Ингибитор сериновых протеаз и фармацевтическая композиция на его основе - RU2232760C2
Код документа: RU2232760C2
Описание
Изобретение относится к ингибитору сериновых протеаз, содержащему заместитель аргинина, связанный с эфиром, к фармацевтической композиции, содержащей ингибитор, а также к применению указанного ингибитора сериновых протеаз для производства лекарственного средства.
Сериновые протеазы являются ферментами, которые играют важную роль в системе свертывания крови. Помимо тромбина и фактора Ха другие примеры данной группы протеаз включают в себя факторы VIIa, IXa, XIa, XIIa и белок С.
Тромбин является конечной сериновой протеазой в каскаде реакций свертывания крови. Главная функция тромбина заключается в расщеплении фибриногена с образованием мономеров фибрина, которые поперечно связываются, чтобы образовать нерастворимый гель. Кроме того, тромбин регулирует свое собственное образование активацией на ранних стадиях каскада факторов V и VIII. Также он выполняет важные функции на клеточном уровне, где он действует на специфические рецепторы, вызывая агрегацию тромбоцитов, активацию эндотелиальных клеток и пролиферацию фибробластов. Таким образом, тромбину принадлежит главная регуляторная роль в гемостазе и образовании тромба. Так как ингибиторы тромбина могут иметь широкую область терапевтического применения, непрерывно предпринимаются попытки открыть новые ингибиторы сериновых протеаз. Трудность заключается в том, чтобы обнаружить соединение, свойства которого сочетают терапевтическую безопасность, избирательность, эффективность и доступность синтеза. В частности, биологическая доступность при пероральном способе введения может оказаться проблематичной для избирательных ингибиторов тромбина.
В международной заявке WO 97/16444 описаны ингибиторы тромбина с модификациями трипептидной последовательности фенилаланил-пролин-аргинин, в которой карбоксильная группа находится на аминоконце и аргинин замещен основным (аминоиминометил)фенилом или основной группой (аминоиминометил) пиридинила, связанных с аналогом пролина эфирной связью.
Цель данного изобретения заключается в обеспечении новыми доступными ингибиторами сериновых протеаз с подходящими фармакологическим и токсикологическим профилями и с достаточной эффективностью при терапевтическом применении.
Заявлено, что данный аспект предусматривает ингибитор сериновых протеаз и, в частности, ингибитор тромбина формулы (I),
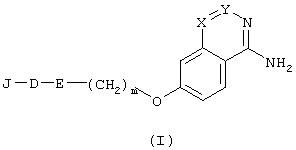
в которой
J является Н, R1, R1-O-C(O)-, R1-C(O)-, R1-SO2-, R3OOC-(CHR2)p-, (R2a, R2b)N-CO-(CHR2)p- или Het-CO-(CHR2)-p;
D является аминокислотой формулы -NH-CHR1-C(О)-, NR4-CH[(CH2)qC(O)OR1 -]-C(O)-, -NR4-CH[(CH2)qC(О)N(R2a,R2b)]-С(О)-, NR4-CH[(CH2)qC(O)Het]-C(O)-, D-1-Tiq, D-3-Tiq, D-Atc, Aic, D-1-Piq или D-3-Piq;
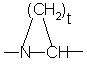
Е представляет собой -NR2-CH2- или фрагмент

необязательно замещенный (1-6С)алкилом, (1-6С)алкокси или бензилокси;
R1 выбран из (1-12С)алкила, (2-12С)алкенила, (2-12С)алкинила, (3-12С)циклоалкила и (3-12С)циклоалкил(1-6С)алкилена, группы которых необязательно могут быть замещены (3-12С)циклоалкилом, (1-6С)алкокси, оксо, ОН, СF3 или галогеном, и из (6-14С)арила, (7-15С)аралкила, (8-16С)аралкенила и (14-20С)(бисарил)алкила, где арильные группы могут быть замещены (1-6С)алкилом, (3-12С)циклоалкилом, (1-6С)алкокси, ОН, СF3 или галогеном;
каждый из R2, R25 и R2b, независимо выбран из Н, (1-8С)алкила, (2-8С)алкенила, (2-8С)алкинила, (3-8С)циклоалкила и (3-6С)циклоалкил(1-4С)алкилена, каждый из которых необязательно может быть замещен (3-6С)циклоалкилом, (1-6С)алкокси, СF3 или галогеном, и из (6-14С)арила и (7-15С)аралкила, где арильные группы могут быть замещены (1-6С)алкилом, (3-6С)циклоалкилом, (1-6С)алкокси, СF3 или галогеном;
R3 имеет значения, определямые для R2, или Het-(1-6С)алкил;
R4 представляет собой Н или (1-3С)алкил;
Х и Y представляют собой СН или N, при условии, что одновременно оба они не являются N.
Het означает 4-, 5- или 6-членный гетероцикл, содержащий один или более гетероатомов, выбранных из О, N и S;
m равно 1 или 2;
р равно 1, 2 или 3;
q равно 1, 2 или 3;
t равно 2, 3 или 4;
и его пролекарство;
и его фармацевтически приемлемые аддитивные соли и/или сольваты.
В частности, соединения согласно изобретению характеризуются улучшенной биодоступностью после перорального введения.
В предпочтительных направлениях данного изобретения соединения имеют формулу (I), в которой
J представляет собой Н, R1, R1-SO2-, R3OOC-(CHR2)p-, (R2a, R2b)N-СО-(CHR2)p- или Het-CO-(CHR2)p-; более предпочтительно представляет собой (3-12С)циклоалкил, необязательно замещенный (1-6С)алкокси; или
D представляет собой аминокислоту формулы -NH-CHR1-C(О)-, -NR4 -CH[(CH2)qC(O)OR1]-C(O)-, -NR4-CH[(CH2)qC(О)N(R2a, R2b)]-C(O)-, -NR4-CH[(СН2)q C(O)Het]-С(О)-; более предпочтительно, представляет собой аминокислоту формулы -NH-CHR1-C(О)- или L-аминокислоту формулы -NR4 -CH[(СН2)2C(О)N(R2a, R2b)]-С(О)-; или
Е представляет собой -N(3-6C)циклоалкил-СН2 или фрагмент

необязательно замещенный (1-6С)алкилом, (1-6С)алкокси или бензилокси; более предпочтительно не имеет замещения; или
R1 выбран из (1-12С)алкила, (3-12С)циклоалкила и (3-12С)циклоалкил(1-6С)алкилена, группы которого необязательно могут быть замещены (3-12С)циклоалкилом, (1-6С)алкокси или оксо, и из (6-14С)арила, (7-15С)аралкила и (14-20С)(бисарил)алкила, где арильные группы необязательно могут быть замещены (1-6С)алкилом, (3-12С)циклоалкилом, (1-6С)алкокси, ОН, СF3 или галогеном; более предпочтительно, выбран из (3-12С)циклоалкила и (3-12С)циклоалкил(1-6С)алкилена, группы которых необязательно могут быть замещены (3-12С)циклоалкилом или (1-6С)алкокси, и из (6-14С)арила, (7-15С)аралкила и (14-20С) (бисарил)алкила, где арильные группы необязательно могут быть замещены (1-6С)алкилом, (3-12С)циклоалкилом, (1-6С)алкокси или галогеном; или
R2 является Н; или
каждый из R2a и R2b независимо выбран из Н, (1-8С) алкила, (3-8С)циклоалкила и (3-6С)циклоалкил(1-4С)алкилена, каждый из которых необязательно замещен (3-6С)циклоалкилом или (1-6С)алкокси, и из (6-14С)арила и (7-15С)аралкила, где арильные группы необязательно замещены (1-6С)алкилом, (3-6С)циклоалкилом, (1-6С)алкокси, СF3 или галогеном; или
R3 выбран из Н, (1-8С)алкила, (3-8С)циклоалкила и (3-6С)циклоалкил(1-4С)алкилена, каждый из которых может быть замещен (3-6С)циклоалкилом или (1-6С)алкокси, и из (7-15С)аралкила, где арильные группы могут быть замещены (1-6С)алкилом, (3-6С)циклоалкилом, (1-6С)алкокси, СF3 или галогеном, и из Het-(1-6C)алкила; более предпочтительно, отбор производится из (1-8С)алкила и (3-8С)циклоалкила, каждый из которых необязательно может быть замещен (3-6С)циклоалкилом или (1-6С)алкокси, и из (7-15С)аралкила, где арильные группы необязательно могут быть замещены (1-6С)алкилом, (3-6С)циклоалкилом, (1-6С)алкокси, СF3 или галогеном, и из Het-(1-6С)алкила; или
R4 представляет собой Н или (1-3С)алкил; или
Х и Y являются СН; или
Het представляет собой 4-, 5- или 6-членный гетероцикл, содержащий один или более гетероатомов, выбранных из О, N и S; или
m равно 2; или
р равно 1; или
q равно 2; или
t равно 3 или 4; более предпочтительно равно 4.
В следующих предпочтительных аспектах D представляет собой аминокислоту формулы -NH-CHR1-C(О)- или глутамил [или его (1-6С)алкилэфир];
R1 выбирают из (3-12С)циклоалкила и (3-12С)циклоалкил(1-6С)алкилена, группы которых могут быть замещены (3-12С)циклоалкилом или (1-6С)алкокси, и из (6-14С)арила, (7-15С)аралкила и (14-20С)(бисарил)алкила, где арильные группы необязательно могут быть замещены (1-6С)алкилом, (3-12С)циклоалкилом, (1-6С)алкокси или галогеном; и
R3 выбирают из (1-8С)алкила и (3-8С)циклоалкила, каждый из которых может быть замещен (3-6С)циклоалкилом или (1-6С)алкокси, и из (7-15С)аралкила, где арильные группы могут быть замещены (1-6С)алкилом, (3-6С)циклоалкилом, (1-6С) алкокси, СF3 или галогеном, и из Het-(1-6C)алкила.
Особенно предпочтительными являются соединения, в которых J представляет собой -СН2СОО(1-6С)алкил, (3-8С)циклоалкил, -SO2 -10-камфору, -СН2СОNНфенил или -CH2 CONH(3-8C)циклоалкил. Другие весьма предпочтительные соединения являются соединениями, в которых D представляет собой D-циклогексилаланинил, D-фенилаланинил, D-дифенилаланинил или глутамил [или его (1-6С)алкилэфир]. Также особо предпочтительными являются соединения, где Е представляет собой фрагмент

