Производные оксазола или их фармацевтически приемлемые катионные соли или кислотно-аддитивные соли - RU2079497C1
Код документа: RU2079497C1
Чертежи



Описание
Данное изобретение относится к некоторым соединениям формул I, II и IV, описанным ниже, полезных в качестве гипогликемических и гипохолестеринемических агентов, методам их использования и фармацевтическим композициям, их содержащим.
Несмотря на ранее открытые инсулина и его последующее широко распространенное применение в лечении диабетов, и более позднее открытие и использование сульфонилкарбамидов (например, хлорпропамид, толбутамид, ацетогексамид, толазамид) и бигуанидов (например, фенформин) в качестве оральных гипогликемических агентов, лечение диабетов остается менее чем успешным. Применение инсулина, необходимое примерно для 10% диабетических пациентов, для которых синтетические гипогликемические является не эффективными (диабеты типа I, инсульнозависимые сахарные диабеты), требует многократных суточных доз, обычно путем самоинъяекций. Установление правильной дозы инсулина требует частых определений сахара в моче или в крови. Прием избыточной дозы инсулина вызывает гипогликемию с эффектами в пределах от слабых аномалий в содержании глюкозы в крови до комы или даже смерти. Лечение не-инсулино зависимых сахарных диабетов (диабеты типа II) обычно состоит из сочетания диеты, физических упражнения, оральных агентов, например сульфонилкарбамидов, и в более тяжелых случаях, инсулина. Однако клинически пригодные гипогликемические агенты к сожалению чреваты другими токсическими проявлениями, которые ограничивают их применение. Во всяком случае при индивидуальном рассмотрении там, где один из этих агентов может не оказывать желаемого действия, другой может иметь успех. Непрерывная потребность в гипогликемических агентах, которые были бы менее токсичны или приносили успех там, где другие не оказывают желаемого действия, несомненно очевидна.
Кроме того, известно, что в Соединенных Штатах и Западной Европе к смертельным исходам приводят атеросклерозы, заболевание артерий. Последовательность патологий, приводящая к атеросклерозам и сердечной недостаточности, была описана детально Ross and Glomset. New England Journal of Medicine. р. 295, 369 377 (1976).
Начальной стадией в этой последовательности является образование "жировых прослоек" в сонной, коронарной и церебральной артериях и в аорте. Эти поражения желтого цвета, обусловленные присутствием липидных отложений, обнаружены преимущественно внутри гладко-мышечных клеток и в макрофагах, внутреннего слоя артерий и аорты. Холестерин и холестериновый эфир составляют в основном этот липид. Кроме того, в качестве постулата принято, что большая часть холестерина, найденного в жировых прослойках, является следствием поглощения из плазмы. Эти жировые прослойки, в свою очередь, приводят к развитию "фиброзной бляшки", которая состоит из аккумулированных внутренних клеток гладкой мышцы, перегруженных липидом и окруженных особым липидом клетчатки, коллагеном, эластином и протеогликанами. Клетки в совокупности с межклеточным веществом ткани формируют фиброзную капсулу, которая включает глубинные отложения осколков клеток и особенный липид клетчатки. Липид представляет собой главным образом свободный и этерифицированный холестерин. Фиброзная бляшка формируется медленно и вероятно со временем становится окаменевшей вследствие отложения известковых солей и некротической, развиваясь до "осложненного поражения", которое объясняет закупорку артерий и тенденцию к тромбозам стенок и спазмам артериальных мышц, что характеризуется наступлением атеросклерозов.
Эпидемиологическое основание имеет твердо установленную гиперлипидемию как первичный фактор риска в вызывании сердечно-сосудистого заболевания (CVD), обусловленного атеросклерозами.
В последние годы ведущие медики вновь обращают внимание на снижение уровней холестерина плазмы и низкий удельный вес липопротеинового холестерина, в частности как существенную ступень в предотвращении СУД. Теперь известно, что верхние пределы "нормального состояния" значительно ниже, чем оценивали прежде. В результате, теперь понятно, что большие группы населений Запада имеют высокую степень риска для развития или прогрессирования СУД вследствие этого фактора. Индивидуумы, которые обладают самостоятельными факторами риска в дополнение к гиперлипидемии, имеют особенно высокую степень риска. Такие самостоятельные факторы риска включают непереносимость глюкозы, повышенное кровяное давление гипертрофированного левого желудочка и принадлежность к мужскому полу. Сердечно-сосудистое заболевание особенно распространено среди диабетических больных, по крайней мере частично, вследствие существования многочисленных самостоятельных факторов риска. Успешное лечение гиперлипидемии у обычного населения и у субъекта с диабетическим заболеванием, в частности, является поэтому исключительно важным в медицине.
Первой ступенью в рекомендуемых терапевтических режимах при гиперлипидемии является диетическое вмешательство. Хотя одна диета вызывает адекватную реакцию у некоторых индивидуумов, у многих других сохраняется высокая степень риска и их нужно лечить далее фармакологическими способами. Новые лекарства для лечения гиперлипидемии обладают по этому высокой потенциальной пользой для большого числа индивидуумов при высокой степени риска развития СУД. Кроме того, особенно желательно успешное лечение как гиперлипидемии, так и гипергликемии, связанных с диабетическим состоянием, простым терапевтическим агентом.
В дополнение к гипогликемическим агентам, названным выше, сообщается о различных других соединениях, обладающих
такого типа активностью, как
рассматривается в обзоре Blank [Burger's Medical Chemistry, Fourth Edition, Part II, John Wiley and Sons, n. y.(1979), p.p.1057 1080] Sehnur, Патент США 4.367.234
раскрывает гипогликемические
оксазолидиндионы формулы
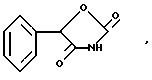
в котором фенильное кольцо является обычно моно- или многократно замещенным в орто/мета положениях. Примечательно, что за исключением 4-фторофенил аналога, перезамещенные производные являются либо неактивными, либо обладают низким уровнем гипогликемической активности.
Schnur, Патент США 4.342,771 раскрывает оксазолидиндион гипогликемические агенты формулы

в которой Y-водород или алкокси, Y'- водород или алкил, и Y"-водород или галоген.
Schnur, Патент США 4.617,312 раскрывает
гипогликемические
тиазолидиндионы формулы

где Rc-низший алкил, Xa-F, Cl или Br, и Ya-водород, хлор, низший алкил или низший алкокси. Примечательно, что соединения требуют орто-замещения алкокси группой, и пара-замещение ограничено по отношению к водороду или галогену.
Kawamatsu и др. Патент США 4.340.605 раскрывают гипогликемические соединения формулы

где Re-связь или низший алкилен и Rd является произвольно замещенной пяти- или шестичленной гетерициклической группой, включающей один или два гетеро-атома, выбранных из N, O и S, L1 и L2, каждая может быть определена как водород. Основанное на отсутствии гипогликемической и плазма триглицеридпониженной активности определенных неэфирных аналогов выдвинуто предположение, что отдельная часть структурной формулы, включающая кислород простого эфира, представляет ценную особенность для полезной активности в этой серии соединений
Sohda и др. Chem. Pharm.Bull. Japan, Vol.30, p.p. 3580 3600 (1982).
Eggler и др. Патент США 4.703,052, раскрывает гипогликемические
тиазолидиндионы формулы

где пунктирная линия представляет произвольную связь, Rf-H, метил или этил, Xb-O, S, SO, SO2, CH2, CO, CHON или NRk,
Rk-H или ацильная группа и многочисленные определения Rg,
Rh и Rj содержат в себе Rg, Rh и Ri как водород или метил,
и Rj как произвольного замещенный фенил, бензил, фенетил или стирол.
Clark и др. Международная Патентная Публикация N WO 89/08651, раскрывает гипогликемические тиазолидиндионы формулы.

