Ингибиторы р38 и способы их применения - RU2357957C2
Код документа: RU2357957C2
Чертежи



































Описание
Эта заявка заявляет приоритет заявки США №10/688849, поданной 15 октября 2003 года, и заявки США №10/378164, поданной 3 марта 2003 года, которые обе включены во всей своей целостности в этот документ посредством ссылки.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Область изобретения
Это изобретение относится к новым ингибиторам МАР-киназы р38 и родственным киназам, фармацевтическим композициям, содержащим эти ингибиторы, и способам получения этих ингибиторов. Они полезны для лечения воспаления, остеоартрита, ревматоидного артрита, псориаза, болезни Крона, воспалительной болезни кишечника, рака, аутоиммунных заболеваний и для лечения других цитокин-опосредованных заболеваний.
Описание предшествующего уровня техники
Целый ряд хронических и острых воспалительных состояний связан со сверхпродукцией провоспалительных цитокинов. Такие цитокины включают в себя, но не ограничиваются ими, фактор некроза опухолей альфа (TNF-α),
интерлейкин-1 бета (IL-1β), интерлейкин-8 (IL-8) и интерлейкин-6 (IL-6). Ревматоидный артрит (РА) представляет собой хроническое заболевание, при котором TNF-α и IL-β вовлечены в появление симптомов заболеваний и в прогрессирование разрушения костей и суставов, наблюдаемого при этом состоянии, подтачивающем здоровье. Недавно одобренные терапевтические способы лечения РА включали в себя растворимый рецептор TNF-α (этанерцепт) и антагонист рецептора IL-1 (анакинра). Эти способы лечения действуют путем блокирования способности их соответствующих цитокинов связываться с их природными рецепторами. В настоящее время исследуются альтернативные способы лечения цитокин-опосредованных заболеваний. Один такой способ включает в себя ингибирование сигнального пути, который регулирует синтез и продукцию провоспалительных цитокинов, подобных р38.
Р38 (также CSBP или RK) представляет собой серин/треонин митоген-активированную протеинкиназу (МАРК), которая, как было показано, регулирует провоспалительные цитокины. Впервые р38 была идентифицирована как киназа, которая после обработки липополисахаридом (ЛПС) становится фосфорилированной по тирозину в мышиных моноцитах. Связь между р38 и реакцией клеток на цитокины впервые была установлена Saklatvala J. с соавт. (Cell, 78: 1039-1049 (1994)), который показал, что IL-1 активирует протеинкиназный каскад, который приводит к фосфорилированию малого белка теплового шока Hsp27, вероятно при помощи митоген-активированной протеин-активированной протеинкиназы 2 (МАРКАР киназы-2). Анализ пептидных последовательностей, происходящих из очищенной киназы, показал, что она относится к МАРК р38, активированной ЛПС в мышиных моноцитах (Han, J., et al., Science, 265: 808-811 (1994)). Одновременно было показано, что МАРК р38 сама активируется киназой, расположенной в каскаде выше, в ответ на множество клеточных стрессов, включая воздействие ультрафиолетового излучения и осмотический шок, и была подтверждена идентичность киназы, которая непосредственно фосфорилирует Hsp27, как МАРКАР киназы-2 (Rouse, J., et al., Cell, 78: 1027-1037 (1994)). Впоследствии сотрудники SmithKline Beecham показали, что МАРК р38 представляла собой молекулярную мишень для ряда пиридинилимидазольных соединений, которые ингибировали продукцию TNF ЛПС-стимулированными моноцитами человека (Lee, J., et al., Nature, 372: 739-746). Это стало ключевым открытием и привело к разработке целого ряда селективных ингибиторов МАРК р38 и выяснению их роли в цитокиновой передаче сигналов.
В настоящее время известно, что многочисленные формы МАРК р38 (α, β, γ, δ), каждая из которых кодируется отдельным геном, образуют часть киназного каскада, вовлеченного в реакцию клеток на различные стимулы, включая осмотический стресс, УФ-свет и цитокин-опосредованные события. Считают, что эти четыре изоформы р38 регулируют различные аспекты внутриклеточной передачи сигналов. Его активация является частью каскада сигнальных событий, которые приводят к синтезу и продукции провоспалительных цитокинов, подобных TNF-α. Р38 действует путем фосфорилирования субстратов, расположенных в каскаде ниже, которые включают в себя другие киназы и транскрипционные факторы. На моделях in vitro и in vivo было показано, что агенты, которые ингибируют киназу р38, блокируют продукцию цитокинов, включая, но не ограничиваясь этим, TNF-α, IL-6, IL-8 и IL-1β (Adams, J. L., et al., Progress in Medicinal Chemistry, 38: 1-60 (2001)).
Было показано, что моноциты периферической крови (МПК), при их стимуляции липополисахаридом (ЛПС) in vitro, экспрессируют и секретируют провоспалительные цитокины. Ингибиторы р38 эффективно блокируют это действие, когда МПК предварительно обрабатывают такими соединениями перед тем, как стимулировать при помощи ЛПС (Lee, J. С., et al., Int. J. Immunopharmacol., 10: 835-843 (1988)). Эффективность ингибиторов р38 на животных моделях воспалительного заболевания ускорила исследование лежащего(их) в основе механизма(ов), который(е) мог(ли) бы объяснить действие этих ингибиторов. Роль р38 в реакции клеток на IL-1 и TNF была исследована на ряде клеточных систем, имеющих отношение к воспалительной реакции, с использованием пиридинилимидазольного ингибитора: эндотелиальные клетки и IL-8 (Hashimoto, S., et al., J. Pharmacol. Exp. Ther., 293: 370-375 (2001)), фибробласты и IL-6/GM-CSF/PGE2 (Beyaert, R., et al., EMBO J., 15: 1914-1923 (1996)), нейтрофилы и IL-8 (Albanyan, E. A., et al., Infect. Immun., 68: 2053-2060 (2000)), макрофаги и IL-1 (Caivano, M. and Cohen, P., J. Immunol., 164: 3018-3025 (2000)) и клетки гладких мышц и RANTES (Maruoka, S., et al., Am. J. Respir. Crit. Care Med., 161: 659-668 (1999)). Разрушительные эффекты многих болезненных состояний вызваны сверхпродукцией провоспалительных цитокинов. Способность ингибиторов р38 регулировать эту сверхпродукцию делает их прекрасными кандидатами для агентов, изменяющих болезнь.
Ингибиторы р38 являются активными в отношении множества общепризнанных моделей заболеваний и демонстрируют положительные эффекты в ряде стандартных животных моделей воспаления, включая коллаген-индуцированный артрит крыс (Jackson, J.R., et al., J.Pharmacol. Exp. Ther., 284: 687-692 (1998)); адъювант-индуцированный артрит крыс (Badger, A.M., et al., Arthritis Rheum., 43: 175-183 (2000); Badger, A.M., et al., J. Pharmacol. Exp. Ther., 279: 1453-1461 (1996)) и каррагенан-индуцированный отек лапы у мышей (Nishikori, Т., et al., Eur. J. Pharm., 451: 327-333 (2002)). В этих животных моделях было показано, что молекулы, которые блокируют действие р38, эффективны при ингибировании резорбции кости, воспалении и других иммунных патологий и воспалительных патологий. Таким образом, безопасный и эффективный ингибитор р38 обеспечит способ лечения заболеваний, подтачивающих здоровье, которые можно регулировать модулированием сигнального пути р38, подобного, но не ограниченного этим, РА.
Ингибиторы р38 хорошо известны специалистам в данной области техники. Обзоры ранних ингибиторов способствовали установлению зависимости активности от структуры, важной для улучшения активности как in vitro, так и in vivo (Salituro, E.G., et al., Current Medicinal Chemistry, 6: 807-823 (1999) и Foster, M.L, et al., Drug News Perspect., 13: 488-497 (2000)). Более современные обзоры сконцентрированы на структурном многообразии новых ингибиторов, исследуемых в качестве ингибиторов р38 (Boehm, J.D. and Adams, J.L., Exp. Opin. Ther. Patents, 10: 25-37 (2000)). В этом изобретении описана новая группа замещенных 2-аза[4.3.0]бициклических гетероароматических соединений в качестве ингибиторов р38, которые полезны для лечения воспаления, остеоартрита, ревматоидного артрита, аутоиммунных заболеваний и для лечения других цитокин-опосредованных заболеваний.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В этом изобретении предложены соединения, способы получения этих соединений и содержащие их фармацевтические композиции, которые ингибируют р38 альфа и ассоциированные р38-опосредованные события, такие как ингибирование продукции цитокинов. Такие соединения, обычно называемые 2-аза[4.3.0]бициклическими гетероароматическими кольцами, полезны в качестве терапевтических агентов в отношении заболеваний, которые можно лечить путем ингибирования сигнального пути р38. В общих чертах, изобретение относится к ингибиторам р38 общей Формулы I
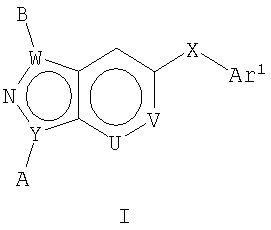
где Y представляет собой С, N;
W представляет собой С, N, S или О, при условии, что W представляет собой N, S или О, когда Y представляет собой С, и W представляет собой С или N, когда Y представляет собой N;
U представляет собой СН или N;
V представляет собой С-Е или N;
Х представляет собой О, S, SO, SO2, NR7, C=O, CHR7, -C=NOR1, -C=CHR1или CHOR1;
R1 представляет собой Н, РО3Н2, SO3Н2, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1 может быть замещенным или незамещенным;
Z представляет собой алкилен, имеющий от 1 до 4 атомов углерода, или алкенилен или алкинилен, каждый из которых имеет от 2 до 4 атомов углерода, где указанный алкилен, алкенилен или алкинилен может быть замещенным или незамещенным;
R7 представляет собой Н или замещенный или незамещенный метил;
Ar1 представляет собой замещенный или незамещенный арил или гетероарил;
А представляет собой Н, ОН, защитную группу для амино, Zn-NR2R3,
Zn-NR2(C=O)R2, Zn-SO2R2, Zn-SOR2, Zn-SR2, Zn-OR2, Zn-(C=O)R2, Zn-(C=O)OR2, Zn-O-(C=O)R2, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или
Zn-Ar1 может быть замещенным или незамещенным;
R2 и R3 независимо представляют собой Н, ОН, защитную группу для амино, защитную группу для спирта, защитную группу для кислоты, защитную группу для тио, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или
Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1 может быть замещенным или незамещенным, или R2 вместе с R3 и N образует насыщенный или частично ненасыщенный гетероцикл, имеющий один или более гетероатомов, где указанный гетероцикл может быть замещенным или незамещенным и где указанный гетероцикл может быть конденсирован с ароматическим кольцом;
В представляет собой Н, NH2 или замещенный или незамещенный метил;
Е представляет собой Н, Zn-NR2R3, Zn-(C=O)R4, Zn-(C=O)R5, Zn-NR5(C=O)R5,
Zn-O(C=O)R5, Zn-OR5, Zn-SOR5, Zn-SOR5, Zn-SR5, Zn-NH(C=O)NHR5или R5;
R4 представляет собой замещенную или незамещенную природную или неприродную аминокислоту, защищенную природную или неприродную аминокислоту, NH(CHR6) (СН2)mOR5, где m означает целое число от 1 до 4, или NR2R3;
R5 представляет собой Н, ОН, защитную группу для амино, защитную группу для спирта, защитную группу для кислоты, защитную группу для тио, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1 может быть замещенным или незамещенным;
R6 представляет собой природную аминокислотную боковую цепь, Zn-NR2R3,
Zn-OR5, Zn-SO2R5, Zn-SOR5 или Zn-SR5 и
n означает 0 или 1,
при условии, что когда В представляет собой Н и А представляет собой CH=CH-R8, где R8 представляет собой замещенный или незамещенный алкил, алкенил, циклоалкил, гетероциклоалкил, арил или гетероарил, тогда Х-Ar1представляет собой заместитель, где Ar1 является отличным от замещенного или незамещенного арила, гетероарила, NH-алкил, NH-циклоалкил, NH-гетероциклоалкил, NH-арил, NH-гетероарил, NH-алкокси или NH-диалкиламид, когда X представляет собой О, S, С=O, S=O, С=СН2, CO2, NH или N(C1-C8-алкил).
Изобретение также направлено на фармацевтически приемлемые пролекарства, фармацевтически активные метаболиты и фармацевтически приемлемые соли соединения Формулы I. Также описаны способы получения соединений Формулы I.
В другом воплощении это изобретение относится к соединениям общей Формулы II
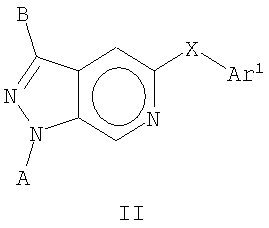
где А, В, Х и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы III

где А, В, X, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей формулы IV

где А, В, X, Е и Ar1 являются такими, как определено выше, при условии, что когда В представляет собой Н и А представляет собой CH=CH-R8, где R8представляет собой замещенный или незамещенный алкил, алкенил, циклоалкил, гетероциклоалкил, арил или гетероарил, тогда Х-Ar1 представляет собой заместитель, где Ar1 является отличным от замещенного или незамещенного арила, гетероарила, NH-алкил, NH-циклоалкил, NH-гетероциклоалкил, NH-арил, NH-гетероарил, NH-алкокси или NH-диалкиламид, когда Х представляет собой О, S, C=O, S=O, C=CH2, CO2, NH или
N(C1-C8-алкил).
В еще одном воплощении это изобретение относится к соединениям общей Формулы V
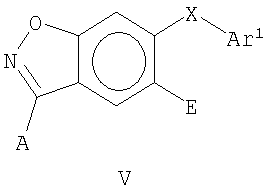
где А, X, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы VI

где А, В, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы VII
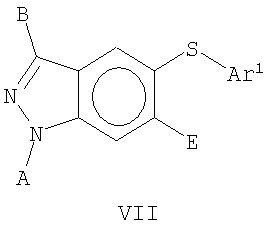
где А, В, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы VIII

где А, В, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы IX

где А, В, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы X

где А, В, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XI
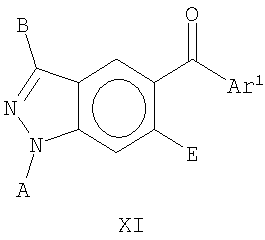
где А, В, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XII

где А, В, Е, R1 и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XIII
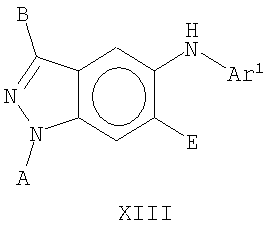
где А, В, Е и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к эфирным соединениям общей Формулы XIV
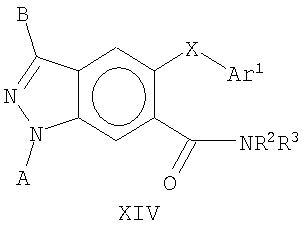
где А, В, X, Ar1, R2 и R3 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XV
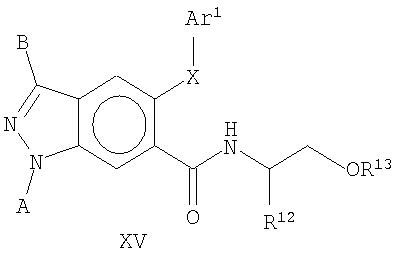
где А, В, X и Ar1 являются такими, как определено выше, и R12 и R13независимо представляют собой алкил, аллил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил или гетероарил, где указанный алкил, аллил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил или гетероарил может быть замещенным или незамещенным.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XVI

где А, В, X, R2, R3 и Ar1 являются такими, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XVII

где Y представляет собой CR1, О, S или NR2;
W представляет собой CR3, N, NR4, S или О при условии, что W представляет собой NR4, S или О, когда Y представляет собой CR1, и W представляет собой CR3 или N, когда Y представляет собой NR2;
R3 представляет собой Н, NH2, F, Cl, метил или замещенный метил;
R4 представляет собой Н, или метил, или замещенный метил;
R1 и R2 независимо представляют собой Н, ОН, защитную группу для амино, Zn-NRaRb, Zn-NRa(C=O)Rb, Zn-SO2Ra, Zn-SORa, Zn-SRa, Zn-ORa, Zn-(C=O)Ra, Zn-(C=O)OR3, Zn-O-(C=O)Ra, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
Ar1 представляет собой арил или гетероарил, каждый из которых может быть замещенным или незамещенным;
Ra и Rb независимо представляют собой Н, ОН, защитную группу для амино, защитную группу для спирта, защитную группу для кислоты, защитную группу для серы, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным,
или Ra и Rb вместе с атомами, к которым они оба присоединены, образуют насыщенное или частично ненасыщенное гетероциклическое кольцо, имеющее один или более гетероатомов в указанном кольце, где указанный гетероцикл может быть замещенным или незамещенным и где указанный гетероцикл может быть конденсирован с ароматическим кольцом;
Z представляет собой алкилен, имеющий от 1 до 4 атомов углерода, или алкенилен или алкинилен, каждый из которых имеет от 2 до 4 атомов углерода, где указанный алкилен, алкенилен или алкинилен может быть замещенным или незамещенным;
n означает 0 или 1;
U представляет собой CRc или N;
V представляет собой CRc или N;
Rc представляет собой Н, F, Cl, метил или замещенный метил;
Х представляет собой О, S, SO, SO2, NR5, C=O, CH2, CH2Zn-OH или С=NORb;
R5 представляет собой Н, метил или замещенный метил;
Rd представляет собой Н, РО3Н2, SO3Н2, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
G, Н, J и Т независимо представляют собой N или CRz, при условии, что когда любые из указанных G, Н, J и Т представляют собой N, тогда общее количество G, Н, J или Т, которые представляют собой N, не превышает 2;
Rz представляет собой Н, F, Cl, Br, CF3, OR6, SR6, низший (С1-С4)алкил, CN или
NR6R7;
R6 и R7 независимо представляют собой Н, CF3, низший (С1-С4)алкил или
низший (С1-С4)гетероалкил;
Q представляет собой -NR8CONH-, -NHCO-, -NR8SO2NH-, -NHSO2-, -CONR11-;
R8 представляет собой Н или низший (С1-С4)алкил;
R11 представляет собой Н или низший (С1-С4)алкил;
Rx представляет собой -(CR9R10)m-, -O(CR9R10)m-, NH(CR9R10)m- или -S(CR9R10)m- при условии, что Q представляет собой -CONR11-, когда Rx представляет собой -O(CR9R10)m-, -NH(CR9R10)m- или -S(SR9R10)m-;
R9 и R10 независимо представляют собой Н или низший алкил, или R9 и R10 вместе с атомами, к которым они оба присоединены, образуют циклоалкильное кольцо, которое может быть насыщенным или частично ненасыщенным;
m означает 1-3;
Ry представляет собой Н, РО3Н, защитную группу для амино, защитную группу для кислорода, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar2, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-Ar2 и Zn-гетероциклоалкил может быть замещенным или незамещенным;
Ar2 представляет собой арил или гетероарил, каждый из которых может быть замещенным или незамещенным, где указанное замещение может осуществляться 1-3 заместителями, независимо выбранными из F, Cl, Br, CF3, CN, алкила, аллила, алкенила, алкинила, гетероалкила, гетероаллила, гетероалкенила, гетероалкинила,
-OR12, -SR12, -SO2R12, -SO2NR13R12, NR13SO2R12, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
R12 и R13 независимо представляют собой Н, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
где когда Ar2 замещен -SO2NR13R12, тогда R12 и R13 могут образовывать циклоалкильное кольцо или гетероциклоалкильное кольцо, которое может быть замещенным или незамещенным, где указанное замещение может осуществляться заместителями, выбранными из алкила, аллила, алкенила, алкинила, гетероалкила, гетероаллила, гетероалкенила, гетероалкинила, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, -COR12, -SO2R12, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
где когда Q представляет собой -CONR11-, тогда Ry вместе с R11дополнительно представляют собой циклоалкильное кольцо или гетероциклоалкильное кольцо, которое может быть замещенным или незамещенным группами, выбранными из алкила, аллила, алкенила, алкинила, гетероалкила, гетероаллила, гетероалкенила, гетероалкинила, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, Zn-Ar1, -COR14 или -SO2R14, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил, Zn-Ar1, -COR14 и -SO2R14 может быть замещенным или незамещенным, и
R14 представляет собой алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным.
В еще одном аспекте настоящего изобретения предложены соединения, которые ингибируют продукцию цитокинов, таких как TNF-α, IL-1, IL-6 и IL-8, включая соединения Формул I-XVII.
В еще одном аспекте настоящего изобретения предложен способ лечения заболеваний или медицинских состояний, опосредованных цитокинами, при котором теплокровному животному вводят эффективное количество соединения Формулы I-XVII, или его фармацевтически приемлемой соли, или его пролекарства, расщепляемого in vivo.
В еще одном аспекте настоящего изобретения предложен способ ингибирования продукции цитокинов, таких как TNF-α, IL-1, IL-6 и IL-8, при котором теплокровному животному вводят эффективное количество соединения Формулы I-XVII, или его фармацевтически приемлемой соли, или его пролекарства, расщепляемого in vivo.
В еще одном аспекте настоящего изобретения предложен способ обеспечения эффекта ингибирования киназы р38, при котором теплокровному животному вводят эффективное количество соединения Формулы I-XVII, или его фармацевтически приемлемой соли, или его пролекарства, расщепляемого in vivo.
В еще одном аспекте настоящего изобретения предложено лечение или предупреждение состояния, опосредованного р38, при котором человеку или животному, нуждающемуся в этом, вводят количество соединения, эффективное для лечения или предупреждения указанного состояния, опосредованного р38, или фармацевтическую композицию, содержащую указанное соединение, где указанное соединение представляет собой соединение Формулы I-XVII, или его фармацевтически приемлемую соль, или его пролекарство, расщепляемое in vivo. Состояние, опосредованное р38, которое можно лечить способами по настоящему изобретению, включает в себя воспалительное заболевание, аутоиммунное заболевание, деструктивное костное нарушение, пролиферативное нарушение, инфекционное заболевание, вирусное заболевание или нейродегенеративное заболевание.
Соединения по этому изобретению также полезны в способах предупреждения клеточной гибели и гиперплазии и, следовательно, их можно применять для лечения или предупреждения реперфузии/ишемии при ударе, сердечных приступах и гипоксии органов. Соединения по этому изобретению также полезны в способах предупреждения тромбин-индуцированной агрегации тромбоцитов.
Соединения по изобретению преимущественно можно применять в комбинации с другими известными терапевтическими агентами.
Изобретение также относится к фармацевтическим композициям, содержащим эффективное количество агента, выбранного из соединений Формул I-XVII или их фармацевтически приемлемого пролекарства, фармацевтически активного метаболита или фармацевтически приемлемой соли.
Дополнительные преимущества и новые отличительные признаки этого изобретения частично будут изложены в описании изобретения, которое следует ниже, а отчасти станут очевидными для специалистов в данной области техники после изучения следующего описания изобретения, или могут стать известными при осуществлении изобретения. Преимущества изобретения могут быть поняты и осуществлены при помощи агентов, комбинаций, композиций и способов, конкретно указанных в прилагаемой формуле изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Прилагаемые схемы, которые включены в этот документ и образуют часть описания изобретения, иллюстрируют неограничивающие воплощения настоящего изобретения и вместе с описанием служат для объяснения принципов изобретения.
На Фиг.1 показана реакционная схема синтеза соединений, имеющих общую структуру 7а.
На Фиг.2 показана реакционная схема синтеза соединения 14а.
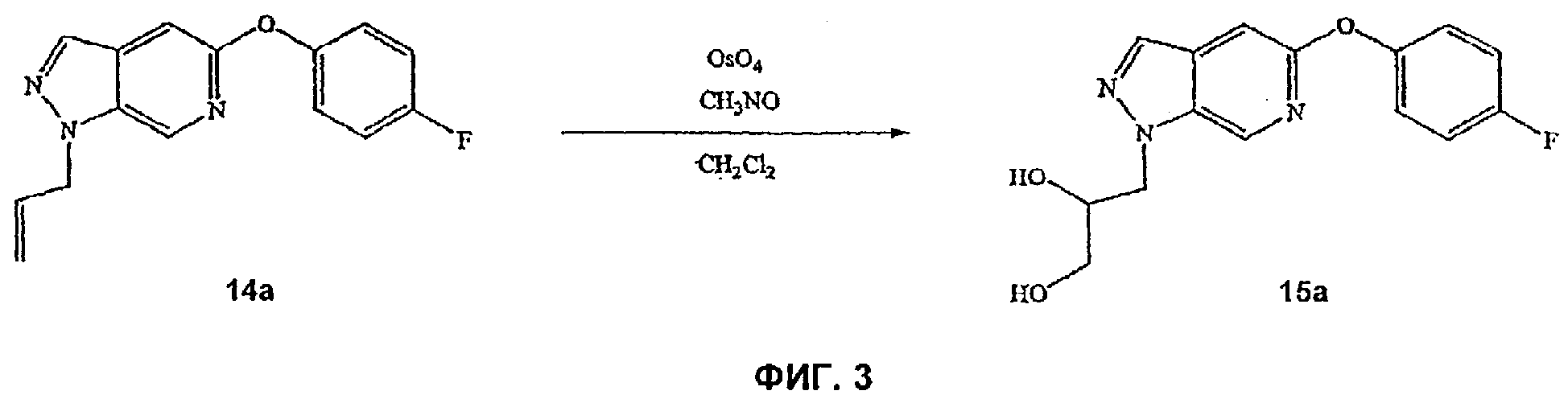
На Фиг.3 показана реакционная схема синтеза соединения 15а.
На Фиг.4 показана реакционная схема синтеза соединения 16а.
На Фиг.5 показана реакционная схема синтеза соединения 17а.
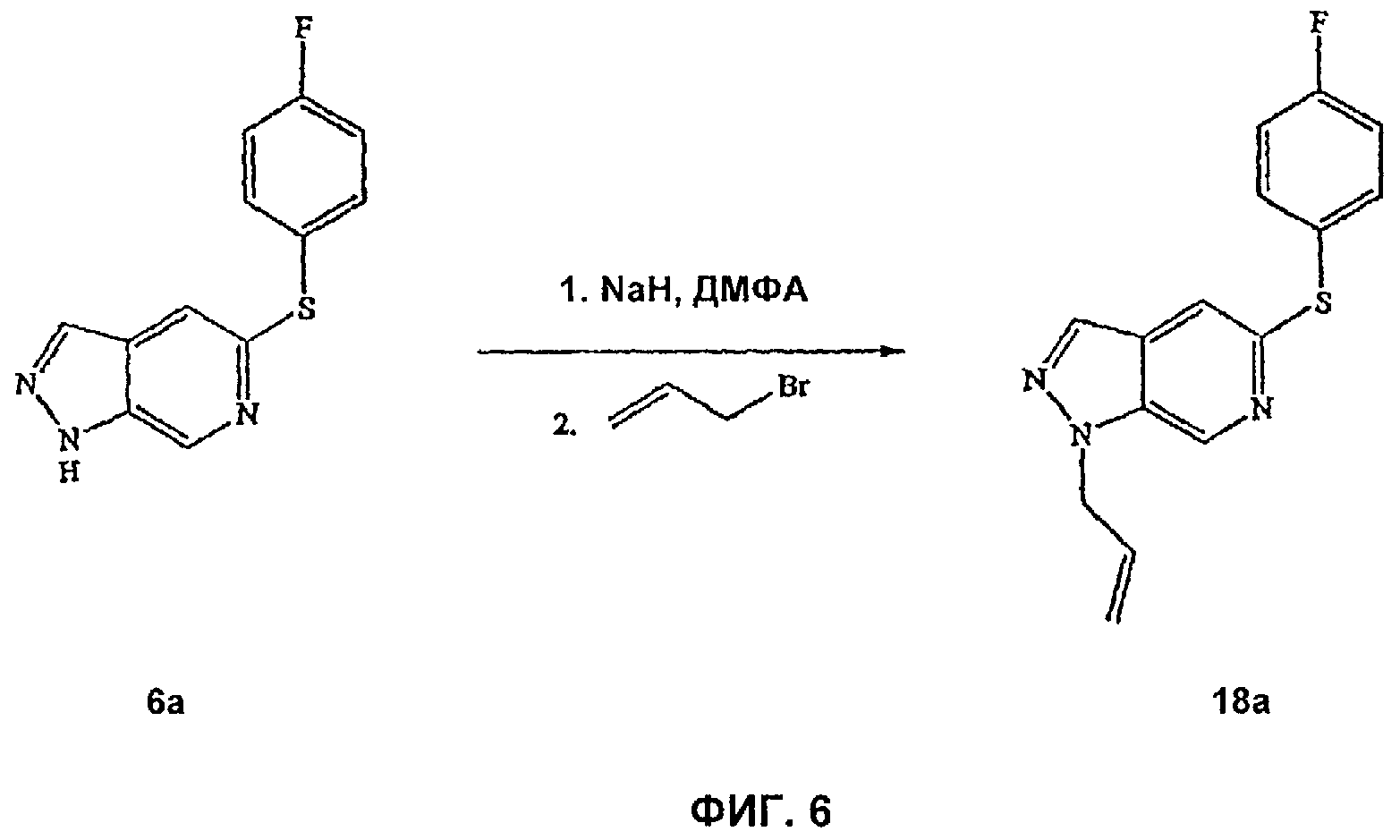
На Фиг.6 показана реакционная схема синтеза соединения 18а.
