Способы получения производных хиназолинона - RU2520098C2
Код документа: RU2520098C2
Описание
[001] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №61/075952, поданной 26 июня 2008 года, содержание которой включено в настоящее описание во всей полноте посредством ссылки.
[002] Настоящее изобретение относится к способам получения производных хиназолинона, подходящих для регулирования экспрессии аполипопротеина А-1 (АпоА-I) и для лечения и профилактики сердечно-сосудистых заболеваний и связанных с ними болезненных состояний, таких как, например, атеросклероз.
[003] Эпидемиологические данные показывают наличие обратной зависимости между содержанием циркулирующего в крови холестерина, транспортируемого липопротеинами высокой плотности (ЛПВП-Х), и частотой клинических проявлений атеросклероза. Полагают, что рост содержания ЛПВП-Х в сыворотке крови на каждый 1 мг/дл вызывает уменьшение риска сердечно-сосудистых заболеваний на 2-3%; уменьшение содержания ЛПНП-Х на 1% уменьшает риск сердечно-сосудистых заболеваний на 2% (Gordon el al. (1997) Am. J. Med. 62, 707-714). Экспериментальные данные также подтверждают защитное действие ЛПВП-Х в отношении сердечно-сосудистых заболеваний. Например, у субъектов с низким содержанием ЛПВП-Х введение гемфиброзила приводило к 6% увеличению содержания ЛПВП-Х и соответствующему уменьшению на 22% риска ишемической болезни сердца (ИБС) (Rubins et al. (1999) N. Engl. J. Med. 341, 410-418). Исследования наследственных нарушений, связанных с низким содержанием ЛПВП-Х вследствие пониженной экспрессии АпоА-I, также показывают связь между повышенным риском заболевания ИБС и низким содержанием ЛПВП-Х.
[004] ЛПВП-Х проявляет свое антиатерогенное действие, по всей видимости, за счет своей роли посредника в обратном транспорте холестерина (ОТХ), в результате которого холестерин привлекается из периферических тканей и транспортируется в печень. Кроме того, ЛПВП-Х также обладает плейотропными биологическими свойствами, которые вносят вклад в его антиатерогенное действие, в частности противовоспалительную, антиоксидантную и противотромботическую активность. ЛПВП-Х существует в двух основных формах, одна из которых содержит как аполипопротеин A-I (АпоА-I), так и аполипопротеин A-II (АпоА-II), а другая содержит АпоА-I без АпоА-II (Schultz et al. (1993) Nature 365, 762-764). Кардиозащитное действие ЛПВП-Х главным образом, но не исключительно, приписывают АпоА-I.
[005] Клинические и экспериментальные данные позволяют предположить, что выработка АпоА-I является важным определяющим фактором содержания ЛПВП-Х в кровотоке. Например, люди с наследственной гиперальфалипопротеинемией (увеличенным содержанием АпоА-I), по всей видимости, защищены от атеросклероза, тогда как люди с недостаточным содержанием АпоА-I (гипоальфалипопротеинемией) проявляют повышенную склонность к сердечно-сосудистым заболеваниям. Кроме того, различные экспериментальные воздействия, направленные на увеличение выработки АпоА-I, связывают с пониженной атерогенностью. Например, человеческий АпоА-I проявляет защитное действие в моделях на трансгенных животных (Shah et al. (1998) Circulation 97, 780-785; Rubin et al. (1991) Nature 353, 265-267), а лечение с помощью АроА- IMilano предотвращает атеросклеротические поражения и приводит к уменьшению атеросклеротических бляшек у пациентов-людей (Nissen et al. (2003) JAMA 290, 2292-2300). Дополнительные направления исследований показывают, что АпоА-I вносит вклад в улучшение обратного транспорта холестерина, уменьшение оксидативного стресса, увеличение активности параоксоназы, увеличение антикоагулянтной активности и усиление противовоспалительной активности (Andersson (1997) Curr. Opin. Lipidol. 8, 225-228). Соответственно, АпоА-I является перспективной мишенью для терапевтического воздействия.
[006] Доступные в настоящее время терапевтические агенты, увеличивающие концентрацию АпоА-I в плазме, например рекомбинантный АпоА-I или пептиды, имитирующие АпоА-I, обладают потенциальными недостатками в отношении, например, стабильности при хранении, доставки активного вещества и периода полувыведения из организма. Таким образом, небольшие молекулярные соединения, обеспечивающие повышающую регуляцию выработки эндогенного АпоА-I, такие как, например, положительные регуляторы экспрессии АпоА-I, могут быть перспективны в качестве новых терапевтических агентов для лечения сердечно-сосудистых заболеваний.
[007] Способы согласно настоящему изобретению обеспечивают улучшенные процедуры получения положительных регуляторов экспрессии АпоА-I. В случае предложенных соединений Формул I, VI и VIII алкилирование исходного фенола этиленкарбонатом, по сравнению с алкилирующими агентами согласно известным способам, является более эффективным и, таким образом, менее дорогостоящим в масштабах производства. Методики проведения сочетания согласно настоящему изобретению, приводящие к получению хиназолинонов, обеспечивают пониженное содержание примесей и увеличенный выход конечных продуктов.
ОПРЕДЕЛЕНИЯ
[008] Следует отметить, что используемые в данном описании и пунктах прилагаемой формулы изобретения формы единственного числа включают указанные объекты во множественном числе, если из контекста явным образом не следует иное. Так, например, ссылка на способ, содержащий «соединение», включает смесь двух или более соединений. Также следует отметить, что термин «или», как правило, используют в значении, включающем «и/или», если из контекста явным образом не следует иное. Если не указано иное, химические группы относятся к соответствующим незамещенным и замещенным формам.
[009] Термины «соединение Формулы I», «соединение Формулы VI» и «соединение Формулы VII» включают любые стереоизомеры, таутомеры и/или фармацевтически приемлемые соли, указанные в настоящем описании. Соединения Формулы I, Формулы VI и Формулы VIII также включают кристаллические и аморфные формы данных соединений, включая, например, полиморфные модификации, псевдополиморфные модификации, сольваты, гидраты, несольватированные полиморфные модификации (включая ангидриды), конформационные полиморфные модификации и аморфные формы соединений, а также их смеси. «Кристаллическая форма», «полиморфная модификация» и «новая форма» в настоящем описании могут быть использованы взаимозаменяемо и включают все кристаллические и аморфные формы соединения, включая, например, полиморфные модификации, псевдополиморфные модификации, сольваты, гидраты, несольватированные полиморфные модификации (включая ангидриды), конформационные полиморфные модификации и аморфные формы, а также их смеси, если не указана конкретная кристаллическая или аморфная форма. Соединения Формулы I, Формулы VI и Формулы VIII также включают фармацевтически приемлемые формы указанных соединений, включая хелаты, нековалентные комплексы, пролекарства и их смеси.
[010] Как отмечено выше, соединения Формулы I, Формулы VI и Формулы VIII также включают пролекарства. В некоторых вариантах реализации «пролекарства» согласно настоящему изобретению включают любые соединения, которые превращаются в соединения Формулы I, Формулы VI и/или Формулы VIII при введении пациенту, например, в ходе метаболического превращения пролекарства. Примеры пролекарств включают производные функциональных групп, таких как карбоксильные группы, в соединениях Формулы I, Формулы VI и/или Формулы VIII. Примеры пролекарств по карбоксильной группе включают, но не ограничиваются ими, эфиры карбоновых кислот, такие как алкиловые эфиры, гидроксиалкиловые эфиры, арилалкиловые эфиры и арилоксиалкиловые эфиры.
[011] «Сольват» образуется при взаимодействии растворителя и соединения. Термины «соединение Формулы I», «соединение Формулы VI» и «соединение Формулы VIII» включают сольваты соединений. Аналогичным образом, «соли» включают сольваты солей. Подходящие сольваты представляют собой фармацевтически приемлемые сольваты, такие как гидраты, включая моногидраты и гемигидраты.
[012] «Хелат» образуется при координации соединения с ионом металла в двух (или более) точках. Термин «соединение» включает хелаты соединений. Аналогичным образом, «соли» включают хелаты солей.
[013] «Нековалентный комплекс» образуется за счет взаимодействия соединения с другой молекулой, при котором между соединением и молекулой не образуется ковалентной связи. Например, комплексообразование может происходить за счет ван-дер-ваальсовых взаимодействий, водородных связей и электростатических взаимодействий (также называемых ионным связыванием). Подобные нековалентные комплексы включены в термин «соединение».
[014] Соединения, предложенные в настоящем изобретении, могут существовать в виде таутомеров, при этом настоящее изобретение включает обе таутомерные формы, даже если изображена лишь одна таутомерная структура. Например, представляется очевидным, что любое упоминание соединения А ниже включает таутомерную структуру В, и наоборот, а также их смесь.