где пунктирная линия представляет связь или отсутствие связи,
V является -CH=CH-, -N=CH-, -CH=N- или,
является CH2 , CHOH, CO, -C=NOR или -CH=CH
X является S, O, NR, -CH=N- или -N=CH-,
Y является CH или N,
Z представляет водород, (C1-C7 алкил или (C3 -C7) циклоалкил, фенил, нафтил, пиридил, фурил, тиенил или фенил моно- или дизамещенный теми же самыми или различными группами, которые представляют (C1-C3) алкил, трифторометил, (C1-C3) алкокси, фторо, хлоро или бромо;
Z' представляет водород или (C1-C3) алкил;
R и R1 каждый независимо водород или метил, и n равно 1, 2 или 3.
Краткое изложение изобретения.
Целью данного изобретения является получение оптически чистых форм спирта, который по существующим ранее ссылкам предварительно был обнаружен только в его рацемической форме. Изобретение обеспечивает каждый спирт в форме существенно свободной от его соответствующего энантиомера.
Данное изобретение направлено на (1S)-5- [4-(3-/5-метил-2-фенил-4-оксазолил)-1-гидроксипропил)бензил] тиазолидин-2,4 -дион (1), где соединение существенно свободно от его соответствующего IR энантиомера.
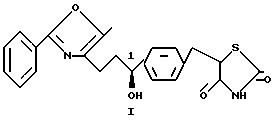
Изобретение также охватывает названный 1R энантиомер, (1R)-5-[4-(3-(5-метил-2-фенил-4-оксазолил) -1- гидроксипропил)бензил] тиазолидин-2,4-дион (II), где названный 1R энантиомер существенно свободен от его соответствующего 1S энантиомера.

Кроме того, в рамках объема изобретения находятся соединения формулы IV,

где Y CHOR (рацемический),
или

R представляет (C1-C4) алкил, (C7-C9) фенилалкил, фенил или алкоксиалкил формулы (CH2)nO(CH2)mCH3;
n равно 2, 3 или 4,
m равно 0, 1, 2, 3 или 4.
Данное изобретение охватывает также фармацевтически приемлемые катионные соли и фармацевтически приемлемые кислотно-аддитивные соли соединений предшествующих двух параграфов.
Подразумевается, что выражение "фармацевтически приемлемые катионные соли" может быть определено, но не ограничено такими солями, как соли щелочных металлов (например, натрия или калия), соли щелочноземельных металлов (например, кальция или магния), соли алюминия, соли аммония и соли с органическими аминами, такие как бензатин (N,N'-дибензилэтилендиамин) колин, диэтаноламин, этилендиамин, меглумин (N-метилглюкамин), бенетамин (N-бензилфенетиламин) диэтиламин, пиперазин, трометамин (2-амино-2-гидроксиметил-1, 3-пропандиол) и процеин. Особенно предпочтительной такой солью является соль натрия.
Подразумевается, что выражение "фармацевтически приемлемые кислотно-аддитивные соли" определяется, но не ограничивается такими солями как хлористоводородная, бромистоводородная, сернокислая, кислая сернокислая, фосфорнокислая, кислая фосфорнокислая (вторичная) кислая фосфорнокислая (первичная) уксуснокислая, янтарной кислоты, лимонной кислоты, метиленсульфоновой кислоты (мезилат) и толуолсульфоновой кислоты (тозилат) соли.
Кроме того, данное изобретение охватывает фармацевтические композиции для использования в лечении гипергликемического млекопитающего или гиперхолестеринемического млекопитающего, которые содержат понижающее содержание глюкозы в крови количество или понижающее содержание холестерина в крови количество соединения формул I, II и VI и фармацевтически-приемлемый носитель. Далее изобретение содержит метод снижения содержания глюкозы в крови у гипергликемического млекопитающего, который включает назначение названному млекопитающему эффективного количества снижающего содержания глюкозы в крови соединения формулы I, II и IV, и метод снижения холестерина в крови у гиперхолестринемического млекопитающего, который включает назначение названному млекопитающему снижающего холестерин крови количества соединения формул I, II и IV.
Также данное изобретение охватывает ключевые промежуточные соединения формулы
III

где X является

Дополнительные промежуточные соединения, которые входят в объем данного изобретения, это соединения формулы V.