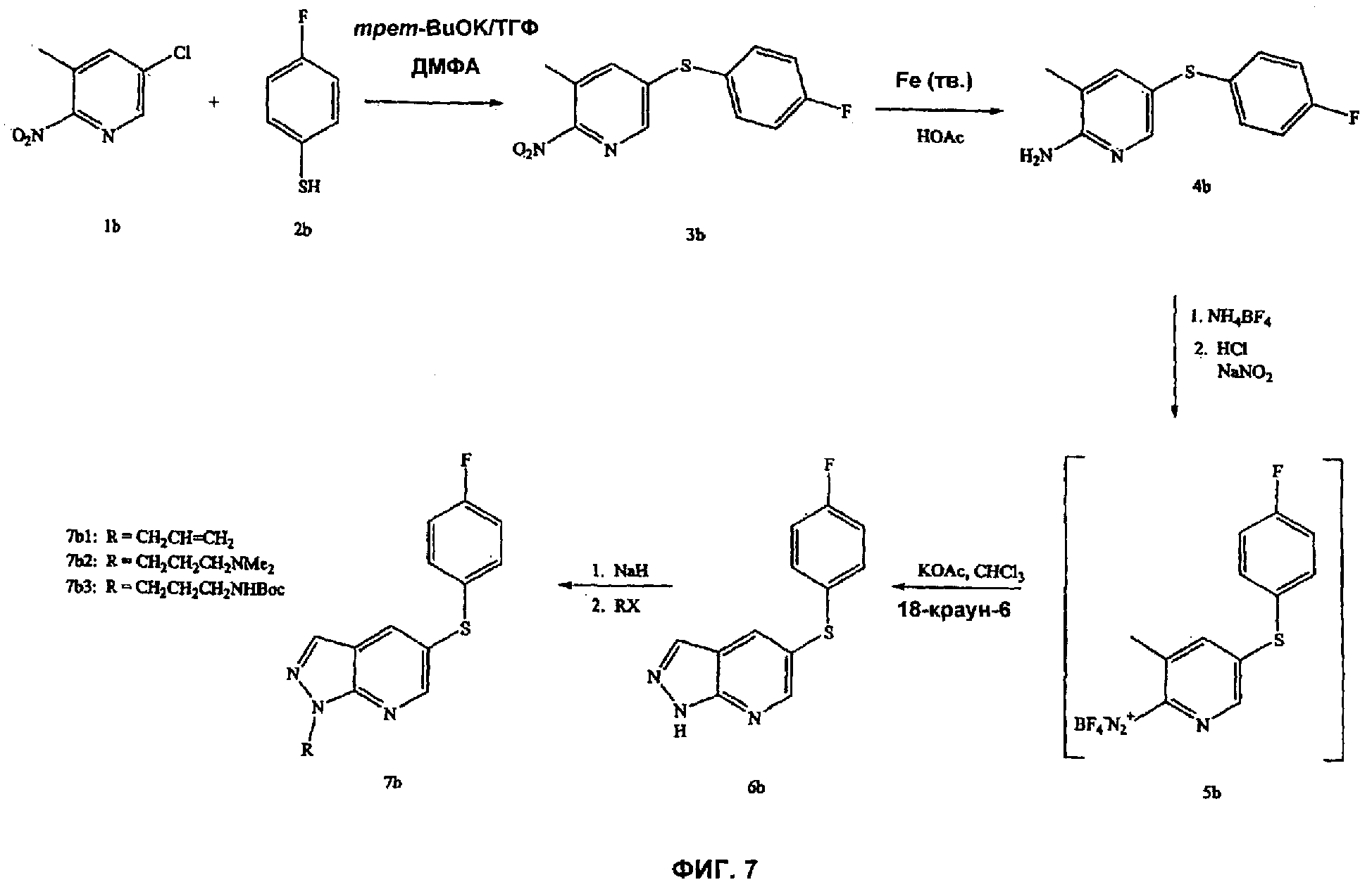
На Фиг.7 показана реакционная схема синтеза соединений, имеющих общую структуру 7b.
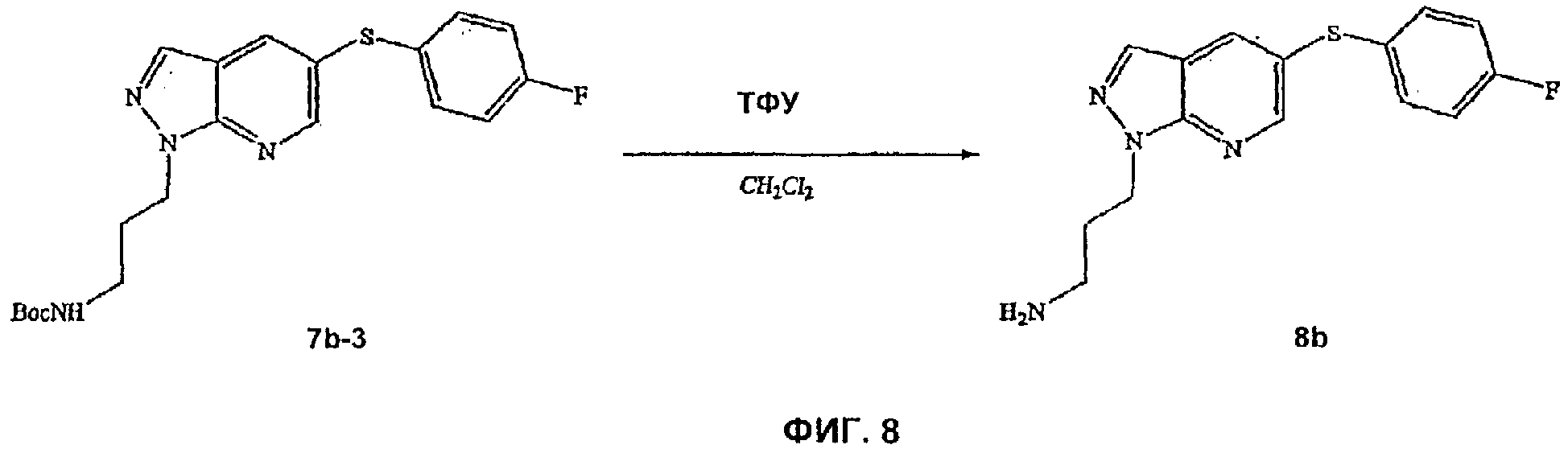
На Фиг.8 показана реакционная схема синтеза соединения 8b.
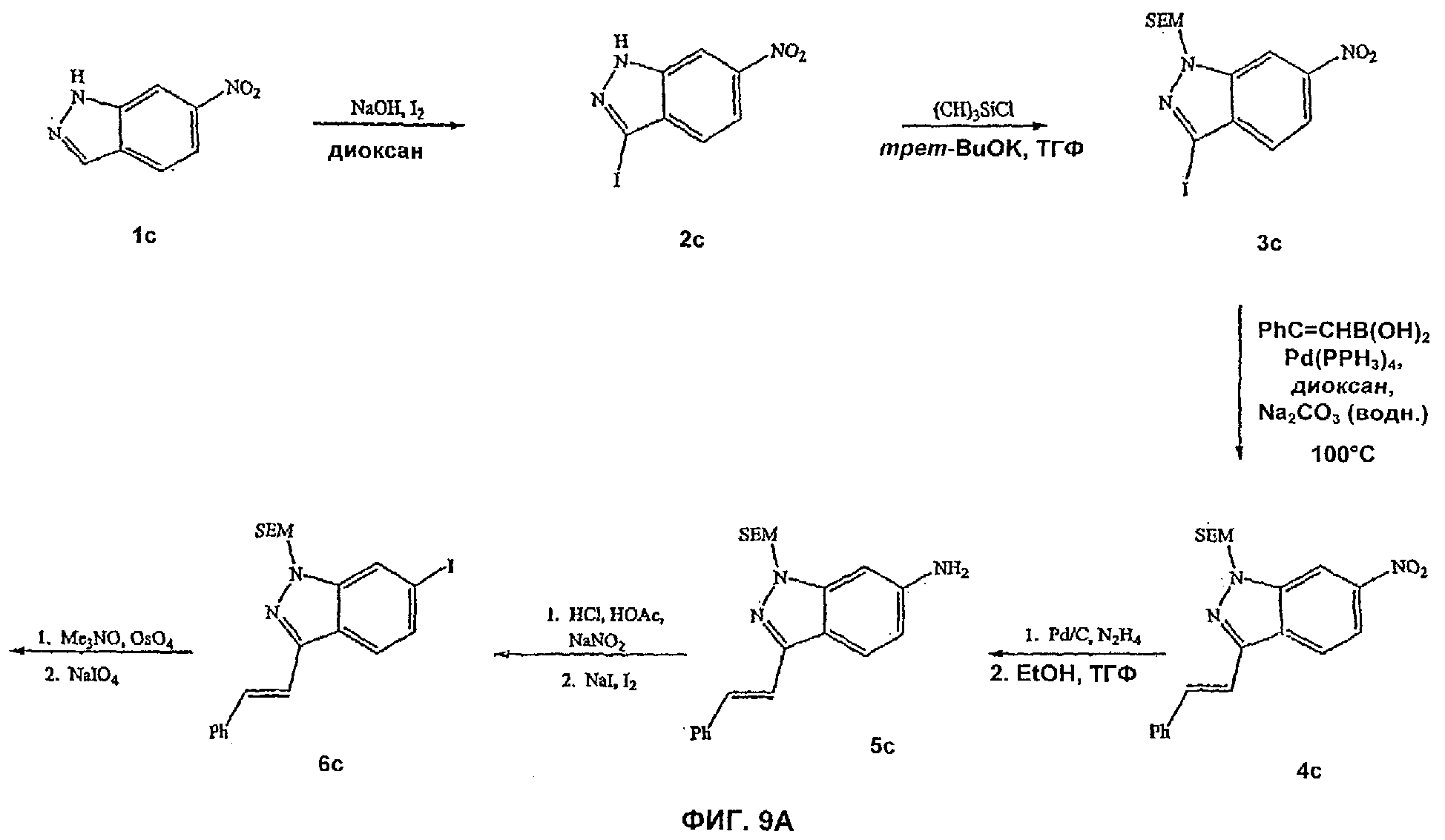
На Фиг.9А-9В показана реакционная схема синтеза соединения 10с.
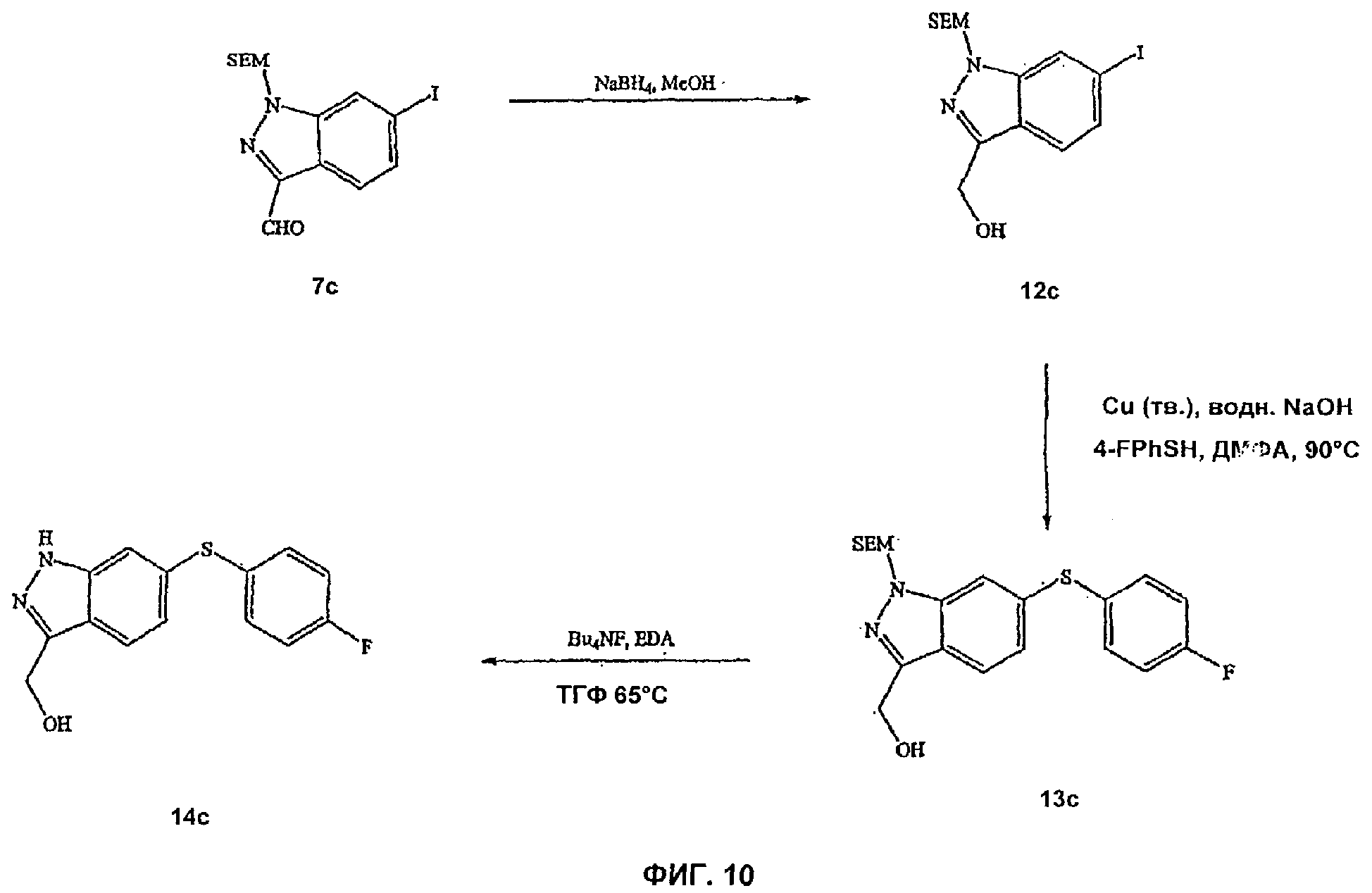
На Фиг.10 показана реакционная схема синтеза соединения 14с.
На Фиг.11 показана реакционная схема синтеза соединения 17с.
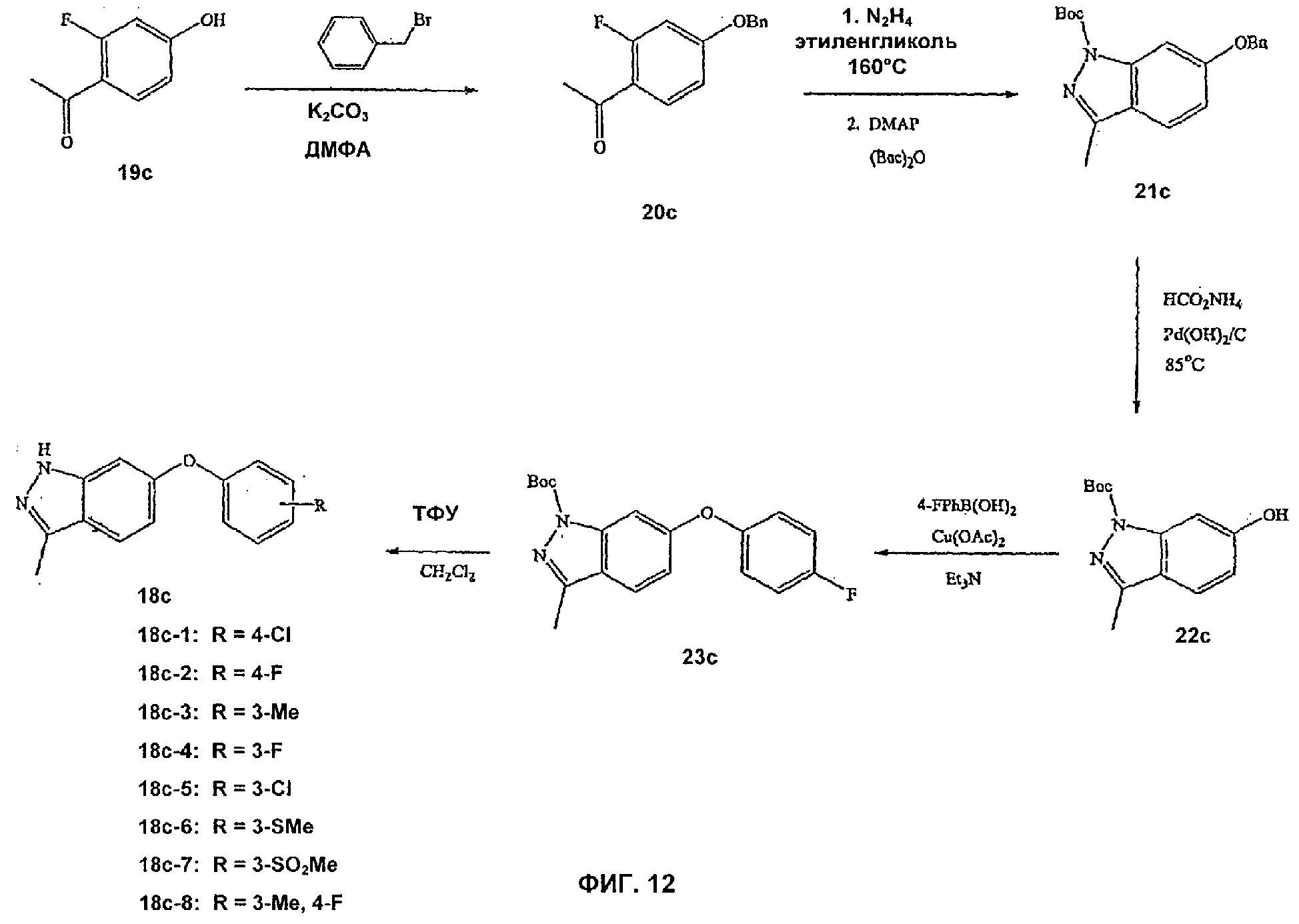
На Фиг.12 показана реакционная схема синтеза соединений, имеющих общую структуру 18с.
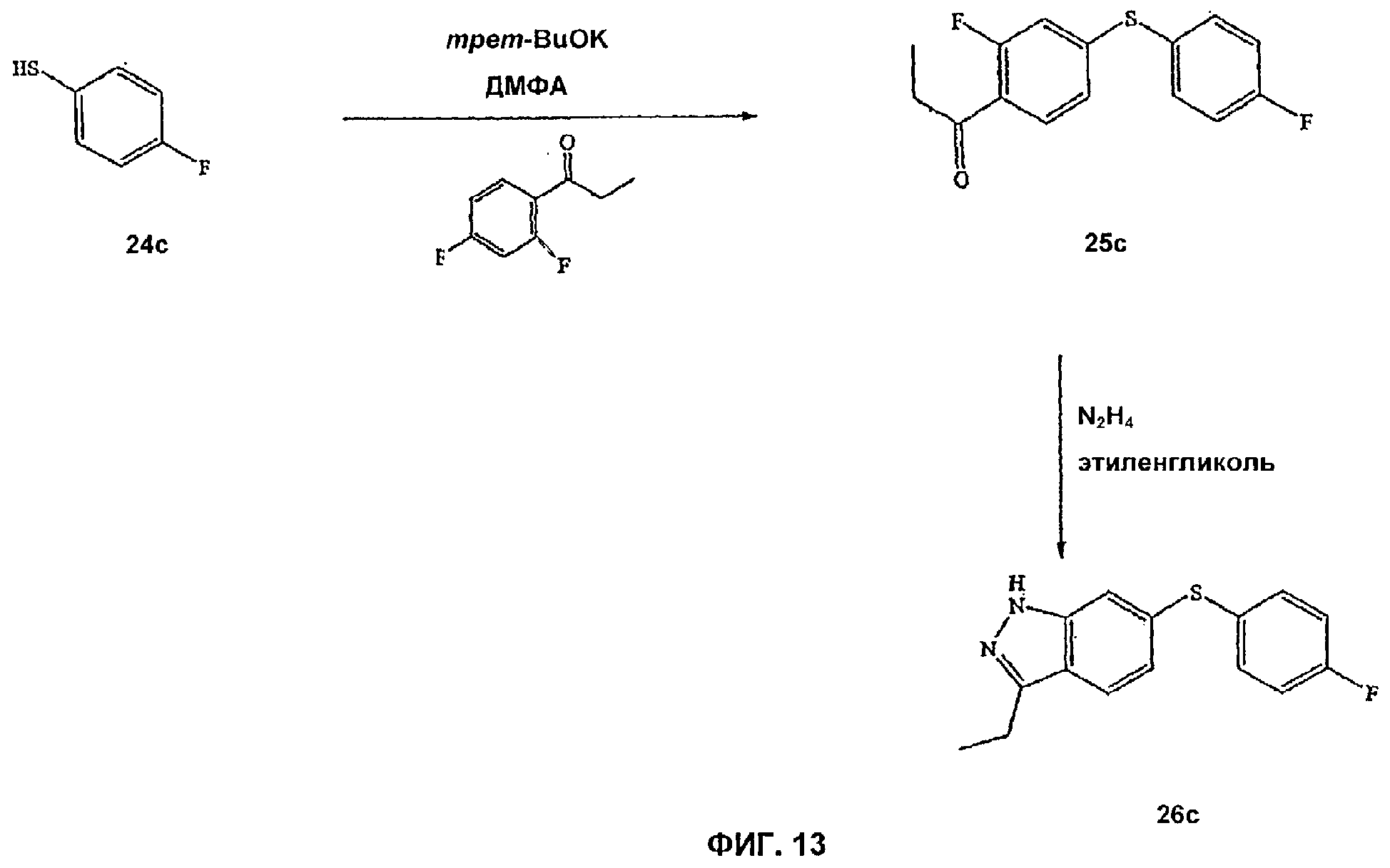
На Фиг.13 показана реакционная схема синтеза соединения 26с.
На Фиг.14А-14В показана реакционная схема синтеза соединения 34с.
На Фиг.15 показана реакционная схема синтеза соединения 38с-1.
На Фиг.16 показана реакционная схема синтеза соединения 39с.
На Фиг.17 показана реакционная схема синтеза соединения 40с.
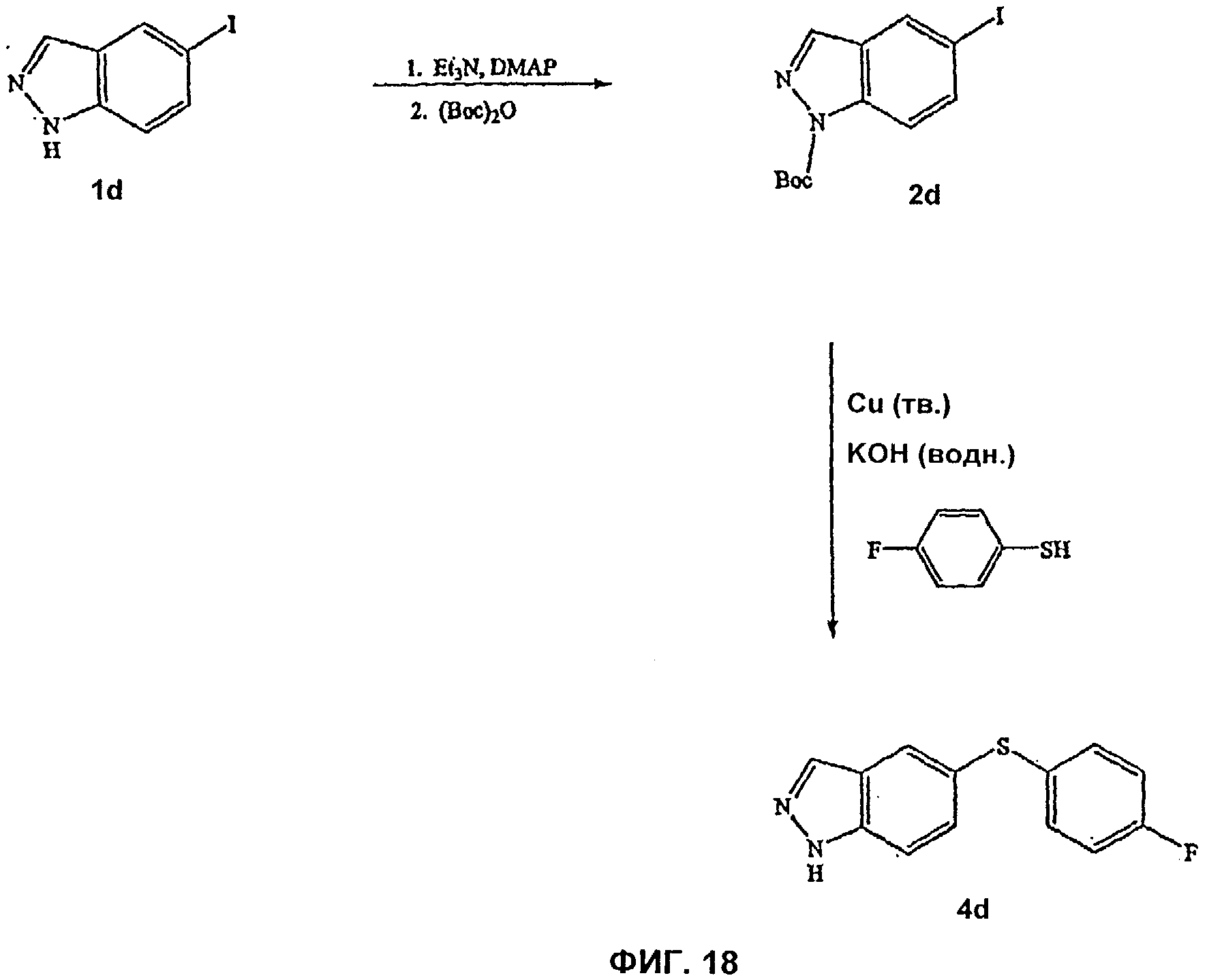
На Фиг.18 показана реакционная схема синтеза соединения 4d.
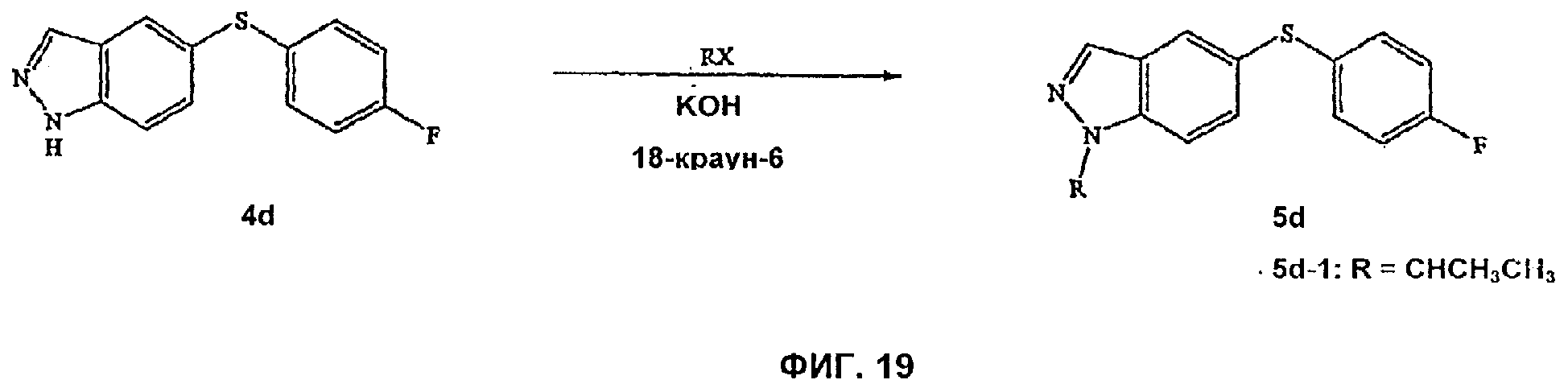
На Фиг.19 показана реакционная схема синтеза соединений, имеющих общую структуру 5d.
