Производные 1,5-бензодиазепина и содержащий их фармацевтический состав - RU2135485C1
Код документа: RU2135485C1
Чертежи
Описание
Настоящее изобретение относится к новым производным 1,5-бензодиазепина и содержащим указанные производные фармацевтическим составам, проявляющим свойства антагонистов холецистокинина, в частности обладающим анксиолитической активностью, для использования в медицине.
Холецистокинины (ХЦК) и гастрин являются структурно близкими пептидами, присутствующими в ткани желудка и кишечника, а также в центральной нервной системе. К числу холецистокининов относятся ХЦК-33 (нейропептид, состоящий в своей естественной форме из 33 аминокислот), сульфат его карбокси-терминального октапептида (который тоже представляет собой естественно встречающийся нейропептид, обозначаемый ХЦК-8), а также 39- и 12-аминокислотные формы. Гастрин встречается в 34-, 17- и 14-аминокислотных формах, причем его минимальная активная последовательность соответствует C-терминальному тетрапептиду Trp-Met-Asp-Phe-NH2 (ХЦК-4), являющемуся общим структурным элементом как для ХЦК, так и для гастрина.
ХЦК и гастрин представляют собой желудочно-кишечные гормоны и нейромедиаторы в центральной и периферической системах, выполняющие свойственные им биологические функции за счет связывания с определенными рецепторами, локализованными в различных участках тела.
Существует по меньшей мере два подтипа рецепторов холецистокинина, обозначаемые ХЦК-А и ХЦК-Б, причем оба указанных подтипа встречаются как в периферической, так и в центральной нервной системе. Описаны антагонисты рецепторов ХЦК и гастрина, предназначенные для лечения заболеваний желудочно-кишечной и центральной нервной систем млекопитающих, в особенности - человека, если указанные заболевания связаны с функцией ХЦК или гастрина.
Известна [патент США N 4988692] группа производных 3- ациламино 1-алкил-5-фенил 1,5-бензодиазепина, проявляющих свойства антагонистов холецистокинина. Согласно описанию к указанному патенту, вышеупомянутые соединения проявляют достоверно более высокое сродство по отношению к рецепторам ХЦК-А, нежели ХЦК-Б.
Настоящее изобретение решает задачу создания новых производных 1,5-бензодиазепина, которые являются высокоэффективными и специфическими антагонистами ХЦК, в особенности антагонистами ХЦК по рецептору ХЦК-Б, и обладают особо благоприятным типом активности, в частности анксиолитической активностью. Изобретение также решает задачу создания новых фармацевтических составов, содержащих такие производные.
В
соответствии с настоящим изобретением предложены производные 1,5-бензодиазепина общей формулы (I)

и их фармацевтически приемлемые соли, где R1 представляет собой фенильную или C1-6-алкильную группу, причем указанная алкильная группа может быть замещена C1-7 - циклоалкильной или C7-11-циклоалкильной мостиковой группой; R2 обозначает фенильную группу, возможно замещенную заместителями, выбранными из числа следующих: атом галогена, C1-4- алкил, трифторметил, или (CH2)nR4, где R4 является гидрокси или C1-4-алкоксигруппой, а n равно 0; R3 представляет собой группу AlkNR8R9, где Alk соответствует прямой С2-6-алкиленовой цепи, возможно замещенной гидроксильной группой; R8 и R9 независимо друг от друга обозначают атом водорода или С1-4-алкил, или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют 5-7-членное насыщенное гетероциклическое кольцо, возможно содержащее дополнительный гетероатом, выбранный из числа следующих: атом кислорода или азота; R10 является атомом водорода или атомом галогена; X соответствует NH; m равно 0 или 1.
Предпочтительными являются производные 1,5-бензодиазепина формулы (I) и их фармацевтически приемлемые соли, в которых R1 представляет собой фенильную, циклогексилметильную, 3-метилбутильную или 1-адамантильную группу, в особенности 1-адамантильную группу; R2 представляет собой фенил, 3-метилфенил, 4-фторфенил или 4-метоксифенил; Alk представляет собой этилен, пропилен или 2-гидроксиметилэтилен; NR8R9 представляет собой амино, диметиламино, диэтиламино, морфолино, пирролидино, пиперидино или гексаметиленимино; R3 представляет собой морфолиноэтил, пирролидиноэтил, пиперидиноэтил, диметиламиноэтил, диэтиламиноэтил, диметиламинопропил, аминопропил или 2-гидроксиметил-2-аминоэтил, в особенности морфолиноэтил; R10 представляет собой атом водорода.
Конкретно, производное по изобретению представляет собой (-) [1-(-адамантилметил)-2, 4-диоксо-5-[2-(N-морфолино) этил] - 2,3,4, 5-тетрагидро-1Н-1,5-бензодиазепин-3-ил] -N'-фенилмочевину и ее физиологически приемлемые соли.
Конкретно, производное по изобретению представляет собой соединение, которое выбрано из группы, включающей в себя: N-[1-(1- адамантилметил)-2,4-диоксо-5-[2-(4-морфолино)этил] - 2,3,4,5-тетрагидро-1H-бензодиазепин-3-ил] -N'-фенилмочевину; N-[1-(1-адамантилметил)-2, 4-диоксо-5-[2-(4-морфолино) этил] -2,3,4,5-тетрагидро-1Н-бензодиазепин-3-ил] -N'-(4- фторфенил)мочевину; N-[2, 4-диоксо-1-(3-метил-1-бутил)-5-[2-(4-морфолино) этил]-2,3,4,5-тетрагидро-1Н-1, 5-бензодиазепин-3-ил] -N'- фенилмочевину; N-[2,4-диоксо-1-(3-метил-1-бутил)-5-[2-(1-пиперидино) этил] -2,3,4, 5-тетрагидро-1Н-1,5-бензодиазепин-3-ил] -N'-фенилмочевину; N-[5-[2-(диметиламино)этил]-2, 4-диоксо-1-(3-метил-1- бутил)-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'- фенилмочевину; N-[5-[2-(диметиламино) этил]-2,4-диоксо-1-(3-метил-1-бутил) 2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил] -N'-(4-метокси- фенил)мочевину; N-[2,4-диоксо-1-(3-метил-1-бутил)-5-[2-(4-морфолино)этил] -2, 3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил] -N'-(4- метоксифенил)мочевину; N-[2, 4-диоксо-1-(3-метил-1-бутил)-5-[2-(4-морфолино) этил] -2,3,4,5-тетрагидро-1Н-1, 5-бензодиазепин-3-ил]-N'-(4- гидроксифенил)мочевину; N-[5-[2-(диэтиламино)этил] -2,4-диоксо-1-(3-метил-1-бутил)-2, 3,4, 5-тетрагидро-1Н-бензодиазепин-3-ил] -N'-фенилмочевину; N-[5-[2-(диэтиламино) этил]-2,4-диоксо-1-(3-метил-1-бутил) -2,3,4,5-тетрагидро-1Н-бензодиазепин-3-ил]-N'-N'-(4- фторфенил)мочевину; N-[(1-адамантилметил)-5-[2-диметиламино)этил]-2,4-диоксо- 1-2,3,4,5-тетрагидро-1Н-1, 5-бензодиазепин-3-ил] -N'-фенилмочевину; N-[1-(1-адамантил)метил-5-[3-(диметиламино)пропил] -2,4- диоксо-2,3,4, 5-тетрагидро-1Н-1,5-бензодиазепин-3-ил] -N'- фенилмочевину; N-[1-(1-адамантилметил)-2, 4-диоксо-5-[3-гидрокси-2(R) аминопропил]-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил] -N'- фенилмочевины гидрохлорид; N-[1-(1-циклогексилметил)-2,4- диоксо-5-[2-(диэтиламино)этил] -2,3,4, 5-тетрагидро-1Н-бензодиазепин-3-ил] -N'- фенилмочевину; N-[1-(1-адамантилметил)-2, 4-диоксо-7-фтор-5-[2-(4- морфолино)-этил]-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N' -фенилмочевину; N-[1-(3-метил-1-бутил)-2,4-диоксо-5-[2-(4-морфолино) этил]-2,3,4, 5-тетрагидро-1Н-бензодиазепин-3-ил] -N'-(4- хлорметил) мочевину; N-[2,4-диоксо-1-(3-метбут-1-ил)-5-[2-(4-морфолино) этил]-2,3,4, 5-тетрагидро-1H-l, 5-бензодиазепин-3-ил] -N'-(4- трифторметил)мочевину; N-[1-(1-адамантилметил)-2,4-диоксо-5-[2-(1- пирролидино)этил] -2,3,4,5-тетрагидро-1Н-бензодиазепин-3-ил] -N'- фенилмочевину; N-[2,4-диоксо-1-[2-(гексаметиленимино) этил] -5- фенил-2,3,4, 5-тетрагидро-1Н-бензодиазепин-3-ил] -N'-(3-толил) мочевину.
Далее, в соответствии с настоящим изобретением предложен фармацевтический состав, проявляющий свойства антагонистов холецистокинина, в частности обладающий анксиолитической активностью, отличающийся тем, что он содержит производное 1,5-бензодиазепина формулы (I) или его фармацевтически приемлемую соль в смеси с одним или несколькими физиологически приемлемыми носителями или эксципиентами.
Очевидно, что производные, отвечающие формуле (I), содержат по меньшей мере один асимметрический атом углерода (а именно атом углерода, занимающий положение 3 диазепинового кольца), и, таким образом, предложенные в соответствии с настоящим изобретением производные включают в себя все стереоизомеры и их смеси, включая рацематы.
К числу физиологически приемлемых солей производных формулы (I) относятся традиционные соли, образуемые фармацевтически приемлемыми неорганическими или органическими кислотами, а также четвертичные соли аммония, получаемые в результате добавления кислоты. Конкретными примерами приемлемых солей служат соли соляной, бромоводородной, серной, фосфорной, азотной, хлорной, фумаровой, уксусной, пропионовой, янтарной, гликолиевой, муравьиной, молочной, яблочной, винной, лимонной, памоевой, молоновой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, фумаровой, толуолсульфоновой, метантолуолсульфоновой, нафталин-2-сульфоновой, бензолсульфоновой и т.п. кислот. Другие, не являющиеся фармацевтически приемлемыми, кислоты, такие как оксалиновая, могут быть использованы при получении солей промежуточных соединений в ходе синтеза предусмотренных настоящим изобретением соединений, а также их фармацевтически приемлемых солей.
Соединения формулы (I) могут образовывать фармацевтически приемлемые соли с соответствующими катионами. К числу приемлемых фармацевтически приемлемых катионов относятся катионы щелочных металлов (в частности, натрия или калия) и щелочноземельных металлов (в частности, кальция и магния).
Соли могут быть также образованы с использованием органических оснований, например N-метилглюкамина.
Кроме того, соединения формулы (I) могут существовать в форме метаболически лабильных эфиров. Примерами метаболически лабильных эфиров соединений формулы (I) могут служить эфиры C1-4-алкилов (например, метиловые или этиловые эфиры); эфиры замещенных или незамещенных аминоалкилов (например, аминометиловые, 2-(N,N- диэтиламин) этиловые или 2-(4-морфолино) этиловые эфиры); либо эфиры ацилоксиалкилов, таких как ацилоксиметил или 1-ацилоксиэтил (например, пивалоилоксиметил, 1-пивалоилоксиэтил, ацетоксиметил, 1-ацетоксиэтил, 1-метокси-1-метил-этилкарбонилоксиэтил, 1-бензоилоксиэтил, изопропоксикарбонилоксиметил, 1-изопропоксикарбонилоксиэтил, циклогексилкарбонилоксиметил, 1-циклогексилкарбонилоксиэтил, циклогексилоксикарбонилоксиметил, 1-циклогексилоксикарбонилоксиэтил, 1-(4-тетрагидропиранилоксикарбонилоксиэтил) или 1-(4-тетрагидропиранилкарбонилокси)этил).
Соединения формулы (I), а также их соли и метаболически лабильные эфиры могут образовывать сольваты, в том числе гидраты.
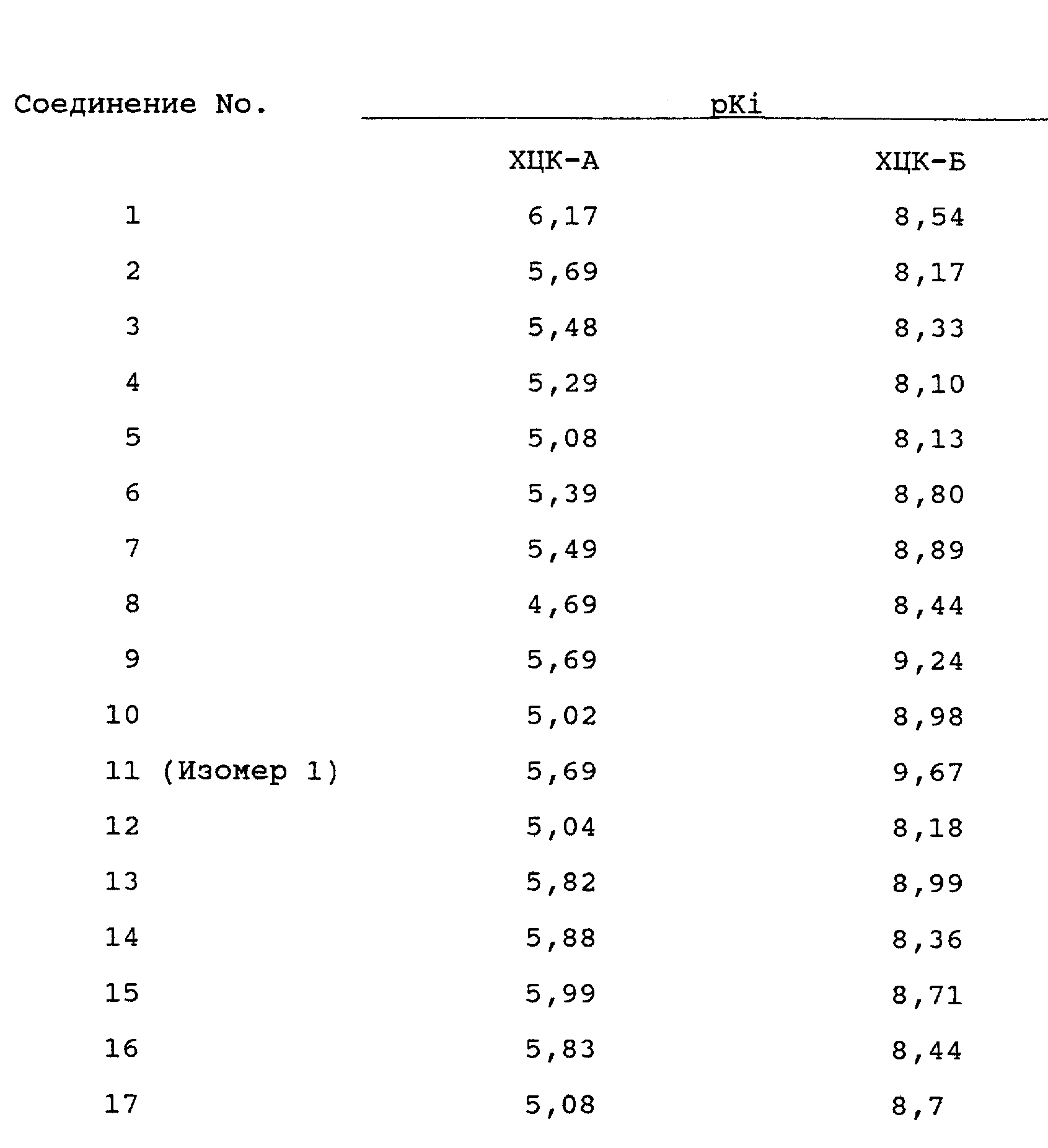
Соединения, предложенные в соответствии с настоящим изобретением, являются эффективными и специфическими антагонистами ХЦК, в особенности по рецептору ХЦК-Б. Это продемонстрировано, например, способностью указанных соединений подавлять вызванное ХЦК-4 сокращение препарата продольного мускульно-миентерического сплетения подвздошной кишки морских свинок в присутствии антагониста рецепторов ХЦК-А.
Кроме того, судя по тому, что соединения по настоящему изобретению способны подавлять вызванное ХЦК-8 сокращение препарата продольного мускульно-миентерического сплетения подвздошной кишки морских свинок, указанные соединения проявляют существенно меньшую активность по отношению к рецепторам ХЦК-А, нежели по отношению к гастрину и/или рецепторам ХЦК-Б.