где t равен 3 или 4.
При этом термины имеют следующий смысл:
(1-12С)алкил, (1-8С)алкил, (1-6С)алкил, (1-3С)алкил означают разветвленную или прямую алкильную группу, имеющую 1-12, 1-8, 1-6 и 1-3 атомов углерода, соответственно, например метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, гексил, октил и тому подобное.
(2-12С)алкенил и (2-8С)алкенил означают разветвленную или прямую алкенильную группу, имеющую 2-12 и 2-8 атомов углерода, соответственно, такую как этенил, 2-бутенил и так далее.
(2-12С)алкинил и (2-8С)алкинил означают разветвленную или прямую алкинильную группу, имеющую 2-12 и 2-8 атомов углерода, соответственно, такую как этинил, пропинил и так далее.
(1-6С)алкокси означает алкоксигруппу, имеющую 1-6 атомов углерода, при этом алкильный фрагмент имеет ранее определенное значение.
(3-12С)циклоалкил, (3-8С)циклоалкил, (3-6С)циклоалкил означают моно- или бициклоалкильную группу, имеющую 3-12, 3-8 и 3-6 атомов углерода, соответственно, подобную циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу, циклооктилу и так далее.
(1-6С)алкилен, (1-4С)алкилен означают разветвленную или прямую группу алкилена, имеющую 1-6 и 1-4 атомов углерода, соответственно, примерами являются -(СН2)а - (где а соответствует числу атомов углерода), -СН(СН3)- и -СН(СН3)-СН2- и так далее.
(2-10С)алкенилен означает разветвленную или прямую группу алкенилена, имеющую 2-10 атомов углерода и одну или более двойных связей, подобную -СН=СН-СН2-, -(СН2)2 -СН=СН-СН(СН3)-, -СН=СН-СН=СН-СН2- и так далее.
(6-14С)арил, (6-12С)арил означают ароматическую углеводородную группу, имеющую от 6 до 14 или 6-12 атомов углерода, соответственно, такую как фенил, нафтил, тетрагидронафтил, инденил и так далее.
(7-15С)аралкил означает аралкильную группу, имеющую от 7 до 15 атомов углерода, где "алкил" представляет собой (1-8С)алкиленовую группу, а арильная группа является (6-14С)арилом, причем обе они такие, как было определено выше.
(8-16С)аралкенил означает аралкильную группу, имеющую от 8 до 16 атомов углерода, где "алкенил" представляет собой (2-10С)алкениленовую группу, а арильная группа является (6-14С)арилом, причем обе они такие, как определено выше.
(14-20С)(бисарил)алкил означает (1-3С)алкильную группу, замещенную по одному и тому же атому углерода или же по разным углеродным атомам двумя независимо выбранными арильными группами, в соответствии с определением термина (6-12С)арила, такими как группа бисфенилметила.
Галоген означает F, Сl, Вг или I.
Tiq означает 1,2,3,4-тетрагидроизохинолин-3-карбоновую кислоту.
Atc подразумевает 2-аминотетралин-2-карбоновую кислоту.
Aic означает 2-аминоиндан-2-карбоновую кислоту.
Piq означает пергидроизохинолил-карбоновую кислоту.
Термин пролекарство подразумевает соединение, которое после введения превращается в одно или более активных соединений формулы I. Приемлемыми пролекарствами являются, например, N-алкоксикарбонил-замещенные (предпочтительно N-этоксикарбонил) производные соединений формулы I.
Так как аминогруппа в аминоизохинолине, аминохиназолине или аминофталазине является менее основной, чем аминогруппа в лизине или аргинине, оказалось неожиданным, что недавно открытые соединения проявляют достаточную эффективность при ингибировании сериновых протеаз.
Как упоминалось, среди соединений данного изобретения имеются ингибиторы сериновых протеаз, вовлеченные в каскад реакций свертывания крови, и в частности, ингибиторы тромбина и/или фактора Ха. Данные соединения могут использоваться в медицинской или ветеринарной терапии, то есть для лечения и предупреждения опосредованных тромбином и связанных с тромбином заболеваний. Данные заболевания включают в себя ряд тромботических и протромботических состояний, при которых активируется система свертывания. Такими заболеваниями и состояниями являются, например, тромбоз глубоких вен, эмболия сосудов легких, тромбофлебит, артериальная окклюзия в результате тромбоза или эмболии, артериальная реокклюзия во время или после ангиопластики или тромболиза, рестеноз после повреждения артерий или инвазивных кардиологических манипуляций, послеоперационный венозный тромбоз или эмболия, острый или хронический атеросклероз, внезапный мозговой удар, инфаркт миокарда, определенные типы рака и метастазы и определенные виды нейродегенеративных заболеваний. Соединения согласно изобретению также могут использоваться in vitro как антикоагулянты или как антикоагулянты в экстракорпоральных системах кровообращения, которые необходимы при диализе и в хирургии.
Согласно следующему аспекту данное изобретение предусматривает способ лечения и/или предупреждения опосредованных тромбином и связанных с тромбином заболеваний у животных или человека, который включает в себя лечение указанного животного или человека терапевтически эффективным количеством соединения в соответствии с данным изобретением.
Соединения согласно изобретению можно вводить энтерально (например, перорально, ректально, через нос или местно) или парентерально (например, внутримышечными, подкожными, внутривенными или внутрибрюшинными инъекциями).
Точная доза и схема приема данных соединений и их композиций обязательно зависят от потребности индивидуального субъекта, которому вводится соединение данного изобретения в виде лекарственного средства, и от степени болезни или потребности и оценки практикующего врача. Вообще парентеральное введение требует более низких дозировок, чем другие способы введения, которые в большей степени зависят от абсорбции. Однако ежедневные дозировки для человека предпочтительно составляют 0,001-100 мг на кг массы тела, более предпочтительно 0,01-10 мг на кг массы тела.
Суточную дозу можно вводить в одной или более стандартных дозах, приемлемых, например, для перорального, ректального, подъязычного или назального способа или через кожу (например, чрезкожный пластырь или в виде крема).
Другой способ введения соединения данного изобретения представляет собой включение его в замкнутую систему (диализ) другими средствами, например инъецированием его - либо постепенно, либо одномоментно - в систему противотока диализной мембраны одновременно с введением крови в кровоток. Кроме того, трубки и/или другое оснащение экстракорпоральной замкнутой системы могут быть снабжены соединением данного изобретения, предпочтительно путем покрытия (но не ограничиваясь данным способом). Альтернативно, соединение данного изобретения может быть адсорбировано на материалах некоторых частей оснащения, например, мембранах, используемых для диализа.
Изобретение предусматривает фармацевтическую композицию для ингибирования снижения агрегации тромбоцитов крови, ингибирования образования фибрина, ингибирования тромбообразования и ингибирования образования эмбола у млекопитающих, содержащую соединение согласно изобретению с приемлемыми вспомогательными средствами. Данные композиции могут заключать в себе антикоагулянты, противотромбоцитарные вещества и тромболитические агенты. Фармацевтические композиции можно вводить в кровь, продукты крови или органы млекопитающих для достижения желаемого ингибирования. Изобретение также предусматривает фармацевтическую композицию для предупреждения или лечения нестабильной стенокардии, резистентной стенокардии, инфаркта миокарда, преходящего нарушения мозгового кровообращения и фибрилляции предсердий, тромботического мозгового удара, эмболии мозга, тромбоза глубоких вен, диссеминированного внутрисосудистого свертывания и реокклюзии или рестеноза реканализированных сосудов у млекопитающих, содержащую соединение данного изобретения в фармацевтической композиции. Данные фармацевтической композиции могут заключать в себе антикоагулянты, противотромбоцитарные вещества и тромболитические агенты. Изобретение также предусматривает способ снижения тромбообразующей способности поверхности у млекопитающих в результате ковалентного или нековалентного присоединения к поверхности соединения согласно изобретению.
Далее изобретение предусматривает вышеописанную фармацевтическую композицию в комбинации с упаковочным материалом, приемлемым для названной композиции, причем упомянутый упаковочный материал содержит инструкции для применения композиции, которая описана выше.
Для создания лекарственных форм, таких как пилюли, таблетки, суппозитории, (микро)-капсулы, порошки, эмульсии, крема, мази, имплантаты, спреи, инъекционные препараты в виде раствора или суспензии, можно использовать подходящие вспомогательные средства, такие как носители, наполнители, связующие вещества, смазочные масла, диспергаторы, эмульгаторы, стабилизаторы, поверхностно-активные вещества, антиоксиданты, красители, консерванты и тому подобное, например, как описано в стандартной ссылке, Gennaro et al., Remington’s Pharmaceutical Sciences, (18th ed., Mack Publishing Company, 1990, см. особо Part 8: Pharmaceutical Preparations and Their Manufacture). Обычно любые фармацевтически допустимые вспомогательные средства, которые не препятствуют функции активных соединений, оказываются подходящими и могут использоваться.
Подходящие носители и наполнители, с которыми можно вводить активное соединение согласно изобретению, заключают в себе, например, агар, спирт, производные целлюлозы, жиры, полисахариды, поливинилпирролидон, диоксид кремния, стерильный физиологический раствор и тому подобное.
Связующие вещества являются агентами, используемыми для придания скрепляющих свойств фармацевтической композиции, полученной с минимальной потерей из фармацевтической композиции во время производства и применения. Связующие вещества представляют собой, например, целлюлозу, крахмалы, поливинилпирролидон и тому подобное.
Подходящая смазка, с которой можно вводить активное вещество согласно изобретению, представляет собой, например, стеарат магния.
Поверхностно-активные вещества являются агентами, способствующими контакту и миграции соединений в различных физических окружающих средах, таких как гидрофильное и гидрофобное окружение. Многие поверхностно-активные вещества известны в области создания фармацевтических композиций, что описано в главе 19 Remington’s Pharmaceutical Sciences (18thedition Editor A.R. Gennaro; Mack Publishing Comp; Easton, Pennsylvania). Поверхностно-активными веществами, которые можно использовать во время процесса приготовления фармацевтической композиции, являются, например, полиэтиленгликоль (PEG) и тому подобное.
Соединения также можно использовать в имплантируемых фармацевтических устройствах, таких как устройства, описанные в патенте США 4767628, содержание которого включено в описание цитированием. В таком случае устройство содержит достаточные количества соединения для медленного высвобождения соединения (например, в течение более месяца).
Соединения формулы (I) могут быть получены из соединения формулы (II) или его производных, в которых аминогруппа у ароматической группы (ариламино) защищена уретаном, таким как Аллок, или амидом, таким как бензоил, где D, Е, X, Y и m имеют определяемые ранее значения, а Pg является N-защитной группой (предпочтительно уретаном, таким как Бок).