где Z-Br,

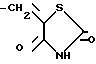
и названное промежуточное соединение по существу свободно от своего соответствующего энантиомера.
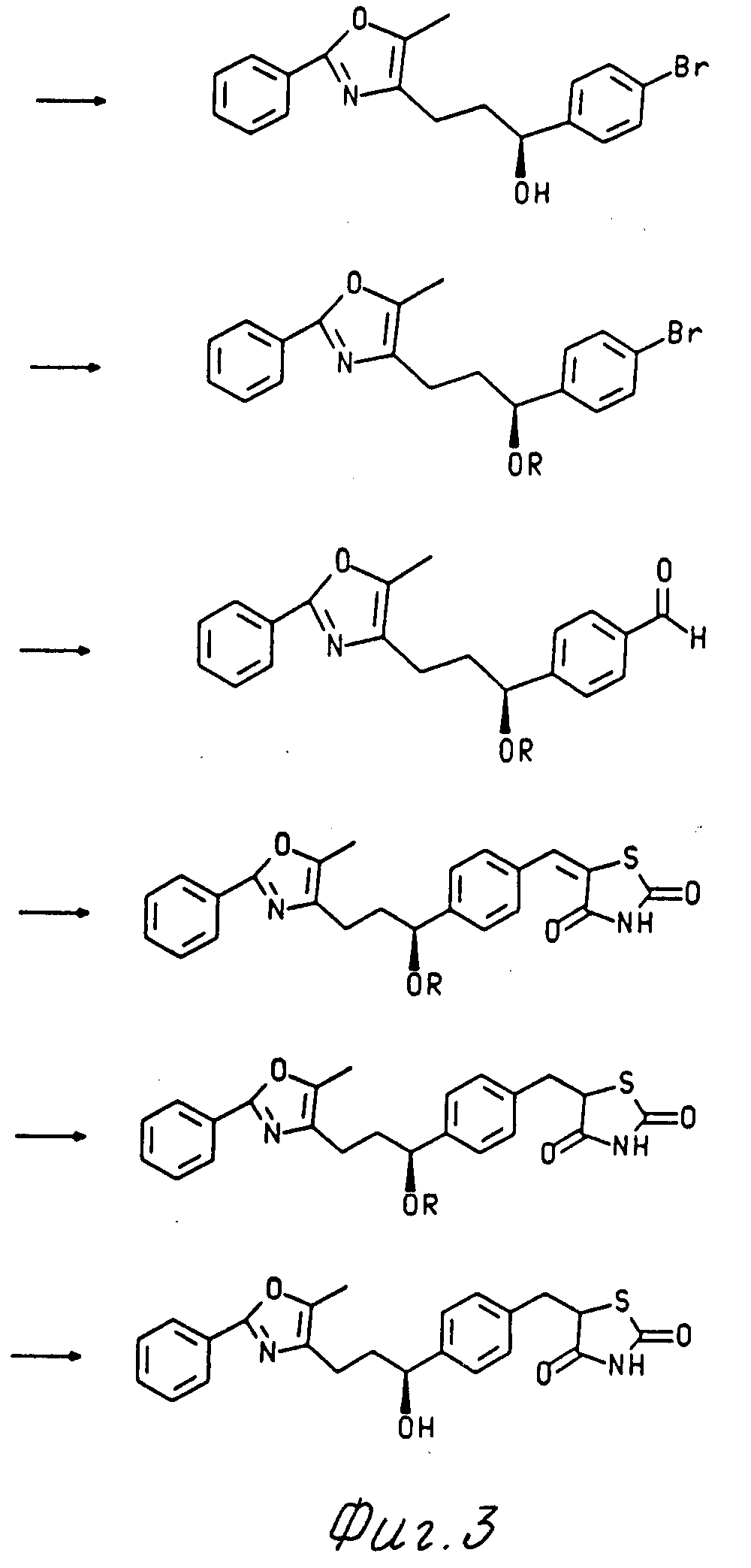
Все сведения, описанные здесь, легко получены в соответствии с реакционной последовательностью, изображенной на схеме 1 и описанной ниже.
Схема 1 изображена на фиг. 1, 2, 3.
п-бромоацетофенон подвергают взаимодействию с гидродом натрия и
диэтилкарбаматом в тетрагидрофуране с целью получения β -кетоэфира, который в дальнейшем подвергают взаимодействию с гидридом
натрия и 5-метил-2-фенил-4-оксазилметил хлоридом в
тетрагидрофуране с последующим гидролизом и декарбоксилированием в нагреваемом до кипения с обратным холодильником растворе уксусной кислоты и
соляной кислоты для получения кетона формулы VI

