Производные имидазола, способы их получения и фармацевтическая композиция на их основе - RU2099342C1
Код документа: RU2099342C1
Чертежи












Описание
Настоящее изобретение относится к области синтеза биологически активных соединений, обладающих фармакологической активностью. Более конкретно, настоящее изобретение относится к новым производным имидазола, к способам их получения и к фармацевтической композиции на их основе.
Новые соединения обладают антагонистической активностью к рецепторам ангиотензина II.
Из патента США N 4880804 известны соединения, близкие по структуре к предложенным соединениям. Однако их биологическая активность недостаточно высока.
Задача изобретения
разработка новых соединений имидазольного типа с более высокой фармацевтической активностью. Эта задача
решается с помощью новых производных имидазола формулы (I):
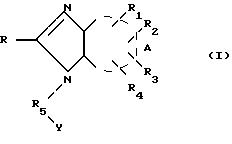
где A фенил, нафтил, пиридил, пиримидинил, тиенил;
R является н-булилом или бутен-1-илом;
R1, R2, R3 и R4 являются такими, что два из них являются атомом водорода, а два других, одинаковые или различные, являются атомом водорода, гидроксилом, алкилом, содержащим не более 4 атомов углерода, карбоксилом, свободным или этерифицированным, линейным или разветвленным алкилом, содержащим не более 4 атомов углерода;
R5 является метиленом;
V является радикалом -Y1-B-Y2, в котором Y1 является фенилом, B является простой углерод-углеродной связью или радикалом -CO-NH-, а Y2 является цианогруппой, карбоксилом, свободным или этерифицированным, линейным или разветвленным алкилом, содержащим не более 4 атомов углерода; индолилом или фенилом, возможно замещенным свободным, этерифицированным или солевым карбоксилом, цианогруппой, формилом, тетразолильным, тетразолилалкильным, тетразолилкарбомоильным радикалом, в которых тетразолильный радикал возможно замещен алкилом, алкенилом или алкоксиалкилом; или радикалом -(CH2)p-SO2-Xa -R14a, в котором p равно 0 или 1, Xa является радикалами -NH-, NHCO-NH-, -NH-CO- или простой связью, а R14a является метилом, этилом, аллилом, пиридилметилом, пиридилэтилом, пиридилом, фенилом или бензилом, с выделением целевого продукта в виде рацемата, энантиомеров диастереоизомеров или солей с минеральными или органическими кислотами или основаниями.
Среди продуктов, являющихся предметом настоящего изобретения, можно, в частности,
назвать:
2-бутил-1-[(4-карбоксифенил)метил] 1Н-бензимидазол-6-карбоновую кислоту,
4-[(2-бутил-1Н-бензимидазол-1-ил]бензойную кислоту,
4-[(2-бутил-1Н-бензимидазол-1-ил)метил]N-(1Н-индол-4-ил)бензамид,
4-[(2-бутил-1Н-нафт(2,3-d)имидазол-1-ил)метил]бензойную кислоту,
4-[(2-бутил-5,6-диметил-1Н-бензимидазол-1-ил)метил]
бензойную кислоту,
4-[(2-бутил-ЗН-имидазо(4,5-c)пиридин-3-ил)метил]бензойную кислоту,
4'-[(2-бутил-3Н-имидазо(4,5,
b)пиридин-3-ил)метил](1,1'-бифенил)2- карбоновую кислоту,
- 2-бутил-1-[(2'-карбокси-(1,1'-бифенил)-4-ил)метил] -6-гидрокси-1Н-тиено(2,3-d) имидазол-5-карбоксилат 1,1-диметилэтила,
и
их соли.
Предметом настоящего изобретения также
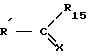
являются способы получения производных имидазола формулы (I). Один из способов заключается в том, что соединение формулы (II):

где X атом кислорода или радикал NH,
R15 означает гидрокси-, алкокси-группу, атом галогена или NH2,
R' имеет значения, указанные для R, где возможные функциональные группы могут быть защищены с помощью защитных групп, вводят в реакцию с соединением формулы (III):
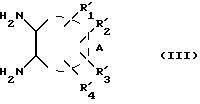
где


где X, R',

получают соединение формулы (IVa):

где R',

Hal R5 Y' (V)
где Hal галоген; Y' имеет значение, указанное для Y, где возможные функциональные группы могут быть защищены с помощью защитных групп, для получения формулы (IX):
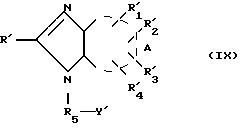
где R',

с последующим снятием защитных групп.
Предпочтительные условия осуществления способа следующие.
Соединение формулы (IVa) может быть получено путем присоединения соединения формулы (II) к свободной аминофункции соединения формулы (III) и циклизацией полученного продукта по второй свободной аминофункции.
Таким образом, соединение формулы (IVa) может быть получено путем взаимодействия соединения формулы (II) с соединением формулы (III) в различных условиях реакции, в частности соединение формулы (II) может конденсироваться с соединением формулы (III), предпочтительно, в органическом растворителе, таком как, например, тетрагидрофуран или диметилформамид в кипящем состоянии или при температуре от 20oC до 200oC.
Соединением формулы (II) может быть валериановая кислота, если X означает атом кислорода, а R15 означает гидроксильный радикал, которая вводится в реакцию с соединением формулы (III), которым может быть, например, производный продукт пиридина, при температуре порядка 170oC, желательно при встряхивании, и реакцию ведут в течение около 18 ч.
Полученные таким образом продукты формулы (IVa) могут конденсироваться с соединением формулы (V) для получения продуктов формулы (IX).
Реакция присоединения продуктов формулы (IVa) к соединению формулы (V), в которой атом галогена означает, предпочтительно, атом брома, может выполняться, например, в органическом растворителе, таком как диметилформамид или тетрагидрофуран: таким образом, галогенсодержащее производное может конденсироваться на анионе имидазола формулы (IVa), приготовленном, например, воздействием сильного основания, такого как гидрат натрия или калия, или же алкоголята натрия или калия, такого как, например, метилат натрия или карбонат калия в диметилформамиде.
Другой способ получения производных имидазола формулы (I) заключается в том, что проводят реакцию соединения общей формулы (VI):

где


где R' и R15 имеют вышеуказанные значения, для получения продукта формулы (X):

который либо восстанавливают до соединения формулы (X'):

которое циклизуют в продукт IVa, охарактеризованный выше, с последующей обработкой полученного продукта IVa, как указано выше, до получения продукта формулы (V):
Hal R5 Y'
где R5 и Y' имеют указанное выше значение, для получения продукта формулы (XI):
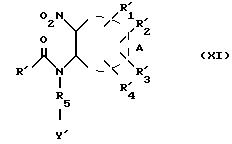
где R',


где R',

Реакция присоединения соединения формулы (II') со свободной аминной группой соединения формулы (VI) для получения продуктов формулы (X) может производиться простым нагревом до температуры примерно от 120oC до 170oC. Если соединением формулы (I') является валериановая кислота, то она используется, главным образом, в избытке по отношению к соединению формулы (VI).
Реакция присоединения соединения формулы (V) к свободной аминогруппе соединения формулы (X) для получения продуктов формулы (XI) может производиться при температуре окружающей среды или нагревом при температуре примерно от 20oC до 150o C, желательно в присутствии основания, такого как триэтиламин, едкий натр, метилат или этилат натрия или же гидрид натрия в растворителе, таком как тетрагидрофуран или диметилформамид.
Восстановление нитро-функции соединения формулы (XI) до амино-функции для получения продуктов формулы (XII), а также восстановление продуктов формулы (X) в продукты формулы (X') может производиться в соответствии с общепринятыми у специалистов методами, в частности путем каталитической гидрогенизации в присутствии гидроокиси палладия в таком растворителе, как, например, этанол, или же цинком в таком растворителе, как, например, уксусная кислота, в присутствии ацетата натрия, или боргидридом натрия.
Реакция циклизации продукта формулы (XII) для получения продуктов формулы (IX) может выполняться простым нагревом или в присутствии катализатора, такого, как, например, хлорид тионила, пентахлорид фосфора или фосфорный ангидрид в таком растворителе, как, например, тетрагидрофуран или диметилформамид.
Третий способ получения производных имидазола формулы (I) заключается в том, что соединения формулы (VI):

где

Hal R5 Y' (V)
где Y' имеет значения Y, где возможные функциональные группы могут быть защищены с помощью защитных групп;
R5 имеет вышеуказанное значение,
с получением промежуточного соединения формулы (VII):
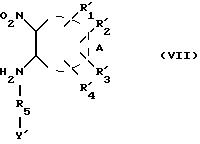
где


где R' и R15 имеют вышеуказанные значения, с получением соединения формулы (XI):
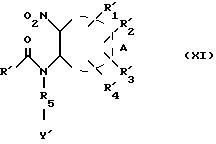
значения радикалов R5, R', Y' и

которое подвергают реакции восстановления для получения соединения формулы (XII):

где радикалы


где радикалы

либо соединение формулы (VII) восстанавливают для получения соединения формулы (VIII):