Термин N-защитная группа, используемый в данном описании, подразумевает группу, обычно используемую в пептидной химии для защиты α-аминогруппы, подобной аллилоксикарбонильной (Аллок; Alloc) группе, трет-бутилоксикарбонильной (Бок) группе, бензилоксикарбонильной (Z) группе, 9-флуоренилметилоксикарбонильной (Fmoc) группе или группе фталоила (Phth). Удаление защитных групп можно осуществлять различными способами, в зависимости от природы данных защитных групп. Рассмотрение защитных аминогрупп и способов их удаления представлено в вышеупомянутом The Peptides, Analysis, Synthesis, Biology, Vol.3, и в T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2nd edition, 1991, John Wiley & Sons, Inc.
Удаление N-защитной группы Pg и необязательная модификация(и) лишенной защиты аминной группы с использованием способов, известных в области пептидного связывания, алкилирования или гидроаминирования, дают соединения формулы (I).
Соединения формулы (II) могут быть получены из соединения формулы (III) или его производных, в которых ариламино защищен уретаном, таким как Аллок, или амидом, таким как бензоил, где Е, X, Y, m и Pg имеют определяемые выше значения. Удаление N-защитной группы Pg в соединениях формулы (III) и пептидное связывание с соединениями формулы Pg-D-OH, в которой D и Pg имеют определяемое выше значение, дают соединения формулы (II).

Альтернативно, соединения формулы (I) могут быть получены непосредственно из соединений формулы (III). Удаление N-защитной группы Pg в соединениях формулы (III) и пептидное связывание с соединениями формулы J-D-OH, в которой J и D имеют определяемые выше значения и, необязательно, могут содержать дополнительную защитную группу, дают соединения формулы (I).
Соединения формул (I), (II) и (III) получают из соединений формулы (IV), в которой Х и Y имеют определяемые выше значения, или из их производных, где ариламино защищен уретаном, таким как Аллок, или амидом, таким как бензоил, с помощью реакции спиртов формулы J-D-E-(CH2) m-OH, Pg-D-E-(СН2 )m-ОН или Рg-Е-(СН2)m-ОН, в которых D, Е, J, m и Рg имеют определяемые выше значения и, необязательно, содержат защитную группу, при стандартных условиях Митсунобу (Mitsunobu) (трибутилфосфин, диалкилазодикарбоксилат) (R.L.Elliot, Н.Kopecka, D.E.Gunn, H.-N. Lin and D.S.Garvey, Bioorg. Med. Chem. Lett., 6, 2283 (1996); K.Wisniewski, A.S.Koldziejczyk and B. Falkiewicz, J.Pept. Sc., 4, 1 (1998)).

Спирты формулы типа Pg-E-(CH2 )m-OH, в которой E=-R3NCH2- и m=2, получают реакцией присоединения соответствующего амина R3NH2 к этилакрилату, восстановлением эфирной функции гидридом лития алюминия и последующим введением N-защитной группы Рg.
Деметилирование метиларильных эфиров формулы (V) до соответствующих фенольных соединений формулы (IV) может быть достигнуто реакцией с ВВr3 [J.F.W.McOmie and D.E.West, Org. Synth., Collect. Vol.V, 412 (1973)] или EtSNa [A.S.Kende and J.P.Rizi, Tetrahedron Lett., 22, 1779 (1981)].

Подходящим исходным материалом для получения соединений формулы (V) являются соединения формулы (VI), в которой Х и Y имеют определяемые выше значения. Группу хлора соединений формулы (VI) можно преобразовать непосредственно в группу амина нагреванием первой с аммиаком под давлением. Альтернативно, группа хлора соединений формулы (VI) может быть преобразована в феноксигруппу в результате реакции с фенолом при щелочных условиях, а обработка впоследствии ацетатом аммония дает группу амина соединений формулы (V). Соединения формулы (V) также можно получить обработкой соединений формулы (VI) азидом натрия и последующим восстановлением арилазида, используя РРh3.

Соединения формулы (VI), в которой Х и Y имеют определяемые выше значения, могут быть получены из соединений формулы (VII) обработкой фосфорилхлоридом. Соединения формулы (VII) описаны в литературе; 7-метокси-3Н-хиназолин-4-он: Chapman et al., J.Chem. Soc. 890 (1947), 6-метокси-2Н-фталазин-1-он: Consonni P. and A.Omodei-Sale, Farmaco, Ed. Sci.76, 691 (1976) (Chem. Abstr. 85-177191). Соединение формулы (VI), в которой Х=СН и Y=CH (1-хлор-6-метокси-изохинолин) можно получить превращением 6-метокси-изохинолина (Hendrickson, J.B.; Rodriguez, С., J.Org. Chem. 1983, 48, 3344-3346) в соль N-оксида, например, с надкислотой, такой как м-хлорнадбензойная кислота, за которым следует обработка НСl и потом реакция данной соли N-оксида с хлорирующим реагентом, подобным фосфорилхлориду (J.Robinson, J.Am.Chem.Soc., 69, 1941 (1939)).