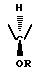
Этот кетон превращают в его S и R спиртовые продукты восстановления, используя один из двух различных методов. Так, восстановление кетона (VI) боргидридом натрия при 0oC в растворе тетрагидрофурана и этанола приблизительно от 20 минут до 8 часов дает рацемический спирт, который является смесью равных количеств соединений формул I и II.
Полученную таким образом рацемическую смесь разделяют на индивидуальные оптически чистые компоненты реакций с хиральным изоцианатом. Названный хиральный изоцианат выбран за его способность давать диастереоизомеры, которые легко разделимы некоторых физическими способами. Таким образом, (R)-(-)-1-(нафтил)этилизоцианат подвергается взаимодействию с рацемическим спиртом в нагреваемом до кипения с обратным холодильником толуоле в течение 17 часов.
Вносят дополнительное количество изоцианата с целью доведения реакции до завершения, и нагревание до кипения с обратным холодильником продолжают в течение 24 часов. Реакция дает два диастереомерных карбамата конфигураций RR и RS.
Различные физические свойства этих диастереомеров приводят к одному, RR изомеру, селективно кристаллизуемому из раствора, содержащего равные количества двух соединений. Раствор, используемый в этом конкретном случае, представляет собой систему диэтиловый эфир/гексан (1/2). Твердый материал, полученный из этой кристаллизации, перекристаллизовывают из этил ацетата для дальнейшей очистки (RR)-диастереомера.
Маточники стадий кристаллизации в перекристаллизации теперь преимущественно содержат (RS)-диастереомер. Удаление растворителей и очистка состава на силикагеле, элюируемым смесью гексан/диэтиловый эфир (1/2) приводит к оптически чистому (RS)-диастереомеру.
Разделенные таким образом стереомеры теперь из карбаматов превращают обратно в спирты по реакции названных карбаматов с трихлорсиланом и триэтиламином в бензоле. Каждый из полученных таким образом спиртов существует в виде отдельного энантиомера по существу свободного от его соответствующего энантиомера.
Вторым методом получения этих спиртов является приготовление их в оптически чистой форме непосредственно из кетонового предшественника через стереоселективный процесс восстановления, устраняющий таким образом необходимость хирального процесса разделения. Это стереоселективное восстановление достигается с помощью боранового восстанавливающего агента, такого как боран метил сульфидный комплекс, катехолборан или боран тетрагидрофуран в присутствии соответствующего хирального оксазаборолидинового катализатора в циклическом эфирном растворителе, таком как диоксан или тетрагидрофуран. Выбор стереохимия катализатора непосредственно влияет на стереохимическую конфигурацию производного спирта. Таким образом, выбор катализатора R-конфигурации приводит к спирту S конфигурации, выбор катализатора S конфигурации приводит к спирту R конфигурации. В частности, предпочтительной системой для получения S-спирта является реакция кетона формулы VI с боран метил сульфидным комплексом в присутствии (R)-тетрагидро -1- метил-3,3-дифенил-1Н, 3-пирроло [1,2-с] [1,3,2]оксазаборола при комнатной температуре в течение приблизительно от 15 минут до 3 часов; предпочтительной системой для получения спирта является реакция кетона формулы VI с боранметил сульфидным комплексом в тетрагидрофуране в присутствии (S)-тетрагидро-1-метил-3,3 дифенил,-1Н, 3Н-пирроло [1,2-с][1,3,2]оксазаборола.
Эти спирты далее перерабатываются до тиазолидиндионовых спиртов и эфиров, как изображено в схеме 1. Все реакции, описанные ниже, равно успешны как с R, так и S конфигурациями спирта формулы III.
Спирт формулы III подвергают взаимодействию с т-бутилдиметилсилилхлоридом и имидазолом в диметилформамиде при комнатной температуре в течение всей ночи для получения О-защищенного спирта. Бромид с защищенной таким образом спиртовой составляющей превращают в альдегид, используя хорошо известные условия н-бутиламин при -78oC, низкотемпературное резкое охлаждение аниона с сухим диметилформамидом и стандартную водную обработку. Под стандартной водной обработкой понижается разбавление реакционной смеси водой и экстракция образующегося водного раствора с достаточным количеством органического растворителя, обычно двумя или тремя порциями, с целью удаления каких-либо органических соединений из водного раствора. Органический растворитель, обычно этил ацетат, удаляют затем при пониженном давлении.
Полученный таким образом альдегид конденсируют с коммерчески доступным 2,4-тиазолидиндионом, используя общепринятые методы нагревания до кипения с обратным холодильником этанола и пиперидиновые катализы с целью получения продукта олефиновой конденсации. Произведенный таким образом олефин гидрируют путем введения водорода в закрытый реакционный сосуд, содержащий олефин, реакционно-инертный растворитель и катализатор. Давление внутри реакционного сосуда можно варьировать от 15 до 50 ф/дм2 (PSi) 1,054-3,515 кг/см2. Гидрирование будет протекать при этих условиях приблизительно от 2 до 48 часов. Предпочтительным катализатором является палладий благодаря его устойчивости к отравлению серой, и палладий, нанесенный на инертный носитель, такой как углерод. Под " реакционным инерционным растворителем" понимают растворитель, который не будет разлагаться или иначе мешать реакции. Реакционные инертные растворители для реакций такого типа включают этанол, метанол или тетрагидрофуран но не ограничиваются этими растворителями. Предпочтительным растворителем в этом случае является тетрагидрофуран.
Защитная группа снимается использованием 3,5% водной хлорной кислотой в тетрагидрофуране при комнатной температуре за приблизительно 12 часов. Конечным результатом этой реакции является спирт формулы 1 или формулы II. Полученный таким образом спирт будет зависеть от того, какой энантиомерный спирт был выбран после того, как названные спирты были получены в оптически чистой форме.
Оптически чистые спирты формулы III также полезны, как промежуточные соединения в приготовлении эфирных производных формулы IV. Так, взаимодействие спирта каждой формулы III с соответствующим основанием и алкил, алкоксиалкил, фенил или аралкил галогенидом формулы RX в реакционном инертном растворителе при температуре в пределах от 0oC до температуры кипения данного растворителя предпочтительно осуществляется от 2 до 48 часов.
R в соединении RX представляет собой (C1-C4) алкил, (C7-C9) аралкил, фенил или алкоксиалкил формулы-(CH2)nO (CH2)m CH3, где n равно 2, 3 или 4 и m равно 0,1,2,3 или 4. Х часть является хлоро, бромо или иодо. Реакционный инертный растворитель для реакций этого типа включает, но не ограничивается ими, диэтиловый эфир, диоксен, диметиксиэтан, тетрагидрофуран и диметилформамид. Предпочтительным растворителем является тетрагидрофуран, тогда как предпочтительным основанием является гидрид натрия. Предпочтительным алкил галогенидами являются метил иодид, этил и бензил бромид.