где радикалы

Реакция присоединения соединения формулы (V) к свободной аминогруппе соединения формулы (VI) для получения продуктов формулы (VII) может производиться в тех же условиях, что и описанные выше для реакции присоединения соединения формулы (V) с продуктами формулы (IVa).
Реакция продукта формулы (VII) с соединением формулы (II') проводится в тех же условиях, что и реакция продукта формулы (VI) с соединением формулы (II').
Восстановление нитрофункции продуктов формулы (VII) до аминофункции для получения продуктов формулы (VIII) может производиться в соответствии с общепринятыми у специалистов методами, в частности в тех же условиях, что и описанные выше для восстановления радикала нитропродуктов формулы (XI) в радикал амино для получения продуктов формулы (XII).
Реакция присоединения соединения формулы (II) к свободной аминофункции продуктов формулы (VIII) с последующей циклизацией полученных таким образом продуктов может производиться в тех же условиях, что и описанная выше реакция взаимодействия соединения формулы (II) с соединением формулы (III).
Ниже приводится неограничивающий перечень примеров защиты
функциональных групп:
гидроксильные группы могут
быть защищены, например, радикалами алкил, триметилсилил, дигидропиран, метоксиметил или тетрагидропиранил;
группы амино могут быть
защищены, например, радикалами ацетил, тритил, бензил,
трет-бутоксикарбонил, фталимидо или другими радикалами, известными в химии пептидов;
ацильные группы, такие как формильная группа,
могут быть защищены, например, в виде циклических или
нециклических кеталей, таких как диметил или диэтилкеталь или диоксикетальный этилен;
кислотные функции, при желании, могут быть
амидированы первичным или вторичным амином, например, в
присутствии метиленхлорида в хлоргидрате 1-этил-3-(диметиламинопропил) карбодиимида при температуре окружающей среды;
кислотные
функции могут быть защищены, например, в виде сложных эфиров,
полученных с помощью легкорасщепляемых сложных эфиров, таких как бензиловые или трет-бутиловые сложные эфиры, или же сложных эфиров,
широко известных в химии пептидов.
Удаление указанных защитных групп производится в соответствии с общепринятыми у специалистов методами, в частности путем кислого гидролиза, выполняемого с использованием такой кислоты, как хлористо-водородная кислота, бензолсульфокислота или паратолуолсульфокислота, муравьиная или трифторуксусная кислота.
Группа фталимидо выделяется гидразином. Перечень различных используемых защитных групп можно найти, например, во французском патенте BF 2499995.
Описанные выше продукты могут быть, при желании, превращены в соль минеральной или органической кислоты в соответствии с общепринятыми у специалистов методами.
Описанные выше продукты, содержащие функциональные карбокси-группы, могут быть превращены в соль неорганическим или органическим основанием или же превращены в сложный эфир: указанные реакции превращения в сложный эфир или соль могут выполняться в соответствии с общепринятыми у специалистов методами.
Соединения формулы (I), а также их аддитивные соли с кислотами обладают фармакологической активностью. Так, соединения формулы (I) обладают антагонистической активностью в отношении рецептора ангиотензина II и являются, таким образом, ингибиторами действия ангиотензина II, в частности ингибируют сосудосуживающее действие, а также трофическое действие на уровне миоцитов.
Предпочтительными соединениями в этом
смысле являются следующие продукты формулы (I):
2-бутил-1-[(4-карбоксифенил)метил]1Н-бензимидазол-6-карбоновая кислота,
4-[(2-бутил-1Н-бензимидазол-1-ил)метил]бензойная кислота,
4-[(2-бутил-1Н-бензимидазол-1-ил)метил]N-(1Н-индол-4-ил)бензамид,
4-[(2-бутил-1Н-нафт(2,3-d)имидазол-1-ил)метил]бензойная кислота,
4-[(2-бутил-5,
6-диметил-1Н-бензимидазол-1-ил)метил]бензойная кислота,
4-[(2-бутил-ЗН-имидазо(4,5-c)пиридин-3-ил)метил]бензойная кислота,
4'-[(2-бутил-3Н-имидазо(4,5-b)пиридин-3-ил)метил] (1,
1'-бифенил)2-карбоновая кислота,
2-бутил-1-[(2'-карбокси-(1,1'-бифенил)-4-ил)метил] -6-гидрокси-1Н-тиено(2,3-d)имидазол-5-карбоксилат 1,1-диметилэтил
также их аддитивные соли с
приемлемыми с фармацевтической точки зрения неорганическими и органическими кислотами.
Указанная выше фармацевтическая активность новых соединений позволяет рекомендовать их при лечении артериальной гипертонии, сердечной недостаточности, почечной недостаточности и для предупреждения рецидива стеноза ангиопластики.
Они могут также использоваться при лечении некоторых желудочно-кишечных и гинекологических нарушений и, в частности, для релаксации на уровне матки.
Изобретение относится также к фармацевтической композиции, включающей в качестве действующего начала соединение формулы (I) и фармацевтически приемлемые целевые добавки.
Указанные фармацевтические композиции могут применяться внутрь, ректальным путем, парентеральным путем или локально, нанесением на кожу или на слизистые оболочки.
Указанные составы могут быть твердыми или жидкими и иметь любую фармацевтическую форму, широко применяемую при лечении человека, как, например, простые или дражевидные таблетки, капсулы, гранулы, свечи, препараты для инъекций, мази, кремы, гели и препараты в аэрозольной упаковке; они производятся обычными методами. Действующее начало вводится в основы, обычно используемые при изготовлении фармацевтических составов, такие как тальк, аравийская камедь, лактоза, амидон, стеарат магния, какао-масло, водные или безводные связующие, жиры животного или растительного происхождения, производные парафина, гликоли, различные увлажняющие, диспергирующие или эмульсионные агенты, консерванты.
Обычно применяемые дозы могут в зависимости от используемого препарата, от особенностей больного и от заболевания составлять от 1 до 100 мг в день для взрослых при приеме внутрь.
Исходные соединения формул (II), (II'), (III), (V), (VI) могут приобретаться в торговой сети или же изготавливаться в соответствии с общепринятыми у специалистов методами.
Некоторые соединения формулы (III) встречаются в торговой сети, например, 3,4-диаминобензоат метила, который можно найти в виде препарата, выпускаемого фирмой "ЛАНКАСТЕР".
В специализированной литературе приводятся многочисленные примеры получения соединений формулы (III), в частности, в следующих статьях: Bull. S.O. C. chim (1957), стр. 2197-2201; Beil. 22, (2), стр. 394; Beil. 22, (2), стр. 395; Beil.24, стр. 395; Beil.13, стр. 270; Beil.13, стр. 207; Beil.13, стр. 179; Beil.25, стр. 481; Beil.13, стр. 1; Beil.24, стр. 469.
Соединения формулы (II) и (II'), представляющие собой валериановую кислоту или пентанимидоат этила, могут быть изготовлены, например, воздействием газообразной хлористо-водородной кислоты в этаноле на нитрил валериановой кислоты, который выпускается фирмой "ЛОНЗА".
Один из примеров получения указанных соединений формулы (II ) приводится, например, в статье: J.A.C.S. (1942), 64, стр. 1827.
Один из методов получения некоторых соединений формулы (V) может состоять в воздействии на соединение формулы (Va):

или иодобензоат метила, который выпускает фирма "ЯНСЕН", соединением формулы (Vb):

или иодотолуол, который выпускается фирмой "ФЛУКА" причем реакция протекает в присутствии порошковой меди при температуре от приблизительно 100oC до 300oC, для получения продукта формулы (Vc):