Пептидное связывание, которое упоминалось как процедурная стадия в вышеописанном способе получения соединений согласно изобретению, можно проводить способами, общеизвестными в области связывания (либо способами конденсации) пептидных фрагментов, такими как азидный способ, способ смешанных ангидридов, способ активированных эфиров, либо, предпочтительно, карбодиимидным способом, особенно с добавлением каталитических и подавляющих рацемизацию соединений, подобных N-гидроксисукцинимиду и N-гидроксибензотриазолу. Подходящие способы описаны в следующих публикациях: The Peptides, Analysis, Synthesis, Biology, Vol.3, E.Gross and J.Meienhofer, eds. (Academic Press, New York, 1981), R.Knorr, A.Trzeciak, W.Bannwarth and D.Gillessen, Tetrahedron Lett., 30, 1927 (1989) и L.A.Carpino, J.Am.Chem.Soc., 115, 4397 (1993).
Соединения согласно изобретению, которые могут быть в виде свободного основания, можно выделить из реакционной смеси в виде фармацевтически приемлемой соли. Фармацевтически приемлемые соли также можно получить обработкой свободного основания формулы I органической или неорганической кислотой, такой как хлористый водород, бромистый водород, йодистый водород, серная кислота, фосфорная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, метансульфоновая кислота, фумаровая кислота, янтарная кислота, винная кислота, лимонная кислота, бензойная кислота и аскорбиновая кислота.
Соединения согласно изобретению содержат один или более хиральных атомов углерода и поэтому могут быть получены в виде чистого энантиомера или в виде смеси энантиомеров, либо в виде смеси, содержащей диастереомеры. Способы получения чистых энантиомеров хорошо известны в данной области, например кристаллизация солей, которые получают из оптически активных кислот и рацемической смеси, или хроматография с использованием хиральных колонок. Для диастереомеров можно использовать колонки с прямой или обращенной фазой.
Изобретение иллюстрируется следующими примерами.
ПРИМЕРЫ
Использованы следующие сокращения:
Аллок: аллилоксикарбонил
Бок: трет-бутоксикарбонил
Элюент: x-y % растворитель А в растворителе В означает, что используют градиент элюента из х% (об./об.) растворителя А в растворителе В до y% (об./об.) растворителя А в растворителе В.
Пример 1
(2S)-1-(N-(-)-Камфорасульфонид-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
1а. 6-Метокси-изохинолин-N-оксид гидрохлорид
При комнатной температуре 133 г м-хлорнадбензойной кислоты (чистота 75%) добавляли порциями к перемешиваемому раствору 6-метокси-изохинолина [Hendrickson, J.B.; Rodriguez, С.; J. Org. Chem. 1983, 48, 3344-3346; 79, 8 г; 500 ммоль] в 1,2 л дихлорметана. Перемешивание продолжали в течение 3 часов, и потом добавляли метанол (1 л). Объем уменьшали до 700 мл, после чего добавляли 800 мл насыщенного раствора хлористого водорода в диэтиловом эфире. Разбавление 1,5 л диэтилового эфира приводило к осаждению желтых кристаллов, которые отделяли фильтрованием, промывали охлажденным диэтиловым эфиром и высушивали в вакууме. Выход: 85 г (80%); белое твердое вещество; точка плавления 189-191°С; (+)-FAB-MC: 176 (МН+-НСl).
1b. 1-Хлор-6-метокси-изохинолин
Гидрохлорид 6-метокси-изохинолин-N-оксида (1а, 85 г; 400 ммоль) осторожно порциями добавляли к фосфорилхлориду (550 мл) при температуре 90°С, после чего смесь перемешивали в течение 6 часов при 90°С. Избыток фосфорилхлорида удаляли в вакууме. Оставшееся белое твердое вещество промывали водой, отфильтровывали и высушивали в вакууме. Выход: 68 г (88%); белое твердое вещество; точка плавления 72-74°С; EI-MC: 193 (М+).
1с. 6-Meтокси-1-фенокси-изохинолин
К смеси 1-хлор-6-метокси-изохинолина (1b, 16,8 г, 87 ммоль) и фенола (67 г) добавляли порошкообразный гидроксид калия (8,4 г). Смесь нагревали в атмосфере азота до 140°С в течение 3 час, оставляли для охлаждения до комнатной температуры и впоследствии разбавляли 280 мл 3N раствора гидроксида натрия и 500 мл дихлорметана. Органический слой промывали 2N гидроксидом натрия, водой и насыщенным раствором соли, высушивали над сульфатом магния и концентрировали при пониженном давлении, получая 21,3 г (98%) белого твердого вещества. ESI-MC: 251,8 (М+Н)+. Rf (фактор удерживания) (силикагель; толуол/этанол, 8/2, об./об.): 0,75.
1d. 1-Амино-6-метокси-изохинолин
Смесь 6-метокси-1-фенокси-изохинолина (1с, 21,3 г, 85 ммоль) и ацетата аммония (55 г) нагревали в атмосфере азота до 150°С и перемешивали в течение ночи. Смесь оставляли для охлаждения до комнатной температуры, после чего добавляли при перемешивании 3N раствор гидроксида натрия (280 мл). Полученный таким образом раствор экстрагировали этилацетатом (2×300 мл), и объединенный органический слой экстрагировали 2N хлористоводородной кислотой (100 мл), содержащей насыщенный раствор соли. Впоследствии рН водного слоя доводили до 12, используя 2N раствор гидроксида натрия. Экстракция этилацетатом (300 мл) давала органический слой, который промывали насыщенным раствором соли (100 мл), высушивали (сульфат магния) и концентрировали при пониженном давлении, получая 11 г белого твердого вещества (75%). ESI-MC: 175,2 (М+Н)+, 349,2 (M+2H)2+. Rf (силикагель; толуол/этанол, 8/2, об./об.): 0, 17.
1е. 1-Амино-6-гидрокси-изохинолин
Раствор трибромида бора (18,2 мл; 370 ммоль) в 20 мл дихлорметана добавляли по каплям к перемешиваемому раствору 1-амино-6-метокси-изохинолина (1d, 11,0 г; 63 ммоль) в 150 мл дихлорметана при 10°С. После перемешивания в течение 4 дней при температуре окружающей среды реакционную смесь помещали в лед, и рН доводили до 9 добавлением концентрированного водного аммиака. Осажденный материал собирали фильтрованием и высушивали в вакууме до получения 8,9 г (88%) названного соединения в виде белого твердого вещества; точка плавления 258-260°С; EI-MC: 160 (М+).1H ЯМР (DMSO-d6): δ 8,11 (д, 1Н), 7,65 (д, 1H), 6,99 (дд, 1Н), 6,86 (д, 1Н), 6,80-6,63 (м, 4Н), 5/2 (шс, 1H).
1f. (2S)-1-трет-Бутоксикарбонил-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
(2S)-1-трет-Бутоксикарбонил-2-(2-гидроксиэтил)пиперидин
[Ikeda, М.; Kugo, Y.; Sato, Т.; J. Chem. Soc. Perkin Trans./1996, 15, 1819-1824; 860 мг, 3,75 ммоль], полученный из растворенного (2S)-2-(2-гидроксиэтил)пиперидина [Beyerman, Н.С.; Recl. Trav. Chim. Pays-Bas 1971, 90, 755-765], 1-амино-6-гидрокси-изохинолина (1е, 480 мг, 3,0 ммоль) и трибутилфосфина (1,5 мл, 6,0 ммоль), растворяли в смеси тетрагидрофуран/N,N-диметилформамид (4:1, об./об, 15 мл). Впоследствии добавляли по каплям раствор диэтилазодикарбоксилата (0,95 мл, 6,0 ммоль) в тетрагидрофуране (5 мл). После перемешивания в течение ночи смесь разбавляли дихлорметаном (100 мл), промывали 2N гидроксидом натрия (2×50 мл), высушивали (сульфат магния) и концентрировали при пониженном давлении. Очистку остатка осуществляли, используя хроматографию на силикагеле (элюент: 2-10% метанол в дихлорметане), получая 827 мг (60%) белой пены. ESI-MC: 372,2 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 9/1, об./об.):0,41. Такую же реакцию также проводили с нерастворенным 1-трет-бутоксикарбонил-2-(2-гидроксиэтил)пиперидином (в количестве 10 ммоль, 63% выход).
1g. (2S)-2-(2-(1-Аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин
К перемешиваемому раствору (2S)-(1-трет-бутоксикарбонил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидина (1f, 371 мг, 1,0 ммоль) в смеси пиридин/дихлорметан (1:2, об./об., 5 мл) добавляли аллилхлорформиат (117 мкл, 1,1 ммоль). После 2 часов перемешивания реакцию гасили добавлением воды, реакционную смесь концентрировали в вакууме, повторно растворяли в дихлорметане (50 мл) и промывали насыщенным раствором двууглекислого натрия (2×25 мл). Высушивание над сульфатом магния и концентрирование при пониженном давлении давало 460 мг (100%) бесцветного масла, которое растворяли в смеси трифторуксусная кислота/-дихлорметан (1:1, об./об., 10 мл) и перемешивали в течение 2 час. Впоследствии реакционную смесь концентрировали в вакууме и хроматографировали на колонке с силикагелем (элюент: 2-12% метанол в дихлорметане), что обеспечивало 285 мг (80%) указанного в заголовке соединения в виде белой пены. ESI-MC: 356,4 (М+Н)+, 281,4 (М+Н-аллок)+. Rf (силикагель; дихлорметан/метанол, 9/1, об./об.): 0,62.
1h. N-(-)-Камфорасульфонил-D-циклогексилаланин
(-)-Камфорасульфонилхлорид (3,0 г, 12 ммоль) по каплям добавляли к перемешиваемой смеси HCl-соли D-циклогексилаланина (2,08 г, 10 ммоль), 1,4-диоксана (20 мл) и насыщенного водного двууглекислого натрия (10 мл). Гетерогенную смесь перемешивали в течение ночи и впоследствии осторожно подкисляли (рН=3), используя IN хлористоводородную кислоту. Экстракция данной смеси дихлорметаном (2×100 мл) с последующим высушиванием над сульфатом магния и концентрированном при пониженном давлении давала 3,22 г (84%) соединения в виде белого твердого вещества. ESI-MC: 386,5 (М+Н)+, 384,5 (М-Н)-. Rf (силикагель; дихлорметан/метанол, 9/1, об./об.): 0,32.
1i. (2S)-1-(N-(-)-Камфорасульфонил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)-пиперидин
2-(1Н-Бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетра-фторборат (TBTU, 321 мг, 1,0 ммоль) добавляли к перемешиваемому раствору (2S)-1-трет-бутоксикарбонил-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидина (1 g, 285 мг/0,80 ммоль), N-(-)-камфорасульфонил-D-циклогексилаланина (1h, 308 мг, 0/80 ммоль) и N, N-диизопропилэтиламина (348 мкл, 2,0 ммоль) в дихлорметане (5 мл). После перемешивания в течение ночи реакционную смесь разбавляли дихлорметаном (50 мл), промывали насыщенным водным двууглекислым натрием (25 мл) и 0,1N хлористоводородной кислотой (25 мл), высушивали (сульфат магния) и концентрировали при пониженном давлении. Очистку остатка осуществляли хроматографией на силикагеле (элюент: 0-5% метанол в дихлорметане), выход 480 мг (83%) белого твердого вещества. ESI-MC: 723,6 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 19/1, об./об.): 0,34.
1j. (2S)-1-(N-(-)-Камфорасульфонил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
В непрерывном потоке сухого азота тетракис-(трифенил-фосфин)палладий(О) (30 мг, 0, 03 ммоль) добавляли к перемешиваемому раствору (2S)-1-(N-(-)-камфорасульфонил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)-пиперидина (1i, 480 мг, 0,66 ммоль) и морфолина (0,26 мл, 3,0 ммоль) в тетрагидрофуране (5 мл). Реакционную смесь перемешивали в течение 2 час и впоследствии концентрировали в вакууме. Остаточный морфолин удаляли выпариванием вместе с 1, 4-диоксаном, и смесь подвергали препаративной ОФ-ВЭЖХ (Delta Pak C18, 100


После сбора соответствующих фракций смесь обессоливали, используя 0,1N хлористоводородную кислоту, и затем лиофилизировали, выход составлял 227 мг (54%) названного соединения в виде белого легкого мелкокристаллического твердого вещества.
ESI-MC: 639,6 (М+Н)+, 637,6 (М-Н)-, 673,4 (М+Сl)-. Аналитическая ВЭЖХ (Supelcosil LC-18-DB 5 um, 250*2/1 мм): Подвижная фаза: А=0,5 М NaH2PO4+H3PO4 pH 2,1; В=Н2О; С=СН3СN/Н2О (3:2, об./об.) (см. таблицу 2).