Эфиры, полученные, как описано в предшествующем параграфе, индивидуально превращают в тиазолидин-2,4 -дионы формулы IV тем же способом, как описано для получения соединений формул 1 и 2. Так, бромо составляющую эфира формулы III подвергают взаимодействию с н-бутиллитием и диметилформамидом в тетрагидрофуране для получения альдегида, который подвергают взаимодействию с 2,4-диазолидиндионом и каталитическим пиперидином в этаноле, получая продукт конденсации в виде олефина. Этот олефин восстанавливают в присутствии палладия на углероде в тетрагидрофуране, получая заданный конечный продукт формулы IV. Специфические подробности реакции, используемых для получения этих эфиров аналогичным подробностям, описанным в предыдущих параграфах относительно приготовления спиртов формул I и II.
Данные соединения формул I, II и IV используются как гипоглицемические или гипохолестеролемические агенты для млекопитающих. Соединения формул I и II дополнительно являются метаболитами своих соответствующих кетонов in vivo (в живом организме). У мужчин преимущественно образуется S-форма спирта. Активность, требуемая для первого из названных клинических применений, определяется путем испытания на гипогликемическое действие на ов/ов мышах следующим способом.
Мыши в возрасте от пяти до восьми недель C57 BL/CJ-ов/ов (полученные из лаборатории Jackson'a, Bartlarbor, Maine) были посажены по пять в клетку при стандартных условиях ухода за животными. Через неделю аклиматизацйионного периода животных взвесили и отобрали для обработки 25 микролитров крови с помощью ocular bleed до любой дальнейшей обработки для отбора крови. Образец крови немедленно разбавили 1 5 физиологическим раствором, содержащим 2,5 мг/мл фторида натрия и 2% гепарина натрия, и держали на льду для метаболических анализов. Затем животных ежедневного в течение пяти дней вводили лекарство (5 50 мг/кг), позитивный контроль (50 мг/кг) циклитазона, Патент США, 4467902, Sohda и др. Chem. Pharm. Bull. vol. 32. p.p. 4460 - 4465, 1984), или связующего вещества. Все лекарства назначались в связующем веществе, состоящем на 25 об. из метилцеллюлозы. На 5 день животных снова взвешивали и отбирали кровь (окулярным путем) для определения уровней метаболитов крови. Свежесобранные образцы центрифугировали две минуты при 10,000 xg (об/мин) при комнатной температуре. Супернатант анализировали на глюкозу, например с помощью ABA 200 Бихроматического Анализатора, используя A-gent глюкозную УФ систему реагентов токсикипазным методом (модификация метода Richterich and Dauwalder, Schweigeriche Mediginische Woshenschrift, 101, 860) (1971), применяя 20, 60 и 100 мг/мл стандарты. Глюкозу плазмы рассчитывали затем по уравнению глюкоза Плазмы (мг/дл) объем образца x 5 x 1,67 8,35 x объем образца, где 5 есть фактор разбавления и 1,67 - регулирование плазмы гематокритом (суммарный гематокрит равен 40%). A registered trademark of Abbot Laboratories, Diagnostics Division, 820 Mission Street, SO, Pasadena, California, 91030.
Животные,
которым давали лекарство с
связующим веществом, сохраняют в основном неизменные гипергликемические уровни глюкозы (например, 250 мг/дл), тогда как животные позитивного контроля имели пониженные
уровни глюкозы (например 130
мг/дл). Испытуемые соединения охарактеризованы в процентах нормализации глюкозы. К примеру, уровень глюкозы, являющийся таким же, как в позитивном контроле, представлен
как 100%
Исследования,
подобные описанным ниже, демонстрируют, что соединения формулы (I) осуществляют снижение уровней холестерина сыворотки у млекопитающих.
Использованы самки мышей (линия C 57 Br/Cd J), полученные из лаборатории Jackson'a, Bar Harbor, Maine, в возрасте 8 12 недель, после 2 4 недельной акклиматизации при свободном доступе к воде и стандартной лабораторной пищи. Животных произвольно разделили на три группы по 6 7 животных. Все три группы перевели на диету, содержащую 0,75% холестерола, 31% сахарозы, 15,5% крахмала, 20% казеина, 17% целлюлозы, 4,5% кукурузного масла, 5% кокосового масла, 0,25% холиевой кислоты, 4% солей и 2% витамина, разрешающую кормить ad. lib (по потребности на выбор) в течение 18 дней, и ежедневно давали лекарство в 9 11 часов a. m. (до полудня) в течение последних 5 дней путем орального введения, контрольной группе с 5 мг/кг связующего вещества (0,1% водной метил целлюлозы) и испытуемым группам с изучаемым соединением при дозах в пределах от 0,1 до 10 мг/кг/день в связующем веществе. После четвертого дня приема, животных не кормил в течение всей ночи, начиная с 5 p.m. (после полудня). На следующее утро испытуемым группам вводилась пятая и последняя доза соединения и, спустя три часа, животных умерщвляли путем обезглавливания. Кровь из главной артерии тела собрали и дали возможность свернуться, и сыворотку анализировали ферментативно, используя Abbott VP автоматический анализатор, на HDL холестерин LDL и VIDL холестерин, и общий холестерин. Независимо от оценки на основании LDL + VLDL уровней холестерола, суммарных уровней холестерина или соотношения LDL + VLDL/HDL, соединения данного изобретения в большинстве случаев показывают благоприятный результат в снижении уровней холестерина. Представленные соединения формул (I, II и IV) могут быть клинически назначены млекопитающим, включая человека, либо оральным, либо парентеральным путем. Предпочтительно введение оральным путем, являющимся более подходящим и избегающим боли и раздражения инъекции. Однако в обстоятельствах, где пациент не может глотать лекарство, или абсорбция следующая за оральным применением ослаблена за счет болезни или другой аномалии, необходимо, чтобы лекарство вводилось парентерально. При обоих путях дозировка находится в пределах приблизительно от 0,10 до 50 мг/кг веса тела субъекта на день, предпочтительно примерно от 0,10 до 10 мг/кг веса тела на день, назначаемая единичной или раздельными дозами. Однако оптимальная дозировка для индивидуального субъекта нуждающегося в лечении, будет определяться лицом ответственным за лечение, обычно первоначально назначаются меньшие дозы и затем производят увеличение относительно этого для определения наиболее подходящей дозы. Она будет изменяться в соответствии с конкретным применяемым соединением и субъектом, подлежащим лечению.
Соединения могут быть использованы в фармацевтических препаратах, содержащих соединение или его фармацевтически приемлемую соль кислоты, в комбинации с фармацевтически-приемлемым носителем или растворителем.
Пригодные фармацевтически-приемлемые носители включают инертные твердые наполнители или растворители и стерильные водные или органические растворы. Активное соединение будет присутствовать в таких фармацевтических композициях в количествах, достаточных, чтобы обеспечить желаемое дозированное количество в пределах, описанных выше. Таким образом, для орального применения соединения могут быть комбинированы с подходящим твердым или жидким носителем или растворителем в форме капсул, таблеток, порошков, сиропов, растворов, суспензий и тому подобных. Фармацевтические композиции могут, при желании, содержать дополнительные компоненты, такие как ароматизаторы, дезодораторы, эксципиенты и тому подобные. Для парентерального применения соединения могут быть комбинированы со стерильными водными или органическими средами для получения инъекционных растворов или суспензий. Например, могут использоваться растворы в кунжутовом или арахисовом масле, водный пропилен гликоль и тому подобные, также как и водные растворы водорастворимых фармацевтически приемлемых дополнительных кислотных солей соединений. Инъекционные растворы, приготовленные таким способом, могут затем применяться внутривенно, внутрибрюшинно, подкожно или внутримышечно, с предпочтительным внутримышечным применением для мужчин.
