Замещенные пиридазин-карбоксамидные соединения в качестве соединений, ингибирующих киназы - RU2526618C2
Код документа: RU2526618C2
Описание
Родственные заявки
Настоящая заявка заявляет приоритет предварительной заявке на патент США №61/132,505, поданной 19 июня 2008 г. Полный текст указанной выше заявки на патент включен в настоящий документ посредством ссылки.
Область техники, к которой относится изобретение
Настоящее изобретение касается новых производных пиридазина, их солей, сольватов, гидратов и полиморфов. В настоящем изобретении также описаны композиции, содержащие соединение по настоящему изобретению и применение таких композиций в способах лечения заболеваний и состояний, связанных с протеинкиназной модуляцией.
Уровень техники
Протеинкиназы представляют собой ферменты, которые катализируют фосфорилирование гидроксильных групп в тирозиновых, сериновых и треониновых фрагментах белков. Многие аспекты жизни клеток (например, рост клеток, дифференциация, разрастание, клеточный цикл и выживание) зависят от активности протеинкиназ. Кроме того, ненормальную активность протеинкиназ связывают с группой заболеваний, таких как рак и воспаление. Поэтому значительные усилия были сконцентрированы на разработке путей модуляции активности протеинкиназ. В частности, предпринимались многочисленные попытки поиска малых молекул, выступающих в роли ингибиторов протеинкиназ.
C-Met протоонкоген кодирует Met-рецепторную тирозинкиназу. Met-рецептор представляет собой гликозилированный димерный комплекс с массой 190 кДа, состоящий из альфа-цепи массой 50 кДа, связанной ди-сульфидным мостиком с бета-цепью массой 145 кДа. Альфа-цепь обнаружена вне клетки, в то время как бета-цепь содержит трансмембранные и цитозольные домены. Met синтезируется в виде предшественника и протеолитически расщепляется, обеспечивая зрелые альфа и бета субъединицы. Наблюдаются структурные сходства с семафоринами и плексинами, семейством лигандов рецепторов, участвующих в межклеточном взаимодействии. Лигандом для Met является фактор роста гепатоцитов (HGF), член семейства факторов роста гепатоцитов, обладающий некоторой гомологией с плазминогеном (Longati, P. et al., Curr. Drug Targets 2001, 2, 41-55); Trusolino, L and Comoglio, P. Nature Rev. Cancer 2002, 2, 289-300].
Met участвует в онкогенезе и развитии метастазов опухолей. Экспрессия Met, наряду с его лигандом HGF, оказывает трансформирующее, опухолеобразующее и метастатическое действие (Jeffers, M. et al., Oncogene 1996, 13, 853-856; Michieli, P. et al., Oncogene 1999, 18, 5221-5231). Сверхэкспрессия MET наблюдается при значительной части видов рака человека, и он амплифицируется во время перехода от первичных опухолей к метастазам. Многочисленные исследования показали корреляцию между экспрессией с-МЕТ и/или HGF/SF и прогрессированием различных типов рака (включая рак легких, ободочной кишки, груди, простаты, печени, поджелудочной железы, мозга, почек, яичников, желудка, кожи и костей). Кроме того, было показано, что сверхэкспрессия с-МЕТ или HGF коррелирует с плохим прогнозом и исходом заболевания при основных видах рака человека, включая рак легких, печени, желудка и груди. с-МЕТ также напрямую связывают с видами рака, для которых не разработаны успешные схемы лечения, такими как рак поджелудочной железы, глиома и гепатоцеллюлярная карцинома.
Met-мутанты, проявляющие повышенную киназную активность, были идентифицированы в наследственной и спорадической формах папиллярного почечноклеточного рака (Schmidt, L. et al., Nat. Genet. 1997, 16, 68-73; Jeffers, M. et al., Proc. Nat. Acad. Sci. 1997, 94, 11445-11500). Было показано, что HGF/Met подавляет аноикоз, обусловленную отрывом от клеток-соседей запрограммированную смерть клеток (апоптоз), у клеток плоскоклеточного рака головы и шеи. Устойчивость к аноикозу или «безъякорная» выживаемость являются отличительной особенностью онкогенных трансформаций эпителиальных клеток (Zeng, Q. et al., J. Biol. Chem. 2002, 277, 25203-25208).
Повышенная экспрессия Met/HGF наблюдается для многих метастатических опухолей, включая рак ободочной кишки (Fazekas, К. et al., Clin. Exp. Metastasis 2000, 18, 639-649), груди (Elliott, В. E. et al., 2002, Can. J. Physiol. Pharmacol. 80, 91-102), предстательной железы (Knudsen, В.S. et al., Urology 2002, 60, 1113-1117), легких (Siegfried, J. M. et al., Ann. Thorac. Surg. 1998, 66, 1915-1918) и желудка (Amemiya, H. et al., Oncology 2002, 63, 286-296). Сигналы HGF-Met также связывают с повышенным риском развития атеросклероза (Yamamoto, Y. et al., J. Hypertens. 2001, 19, 1975-1979; Morishita, R. et al., Endocr. J. 2002, 49, 273-284) и усилением фиброза легких (Crestani, В. et al., Lab. Invest. 2002, 82, 1015-1022).
Киназа анапластической лимфомы (ALK) принадлежит к надсемейству рецепторных тирозинкиназ (RTK) протеинкиназ. Экспрессия ALK в нормальных тканях взрослого человека ограничена клетками эндотелия, периваскулярными клетками и редко нервными клетками. Онкогенные, конститутивно активные химерные белки ALK экспрессируются при анапластической крупноклеточной лимфоме (ALCL) и воспалительных миофибробластических опухолях (ШМТ) вследствие t2; хромосомных транслокаций. Также недавно было показано, что ALK выступает в роли онкогена в небольшой части случаев немелкоклеточного рака легких и нейробластомы (Choi et al, Cancer Res 2008; 68: (13); Webb et al, Expert Rev. Anticancer Ther. 9(3), 331-356, 2009).
Анапластические крупноклеточные лимфомы (ALCL) представляют собой подтип семейства низкодифференцированных неходжкинских лимфом с четко выраженной морфологией, иммунофенотипом и прогнозом. Постулируется, что ALCL развиваются из Т-клеток и, в редких случаях, обладают также фенотипом В-клеток. Кроме того, в 40% случаев клетки-предшественники остаются неизвестными и классифицируются как «ноль». Впервые описанная как гистологическая единица Штейном с соавторами на основе экспрессии CD-30 (Ki-1), ALCL представляет собой системное заболевание, поражающее кожу, кости, мягкие ткани и другие органы, с участием или без участия лимфоузлов. ALCL можно разделить по меньшей мере на два подтипа, характеризующиеся наличием или отсутствием хромосомных перестановок между генным локусом киназы анапластической лимфомы (ALK) и различными химерными партнерами, такими как нуклеофосмин (NPM). Примерно 50-60% случаев ALCL связано с хромосомной транслокацией t(2;5;)(p23;q35), в результате которой создается гибридный ген, состоящий из внутриклеточного домена рецептора ALK тирозинкиназы, располагающегося рядом с NPM. Получающийся химерный белок NPM-ALK обладает конститутивной тирозинкиназной активностью, и было показано, что он трансформирует различные типы кровеобразующих клеток in vitro и способствует образованию опухолей in vivo. Другие менее распространенные ALK химерные партнеры, например тропомиозин-3 и тяжелая цепь клатрина, также были идентифицированы в ALCL и в CD30-негативной крупноклеточной лимфоме. Несмотря на небольшие различия в передаче сигнала и некоторых биологических функциях, все химерные продукты, по-видимому, трансформируются в фибробласты и кровеобразующие клетки. ALK химерные белки также были зафиксированы в редкой форме злокачественной воспалительной миофибробластической опухоли. При интенсивном анализе лейкемогенного потенциала NPM-ALK в животных моделях были найдены дополнительные доказательства важности NPM-ALK и других перестановок ALK для развития ALK-положительной ALCL и других заболеваний.
Сообщалось о 2-аминопиридинах, таких как PF-2341066, как потенциальных ингибиторах HGF рецепторной тирозинкиназы (c-Met) и ALK (J.G.Christensen, et al. Abstract LB-271, AACR 2006 meeting; H.Y.Zou et al. Cancer Res 2007; 67: 4408; патенты: WO 2004076412, WO 2006021881, WO 2006021886).
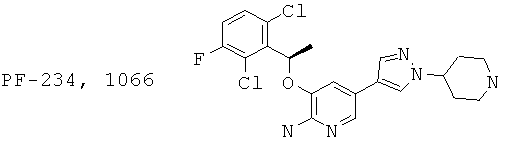
Поскольку все еще сохраняется неудовлетворенная потребность в средствах лечения заболеваний, опосредуемых киназами, является желательным создание новых и альтернативных подходов к лечению и профилактике таких заболеваний, нарушений или их симптомов.
Краткое описание изобретения
Настоящее изобретение касается соединений, являющихся производными пиридазина, композиций, содержащих указанные соединения, и способов применения указанных соединений и композиций соединений. Указанные соединения и содержащие их композиции могут применяться для лечения или профилактики заболеваний или симптомов заболеваний, включая заболевания, опосредуемые или вызываемые модулирующей активностью протеинкиназ.
Настоящее изобретение решает описанные выше задачи с помощью выделенного соединения формулы I
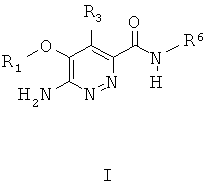
или его соли, или пролекарства, или соли его пролекарства, или его гидрата, сольвата или полиморфа, в котором:
R1 представляет собой арилалкил или гетероарилалкил, каждый из которых необязательно замещен 1-4 независимо выбранными Z1;
R3 представляет собой атом водорода, гидроксил, алкокси или алкиламиногруппу;
R6 представляет собой необязательно замещенный арил или гетероарил, насыщенный или ненасыщенный гетероциклил, где R6 необязательно замещен 1-3 группами, независимо выбранными из алкила, циклоалкила, гетероциклила, алкокси, гидроксиалкила, -C(O)NR7R8 и Z1; где каждый из перечисленных необязательно может иметь дополнительные заместители;
R7 и R8 каждый независимо выбраны из Н, алкила, циклоалкила, алкенила, алкинила, арила, гетероциклила, гетероарила, или R7 и R8 вместе с атомом азота образуют гетероциклил или гетероарил;
каждый Z1 представляет собой атом галогена, CN, NO2, OR15, SR15, S(O)2OR15, NR15R16, C1-C2 перфторалкил, C1-C2 перфторалкокси, 1,2-метилендиокси, C(O)OR15, C(O)NR15R16, OC(O)NR15R16, NR15C(O)NR15R16, C(NR16)NR15R16, NR15C(NR16)NR15R16, S(O)2NR15R16, R17, C(O)R17, NRC(O)R17, S(O)R17, S(O)2R17, R16, оксо, C(O)R16, C(O)(CH2)nOH, (CH2)nOR15, (CH2)nC(O)NR15R16, NR15S(O)2R17, где n независимо равен 0-6 включительно;
каждый R15 независимо представляет собой атом водорода, C1-C4 алкил или С3-С6 циклоалкил;
каждый R16 независимо представляет собой атом водорода, алкенил, алкинил, С6-С6 циклоалкил, арил, гетероциклил, гетероарил, С1-С4 алкил или C1-C4 алкил, замещенный С3-С6 циклоалкилом, арилом, гетероциклилом или гетероарилом;
каждый R17 независимо представляет собой С3-С6 циклоалкил, арил, гетероциклил, гетероарил, C1-C4 алкил или C1-C4 алкил, замещенный С3-С6 циклоалкилом, арилом, гетероциклилом или гетероарилом.
Соединения по настоящему изобретению и содержащие их композиции могут применяться для лечения или уменьшения тяжести модулируемых протеинкиназами заболеваний, нарушений или их симптомов, т.е. нарушений, подлежащих эффективному лечению ингибиторами протеинкиназ, например, c-met, ron, ALK и их химерных белков, таких как EML4-ALK и NPM-ALK.
Другой объект настоящего изобретения относится к способу лечения заболевания или симптомов заболевания у нуждающегося в таком лечении пациента, включающему введение пациенту эффективного количества соединения любой указанной в настоящей заявке формулы или его фармацевтически приемлемой соли, сольвата или гидрата (или их композиций). Заболевание или симптом заболевания могут быть любым из модулируемых протеинкиназами (например, c-met, ron, ALK и их химерные белки, такие как EML4-ALK и NPM-ALK). Заболевание или симптом заболевания могут представлять собой, например, рак или пролиферативное заболевание или нарушение (например, включая указанные в настоящей заявке).
Подробное описание изобретения
Определения
Термины «облегчать» и «лечить» применяются взаимозаменяемо и оба означают уменьшение, подавление, ослабление, остановку или стабилизацию развития или прогрессирования заболевания (например, указанного в настоящей заявке заболевания или нарушения).
Под термином «заболевание» понимают любое состояние или нарушение, которое нарушает или мешает нормальной работе клетки, ткани или органа.
Под «маркером» понимают любое изменение, связанное с заболеванием или нарушением. Например, любой белок или полинуклеотид, изменение уровня экспрессии или активности которого связано с заболеванием или нарушением.
В настоящем описании термины «содержит», «содержащий» и «имеющий» и т.п. имеют значение, описанное для них в патентном законодательстве США, и могут означать «включает», «включающий» и т.п.; «состоящий преимущественно из» и «состоящий преимущественно» также имеют значение, описанное для них в патентном законодательстве США, и данный термин допускает изменения, допуская наличие выходящего за рамки перечисленного, при условии, что основные или новые характеристики перечисленного не изменяются в случае наличия выходящего за рамки перечисленного, но исключают описанные в предшествующем уровне техники варианты.
Термин «соединение» при использовании в настоящей заявке также включает соли, пролекарства и соли пролекарства соединения, отвечающего представленным в настоящей заявке формулам. Данный термин также включает любые сольваты, гидраты и полиморфы любого из описанных выше. Специальное перечисление «пролекарства», «соли пролекарства», «сольвата», «гидрата» или «полиморфа» в некоторых аспектах настоящего изобретения, описанных в данной заявке, не должно интерпретироваться как намеренный пропуск данных форм в других аспектах настоящего изобретения, в которых термин «соединение» используется без перечисления других указанных форм.
Соль соединения по настоящему изобретению образуется между кислотой и основной группой соединения, такой как аминогруппа, или основанием и кислотной группой соединения, такой как карбоксильная группа. Согласно другому предпочтительному варианту выполнения, соединение представляет собой фармацевтически приемлемую аддитивную соль с кислотой.
При использовании в настоящей заявке, и если не указано иное, термин «пролекарство» означает производное соединения, которое может гидролизоваться, окисляться или иным образом вступать в реакцию в биологических условиях (in vitro или in vivo), давая соединение по настоящему изобретению. Пролекарства могут становиться активными только после такой реакции в биологических условиях, или они могут обладать активностью в своей непрореагировавшей форме. Примеры пролекарств, охватываемых настоящим изобретением, включают (но не ограничены только ими) аналоги или производные соединений, отвечающих любой из описанных в данном тексте формул, которые содержат биогидролизуемые фрагменты, такие как амиды, сложные эфиры, карбаматы, карбонаты и фосфатные аналоги. Пролекарства обычно можно получить с использованием хорошо известных методов, таких как описанные в Burger's Medicinal Chemistry and Drug Discovery (1995) 172-178, 949-982 (Manfred E.Wolff ed., 5th ed); см. также Goodman and Oilman's, The Pharmacological basis of Therapeutics, 8th ed., McGraw-Hill, Int. Ed. 1992, «Biotransformation of Drugs».
При использовании в настоящей заявке, и если не указано иное, термин «биогидролизуемый фрагмент» означает функциональную группу (например, амид, сложный эфир, карбамат, карбонат или фосфатный аналог), которая либо 1) не нарушает биологическую активность соединения и придает данному соединению полезные свойства in vivo, такие как всасываемость, длительность действия или скорость начала действия; либо 2) сама по себе не обладает биологической активностью, но in vivo превращается в биологически активное соединение.
Соль пролекарства представляет собой соединение, образуемое кислотой и основной группой пролекарства, такой как аминогруппа, или основанием и кислотной группой пролекарства, такой как карбоксильная группа. В одном варианте выполнения, соль пролекарства представляет собой фармацевтически приемлемую соль.
Особенно предпочтительными пролекарствами и солями пролекарств являются те, которые увеличивают биодоступность соединения по настоящему изобретению при введении таких соединений млекопитающим (например, улучшая поступление в кровь для перорально вводимых соединений), или которые улучшают поступление действующего вещества в участок организма (например, в мозг или центральную нервную систему) по сравнению с самим действующим веществом. Предпочтительные пролекарства включают производные, в которых группа, улучшающая растворимость в воде или активный транспорт через стенки желудочно-кишечного тракта, присоединена к структуре, имеющей описанную в настоящей заявке формулу. Смотри, например, Alexander, J. et al. Journal of Medicinal Chemistry 1988, 31, 318-322; Bundgaard, H. Design of Prodrugs; Elsevier: Amsterdam, 1985; pp 1-92; Bundgaard, H.; Nielsen, N.M. Journal of Medicinal Chemistry 1987, 30, 451-454; Bundgaard, H. A Textbook of Drug Design and Development; Harwood Academic Publ.: Switzerland, 1991; pp 113-191; Digenis, G.A. et al. Handbook of Experimental Pharmacology 1975, 28, 86-112; Friis, G.J.; Bundgaard, H. A Textbook of Drug Design and Development; 2 ed.; Overseas Publ.: Amsterdam, 1996; pp 351-385; Pitman, I.H. Medicinal Research Reviews 1981, 1, 189-214.
Термин «фармацевтически приемлемый», при использовании в настоящей заявке, относится к компоненту, который, в рамках профессиональных медицинских представлений, подходит для использования в контакте с тканями человека и других млекопитающих, без проявления ненужной токсичности, раздражения, аллергической реакции и т.п., и отвечает разумному соотношению польза/риск. «Фармацевтически приемлемая соль» означает любую нетоксичную соль, которая при введении пациенту дает, напрямую или косвенно, соединение или пролекарство соединения по настоящему изобретению.
Кислоты, обычно применяемые для получения фармацевтически приемлемых солей, включают неорганические кислоты, такие как бисульфид водорода, хлороводородная, бромоводородная, иодоводородная, серная и фосфорная кислота, а также органические кислоты, такие как пара-толуолсульфокислота, салициловая, винная, дивинная, аскорбиновая, малеиновая, безиловая, фумаровая, глюконовая, глюкуроновая, муравьиная, глутаминовая кислота, метансульфокислота, этансульфокислота, бензол-сульфокислота, молочная, щавелевая кислота, пара-бромфенилсульфокислота, угольная, янтарная, лимонная, бензойная и уксусная кислота, и родственные им неорганические и органические кислоты. Таким образом, указанные фармацевтически приемлемые соли включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, иодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капраты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацинаты, фумараты, малеаты, бутил-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, терефталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, β-гидроксибутираты, гликолаты, малеаты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты, манделаты и подобные им соли. Предпочтительные соли, образующиеся при добавлении кислоты, включают соли, образующиеся с неорганическими кислотами, такими как хлороводородная кислота и бромоводородная кислота, и особенно соли, образующиеся с органическими кислотами, такими как малеиновая кислота.