На Фиг.20 показана реакционная схема синтеза соединения 8d.
На Фиг.21 показана реакционная схема синтеза соединения 10d-1.
На Фиг.22 показана реакционная схема синтеза соединения 11d-1.
На Фиг.23 показана реакционная схема синтеза соединения 13d.
На Фиг.24А-24В показана реакционная схема синтеза соединения 8е-1.
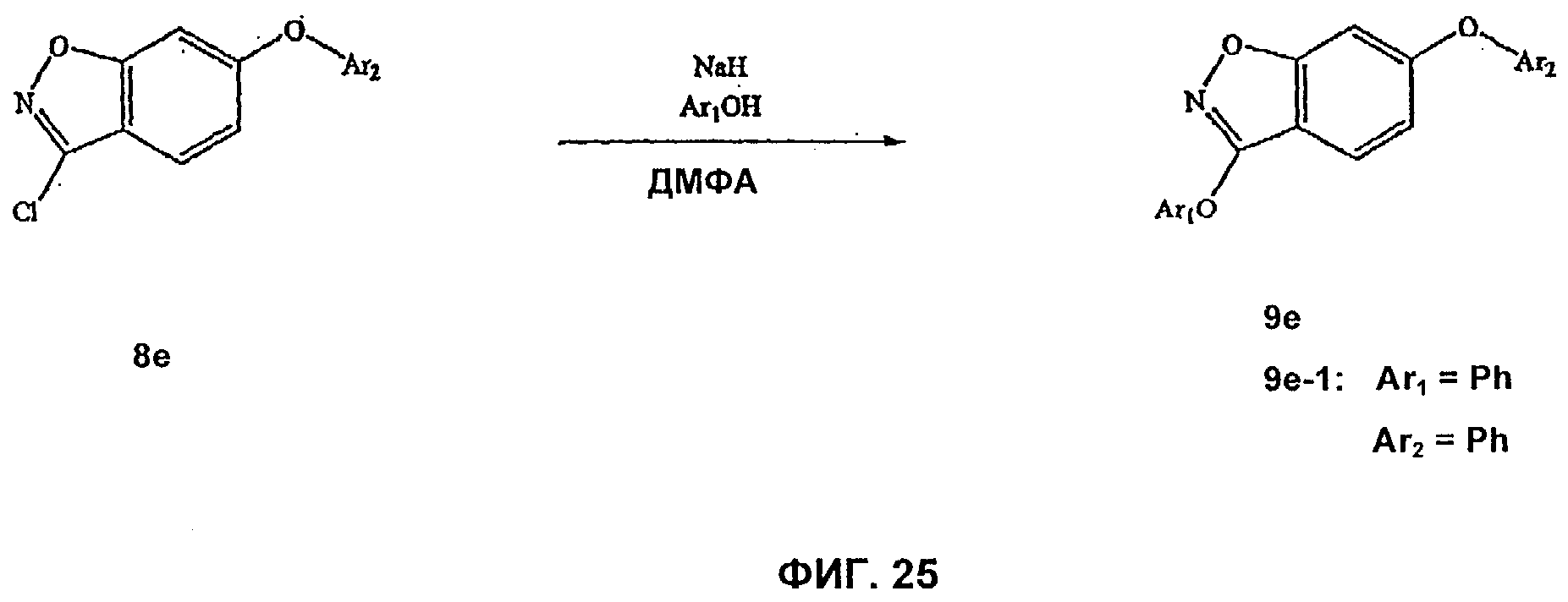
На Фиг.25 показана реакционная схема синтеза соединения 9е.
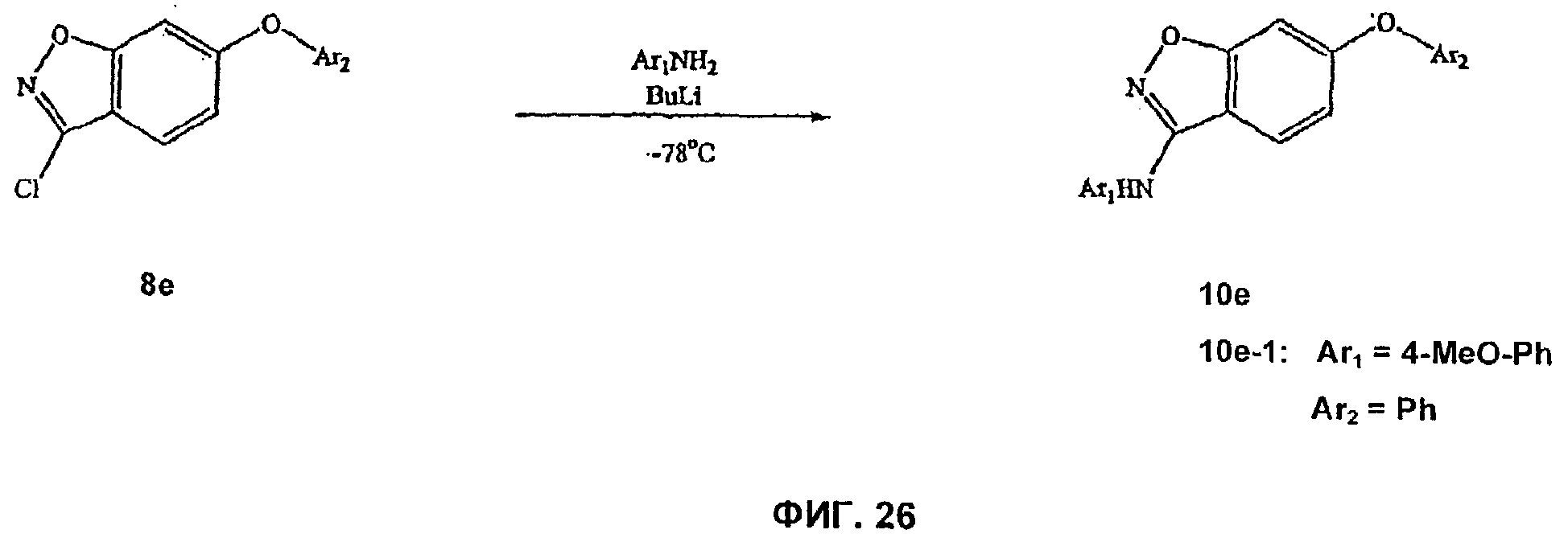
На Фиг.26 показана реакционная схема синтеза соединения 10е-1.
На Фиг.27 показана реакционная схема синтеза соединений, имеющих общую структуру 7f.
На Фиг.28 показана альтернативная реакционная схема синтеза соединений, имеющих общую структуру 7f.
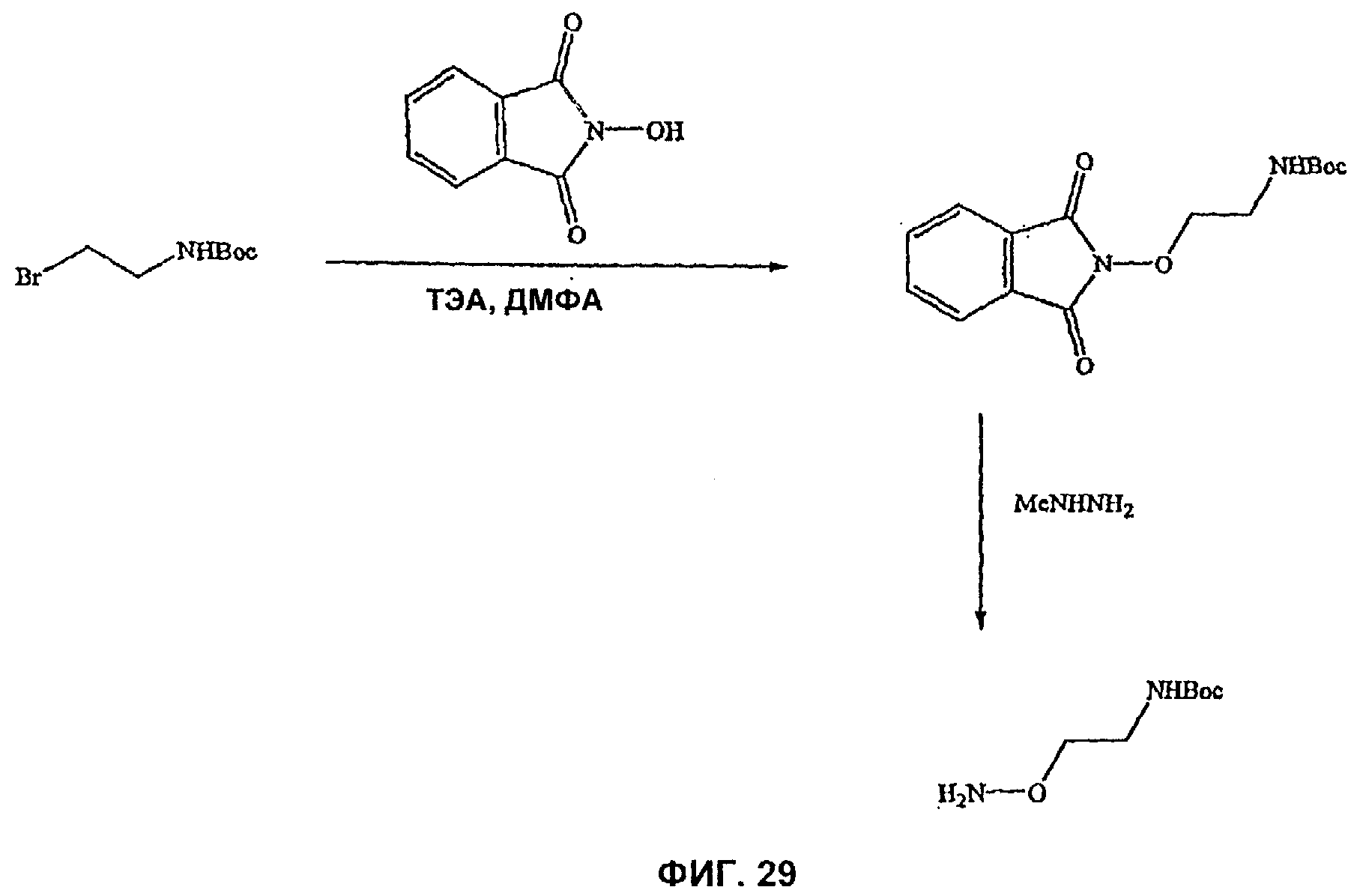
На Фиг.29 показана реакционная схема синтеза промежуточного карбоксамида, используемого в синтезе соединения 7f-5 и 7f-6.
На Фиг.30А-30С показана реакционная схема синтеза соединений, имеющих общую структуру 1g.
На Фиг.31 показана реакционная схема синтеза соединений, имеющих общую структуру 4f.
На Фиг.32 показана реакционная схема синтеза соединений, имеющих общую структуру 5f.
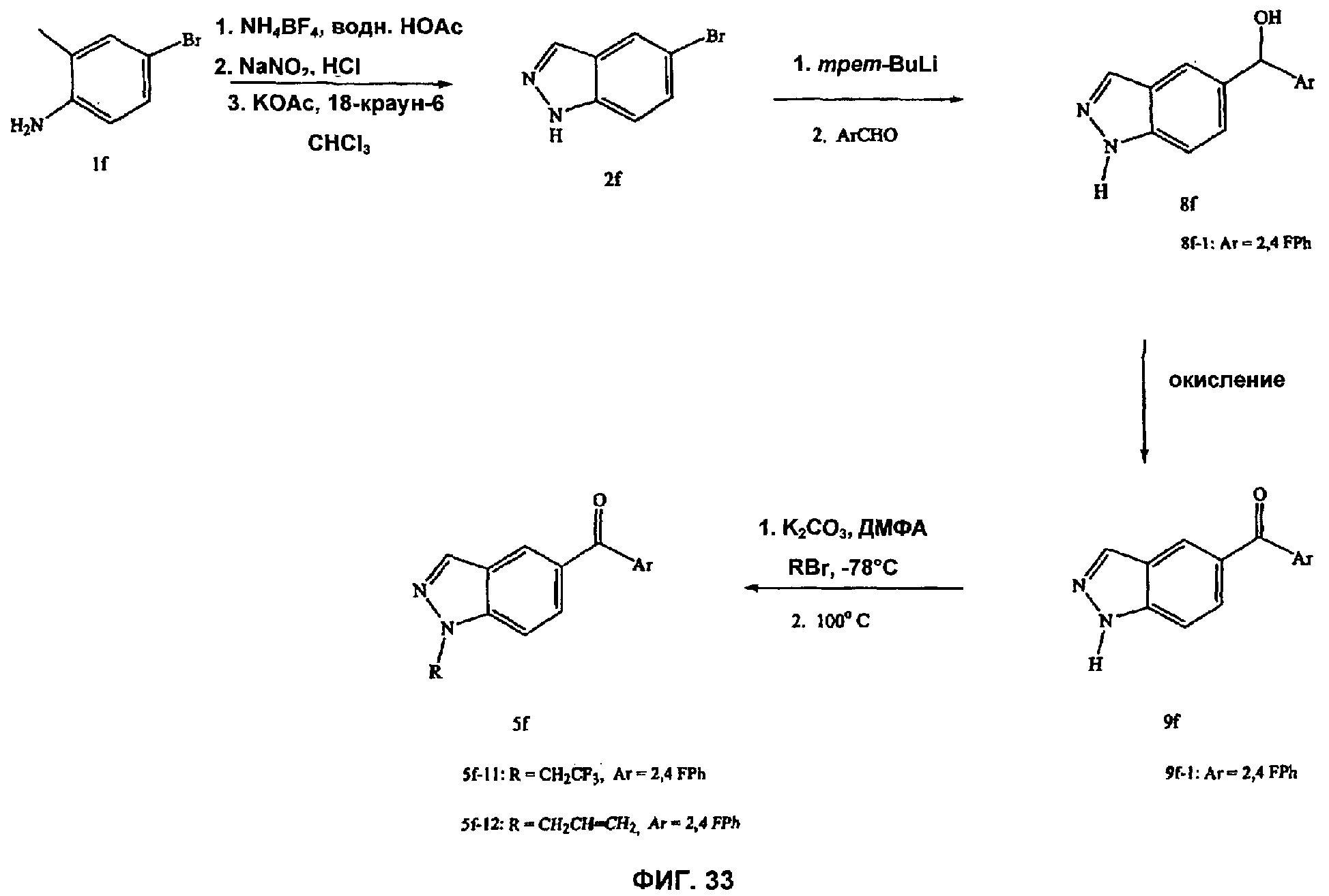
На Фиг.33 показана альтернативная реакционная схема синтеза соединений, имеющих общую структуру 5f.
На Фиг.34 показана реакционная схема синтеза соединений, имеющих общую структуру 2h.
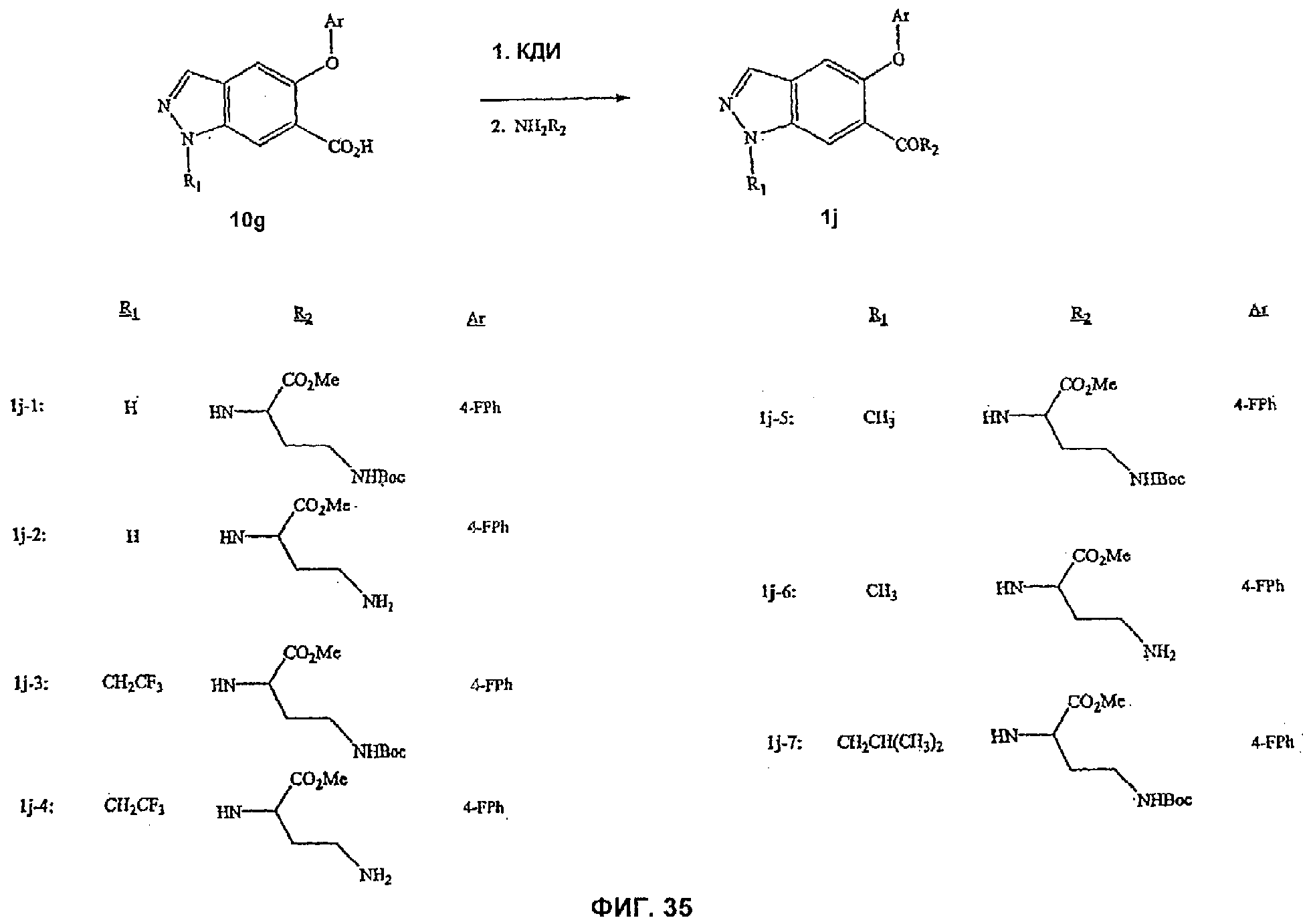
На Фиг.35 показана реакционная схема синтеза соединений, имеющих общую структуру 1j.
На Фиг.36 показана реакционная схема синтеза соединений, имеющих общую структуру 1k.
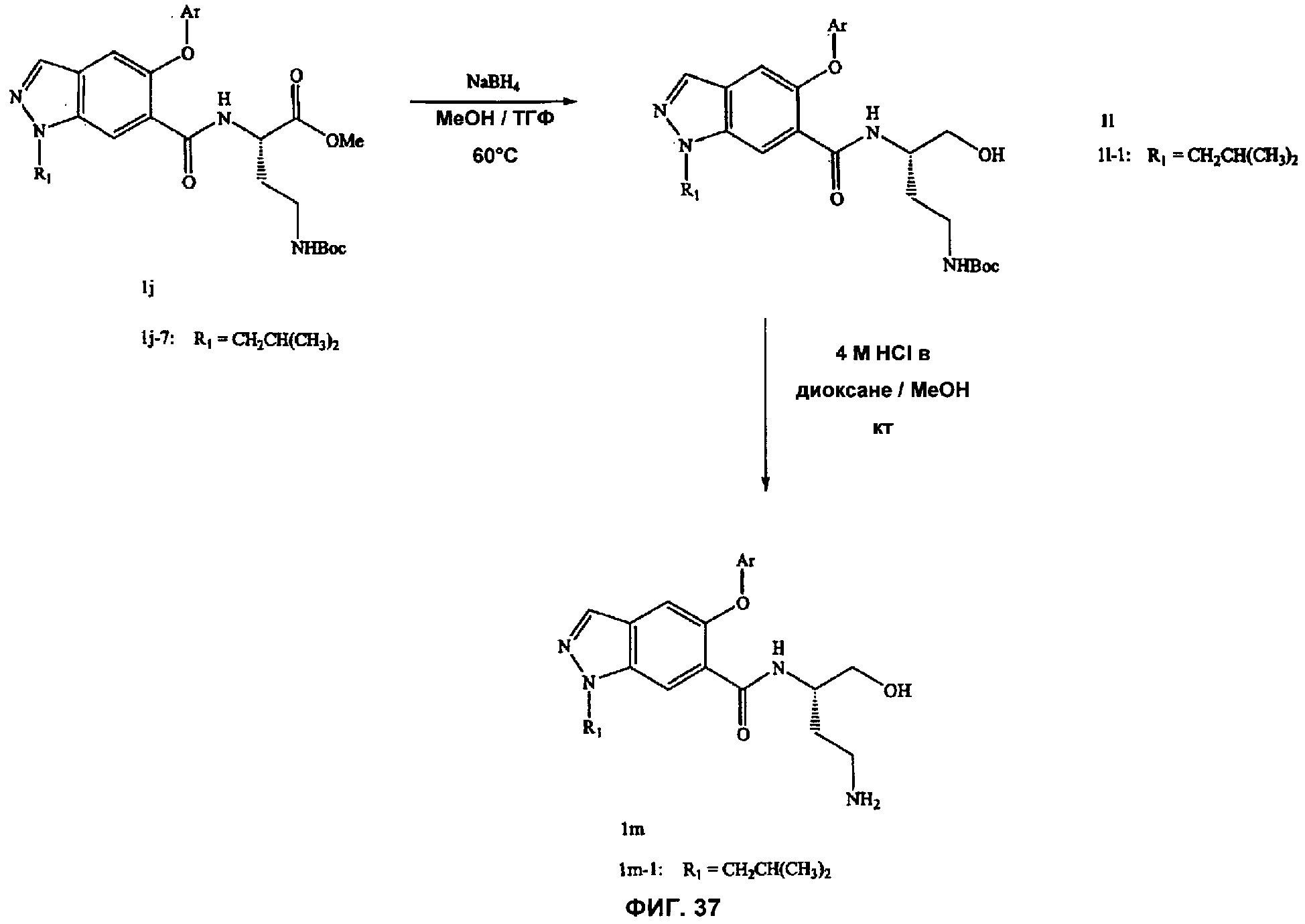
На Фиг.37 показана реакционная схема синтеза соединений, имеющих общую структуру 1m.
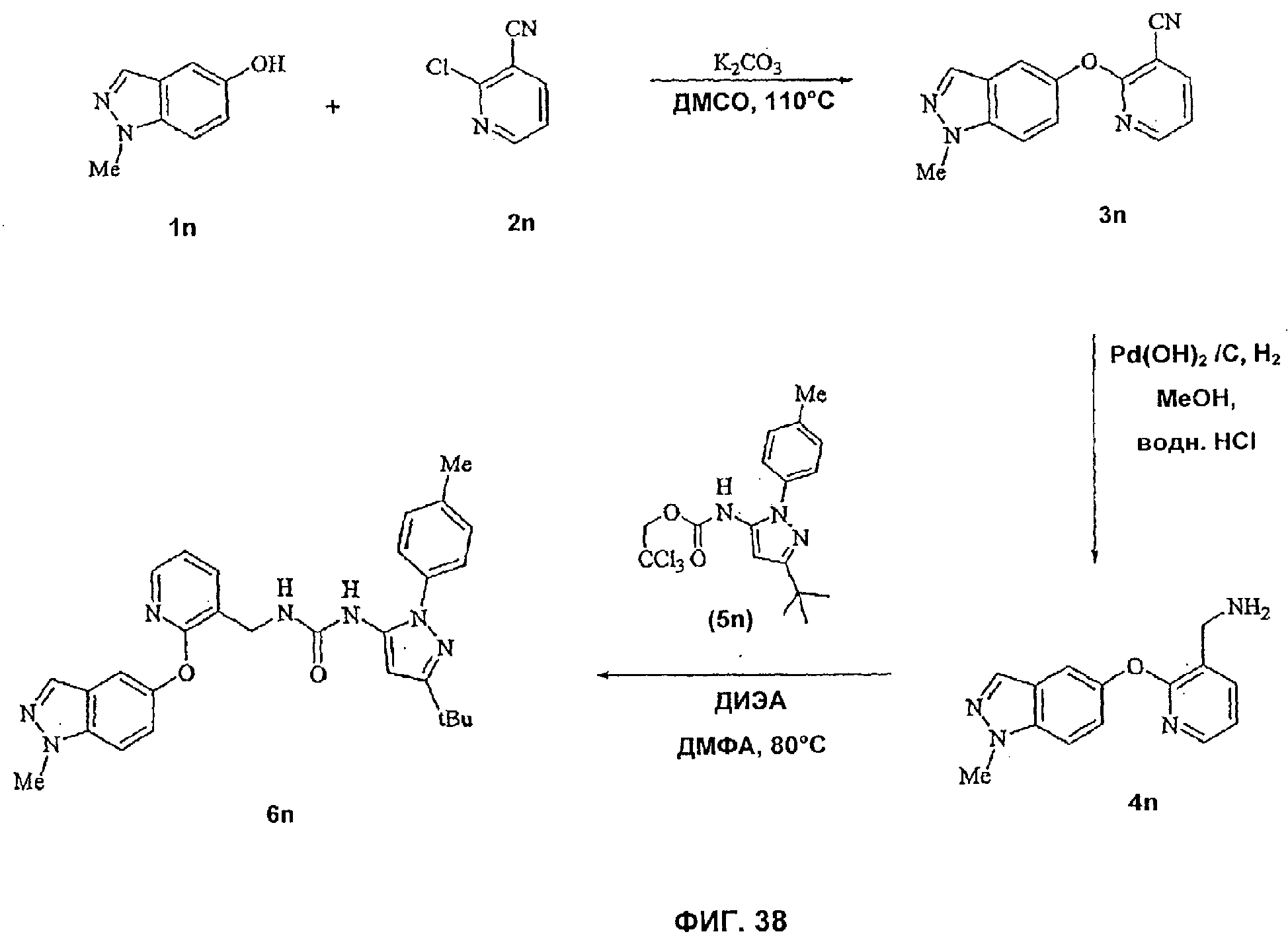
На Фиг.38 показана реакционная схема синтеза соединения 6n.
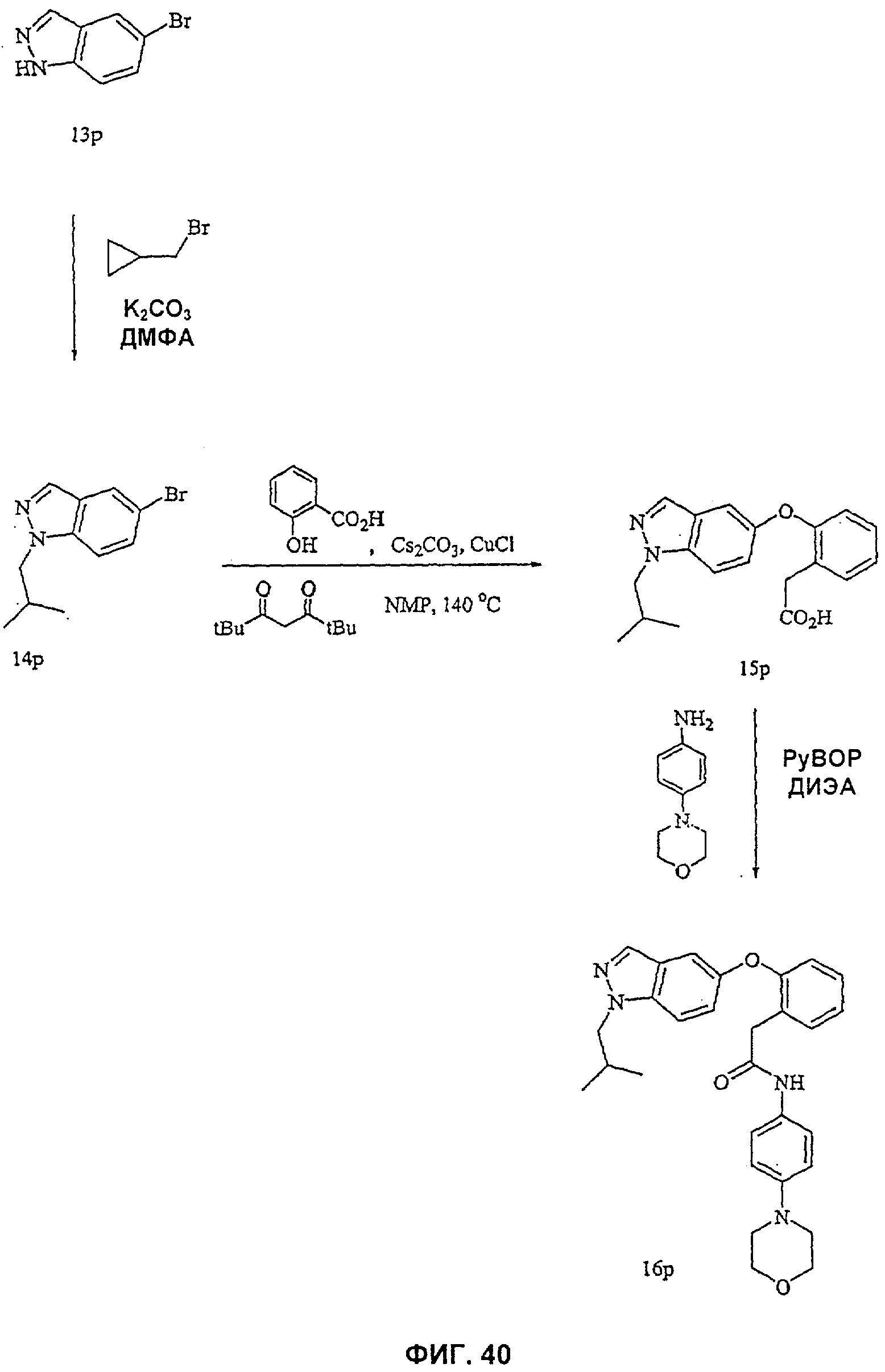
На Фиг.39 показана реакционная схема синтеза соединения 13р.