Данное изобретение иллюстрируется последующими примерами. Однако, должно быть понятно, что изобретение не ограничивается специфическими деталями этих примеров. Все реакции проводятся в инертной атмосфере, такой как азот, если не указано особо. Сокращения ТГФ и ДМФ, где используются, относятся к тетрагидрофурану и диметилформамиду соответственно. Предполагается, что такие растворители, содержащие достаточно небольшое количество воды, удовлетворяют тому, что названная вода не мешает протеканию названных реакций. Использованная здесь номенклатура основана на Rigandy and Klesney, IUPAC Nomenclature of Organic Chemistry, 1979 Ed. Pergamon Press, Hew York, 1979.
Пример 1
Этил-4-бромобензоилацетат
Гидрид натрия (5,2 г, 0,21 моль)
суспендировали в сухом этиловом эфире и охладили до 0oC. Добавили диэтил карбонат (17,7 г, 0,15 моля)
и содержимое перемешивали в течение десяти минут, тогда как добавления по каплям
п-бромацетофенона (19,9 г. 0,1 моля) в диэтиловом эфире (50 мл) и этанола (0,2 мл) инициировало реакцию. Прибавление
продолжали в течение двадцати минут, раствор нагревали до кипения с обратным
холодильником в течение трех часов, охладили до комнатной температуры и вылили в охлажденную 10% водную соляную кислоту
(250 мл). Водный раствор дважды экстрагировали диэтиловым эфиром (750 мл) и
объединенные экстракты
промывали последовательно водой (250 мл), раствором соли (250 мл) и высушили (MgSO4).
Растворитель отогнали при пониженном давлении и остаток очистили на силикагеле,
элюируя смесью гексан/этил ацетат (4,1), получив 20,2 г (74%) названного в заглавии соединения в виде масла. ПМР
(60MH2, ClDCl3): δ.1,1(t, 3H) 3,9(S, 2H), 4,1(q, 2H), 4,
1(q, 2H), 7,55(d, J 7H, 2H), 7,75(d, J 2H2, 2H).
Здесь и далее по тексту в спектрах ПМР (1H ЯМР) d дуплет, S - синглет, t триплет, m мультиплет, q квартет, d d двойной дупдет, b - уширенный сигнал.
Пример 2
4-[3-(5-метил-2-фенил-4-оксазолил)пропионил]бромобензол
Гидрид натрия (1,3 г, 55 ммолей) суспендировали в ТГФ (75 мл) и
охладили до 0oC. Названное в заглавии соединение примера 1
(14,9 г, 55 ммолей) растворили в ТГФ (75 мл) и добавили по каплям к суспензии за 30 минут. Образовавшийся раствор перемешивали
дополнительно 30 минут после этого времени добавили порциями за 5 минут
твердый 5-метил-2-фенил-4-оксазолил метил хлорид (10,0 г, 48 ммолей). Реакционную смесь нагревали до кипения с обратным
холодильником в течение 48 часов, охладили до комнатной температуры и
сконцентрировали при пониженном давлении. Остаток растворили в уксусной кислоте (120 мл) и концентрированной HCl (30 мл), и
нагревали до кипения с обратным холодильником пять часов. Реакционную смесь
охладили до комнатной температуры и вылили в ледяную воду (300 мл). Водный раствор экстрагировали дважды этилацетатом (500
мл) и органические экстракты объединили и промыли раствором соли (250 мл),
высушили (MgSO4) и сконцентрировали при пониженном давлении. Очистка на силикагеле, элюируемом смесью гексан/этил
ацетат (4/1), привела к сырому твердому продукту, который был в дальнейшем
очищен перекристаллизацией из гексана для получения названного в заглавии соединения (11,5 г, 65%) в виде белых кристаллов с
Т. пл. 80 81oC. ПМР (60 MH2, CDCl3); d 2,
2 (S, 3H), 2,8(т, 2H), 3,2(т, 2H), 7,2 - 8,0 (т, 9H).
Пример 3
(S)-4-[3-(5-метил-2-фенил-4-оксазолил)-1-гидроксипропил]бромбензол
Названное в заглавии предыдущего
примера соединение (20 г, 54 ммоля) растворили ТГФ (200 мл) при температуре окружающей
среды и обработали 4A молекулярными ситами (10 г, предварительно высушенными в глубоком вакууме при 150oC, в течение ночи). После выдерживания в течение ночи, раствор декантировали с ситов
и было определено содержание воды 0,0092% (анализами Карла Фишера). Добавили (R)-Тетрагидро-1- метил-3,
3-дифенил-1H, 3H-пирроло[1,2-c] [1,3,2] оксазоборол (748 мг, 2,7 ммоля) при температуре
окружающей среды и раствор обработали боран метил сульфидным комплексом (2M, в ТГФ, 76 мл. 152 ммоля)
добавлением по каплям в течение 75 минут. Реакционную смесь перемешивали дополнительно 15 минут,
охладили до 0oC и погасили реакцию путем добавления по каплям холодного метанола (280 мл).
Раствор после гашения перемешивали 18 часов при температуре окружающей среды. Растворители
отогнали при пониженном давлении и остаток растворили в метилен хлориде (200 мл) и промыли последовательно
водным фосфатным буфером с pH 4 (200 мл) и высушили (MgSO4). Органический слой
перегнали при атмосферном давлении до остаточного объема 100 мл, добавили гексан и продолжили перегонку до
достижения дистиллятом температуры 62oC. Убрали источник тепла и остаток
кристаллизовали и гранулировали в течение 16 часов. Белый твердый продукт собрали вакуумной фильтрацией и высушили
в глубоком вакууме для получения названного в заглавии соединения (17,46 г. 87%
>99% избыток энантиомера).
Пример 4
4-[3-(5-метил-2-фенил-4-оксазолил)-1гидроксипропил]бромобензол
Названное в заглавии примера 2 соединение (5,0 г, 13 ммолей)
растворили в ТГФ (75 мл) и добавили по каплям в течении 20 минут к
суспензии боргидрида натрия (513 мг, 13 ммолей) в 75 мл этанола при 0oC и реакционную смесь перемешивали в течение 3 часов
при 0oC. Реакционную смесь вылили в ледяную воду (500
мл) и дважды экстрагировали диэтиловым эфиром (700 мл). Органические экстракты объединили и промыли водой (250 мл) раствором соли (250
мл) и высушили (MgSO4). Растворитель удалили при
пониженном давлении и остаток перекристаллизовали из гексана для получения 4,4 г (92%) рацемического названного в заглавии соединения
Т. пл. 82 83oC. ПМР (60 MH2,
CDCl3); d 2,0 (m, 2H), 2,2(S, 3H), 2,5 (t, J 6H2, 2H), 4,6 (m, 1H), 4,7 (уширенный S, 1H, гидроксил протон), 7,1 7,5 (m,
7H), 7.8 8,0 (m, 2H).
Пример 5
(RR)-4-[3-[5-метил-2-фенил-4-оксазолил)-1-(1-нафтил) этиламинокарбонилокси)пропил]бромобензол.
Названное в заглавии примера 4 соединение (1,8 г. 5 ммолей) обработали (R)-(-)-1-(нафтил)этилизоцианатом (1,0 г, 5 ммоля) в толуоле (100 мл) и образовавшийся раствор нагревали до кипения с обратным холодильником в течение 17 часов. Прибавили дополнительный 1 г изоцианата и нагревание до кипения с обратным холодильником продолжали дополнительно 24 часа. Растворитель удалили при пониженном давлении и остаток перекристаллизовывали из смеси диэтиловый эфир/гексан (1/2) для получения 1,1 г (37%) твердого продукта. Перекристаллизация из этил ацетата привела к 570 мг (20%) чистого более полярного соединения, названного в заглавии с Т.пл. 185 186oC.

