качестве Q1 и Q2 водород, используются преимущественно как производные.
При введении в организм соединения формулы (1) независимо от того новые они или нет способны образовывать устойчивые комплексы с ионами радиоактивных металлов, прежде всего с радиоактивными стронцием и цезием, присутствующими в крови и внеклеточном пространстве, которые затем выводятся из организма. Новые соединения из числа соединений с фоpмулой (1) содержат группу с формулой (2), в которой R либо не является водородом, либо, если R представляет собой водород, то q равно 1. Иными словами эти соединения являются комплексами, в которых М и N, кроме того, отличаются от натрия или лития, если q равно 0.
Таким образом, с одной стороны, изобретение относится к соединениям формулы (1), в которой Q1 и Q2 -водород или группа формулы (2 ) при условии, что по меньшей мере один из них не является водородом и что в группах формулы (2) в качестве R могут использоваться водород, С1-5алкенильная группа с прямой или разветвленной цепью, С2-5 алкенильная группа с прямой или разветвленной цепью, фенильная или фенил С1-5алкильная группа, причем две последние могут быть в случае необходимости заменены по ароматическому компоненту одним или более галогенами, С2-5алкильной, С1-5 алкокси, циано- или нитpогpуппами. Ме представляет собой щелочной металл или щелочной редкоземельный металл, или переходный металлический ион; q-0 или 1; М и N независимо друг от друга представляют собой водород, щелочной металл, щелочной редкоземельный металл или в случае необходимости замещенный ион аммония, m, n и р эквиваленты зарядов соответственно N, M или Ме; s и r независимо друг от друга 0, 1, 2 и 3 или 4 при условии, что r, s и q одновременно не могут быть равны 0, количество водородных атомов в М и N может составлять 0, 1 или 2, q представляет собой 1, если R представлен водородом, и М и N не являются ионами натрия или лития, если q равно 0.
Из числа заместителей в соединениях с формулой (1) R в качестве С1-5 алкильной группы может быть представлен прямой или разветвленной цепью, например метильной, этильной, n-пропильной, изопропильной, n-бутильной, втор-бутильной, трет-бутильной, n-пентильной или изопентильной группой, пpедпочтительно метильной или этильной группой. R в качестве С2-5 алкенильной группы может быть, в частности, представлен винильной или пропенильной группой, а R в качестве фенил С1-5 алкильной группы может содержать одну из вышеуказанных алкильных цепей, предпочтительно метильную.
Ме в качестве иона щелочного металла должен быть предпочтительно представлен ионами натрия или калия, в качестве иона щелочноземельных металлов ионами кальция или магния, а в качестве переходного металлического иона ионом металлов, принадлежащих к третьей, четвертой или пятой группе предпочтительно ионом двухвалентного железа или цинка. М и N в качестве ионов щелочных или щелочноземельных металлов представлен предпочтительно вышеуказанными ионами, тогда как замещенный ион аммония содержит 1, 2, 3 или 4 вышеуказанных алкильных, фенильных или фенилалкильных групп (при этом предполагается отсутствие стерических затруднений). Соединения, содержащие ион четырехметильного аммония, нельзя использовать для введения в живые организмы из-за их токсичности.
Известно, что при ядерных взрывах или при авариях на ядерных реакторах в атмосферу поступают чрезвычайно опасные радиоактивные изотопы, такие как йод-131 (131I), стронций-89 и 90 (89Sr и90Sr), а также цезий-134 и 137 (134 Cs и137Cs) и церий-141 и 144 (141Се и144Се) (см. например, Nuclear and Radiochemistry, John Willy and Jons, 1981, рр. 158-166). При попадании этих изотопов в легкие в процессе дыхания или в пищеварительный тракт с загрязненными пищей и жидкостями, а также в кровеносную и лимфатическую систему в результате ресорбции через кожные покровы они откладываются и накапливаются в тканях, что, в конечном счете, приводит к тяжелым последствиям для здоровья (см. Summary Report on the Post Recident Review Meеting on the Chernobyl Quident, Safety Series N 75, IAEA, Vienna, 1986).
В случае радиоактивного поражения стронций уже через несколько часов обнаруживается в костной ткани, и его удаление из организма не представляется возможным. Это чрезвычайно затрудняет задачу защиты организма от радиоактивного стронция.
Единственная возможность такой защиты состоит в предотвращении фиксации стронция в ткани, прежде всего костной, посредством введения в организм соответствующих соединений, образующих специфические комплексы со стронцием. Таким путем осуществляются связывание изотопа, присутствующего в крови или внеклеточном пространстве, в стабильной форме и выведение его из организма.
Решение этой проблемы усложняется тем, что описанные в литературе комплексообразующие соединения, например этилендиамин-четырехуксусная кислота или диэтилентриаминпентауксусная кислота, образуют с кальцием значительно более стабильные комплексы, чем со стронцием (Catsch A. Radioaсtive Metal Mobilization in Medicine, Ed. Charles C.Thomas, Springfield, Illinois, 1964; Catsch A. Dekorporierung radioaktiver und stabiler Mettallionen, Therapeutische Grundlagen, Ed. Thiemig, Munich, 1968; Catsch A. Removal of Transuranium Elements by Chelating Agents m: Diagnosis and Treatment of Incorporated Radionuclides, IAEA Publication N ST1/PUB/411, IAEA, Vienna, 1976, p.295).
Новые перспективы для исследования в этом напpавлении открылись после синтеза кронэфиров и криптандовых молекул. В этом случае механизм комплексообразования отличается от известного ранее, так как благодаря структуре новых комплексирующихся молекул металлические ионы локалиуются в ячейках строго определенного размера, в связи с чем стабильность комплексов зависит прежде всего от размеров данного типа ионов.
Первые обнадеживающие результаты были получены при исследованиях 4, 7, 13, 16, 21, 24-гексаокса-1, 10-диазабицикло (8,8,8)гексакозана, образующего со стронцием комплекс, показатель стабильности которого на несколько порядков выше чем у комплекса его с кальцием (см. Coordination Chemistry of Macrocychie, Compounds Ed. G. A. Melson, Plenum Press, 1979). Однако при изучении этого соединения в экспериментах на животных удалось доказать лишь то, что образующийся вне организма комплекс с лигандой не подвергался диссоциации после его введения в организм. В то же время не были получены данные о возможности выведения радиоактивного стронция из организма в форме стабильного комплекса с лигандой. Кроме того, лиганда оказалась чрезвычайно токсичной (W.H.Muller, Naturwiss, 57, 2248, 1970; W.H.Muller and W.A.Muller, Naturwiss, 61, 455, 1974; W.H.Muller et al; Naturwiss, 64, 96, 1977; J.Knajfe et al, 12th Ann. Meeting of ESRB, Budapest, 1976; J. Batsch et al. Nukleonika, 23, 305, 1978).
Соединения формулы (1), представляющие собой соли в случае q, равном 0, и комплексы при q, равном 1, обладают специфическими комплексообразующими свойствами, которые позволяют им связывать и выводить металлические ионы, опасные для организма, особенно радиоактивный стронций и церий, которые проникают в живой организм и поступают в кровяное русло и/или внеклеточное пространство. При введении человеку или животным фармацевтических композиций, содержащих соединения формулы (1) в качестве активных ингредиентов, удается предотвратить накопление в тканях радиоактивного стронция, что в свою очередь позволяет избежать или уменьшить тяжелое вредоносное действие радиоактивной нагрузки на состояние здоровья.
F. de Jond et al, предложили способ получения N,N'-bis(дикарбоксиметил)-1,4,10,13-тетраокса-7, 16-диазациклооктадекана в форме четырехлитиевой соли (Rec. Trav. Chim. Pays-Bas, 102, 164, 1983). В соответствии с этой методикой требуемый криптанд реагирует с метил-2-бромомалонатом и полученное эфирное производное подвергается гидролизу до образования литиевой соли с выходом не более 15 Согласно патенту Великобритании N 2024822 получаемая таким образом литиевая соль может быть использована в форме композиции, повышающей растворимость сульфата бария, в нефтеперерабатывающей промышленности. В том же описании содержится упоминание о соответствующей четырехнатриевой соли, хотя ее применение не описано в специальных примерах. Двойная соль четырехнатриевой соли с бромидом натрия, а также пригодность такой двойной соли для терапевтического применения описаны в заявке на Венгерский патент N 2614/89.
Растворимые в воде соли
и комплексы с формулой (1) согласно изобретению, в которых Q1, Q2, R, Me, M, N, m, n, p, s, r и q определены
выше, можно получать по аналогии с описанной в предшествующем
изложении реакцией, предполагающей взаимодействие соответствующей галогенизированной дикарбоксильной кислоты формулы (3)
X-
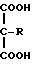
По другому способу растворимые в воде соли и комплексы соединений с формулой (1), в которых М и N представлены водородом или ионами щелочных или щелочноземельных металлов, а Ме ионом щелочноземельного металла, также могут быть синтезированы посредством реакции между 1,4,10,13-тетраокса-7, 16-диазациклооктадеканом и 2-галодикарбоксильной кислотой формулы (3), предпочтительно 2-бромомалоновой кислотой, в водной среде с рН 6 13 в присутствии гидроокиси щелочного или щелочноземельного металла, соответствующей соли, которую требуется получить.