На Фиг.40 показана реакционная схема синтеза соединения 16р.
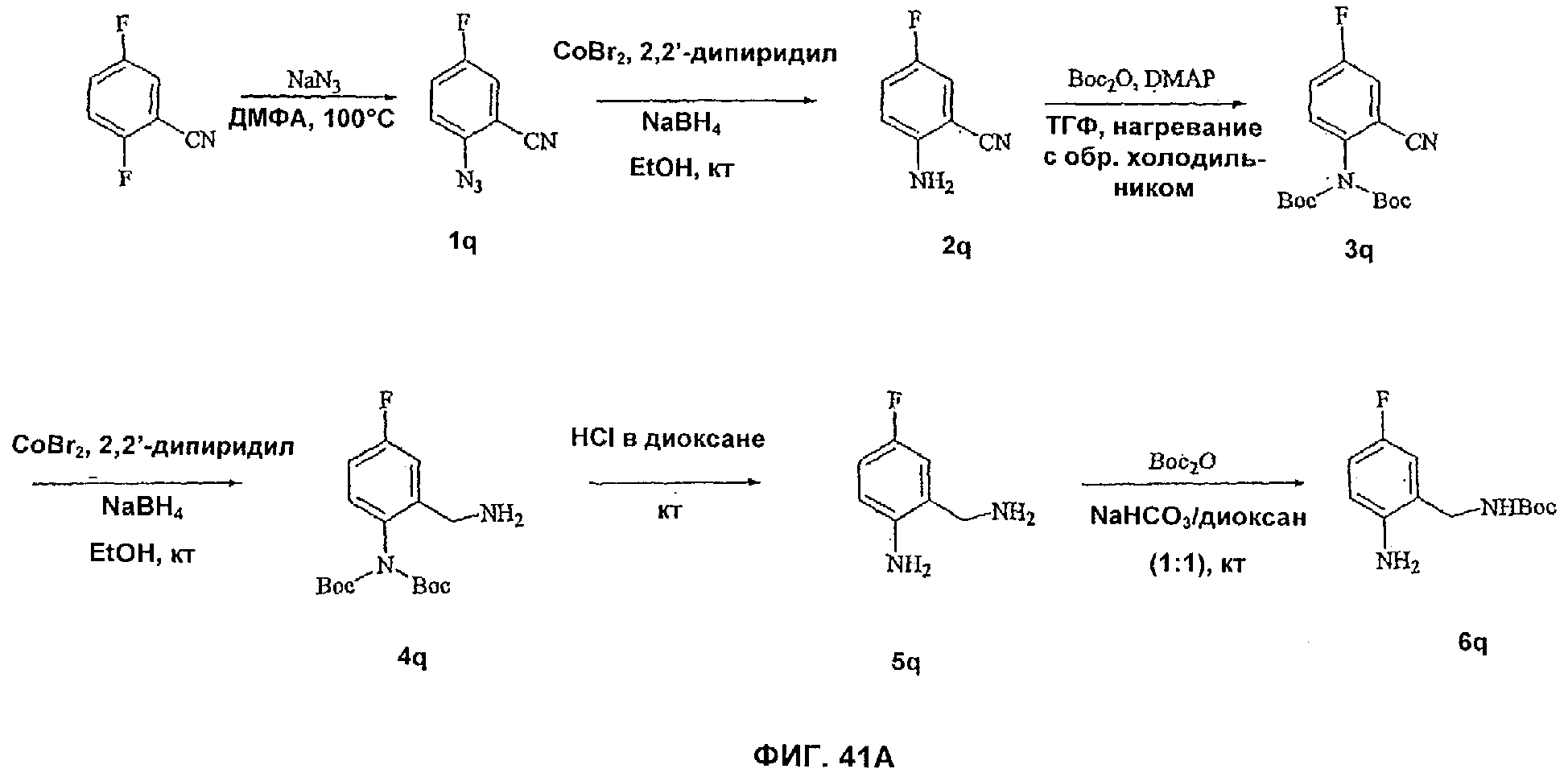
На Фиг.41А-В показана реакционная схема синтеза соединений 9q-1 и 9q-2.
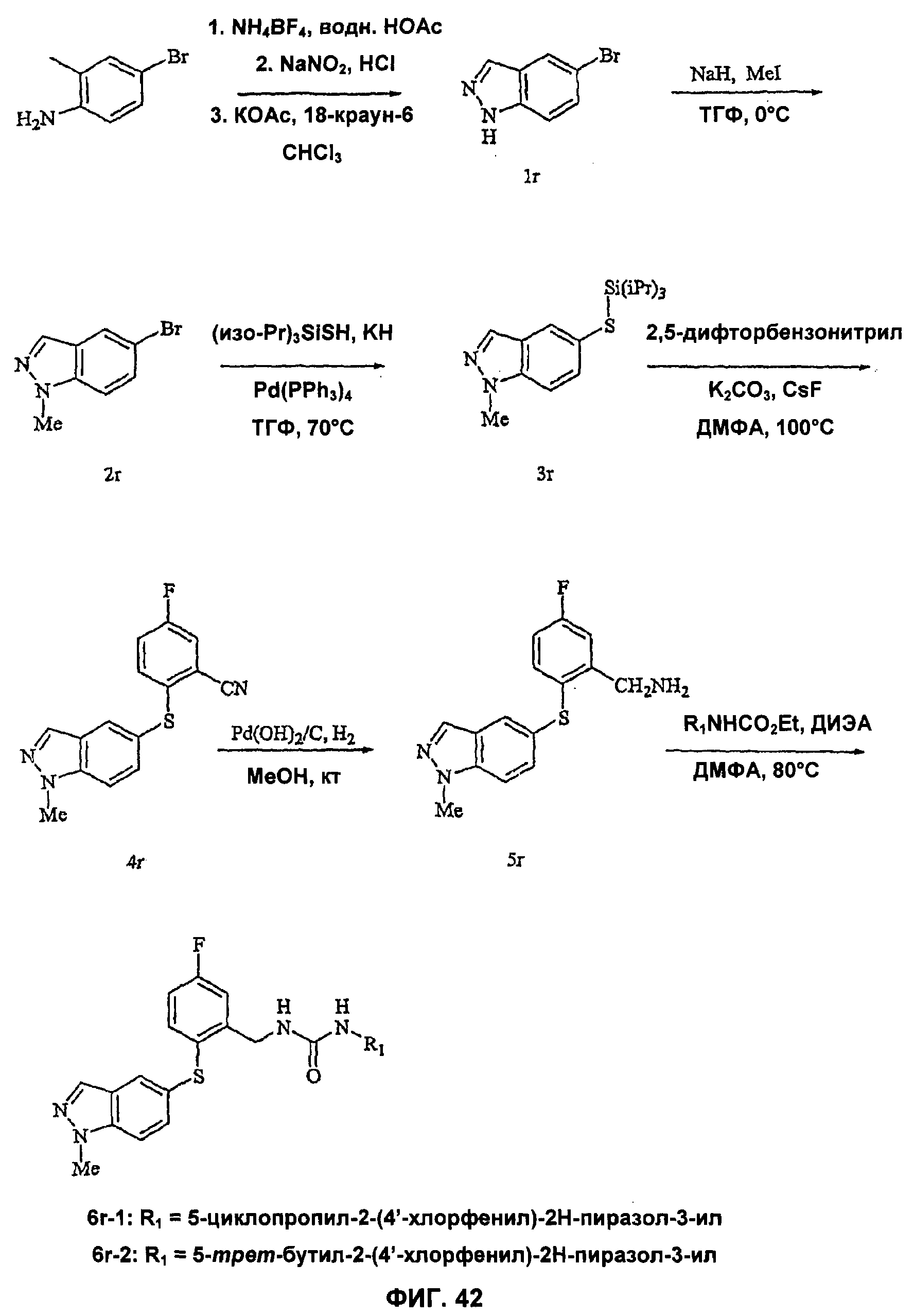
На Фиг.42 показана реакционная схема синтеза соединения 6r-2.
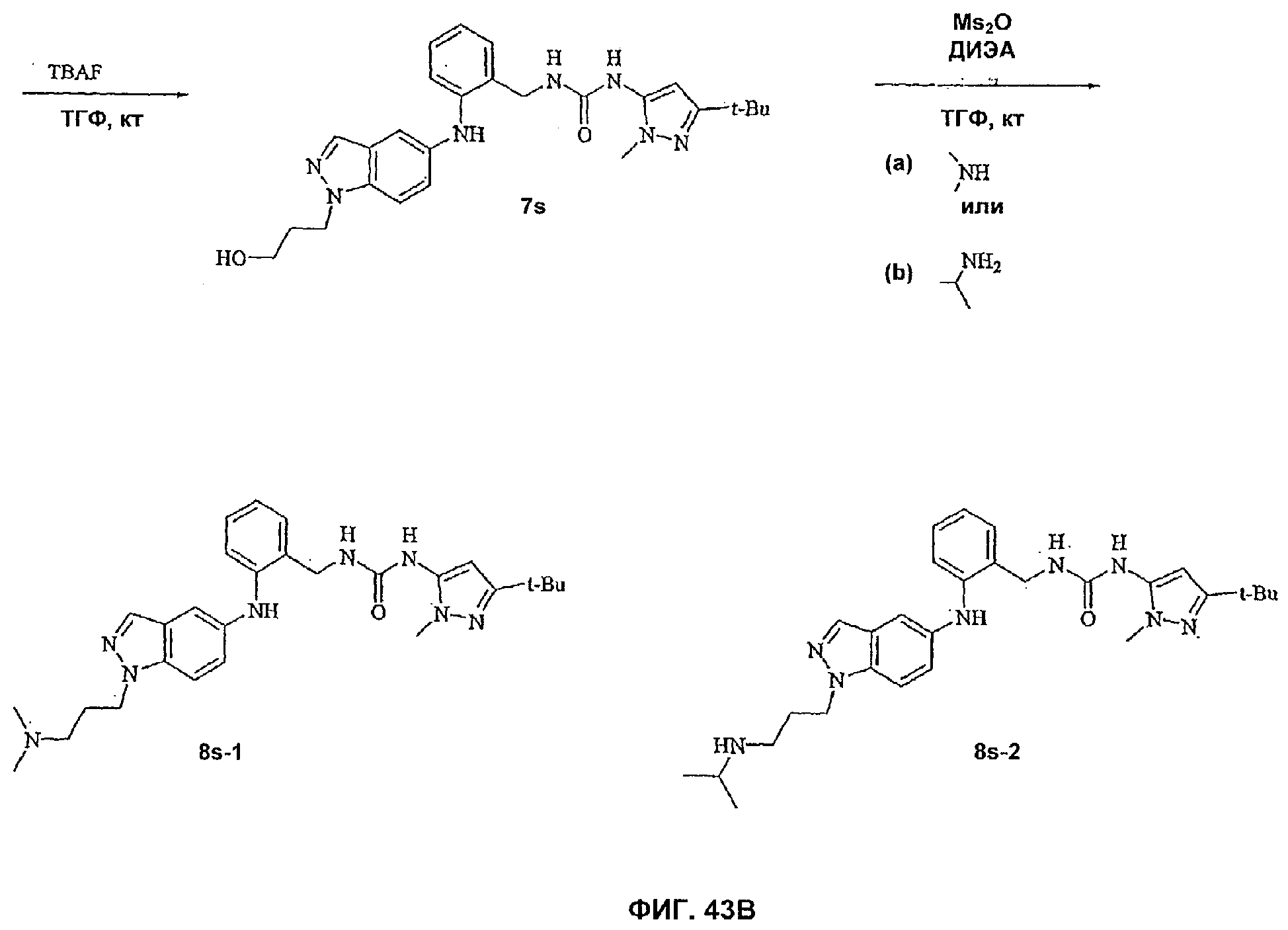
На Фиг.43А-В показана реакционная схема синтеза соединения 8s-2.
На Фиг.44 показана реакционная схема синтеза соединения 7t-2.
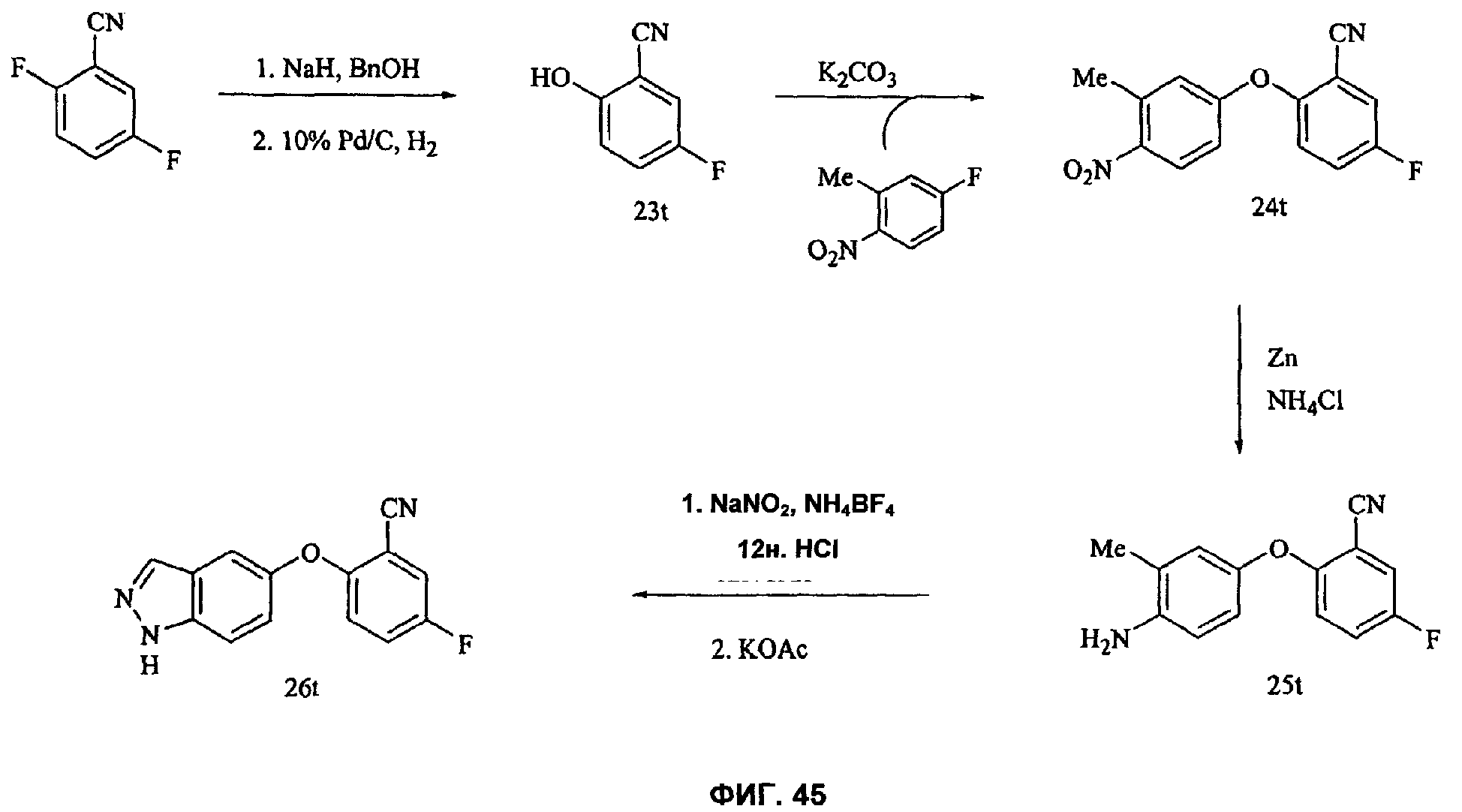
На Фиг.45 показана реакционная схема синтеза соединения 26t.
На Фиг.46 показана реакционная схема синтеза соединения 28t.
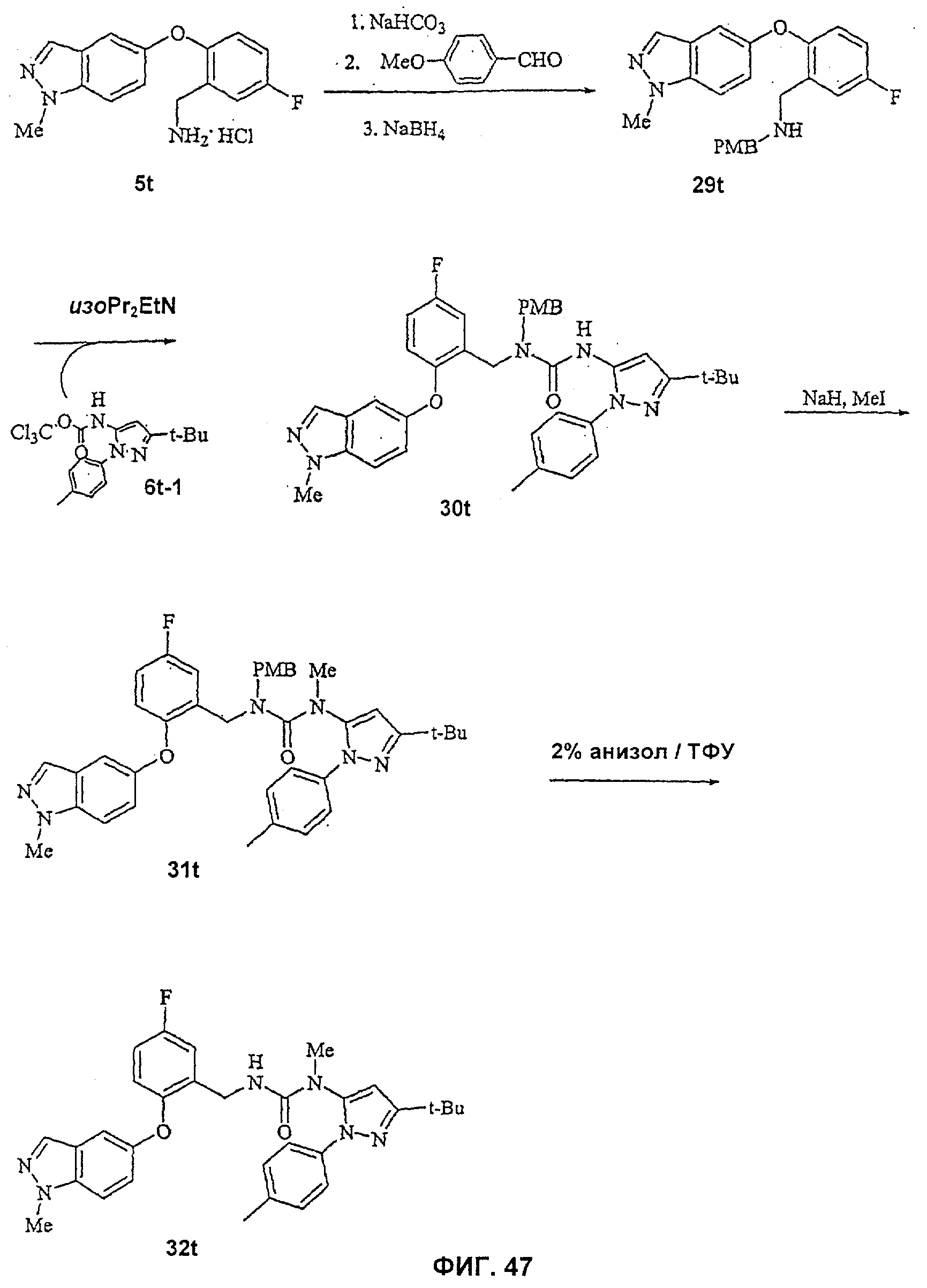
На Фиг.47 показана реакционная схема синтеза соединения 32t.
На Фиг.48 показана реакционная схема синтеза соединения 4u.
На Фиг.49 показана реакционная схема синтеза соединений 7v и 8v.
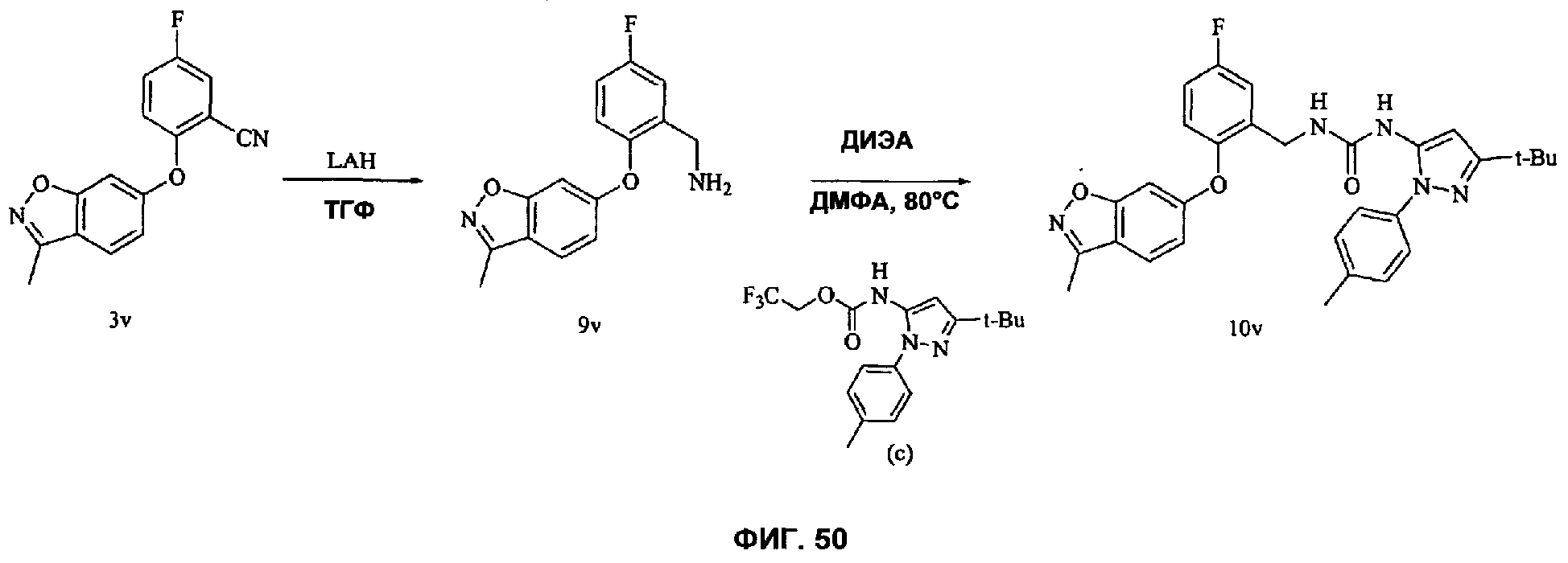
На Фиг.50 показана реакционная схема синтеза соединения 10v.
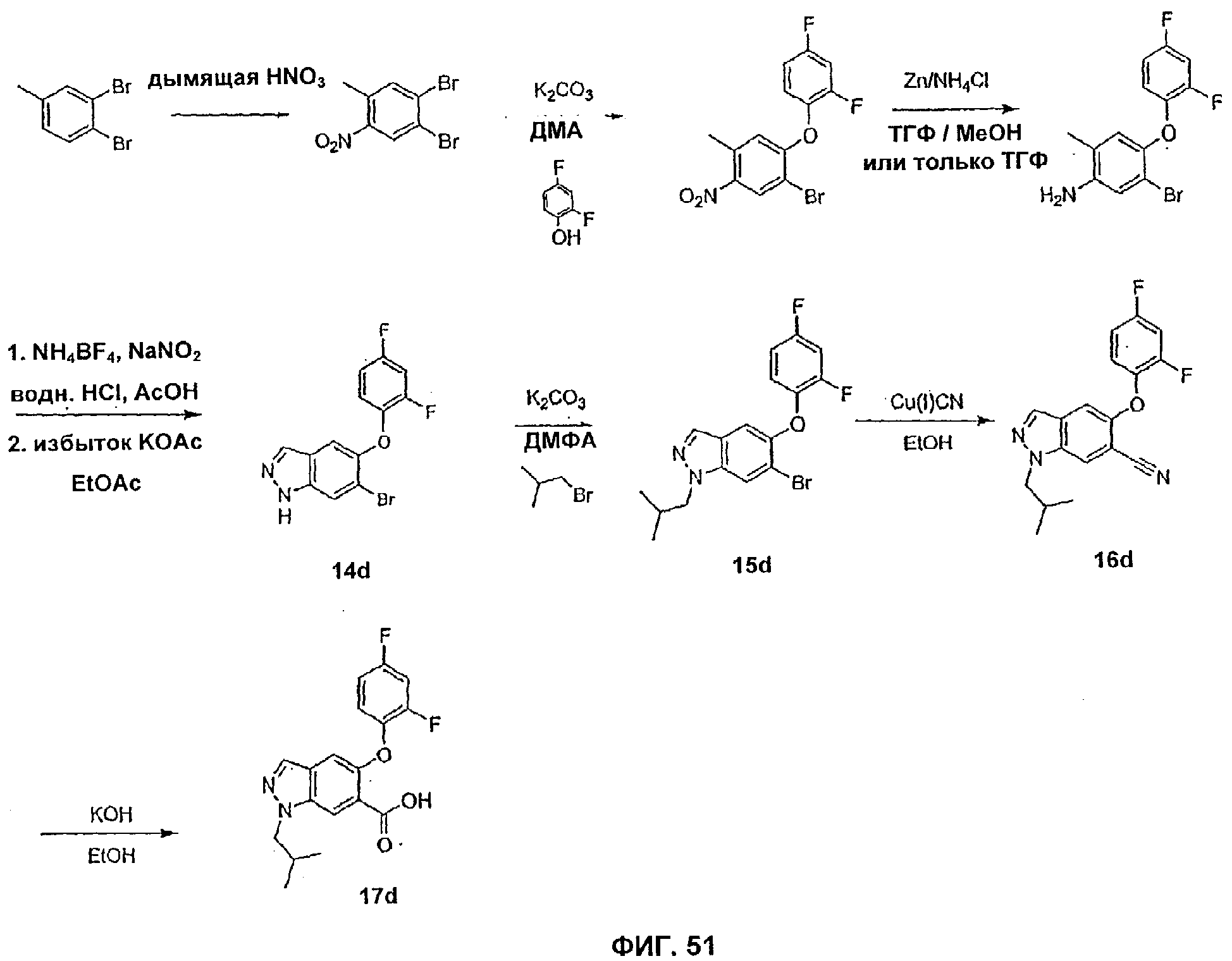
На Фиг.51 показана реакционная схема синтеза соединения 17d.
На Фиг.52 показана реакционная схема синтеза соединения 20d.