Выделение и использование препарата продольного мускульно- миентерического сплетения подвздошной кишки морских свинок описано K-H Buchheit et al [Nauyn-Schmeideberg's Arch.Pharmacol. (1985), 329, p.36-41] и V.L.Lucaites et al [J.Pharmacol.Exp.Ther.(1991), 256, 695-703].
Соединения по настоящему изобретению являются также антагонистами гастрина, что показано их способностью подавлять вызванное пентагастрином выделение желудочного сока, обнаруживаемое на препаратах слизистой желудка с использованием методики, описанной J.J. Reeves and R.Stables [Br.J.Pharmac., 1985, 86, p.677-684].
Более высокое сродство соединений по настоящему изобретению по отношению к рецепторам ХЦК-Б, нежели ХЦК-А, было также установлено посредством анализов по связыванию рецепторов ХЦК, описанных G. Dal Forno et al. [J.Pharmcol. Exp & Ther., 261, 1056-1063].
Как показано с использованием стандартных фармакологических тестов, предусмотренные настоящим изобретением соединения проявляют анксиолитическую активность. В частности, этот результат был получен в тесте на мышах с использованием черно-белой камеры, а также в тесте на определение агрессивности у мармозеток.
Таким образом, производные 1,5-бензодиазепина по настоящему изобретению или их фармацевтически приемлемые соли могут применяться в терапии, в частности в медицине.
Соединения по изобретению пригодны для лечения и/или предупреждения заболеваний млекопитающих, в особенности человека, при которых модификация действия ХЦК и/или гастрина оказывает благоприятный терапевтический эффект. Поэтому предусмотренные настоящим изобретением производные 1, 5-бензодиазепина пригодны для лечения заболеваний центральной нервной системы, при которых оказывается затронутой функция ХЦК и/или гастрина. Примерами указанных заболеваний служат состояния тревоги (в том числе паническое состояние, агарофобия, социальная фобия, простая фобия, навязчивые идеи, посттравматический стресс и общая тревога), сниженная подвижность, депрессия, болезнь Паркинсона или психоз. Кроме того, предусмотренные настоящим изобретением соединения пригодны для лечения желудочно-кишечных заболеваний, в особенности таких, при которых желательно понижение кислотности желудка. К числу указанных заболеваний относятся пептическая язва, воспаление пищевода и синдром Золлингера-Эллисона. Кроме того, рассматриваемые соединения могут быть пригодны для лечения желудочно-кишечных заболеваний, таких как синдром раздражения кишечника, повышенная секреция поджелудочной железы, острый панкреатит, нарушения перистальтики, гиперплазия антральных G-клеток, гиперплазия фундальной слизистой или желудочно-кишечные новообразования. Указанные соединения также могут найти применение при лечении зависимости от антибиотиков или агентов, предназначенных для временного применения; синдрома Жилля де ла Турэ; нарушений систем регуляции аппетита; а кроме того - при лечении определенных типов опухолей глотки, нижних отделов пищевода, поджелудочной железы, желудка, тонкого кишечника и прямой кишки. Предусмотренные настоящим изобретением соединения также пригодны для местного обезболивания, для усиления обезболивания, вызванного наркотиками или другими агентами, а кроме того - для анестезии или снятия чувствительности к боли.
Специалистам в данной области будет очевидно, что в настоящем описании понятие "лечение" охватывает как профилактику, так и лечение уже развившихся заболеваний или симптомов.
Соединения по изобретению могут, соответственно, применяться в производстве лекарственного средства, предназначенного для лечения заболеваний, при которых модификация действия ХЦК и/или гастрина оказывает благоприятный терапевтический эффект.
Следует иметь в виду, что необходимое для лечения количество предусмотренного настоящим изобретением соединения должно варьироваться в зависимости от заболевания, а также от возраста и состояния пациента, и должно определяться исключительно лечащим врачом или ветеринаром. Вместе с тем, дозы, применяемые при лечении взрослого человека, обычно должны находиться в пределах 0,01 - 2000 мг в день, в том числе 0,01 - 500 мг в день.
Желаемая доза может применяться традиционным образом в виде одной дозы или нескольких доз, вводимых через определенные промежутки времени, например, в виде двух, трех, четырех или большего числа субдоз в день.
Так как предусмотренные настоящим изобретением соединения являются для животных функциональными антагонистами ХЦК, указанные соединения при использовании в дозах приблизительно от 1 мг/кг до 10 мг/кг могут быть использованы также в качестве пищевых добавок, способствующих повышению аппетита животного.
Несмотря на то, что предусмотренные настоящим изобретением соединения могут быть использованы в терапевтических целях в химически чистом виде, предпочтительно использовать указанный активный ингредиент в форме фармацевтического состава.
Фармацевтический состав по изобретению, содержащий производные формулы (I) или их фармацевтически приемлемые соли вместе с одним или несколькими физиологически приемлемыми носителями или эксципиентами, может также содержать другие терапевтические и/или профилактические ингредиенты. Вышеуказанные носители должны быть "приемлемыми" в смысле их совместимости с другими ингредиентами соответствующего состава, а также безвредности по отношению к их реципиенту.
Примерами фармацевтических составов по изобретению могут служить такие составы, которые специально предназначены для перорального, трансбуккального, парэнтерального, имплантационного или ректального введения. Предпочтительным является пероральное введение.
Таблетки и капсулы, предназначенные для перорального введения, могут включать в себя традиционные эксципиенты, такие как связывающие агенты (например, сироп, гуммиарабик, желатин, сорбитол, трагакант, крахмальный клей или поливинилпирролидон), наполнители (например, лактозу, сахар, микрокристаллическую целлюлозу, кукурузный крахмал, фосфат кальция или сорбитол), способствующие скольжению агенты (например, гидрированные растительные масла, стеарат магния, стеариновая кислота, тальк, полиэтилен гликоль или окись кремния), дезинтегрирующие агенты (например, картофельный крахмал или крахмальный гликоллат натрия) или смачивающие агенты (в частности, лаурил сульфат натрия). Указанные таблетки могут быть покрыты оболочкой в соответствии с хорошо известными способами. Жидкие составы, предназначенные для перорального введения, могут находиться в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров, либо могут представлять собой капли для перорального введения или сухой продукт, предназначенный для смешивания с водой или другими приемлемыми носителями перед употреблением. Указанные препараты могут содержать традиционные добавки, такие как агенты, способствующие образованию суспензий (в том числе, сироп сорбита, метилцеллюлоза, глюкозо/сахарный сироп, желатин, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза, алюминий- стеаратный гель или гидрированные пищевые жиры); агенты, способствующие образованию эмульсий (в том числе, лецитин, моноолеат сорбитана или гуммиарабик); неводные носители, которые могут включать в себя пищевые масла (в том числе, миндальное масло, фракционированное кокосовое масло, масляные эфиры, пропилен гликоль или этиловый спирт); а также консерванты (в том числе, метил или пропил п-гидроксибензоаты или рябиновая кислота). Указанные препараты могут быть составлены также в виде суппозиториев, т.е. могут содержать традиционные основы суппозиториев, такие как масло какао или другие глицериды.
При трансбуккальном введении указанный состав может иметь форму таблеток или лепешек, полученных традиционным образом.
Предусмотренные настоящим изобретением составы могут быть предназначены для парэнтерального введения с помощью инъекции или продолжительного вливания. Составы, предназначенные для инъекций, могут быть представлены в виде единиц дозы, находящихся в предварительно стерилизованных шприцах, пузырьках и ампулах, либо в виде многодозовых контейнеров, содержащих добавленный консервант. Указанные составы могут иметь форму суспензий, растворов или эмульсий в масляных или водных носителях, а также могут включать в себя дополнительные агенты, такие как способствующие образованию суспензий, стабилизирующие и/или способствующие измельчению агенты. В альтернативном случае указанный активный ингредиент может представлять собой порошок, полученный путем низкотемпературной сушки и предназначенный для смешивания с приемлемым носителем (например, со стерильной, не содержащей пирогена водой) перед употреблением.
Предусмотренный настоящим изобретением состав может быть создан в форме депонированого препарата. Указанные составы, служащие в течение длительного промежутка времени, могут вводиться посредством имплантации (например, подкожно или внутримышечно), либо с помощью внутримышечной инъекции. В соответствии с этим, предусмотренные настоящим изобретением соединения могут предназначаться к использованию вместе с приемлемыми полимерными или гидрофобными веществами (такими, как, например, эмульсия в приемлемом масле), ионообменными смолами, либо их слаборастворимыми производными, в том числе, например, слаборастворимыми солями.
Предусмотренные настоящим изобретением составы могут содержать от 0,1 до 99% соответствующего активного ингредиента, обычно - от 30 до 95% в случае таблеток и капсул, а также от 3 до 50% в случае жидких препаратов.
Соединения формулы (I), а также их соли могут быть получены с помощью описанных ниже общих способов. Если это не оговорено особо, использованные в нижеследующем описании группы R1-R10 соответствуют таковым в случае соединений, предусмотренных формулой (I).
В соответствии с первым
способом (А), соединения, отвечающие формуле (I), в которой X обозначает группу NH, можно получить, приводя соединение,
описываемое формулой (II)

в которой Y представляет собой остаток NHCOR11, где R11 соответствует возможно замещенной феноксигруппе или 1-имидазольной группе, с амином, предусмотренным формулой (III)
H2NR2
возможно в присутствии основания, такого как третичный амин (например, триэтиламин). Обычно указанную реакцию осуществляют в приемлемом растворителе, таком как галогенированный углеводород (например дихлорметан), эфир (например тетрагидрофуран) или амид (например N,N-диметилформамид), возможно при температуре в пределах от комнатной до температуры дефлегмации соответствующего растворителя.
В
конкретном варианте способа (А), при котором Y представляет собой остаток
NHCOR11, а радикал R11 обозначает 1-имидазольную группу, соответствующий имидазолид (II) может быть
получен in situ; в этом случае амин, отвечающий формуле (III), смешивают с
соединением, предусмотренным формулой (IV)

в присутствии карбонилдиимидазола при вышеописанных условиях. В случае процесса (А), при котором Y представляет собой остаток NHCOR11, а радикал R11 обозначает возможно замещенную феноксигруппу, указанную реакцию с первичным амином (III) предпочтительно осуществляют в присутствии основания, такого как третичный амин (например триэтиламин).
Соединения, отвечающие формуле (III), в которой радикал R11 соответствует возможно замещенной феноксигруппе, можно получить из первичного амина (IV), приводя его во взаимодействие с соответствующим возможно замещенным фенил хлороформатом в присутствии основания, такого как пиридин. Указанная реакция может быть осуществлена в растворителе, таком как галогенированный углеводород (в частности, дихлорметан) при температуре в пределах от 0 до 50oC.
Соединения, отвечающие формуле (II), в которой радикал R11 соответствует 1-имидазольной группе, можно получить, приводя соединение, описываемое формулой (IV), во взаимодействие с карбонилдиимидазолом в присутствии приемлемого растворителя, такого как галогенированный углеводород (например, дихлорметан) или эфир (например, тетрагидрофуран) при температуре в пределах от 0 до 50oC (обычно при комнатной температуре).
В
соответствии с еще одним общим способом (Б), соединения, предусмотренные формулой (I), можно
получить, приводя соединение, отвечающее формуле (IV), во взаимодействие с изоцианатом, описываемым
формулой (V)
O=C=N-R2
или ацил хлоридом, соответствующим формуле (VI),
ClCO(X)R2
Указанную реакцию обычно проводят в присутствии приемлемого
растворителя, такого как галогенированный углеводород (например дихлорметан), эфир (например
тетрагидрофуран) или нитрил (например ацетонитрил), либо смесь указанных растворителей при температуре в
пределах от 0 до 80oC.
Соединения, предусмотренные формулой (IV),
можно получить, восстанавливая соединения, отвечающие формуле (VII),

в которой Ra3 представляет собой группу R3 по формуле (I).
Восстановление соединений, описываемых формулой (VII), до соединений, соответствующих формуле (VI), можно получить в результате реакции между цинком и уксусной кислотой. Указанную реакцию проводят при температуре в пределах между 0 и 50oC. В альтернативном случае вышеописанное восстановление осуществляют с использованием палладиево-угольного катализатора и формата аммония в растворителе, таком как метанол.
Соединения, отвечающие формуле (VII), можно получить, приводя ортофенилендиамин (VIII) во взаимодействие с хлоридом диацида (IX) в растворителе, таком как простой эфир (в частности, тетрагидрофуран) или сложный эфир (в частности, этилацетат).

Соединения, предусмотренные формулой (VIII), либо являются известными, либо могут быть получены с помощью аналогичных методов. Так, соединение, описываемое формулой (VIII), можно получить, например, алкилируя амин (X).

Итак, амин (X) можно привести во взаимодействие с соединением R1L, где L представляет собой отщепляемую группу (например атом хлора или брома), возможно в присутствии иодида натрия в растворителе, таком как N,N-диметилформамид. Альтернативный вариант введения группы R1 можно осуществить в том случае, если амин (X) приводят во взаимодействие с соответствующим альдегидом при стандартных условиях восстановительного алкилирования.
Обычно соединения, предусмотренные формулами (III), (V) и (VI), либо являются известными, либо могут быть получены с помощью методов, используемых для синтеза известных соединений.
В соответствии с еще одним способом (В), соединение, описываемое формулой (1),
можно получить, приводя соединение, отвечающее формуле (XI),
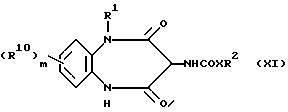
во взаимодействие с алкилирующим агентом R8R9N-Alk-L, где L представляет собой отщепляемую группу, в том числе атом галогена.
Указанный процесс обычно осуществляют в присутствии сильного основания, такого как карбонат щелочного металла (например, карбонат калия) или гидрид натрия в апротонном растворителе, таком как N,N-диметилформамид.
Соединения, предусмотренные
формулой (XI), можно получить из амина (XII)
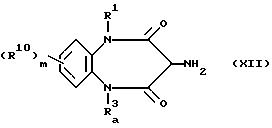
где R1, R10 и m соответствуют обозначениям, использованным в формуле (I); Ra3 представляет собой атом водорода или группу, защищающую атом азота; приводя его во взаимодействие с изоцианатом (V) или хлоридом диацида (IV), после чего при необходимости удаляют защитную группу Ra3.
Соединения, описываемые формулой (XII), могут быть получены из соединения, отвечающего формуле (VII), где Ra3 обозначает группу, защищающую атом азота (например, бензильную или п-метоксибензильную группу). Таким образом, соединение, предусмотренное формулой (VII), можно восстановить с использованием палладиево-угольного катализатора в присутствии формата аммония с образованием желаемого соединения (XII), где Ra3 обозначает атом водорода. В альтернативном случае желаемое соединение (XII) можно получить, восстанавливая соединение, описываемое формулой (VII), в которой Ra3 является группой, защищающей атом азота, после чего при желании указанную защитную группу Ra3 удаляют с помощью традиционных методов.
Так, восстановление соответствующего гидразона можно получить, осуществляя реакцию между цинком и уксусной кислотой, а затем при желании удаляя указанную защитную группу с помощью гидрогенолиза или реакции с использованием нитрата церия-аммония.
Амины, предусмотренные формулой (VIII, где Ra3 обозначает атом водорода) или формулой (XI), либо являются известными соединениями, либо могут быть получены из соответствующих нитропроизводных (XIII) или (XIV).

Приводя нитросоединения (XIII) или (XIV) во взаимодействие с Na2S2O4, получают соответствующий первичный амин, который можно алкилировать традиционным образом с выходом желаемого диамина (VIII).
Соединения, описываемые формулой (XII), где Ra3 представляет собой атом водорода,
могут быть получены из соединений, отвечающих формуле (XV)

посредством удаления защитной N-бензилоксикарбонильной группы с использованием стандартных методов, в том числе гидрирования в присутствии водорода и палладиевого катализатора.
Соединения, предусмотренные формулой
(XV), можно получить, обрабатывая соединения (XVI)

приемлемой кислотой, например, соляной кислотой.
Соединения, отвечающие формуле (XVI), можно получить, приводя защищенный диамин (XVII)
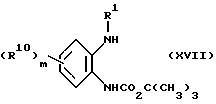

во взаимодействие с кислотой (XVIII) в присутствии дициклогексилкарбодиимида.
Защищенный диамин (XVII) может быть получен из соответствующего нитропроизводного (XIII) с использованием стандартных методов. Так, нитрогруппу можно восстановить с помощью Na2S2O4, а образующийся при этом амин посредством стандартных методов превратить в соответствующее N-тбутоксикарбокси- производное.