Соединения с формулой (1), в которой q равно 1, можно получить посредством реакции соли щелочного металла формулы (1), в которой q равно 0, а М, также как N, представляют собой ионы щелочного металла, желательно четырехнатриевой соли, с эквивалентным количеством комплексообразующего металлического галида (допустимо хлорида металла).
Действие соединений формулы (1) согласно изобретению, которое проявляется в усилении выведения металлических ионов, поражающих живой организм, изучали в условиях загрязнения мышей линии Суисс и крыс линии Уистар обоего пола ионами радиоактивного стронция или церия.
Выведение радиоизотопов изучали после их введения разными способами в различные ткани и участки тела, в частности в кровяное русло, брюшную полость, легкие, мышцы или подкожную соединительную ткань подопытных животных. Соединение, способствовавшее выведению радиоактивных изотопов, вводили ежедневно один или два раза в день посредством инъекций, в форме порошка, жидкого аэрозоля или пластыря. После этого определяли общую радиоактивность организма и получали кривые задержки, которые затем сравнивали с аналогичными параметрами, полученными на контрольных животных.
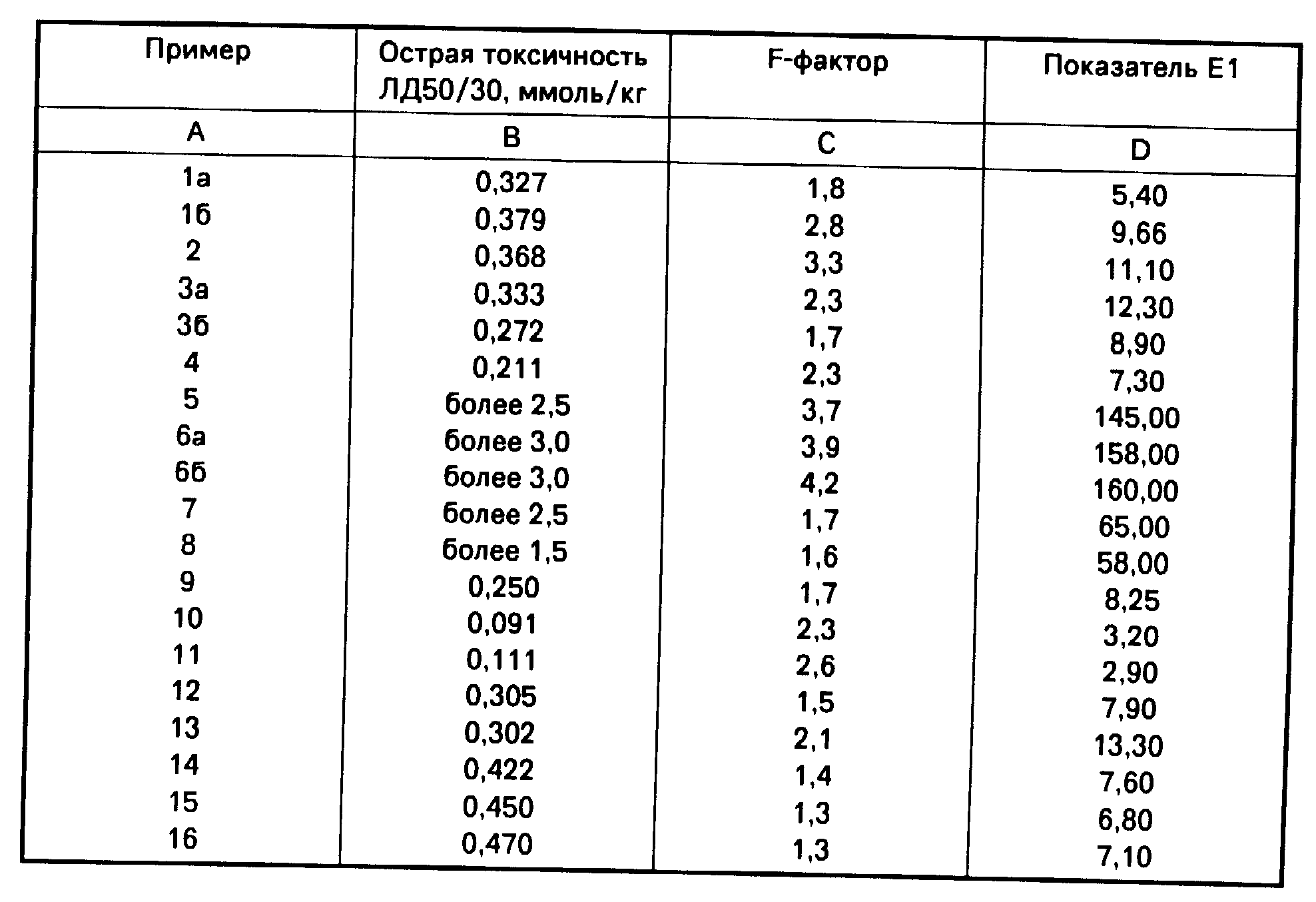
Кривые задержки анализировали с помощью компьютера, используя для этой цели программу, известную как Nonlinear Regression by the Code of BMDP-3R(BMDP Statistical Software Manual, UCLA, Los Angeles, 1990, Chief Editor, W, J. Dixon). На основании полученных результатов можно сделать вывод о том, что кривые описываются двухкомпонентной нисходящей экспоненциальной функцией. Для расчета эффективности использовали два показателя. Одним из них служил так называемый Е-фактор, характеризующий усиление выведения радиоактивности в сопоставлении с наблюдавшимся у контрольных животных, который отражает увеличение выведения радиоизотопа под воздействием тестируемого соединения по сравнению с его интенсивностью у животных, не получавших это соединение (см. колонку С в таблице).
Для более наглядной сравнительной характеристики соединений, являющихся предметом изобретения, приводятся так называемые показатели Е1, которые получали посредством перемножения интенсивности выведения радиоизотопа в процентах к наблюдавшемуся в контрольной группе (эффективность Е) и показателя острой токсичности (ЛД50/30, безвредный, 1), приведенного в колонке D таблицы. Хотя показатель Е1 в цифровом выражении и не идентичен показателю терапевтической безопасности соединения, он во всех случаях характеризует слабую или высокую активность тестируемого продукта.
Важным экспериментальным результатом считают отсутствие определяемого с помощью использованного метода радиоактивного стронция в костях животных, которым вводили активное соединение согласно изобретению. В мягких тканях и печени животных остаточная радиоактивность составляла 5-10% ее общей задержки, тогда как у контрольных животных основная часть задержки была обусловлена накоплением радиоактивности в костях (65 70).
Сходные результаты были получены при различных путях введения тестируемых соединений в организм животных. Показано, что очень высокой эффективностью обладали соединения с общей формулой (1), в которых Q1 и Q2 представлены одним и тем же заместителем, а R является водородом. Из числа этих соединений особенно замечательными свойствами обладал N,N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекан-кальц иевыкомплекс, двойная соль. Ценность высоко эффективных соединений с формулой (1) обусловлена также их очень хорошими показателями терапевтической безопасности. Вышеуказанная двунатриевая соль кальция особенно предпочтительна благодаря ее низкой токсичности и выраженной способности усиливать выведение радиоактивных изотопов из живого организма.
Соединения с формулой (1) можно использовать в составе лекарственных средств в комбинации с обычными носителями и другими хорошо известными вспомогательными материалами. Подходящие для этой цели носители и другие добавки подробно описаны в многочисленных руководствах по данному вопросу.
Изучение эффективности соединений, описанных в примерах 1 16, показало, что после их введения происходит абсорбция активного ингредиента, который затем обеспечивает выведение радиоактивных изотопов. Введение осуществляется в составе инъецируемых растворов или в форме таблеток, помещаемых под язык, драже, капсул, таблеток для приема внутрь, порошков, жидких аэрозолей и накожных повязок. Эффективные дозы составляют 1 200 мкмоль/кг веса тела, предпочтительно 10 100 мкг/кг. Введение производится в форме одной или нескольких частей данной общей дозировки, желательно в виде двух субдозировок.
Лекарственные средства, содержащие в качестве активного ингредиента соединения формулы (1), пригодны также для предотвращения накопления в живом организме вредных для него металлических ионов.
П р и м е р 1. Получение N,N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7, 16-диа-зациклооктадена, тетранатриевой соли.