Подходящие основания для образования фармацевтически приемлемых солей с кислотными функциональными группами пролекарств по настоящему изобретению включают (но не ограничиваются только ими) гидроксиды щелочных металлов, таких как натрий, калий и литий; гидроксиды щелочноземельных металлов, таких как кальций и магний; гидроксиды других металлов, таких как алюминий и цинк; аммиак и органические амины, такие как незамещенные или гидрокси-замещенные моно-, ди- или триалкиламины; дициклогексиламин; трибутиламин; пиридин; N-метил, N-этиламин; диэтиламин; триэтиламин; моно-, бис- или трис-(2-гидрокси-низший алкил амины), такие как моно-, бис- или трис-(2-гидроксиэтил)амин, 2-гидрокси-трет-бутиламин или трис-(гидроксиметил)метиламин, N,N-ди-низший алкил-N-(гидрокси низший алкил)-амины, такие как N,N-диметил-N-(2-гидроксиэтил)амин или три-(2-гидроксиэтил)амин; N-метил-D-глюкамин; и аминокислоты, такие как аргинин, лизин и т.п.
При использовании в настоящей заявке термин «гидрат» означает соединение, которое дополнительно содержит стехиометрическое или нестехиометрическое количество воды, связанное нековалентными межмолекулярными силами.
При использовании в настоящей заявке термин «сольват» означает соединение, которое дополнительно содержит стехиометрическое или нестехиометрическое количество растворителя, такого как вода, ацетон, этанол, метанол, дихлорметан, 2-пропанол и т.п., связанное нековалентными межмолекулярными силами.
При использовании в настоящей заявке термин «полиморф» означает твердые кристаллические формы соединения или их комплекс, которые можно охарактеризовать физическими методами, такими как, например, рентгеноструктурный анализ порошка или инфракрасная спектроскопия. Разные полиморфы одного и того же соединения могут обладать разными физическими, химическими и/или спектроскопическими свойствами. Разные физические свойства включают (но не ограничиваются только ими) устойчивость (например, к нагреванию, свету или влаге), сжимаемость и плотность (важно для производства готовых лекарственных форм), гигроскопичность, растворимость и скорость растворения (которые могут влиять на биодоступность). Различия в устойчивости могут быть результатом изменений химической реакционной способности (например, различная окисляемость, в результате которой дозированная форма теряет цвет быстрее, когда она изготовлена из одного полиморфа, чем в случае, когда она изготовлена из другого полиморфа), или механических характеристик (например, таблетки крошатся при хранении по мере того, как кинетически выгодный полиморф превращается в термодинамически более устойчивый полиморф), или по обеим причинам (например, таблетки из одного полиморфа более подвержены разрушению при высокой влажности). Различные физические свойства полиморфов могут оказывать влияние на работу с ними. Например, один полиморф может быть более склонен к образованию сольватов, или труднее фильтроваться, или его сложнее отмыть от примесей, чем другой, вследствие, например, формы его частиц или распределения частиц по размеру.
Термин «практически не содержит других стереоизомеров» при использовании в настоящей заявке означает присутствие меньше 25% других стереоизомеров, предпочтительно меньше 10% других стереоизомеров, более предпочтительно меньше 5% других стереоизомеров, и наиболее предпочтительно меньше 2% других стереоизомеров или меньше «Х» % других стереоизомеров (где Х - это число от 0 до 100 включительно).
Способы получения и синтеза диастереомеров хорошо известны в данной области техники и могут использоваться для получения конечных соединений, или исходных соединений, или промежуточных соединений. В других вариантах выполнения соединение представляет собой выделенное соединение. Термин «энантиомерный избыток по меньшей мере Х %» при использовании в настоящей заявке означает, что по меньшей мере Х% соединения представляет собой единственную энантиомерную форму, где Х - это число от 0 до 100 включительно.
Термин «устойчивые соединения», при использовании в настоящей заявке, относится к соединениям, которые обладают достаточной устойчивостью для их производства, и которые сохраняют целостность соединения на протяжении периода времени, достаточного для достижения указанных в настоящей заявке целей (например, введение в состав терапевтических продуктов, промежуточных соединений для использования в производстве терапевтических соединений, промежуточных соединений, которые можно выделить и хранить, лечение заболевания или состояния, восприимчивого к терапевтическим средствам).
Термин «стереоизомер» относится и к энантиомерам, и к диастереомерам.
При использовании в настоящей заявке термин «галоген» относится к любому радикалу фтора, хлора, брома или иода.
Термины «алк» или «алкил» относятся к углеводородным группам с линейной или разветвленной цепь, содержащим от 1 до 12 атомов углерода, предпочтительно от 1 до 8 атомов углерода. Выражение «низший алкил» относится к алкильным группам, содержащим от 1 до 4 атомов углерода (включительно).
Термин «арилалкил» относится к фрагменту, в котором атом водорода алкила заменен на арильную группу.
Термин «алкенил» относится к углеводородным группам с линейной или разветвленной цепь, содержащим от 2 до 12 атомов углерода, предпочтительно от 2 до 4 атомов углерода, содержащим по меньшей мере одну двойную связь. В случаях, когда алкенильная группа связана с атомом азота, предпочтительно, чтобы такая группа не была связана непосредственно через атом углерода двойной связи.
Термин «алкокси» относится к радикалу -O-алкил. Термин «алкилендиоксо» относится к двухвалентному фрагменту структуры -O-R-O-, в котором R представляет собой алкилен.
Термин «алкинил» относится к углеводородным группам с линейной или разветвленной цепью, содержащим от 2 до 12 атомов углерода, предпочтительно от 2 до 4 атомов углерода, содержащим по меньшей мере одну тройную связь. В случаях, когда алкинильная группа связана с атомом азота, предпочтительно, чтобы такая группа не была связана непосредственно через атом углерода тройной связи.
Термин «алкилен» относится к двухвалентному мостику с линейной цепью, содержащей от 1 до 5 атомов углерода, соединенных простыми связями (например, -(СН2)x-, где x равен 1-5), который может быть замещен 1-3 низшими алкильными группами.
Термин «алкенилен» относится к мостику с линейной цепью, включающей одну или две двойные связи, содержащей от 2 до 5 атомов углерода, соединенных простыми связями, который может быть замещен 1-3 низшими алкильными группами. Примерами алкениленовых групп являются -СН=СН-СН=СН-, -СН2-СН=СН-, -СН2-СН=СН-СН3-, -С(СН3)2СН=СН- и -СН(С2Н5)-СН=СН-.
Термин «алкинилен» относится к мостику с линейной цепью, включающей тройную связь, содержащей от 2 до 5 атомов углерода, соединенных простыми связями, который может быть замещен 1-3 низшими алкильными группами. Примерами алкиниленовых групп являются -C≡C-, -CH2-C≡C-, -СН(СН3)С≡С- и -С≡С-СН(С2Н5)СН2-.
Термины «циклоалкил» и «циклоалкенил» при использовании в настоящей заявке включают, соответственно, насыщенные и частично ненасыщенные циклические углеводородные группы, содержащие от 3 до 12 атомов углерода, предпочтительно от 3 до 8 атомов углерода, и более предпочтительно от 3 до 6 атомов углерода.
Термины «Ar» или «арил» относятся к ароматическим циклическим группам (например, 6-членным моноциклическим, 10-членным бициклическим или 14-членным трициклическим системам), которые содержат от 6 до 14 атомов углерода. Примеры арильных групп включают фенил, нафтил, дифенил и антрацен.
Термин «гетероарил» относится к моноциклической группе или группе с конденсированными кольцами (т.е. циклами, для которых является общей пара соседних атомов углерода), состоящей из 5-12 атомов в цикле и содержащей один, два, три или четыре гетероатома в цикле, выбранных из N, О или S, при этом остальными атомами являются С, и, кроме того, имеющей полностью сопряженную π-электронную систему, где 0, 1, 2, 3 или 4 атома в каждом кольце могут иметь заместитель. Примерами (неограничивающими) гетероарильных групп являются пиррол, фуран, тиофен, имидазол, оксазол, тиазол, пиразол, пиридин, пиримидин, хинолин, хиназолин, изохинолин, пурин и карбазол.
Термины «гетероцикл», «гетероциклический» или «гетероцикло» относятся к полностью насыщенным или частично ненасыщенным циклическим группам, например 3-7-членным моноциклическим, 7-12-членным бициклическим или 10-15-членным трициклическим системам, которые содержат по меньшей мере один гетероатом по меньшей мере в одном кольце, где 0, 1, 2 или 3 атома в каждом кольце могут иметь заместитель. Каждое кольцо гетероциклической группы, содержащей гетероатом, может иметь 1, 2, 3 или 4 гетероатома, выбранных из атомов азота, атомов кислорода и/или атомов серы, где гетероатомы азота и серы необязательно могут быть окислены, и гетероатомы азота необязательно могут быть кватернизованы. Гетероциклическая группа может быть присоединена через любой гетероатом или атом углерода в цикле или циклической системе.
Термин «гетероциклил» относится к полностью насыщенным или частично ненасыщенным циклическим группам, например 3-7-членным моноциклическим, 7-12-членным бициклическим или 10-15-членным трициклическим системам, которые содержат по меньшей мере один гетероатом по меньшей мере в одном кольце, где 0, 1, 2 или 3 атома в каждом кольце могут иметь заместитель. Каждое кольцо гетероцикл ильной группы, содержащей гетероатом, может иметь 1, 2, 3 или 4 гетероатомов, выбранных из атомов азота, атомов кислорода и/или атомов серы, где гетероатомы азота и серы необязательно могут быть окислены, и гетероатомы азота необязательно могут быть кватернизованы. Гетероциклильная группа может быть присоединена через любой гетероатом или атом углерода в цикле или циклической системе.
Термин «заместители» относится к группе, являющейся «заместителем» в любой функциональной группе, указанной в настоящей заявке, например в алкильной, алкенильной, алкинильной, циклоалкильной, циклоалкенильной, арильной, гетероциклильной или гетероарильной группе, при любом атоме в данной группе. Подходящие заместители включают, без ограничений, галоген, CN, NO2, OR15, SR15, S(O)2OR15, NR15R16, C1-C2 перфторалкил, C1-C2 перфторалкокси, 1,2-метилендиокси, C(O)OR15, C(O)NR15R16, OC(O)NR15R16, NR15C(O)NR15R16, C(NR16)NR15R16, NR15C(NR16)NR15R16, S(O)2NR15R16, R17, C(O)R17, NR15C(O)R17, S(O)R17, S(O)2R17, R16, оксо, C(O)R16, C(O)(CH2)nOH, (CH2)nOR15, (CH2)nC(O)NR15R16, NR15S(O)2R17, где n независимо равен 0-6 включительно. Каждый R15 независимо представляет собой атом водорода, C1-C4 алкил или С3-С6 циклоалкил. Каждый R16 независимо представляет собой атом водорода, алкенил, алкинил, С3-С6 циклоалкил, арил, гетероциклил, гетероарил, C1-C4 алкил или C1-C4 алкил, замещенный С3-С6 циклоалкилом, арилом, гетероциклилом или гетероарилом. Каждый R17 независимо представляет собой С3-С6 циклоалкил, арил, гетероциклил, гетероарил, C1-C4 алкил или C1-C4 алкил, замещенный С3-С6 циклоалкилом, арилом, гетероциклилом или гетероарилом. Каждый С3-С6 циклоалкил, арил, гетероциклил, гетероарил и C1-C4 алкил в каждом R15, R16 и R17 может необязательно иметь в качестве заместителя галоген, CN, C1-C4 алкил, ОН, C1-C4 алкокси, NH2, C1-C4 алкиламино, C1-C4 диалкиламино, C1-C2 перфторалкил, C1-C2 перфторалкокси или 1,2-метилендиокси.
Термин «оксо» относится к атому кислорода, который образует карбонил, когда присоединен к атому углерода, N-оксид, когда присоединен к атому азота, и сульфоксид или сульфон, когда присоединен к атому серы.
Термин «ацил» относится к алкилкарбонильному, циклоалкилкарбонильному, арилкарбонильному, гетероциклилкарбонильному или гетероарилкарбонильному заместителю, каждый из которых может иметь дополнительные заместители.
Перечисление химических групп в любом определении переменной величины в настоящей заявке включает определения данной переменной как любой отдельной группы или комбинации перечисленных групп. Перечисление вариантов выполнения для переменной величины в настоящей заявке включает варианты выполнения как любой отдельный вариант выполнения или в комбинации с любыми другими вариантами выполнения или их частями.
Соединения по настоящему изобретению могут содержать один или более асимметрических центров, и, таким образом, представлять собой рацематы и рацемические смеси, единственные энантиомеры, индивидуальные диастереомеры и смеси диастереомеров. Все такие изомерные формы указанных соединений включены в настоящее изобретение. Соединения по настоящему изобретению могут также присутствовать в виде нескольких таутомерных форм, в таких случаях настоящее изобретение включает все таутомерные формы описанных в настоящей заявке соединений. Все такие изомерные формы указанных соединений включены в настоящее изобретение. Все кристаллические формы описанных в настоящей заявке соединений включены в настоящее изобретение.
Соединения по настоящему изобретению
Объектом настоящего изобретения является соединение формулы
или его соль, или пролекарство, или соль его пролекарства, или его гидрат, сольват или полиморф, в котором:
R1 представляет собой арилалкил или гетероарилалкил, каждый из которых необязательно замещен 1-4 независимо выбранными Z1;
R3 представляет собой атом водорода, гидроксил, алкокси или алкиламиногруппу;
R6 представляет собой необязательно замещенный арил или гетероарил, насыщенный или ненасыщенный гетероциклил, где R6 необязательно замещен 1-3 группами, независимо выбранными из алкила, циклоалкила, гетероциклила, алкокси, гидроксиалкила, -C(O)NR7R8 и Z1; каждая из которых может независимо быть дополнительно замещенной;
R7 и R8 каждый независимо выбраны из Н, алкила, циклоалкила, алкенила, алкинила, арила, гетероциклила, гетероарила или R7 и R8 вместе с атомом азота образуют гетероциклил или гетероарил;
каждый Z1 представляет собой атом галогена, CN, NO2, OR15, SR15, S(O)2OR15, NR15R16, C1-C2 перфторалкил, C1-C2 перфторалкокси, 1,2-метилендиокси, C(O)OR15, C(O)NR15R16, OC(O)NR15R16, NR15C(O)NR15R16, C(NR16)NR15R16, NR15C(NR16)NR15R16, S(O)2NR15R16, R17, C(O)R17, NR15C(O)R17, S(O)R17, S(O)2R17, R16, оксо, C(O)R16, C(O)(CH2)nOH, (CH2)nOR15, (CH2)nC(O)NR15R16, NR15S(O)2R17, где n независимо равен 0-6 включительно;
каждый R15 независимо представляет собой атом водорода, C1-C4 алкил или С3-С6 циклоалкил;
каждый R16 независимо представляет собой атом водорода, алкенил, алкинил, С3-С6 циклоалкил, арил, гетероциклил, гетероарил, C1-C4 алкил или C1-C4 алкил, замещенный С3-С6 циклоалкилом, арилом, гетероциклилом или гетероарилом;
каждый R17 независимо представляет собой С3-С6 циклоалкил, арил, гетероциклил, гетероарил, C1-C4 алкил или C1-C4 алкил, замещенный С3-С6 циклоалкилом, арилом, гетероциклилом или гетероарилом.
В одном варианте выполнения в настоящем изобретении описано соединение, где R6 представляет собой необязательно замещенный арил или гетероарил, насыщенный или ненасыщенный гетероциклил, где R6 замещен алкилом или группой -C(O)NR7R8. В другом варианте выполнения R6 представляет собой замещенный гетероциклил, где R6 замещен C1-C4 алкилом или C1-C4 алкоксиалкилом.
В другом варианте выполнения R6 представляет собой замещенный арил, где R6 замещен группой -C(O)NR7R8. В другом варианте выполнения R6представляет собой замещенный гетероарил, где R6 замещен группой -C(O)NR7R8.
В одном варианте выполнения в настоящем изобретении описано соединение, в котором R1 представляет собой арилалкил, необязательно замещенный 1-4 независимыми Z1.
В другом варианте выполнения каждый Z1 независимо представляет собой галоген.
В другом варианте выполнения в настоящем изобретении описано соединение, в котором R3 представляет собой Н.
В некоторых вариантах выполнения в настоящем изобретении описано соединение формулы II:
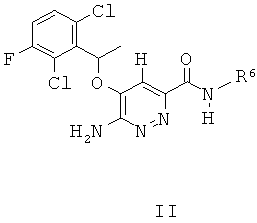
или его соль, или пролекарство, или соль его пролекарства, или его гидрат, сольват или полиморф, в котором:
R6 представляет собой необязательно замещенный арил или гетероарил, насыщенный или ненасыщенный гетероциклил, где R6 необязательно замещен алкилом, циклоалкилом, гетероциклилом, алкокси, гидроксиалкилом или группой -C(O)NR7R8; и
R7 и R8 каждый независимо выбраны из Н, алкила, циклоалкила, алкенила, алкинила, арила, гетероциклила, гетероарила, или R7 и R8 вместе с атомом азота образуют гетероциклил или гетероарил.
Примерные соединения по настоящему изобретению изображены в Таблице 1. В данных примерах стереохимия хиральных атомов углерода независимо соответствует RS, R или S. Изображенные в настоящей заявке структуры, включая структуры в Таблице 1, могут содержать определенные -NH-, -NH2 (амино) и -ОН (гидроксильные) группы, в которых соответствующие атомы водорода не изображены, однако их следует читать как -NH-, -NH2 или -ОН. В некоторых структурах изображена связь в виде черточки, которая обозначает метильную группу.