В соответствии с еще одним способом (Г), соединения, предусмотренные формулой (I), можно получать в результате реакции между соединением, описываемым
формулой (XIX),

в которой R1, R2, R10, m и Alk соответствуют обозначениям, использованным выше в формуле (I); а L представляет собой отщепляемую группу; и амином Ra8Rb9NH, где радикалы Ra8 и/или Rb9 соответствуют обозначениям, использованным выше применительно к остаткам R8 и R9; либо каждый из указанных радикалов независимо друг от друга является группой, защищающей атом азота (в том числе арилметильной группой, например, бензилом); после чего при необходимости или желании удаляют любую из вышеупомянутых групп, защищающих атом азота. Рассматриваемую реакцию можно осуществлять в отсутствие или в присутствии дополнительного растворителя (в частности эфира, такого как тетрагидрофуран). Обычно вышеупомянутая отщепляемая группа L представляет собой атом галогена (например атом хлора, брома или иода), либо сульфонилокси, в том числе алкилсульфонилокси (например метансульфонилокси) или арилсульфонилокси (например фенилсульфонилокси). Удаление указанной группы Ra8 или Rb9защищающей атом азота, можно осуществить с помощью традиционных методов, например, посредством гидрогенолиза.
Соединения, отвечающие формуле (XIX), можно традиционным образом получить из соответствующих соединений, предусмотренных формулой (XIX), в которой L представляет собой гидроксильную группу. Так, соединения, описываемые формулой (XIX, где L обозначает сульфонильную группу), можно синтезировать в результате реакции между соответствующим гидроксисоединением и необходимым алкил- или арилсульфонилхлоридом в присутствии третичного органического основания.
Соединения, предусмотренные формулой (XIX), в которой L является атомом галогена, можно получить из соответствующего гидроксисоединения с помощью стандартных методов, используемых для превращения гидроксильных групп в остаток галогена. Так, соединения, описываемые формулой (XIX), где L обозначает атом брома, можно получить, обрабатывая соответствующее гидроксисоединение тетрабромидом углерода и трифенилфосфином.
Соединения, отвечающие формуле (XIX), в которой остаток L представляет собой гидроксильную группу, могут быть получены из соответствующего амина (VIII, где радикал Ra3 обозначает защищенную гидроксиалкильную группу) посредством его взаимодействия с хлоридом диацида (IX), после чего восстанавливают образовавшийся при этом гидразон с получением амина (XII, где радикал Ra3 является гидроксиалкильной группой или ее защищенным производным).
Приводя образовавшийся таким образом амин (XII) во взаимодействие с изоцианатом (V) или ацил хлоридом (VI), получают желаемое соединение (XIX), в котором радикал L представляет собой гидроксильную группу.
В соответствии с еще одним способом (Д), соединение, предусмотренное формулой (I), можно традиционным образом превратить в другое соединение, отвечающее формуле (I).
Так, соединение, описываемое формулой (I), где радикал R2 обозначает фенил, замещенный гидроксильной группой, можно получить из соединения, в котором остаток R2 является фенилом, замещенным метоксигруппой, с помощью традиционных методов, в частности, реакции с иодидом алюминия. Аналогичным образом соединения, отвечающие формуле (I), где радикал R2 представляет собой фенильную группу, замещенную карбоксильной группой, можно получить в результате гидролиза соответствующего соединения, предусмотренного формулой (I), в которой остаток R2 обозначает фенильную группу, замещенную алкоксикарбонильной группой.
В случае вышеописанного способа группы R1, R2 и R3, входящие в состав промежуточных соединений II, III, V и VI, могут представлять собой соответствующие группы, описанные в формуле (I), либо остатки, которые можно превратить в указанные группы.
Метаболически лабильные эфиры соединений, описываемых формулой (I), можно традиционным образом получить из соответствующих карбоновых кислот. Соли, образующиеся в результате добавления кислот к соединениям, предусмотренным формулой (I), можно синтезировать традиционным образом. Так, соединение, отвечающее формуле (I), можно обработать желаемой кислотой обычно в присутствии растворителя (например, спирта) с получением раствора желаемой соли, которую можно выделить традиционным образом.
Соединения, предусмотренные формулой (I), содержат по меньшей мере один асимметрический атом углерода, а именно атом углерода, входящий в состав диазепинового кольца, к которому присоединен остаток NHCOXR2. Конкретные энантиомеры соединений, описываемых формулой (I), можно выделить путем разделения рацемических соединений с использованием традиционных методов, таких как хиральная HPLC. В альтернативном случае отдельные энантиомеры, отвечающие формуле (I) можно получить из соответствующих энантиомеров соединений (IV) с помощью вышеописанных способов, которые предназначены для синтеза соединений, предусмотренных настоящим изобретением, из соединений, отвечающих формуле (IV).
Отдельные энантиомеры соединений (IV) можно получить с помощью традиционных методов. Так, рацемат амина (IV) можно привести во взаимодействие с оптически активным реагентом, таким как производное фенилаланина или миндальной кислоты, а образующиеся при этом диастереоизомеры - разделить с помощью традиционных методов. Затем желаемый энантиомерный амин (IV) можно получить из выделенного диастереоизомера, удаляя остаток фенилаланина или миндальной кислоты с помощью традиционных методов.
Следующие ниже примеры не имеют ограничивающего характера и призваны проиллюстрировать настоящее изобретение.
Если это не оговорено особо, в примерах приведены неуточненные точки плавления (т. пл. ), определенные с помощью аппарата Бухи. Все температуры соответствуют температурам по шкале Цельсия. Определение инфракрасных спектров проводили в растворах хлороформа- d1 на приборе FT-IR. Спектры ядерного магнитного резонанса (1H-ЯМР) регистрировали при 300 мГц в растворах хлороформа-di. Химические сдвиги выражали во /ооо/оо по отношению к Me4Si, служащему в качестве внутреннего стандарта, и представляли в виде синглетов (s), дуплетов (d), двойных дуплетов (dd) или мультиплетов (т). Хроматографию на колонке осуществляли с использованием силикагеля (Merck AG Darmstadt, Germany). Высушивание растворов проводили над безводным сульфатом натрия. Обозначение "петрол" соответствует петролейному эфиру, т. кип. 40-6ОoC. Использовали: дихлорметан, повторно перегнанный над гидридом кальция; тетрагидрофуран, повторно перегнанный над натрием; этиловый эфир, повторно перегнанный натрием; а также этилацетат, высушенный над активированными молекулярными ситами. В тексте использованы следующие обозначения: ЭА - этилацетат, ЦГ - циклогексан, П - петролейный эфир 40-60oC, ТГФ - тетрагидрофуран, ДХМ -дихлорметан, ЭЭ - этиловый эфир, ДМФ - N,N-диметилформамид. ТЛХ соответствует тонкослойной жидкостной хроматографии на пластинах силикагеля. Если это не оговорено особо, все соединения представляют собой рацемические смеси.
Промежуточное соединение 1
N-(4-Метоксифенилметил)-2-нитроанилин
Смесь
1-фтор-2-нитробензола
(20 г) и 4-метоксибензоламина (18,52 мл) в сухом тетрагидрофуране (100 мл) перемешивали в атмосфере азота при температуре 23oC в течение 18 ч. Фильтровали полученную
смесь, после чего
органическую фазу концентрировали в вакууме с получением масла. К указанному маслу добавляли этанол (50 мл) и выделяли образовавшееся твердое вещество. После фильтрации выделяли
вышепоименованное
соединение в виде оранжевого твердого вещества (16,35 г). Соответствующий фильтрат концентрировали в вакууме, а полученный остаток обрабатывали еще одной порцией этанола (10 мл) с
выходом
дополнительного количества вышепоименованного соединения (7,9 г). Т.пл. 81-82oC. Т.Ж.X. ЦГ-ЭА (в соотношении 8:2): Rf 0,55.
Промежуточное соединение 2
N-(4-Метоксифенилметил)-1,2-фенилендиамин
К суспензии Промежуточного соединения 1 (24 г) в смеси 95%-ного этанола (500 мл) и воды (500 мл) добавляли карбонат калия (96,95 г) и
гидросульфит
натрия (80,96 г). Образовавшуюся смесь перемешивали при температуре 23oC в течение 1 ч, после чего подкисляли концентрированной соляной кислотой (150 мл) до pH 3. Указанную
смесь
концентрировали в вакууме до половины объема, а выделенный остаток подщелачивали раствором 10%-ного гидроксида натрия (900 мл) до pH 10. Полученную смесь экстрагировали этилацетатом (1200 мл).
Соответствующую органическую фазу промывали солевым раствором (600 мл), высушивали и концентрировали в вакууме с образованием коричневого твердого вещества. Указанное вещество растирали с диэтиловым
эфиром с получением вышепоименованного соединения в виде бежевого твердого вещества (17,7 г).
Т.пл. 91-92oC. Т.Ж.X. ЦГ-ЭА (в соотношении 8:2): Rf 0,22.
Промежуточное соединение 3
N-(4-Метоксифенилметил)-N'-(3-метил-1-бутил)-1,2-фенилендиамин
К раствору Промежуточного соединения 2 (10,68 г) и иодида натрия (12,8 г) в
диметилформамиде (400 мл) в атмосфере азота добавляли бром-3-метилбутан (10,68 мл). Образовавшийся раствор выдерживали в атмосфере азота при температуре 80oC в течение 4 ч, после чего
охлаждали до комнатной температуры, разбавляли водой (300 мл) и экстрагировали диэтиловым эфиром (2 х 700 мл). Объединяли соответствующие органические экстракты, промывали их солевым раствором (1000
мл), высушивали и концентрировали в вакууме с получением масла, которое очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 9: 1) с выходом вышепоименованного соединения в
виде желтого масла (14,0 г). Т.Ж.X. ЦГ-ЭА (В соотношении 9:1): Rf 0,42.
ИК: 1610 и 1601 (С=С) см-1.
1H-ЯМР: 7,31 (d); 6,89 (d); 6,84-6, 74 (m); 4,22 (s); 3,81 (s); 3,10 (t); 1,75 (m); 1,6-1,5 (m); 0,94 (d).
Промежуточное соединение 4
2,4-Диоксо-5-(4-метоксифенилметил)-1-(3-метил-1-бутил) - 3-фенил-гидразоно-2,
3,4,5-тетрагидро-1Н-1,5-бензодиазепин
В 100 мл ТГФ вводили Промежуточное соединение 3 (14,0 г) и 2-фенилгидразономалонилдихлорид (13,8 г), а образовавшуюся смесь в атмосфере азота по каплям
добавляли в колбу, содержащую ТГФ (100 мл). По завершении добавления полученный раствор выдерживали при температуре 50oC в течение 2 ч. Затем указанный раствор концентрировали в вакууме, а
выделенный остаток растирали с диэтиловым эфиром с выходом вышепоименованного соединения в виде желтого твердого вещества (8,9 г). Соответствующий фильтрат концентрировали в вакууме и очищали с
помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 8:2) с получением дополнительного количества вышепоименованного соединения (6,35 г).
Т.пл. 189-191oC. Т.Ж.X. ЦГ-ЭА (В соотношении 8:2): Rf 0,30.
Промежуточное соединение 5
3-Амино-2,4-диоксо-5-(4-метоксифенилметил)-1-(3-метил-1- бутил)-2,3,4,5-тетрагидро-1Н-1,
5-бензодиазепин
К суспензии Промежуточного соединения 4 (15,25 г) в уксусной кислоте (150 мл) добавляли цинковую пыль (15,8 г). Образовавшуюся смесь выдерживали при температуре 40o
C в течение 2 ч, после чего отделяли от использованного цинка. Полученный фильтрат подщелачивали 10%-ным раствором гидроксида натрия (2000 мл) до pH 10, а указанную смесь экстрагировали этилацетатом
(2000 мл). Объединяли соответствующие органические экстракты, промывали их солевым раствором (1000 мл), высушивали и концентрировали в вакууме с получением масла, которое очищали с помощью импульсной
хроматографии (элюция ЭА) с выходом вышепоименованного соединения в виде белого твердого вещества (8,24 г).
Т.пл. 115-116oC. Т.Ж.X. ЦГ-MeOH (в соотношении 95:5): Rf 0,25.
Промежуточное соединение 6
3-Амино-2,4-диоксо-1-(3-метил-1-бутил)-2,3,4,5-тетрагидро-1Н- 1,5-бензодиазепин
К раствору Промежуточного соединения 5 (3,0
г) в смеси ацетонитрила (90 мл) и воды (10 мл) добавляли нитрат аммония и церия (IV) (17,45 г). Образовавшийся раствор перемешивали при температуре 23oC в течение 36 ч, после чего
концентрировали в вакууме с получением массы твердого вещества. Выделенное вещество разбавляли 10%-ным раствором гидроксида натрия (150 мл), перемешивали при температуре 23oC в течение 30
мин, после чего отфильтровывали неорганические соли. Полученный водный раствор экстрагировали этилацетатом (4 х 100 мл). Объединяли соответствующие органические экстракты, промывали их солевым
раствором (300 мл), высушивали и концентрировали в вакууме с получением масла, которое очищали с помощью импульсной хроматографии (элюция смесью ДХМ и MeOH в соотношении 95:5) с выходом
вышепоименованного соединения в виде белого твердого вещества (0,6 г).
Т.пл. 148-150oC. Т.Ж.X. ДХМ-MeOH (в соотношении 95:5): Rf 0,38.
Промежуточное соединение 7
N-[2,4-Диоксо-1-(3-метил-1-бутил)-2,3,4,5-тетрагидро-1Н-1,5- бензодиазепин-3-ил]-N'-фенилмочевина
К раствору Промежуточного соединения 6 (0,4 г) в сухом
дихлорметане (5 мл) в атмосфере азота добавляли фенилизоцианат (0,4 г). Образовавшуюся смесь перемешивали при температуре 23oC в течение 30 мин, после чего концентрировали в вакууме.
Выделенный остаток растирали с диэтиловым эфиром с выходом вышепоименованного соединения в виде белого твердого вещества (0,478 г).
Т.пл. 249-250oC. Т.Ж.X. ДХМ-MeOH (в соотношении 95:5): Rf 0,38.
Промежуточное соединение 8
N-[2,4-Диоксо-1-(3-метил-1-бутил)-2,3,4,5-тетрагидро-1Н-1,5- бензодиазепин-3-ил]-N'-(4-метоксифенил)
мочевина
К раствору Промежуточного соединения 6 (0,19 г) в сухом дихлорметане (5 мл) в атмосфере азота добавляли 4-метоксифенилизоцианат (0,1 мл). Образовавшуюся смесь перемешивали при
температуре 23oC в течение 30 мин, после чего концентрировали в вакууме. Выделенный остаток растирали с диэтиловым эфиром с выходом вышепоименованного соединения в виде белого твердого
вещества (0,197 г).
Т.пл. 150-152oC. Т.Ж.X. ЭА-MeOH (в соотношении 95:5): Rf 0,78.
Промежуточное соединение 9
N-[2,
4-Диоксо-5-(4-метоксифенилметил)-1-(3-метил-1-бутил) -2,3,4,5-тетрагидро-1Н-1.5-бензодиазепин-3-ил]-N'-фенилмочевина
К раствору Промежуточного соединения 5 (0,05 г) в ацетонитриле добавляли
фенилизоцианат (0,017 мл). Образовавшуюся смесь перемешивали при температуре 23oC в течение 30 мин, после чего фильтровали с выходом вышепоименованного соединения в виде белого твердого
вещества (0,062 г).
Т.пл. 206-208oC. Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,4.
Промежуточное соединение 10
1-(3-метил-1-бутил)амино-2-нитробензол
К раствору 2-фторнитробензола (2,4 г) в ТГФ (20 мл) в атмосфере азота по каплям добавляли раствор амино 3-метилбутана (1,5 г) в ТГФ (20 мл).
Образовавшуюся смесь перемешивали при температуре 23oC в течение 3 ч, после чего выдерживали при температуре дефлегмации в течение 1,5 ч. Охлаждали указанный раствор до температуры 23oC, после чего концентрировали его в вакууме с выходом грубо очищенного соединения, которое очищали с помощью импульсной хроматографии на силикагеле (элюция смесью ЦГ и ЭА в соотношении 9:1) с
получением вышепоименованного соединения в виде желтого масла (2,12 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 8:2): Rf 0,79.
ИК: 3383 (NH); 1620 (C=C) см-1 .