в котором радикал карбокси, превращенный в сложный эфир, может, при желании, быть освобожден от алкильного радикала общепринятыми у специалистов методами, или, например, путем кислого или щелочного гидролиза, с выполнением реакции бромирования на метиловом радикале общепринятыми у специалистов методами, например, действием н-бромсукцинимида в тетрахлориде углерода.
Примеры получения соединений формулы (VI) описаны в специализированной литературе и приводятся, в частности, в патенте США N 4880804 и в патентной заявке ЕЭС EP N 0400974.
Приводимые далее примеры иллюстрируют изобретение, вместе с тем не ограничивая его.
Пример 1. 2-Бутил-1[(4-цианофенил)метил] 1Н-бензимидазол-6-карбоксилат метила и его гомолог 5-карбоксилат метила
Этап А.
2-Бутил-1Н-бензимидазол-5-карбоксилат метила
В раствор 5 г 3,4-диаминобензоата метила в 60 см3 тетрагидрофурана вводят 5,47 г хлоргидрата пентанэтилимидоата (полученного в
соответствии с указаниями J.A.C.S. 64, 1827 (1942)) и взбалтывают в течение 3 ч при температуре от 30 до 90oC. Затем производят выпаривание тетрагидрофурана, добавляют 50 см насыщенного
раствора бикарбоната натрия и экстрагируют с использованием метиленхлорида. Затем производят промывку водой, сушку и выпаривание досуха при пониженном давлении. Таким образом получают 9 г продукта,
который подвергают кристаллизации в простом изопропиловом эфире. В результате получают 6,65 г искомого продукта. Т.пл. 114oC (см. табл.1).
Этап Б.
2-Бутил-1-[(4-цианофенил)метил]1Н-бензимидазол 6 карбоксилат метила (продукт А) и его изомер 5-карбоксилат метила (продукт Б)
В раствор 4,64 г продукта, полученного на описанном выше этапе А,
в 46 см3 диметилформамида добавляют 960 мг в 50%-ной дисперсии гидрида натрия в масле. Затем смесь взбалтывают в течение 1 ч 30 мин при комнатной температуре и добавляют 4,4 г
4-бромметилбензонитрила. Затем взбалтывают в течение 30 мин и медленно добавляют 100 см3 воды, взбалтывают 30 мин, после чего выполняются центрифугирование, промывка водой и высушивание
при
температуре 100oC при пониженном давлении. В результате получают 7,4 г сырого продукта. Т.пл. 140oC.
Выделение продукта А.
7,4 г указанного выше продукта растворяют в 400 см3 этилацетата в состоянии кипения (с рефлюксом), после чего горячий раствор фильтруют и концентрируют до получения общего объема 100 см3. Затем взбалтывают в течение 1 ч при комнатной температуре. В результате, после центрифугирования получают 3,4 г искомого продукта. Т.пл. 200 205oC.
Выделение продукта Б.
Маточный раствор продукта А выпаривают досуха и подвергают кристаллизации в простом изопропиловом эфире. В результате получают 3,35 г целевого продукта. Т.пл. 120oC (см. табл. 2).
Пример 2. 2-Бутил-1-[(4-карбоксифенил)метил]1Н-бензимидазол-6-карбоновая кислота
В течение 19 ч производится перемешивание кипящего раствора 500 мг продукта А,
полученного
в примере 1, в 2,5 см3 раствора, состоящего из равных частей серной кислоты, уксусной кислоты и воды. Затем раствор охлаждают, добавляют 30 г льда и подщелачивают натровым
щелоком. Затем с
помощью уксусной кислоты pH доводят до 6, взбалтывают в течение 15 мин при комнатной температуре, после чего выполняются центрифугирование, промывка водой и высушивание при
температуре 90oC
при пониженном давлении. В результате получают 480 мг продукта. Т.пл. 290oC.
Очистка: 556 мг продукта, полученного как указано выше, растворяют в 30 см3 метанола в состоянии кипения (с рефлюксом), после чего раствор концентрируют до 10 см3, добавляют 2 см3 воды и, после начала кристаллизации, добавляют 20 см3 воды. Через 3 ч выполняют центрифугирование, трехкратную промывку в 5 см3 смеси метанол-вода (1 - 1). В результате получают 490 мг продукта. Т.пл. 290oC.
Эти 490 мг продукта растворяют в 40 см3 изопропанола в состоянии кипения (с рефлюксом) и концентрируют до 15 см3. Затем выдерживают в течение 16 ч при комнатной температуре, подвергают центрифугированию и в результате получают 430 мг искомого продукта. Т.пл. 290oC (см. табл. 3).
Пример 3.
2-Бутил-1-[(4-карбоксифенил)метил]1Н-бензимидазол-5-карбоновая
кислота
Используя 500 мг продукта Б, полученного в примере 1, выполняют операции, как указано в примере 2. В результате
получают 390 мг сырого продукта. Т.пл. 200oC.
Сырой продукт растворяют в 10 см3 этанола при температуре 60oC, добавляют 10 см3 воды и выстаивают в течение 2 ч при комнатной температуре. Затем раствор подвергают центрифугированию и получают 260 мг продукта, который растворяют в 1 50 см3 этилацетата в состоянии кипения (с рефлюксом), концентрируют его до общего объема в 30 см3, выстаивают в течение 16 ч при комнатной температуре и подвергают центрифугированию. В результате получают 220 мг целевого продукта. Т.пл. ≈ 225oC.
Химический состав для C20H20N2O4 352,38
расчетный C 68,17; H 5,72; N 7,95
полученный C
68,2; H 5,7; N 7,9
Инфракрасный спектр: С 0
Вазелиновое масло
1717 и 1685 см-1
Пример 4. 4-[(2-Бутил-1-Н-бензимидазол-1-ил)метил]бензонитрил
Операции
выполняются, как на этапе Б примера 1, но исходят из 3,5 г 2-бутил-1Н-бензимидазола
(полученного в соответствии с POOL и Al.Am. Soc. 59, 178 (1937)) и 4,3 г 4-бромметилбензонитрила. В результате
получают 6,8 г искомого продукта, Т.пл. 130oC, который растворяют в
метиленхлориде, обрабатывают активированным углем, фильтруют и выпаривают досуха, в результате чего получают 6 г
продукта Т.пл. 130oC, который подвергается рекристаллизации в простом
изопропиловом эфире. Таким образом получают 4,7 г целевого продукта Т.пл. 148oC; проба для анализа была
получена после двух последовательных рекристаллизаций 1,2 г указанного выше продукта
в этиловом эфире для получения 700 мг очищенного продукта, Т.пл. 150oC.
Xимический
состав для С19H19N3 289,381
расчетный C 78,86; H
6,62; N 14,52
полученный C 79,0; H 6,6; N 14,6
Пример 5.
4-[(2-Бутил-1-Н-бензимидазол-1-ил)метил]бензойная кислота
Операции выполняются, как в примере 2, исходя из 1 г
продукта, полученного в примере 4. В результате получают 1 г искомого продукта,
который подвергается рекристаллизации в 10 см3 этилацетата. В результате получают 900 мг целевого продукта.
Т.пл. 235oC.
Проба после анализа была получена после двух последовательных рекристаллизаций в изопропаноле, в результате чего получают 500 мг чистого продукта. Т.пл. 235oC (см. табл. 4).
Пример 6.
4-[(2-Бутил-1-Н-бензимидазол-1-ил)метил] N-(1Н-индол-4-ил)бензамид
В суспензию 180 г продукта, полученного в примере 5, в 4 см3
метиленхлорида добавляют 192 мг хлоргидрата
1-этил-З-(3-диметиламинопропил) карбодиимида. Затем производят взбалтывание в течение 10 мин при комнатной температуре и добавляют 53 мг 4-аминоиндола.
Затем взбалтывают в течение 18 ч, дважды
экстрагируют с помощью 25 см3 метиленхлорида, промывают водой, высушивают и выпаривают досуха при пониженном давлении. Таким образом получают 230 мг
остатка, который подвергается
хроматографии на двуокиси кремния (элюент: метиленхлорид-метанол (95:5)). В результате получают 115 мг искомого продукта. Т.пл. 196oC (см. табл. 5).
Пример 7.
2-Бутил-1-[(4-цианофенил)метил]1Н-бензимидазол-4-карбоксилат метила
Этап А. 2-Бутил-1Н-бензимидазол-4-карбоксилат метила
Операции выполняют, как на этапе А
примера 1, но используют
2,5 г 2,3-диаминометилбензоата (приготовленного согласно J.Chem.Soc. 117 (1920), стр. 775 и CAN. J. Chem. 55 (1977), стр. 1653-1657) и вводят 2,73 г хлоргидрата
пентанэтилимидоата (приготовленного в
соответствии с J.A.C.S. 64, 1827, (1942)). В результате получают 3,05 г искомого продукта, кристаллизованного в бензине G. Т.пл. ≈ 85oC. Пробу
для анализа получают после двух
последовательных рекристаллизаций 280 мг продукта в простом изопропиловом эфире. В результате получают 100 мг продукта. Т.пл. 97oC (см. табл. 6).
Этап Б.
2-Бутил-1-[(4-цианофенил)метил] 1Н-бензимидазол-4-карбоксилат метила
Операции выполняют, как на этапе Б примера 1, но исходят из 1,86 г продукта, полученного на
предшествующем этапе А, с
использованием 1,88 г 4-бромметилбензонитрила. Таким образом получают 3,5 г продукта, который подвергают хроматографии на двуокиси кремния (элюант: этилацетат-циклогексан
(6:4)). В результате
получают 2,05 г искомого продукта, рекристаллизованного в растворе этилового эфира. Т.пл. 113oC.
Спектр ЯМР (CDCl3, 250 МГц)
CH3-(CH2)2 0,92 млн-1(t)
CH2 центральн. 1,43-1,82 млн-1(m)