а) 2,74 г (14,98 ммоль) 2-броммомалоновой кислоты нейтрализуют в 1 мл воды, добавляя в нее раствор гидроокиси натрия в концентрации 7,410 моль/л в присутствии индикатора фенолфталеина. Затем в полученную смесь добавляют 1,75 г (3,69 ммоль) N-дикарбоксиметил-1,4,10, 13-тетраокса-7,16-диазациклооктадекана (двунатриевой соли), содержащего 14,10 мас. бромида натрия (промежуточного), полученного в предшествующей реакции, и 1,95 г (7,43 ммоль) 1,4,10,13-тетраокса-7, 16-диазациклооктадекана. Реакционную смесь оставляют при 60оС на 10-11 ч, добавляя в нее раствор гидроокиси натрия в концентрации 7,410 моль/л (14,98 ммоль) порциями по 0,1 мл общим количеством 2,02 мл. После завершения реакции смесь при необходимости отфильтровывают добавляют бромид натрия, упаривают, высушивают при пониженном давлении и экстрагируют несколькими порциями метиленхлорида, общий объем которого составляет 20-25 мл. Экстракт упаривают до сухого остатка, добавляют к последнему 10 15 мл петролейного эфира, а полученный после фильтрации осадок высушивают в токе азота. 2,27 г (4,82 ммоль) полученного таким образом продукта, содержащего 13,3 мас. бромида натрия (промежуточного), используют при изготовлении следующей партии искомого продукта.
Идентификационные показатели промежуточно:1Н-ЯМР-спектр (200 мГц, D2O), δ частей/млн.): 3,87 (1Н, S), 3,67 (18H, m), 2,78-2,92 (8H, m).
Остаток,
образующийся после экстракции метиленхлоридом, экстрагируют 60 мл безводного этанола до
тех пор, пока в экстракте практически не остается твердого материала. Остаток после конечной экстракции
разводят в 6 7 мл воды и после добавления бромида натрия упаривают и высушивают до сухого
остатка. Последний экстрагируют 30 мл безводного этанола, как описано выше. Оба этанольных экстракта
объединяют и упаривают до сухого остатка, который содержит 4,89 г двойной соли, в которой имеется
22,71 моль бромида натрия. Выход для используемого макроцикла составляет 94,1% материала. Остаток
после конечной экстракции разводят в 6 7 мл воды и после добавления бромида натрия упаривают и
высушивают до сухого остатка. Последний экстрагируют 30 мл безводного этанола, как описано выше. Оба
этанольных экстракта объединяют и упаривают до сухого остатка, который содержит 4,89 г двойной соли,
в которой имеется 2,71 моль бромида натрия. Выход для используемого макроцикла составляет 94,1%
Идентификационные показатели для двойной соли: ИК-спектр (KBr, см-1): 2950, 2868 (m,
γ CH), 1605 (VS, COO/as), 1430 (m, γ COO/S). Другие характерные, но не
идентифицированные частоты: 1350 (S), 1320 (S), 1095 (S), 928 (W).1Н-ЯМР-спектр (200 мГц, D2
О, δ частей/млн.): 54,00 (2H,S) 3,70 (8H, S), 3,63 (8H, t), 2,92 (8H, t).
б) Бромид натрия удаляют из двойной соли посредством экстракции 50 мл 95%-ного (по весу) этанола.
Образующийся остаток высушивают и освобождают от этанола упариванием при пониженном
давлении с образованием 3,22 г конечного продукта. Выход для данного макроцикла составляет 93,2
Идентификационные показатели для последнего:1Н-ЯМР-спектр (200 мГц, D2О,
δ частей/млн.): 3,95 (2H, s), 3,64 (8H, s), 3,60 (8H, t), 2,85 (8H, t).13С-ЯМР-спектр
(50 мГц, D2О, δ частей/млн.): 179,95 (C 0), 76,45 (N-CH-(COO)2),
71,66 и 70,84 (O-CH2), 54,06 (N-CH2).
П р и м е р 2. Получение N, N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диа-зациклооктадекана, тетранатриевой соли, содержащей двунатриевый оксималонат.
В соответствии с процедурой, описанной в примере 1,2, 84 г (15,54 ммоль) 2-бромомалоновой кислоты, 2,02 г (4,11 ммоль) N-дикарбоксиметил-1,4,10, 13-тетраокса-7, 16-диазациклооктадекана двунатриевой соли, содержащей 16,87 мас. бромида натрия и 2,01 г (7,66 ммоль) 1,4,10,13-тетраокса-7,16-диазациклооктадекана, смешивают в растворе, но реакционную смесь оставляют при 50оС, а гидроокись натрия отдельными порциями добавляют на протяжении 10 ч. В результате экстракции метиленхлоридом получают однозамещенное производное 2,17 г (4,41 ммоль). Оно содержит 16,93 мас. бромида натрия. Вес конечного продукта 3,92 г, выход 96,5 (при расчете на использованный макроцикл). Продукт содержит 1,6 мас. динатриевого оксималоната.
Спектр1Н-ЯМР (200 мГц, D2О, δ частей/млн.) соответствует спектру продукта, полученного по примеру 1, за исключением того, что он дает также резонансный сигнал при 4, 31 (S), что характерно для оксималоната.
П р и м е р 3. Получение N,N'-бис(дикарбоксиметил)-1, 4,10,13-тетраокса-7,16-диа-зациклооктадекана, тетракалиевой соли.
а) 1,32 г (7,22 ммоль) 2-бромомалоновой кислоты нейтрализуют в 1 мл воды, добавляя раствор гидроокиси калия в концентрации 5,760 моль/ в присутствии индикатора фенолфталеина. В полученный таким образом раствор добавляют 1,25 г (4,77 ммоль) 1,4,10,13-тетраокса-7,16-диазациклоокста-декана. Реакционную смесь нагревают до 50оС в течение 26 ч, добавляя в нее порциями эквивалентное количество раствора гидроокиси калия в концентрации 5,760 моль/л. После упаривания смеси твердый остаток высушивают при пониженном давлении, а затем экстрагируют метиленхлоридом общим объемом 20 мл в несколько приемов. После упаривания остаток высушивают с образованием 0,85 г (1,73 ммоль) N-дикарбоксиметил-1,4,10, 13-тетраокса-7,16-диазациклоктадена, дикалиевой соли, содержащей 16,7 мас. бромида калия (промежуточного). Этот продукт может быть использован для приготовления следующей партии требуемого продукта.
Остаток после экстракции метиленхлоридом экстрагируют 60 мл безводного этанола и после упаривания раствора остаток высушивают с образованием 2,18 г искомого продукта, т.е. с выходом 94 при расчете на использованный макроцикл.
Продукт представляет собой двойную соль с бромидом калия, которая содержит 29,97 мас. бромида калия.
1 Н-ЯМР-спектр промежуточного соединения (200 мГц, D2О, δ частей/млн.): 3,86 (1H, S) 3,63 (16H, m), 2,89 (4H, t), 2,78 (4H, m).
1Н-ЯМР-спектр двойной соли, образованной бромидом калия, (200 мГц, D2О, δ частей/млн.): 3,99 (2H, s), 3,69 (8H, s), 2,63 (8H, t), 2,86 (8H, t).
б) Чистый, свободный от бромида калия, продукт может быть получен, как описано выше с применением 97 об. этанола. В результате образуется 1,16 г искомого продукта, т.е. выход составляет 75,6 при пересчете на использованный макроцикл.
1Н-ЯМР-спектр искомого продукта (200 мГц, D2О, δ частей/млн.): 4,00 (2Н, широкий s), 3,70 (8Н, широкий s), 3,65 (8Н, широкий), 2,88 (8Н, широкий).
П р и м е р 4. Получение N,N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7, 16-диа-зациклооктадекан-магн иевокомплекса, динатриевой соли.
0,30 г (1,46 ммоль) гексагидрата хлористого магния, разведенного в 2 мл воды, добавляют в раствор, содержащий 0,81 г (1, 46 ммоль) продукта, полученного, как описано в примере 1б, в 3 мл воды. Спустя 30 мин, раствор упаривают при пониженном давлении и остаток высушивают с образованием 0,93 г (97,8) искомого продукта, содержащего 17,99 мас. хлористого натрия.
1Н-ЯМР-спектр конечного продукта (200 мГц, D2О в присутствии NaOD, δ частей/млн.): 4,00 (2H, s), 3,67 (8H, s), 3, 62 (8H, широкая), 2,88 (8Н, широкая).13Н-ЯМР-спектр (50 мГц, D2О, δ частей/млн): 179,71 (C=O) 71,88 и 71,05 (О-СН2), 54,51 (N-CH2).
Примечание: резонансный сигнал CH(COO)2 отсутствует из-за дейтерирования.
П р и м е р 5. Получение N, N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7, 16-диа-зациклооктадекан-каль циевкомплекса, динатриевой соли, содержащей хлористый натрий.
Используют процесс, описанный в примере 4, с 0, 92 г (1,65 ммоль) продукта, полученного в соответствии с примером 1б, и 0,25 г (1,65 ммоль) дигидрада хлористого кальция, что дает 1,08 г (98,2) искомого продукта, содержащего 17,57 мас. хлористого натрия.