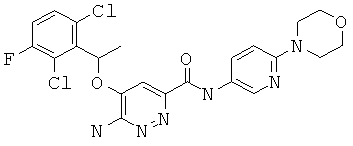
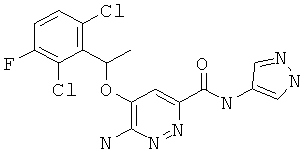
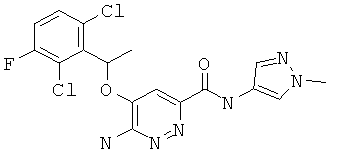
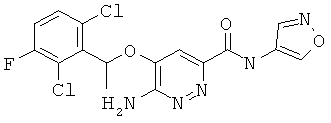
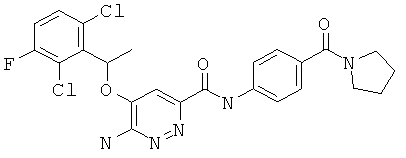
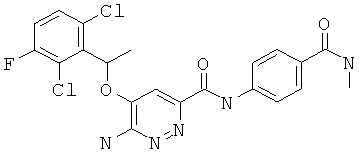
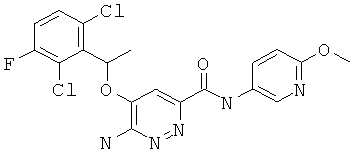
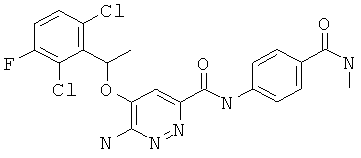
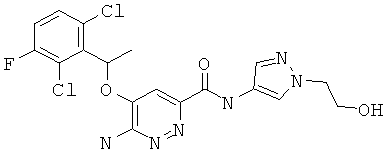
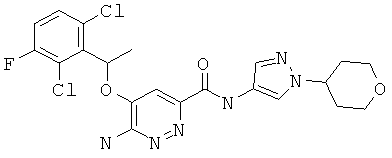
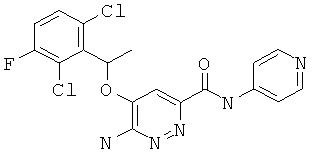
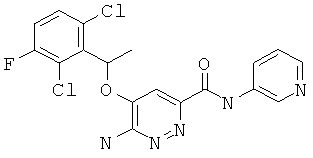
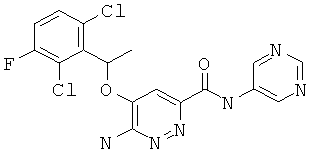
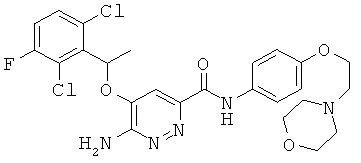
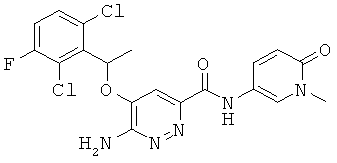
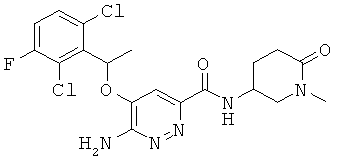
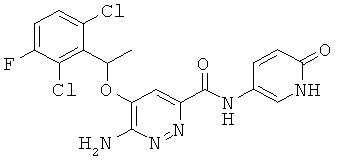
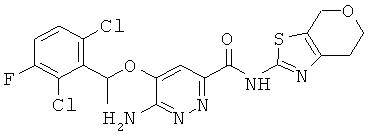
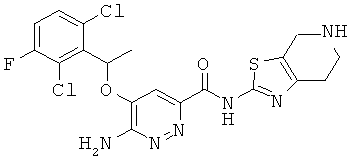
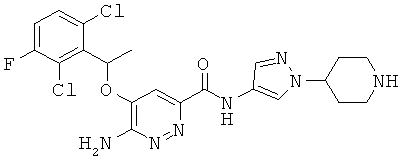
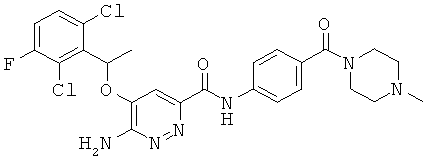
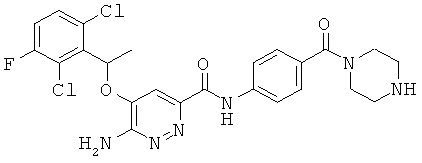
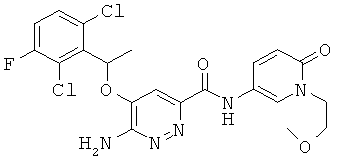
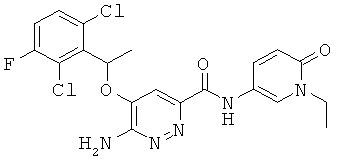
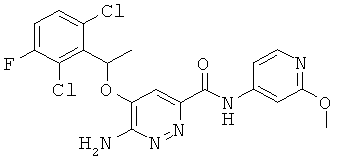
Примерные соединения по настоящему изобретению перечислены ниже:
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-6-морфолин-4-ил-пиридин-3-ил-карбоксамид (1);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-пиразол-4-илкарбоксамид (2);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-метилпиразол-4-ил)карбоксамид (3);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-изоксазол-4-илкарбоксамид (4);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(пирролидинилкарбонил)фенил]карбоксамид (5);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(N-метилкарбамоил)фенил]карбоксамид (6);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6-метокси(3-пиридил))карбоксамид (7);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[6-(N-метилкарбамоил)(3-пиридил)]карбоксамид (8);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[1-(2-гидроксиэтил)пиразол-4-ил]карбоксамид (9);
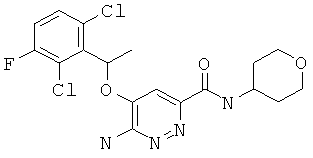
N-(1-(2Н-3,4,5,6-тетрагидропиран-4-ил)пиразол-4-ил){6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}карбоксамид (10);
6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-пиридазин-3-ил-N-(пиридин-4-ил)-карбоксамид (11);
6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-пиридазин-3-ил-N-(пиридин-3-ил)-карбоксамид (12);
6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-пиридазин-3-ил-N-(пиримидин-5-ил)-карбоксамид (13);
6-амино-5-[1-(2,6-дихлор-3-фторфенил)-этокси]-пиридазин-3-карбоновой кислоты (тетрагидро-пиран-4-ил)-амид (14);
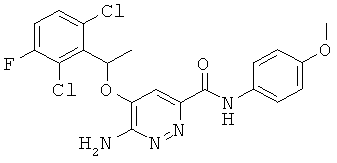
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(4-метоксифенил)карбоксамид (15);
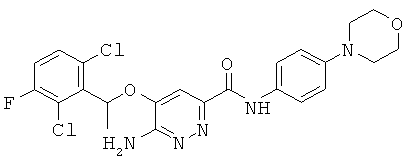
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(4-морфолин-4-илфенил)карбоксамид (16);
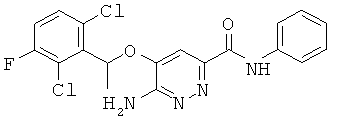
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-бензамид (17);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(2-морфолин-4-илэтокси)фенил]карбоксамид (18);
6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-метил-6-оксо-1,6-дигидро-пиридин-3-ил))карбоксамид (19);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-метил-6-оксо(3-пиперидил))карбоксамид (20);
6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6-оксо-1,6-дигидропиридин-3-ил))карбоксамид(21);
6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6,7-дигидро-4Н-пирано[4,3^]1,3-тиазол-2-ил)карбоксамид (22);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(4,5,6,7-тетрагидро-1,3-тиазоло[5,4-с]пиридин-2-ил)карбоксамид (23);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-(4-пиперидил)пиразол-4-ил)карбоксамид(24);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-{4-[(4-метилпиперазинил)карбонил]фенил}карбоксамид (25);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(пиперазинилкарбонил)фенил]карбоксамид (26);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[1-(2-метоксиэтил)-6-оксо-1,6-дигидро-пиридин-3-ил)]карбоксамид (27);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-этил-6-оксо-1,6-дигидро-пиридин-3-ил))карбоксамид (28); и
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(2-метокси(4-пиридил))карбоксамид (29).
Синтез соединений, соответствующих приведенным в настоящей заявке формулам, может быть легко осуществлен химиками-синтетиками, обладающими обычными для данной области техники навыками. Соответствующие методики и промежуточные продукты описаны, например, в настоящей заявке. Каждый из упомянутых в настоящей заявке патентов, заявок на патент и публикаций, опубликованных в традиционных журналах или доступных только в Интернете, включен в настоящий текст в полном объеме посредством ссылки.
Другие подходы к синтезу соединений, соответствующих приведенным в настоящей заявке формулам, могут быть разработаны на основе процитированных в настоящей заявке ссылок. Варьирование указанных методик и их оптимизация находятся в рамках навыков квалифицированного специалиста в данной области.
Конкретные подходы и соединения, приведенные выше, не являются ограничивающими. В химических структурах на приведенных в настоящей заявке схемах изображены переменные, значения которых соответствуют определениям химических групп (фрагменты, атомы и т.д.) в соответствующих положениях, приведенных в настоящей заявке формул соединений, обозначаемым одинаковыми (например, R1, R2, R, R', Х и т.д.) или разными переменными. В рамках навыков квалифицированного специалиста в данной области находится определение того, подходит ли химическая группа в структуре соединения для использования в синтезе другого соединения. В арсенал квалифицированного специалиста в данной области входят дополнительные методы синтеза соединений, отвечающих приведенным в настоящей заявке формулам, и их предшественников, включая и те, которые соответствуют методам, не представленным в явном виде на схемах в данном тексте. Методы оптимизации условий реакций, при необходимости с минимизацией количеств побочных продуктов, известны в данной области техники. Описанные в настоящей заявке способы могут также дополнительно включать стадии до или после описанных в настоящей заявке стадий, служащие для введения или удаления подходящих защитных групп, применение которых позволяет получать описанные в настоящей заявке соединения. Кроме того, различные стадии синтеза могут осуществляться в другой последовательности или другом порядке, с получением целевых соединений. Синтетические трансформации и методология использования защитных групп (введение защиты и снятие защиты), которые могут применяться для синтеза целевых соединений, известны в данной области техники и включают, например, описанные в книгах R.Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W.Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons (1999); L.Fieser and M.Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); и L.Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995), и в их более поздних изданиях.
Описанные в настоящей заявке способы охватывают превращение соединений одной формулы в соединения другой формулы. Процесс превращения относится к одной или более химическим трансформациям, которые могут осуществляться in situ или с выделением промежуточных соединений. Трансформации могут включать взаимодействие исходных соединений или промежуточных соединений с дополнительными реагентами с использованием известных в данной области техники методик, включая описанные в процитированных источниках. Промежуточные соединения можно использовать после очистки или без очистки (например, фильтрование, перегонка, сублимация, кристаллизация, растирание, твердофазная экстракция и хроматография).
Предусматриваемыми в настоящем изобретении комбинациями заместителей и переменных являются только такие, которые приводят к образованию устойчивых соединений.
В настоящем изобретении также описаны композиции, содержащие эффективное количество соединения, отвечающего любой из приведенных в настоящей заявке формул, или фармацевтически приемлемой соли, соль-вата, гидрата, полиморфа или пролекарства (если применимо) указанного соединения; и приемлемый носитель. Предпочтительно, композиция по настоящему изобретению имеет вид препарата для фармацевтического применения («фармацевтическая композиция»), где носитель представляет собой фармацевтически приемлемый носитель. Носитель (или носители) должен быть «приемлемым» в смысле совместимости с остальными ингредиентами препарата и, в случае фармацевтически приемлемого носителя, не наносить вреда реципиенту в количествах, обычно применяемых в медикаментах.
Фармацевтически приемлемые носители, адъюванты и растворители, которые могут применяться в составе фармацевтических композиций по настоящему изобретению, включают (но не ограничиваются только ими) ионообменные материалы, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные соединения, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, динатрия гидрофосфат, натрия дигидрофосфат, хлорид натрия, соли цинка, коллоидный оксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрия карбоксиметилцеллюлозу, полиакрилаты, воски, полиэтилен-полиоксипропилен блок-полимеры, полиэтиленгликоль и ланолин.
Фармацевтические композиции по настоящему изобретению включают композиции для перорального, ректального, назального, местного (включая буккальное и прием под язык), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения. В некоторых вариантах выполнения настоящего изобретения соединения, отвечающие приведенным в настоящей заявке формулам, вводят чрескожно (например, с использованием чрескожного пластыря). Другие лекарственные формы удобно применять в виде дозированных форм, например таблеток и капсул с замедленным высвобождением, а также липосом, и они могут быть приготовлены любыми хорошо известными в фармацевтике методами. Смотрите, например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Philadelphia, PA (17th ed. 1985).
Такие способы приготовления включают стадию объединения с вводимой молекулой таких ингредиентов, как носитель, который содержит один или более вспомогательных ингредиентов. В целом, композиции получают однородным и аккуратным смешиванием действующих веществ с жидкими носителями, липосомами или тонко измельченными твердыми носителями, затем при необходимости придают продукту нужную форму.
В некоторых предпочтительных вариантах выполнения настоящего изобретения соединение вводят перорально. Композиции по настоящему изобретению, пригодные для перорального применения, могут иметь вид отдельных единиц, таких как капсулы, саше или таблетки, каждая из которых содержит определенное количество действующего вещества в виде порошка или гранул; в виде раствора или суспензии в водном растворе или неводном растворе; или в виде эмульсии типа масло-в-воде или вода-в-масле, или упакованное в липосомы, или в виде болюса и т.д. В качестве контейнера для таких суспензий могут использоваться мягкие желатиновые капсулы, которые могут обладать дополнительным преимуществом, состоящим в повышении скорости всасывания соединения.
Таблетку можно изготавливать методом прессовки или формования, при необходимости с одним или более вспомогательными ингредиентами. Прессованные таблетки можно изготавливать прессовкой в соответствующем устройстве действующего вещества в сыпучем виде, таком как порошок или гранулы, при желании смешанного со связующим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим средством. Формованные таблетки можно изготавливать путем формования в соответствующем устройстве смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки при необходимости можно покрывать оболочкой или делать шероховатыми, а также изготавливать таким образом, чтобы обеспечить замедленное или контролируемое высвобождение действующего вещества. Способы получения таких композиций с замедленным или контролируемым высвобождением фармацевтически активных ингредиентов, таких как описанные в настоящей заявке и другие известные в данной области соединения, известны и описаны в нескольких патентах США, в число которых входят (но ими не ограничиваются) патент US №4,369,172; и 4,842,866, и процитированные в них ссылки. Покрытия могут применяться для доставки соединений в кишечник (смотрите, например, патент США 6,638,534, 5,217,720, и 6,569,457, 6,461,631, 6,528,080, 6,800,663, и процитированные в них ссылки). Пригодной для применения готовой лекарственной формой соединений по настоящему изобретению является форма энтеросолюбильных гранул, в которых оболочка содержит гидроксипропилметилцеллюлозы ацетата сукцинат.
В случае таблеток для перорального применения обычно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие средства, такие как стеарат магния. Для перорального применения в форме капсул подходящие разбавители включают лактозу и высушенный кукурузный крахмал. При пероральном введении водных суспензий действующее вещество объединяют с эмульгирующими и суспендирующими средствами. При желании можно добавлять определенные подсластители, и/или ароматизаторы, и/или красители.
Композиции, подходящие для местного применения, включают ромбовидные пастилки, содержащие ингредиенты в ароматизированной основе, обычно сахарозе и смоле акации или трагакантовой камеди; и пастилки, содержащие действующее вещество в инертной основе, такой как желатин и глицерин, или сахароза и смола акации.
Композиции, подходящие для парентерального применения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферные вещества, бактериостатики и растворенные вещества для того, чтобы сделать препарат изотоничным крови реципиента; и водные и неводные стерильные суспензии, которые могу включать суспендирующие средства и загустители. Препараты могут выпускаться в контейнерах, содержащих одну дозу или несколько доз, например запаянных ампулах и пузырьках, и могут храниться в лиофилизованном состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Растворы для немедленного введения путем инъекции можно готовить из стерильных порошков, гранул и таблеток.
Такие растворы для инъекций могут иметь вид, например, стерильной инъецируемой водной или масляной суспензии. Такую суспензию можно готовить согласно известным в данной области техники методикам, с использованием подходящих диспергирующих или увлажняющих средств (таких как, например, Tween 80) и суспендирующих средств. Стерильным инъецируемым препаратом также может быть стерильный инъецируемый раствор или суспензия в нетоксичном парентерально-приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут применяться, находятся маннит, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно применяются стерильные нелетучие масла. Для этой цели может использоваться любое обычное нелетучее масло, включая синтетические моно- и диглицериды. Жирные кислоты, такие как олеиновая кислота, и их глицеридные производные могут применяться для приготовления инъецируемых растворов, наряду с природными фармацевтически приемлемыми маслами, такими как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных формах. Данные масляные растворы или суспензии могут также содержать длинноцепочечные спирты в качестве разбавителей или диспергаторов.
Фармацевтические композиции по настоящему изобретению могут вводиться в виде суппозиториев для ректального введения. Данные композиции можно приготовить смешиванием соединения по настоящему изобретению с подходящим нераздражающим эксципиентом, твердым при комнатной температуре, но жидким при ректальной температуре, который плавится в прямой кишке, высвобождая действующие вещества. Такие материалы включают (но не ограничиваются только ими) масло какао, пчелиный воск и полиэтиленгликоли.
Фармацевтические композиции по настоящему изобретению можно вводить в виде назальных аэрозолей или путем ингаляции. Такие композиции готовят согласно хорошо известным в данной области техники методикам, и их можно приготовить в виде раствора в физрастворе с применением бензилового спирта или других подходящих консервантов, усилителей всасывания для повышения биодоступности, фтороуглеродов и/или других солюбилизирующих или диспергирующих средств, известных в данной области техники.
Местное нанесение фармацевтических композиций по настоящему изобретению особенно уместно в тех случаях, когда необходимо обработать области или органы, легко доступные для местного нанесения. Для местного нанесения на кожу фармацевтические композиции получают в виде подходящих мазей, содержащих действующие вещества, суспендированные или растворенные в носителе. Носители для местного нанесения соединений по настоящему изобретению включают (но не ограничиваются только ими) минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен-полиоксипропилен соединение, эмульгированный воск и воду. Альтернативно, фармацевтическая композиция может быть получена в виде подходящего лосьона или крема, содержащего действующее вещество, суспендированное или растворенное в носителе. Подходящие носители включают (но не ограничиваются только ими) минеральное масло, моностеарат сорбитана, полисорбат 60, воск из цетиловых сложных эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Фармацевтические композиции по настоящему изобретению можно также местно применять в нижнем отделе желудочно-кишечного тракта с помощью ректальных суппозиториев или в виде соответствующих препаратов для клизмирования. Настоящее изобретение также охватывает трансдермальные пластыри и ионофоретическое введение.