Промежуточное соединение 11
2-(3-Метил-1-бутил)амино-анилин
К суспензии Промежуточного соединения 10 (2,12 г) в смеси этанола (30 мл) и воды (70 мл) добавляли
раствор карбонат калия (9,1 г) и гидросульфита натрия (8 г) в воде (50 мл). Образовавшуюся смесь перемешивали при температуре 23oC в течение 1 ч, после чего подкисляли концентрированной
соляной кислотой до pH 3. Указанную смесь подщелачивали раствором 10%-ного гидроксида натрия до pH 10 и экстрагировали этилацетатом (2 х 100 мл); объединяли соответствующие экстракты, промывали их
солевым раствором (150 мл), высушивали и концентрировали в вакууме с получением вышепоименованного соединения в виде коричневого твердого вещества (1,8 г).
Т.Ж. X. ЦГ-ЭА (в соотношении 8:2): Rf 0,36.
ИК: 3420 (NH); 1620 (C=C) см-1.
Промежуточное соединение 12
N-(2,
2-Диметилэтоксикарбонил)-N'-(3-метил-1-бутил)- 1,
2-фенилендиамин
К раствору Промежуточного соединения II (3 г) в смеси ТГФ (50 мл) и воды (40 мл) добавляли ди-т-бутил дикарбонат (2,44 г) и
кислый карбонат натрия (1,42 г), а образовавшуюся
смесь перемешивали при температуре 30oC в течение 1,5 ч и концентрировали в вакууме. Выделенный остаток разбавляли этилацетатом (150 мл),
а также промывали водой (50 мл) и солевым раствором
(50 мл). Высушивали соответствующую органическую фазу и концентрировали ее в вакууме с получением масла, которое очищали с помощью импульсной
хроматографии (элюция смесью ЦГ и ЗА в соотношении 9:1) с
выходом вышепоименованного соединения в виде воскообразного вещества (3,1 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 9:1): Rf 0,37.
ИК: 3420 (NH); 1722-1697 (C=O) см-1.
Промежуточное соединение 13
N-(2,
2-Диметилэтоксикарбонил)-N'-[2-(1- бензилоксикарбониламино-1-этоксикарбонил)-2-оксоэтил]-N'-(3- метил-1-бутил)-1,
2-фенилендиамин
К раствору моноэтилового эфира бензилоксикарбониламино
малоновой (0,90 г) в этилацетате (40 мл) добавляли N,N'-дицик- логексилкарбодиимид (0,76 г) и гидрат
1-гидроксибензотриазола (0,55 г). По завершении добавления полученную смесь перемешивали при
температуре 20oC в течение 1 ч, после чего добавляли раствор Промежуточного соединения 2 (0,88
г) в этилацетате (20 мл) и продолжали перемешивание в течение еще 2 ч. Затем образовавшуюся
реакционную смесь выдерживали при температуре дефлегмации в течение 4 ч, после чего оставляли на 20 ч при
температуре 20 с, фильтровали и промывали водой (50 мл) и солевым раствором (50 мл).
Высушивали соответствующую органическую фазу, концентрировали ее в вакууме, а полученный остаток очищали с помощью
импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 9:1) с выходом
вышепоименованного соединения в виде масла (0,64 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 8:2): Rf 0, 33.
ИК: 3500-3300 (NH); 1726-1672 (C=O) см-1.
Промежуточное соединение 14
1-(3-Метил-1-бутил)-3-бензилоксикарбониламино-2,4-диоксо- 2,3,4,
5-тетрагидро-1Н-1,5-бензодиазепин
К суспензии Промежуточного соединения 13 (0,
64 г) в этаноле (15 мл) добавляли концентрированную соляную кислоту (5 мл). Образовавшуюся смесь перемешивали при
температуре 23oC в течение 2 ч, разбавляли этилацетатом, промывали водой,
высушивали и концентрировали в вакууме с получением масла, которое очищали с помощью импульсной хроматографии с
выходом вышепоименованного соединения в виде белой пены (0,23 г).
Т.Ж.X. ЭА-ЦГ (в соотношении 1:1): Rf 0,59.
ИК: 3431, 3256 (NH); 1734, 1717 (C=O) см-1.
Промежуточное соединение 15
5-[2- (Диэтиламино)этил] -1-(3-метил-1-бутил)-3- бензилоксикарбониламино-2,4-диоксо-2,3,4,5-тетрагидро-1Н-1,5- бензодиазепин
Суспензию Промежуточного соединения 14 (0,22 г) и карбоната калия
(0,24 г) в смеси ацетона (30 мл) и воды (1 мл) перемешивали при температуре дефлегмации в течение 6 ч. Образовавшийся раствор
концентрировали в вакууме, а выделенный остаток разбавляли этилацетатом
(150 мл), после чего промывали водой (50 мл) и солевым раствором (50 мл). Высушивали соответствующую органическую фазу и
концентрировали ее в вакууме с получением масла, которое очищали с помощью
импульсной хроматографии (элюция смесью ЭА и MeOH в соотношении 9:1) с выходом вышепоименованного соединения в виде масла (0,
194 г).
Т.Ж.X. ЭА-ЦГ (в соотношении 3:1): Rf 0, 23.
ИК: 3400 (NH); 1697-1663 (C=O) см-1.
Промежуточное соединение 16
3-Амино-5-[2-(диэтиламино)этил] -1-(3-метил-1-бутил)- 2,4-диоксо-2,3,4,
5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного соединения 15 (0,18 г) в метаноле (20 мл) добавляли 5%-ный
палладиево-угольный катализатор (0,04 г), а образовавшуюся смесь в
течение 2 ч подвергали гидрированию при давлении 1 атм. С помощью фильтрации через целит удаляли использованный катализатор,
соответствующий фильтрат концентрировали в вакууме, а полученный остаток
растворяли в этилацетате (100 мл), промывали водой (100 мл) и солевым раствором (10 мл), высушивали и концентрировали в вакууме
с выходом вышепоименованного соединения в виде воскообразного вещества
(0,27 г).
Т.Ж.Х. ЭА-MeOH (в соотношении 1:1): Rf 0,3.
ИК: 1680-1651 (C=O) см-1.
Промежуточное соединение 17
N-(1-Адамантилкарбонил)-N'-(4-метоксифенилметил)-1,2- фенилендиамин
К раствору Промежуточного соединения 2 (1,83 г) и
триэтиламина (1,45 мл) в сухом ТГФ (100 мл) при температуре 23oC в атмосфере азота по каплям добавляли хлорид 1-адамантанкарбонила (1,91 г). Образовавшуюся смесь перемешивали при
температуре дефлегмации в течение 3 ч, позволяли ей остыть до температуры
23oC, после чего разбавляли этилацетатом (120 мл), промывали солевым раствором (150 мл), высушивали и
концентрировали в вакууме. Выделенное при этом вещество кристаллизовали из смеси ДХМ и
ЦГ с выходом вышепоименованного соединения в виде белого твердого вещества (2,3 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 8:2): Rf 0,34.
ИК: 3393, 3304 (NH) см-1.
Промежуточное соединение 18
N-(1-Адамантилметил)-N'-(4-метоксифенилметил)-1,
2-фенилендиамин
К раствору Промежуточного соединения 17 (2,3 г) в сухом ТГФ
(70 мл), предварительно нагретому до температуры дефлегмации, в атмосфере азота по каплям добавляли
бор-диметилсульфидный комплекс (10 М раствор; 15 мл). Отгоняли диметилсульфид и ТГФ (50 мл),
образовавшейся смеси позволяли остыть до комнатной температуры, после чего добавляли 10%-ный раствор
карбоната калия (30 мл) и перемешивали указанную смесь при температуре 20oC в течение
40 мин. В дальнейшем рассматриваемую смесь разбавляли метанолом (20 мл) и перемешивали при температуре
дефлегмации в течение 3 ч, а затем - при температуре 20oC в течение 20 ч. Добавляли
этилацетат (100 мл), разделяли образовавшиеся фазы, соответствующую органическую фазу промывали солевым
раствором (2 х 50 мл), высушивали и концентрировали в вакууме с получением масла, которое
очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 9: 1) с выходом
вышепоименованного соединения (0,2 г) в виде белого твердого вещества.
Т.пл. 132-134oC. Т.Ж.X. ЦГ-ЭА (в соотношении 9:1): Rf 0,63.
Промежуточное
соединение 19
1-(1-Адамантилметил)-диоксо-5-(4-метоксифенилметил)-3- фенилгидразоно-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
В 40 мл ТГФ вводили Промежуточное соединение 18 (1,22 г) и
2-фенилгидразономалонилдихлорид (0,940 г), а образовавшуюся смесь в атмосфере азота по каплям добавляли в колбу, содержащую ТГФ (40 мл). По завершении добавления полученный раствор выдерживали при
температуре 50oC в течение 4 ч. Затем указанный раствор разбавляли этилацетатом (10 мл), после чего промывали насыщенным раствором кислого карбоната натрия (2 х 100 мл) и солевым раствором
(2 х 80 мл), высушивали и концентрировали в вакууме с выходом вышепоименованного соединения в виде желтой пены (1,64 г).
Т.пл. 170-188oC. Т.Ж.X. ЦГ-ЭА (в соотношении 8:2): Rf 0,60.
ИК: 3441 (NH); 1661, 1653 (C=O) см-1.
Промежуточное соединение 20
1-(1-Адамантилметил)-3-амино-2,
4-диоксо-5-(4- метоксифенилметил)-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К суспензии Промежуточного соединения 19 (1,36 г) в уксусной кислоте (30 мл) добавляли цинковую пыль (1,46 г).
Образовавшуюся смесь перемешивали при температуре 20oC в течение 18 ч, после чего добавляли дополнительное количество цинковой пыли (0,30 г) в уксусной кислоте (3 мл) и продолжали
перемешивание в течение еще 1 ч. Затем указанную смесь отделяли от использованного цинка, разбавляли этилацетатом (150 мл), промывали насыщенным раствором кислого карбоната натрия (2 х 150 мл) и
солевым раствором (200 мл), высушивали и концентрировали в вакууме с получением масла (1,31 г), которое очищали с помощью импульсной хроматографии (элюция ЭА, а затем - смесью ЭА и метанола в
соотношении 95:5) с выходом вышепоименованного соединения в виде светло-желтого твердого вещества (0,90 г).
Т.пл. 223-225oC. Т.Ж.X. ДХМ-MeOH (в соотношении 95:5): Rf 0,34.
ИК: 1700 и 1670 (С=O) см-1.
Промежуточное соединение 21
1-(1-Адамантилметил)-3-амино-2,4-диоксо-2,3,4,5- тетрагидро-1Н-1,
5-бензодиазепин
К раствору Промежуточного соединения 20 (3,85 г) в смеси ацетонитрила (125 мл) и воды (5 мл) добавляли нитрат аммония и церия (IV) (3,85 г). Указанный раствор перемешивали
при
температуре 23oC в течение 36 ч, после чего к образовавшейся суспензии добавляли воду (5 мл) и продолжали перемешивание в течение еще 30 ч при температуре 50oC. Полученный
раствор концентрировали в вакууме с получением массы твердого вещества. Выделенное вещество разбавляли 10%-ным раствором гидроксида натрия (250 мл) и перемешивали при температуре 23oC в
течение 1 ч, после чего отфильтровывали неорганические соли. Оставшийся водный раствор экстрагировали этилацетатом (2 х 150 мл), объединяли соответствующие органические экстракты, промывали их
солевым
раствором (300 мл), высушивали и концентрировали в вакууме с получением масла (0,750 г), которое очищали с помощью импульсной хроматографии (элюция смесью ДХМ и MeOH в соотношении 95: 5) с
выходом
вышепоименованного соединения в виде белого твердого вещества (0,500 г).
Т.Ж.X. ДХМ-MeOH (в соотношении 95:5): Rf 0,34.
ИК: 3213-3126 (NH И NH2); 1705, 1668 и 1660 (С=O) см-1.
Промежуточное соединение 22
N-[(1-Адамантилметил)-2,4-диоксо-2,3,4,5-тетрагидро-1Н-1,
5- бензодиазепин-3-ил]-N'-фенилмочевина
К раствору Промежуточного соединения 21 (0,14 г) в сухом ацетонитриле (10 мл) в атмосфере азота добавляли фенилизоцианат (0,05 ил). Полученную смесь
перемешивали при температуре 23oC в
течение 30 мин, после чего отфильтровывали образовавшееся твердое вещество и промывали его ацетонитрилом с выходом вышепоименованного соединения в виде
белого твердого вещества (0,146 г).
Т.пл. > 280oC. Т.Ж.X. ДХМ-MeOH (в соотношении 95:5): Rf 0,46.
ИК: 3383, 3215 (NH); 1697, 1676 и 1665 (C=O); 1597 (С=С) см-1.
Промежуточное соединение 23
1-(1-Адамантилкарбониламино)-2-нитробензол
К раствору 2-нитроанилина (10,4 г) и триэтиламина (12,6 мл)
в ацетоне (50 мл) в атмосфере азота по
каплям добавляли раствор хлорида 1-адамантанкарбонила (17,95 г). Образовавшуюся смесь перемешивали при температуре 23oC в течение 22 ч, после чего
добавляли дополнительное количество
ацетона (50 мл). Полученную смесь выдерживали при температуре 70oC в течение 3 ч. Затем указанной смеси позволяли остыть до температуры 23oC,
после чего фильтровали; выделенное
при этом коричневое твердое вещество кристаллизовали из ацетона с выходом вышепоименованного соединения в виде желтого твердого вещества (17,3 г).
Т.пл. 111-114oC. Т.Ж.X. ЦГ-ЭА (В соотношении 10:2): Rf 0,67.
Промежуточное соединение 24
1-(1-Адамантилметиламино)-2-нитробензол
К раствору
Промежуточного соединения 23 (13,5 г)
в сухом толуоле (160 мл), предварительно охлажденному до температуры 10oC, в атмосфере азота по каплям добавляли бор-диметилсульфидный комплекс (10 М
раствор; 6,0 мл). Образовавшийся
раствор перемешивали при температуре 10oC в течение 15 мин, после чего выдерживали при температуре 110oC в течение 1 ч. Указанной смеси
позволяли остыть до комнатной температуры,
после чего добавляли 10%-ный раствор карбоната калия (50 мл) и перемешивали полученную смесь при температуре 23oC в течение 40 мин. Разделяли
образовавшиеся фазы, соответствующий
органический экстракт промывали солевым раствором (50 мл), высушивали и концентрировали в вакууме с получением массы твердого вещества, которую очищали с
помощью
импульсной хроматографии (элюция
смесью ЦГ и ЭА в соотношении 10: 1) с выходом вышепоименованного соединения в виде оранжевого твердого вещества (7,0 г).
Т.пл. 106-109oC. Т.Ж.X. ЦГ-ЭА (в соотношении 10:1): Rf 0,68.
Промежуточное соединение 25
2-(1-Адамантилметиламино)-анилин
К раствору Промежуточного соединения 24 (6,9 г)
в смеси этанола (50 мл) и воды (130
мл) добавляли раствор карбонат калия (23,2 г) и гидросульфита натрия (20,9 г) в воде (150 мл). Образовавшуюся смесь перемешивали при температуре 23oC в
течение 30 мин, после чего подкисляли
концентрированной соляной кислотой до pH 3. Затем указанную смесь подщелачивали раствором 10%-ного гидроксида натрия до pH 10 и концентрировали до половины
объема. Полученный остаток экстрагировали
этилацетатом (2 х 300 мл); объединяли соответствующие экстракты, промывали их солевым раствором (150 мл), высушивали и концентрировали в вакууме с выделением
остатка, который очищали с помощью
импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 10:2) с выходом вышепоименованного соединения в виде серого твердого вещества (5,0 г).
Т.пл. 101-104oC. Т.Ж.X. ЦГ-ЭА (в соотношении 10:2): Rf 0,36.
Промежуточное соединение 26
N-1-Адамантилметил-N'-[2-(4-морфолино)этил]-1,
2-фенилендиамин
Раствор Промежуточного
соединения 25 (1,3 г), иодида натрия (0,76 г) и гидрохлорида 2-(4-морфолино)этил хлорида (0,94 г) в сухом диметилформамиде (40 мл) в течение 4 ч
выдерживали при температуре 160oC в
атмосфере азота. Затем указанную смесь охлаждали до температуры 23oC и концентрировали в вакууме. Полученный остаток разбавляли этилацетатом
(150 мл), после чего промывали 10%-ным раствором
гидроксида натрия (50 мл) и солевым раствором (150 мл). Высушивали соответствующую органическую фазу и концентрировали ее в вакууме с получением масла,
которое очищали с помощью импульсной
хроматографии (элюция смесью ЦГ и ЭА в соотношении 6: 4) с выходом вышепоименованного соединения в виде коричневого масла (0,55 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0, 39.