1 Н-ЯМР-спектр продукта (200 МГц, D2О, δ частей/млн.): 3,90 (4Н, широкая), 3,53 (14Н, широкая), 2,92 (4Н, коалесцентная t), 2,72 (4Н, коалесцентная t).13С-ЯМР-спектр (50 мГц, D2О, δ частей/млн. ): 179,23 (С=0), 82,38 (СН/COOH/2), 71,62 (О-СН2), 55,63 (N-CH2).
П р и м е р 6. Получение N, N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диа-зациклооктадекан-каль циевкомплекса, динатриевой соли, содержащей динатриевый оксималонат и хлористый натрий.
3,44 г (18,825 ммоль) 2-броммалоновой кислоты нейтрализуют в 1 мл воды, добавляя раствор гидроокиси натрия в концентрации 8,360 моль/л в присутствии индикатора фенолфталеина. Затем добавляют 2,010 г (7,65 ммоль) 1, 4,10,13-тетраокса-7,16-диазациклооктадекана. Реак- ционную смесь нагревают при 30 45оС на протяжении 43 48 ч, добавляя 1,83 мл раствора гидроокиси натрия, который необходим для образования двузамещенного соединения. После этого реакционную смесь оставляют на 22 25 ч при 60оС, добавляя отдельными порциями раствор гидроокиси натрия в количествах, необходимых для гидролизации непрореагировавшего 2-броммалоната. По окончании реакции раствор упаривают и в дальнейшем действуют, как описано в примере 1 б, с 4,720 г сухого неочищенного продукта, который содержит 12 мас. динатриевого малоната и практически не содержит бромида натрия. Затем полученный продукт используют для осуществления одной из следующих процедур.
а) Сырой продукт разводят в смеси из 3 мл воды и 7,5 мл раствора хлористого кальция, имеющего концентрацию 1000 моль/л. Добавляют 12 мл (99,7 об.) этанола и 0,85 мл хлористого кальция при непрерывном помешивании. После этого содержание этанола в смеси доводят до 90 добавляя 105 мл (99,7) этанола. Полученную суспензию энергично перемешивают в течение 30 60 мин при нагревании, после чего отфильтровывают твердые частицы, фильтрат упаривают при пониженном давлении и наполовину сухой продукт продолжают подсушивать при 75 85оС и пониженном давлении до получения 4,48 г (90,6) требуемого вещества, содержащего 14 мас. хлористого натрия и 1,08 мас. динатриевого оксималоната.
1Н-ЯМР-спектр полученного соединения (200 мГц, D2О, δ частей/млн.) соответствует спектру продукта, получаемого по примеру 5, за исключением того, что резонансный сигнал появляется также при 4,31 частях/млн. (1Н, S), что характерно для оксималоната.
б) Повторяют процедуру, описанную в разделе а), с той разницей, что после добавления первой порции раствора хлористого кальция (7,50 мл) добавляют еще одну порцию объемом 3,10 мл с концентрацией хлористого кальция 1000 моль/л. Содержание этанола в реакционной смеси доводят до 90 об. добавляя 114 мл этанола, после чего продукт высушивают в токе азота до получения вещества 5 г (94,9), в котором содержится 19,5 моль. соли кальция кальциевого комплекса (при расчете на общий макроцикл), а также динатриевая соль кальциевого комплекса. Конечный продукт содержит 13,2 мас. хлористого натрия, 7,35 мас. воды и незначительное количество двунатриевого оксималоната.
1Н-ЯМР-спектр продукта (200 мГц, D2О δ частей/млн.) соответствует спектру продукта, полученного в примере 5.
П р и м е р 7. Получение N,N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диа-зациклооктадекан-каль циевдиаммония.
0,5 мл воды, 2,04 мл раствора кальция в концентрации 0, 998 моль/л и затем 30 мл безводного этанола добавляют к 0,554 г (1000 ммоль) тетранатриевой соли, полученной в соответствии с примером 1 б. Раствор упаривают приблизительно до трети первоначального объема и к оставшемуся его количеству добавляют этанол в количестве, достаточном для доведения его концентрации в растворе до 95 96 об. Затем раствор нагревают, перемешивают на протяжении 30 мин, осадившийся хлористый натрий отфильтровывают и промывают безводным этанолом. К объединенному фильтрату добавляют 0,554 г (1000 ммоль) тетранатриевой соли и 0,214 г (4000 ммоль) хлористого аммония, а затем воду в количестве, достаточном для растворения твердого материала. После упаривания раствора остаток обезвоживают, нагревая при 75-80оС и пониженном давлении, до образования 1,452 г (98,1%) искомого продукта, содержащего 19,91 мас. хлористого натрия и 7,33 мас. воды. Очищенный от хлорида конечный продукт получают дальнейшей очисткой безводным этанолом.
1 Н-ЯМР-спектры свободного от хлорида и содержащего хлористый натрий продуктов (200 мГц, D2О, δ частей/млн), идентичны: 3,90 (4Н, широкий, m), 3,73 (14Н, широкий m), 2,92 (4Н, широкий t), 2,72 (4Н, широкий t).
П р и м е р 8. Получение N,N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диа-зациклооктадекан-каль циевкомплекса, кальциевой соли.
После
нейтрализации 2,743 г (9,53 ммоль) 2-броммалоновой кислоты в 2 мл воды путем добавления отдельными порциями гидроокиси кальция в присутствии индикатора фенолфталеина в полученный
раствор вводят 1000
г (3,81 ммоль) 1,4,10,13-тетраокса-7,16-диазациклооктадекана. Реакционную смесь нагревают при 40, 45 и, наконец, 50оС на протяжении в общей сложности 72 ч, а затем до
60оС в
течение 24 ч при добавлении отдельными порциями 0,85 г (11,47 ммоль) гидроокиси кальция в условиях энергичного перемешивания. После этого осадок, основную часть которого составляет
оксималонат
кальция, отфильтровывают и промывают тремя порциями воды по 4 5 мл каждая. Объединенный фильтрат упаривают при пониженном давлении, после чего из образующегося остатка дважды отгоняют по
35 мл
метилен-хлорида, получая твердый продукт, который подсушивают при 75-85оС и пониженном давлении. Таким образом, получают искомое вещество 2,710 г с выходом 89,7
1
Н-ЯМР-спектр этого соединения (200 мГц, D2О, δ частей/млн.) идентичен спектру продукта, получаемого как описано в примере 5. ИК-спектр (KBr, см-1): 2920, 2880 (m, γ
C-H), 1615 (vs, γ COO/as), 1450 (m γ COO/s). Другие характерные, но не идентифицированные частоты: 1355, 1290 (m), 1250 (m), 1085 (vs), 950 (m).
П р и м е р 9. Получение N,N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диа-зациклооктадекан-желе зист(II) комплекса, динатриевой соли.
Используют процесс, описанный в примере 4, 0,747 г (1,35 ммоль) продукта, полученного в соответствии с разделом б) примера 1, и 0,268 г (1,35 ммоль) тетрагидрата хлористого железа. В отличие от ранее описанной процедуры окисление двухвалентного железа до трехвалентного предотвращают, осуществляя реакцию в атмосфере азота. С помощью этого способа получают искомое соединение с выходом 91 в количестве 0,835 г при содержании в нем хлористого натрия 17,16 мас.
1Н-ЯМР-спектр конечного продукта не поддается определению из-за присутствия парамагнитных ионов железа. ИК-спектр (KBr, см-1): 2910, 2880 (m, γ C-H), 1630 (vs, γ COO/as), 1400 (s, γ COO/s). Другие характерные, но не идентифицированные частоты: 1355 (m), 1330 (m), 1100 (s), 930 (m).
П р и м е р 10. Получение N, N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16- диазациклооктадекан-цинкового комплекса, динатриевой соли.
Используют 0,735 г (1,32 ммоль) продукта, полученного как описано в примере 1 б, и 0,180 г (1,32 ммоль) безводного хлористого цинка, из которых по способу, представленному в примере 4, получают 0,89 г требуемого продукта с выходом 97,5 который содержит 16,92 мас. хлористого натрия.
1Н-ЯМР-спектр (200 мГц, D2О, δ частей/млн.): 3,6 4,2 (18Н, широкая система полос t), 3,10 (8Н, широкая t).
П р и м е р 11. Получение N,N'-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диа- зациклооктадекана, трехнатриевой соли.
1,72 мл раствора соляной кислоты в концентрации 1,048 моль/л добавляют к раствору 1000 г (1,804 моль) тетранатриевой соли, полученной, как описано в примере 1 б, в 5 мл воды в условиях охлаждения (0 5оС). После этого раствор упаривают, остаток обезвоживают метилен-хлоридом и подсушивают при 60оС и пониженном давлении до получения 1,069 г конечного продукта с выходом 96,7 который содержит 9,5 мас. хлористого натрия.
1 Н-ЯМР-спектр (200 мГц, D2О, δ частей/млн.): 4,18 (2H, s), 3,79 (16Н, широкая m), 3,29 (8Н, широкая s). Спектр продукта в D2О в присутствии NaOD соответствует спектру, приведенному в примере 1 б. ИК-спектр (KBr, см-1): 2940, 22850 (m, γ C-H), 1655, 1605 (vs, γ COO/as), 1400 (m. γ COO/s). Другие характерные, но неидентифицированные частоты: 1345 (s), 1320 (s), 1120 (s), 1100 (s), 930 (m).