Особенно предпочтительными производными и пролекарствами являются те, которые увеличивают биодоступность соединений по настоящему изобретению, в случае, когда такие соединения вводятся млекопитающим (например, делая перорально вводимое соединение более легко всасываемым в кровь), или те, которые улучшают доставку действующего вещества в биологический отдел (например, в мозг или центральную нервную систему), по сравнению с самими действующими веществами. Предпочтительные пролекарства включают производные, в которых группа, улучшающая растворимость в воде или активный транспорт через стенку кишечника, присоединена к структуре соответствующей формулам, отвечающим описанным в настоящей заявке. Смотри, например, работы Alexander, J. et al. Journal of Medicinal Chemistry 1988, 31, 318-322; Bundgaard, H. Design of Prodrugs; Elsevier: Amsterdam, 1985; pp 1-92; Bundgaard, H.; Nielsen, N.M. Journal of Medicinal Chemistry 1987, 30, 451-454; Bundgaard, H. A Textbook of Drug Design and Development, Harwood Academic Publ.: Switzerland, 1991; pp 113-191; Digenis, G.A. et al. Handbook of Experimental Pharmacology 1975, 28, 86-112; Friis, G.J.; Bundgaard, H. A Textbook of Drug Design and Development, 2 ed.; Overseas Publ.: Amsterdam, 1996; pp 351-385; Pitman, I.H. Medicinal Research Reviews 1981, 1, 189-214.
Введение рассматриваемых терапевтических средств может быть локальным, то есть введение может осуществляться в целевую область. Можно использовать различные методики для введения обсуждаемых композиций в целевые области, такие как инъекции, применение катетеров, трокаров, впрыскиваемых композиций, плюронилового геля, стентов, полимеров с замедленным высвобождением лекарственного средства или других приспособлений для внутреннего доступа.
Согласно другому варианту выполнения в настоящем изобретении описан способ импрегнирования имплантируемого приспособления для высвобождения лекарственного средства, включающий стадию контакта указанного приспособления для высвобождения лекарственного средства с соединением или композицией по настоящему изобретению. Имплантируемые приспособления для высвобождения лекарственного средства включают (но не ограничиваются только ими) капсулы или патроны из биоразлагаемого полимера, капсулы из неразлагаемого проницаемого полимера и пластины из биоразлагаемого полимера.
Согласно другому варианту выполнения в настоящем изобретении описано такое имплантируемое медицинское приспособление, покрытое соединением или композицией, содержащей соединение по настоящему изобретению, которое обладает терапевтической активностью.
В другом варианте выполнения композиция по настоящему изобретению дополнительно содержит второе терапевтическое средство. Второе терапевтическое средство включает любое соединение или терапевтическое средство, имеющее или проявляющее благоприятные свойства при введении отдельно или совместно с соединением, отвечающим любой из приведенных в настоящей заявке формул. Соединения, которые можно успешно комбинировать с указанными соединениями, включают другие ингибиторы киназ и/или другие химиотерапевтические средства для лечения заболеваний и нарушений, описанных выше.
Такие средства подробно описаны в данной области техники. Предпочтительно второе терапевтическое средство представляет собой средство, применяющееся при лечении или профилактике заболевания или состояния, выбранного из рака.
Еще более предпочтительно второе терапевтическое средство, вводимое в состав препарата с соединением по настоящему изобретению, представляет собой средство, применяемое для лечения заболеваний/нарушений, опосредуемых c-met, ron или ALK и их химерными белками, такими как EML4-ALK и NPM-ALK.
В другом варианте выполнения настоящего изобретения настоящее изобретение обеспечивает отдельные дозированные формы соединения по настоящему изобретению и второго терапевтического средства, ассоциированные друг с другом. Термин «ассоциированные друг с другом» при использовании в настоящей заявке означает, что указанные отдельные дозированные формы упакованы вместе или каким-либо другим образом соединены одна с другой, так что очевидно, что эти отдельные дозированные формы должны продаваться и вводиться совместно (с интервалом между ними меньше 24 часов, поочередно или одновременно).
В фармацевтических композициях по настоящему изобретению соединение по настоящему изобретению присутствует в эффективном количестве. При использовании в настоящей заявке термин «эффективное количество» относится к количеству, которого при правильном режиме дозирования достаточно для уменьшения или облегчения тяжести, продолжительности или прогресса подвергающегося лечению расстройства, предотвращения развития подвергающегося лечению расстройства, для регрессии подвергающегося лечению расстройства или для усиления профилактического или терапевтического эффекта другой терапии.
Взаимосвязь дозировок для животных и людей (в миллиграммах на квадратный метр поверхности тела) описана в работе Freireich et al., (1966) Cancer Chemother Rep 50: 219. Площадь поверхности тела можно приблизительно определить исходя из веса и роста пациента. Смотри, например, Scientific Tables, Geigy Pharmaceuticals, Ardley, N.Y., 1970, 537. Эффективное количество соединения по настоящему изобретению может варьировать от около 0.001 мг/кг до около 500 мг/кг, более предпочтительно от 0.1 мг/кг до около 2.5 мг/кг. Эффективные дозировки также варьируются, как известно квалифицированным специалистам в данной области, в зависимости от подвергающегося лечению заболевания, тяжести заболевания, способа введения, пола, возраста и общего состояния здоровья пациента, применения добавок, возможности совместного использования других способов лечения, таких как другие лекарственные средства, и от мнения лечащего врача.
Для фармацевтических композиций, содержащих второе терапевтическое средство, эффективное количество второго терапевтического средства составляет от около 20% до 100% дозировки, обычно применяемой в случае монотерапии при применении данного средства. Предпочтительно эффективное количество составляет от около 70% до 100% от обычной дозировки при монотерапии. Обычные дозировки вторых терапевтических средств при монотерапии хорошо известны в данной области техники. Смотри, например, книги Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), каждая из которых полностью включена в настоящий текст посредством ссылки.
Ожидается, что некоторые из вторых терапевтических средств, упомянутых выше, будут работать синергично с соединениями по настоящему изобретению. Это позволит снизить эффективную дозировку второго терапевтического средства и/или соединения по настоящему изобретению по сравнению с дозировками, необходимыми при монотерапии. Преимущество такого подхода состоит в минимизации токсичных побочных эффектов второго терапевтического средства или соединения по настоящему изобретению, в синергетическом улучшении действия, повышении легкости введения или применения, и/или в уменьшении общего расхода препарата или композиции.
Способы лечения
Согласно другому варианту выполнения в настоящем изобретении описан способ лечения субъекта, страдающего, или подверженного заболеванию, или нарушению или его симптому (например, описанные в настоящей заявке), включающий стадию введения указанному субъекту эффективного количества соединения или композиции по настоящему изобретению. Такие заболевания хорошо известны в данной области техники, а также описаны в настоящей заявке.
В одном варианте выполнения настоящего изобретения указанный способ лечения включает лечение нарушения, которое опосредовано протеинкиназой, например, c-met, ron.
В другом варианте настоящее изобретение обеспечивает способ лечения заболевания у пациента, включающий введение пациенту соединения формулы I.
В другом варианте настоящее изобретение обеспечивает способ лечения заболевания у пациента, включающий введение пациенту композиции, содержащей соединение формулы I.
В некоторых вариантах выполнения указанное заболевание опосредовано c-met или ron киназами.
В другом варианте выполнения указанное заболевание представляет собой рак или пролиферативное заболевание.
В другом варианте выполнения указанное заболевание представляет собой рак легких, ободочной кишки, груди, предстательной железы, печени, поджелудочной железы, мозга, почки, яичника, желудка, кожи или костей, рак желудка, груди, поджелудочной железы, глиому и гепатоклеточную карциному, папиллярный почечно-клеточный рак или плоскоклеточный рак органов головы и шеи.
В одном варианте выполнения способ по настоящему изобретению применяется для лечения субъекта, страдающего или подверженного заболеванию или состоянию. Такие заболевания, нарушения или их симптомы включают, например, модулируемые протеинкиназой (например, c-met, ron, ALK и их химерными белкам, такими как EML4-ALK и NPM-ALK). Заболевание или симптом заболевания могут представлять собой, например, рак или пролиферативное заболевание или нарушение. Заболевание или симптом заболевания могут представлять собой рак легких, ободочной кишки, груди, предстательной железы, печени, поджелудочной железы, мозга, почки, яичника, желудка, кожи или костей, рак желудка, груди, поджелудочной железы, глиому, гепатоклеточную карциному, папиллярный почечно-клеточный рак или плоскоклеточный рак органов головы и шеи. Способы, описанные в настоящем изобретении, включают такие, в которых субъект идентифицируется как нуждающийся в указанном лечении. Идентификация субъекта, как нуждающегося в таком лечении, находится в рамках квалификации субъекта или профессионального медика и может быть субъективной (например, представлять собой суждение) или объективной (например, количественно измеряемой с помощью теста или диагностического метода).
В другом варианте выполнения в настоящем изобретении описан способ модуляции активности протеинкиназы (например, протеин-тирозинкиназа, перечисленные в настоящей заявки киназы) в клетках, включающий контакт клетки с одним или более соединениями, отвечающими любой из приведенных в настоящей заявке формулам.
В другом варианте выполнения, описанный выше способ лечения включает дополнительную стадию совместного введения указанному пациенту одного или более вторых терапевтических средств. Выбор второго терапевтического средства может осуществляться из любых вторых терапевтических средств, применение которых известно для описанных в настоящей заявке показаний. Дополнительные терапевтические средства включают (но не ограничены только ими) средства для лечения заболеваний, нарушений или их симптомов, включая, например, противораковые средства, антипролиферативные средства, антинеопластические средства, противоопухолевые средства, антинеопластические средства антиметаболитного типа/ингибиторы тимидилат-синтазы, антинеопластические средства алкилирующего типа, антинеопластические средства типа антибиотиков или любое другое средство, обычно вводимое в качестве первичного или дополнительного средства согласно протоколам противоопухолевой терапии (например, средства против тошноты, против анемии и т.д.), включая, например, винбластин сульфат, винкристин, виндезин, винестрамид, винорелбин, винтриптол, винзолидин, тамоксифен, торемифен, ралоксифен, дролоксифен, иодоксифен, мегестрол ацетат, анастрозол, летразол, боразол, эксеместан, флутамид, нилутамид, бикалутамид, ципротерон ацетат, госерелин ацетат, лупролид, финастерид, герцептин, метотрексат, 5-фторурацил, цитозин арабинозид, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин, митрамицин, цисплатин, карбоплатин, мелфалан, хлорамбуцил, бусульфан, циклофосфамид, ифосфамид, нитрозомочевины, тиотефан, винкристин, таксол, таксотер, этопозид, тенипозид, амсакрин, иринотекан, топтекан, эпотилон, Iressa, Avastin, OSI-774, ингибиторы ангиогенеза, ингибиторы EGF, ингибиторы МЕК, ингибиторы VEGF, ингибиторы CDK, ингибиторы Her1 и Her2 и моноклональные антитела.
Термин «совместное введение» при использовании в настоящей заявке означает, что второе терапевтическое средство может вводиться совместно с соединением по настоящему изобретению как часть единой лекарственной формы (например, в виде композиции по настоящему изобретению, содержащей соединение по настоящему изобретению и второе терапевтическое средство, как описано выше) или в виде отдельных лекарственных форм. Альтернативно, дополнительное средство может вводиться до введения соединения по настоящему изобретению, последовательно с ним или после него. При такой комбинированной терапии и соединения по настоящему изобретению, и второе терапевтическое средство (средства) вводят общепринятыми способами. Введение субъекту композиции по настоящему изобретению, содержащей и соединение по настоящему изобретению, и второе терапевтическое средство, не исключает отдельного введения того же терапевтического средства, любого другого второго терапевтического средства или любого соединения по настоящему изобретению указанному субъекту в другое время в ходе лечения.
Эффективные количества указанных вторых терапевтических средств хорошо известны квалифицированным специалистам в данной области, и указания по дозировке можно найти в патентах и опубликованных заявках на патенты, ссылки на которые приведены в настоящей заявке, а также в книгах Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishin r, Loma Linda, Calif. (2000), и других медицинских текстах. Однако определение оптимального диапазона эффективных количеств второго терапевтического средства находится в рамках умений квалифицированного специалиста.
В одном варианте выполнения настоящего изобретения, в котором субъекту вводят второе терапевтическое средство, эффективное количество соединения по настоящему изобретению меньше его эффективного количества, которое вводилось бы в случае отсутствия введения второго терапевтического средства. В другом варианте выполнения эффективное количество второго терапевтического средства меньше его эффективного количества, которое вводилось бы в случае отсутствия введения соединения по настоящему изобретению. Таким образом, могут быть сведены к минимуму нежелательные побочные эффекты, связанные с высокими дозировками какого-либо из применяемых средств. Другие потенциальные преимущества (включая, без ограничения, улучшенные режимы дозирования и/или снижение стоимости лекарств) будут очевидны квалифицированным специалистам в данной области.
В другом варианте в настоящем изобретении описано использование соединения, отвечающего любой из приведенных в настоящей заявке формул, отдельно или совместно с одним или более из описанных выше вторых терапевтических средств, для получения лекарственного средства, либо в виде единой композиции, либо в виде отдельных лекарственных форм, для лечения или профилактики у пациента указанного выше заболевания, нарушения или симптома. Другим объектом настоящего изобретения является соединение, отвечающее приведенным в настоящей заявке формулам, предназначенное для лечения или профилактики у пациента описанного в настоящей заявке заболевания, нарушения или их симптома.
В других вариантах выполнения настоящего изобретения описанные в настоящей заявке способы включают способы, дополнительно включающие мониторинг ответа субъекта на применяемое лечение. Такой мониторинг может включать периодический забор образцов тканей субъекта, его жидких сред, проб, клеток, белков, химических маркеров, генетического материала и т.д. как маркеров или индикаторов для применяемого режима лечения. В других способах субъект подвергается предварительному скринингу или идентифицируется как нуждающийся в таком лечении путем анализа соответствующих маркеров или индикаторов, свидетельствующих о необходимости такого лечения.
В одном варианте выполнения в настоящем изобретении описан способ мониторинга прогресса лечения. Данный способ включает стадию определения уровня диагностического маркера (Маркер) (например, любая описанная в настоящей заявке мишень или тип клеток, модулируемые описанным в настоящей заявке соединением) или проведения диагностического замера (например, скрининг, анализ), осуществляемую в отношении пациента, страдающего или подверженного перечисленным в настоящей заявке нарушениям или их симптомам, когда субъекту вводится терапевтическое количество описанного в настоящей заявке соединения, достаточное для лечения заболевания или его симптомов. Уровень Маркера, определяемый в данном способе, можно сравнить с известными уровнями Маркера у контрольных здоровых представителей или у других больных пациентов для определения статуса заболевания у пациента. В предпочтительных вариантах выполнения второй уровень Маркера у субъекта определяют позже, чем определение первого уровня Маркера, и сравнивают полученные два значения для мониторинга течения заболевания или эффективности терапии. В некоторых предпочтительных вариантах выполнения уровень Маркера у пациента до лечения определяют до начала лечения в соответствии с настоящим изобретением; полученный уровень Маркера до лечения затем можно сравнить с уровнем Маркера у субъекта после лечения для определения эффективности лечения.
В некоторых вариантах описанного способа уровень Маркера или активность Маркера у пациента определяют по меньшей мере один раз. Сравнение уровней Маркера, например, с другими значениями уровня Маркера, полученными раньше или позже для того же пациента, для другого пациента или для субъекта, соответствующего норме, может использоваться для определения, оказывает ли терапия согласно настоящему изобретению желаемый эффект, что позволяет соответствующим образом регулировать уровень дозировки. Определение уровней Маркера можно осуществлять с помощью любого подходящего метода забора образцов и анализа экспрессии, известного в данной области техники или описанного в настоящей заявке. Предпочтительно, сначала проводят забор образца ткани или жидкой среды субъекта. Примеры подходящих образцов включают кровь, мочу, ткань, клетки рта или щеки и образцы волос, содержащие корни. Другие подходящие образцы известны квалифицированным специалистам. Определение уровней белка и/или уровней мРНК (например, уровней Маркера) в образце можно осуществлять согласно любой подходящей методике, известной в данной области техники, включая (но не ограничиваясь только ими) иммуноферментный анализ, ELISA (ферментсвязанный иммуносербентный анализ, методики введения радиоактивной метки и последующего анализа, методы блоттинга и хемолюминисценции, ПЦР в режиме реального времени и т.п.
В настоящем изобретении также описаны наборы для применения в лечении заболеваний, нарушений или их симптомов, включая описанные в настоящей заявке. Данные наборы содержат: а) фармацевтическую композицию, содержащую соединение, соответствующее любой из приведенных в настоящей заявке формул, или его соль, или пролекарство, или соль его пролекарства, или его гидрат, сольват или полиморф, где указанная фармацевтическая композиция находится в контейнере; и b) инструкцию, описывающую способ применения фармацевтической композиции для лечения заболеваний, нарушений или их симптомов, включая описанные в настоящей заявке.
Контейнером может служить любой сосуд или другое герметично закрытое или герметично закрывающееся устройство, в которое может помещаться указанная фармацевтическая композиция. Примеры включают сосуды, сосуды с разделенными или несколькими отделами, где каждый отдел или ячейка содержит однократную дозу указанной композиции, разделенную на ячейки упаковку из фольги, в которой каждая ячейка содержит однократную дозу указанной композиции, или дозирующее устройство, отмеряющее однократные дозы указанной композиции. Контейнер может иметь любую общепринятую форму или известную в данной области форму и изготавливается из фармацевтически приемлемого материала, например: бумажная или картонная коробка, стеклянный или пластиковый сосуд или банка, герметично закрывающийся пакет (например, для хранения запаса таблеток, помещаемых в другой контейнер) или блистер с отдельными дозами, которые выдавливаются из упаковки в соответствии с режимом приема. Применяемый тип контейнера может зависеть от конкретной применяемой лекарственной формы, например обычная картонная коробка в целом непригодна для упаковки жидкой суспензии. Допустимо использование более одного контейнера в одной упаковке при производстве лекарственных форм. Например, таблетки могут помещаться в сосуд, который в свою очередь находится в коробке. Предпочтительным контейнером является блистер.
Набор может дополнительно содержать информацию и/или инструкции для врача, фармацевта или субъекта. Такое мнемоническое представление информации включают номера, напечатанные на каждой ячейке или отделении, содержащем дозу, которые соответствуют дням приема, в которые следует принимать обозначенные таким образом таблетки или капсулы, или дни недели, напечатанные на каждой ячейке или отделении, или карточку, содержащую информацию такого типа.
Биологическую активность описанных в настоящей заявке соединений можно оценить по известным в данной области техники методикам, включая, например, описанные в настоящей заявке. Некоторые из описанных в настоящей заявке соединений демонстрируют неожиданно превосходные свойства (например, ингибирование Р450, Met, Ron и т.д.; фармакокинетические свойства и т.д.), которые делают их превосходными кандидатами в качестве потенциальных терапевтических средств.
Все процитированные в настоящей заявке ссылки, относящиеся к печатным изданиям, электронным базам данных, или доступные с помощью компьютера или в других формах, включены в настоящую заявку в полном объеме посредством ссылки, включая (но не ограничиваясь только ими) рефераты, статьи, журналы, публикации, тексты, монографии, таблицы технических данных, веб-сайты, доступные в Интернете, базы данных, патенты, заявки на патенты и патентные публикации.