[015] Используемые в настоящем описании термины имеют значения, приведенные в публикации патента США №2006/0205767 в пп.3-7, содержание которого включено в настоящее описание посредством ссылки. Термин «радикал», используемый в данных определениях, относится к группе-заместителю или к изменяемым группам.
[016] Кроме того, термин «имидо» относится к группе, обладающей структурой -C(O)NC(O)-R2, где R2 может быть выбран из алкила, алкенила, алкинила, арила, арилалкила, циклоалкила, галоалкила, гетероарила и гетероциклила.
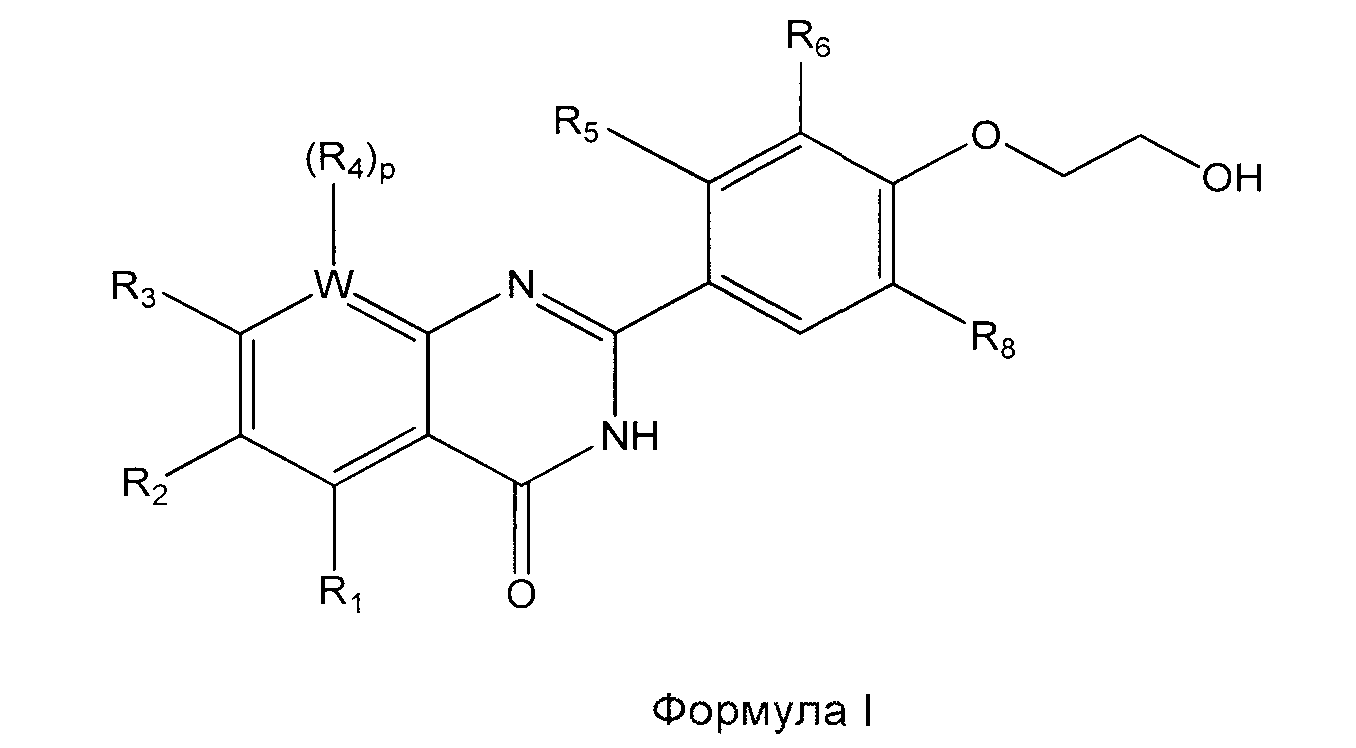
[017] В одном из вариантов реализации изобретения предложен способ получения соединения Формулы I:
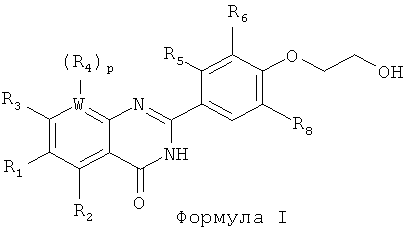
и сольватов, гидратов, таутомеров и фармацевтически приемлемых солей указанного соединения, где:
каждый из R1, R2, R3 и R4 независимо выбран из алкокси, алкила, амидо, арилокси, циклоалкила, галогена, гетероциклила, водорода и нитро;
R6 выбран из алкила, алкокси и галогена;
R5 представляет собой водород, или R5 и R6 совместно с атомами углерода, к которым они присоединены, образуют кольцо, выбранное из арила, циклоалкила и гетероциклила;
R8 выбран из алкила, алкокси, галогена и водорода;
W представляет собой С или N, где в случае если W представляет собой N, р равно 0, а в случае если W представляет собой С, р равно 1;
включающий:
a) взаимодействие альдегида Формулы II:

где R5, R6 и R8 определены выше, с этиленкарбонатом с образованием соединения Формулы III:

и
b) взаимодействие соединения Формулы III с соединением Формулы IV:

где R1, R2, R3 и R4 определены выше, с образованием соединения Формулы I.
[018] В одном из вариантов реализации каждый из R1 и R3 может быть независимо выбран из алкокси, алкила, галогена и водорода. В другом варианте реализации каждый из R1 и R3 может быть независимо выбран из хлора, водорода, метокси и метила. В другом варианте реализации каждый из R1 и R3 может представлять собой метокси.
[019] В одном из вариантов реализации R2 может быть выбран из брома, водорода, метокси и метиламидо. В другом варианте реализации R2 может представлять собой водород. В одном из вариантов реализации W может представлять собой N. В другом варианте реализации W может представлять собой С, a R4 может представлять собой водород.
[020] В одном из вариантов реализации R6 может быть выбран из хлора, метокси и метила. В другом варианте реализации R6 может представлять собой метил. В другом варианте реализации каждый из R6 и R8 независимо может быть выбран из алкила и галогена. В одном из вариантов реализации R8 может быть выбран из хлора, водорода, метокси и метила. В другом варианте реализации каждый из R6 и R8 может представлять собой метил.
[021] В одном из вариантов реализации соединение Формулы I может быть выбрано из:
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-она;
2-(3-хлор-4-(2-гидроксиэтокси)фенил)-5,7-диметоксихиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3-метоксифенил)-5,7-диметоксихиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметоксихиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-6,7-диметоксихиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметоксипиридо[2,3-d] пиримидин-4(3Н)-она;
N-(2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)4-оксо-3,4-дигидрохиназолин-6-ил)ацетамида;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметилхиназолин-4(3Н)-она;
5,7-дихлор-2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-6-метоксихиназолин-4(3H)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5-метоксихиназолин-4(3Н)-она;
6-бром-2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-она; и
2-(4-(2-гидроксиэтокси)-3-метилфенил)-5,7-диметоксихиназолин-4(3Н)-она,
и сольватов, гидратов, таутомеров и фармацевтически приемлемых солей указанных соединений.
[022] В другом варианте реализации соединение Формулы I представляет собой 2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметоксихиназолин-4(3Н)-он или сольват, гидрат, таутомер или фармацевтически приемлемую соль указанного соединения.
[023] В одном из вариантов реализации стадию проведения взаимодействия можно осуществлять в больших масштабах. В одном из вариантов реализации «большие масштабы» относятся к применению по меньшей мере 50 граммов исходного вещества, промежуточных веществ или реагентов, например, применению по меньшей мере 100 граммов, по меньшей мере 500 граммов, по меньшей мере 1 кг, по меньшей мере 10 кг, по меньшей мере 25 кг, по меньшей мере 50 кг, по меньшей мере 100 кг, по меньшей мере 250 кг или по меньшей мере 500 кг.
[024] В одном из вариантов реализации альдегид Формулы II:
может быть объединен с этиленкарбонатом в растворителе, таком как диметилформамид, дихлорметан, изопропанол, метанол, тетрагидрофуран, толуол, ксилол и вода, и перемешан при повышенной температуре, такой как 110°С, с образованием алкилированного соединения Формулы III. Данное соединение может быть очищено посредством кристаллизации из, например, смеси дихлорметан/гептан. Применение этиленкарбоната обеспечивает улучшенный контроль за протеканием процесса алкилирования и является значительно более экономически эффективным по сравнению с другими известными способами.
[025] Соединение Формулы III затем может быть объединено с соединением Формулы IV в N,N-диметилацетамиде (DMAC). Другие подходящие растворители включают ацетонитрил, бензол и метанол.
[026] Бисульфит натрия затем может быть добавлен по частям, таким как одна третья часть, при нагревании, например, при примерно 115°С. Кислоту, такую как моногидрат п-толуолсульфокислоты, можно добавить с первой порцией бисульфита натрия. Реакционную смесь можно перемешивать в течение по меньшей мере примерно 90 минут, например 90-105 минут, между добавлениями порций. Постепенное добавление порций в течение по меньшей мере 4 часов значительно уменьшает количество примесей, присутствующих в конечном соединении Формулы I, некоторые из которых в противном случае было бы сложно удалить на стадии очистки.
[027] После завершения реакции реакционную смесь можно охладить и полученный продукт можно перекристаллизовать из, например, смеси DMAC/гептан. Альтернативно, неочищенный продукт можно растереть с ацетоном. Согласно другому варианту реализации первую стадию очистки можно не проводить. После первой стадии очистки продукт может быть перекристаллизован из смеси этанол/вода с получением соединения Формулы I.
[028] Согласно другому варианту реализации соединение Формулы I может быть обработано изоцианатом Формулы V
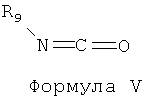
и основанием, таким как триэтиламин или основание Хюнига, с образованием карбамата Формулы VI.