CO2CH3 4,04 млн-1(s)
N-CH2-C6H5 5,44 млн-1
Бензонитрил 7,09-7, 61 млн-1(d)
H5 7,95 млн-1 (dd)
H6, H7 ≈ 7,27 млн-1(m)
Пример 8. 2-Бутил 1-[(4-карбоксифенил)метил]1Н-бензимидазол 4-карбоновая кислота
Операции выполняются, как в примере 2, но исходят из 1 г продукта, полученного в примере 7. В результате получают 1 г искомого продукта. Т.пл. 250oC.
Пробу для анализа получают после двух последовательных рекристаллизаций в смеси метиленхлорид-метанол с получением 345 мг продукта. Т.пл. 250oC (см. табл. 7).
Пример 9. 4-[(2-Бутил-1Н-нафт(2,3-d)имидазол-1-ил)метил]бензонитрил
Этап А. 2-Бутил-1Н-нафт(2,3-d)имидазол
Операции выполняют, как на этапе А
примера 1, но исходят из 2,7 г 2,3-диаминонафталина с использованием 3,4 хлоргидрата пентанэтилимидоата (приготовленного в соответствии с J.A.C.S. 64, 1827 (1942)) и дихлорэтана в качестве
растворителя. Таким образом получают 4,5 г сырого продукта, который растворяют в 100 см3 этилацетата в состоянии кипения (с рефлюксом), концентрируют его до общего объема в 30 см3
, выстаивают в течение 16 ч и подвергают центрифугированию. В результате получают 2,37 г искомого продукта. Т.пл. 188oC.
Пробу для анализа получают последовательными рекристаллизациями 500 мг полученного выше продукта в этилацетате, а затем в изопропаноле. В результате получают 280 мг целевого продукта. Т.пл. 190oC (см. табл. 8).
Этап
Б.
4-[(2-Бутил-1Н-нафт(2,3 d)ииидазол-1-ил)метил]бензонитрил
Операции выполняют, как на этапе Б примера 1, но исходят из 449 мг продукта, полученного на предшествующем этапе А, с
использованием
392 мг 4-бромметилбензонитрила. Таким образом получают 460 мг продукта, Т.пл. 138oC, после рекристаллизации в этиловом эфире.
Пример 10.
4-[(2-Бутил-1-Н-нафт(2,
3-d)имидазол-1-ил)метил] бензойная кислота
В течение 21 ч доводят до температуры кипения (с рефлюксом) суспензию 420 мг соединения, полученного в примере 9, с 4 см3 этанола и 7,5
см3 едкого натра 2N. Затем производят охлаждение, добавляют 5 см3 льда и нейтрализуют до pH 7 с помощью 7,5 см3 хлористоводородной кислоты (2N),
после чего добавляют
30 см3 метанола, нагревают до температуры кипения (с рефлюксом) и концентрируют до 20 см3. После центрифугирования получают 420 кг искомого продукта.
Затем 420 кг указанного выше продукта подвергают рекристаллизации сперва в изопропаноле, а затем в этилацетате, в результате чего получают 260 мг продукта. Т.пл. 220oC (см. табл. 9).
Пример 11. 4[(2-Бутил-3Н-имидазо(4,5-d)пиримидин-3-ил)-метил]бензонитрил (продукт А) и 4-[(2-бутил-3Н-имидазо(5,6-d)пиримидин-3-ил)метил]бензонитрил (продукт Б)
Этап А.
2-Бутил-3Н-имидазо (4,
5-d)пиримидин
Операции выполняют, как в примере 1, но исходят из 5 г 4,5-диаминопиримидина с использованием 10 г хлоргидрата пентанэтилимидоата и диметилформамида в
качестве растворителя.
Таким образом получают 2,1 г искомого продукта, кристаллизованного из раствора этилацетата. Т.пл. 166oC. Затем выполняют рекристаллизацию указанного выше продукта в
этилацетате и получают 1,
8 г целевого продукта, Т.пл. 168oC. 200 мг пробы для анализа получены дополнительной рекристаллизацией в этилацетате 300 мг указанного выше продукта. Т.пл. 168oC (см. табл.
10).
Этап Б. 4-[(2-Бутил-3Н-имидазо(4,5-d)пиримидин-3-ил)метил] бензонитрил (продукт А) и 4-[(2-бутил-3Н-имидазо(5,6-d)-пиримидин-3-ил)метил]бензонитрил (продукт
Б)
Операции
выполняют, как на этапе Б примера 1, но исходят из 352 мг соединения, полученного на предшествующем этапе А, с использованием 431 мг 4-бромметилбензонитрила. Таким образом
получают 630 мг сырого
продукта, который подвергается хроматографии на двуокиси кремния (элюент: метиленхлорид-метанол (9:1)). В результате получают 130 мг продукта А, кристаллизованного из раствора
в этиловом эфире, Т.пл.
112oC и 35 мг продукта Б, Т.пл. 85oC, кристаллизованного из смеси метилэтилкетона и простого эфира (см. табл. 11).
Пример 12.
4[(2-Бутил-3Н-имидазо(4,
5-d)пиримидин-3-ил)-метил]бензойная кислота
В течение 2 ч перемешивают при температуре кипения (с рефлюксом) 400 мг продукта А, полученного, как указано в примере 11,
с 8 см3
этанола, содержащего 10% воды в 1 см3 натрового щелока. Затем добавляют 20 см3 воды, нейтрализуют уксусной кислотой до pH 5-6, экстрагируют метиленхлоридом,
промывают, высушивают
и выпаривают досуха при пониженном давлении. Таким образом получают 280 мг целевого продукта, кристаллизованного из раствора в смеси "этилацетат-эфир". Т.пл. 165o
C.
Затем полученный продукт подвергают двум последовательным рекристаллизациям в метилэтилкетоне. В результате получают 185 мг искомого соединения, Т. пл. 173oC (см. табл. 12).
Пример 13. 4-[(2-Бутил-3Н-имидазо(4,5-d)пиримидин-3-ил)-метил]бензойная кислота
Путем выполнения операций, как указано в примере 12, но исходя из продукта В, полученного
в примере 11,
получают целевой продукт. Т.пл. 180oC.
Пример 14. 4-[(2-Бутил-5,6-диметил-1Н-бензимидазол-1-ил)-метил]бензонитрил
Этап А. 2-Бутил-5,
6-диметил-1Н-бензимидазол
Операции выполняют, как на этапе А примера 1, но исходят из 2,04 г 4,5-диметил-1,2-фенилендиамина с использованием 3,73 г хлоргидрата пентанэтилимидоата
(приготовленного в соответствии с
J.A.C.S. 64, 1827 (1942)). Таким образом получают 3,48 г сырого продукта, который подвергается хроматографии на двуокиси кремния (элюент: метиленхлорид-метанол
(9:1)). В результате получают 2,27 г
целевого продукта. Т.пл. 110oC.
Проба для анализа была получена в результате рекристаллизации 120 мг указанного выше продукта в простом изопропиловом эфире. В результате получают 88 мг продукта. Т.пл. 110oC (см. табл. 13).
Этап Б. 4-[(2-Бутил-5,6-диметил-1Н-бензимидазол-1-ил)метил]-бензонитрил
Операции выполняют, как на этапе Б
примера 1, но исходят из 2,02 г продукта, полученного на приведенном выше этапе А, с использованием 2,21 г 4-бромметилбензонитрила. Таким образом получают 2,88 г
искомого соединения,
кристаллизованного из простого эфира. Т.пл. 159oC.
Пробу для анализа получают после рекристаллизации 476 мг продукта в изопропаноле, а затем в этилацетате. В результате получают 132 мг целевого продукта, Т.пл. 160oC (см. табл. 14).
Пример 15. 4-[(2-Бутил-5,6-диметил-1Н-бензимидазол-1-ил)-метил]бензойная кислота
Операции выполняют,
как в примере 12, но исходят из 395,7 мг продукта, полученного в приведенном выше примере 14. Это дает возможность получить 367 мг целевого продукта. Т.пл. 220o
C.
Проба для анализа была получена в результате двойной рекристаллизации указанного выше продукта в изопропаноле. В результате получают 198 мг чистого продукта. Т.пл. 254oC (см. табл. 15).
Пример 16. 1Н-Бензимидазол-метил-бензонитрил
Операции выполняют, как на этапе Б примера 1, но исходят из 951 мг бензимидазола с использованием 1,97 г
4-бромметилбензонитрила в
диметилформамиде. Таким образом получают 2,48 г сырого продукта, который подвергается хроматографии на двуокиси кремния (элюент: метиленхлорид-метанол (95:5)). В результате,
после кристаллизации из
эфира, получают 1,34 г искомого продукта. Т.пл. 94oC.
Проба для анализа была получена в результате двух последовательных рекристаллизаций 422 мг
полученного выше продукта в
эфире, а затем в простом изопропиловом эфире. В результате получают 167 мг очищенного продукта, Т.пл. 94oC. (см. табл. 16)
Пример 17.
4-[(1Н-Бензимидазол-1-ил)метил]бензойная
кислота
Операции выполняют, как в примере 12, но исходят из 921 мг соединения, полученного в приведенном выше примере 16. Это дает возможность
получить 888 мг искомого продукта. Т.пл. >
260oC.
Проба для анализа была получена в результате двойной рекристаллизации указанного выше продукта в изопропаноле, а затем в этилацетате. В результате получают 390 мг чистого продукта. Т.пл. > 260oC (см. табл. 17).
Пример 18. 4-[(2-Бутил-1Н-имидазо(4,
5-c)пиридин-1-ил)-метил]бензонитрил (продукт А) и 4-[(2-бутил-1Н-имидазо(3,
4-c)пиридин-1-ил)метил]бензонитрил (продукт Б)
Этап А. 2-Бутил-1Н-имидазо(4,5-c)пиридин
Смесь 3 г 3,
4-диаминопиридина и 8,28 г валериановой кислоты нагревают в течение 18 ч при
температуре 170oC. Затем реакционную среду подвергают хроматографии на двуокиси кремния (элюент:
этилацетат-метанол (8:2)).
В результате получают 4,8 г целевого продукта, используемого в чистом виде на следующем этапе.
Этап Б. 4-[(2-Бутил-1Н-имидазо(4,
5-c)пиридин-1-ил)метил] -бензонитрил (продукт А) и 4-[(2-бутил-1Н-имидазо(3,
4-c)пиридин-1-ил)метил]бензонитрил (продукт Б)
Операции выполняют, как на этапе Б примера 1, но исходят из 4,09 г
продукта, полученного на предшествующем этапе А, с использованием 4,58 г
4-бромметилбензонитрила. Таким образом получают 8,5 г сырого продукта, который подвергается хроматографии на двуокиси кремния
(элюент: метиленхлорид-метанол (9:1)). В результате получают 290 мг
продукта Б (кристаллизованного в эфире). Т.пл. 140oC. И 380 мг продукта А (кристаллизованного в эфире). Т.пл. 164oC (см. табл. 18).
Пример 19.
4-[(2-Бутил-1Н-имидазо(4,5-c)пиридин-1-ил)метил]бензойная кислота
Операции выполняют, как в примере 12, но исходят из 320 мг продукта А,
полученного в примере 18. Это дает возможность
получить 320 мг сырого продукта, Т.пл. ≈ 210oC. Затем этот продукт подвергается рекристаллизации в изопропаноле с 40% воды, в
результате чего получают 250 мг искомого продукта, Т.
пл. 246oC. Данный продукт вновь подвергается рекристаллизации в 10 см3 изопропанола. В конечном итоге получают 180 мг
искомого соединения. Т.пл. 246oC (см. табл.
19).
Пример 20. 4-[(2-Бутил-3Н-имидазо(4,5-c)пиридин-3-ил)метил] бензойная кислота
Операции выполняют, как в примере 12,
но исходят из продукта Б, полученного в примере 18.
Получают 170 мг продукта, Т.пл. 200oC. После рекристаллизации в изопропаноле получают продукт, Т.пл. 216oC (см. табл.
20).
Пример 21.
4-[(2-(1-Бутенил)1Н-бензимидазол-1-ил)метил]-бензонитрил
Этап А. 4-[(2-(1-бромбутил)1Н-бензимидазол-1-ил)метил]бензонитрил
В течение 2 ч под лампой
мощностью 60 Вт взбалтывают при
температуре 60oC суспензию, состоящую из 1,16 г продукта, полученного в примере 4, 36 см3 тетрахлорметана, 712 мг N-бромсукцинимида и нескольких
кристаллов перекиси бензила.
Затем смесь охлаждают, экстрагируют метиленхлоридом, промывают насыщенным раствором бикарбоната натрия, а затем водой, высушивают и выпаривают досуха при пониженном
давлении. Таким образом получают 2,
1 г остатка, который подвергают хроматографии на двуокиси кремния (элюент: этилацетат-циклогексан (1: 1)). Таким образом получают 600 мг искомого продукта. Т.пл.
132oC.
Пробы для анализа была получена в результате рекристаллизации 100 мг полученного выше продукта в 20 см3 эфира. В результате получают 58 мг целевого продукта. Т.пл. 135oC (см. табл. 21).
Этап Б. 4-[(2-(1-Бутенил)1Н-бензимидазол-1-ил)метил]бензонитрил
В раствор 1,10 г продукта, полученного, как на этапе А, в 5
см3 диметилформамида
добавляют 1,33 г карбоната лития и 1,56 г бромида лития. Затем взбалтывают в течение 15 мин в состоянии кипения (с рефлюксом). После этого диметилформамид выпаривают,
а остаток подвергают
хроматографии на двуокиси кремния (элюент: циклогексан-этилацетат (1:1)). В результате получают 480 мг целевого продукта, Т.пл. 125oC, кристаллизованного в эфире (см.
табл. 22).
Пример 22. 4-[(2-(1-Бутенил)1Н-бензимидазол-1-ил)метил]-бензойная кислота
Операции выполняют, как в примере 12, но исходят из 430 мг продукта, полученного в примере
21. Это дает
возможность получить 450 мг искомого продукта, Т.пл. 210oC, а затем 225oC.
Проба для анализа была получена в результате последовательных рекристаллизаций в водном метаноле, этилацетате, наконец, в водном метаноле, слегка подкисленном уксусной кислотой. В результате получают 150 мг целевого продукта. Т.пл. 240oC (см. табл. 23).
Пример
23. 4-[(2-Бутил-3Н-имидазо(4,5-b)пиридин-1-ил)метил]бензонитрил (продукт А) и 4-[(2-бутил-3Н-имидазо(4,5-b)пиридин-3-ил)метил]бензонитрил (продукт Б)
Этап А.
2-Бутил-3Н-имидазо(4,
5-b)пиридин
Операции выполняют, как на этапе А примера 1, но исходят из 3,27 г 2,3-диаминопиридина с использованием 6,5 см3 валериановой кислоты. В результате
хроматографии на
двуокиси кремния (элюент: этилацетат-метанол (8:2)) получают 5,7 г продукта, который подвергается обработке активированным углем в этиловом эфире и кристаллизации в 20 см3