Промежуточное соединение 27
1-(1-Адамантилметил)-2,4-диоксо-5-[2-(4 - морфолино)этил]-3-фенил-гидразоно-2,3,4,5-тетрагидро-1Н-1,
5- бензодиазепин
К раствору
2-фенилгидразономалонилдихлорида (0,4 г) в этилацетате (30 мл) в атмосфере азота по каплям добавляли раствор Промежуточного соединения 26 (0,5 г) в этилацетате
(20 мл). По завершении добавления
полученный раствор выдерживали при температуре 80oC в течение 1 ч. Указанный раствор охлаждали до температуры 23oC, а затем промывали его
10%-ным раствором гидроксида натрия (30
мл) и солевым раствором (50 мл). Высушивали соответствующую органическую фазу, концентрировали ее в вакууме, а выделенный остаток очищали с помощью импульсной
хроматографии (элюция смесью ЦГ и ЭА в
соотношении 1:5) с выходом вышепоименованного соединения в виде желтого твердого вещества (0,45 г).
Т.пл. 110-120oC. Т.Ж.X. ЦГ-ЭА (в соотношении 1:5): Rf 0, 48.
Промежуточное соединение 28
1-(1-Адамантилметил)-3-амино-2,4-диоксо-5-[2-(4-морфолино) этил-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного
соединения 27 (0,45 г) в ледяной уксусной кислоте (20 мл) добавляли цинковую пыль (0,4 г). Образовавшуюся смесь перемешивали при температуре 23oC в течение
1,5 ч, после чего отделяли ее от
использованного цинка. Соответствующую органическую фазу концентрировали в вакууме, разбавляли этилацетатом (50 мл), а затем промывали 10%-ным раствором гидроксида
натрия (20 мл) и солевым раствором
(20 мл). Высушивали соответствующую органическую фазу и концентрировали ее в вакууме с получением желтого масла (1,31 г), которое очищали с помощью импульсной
хроматографии (элюция смесью ЭА и MeOH в
соотношении 10:1) с выходом вышепоименованного соединения в виде белого твердого вещества (0,25 г).
Т.пл. 75-80oC. Т.Ж.X. ЭА-MeOH (в соотношении 10:1): Rf 0, 11.
Промежуточное соединение 29
N-[1-Адамантилметил)-N'-[2-(1-пирролидино)этил]-1,2-фенилендиамин
Раствор Промежуточного
соединения 26 (2,0 г), иодида натрия (1,17
г) и гидрохлорида 2-(1-пирролидино) этил хлорида (1,33 г) в сухом диметилформамиде (40 мл) в течение 4 ч выдерживали при температуре 160oC в
атмосфере азота. Затем указанный раствор
охлаждали до температуры 23oC и концентрировали в вакууме. Полученный остаток разбавляли этилацетатом (150 мл), после чего промывали 10%-ным
раствором гидроксида натрия (50 мл) и солевым
раствором (50 мл). Высушивали соответствующую органическую фазу и концентрировали ее в вакууме с получением масла, которое очищали с помощью импульсной
хроматографии (элюция смесью ЭА и MeOH в
соотношении 95:5) с выходом вышепоименованного соединения в виде коричневого масла (0,54 г). Т.Ж.X. ЭА-MeOH (в соотношении 9:1): Rf 0,33.
Промежуточное соединение 30
1-(1-Адамантилметил)-2,4-диоксо-5-[2-(1-пирролидино) этил] -3-фенилгидразоно-2,3,4,5-тeтpaгидpo-1H-1,5-бензодиазепин
К раствору
2-фенилгидразономалонилдихлорида (0,416 г) в этилацетате
(30 мл) при температуре 23oC в атмосфере азота по каплям добавляли раствор Промежуточного соединения 29 (0,5 г) в этилацетате (20
мл). По завершении добавления полученный раствор
выдерживали при температуре 80oC в течение 3 ч. Указанный раствор охлаждали до температуры 23oC, а затем промывали его 9 М
раствором гидроксида аммония (25 мл) и солевым
раствором (50 мл). Высушивали соответствующую органическую фазу, концентрировали ее в вакууме, а выделенный остаток очищали с помощью импульсной
хроматографии (элюция смесью ЭА и MeOH в соотношении
10:1) с выходом вышепоименованного соединения в виде желтого твердого вещества (0,27 г).
Т.пл. 105-110oC. Т.Ж.X. ЭА-MeOH (в соотношении 10:1): Rf 0,44.
Промежуточное соединение 31
1-(1-Адамантилметил)-3-амино-2,4-диоксо-5-[2-(1- пирролидино)-этил] -2,3,4,5-тетрагидро-1Н-1,
5-бензодиазепин
К раствору Промежуточного
соединения 30 (0,25 г) в ледяной уксусной кислоте (20 мл) добавляли цинковую пыль (0,23 г). Образовавшуюся смесь перемешивали при температуре 23oC в течение 15 мин. Указанную смесь отделяли
от использованного цинка, разбавляли этилацетатом (25 мл), после чего промывали 10%-ным раствором гидроксида натрия (20 мл) и солевым раствором (50
мл). Высушивали соответствующую органическую фазу и
концентрировали ее в вакууме с получением желтого масла, которое очищали с помощью импульсной хроматографии (элюция градиентом смеси ЭА и MeOH в
соотношении от 9:1 до 7:3) с выходом вышепоименованного
соединения в виде белой пены (0,15 г).
Т.Ж.Х. ЭА-MeOH (в соотношении 7:3): Rf 0,3.
Промежуточное
соединение 32
Диамин
N-(1-адамантилметил)-N'-[3'- (1,1-диметилэтилоксикарбонил)-2', 2'-диметил-4'-метилен-1,2- фенилена
К раствору Промежуточного соединения 25 (0,100 г) и 1,
1-диметилэтил (R)-4-формил-2,
2-диметил-3-оксазолидинкарбоксилата (0,125 г) в метаноле (10 мл) добавляли уксусную кислоту (0,027 мл) и цианоборогидрид натрия (0,050 г). Образовавшийся раствор
перемешивали при температуре 23oC в течение 1 ч, после чего добавляли насыщенный раствор кислого карбоната натрия (50 мл), а полученную реакционную смесь экстрагировали этилацетатом (50
мл). Соответствующие органические
экстракты промывали солевым раствором (50 мл), высушивали и концентрировали в вакууме с получением масла (1,31 г), которое очищали с помощью импульсной хроматографии
(элюция смесью ЦГ и ЭА в
соотношении 9:1) с выходом вышепоименованного соединения в виде пены (0,100 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 5:25): Rf 0,4.
Промежуточное соединение 33
1-(1-Адамантилметил)-2,4-диоксо-5-[3'-(1,1-диметил-этилокси- карбонил)-2', 2'-диметил-4'-метилен-оксазолидин] -3- фенилгидразоно-2,3,4,5-тетрагидро-1Н-1,
5-бензодиазепин
К суспензии
карбоната калия в ТГФ (20 мл) при температуре 23oC в атмосфере азота по каплям добавляли раствор Промежуточного соединения 32 (5,8 г) в ТГФ (20 мл) и
раствор
2-фенилгидразономалонилдихлорида (4,5 г) в ТГФ (50 мл). По завершении добавления полученный раствор выдерживали при температуре 80oC в течение 2 ч. Указанный раствор охлаждали до
температуры 23oC, а затем промывали его 10%-ным раствором гидроксида натрия (25 мл) и солевым раствором (50 мл). Высушивали соответствующую органическую фазу, концентрировали ее в вакууме,
а выделенный остаток очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 8:2) с выходом вышепоименованного соединения в виде пены (4,9 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 2:1): Rf 0,8.
Промежуточное соединение 34
1-(1-Адамантилметил)-3-амино-2,4-диоксо-5-[3'-(1,1-диметил- этилокси-карбонил)-2',
2'-диметил-4'-метилен-оксазолидин 1 - 2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного соединения 33 (1,0 г) в метаноле (100 мл) добавляли 10%-ный палладиево-угольный
катализатор (0,33 г) и п-толуолсульфоновую кислоту (0,325 г). Образовавшуюся смесь в течение 1 ч подвергали гидрированию при давлении 4 атм. и температуре 23oC, после чего фильтровали
через
целит и концентрировали в вакууме с выходом вышепоименованного соединения в виде белого твердого вещества (0,25 г).
Т.Ж.X. ЭА-MeOH (в соотношении 26:4): Rf 0,4.
Промежуточное соединение 35
N-[1-(1-Адамантилметил)-2,4-диоксо-5-[3-гидрокси-2(R) -(диметил-этилокси-карбонил)амино-1-пропил] -2,3,4,5-тетрагидро-1Н- 1,5-бензодиазепин-1-ил]
-N'-фенилмочевина. Изомер 1 и Изомер 2
Раствор Промежуточного соединения 34 (3,30 г) в смеси трифторуксусной кислоты и дихлорметана (50 мл; 0,5 М) перемешивали при температуре 20o
C
в течение 1 ч; затем добавляли 5%-ный раствор кислого карбоната натрия (100 мл), отделяли органическую фазу, промывали ее солевым раствором (100 мл), высушивали и концентрировали в вакууме с
выделением остатка, который отбирали в ацетонитрил (50 мл). К полученному раствору добавляли фенилизоцианат (0,072 мл); образовавшуюся реакционную смесь перемешивали при температуре 23oC в
течение 15 ч, концентрировали в вакууме с выделением поименованного соединения в виде Изомеров 1 и 2. Указанную смесь разделяли посредством очистки с помощью импульсной хроматографии на силикагеле
(элюция смесью ЦГ и ЭД в соотношении 9:1) с выходом вышепоименованного Изомера 1 в виде белого твердого вещества (0,6 г).
Т.пл. 188oC. Т.Ж.X. ЦГ-ЭА (В соотношении 1:1): Rf 0,6.
ИК: 3431 (NH, ОН); 1699 (C=O) см-1.
1H-ЯМР: 7,66 (m); 7,46-7,42 (m); 7,05 (m); 6,99 (s); 6,34 (d); 5,42 (d); 5,16 (d); 4,39 (d); 4,30-4,18 (m); 3,96-3,80 (m); 3,66 (t); 3,49 (dd); 3,19 (d); 1,83 (m); 1,66-1,10 (m); 1,44 (s).
Продолжая элюцию, получали несколько смешанных фракций (0,34 г), после чего
элюировали вышепоименованный Изомер 2 (0,9 г)
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,6.
ИК: 3364 (NH, ОН); 1697 (C=O) см-1.
1 H-ЯМР: 7,70-7,10 (m); 6,92 (s); 6,69 (d); 5,37 (bd); 5,16 (d); 4,38 (d); 4,24-4,0 (m); 3,96-3,68 (m); 3,70-3,44 (m); 3,35 (d); 1,85 (m); 1,68-1,20 (m); 1,41 (s).
Промежуточное
соединение 36
N-Циклогексилметил-1,2-фенилендиамин
Раствор 1,2-фенилендиамина (5,0 г), циклогексилметил бромида (7,0 г) и иодида натрия (7,0 г) в сухом диметилформамиде (250 мл) в
течение 24 ч перемешивали при температуре 32oC в атмосфере азота. Затем указанный раствор разбавляли водой (200 мл) и экстрагировали этилацетатом (150 мл); объединяли соответствующие
органические фракции, промывали их солевым раствором (500 мл), высушивали и концентрировали в вакууме с получением масла, которое очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в
соотношении 8:2) с выходом вышепоименованного соединения в виде белого твердого вещества (1,8 г). Т.Ж.Х. ЦГ-ЭА (в соотношении 1:1): Rf 0,55.
ИК: 3400, 3371 и 3271 (NH2 и NH) см-1.
Промежуточное соединение 37
N-Циклогексилметил-N'-[2-(диэтиламино)этил]-1,2-фенилендиамин
Раствор Промежуточного соединения 36 (2,42
г), иодида натрия (1,22 г) и гидрохлорида 2-диэтиламиноэтил хлорида (2,0 г) в сухом диметилформамиде (100 мл) в течение 4 ч выдерживали при температуре 160oC в атмосфере азота. Указанный
раствор охлаждали до температуры 23oC и концентрировали в вакууме. Полученный остаток разбавляли 10%-ным раствором гидроксида натрия (100 мл) и экстрагировали этилацетатом (200 мл).
Высушивали соответствующую органическую фазу, промывали ее солевым раствором (150 мл) и концентрировали в вакууме с получением масла, которое очищали с помощью импульсной хроматографии (элюция смесью
ЦГ и ЭА в соотношении 4:6) с выходом вышепоименованного соединения в виде коричневого масла (1,7 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,26.
ИК: 1601 (С=С) см-1.
Промежуточное соединение 38
1-Циклогексилметил-2,4-диоксо-5-[2-(диэтиламино)этил] -3- фенил-гидразоно-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору 2-фенилгидразономалонилдихлорида (1,37 г) в этилацетате (50 мл) при температуре 23oC в атмосфере азота по каплям добавляли раствор Промежуточного соединения 37 (1,7 г) в
этилацетате (50 мл). По завершении добавления полученный раствор выдерживали при температуре 80oC в течение 4 ч. Указанный раствор охлаждали до температуры 23oC, а затем
промывали его 10%-ным раствором гидроксида натрия (50 мл) и солевым раствором (50 мл). Высушивали соответствующую органическую фазу, концентрировали ее в вакууме, а выделенный остаток очищали с
помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 6: 4) с выходом вышепоименованного соединения в виде желтой пены (1,0 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,34.
ИК: 3441-3186 (NH); 1661 (C=O) см-1.
Промежуточное соединение 39
3-Амино-1-циклогексилметил-5-[2-(диметиламино)этил] -2,
4- диоксо-2,
3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного соединения 38 (0,38 г) в ледяной уксусной кислоте (5 мл) добавляли цинковую пыль (0,3 г). Образовавшуюся смесь
перемешивали при
температуре 23oC в течение 3 ч, после чего отделяли от использованного цинка. Полученный фильтрат подщелачивали 10%-ным раствором гидроксида натрия до pH 10 и
экстрагировали этилацетатом (2
х 40 мл). Соответствующие органические экстракты промывали солевым раствором (60 мл), высушивали и концентрировали ее в вакууме с получением масла, которое очищали с
помощью импульсной хроматографии
(элюция смесью ДХМ и MeOH в соотношении 9: 1) с выходом вышепоименованного соединения в виде желтого твердого вещества (0,17 г).
Т.пл. 94-95oC. Т.Ж.X. ДХМ-MeOH (в соотношении 85:15): Rf 0,78.
ИК: 1695 и 1664 (C=O) см-1.
Промежуточное соединение 40
5-фтор-N-[2-(4-морфолино)этил]-2-нитроаналин
К раствору 2,4-дифторнитробензола (3,0 г) в тетрагидрофуране (30 мл) по каплям добавляли раствор 2-(N-морфолино) этиламина (2,45 г) в
тетрагидрофуране (10 мл), а образовавшуюся смесь
перемешивали при температуре 23oC в течение 1,5 ч. Указанную смесь концентрировали в вакууме, кристаллизовали из смеси ДХМ и Петрола, после
чего очищали с помощью импульсной хроматографии
(элюция смесью ЦГ и ЭА в соотношении 1:1) с выходом вышепоименованного соединения в виде желтого твердого вещества (3,0 г).
Т.пл. 103-104oC. Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,39.
ИК (nujol): 3400 (N-H); 1632 (C=C); 1570, 1312 (NO2) см-1.
Промежуточное соединение 41
5-фтор-N'-[2-(4-морфолино)этил]-1,2-бензолдиамин
К суспензии Промежуточного соединения 40 в смеси этанола и воды (150 мл в соотношении 1: 1) добавляли
карбонат калия (9,7 г) и гидросульфит
натрия (8,4 г), а образовавшуюся смесь перемешивали при температуре 23oC в течение 1 ч. Указанную смесь концентрировали в вакууме, подкисляли
концентрированной соляной кислотой до pH 3 и
экстрагировали этилацетатом (150 мл). Соответствующую водную фазу подщелачивали 10%-ным раствором гидроксида натрия, после чего экстрагировали
этилацетатом (2х 150 мл). Соответствующую органическую
фазу промывали солевым раствором (200 мл), высушивали и концентрировали в вакууме с выделением вышепоименованного соединения в виде коричневого
масла (1,9 г).
Т.Ж.X. ЭА-MeOH (в соотношении 9:1): Rf 0,28.
ИК (nujol): 3337 (N-H); 1614 (C=C) см-1.