[029] В одном из вариантов реализации R9 может быть выбран из алкила, арила, циклоалкила, гетероарила и гетероциклила. В другом варианте реализации R9 может представлять собой арил, замещенный одной или более группами, выбранными из алкокси, алкила и галогена. Согласно другому варианту реализации соединение Формулы VI представляет собой 2-(4-(5,7-диметокси-4-оксо-3,4-дигидрохиназолин-2-ил)-2,6-диметилфенокси)этилциклогексилкарбамат или сольват, гидрат, таутомер или фармацевтически приемлемую соль указанного соединения.
[030] В другом варианте реализации соединение Формулы I может быть обработано реагентом с образованием уходящей группы R10, показанной в Формуле VII. Уходящая группа R10 может быть выбрана из галогена, сулъфонила и фосфония, таких как хлорид, метансульфонил, п-толуолсульфонил и трифенилфосфоний. Реагент может быть выбран из тионилхлорида, метансульфонилхлорида, п-толуолсульфонилхлорида и PPh3/диэтилазодикарбоксилата.

[031] Соединение Формулы VII затем может быть обработано нуклеофильным реагентом, таким как алкоксид, амин или гетероцикл, содержащий по меньшей мере один атом азота, включая имиды, с образованием соединения Формулы VIII. Альтернативно, в случае если R10 представляет собой трифенилфосфоний, соединение Формулы VII может быть обработано in situ HN3 с последующим восстановлением реагентами, такими как Pd-С/Н2 с образованием промежуточного амина, который затем может быть обработан ацилирующим агентом с образованием соединения Формулы VIII, содержащего амидогруппу или имидогруппу. Как показано в Формуле VIII, R11 может быть выбран из алкокси, амидо, амино, имидо и гетероциклила. В одном из вариантов реализации R11 выбран из метокси, метиламино, морфолино, пиперазино и пиперидино.
[032] Согласно одному из вариантов реализации соединение Формулы I может быть обработано метансульфонилхлоридом и триэтиламином в дихлорметане с образованием соответствующего мезилата. Мезилат затем может быть обработан амином, таким как метиламин, в кипящем этаноле с образованием соединения Формулы VIII. Согласно другому варианту реализации соединение Формулы VIII представляет собой 2-(3,5-диметил-4-(2-(метиламино)этокси)фенил)-5,7-диметоксихиназолин-4(3Н)-он или сольват, гидрат, таутомер или фармацевтически приемлемую соль указанного соединения.
[033] Согласно другому варианту реализации соединение Формулы VIII может быть получено посредством взаимодействия альдегида Формулы II с этиленкарбонатом с образованием соединения Формулы III. Соединение Формулы III затем может взаимодействовать с реагентом с образованием уходящей группы R12 в соединении Формулы IX.

[034] В одном из вариантов реализации R12 может быть выбран из галогена, сульфонила и фосфония, таких как хлорид, метансульфонил, п-толуолсульфонил и трифенилфосфоний. Согласно другому варианту реализации изобретения реагент выбран из тионилхлорида, метансульфонилхлорида, п-толуолсульфонилхлорида и PPh3/диэтилазодикарбоксилата.
[035] Соединение Формулы IX может быть обработано нуклеофильным реагентом; таким как алкоксид, амин или гетероцикл, содержащий по меньшей мере один атом азота, включая имиды, с образованием соединения Формулы X. Альтернативно, в случае когда R12 представляет собой трифенилфосфоний, соединение Формулы VII может быть обработано in situ NH3 с последующим восстановлением реагентами, такими как Pd-C/H2 с образованием промежуточного амина, который может быть обработан ацилирующим агентом с образованием соединения Формулы VIII, содержащего амидогруппу или имидогруппу.