П р и м е р 12. Получение N-дикарбоксиметил-N'-(1,1-дикарбоксиэтил)-1,4,10,13-тетраокса-7,16-диазацикл оокттетранатриевой соли.
После нейтрализации 3,003 г (15,25 ммоль) бромметилмалоновой кислоты в 0,5 мл воды добавлением раствора гидроокиси натрия в концентрации 8,360 моль/л при 0 5оС в присутствии индикатора фенолфталеина в раствор добавляют 2000 г (7,62 ммоль) 1,4,10,13-тетраокса-7,16-диазациклооктадекана. Полученную смесь оставляют на 8-10 дн при 20-25оС, добавляя порциями 1,82 мл (15,25 ммоль) раствора гидроокиси натрия концентрацией 8,360 моль/л. По окончании реакции смесь перемешивают в течение 30 мин при 55 60оС, а затем упаривают. Сухой остаток экстрагируют несколькими порциями метилен-хлорида общим объемом 25 30 мл. После упаривания экстракта остаток обрабатывают эфиром и твердый осадок отфильтровывают. Остающийся материал (после экстракции эфиром) содержит небольшое количество 1,4,10,13-тетраокса-7,16-диазациклооктадекана, который можно удалить, разводя продукт метилен-хлоридом и осаждая эфиром. Таким способом получают очищенный N-(1,1-дикарбоксиэтил)-1, 4,10,13-тетраокса-7,16-диазациклооктадекан, двунатриевую соль, содержащую 13,81 мас. бромида натрия (промежуточного). Выход конечного продукта составляет 1,390 г (37,2%).
После
упаривания эфирного экстракта получают 1,159 г очищенного 2,4,10,13-тетраокса-7,16-диазациклооктадекана. Таким образом, конечный выход продукта достигает 38,5
1Н-ЯМР-спектр
промежуточного соединения (200 мГц, D2О, δ частей/млн.) 3,66 (16H, m), 2,82 (4H, t), 2,72 (4H, t), 1,36 (3H, s).
1Н-ЯМР-спектр (200 мГц, D2О, δ частей/млн.): 4,18 (2H, s), 3,79 (16Н, широкая m), 3,29 (8Н, широкая s). Спектр продукта в D2О в присутствии NaOD соответствует спектру, приведенному в примере 1 б. ИК-спектр (KBr, см-1): 2940, 2850 (m, γ C-H), 1655, 1605 (vs, γ COO/as), 1400 (m. γ COO/s). Другие характерные, но неидентифицированные частоты: 1345 (s), 1320 (s), 1120 (s), 1100 (s), 930 (m).
П р и м е р 12. Получение N-дикарбоксиметил-N'-(1,1-дикарбоксиэтил)-1,4,10,13-те- траокса-7,16-диазациклооктадекана, тетранатриевой соли.
После нейтрализации 3, 003 г (15,25 ммоль) бромметилмалоновой кислоты в 0,5 мл воды добавлением раствора гидроокиси натрия в концентрации 8,360 моль/л при 0 5оС в присутствии индикатора фенолфталеина в раствор добавляют 2000 г (7,62 ммоль) 1,4,10,13-тетраокса-7,16-диазациклооктадекана. Полученную смесь оставляют на 8-10 дн при 20-25оС, добавляя порциями 1,82 мл (15,25 ммоль) раствора гидроокиси натрия концентрацией 8,360 моль/л. По окончании реакции смесь перемешивают в течение 30 мин при 55 60оС, а затем упаривают. Сухой остаток экстрагируют несколькими порциями метилен-хлорида общим объемом 25 30 мл. После упаривания экстракта остаток обрабатывают эфиром и твердый осадок отфильтровывают. Остающийся материал (после экстракции эфиром) содержит небольшое количество 1,4,10, 13-тетраокса-7,16-диазациклоокта-декана, который можно удалить, разводя продукт метилен-хлоридом и осаждая эфиром. Таким способом получают очищенный N-(1, 1-дикарбоксиэтил)-1,4,10,13-тетраокса-7, 16-диазациклооктадекан, двунатриевую соль, содержащую 13,81 мас. бромида натрия (промежуточного). Выход конечного продукта составляет 1,390 г (37,2%).
После упаривания эфирного
экстракта получают 1,159 г очищенного 2,4,10,13-тетраокса-7,16-диазациклооктадекана. Таким образом, конечный выход продукта достигает 38,5
1
Н-ЯМР-спектр промежуточного соединения
(200 мГц, D2О, δ частей/млн.) 3,66 (16H, m), 2,82 (4H, t), 2,72 (4H, t), 1,36 (3H, s).
Используя это промежуточное соединение получают двузамещенное производное по описанному ниже способу.
После нейтрализации 0,483 г (2,64 ммоль) 2-броммалоновой кислоты в 0,5 мл воды посредством добавления раствора
гидроокиси натрия в концентрации 8,360 моль/л в
присутствии индикатора фенолфталеина добавляют 1,002 г (2,04 ммоль) вышеописанного промежуточного соединения, содержащего 13,81 мас. бромида натрия.
После этого реакционную смесь оставляют на 72 ч при
30-45оС, а затем еще на 24 ч при 50-60оС, добавляя по порциям 0,32 мл (2,64 ммоль) раствора гидроокиси натрия в концентрации
8,360 моль/л. По окончании реакции раствор упаривают,
а остаток высушивают при 75 80оС и пониженном давлении. Сухой остаток экстрагируют безводным этанолом, этанольный экстракт упаривают
досуха при пониженном давлении и высушивают с
образованием 1,323 г двойной соли, содержащей 2,06 моль бромида натрия, с выходом 83%
Продукт, свободный от бромида, можно получить посредством
экстракции двойной соли 96 об. этанолом, как
описано в примере 1 б. В результате образуется 0,532 г требуемого вещества с выходом 54,5
1Н-ЯМР- и ИК-спектры двойной соли практически идентичны
спектрам четырехнатриевой соли. ИК-спектр
(KBr, см-1): 2960, 2870 (m, γ C-H), 1645, 1600 (vs, γ COO/as), 1405, 1440 (m. γ COO/s). Другие характерные, но
неидентифицированные частоты: 1355 (s), 1315 (s), 10995
(s), 930 (m).1Н-ЯМР-спектр (200 мГц, D2О, δ частей/млн.): 3,89 (1H, s), 3,68 (16H, m), 2,92 (4H, t), 2,78 (4H, t),
1,41 (3H, s).
П р и м е р 13. Получение N-дикарбоксиметил-N'-(1,1-дикарбоксипропил)-1,4,10,13-тетраокса-7,16-диазаци клоотетранатриевой соли, двойной соли натрия бромида.
Получение осуществляют по способу, описанному в
примере 12, используя 1,608 г (7,62 ммоль) 2-бромоэтилмалоновой кислоты и 1000 г (3,81 ммоль) 1,4,10,13-тетраокса-7,16-диазациклооктадекана,
что дает 0,517 г N-(1,1-дикарбоксипропил)-1,4,10,
13-тетра-окса-7,16-диазациклооктадекана, двунатриевой соли, которая содержит 15,61 мас. бромида натрия. После упаривания эфирного экстракта получают 0,
508 г 1,4,10,13-тетраокса-7,
16-диазациклооктадекана. Таким образом, истинный выход искомого продукта составляет 53,3
1Н-ЯМР-спектр промежуточного соединения (200 мГц, D2
O, δ частей/млн.): 3,
68 (16H, m), 2,88 (4H, t), 2,82 (4H, t), 1,84 (2H, q), 0,90 (3H, t).
Искомая двойная соль, содержащая 3,10 моль бромида натрия, образуется с выходом 52,7 в количестве 0,475 г при использовании 0,517 г (1,00 ммоль) вышеуказанного промежуточного соединения, содержащего 15,61 мас. бромида натрия и 0,229 г (1,20 ммоль) 2-броммалоновой кислоты.
1Н-ЯМР-спектр искомой двойной соли (200 мГц, D2О, δ частей/млн.), 3,88 (1H, s), 3,67 (16H, m), 2,91 (4H, t), 2,86 (4H, t), 1,84 (2H, q), 0,88 (3H, t).
П р и м е р 14. Получение N-дикарбоксиметил-N'-(бензил-дикарбоксиметил)-1,4,10, 13-тетраокса-7,16-диазациклооктадекана, четырехнатриевой соли, содержащей двунатриевый оксималонат.