Время удерживания: 39,27 мин (96,0% чистоты).
Пример 2
N-(N’-Пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклогексиламин
2а. Этил N-трет-бутоксикарбонил-3-циклогексиламинопропаноат
Этилакрилат (1,09 мл, 10 ммоль) добавляли к перемешиваемому раствору циклогексиламина (1,14 мл, 10 ммоль) в смеси этанол/тетрагидрофуран (1:1, об./об., 30 мл). После перемешивания в течение ночи последовательно добавляли пиридин и ди-трет-бутилдикарбонат, и смесь перемешивали в течение дополнительных 5 час. Концентрирование реакционной смеси с последующей очисткой остатка хроматографией на силикагеле (элюент: смесь этилацетат/гептан, 1:4, об./об.) обеспечивало 2,18 г (73%) названного соединения в виде белой пены. ESI-MC: 300,2 (М+Н)+, 244,2 (М+Н-С4Н8)+, 200,2 (М+Н-Вос)+. Rf (силикагель; этилацетат/гептан, 1:4, об./об.): 0,51.
2b. N-трет-Бутоксикарбонил-3-циклогексиламинопропанол
К перемешиваемому раствору этил N-трет-бутоксикарбонил-3-циклогексиламинопропаноата (2а, 2,18 г, 7,3 ммоль) в тетрагидрофуране (20 мл) добавляли гидрид лития алюминия (1,0 М раствор в тетрагидрофуране, 10 мл). Реакционную смесь перемешивали в течение 1 часа, после чего медленно добавляли этилацетат (5 мл). Потом добавляли водную лимонную кислоту (0,5 М, 50 мл) и гетерогенную смесь экстрагировали Et2O (2×100 мл). Органический слой промывали водным двууглекислым натрием (1N, 25 мл). Высушивание над сульфатом магния, концентрирование при пониженном давлении и очистка хроматографией на силикагеле (элюент: этилацетат/гептан, 3:7, об./об.) обеспечивало 1,50 г (80%) соединения в виде белой пены. ESI-MC: 258,2 (М+Н)+, 280,3 (M+Na)+, 202,2 (М+H-С4Н8)+, 158,2 (М+Н-Бoк)+. Rf (силикагель; этилацетат/гептан, 1:4, об./об.): 0,31.
2с. N-трет-Бутоксикарбонил-N-(3-(1-аминоизохинолин-6-окси)пропил)циклогексиламин
Данное соединение получали из N-трет-бутоксикарбонил-3-циклогексиламинопропанола (2b, 257 мг, 1,0 ммоль) и 1-амино-6-гидроксиизохинолина (1е, 160 мг, 1,0 ммоль) по способу Митсунобу (Mitsunobu), описанному в примере If. Выход: 260 мг (65%). ESI-MC: 400,1 (М+Н)+, 344,1 (М+Н-C4 Н8)+, 300,1 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,28.
2d. N-(3-(1-Аллилоксикарбониламино-изохинолин-6-окси-пропил))циклогексиламин.
Названное соединение получали осуществлением Аллокзащиты и последующего удаления Бок-защиты в N-трет-бутоксикарбонил-3-(1-амино-изохинолин-6-окси) пропил) циклогексиламине (2с, 260 мг, 0,65 ммоль) согласно способу, описанному в примере 1g. Выход: 177 мг (71%). ESI-MC: 384,2 (М+Н)+, 300,2 (М+Н-Аллок)+. Rf (силикагель; дихлорметан/метанол, 17:3, об./об.): 0,47.
2е. Гидрохлорид D-циклогексилаланинбензилового эфира
К перемешиваемому раствору N-трет-бутоксикарбонил-D-циклогексилаланина (2,71 г, 10 ммоль) в метаноле (50 мл), содержащему 1 мл воды, добавляли карбонат цезия (1,63 г, 5,0 ммоль). После перемешивания в течение 2 часов реакционную смесь концентрировали в вакууме и повторно растворяли в N,N-диметилформамиде (50 мл). Впоследствии добавляли бензилбромид (2,38 мл, 20 ммоль) и реакционную смесь перемешивали в течение ночи. Смесь разбавляли этилацетатом (200 мл), промывали водным двууглекислым натрием (1М, 2×50 мл), высушивали (сульфат магния) и концентрировали при пониженном давлении. Очистку остатка осуществляли хроматографией на силикагеле (элюент: 0-30% этилацетата в гептане). Объединенные фракции концентрировали в вакууме и потом обрабатывали 3 М хлористым водородом в 1,4-диоксане (50 мл). После перемешивания в течение ночи реакционную смесь концентрировали при пониженном давлении, выход 2,94 г (74%) названного соединения в виде белой пены. ESI-MC: 262,4 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 19/1, об./об.): 0,41.
2f. N-трет-Бутоксикарбонил-N-пропоксикарбонилметил-D-циклогексилаланинбензиловый эфир
н-Пропилбромацетат (1,05 мл, 8,1 ммоль) добавляли к перемешиваемому раствору D-циклогексилаланинбензилового эфира - НСl (2,94 г, 7,4 ммоль) и N,N-диизопропилэтиламина (3,5 мл, 20 ммоль) в ацетонитриле (20 мл). Реакционную смесь оставляли для перемешивания в течение 6 дней и потом концентрировали при пониженном давлении. Остаток растворяли в дихлорметане (100 мл) и промывали водным двууглекислым натрием (1 М, 50 мл), высушивали (сульфат магния) и концентрировали в вакууме. Неочищенное вещество повторно растворяли в дихлорметане (20 мл) и затем обрабатывали N,N-диизопропилэтиламином (3,5 мл, 20 ммоль) и ди-трет-бутилдикарбонатсм (1,74 г, 8,0 ммоль). После перемешивания в течение 4 дней реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на силикагеле (элюент: 0-50% этилацетат в гептане), выход 2,39 г (70%) названного соединения в виде бесцветного масла. ESI-MC: 461,5 (М+Н)+, 407,5 (M+H-C4H8)+ 361,5 (М+Н-Бок)+ . Rf (силикагель; этилацетат/гептан, 1/3, об./об.): 0,27.
2q. N-трет-Бутоксикарбонил-N-пропоксикарбонилметил-D-циклогексилаланин
Раствор N-трет-бутоксикарбонил-N-пропоксикарбонилметил-D-циклогексилаланинбензилового сложного эфира (2f, 2,39 г, 5,2 ммоль) в N,N-диметилформамиде (25 мл) обрабатывали палладием на активированном угле (10% Pd, 250 мг). Водородный газ барботировали через последний раствор при атмосферном давлении в течение 2 час. Впоследствии реакционную смесь фильтровали через Целит, и фильтрат упаривали при пониженном давлении, обеспечивая 1,90 г (93%) названного соединения в виде белого твердого вещества. ESI-MC: 371,2 (М+Н)+, 369,2 (М-Н)-, 315,2 (M+H-C4H8)+ 271,5 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9/1, об./об.): 0,42.
2h. N-(N’-трет-Бутоксикарбонил-N’-пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-аллилоксикарбониламио-изохинолин-6-окси)пропил))циклогексиламин
Данное соединение получали из N-(3-(1-аллилоксикарбонил-амино-изохинолин-6-окси)пропил))циклогексиламина (2d, 192 мг, 0,50 ммоль) и N-трет-бутоксикарбонил-N-пропоксикарбонилметил-D-циклогексилаланина (2g, 185 мг, 0,50 ммоль) с помощью реакции связывания пептидов, описанной в примере 1i. Выход: 221 мг (60%). ESI-MC: 737,6 (М+Н)+, 681,5 (М+Н-C4Н8)+, 637,6 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,68.
2i. N-(N’-Пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклогексиламин
Названное соединение получали из N-(N’-трет-бутоксикарбонил-N’-пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))циклогексиламина (2h, 221 мг, 0,30 ммоль) согласно способу, описанному в примере 1i для удаления группы аллилоксикарбонила. Потом неочищенный продукт обрабатывали смесью дихлорметан/трифторуксусная кислота (1:1, об./об., 10 мл) и перемешивали в течение 2 час, после чего реакционную смесь концентрировали в вакууме. Очистку остатка осуществляли методом препаративной ВЭЖХ, описанным в примере lj. Обессоливание с использованием 0, 1N хлористоводородной кислоты и последующая лиофилизация давали 67 мг (41%) названного соединения в виде белого легкого мелкокристаллического твердого вещества. ESI-MC: 553,6 (М+Н)+, 587,9 (М+Сl). Время удерживания при аналитической ВЭЖХ (градиентный пример 1j): 27,91 мин (95,8% чистоты).
Пример 3
N-(N’-Пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклопентиламин
3а. Этил N-трет-бутоксикарбонил-3-циклопентиламинопропаноат
Названное соединение получали из этилакрилата (1,09 мл, 10 ммоль) и циклопентиламина (0,99 мл, 10 ммоль) согласно примеру 2а. Выход: 2,28 г (80%). ESI-MC: 286,2 (М+Н)+, 230,2 (M+H-C4H8)+, 186,2 (М+Н-Бок)+, Rf (силикагель; этилацетат/-гептан, 1:4, об./об.): 0,45.
3b. N-трет-Бутоксикарбонил-3-циклопентиламинопропанол
Данное соединение получали из N-трет-бутоксикарбонил-3-циклопентиламинопропаноата (3а, 2,28 г, 8,0 ммоль), используя способ, описанный в примере 2b. Выход: 1,34 г (69%). ESI-MC: 244,2 (М+Н)+, 266,3 (M+Na)+, 188,2 (M+H-C4H8)+, 144,2 (М+Н-Бок)+. Rf (силикагель; этилацетат/гептан, 1:4, об./об.): 0,30.
3с. N-трет-Бутоксикарбонил-N-(3-(1-амино-изохинолин-6-окси)пропил))циклопентиламин
Данное соединение получали из N-трет-бутоксикарбонил-3-циклопентиламинопропанола (3b, 243 мг, 1,0 ммоль) и 1-амино-6-гидрокси-изохинолина (1е, 160 мг, 1,0 ммоль) по способу Митсунобу, описанному в примере 1f. Выход 262 мг (68%). ESI-MC: 386,1 (М+Н)+, 330,1 (M+H-C4H8 )+, 286,1 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,25.
3d. N-(3-(1-Аллилоксикарбониламино-изохинолин-6-окси)-пропил))циклопентиламин
Названное соединение получали проведением Аллок-защиты и последующего удаления Бок-защиты в N-трет-бутоксикарбонил-3-(1-амино-изохинолин-6-окси)пропил)циклопентиламине (3с, 262 мг, 0,68 ммоль) согласно способу, описанному в примере 1g. Выход: 171 мг (68%). ESI-MC: 370,2 (М+Н)+, 286,2 (М+Н-Аллок)+. Rf (силикагель; дихлорметан/метанол, 17:3, об./об.): 0,44.
3е. N-(N’-трет-Бутоксикарбонил-N’-пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-аллилоксикарбониламино-изокинолин-6-окси)пропил))циклопентиламин
Названное соединение получали из N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))циклопентиламина (3d, 185 мг, 0,50 ммоль) и N-трет-бутоксикарбонил-N-пропоксикарбонилметил-D-циклогексилаланина (2g, 185 мг, 0,50 ммоль) с помощью реакции связывания пептидов, описанной в примере 1i. Выход: 256 мг (71%). ESI-MC: 723,6 (М+Н)+, 667,5 (M+H-C4H8)+, 623,6 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,64.
3f. N-(N’-Пропоксикарбонилметил-D-циклогексилаланинил-N-(3-(1-амино-изохинолин-6-окси)пропил))циклопентиламин
Названное соединение получали из N-(N’-трет-бутоксикарбонил-N’-пропоксикарбонилметил-D-циклогексилаланинил-N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))циклопентиламина (3е, 256 мг, 0,35 ммоль) согласно способу, описанному в примере 2i, для удаления Аллок- и Бок-защитных групп.
Очистка остатка осуществлена по способу препаративной ВЭЖХ, описанному в примере 1j. Обессоливание с использованием 0,1N хлористоводородной кислоты, и последующая лиофилизация давали выход 113 мг (59%) названного соединения в виде белого легкого мелкокристаллического твердого вещества. ESI-MC: 539,5 (М+Н)+, 573,9 (М+Сl). Время удерживания при аналитической ВЭЖХ (градиентный пример 1j): 25,84 мин (90,1% чистоты).
Пример 4
N-(N’-Пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклобутиламин
4а. Этил N-трет-бутоксикарбонил-3-циклобутиламинопропаноат
Названное соединение получали из этилакрилата (1,09 мл, 10 ммоль) и циклобутиламина (0,85 мл, 10 ммоль) согласно примеру 2а. Выход: 1,71 г (63%). ESI-MC: 272,2 (М+Н)+, 216,2 (M+H-C4H8)+, 172,2 (М+Н-Бок)+. Rf (силикагель; этилацетат/гептан, 1:4, об./об.): 0,44.
4b. N-трет-Бутоксикарбонил-3-циклобутиламинопропанол
Названное соединение получали из N-трет-бутоксикарбонил-3-циклобутиламинопропаноата (4а, 1,71 г, 6,3 ммоль), используя способ описанный в примере 2b. Выход: 0,94 г (65%). ESI-MC: 230,2 (М+Н)+, 252,3 (M+Na)+, 174,2 (M+H-C4H8)+. Rf (силикагель; этилацетат/гептан, 1:4, об./об.): 0,37.
4с. N-трет-Бутоксикарбонил-N-(3-(1-амино-изохинолин-6-окси)пропил))циклобутиламин
Названное соединение получали из N-трет-бутоксикарбонил-3-циклобутиламинопропанола (4b, 229 мг, 1,0 ммоль) и 1-амино-6-гидрокси-изохинолина (1е, 160 мг, 1,0 ммоль) по способу Митсунобу, описанному в примере 1f. Выход: 230 мг (62%). ESI-MC: 372,1 (М+Н)+, 316,1 (M+H-C4H8)+, 272,1 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,26.
4d. N-(3-(1-Аллилоксикарбониламино-изохинолин-6-окси)-пропил))циклобутиламин
Названное соединение получали осуществлением аллок-защиты и последующего удаления Бок-защиты в N-трет-бутоксикарбонил-3-(1-амино-изохинолин-6-окси)пропил)циклобутиламине (4с, 230 мг, 0,62 ммоль) согласно способу, описанному в примере 1g. Выход: 166 мг (75%). ESI-MC: 356,2 (М+Н)+, 286,2 (М+Н-Аллок)+. Rf (силикагель; дихлорметан/метанол, 17:3, об./об.): 0,44.
4е. N-(N’-трет-Бутоксикарбонил-N’-пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))циклобутиламин
Данное соединение получали из N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))циклобутиламина (4d, 142 мг, 0,40 ммоль) и N-трет-бутоксикарбонил-N-пропоксикарбонил-метил-D-циклогексилаланина (2g, 148 мг, 0,40 ммоль) с помощью реакции связывания пептидов, описанной в примере 1i. Выход: 207 мг (73%). ESI-MC: 709,5 (М+Н)+, 653,5 (M+H-C4H8)+, 609,6 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,61.
4f. N-(N’-Пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил)циклобутиламин
Названное соединение получали из N-(N’-трет-бутоксикарбонил-N’-пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))-циклобутиламина (4е, 207 мг, 0,29 ммоль) согласно способу, описанному в примере 2i для удаления аллок- и Бок-защитных групп. Очистку остатка осуществляли способом препаративной ВЭЖХ, описанной в примере 1j. Обессоливание с использованием 0,1 N хлористоводородной кислоты и последующая лиофилизация давали выход 36 мг (24%) названного соединения в виде белого легкого мелкокристаллического твердого вещества. ESI-MC: 525,5 (М+Н)+, 559,9 (М+Сl). Время удерживания при аналитическая ВЭЖХ (градиентный пример 1j): 25,10 мин (97,5% чистоты).
Пример 5
N-(N’-Пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклопропиламин
5а. Этил N-трет-бутоксикарбонил-3-циклопропиламинопропаноат
Названное соединение получали из этилакрилата (1,09 мл, 10 ммоль) и циклопропиламина (0,69 мл, 10 ммоль) согласно примеру 2а. Выход: 2,06 г (80%). ESI-MC: 258,2 (М+Н)+, 202,2 (M+H-C4H8)+, 158,2 (М+Н-Бок)+. Rf (силикагель; этилацетат/-гептан, 1:4, об./об.): 0,38.
5b. N-трет-Бутоксикарбонил-3-циклопропиламинопропанол
Данное соединение получали из N-трет-бутоксикарбонил-3-циклопропиламинопропаноата (5а, 2,06 г, 8,0 ммоль), используя способ, описанный в примере 2b. Выход: 1,22 г (71%). ESI-MC: 216,2 (М+Н)+, 160,2 (М+Н-C4Н8)+. Rf (силикагель; этилацетат/гептан, 1:4, об./об.): 0,21.
5с. N-трет-Бутоксикарбонил-N-(3-(1-амино-изохинолин-6-окси)пропил))циклопропилaмин
Данное соединение получали из N-трет-бутоксикарбонил-3-циклопропиламинопропанола (5b, 215 мг, 1,0 ммоль) и 1-амино-6-гидрокси-изохинолина (1е, 160 мг, 1,0 ммоль) по способу Митсунобу, описанному в примере 1f. Выход: 226 мг (63%). ESI-МС: 358,1 (М+Н)+, 302,1 (M+H-C4H8 )+, 258,1 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,19.
5d. N-(3-(1-Аллилоксикарбониламино-изохинолин-6-окси)-пропил))циклопропиламин
Названное соединение получали с применением Аллок-защиты и последующего удаления Бок-защиты из N-трет-бутоксикарбонил-3-(1-амино-изохинолин-6-окси)пропил)циклопропиламина (5с, 226 мг, 0, 63 ммоль) согласно способу, описанному в примере 1g. Выход: 148 мг (69%). ESI-MC: 342,2 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 17:3, об./об.): 0,36.
5е. N-(N’-трет-Бутоксикарбонил-N’-пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))циклопропиламин
Данное соединение получали из N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))циклопропиламина (5d, 136 мг, 0,40 ммоль) и N-трет-бутоксикарбонил-N-пропоксикарбонилметил-D-циклогексилаланина (2g, 148 мг, 0,40 ммоль) реакцией связывания пептидов, описанной в примере 1i. Выход: 221 мг (78%). ESI-MC: 695,3 (М+Н)+, 639,5 (M+H-C4H8)+, 595,3 (М+Н-Бок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,49.
5f. N-(N’-Пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклопропиламин
Названное соединение получали из N-(N’-трет-бутоксикарбонил-N’-пропоксикарбонилметил-D-циклогексилаланинил)-N-(3-(1-аллилоксикарбониламино-изохинолин-6-окси)пропил))циклопропиламина (5е, 221 мг, 0,31 ммоль) согласно способу, описанному в примере 2i для удаления Аллок- и Бок-защитных групп. Очистку остатка проводили способом препаративной ВЭЖХ, описанным в примере 1j. Обессоливание с использованием 0,1 N хлористоводородной кислоты и последующая лиофилизация давали выход 87 мг (55%) названного соединения в виде белого легкого мелкокристаллического твердого вещества. ESI-MC: 511,3 (М+Н)+ , 545,9 (М+Сl). Время удерживания при аналитической ВЭЖХ (градиентный пример 1j): 24,02 мин (96,0% чистоты).
Пример 6
(2S)-1-(N-Пропоксикарбонилметил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пирролидин
6а. (2S)-1-трет-Бутоксикарбонил-2-(2-(1-амино-изохинолин-6-окси)этил)пирролидин
Данное соединение получали из N-трет-бутоксикарбонил-L-β-гомопролинола [Leyendecker, F., Jesser, F.; Laucher, D.;
Tetrahedron Lett. 1983, 24, 3513-3516; 590 мг, 2,75 ммоль] и 1-амино-6-гидрокси-изохинолина (1е, 320 мг, 2,0 ммоль) по способу Митсунобу, описанному в примере 1f. Выход: 650 мг (91%). ESI-MC: 358,0 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,7.
6b. (2S)-2-(2-(1-Аллилоксикарбониламино-изохинолин-6-окси)этил)пирролидин
Данное соединение получали из (2S)-1-трет-бутоксикарбонил-2-(2-(1-амино-изохинолин-6-окси)этил)пирролидина (6а, 650 мг, 1,8 ммоль) осуществлением Аллок-защиты и удаления Бок-защиты, описанных в примере 1g. Выход: 558 мг (90%). ESI-MC: 342,2 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,22.
6с. (2S)-1-(N-Пропоксикарбонилметил-N-трет-бутоксикарбонил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пирролидин
Проводили реакцию N-пропоксикарбонилметил-N-трет-бутоксикарбонил-D-циклогексилаланина (2g, 925 мг, 2,5 ммоль) и (2S)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)-пирролидина (6b, 558 мг, 1,63 ммоль) с использованием протокола реакции связывания пептидов, описанного в примере 1i. Выход: 590 мг (52%) названного соединения в виде бесцветного масла. ESI-MC: 695,6 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,41.
6d. (2S)-1-(N-Пропоксикарбонилметил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пирролидин
Названное соединение получали из (2S)-1-(N-пропоксикарбонилметил-N-трет-бутоксикарбонил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пирролидина (6с, 590 мг, 0,85 ммоль) согласно способу, описанному в примере 2i для удаления Аллок- и Бок-защитных групп. Очистку остатка выполняли способом препаративной ВЭЖХ, списанным в примере 1j. Обессоливание с использованием 0,1N хлористоводородной кислоты и последующая лиофилизация давали 107 мг (25%) названного соединения в виде белого легкого мелкокристаллического твердого вещества. ESI-MC: 511,6 (М+Н)+. Время удерживания при аналитической ВЭЖХ (градиентный пример 1j): 42,96 мин (96,4% чистоты).
Пример 7
1-(Т-Пропоксикарбонилметил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
7а. 1-(N-пропоксикарбонилметил-N-трет-бутоксикарбонил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин
2-(2-(1-Аллилоксикарбониламино-изохинолин-6-окси)этил)-пиперидин (1g, 330 мг, 0,75 ммоль) и N-пропоксикарбонилметил-N-трет-бутоксикарбонил-D-циклогексилаланин (2g, 273 мг, 0,75 ммоль) конденсировали, используя протокол реакции связывания пептидов, описанный в примере 1i, получая 239 мг (45%) названного соединения в виде бесцветного масла. ESI-MC: 709,7 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.):0,56.
7b. 1-(N-Пропоксикарбонилметил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
Названное соединение получали снятием Аллок-защиты и последующего удаления Бок-защиты 1-(N-пропоксикарбонилметил-N-трет-бутоксикарбонил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидина (7а, 239 мг, 0,34 ммоль) согласно способу, описанному в примере 1j. Очистку остатка проводили способом препаративной ВЭЖХ, описанным в примере 1j. Обессоливание с использованием 0,1N хлористоводородной кислоты и последующая лиофилизация давали выход 87 мг (49%) названного соединения (2 диастереомера) в виде белого легкого мелкокристаллического твердого вещества. ESI-MC: 525,4 (М+Н)+. Время удерживания при аналитической ВЭЖХ (градиентный пример 1j): 25,88 мин (32,4%) и 27,52 мин (66,6%).
Пример 8
1-(N-Циклооктил-γ-трет-бутил-L-глутамил)-2-(2-(1-аминоизохинолин-6-окси)этил)пиперидин
8а. N-Циклооктил-γ-трет-бутил-L-глутаминовая кислота
К перемешиваемой суспензии γ-трет-бутил-L-глутаминовой кислоты (4,06 г, 20,0 ммоль) и циклооктанона (3,15 г, 25 ммоль) в смеси N,N-диметилформамид/уксусная кислота (99:1, об./об., 50 мл) небольшими порциями добавляли триацетоксиборгидрид натрия (6,36 г, 30,0 ммоль), и смесь перемешивали в течение ночи. После упаривания растворителя остаток растворяли в воде (50 мл). рН доводили до 9, используя 2N раствор гидроксида натрия, с последующей экстракцией диэтиловым эфиром (50 мл). Впоследствии рН водного слоя осторожно доводили до 2,5, используя 1,0N хлористоводородную кислоту. Экстракция дихлорметаном давала органический слой, который высушивали (сульфат магния) и концентрировали при пониженном давлении, с выходом 4,82 г (77%) названного соединения в виде белого твердого вещества. ESI-MC: 314,2 (М+Н)+, 336,2 (M+Na)+. Rf (силикагель; этилацетат/пиридин/уксусная кислота/вода 63:20:6:11, об./об.):0,80.
8b. 1-(N-Циклооктил-γ-трет-бутил-L-глутамил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин
N-Циклооктил-γ-трет-бутил-L-глутаминовую кислоту (8а, 344 м.г, 1,1 ммоль) и 2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин (1g, 391 мг, 1,1 ммоль) связывали, используя способ, описанный в примере 1i, получая 380 мг (58%) названного соединения в виде бесцветного масла. ESI-MC: 651,3 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об.): 0,23.
8с. 1-(N-Циклооктил-γ-трет-бутил-L-глутамил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
Данное соединение получали из 1-(N-циклооктил-γ -трет-бутил-L-глутамил)-2-(2-(1-аллилоксиамино-изохинолин-6-окси)-этил)пиперидина (8b, 380 мг, 0,58 ммоль), используя удаление аллок-защиты и способ очистки, описанный в примере 1j. Выход: 81 мг (25%, 2 диастереомера по С-2 пиперидину). ESI-MC: 567,4 (М+Н)+. Время удерживания при аналитической ВЭЖХ (градиентный пример 1j): составило 26,20 мин (47,2% чистоты) и 27,07 мин (52,0% чистоты).
Пример 9
1-(N-Циклопентиламинокарбонилметил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
9а. N-Аллилоксикарбонил-N-трет-бутоксикарбонилметил-D-циклогексилаланин.
К перемешиваемому раствору N-трет-бутоксикарбонилметил-D-циклогексилаланина [Hamada, Y., Shibata, M., Sugiura, Т., Kato, S., Shioiri, Т., J.Org.Chem. 1987, 52, 1252-1255; 2,85 г, 10 ммоль] в 1,4-диоксане (50 мл) последовательно добавляли насыщенный водный двууглекислый натрий (25 мл) и аллилхлорформиат (1,17 мл, 11 ммоль). После перемешивания в течение 3 дней реакционную смесь осторожно подкисляли (рН=3), используя 1N хлористоводородную кислоту, и потом экстрагировали дихлорметаном. Высушивание над сульфатом магния и концентрирование при пониженном давлении давало намеченное соединение в виде белого твердого вещества (2,41 г, 65%). ESI-MC: 370,4 (М+Н)+, 314,4 (М+Н-С4Н8 )+, 230,3 (М+Н-C4Н8-Аллок)+. Rf (силикагель; этилацетат/пиридин/уксусная кислота/вода, 63/20/10/7, об./об.): 0,69.
9b. 1-(N-Аллилоксикарбонил-N-трет-бутоксикарбонилметил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин
2-(1Н-Бензотриазол-1-ил)-1,1,3, 3-тетраметилуроний тетра-фторборат (TBTU, 616 мг, 1,9 ммоль) добавляли к перемешиваемому раствору 2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидина (1g, 676 мг, 1,9 ммоль) и N-аллилоксикарбонил-N-трет-бутоксикарбонилметил-D-циклогексилаланина (9а, 709 мг, 1,9 ммоль) в N,N-диметилформамиде (15 мл) при 0°С. рН приводили к 8, используя N,N-диизопропилэтиламин. После перемешивания в течение ночи при комнатной температуре реакционную смесь концентрировали в вакууме. Остаток разбавляли этилацетатом (100 мл), промывали 5% (мас./мас.) водным двууглекислым натрием (2×50 мл) и насыщенным раствором соли (50 мл), высушивали (сульфат магния) и сконцентрировали при пониженном давлении. Очистку остатка проводили хроматографией на силикагеле (элюент: 33-50% этилацетат в гептане), выход 825 мг (57%) названного соединения в виде белой пены. ESI-MC: 707,4 (М+Н)+. Rf (силикагель; этилацетат/пиридин/уксусная кислота/вода, 232/31/18/7, об./об.): 0, 91.
9с. 1-(N-Аллилоксикарбонил-N-карбоксиметил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин
Раствор 1-(N-аллилоксикарбонил-N-трет-бутоксикарбонил-метил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидина (9b, 825 мг, 1,3 ммоль) в смеси трифторуксусная кислота/дихлорметан (2/3, об./об.) перемешивали в течение 5 час при комнатной температуре. Реакционную смесь концентрировали в вакууме с выходом 0,76 г (100%) коричневатого твердого вещества. ESI-MC: 651,4 (М+Н)+, 649,4 (М-Н)+, Rf (силикагель; этилацетат/пиридин/уксусная кислота/вода, 232/31/18/7, об./об.): 0,31.
9d. 1-(N-Аллилоксикарбонил-N-циклопентиламинокарбонил-метил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин
1-(N-Аллилоксикарбонил-N-карбоксилметил-D-циклогексил-аланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)-этил)пиперидин (9с, 189 мг, 0,29 ммоль) и циклопентиламин (40,3 мкл, 0,41 ммоль) конденсировали, используя способ, описанный в примере 9b, с выходом 187 мг (90%) названного соединения. ESI-MC: 718,4 (M+H)+, 716,4 (М-Н)-, Rf (силикагель; дихлорметан/метанол, 95/5, об./об.): 0,72.
9е. 1-(N-Циклопентиламинокарбонилметил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
Аллок-защитные группы в 1-(N-аллилоксикарбонил-N-циклопентиламинокарбонилметил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидине (9d, 187 мг, 0,26 ммоль) удаляли согласно способу, описанному в примере 1j (10 моль% Pd, 10 эквивал. морфолина). Очистку остатка осуществляли способом препаративной ВЭЖХ, описанным в примере 1j. Обессоливание с использованием 0,1N хлористоводородной кислоты и последующая лиофилизация давали выход 102 мг (66%) названного соединения (2 диастереомера) в виде белого легкого мелкокристаллического твердого вещества. ESI-МС: 550,4 (М+Н)+, 272 (C16H22N3O)+, 251 (C15H27N2O)+, 548,4 (М-Н)-, 584 (М+Сl)-. Время удерживания при аналитической ВЭЖХ (градиентный пример 1j): составило 31,19 мин (44,4%) и 33,31 мин (55,6%).
Пример 10
1-(N-Анилинокарбонилметил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
10a. 2-(N-Аллилоксикарбонил-N-анилинокарбонилметил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин
Используя способ, описанный в примере 9b, проводили реакцию связывания 1-(N-аллилоксикарбонил-N-гидроксикарбонилметил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидина (9с, 189 мг, 0,29 ммоль) и анилина (38 мкл, 0, 41 ммоль). Выход: 206 мг (98%). ESI-MC: 726,4 (М+Н)+. Rf (силикагель; дихлорметан/метанол, 95/5, об./об.): 0,73.
10b. 1-(N-Анилинокарбонилметил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
Аллок-защитные группы в 1-(N-аллилоксикарбонил-N-анилинокарбонилметил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидине (10а, 206 мг, 0,28 ммоль) удаляли по способу, списанному в примере 1j (10 моль% Pd, 10 эквивал. морфолина). Очистку остатка осуществляли способом препаративной ВЭЖХ, описанным в примере 1j. Обессоливание с использованием 0,1N хлористоводородной кислоты и последующая лиофилизация давала 80 мг (51%) названного соединения (2 диастереомера) в виде белого легкого мелкокристаллического твердого вещества. ESI-MC: 558,0 (М+Н)+, 580,1 (M+Na)+, 272,2 (С16H22N3О)+, 259,3 (C16H23N2O)+, 556,0 (М-Н)-, 592,3 (М+Сl)-, Rf (силикагель; дихлорметан/метанол, 95/5, об./об.): 0,32.
Время удерживания при аналитической ВЭЖХ (градиентный пример 1j) составило 32,76 мин (43,5%) и 34,52 мин (56,5%).
Пример 11
1-(N-Циклогексил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
11а. N-Циклогексил-D-циклогексилаланин
Циклогексанон (1,55 мл, 15 ммоль) добавляли к перемешиваемой суспензии HCl-соли D-циклогексилаланина (2,08 г, 10 ммоль) в N,N-диметилформамиде (10 мл), содержащем 0,1 мл уксусной кислоты. Потом добавляли триацетоксиборгидрид натрия (3,18 г, 15 ммоль) и реакционную смесь перемешивали в течение ночи. Через 17 часов прозрачный раствор концентрировали при пониженном давлении и суспендировали в воде (15 мл). После подкисления (1N хлористоводородная кислота) гетерогенной смеси до рН=3,0, добавляли дихлорметан (150 мл), и смесь перемешивали механически в течение 30 минут. Органический слой высушивали (сульфат магния) и концентрировали при пониженном давлении, обеспечивая 1,57 г (63%) названного соединения в виде белого твердого вещества. ESI-MC: 254,2 (М+Н)+, 252,1 (М-Н)-. Rf (силикагель; дихлорметан/метанол, 8:2, об./об.):0,29.
11b. 1-(N-Циклогексил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидин
Данное соединение получали из N-циклогексил-D-циклогексилаланина (11а, 127 мг, 0,50 ммоль) и 2-(2-(1-аллилоксикарбониламино)изохинолин-6-окси)этил)пиперидина (1g, 178 мг, 0,50 ммоль) в результате реакции связывания пептидов, описанной в примере 1i. Выход: 224 мг (76%). ESI-MC: 591,4 (М+Н)+, 589,4 (М-Н)-, 507,3 (М+Н-аллок)+. Rf (силикагель; дихлорметан/метанол, 9:1, об./об): 0, 69/0,72 (2 диастереомера по С-2 пиперидина).
11с. 2-(N-Циклогексил-D-циклогексилаланинил)-2-(2-(1-амино-изохинолин-6-окси)этил)пиперидин
Данное соединение получали из 1-(N-циклогексил-D-циклогексилаланинил)-2-(2-(1-аллилоксикарбониламино-изохинолин-6-окси)этил)пиперидина (11b, 224 мг, 0,38 ммоль) по способу, описанному в примере 1j для снятия Аллок-защитной группы. Очистку остатка проводили способом препаративной ВЭЖХ, описанным в примере 1j. Обессоливание с использованием 0,1N хлористоводородной кислоты и последующая лиофилизация давали выход 102 мг (53%) названного соединения в виде белого легкого мелкокристаллического твердого вещества (2 диастереомера по С-2 пиперидину). ESI-MC: 507,3 (М+Н)+, 623,4 (М+Сl). Выход: 224 мг (76%). ESI-MC: 507, 3 (М+Н)+. Время удерживания при аналитической ВЭЖХ (градиентный пример 1j) составило 30,94 мин (41,2%); 32,20 мин (53,4%).
Пример 12
N-(N’-изо-Пропоксикарбонилметил-D-дифенилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклогексиламин
В соответствии со способами, описанными в предшествующих примерах, получен N-(N’-изо-пропоксикарбонилметил-D-дифенилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил)) циклогексиламин. Данное соединение (1,11 г) смешивали с 1 мл дихлорметана и 9 мл трифторуксусной кислоты. После смешивания при комнатной температуре в течение 16 часов реакционную смесь концентрировали, обрабатывали толуолом и снова концентрировали. Остаток обрабатывали смесью трет-бутанола и воды, промывали простым эфиром и концентрировали. Добавление к остатку этанола, фильтрование и удаление этанола из фильтрата давало 0,72 г N-(N’-гидрокси-карбонилметил-D-дифенилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклогексиламина. К 0,34 г данного соединения добавляли 10 мл 2-пропанола и 0,16 мл тионилхлорида. После перемешивания в течение 18 часов при 60°С реакционную смесь концентрировали. Остаток дважды хроматографировали на колонке с силикагелем (силикагель, первая колонка: дихлорметан/метанол = 9/1 (об./об.); вторая колонка: толуол/этанол = 98/2 градиент до 95/5 (об./об.). Неочищенный продукт растирали в порошок с простым эфиром, чтобы получить 45 мг названного соединения. ESI-MC: 595 (М+Н)+, 629 (М+Сl)-. Аналитическая ВЭЖХ (Supelcosil LC-18-DB 5 um, 250*2,1 мм): Подвижная фаза: А=0,5 М NaH2PO4+Н3РO4 рН 2,1; В=Н2О; С=СН3СN/Н2О (3:2, об./об.) (см. таблицу 3).