[036] Как показано в Формуле X, R13 может быть выбран из алкокси, амидо, амино, имидо и гетероциклила. В одном из вариантов реализации R13 выбран из метокси, метиламино, морфолино, пиперазино и пиперидино.
[037] Соединение Формулы Х затем может быть подвергнуто конденсации с соединением Формулы IV с образованием соединения Формулы VIII. В одном из вариантов реализации каждый из R6, и R8 представляет собой метил. Согласно другому варианту реализации каждый из R1 и R3 представляет собой водород. В другом варианте реализации соединение Формулы VIII представляет собой 2-(3,5-диметил-4-(2-морфолиноэтокси)фенил)хиназолин-4(3Н)-он или сольват, гидрат, таутомер или фармацевтически приемлемую соль указанного соединения.
ПРИМЕРЫ
[038] Изобретение далее проиллюстрировано следующими неограничивающими примерами, в которых следующие аббревиатуры имеют следующие значения. В случае если аббревиатура приведена без определения, данная аббревиатура имеет соответствующее общепринятое значение.
[039] Аббревиатуры, используемые в настоящем описании, обозначают следующие соединения, реагенты и заместители: ацетонитрил (MeCN); диизопропилэтиламин (DIPEA); N,N-диметилацетамид (DMAC); диметилформамид (DMF); этилацетат (EtOAc); метансульфонилангидрид (MS2O); метансульфонилхлорид (MsCl); п-толуолсульфокислота (п-TsOH) и триэтиламин (Et3N).


2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметоксихиназолин-4(3Н)-он (4)
[040] Исходный 4-гидрокси-3,5-диметилбензальдегид (1; 70 кг), К2СО3 (9,8 кг) и DMF (133 кг) смешали и перемешивали при 110°С в атмосфере азота. Этиленкарбонат (45,6 кг) в DMF (46 кг) добавили к смеси в течение 4 часов с использованием диафрагменного насоса. Реакционную смесь перемешивали при 110°С в течение 12 часов до тех пор, пока не осталось менее 5% исходного вещества 1. Реакционную смесь охладили до 25°С и добавили воду (1300 кг) с последующим добавлением смеси дихлорметана и гептана (3/2 по объему; 1300 кг). Смесь перемешивали в течение 30 минут. Органический слой отделили, водный слой экстрагировали смесью дихлорметана и гептана (3/2 по объему, 1300 кг). Объединенные органические слои промыли водным раствором гидроксида натрия (3 М, 460 кг) с последующим трехкратным промыванием водой (3×710 кг) и осушили над сульфатом натрия (60 кг). Дихлорметан удалили из осушенного органического слоя посредством перегонки при поддержании температуры ниже 40°С. Гептан (260 кг) и затравочные кристаллы добавили для инициирования кристаллизации и смесь перемешивали при 20°С в течение 2 часов. Смесь отфильтровали, промыли гептаном (60 кг) и осушили под вакуумом до достижения постоянной массы с получением промежуточного соединения 2 (71,3 кг, 78,8%).1Н-ЯМР (ДМСО-d6): δ 9,82 (1Н), 7,54 (2H), 4,96 (1Н), 3,85 (2H), 3,74 (2Н), 2,29 (6Н).
[041] Промежуточное соединение 2 (58,74 кг), N,N-диметилацетамид (280 кг) и исходное вещество 3 (56,00 кг) объединили и добавили моногидрат п-толуолсульфокислоты (5,90 кг) и 1/3 требуемого количества бисульфита натрия (24,1 кг). Смесь нагрели до 115°С и перемешивали в течение 90-105 минут перед добавлением второй порции 1/3 требуемого количества бисульфита натрия (24,1 кг). Оставшийся бисульфит натрия (24,1 кг) добавили через следующие 90-105 минут. Реакционную смесь перемешивали при 115°С до завершения реакции, определенного посредством ВЭЖХ (приблизительно в течение 1 часа, осталось менее 4% промежуточного соединения 2). Реакционную смесь охладили до 25°С и добавили в воду (1770 кг). Смесь перемешивали при 20°С в течение 6 часов для завершения кристаллизации. Неочищенное вещество отделили фильтрованием, промыли водой (234 кг) и осушили в вакууме до постоянного веса. Неочищенное вещество растворили в N,N-диметилацетамиде (252 кг) при 80°С до полного растворения вещества. Раствор охладили до 60°С и медленно в течение 1 часа добавили гептан (918 кг), поддерживая температуру выше 35°С. Раствор охладили до 35°С и перемешивали при 35°С в течение как минимум 1 часа. Твердое вещество отделили фильтрованием, промыли гептаном (250 кг) и осушили до постоянной массы в вакууме. Выход: 92,5%, чистота 98,6%. Сухое твердое вещество (83,1 кг) добавили к 1:1 смеси этанола и воды (1/1 по объему, 1670 кг), смесь нагрели до примерно 84°С (кипячение с обратным холодильником) до полного перехода вещества в раствор. Раствор охладили до 70°С и отфильтровали, затем охладили до 30°С в течение 2 часов. Раствор охладили до 0°С. Смесь перемешивали при 0°С в течение 1 часа, перед отделением вещества посредством фильтрования, промыли смесью этанол/вода (1/1 по объему, 33 кг) и осушили в вакууме до постоянной массы. Вещество просеяли через сито 60 меш и получили 2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметилхиназолин-4(3Н)-он (4). Выход: 66,4 кг, 79,9%.
ПРИМЕР 2
Соединения, которые могут быть получены аналогично Примеру 1
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-он;
2-(3-хлор-4-(2-гидроксиэтокси)фенил)-5,7-диметилхиназолин-4(3Н)-он;
2-(4-(2-гидроксиэтокси)-3-метоксифенил)-5,7-диметилхиназолин-4(3Н)-он;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-6,7-диметоксихиназолин-4(3Н)-он;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметоксипиридо[2,3-d]пиримидин-4(3H)-OH;
N-(2-(4-(2-гидроксиэтокси)3,5-диметилфенил)4-оксо-3,4-дигидрохиназолин-6-ил)ацетамид;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметилхиназолин-4(3Н)-он;
5,7-дихлор-2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-он;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-6-метоксихиназолин-4(3Н)-он;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5-метоксихиназолин-4(3Н)-он;
6-бром-2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-он и
2-(4-(2-гидроксиэтокси)-3-метилфенил)-5,7-диметоксихиназолин-4(3Н)-он.
ПРИМЕР 3