простого
изопропилового эфира. Таким образом получают 3,9 г искомого продукта. Т.пл. 104oC.
Проба для анализа была получена путем рекристаллизации 300 мг указанного выше продукта в этиловом эфире, в результате чего получают 240 мг искомого продукта. Т.пл. 104oC.
Химический состав для C10H13N3 175,235
расчетный
C 68,54; H 7,48; N 23,98
полученный C 68,3; H 7,5; N 23,7
Этап Б. 4-[(2-Бутил-3Н-имидазо(4,5-b)пиридин-1-ил)метил] бензонитрил (продукт А) и 4-[(2-бутил-3Н-имидазо(4,
5-b)-пиридин-3-ил)метил]бензонитрил (продукт Б)
Операции выполняют, как на этапе Б примера 1, но исходят из 700 мг продукта, полученного на предыдущем этапе А, с использованием 200 мг
50%-ной
дисперсии гидрида натрия в масле и 800 мг 4-бромбензонитрила.
В результате хроматографии на двуокиси кремния (элюент: метиленхлорид-метанол(9:1)) получают:
фракция А:
510 мг
продукта, кристаллизованного из этилового эфира (Т.пл. 103oC) подвергают рекристаллизации в 3 см3 этилацетата для получения 280 мг искомого продукта, Т.пл. 130o
C.
фракция Б: 170 мг продукта, кристаллизованного из этилового эфира (Т.пл. 130oC), перекристаллизовывают в 3 см3 этилового эфира и получают 155 мг искомого продукта, Т.пл. 103oC.
Химический состав фракции А для C18H18N4 290,37
расчетный C 74,46; H 6,25; N 19,29
полученный C 74,
2; H 6,2;
N 19,1
Химический состав фракции Б для C18H18N4 290,37
расчетный C 74,46; H 6,25; N 19,29
полученный C 74,4; H 6,2; N 19,2
Пример
24. 4-[(2-Бутил)1Н-имидазо(4,5-b)пиридин-1-ил)-метил] бензойная кислота
Операции выполняют, как в примере 12, но исходят из 250 мг продукта, полученного на предыдущем этапе,
фракция А, с
использованием 0,7 см3 гидроокиси натрия. В результате получают 210 мг целевого продукта, Т.пл. 190oC, который подвергается двойной рекристаллизации в изопропаноле
для получения
140 мг искомого продукта. Т.пл. 200oC (см. табл. 24).
Пример 25. 4-[(2-Бутил)3Н-имидазо(4,5-b)пиридин-3-ил)-метил] бензойная кислота
Операции
выполняют, как в
примере 12, но исходят из 130 мг фракции Б, полученной в примере 23. Таким образом получают 110 мг целевого продукта, Т. пл. 180oC, кристаллизованного из смеси
"изопропанол-вода". После
рекристаллизации данного продукта в 2 см3 этилового эфира получают 80 мг искомого продукта. Т.пл. 180oC (см. табл. 25).
Пример 26.
4-[(2-Бутил-3Н-имидазо(4,
5-b)пиридин-3-ил)-метил](1,1'-бифенил)2-карбоксилат метила (фракция А) и 4'-[(2-бутил-1Н-имидазо(4,5-b)пиридин-3-ил)метил-1,1'-бифенил]2-карбоксилат метила (фракция Б)
Операции выполняют, как
на этапе Б примера 1, но исходят из 876 мг продукта, полученного на предшествующем этапе А примера 23, с использованием 250 мг 50% -ной взвеси гидрида натрия в масле и 1,
53 г 4'-бром(1,
1'-бифенил)2-карбоксилат метила. После хроматографии на двуокиси кремния (элюент: метиленхлорид-метанол (9: 1)) получают 1,2 г соединения фракции А, Т.пл. 135oC, и 450 мг
соединения фракции
Б.
Затем выполняют рекристаллизацию 100 мг соединения А в этиловом эфире, в результате чего получают 80 мг продукта, Т.пл. 140oC (см. табл. 26).
Пример 27.
4'-[(2-Бутил-1Н-имидазо(4,5-b)пиридин-1-ил)-метил](1,1'-бифенил)2-карбоновая кислота
Операции выполняют, как в примере 12, но исходят из 680 мг продукта фракции А,
полученного в примере 26, с
использованием 0,7 см3 концентрированной гидроокиси натрия. Это дает возможность получить 650 мг искомого продукта. Т.пл. 170oC. 900 мг продукта,
полученного, как указано выше,
растворяют в 200 см3 изопропанола при температуре кипения (с рефлюксом), концентрируют до 20 см3, и добавляют 30 см3 воды. После
центрифугирования получают 810 мг
продукта, Т.пл. 205oC, который вновь подвергают рекристаллизации в 5 см3 этанола. В результате получают 640 мг целевого продукта. Т.пл. 205oC (см. табл. 27).
Пример 28. 4'-[(2-Бутил-3Н-имидазо(4,5-b)пиридин-3-ил)-метил](1,1'-бифенил)2-карбоновая кислота
Операции выполняют, как в примере 12, но исходят из
410 мг продукта фракции Б,
полученного в примере 26, с использованием 0,4 см3 натрового щелока.
В результате получают 320 мг целевого продукта, Т.пл. 185oC, которые подвергают рекристаллизации в 10 см3 смеси "изопропанол-вода" (1:1). Таким образом получают 210 мг продукта, Т.пл. 190oC. Полученный продукт в виде раствора обрабатывают в этилацетате с активированным углем и концентрируют, после фильтрации до 3 см3. После центрифугирования получают 140 мг искомого продукта. Т.пл. 190oC (см. табл. 28).
Приготовление исходного соединения, используемого в примере 29 и 30
Этап А. 2-Нитро-3-тиофенамин
При комнатной температуре растворяют 12,8 г 85-ного 2-нитротиофена (фирма
"ОЛДРИХ"),
33,6 г 4-амино-4Н-1,2,4-триазола (1-1, 3-4) (фирма "ФЛУКА") в 100 см3 безводного диметилсульфоксида.
Затем охлаждают до 5oC и добавляют примерно за 15 мин раствор 22,4 г трет-бутилата калия в 100 см3 безводного диметилсульфоксида.
Полученную суспензию взбалтывают примерно 15 мин при комнатной температуре, а затем вливают в 0, 6 л насыщенного раствора хлорида аммония.
Затем трижды выполняют экстрагирование с помощью 500 см3 этилацетата, тройную промывку в 400 см3 воды, высушивание, фильтрацию и выпаривание досуха.
Полученный продукт растворяют в 800 см3 метиленхлорида. Затем фильтруют и выпаривают досуха.
Полученный продукт кристаллизуют в 20 см3 простого изопропилового эфира. Затем выполняют центрифугирование, промывку простым изопропиловым эфиром и сушку при 80oC при пониженном давлении.
В результате получают 5,1 г целевого продукта, Т.пл. 159oC.
Химический состав для C4H4N2O2S 144,15
расчетный
C 33,33; H 2,80; N 19,43; S 22,24
полученный C 33,0; H 2,7; N 19,2; S 22,2
Инфракрасный спектр: (CHCl3)
-NH2 3510 см-1 +
3380 см-1
1608 см-1
-NO2 1554 см-1 1328 см-1
Этап Б 3-Трет-бутилкарбонат амино-2-нитротиофена
При комнатной
температуре растворяют 1,3 г
продукта, полученного на этапе А, в 15 см3 безводного тетрагидрофурана. Затем охлаждают до +4oC и добавляют за 10 мин раствор 2 г ди-трет-бутил
дикарбоната (фирма "ФЛУКА") и 110
мг диметиламинопиридина в 15 см3 безводного тетрагидрофурана.
Затем взбалтывают еще 30 мин и выпаривают растворители.
Полученное масло подвергают очистке методом хроматографии на двуокиси кремния (элюент: этилацетат-флуген (2:8)).
Полученный продукт кристаллизуют в 3 см3 простого изопропилового эфира.
Затем выполняют центрифугирование и промывку в простом изопропиловом эфире и высушивают при пониженном давлении.
Таким образом получают 2,13 г целевого продукта, Т.пл. 80oC.
Химический состав для C9H12N2O4S=244,27
расчетный C 44,25; H 4,95; N 11,47; S 13,13
полученный C 44,4; H 4,9; N 11,5; S 13,
2
Этап В. 3-Трет-бутилкарбонат амино-2-валериламинотиофена
При комнатной температуре смешивают 45 г никеля Ренея (фирма "ПРОЛАБО"),
предварительно промытого водой, 50 см3
валерианового ангидрида (фирма "МЕРК") и 15 г продукта, полученного на этапе Б.
Затем выполняют гидрогенизацию при комнатной температуре в течение 6 ч, фильтрацию, промывку метиленхлоридом и высушивание досуха. Затем добавляют 150 см3 бензина G, затравливают кристаллизацию, охлаждают за 1 ч примерно до -10oC, центрифугируют и промывают бензилом G.
Затем выполняют сушку примерно при 80oC при пониженном давлении. Вес: 12,5 г, Т.пл. 122-124oC.
После кристаллизации в этиловом эфире получают 30 мг целевого продукта, исходя из 40 мг указанного выше продукта, Т.пл. 128oC (см. табл. 29).
Этап Г. Трифторацетат
2-трет-бутилкарбонат амино-3-аминотиофена
В 25
см3 трифторуксусной кислоты (фирма "ФЛУКА") вводят за 10 мин при температуре около +5oC 8,5 г продукта, полученного на
этапе В, после чего взбалтывают при комнатной температуре в
течение 50 мин.
Затем выпаривают досуха.
После этого полученную вязкую жидкость растворяют в 500 см3 этилового эфира в состоянии кипения (с рефлюксом), обрабатывают активированным углем и фильтруют. Затем концентрируют примерно до 30 см3, добавляют 30 см3 простого изопропилового эфира, концентрируют примерно до 40 см3, кристаллизуют, центрифугируют, промывают простым изопропиловым эфиром, а затем метиленхлоридом.
Затем производится сушка при пониженном давлении при температуре около 70o C. Вес: 5,13 г, Т.пл. 100oC.
После рекристаллизации в этиловом эфире 370 мг указанного выше продукта получают 240 мг целевого продукта, Т.пл. 100oC (см. табл. 30).
Этап Д. 2-Бутилтиено (2,3)имидазол
При комнатной температуре растворяют 940 мг продукта,
полученного на этапе Г, в 20 см3 воды. Затем добавляют 40 см3 насыщенного раствора карбоната натрия. После этого трижды экстрагируют с помощью 60 см3 метиленхлорида,
высушивают, фильтруют и выпаривают. Затем растворяют полученную вязкую
жидкость при комнатной температуре в 3 см3 оксихлорида фосфора и взбалтывают в течение 10 мин при комнатной
температуре, после чего нагревают в течение 1 ч до температуры кипения (с
рефлюксом) при внешней температуре около 115oC.
Затем избыток реактивы выпаривают, добавляют 30 см3 насыщенного раствора бикарбоната натрия, трижды экстрагируют с помощью 60 см3 сухого метиленхлорида, обрабатывают активированным углем и выпаривают досуха.
Полученный продукт растворяют в 300 см3 этилового эфира в состоянии кипения (с рефлюксом), фильтруют и концентрируют до 5 см3. После кристаллизации выполняют центрифугирование, промывку несколькими каплями этилового эфира и сушку при пониженном давлении при температуре около 100oC.
В результате получают 400 мг целевого продукта. Т.пл. 160oC.
Химический состав для C9H12N2S= 180,27
расчетный C 59,96; H 6,71; N 15,54; S 17,79
полученный C 60,1; H 6,8; N 15,5; S 17,
8
Пример 29. 4'-[(2-Бутил-1Н-тиено(2,
3-d)имидазол-1-ил)-метил]-(1,1'-бифенил)-2-карбоксилат метила (изомер А)
Пример 30. 4'-[(2-Бутил-3Н-тиено(2,3-d)имидазол-3-ил)метил]-(1,
1'-бифенил)-2-карбоксилат метила (изомер Б)
При комнатной температуре растворяют 360 мг продукта, полученного на этапе Д, исходного для приготовления примеров 29 и 30, в 7 см3
безводного тетрагидрофурана.
При комнатной температуре дважды добавляют 96 мг 50%-ной взвеси гидрида натрия в масле.
После выделения водорода взбалтывают еще 10 мин в атмосфере азота, а затем добавляют разом 610 мг бромметил-(1,1'-бифенил) 2-карбоксилата метила (приготовленного согласно патентной заявки ЕЭС EP N 0253310).
Затем взбалтывают при комнатной температуре в течение около 35 мин, а затем выпаривают растворитель в атмосфере азота. После этого добавляют 20 см3 воды, трижды экстрагируют с помощью 60 см3 метиленхлорида, дважды промывают в 10 см3 воды, высушивают органическую фазу, фильтруют и выпаривают.
Путем хроматографии на двуокиси кремния (элюент: этилацетат-циклогексан (50:50)) получают оба целевых изомера, которые являются предметом примера 29 (изомер А) и примера 30 (изомер Б).
Таким образом получают 600 мг изомера А (Rf 0,40) и 250 мг изомера Б (Rf 0,28).
Спектр
ЯМР (изомер А) (CDCl3, 250 МГц млн-1)
CH3 0,96 (t)
CH3
-CH2-(CH2)2- 1,44 (m)
CH3
-CH2-CH2-CH2 1,80 (m)
CH3-(CH2)2-CH2
2,82 (t)
CO2-CH3 3,63 (s)
N-CH2-C6H4 5,29 (s)
S-CH2-CH2 6,96 (d)
S-CH2
-CH2 6,64 (d)
Спектр ЯМР (изомер Б) (CDCl3, 250
МГц млн-1)
CH3 0,96 (t)
CH3-CH2(CH2)2
- 1,47 (m)
CH3-CH2-CH2-CH2
1,84 (m)
CH3-(CH2)2-CH2 2,87 (t)
CO2-CH3 3,63 (s)
N-CH2-C6H4 5,24 (s)
S-CH2-CH2 7,11 (d)
S-CH2-CH2 6,87 (d)
Пример 31.
4'-[(2-Бутил-1Н-тиено(2,3-d)имидазол-1-ил)-метил]-(1,1'-бифенил)-2-карбоновая
кислота
600 мг продукта из примера 29 (изомер А) растворяют при комнатной температуре в 7 см3
метанола. Затем добавляют 0,5 см3 натрового щелока (10 N). После этого
нагревают в течение 1 ч до температуры кипения (с рефлюксом), выпаривают растворитель и растворяют сухой экстракт в
15 см3 воды.
Затем слегка охлаждают в водяной бане и добавляют ледяную уксусную кислоту до получения pH от 4 до 5.
После этого выполняют центрифугирование, промывку водой и высушивание при пониженном давлении при температуре около 100oC.
В результате получают 400 мг целевого продукта, Т.пл. 203oC.
Химический состав для C23H22N2OS 390,506
расчетный C 70,74; H 5,68; N 7,17; S 8,21
полученный C 70,8; H 5,6; N 7,1; S 8,2
Инфракрасный
спектр: Вазелиновое масло
Кислота