Пример 13
N-(N’-Циклопентиламинокарбонилметил-D-дифенилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклогексиламин
Названное соединение (56 мг) получали согласно способам, описанным в предшествующих примерах, из N-(N’-гидроксикарбонилметил-D-дифенилаланинил)-N-(3-(1-амино-изохинолин-6-окси)пропил))циклогексиламина (380 мг). ESI-MC: 620 (М+Н)+, 654 (М+Сl)-. Время удерживания при аналитической ВЭЖХ (градиентный пример 12): 37,2 мин.
Пример 14a-f
Следующие соединения получали согласно способам, описанным в предшествующих примерах:




Пример 15
Биологические активности соединений данного изобретения определяли с помощью следующих способов тестирования.
Исследование антитромбина
Тромбин (Фактор IIа) является фактором каскада реакций свертывания крови.
Антитромбиновую активность соединений данного изобретения определяли спектрофотометрическим измерением скорости гидролиза хромогенного субстрата S-2238, расщепляемого тромбином. Данное исследование антитромбиновой активности в буферной системе использовали для оценки величин IC50 исследуемых соединений.
Тест-среда: Буфер трометамин-NaCl-полиэтиленгликоль 6000 (TNP) буфер
Стандартное соединение: 12581 (Kabi)
Наполнитель: буфер TNP
Растворимости способствуют диметилсульфоксид, метанол, этанол, ацетонитрил или трет-бутиловый спирт, которые не проявляют неблагоприятного эффекта в концентрациях вплоть до 2,5% в конечной реакционной смеси.
Используемые реагенты (Все используемые ингредиенты являются аналитически чистыми).
1. Буфер трометамин-NaCl (TN)
Состав буфера:
Трометамин (Трис) 6,057 г (50 ммоль)
NaCl 5,844 г (100 ммоль)
Вода до 1 л
рН раствора доводили до 7,4 при 37°С, используя НСl (10 ммоль. л-1).
2. Буфер TNP
Полиэтиленгликоль 6000 растворяли в буфере TN с получением концентрации 3 г.л-1
3. Раствор S-2238
Одну ампулу S-2238 (25 мг; Kabi Diagnostica, Sweden) растворяли в 20 мл буфера TN с получением концентрации 1,25 мг.мл-1 (2 ммоль. л-1).
4. Раствор тромбина
Тромбин человека (16000 нКат.ампулу-1; Centraal Laboratorium voor Bloedtransfusie, Amsterdam, The Netherlands) растворяли в TNP-буфере с получением маточного раствора 835 нКат.мл-1.
Непосредственно перед использованием данный раствор разбавляли буфером TNP до получения концентрации 3,34 нКат.мл-1.
- Для водных растворов использовали сверхчистую воду (качество Milli-Q).
Приготовление растворов исследуемых и стандартных соединений
Исследуемые и стандартные соединения растворяли в воде Milli-Q, получая концентрации образцов 10-2 мол.л-1. Каждую концентрацию постепенно разбавляли наполнителем с получением концентраций 10-3, 10-4 и 10-5 мол.л-1. Разбавления, включая раствор образца, использовали в исследовании (конечные концентрации в реакционной смеси: 3.10-3; 10-3; 3.10-4; 10-4; 3.10-5; 10-5; 3.10-6 и 10-6 мол.л-1, соответственно).
Способ
При комнатной температуре по 0,075 мл и 0,025 мл растворов исследуемых соединений или стандартных соединений, или наполнителя поочередно раскапывали в лунки титрационного микропланшета и данные растворы разбавляли 0,115 мл и 0,0165 мл TNP-буфера, соответственно. Аликвоту 0,030 мл раствора S-2238 добавляли в каждую лунку, и планшет предварительно нагревали и преинкубировали в термостате (Amersham) при встряхивании в течение 10 мин при 37°С. После преинкубации гидролиз S-2238 начинали добавлением 0,030 мл раствора тромбина в каждую лунку. Планшет инкубировали (при встряхивании в течение 30 сек) при 37°С. Начиная с 1 мин инкубации, измеряли поглощение каждого образца при 405 нм каждые 2 мин в течение периода 90 мин, используя кинетический микропланшет-ридер (Twinreader plus. Flow Laboratories).
Все данные регистрировали в персональном компьютере IBM, используя программу LOTUS-MEASURE. Для каждой концентрации соединения (выраженной в мол.л-1 реакционной смеси) и для контроля вычерчивали диаграмму поглощения в зависимости от времени реакции в мин.
Оценка реакции Для каждой конечной концентрации максимальное поглощение рассчитывали из графика исследования. Значение IC50 (конечная концентрация, выраженная в мкмол.л-1, вызывающая 50% ингибирование максимального поглощения контроля) рассчитывали с использованием анализа logit-преобразования согласно Hafner et al. (Arzneim.-Forsch./Drug Res. 1977; 27 (II): 1871-3). Результаты представлены в табл.4.