2-(4-(5,7-диметокси-4-оксо-3,4-дигидрохиназолин-2-ил)-2,6-диметилфенокси)этил-4-фтор-фенилкарбамат(5)
[042] Смесь 4-фторфенилизоцианата (0,138 мл, 1,14 ммоль), Et3N (0,185 мл, 1,32 ммоль) и 4 (0,0700 г, 0,189 ммоль) в ТГФ (1,00 мл) нагревали с обратным холодильником в течение 8 часов. Смесь охладили, разбавили EtOAc (200 мл), промыли и разбавили водным NH4Cl (3×75 мл), солевым раствором (75 мл), осушили над Na2SO4, отфильтровали и концентрировали в вакууме. Остаток очистили на силикагеле (12 г, CH2Cl2/MeOH) и продукт подвергли лиофилизации из MeCN/H2O с образованием 2-(4-(5,7-диметокси-4-оксо-3,4-дигидрохиназолин-2-ил)-2,6-диметоксифенокси)этил-4-фторфенилкарбамата (5) (0,0710 г, 74%) в виде белого твердого вещества:1Н-ЯМР (300 МГц, ДМСО-d6) δ 11,83 (s, 1 Н), 9,78 (s, 1 H), 7,91 (s, 2H), 7,54-7,44 (m, 2H), 7,18-7,08 (m, 2H), 6,73 (d, J=2,31 Гц, 1Н), 6,51 (d, J=2,31 Гц, 1Н), 4,47-4,38 (m, 2H), 4,12-4,03 (m, 2H), 3,89 (s, 3Н), 3,84 (s, 3Н), 2,31 (s, 6H); MS (APCI) m/z 508 [C27H26FN3O6+Н]+.
ПРИМЕР 4
[043] 2-(4-(5,7-диметокси-4-оксо-3,4-дигидрохиназолин-2-ил)-2,6-диметилфенокси)-этилциклогексилкарбамат может быть получен с использованием способа, аналогичного Примеру 3.
ПРИМЕР 5
2-(3,5-диметил-4-(2-(метиламино)этокси)фенил)-5,7-диметокси-хиназолин-4(3H)-он (7)
[044] К смеси соединения 4 (2,00 г, 5,40 ммоль) и Et3M (0,977 мл, 7,02 ммоль) в CH2Cl2) (27,0 мл) при комнатной температуре медленно добавили MsCl (0,543 мл, 7,02 ммоль). Через 1 день к смеси добавили дополнительное количество Et3N (0,977 мл, 7,02 ммоль) и MsCl (0,543 мл, 7,02 ммоль) и смесь перемешивали в течение 2 часов, затем разбавили EtOAc (300 мл) и промыли 10% водной лимонной кислотой (3×75 мл), разбавили водным NaHCO3 (75 мл) и солевым раствором (75 мл). Нерастворимое твердое вещество собрали посредством фильтрования, получив мезилат 6 (0,890 г, 37%):1Н-ЯМР (300 МГц, ДМСО-d6) δ 11,84 (s, 1Н), 7,91 (s, 2H), 6,74 (d, J=2.32 Гц, 1Н), 6,52 (d, J=2.32 Гц, 1Н), 4,59-4,48 (m, 2H), 4,15-4,04 (m, 2H), 3,89 (s, 3Н), 3,84 (s, 3H), 3,25 (s, 3H), 2,32 (s, 6H).
[045] Мезилат (6) (0,200 г, 0,446 ммоль) и 33% CH3NH2 в EtOH (5,00 мл) нагревали с обратным холодильником в течение ночи. Растворитель удалили в вакууме, осадок очистили на силикагеле (12 г, CH2Cl2/СН3ОН) и продукт подвергли лиофилизации из MeCN/H2O с получением 2-(3,5-диметил-4-(2-(метиламино)этокси)фенил)-5,7-диметоксихиназолин-4(3Н)-она (7) (0,0968 г, 57%) в виде желтого твердого вещества:1Н-ЯМР (300 МГц, ДМСО-d6) δ 7,90 (s, 2H), 6,73 (d, J=2,29 Гц, 1Н), 6,52 (d, J=2.29 Гц, 1Н), 3,94-3,80 (m, 8H), 2,98 (t, J=5.46 Гц, 2H), 2,45 (s, 3Н), 2,33-2,28 (m, 8H); MS (APCI) m/z 384 [C21H25N3O4+Н]+.
ПРИМЕР 6
[046] 2-(3,5-диметил-4-(2-(метокси)этокси)фенил)-5,7-диметоксихиназолин-4(3Н)-он может быть получен с применением способа, аналогичного Примеру 5, где мезилат (6) обрабатывают NaOMe в МеОН или МеОН/К2СО3 вместо CH3NH2.
ПРИМЕР 7
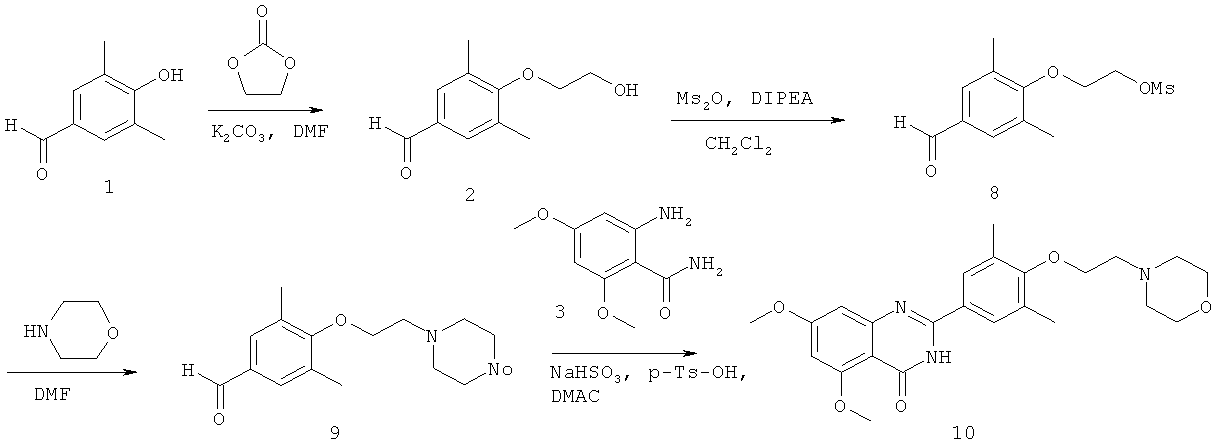
[047] Промежуточное соединение 2 получали из 1 в соответствии со способом согласно Примеру 1. К смеси 2 и диизопропилэтиламина в CH21Cl2 при 0°C добавляли Ms2O. Реакционную смесь перемешивали до завершения реакции, определяемого с помощью тонкослойной хроматографии (ТСХ). Реакционную смесь разбавляли этилацетатом и промывали охлажденным насыщенным NaHCO3 и солевым раствором. Органический слой осушали над Na2SO4, отфильтровывали и концентрировали с образованием мезилата 8.
[048] Смесь соединения 8 и морфолина в ДМФ нагревали до 50°С. Реакционную смесь перемешивали до завершения реакции, определяемого с помощью ТСХ. Реакционную смесь охлаждали до комнатной температуры и добавляли этилацетат. Смесь промывали водой, осушали над Na2SO4, фильтровали и концентрировали, получая неочищенное промежуточное соединение 9. Неочищенный продукт очищали с помощью колоночной хроматографии с образованием чистого промежуточного соединения 9.
[049] Исходное вещество 3 и промежуточное соединение 9 объединяли в DMAC с последующим добавлением п-TsOH и бисульфита натрия. Реакционную смесь нагревали до 115°С. Реакционную смесь перемешивали до завершения реакции, определяемого с помощью ТСХ. Реакционную смесь охлаждали до комнатной температуры и добавляли воду. Смесь трижды экстрагировали CH2Cl2. Объединенные органические слои промывали водой, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищают посредством хроматографии с получением 2-(3,5-диметил-4-(2-(морфолино)этокси)фенил)-5,7-диметоксихиназолин-4(3Н)-она (1.0).
[050] Содержание всех документов, на которые сделаны ссылки в настоящей заявке, включено в настоящее описание во всей полноте посредством ссылки. Другие варианты реализации настоящего изобретения будут очевидны для специалиста в данной области техники после ознакомления с настоящим описанием и примерами практической реализации изобретения, предложенного в настоящей заявке. Предполагается, что описание изобретения и примеры являются лишь иллюстративными, тогда как объем и сущность изобретения определяются приведенной далее формулой изобретения.
Реферат
Изобретение относится к улучшенному способу получения производных хиназолинов структурной формулы I:где значения радикалов R-R, R, W, Р приведены в формуле изобретения. Способ включает взаимодействие альдегида формулы II:с этиленкарбонатом с образованием соединения формулы III:и взаимодействие соединения формулы III с соединением формулы IV:с образованием соединения формулы I. Изобретение также относится к способам получения производных соединения формулы I структурных формул VI и VIII. Указанные способы позволяют получать продукты с пониженным содержанием примесей и повышенным выходом. 4 н. и 19 з.п. ф-лы, 7 пр.
Формула