Примеры
Синтез 5-[(2,6-дихлор-3-фторфенил)этокси]-6-{(трет-бутокси)-N-[(трет-бутил)оксикарбонил]карбониламино}пиридазин-3-карбоновой кислоты (А)
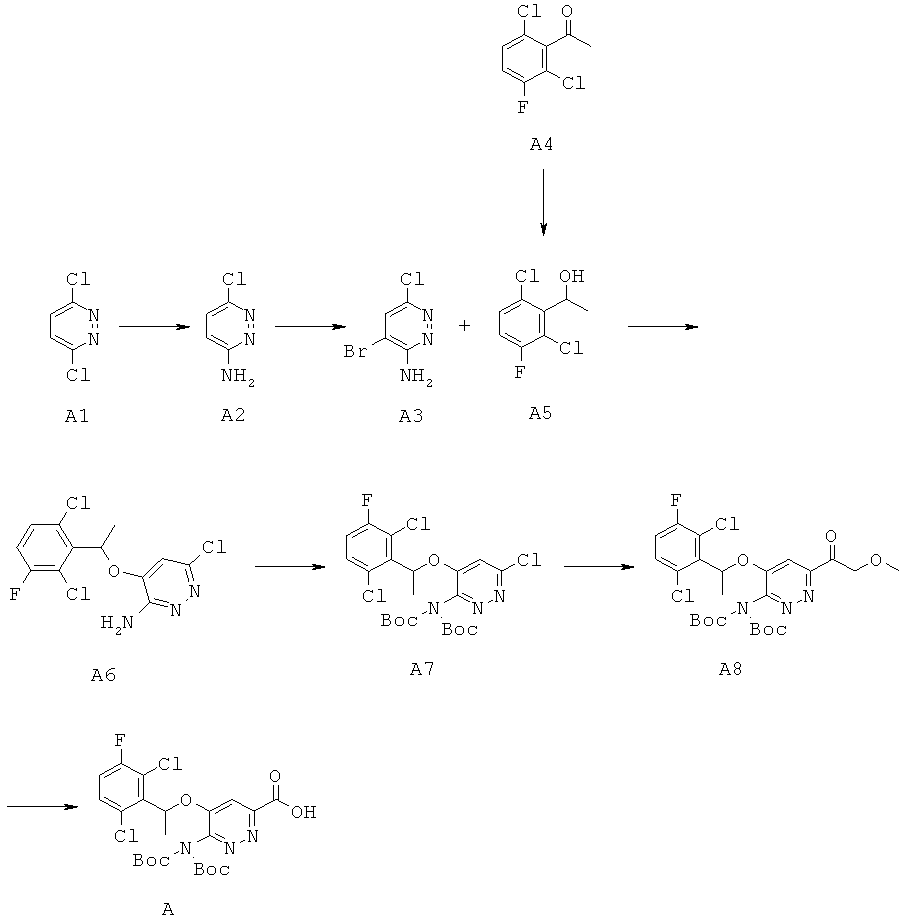
Стадия 1: Суспензию А1 (400 г, 2.68 моль) в 25%-ном растворе гидроксида аммония (3 л) нагревали до 130°С в течение 12 ч в герметично закрытом сосуде. После охлаждения реакционного сосуда до 0°С смесь отфильтровали. Полученный твердый осадок несколько раз промывали водой и сушили в вакууме, получая А2 (284 г, 82%).
Стадия 2: К раствору А2 (284 г, 2.19 моль) в метаноле (3.5 л) добавляли NaHCO3 (368.4 г, 4.38 моль) при комнатной температуре, затем прикалывали бром (350 г, 2.19 моль). По окончании добавления реакционную смесь перемешивали в течение 20 ч, затем фильтровали и промывали несколько раз метанолом. Фильтрат упаривали, остаток растворяли в воде (2 л) и экстрагировали этилацетатом (2 л × 3). Объединенные органические фазы промывали 10%-ным водным раствором тиосульфата натрия (2 л), насыщенным водным раствором бикарбоната натрия (2 л) и насыщенным водным раствором хлорида натрия (2 л), сушили над безводным сульфатом магния и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат: петролейный эфир=2:1), получая A3 (159.8 г, 35%).
Стадия 3: К раствору А4 (150 г, 0.72 моль) в метаноле (800 мл), охлажденному до 0°С, порциями добавляли NaBH4 (66 г, 1.74 моль). Полученную смесь перемешивали при комнатной температуре примерно 1 час, после чего упаривали. К остатку добавляли воду (1 л) при 0°С, затем 3 н. раствор HCl до рН=6. Полученную смесь экстрагировали этилацетатом (1 л × 2). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и упаривали, получая А5 (148.6 г, 98%).
Стадия 4: К раствору А5 (147.6 г, 0.71 моль) в ТГФ (3 л) добавляли 60%-ный NaH (28.4 г, 0.71 моль) при 0°С, полученную смесь перемешивали при той же температуре 30 минут, затем быстро добавляли A3 (147 г, 0.71 ммоль). Полученную смесь нагревали с возвратом флегмы в течение ночи и упаривали. Остаток очищали методом колоночной хроматографии (петролейный эфир: этилацетат=4:1), получая промежуточный продукт А6 (89.3 г, 37.6%).
Стадия 5: К раствору А6 (97 г, 0.288 моль) в ДМФА (1 л) добавляли Boc2O (113 г, 0.519 моль) и DMAP (7 г, 58 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи и упаривали. Остаток очищали методом колоночной хроматографии (петролейный эфир: этилацетат=10:1), получая А7 (136 г, 88%).
Стадия 6: Ацетат натрия (41 г, 0.50 моль) добавляли к раствору А7 (136 г, 0.25 моль) в смеси этанол/ДМФА [(5:1) (1200 мл)]. Полученную смесь дегазировали, после чего добавляли Pd(dppf)Cl2.CH2Cl2 (18.63 г, 22.5 ммоль). Полученную смесь нагревали в атмосфере СО при 90°С в течение 1.5 часов, после чего упаривали. Остаток очищали методом колоночной хроматографии (петролейный эфир:этилацетат=1:4), получая А8 (141 г, 97%).
Стадия 7: К раствору А8 (141 г, 0.246 моль) в ТГФ (650 мл) добавляли 1 н. водный раствор LiOH. (390 мл). Полученную смесь перемешивали при комнатной температуре в течение двух дней, затем подкисляли добавлением 2 н. раствора HCl до рН=5, экстрагировали этилацетатом (300 мл × 5). Объединенные органические фазы сушили над Na2SO4, фильтровали и упаривали, получая А (134 г, 99%).
ПРИМЕР 1: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-6-морфолин-4-ил-пиридин-3-ил карбоксамид (1)
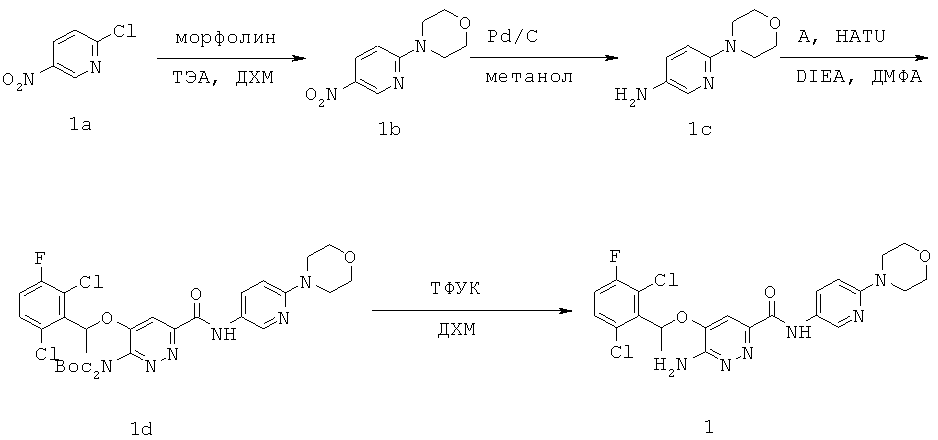
Стадия 1: Смесь 1а (5.0 г, 29.3 ммоль), морфолина (12.8 г, 146.6 ммоль) и ТЭА (10 мл) в ДХМ (30 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (30 мл) и разделяли два слоя. Водный слой экстрагировали ДХМ (30 мл × 2). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали, получая 1b (6.38 г, 98%) в виде желтого твердого вещества.
Стадия 2: Смесь 1b (300 мг, 1.36 ммоль) и Pd/C (10%, 300 мг) в метаноле гидрировали при атмосферном давлении при комнатной температуре в течение 2.5 ч, фильтровали и упаривали, получая 1с (258 мг, 100%).
Стадия 3: К раствору А (200 мг, 0.37 ммоль) в ДМФА (10 мл) добавляли HATU (209 мг, 0.55 ммоль), затем DIEA (95 мг, 0.73 ммоль). Полученную смесь перемешивали при комнатной температуре 30 минут, добавляли 1 с (105 мг, 0.55 ммоль). После перемешивания при комнатной температуре в течение 1.5 часов растворители упаривали и остаток очищали методом колоночной хроматографии (петролейный эфир: этилацетат=1:2), получая 1d (185 мг, 71%).
Стадия 4: К раствору 1d (185 мг, 0.26 ммоль) в ДХМ (3 мл) добавляли ТФУК (1 мл), полученную смесь перемешивали при комнатной температуре 1 час, упаривали и подщелачивали насыщенным раствором Na2CO3 до рН=9, экстрагировали ДХМ (5 мл × 4). Объединенные органические слои сушили и упаривали. Остаток очищали методом колоночной хроматографии (ДХМ: метанол=1:2) и растирали в метаноле, получая 1 (54 мг, 40.7%). 1Н-ЯМР (300 МГц, CDCl3): δ=9.64 (с, 1Н), 8.34 (д, 1Н), 8.09 (дд, 1Н), 7.39 (с, 1Н), 7.31-7.36 (м, 1Н), 7.06-7.12 (м, 1Н), 6.65 (д, 1Н), 6.23-6.26 (м, 1Н), 5.34 (с, 1Н), 3.81-3.84 (м, 4Н), 3.44-3.48 (м, 4Н), 1.89 (д, 3Н). LC-MS [М+Н]+: 507.0
ПРИМЕР 2: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-пиразол-4-илкарбоксамид 2
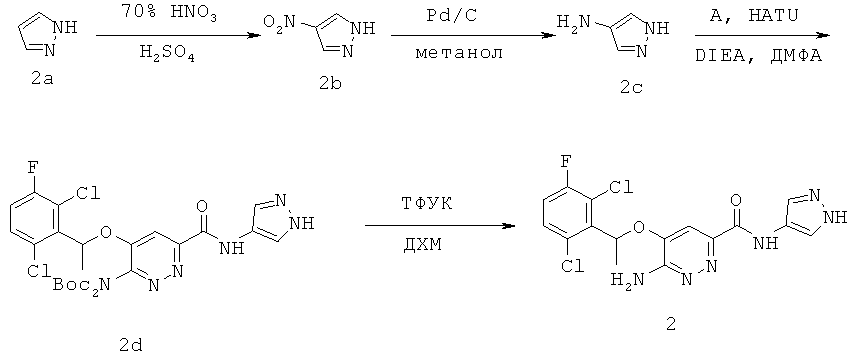
Стадия 1: 2a (5.0 г, 73.5 ммоль) добавляли порциями в H2SO4 (35 мл), поддерживая температуру ниже 40°С, затем прикалывали 70%-ную HNO3 (5.06 мл, 80.6 ммоль), поддерживая температуру ниже 55°С. Полученную смесь нагревали при 55°С в течение 5 часов и охлаждали до 0°С. Полученную смесь нейтрализовывали 50%-ным раствором NaOH, и полученную суспензию разбавляли этилацетатом. Полученный осадок отфильтровывали. Фильтрат отделяли, и органическую фазу промывали водой и насыщенным водным раствором хлорида натрия, сушили над MgSO4 и упаривали в вакууме. Остаток перекристаллизовывали из этанола, получая 2b (7.1 г, 85.5%).
Стадия 2: Методика превращения 2b в 2 аналогична таковой для превращения 1b в 1, при этом было получено вещество 2 (6.9 мг, выход 2 из А равен 2.6%). 1Н-ЯМР (300 МГц, CD3OD): δ=7.87 (д, 2Н), 7.45-7.50 (м, 1Н), 7.27 (т, 1Н), 7.17 (с, 1Н), 6.25-6.32 (м, 1Н), 1.88 (д, 3Н). LC-MS [М+Н]+: 411.0.
ПРИМЕР 3: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-метилпиразол-4-ил)карбоксамид 3
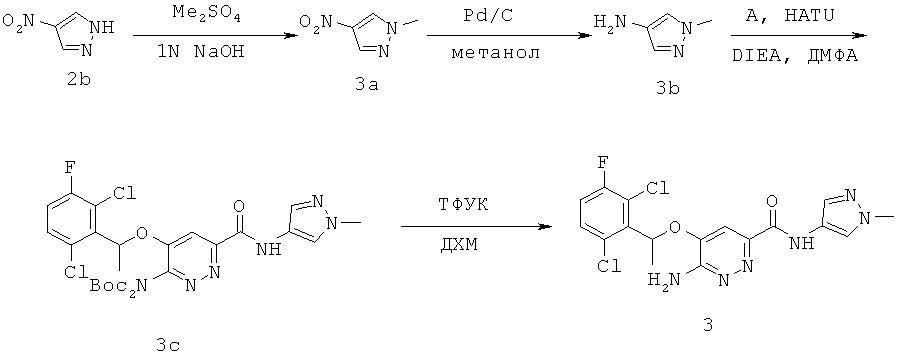
Стадия 1: Диметилсульфат (3.33 г, 26.4 ммоль) медленно добавляли к перемешиваемому раствору 2b (1.0 г, 8.85 ммоль) в 1 н. растворе NaOH (10 мл), нагретому до 30°С. После перемешивания при комнатной температуре в течение 3.5 часов, реакционную смесь экстрагировали этилацетатом (10 мл × 4), объединяли органические фазы, промывали насыщенным водным раствором хлорида натрия (20 мл), сушили над MgSO4, фильтровали и упаривали. Остаток растирали в петролейном эфире и фильтровали, получая 3а (0.98 г, 87%) в виде белого твердого вещества.
Стадия 2: Методика превращения 3а в 3 аналогична таковой для превращения 1b в 1, при этом было получено вещество 3 (133 мг, выход 3 из А составил 42.7%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=10.76 (с, 1Н), 8.02 (с, 1Н), 7.64 (с, 1Н), 7.56-7.61 (м, 1Н), 7.47 (т, 1Н), 7.01 (с, 1Н), 6.82 (с, 2Н), 6.15-6.22 (м, 1Н), 3.78 (с, 3Н), 1.81 (д, 3Н). LC-MS [М+Н]+: 424.9.
ПРИМЕР 4: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-изоксазол-4-илкарбоксамид 4
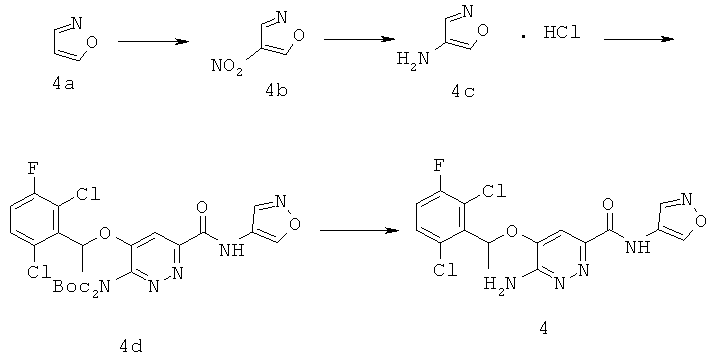
Стадия 1: К раствору 4а (1 г, 14.5 ммоль) в трифторуксусном ангидриде (7 мл, 50.7 ммоль) добавляли нитрат аммония (1.8 г, 22.5 ммоль) порциями по 0.3 г, поддерживая температуру реакции в интервале 25-30°С. По окончании добавления смесь выливали в ледяную воду и экстрагировали ДХМ (15 мл × 4). Экстракт промывали водой, и водный слой экстрагировали ДХМ. Объединенные метиленхлоридные экстракты сушили над MgSO4, фильтровали и упаривали, получая желто-зеленое масло. Полученное масло растирали в гексане (охлажденном до 5°С), получая твердое вещество, которое отфильтровывали, получая 4b (0.72 г, 44%).
Стадия 2: К раствору 4b (200 мг, 1.75 ммоль) в конц. HCl (9 мл) добавляли SnCl2×2H2O (1.98 г, 8.77 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1.5 ч, затем добавляли насыщенный водный раствор Na2CO3 до рН=8-9 и фильтровали. Водную фазу экстрагировали этилацетатом (30 мл × 4), и объединенные экстракты сушили над безводным Na2SO4 и фильтровали. Добавляли к фильтрату 50 мл раствора HCl/Et2O и перемешивали 30 мин, после чего упаривали досуха, получая 4с (125 мг, 59%).
Стадия 3: Превращение 4 с в 4 аналогично превращению 1с в 1, при этом было получено вещество 4 (99 мг, 44%). 1Н-ЯМР (300 МГц, ДМСО): δ=11.13 (с, 1Н), 9.22 (с, 1Н), 8.78 (с, 1Н), 7.57-7.61 (м, 1Н), 7.44-7.49 (м, 1Н), 7.01 (с, 2Н), 6.98 (с, 1Н), 6.15-6.21 (м, 1Н), 1.80 (д, 3Н). LC-MS [М+Н]+: 411.9.
ПРИМЕР 5: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(пирролидинилкарбонил)фенил]карбоксамид 5
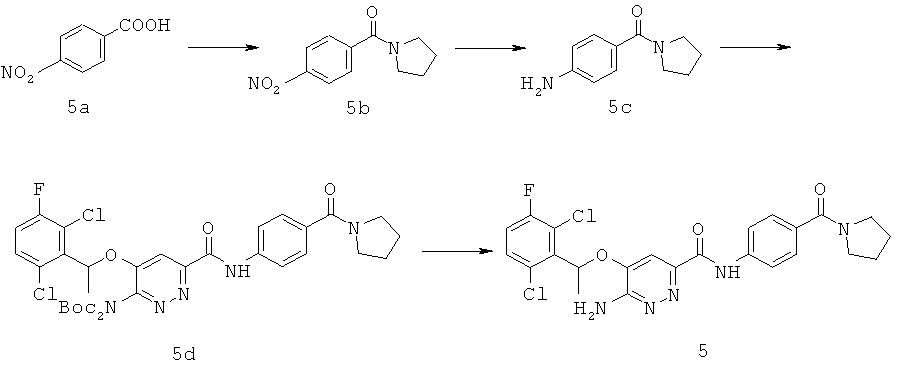
Стадия 1: К раствору 5а (500 мг, 3 ммоль), HATU (1.71 г, 4.5 ммоль) и DIEA (1.16 г, 9 ммоль) в ДМФА добавляли пирролидин (320 мг, 4.5 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре. После упаривания остаток очищали методом колоночной хроматографии (этилацетат:МеОН=4:1), получая 5b (0.52 г, 79%).