Используют процедуру,
описанную в примере 12, с той разницей, что реакцию осуществляют в смеси воды с этанолом (1:1), в которую добавляют 2,081 г (7,62 ммоль) 2-бромо-2-бензилмалоновой кислоты и 1000
г (3,81 ммоль) 1,4,10,
13-тетраокса-7,16-диазациклооктадекана. Таким путем получают 0,372 г N-(бензил-дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклоокта-декана, содержащего 11,21 мас. бромида
натрия (промежуточного
соединения) с выходом 17,4 После упаривания эфирного экстракта получают 0,714 г 1,4,10,13-тетраокса-7,16-диазациклооктадекана. Таким образом, истинный выход достигает 60,9%
1
Н-ЯМР-спектр промежуточного соединения (200 мГц, D2О, δ частей/млн.): 7,45 (2Н, d), 7,29 (3H, m), 3,68 (12H, m), 3,57 (4H, t), 3,47 (2H, s), 2,78 (8H, m).
Искомую
двойную соль, содержащую 0,42 моль двунатриевого оксималоната, получают, используя 0,372 г (0,662 ммоль) вышеуказанного промежуточного соединения, содержащего 11,21 мас. бромида
натрия и 0,161 г (0,
880 моль) 2-броммалоновой кислоты. Для получения конечного продукта сначала проводят экстракцию метилен-хлоридом, а затем абсолютным этанолом, что дает 0,432 г искомой соли с
выходом 91,4
1Н-ЯМР-спектр конечного продукта (200 мГц, D2О, δ частей/млн.): 7,43 (2H, d), 7,29 (3H, m), 3,90 (1H, s) 3,64 (16H, широкая), 3,34 (2H, s), 2,91 (4H,
t), 2,80 (4H, t).
П р и м е р 15. Получение N,N'-бис(1,1-дикарбоксиэтил)-1,4,10,13-тетраокса-7,16-диазациклооктадекана, четырехнатриевой соли.
После нейтрализации 3,11 г (14,79 ммоль) бромметилмалоновой кислоты, разведенной в 1 мл, путем добавления раствора гидроокиси натрия в концентрации 8,360 моль/л при 0 5оС в присутствии индикатора фенолфталеина добавляют 3,52 г (6, 565 ммоль) N-(1,1-дикарбоксиэтил)1,4,10,13-тетраокса-7,16-диазациклооктадекана, двунатриевой соли, содержащей 18,6 мас. бромида натрия (промежуточного). Затем реакционную смесь нагревают до 20 25оС и оставляют на 10-12 дн, добавляя порциями 1,77 мл (14,79 ммоль) раствора гидроокиси натрия в концентрации 8,360 моль/л. Перед окончанием реакции смесь в течение 30 мин прогревают при 55 60оС и упаривают. Сухой остаток экстрагируют несколькими порциями метилен-хлорида общим объемом 45 50 мл. Экстракт упаривают, остаток обрабатывают эфиром, преципитат отфильтровывают и высушивают. Таким способом получают 2,08 г (4,29 ммоль) промежуточного соединения, содержащего 14 мас. бромида натрия, который можно использовать при приготовлении следующей партии продукта.
Твердый материал, остающийся после экстракции метиленхлоридом, высушивают при 75-80оС и пониженном давлении. После экстракции сухого остатка 55 60 мл безводного
этанола полученный
экстракт упаривают до сухого остатка при пониженном давлении и высушивают до образования 3,90 г двойной соли, содержащей 78,2 мас. бромида натрия при выходе 22,2 Расчетный выход
промежуточного
продукта, используемого в реакции, составляет 64,3
1Н-ЯМР-спектр двойной соли (200 мГц, D2О, δ частей/млн.): 3,67 (8H, s), 3,64 (8H, t), 2,73 (8H,
t), 1,36 (6H,
s).
Продукт, не содержащий бромида натрия, получают из 3,90 г двойной соли, содержащей 78,2 мас. бромида натрия, которую экстрагируют 96 об. этанолом, как описано в
примере 1 б. Этим
способом получают 0,489 г требуемого продукта с выходом 57,5
После упаривания, высушивания и повторной экстракции безводным этанолом получают двойную соль, содержащую 80 90
мас. бромида
натрия (из фильтрата с 96%-ным этанолом). Эту двойную соль можно использовать при получении следующей партии продукта.
1Н-ЯМР-спектр искомого продукта (200 мГц, D2 О, δ частей/млн.): 3,70 (16Н, широкая m), 2,71 (8Н, коалесцентная t), 1,34 (6Н, расширенная s).
П р и м е р 16. Получение N,N'-бис(бензил-дикарбоксиметил)-1,4,10, 13-тетраокса-7, 16-диазациклооктадека на, тетранатриевой соли, двойной соли бромида натрия.
Используют процедуру, описанную в примере 15. Реакцию осуществляют с 0,362 г (1,324 ммоль)
2-бромо-2-бензилмалоновой кислоты и 0,372 г (0,662 ммоль) однозамещенного промежуточного соединения, содержащего 11,21 мас. бромида натрия в смеси этанола с водой при объемном соотношении 1:1. Таким
путем получают 0,085 г двойной соли, содержащей 4,2 моль бромида натрия, с выходом 11,1%
В ходе описанной процедуры получают 0,296 г промежуточного продукта, который содержит 11,21 мас.
бромида натрия. Таким образом, расчетный выход для используемого в реакции промежуточного продукта составляет 54,2%
1Н-ЯМР-спектр искомого соединения (200 мГц, D2О, δ
частей/млн.): 7,64 (4H, d), 7,45 (6H, m), 3,65 (12H, m), 3,65 (12H, m), 3,52 (4H, t), 3,31 (4H, s), 2,68 (8H, t).
П р и м е р 17. Показатели острой токсичности продуктов, получаемых согласно примерам 1 16, определяли на лабораторных мышах и крысах по нижеописанной методике.
Растворы, содержащие тестируемые соединения в различных концентрациях, готовили на основе физиологического раствора или 5%-ного (по весу) раствора глюкозы. Активные соединения в разных концентрациях вводили в кровяное русло животных, применяя для этой цели медленные, на протяжении 3 5 мин, инъекции. Для тестирования каждой дозировки использовали группы мышей линии Суисс и крыс линии Уистар по 6-10 особей в каждой. После инъекций наблюдения за животными продолжались в течение 30 дн. Показатели ЛД 50/30, т.е. дозы, вызывавшие гибель 50 животных на протяжении 30 дн, определяли по числу умерших в течение этого периода мышей и крыс с использованием пробит-анализа, как описано D.J.Finney (Probit Analysis, Ind ed. Cambridge University Press, 1952). Эти показатели выражали в ммоль/кг веса тела. Результаты оценки токсичности соединений, полученных согласно изобретению, суммированы в колонке В таблицы.
В таблице приведены основные характеристики продуктов согласно изобретению.
П р и м е р 18. Описан способ оценки стимулирующего действия соединений, получаемых согласно примерам 1 16, на выведение радиоактивных изотопов из организма мышей.
В брюшную полость животных вводили радиоактивные стронций (85 SrCl2) или церий (144СeCl3) с активностью от 37 до 74 кБк (1 2 мкКи). Затем животных подразделяли на группы по 5 0 особей каждая. Спустя 30 60 мин после введения радиоизотопов одной группе животных внутривенно инъекцировали активное соединение в количестве, обеспечивавшем его концентрацию в организме от 50 до 100 мкмоль/кг веса тела. Животным контрольной группы аналогично вводили носитель (стерильный физиологический раствор или 5%-ный раствор глюкозы), не содержащий тестируемый препарат.