и таутомеров и фармацевтически приемлемых солей указанного соединения, где:
каждый из R1, R2, R3 и R4 независимо выбран из алкокси, алкила, амидо, арилокси, циклоалкила, галогена, гетероциклила, водорода и нитро;
R6 выбран из алкила, алкокси и галогена;
R5 представляет собой водород или R5 и R6 совместно с атомами углерода, к которым они присоединены, могут образовывать кольцо, выбранное из арила, циклоалкила и гетероциклила;
R8 выбран из алкила, алкокси и галогена;
W представляет собой С или N, где в случае если W представляет собой N, р равно 0, а в случае если W представляет собой С, р равно 1;
включающий
а) взаимодействие альдегида Формулы II:

Формула II
где R5, R6 и R8 определены выше, с этиленкарбонатом с образованием соединения Формулы III:

и
b) взаимодействие соединения Формулы III с соединением Формулы IV

где R1, R2, R3 и R4 определены выше, с образованием соединения Формулы I.
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметоксихиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-6,7-диметоксихиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметоксипиридо[2,3-d]пиримидин-4(3Н)-она;
N-(2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-4-оксо-3,4-дигидрохиназолин-6-ил)ацетамида;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5,7-диметилхиназолин-4(3Н)-она;
5,7-дихлор-2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-6-метоксихиназолин-4(3Н)-она;
2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)-5-метоксихиназолин-4(3Н)-она; и
6-бром-2-(4-(2-гидроксиэтокси)-3,5-диметилфенил)хиназолин-4(3Н)-она
и таутомеров и фармацевтически приемлемых солей указанных соединений.