Реферат
Изобретение относится к новому ингибитору сериновых протеаз формулы (I):
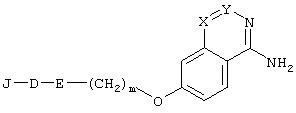
в которой J является R1, R1-SO2-, R3 OOC-(CHR2)p- или (R2aR2b)N-CO-(CHR2)P-;D является аминокислотой формулы -NH-CHR1-C(О)- или -NR4 -CH[(CH2)qC(O)OR1]-C(O)-; Е представляет -NR2-CH2- или фрагмент
R1 выбирают из (1-12С)алкила, (3-12С)циклоалкила и (3-12С)циклоалкил(1-6С)алкилена, группы которых необязательно замещены (3-12С)циклоалкилом, и из (14-20С)(бисарил)алкила; каждый из R2, R2а и R2b независимо выбран из Н, (1-8С)алкила, (3-8С)циклоалкила и (6-14С)арила; R3 имеет те же значения, какие определены для R2; R4 представляет собой Н; Х и Y представляют собой СН; m равно 1 или 2; р равно 1, 2 или 3; q равно 1, 2 или 3; t равно 2, 3 или 4; или к его N-алкоксикарбонил-замещенному производному; и/или к его фармацевтически приемлемой аддитивной соли и/или сольвату. Изобретение относится также к фармацевтической композиции, обладающей тромбин-ингибирующей активностью. Технический результат – получение новых соединений и лекарственных средств на их основе для лечения или предупреждения опосредованных тромбином и связанных с тромбином заболеваний. 2 н. и 6 з.п. ф-лы, 3 табл.
Формула




Комментарии