Пример 6
(RS)-4-[3-(5-метил-2-фенил-4-оксазолил)-3-(1
-нафтил)этиламинокарбонилокси] пропил бромобензол
Маточник от стадий кристаллизации и перекристаллизации предыдущего примера сконцентрировали при
пониженном давлении и очистили на силикагеле,
элюируя смесью гексан/диэтиловый эфир (1/2) для получения 630 мг (22%) чистого менее полярного диастереомера. Т.пл. 120 125oC [α]D- 39,55 (C 0,31, МДСО).
Пример 7
(S)-4-[3-(5-метил-2-фенил-4- оксазолил)-1-гидроксипропил]бромобензол
Названное в заглавии примера 6 соединение (1,56 г, 2,7
ммоля) растворили в бензоле (65 мл), обработали
трихлорсиланом (1,4 мл) и триэтиламином (1,9 мг) и образовавшийся раствор перемешивали при температуре окружающей среды в течение 18 часов. Реакционную
смесь разбавили водой (250 мг) и этилацетатом
(250 мл) и перемешивали десять минут. Слои разделили и водный слой экстрагировали этил ацетатом (250 мл). Органические экстракты объединили, промыли
насыщенным водным бикарбонатом натрия (100 мл),
водой (100 мл), раствором соли (100 мл) и высушивали (MgSO4). Растворители удалили при пониженном давлении и остаток очистили на силикагеле,
элюируя смесью гексан/диэтиловый эфир (1/1),
получив чистый S-спирт в виде смолы (820 мг). ПМР спектр показал, что это рацемическая смесь.
Пример 8
(S)-4-[1-(т-бутилдиметилсилилокси)-3-(5-метл-2-фенил)-4- оксазолил)пропил]бромобензол
Названное в заглавии примера 7 соединение (769 мл, 2,0 ммоля) T-бутилдиметилсилилхлорид (377 мг, 2,5
ммоля) и имидазол (340 мг, 5,0 ммоля) объединил в ДМФ (10 мл) и перемешивали при комнатной температуре 24 часа. Реакционную смесь разбавили водой (100 мл) и экстрагировали этилацетатом (2х100 мл).
Органические слои объединили, промыли водой (100 мл), насыщенным водным бикарбонатом натрия (100 мл), раствором соли (100 мл) и высушили (MgSO4). Растворитель удалили при пониженном
давлении для получения названного в заглавии соединения в виде смолы (860 мг, 85%). ПМР (60 MH2, CDCl3) δ 0,5 (d, 6Н), 1,0 (s, 9Н), 2,0 2,7 (m, 4Н), 2,3 (s, 3Н), 4,8 (t,
J
5Н2, J Н), 7,1 7,6 (m, 7Н), 7,9 8,1 (m, 2Н).
Пример 9
(S)-4-[1-(т-бутилдиметилсилилокси)-3-(5-метил-2-фенил-4-оксазолил) пропил]бензальдегид.
н-Бутиллитий (1,6 М в гексане, 1,3 мл) добавили в течение десяти минут к названному в заглавии примера 8 соединению (780 мг, 1,6 ммоля) в ТГФ (60 мл). Реакционную смесь перемешивали при 78oC дополнительно 50 минут и добавили сухой ДМФ (152 мг, 2,0 ммоля). Реакционную смесь перемешивали дополнительно 1,5 часа при 78oC и затем 1,5 часа при комнатной температуре. Реакционную смесь разбавили этилацетатом (200 мл) и промыли водой (50 мл), 10% водным насыщенным бикарбонатом натрия (50 мл), раствором соли (50 мл) и высушили (MgSO4). Растворитель удалили при пониженном давлении и остаток очистили на на силикагеле, элюируя смесью гексан/диэтиловый эфир (4/1), для получения названного в заглавии альдегида (650 мг, 93%). ПМР (60 MH2, CDCl3), d 0, 5 (d, 6Н), 1,0 (S, 9Н), 2,0 2,7 (m,4Н), 2,3 (S, 3Н), 4,9 (dd, J 6НZ, 12НZ, 1), 7,2 8,0 (m, 9Н), 10,1 (S, 1Н).
Пример 10
(S)-5-[4(1-т-бутилдиметилсилокси)-3-(5-метил-2-фенил
-4-ксазолил)попил)фенилметилен]тиазолидин-2,4-дион.
Названное в заглавии примера 9 соединение (341 мг, 0,78 ммоля), 2,4-тиазолидиндион (183, мг, 1,56 ммоля) и пиперидин (14 мг, 0,15 ммоля) объединили в этаноле (10 мл) и нагревали до кипения с обратным холодильником в течение 18 часов. Реакционную смесь охладили до комнатной температуры и сконцентрировали при пониженном давлении. Остаток очистили на силикагеле, элюируя смесью гексан/этил ацетат/ уксусная кислота (16/4/1), для получения твердого продукта, который растерли в гексане и получили названное в заглавии соединение в виде твердого белого продукта (163 мг, 39%) Т.пл. 158-160oC. ПМР (300 МН2, CD Cl3): d -0,5 (d, 6H), 1,0 (S,9H), 2,0-2,7 (m, 4H), 2,3 (S,3H), 4,9 (m,1H), 7,6-7,7 (m,7H), 7,8 (S,1H), 8,0 (m, 2H).
Пример 11
(S)-5-[4-(1-(т-бутилдиметилсилилокси)-3-(5-метил-2-фенил -4-оксазолил)протил)бензил] тиазолидин-2,4-дион.
Названное в заглавии примера 10 соединение (160 мл. 0,3 ммоля) и 10% палладий на углероде (160 мл) объединили в ТГФ (10 мл) и восстанавливали на вибраторе Парра при 50 psi и комнатной температуре в течение 22 часов. Суспензию профильтровывали через инфузорную землю и растворитель удалили при пониженном давлении для получения названного в заглавии соединения в виде смолы (180 мг). ПМР (300 МН2, CDCl3): d 0,5 (d, 6H), 1,0 (S, 9H), 2,0-2,2 (m, 2H), 2,3 (S,3H), 2,4-2,6(m, 2H), 3,4 (d d, 1H), 4,3 (d d,1H), 4,7 (d d, 1H), 7,0-7,3 (m, 7H), 7,8 (m, 2H).
Пример 12
Натриевая соль
(S)-5-[4-(3-(5-метил-2-фенил-4-оксазолил) -1-гидроксипропилбензил]тизолидин-2,4-диона
Названное в заглавии примера 11 соединение (160 мг, 0,3 ммоля) растворили в ТГФ (5 мл) и обработали 3,5%
водной хлорной кислотой (3 мл). Реакционную смесь перемешивали при комнатной температуре 12 часов, разбавили этилацетатом (25 мл), промыли водой (25 мл) раствором соли (25 мл) и высушили (MgSO4). Растворитель удалили при пониженном давлении, остаток очистили на силикагеле, элюируя смесью гексан-этил ацетат/уксусная кислота (66/33/1), для получения) 115 мг свободного основания в виде
смолы. Смолу растворили в метаноле (10 мл), обработали метилатом натрия (15 мг, 0,3 ммоля) и перемешивали при комнатной температуре 2,5 часа. Растворитель удалили при пониженном давлении и остаток
растворили с диэтиловым эфиром для получения названного в заглавии соединения в виде твердого продукта. (79 мг, 60%). Т.пл 235-240oC. ПМР (300 MH2), ДМСО d6): d 1,9
(m,
2H), 2,3(S, 3H), 2,5(m, 2H), 2,7 (d d, 1H), 3,4 (d d, 1H) 4,1(d d, 1H), 4,5(m, 1H), 5,2(d, 1H гидроксил протон), 7,1 (d,2H), 7,5 (m,3H), 7,9 (m,2H).
Пример 13
Натриевая
соль