Ароматическое соединение 1595 см-1
Гетероароматические соединения 1504 см-1 1516 см-1 1482 см-1
Пример 32. 4'-[(2-Бутил-3Н-тиено(2,3-d)имидазол-3-ил)-метил]-(1, 1'-бифенил)-2-карбоновая кислота
Операции выполняют, как в примере 31, но исходят из 250 мг продукта примера 30 (изомер Б) в 4 см3 метанола, после чего добавляют 0,3 см3 едкого натра (10 N).
В результате получают 160 мг целевого продукта, Т.пл. 203oC (см. табл. 31).
Приготовление исходного соединения для примера 33
Этап А. 3-Амино-4-валериламинотеофен
При комнатной температуре смешивают 3,06 г 3,
4-диаминотиофена (фирма "ЯНСЕН") в 45 см3 метиленхлорида и 9 см3
триэтиламина.
При температуре 20oC добавляют примерно за 1 ч раствор 3,6 г хлорида валероила (фирма "ФЛУКА") в 45 см3 метиленхлорида. Затем взбалтывают еще 30 мин и выпаривают досуха. После хроматографического разделения (элюент: метанол-метиленхлорид (5: 95)) получают 3, 2 г целевого моноамида, который подвергают кристаллизации в 15 см3 смеси 50:50 простого изопропилового эфира и циклогексана, центрифугируют, промывают этой смесью и высушивают при пониженном давлении при температуре около 100oC.
В результате получают 2,14 г целевого продукта. Т.пл. 110oC (см. табл. 32).
Этап Б.
2-Бутилтиено[3,4]имидазол
При комнатной температуре и при взбалтывании растворяют 1 г
продукта, полученного на этапе А, в 3 см3 оксихлорида фосфора, после чего нагревают до
температуры кипения (с рефлюксом) в бане при температуре примерно 120oC в течение около 1
ч.
Затем выпаривают досуха, добавляют 100 см3 насыщенного водного раствора бикарбоната натрия, взбалтывают в течение 20 мин при комнатной температуре, выполняют двукратное экстрагирование с помощью 100 см3 метиленхлорида, промывают 50 см3 раствора бикарбоната натрия, высушивают органическую фазу, фильтруют и выпаривают.
Полученную вязкую жидкость растворяют в 600 см3 этилового эфира в состоянии кипения (с рефлюксом). Затем фильтруют и выпаривают досуха. Таким образом получают 570 мг продукта, который подвергают кристаллизации в 2 см3 изопропилового эфира. В результате получают 500 мг целевого продукта, Т.пл. 118oC.
Спектр ЯМР (CDCl3 60 МГц млн-1
)
S-CH2 6,71
-CH2 2,68-2,80-2,91
CH2
центральн. 1,1 по 2,13
CH3 0,83-0,93-1,03
Пример 33.
4'-[(2-Бутил-1Н-тиено(3,4-d)имидазол-1-ил)-метил]-(1,1'-бифенил)-2-карбоксилат метила
При комнатной
температуре растворяют 500 мг продукта, полученного на этапе Б приготовления исходного
соединения для примера 33, в 10 см3 безводного тетрагидрофурана.
Затем добавляют 120 мг 50%-ной взвеси гидрида натрия в масле, взбалтывают до окончания выделения водорода, а затем в атмосфере азота в течение 10 мин и добавляют 750 мг бромметил(1,1'-бифенил)2-карбоксилата метила (приготовленного согласно патентной заявке ЕЭС EP N 0253310).
После этого взбалтывают при комнатной температуре в течение примерно 1 ч, выпаривают досуха. Затем выполняют тройное экстрагирование со 100 см3 метиленхлорида, двойную промывку в 50 см3 воды, высушивание, фильтрацию и выпаривание досуха. После хроматографии на 400 г двуокиси кремния (элюент: этилацетат-циклогексан (5:5)) получают 1,14 г целевого продукта.
Полученную вязкую жидкость кристаллизуют в 2 см3 простого изопропилового эфира. Затем выполняют центрифугирование, промывку несколькими каплями простого изопропилового эфира и сушку при пониженном давлении при температуре около 70oC.
Путем двух последовательных рекристаллизаций в простом изопропиловом эфире получают 70 мг целевого продукта, Т.пл. 120o C.
Химический состав для C9H12N2S 180,26
расчетный C 59,96; H 6,71; N 15,54 S 17,79
полученный C 60,0; H 6,8; N 15,5 S 17,6
Пример 34. 4'[(2-Бутил-1Н-тиено(3,4-d)имидазол-1-ил)-метил]-(1,1'-бифенил)-2-карбоновая кислота
При комнатной температуре и при взбалтывании растворяют 1,14 г продукта, полученного в примере
33, в 25 см3 метанола. Затем добавляют 1 см3 натрового щелока и нагревают до
температуры кипения (с рефлюксом) в течение 1 ч.
Затем метанол выпаривают, растворяют полученный сухой экстракт в 75 см3 воды и добавляют ледяную уксусную кислоту до получения pH 6-5.
После этого взбалтывают еще 15 мин, подвергают центрифугированию, промывают водой и высушивают при пониженном давлении при температуре около 80oC. Таким образом получают 870 мг целевого продукта, Т.пл. 168oC.
1,02 г продукта, полученного, как указано выше, растворяют в 50 см3 этанола в состоянии кипения (с рефлюксом). После этого добавляют 1 каплю уксусной кислоты, а затем 40 см3 воды. Затем выполняют кристаллизацию, центрифугирование, промывку смесью воды и спирта (50:50) и высушивают при пониженном давлении при температуре около 100oC. Таким образом получают 900 мг искомого продукта, Т.пл. 168oC.
Полученный выше продукт растворяют в 100 см3 этилацетата в состоянии кипения (с рефлюксом), фильтруют, концентрируют примерно
до 50 см3, кристаллизуют, центрифугируют, промывают этилацетатом и высушивают при пониженном
давлении при температуре около 100oC. В результате получают 700 мг целевого продукта,
Т.пл. 168oC. (см. табл. 33)
Приготовление исходного соединения для примера 36
Этап А. 5-бром-2-бутил-1Н-имидазол-4-метанол
10 г 2-бутил-1Н-имидазол-4-метанола
(приготовленного согласно патентной заявке ЕЭС EP N 0253310) вводят, взбалтывая, в 150 см3
диоксана и 150 см3 2-метоксиэтанола. Затем добавляют 12,7 г Н-бромсукцинимида и
продолжают взбалтывание при температуре 40oC в течение около 2 ч.
Затем реакционной среде дают остыть при комнатной температуре в течение 1 ч 30 мин. После этого выполняют выпаривание, и кристаллы поглощают в 250 см3 этилацетата и 150 см3 воды. Затем выполняют двойное экстрагирование с помощью 100 см3 этилацетата.
После этого выполняют двойную промывку в 100 см3 воды, высушивают, фильтруют, добавляют к раствору активированного угля и взбалтывают в течение примерно 10 мин. Затем фильтруют и выпаривают. Кристаллы растворяют при повышенной температуре в 64 см3 этилацетата, после чего раствор выстаивают в течение 1 ч 30 мин при температуре около 0oC.
Затем производят центрифугирование, три раза промывают кристаллы в 6 см3 этилацетата и три раза в 6 см3 метиленхлорида, после чего центрифугируют и высушивают. В результате получают 4, 39 г целевого продукта, Т.пл. 160oC (см. табл. 34).
Этап Б.
2-Бромо-2-бутил-1Н-имидазол-4-карбоксальдегид
4,37 г продукта, полученного на этапе А, вводят в 130
см3 диоксана, после чего добавляют, взбалтывая, 16,32 г двуокиси марганца.
Смесь нагревают при температуре ≈ 100oC примерно 2 ч. Затем смесь фильтруют, ополаскивают диоксаном и выпаривают. В результате получают 3,72 г целевого продукта, Т.пл. 113oC (см. табл. 35).
Этап В.
4'-[(4-Бромо-2-бутил-5-формил-1Н-имидазол-1-ил)метил]-(1,1'-бифенил)-2-карбоксилат метила
3,7 г продукта, полученного на этапе Б,
вводят в 50 см3 диметилформамида. При
взбалтывании добавляют 2,465 г бикарбоната калия и выдерживают примерно 5 мин. Затем добавляют 5,86 г бромметил(1,1'-бифенил)2-карбоксилата метила
(приготовленного согласно патентной заявке ЕЭС EP N
0253310) в 55 см3 диметилформамида.
Смесь выдерживают примерно 3 сут при комнатной температуре. Затем реакционную среду гидролизуют в 100 см3 воды и трижды экстрагируют с помощью 50 см3 этилацетата.
Затем выполняют промывку в 100 см3 воды, насыщенной хлоридом натрия, высушивание, фильтрацию и выпаривание. После хроматографии (элюент: этилацетат/метиленхлорид (5:95)) получают 6,36 г целевого продукта (см. табл. 36).
Пример 35.
2-Бутил-1-[(2'-карбокси-(1,1'-бифенил)-4-ил)-метил]-1Н-тиено(2,
3-d)имидазол-5-карбоксилат этила
Этилат натрия (149,5 мг х 2) вводят в 5 см3 этанола и добавляют, взбалтывая, (264 мг
х 2) меркаптоэтилацетат (приготовленный согласно 0153229
- патент "ТЭЧ АСЗОДИ" 85211781) в 2 см3 этанола.
Затем добавляют 500 мг продукта, полученного на этапе В, в 12 см3 этанола. Нагревают до состояния кипения (с рефлюксом) около 78oC, взбалтывая, в течение 3 ч. Затем удаляют растворители, добавляют 400 см3 воды и 5 раз экстрагируют в 100 см3 метиленхлорида. После этого выполняют высушивание, фильтрацию и выпаривание. После хроматографии (элюент: этилацетат/метиленхлорид (5:95)) получают 322,5 мг целевого продукта (см. табл. 37).
Пример 36.
2-Бутил-1-[2'-карбокси-(1,1'-бифенил)-4-ил)-метил]-1Н-тиено(2,3-d)имидазол-5-карбоновая кислота
295,4 мг продукта, полученного в примере
35, вводят в 10 см3 этанола. Затем
прикапывают 1,55 см3 едкого натра (2 Н) и выдерживают смесь при комнатной температуре, взбалтывая, в течение примерно 5 сут. Затем выпаривают
при пониженном давлении и поглощают в 8 см3 воды.
Затем добавляют 1,55 см3 хлористоводородной кислоты (2N), фильтруют и высушивают при пониженном давлении. Затем продукт поглощают в 9 см3 горячего изопропанола. Добавляют 4 см3 воды, фильтруют и высушивают. В результате получают 140 мг целевого продукта. Т.пл. 264oC (см. табл. 38).
Приготовление исходного
соединения для примера 37
Этап А. Меркаптоацетат 1,1-диметилэтила
Этап АЛЬФА. Тиоацетат 1,1-диметилэтила
Смешивают 35,24 г
0-этилдитиокарбоната калия в 150 см3
ацетона при температуре около 0 до 2oC и добавляют за 10 мин 39,16 г бромацетата 1,1-диметилэтила. Смесь взбалтывают при комнатной
температуре, а затем вливают в 800 см3 эфира,
концентрируют и фильтруют.
Остаток поглощают в 400 см3 эфира, фильтруют и вновь выпаривают и получают 49,58 г целевого продукта.
Этап БЕТА. Меркаптоацетат
1,1-диметилэтила
Продукт, полученный на этапе АЛЬФА, охлаждают до температуры около 0oC и прикапывают 6,48 г 1,
2-диаминоэтана.
Затем взбалтывают примерно 2 ч при комнатной температуре и добавляют 200 см3 гексана, взбалтывают примерно 10 мин и экстрагируют остаток гексаном. Затем гексановый раствор промывают с 200 см3 хлористоводородной кислоты (0,1N), а затем с 200 см3 раствора бикарбоната натрия, после чего высушивают и концентрируют.
Остаток дистиллируют при пониженном давлении и получают 21,71 г целевого продукта.
Точка кипения при давлении 21 мм рт.ст. 72oC.
Этап Б. 3-Амино-3[(2(1,
1-диметилэтокси)-2-оксоэтил)тио]-2-[(1-оксопентил)амино]пропеноат
этила
5 г 2-[(1-оксопентил)амино]2-цианоэтаноат этила смешивают в 45 см3 метиленхлорида, а затем добавляют 0,33
см3 триэтиламина и 2,15 г продукта, полученного на этапе
А. Затем взбалтывают при комнатной температуре в течение примерно 24 ч и получают 8,49 г целевого продукта.
Этап В.
2-Бутил-5-[(2(1,
1-диметилэтокси)-2-оксоэтил)тио]-1Н-имидазол-4-карбоксилат этила
Смешивают 127 см3 метиленхлорида и 9,81 г пентахлорида фосфора. Смесь доводят примерно до -78oC и
прикапывают 6,33 г диметиламинопиридина, растворенного в 59 см3 метиленхлорида. Затем взбалтывают в течение примерно 5 мин и добавляют 8,49 г продукта, полученного на этапе Б,
после чего
вновь взбалтывают около 5 мин при температуре -78oC.
Реакционной смеси дают вернуться к комнатной температуре и взбалтывают в течение около 24 ч. После этого реакционную среду вливают в 400 см3 бикарбоната натрия и взбалтывают 20 мин, после чего декантируют и трижды экстрагируют водную фазу с помощью 300 см3 этилацетата.
Органические фазы промывают в 500 см3 воды, высушивают, фильтруют и выпаривают при пониженном давлении. Таким образом получают 7,61 г вязкой жидкости, которую подвергают очистке методом хроматографии (элюент: этилацетат/метиленхлорид (10:90)). В результате получают 3,49 г целевого продукта. (P 0,26). (Табл. 39).
Этап Г. 2-Бутил-1-[(2'-(метоксикарбонил) (1,
1'-бифенил)-4-ил)метил]5-[(2(1,1-диметилэтокси)2-оксоэтил)тио]-1Н-имидазол-5-карбоксилат этила
1 г продукта, полученного на этапе В, смешивают в 6 см3 диметилформамида и добавляют
450 мг бикарбоната калия. Затем взбалтывают примерно 5 мин при комнатной температуре и вводят 1,07 г бромметил-(1,1'-бифенил)-2-карбоксилат метила (приготовленного согласно патентной заявке ЕЭС EP N
0253310), растворенного в 6 см3 диметилформамида. Смесь выдерживают со взбалтыванием при комнатной температуре примерно в течение 3 сут.
Реакционную среду подвергают гидролизу с помощью 500 см3 воды, а затем трижды экстрагируют с помощью 200 см3 этилацетата. Органические фазы промывают в 200 см3 воды, а затем в 200 см3 воды, насыщенной хлоридом натрия, а затем высушивают, фильтруют и выпаривают. Таким образом получают 1,785 г вязкой жидкости, которую очищают методом хроматографии (элюент: этилацетат/метиленхлорид (2:98)). В результате получают 1,381 г целевого продукта. (Rf 0,26) (см. табл. 40).
Пример 37. 1,1-Диметилэтиловый эфир 2-бутил-6-гидрокси-1-[(2'-(метоксикарбонил)-(1,
1'-бифенил)-4-ил)метил]1Н-тиено[4,5-b]имидазол-5-карбоновой кислоты
47 мг продукта, полученного на этапе Г, вводят в 1 см3 тетрагидрофурана, затем раствор доводят примерно до
температуры -78oC и добавляют раствор биотриметилсилиламида лития (1 моль/л) в 0,42 см3 тетрагидрофурана.
Затем реакционную среду выдерживают около 2 ч в условиях взбалтывания при температуре -78oC, после чего доводят до комнатной температуры примерно за 2 ч. После этого реакционную среду охлаждают до примерно -78oC и затем гидролизуют с помощью 10%-ного раствора уксусной кислоты в тетрагидрофуране.
После этого раствор доводят до комнатной температуры, выпаривают при пониженном давлении и поглощают в 30 см3 этилацетата. Затем органическую фазу промывают водой, а затем водой, насыщенной хлоридом натрия. После этого органическую фазу высушивают, фильтруют и выпаривают. Таким образом получают 40 мг продукта, который очищают методом хроматографии (элюент: этилацетат/метиленхлорид (2:98)), в результате чего получают 16,5 мг целевого продукта (см. табл. 41).
Пример
38. 1,
1-Диметиловый эфир 2-бутил-1-[(2'-карбокси-(1,1'-бифенил)-4-ил)метил]-6-гидрокси-1Н-тиено(2,3-d)имидазол-5-карбоновой кислоты
208,9 мг продукта, полученного в примере 37, смешивают в
12
см3 этанола и прикапывают 2 см3 едкого натра 2N.
Затем смесь выдерживают около 3 сут в условиях взбалтывания при комнатной температуре, после чего реакционную среду выпаривают и поглощают в 10 см3 горячей воды.
После этого добавляют 2 см3 хлористо-водородной кислоты (2N), фильтруют и высушивают при пониженном давлении в течение около 24 ч. Таким образом получают 185,6 мг порошкообразного продукта, который подвергают рекристаллизации путем растворения в 14 см3 горячего изопропанола, после чего добавляют 6 см3 воды и охлаждают.
Полученные кристаллы подвергают центрифугированию, промывают водой и высушивают при пониженном давлении при температуре около 40oC в течение примерно 14 ч. В результате получают 137,5 мг целевого продукта, Т.пл. 211oC-222oC.
Пример 39. 2-Бутил-1-[(2'-карбокси-(1,
1'-бифенил)-4-ил)-метил]-6-гидрокси-1Н-тиено(2,3-d)имидазол-5-карбоновая кислота
Данная кислота может быть получена из продукта примера 38 путем омыления сложноэфирной функциональной группы
CO2 трет.Бу с получением COOH в положении 5 цикла тиено.
Пример 40. 1,1-Диметилэтиловый эфир 2-бутил-1-[(2'-карбокси-(1,1'-бифенил)-4-ил)метил]-1Н-тиено(2,
3-d)-имидазол-6-карбоновой кислоты
Данный продукт может быть получен, как в примере 37.
Пример 41. 1,1'-Диметилэтиловый эфир 2-бутил-1-[(2'-(метоксикарбонил)-(1,
1'-бифенил)-4-ил)метил]-1Н-тиено(4,5-b)имидазол-6-карбоновой кислоты
Данный продукт может быть получен, как в примере 38.
Пример 42. 2-Бутил-1-[(2'-карбокси-(1,
1'-бифенил)-4-ил)-метил]-1Н-тиено(2,3-d)имидазол-6-карбоновая кислота
Данная кислота может быть получена, как в примере 39.
Пример 43. Фармацевтическая композиция
Получают таблетки следующего состава:
Продукт примера 10 10 мг
Основа для готовой таблетки 100 мг
(конкретные компоненты основы: лактоза, тальк, крахмал, стеарат
магния).
Фармакологические результаты
1. Тест на рецепторе ангиотензина II
Для данного теста используют свежий мембранный препарат, полученный из печени крысы. Ткань
измельчают
политроном в буферном растворе Tris 50 ммоль, pH 7,4, после чего выполняют 3 центрифугирования при 30000 g в течение 15 мин с промежуточным отбором осадков буферным раствором Tris pH 7,
4.
Последние осадки суспензируют в инкубационном буферном растворе (Tris 20 ммоль, NaCl 135 ммоль, KCl 10 ммоль, глюкоза 5 ммоль, MgCl2 10 ммоль, PMSF 0,3 ммоль, бацитрацин 0,1 ммоль, BSA 0,2%).
Затем аликвотные доли по 2 см3 распределяют в гемолизные пробирки и добавляют125 I ангиотензина II (25000 DPM на пробирку) и исследуемый
продукт. Вначале продукт трижды тестируют при 10-5M. Если исследуемый продукт смещает более 50% радиоактивности, специфически связанной с рецептором, то его вновь тестируют согласно серии
из 7 концентраций для определения концентрации, которая ингибирует 50% радиоактивности, специфически связанной с рецептором. Таким образом определяют ингибирующую концентрацию 50%
Неспецифическая связь определяется добавлением продукта примера 94 патент ЕЭС N 0253310 при 10-5М (трижды). Затем производят инкубацию при 25oC в течение 150 мин, устанавливают
на 5 мин в водяную баню при 0oC, выполняют вакуумную фильтрацию, ополаскивают буферным раствором Tris pH 7,4 и измеряют радиоактивность в присутствии сцинтилятора Тритон.
Результат непосредственно выражается в ингибирующей концентрации 50% (CI50), то есть в концентрации исследуемого продукта, выраженной в нмоль, необходимой для смещения 50% удельной радиоактивности, зафиксированной на исследуемом рецепторе.
Полученные результаты:
Продукт CI50 в наномолях
34 789
5 1440
10 2270
2 2350
20 2715
15 3610
6 3460
27 89
25 440
28 217
31 140
32 298
38 54
2. Выявление антагонистической
активности ангиотензина II на изолированной воротной вене
Воротная вена берется у самцов крыс Уистар (весом около 350 г) (IFFA Credo France) после раздробления шейного позвонка и быстро
погружается в физиологический раствор (см. ниже) при комнатной температуре. Кольцо примерно в 1 мм устанавливают в ванну с изолированным органом, содержащую 20 см3 следующего
физиологического раствора (состав в ммоль: NaCl 118,3 KCl 4,7 - MgSO4 1,2 KH2PO4 1,2 NaHCO3 25 глюкоза 11,1 CaCl2 2,5), при этом среда
выдерживается при температуре 37oC и насыщается кислородом смесью O2 (95%), CO2 (2%). Начальное натяжение составляет 1 г, кольца оставляют в покое от 60 до 90 мин.
Для
предотвращения самопроизвольного сокращения в инкубационную ванну добавляют верапамил (1.10-6М).
В конце периода покоя ангиотензин II (гипертензин Циба) 3.10-8М добавляют в инкубационную ванну и оставляют в контакте с препаратом в течение 1 мин. Эту операцию повторяют каждые 30 мин, причем между двумя стимуляциями ангиотензином ткань 3-4 раза промывается. Исследуемое соединение вводится в ванну за 15 мин до новой стимуляции ангиотензином. Применяя все более высокие концентрации соединения, можно определить CI50 (концентрации продукта, вызывающей ингибирование 50% реакции на ангиотензин), выраженный в молях.
Полученные результаты:
Продукт CI50 в наномолях
10 3280
2
3400
5 3600
25 830е
Реферат
Новые производные имидазолов, способ их получения; новые полученные промежуточные продукты, их применение в качестве медикаментов и содержащие их фармацевтические соединения. Предложены новые производные имидазолов, способ их получения и их использование в качестве лекарственных средств, использующихся в качестве ингибиторов действия ангиотензина II. Также предложен способ приготовления лекарственного средства. 3 с. и 2 з.п. ф-лы, 41 табл.
Формула