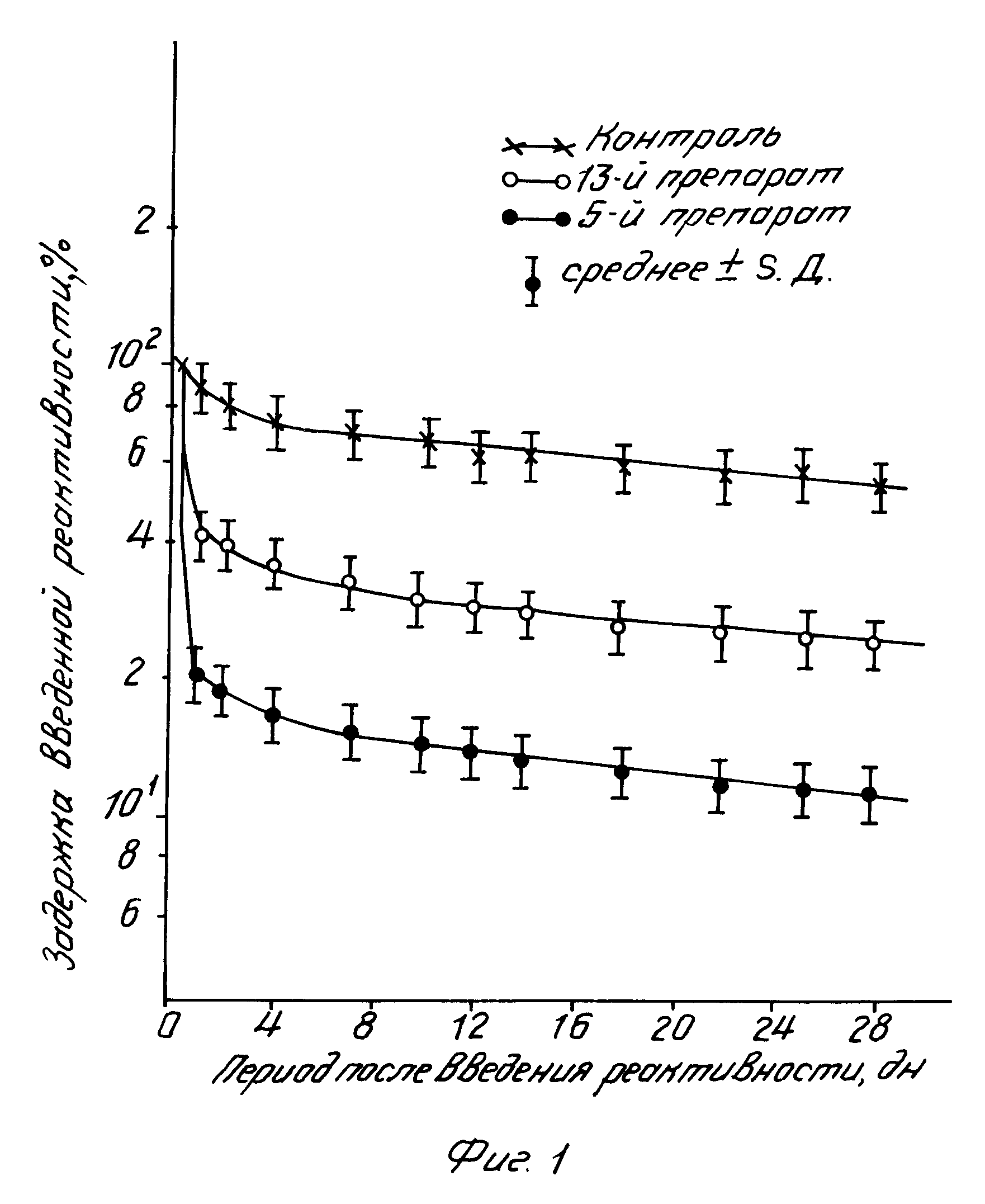
Количество радиоактивности в организме определяли сразу после введения изотопа, а затем повторяли измерения ежедневно или через каждые два или три дня в особом аппарате, сконструированном для установления содержания радиоактивности в теле мелких животных. Полученные результаты сравнивали с фоновой активностью в день 0 и рассчитывали так называемое время задержки, устанавливая его корреляцию с продолжительностью периода после введения радиоизотопа. Изменения содержания радиоактивности в организме животных со временем (в днях) показаны на фиг.1, на котором по ординате отложена задержка введенной радиоактивности (в процентах от ее исходного количества), а по абсциссе период после введения радиоактивности. Из фиг.1 следует, что скорость выведения стронция-85 после его введения в брюшную полость животных контрольной группы была невысокой: в течение первого дня выводилось только 15 в течение 4 дн 25 а на протяжении 7 дн 30 общего количества радиоактивности. В последующем скорость экскреции еще более замедлялась. С другой стороны, после однократного введения активного соединения, полученного как описано в примере 13, в дозе 100 мкмоль/кг в течение первого дня выводилось 40 всего количества радиоактивности, в течение 4 дн 65 а в течение 7 дн 67 Еще более впечатляющие результаты были получены при использовании соединения, полученного, как описано в примере 5. Выведение радиоактивности в указанные сроки составляло в этом случае соответственно 81, 84 и 85% На основании статистического анализа вышеуказанным методом было установлено, что кривые задержки активных соединений согласно изобретению описываются двукомпонентной нисходящей экспоненциальной функцией. Факторы Е и показатели Е1, характеризующие продукты, получаемые согласно изобретению, представлены в колонках С и D таблицы. Очевидно, что эффективность отдельных препаратов с точки зрения их стимулирующего действия на выведение радиоактивности из организма подопытных животных значительно отличается, особенно если основываться при ее оценке на показателях Е1. Соединения, имеющие показатель Е1 между 0 и 5, являются слабо активными, соединения с показателями Е1 от 5 до 50 могут оцениваться как обладающие средней эффективностью, а соединения, имеющие показатели Е1 от 50 до 100 или более высокие, должны рассматриваться в качестве высоко эффективных для данной цели препаратов.
П р и м е р 19.
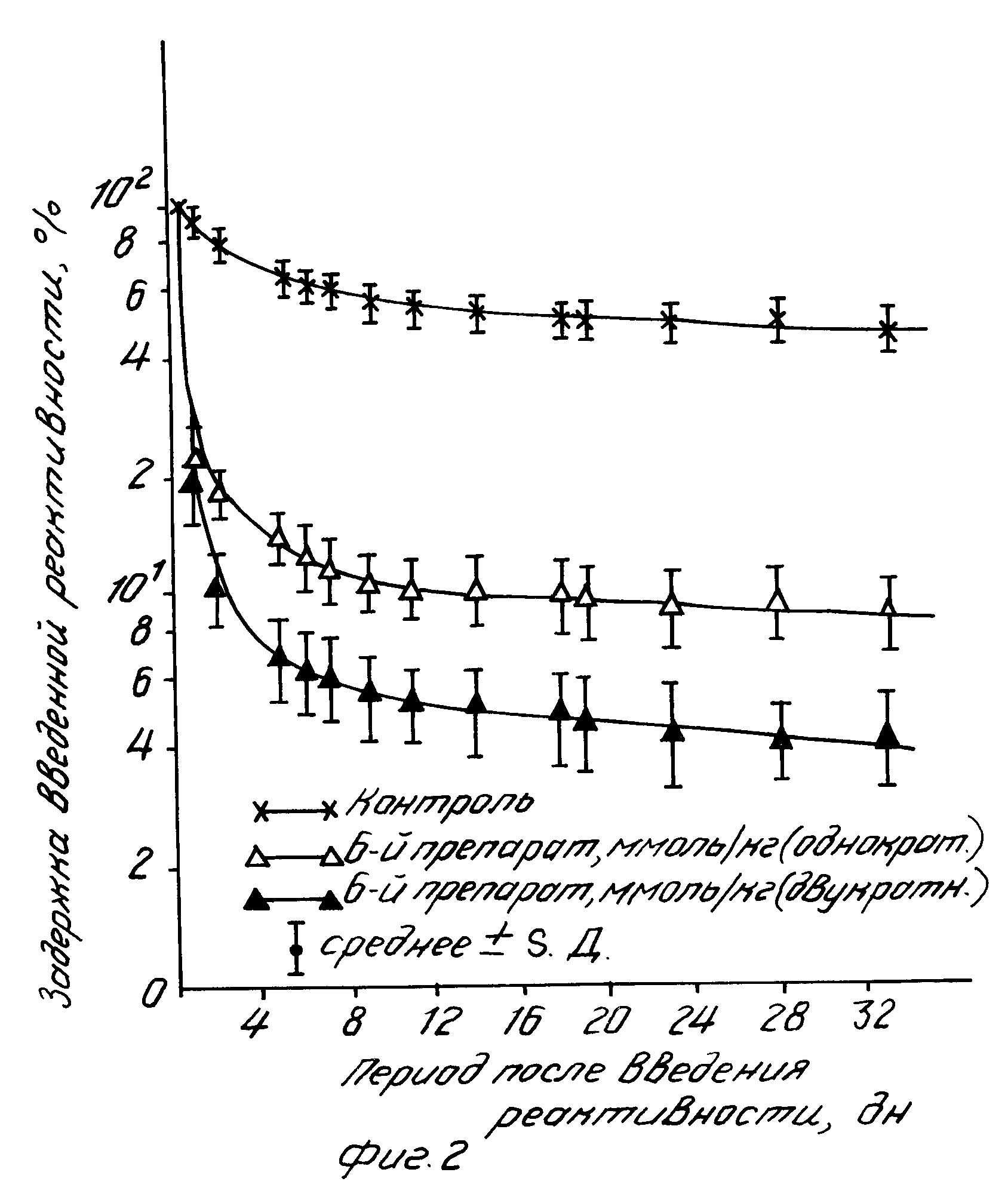
а) Кривые задержки радиоактивности, представленные на фиг.2, иллюстрируют выведение радиоактивного стронция, который вводили внутритрахейно в легкие крыс линии Уистар после внутрибрюшной инъекции соединения, описанного в примере 6б. Характер верхней кривой на фиг.2 свидетельствует о том, что радиоактивный стронций слабо выводится из организма контрольных животных, которым инъекциировали растворитель, не содержащий тестируемого соединения. На протяжении нескольких дней после радиоактивного загрязнения из организма выводилось не более 30 35 исходного количества радиоактивности. В то же время уровень радиоактивного загрязнения организма в целом уменьшался с 90 (в контроле) до 20 у животных, которым вводили однократно 50 мкмоль/кг веса тела соединения согласно изобретению (средняя кривая) или которым вводили его дважды на протяжении суток после загрязнения с 3-часовым интервалом (нижняя кривая). Быстрое удаление радиоизотопа продолжалось на протяжении всего периода наблюдений и достигало 88 90 при однократном введении препарата и до 94 96 после его двукратного введения. Важно подчеркнуть, что в ходе эксперимента радиоактивный стронций практически отсутствовал в костной ткани животных, которым вводили одно из соединений согласно изобретению (по данным обследования после забоя в конце эксперимента). Остаточная радиоактивность в мягких тканях и печени составляла 5-10% тогда как в контроле основная часть метки (65 70) задерживалась в костях. Аналогичные результаты были получены при внутривенных инъекциях тестируемых соединений или при их введении в подкожную соединительную ткань.