(R)-5-[4-(3-(5-метил-2-фенил-4-оксазолил) -1-гидроксипропил)бензил]тиазолидин-2,4-дион
Названное в заглавии соединение этого примера приготовлено в основном выполнением последовательных
стадий, описанных в примерах 7-12, начиная с соединения названного в заглавии примера 5. Т.пл. 2450-250oC. ПМР (300 MH2, ДМСО d6) d 1,9 (m,2H), 2,3 (S,3E), 2,5 (m,2H), 2,7 (d d,
1H), 3,4 (d d,1H), 4,1 (d d,1H), 4,5 (m,1H), 5,2 (d,1H гидроксил протон), 7,1 (d,2H), 7,5 (m, 3H), 7,9 (m,2H).
Пример 14
(S)-4-[3-(5-метил-2-фенил-4-оксазолил)-1-этоксипропил]
бромобензил
Названное в заглавии примера 7 соединение (1,0 г, 2,7 ммоля) и гидрид натрия (324 мл, 6,7 ммоля) растворили в ТГФ (30 мл)
при 0oC. Реакционную смесь обработали этил
иодидом (1,0 г, 6,7 ммоля) и содержимое нагревали до кипения с обратным холодильником в течение 18 часов. Реакционную смесь охладили до комнатной
температуры и сконцентрировали при пониженном давлении,
растворили в воде (25 мл) и экстрагировали дважды этил ацетатом (50 мл). Органические экстракты объединили, промыли водой (25 мл), раствором
соли (25 мл) и высушили (MgCO4). Растворитель
удалили при пониженном давлении и очистили остаток на силикагеле, элюируя смесью гексан/этил ацетат /3/1/, получив названное в заглавии
соединение в виде смолы (1,1 г. 90) ПМР (300 МН2, CDCl3) d 1,15 (t,3H), 2,0 (m,2H), 2,3 (S,3H), 2,5 (t,2H), 3,2-3,4 (m,2H), 4,2 (d d,1H), 7,2 (d,2H), 7,4 (m,5H), 7,9 (d,2H).
Пример 15
Следующие оптически чистые эфирные
производные приготовлены взаимодействием соответствующего алкил галогенида (RX) с требуемым оптическим чистым спиртом указанной стериохимии,
использованием в основном того же приема, что описан в
примере 14 (фиг. 4).
Пример 16
(S)-4-[(5-метил-2-фенил-4-оксазолил)-1-метоксипропил]бензальдегид
(S)-4-[3-(5-метил-2-фенил-4-оксазолил)-3-метоксипропил] бромобензол
/1,1 г. 2,8 ммоля, приготовленный как описано в примере 15/ растворили в ТГФ (30 мл), охладили до -78oC и обработали
н-бутиллитием (2,5 М в ТГФ, 1,2 мл, 3,0 ммоля), прибавляемым по каплям
через шприц. После прибавления реакционную смесь перемешивали при -78oC дополнительный час и обработали сухим ДМФ
(220 мг 30 ммолей). Реакционную смесь перемешивали при -78oC в
течение 90 минут и при температуре окружающей среды 24 часа. Реакционную смесь разбавили этил ацетатом (200 ил) промыли водой
(50 мл), 10% водной соляной кислотой (50 мл), водой (50 мл), раствором
соли (50 мл) и высушили (MgSO4). Растворитель удалили при пониженном давлении и остаток очистили на силикагеле,
элюируя смесью гексан/этил ацетат (3/1), получив вязкое масло (580 мг. 62%)
ПМР (300 МН2, CD Cl3): d 2,0 (m, 2H), 2,3 (S, 3H), 2,5 (t, 2H), 3,2 (S, 3H), 4,15 (d d, 1H), 7,3 (m, 3H), 7,4 (d,
2H), 7,8 (d, 2H), 7,9 (m, 2H), 9,9 (S, 1H).
Пример 17
(S)-5-[4-(3-(5-метил-2-фенил-4-4оксазолил) -1-метоксипропил)фенилметилен]тиазллизин-2,4-дион
Названное в
заглавии примера 16 соединение (580 мг. 1,7 ммоля) пиперидин (30 мг, 0,34 ммоля)
и 2,4-тиазолидиндион (405 мг, 3,4 ммоля) объединили в эталоне (20 мл) и образовавшийся раствор нагревали до кипячения
с обратным холодильником всю ночь. Растворитель удалили при пониженном давлении и
остаток очистили на силикагеле, элюируя смесью гексан/этил/ацетат /3/1/ плюс 5% уксусная кислота, получив названное в
заглавии соединение в виде твердого продукта (640 мг, 87%). Т. пл. 205 206oC. ПМР (300 МН2, ДМСО-d6), d 2,0 (m, 2H), 2,3 (S, 3H), 2,4 (t, 2H), 3,1 (S, 3H), 4,2 (d d, 1H), 7,4 (m,
5H), 7,6 (d, 2H), 7,7 (S, 1H), 7,9 (m, 2H).
Пример 18
(S)-5-[4-(3-(5-метил-2-фенил-4-оксазолил)-1- метоксипропил)бензил]тиазолидин-2,4-дион
Названное в заглавии
примера 17 соединение (640 мг, 1,5 ммоля) растворили в ТГФ (50 мл) и гидрировали в
присутствии сера-устойчивого 10% палладия на углерода (640 мг) на Вибраторе Парра при 50 Psi в течение 20 часов.
Катализатор удалили путем фильтрации через инфузорную землю и фильтрат сконцентрировали
при пониженном давлении. Остаток очистили на силикагеле, элюируя смесью гексан-этил ацетат (3/1) плюс 5%
уксусная кислота, получив сырой материал, который был далее очищен растворением остатка в 50 мл
этил ацетата, промыванием водой (25 мл), насыщенным водным бикарбонатом натрия (25 мл), раствором соли
(25 мл) и высушиванием (MgSO4), что привело к бесцветной смоле (229 мг, 35%) ПМР (300
МН2, CDCl3): d 2,0 (m, 2H), 2,3 (S, 3H), 2,5 (t, 2h), 3,1 (d d, 1H), 3,2 (S, 3H), 3,5 (d d,
1H), 4,1 (d d, 1H), 4,4 (d d, 1H, 7,2 (m 4H), 7,4 (m, 3H), 7,9 (m, 2H), 8,1 (bs, 1H, NH)
Пример 19
Следующие эфирные производные приготовлены в основном выполнением последовательных
стадий, описанных в примерах с 16 по 18, и начиная с приготовления эфира, как описано в примерах
14 и 15 (фиг. 5).
Реферат
Оптически чистые тиазолидиндионовые спирты и эфиры и синтетические промежуточные соединения для получения названных спиртов и эфиров. Эти соединения имеют применение в качестве гипогликемических и гипохолестеринемических агентов. 3 с. и 12 з.п. ф-лы, 5 ил.

Формула
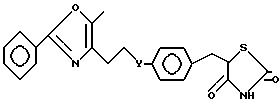
где Y группа CHOR, где R водород, С1-С4-алкил, С7-С9-фенилалкил, фенил или алкоксиалкил формулы (CH2)nO(CH2)mCH3, где n 0, 1, 2, 3 или 4; m 2, 3 или 4, при условии, что когда R водород, соединение I представляет собой чистый 1R или 1S энантиомер, по существу свободный от соответствующего энантиомера, или их фармацевтически приемлемые катионные соли или кислотно-аддитивные соли.
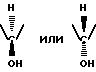
и по существу свободное от соответствующего энантиомера.

где Z-

11. Соединение по п.10, где Z Br
12. Соединение по п.10, где Z
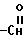
13. Соединение по п.10, где Z

14. Соединение по п.10, где Z

15. Соединение формулы

где X -

и по существу свободно от соответствующего энантиомера.
Комментарии