Стадия 2: К раствору 5b (370 мг) в МеОН добавляли 10% Pd/C (200 мг). Полученную смесь гидрировали при комнатной температуре в течение 1 ч. Реакционную смесь фильтровали, и фильтрат упаривали, получая 5с (276 мг, 86.5%).
Стадия 3: Превращение 5 с в 5 аналогично превращению 1 с в 1, при этом было получено вещество 5 (54.5 мг, 29%). 1Н-ЯМР (300 МГц, ДМСО): δ=10.69 (с, 1Н), 7.89 (с, 1Н), 7.87 (с, 1Н), 7.56-7.61 (м, 1Н), 7.44-7.50 (м, 3Н), 7.02 (с, 1Н), 6.98 (с, 2Н), 6.19-6.21 (м, 1Н), 3.39-3.46 (м, 4Н), 1.80-1.87 (м, 7H). LC-MS [M+H]+: 518.2.
ПРИМЕР 6: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(N-метилкарбамоил)фенил]карбоксамид 6
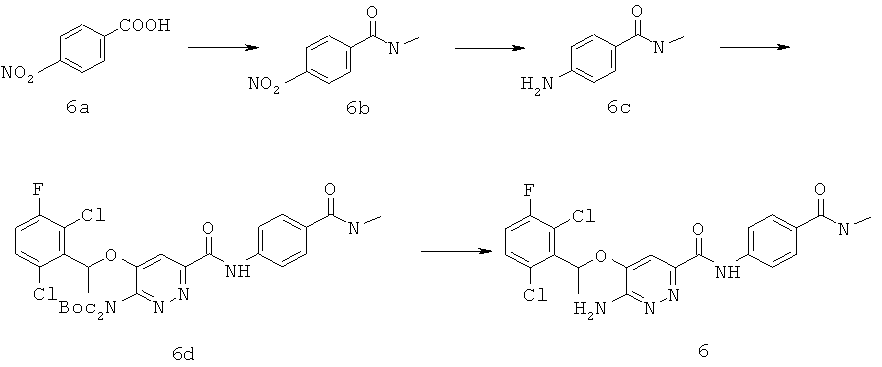
Превращение 6а в 6 аналогично превращению 5а в 5, при этом было получено вещество 6 (53 мг, 44%). 1Н-ЯМР (300 МГц, ДМСО): δ=10.69 (с, 1Н), 8.35-8.39 (м, 1Н), 7.90 (д, 2Н), 7.79 (д, 2Н), 7.57-7.61 (м, 1Н), 7.47-7.50 (м, 1Н), 7.01 (с, 1Н), 6.99(с, 2Н), 6.16-6.22 (м, 1Н), 2.76 (д, 3Н), 1.81 (д, 3Н). LC-MS [М+Н]+: 478.0.
ПРИМЕР 7: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6-метокси(3-пиридил))карбоксамид 7
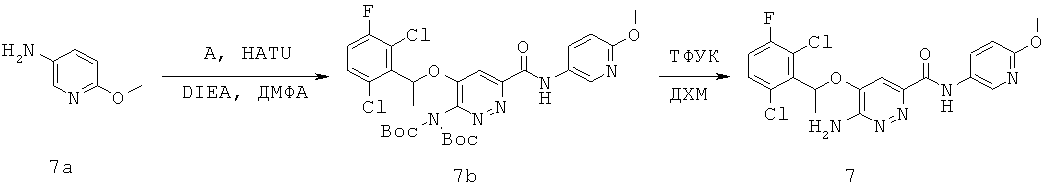
Синтез 7 из 7а проводили аналогично превращению 1с в 1, получая 7 (75 мг, 33%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=10.62 (с, 1Н), 8.54 (д, 1Н), 8.08-8.13 (дд, 1Н), 7.56-7.61 (м, 1Н), 7.44-7.49 (м, 1Н), 7.02 (с, 1Н), 6.94 (с, 2Н), 6.78 (д, 1Н), 6.16-6.22 (м, 1Н), 3.81 (с, 3Н), 1.81 (д, 3Н). LC-MS [М+Н]+: 452.0
ПРИМЕР 8: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[6-(N-метилкарбамоил)(3-пиридил)]карбоксамид 8
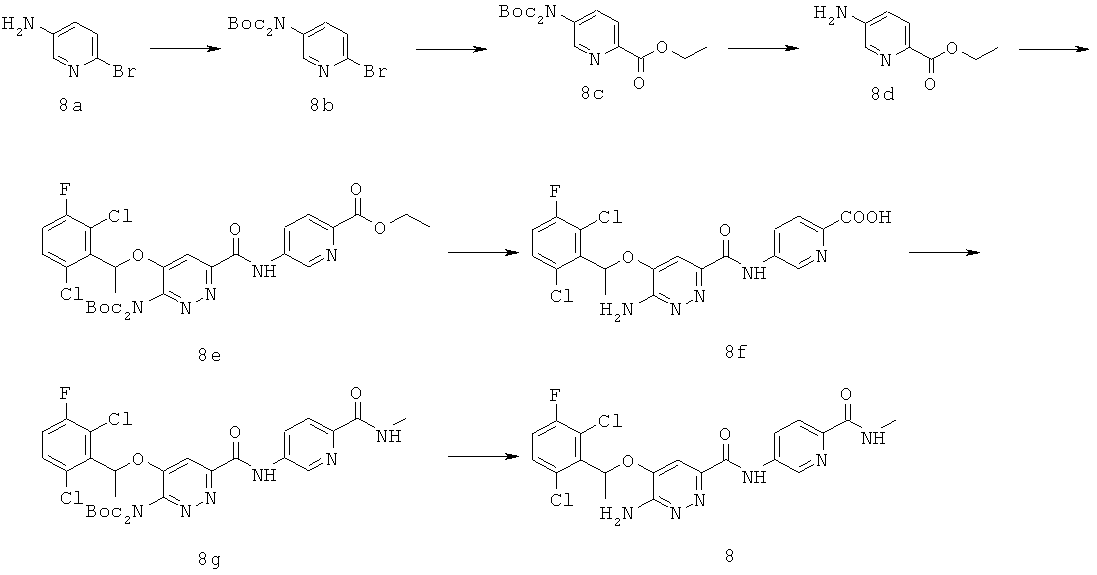
Стадия 1: К раствору 8а (1.13 г, 6.5 ммоль) и Boc2O (2.8 г, 12.8 ммоль) в ДМФА (30 мл) добавляли DMAP (159 мг, 1.3 ммоль) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение ночи и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат: петролейный эфир=1:10), получая 8b (1.05 г, 43%).
Стадия 2: Ацетат натрия (373 мг, 4.55 моль) добавляли к раствору 8b (849 мг, 2.276 моль) в смеси этанол/ДМФА [(5:1) (84 мл)]. Полученную смесь дегазировали, затем добавляли Pd(dppf)Cl2×CH2Cl2 (186 мг, 0.228 ммоль). Полученную смесь нагревали в атмосфере СО при 90°С в течение 1.5 ч, после чего упаривали. Остаток очищали методом колоночной хроматографии (петролейный эфир: этилацетат=10:1), получая 8с (0.7 г, 84%).
Стадия 3: К раствору 8с (350 мг, 0.956 ммоль) в 5 мл ДХМ добавляли ТФУК (1.1 мл, 14.34 ммоль). Полученную смесь перемешивали в течение 2 ч и упаривали досуха, получая 8d.
Стадия 4: Синтез 8е из 8d проводили аналогично превращению А в 5d, получая 8е (390 мг, 70.3%).
Стадия 5: К раствору 8е (390 мг, 0.562 ммоль) в 4 мл ТГФ добавляли 2 мл 1 н. раствора LiOH. Полученную смесь перемешивали в течение 3 часов при комнатной температуре, после чего упаривали большую часть растворителя. Остаток подкисляли до рН=3-4 и экстрагировали ДХМ (20 мл × 3), сушили над Na2SO4 и упаривали, получая 8f (330 мг, 88.2%).
Стадия 6: Синтез 8g из 8f проводили аналогично превращению А в 5d, получая 8g (147 мг, 80.3%).
Стадия 7: Синтез 8 из 8g проводили аналогично превращению 1d в 1, получая 8 (18.5 мг, 18%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=11.04 (с, 1Н), 9.04 (д, 1Н), 8.63-8.67 (м, 1Н), 8.46-8.49 (дд, 1Н), 7.97 (д, 1Н), 7.58-7.62 (м, 1Н), 7.45-7.51 (м, 1Н), 7.06 (с, 3Н), 6.18-6.22 (м, 1Н), 2.77-2.81 (д, 3Н), 1.82 (д, 3Н). LC-MS [М+Н]+: 479.0.
ПРИМЕР 9: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[1-(2-гидроксиэтил)пиразол-4-ил]карбоксамид 9
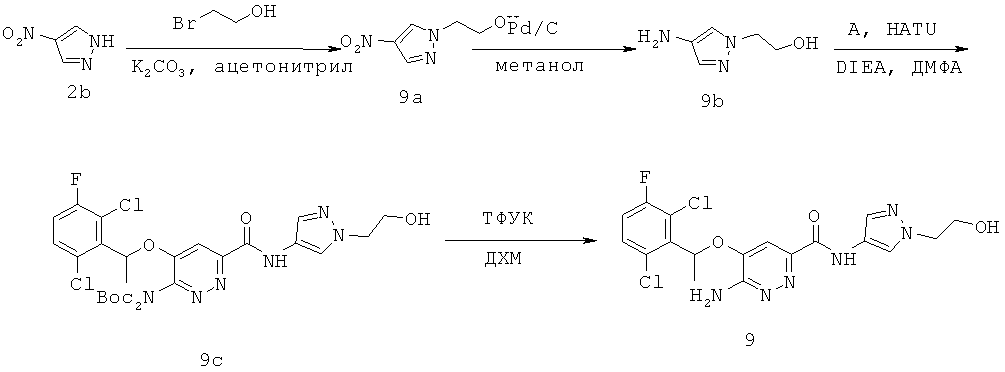
Стадия 1: Раствор 2b (0.8 г, 7.08 ммоль), 2-бромэтан-1-ола (0.97 г, 7.76 ммоль) и К2СО3(1.46 г, 10.56 ммоль) в ацетонитриле (15 мл) нагревали до 60°С в течение 6 часов, затем растворитель упаривали, и к остатку добавляли воду (15 мл), экстрагировали этилацетатом (10 мл × 3), сушили над MgSO4 и упаривали, получая 9a (1.05 г, 94%) в виде белого твердого вещества.
Стадия 2: Методика превращения 9a в 9 аналогична таковой для превращения 1b в 1, при этом было получено вещество 9 (230 мг, выход 9 из А составил 69%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=10.77 (с, 1Н), 8.06 (с, 1Н), 7.66 (с, 1Н), 7.57-7.62 (м, 1Н), 7.47 (т, 1Н), 7.01 (с, 1Н), 6.86 (с, 2Н), 6.17-6.20 (м, 1Н), 4.84 (т, 1Н), 4.08 (т, 2Н), 3.66-3.72 (м, 2Н), 1.81 (д, 3Н). LC-MS [М+Н]+: 454.9.
ПРИМЕР 10: N-(1-(2Н-3,4,5,6-тетрагидропиран-4-ил)пиразол-4-ил){6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}карбоксамид 10
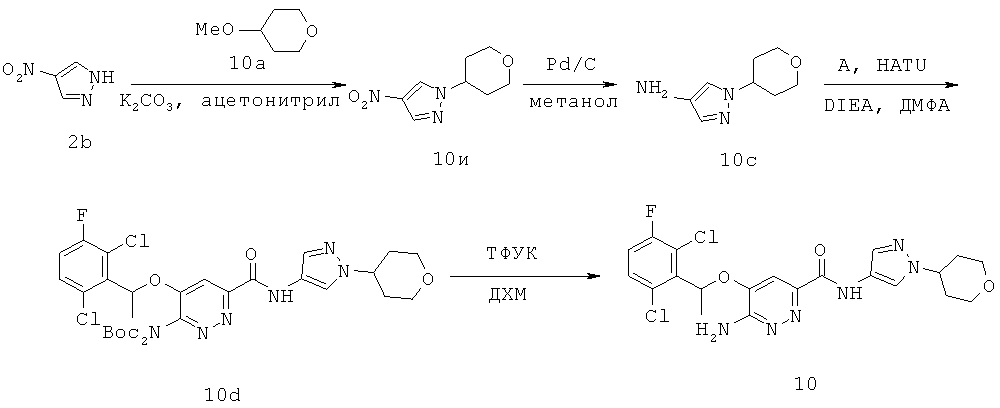
Стадия 1: К раствору 2b (1.0 г, 8.85 ммоль) в ДМФА (30 мл) добавляли NaH (60%, 0.71 г, 10.6 ммоль) при 0°С, и перемешивали при той же температуре 1 час, затем добавляли 10a (2.23 г, 12.4 ммоль). Полученную смесь нагревали до 100°С в течение двух дней, упаривали и очищали колоночной хроматографией, получая 10b (0.822 г, 55.5%).
Стадия 2: Методика превращения 10a в 10 аналогична таковой для превращения 1b в 1, при этом было получено вещество 10 (18.5 мг, выход 10 из А составил 7.5%). 1Н-ЯМР (300 МГц, CDCl3): δ=9.64 (с, 1Н), 8.07 (с, 1Н), 7.55 (с, 1Н), 7.37 (с, 1Н), 7.31-7.36 (м, 1Н), 7.06-7.12 (м, 1Н), 6.23-6.26 (м, 1Н), 5.35 (с, 2Н), 4.28-4.34 (м, 1Н), 4.08-4.13 (м, 2Н), 3.49-3.57 (м, 2Н), 2.05-2.12 (м, 4Н), 1.88 (д, 3Н). LC-MS [М+Н]+: 495.0.
Пример 11: 6-амино-5-[1-(2,6-дихлор-3-фторфенил)-этокси]-пиридазин-3-карбоновой кислоты пиридин-4-иламид
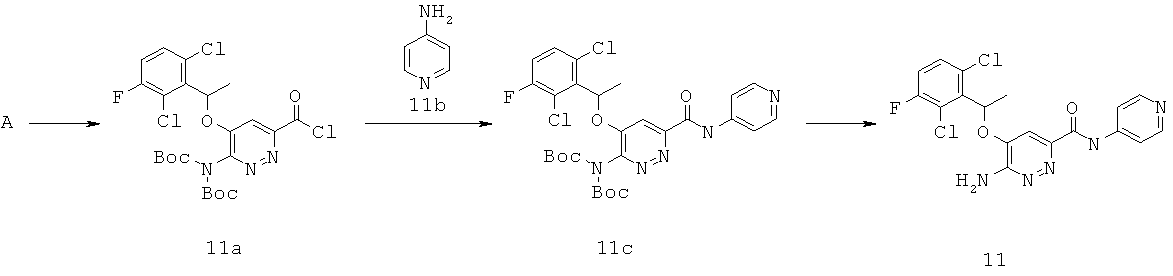
К смеси А (50 мг, 0.092 ммоль) и ТЭА (19 мг, 0.18 ммоль) в ДХМ (5 мл) по каплям добавляли оксалилхлорид (23 мг, 0.18 ммоль) при 0°С. По окончании добавления смесь перемешивали при комнатной температуре 2 часа и упаривали. Остаток растворяли в ДХМ (2 мл) и добавляли к смеси 11b (17 мг, 0.18 ммоль) и ТЭА (46 мг, 0.46 ммоль) в ДХМ (4 мл) по каплям при 0°С. По окончании добавления смесь перемешивали при комнатной температуре 2 дня, после чего упаривали. Остаток растворяли в смеси ДХМ (3 мл) и ТФУК (1 мл), перемешивали при комнатной температуре 2 часа и упаривали. Остаток подщелачивали насыщенным водным раствором Na2CO3 до рН=8 и экстрагировали этилацетатом (10 мл × 5). Объединенные органические фазы сушили над MgSO4 и упаривали. Остаток очищали препаративной ТСХ, получая указанное в заголовке соединение (5.1 мг, 13%). 1Н-ЯМР (300 МГц, CDCl3): δ=9.94 (с, 1Н), 8.52-8.54 (д, 2Н), 7.62-7.64 (дд, 2Н), 7.33-7.38 (м, 2Н), 7.07-7.13 (м, 1Н), 6.24-6.27 (м, 1Н), 5.43 (с, 2Н), 1.89-1.92 (д, 3Н). LC-MS [М+Н]+: 422.0.
Пример 12: 6-амино-5-[1-(2,6-дихлор-3-фторфенил)-этокси]-пиридазин-3-карбоновой кислоты пиридин-3-иламид
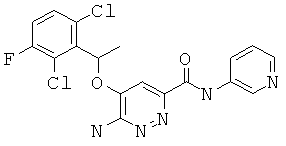
Синтез проводили аналогично описанному в Примере 11 (36 мг, 32% для конечной стадии сочетания). 1Н-ЯМР (300 МГц, CDCl3): δ=9.85 (с, 1Н), 8.79-8.80 (д, 1Н), 8.36-8.38 (дд, 1Н), 8.24-8.28 (м, 1Н), 7.40 (с, 1Н), 7.30-7.40 (м, 1Н), 7.07-7.13 (кв, 1Н), 6.23-6.29 (кв, 1Н), 5.41 (с, 2Н), 1.89-1.91 (д, 3Н). LC-MS [M+H]+: 422.0.
Пример 13: 6-амино-5-[1-(2,6-дихлор-3-фторфенил)-этокси]-пиридазин-3-карбоновой кислоты пиримидин-5-иламид
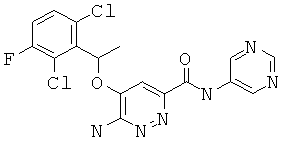
Синтез проводили аналогично описанному в Примере 11. 1Н-ЯМР (300 МГц, CDCl3): δ=9.86 (с, 1Н), 9.16 (с, 2Н), 8.99 (с, 1Н), 7.34-7.39 (м, 2Н), 7.08-7.14 (кв, 1Н), 6.22-6.27(кв, 1Н), 5.47 (с, 2Н), 1.89-1.92 (д, 1Н). LC-MS [M+H]+: 423.0.
Пример 14: 6-амино-5-[1-(2,6-дихлор-3-фторфенил)-этокси]-пиридазин-3-карбоновой кислоты (тетрагидро-пиран-4-ил)-амид
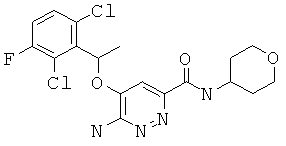
Синтез проводили аналогично описанному в Примере 11 (1.0 мг, 13% для конечной стадии). 1Н-ЯМР (300 МГц, CDCl3): δ=7.30-7.35 (м, 1Н), 7.06-7.13 (м, 1Н), 6.79 (с, 1Н), 6.13-6.19 (м, 1Н), 5.16 (с, 2Н), 4.16-4.26 (м, 1Н), 3.48-3.78 (м, 2Н), 1.83-1.85 (д, 3Н), 1.60-1.60 (м, 6Н). LC-MS [M+H]+: 429.1.