где А фенил, нафтил, пиридил, пиримидинил, тиенил;
R н-бутил или бутен-1-ил;
R1, R2, R3 и R4 являются такими, что два из них являются атомом водорода, а два других, одинаковые или различные, являются атомом водорода, гидроксилом, алкилом, содержащим не более 4 атомов углерода, карбоксилом, свободным или этерифицированным, линейным или разветвленным алкилом, содержащим не более 4 атомов углерода;
R5 метилен;
Y радикал -Y1 -B Y2, в котором Y1 фенил; B - простая углерод-углеродная связь или радикал -CO NH-, а Y2 является цианогруппой, карбоксилом, свободным или этерифицированным линейным или разветвленным алкилом, содержащим не более 4 атомов углерода; индолилом или фенилом, возможно замещенным свободным, этерифицированным или солевым карбоксилом, цианогруппой, формилом, тетразолильным, тетразолилалкильным, тетразолилкарбомоильным радикалом, в которых тетразолильный радикал возможно замещен алкилом, алкенилом или алкоксиалкилом; или радикалом - (CH2)p SO2 Xa R14a,
в котором p 0 или 1; Xa радикалы -NH-, NHCO NH-, -NH CO- или простая связь, а R14а метил, этил, аллил, пиридилметил, пиридилэтил, пиридил, фенил или бензил,
с выделением целевого продукта в виде рацемата, энантиомеров диастереоизомеров или солей с минеральными или органическими кислотами или основаниями.
где X атом кислорода или радикал NH;
R15 гидрокси-, алкоксигруппа, атом галогена или NH2;
R1 имеет значения, указанные для R,
где возможные функциональные группы могут быть защищены с помощью защитных групп,
вводят в реакцию с соединением формулы III