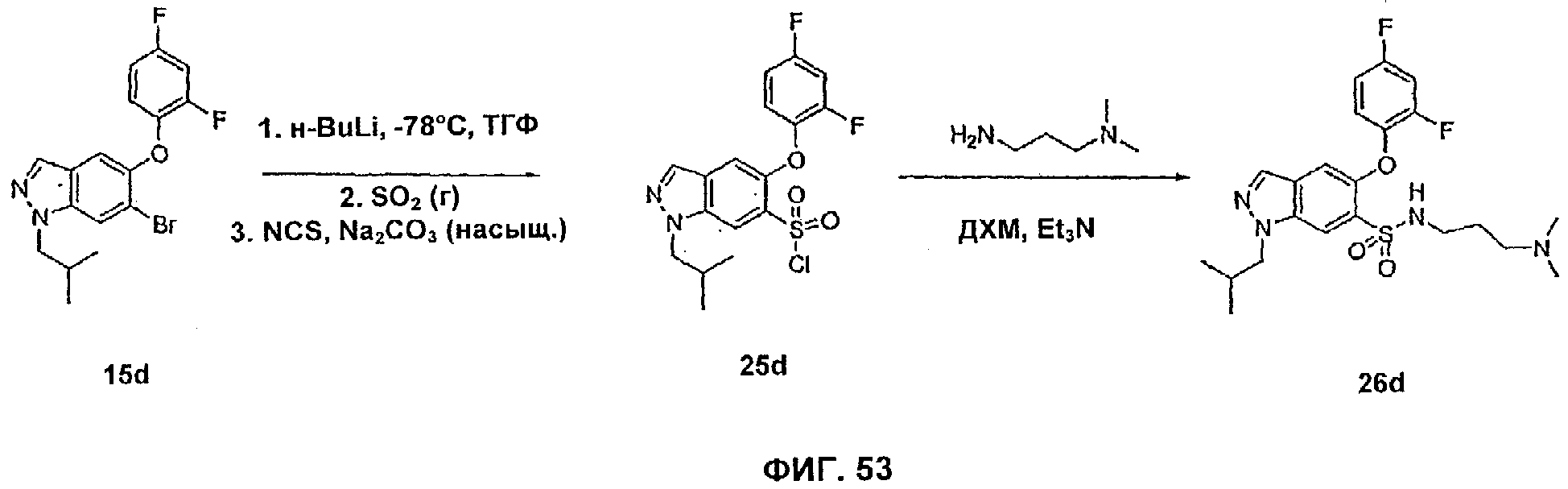
На Фиг.53 показана реакционная схема синтеза соединения 26d.
На Фиг.54 показана реакционная схема синтеза соединения 47d.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения Формул I-XVII по изобретению полезны для ингибирования р38 альфа и ассоциированных р38-опосредованных событий, таких как продукция цитокинов. Такие соединения обладают эффективностью в качестве терапевтических агентов в отношении заболеваний, которые можно лечить путем ингибирования сигнального пути р38. В общих чертах, изобретение относится к соединениям общей Формулы I
где Y представляет собой С, N;
W представляет собой С, N, S или О при условии, что W представляет собой N, S или О, когда Y представляет собой С, и W представляет собой С или N, когда Y представляет собой N;
U представляет собой СН или N;
V представляет собой С-Е или N;
Х представляет собой О, S, SO, SO2, NR7, C=O, CHR7, -C=NOR1, -C=CHR1или CHOR1;
R1 представляет собой H, РО3Н2, SO3Н2, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-тетероциклоалкил или Zn-Ar1 может быть замещенным или незамещенным;
Z представляет собой алкилен, имеющий от 1 до 4 атомов углерода, или алкенилен или алкинилен, каждый из которых имеет от 2 до 4 атомов углерода, где указанный алкилен, алкенилен или алкинилен может быть замещенным или незамещенным;
R7 представляет собой Н или замещенный или незамещенный метил;
Ar1 представляет собой замещенный или незамещенный арил или гетероарил;
А представляет собой Н, ОН, защитную группу для амино, Zn-NR2R3,
Zn-NR2(C=O)R2, Zn-SO2R2, Zn-SOR2, Zn-SR2, Zn-OR2, Zn-(C=O)R2, Zn-(C=O)OR2, Zn-O-(C=O)R2, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или
Zn-Ar1 может быть замещенным или незамещенным;
R2 и R3 независимо представляют собой Н, ОН, защитную группу для амино, защитную группу для спирта, защитную группу для кислоты, защитную группу для тио, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или
Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1 может быть замещенным или незамещенным, или R2 вместе с R3 и N образует насыщенный или частично ненасыщенный гетероцикл, имеющий один или более гетероатомов, где указанный гетероцикл может быть замещенным или незамещенным и где указанный гетероцикл может быть конденсирован с ароматическим кольцом;
В представляет собой Н, NH2 или замещенный или незамещенный метил;
Е представляет собой Н, Zn-NR2R3, Zn-(C=O)R4, Zn-(C=O)R5, Zn-NR5(C=O)R5, Zn-O(C=O)R5, Zn-OR5, Zn-SO2R5, Zn-SOR5, Zn-SR5, Zn-NH(C=O)NHR5или R5;
R4 представляет собой замещенную или незамещенную природную или неприродную аминокислоту, защищенную природную или неприродную аминокислоту, NH(CHR6) (CH2)mOR5, где m означает целое число от 1 до 4, или NR2R3;
R5 представляет собой Н, ОН, защитную группу для амино, защитную группу для спирта, защитную группу для кислоты, защитную группу для тио, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1 может быть замещенным или незамещенным;
R6 представляет собой природную аминокислотную боковую цепь, Zn-NR2R3,
Zn-OR5, Zn-SO2R5, Zn-SOR5 или Zn-SR5 и
n означает 0 или 1,
при условии, что когда В представляет собой Н, и А представляет собой CH=CH-R8, где R8 представляет собой замещенный или незамещенный алкил, алкенил, циклоалкил, гетероциклоалкил, арил или гетероарил, тогда Х-Ar1представляет собой заместитель, где Ar1 является отличным от замещенного или незамещенного арила, гетероарила, NH-алкил, NH-циклоалкил, NH-гетероциклоалкил, NH-арил, NH-гетероарил, NH-алкокси или NH-диалкиламид, когда Х представляет собой О, S,
C=O, S=O, C=CH2, CO2, NH или N(C1-C8-алкил).
Используемый здесь термин "алкил" относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу длиной от одного до двенадцати атомов углерода, где алкильный радикал может быть возможно независимо замещен одним или более чем одним заместителем, описанным ниже. Примеры алкильных групп включают в себя, но не ограничиваются ими, метил, этил, н-пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, трет-пентил, гексил, изогексил и тому подобное.
"Алкилен" означает линейный или разветвленный насыщенный двухвалентный углеводородный радикал длиной от одного до двенадцати атомов углерода, например метилен, этилен, пропилен, 2-метилпропилен, пентилен и тому подобное.
Термин "алкенил" относится к линейному разветвленному одновалентному углеводородному радикалу длиной от двух до двенадцати атомов углерода, содержащему по меньшей мере одну двойную связь, например, к этенилу, пропенилу и тому подобному, где алкенильный радикал может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь, и включает в себя радикалы, имеющие цис- и транс- ориентации или, альтернативно, Е- и Z- ориентации.
Термин "алкенилен" относится к линейному или разветвленному двухвалентному углеводородному радикалу длиной от двух до двенадцати атомов углерода, содержащему по меньшей мере одну двойную связь, где алкениленовый радикал может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь. Примеры включают в себя, но не ограничиваются ими, этенилен, пропенилен и тому подобное.
Термин "алкинил" относится к линейному или разветвленному одновалентному углеводородному радикалу длиной от двух до двенадцати атомов углерода, содержащему по меньшей мере одну тройную связь. Примеры включают в себя, но не ограничиваются ими, этинил, пропинил и тому подобное, где алкинильный радикал может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь.
Термин "алкинилен" относится к линейному или разветвленному двухвалентному углеводородному радикалу длиной от двух до двенадцати атомов углерода, содержащему по меньшей мере одну тройную связь, где алкиниленовый радикал может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь.
Термин "аллил" относится к радикалу, имеющему формулу RC=CHCHR, где R представляет собой алкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил или любой заместитель, как определено здесь, где аллил может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь.
Термин "циклоалкил" относится к насыщенному или частично ненасыщенному циклическому углеводородному радикалу, имеющему от трех до двенадцати атомов углерода, где циклоалкил может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь. Более того, термин "циклоалкил" включает в себя бициклические и трициклические циклоалкильные структуры, где бициклические и трициклические структуры могут включать в себя насыщенный или частично ненасыщенный циклоалкил, конденсированный с насыщенным или частично ненасыщенным циклоалкильным или гетероциклоалкильным кольцом, или арильным или гетероарильным кольцом. Примеры циклоалкильных групп включают в себя, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и тому подобное.
Термин "гетероалкил" относится к насыщенному линейному или разветвленному одновалентному, углеводородному радикалу длиной от одного до двенадцати атомов углерода, где по меньшей мере один атом углерода заменен гетероатомом, выбранным из N, О или S, и где радикал может представлять собой углеродный радикал или гетероатомный радикал (т.е. гетероатом может находиться в середине или в конце этого радикала). Гетероалкильный радикал может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь. Термин "гетероалкил" охватывает алкокси- и гетероалкоксирадикалы.
Термин "гетероциклоалкил" относится к насыщенному или частично ненасыщенному циклическому радикалу длиной от 3 до 8 кольцевых атомов, в котором по меньшей мере один атом кольца представляет собой гетероатом, выбранный из азота, кислорода и серы, при этом остальные атомы кольца представляют собой С, где один или более атомов кольца могут быть возможно независимо замещены одним или более чем одним заместителем, описанным ниже, и где гетероциклоалкильное кольцо может быть насыщенным или частично ненасыщенным. Радикал может представлять собой углеродный радикал или гетероатомный радикал. "Гетероциклоалкил" также включает в себя радикалы, где гетероциклические радикалы конденсированы с ароматическими или гетероароматическими кольцами. Примеры гетероциклоалкильных колец включают в себя, но не ограничиваются ими, пирролидин, пиперидин, пиперазин, тетрагидропиранил, морфолин, тиоморфолин, гомопиперазин, фталимид и их производные.
Термин "гетероалкенил" относится к линейному или разветвленному одновалентному углеводородному радикалу длиной от двух до двенадцати атомов углерода, содержащему по меньшей мере одну двойную связь, например к этенилу, пропенилу и тому подобному, где по меньшей мере один из атомов углерода заменен гетероатомом, выбранным из N, О или S, и где радикал может представлять собой углеродный радикал или гетероатомный радикал (т.е. гетероатом может находиться в середине или в конце этого радикала). Гетероалкенильный радикал может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь, и включает в себя радикалы, имеющие цис- и транс- ориентации или, альтернативно, Е- и Z- ориентации.
Термин "гетероалкинил" относится к линейному или разветвленному одновалентному углеводородному радикалу длиной от двух до двенадцати атомов углерода, содержащему по меньшей мере одну тройную связь. Примеры включают в себя, но не ограничиваются ими, этинил, пропинил и тому подобное, где по меньшей мере один атом углерода заменен гетероатомом, выбранным из N, О или S, и где радикал может представлять собой углеродный радикал или гетероатомный радикал (т.е. гетероатом может находиться в середине или в конце этого радикала). Гетероалкинильный радикал может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь.
Термин "гетероаллил" относится к радикалам, имеющим формулу RC=CHCHR, где R представляет собой алкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил или любой заместитель, как определено здесь, где по меньшей мере один атом углерода заменен гетероатомом, выбранным из N, О или S и где радикал может представлять собой углеродный радикал или гетероатомный радикал (т.е. гетероатом может находиться в середине или в конце этого радикала). Гетероаллил может быть возможно независимо замещен одним или более чем одним заместителем, описанным здесь.
"Арил" означает одновалентный ароматический углеводородный моноциклический радикал из 6-10 кольцевых атомов или полициклический ароматический углеводород, возможно независимо замещенный одним или более чем одним заместителем, описанным здесь. Более конкретно термин "арил" включает в себя, но не ограничивается этим, фенил, 1-нафтил, 2-нафтил и их производные.
"Гетероарил" означает одновалентный моноциклический ароматический радикал из 5-10 кольцевых атомов или полициклический ароматический радикал, содержащий один или более чем один гетероатом в кольце, выбранный из N, О или S, при этом остальные атомы кольца представляют собой С. Ароматический радикал возможно независимо замещен одним или более чем одним заместителем, описанным здесь. Примеры включают в себя, но не ограничиваются ими, фурил, тиенил, пирролил, пиридил, пиразолил, пиримидинил, имидазолил, пиразинил, индолил, тиофен-2-ил, хинолил, бензопиранил, тиазолил и их производные.
Термин "галогено" представляет собой фторо, хлоро, бромо или йодо.
Термин "защитные группы для амина" относится к органическим группам, предназначенным для защиты атомов азота от нежелательных реакций в процессах синтеза, и включает в себя, но не ограничивается этим, бензил, бензилоксикарбонил (CBZ), трет-бутоксикарбонил (Вое), трифторацетил и тому подобное.
Термин "защитные группы спирта" относится к органическим группам, предназначенным для защиты спиртовых групп или заместителей от нежелательных реакций в процессах синтеза, и включает в себя, но не ограничивается этим, (триметилсилил)этоксиметил (SEM), трет-бутил, метоксиметил (MOM) и тому подобное.
Термин "защитные группы серы" относится к органическим группам, предназначенным для защиты серосодержащих групп или заместителей от нежелательных реакций в процессах синтеза, и включает в себя, но не ограничивается этим, бензил, (триметилсилил)этоксиметил (SEM), трет-бутил, тритил и тому подобное.
Термин "защитные группы кислоты" относится к органическим группам, предназначенным для защиты кислотных групп или заместителей от нежелательных реакций в процессах синтеза, и включает в себя, но не ограничивается этим, бензил, (триметилсилил)этоксиметил (SEM), метилэтил и трет-бутиловые сложные эфиры и тому подобное.
В общих чертах, различные группировки или функциональные группы соединений Формул I-XVII могут быть возможно замещены одним или более чем одним заместителем. Примеры заместителей, подходящих для целей этого изобретения, включают в себя, но не ограничиваются ими, галогено, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил, Zn-OR, Zn-NO2, Zn-CN, Zn-CO2R, Zn-(C=O)R, Zn-O(C=O)R, Zn-O-алкил, Zn-OAr, Zn-SH, Zn-SR, Zn-SOR, Zn-SO2R, Zn-S-Ar, Zn-SOAr, Zn-SOzAr, арил, гетероарил, Zn-Ar, Zn-(C=O)NR2R3, Zn-NR2R3, Zn-NR(C=O)R, Zn-SO2NR2R3, РО3Н2, SO3Н2, защитные группы для амина, защитные группы для спирта, защитные группы для серы или защитные группы для кислоты, где
Z представляет собой алкилен, имеющий от 1 до 4 атомов углерода, или алкенилен или алкинилен, каждый из которых имеет от 2 до 4 атомов углерода, где указанный алкилен, алкенилен или алкинилен может быть замещенным или незамещенным;
n означает 0 или 1,
R1, R2 и R3 представляют собой алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил или Zn-гетероциклоалкил и
Ar представляет собой арил или гетероарил, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил, Ar, R1, R2 и R3могут быть дополнительно замещенными или незамещенными.
Соединения по этому изобретению могут обладать одним или более чем одним асимметрическим центром; следовательно, такие соединения могут быть получены в виде индивидуальных (R)- или (S)-стереоизомеров или в виде их смесей. Если не указано иначе, то подразумевают, что описание или наименование конкретного соединения в описании и формуле изобретения включает как индивидуальные энантиомеры, так и их смеси, рацемические или иные. Соответственно, это изобретение также включает в себя рацематы и разделенные энантиомеры и диастереомеры соединений Формул I-XVII. Способы определения стереохимии и разделения стереоизомеров хорошо известны из уровня техники (см. обсуждение в Chapter 4 of "Advanced Organic Chemistry", 4 th edition J. March, John Wiley and Sons, New York, 1992).
Кроме соединений Формул I-XVII изобретение также включает в себя сольваты, фармацевтически приемлемые пролекарства, фармацевтически активные метаболиты и фармацевтически приемлемые соли таких соединений.
Термин "сольват" относится к агрегату молекулы с одной или более молекулами растворителя.
"Фармацевтически приемлемое пролекарство" представляет собой соединение, которое может быть превращено в физиологических условиях или посредством сольволиза в указанное соединение или в фармацевтически приемлемую соль такого соединения.
"Фармацевтически активный метаболит" представляет собой фармакологически активный продукт, полученный в результате метаболизма в организме определенного соединения или его соли. Метаболиты соединения могут быть идентифицированы с использованием общепринятых методик, известных из уровня техники, и их активности могут быть определены с использованием тестов, таких как тесты, описанные здесь.
Пролекарства и активные метаболиты соединения могут быть идентифицированы с использованием общепринятых методик, известных из уровня техники. Различные формы пролекарств известны из уровня техники. Примеры производных таких пролекарств описаны, например, в a) Design of Prodrugs, edited by H.Bundgaard, (Elsevier, 1985) и Methods in Enzymology, Vol.42, p.309-396, edited by K.Widder, et al. (Academic Press, 1985); б) A Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H.Bundgaard, Chapter 5 "Design and Application of Prodrugs", by H.Bundgaard p.113-191 (1991); в) H.Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992); г) H.Bundgaard, et al., Journal of Pharmaceutical Sciences, 77:285 (1988), и д) N. Kakeya, et al., Chem. Pharm, Bull., 32: 692 (1984), каждый из которых включен здесь посредством ссылки.
"Фармацевтически приемлемая соль" представляет собой соль, которая сохраняет биологическую эффективность свободных кислот и оснований указанного соединения и которая не является биологически или в другом отношении нежелательной. Соединение по изобретению может обладать достаточно кислотными, достаточно основными или и теми и другими функциональными группами и соответственно взаимодействовать с любым из ряда неорганических или органических оснований и неорганических и органических кислот с образованием фармацевтически приемлемой соли. Примеры фармацевтически приемлемых солей включают в себя соли, полученные взаимодействием соединений по настоящему изобретению с минеральной или органической кислотой или неорганическим основанием, при этом такие соли включают в себя сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себакаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, γ-гидроксибутираты, гликолаты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты и манделаты.
Если соединение по изобретению представляет собой основание, то желательная фармацевтически приемлемая соль может быть получена любым подходящим способом, доступным из уровня техники, например обработкой свободного основания неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидильная (pyranosidyl) кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота, или тому подобное.
Если соединение по изобретению представляет собой кислоту, то желательная фармацевтически приемлемая соль может быть получена любым подходящим способом, например обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксидом щелочного металла или гидроксидом щелочноземельного металла, или тому подобного. Иллюстративные примеры подходящих солей включают в себя, но не ограничиваются ими, органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Соединения по изобретению могут быть получены с использованием реакционных путей и схем синтезов, описанных ниже, с помощью методик, имеющихся в уровне техники, с использованием исходных веществ, которые легко доступны.
Кроме соединений общей Формулы I, это изобретение дополнительно включает в себя соединения общей Формулы II
где А, В, Х и Ar1 являются такими, как определено выше.
На Фиг.1-6 представлены примеры синтеза конкретных соединений, имеющих общую Формулу II. В одном общем способе синтеза пиразольные соединения Формулы II получают следующим образом. 2-Хлор-4-метил-5-нитропиридин в подходящем безводном растворителе обрабатывают арил- или гетероарилфенолом или тиофенолом и основанием, таким как NaH. Через соответствующий период времени реакционную смесь распределяют между органическим растворителем и водой и из органического слоя выделяют 2-O-арил- или 8-арилзамещенное-4-метил-5-нитропиридиновое промежуточное соединение. Заместитель NO2 затем восстанавливают, например, путем обработки порошком железа в уксусной кислоте при нагревании в течение периода времени с последующей обработкой подходящим основанием, таким как NaOH. Полученное анилиновое промежуточное соединение выделяют путем экстракции реакционной смеси органическим растворителем. Затем промежуточное анилиновое соединение объединяют с тетрафторборатом аммония с последующим добавлением основания, такого как КОАс, и катализатора фазового переноса (например, 18-краун-6) и получают бициклическое пиразольное соединение Формулы II, где А представляет собой водород. Для получения 1-N-замещенных пиразольных соединений Формулы II, где А является отличным от водорода, пиразольное соединение подвергают взаимодействию с подходящим основанием и соединением формулы RX, где Х представляет собой галоген и R представляет собой алкил, аллил, алкенил, алкинил, аллил, циклоалкил, гетероциклоалкил, бензил или СН2-гетероарил, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы III
где А, В, X, Е и Ar1 являются такими, как определено выше.
На Фиг.7-8 представлены примеры синтеза конкретных соединений, имеющих общую Формулу III. В одном общем способе синтеза соединения Формулы III получают следующим образом. Арилтиофенол или арилфенол добавляют к сильному основанию в безводном растворителе, а затем подвергают взаимодействию с 5-хлор-3-метил-2-нитропиридином, получая 6-S-арил- или 6-O-арилзамещенное 2-метил-З-нитропиридиновое промежуточное соединение. NO2-заместитель восстанавливают, например, путем обработки порошком железа в уксусной кислоте при нагревании в течение периода времени с последующей обработкой подходящим основанием, таким как NaOH. Полученное анилиновое промежуточное соединение выделяют путем экстракции реакционной смеси органическим растворителем. Промежуточное анилиновое соединение затем обрабатывают тетрафторборатом аммония с последующим добавлением основания, такого как КОАс, и катализатора фазового переноса (например, 18-краун-6) и получают бициклическое азаиндазольное соединение Формулы III, где А представляет собой водород. Для получения 1-N-замещенных азаиндазольных соединений Формулы III, где А является иным, чем водород, азаиндазольное соединение подвергают взаимодействию с подходящим основанием и соединением формулы RX, где Х представляет собой галоген, и R представляет собой алкил, аллил, алкенил, алкинил, циклоалкил, гетероциклоалкил, бензил или СН2-гетероарил, как определено выше.
В еще одном воплощении это изобретение относится к соединениям общей Формулы IV
где А, В, X, Е и Ar1 являются такими, как определено выше, при условии, что когда В представляет собой Н и А представляет собой CH=CH-R8, где R8представляет собой замещенный или незамещенный алкил, алкенил, циклоалкил, гетероциклоалкил, арил или гетероарил, тогда Х-Ar1 представляет собой заместитель, где Ar1 является отличным от замещенного или незамещенного арила, гетероарила, NH-алкил, NH-циклоалкил, NH-гетероциклоалкил, NH-арил, NH-гетероарил, NH-алкокси или NH-диалкиламид, когда Х представляет собой О, S, С=O, S=O, С=СН2, CO2, NH или
N(C1-C8-алкил).
На Фиг.9-13 представлены примеры синтеза конкретных соединений, имеющих общую Формулу IV. В одном общем способе синтеза соединения Формулы IV получают следующим образом. 6-Нитроиндол обрабатывают основанием и йодом и полученный 3-йод-6-нитроиндол обрабатывают основанием и агентом с защитной группой для амино, таким как триметилсилилэтоксиметилхлорид (SEM-Cl). Обработка защищенного 6-нитроиндольного соединения транс-2-фенилвинилбороновой кислотой и подходящим катализатором, таким как Pd(PPh3)4, дает 1-N-фенилвинил-6-нитроиндольное промежуточное соединение. Восстановление 6-NO2 заместителя восстанавливающим агентом, таким как гидразин, и подходящим катализатором (например, палладием на угле) дает l-N-замещенное 6-аминоиндольное производное. Обработка этого производного нитритом натрия с последующим добавлением йодида натрия и йода дает 1-N-замещенное-3-фенилвинил-6-йодиндазольное производное. Обработка этого производного окисляющим(и) агентом(ами), таким как тетроксид осмия и периодат натрия, дает 1-N-защищенное 3-карбальдегид-6-йодиндазольное производное. Это производное затем может быть использовано в некоторых способах синтеза с получением различных индазольных соединений по этому изобретению, таких, как описано в Примерах.
В альтернативном способе синтеза 6-OAr-замещенные соединения Формулы IV получают следующим образом. Обработка 2-фтор-4-гидроксиацетофенона подходящим реагентом с защитной группой для фенола с последующим добавлением гидразина при нагревании для индукции циклизации дает индазольное соединение. Индазольное соединение защищают в положении 1-N подходящим реагентом с защитной группой для амино. Удаление защитной группы для фенола и обработка арилбороновой кислотой с последующим удалением защитной группы для амино дает 6-OAr-замещенное соединение Формулы IV.
В альтернативном способе синтеза 6-SAr-замещенные соединения Формулы IV получают следующим образом. 4-Фтортиофенол обрабатывают сильным основанием, таким как трет-бутоксид калия, и к полученному феноксиду добавляют 2,4-дифторпропиофенон. Добавление гидразина к полученному промежуточному соединению с последующим нагреванием для индукции циклизации дает 6-SAr-замещенное соединение Формулы IV.
В альтернативном способе синтеза 5-OAr- и 5-SAr-замещенные соединения Формулы IV получают следующим образом. Этерификация 5-фтор-2-нитробензойной кислоты с последующей обработкой полученного сложного эфира смесью либо ArOH, либо ArSH и сильного основания дает 5-XAr-замещенный метиловый эфир 2-нитробензойной кислоты, где Х представляет собой О или S. Омыление этого сложного эфира с последующим добавлением гидроксида аммония дает 2-нитробензамидное промежуточное соединение. 2-Нитробензамид превращают в 2-нитробензонитрильное промежуточное соединение путем обработки оксалилхлоридом. Восстановление нитрозаместителя с последующим добавлением нитрита натрия дает 3-амино-5-XAr-замещенное индазольное соединение Формулы IV, где Х представляет собой О или S.
В альтернативном способе синтеза 6-OAr-замещенные соединения Формулы IV получают следующим образом. 2-Фтор-4-гидроксибензонитрил объединяют с арилбороновой кислотой, ацетатом меди и основанием, получая 2-фтор-4-арилоксибензонитрильное промежуточное соединение.
Перемешанный раствор этого промежуточного соединения с гидразином нагревают с обратным холодильником, получая 3-амино-6-арилоксииндазольное соединение. Это соединение может быть использовано в качестве исходного вещества для синтеза 3-амидиндазольных производных с использованием стандартной химии синтеза амидов, известной специалистам в данной области техники.
В еще одном воплощении это изобретение относится к соединениям общей Формулы V
где А, X, Е и Ar являются такими, как определено выше.
На Фиг.24-26 представлены примеры синтеза конкретных соединений, имеющих общую Формулу V. В одном общем способе синтеза соединения Формулы V получают следующим образом. 4-Фтор-2-гидроксибензойную кислоту подвергают этерификации и 2-гидроксигруппу защищают подходящей защитной группой для спирта. Замещение фторогруппы группой O-Ar или S-Ar осуществляют путем обработки основанием и ArOH или ArSH, где Ar представляет собой арил или гетероарил, как определено выше. Удаление защитной группы для спирта и омыление сложного эфира с последующей обработкой карбонилдиимидазолом для осуществления циклизации дает 6-OAr- или 6-SAr-3-гидроксибензизоксазольное соединение. 3-Гидроксибензизоксальное соединение превращают в 3-хлорбензизоксазольное производное путем обработки POCl3 и основанием. Этот продукт затем может быть использован для получения 3-O-Ar- или 3-NH-Ar-замещенных бензизоксазольных соединений по этому изобретению. Например, 6-замещенное-3-хлорбензизоксазольное соединение может быть добавлено к смеси ArOH и сильного основания (например, NaH) с получением 6-замещенного-3-О-Ar-бензизоксального производного. В альтернативном способе синтеза 6-замещенное-3-хлорбензизоксазольное соединение может быть добавлено к смеси ArNH2 и сильного основания с получением 6-замещенного-3-NHAr-бензизоксазольного производного.
В еще одном воплощении это изобретение относится к соединениям общих Формул VI и VII
где А, В, Е и Ar1 являются такими, как определено выше.
На Фиг.14-15 представлены примеры синтеза конкретных соединений, имеющих общую Формулу VI, а на Фиг.18, 19 и 23 представлены примеры синтеза конкретных соединений, имеющих общую Формулу VII. В одном общем способе синтеза соединения Формул VI и VII получают следующим образом. 5-Йод-1Н-индазол получают путем обработки 5-амино-1Н-индазола раствором NaNO2 в воде с последующим добавлением KI. После выделения продукта путем экстракции реакционной смеси органическим растворителем, этот продукт может быть затем использован в различных способах синтеза для получения индазольных соединений по этому изобретению. В одном из способов 1-аминогруппу 5-йод-1 Н-индазола защищают подходящей защитной группой для амино и защищенный 5-йодиндазол обрабатывают основанием, порошком меди и арилфенолом или арилтиофенолом, получая 5-O-арилзамещенный индазол (Формула VI) или 5-3-арилзамещенный индазол (Формула VII). Удаление защитной группы для амино дает соединение по этому изобретению, имеющее Формулу VI или VII.