ПРИМЕР 15: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(4-метоксифенил)карбоксамид
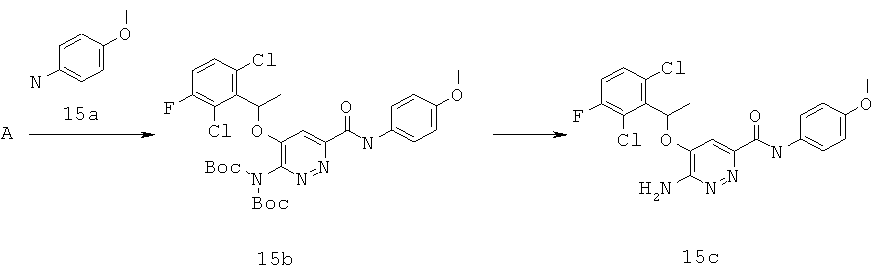
Стадия 1: Смесь А (300 мг, 0.55 ммоль), HATU (313 мг, 0.82 ммоль) и DIEA (142 мг, 1.10 ммоль) в ДМФА (15 мл) перемешивали при комнатной температуре 0.5 часа, затем добавляли 15а (74 мг, 0.60 ммоль). Полученную смесь перемешивали при комнатной температуре 0.5 часа и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат: петролейный эфир=1:4), получая 15b (196 мг, 55%).
Стадия 2: 15b (196 мг, 0.30 ммоль) растворяли в смеси ДХМ (5 мл) и ТФУК (1.5 мл), перемешивали при комнатной температуре 2 часа и упаривали. Остаток доводили добавлением насыщенного водного раствора Na2CO3 до рН=8 и экстрагировали этилацетатом (10 мл × 5). Объединенные органические фазы сушили над MgSO4 и упаривали. Остаток растирали в метаноле и фильтровали, получая 15 (114 мг, 84%). 1Н-ЯМР (300 МГц, CDCl3): 5=1.88 (д, 3Н), 3.80 (с, 3Н), 5.34 (с, 2Н), 6.21-6.29 (м, 1Н), 6.87-6.90 (м, 2Н), 7.06-7.11 (м, 1Н), 7.31-7.36 (м, 1Н), 7.42 (с, 1Н), 7.58-7.62 (м, 2Н), 9.69 (с, 1H). LC-MS [M+H]+: 450.9.
ПРИМЕР 16: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(4-морфолин-4-илфенил)карбоксамид

Синтез проводили аналогично описанному в Примере 15 (95 мг, 51% для последней стадии). 1Н-ЯМР (300 МГц, CDCl3): δ=1.88 (д, 3Н), 3.12 (т, 4Н), 3.86 (т, 4Н), 5.33 (с, 2Н), 6.24-6.26 (м, 1Н), 6.90 (д, 2Н), 7.05-7.11 (м, 1Н), 7.31-7.36 (м, 1Н), 7.41 (с, 1Н), 7.60 (д, 2Н), 9.68 (с, 1Н). LC-MS [M+H]+: 505.9.
ПРИМЕР 17: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-бензамид
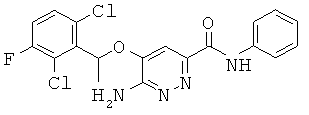
Синтез проводили аналогично описанному в Примере 15 (50 мг, 32% для последней стадии). 1Н-ЯМР (300 МГц, CDCl3): δ=1.90 (д, 3Н), 5.34 (с, 2Н), 6.23-6.29 (м, 1Н), 7.06-7.15 (м, 2Н), 7.32-7.38 (м, 3Н), 7.43 (с, 1Н), 7.68-7.71 (м, 2Н), 9.79 (с, 1Н). LC-MS [M+H]+: 420.9.
ПРИМЕР 18: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил)-N-[4-(2-морфолин-4-илэтокси)фенил]карбоксамид
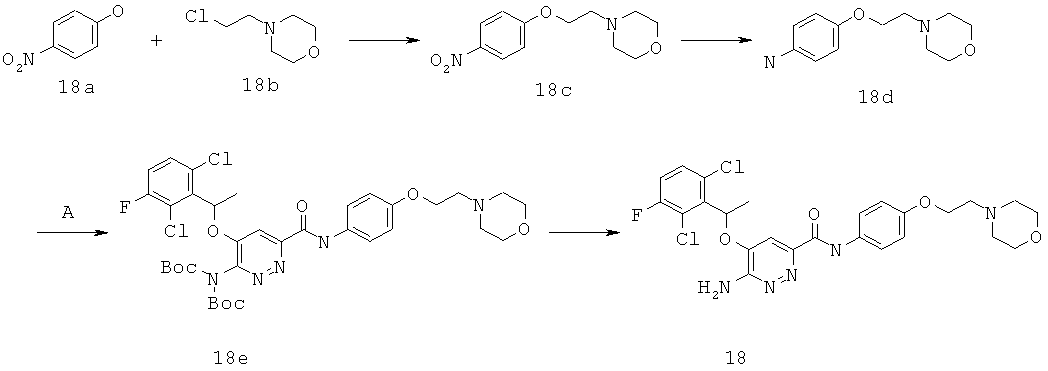
Стадия 1: Смесь 18a (3.04 г, 22 ммоль), 18b (3.72 г, 20 ммоль) и К2СО3 (2.07 г, 60 ммоль) в CH3CN (80 мл) и нагревали с возвратом флегмы 2.5 часа. Твердый осадок отфильтровывали, и фильтрат упаривали в вакууме. Остаток очищали методом колоночной хроматографии (этилацетат: петролейный эфир=1:2), получая 18c (4.58 г, 91%).
Стадия 2: К раствору 18 с (160 мг, 0.63 ммоль) в метаноле (10 мл) добавляли 10% Pd/C (140 мг). Полученную смесь гидрировали в атмосфере H2 в течение ночи. Pd/C отфильтровывали, и фильтрат упаривали, получая неочищенный 18d (135 мг, 96%), который использовали в следующей стадии без дополнительной очистки.
Стадия 3: Методика превращения 18d в 18 аналогична описанной в Примере 15 (131 мг, 44% из А). 1Н-ЯМР (300 МГц, CDCl3): δ=1.89 (д, 3Н), 2.57 (т, 4Н), 2.79 (т, 2Н), 3.73 (т, 4Н), 4.10 (т, 2Н), 5.34 (с, 2Н), 6.22-6.28 (м, 1Н), 6.87-6.92 (м, 2Н), 7.06-7.08 (м, 1Н), 7.31-7.36 (м, 1Н), 7.41 (с, 1Н), 7.57-7.62 (м, 2Н), 9.69 (с, 1Н). LC-MS [M+H]+: 550.0.
ПРИМЕР 19: 6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-метил-6-оксо-1,6-дигидро-пиридин-3-ил))карбоксамид

Стадия 1: К раствору 19a (1.0 г, 7.14 ммоль) в ДМФА (30 мл) добавляли NaH (0.34 г, 8.57 ммоль). Суспензию перемешивали при 0°С в течение 0.5 ч и прикапывали CH3I (1.1 г, 7.86 ммоль) при 0°С. Полученную смесь оставляли нагреваться до комнатной температуры в течение 1 часа и упаривали. К остатку добавляли насыщенный раствор NaHCO3 (5 мл) и воду (5 мл). Суспензию экстрагировали ДХМ (15 мл) дважды. Объединенные экстракты промывали водой, сушили над MgSO4 и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат: петролейный эфир=1:20), получая 19b (693 мг, 63%).
Стадия 2: Порошок восстановленного железа (129 мг, 2.30 ммоль) и 2 н. раствор HCl (0.07 мл) добавляли к перемешиваемому раствору 19b (113 мг, 0.33 ммоль) в этаноле (3 мл) при 0°С. Полученную смесь нагревали с обратным холодильником 2 часа и фильтровали. Коричневый осадок промывали несколько раз этанолом. Объединенные этанольные фазы упаривали, остаток растворяли в этилацетате (15 мл) и промывали 1.5 н. водным раствором Na2CO3 (20 мл). Двухфазную смесь разделяли, и водную фазу повторно экстрагировали этилацетатом (15 мл × 3). Объединенные органические фазы сушили над MgSO4, фильтровали и упаривали, получая 19с (205 мг, около 100%).
Стадия 3: Методика превращения 19с в 19 аналогична описанной в Примере 15 (70 мг, 42% из А). 1Н-ЯМР (300 МГц, CDCl3): δ=1.89 (д, 3Н), 3.57 (с, 3Н), 5.40 (с, 2Н), 6.21-6.27 (м, 1Н), 6.59 (д, 1Н), 7.06-7.12 (м, 1Н), 7.26-7.37 (м, 3Н), 8.28 (д, 1Н), 9.40 (с, 1Н). LC-MS [M+H]+: 451.9.
ПРИМЕР 20: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-метил-6-оксо(3-пиперидил))карбоксамид
Стадия 1: Методика превращения 19b в 20а аналогична таковой для превращения 18с в 18d, при этом было получено вещество 20а (92 мг, 91% из).
Стадия 2: Методика превращения 20а в 20 аналогична описанной в Примере 15 (131 мг, 21% из А). 1Н-ЯМР (300 МГц, CDCl3): δ=1.88 (д, 3Н), 1.92-2.08 (м, 2Н), 2.47-2.54 (м, 2Н), 2.92 (д, 3Н), 3.20-3.27 (м, 1Н), 3.59-3.65 (м, 1Н), 4.39-4.42 (м, 1Н), 5.37 (с, 2Н), 6.18-6.24 (м, 1Н), 7.06-7.11 (м, 1Н), 7.31-7.36 (м, 2Н), 7.95 (д, 1Н). LC-MS [M+H]+: 457.1.
ПРИМЕР 21: 6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6-оксо-1,6-дигидропиридин-3-ил))карбоксамид
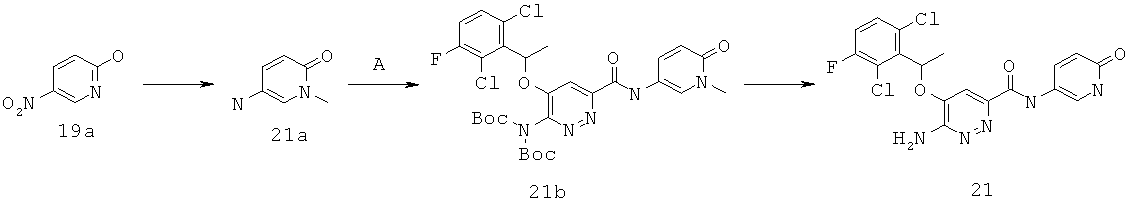
Стадия 1: Методика превращения 19а в 21а аналогична таковой для превращения 19b в 19с, при этом было получено вещество 21а, которое использовали в следующей стадии без дополнительной очистки.
Стадия 2: Методика превращения 21а в 21 аналогична описанной в Примере 15 (6.8 мг, 4.2% из 39 с). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.82 (д, 3Н), 6.14-6.21 (м, 1Н), 6.32 (д, 1Н), 6.89 (с, 2Н), 6.99 (с, 1Н), 7.47 (т, 1Н), 7.56-7.61 (м, 1Н), 7.76-7.80 (м, 1Н), 7.93 (с, 1Н), 10.40 (с, 1Н), 11.41 (ушир.с, 1Н). LC-MS [М+Н]+: 437.9.
ПРИМЕР 22: Синтез 6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6,7-дигидро-4Н-пирано[4,3-d]1,3-тиазол-2-ил)карбоксамида
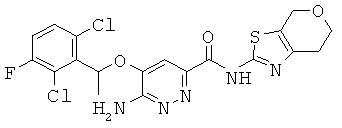
Синтез проводили аналогично описанному в Примере 15 (126 мг, 36% для последней стадии). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.83 (д, 3Н), 2.64-2.73 (м, 2Н), 3.92 (т, 2Н), 4.68 (с, 2Н), 6.18-6.24 (м, 1Н), 6.98-7.12 (м, 3Н), 7.46 (т, 1Н), 7.58-7.62 (м, 1Н), 11.69 (с, 1Н). LC-MS [М+Н]+: 484.1.
ПРИМЕР 23: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил)-N-(4,5,6,7-тетрагидро-1,3-тиазоло[5,4-с]пиридин-2-ил)карбоксамид
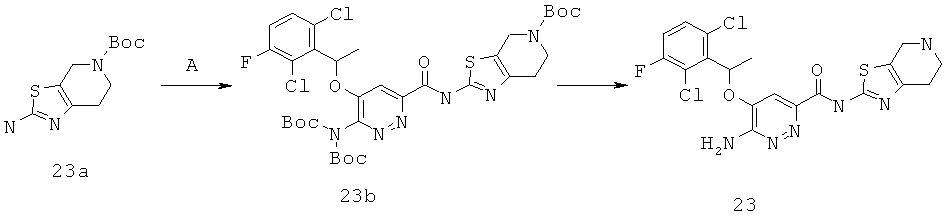
Синтез проводили аналогично описанному в Примере 15 (102 мг, 42% для последней стадии). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.82 (д, 3Н), 1.97-2.03 (м, 1Н), 2.51-2.58 (м, 2Н), 2.96 (т, 2Н), 3.80 (с, 2Н), 6.18-6.24 (м, 1Н), 6.99 (с, 1Н), 7.07 (ушир.с, 2Н), 7.48 (т, 1Н), 7.58-7.63 (м, 1Н). LC-MS [M+H]+: 482.9.
ПРИМЕР 24: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-(4-пиперидил)пиразол-4-ил)карбоксамид
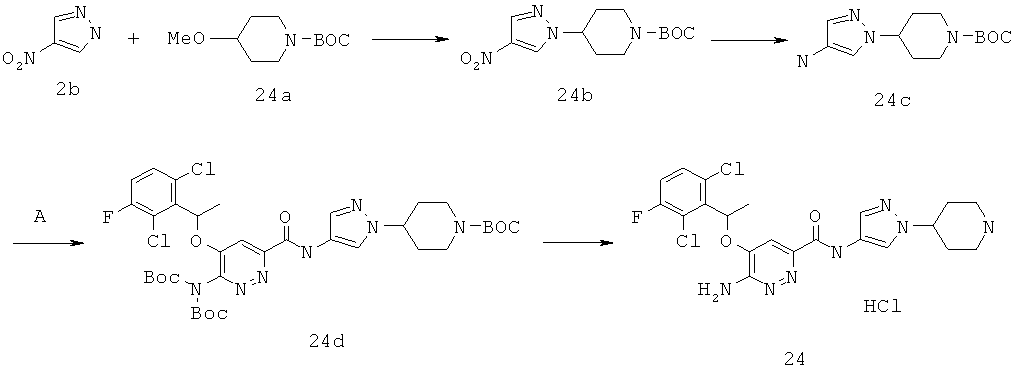
Синтез проводили аналогично описанному в Примере 10 (6.8 мг). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.87 (д, 3Н), 2.14-2.19 (м, 4Н), 2.99-3.06 (м, 2Н), 3.32-3.44 (м, 2Н), 4.42-4.50 (м, 1Н), 6.28-6.34 (м, 1Н), 7.14 (с, 1Н), 7.51 (т, 1Н), 7.61-7.66 (м, 1Н), 7.70 (с, 1Н), 8.07 (с, 1Н), 8.69 (ушир.с, 1Н), 9.16-9.18 (м, 1Н), 9.39-9.42 (м, 1Н), 10.93 (с, 1Н). LC-MS [M+H]+: 494.0
ПРИМЕР 25: {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-{4-[(4-метилпиперазинил)карбонил]фенил}карбоксамид
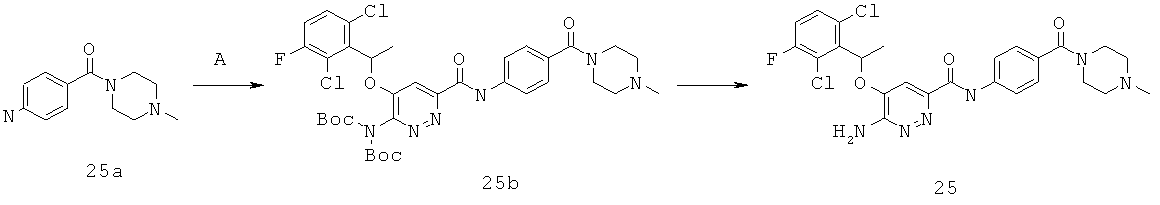
Синтез проводили аналогично описанному в Примере 15 (102 мг, 42% для последней стадии). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=1.84 (д, 3Н), 2.78 (д, 3Н), 3.02-3.11 (м, 2Н), 3.35-3.43 (м, 4Н), 3.77-3.96 (м, 2Н), 6.20-6.27 (м, 1Н), 7.06 (с, 1Н), 7.42-7.63 (м, 5Н), 7.94 (д, 2Н), 10.59 (ушир.с, 1Н), 10.75 (с, 1Н). LC-MS [M+H]+: 547.1.
ПРИМЕР 26: Синтез {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(пиперазинилкарбонил)фенил]карбоксамид
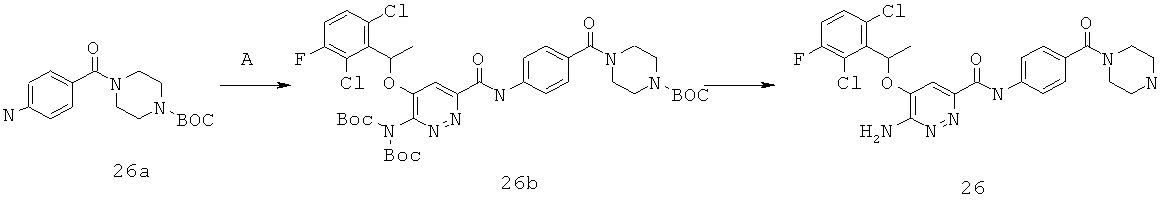
Синтез проводили аналогично описанному в Примере 15 (139 мг, 70% из А). 1Н-ЯМР (300 МГц, CDCl3): δ=0.85-0.86 (м, 1Н), 1.90 (д, 3Н), 2.88 (м, 4Н), 3.56 (ушир.с, 4Н), 5.40 (с, 2Н), 6.22-6.29 (м, 1Н), 7.06-7.12 (м, 1Н),7.32-7.37 (м, 2Н), 7.40-7.43 (м, 2Н), 7.72-7.75 (м, 2Н), 9.89 (с, 1Н). LC-MS [M+H]+: 533.0.