Промежуточное соединение
42а
N-(1-Адамантилкарбонил)-4-фтор-N'-[2-(4-морфолино) этил] -1,2-бензолдиамин
К смеси Промежуточного соединения 41 (1,9 г) и триэтиламина (1,36 мл) в сухом ТГФ (70 мл) по каплям
добавляли
раствор 1-адамантанкарбонил хлорида (1,78 г) в сухом ТГФ (30 мл). Образовавшуюся смесь в течение 1,5 ч выдерживали при температуре 60oC, после чего в вакууме выпаривали
использованные
растворители. Полученный остаток отбирали в этилацетат (200 мл), промывали водой (100 мл) и солевым раствором (50 мл), высушивали и концентрировали в вакууме с выходом
вышепоименованного соединения в
виде белого твердого вещества (3,23 г).
Т.пл. 172-174oC. Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,31.
ИК (nujol): 3375, 3314 (NH); 1647 (C=O); 1618, 1600 (C=C) см-1.
Промежуточное соединение 42б
N-(1-Адамантилметил)-4-фтор-N'-[2-(4-морфолино)этил]- 1,2-бензолдиамин
К
охлажденной (0oC)
суспензии Промежуточного соединения 42а (3,32 г) в толуоле (40 мл) в течение 15 мин по каплям добавляли раствор Витрида [дигидро-бис(2-метоксиэтокси) алюмината натрия] (5,
7 г) в толуоле (10 мл).
Образовавшуюся смесь перемешивали в течение 10 мин при температуре 0oC, а затем - в течение 30 мин при температуре 23oC. Для прекращения реакции в
течение 15 мин добавляли
этилацетат (20 мл) при температуре 0oC. Еще через 15 мин к указанной смеси добавляли дополнительное количество этилацетата (100 мл) и промывали водой (3 х 100 мл),
соответствующую водную
фазу экстрагировали этилацетатом (200 мл), объединяли полученные органические экстракты, промывали их солевым раствором (150 мл), высушивали и концентрировали в вакууме.
Выделенный остаток очищали с
помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 7:3) с выходом вышепоименованного соединения в виде воскообразного вещества (2,2 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,49.
ИК (nujol): 3300 (NH); 1612 (С=С) см-1.
Промежуточное соединение 43
1-Адамантилметил-2,
4-диоксо-7-фтор-5-[2-(4-морфолино)этил] -3-фенилгидразоно-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного соединения 42б (2,05 г) в этилацетате (30 мл) по каплям добавляли
раствор 2-фенилгидразономалонилдихлорида (1,43 г) в этилацетате (50 мл), а полученную смесь выдерживали при температуре 60oC в течение 3 ч. Указанную смесь разбавляли этилацетатом (150 мл)
и промывали 5%-ным раствором гидроксида натрия (100 мл); соответствующую водную фазу повторно экстрагировали этилацетатом (100 мл), объединяли полученные органические фракции, промывали их солевым
раствором (100 мл), высушивали и концентрировали в вакууме. Выделенный остаток очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 7:3) с выходом вышепоименованного
соединения в виде желтой пены (2,05 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,42.
ИК (nujol): 1663 (C=O); 1603, 1590 (С=С) см-1.
Промежуточное соединение 44
1-Адамантилметил-3-амино-2,4-диоксо-7-фтор-5-[2-(4- морфолино)-этил] -2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного
соединения 43 (2,05 г) в ледяной уксусной кислоте (30 мл) добавляли цинковую пыль (1,76 г) и перемешивали указанную смесь при температуре 23oC в течение 4 ч, после чего фильтровали через
целит; выделенное твердое вещество промывали этилацетатом, а полученный фильтрат подщелачивали 10%-ным раствором гидроксида натрия. Разделяли образовавшиеся фазы, соответствующую водную фазу
экстрагировали этилацетатом (2 х 50 мл), объединяли полученные органические экстракты, промывали их солевым раствором (50 мл), высушивали и концентрировали в вакууме. Выделенный остаток очищали с
помощью импульсной хроматографии (элюция смесью ЭА и MeOH в соотношении 9: 1) с выходом вышепоименованного соединения в виде белой пены (1,1 г).
Т.Ж.Х. ЭА-MeOH (в соотношении 8:2): Rf 0,5.
ИК (nujol): 3400 (NH),- 1693-1663 (CO); 1605 (С=С) см-1.
Промежуточное соединение 45
N-(3-Метил-1-бутил)-N'-[2-(4-морфолино)этил]-1,2-бензолдиамин
К раствору 2-[2-(4-морфолиноэтил)амино] анилина (5,7 г) и 3-метилбутиральдегида (2,7 мл) в метаноле (100 мл) добавляли ледяную
уксусную кислоту (1,5 мл). Образовавшуюся смесь перемешивали при температуре 23oC в течение 10 мин, после чего порциями добавляли цианоборогидрид натрия (3,5 г). Перемешивание продолжали в
течение еще 3 ч, затем концентрировали указанную смесь в вакууме, выделенный остаток разбавляли этилацетатом (500 мл), промывали 5%-ным раствором бикарбоната натрия (2 х 100 мл) и солевым раствором,
высушивали и выпаривали в вакууме использованные растворители. Выделенное грубо очищенное вещество очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 55:45) с выходом
вышепоименованного соединения в виде бесцветного масла (3,3 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,33.
ИК: 1601 (С=O) см-1.
1H-ЯМР: 6,79; 6,66 (m); 3,71; 3,13 (m); 2,69; 2,49; 1,79; 1,58; 0,97.
Промежуточное соединение 46
2,4-Диоксо-1-(3-метил-1-бутил)-5-[2-(4-морфолино)этил]
-3- фенилгидразоно-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного соединения 45 (3,3 г) в этилацетате (150 мл) по каплям добавляли раствор 2-фенилгидразономалонилдихлорида
(3,6 г) в этилацетате (250 мл), а полученную смесь выдерживали при температуре дефлегмации в течение 2 ч. Указанную смесь концентрировали в вакууме, а выделенный остаток очищали с помощью импульсной
хроматографии (элюция ЭА) с выходом вышепоименованного соединения в виде желтого твердого вещества (3,6 г).
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,13.
ИК: 1653 и 1626 (С=O) см-1.
Промежуточное соединение 47
3-Амино-2,4-диоксо-(3-метил-1-бутил)-5-[2-(4-морфолино)этил] - 2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного соединения 46 (3,6 г) в ледяной уксусной кислоте (60 мл) добавляли металлический цинк (3,8 г) и перемешивали указанную смесь при температуре 23oC в течение 15
мин,
после чего разбавляли этилацетатом (150 мл) и фильтровали; выделенное твердое вещество промывали этилацетатом и 10%-ным раствором гидроксида натрия (20 мл). К полученному фильтрату добавляли
дополнительное количество 10%-ного раствора гидроксида натрия (150 мл) до pH 10, после чего образовавшийся раствор экстрагировали этилацетатом (150 мл). Полученную органическую фракцию промывали
солевым раствором (100 мл), высушивали и концентрировали в вакууме. Выделенный остаток очищали с помощью импульсной хроматографии (элюция смесью ЭА и MeOH в соотношении 10: 1) с выходом
вышепоименованного соединения в виде светло-желтого твердого вещества (1,5 г).
Т.пл. 117-119oC. Т.Ж.X. ЭА-MeOH (в соотношении 10:1): Rf 0,37.
ИК: 1691 (С=O) см-1.
Промежуточное соединение 48
N-[2-(Гексаметиленимино)этил]-N'-фенил-1,2-бензолдиамин
К раствору карбоната калия (4,48 г) и иодида
калия (2,16 г) в сухом ксилоле добавляли N-фенил-1,2-бензолдиамин (2,0 г) и гидрохлорид 2-(гексаметиленимино)-этилхлорида (2,57 г), а образовавшуюся смесь в течение 2 ч подвергали дефлегмации в
атмосфере азота. Указанный раствор разбавляли метилен хлоридом (100 мл), промывали 5%-ным нашатырном спиртом (50 мл), водой (50 мл) и солевым раствором (70 мл), высушивали и выпаривали в вакууме
использованные растворители. Полученное грубо очищенное вещество очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 6:4) с выходом вышепоименованного соединения (2,09 г)
в
виде темного масла.
Т.Ж.X. ЦГ-ЭА (в соотношении 4:6): Rf 0,45.
ИК: 3252 (NH); 1597 (C=C) см-1.
Промежуточное соединение
49
2,4-Диоксо-1-[2-(гексаметиленимино)этил] -5-фенил-3- фенилгидразоно-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
К раствору Промежуточного соединения 48 (2,08 г) в этилацетате (115 мл)
по
каплям добавляли раствор 2-фенилгидразономалонилдихлорида (1,81 г) в этилацетате (115 мл), а полученную смесь перемешивали при температуре 23oC в течение 1 ч, а затем при температуре
50oC в течение 1 ч. Указанную смесь промывали 10%-ным раствором гидроксида натрия (100 мл) и солевым раствором (100 мл), высушивали и концентрировали в вакууме. Выделенный остаток очищали
с
помощью импульсной хроматографии (элюция ЭА) с выходом вышепоименованного соединения в виде желтой пены (2,03 г).
Т.Ж.Х. ЭА: Rf 0,34.
ИК: 1664 (C=O); 1591 (C=C) см-1.
Промежуточное соединение 50
3-Амино-2,4-диоксо-5-[2-(гексаметиленимино)этил] -5- фенил-2,3,4,5-тетрагидро-1H-1,5-бензодиазепин
К
раствору
Промежуточного соединения 49 (0,50 г) в сухом метаноле (20 мл) добавляли аммоний формат (0,656 г) и 10%-ный палладиево-угольный катализатор (0,463 г). Указанную смесь подвергали дефлегмации
в течение
30 мин, после чего охлаждали до температуры 23oC и фильтровали через целит. Полученный фильтрат концентрировали в вакууме, а выделенный остаток отбирали в диэтиловый эфир (50 мл)
и
экстрагировали 10%-ным раствором соляной кислоты (50 мл). Соответствующую водную фазу нейтрализовали твердым бикарбонатом натрия, после чего экстрагировали этилацетатом (2 х 50 мл). Полученную
органическую фракцию промывали солевым раствором (50 мл), высушивали и концентрировали в вакууме с выходом вышепоименованного соединения в виде оранжевой пены (0,36 г).
Т.Ж.X. ДХМ-MeOH (в соотношении 95:5): Rf 0,42.
Промежуточное соединение 51
Метиловый эфир (S)-(+)-2-(4-толуолсульфонилокси)-фенилуксусной кислоты
Способ A:
(S)-(+)-метил манделат (3,0 г) и триэтиламин (6,13 мл) растворяли в сухом дихлорметане (40 мл). Полученную смесь охлаждали до температуры 0oC, после чего при перемешивании добавляли
4-толуолсульфонил хлорид (6,87 г). Образовавшийся раствор выдерживали при указанной температуре в течение 40 мин, а затем в течение 20 мин позволяли нагреться до температуры 23oC. В
дальнейшем рассматриваемую смесь разбавляли дихлорметаном (20 мл), промывали солевым раствором (50 мл), высушивали и концентрировали в вакууме. Выделенное грубо очищенное вещество очищали с помощью
импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 5:1, а затем - 2:1) с выходом вышепоименованного соединения в виде белого воскообразного вещества (5,75 г).
Т.пл. 57-58oC. Т.Ж.X. ЦГ-ЭА (в соотношении 2:1): Rf 0,54.
HPLC: (+)/(-) = 91,4/8,6 e.e. = 82,8%.
Способ Б: (S)-(+)-метил манделат (3,0 г) и пиридин (2, 9 мл) растворяли в сухом дихлорметане (40 мл). Полученную смесь охлаждали до температуры 0oC, после чего при перемешивании добавляли 4-толуолсульфонил хлорид (6,87 г). Образовавшийся раствор выдерживали при указанной температуре в течение 15 мин, а затем позволяли нагреться до температуры 23oC. В дальнейшем рассматриваемую смесь разбавляли дихлорметаном (20 мл), промывали 5%-ным раствором соляной кислоты (80 мл), солевым раствором (80 мл), высушивали и концентрировали в вакууме. Выделенное грубо очищенное вещество очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 4:1, а затем - 2:1) с выходом бесцветного масла, которое еще раз очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в соотношении 5:1) с выходом вышепоименованного соединения в виде белого воскообразного вещества (2,2 г).
Т.пл. 57-58oC. Т.Ж.X. ЦГ-ЭА (в соотношении 2:1): Rf 0,54.
HPLC: (+)/(-) = 99,8/0,2 e.e. = 99,6%.
Промежуточное соединение 52
Метиловый эфир [1-(адамантилметил)-2,4-диоксо-5-[2-(4-морфолино) этил] -2,3,4,5-тетрагидро-1Н-1,
5-бензодиазепин-3-ил] амино фенилуксусной кислоты (Изомер 1 и Изомер 2)
К смеси Промежуточного соединения 28 (0,905 г) и Промежуточного соединения 51 (1,28 г) в сухом тетрагидрофуране (30
мл)
добавляли диизопропиланин (0,348 мл). Указанную смесь подвергали дефлегмации в течение 4 ч, после чего разбавляли дихлорметаном (100 мл), промывали насыщенным раствором хлорида аммония (100 мл) и
солевым раствором (100 мл), высушивали и концентрировали в вакууме. Выделенное грубо очищенное вещество дважды очищали с помощью импульсной хроматографии (элюция градиентом смеси ЦГ и ЭА в
соотношении
от 1:3 до 1:9, а затем - смесью ЭА и MeOH в соотношении 4 :1) с выходом:
- вышепоименованного Изомера 1 (0,335 г) в виде белой пены.
Т.Ж.X. ЭА-MeOH (в соотношении 24:1): Rf 0,65.
ИК: 1742, 1700, 1666 (C=O) см-1.
- вышепоименованного Изомера 2 (0,081 г) в виде белой пены.
Т.Ж.X. ЭА-MeOH (в соотношении 24:1): Rf 0,61.
HPLC: e.e. = 90,6%.
Промежуточное соединение 53
N-(трет-бутоксикарбонил)-D-фенилаланин
К
раствору
D-фенилаланина (3 г) в смеси диоксана и воды (54 мл в соотношении 2:1), а также 1 н. раствора гидроксида натрия (18 мл), добавляли ди-трет-бутилдикарбонат (4,32 г). Полученную смесь
перемешивали при
температуре 23oC в течение 3 ч, выпаривали диоксан в вакууме, охлаждали соответствующую водную фазу и экстрагировали ее этилацетатом (30 мл). Указанный водный раствор
подкисляли твердой
лимонной кислотой до pH 3 и экстрагировали этилацетатом (2 х 30 мл). Полученную органическую фракцию промывали солевым раствором (30 мл), высушивали и выпаривали с выходом грубо
очищенного
вышепоименованного соединения в виде масла (4,3 г).
1H-ЯМР (CDCl3): 7,4-7,1 (m); 5,0-4,5 (bm); 3,3-2,7 (bm); 1,4 (s).
Промежуточное
соединение
54
N-(1-Адамантилметил-5-[2-(4-морфолино)этил] -2,4-диоксо- 2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил)-2-D-(3-трет- бутоксикарбонил)-фенилпропионамид
К раствору
Промежуточного
соединения 53 в этилацетате (150 мл) добавляли N, N'-дициклогексилкарбодиимид (0,784 г) и 1-гидроксибензотриазол (0,565 г). Полученную смесь перемешивали при температуре 23o
C в течение 2 ч,
после чего добавляли раствор Промежуточного соединения 28 (1,5 г) в этилацетате (10 мл). Образовавшийся раствор перемешивали при температуре 23oC в течение 3 ч, а затем
фильтровали и
концентрировали в вакууме. Выделенный остаток очищали с помощью импульсной хроматографии (элюция градиентом смеси ЭА и ЦГ в соотношении от 1:3 до чистого ЭА) с выходом
вышепоименованного соединения (1,
84 г) в виде пены.
Т.Ж.X. ЭА-ЦГ (в соотношении 2:1): Rf 0,2.
ИК: 3400 (N-H); 1707, 1672 (С=O) см-1.
Промежуточное соединение
55
N-(1-Адамантилметил-5-[2-(4-морфолино)этил] -2,4-диоксо- 2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил)-2-D-амино-3- фенилпропионамид (Изомер 1 и Изомер
2)
Промежуточное соединение
54 (1,84 г) растворяли в смеси трифторуксусной кислоты (6 мл) и дихлорметана (6 мл), а полученную смесь перемешивали при температуре 23oC в течение 30
мин. Образовавшуюся реакционную среду
концентрировали в вакууме и растирали с диэтиловым эфиром с выходом трифторуксусной соли вышепоименованного соединения, которую отфильтровывали и высушивали.