В альтернативном способе 5-йод-1 Н-индазол обрабатывают основанием и RX или Ar1СН2Х, где R представляет собой алкил или аллил и Ar1представляет собой арильную или гетероарильную группу, как определено выше, и Х представляет собой галоген или другую подходящую уходящую группу. 1-N-Замещенный 5-йодиндазол затем обрабатывают основанием, порошком меди и арилтиофенолом или арилфенолом, получая 5-O-арилзамещенное индазольное (Формула VI) или 5-S-арил-1-N-замещенное индазольное (Формула VII) соединение по этому изобретению.
В еще одном воплощении это изобретение относится к соединениям общей Формулы VIII
где А, В, Е и Ar1 являются такими, как определено выше.
На Фиг.22 представлен пример синтеза конкретного соединения, имеющего общую Формулу VIII. В одном общем способе синтеза соединения Формулы VIII получают окислением соединения Формулы VII окисляющим агентом, который будет окислять арилсульфид до соответствующего арилсульфинильного производного.
В еще одном воплощении это изобретение относится к соединениям общей Формулы IX
где А, В, Е и Ar1 являются такими, как определено выше.
На Фиг.21 представлен пример синтеза конкретного соединения, имеющего общую Формулу IX. В одном общем способе синтеза соединения Формулы IX получают путем окисления соединения Формулы VII окисляющим агентом, который будет окислять арилсульфид до соответствующего арилсульфонильного производного.
В еще одном воплощении это изобретение относится к соединениям общей Формулы X
где А, В, Е и Ar1 являются такими, как определено выше.
На Фиг.31 представлен пример синтеза конкретного соединения, имеющего общую Формулу X. В одном общем способе синтеза соединения Формулы Х получают следующим образом. К смеси тетрафторбората аммония и уксусной кислоты добавляют 4-бром-2-метиланилин. Спустя некоторое время к этой смеси добавляют нитрит натрия, а затем основание, такое как ацетат калия, и катализатор фазового переноса, такой как 18-краун-6, получая 5-броминдазол. Броминдазол обрабатывают RBr в присутствии основания, получая 1-N-замещенное 5-броминдазольное производное, где R представляет собой А, как определено выше для Формулы X, за исключением водорода. Обработка 1-N-замещенного производного с помощью Ar1СНО в присутствии сильного основания, такого как бутиллитий, где Ar1 является таким, как определено выше, дает спиртовое соединение Формулы X.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XI
где А, В, Е и Ar1 являются такими, как определено выше.
На Фиг.32 представлен пример синтеза конкретного соединения, имеющего общую Формулу XI. В одном общем способе синтеза соединения Формулы XI получают следующим образом. К смеси тетрафторбората аммония и уксусной кислоты добавляют 4-бром-2-метиланилин. Спустя некоторое время к этой смеси добавляют нитрит натрия, а затем основание, такое как ацетат калия, и катализатор фазового переноса, такой как 18-краун-6, получая 5-броминдазол. Броминдазол обрабатывают RBr в присутствии основания, получая 1-N-замещенное 5-броминдазольное промежуточное соединение, где R представляет собой А, как определено выше для Формулы XI, за исключением водорода. Обработка 1-N-замещенного промежуточного соединения с помощью Ar1СНО в присутствии сильного основания, такого как бутиллитий, где Ar1 является таким, как определено выше, с последующей обработкой подходящим окисляющим агентом дает 1-N-замещенное соединение Формулы XI. Альтернативный способ синтеза соединения Формулы XI показан на Фиг.33.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XII
где А, В, Е, R1 и Ar1 являются такими, как определено выше.
На Фиг.27 представлен пример синтеза конкретного соединения, имеющего общую Формулу XII. В одном общем способе синтеза соединения Формулы XII получают следующим образом. К смеси тетрафторбората аммония и уксусной кислоты добавляют 4-бром-2-метиланилин. Спустя некоторое время к этой смеси добавляют нитрит натрия, а затем основание, такое как ацетат калия, и катализатор фазового переноса, такой как 18-краун-6, получая 5-броминдазол. Броминдазол обрабатывают RBr в присутствии основания, получая 1-N-замещенное 5-броминдазольное производное, где R представляет собой алкил, аллил, ArCH2 или гетероарил-СН2, как определено выше. Обработка 1-N-замещенного производного с помощью Ar1СНО в присутствии сильного основания, такого как бутиллитий, где Ar1 является таким, как определено выше, с последующей обработкой подходящим окисляющим агентом дает 1-N-замещенное 5-C=OR-производное. Добавление NH2OR1 к этому производному в пиридине, где R6 является таким, как определено выше, дает оксим Формулы XII. Альтернативный способ синтеза соединений Формулы XII показан на Фиг.28.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XIII
где А, В, Е и Ar1 являются такими, как определено выше.
На Фиг.34 представлен пример синтеза конкретного соединения, имеющего общую Формулу XIII. В одном общем способе синтеза соединения Формулы XIII получают следующим образом. К смеси тетрафторбората аммония и уксусной кислоты добавляют 4-бром-2-метиланилин. Спустя некоторое время к смеси добавляют нитрит натрия, а затем основание, такое как ацетат калия, и катализатор фазового переноса, такой как 18-краун-6, получая 5-броминдазол. Броминдазол обрабатывают RBr в присутствии основания, получая 1-N-замещенное 5-броминдазольное промежуточное соединение, где R представляет собой А, как определено выше для Формулы XIII, за исключением водорода. Обработка 1-N-замещенного промежуточного соединения сильным основанием, таким как трет-бутиллитий, с последующим добавлением триметилбората дает индазольное промежуточное соединение 5-бороновой кислоты. Добавление медного(II) катализатора с последующим добавлением замещенного или незамещенного анилина дает соединение Формулы XIII.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XIV
где А, В, X, Ar1, R2 и R3 являются такими, как определено выше.
На Фиг.30А-30С представлен пример синтеза конкретного соединения, имеющего общую Формулу XIV. В одном общем способе синтеза соединения Формулы XIV получают следующим образом. 1-Фтор-3-метилбензол подвергают реакции присоединения с образованием 2-фтор-4-метилбензойной кислоты, а затем нитрованию с получением 2-фтор-4-метил-5-нитробензойной кислоты. Кислотную группу этерифицируют, а затем фторогруппу замещают ArO- после обработки ArOH и сильным основанием. Восстановление нитрогруппы с последующим диазотированием и циклизацией дает 5-OAr-6-СО2Ме индазольное производное, которое затем обрабатывают RBr в присутствии основания, получая 1-N замещенное производное. Гидролиз сложноэфирной группы с последующим амидированием дает 6-амид индазольное производное, имеющее Формулу XIV.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XV
где А, В, X и Ar1 являются такими, как определено выше, и R12 и R13независимо представляют собой алкил, аллил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил или гетероарил, где указанный алкил, аллил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил или гетероарил может быть замещенным или незамещенным.
На Фиг.34 представлен пример синтеза конкретного соединения, имеющего общую Формулу XV. В одном общем способе синтеза соединения Формулы XV получают следующим образом. 5-OAr-6-СО2Ме индазольное производное получают, как описано выше для синтеза соединений Формулы XIV, а затем обрабатывают RBr в присутствии основания, получая 1-N замещенное производное. Гидролиз сложноэфирной группы с последующей обработкой карбонилдиимидазолом и аминокислотой дает 6-замещенное индазольное производное, имеющее Формулу XV.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XVI
где А, В, X, R2, R3 и Ar1 являются такими, как определено выше.
В одном общем способе синтеза соединения Формулы XVI получают следующим образом. 5-OAr-6-СО2Ме индазольное производное получают, как описано выше для синтеза Формулы XIV, а затем восстанавливают, например, обработкой ВН3 в ТГФ. Очистка дает соединение Формулы XVI.
В еще одном воплощении это изобретение относится к соединениям общей Формулы XVII
где Y представляет собой CR1, О, S или NR2;
W представляет собой CR3, N, NR4, S или О при условии, что W представляет собой NR4, S или О, когда Y представляет собой CR1, и W представляет собой CR3 или N, когда Y представляет собой NR2;
R3 представляет собой Н, NH2, F, Cl, метил или замещенный метил;
R4 представляет собой Н или метил или замещенный метил;
R1 и R2 независимо представляют собой H, ОН, защитную группу для амино, Zn-NRaRb, Zn-NRa(C=O)Rb, Zn-SO2Ra, Zn-SORa, Zn-SRa, Zn-ORa, Zn-(C=O)Ra, Zn-(C=O)ORa, Zn-O-(С=O)Ra, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
Ar1 представляет собой арил или гетероарил, каждый из которых может быть замещенным или незамещенным;
R3 и R13 независимо представляют собой Н, ОН, защитную группу для амино, защитную группу для спирта, защитную группу для кислоты, защитную группу для серы, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным,
или Ra и Rb вместе с атомами, к которым они оба присоединены, образуют насыщенное или частично ненасыщенное гетероциклическое кольцо, имеющее один или более гетероатомов в указанном кольце, где указанный гетероцикл может быть замещенным или незамещенным и где указанный гетероцикл может быть конденсирован с ароматическим кольцом;
Z представляет собой алкилен, имеющий от 1 до 4 атомов углерода, или алкенилен или алкинилен, каждый из которых имеет от 2 до 4 атомов углерода, где указанный алкилен, алкенилен или алкинилен может быть замещенным или незамещенным;
n означает 0 или 1;
U представляет собой CRc или N;
V представляет собой CRc или N;
Rc представляет собой Н, F, Cl, метил или замещенный метил;
Х представляет собой О, S, SO, SO2, NR5, C=O, CH2, CH2Zn-OH или С=NORd;
R5 представляет собой Н, метил или замещенный метил;
Rd представляет собой Н, РО3Н2, SO3Н2, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
G, Н, J и Т независимо представляют собой N или CRz при условии, что когда любые из указанных G, Н, J и Т представляют собой N, тогда общее количество G, Н, J или Т, которые представляют собой N, не превышает 2;
Rz представляет собой Н, F, Cl, Br, CF3, OR6, SR6, низший (С1-С4)алкил, CN или
NR6R7;
R6 и R7 независимо представляют собой Н, CF3, низший (С1-С4)алкил или
низший (С1-С4)гетероалкил;
Q представляет собой -NR8ONH-, -NHCO-, -NR8SO2NH-, -NHSO2-, -CONR11-;
R8 представляет собой Н или низший (С1-С4)алкил;
R11 представляет собой Н или низший (С1-С4)алкил;
Rx представляет собой -(CR9R10)m-, -O(CR9R10)m-, NH(CR9R10)m- или -S(CR9R10)m- при условии, что Q представляет собой -CONR11-, когда Rxпредставляет собой
O(CR9R10)m-, -NH(CR9R10)m- или -S(CR9R10)m-;
R9 и R10 независимо представляют собой Н, или низший алкил, или R9 и R10 вместе с атомами, к которым они оба присоединены, образуют циклоалкильное кольцо, которое может быть насыщенным или частично ненасыщенным;
m означает 1-3;
Ry представляет собой Н, РО3Н, защитную группу для амино, защитную группу для кислорода, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar2, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-Ar2 и Zn-гетероциклоалкил может быть замещенным или незамещенным;
Ar2 представляет собой арил или гетероарил, каждый из которых может быть замещенным или незамещенным, где указанное замещение может осуществляться 1-3 заместителями, независимо выбранными из F, Cl, Br, CF3, CN, алкила, аллила, алкенила, алкинила, гетероалкила, гетероаллила, гетероалкенила, гетероалкинила,
-OR12, -SR12, -SO2R12, -SO2NR13R12, NR13SO2R12, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
R12 и R13 независимо представляют собой Н, алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
где когда Ar2 замещен -SO2NR13R12, тогда R12 и R13 могут образовывать циклоалкильное кольцо или гетероциклоалкильное кольцо, которое может быть замещенным или незамещенным, где указанное замещение может осуществляться заместителями, выбранными из алкила, аллила, алкенила, алкинила, гетероалкила, гетероаллила, гетероалкенила, гетероалкинила, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, -COR12, -SO2R12, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным;
где когда Q представляет собой -CONR11-, тогда Ry вместе с R11дополнительно представляют собой циклоалкильное кольцо или гетероциклоалкильное кольцо, которое может быть замещенным или незамещенным группами, выбранными из алкила, аллила, алкенила, алкинила, гетероалкила, гетероаллила, гетероалкенила, гетероалкинила, алкокси, гетероалкокси, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, Zn-Ar1, -COR14 или -SO2R14, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил, Zn-Ar1, -COR14 и -SO2R14 может быть замещенным или незамещенным, и
R14 представляет собой алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, Zn-циклоалкил, где указанный циклоалкил является насыщенным или частично ненасыщенным, Zn-гетероциклоалкил, где указанный гетероциклоалкил является насыщенным или частично ненасыщенным, или Zn-Ar1, где указанный алкил, аллил, алкенил, алкинил, гетероалкил, гетероаллил, гетероалкенил, гетероалкинил, алкокси, гетероалкокси, Zn-циклоалкил, Zn-гетероциклоалкил и Zn-Ar1 может быть замещенным или незамещенным.
На Фиг.38-50 представлены примеры синтеза конкретных соединений, имеющих общую Формулу XVII.
Терапевтически эффективные количества соединений по изобретению могут быть использованы для лечения заболеваний, опосредованных модулированием или регулированием протеинкиназ. "Эффективное количество" предназначено для обозначения количества соединения, которое при введении млекопитающему, нуждающемуся в таком лечении, является достаточным для осуществления лечения заболевания, опосредованного активностью одной или более чем одной протеинкиназы, такой как р38 альфа, и ассоциированных р38-опосредованных событий, таких как продукция цитокинов. Так, например, терапевтически эффективное количество соединения, выбранного из Формул I-XVII, или его соли, активного метаболита или пролекарства, представляет собой количество, достаточное для модулирования, регулирования или ингибирования активности одной или более чем одной протеинкиназы таким образом, что болезненное состояние, которое опосредуется этой активностью, устраняется или облегчается.
Количество данного агента, которое будет соответствовать такому количеству, будет варьировать в зависимости от факторов, таких как конкретное соединение, болезненное состояние и его тяжесть, особенностей (например, веса) млекопитающего, нуждающегося в лечении, но тем не менее может быть стандартно определено специалистом в данной области техники. "Лечение" предназначено для обозначения по меньшей мере смягчения болезненного состояния у млекопитающего, такого как человек, которое находится под влиянием, по меньшей мере частично, активности одной или более чем одной протеинкиназы, такой как р38, и включает в себя, но не ограничивается этим, профилактику возникновения болезненного состояния у млекопитающего, особенно когда обнаружено, что это млекопитающее предрасположено к тому, чтобы иметь это болезненное состояние, но которое у него все же не выявлено; модулирование и/или ингибирование болезненного состояния; и/или облегчение болезненного состояния.
Для применения соединения Формулы I-XVII , или его фармацевтически приемлемой соли, или его пролекарства, расщепляемого in vivo, для терапевтического лечения (включая профилактическое лечение) млекопитающих, в том числе человека, его обычно приготавливают в виде фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Согласно этому аспекту изобретения предложена фармацевтическая композиция, которая содержит соединение Формулы I-XVII , или его фармацевтически приемлемую соль, или его пролекарство, расщепляемое in vivo, как определено выше, вместе с фармацевтически приемлемыми разбавителем или носителем.
Композиции по изобретению могут находиться в форме, подходящей для перорального применения (например, в виде таблеток, лепешек, твердых или мягких капсул, водных или масляных суспензий, эмульсий, дисперсных порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов, или суспензий), для введения путем ингаляции (например, в виде мелко измельченного порошка или жидкого аэрозоля), для введения путем инсуффляции (например, в виде мелко измельченного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного или внутримышечного дозирования или в виде суппозитория для ректального дозирования). Например, композиции, предназначенные для перорального применения, могут содержать, например, один или более чем один краситель, подсластитель, корригент и/или консервант.
Подходящие фармацевтически приемлемые эксципиенты для приготовления таблеток включают в себя, например, инертные разбавители, такие как лактоза, карбонат натрия, фосфат кальция или карбонат кальция, гранулирующие и разрыхляющие агенты, такие как кукурузный крахмал или альгиновую кислоту; связующие агенты, такие как крахмал; смазывающие агенты, такие как стеарат магния, стеариновая кислота или тальк; консерванты, такие как этил- или пропил-п-гидроксибензоат, и антиоксиданты, такие как аскорбиновая кислота. Препараты в форме таблеток могут быть непокрытыми или покрытыми либо для модификации их дезинтеграции и последующей абсорбции активного ингредиента в желудочно-кишечном тракте, либо для улучшения их стабильности и/или внешнего вида, в любом случае с использованием традиционных покрывающих агентов и методик, хорошо известных из уровня техники.
Композиции для перорального применения могут находиться в форме твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в форме мягких желатиновых капсул, в которых активный ингредиент смешан с водой или маслом, таким как арахисовое мало, вазелиновое масло или оливковое масло.
Водные суспензии обычно содержат активный ингредиент в виде тонкого порошка вместе с одним или более чем одним суспендирующим агентом, таким как натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и гуммиарабик; диспергирующим или увлажняющим агентом, таким как лецитин, или продукты конденсации алкиленоксида с жирными кислотами (например, полиоксиэтиленстеарат), или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксикетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, происходящими из жирных кислот и гексита, такие как полиоксиэтиленсорбит моноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, происходящими из жирных кислот и ангидридов гексита, например полиэтиленсорбит моноолеат. Водные суспензии также могут содержать один или более чем один консервант (такой как этил- или пропил-п-гидроксибензоат), антиоксидант (такой как аскорбиновая кислота), краситель, корригент и/или подсластитель (такой как сахароза, сахарин или аспартам).
Масляные суспензии могут быть приготовлены путем суспендирования активного ингредиента в растительном масле (таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло) или в минеральном масле (таком как вазелиновое масло). Масляные суспензии могут также содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Для обеспечения приятного вкуса перорального препарата могут быть добавлены подсластители, такие как указаны выше, и корригенты. Эти композиции могут быть сохранены путем добавления антиоксиданта, такого как аскорбиновая кислота.
Дисперсные порошки и гранулы, подходящие для получения водной суспензии путем добавления воды, обычно содержат активный ингредиент вместе с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или более чем одним консервантом. Примерами подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов являются такие, которые уже упомянуты выше. Также могут присутствовать дополнительные эксципиенты, такие как подсластители, корригенты или красители.
Фармацевтические композиции по изобретению также могут находиться в форме эмульсий масло-в-воде. Масляная фаза может представлять собой растительное масло, такое как оливковое масло или арахисовое масло, или минеральное масло, такое как, например, вазелиновое масло, или смесь любых этих масел. Подходящие эмульгаторы могут представлять собой, например, природные смолы, такие как гуммиарабик или трагакантовая камедь, природные фосфатиды, такие как соя культурная, лецитин, сложные эфиры или неполные сложные эфиры, происходящие из жирных кислот и ангидридов гексита (например, сорбит моноолеат), и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, такие как полиоксиэтиленсорбит моноолеат. Также эмульсии могут содержать подсластители, корригенты и консерванты.
Сиропы и эликсиры могут быть приготовлены с подсластителями, такими как глицерин, пропиленгликоль, сорбит, аспартам или сахароза, и также могут содержать успокаительное средство, консервант, корригент и/или краситель.
Фармацевтические композиции также могут находиться в форме стерильной инъекцируемой водной или масляной суспензии, которая может быть приготовлена в соответствии с известными методиками с использованием одного или более чем одного подходящего диспергирующего или увлажняющего агента и суспендирующего агента, которые упомянуты выше. Стерильный препарат для инъекции также может представлять собой стерильный раствор или суспензию для инъекции в нетоксичном, приемлемом для парентерального применения разбавителе или растворителе, например раствор в 1,3-бутандиоле.
Суппозиторные препараты могут быть приготовлены путем смешивания активного ингредиента с подходящим нераздражающим эксципиентом, который является твердым при обычных температурах, но становится жидкостью при ректальной температуре, и который, следовательно, будет плавиться в прямой кишке с высвобождением лекарства. Подходящие эксципиенты включают в себя, например, какао-масло и полиэтиленгликоли.
Местные препараты, такие как кремы, мази, гели и водные или масляные растворы или суспензии, обычно могут быть получены путем приготовления активного ингредиента с традиционным, приемлемым для местного применения растворителем или разбавителем с использованием традиционных методик, хорошо известных в данной области техники.
Композиции для введения путем инсуффляции могут находиться в форме мелко измельченного порошка, содержащего частицы со средним диаметром, например, 30 мкм или много меньше, при этом сам порошок включает либо только активный ингредиент, либо активный ингредиент, разбавленный одним или более чем одним физиологически приемлемым носителем, таким как лактоза. Порошок для инсуффляции удобно хранить в капсуле, содержащей, например, от 1 до 50 мг активного ингредиента, для применения с турбоингаляторным устройством, таким, которое применяется для инсуффляции известного агента кромогликата натрия.
Композиции для введения путем ингаляции могут находиться в форме традиционного аэрозоля под давлением, приспособленного для высвобождения активного ингредиента, либо в форме аэрозоля, содержащего тонко измельченные твердые вещества или жидкие капли. Могут быть использованы традиционные аэрозольные пропелленты, такие как летучие фторированные углеводороды или углеводороды, и аэрозольное устройство удобно приспособлено для высвобождения дозированного количества активного ингредиента.
Для дополнительной информации о фармацевтических препаратах см. главу 25.2 в Comprehensive Medicinal Chemistry, Volume 5, Corwin Hansch; Chairman of Editorial Board, Pergamon Press 1990, которая специально включена здесь посредством ссылки.
Количество соединения по этому изобретению, которое объединяют с одним или более чем одним эксципиентом для получения унифицированной лекарственной формы, обязательно будет варьировать в зависимости от организма "хозяина", которого лечат, и конкретного пути введения. Например, препарат, предназначенный для перорального введения человеку, будет содержать, например, от 0,5 мг до 2 г активного ингредиента с подходящим и удобным количеством эксципиентов, которое может варьировать от примерно 5 до примерно 98 мас.% общей композиции. Дозированные лекарственные формы обычно будут содержать от примерно 1 мг до примерно 50 мг активного ингредиента. Для дополнительной информации о путях введения и дозовых режимах см. главу 25.3 в Comprehensive Medicinal Chemistry, Volume 5, Corwin Hansch; Chairman of Editorial Board, Pergamon Press 1990, которая специально включена здесь посредством ссылки.
Размер дозы соединения Формулы I-XVII для терапевтических или профилактических целей будет естественно варьировать в зависимости от природы и тяжести состояний, возраста и пола животного или пациента и пути введения, в соответствии с правилами, хорошо известным в медицине.
В одном аспекте этого изобретения соединения по этому изобретению или их фармацевтически приемлемые соли или пролекарства могут быть приготовлены в форме фармацевтических композициях для введения животным или человеку для лечения или профилактики состояния, опосредованного р38. Термин "состояние, опосредованное р38", используемый здесь, означает любое заболевание или другое опасное состояние, в которых р38, как известно, играет роль. Он включает в себя состояния, которые, как известно, вызваны сверхпродукцией IL-1, TNF, IL-6 или IL-8. Такие состояния включают в себя, без ограничения, воспалительные заболевания, аутоиммунные заболевания, деструктивные костные нарушения, пролиферативные нарушения, инфекционные заболевания, вирусное заболевание и нейродегенеративные заболевания.
Воспалительные заболевания, которые можно лечить или предотвращать, включают в себя, но не ограничиваются ими, острый панкреатит, хронический панкреатит, астму, аллергии и респираторный дистресс-синдром взрослых.
Аутоиммунные заболевания, которые можно лечить или предотвращать, включают в себя, но не ограничиваются ими, гломерулонефрит, ревматоидный артрит, системную красную волчанку, склеродерму, хронический тиреоидит, болезнь Грейвса, аутоиммунный гастрит, инсулинзависимый сахарный диабет (тип I), аутоиммунную гемолитическую анемию, аутоиммунную нейтропению, тромбоцитопению, атопический дерматит, хронический активный гепатит, астенический бульбарный паралич, рассеянный склероз, воспалительную болезнь кишечника, язвенный колит, болезнь Крона, псориаз или болезнь "трансплантат против хозяина".
Деструктивные костные нарушения, которые можно лечить или предотвращать, включают в себя, но не ограничиваются ими, остеопороз, остеоартрит и костные нарушения, связанные с множественной миеломой.
Пролиферативные заболевания, которые можно лечить или предотвращать, включают в себя, но не ограничиваются ими, острую миелоидную лейкемию, хроническую миелоидную лейкемию, метастатическую меланому, саркому Калоши и множественную миелому.
Инфекционные заболевания, которые можно лечить или предотвращать, включают в себя, но не ограничиваются ими, сепсис, септический шок и шигеллез.
Вирусные заболевания, которые можно лечить или предотвращать, включают в себя, но не ограничиваются ими, острый инфекционный гепатит (включая гепатит А, гепатит В и гепатит С), ВИЧ-инфекцию и ЦМВ (цитомегаловирусный) ретинит.
Дегенеративные состояния или заболевания, которые можно лечить или предотвращать соединениями по этому изобретению, включают в себя, но не ограничиваются ими, болезнь Альцгеймера, болезнь Паркинсона, церебральную ишемию и другие нейродегенеративные заболевания.
"Состояния, опосредованные р38" также включают в себя ишемию/реперфузию при ударе, сердечные приступы, ишемию миокарда, гипоксию органов, сосудистую гиперплазию, гипертрофию сердца и тромбин-индуцированную агрегацию тромбоцитов.
Кроме того, ингибиторы р38 по этому изобретению также способны ингибировать экспрессию индуцибельных провоспалительных белков, таких как простагландин-эндопероксид-синтаза-2 (PGHS-2), также известная как циклооксигеназа-2 (СОХ-2). Поэтому другие "состояния, опосредованные р38", представляют собой отек, аналгезию, лихорадку и боль, такую как нервно-мышечная боль, головная боль, раковая боль, зубная боль и артритная боль.
Состояния и болезни, которые можно лечить или предотвращать ингибиторами р38 по этому изобретению, также могут быть удобно сгруппированы в соответствии с цитокином (например, IL-1, TNF, IL-6, IL-8), который, как предполагают, отвечает за это заболевание.
Так, болезнь или состояние, опосредованная(ое) IL-1, включает в себя ревматоидный артрит, остеоартрит, удар, эндотоксемию и/или токсический шок, воспалительную реакцию, вызванную эндотоксином, воспалительную болезнь кишечника, туберкулез, атеросклероз, мышечную дистрофию, кахексию, псориатический артрит, синдром Рейтера, подагру, травматический артрит, артрит, вызванный вирусом краснухи, острый синовит, диабет, заболевание панкреатических β-клеток и болезнь Альцгеймера.
Болезнь или состояние, опосредованная(ое) TNF, включает в себя ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие артритные состояния, сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, токсический шок, респираторный дистресс-синдром взрослых, церебральную малярию, хроническое обструктивное заболевание легких, силикоз, легочный саркоидоз, резорбцию кости, реперфузионное повреждение, реакцию "трансплантат против хозяина", отторжения аллотрансплантата, лихорадку и миалгии вследствие инфекции, кахексию, вторичную по отношению к инфекции, СПИД, СПИД-ассоциированный комплекс или злокачественность, келоидное образование, образование рубцовой ткани, болезнь Крона, язвенный колит или гипертермию. Болезни, опосредованные TNF, также включают в себя вирусные инфекции, такие как ВИЧ, ЦМВ, грипп и герпес, и вирусные инфекции в ветеринарии, такие как лентивирусные инфекции, включая, но не ограничиваясь ими, вирус конской инфекционной анемии, вирус артрита коз, вирус висна или вирус маэди (maedi), или ретровирусные инфекции, включая вирус иммунодефицита кошек, вирус иммунодефицита коров или вирус иммунодефицита собак.
Болезнь или состояние, опосредованная(ое) IL-8, включает в себя заболевания, характеризующиеся массивной инфильтрацией нейтрофилов, такие как псориаз, воспалительная болезнь кишечника, астма, реперфузионное повреждение сердца и почек, респираторный дистресс-синдром взрослых, тромбоз и гломерулонефрит.