ПРИМЕР 27: Синтез {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[1-(2-метоксиэтил)-6-оксо-1,6-дигидро-пиридин-3-ил)]карбоксамида
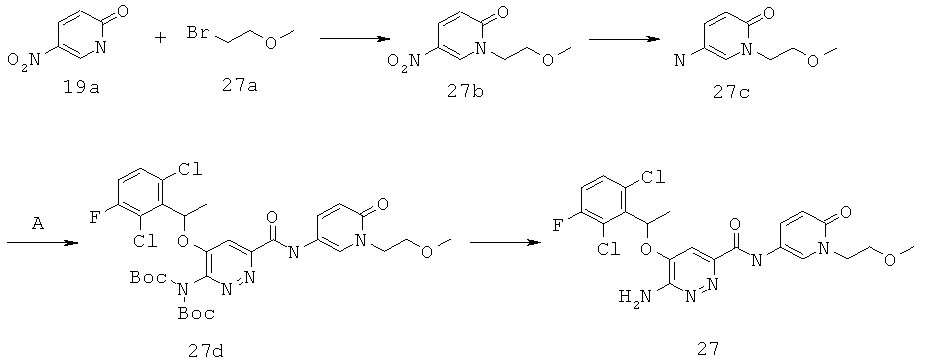
Синтез проводили аналогично описанному в Примере 19 (157 мг, 56% из А). 1Н-ЯМР (300 МГц, CDCl3): δ=1.89 (д, 3Н), 3.32 (с, 3Н), 3.69 (т, 2Н), 4.10-4.15 (м, 2Н), 5.38 (с, 2Н), 6.23-6.27 (м, 1Н), 6.58 (д, 1Н), 7.07-7.12 (м, 1Н), 7.32-7.44 (м, 3Н), 8.13 (д, 1Н), 9.39 (с, 1Н). LC-MS [М+Н]+: 496.0.
ПРИМЕР 28: Синтез {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-этил-6-оксо-1,6-дигидро-пиридин-3-ил))карбоксамида.
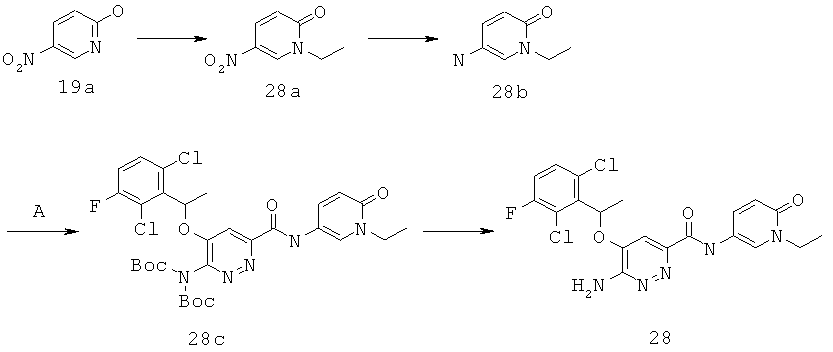
Стадия 1: Гидрид натрия (0.63 г 60%-ной дисперсии в минеральном масле, 15.8 ммоль) добавляли к раствору соединения 19а (2 г, 14.4 ммоль) в ДМФА (20 мл) при комнатной температуре и перемешивали 30 минут. В реакционную смесь добавляли этилиодид (2.2 г, 14.4 ммоль) и перемешивали 16 часов при комнатной температуре. Реакционную смесь разбавляли этилацетатом, промывали водой, сушили над сульфатом натрия и упаривали в вакууме, получая соединение 28а (2 г, 60%).
Стадия 2: Смесь соединения 28а (5 г, 29.7 ммоль) и Fe (6.7 г, 119 ммоль) в АсОН (5 мл), воде (50 мл) и МеОН (50 мл) нагревали с возвратом флегмы 30 минут. Растворитель удаляли в вакууме, и остаток очищали методом колоночной хроматографии, получая соединение 28b (2.5 г, 60%).
Стадия 3: К раствору соединения 28b (1 г, 7.25 ммоль) в ДМФА (30 мл) добавляли HATU (4.13 г, 10.87 ммоль), соединение А (20 мг, 163 ммоль), DIEA (3.8 мл, 21.74 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь обрабатывали водой и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над MgSO4 и упаривали при пониженном давлении, и полученный сырой продукт очищали методом флэш-хроматографии (ДХМ:МеОН=10:1), получая соединение 28с (3.2 г, 66%).
Стадия 4: К раствору соединения 28 с (2 г, 3 ммоль) в ДХМ (5 мл) добавляли ТФУК (3 мл). Полученную смесь перемешивали при комнатной температуре 4 часа и упаривали. Остаток очищали методом колоночной хроматографии (ДХМ:МеОН=20:1), получая 28 (700 мг, 50%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=10,04 (с, 1Н), 8.23-8.24 (д, 1Н), 7.69-7.73 (дд, 1Н), 7.56-7.61 (м, 1Н), 7.44-7.50 (т, 1Н), 6,97 (с, 1Н), 6.92 (с, 2Н), 6.33-6.37 (д, 1Н), 6.15-6.18 (kb, 1Н), 3.85-3.92 (кв, 2Н), 1.80-1.82 (д, 3Н), 1.17-1.22 (т, 3Н). LC-MS [M+H]+: 467.0.
ПРИМЕР 29: Синтез {6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(2-метокси(4-пиридил))карбоксамида.
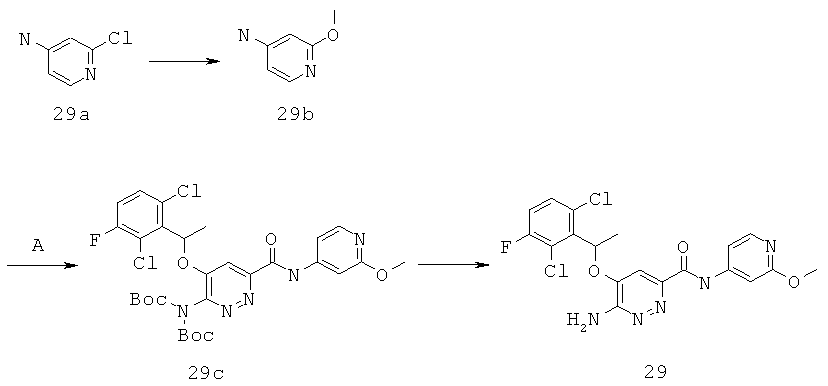
Стадия 1: 4-амино-2-хлорпиридин (15 г, 117 ммоль, 1.0 экв.) растворяли в 100 мл ТГФ. Добавляли раствор метоксида натрия в метаноле (1.0 М, 234 мл, 234 ммоль, 2.0 экв.) и полученный раствор нагревали с возвратом флегмы в герметично закрытом сосуде 16 часов при 140°С. Реакционную смесь выливали в 500 мл интенсивно перемешиваемого насыщенного раствора бикарбоната натрия. Добавляли 500 мл этилацетата и разделяли слои. Органический слой сушили над сульфатом натрия, декантировали и упаривали в вакууме. Хроматографическая очистка на SiO2 (30% этилацетата в гексанах) дала 29b в виде желтого твердого вещества (2.1 г, 14%).
Стадия 2-3: Дальнейший синтез аналогичен описанному в Примере 28 (700 мг, 67% для последней стадии). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=10.83 (с, 1Н), 8.00-8.02 (д, 1Н), 7.57-7.61 (м, 1Н), 7.44-7.50 (м, 2Н), 7.35-7.36 (д, 1Н), 7.02 (м, 3Н), 6.19-6.21 (кв, 1Н), 3.81 (с, 3Н), 1.80-1.83 (д, 3Н). LC-MS [М+Н]+: 453.0.
ПРИМЕР 30: Биологические данные
Биохимические испытания на c-Met и ALK
Испытания с киназами. Испытания проводили по методикам, описанным в Fabian et al. (2005) Nature Biotechnology, vol.23, p.329 и в Karaman et al. (2008) Nature Biotechnology, vol.26, p.127.
Для большинства тестов меченые киназой штаммы фагов Т7 выращивали параллельно в 24-луночных планшетах, применяя в качестве хозяина Е. coli из штамма BL21. Е. coli выращивали до фазы логарифмического роста и инфицировали фагом Т7 из замороженного стокового раствора (множественность заражения ~0.1), и инкубировали при встряхивании при 32°С до лизиса (~90 минут). Лизаты центрифугировали (6000 g) и отфильтровали (0.2 мм) для удаления остатков клеток. Оставшиеся киназы продуцировали в клетках НЕК-293 и затем вводили ДНК-метки для детектирования методом количественного ПЦР. Покрытые стрептавидином магнитные бусины обрабатывали биотинилированными низкомолекулярными лигандами в течение 30 минут при комнатной температуре, получая аффинные смолы для испытаний с киназами. Бусины с присоединенными лигандами блокировали избытком биотина и промывали блокирующим буфером (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 мМ DTT) для удаления несвязанного лиганда и уменьшения неспецифичного связывания фага. Реакции связывания проводили комбинированием киназ, аффинных бусин со связанными лигандами и испытуемых соединений в 1х буфере для связывания (20% SeaBlock, 0.17х PBS, 0.05% Tween 20, 6 мМ DTT). Испытуемые соединения готовили в виде 40х стоковые растворы в 100% ДМСО и разбавляли непосредственно в растворе для анализа. Все реакции проводили в полипропиленовых 384-луночных планшетах в конечном объеме 0.04 мл. Планшеты инкубировали при комнатной температуре при встряхивании в течение 1 часа, и аффинные бусины промывали отмывочным буфером (1х PBS, 0.05% Tween 20). Затем бусины заново суспендировали в элюирующем буфере (1х PBS, 0.05% Tween 20, 0.5 мМ небиотинилированного аффинного лиганда) и инкубировали при комнатной температуре при встряхивании в течение 30 минут. Концентрацию киназы в элюатах определяли методом количественного ПЦР.
Для большинства соединений были получены значения IC50 менее 100 нМ в анализе с MET, и для некоторых соединений были получены значения IC50 менее 100 нМ в анализе с ALK.
Биохимический анализ Ron
Биологическую активность соединений в целом оценивали согласно следующей методике. В конечном реакционном объеме 25 мкл, Ron (h) (5-10 mU) инкубировали с 8 мМ MOPS pH 7.0, 0.2 мМ ЭДТА, 250 мкМ KKSRGDYMTMQI Г, 10 мМ ацетата магния и [(γ-33Р-АТР] (удельная активность около 500 импульсов в минуту/пмоль, концентрация, как требуется). Реакцию инициировали добавлением смеси MgATP. После инкубирования в течение 40 минут при комнатной температуре, реакцию останавливали добавлением 5 мкл 3%-ного раствора фосфорной кислоты. Затем 10 мкл реакционного раствора помещали в плоский фильтр Р30 и промывали три раза в течение 5 минут в растворе фосфорной кислоты (75 мМ) и один раз в метаноле, после чего сушили и проводили сцинтилляционные измерения.
Анализ фосфорилирования c-Met рецептора
В данном анализе использовали клетки А549. Засеивание клеток проводили с плотностью 40000 клеток/лунку в среде для выращивания (RPMI + 10% FBS) в 24-луночных планшетах и культивировали в течение ночи при 37°С для присоединения. Клетки обрабатывали минимальной средой (RPMI + 1% BSA). Растворы испытуемых соединений добавляли в лунки планшета и инкубировали при 37°С в течение 1 часа. Затем клетки охлаждали до комнатной температуры в течение 15 минут, затем стимулировали добавлением HGF (40 нг/мл) в течение 15 минут. Клетки промывали один раз ледяным фосфатно-солевым буфером, после чего лизировали лизирующим буфером (110 мкл/лунку) (Cell Signaling #9803+0.2% ингибитора протеаз, Sigma P1860) в течение 1 часа при 4°С. Лизаты клеток переносили в микроцентрифужные пробирки и центрифугировали при 10000 об/мин в течение 10 минут при 4°С, и проводили количественное определение фосфорилированного HGFR с помощью набора Human Phospho-HGF R/c-Met ELISA kit (R&D, DYC2480) согласно инструкциям производителя.
Включение посредством ссылки
Каждая из заявок и патентов, процитированных в настоящей заявке, а также каждый документ или ссылка, процитированные в этих заявках и патентах (включая документы, поступающие в ходе рассмотрения заявки; «процитированные в заявке документы»), и каждая из РСТ и иностранных заявок или патентов, соответствующая приоритету и/или упомянутая в приоритете любой из указанных заявок и патентов, и каждый из документов, процитированных или указанных в качестве ссылки в каждом из процитированных в заявке документов, включены в настоящую заявку посредством ссылки. В общем можно отметить, что документы и ссылки цитируются в настоящей заявке либо в Списке Ссылок, либо в самом тексте; и каждый из указанных документов или ссылок («процитированных в настоящей заявке ссылок»), а также каждый документ или ссылка, процитированные в каждой из процитированных в настоящей заявке ссылок (включая любые спецификации, инструкции производителей и т.д.), включены в настоящую заявку посредством ссылки.
Реферат
Изобретение относится к производным пиридазина формулы II, в которой радикалы и символы имеют определения, указанные в формуле изобретения, или к их фармацевтически приемлемым солям. Соединения формулы II проявляют ингибирующее действие в отношении протеинкиназ, таких как c-met, ron, или ALK, или химерные белки, и могут быть полезны для лечения нарушений, связанных с ненормальной активностью протеинкиназ, таких как рак. 3 н. и 4 з.п. ф-лы, 1 табл., 30 пр.
Формула
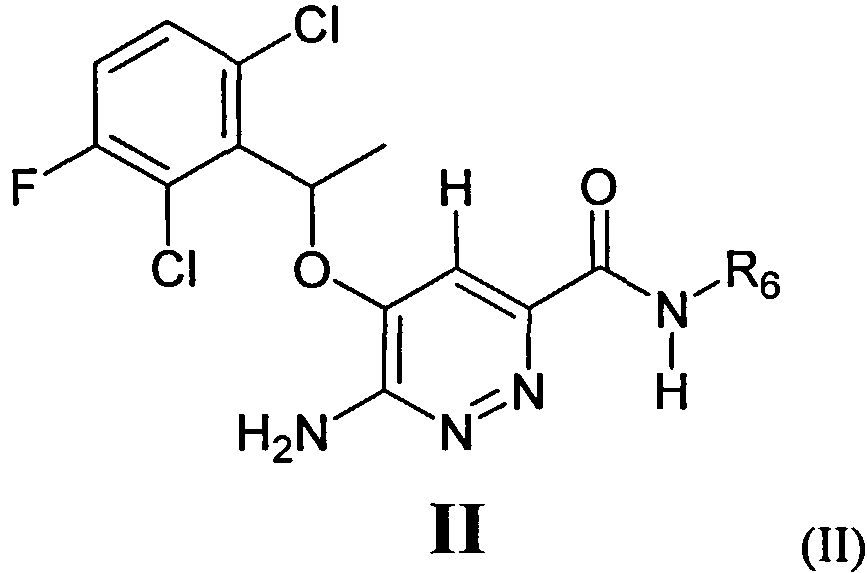
или его фармацевтически приемлемая соль, в котором:
R6 представляет собой:
i. фенил, необязательно замещенный:
a. -C(O)NR7R8;
b. C1-C6 алкокси, необязательно замещенным гетероциклилом, содержащим 5-6 кольцевых атомов, где 1-2 указанных кольцевых атомов независимо представляют собой N или О, где указанный гетероциклил необязательно замещен C1-C6 алкилом; или
c. гетероциклилом, содержащим 5-6 кольцевых атомов, где 1-2 указанных кольцевых атомов независимо представляют собой N или O, где указанный гетероциклил необязательно замещен C1-C6 алкилом;
ii. гетероарил, содержащий 5-6 кольцевых атомов, где 1-2 указанных кольцевых атомов независимо представляют собой N, где указанный гетероарил необязательно замещен:
a. гетероциклилом, содержащим 5-6 кольцевых атомов, где 1-2 указанных кольцевых атомов независимо представляют собой N или О;
b. C1-C6 алкилом;
c. C1-C6 алкокси;
d. -C(O)NR7R8; или
e. C1-C6 alkyl-OH;
iii. ненасыщенный гетероциклил, содержащий шесть кольцевых атомов, где один из указанных кольцевых атомов представляет собой N, необязательно замещенный C1-C6 алкилом или оксо, где указанный C1-C6 алкил необязательно замещен C1-C6 алкокси;
iv. 6,7-дигидропирано[4,3-d]тиазол; или
v. 4,5,6,7-тетрагидротиазол[5,4-c]пиридин;
R7 и R8 каждый независимо выбираются из H, C1-C6 алкила, или R7 и R8 вместе с атомом азота образуют гетероциклил, содержащий 5-6 кольцевых атомов, где 0-1 указанных кольцевых атомов представляют собой N, где указанный гетероциклил необязательно замещен C1-C6 алкилом, и
где указанный гетероарил не представляет собой незамещенный пиридин-3-ил, незамещенный пиридин-4-ил или незамещенный пиримидин-5-ил.
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-6-морфолин-4-илпиридин-3-ил-карбоксамид (1);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-пиразол-4-илкарбоксамид (2);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-метилпиразол-4-ил)карбоксамид (3);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-изоксазол-4-илкарбоксамид (4);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(пирролидинилкарбонил)фенил]карбоксамид (5);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(N-метилкарбамоил)фенил]карбоксамид(6);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6-метокси(3-пиридил))карбоксамид (7);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[6-(N-метилкарбамоил)(3-пиридил)]карбоксамид (8);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[1-(2-гидроксиэтил)пиразол-4-ил]карбоксамид (9);
N-(1-(2H-3,4,5,6-тетрагидропиран-4-ил)пиразол-4-ил){6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}карбоксамид (10);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(4-метоксифенил)карбоксамид (15);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(4-морфолин-4-илфенил)карбоксамид (16);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-бензамид(17);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(2-морфолин-4-илэтокси)фенил]карбоксамид (18);
6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-метил-6-оксо-1,6-дигидро-пиридин-3-ил))карбоксамид (19);
6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6-оксо-1,6-дигидропиридин-3-ил))карбоксамид (21);
6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(6,7-дигидро-4Н-пирано[4,3-d]1,3-тиазол-2-ил)карбоксамид (22);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(4,5,6,7-тетрагидро-1,3-тиазоло[5,4-c]пиридин-2-ил)карбоксамид (23);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-(4-пиперидил)пиразол-4-ил)карбоксамид (24);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-{4-[(4-метилпиперазинил)карбонил] фенил} карбоксамид (25);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[4-(пиперазинилкарбонил)фенил]карбоксамид (26);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-[1-(2-метоксиэтил)-6-оксо-1,6-дигидро-пиридин-3-ил)]карбоксамид (27);
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(1-этил-6-оксо-1,6-дигидро-пиридин-3-ил))карбоксамид (28); и
{6-амино-5-[(2,6-дихлор-3-фторфенил)этокси]пиридазин-3-ил}-N-(2-метокси(4-пиридил))карбоксамид (29).
Комментарии