где R, R, R и R имеют приведенные выше значения соответственно для R1, R2, R3 и R4, где возможные функциональные группы могут быть защищены с помощью защитных групп,
и после возможного выделения промежуточного продукта формулы IV

где X, R1, R, R, R и R имеют вышеприведенные значения,
получают соединение формулы IV а

где R1, R, R, R и R имеют вышеприведенные значения,
которое вводят в реакцию с соединением формулы V
Hal R5 Y1,
где Hal галоген;
Y1 имеет значения, указанное для Y,
где возможные функциональные группы могут быть защищены с помощью защитных групп
для получения продуктов формулы IX

где R1, R, R, R и R R5 и Y1 имеют вышеприведенные значения,
с последующим снятием защитных групп.

где R, R, R и R имеют вышеприведенные значения для R1, R2, R3 и R4, где возможные функциональные группы могут быть защищены с помощью защитных групп,
с соединением формулы II'

где R1 и R15 имеют приведенные в п.2 значения для получения продукта формулы Х

который либо восстанавливают до соединения формулы Х'

которое циклизуют в продукт IVа, определенный в п.2, с последующей обработкой полученного продукта IVа, как указано в п.2, до получения продукта формулы IX,
либо подвергают взаимодействию с соединением формулы V
Hal R5 Y1,
где R5 и Y1 имеют указанное в п.2 значение,
для получения продукта формулы XI

где R1, R, R, R, R R5 и Y1 имеют указанное выше значение,
которое подвергают селективному восстановлению нитрофункции с получением соединения формулы XII

где R1, R, R, R, R R5 и Y1 имеют указанное выше значение,
которое подвергают циклизации с получением продукта формулы IX с последующим удалением защитных групп.

где R, R, R и R имеют вышеприведенные значения для R1, R2, R3 и R4, где возможные функциональные группы могут быть защищены с помощью защитных групп,
подвергают взаимодействию с соединением формулы V
Hal R5 Y1,
где Y1 имеет значения Y, где возможные функциональные группы могут быть защищены с помощью защитных групп;
R5 имеет вышеуказанное значение,
с получением промежуточного соединения формулы VII

где R - R R5 и Y1 имеют вышеуказанные значения,
и полученное промежуточное соединение формулы VII либо подвергают взаимодействию с соединением формулы II'

где R1 и R15 имеют вышеуказанные значения,
с получением соединения формулы XI
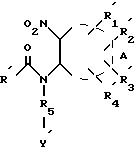
значения радикалов R5, R1, Y1 и R - R имеют вышеуказанные значения,
которые подвергают реакции восстановления для получения соединения формулы XII

где радикалы R- R, R1, R5 и Y1 имеют вышеуказанные значения,
и полученное соединение подвергают реакции циклизации для получения соединения IX
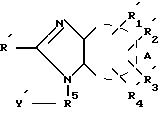
где радикалы

с последующим снятием защитных групп, либо соединение формулы VII восстанавливают для получения соединения формулы VIII

где радикалы R - R, R5 и Y1 имеют указанные в п.1 значения,
которые вводят во взаимодействие с соединением формулы II для получения соединения формулы IX с последующим снятием защитных групп.
Комментарии