Кроме того, соединения по этому изобретению могут быть использованы местно для лечения или профилактики состояний, вызванных или обостренных IL-1 или TNF. Такие состояния включают в себя воспаленные суставы, экзему, псориаз, воспалительные состояния кожи, такие как солнечный ожог, воспалительные состояния глаз, такие как конъюктивит, гипертермия, боль и другие состояния, связанные с воспалением.
Соединения по этому изобретению можно применять в комбинации с другими лекарствами и терапиями, используемыми для лечения болезненных состояний, для которых было бы полезно ингибирование цитокинов, в частности IL-1, TNF, 1L-6 или IL-8.
Например, благодаря своей способности ингибировать цитокины соединения Формулы I-XVII имеют большое значение при лечении некоторых воспалительных и невоспалительных заболеваний, которые в настоящее время лечат нестероидным противовоспалительным лекарством (NSAID), ингибирующим циклооксигеназу, таким как индометацин кеторолак, ацетилсалициловая кислота, ибупрофен, сулиндак, толметин и пироксикам. Совместное введение соединения Формулы I-XVII с NSAID может привести к снижению количества последнего агента, необходимого для достижения терапевтического эффекта, и соответственно к уменьшению вероятности неблагоприятных побочных эффектов от введения NSAID, таких как желудочно-кишечные эффекты. Таким образом, согласно еще одному признаку изобретения предложена фармацевтическая композиция, содержащая соединение Формулы I-XVII, или его фармацевтически приемлемую соль, или его сложный эфир, расщепляемый in vivo, в сочетании или в смеси с нестероидным противовоспалительным агентом, ингибирующим циклооксигеназу, и фармацевтически приемлемый разбавитель или носитель.
Соединения Формулы I-XVII также могут быть использованы в лечении состояний, таких как ревматоидный артрит, в комбинации с противоартритными агентами, такими как золото, метотрексат, стероиды и пенициллинамин, и таких состояний, как остеоартрит, в комбинации со стероидами.
Соединения по настоящему изобретению также можно вводить при деградационных заболеваниях, например остеоартрите, с хондрозащитными, противодеградационными и/или репаративными агентами, такими как диацерин (Diacerhein), препаратами гиалуроновой кислоты, такими как Гиалан, Румалон, Артепарон, и глюкозаминовыми солями, такими как Антрил.
Соединения Формулы I-XVII также можно применять при лечении астмы в комбинации с противоастматическими агентами, такими как бронходилататоры и антагонисты лейкотриенов.
Несмотря на то, что соединения Формулы I-XVII главным образом имеют большое значение в качестве терапевтических агентов для применения у теплокровных животных (включая человека), они также полезны всякий раз, когда требуется ингибировать эффекты цитокинов. Таким образом, они полезны в качестве фармакологических стандартов для применения при разработке новых биологических тестов и в поиске новых фармакологических агентов.
Активность соединений по этому изобретению может быть проанализирована в отношении ингибирования р38 in vitro, in vivo или в клеточной линии. Исследования in vitro включают в себя исследования, которые определяют ингибирование или киназную активность, или АТФазную активность активированной р38. Альтернативные исследования in vitro количественно определяют способность ингибитора связываться с р38 и могут быть измерены либо путем радиоактивного мечения ингибитора перед связыванием, выделением комплекса ингибитор/р38 и определением количества, связанного с радиоизотопом, либо путем проведения конкурентного эксперимента, в котором новые ингибиторы инкубируют с р38, связанной с известными радиолигандами. Эти и другие исследования полезны в анализах in vitro и в клеточных культурах и хорошо известны специалистам в данной области техники.
Анализы ингибиторного эффекта соединений по этому изобретению на клеточных культурах могут быть использованы для определения количеств TNF-α, IL-1, IL-6 или IL-8, продуцируемых в цельной крови или ее клеточных фракциях, в клетках, обработанных ингибитором, по сравнению с клетками, обработанными отрицательными контрольными веществами. Уровень этих цитокинов может быть определен путем использования имеющихся в продаже ELISA или, как описано в разделе Биологические Примеры, приведенном ниже.
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Биологические активности соединений по изобретению были продемонстрированы следующими анализами in vitro.
Биохимический анализ р38
Активность р38 анализировали при комнатной температуре в 100 мкл реакционной смеси, содержащей 5 нМ активированного фермента р38а и 1 мкМ ATF-2 (слитого белка активизирующего фактора транскрипции 2) в качестве субстрата в 25 мМ HEPES (рН 7,4), 100 мкМ ванадата, 1 мМ DTT, 10 мМ MgCl2 и 10 мкМ [□-33P]-АТФ (примерно 0,1 мкКи Р33/реакцию). Реакцию прерывали через 30-40 минут добавлением 25%-ной ТХУ, оставляли стоять в течение 5 минут и затем переносили непосредственно на GF-B мембранный фильтрационный планшет. Фильтр дважды промывали в течение 30 секунд 0,5%-ной фосфорной кислотой с использованием Tomtec Mach III Automated Harvester. После промывки вакуум поддерживали в течение 30 секунд для того, чтобы высушить фильтр. В каждую лунку фильтрационного планшета добавляли примерно 30 мкл сцинцилляционной жидкости и затем снимали показания в жидкостном сцинцилляционном счетчике (Packard TopCount HTS).
Анализ РВМС
Способность соединений по этому изобретению ингибировать продукцию TNF-α оценивали, используя мононуклеарные клетки периферической крови человека (РВМС), которые синтезируют и секретируют TNF-α при стимуляции липополисахаридом.
Растворы исследуемого соединения готовили, делая 5-кратные серийные разведения в ДМСО, которые затем разводили до 5-кратных маточных растворов путем разведения MEM, 2%-ной инактивированной нагреванием фетальной телячьей сывороткой (FBS), 20 мМ HEPES, 2 мМ L-глутамина и 1% пенициллин/стрептомицина.
РВМС выделяли из человеческой крови, как описано ниже. Образцы цельной крови собирали от волонтеров в Vacutainer™ CPT (Becton Dickinson). Содержимое пробирок, перемешивали и центрифугировали при комнатной температуре (18-25°С) в горизонтальном роторе в течение минимум 15 минут при 1500-1800 RCF (относительная центробежная сила). Для каждого донора слои лейкоцитов объединяли в отдельную пробирку и дважды промывали фосфатно-солевым буфером (PBS). Клеточный осадок ресуспендировали в MEM, 2%-ной инактивированной нагреванием фетальной телячьей сыворотке (FBS), 20 мМ HEPES, 2 мМ L-глутамина и 1% пенициллин/стрептомицина.
Общее количество клеток определяли с использованием гемоцитометра и концентрацию клеточной суспензии доводили до 2×106 клеток/мл.
В каждую лунку 96-луночного планшета для клеточных культур добавляли 0,1 мл клеточной суспензии. Добавляли 30 мкл раствора исследуемого соединения и клетки инкубировали при 37°С в СО2-инкубаторе (5% CO2) в течение 1 часа. Затем в каждую лунку добавляли 20 мкл 7,5 нг/мл липополисахарида (ЛПС из Е.coli К-235) и клетки возвращали в инкубатор с 5% СО2 при 37°С на 16-20 часов. Клетки центрифугировали в течение 15 минут при 1100 RCF. Примерно 0,12 мл супернатанта переносили в чистый 96-луночный полипропиленовый планшет. Образцы анализировали либо сразу, либо хранили их при -80°С до тех пор, пока их не использовали для анализа. Уровни TNF-α определяли в каждом образце с использованием иммуноферментного анализа (ELISA) TNF-α человека, как описано ниже.
Уровни TNF-α определяли, используя следующую методику. Планшеты, покрытые антителами к TNF-α, приготавливали путем добавления в лунки 96-луночного планшета Immunol 4 (Immulon 4 ELISA Flat Bottom Plate; Dynex, номер по каталогу 011-010-3855) 150 мкл 2 мкг/мл очищенных мышиных моноклональных анти-TNF-α IgG в карбонат-бикарбонатном буфере (ВирН™ Carbonate-Bicarbonate Buffer Pack) и инкубировали в течение ночи при 2-8°С. Покрывающий раствор удаляли и добавляли 200 мкл "блокирующего буфера" (20 мМ HEPES, рН 7,4, 150 мМ NaCl, 2% БСА), планшеты хранили при 2-8°С до до использования. Десятиточечную стандартную кривую для рекомбинантного человеческого TNF-α получали путем серийных разведении (1:2) в "разбавителе для образцов" (20 мМ HEPES, рН 7,4, 150 мМ NaCl, 2 мМ MgCl2, 1% БСА) с максимальной концентрацией 6000 пг/мл.
Блокирующий раствор удаляли из планшетов для иммуноферментного анализа TNF-α путем 5-кратной промывки по 300 мкл "буфера для промывки" (20 мМ HEPES, рН 7,4, 150 мМ NaCl, 2 мМ MgCl2, 0,02% Tween-20). Во все лунки добавляли 50 мкл "разбавителя для образцов", а затем во все лунки добавляли или 50 мкл стандартного раствора TNF-α, или супернатант исследуемого соединения. Планшет инкубировали при комнатной температуре в течение 1 часа при перемешивании (300 об/мин). Планшет промывали 5 раз по 300 мкл "буфера для промывки". В каждую лунку добавляли 100 мкл 0,2 мкг/мл биотинилированного козлиного анти-TNF-α человека в "разбавителе антител" (20 мМ HEPES, рН 7,4, 150 мМ NaCl, 2 мМ MgCl2, 1% БСА, 0,02% Tween-20) и планшет инкубировали при комнатной температуре в течение 1 часа при перемешивании (300 об/мин). Планшет промывали 5 раз по 300 мкл "буфера для промывки" на лунку. В каждую лунку добавляли по 100 мкл 0,02 мкг/мл стрептавидин-щелочной фосфатазы в "разбавителе для антител", и планшет инкубировали при комнатной температуре в течение 1 часа при перемешивании (300 об/мин). Планшет промывали 5 раз по 300 мкл "буфера для промывки" на лунку. В каждую лунку добавляли по 200 мкл 1 мг/мл pNPP (п-нитрофенилфосфата) в диэтаноламиновом буфере с 0,5 мМ MgCl2 и планшет инкубировали в течение 30-45 минут при комнатной температуре и перемешивании (300 об/мин). Ход реакции контролировали путем определения оптической плотности (OD): когда верхняя стандартная величина достигала значения OD между 2,0 и 3,0, в каждую лунку добавляли по 50 мкл 2 н. NaOH. Оптическую плотность в каждой лунке определяли в течение 30 минут, используя микротитрационный планшет-ридер, установленный на 405 нм. Данные анализировали в XL fit с использованием 4-параметрической аппроксимации кривой.
В описанных выше исследованиях использовали следующие реагенты: фосфатно-солевой буфер Дульбекко без кальция или магния (Gibco, № по каталогу 14190); Минимальная поддерживающая среда Игла (MEM; Gibco, № по каталогу 11090); пенициллин-стрептомицин (Gibco, № по каталогу 15140); L-глутамин, 200 мМ (Gibco, № по каталогу 25030); HEPES, 1 М (Gibco, № по каталогу 15630); фетальная телячья сыворотка (FBS; HyClone, № по каталогу SH30070.03); липополисахариды из Escherichia coli K-235 (ЛПС; Sigma, № по каталогу L2018); анти-TNF-α, очищенные мышиные моноклональные IgG (R&D Systems, № по каталогу МАВ210); упаковка ВирН™ карбонат-бикарбонатного буфера (Pierce, № по каталогу 28382); HEPES (FW 238.3; Sigma, № по каталогу Н3575); NaCl (Sigma, № по каталогу S7653); бычий сывороточный альбумин (БСА; Jackson ImmunoReseach, № по каталогу 001-000-162); полиоксиэтилен 20 сорбитан монолаурат (Sigma, № по каталогу Р2287); гексагидрат хлорида магния (Sigma, № по каталогу М2670); рекомбинантный TNF-α человека (R&D Systems, № по каталогу 210ТА010); биотинилированные, аффинно очищенные, козлиные IgG против TNF-α (R&D Systems, № по каталогу BAF210); стрептавидин-щелочная фосфатаза (Jackson ImmunoResearch, № по каталогу 016-050-084); диэтаноламиновый субстратный буфер (Pierce, № по каталогу 34064); п-нитрофенилфосфат (Sigma, № по каталогу N2765).
В Таблице 3 показаны результаты ингибирования р38 и ингибирования ЛПС-индуцированной секреции TNF-α из мононуклеарных клеток периферической крови человека (РВМС). "Активное" соединение определено, как соединение, имеющее IC50 ниже 500 нМ.
Исследование мышей
Мышиная модель ЛПС-индуцированной продукции TNF-α
TNF-α индуцировали в мышах-самцах породы DBA-2J (из Jackson Laboratories) инъекцией 2 мг/кг липополисахарида (Sigma, St. Louis) в хвостовую вену. Через девяносто минут мышей, анестезированных изофлураном, обескровливали сердечной пункцией. Затем образцы крови оставляли свертываться в течение двух часов при 4°С и центрифугировали. Сыворотку отделяли в пробирки типа "эппендорф" для последующего анализа TNF-α. Анализ TNF-α проводили, используя набор для ELISA (Quantikine, MN), в соответствии с инструкциями, прилагаемыми к набору.
Соединение AR-00112190 получали с 10% ДМСО и 90% 20%-ного 2-гидроксил-п-циклодекстрина (HPCD). Соединение AR-00112190 является производным соединения 14д (см. Фиг.3), где А представляет собой изобутил. Затем соединение последовательно разбавляли растворителем (10% ДМСО, 90% 20%-ного HPCD) для получения концентраций, необходимых для меньших дозовых уровней. Соединение переходило в раствор с добавлением ДМСО, а затем "выходило" из раствора при добавлении 20%-ного HPCD. Поэтому соединения дозировали в виде суспензий. За 30 минут до инъекции ЛПС соединение AR-00112190 (10, 30 и 100 мг/кг) перорально вводили семи группам мышей-самцов породы DBA-2J (семь животных в группе).
Обработка соединением AR-00112190 (10, 30 и 100 мг/кг) также значительно уменьшала уровни TNF-α. AR-00112190 показало аналогичное ингибирование (42%), наблюдаемое в дозе 100 мг/кг (Таблица 4).
Результаты этого анализа продемонстрировали существенные положительные эффекты при 10, 30 и 100 мг/кг AR-00112190 (29%, 44% и 42%).
ПРЕПАРАТИВНЫЕ ПРИМЕРЫ
Следующие примеры включены для иллюстрации изобретения. Однако следует понимать, что эти примеры не ограничивают изобретение и предназначены только для того, чтобы предложить способ практического осуществления изобретения. Специалисты в данной области техники поймут, что описанные химические реакции могут быть легко адаптированы для получения целого ряда других ингибиторов р38 по изобретению, и альтернативные способы получения соединений по этому изобретению рассматриваются, как находящиеся в рамках изобретения. Например, синтез соединений по изобретению, для которых не приведены примеры, может быть успешно осуществлен посредством модификаций, очевидных для специалистов в данной области техники, например при помощи соответствующих защитных мешающих групп или путем использования других подходящих реагентов, известных в данной области техники и отличающихся от тех, которые описаны, и/или путем осуществления стандартных изменений реакционных условий. Альтернативно другие реакции, раскрытые здесь или известные из уровня техники, будут признаны применимыми для получения других соединений по изобретению.
ПРИМЕРЫ
В примерах, описанных ниже, если не указано иначе, все температуры определены в градусах Цельсия. Реагенты были приобретены у таких коммерческих поставщиков, как Aldrich Chemical Company, Lancaster, TCI или Maybridge, и, если не указано иначе, были использованы без дополнительной очистки. Тетрагидрофуран (ТГФ), N,N-диметилформамид (ДМФА), дихлорметан (ДХМ), толуол, диоксан и 1,2-дифторэтан были приобретены у фирмы Aldrich в надежно запечатанных бутылках и использованы в том виде, как они были получены.
Изложенные ниже реакции обычно проводили при положительном давлении азота или аргона или с использованием осушительного патрона (если не указано иначе) в безводных растворителях; реакционные колбы обычно были снабжены резиновыми мембранами для введения субстратов и реагентов через шприц. Стеклянную посуду сушили в сушильном шкафу и/или нагреванием.
Колоночную хроматографию выполняли на системе Biotage (производитель: Dyax Corporation), имеющей колонку с силикагелем, или на картридже с силикагелем SepPak (Waters).
Спектры1Н-ЯМР регистрировали на приборе Bruker, работающем при 300 МГц, или на приборе Varian, работающем при 400 МГц. Спектры1Н-ЯМР получали из
CDCl3 растворов (в млн-1) с использованием хлороформа в качестве эталона (7,25 млн-1). Другие растворители для ЯМР использовали в случае необходимости. Когда сообщается о мультиплетности пика, то используются следующие сокращения: s (синглет), d (дублет), t (триплет), m (мультиплет), br (уширенный), dd (дублет дублетов), dt (дублет триплетов). В тех случаях, когда приведены константы взаимодействия, они представлены в герцах (Гц).
Пример 1
Получение 5-(4-фторфенилсульфанил)-1-(4-метоксибензил)-1H-пиразоло[3,4-с]пиридина (7а)
На Фиг.1 показана реакционная схема синтеза соединений 7а, имеющих общую Формулу II. В этом примере описан синтез соединения 7а, где R представляет собой 4-метоксибензил и X представляет собой серу.
Стадия А: 1,285 г 2-хлор-4-метил-5-нитропиридина (соединения 1а) и 1,023 г 4-фторбензолтиола растворяли в 15 мл безводного ТГФ в атмосфере сухого азота. К этому раствору медленно добавляли 207 мг гидрида натрия (95% в масле). Затем реакционную смесь распределяли между EtOAc и 0,1 н. водным NaOH (для удаления любого количества непрореагировавшего тиола), а затем органический слой промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, элюируя градиентом: от 1:1 гексан/СН2Cl2 до 100% CH2Cl2 и получая 1,90 г соединения 2а.
Стадия В: Примерно 1,90 г соединения 2а и 1,88 г порошка железа добавляли к 20 мл уксусной кислоты в атмосфере сухого азота. Затем реакционную смесь нагревали до 90°С в течение примерно 45 минут с получением промежуточного продукта 3а. Примерно 1,90 г промежуточного продукта 3а и 1,160 г NaOH растворяли в 20 мл метанола в атмосфере сухого азота в течение примерно 3,5 часов, затем реакционную смесь охлаждали до температуры окружающей среды и перемешивали при температуре окружающей среды в течение 12 часов. Реакционную смесь концентрировали под вакуумом и затем распределяли между CH2Cl2 и водой. Затем слой CH2Cl2 промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом, получая соединение 4а.
Стадия С: 1,54 г соединения 4а (без дополнительной очистки) и 896 мг тетрафторбората аммония переносили в 10 мл раствора ацетона и воды (1:1). Затем реакционную смесь помещали на ледяную баню (0°С), к которой добавляли 600 мкл концентрированной HCl, а затем 514 мг нитрита натрия. Затем реакционную смесь перемешивали в течение примерно 45 минут, после чего формировался осадок промежуточного соединения 5а. Осадок собирали, сушили на воздухе и затем дополнительно сушили путем азеотропной перегонки из этанола и толуола, получая примерно 800 мг соединения 5а. Примерно 800 мг соединения 5а (без дополнительной очистки), 312 мг ацетата калия и 190 мг 18-краун-6 растворяли/суспендировали в 5 мл хлороформа в атмосфере сухого азота. Затем реакционную смесь распределяли между CH2Cl2 и водой. Слой CH2Cl2 промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая 388 мг соединения 6а.
Стадия D: 173,3 мг соединения 6а, 195 мг карбоната калия, 110 мкл 4-метоксибензилхлорида и 10,5 мг йодида натрия растворяли/суспендировали в 1 мл безводного ДМФА в атмосфере сухого азота. Реакционную смесь нагревали до 85°С в течение примерно 1,5 часов и затем охлаждали до температуры окружающей среды. Реакционную смесь распределяли между CH2Cl2 и водой, слой CH2Cl2 промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая примерно 100 мг соединения 7а.
Пример 2
Получение 1-аллил-5-(4-фторфенокси)-1Н-пиразоло[3,4-с]пиридина (14а)
На Фиг.2 показана реакционная схема синтеза соединения 14а, имеющего общую Формулу II.
Стадия А: В круглодонной колбе 4-фторфенол (соединение 8а; 1,3 мл, 2,0 ммоль) разбавляли 25 мл безводного ТГФ и реакционную смесь охлаждали на ледяной бане, пока медленно добавляли трет-бутоксид калия (12,0 мл, 12,0 ммоль). Затем добавляли 2-хлор-4-метил-5-нитропиридин (соединение 1а; 2,23 г, 12,5 ммоль), реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали и остаток разбавляли CH2Cl2. Органический слой промывали 1 н. раствором NaOH и рассолом, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали до темного остатка, который очищали на колонке с диоксидом кремния Biotage 40 М, элюируя смесью CH2Cl2/гексаны (50:50) и получая 2,84 г соединения 10а в виде белого твердого вещества.
Стадия В: В круглодонной колбе соединение 10а (2,6 г, 11 ммоль) разбавляли 40 мл EtOH, а затем добавляли Pd(OH)2 (230 мг, 2 ммоль) с последующим добавлением формиата аммония (3,3 г, 53 ммоль). Реакционную смесь нагревали до 80°С до тех пор, пока исходное вещество 10а не израсходуется, что определяли посредством ВЭЖХ. Реакционную смесь фильтровали через бумагу из стекловолокна и фильтрат концентрировали. Остаток разбавляли CH2Cl2, органический слой промывали насыщенным NaHCOз и рассолом, сушили над Na2SO4, фильтровали и концентрировали, получая 1,92 г соединения 11а в виде белого твердого вещества.
Стадия С: Соединение 11а превращали в соединение 13а согласно способу, описанному в стадии С Примера 1. Соединение 11а и тетрафторборат аммония переносили в раствор ацетона и воды (1:1). Затем реакционную смесь помещали на ледяную баню (0°С), добавляли концентрированную HCl, затем нитрит натрия, и формировался осадок. Осадок собирали, сушили на воздухе и затем дополнительно сушили путем азеотропной перегонки из этанола и толуола, получая промежуточное соединение 12а. Соединение 12а, ацетат калия и 18-краун-6 растворяли/суспендировали в хлороформе в атмосфере сухого азота. Затем реакционную смесь распределяли между CH2Cl2 и водой. Слой CH2Cl2 промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая соединение 13а.
Стадия D: В круглодонной колбе соединение 13а разбавляли 4 мл ДМФА, затем добавляли 22 мг NaH, и начиналось выделение пузырьков. После стабилизации добавляли 0,8 мл аллилбромида и смесь перемешивали в атмосфере азота при комнатной температуре. Реакционную смесь гасили водой и затем концентрировали. Остаток разбавляли CH2Cl2, органический слой промывали насыщенным бикарбонатом натрия и рассолом, концентрировали до пленки и сушили. Полученный остаток очищали на колонке Biotage 12 М с диоксидом кремния, элюируя смесью 4% EtOAc:CH2Cl2 и получая соединение 14а.
Пример 3
Получение 3-[5-(4-фторфенилокси)пиразоло[3,4-с]пиридин-1-ил]пропан-1,2-диола (15а)
На Фиг.3 показана реакционная схема синтеза соединения 15а, имеющего общую Формулу II. В круглодонной колбе 79 мг (0,3 ммоль) соединения 14а, полученного согласно Примеру 2, разбавляли 2 мл безводного CH2Cl2. В атмосфере азота добавляли триметиламин-N-оксид (27 мг, 0,35 ммоль). После того, как все твердые вещества растворялись, добавляли OsO4(11 мг, 0,04 ммоль) и реакционную смесь перемешивали при комнатной температуре. Затем реакционную смесь распределяли между CH2Cl2 и водой. Органический слой сушили над Na2SO4, фильтровали и затем концентрировали до пленки. Пленку очищали на колонке Biotage 12 М с диоксидом кремния, элюируя EtOAc и получая 82 мг соединения 15а.
Пример 4
Получение [5-(4-фторфенилокси)пиразоло[3,4-с]пиридин-1-ил]ацетальдегида (16а)
На Фиг.4 показана реакционная схема синтеза соединения 16а. 0,3 М раствор NaIO4 (2 мл) объединяли с 1 г силикагеля с получением суспензии. Суспензию разбавляли 3 мл СН2Cl2 и добавляли 82 мг (0,3 ммоль) соединения 15а, полученного согласно Примеру 3, в суспензию с 1 мл CH2Cl2, и эту суспензию перемешивали в течение 2 часов. Через 3 часа реакционную смесь фильтровали и наполнитель промывали CH2Cl2. Фильтрат концентрировали, получая 35 мг соединения 16а в виде коричневой пленки.
Пример 5
Получение 5-(4-фторфенилокси)-1-оксазол-5-илметил-1Н-пиразоло[3,4-с]пиридина (17а)
На Фиг.5 показана реакционная схема синтеза соединения 17а, имеющего общую Формулу II. В круглодонной колбе соединение 16а (32 мг, 0,11 ммоль), полученное согласно Примеру 4, объединяли с МеОН (2 мл) и К2СО3(32 мг, 0,2 ммоль), затем добавляли тозилметилизоцианид (25 мг, 0,13 ммоль) и реакционную смесь нагревали до температуры дефлегмации. Затем реакционную смесь концентрировали, остаток разбавляли CH2Cl2. СН2Cl2промывали водой и 1 н. HCl, отделяли и концентрировали. Полученный остаток очищали на колонке с диоксидом кремния, элюируя смесью 80% EtOAc/CH2Cl2 и получая соединение 17а.
Пример 6
Получение 1-аллил-5-(4-фторфенилсульфанил)-1Н-пиразоло[3,4-с]пиридина (18а)
На Фиг.6 показана реакционная схема синтеза соединения 18а, имеющего общую Формулу II. В круглодонной колбе с входным отверстием для азота соединение 6а, полученное согласно Примеру 1, разбавляли 4 мл ДМФА, затем добавляли 22 мг NaH, и начиналось выделение пузырьков. После стабилизации добавляли аллилбромид (0,8 мл) и реакцию перемешивали в атмосфере азота при комнатной температуре. Реакционную смесь гасили водой и затем концентрировали. Остаток переносили в CH2Cl2 и промывали насыщенным раствором бикарбоната натрия и рассолом, затем сушили до оранжевой пленки. Пленку очищали на колонке Biotage 12 М с диоксидом кремния, элюируя смесью 4% EtOAc/СН2Cl2 и получая соединение 18а.
Пример 7
Получение 1-N-замещенных 7-азаиндазолов (7b)
На Фиг.7 показана реакционная схема синтеза соединения 7b, имеющего общую Формулу III.
Стадия А: В круглодонной колбе 4-фторбензолтиол разбавляли безводным ТГФ. Реакционную смесь охлаждали до 0°С на ледяной бане и затем к реакционной смеси медленно добавляли 1,0 М трет-бутоксид калия в ТГФ. Реакционную смесь перемешивали при 0°С в течение 10 минут, затем добавляли 5-хлор-3-метил-2-нитропиридин (соединение 1b), реакционную смесь перемешивали при 0°С в течение 10 минут и затем нагревали до комнатной температуры. Реакционную смесь концентрировали, остаток разбавляли СН2Cl2. CH2Cl2 промывали 1 н. раствором NaOH и рассолом, сушили над Na2SO4, фильтровали и фильтрат концентрировали до желтого масла. Полученный остаток очищали на колонке Biotage 40 М, элюируя смесью гексан/СН2Cl2 (50:50) и получая соединение 3b.
Стадия В: Соединение 3b восстанавливали порошком железа и уксусной кислотой, как описано в Примере 1, стадия В, получая соединение 4b.
Стадия С: Соединение 4b затем обрабатывали тетрафторборатом аммония с последующей обработкой концентрированной HCl и нитритом натрия, как описано в Примере 1, стадия С, получая промежуточное соединение 5b. Без дополнительной очистки соединение 5b объединяли с ацетатом калия и 18-краун-6, как описано в Примере 1, стадия С, получая соединение 6b.
Стадия D: Каждое из соединений 7b-1, 7b-2 и 7b-3 получали из соединения 6b, как показано на Фиг.7. Для получения соединения 7b-1 соединение 6b обрабатывали NaH и аллилбромидом, как описано в Примере 6.