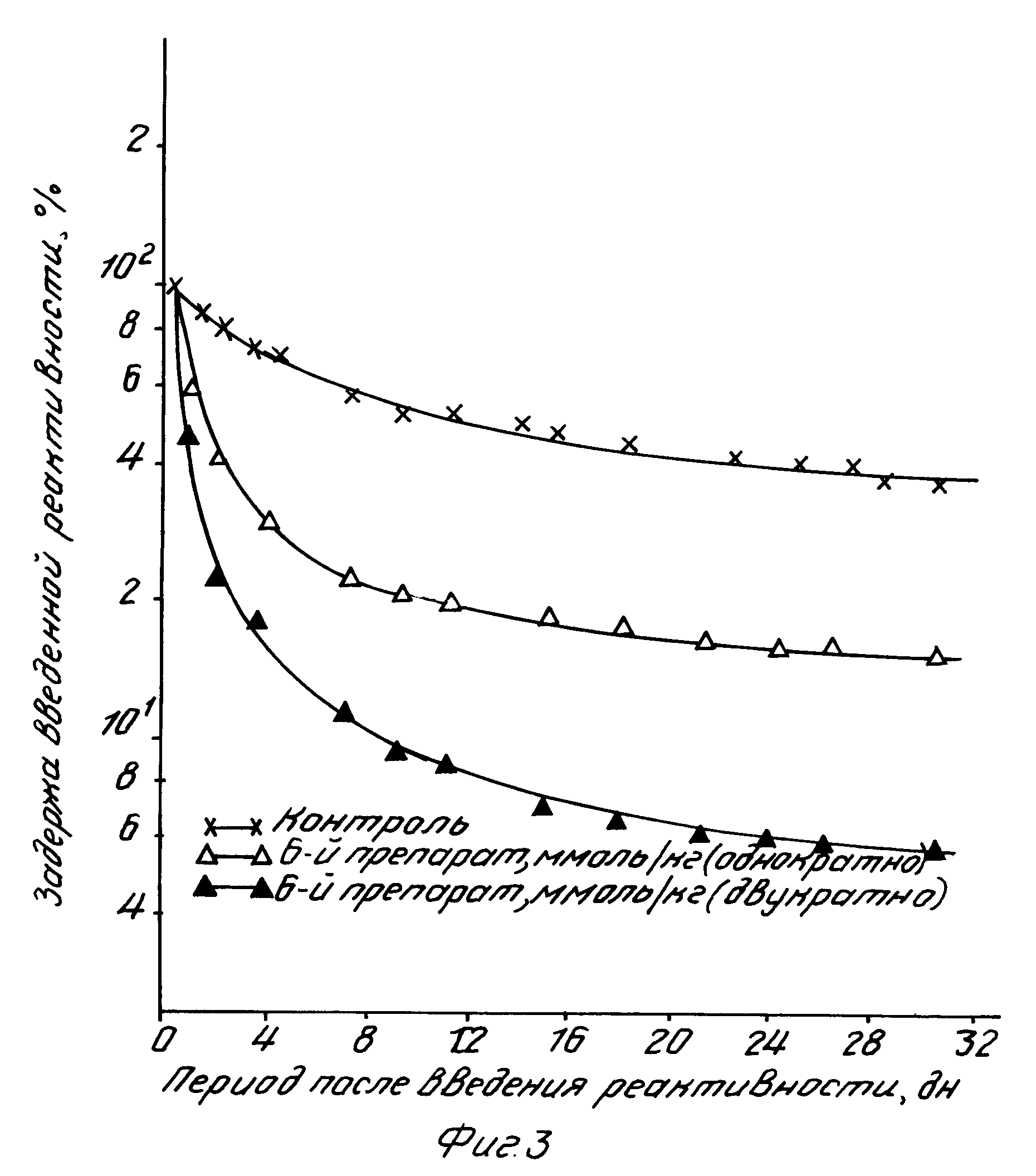
б) Соединения согласно изобретению, усиливающие выведение радиоактивности, испытывали также на способность стимулировать экскрецию других радиоактивных металлов, в частности церия-144, который относится к группе редкоземельных элементов. В данном примере иллюстрируется выведение144CeCl3 после его введения в легкие самок крыс линии Уистар как функция времени, прошедшего после однократной дозы или после повторного введения с интервалом в 24 ч (см. фиг.3). Полученные результаты показывают, что соединения согласно изобретению можно использовать для усиления выведения из организма радиоактивных материалов с относительно слабой растворимостью в биологических жидкостях (при загрязнении легких). К концу эксперимента, т.е. на 30 дн, у контрольных животных в легких сохранялось до 40 исходного количества радиоактивности, тогда как у животных, получавших однократную внутрибрюшинную инъекцию одного из активных соединений изобретению, этот показатель не превышал 14 Общая задержка радиоактивности под воздействием тестируемого соединения спустя 60 мин уменьшилась до 5,6 и продолжала уменьшаться на протяжении последующих суток. Ее резкое снижение в самом начале эксперимента и замедление в более поздние сроки были, по-видимому, обусловлены характером растворимости радиоактивного загрязняющего материала и особенностями элиминирования в составе комплекса.
П р и м е р 20. В примерах 17 19 описано действие соединений согласно изобретению на удаление радиоизотопов после введения в кровяное русло, брюшную полость и подкожную соединительную ткань. С точки зрения терапевтической эффективности у людей и, что более важно, с точки зрения быстрой и эффективной защиты крупных континентов населения представлялось важным показать, что соединения согласно изобретению можно использовать для ускорения удаления радиоактивных изотопов и при других путях их введения в организм. С этой целью были проведены эксперименты на крысах линии Уистар. Части животных радиоактивный стронций вводили в брюшную полость, а спустя 60 мин им внутритрахейно вводили активные соединения, полученные согласно изобретению. В качестве таких соединений использовали препараты с показателем Е1 более 100, которые показаны в таблице. После введения на протяжении 30 дн определяли общую радиоактивность организма крыс. При анализе кривых задержки радиоактивности оказалось, что в случае их построения обычным способом соединения согласно изобретению обладали высокой эффективностью с точки зрения элиминирования метки после их вдыхания в форме порошка или аэрозоля. У контрольных животных задержка достигала 91 а у крыс, которым вводили препарат согласно изобретению, она в первый же день уменьшалась до 15 а на третий день не превышала 10 Это наблюдение подтвердилось после расчета показателя Е1 (164) и фактора F (7,9)(сравни с таблицей).
В дополнительных экспериментах изучали возможность адсорбции активных соединений согласно изобретению с поверхности эпителиальных тканей. На спине крыс выбривали участок размером 3 х 3 см. После введения в их легкие внутритрахейным способом радиоактивного стронция (в условиях анестезии) определяли общий уровень радиоактивности в организме. После этого раствор соединения согласно изобретению наносили на подготовленный участок кожи и закрывали последний липким пластырем. На основании ежедневных измерений общего уровня радиоактивности в организме установлено, что соединение согласно изобретению обладало способностью усиливать экскрецию радиоактивности и при действии через кожу. Исходя из результатов измерений были рассчитаны фактор F и показатель Е1, которые оказались равными соответственно 105 и 110.
Полученные экспериментальные результаты показывают, что активные соединения, получаемые согласно изобретению, можно использовать в составе лекарственных средств в форме таблеток для помещения под язык, свечей, растворимых в пищеварительном тракте, драже, капсул или накожного пластыря.
Реферат
Использование: для выведения поражающих живые организмы металлических ионов, особенно радиоактивных изотопов. Изобретение относится к частично новым металлическим комплексам, солям и двойным солям 1, 4, 10, 13-тетраокса-7, 16-диазациклооктадекановых соединений первой формулы, приведенной в формуле изобретения, обладающим свойством выводить радиоактивные изотопы из живого организма, а также к фармацевтическим композициям, содержащим соединения указанной формулы. 6 з. п. ф-лы, 3 ил., 1 табл.
Формула
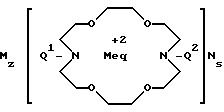
где Q1 и Q2 имеют одинаковые или различные значения и обозначают группу
где R водород, C1-C5-алкил с прямой или разветвленной цепью, фенил, фенил-C1-C5-алкил;
Me Mg, Ca, Fe (II), Zn; M N Na, K;
g 0,1;
z и s 1,2,
или M и N вместе Ca; z и s 0, при условии, что g 0, то M N ≠ Na, или g, z, s не могут одновременно равны 0.
Комментарии