Указанную соль суспендировали в
этилацетате (50 мл) и экстрагировали 5%-ным раствором нашатырного спирта (70 мл). Соответствующую органическую фракцию промывали солевым раствором, высушивали и
концентрировали в вакууме с выходом
белой пены (1,24 г). Разделение двух вышеупомянутых диастереоизомеров осуществляли с помощью импульсной хроматографии (элюция градиентом смеси ЭА и MeOH в
соотношении от 98:2 до 95:5) с выходом:
- вышепоименованного Изомера 1 (0,618 г) в виде белой пены.
Т.Ж.X. ЭА-MeOH (в соотношении 9,26:0,75): Rf 0,38.
- вышепоименованного Изомера 2 (0,440 г) в виде белой пены.
Т.Ж.X. ЭА-MeOH (в соотношении 9,25:0,25): Rf 0,22.
ИК: 1705, 1666 (С=O) см-1.
Промежуточное соединение
56
N-(1-Адамантилметил-5-[2-(4-морфолино)этил] -2,4-диоксо- 2,3,4,5-тетрагидро-1H-1,
5-бензодиазепин-3-ил)-3-фенил-2-D-(3- фенилтиомочевина)-пропионамид
К раствору Промежуточного
соединения 55 (Изомер 1; 0,61 г) в дихлорметане (50 мл) добавляли фенилизотиоцианат (0,149 г).
Полученную смесь перемешивали при температуре 23oC в течение 3 ч, а затем - при температуре
50oC в течение 30 мин. Выпаривали использованный растворитель, а выделенный остаток
очищали с помощью импульсной хроматографии (элюция смесью ЭА и ЦГ в соотношении 1: 1) с выходом
вышепоименованного соединения в виде белой пены (0,66 г).
Т.Ж.X. ЭА-ЦГ (в соотношении 1:1): Rf 0,38.
ИК: 1705, 1666 (С=O) см-1.
Промежуточное соединение 57
(-)3-Амино-1-(1-адамантилметил)
-2.4-диоксо-5-[2-(4- морфолино)-этил-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин
Способ А:
К раствору Промежуточного
соединения 52 (Изомер 1; 0,187 г) в метаноле (10 мл) добавляли
20%-ный гидроксид палладия (II) на активированном угле (0,218 г). Указанную смесь в течение 5 ч подвергали гидрированию при атмосферном
давлении, после чего фильтровали через целит. Выпаривали
использованные растворители, а выделенное грубо очищенное вещество очищали с помощью импульсной хроматографии (элюция смесью ЦГ и ЭА в
соотношении 1:1, а затем - смесью ЭА и МеоН в соотношении 3:2) с
выходом вышепоименованного соединения в виде белого твердого вещества (0,140 г).
Т.пл. 180-185oC. Т.Ж.X. ЭА-MeOH (в соотношении 1:1): Rf 0,44.
α D = -36o.
ИК: 3480-3350 (N-H); 1695, 1664 (C=O) см-1.
Способ Б:
Промежуточное соединение 57 (0,65 г) растворяли в
трифторуксусной кислоте (15 мл) и перемешивали при температуре 60oC в течение 30 мин. Образовавшийся раствор концентрировали в
вакууме, выделенный остаток разбавляли этилацетатом (60 мл),
после чего промывали 5%-ным раствором бикарбоната натрия (20 мл) и солевым раствором. Высушивали соответствующую органическую фазу и
концентрировали ее в вакууме, полученный остаток очищали с помощью
импульсной хроматографии (элюция смесью ЭА и ЦГ в соотношении 1:1, а затем - смесью ЭА и MeOH в соотношении 9:1) с выходом
вышепоименованного соединения (0,05 г), а также исходного вещества (0,508
г).
Продукт, полученный в результате взаимодействия между вышепоименованным соединением и фенилизоцианатом, характеризовался тем же временем удержания (5,2 мин), что и Изомер 1 из примера 11 (колонка Pirkle D-DNBPG С5 (25х2,4), элюция смесью ДХМ и ИПС в соотношении 93:7).
Не вступившее в реакцию исходное вещество (0,196 г) вновь вовлекли в описанную процедуру, в течение 22 ч перемешивая его трифторуксусной кислотой (10 мл) при температуре 40oC. Проведя все необходимые этапы, получили дополнительное количество вышепоименованного соединения (0,050 г; чистота энантиомера 97:3).
Пример 1
N-[2,4-Диоксо-1-(3-метил-1-бутил)-5-[2-(4-морфолино) этил] -2,
3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'- фенилмочевина
Способ A:
К раствору Промежуточного соединения 7 (110 мг) в сухом ДМФ (5 мл) в атмосфере азота добавляли гидрид натрия
(19,8 мг). Образовавшуюся смесь перемешивали при температуре 23oC в течение 30 мин, после чего добавляли гидрохлорид 4-(2-хлорэтил)морфолина (67,6 мг). Указанную смесь выдерживали при
температуре 70oC в течение 5 ч, затем охлаждали до
температуры 23oC, разбавляли 5%-ным раствором кислого карбоната натрия (30 мл) и экстрагировали этилацетатом (3 х 30 мл).
Объединяли соответствующие органические экстракты, промывали их
солевым раствором (60 мл), высушивали и концентрировали в вакууме с получением масла. Указанное масло очищали с помощью импульсной
хроматографии (элюция ЭА), а выделенное при этом твердое вещество
вновь очищали, растирая его с диэтиловым эфиром с выходом вышепоименованного соединения в виде белого твердого вещества (78 мг).
Т.пл. 129-130oC. Т.Ж.X. ЭА-MeOH (в соотношении 95:5): Rf 0,46.
Способ Б:
Смесь Промежуточного соединения 7 (50 мг), карбоната калия (54 мг),
гидрохлорида 4-(2-хлорэтил)морфолина (26,6 мг), ацетона
(10 мл) и воды (1 мл) перемешивали при температуре 75oC в течение 17 ч. Затем указанную суспензию охлаждали до температуры 23oC, отфильтровывали неорганические соединения, а
соответствующий фильтрат концентрировали в вакууме. Полученный остаток растирали с ацетонитрилом с выходом вышепоименованного соединения в виде
белого твердого вещества (45 мг).
Т.пл. 129-130oC. Т.Ж.X. ЭА-MeOH (в соотношении 95:5): Rf 0,46.
ИК: 3400 (N-H); 1695 и 1637 (С=O); 1601 и 1558 (C=C) см-1.
1 H-ЯМР: 7,6-7,54 (m); 7,46-7,24 (m); 7,05 (t); 6,75 (s); 6,22 (d); 5,09 (d); 4,4-4,2 (m); 3,8-3,6 (m); 2,6-2,35 (m); 1,6-1,35 (m); 0,88 (d); 0,86 (d).
Пример 2
N-[2,
4-Диоксо-1-(3-метил-1-бутил)-5-[2-(1-пиперидино)этил] - 2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'- фенилмочевина
Смесь Промежуточного
соединения 7 (50 мг), карбоната калия (54 мг),
гидрохлорида 4-(2-хлорэтил) пиперидина (26,33 мг), ацетона (10 мл) и воды (1 мл) перемешивали при температуре 75oC в течение 18 ч. Затем
указанную суспензию охлаждали до температуры 23oC, отфильтровывали неорганические соединения, а соответствующий фильтрат концентрировали в вакууме. Полученный остаток растирали с диэтиловым
эфиром с выходом вышепоименованного соединения в
виде белого твердого вещества (45 мг).
Т.пл. 107-109oC. Т.Ж.X. ЭА-MeOH (в соотношении 95:5): Rf 0,2.
ИК: 3429 И 3192 (N-H); 1699 И 1647 (C=O); 1601 (С=С) см-1.
1H-ЯМР: 7,6 (m); 7,44-7,30 (m); 7,05 (t); 6,75 (s); 6,22 (d); 5,08 (d); 4,4-4,2 (m); 3,8-3, 65 (m); 2,5-2,3 (m); 1,56-1,3 (m); 0,87 (d); 0,84 (d).
Пример 3
N-[5-[2-(Диметиламино)этил] -2,4-диоксо-1-(3-метил-1-бутил)- 2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'
- фенилмочевина
Смесь Промежуточного
соединения 7 (50 мг), карбоната калия (54 мг), гидрохлорида 2-диметиламиноэтилхлорида (20,6 мг), ацетона (10 мл) и воды (1 мл) перемешивали при
температуре 75oC в течение 20 ч. Затем
указанную суспензию охлаждали до температуры 23oC, отфильтровывали неорганические соединения, а соответствующий фильтрат концентрировали в
вакууме. Полученный остаток очищали с помощью
импульсной хроматографии (элюция смесью ЭА и MeOH в соотношении 95: 5) с выходом вышепоименованного соединения в виде белого твердого вещества (45 мг).
Т.пл. 159-161oC. Т.Ж.X. ЭА-MeOH (в соотношении 95:5): Rf 0,39.
ИК: 3350 (N-H); 1695 и 1641 (С=O); 1601 (C=C) см-1.
1H-ЯМР: 7,54-7,2 (m); 7,055 (t); 6, 71 (s); 6,20 (d); 5,08 (d); 4,4-4,25 (m); 3,8-3,6 (m); 2,5-2,3 (m); 2,17 (s); 1,5 (m); 1,45-1,35 (m); 0,87 (d); 0,85 (d).
Пример 4
N-[2,
4-Диоксо-1-(3-метил-1-бутил)-5-[2-(4-морфолино) этил] -2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'-(4- метоксифенил)мочевина
Смесь Промежуточного соединения 8 (0,175 г), карбоната
калия
(0,178 г), гидрохлорида 4-(2-хлорэтил)морфолина (0,087 г), ацетона (20 мл) и воды (2 мл) перемешивали при температуре 75oC в течение 18 ч. Затем указанную суспензию охлаждали до
температуры 23oC, отфильтровывали неорганические соединения, а соответствующий фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью импульсной хроматографии (элюция
смесью ЭА и MeOH в соотношении 98: 2) с выходом вышепоименованного соединения в виде белого твердого вещества (45 мг).
Т.пл. 161-163oC. Т.Ж.X. ЭА-MeOH (в соотношении 9:1): Rf 0,53.
ИК: 3349 (N-H); 1728, 1700 И 1653 (С=O) см-1.
1H-ЯМР: 7,54 (m); 7,42-7,32 (m); 7,26 (d); 6,86 (d); 6,43 (s); 6,06 (d); 5,06 (d); 4,4-4,1 (m); 3,78 (m); 3,8-3,6 (m); 2,6-2,2 (m); 1,6-1,3 (m); 0,87 (d); 0,84 (d).
Пример 5
N-[2,4-Диоксо-1-(3-метил-1-бутил)-5-[2-(4-морфолино)этил] - 2,3,4,
5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'-(4- гидроксифенил)мочевина
К раствору соединения Примера 4 (0,05 г) в сухом ацетонитриле (20 мл) в атмосфере азота добавляли иодид алюминия (0,194
г). Образовавшийся раствор выдерживали при температуре 90oC в течение 24 ч. Добавляли еще одну порцию иодида алюминия (0,194 г) и выдерживали указанную смесь при температуре 90o
C
в течение еще 24 ч. Полученную смесь охлаждали до температуры 23oC, разбавляли водой (5 мл) и 5%-ным раствором тиосульфата натрия (25 мл), после чего экстрагировали этилацетатом (2 х 30
мл). Объединяли соответствующие органические экстракты, промывали их солевым раствором (50 мл), высушивали и концентрировали в вакууме с получением остатка, который растирали с диэтиловым эфиром с
выходом вышепоименованного соединения в виде белого твердого вещества (0,030 г).
Т.пл. 104-105oC. Т.Ж.X. ЭА-MeOH (в соотношении 95:5): Rf 0,2.
ИК: 3450 И 3340 (NH и ОН); 1697 и 1663 (С=O) см-1.
1H-ЯМР: 7,55 (m); 7,46-7,32 (m); 7,07 (m); 6,62 (m); 6,46-6,10 (m); 5,07 (d); 4,34 (m); 4,20 (m); 3,9-3,6 (m); 2,7-2,3 (m); 1,8-1,3 (m); 0,86 (d); 0,84 (d).
Пример 6
N-[5-[2-(Диэтиламино)этил] -2,4-диоксо-1-(3- метил-1-бутил)-2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]
-N'-фенилмочевина
К смеси Промежуточного соединения 16 (0,07 г) в ацетонитриле (3,5 мл) добавляли фенилизоцианат (0,022 мл). Образовавшуюся реакционную среду перемешивали при температуре
23oC в течение 20 мин, после чего отфильтровывали твердое вещество и промывали его ацетонитрилом с выходом вышепоименованного соединения в виде белого твердого вещества (0,066 г).
Т.пл. 147oC. Т.Ж.X. ЭА-MeOH (в соотношении 8:2): Rf 0,64.
ИК: 3315 (N-H); 1703 и 1666 (C=O) см-1.
1H-ЯМР: 7,57 (m); 7,39 (m); 7,37-7,29 (m); 7,04 (t); 6,84 (bs); 6,26 (d); 5,08 (d); 4,36-4,2 (m); 3,74 (m); 3,62 (m); 2,63 (m); 2,49 (q); 1,48 (m); 1,38 (m); 0,94 (t); 0,86 (d); 0,83 (d).
Пример
7
N-[1-[1-(Адамантилметил)-5-[2-(диметиламино)этил] -2,4-диоксо- 2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'-фенилмочевина
Смесь Промежуточного соединения 22 (0,134 г) и
80%-ной
масляной суспензии гидрида натрия (0,020 г) в сухом ДМФ (5 мл) перемешивали при температуре 20oC в течение 15 мин, после чего добавляли гидрохлорид 2-диметиламиноэтилхлорида (0,052
мг), а
полученную смесь в течение 4 ч выдерживали при температуре 80oC. Затем указанную смесь охлаждали до температуры 23oC, разбавляли этилацетатом (50 мл) и насыщенным
раствором
кислого карбоната натрия (50 мл), объединяли соответствующие органические фазы, промывали их солевым раствором, высушивали и концентрировали в вакууме. Полученный остаток очищали с помощью
импульсной
хроматографии (элюция смесью ЭА и MeOH в соотношении 9:1) с выходом вышепоименованного соединения в виде белого твердого вещества (0,13 г).
Т.пл. 150-oC. Т.Ж.X. ЭА-MeOH (в соотношении 9:1): Rf 0,31.
ИК: 3400 (N-H); 1697 и 1666 (C=O) см-1.
1H-ЯМР: 7,66-7,41 (m); 7,40-7,20 (m); 7,07 (m); 6,50 (bs); 6,08 (d); 5,10 (d); 4,39 (d); 4,11-3,79 (m); 3,23 (d); 2,9-2,66 (m); 2,33 (s); 1,83 (та); 1,66-1,40 (m); 1,25 (m).
Пример 8
N-[1-[1-(Адамантилметил)-5-[3-(диметиламино)пропил] - 2,
4-диоксо-2,3,4,5-тетрагидро-1Н-1, 5-бензодиазепин-3-ил]-N'- фенилмочевина
Смесь Промежуточного соединения 22 (0,103 г) и 80%-ной
масляной суспензии гидрида натрия (0,017 г) в сухом ДМФ (5 мл)
перемешивали при температуре 20oC в течение 15 мин, после чего добавляли гидрохлорид 3-(диметиламино)пропилхлорида (0,052 мг),
а полученную смесь в течение 4 ч выдерживали при температуре
80oC. Затем указанную смесь охлаждали до температуры 23oC, разбавляли этилацетатом (50 мл) и насыщенным раствором
кислого карбоната натрия (50 мл), объединяли соответствующие
органические фазы, промывали их солевым раствором, высушивали и концентрировали в вакууме. Полученный остаток очищали с помощью импульсной
хроматографии (элюция смесью ЭА и MeOH в соотношении 9:1) с
выходом вышепоименованного соединения в виде белого твердого вещества (0,09 г).
Т.пл. 138-140oC. Т.Ж.X. ЭА-MeOH (в соотношении 9:1): Rf 0,24.
ИК: 3315 (N-H); 1699 и 1639 (C=O) см-1.
1H-ЯМР: 7,52-7,24 (m); 7,04 (t); 6,87 (bs); 6,27 (d); 5,09 (d); 4,39 (d); 4,08-3,80 (m); 3,24 (d); 2,45 (m); 2,28 (s); 2, 2-2,0 (m); 1,98-1,70 (m); 1,6-1,2 (m).