Формула VI
и таутомеров и фармацевтически приемлемых солей указанного соединения,
где
каждый из R1, R2, R3 и R4 независимо выбран из алкокси, алкила, амидо, арилокси, циклоалкила, галогена, гетероциклила, водорода и нитро;
R6 выбран из алкила, алкокси и галогена;
R5 представляет собой водород или R5 и R6 совместно с атомами углерода, к которым они присоединены, могут образовывать кольцо, выбранное из арила, циклоалкила и гетероциклила;
R8 выбран из алкила, алкокси и галогена;
W представляет собой С или N, где в случае если W представляет собой N, р равно 0, а в случае если W представляет собой С, р равно 1;
R9 выбран из алкила, арила, циклоалкила, гетероарила и гетероциклила,
включающий взаимодействие соединения формулы I

и таутомеров и фармацевтически приемлемых солей указанного соединения, где:
R1, R2, R3, R4, R5, R6, R8, W и р определены выше,
с изоцианатом формулы V:

где R9 выбран из группы, включающей алкил, арил, циклоалкил, гетероарил и гетероциклил,
с образованием соединения формулы VI.

и таутомеров, и фармацевтически приемлемых солей указанного соединения,
в котором:
каждый из R1, R2, R3 и R4 независимо выбран из алкокси, алкила, амидо, арилокси, циклоалкила, галогена, гетероциклила, водорода и нитро;
R6 выбран из алкила, алкокси и галогена;
R5 представляет собой водород или R5 и R6 совместно с атомами углерода, к которым они присоединены, могут образовывать кольцо, выбранное из арила, циклоалкила и гетероциклила;
R8 выбран из алкила, алкокси и галогена;
W представляет собой С или N, где в случае если W представляет собой N, р равно 0, а в случае если W представляет собой С, р равно 1;
R11 выбран из алкокси, амидо, амино, имидо и гетероциклила;
включающий
а) взаимодействие соединения Формулы I
и таутомеров и фармацевтически приемлемых солей указанного соединения, где
R1, R2, R3, R4, R5, R6, R8, W и р определены выше
с реагентом для получения уходящей группы R10 с образованием соединения Формулы VII:

и таутомеров и фармацевтически приемлемых солей указанного соединения,
где R10 выбран из галогена, сульфонила и фосфония; и
R1, R2, R3, R4, R5, R6, R8, W и р определены выше; и
b) взаимодействие соединения Формулы VII с нуклеофильным реагентом с образованием соединения Формулы VIII.
метансульфонила, п-толуолсульфонила и трифенилфосфония.

и таутомеров и фармацевтически приемлемых солей указанного соединения, где:
каждый из R1, R2, R3 и R4 независимо выбран из алкокси, алкила, амидо, арилокси, циклоалкила, галогена, гетероциклила, водорода и нитро;
R6 выбран из алкила, алкокси и галогена;
R5 представляет собой водород или R5 и R6 совместно с атомами углерода, к которым они присоединены, образуют кольцо, выбранное из арила, циклоалкила и гетероциклила;
R8 выбран из алкокси, алкила и галогена;
R11 выбран из алкокси, амидо, амино, имидо и гетероциклила;
W представляет собой С или N, где в случае если W представляет собой N, р равно 0, а в случае если W представляет собой С, р равно 1;
включающий
а) взаимодействие альдегида Формулы II:

где R5, R6 и R8 определены выше, с этиленкарбонатом с образованием соединения Формулы III:

b) взаимодействие соединения Формулы III с реагентом для получения уходящей группы R12 с образованием соединения Формулы IX:

где R12 выбран из галогена, сульфонила и фосфония;
с) взаимодействие соединения Формулы VIII с нуклеофильным реагентом с образованием соединения Формулы X:
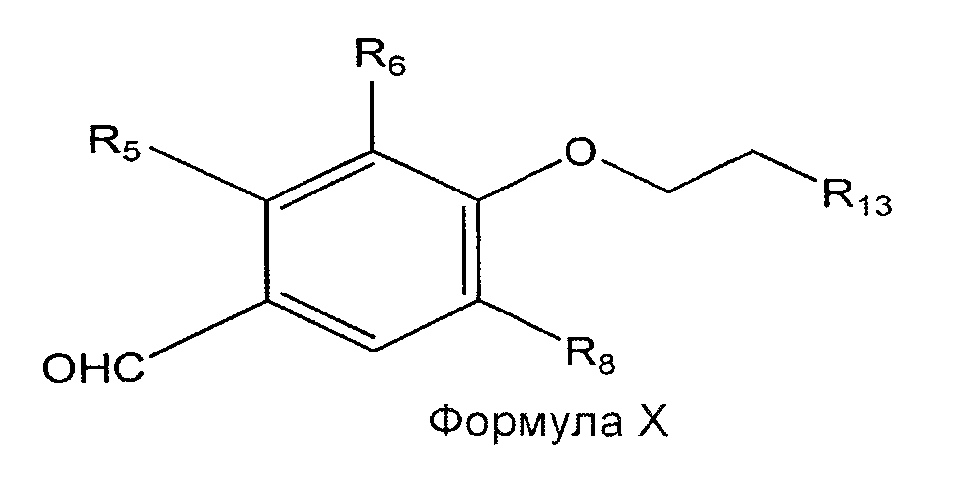
где R13 выбран из алкокси, амидо, амино, имидо и гетероциклила; и
d) взаимодействие соединения Формулы Х с соединением Формулы IV:

где R1, R2, R3 и R4 определены выше, с образованием соединения Формулы VIII.
Комментарии