Пример 9
N-[1-(1-Адамантилметил)-2,4-диоксо-5-[2-(4-морфолино)этил] - 2,3,4,
5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'-фенилмочевина
К раствору Промежуточного соединения 28 (0,16 г) в ацетонитриле (7 мл) добавляли фенилизоцианат (0,041 мл). Образовавшуюся реакционную
среду перемешивали при температуре 23oC в
течение 30 мин, после чего концентрировали в вакууме. Выделенный остаток растирали с диэтиловым эфиром с выходом вышепоименованного соединения в
виде белого твердого вещества (0,1 г).
Т.пл. 188-190oC. Т.Ж.X. ЭА-MeOH (в соотношении 1:1): Rf 0,18.
ИК: 3317 (N-H); 1699 и 1666 (C=O) см-1.
1H-ЯМР: 7,80 (m); 7, 5-7,2 (m); 7,05 (t); 6,81 (s); 6,24 (d); 5,12 (d); 4,39 (d); 4,14 (m); 3,9-3,6 (m); 3,23 (d); 2,94-2,76 (m); 1,83 (s); 1,7-1,1 (m).
Пример 10
N-[1-(1-Адамантилметил)-2,
4-диоксо-5-[2-(пирролидино) этил] -2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N' - фенилмочевина
К раствору Промежуточного
соединения 31 (0,1 г) в дихлор-метане (7 мл) добавляли
фенилизоцианат (0,026 мл). Образовавшуюся реакционную среду перемешивали при температуре 23oC в течение 30 мин, после чего
концентрировали в вакууме. Выделенный остаток растирали с
диэтиловым эфиром с выходом вышепоименованного соединения в виде белого твердого вещества (0,08 г).
Т.пл. 155-160o C. Т.Ж.X. ЭА-MeOH (в соотношении 10:1): Rf 0, 43.
ИК: 3200 (N-H); 1695 и 1664 (С=O) см-1.
1H-ЯМР: 7,65 (m); 7,4-7,28 (m); 7,03 (t); 6, 91 (s); 6,31 (d); 5,11 (d); 4,38 (d); 4,15 (m); 3,88 (m); 3,22 (d); 2,94 (m); 2,66-2,54 (m); 1,86-1,76 (m); 1,46 (d); 1,25 (d); 1,20 (d).
Пример 11
N-[1-(1-Адамантилметил)-2,
4-диоксо-5-[2-(4-морфолино)этил] - 2,3,4,5-тетрагидро-1Н-1,
5-бензодиазепин-3-ил]-N'-фенилмочевина (Изомер 1 и Изомер 2)
Способ А:
Соединение из Примера 9 разделяли на чистые
энантиомеры (Изомер 1 и Изомер 2) с помощью препаративной HPLC
(колонка Pirkle D-DNBPG 25х2,4 см; элюция смесью ДХМ и ИПС в объемном соотношении 93:7).
Вышепоименованное соединение,
Изомер 1:
Время удержания 5,2 мин, α D = -50,
0o (CHCl3)
Вышепоименованное соединение. Изомер 2
Время удержания 7,8 мин, α D = +42,0o (CHCl3).
Способ Б:
К раствору Промежуточного соединения 57 (0,1 г; получено в соответствии со способом А) в сухом ацетонитриле (5 мл) добавляли
фенилизоцианат (0,049 мл), а образовавшуюся смесь перемешивали при
температуре 23oC в течение 5 мин. Отфильтровывали твердое вещество, промывали его ацетонитрилом и растирали со смесью ЭЭ
и ЦГ в соотношении 1:1 с выходом Изомера 1 вышепоименованного
соединения (0,094 г) в виде белого твердого вещества.
Т.пл. 157-159oC. Т.Ж.X. ЭА-ЦГ (в соотношении 1:1): Rf 0,30.
e.e. = 94%. α D = -40o.
ИК: 3400 (N-H); 1699, 1668 и 1641 (С=O) см-1.
1H-ЯМР: 7,80 (m); 7, 44-7,24 (m); 7,05 (t); 6,75 (bs); 6,21 (bd); 5,12 (d); 4,39 (d); 4,14 (m); 3,78 (m); 3,75 (m); 3,22 (d); 2,93 (m); 2,75 (m); 2,57 (m); 1,83 (m); 1,64-1,18 (т).
Пример 12
Гидрохлорид N-[1-(1-адамантилметил)-2,
4-диоксо-5-[(3- гидрокси-2(R)амино)-пропил] -2,3,4,5-тетрагидро-1Н-1,5- бензодиазепин-3-ил] -N'-фенилмочевина
Рацемический раствор Промежуточного
соединения 35 (0,34 г) в метаноле (50 мл),
предварительно насыщенном соляной кислотой, перемешивали при температуре 20oC в течение 4 ч, после чего образовавшуюся реакционную смесь
концентрировали в вакууме, отбирали в диэтиловый
эфир и кристаллизовали из смеси метанола и диэтилового эфира с выходом рацемического вышепоименованного соединения в виде белого твердого вещества (0,
175 г).
Т.пл. > 240oC (с разложением).
ИК: 3440-2500 (NH, ОН, NH3+); 1697 и 1684 (С=O) см-1.
1H-ЯМР: 9,20 (bs); 8,36 (bs); 8,09 (bs); 7,74 (m); 7,46 (m); 7,33 (m); 7,21 (m); 6,90 (m); 5,69 (t); 5,41 (t); 4,92 (d); 4,21 (m); 4,04-3,82 (m); 3,82-3,56 (m); 3,40 (m); 1,79 (m); 1,64-1,08 (m).
Пример 13
N-[1-(Циклогексилметил)-2,4-диоксо-5-[(2-диэтиламино) этил] -2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил]-N'- фенилмочевина
К
раствору Промежуточного соединения 39 (0,057 г) в сухом
ацетонитриле (2 мл) добавляли фенилизоцианат (0,018 мл). Образовавшуюся смесь перемешивали при температуре 23oC в течение 30 мин,
после чего фильтровали с выходом вышепоименованного
соединения в виде белого твердого вещества (0,05 г).
Т.пл. 186-188oC. Т.Ж.X. ДХМ-MeOH (в соотношении 9:1): Rf 0,8.
ИК: 3400 (N-H); 1699, 1666 и 1641 (С=O) см-1.
Пример 14
N-[1-(1-Адамантилметил)-2,4-диоксо-7-фтор-5-[2-(4- морфолино)-этил] -2,3,4,
5-тетрагидро-1Н-1,5-бензодиазепин-3-ил] -N'-фенилмочевина
К раствору Промежуточного соединения 44 (0,091 г) в ацетонитриле (4 мл) добавляли фенилизоцианат (0,23 мл), а образовавшуюся смесь
перемешивали при температуре 23oC в течение 30 мин.
Указанную смесь концентрировали в вакууме, а выделенный остаток очищали с помощью импульсной хроматографии (элюция ДХМ), после чего
растирали с петролом с выходом вышепоименованного соединения (0,074
г) в виде белого твердого вещества.
Т.Ж.X. ЦГ-ЭА (в соотношении 1:1): Rf 0,37.
ИК: 3327 (N-H) 7 1695-1660 (С=O) см-1.
Соединение из Примера 14 разделяли на энантиомеры (Изомер 1 и Изомер 2) с помощью хиральной HPLC (с использованием колонки Pirkle D-DNBPG 25х2,4 см; скорость потока 1,0 мл/мин; 235 нм (детектор УФ); элюция смесью ДХМ и ИПС в объемном соотношении 93:7).
Изомер 1 (0,048 г) в виде белого твердого вещества;
HPLC: время удержания 4,4 мин, 100%-ное преобладание энантиомера. ИК
(nujol): 3327 (NH); 1695-1660 (СО) см-1.
Изомер 2 (0,045 г) в виде белого твердого вещества;
HPLC: время удержания 6,0 мин, 96%-ное преобладание энантиомера.
α D = +31,3. ИК (nujol): 3327 (NH); 1695-1660 (CO) см-1
Пример 15
N-[1-(3-Метил-1-бутил)-2,
4-диоксо-5-[2-(4-морфолино)этил] -2,3,4,5-тетрагидро-1Н-1,
5-бензидиазепин-3-ил]-N'-(4-хлорофенил) мочевина
К раствору Промежуточного соединения 47 (0,05 г) в ацетонитриле (2 мл) добавляли
4-хлорфенилизоцианат (0,019 мл). Образовавшуюся реакционную
смесь перемешивали при температуре 23oC в течение 30 мин, после чего выпаривали растворители в вакууме. Выделенный остаток
растирали с диэтиловым эфиром с выходом вышепоименованного
соединения в виде белого твердого вещества (0,046 г).
Т.пл. 210-212oC. Т.Ж.X. ЭА-MeOH (в соотношении 95:5): Rf 0,53.
ИК: 1693-1641 (С=0) см-1.
Пример 16
N-[2,4-Диоксо-1-(3-Метил-1-бутил)-5-[2-(4-морфолино)этил] - 2,3,4,5-тетрагидро-1Н-1,
5-бензодиазепин-3-ил]-N'-(4-трифторметил) фенилмочевина
К
раствору Промежуточного соединения 47 (0,090 г) в сухом ацетонитриле (2 мл) добавляли 4-трифторметилфенилизоцианат (0,021 мл).
Отфильтровывали образовавшееся твердое вещество, промывали его диэтиловым
эфиром с выходом вышепоименованного соединения (0,06 г) в виде белого твердого вещества.
Т.пл. 208-210oC. Т.Ж.X. ЭА-MeOH (в соотношении 19:1): Rf 0,65.
Пример 17
N-{ 2,4-Диоксо-1-[(гексаметиленимино)этил]-5-фенил-2,3,4,5- тетрагидро-1H-1,
5-бензодиазепин-3-ил]-N'-(3-толил)мочевина
К раствору Промежуточного соединения 50 (0,
154 г) в сухом ацетонитриле (7 мл) добавляли 3-толилизоцианат (0,040 мл). Указанную реакционную смесь
перемешивали при температуре 23oC в течение 10 мин, после чего отфильтровывали
образовавшееся твердое вещество и высушивали его с выходом вышепоименованного соединения в виде белого
твердого вещества (0,130 г).
Т.пл. 120-101oC. Т.Ж.X. ЭА-MeOH (в соотношении 95:5): Rf 0,53.
ИК: 1703, 1643 (С=0) см-1.
Пример 18
Гидрохлорид (-){1-(1-адамантилметил)-2,
4-диоксо-5-[2-(4- морфолино)этил] -2,3,4,5-тетрагидро-1Н-1,5-бензодиазепин-3-ил} - N'-фенилмочевины
Изомер 1 из Примера
11 (0,089 г) растворяли в метаноле (15 мл), предварительно насыщенном
газообразной соляной кислотой, а образовавшуюся смесь перемешивали при температуре 0oC в течение 2 ч. Указанную смесь
концентрировали в вакууме и выпаривали вместе с дихлорметаном и
диэтиловым эфиром. Полученный остаток растирали с диэтиловым эфиром с выходом вышепоименованного соединения в виде белого твердого
вещества (0,065 г).
Т.пл. 260-263oC. Т.Ж.X. ЭА-MeOH (в соотношении 9:1): Rf 0,66.
ИК (CHCl3): 3300-2500 (NH); 1701, 1666 (C=O); 1599 (C=C) см-1.
1H-ЯМР: 13,2 (b); 7,56 (m); 7,45-7,20 (m); 7,06 (m); 6,44 (bd); 5,03 (d); 4,51 (m); 4,36 (d); 4,20 (m); 3,98 (m); 3,52 (m); 3,22 (d); 3,01 (m); 1,85 (m); 1,70-1,10 (m).
Примеры фармацевтического
использования
Капсулы или таблетки - мг/единицу дозы
Активный ингредиент - 0,1
Полиэтиленгликоль - 15,0
Лактоза - 52,4
Крахмал - 30,0
Стеарат
магния - 0,5
Диоксид силикона - 1,0
Лаурил сульфат натрия - 1,0
Итого - 100,0
Активный ингредиент
диспергируют в приемлемом растворителе (например, этаноле) вместе
с полиэтиленгликолем. Удаляют использованный растворитель. Полученный таким образом порошок смешивают с другими эксципиентами.
Указанная смесь может быть использована для наполнения желатиновых капсул
или прессоваться с помощью соответствующих прессов. Используя традиционный метод, полученные таблетки можно покрывать
оболочкой.
Активный ингредиент - 0,1
Повидон - 15,4
Лактоза - 74,0
Гидрированные растительные масла - 3,0
Диоксид силикона - 1,0
Лаурил
сульфат натрия - 1,5
Кросповидон - 5,0
Итого - 100,0
Активный ингредиент диспергируют в приемлемом растворителе (например, этаноле) вместе с повидоном. Образовавшимся
раствором опрыскивают лактозу, после чего удаляют использованный растворитель.
Полученный таким образом порошок смешивают с другими эксципиентами. Указанная смесь может быть использована для
наполнения желатиновых капсул или прессоваться с помощью соответствующих прессов.
Используя традиционный метод, полученные таблетки можно покрывать оболочкой.
Жидкость для
перорального введения
Активный ингредиент - 70-100 мкг/дозу
Этанол
- 5-15%
Сахаринат натрия - 0,1-1%
Пропилен гликоль - 10-100%
Очищенная вода - до
100%
Упаковка: пластиковая или стеклянная бутылочка, либо другая приемлемая
упаковка.
Состав для инъекций
Активный ингредиент - 0,1-100 мкг
Фосфат натрия
- 1,5 мг/мл
NaOH - до желаемого pH (3-9)
Пропилен гликоль - 10-500
мг/мл
Вода для инъекции - До 0,5-10 мл
Упаковка: стеклянная (ампулы) с притертой пробкой
(пузырьки, шприцы), запаянная пластиковая или металлическая (только пузырьки), либо другая
приемлемая упаковка. В верхнюю часть контейнера может быть введен инертный газ (например, азот).
Связывание ХЦК-рецептора
Для определения степени сродства соединений,
предусмотренных настоящим изобретением, по отношению к рецепторам ХЦК-А (тест на поджелудочной железе) и
рецепторам ХЦК-Б (тест на коре полушарий головного мозга морских свинок) использовали метод,
описанный в [5]. Величины pKi для следующих соединений приведены в таблице.
Предусмотренные настоящим изобретением соединения практически нетоксичны при использовании в фармацевтически приемлемых дозах. Так, не было обнаружено каких-либо нежелательных эффектов при пероральном введении мышам и крысам свободного основания соединения из Примера 18 в дозах, обеспечивающих анксиолитическую активность.
Реферат
Описываются новые производные 1, 5-бензодиазепина общей формулы (I) и их фармацевтически приемлемые соли, где R1 - фенильная или C1-6-алкильная группа, причем указанная алкильная группа может быть замещена С3-7-циклоалкильной или С7-11-циклоалкильной (мостиковой) группой; R2 обозначает фенильную группу, возможно замещенную заместителями, выбранными из числа следующих: атом галогена, С1-4-алкил, трифторметил или (СH2)n R4, где R4 является гидрокси или С1-4-алкоксигруппой, а n равно 0; R3 представляет собой группу AlkNR8R9, где Alk соответствует прямой С2-6-алкиленовой цепи, возможно замещенной гидроксильной группой; R8 и R9 вместе с атомом азота, к которому они присоединены, образуют 5-7 - членное насыщенное гетероциклическое кольцо, возможно содержащее дополнительный гетероатом, выбранный из числа следующих: атом кислорода или азота; R10 является атомом галогена; Х соответствует остатку NH; m равно 0 или 1. Вышеуказанные соединения являются антагонистами гастрина и ХЦК. 2 с. и 10 з.п. ф-лы, 1 табл.
Формула

и их фармацевтически приемлемые соли, где R1 представляет собой фенильную или С1-6-алкильную группу, причем указанная алкильная группа может быть замещена С3-7-циклоалкильной или С7-11-циклоалкильной мостиковой группой; R2 обозначает фенильную группу, возможно замещенную заместителями, выбранными из числа следующих: атом галогена, С1-4-алкил, трифторметил или (CH2)nR4, где R4 является гидрокси или С1-4-алкоксигруппой, а n равно 0; R3 представляет собой группу AlkNR8R9, где Аlk соответствует прямой С2-6-алкиленовой цепи, возможно замещенной гидроксильной группой; R8 и R9 независимо друг от друга обозначают атом водорода или С1-4-алкил, или R8 и R9 вместе с атомом азота, к которому они присоединены, образуют 5-7-членное насыщенное гетероциклическое кольцо, возможно содержащее дополнительный гетероатом, выбранный из числа следующих: атом кислорода или азота; R10 является атомом водорода или атомом галогена; X соответствует остатку NH; m равно 0 или 1.
Комментарии