Пример 8
Получение 3-[5-(4-фторфенилсульфанил)пиразоло[4,3-b]пиридин-1-ил]пропиламина (8b)
На Фиг.8 показана реакционная схема синтеза соединения 8b, имеющего общую Формулу III. В круглодонной колбе соединение 7b-3, полученное согласно Примеру 7, разбавляли CH2Cl2 и трифторуксусной кислотой. Реакционную смесь перемешивали до тех пор, пока исходное вещество не израсходуется, что определяли методом ТСХ, и затем концентрировали, полученный остаток разбавляли СН2Cl2. СН2Cl2 промывали 1 н. NaOH и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученный остаток очищали на колонке Biotage 12 М с диоксидом кремния, элюируя смесью 10% MeOH/CH2Cl2/NH4OH и получая соединение 8b.
Пример 9
Получение 6-(4-фторфенилсульфанил)-3-(4-метоксибензил)-1Н-индазола (10с)
На Фиг.9 показана реакционная схема синтеза соединения 10с, имеющего общую Формулу IV.
Стадия А: В круглодонной колбе 6-нитроиндол (соединение 1с; 15,5 г, 95 ммоль) растворяли в 1,4-диоксане (400 мл). Добавляли NaOH (3,8 г, 95 ммоль) и реакционную смесь перемешивали в течение 10 минут.Затем к реакционной смеси добавляли 266 мл 2 н. NaOH с последующим добавлением кристаллов йода (две порции, 54,4 г для общего добавления 214 ммоль) и реакционную смесь перемешивали в течение 12 часов. Реакционную смесь гасили 10%-ной лимонной кислотой и разбавляли EtOAc. Органический слой промывали 10%-ным NaHSO3, NaHCO3 и рассолом, сушили над Na2SO4, фильтровали и концентрировали, получая 27,5 мг соединения 2с в виде оранжевого твердого вещества.
Стадия В: Соединение 2с (5,18 г) растворяли в 50 мл безводного ТГФ в атмосфере сухого азота. К этому раствору добавляли 18,8 мл 1,0 М раствора трет-бутоксида калия в ТГФ. Реакционную смесь перемешивали в течение примерно 15 минут, после чего добавляли 3,20 мл хлортриметилсилана. Затем реакционную смесь распределяли между EtOAc и насыщенным водным NaHCO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая 3,85 г соединения 3с в виде желтого твердого вещества.
Стадия С: Соединение 3с (3,85 г), 7,66 г транс-2-фенилвинилбороновой кислоты, 531 мг Pd(PPh3)4 и 14,20 мл 2,0 М Na2CO3 растворяли/суспендировали в 50 мл диоксана в атмосфере сухого азота. Реакционную смесь нагревали до температуры дефлегмации в течение ночи, затем охлаждали до температуры окружающей среды и концентрировали под вакуумом. Полученный остаток распределяли между CH2Cl2 и водой. Слой CH2Cl2 сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая соединение 4с.
Стадия D: Соединение 4с (573 мг) и 103 мг 10% Pd/C растворяли/суспендировали в 10 мл раствора EtOH/ТГФ (3:1) в атмосфере сухого азота. К этому раствору добавляли 500 мкл гидразина и реакционную смесь перемешивали в течение 2 часов при температуре окружающей среды. Затем реакционную смесь фильтровали через целит, целит промывали EtOH и CH2Cl2, фильтрат концентрировали под вакуумом. Полученный остаток распределяли между CH2Cl2 и водой. Слой CH2Cl2 промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом, получая соединение 5с.
Стадия Е: Соединение 5с (2,51 г) растворяли в растворе 30 мл уксусной кислоты и 6 мл воды в атмосфере сухого азота. К этой реакционной смеси добавляли 3,2 мл концентрированной HCl. Реакционную смесь затем охлаждали до 0°С и добавляли 535 мг нитрита натрия. Затем реакционную смесь перемешивали в течение примерно 30 минут, после чего к реакционной смеси добавляли 4,0 мл водного раствора 1,23 мг йодида натрия и 885 мг йода. Примерно через 4 часа реакционную смесь гасили водным насыщенным NaHCO3 (медленное добавление) и затем распределяли между
CH2Cl2 и водой. Слой CH2Cl2 сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая 1,90 г соединения 6с.
Стадия F: Соединение 6с (1,90 г) и 509 мг дигидрата триметиламин-N-оксида растворяли в 30 мл CH2Cl2 в атмосфере сухого азота. К этой реакционной смеси добавляли 51 мг тетроксида осмия. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Добавляли периодат натрия (1,71 г), растворенный примерно в 30 мл воды, и реакционную смесь перемешивали в течение 1 часа. Затем реакционную смесь распределяли между EtOAc и водой. Слой EtOAc промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая 889 мг соединения 7с.
Стадия G: Соединение 7с (460 мг) добавляли к 10 мл безводного ТГФ в атмосфере сухого азота. Смесь охлаждали до -78°С и затем добавляли 2,80 мл 4-метоксифенилмагнийбромида в ТГФ (0,5 М). Реакционную смесь медленно нагревали до комнатной температуры, гасили водой и распределяли между EtOAc и насыщенным водным NaHCO3. Органический слой сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая 320 мг промежуточного продукта. Промежуточный продукт (151 мг) растворяли в 1 мл CH2Cl2 и 60 мкл триэтилсилана в атмосфере сухого азота. К этой реакционной смеси добавляли 1 мл трифторуксусной кислоты. Затем реакционную смесь концентрировали под вакуумом и остаток распределяли между CH2Cl2 и водным насыщенным NaHCO3. Слой CH2Cl2сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, элюируя градиентом: от 10:1 гексан/СН2Cl2 до 100% CH2Cl2, и получая 76,6 мг соединения 8с.
Стадия Н: Соединение 8с (151 мг), 80 мкл 4-фторфенилтиола, 12,0 мг порошка меди и 300 мкл 5,0 М водного NaOH добавляли к 1 мл безводного ДМФА в запаянной пробирке и затем нагревали до 90°С в течение 16 часов. Реакционную смесь распределяли между CH2Cl2 и 1,0 М водным NaOH. Слой СН2Cl2 промывали 1,0 М водным NaOH, 3,0 н. водным NH4OH и рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали на колонке Biotage, получая 76,6 мг соединения 9с.
Стадия I: Соединение 9с (76,6 мг) и 100 мкл этилендиамина растворяли в 1,6 мл 1,0 М раствора фторида тетрабутиламмония в ТГФ в атмосфере сухого азота. Реакционную смесь нагревали до температуры дефлегмации в течение примерно 12 часов. Затем реакционную смесь охлаждали до комнатной температуры и распределяли между CH2Cl2 и водой. Слой СН2Cl2 промывали 10%-ной водной лимонной кислотой и насыщенным водным NaHCO3, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали на колонке Biotage, элюируя градиентом: от 5:1 гексан/СН2Cl2 до 100% СН2Cl2, получая 25 мг соединения 10с.
Пример 10
Получение [6-(4-фторфенилсульфанил)-1Н-индазол-3-ил]метанола (14с)
На Фиг.10 показана реакционная схема синтеза соединения 14с, имеющего общую Формулу IV.
Стадия А: Соединение 7с (520 мг), полученное согласно Примеру 9, растворяли в 5 мл метанола в атмосфере сухого азота. К этому раствору добавляли 98,3 мг борогидрида натрия. Примерно через 30 минут реакционную смесь концентрировали под вакуумом и затем распределяли между СН2Cl2 и водой. Слой CH2Cl2 сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая соединение 12с.
Стадия В: Соединение 12с (151 мг), метанол, 100 мкл 4-фторфенилтиола, 6,0 мг порошка меди и 250 мкл 5,0 М водного NaOH добавляли к 1 мл безводного ДМФА в запаянной пробирке и затем нагревали до 90°С в течение примерно 30 часов, после чего реакционную смесь охлаждали до температуры окружающей среды и распределяли между CH2Cl2 и 1,0 М водным NaOH. Слой CH2Cl2 промывали 1,0 М водным NaOH, 3,0 н. водным NH4OH и рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, элюируя смесью СН2Cl2/EtOAc (5:1) и получая 67,9 мг соединения 13с.
Стадия С: Соединение 13с (67,9 мг) и 100 мкл этилендиамина растворяли в 1,5 мл фторида тетрабутиламмония в ТГФ (1,0 М) в атмосфере сухого азота. Реакционную смесь нагревали до температуры дефлегмации в течение примерно 12 часов, затем охлаждали до комнатной температуры и распределяли между CH2Cl2 и водой. Слой
CH2Cl2 промывали 10%-ной водной лимонной кислотой и насыщенным водным NaHCO3, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая 18 мг соединения 14с.
Пример 11
Получение 6-(4-фторфенилсульфанил)-3-метоксиметил-1Н-индазола (17с)
На Фиг.11 показана реакционная схема получения соединения 17с, имеющего общую Формулу IV.
Стадия А: Соединение 12с (186 мг), полученное согласно Примеру 10, стадия А, растворяли в 5 мл безводного ТГФ в атмосфере сухого азота. К этому раствору добавляли 36,8 мг гидрида натрия (60% в масле), реакционную смесь перемешивали в течение примерно 15 минут и затем к реакционной смеси добавляли 60 мкл метилйодида. Примерно через 1 час реакционную смесь гасили водой и распределяли между СН2Cl2 и водным насыщенным NaHCO3. Слой CH2Cl2 сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, элюируя смесью СН2Cl2/EtOAc (100:1) и получая 76,1 мг соединения 15с.
Стадия В: Соединение 15 с подвергали взаимодействию с 4-фтортиофенолом, порошком меди и водным NaOH в ДМФАтем же способом, который описан в стадии В Примера 10, получая соединение 16с с выходом 22%.
Стадия С: Соединение 16с подвергали взаимодействию с фторидом тетрабутиламмония и этилендиамином в ТГФ тем же способом, который описан в стадии С Примера 10, получая соединение 17с с выходом 53%.
Пример 12
Получение 6-(4-фторфенокси)-3-метил-1Н-индазола (18с-2)
На Фиг.12 показана реакционная схема синтеза соединений, имеющих общую структуру 18с и общую Формулу IV. В этом примере описан синтез соединения 18с-2, где Ar представляет собой 4-фторфенил.
Стадия А: 2-Фтор-4-гидроксиацетофенон (соединение 19с; 1,42 г) и 1,40 г карбоната калия растворяли/суспендировали в 30 мл безводного ДМФА в атмосфере сухого азота. К этой реакционной смеси добавляли 1,20 мл бензилбромида. Примерно через 90 минут реакционную смесь нагревали до 65°С в течение примерно 45 минут и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали под вакуумом, остаток распределяли между СН2Cl2 и водой. Слой CH2Cl2 промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом, получая соединение 20с.
Стадия В: Соединение 20с (1,87 г) добавляли к 20 мл этиленгликоля в атмосфере сухого азота. К этой реакционной смеси добавляли 250 мкл безводного гидразина. Смесь перемешивали в течение 1 часа при комнатной температуре и затем нагревали до 160°С в течение примерно 7 часов. Затем реакционную смесь охлаждали до комнатной температуры и гасили водой. Выпавшую в осадок соль собирали и сушили на воздухе, а затем дополнительно сушили путем азеотропной перегонки из этанола и толуола. Выпавшую в осадок соль разбавляли безводным ацетонитрилом и затем добавляли 500 мг диметиламинопиридина и 311 мг ди-трет-бутилдикарбоната (Вос-ангидрида). После растворения всех твердых веществ реакционную смесь концентрировали под вакуумом и полученный остаток очищали на колонке Biotage, получая 710 мг соединения 21с.
Стадия С: Соединение 21с (710 мг), 662 мг формиата аммония и 223 мг катализатора Перлмана (Pearlman, Pd(OH)2/C) растворяли/суспендировали в 20 мл этанола в атмосфере сухого азота. Реакцию нагревали до 85°С в течение примерно 30 минут и затем фильтровали через целит. Целит промывали EtOH и объединенные фильтраты концентрировали под вакуумом. Полученный остаток распределяли между CH2Cl2 и насыщенным водным NaHCO3, сушили над Na2SO4, фильтровали и концентрировали под вакуумом, получая соединение 22с.
Стадия D: Соединение 22с (103 мг), 174 мг 4-фторфенилбороновой кислоты, 75 мг ацетата меди (II) и 300 мкл триэтиламина растворяли/суспендировали в 2 мл безводного СН2Cl2, и к этому раствору добавляли 4Å молекулярные сита. Реакционную смесь подвергали воздействию воздуха в течение примерно 5 часов и затем фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, элюируя СН2Cl2 и получая 85 мг соединения 23с.
Стадия Е: Соединение 23с (85 мг) растворяли в 2 мл раствора СН2Cl2/ТФУ (1:1) в атмосфере сухого азота. Реакционную смесь перемешивали в течение примерно 30 минут, после чего ее концентрировали под вакуумом. Полученный остаток распределяли между CH2Cl2 и водным насыщенным NaHCO3. Слой CH2Cl2 сушили над Na2SO4, фильтровали и концентрировали под вакуумом, получая 18с-2.
Для получения других соединений, имеющих общую структуру 18с, соединение 22с подвергали взаимодействию с фенилборатом или соответствующим образом замещенным фенилборатом, как описано в стадии D, а затем обрабатывали, как описано в стадии Е.
Пример 13
Получение 3-этил-6-(4-фторфенилсульфанил)-1Н-индазола (26с)
На Фиг.13 показана реакционная схема синтеза соединения 26с, имеющего общую Формулу IV.
Стадия А: 4-Фтортиофенол (соединение 24с; 900 мкл) растворяли в 40 мл безводного ТГФ в атмосфере сухого азота. К этому раствору добавляли 8,40 мл трет-бутоксида калия в ТГФ (1,0 М), а затем 10 мл безводного ДМФА. Реакционную смесь перемешивали при температуре окружающей среды в течение 10 минут, после чего добавляли 1,43 г 2,4-дифторпропиофенона и смесь оставляли реагировать в течение примерно 12 часов при комнатной температуре. Затем реакционную смесь распределяли между Et2O и водой. Слой Et2O промывали насыщенным водным
NaHCO3, сушили над Na2SO4, фильтровали и концентрировали под вакуумом, получая соединение 25с.
Стадия В: Соединение 25с (2,34 г) и 260 мкл безводного гидразина суспендировали/растворяли в этиленгликоле в атмосфере сухого азота. Затем реакционную смесь нагревали до примерно 70°С в течение примерно часа, а затем до примерно 160°С в течение примерно 12 часов. Реакционную смесь охлаждали до комнатной температуры и гасили примерно 100 мл воды, затем распределяли между CH2Cl2 и водой. Слой CH2Cl2 промывали водой и водным насыщенным NaHCO3, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая 770 мг соединения 26с.
Пример 14
Получение 5-(4-фторфенокси)-1Н-индазол-3-иламина (34с)
На Фиг.14 показана реакционная схема синтеза соединения 34с, имеющего общую Формулу VI.
Стадия А: В круглодонной колбе к 5-фтор-2-нитробензойной кислоте (соединение 27с; 10,0 г, 54,0 ммоль) добавляли 50 мл МеОН и 200 мл толуола. Добавляли примерно 41 мл триметилсилилдиазометана (2,0 М) медленно, при перемешивании. После прекращения выделения пузырьков реакцию гасили 1 мл уксусной кислоты. Реакционную смесь концентрировали в вакууме, получая соединение 28с.
Стадия В: В круглодонной колбе 4-фторфенол (4,0 г, 35 ммоль) разбавляли 100 мл безводного ТГФ. Реакционную смесь охлаждали до 0°С на ледяной бане и затем медленно добавляли 1,0 М трет-бутоксид калия в ТГФ (35 мл, 35 ммоль). Реакционную смесь перемешивали в течение 10 минут и затем добавляли соединение 28с (7,4 г, 37 ммоль) в 50 мл ТГФ. Реакционную смесь перемешивали при 0°С в течение 10 минут и затем нагревали до комнатной температуры и перемешивали в течение примерно 12 часов. Реакционную смесь концентрировали, остаток разбавляли CH2Cl2. CH2Cl2промывали 1 н. NaOH и рассолом, сушили над Na2SO4, фильтровали и концентрировали до масла. Масло очищали на колонке Biotage 40 М, элюируя смесью гексан/СН2Cl2 (50:50) и получая соединение 29с в виде масла.
Стадия С: В круглодонной колбе соединение 29с (40 г, 13 ммоль) добавляли к 60 мл МеОН с последующим добавлением 6 н. NaOH (4,3 мл, 26 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем концентрировали и полученный остаток разбавляли 50 мл воды. Добавляли примерно 5 мл 2 н. раствора HCl (рН равно 2,0), и из раствора осаждалось твердое вещество. Твердое вещество растворяли в CH2Cl2, органический слой промывали рассолом, сушили над Na2SO4, фильтровали и затем концентрировали в толуоле, получая соединение 30с в виде белого твердого вещества.
Стадия D: В круглодонной колбе соединение 30с растворяли в 40 мл тионилхлорида и нагревали до 90°С в течение 2 часов. Реакционную смесь охлаждали и затем концентрировали до желтоватого твердого вещества. Твердое вещество растворяли в 20 мл ацетона и охлаждали до 0°С на ледяной бане, затем очень медленно добавляли 10 мл NH4OH. Реакционную смесь гасили водой и затем концентрировали. Полученный остаток экстрагировали CH2Cl2, СН2Cl2 сушили над Na2SO4 и концентрировали, получая соединение 31с.
Стадия Е: В круглодонной колбе соединение 31с (3,4 г, 12,3 ммоль) растворяли в 100 мл дихлорэтана, затем добавляли оксалилхлорид (5,4 мл, 62 ммоль) и реакционную смесь нагревали до 55°С в течение 2 часов. Реакционную смесь концентрировали, полученное масло перемешивали в воде (50 мл) и затем охлаждали до примерно 0°С на ледяной бане (по мере того, как медленно добавляли NH4OH для гашения избытка оксалилхлорида). Реакционную смесь экстрагировали CH2Cl2, органический слой сушили над Na2SO4, фильтровали и концентрировали, получая соединение 32с в виде темного масла.
Стадия F: В круглодонной колбе соединение 32с (2,21 г, 8,5 ммоль) разбавляли 100 мл EtOH и затем добавляли Pd(OH)2 (300 мг) с последующим добавлением формиата аммония (2,7 г, 43 ммоль). Реакционную смесь нагревали до температуры дефлегмации в течение 18 часов, фильтровали через бумагу из стекловолокна для удаления Pd и бумагу промывали EtOH. Фильтрат концентрировали, полученный остаток переносили в CH2Cl2 и промывали насыщенным бикарбонатом натрия и рассолом, сушили над Na2SO4, фильтровали и концентрировали, получая соединение 33с в виде желтого твердого вещества.
Стадия G: Соединение 33с (280 мг, 1,3 ммоль) помещали в круглодонную колбу на баню с ледяной водой и добавляли 5 мл НОАс и 2,5 мл Н2О. Реакционную смесь поддерживали при температуре 0°С, добавляли HCl (0,35 мл, 6 ммоль) и через 5 минут добавляли NaNO2 (93 мг, 1,3 ммоль). Примерно через 1 час добавляли дигидрат хлорида олова (II) (554 мг, 2,5 ммоль) и реакционную смесь перемешивали в течение 30 минут. Затем реакционную смесь нагревали до комнатной температуры, концентрировали и остаток переносили в СН2Cl2. Органический слой промывали водой и рассолом, фильтровали, сушили над Na2SO4, фильтровали и концентрировали до пленки. Пленку растирали с CH2Cl2 и твердые вещества собирали. Эти твердые вещества затем нагревали в 1-бутаноле (120°С) в трубке под давлением в течение 12 часов для индукции циклизации, затем реакцию охлаждали и твердое вещество собирали фильтрацией с получением соединения 34с.
Пример 15
Получение N-[6-(4-фторфенокси)-1Н-индазол-3-ил]ацетамида (38с-1)
На Фиг.15 показана реакционная схема синтеза соединений 38с, имеющих общую Формулу IV. В этом примере описан синтез соединения 38с-1, где X представляет собой кислород.
Стадия А: 2-Фтор-4-гидроксибензонитрил (соединение 35с-1; 1,40 г), 2,86 г 4-фторфенилбороновой кислоты, 1,86 г ацетата меди (II) и 7,20 мл триэтиламина растворяли/суспендировали в 100 мл безводного CH2CI2, к этой реакционной смеси добавляли 4Å молекулярные сита. Реакционную смесь подвергали воздействию воздуха через осушительный патрон и перемешивали при температуре окружающей среды в течение 16 часов. Реакционную смесь фильтровали, фильтрат промывали 10%-ным водным NaHSO4, 1 н. водным NaOH и рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом, получая 530 мг соединения 36с-1.
Стадия В: Соединение 36с-1 (208 мг) и 150 мкл безводного гидразина растворяли в 5 мл бутанола. Реакционную смесь нагревали до температуры дефлегмации в атмосфере сухого азота в течение 15 часов, затем охлаждали до температуры окружающей среды, концентрировали под вакуумом и растирали с этиловым эфиром. Полученное розовое твердое вещество (соединение 37с-1) собирали фильтрацией, промывали этиловым эфиром и затем сушили на воздухе.
Стадия С: Соединение 37с-1 (97 мг) и 40 мкл уксусного ангидрида суспендировали/растворяли в дихлорэтане в атмосфере сухого азота. Реакционную смесь нагревали до 60°С в течение примерно 1 часа, затем охлаждали до комнатной температуры и перемешивали в течение 12 часов. Белый осадок (соединение 38с-1) собирали фильтрацией в вакууме и затем сушили на воздухе.
Пример 16
Получение 2-[6-(4-фторфенокси)-1H-индазол-3-ил]изоиндол-1,3-диона (39с)
На Фиг.16 показана реакционная схема синтеза соединения 39с, имеющего общую Формулу VI.
Соединение 37с-1 (660 мг) и 654 мг N-карбоэтоксифталимида суспендировали/растворяли в 15 мл дихлорэтана в атмосфере сухого азота при комнатной температуре в течение примерно 13 часов. Примерно через 20 минут реакционную смесь нагревали до 65°С в течение примерно 5,5 часов, после чего ее охлаждали до комнатной температуры и фильтровали. Белый осадок (соединение 39с) промывали дихлорэтаном и затем сушили на воздухе.
Пример 17
Получение 3-(1,3-дигидроизоиндол-2-ил)-6-(4-фторфенокси)-1Н-индазола (40с)
На Фиг.17 показана реакционная схема синтеза соединения 40с, имеющего общую Формулу VI. Соединение 39с (25 мг), полученное согласно Примеру 16, суспендировали в 1 мл безводного ТГФ в атмосфере сухого азота. К этому раствору добавляли 1,0 мл 1,0 М раствора ВН3 в ТГФ. Реакционную смесь перемешивали при комнатной температуре в течение примерно 1 часа и затем нагревали до температуры дефлегмации в течение 2 часов. Затем реакционную смесь охлаждали до комнатной температуры и осторожно добавляли 2,0 мл метанола. Смесь перемешивали в течение примерно 10 минут и затем концентрировали под вакуумом. Полученный остаток очищали на колонке Biotage, получая 5 мг соединения 40с.
Пример 18
Получение 5-(4-фторфенилсульфанил)-1Н-индазола (4d)
На Фиг.18 показана реакционная схема синтеза соединения 4d, имеющего общую Формулу VII.
Стадия А: Смесь 6-йод-1Н-индазола (соединение 1d) в CH3CN (11 мл) обрабатывали триэтиламином и диметиламинопиридином. После охлаждения до 0°С добавляли по каплям раствор ди-трет-бутилдикарбоната (Вос-ангидрида) в CH3CN (10 мл). После перемешивания при комнатной температуре в течение 3 часов реакционную смесь концентрировали в вакууме, полученный остаток распределяли между Н2О и эфиром. рН доводили 1 н. HCl до 2, органическую фазу отделяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме, получая соединение 2d в виде масла.
Стадия В: Смесь соединения 2d в ДМФА (25 мл) обрабатывали 5 н. КОН, порошком Cu и ArSH. В этом примере ArSH представлял собой 4-фтортиофенол. Реакционную смесь нагревали при 110°С в течение 48 часов, затем охлаждали до комнатной температуры, концентрировали в вакууме, подкисляли 1 н. HCl и экстрагировали в CH2Cl2. Органический слой фильтровали через бумагу 1PS, концентрировали в вакууме, полученный остаток очищали на колонке Biotage, элюируя 100% CH2Cl2, 5% Et2O/CH2Cl2 и затем 10% Et2O/CH2Cl2, получая соединение 4d.
Пример 19
Получение 5-(4-фторфенилсульфанил)-1-изопропил-1Н-индазола (5d-1)
На Фиг.19 показана реакционная схема синтеза соединений, имеющих общую структуру 5d и имеющих общую Формулу VII. В этом примере описан синтез соединения 5d-1, где R представляет собой изопропил.
Раствор соединения 4d, полученного согласно Примеру 18, в ТГФ (1 мл) обрабатывали порошкообразным КОН с последующим добавлением 18-краун-6 и RX. В этом примере RX представлял собой изопропилйодид. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере азота. Затем реакционную смесь разбавляли CH2Cl2 и фильтровали, фильтрат концентрировали в вакууме и остаток разбавляли CH2Cl2. Органический слой промывали насыщенным водным NaHCO3, фильтровали через бумагу 1PS и концентрировали в вакууме. Полученный остаток очищали на колонке Biotage, элюируя смесью гексан/Et2O (4:1) и получая соединение 5d-1 в виде желтого масла.
Пример 20
Получение 5-йод-1-(4-метоксибензил)-1Н-индазола (8d-1)
На Фиг.20 показана реакционная схема синтеза соединений 8d. В этом примере описан синтез соединения 8d-1, где Ar1 представляет собой 4-метоксифенил.
Стадия А: Суспензию 5-аминоиндазола (соединения 6d) в 6 М растворе HCl (150 мл) охлаждали до 0°С и обрабатывали по каплям раствором NaNO2 в воде (15 мл). После перемешивания при 0°С в течение 30 минут реакционную смесь добавляли к холодному раствору KI в воде (105 мл). Смесь оставляли нагреваться до комнатной температуры, и перемешивание продолжали при комнатной температуре в течение 18 часов. Смесь гасили 10%-ным Na2S2O3 и экстрагировали Et2O. Двухфазную смесь фильтровали, нерастворимые твердые вещества промывали водой и сушили в вакууме в течение ночи. Органическую фазу отделяли и дополнительно промывали водным насыщенным NaHCO3, водой, фильтровали через бумагу 1PS, упаривали в вакууме до розового остатка.
Стадия В: Раствор соединения 1d в ДМФА обрабатывали К2СО3 с последующим добавлением замещенного или незамещенного бензилгалогенида при комнатной температуре в атмосфере азота. В этом примере бензилгалогенид представлял собой 4-метоксибензилхлорид. Смесь нагревали при 100°С в течение 48 часов атмосфере азота. Смесь обрабатывали 0,2 экв. NaI (123 мг) и нагревание продолжали в течение 18 часов. Растворитель выпаривали в вакууме и остаток переносили в CH2Cl2 и 1 н. HCl. Органический слой отделяли, промывали водным насыщенным NaHCOз и концентрировали с получением масла. Масло очищали на колонке Biotage, элюируя градиентом гексан/Et2О (от 3:1 до 3:2) и получая соединение 8d-1.
Пример 21
Получение 5-(4-фторбензолсульфонил)-1-(4-метоксибензил)-1Н-индазола (10d-1)
На Фиг.21 показана реакционная схема синтеза соединений 10d, имеющих общую Формулу IX. В этом примере описан синтез соединения 10d-1, где Ar1 представляет собой 4-метоксифенил и Ar2 представляет собой 4-фторфенил.
Стадия А: Смесь соединения 8d, 5 н. КОН, порошка меди и Ar2SH в растворе воды и ДМФА нагревали с обратным холодильником в течение примерно 18 часов. В этом примере Ar2SH представлял собой 4-фтортиофенол. Затем смесь охлаждали до комнатной температуры, подкисляли 1 н. HCl и экстрагировали CH2Cl2. Органический слой фильтровали через бумагу 1PS, концентрировали в вакууме, полученный остаток очищали на картридже с силикагелем SepPak, элюируя смесью гексан/Et2O (4:1) и получая соединение 9d.
Стадия В: Раствор соединения 9d в ацетоне (0,2 мл), содержащий MgSO4, обрабатывали раствором NaIO4 и KMnO4 в воде (0,2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Затем реакционную смесь обрабатывали водным бисульфитом натрия и экстрагировали СН2Cl2. Органический слой фильтровали через бумагу 1PS и концентрировали в вакууме, получая 2,1 мг соединения 10d в виде желтого масла.
Пример 22
Получение 5-(4-фторбензолсульфинил)-1-(4-метоксибензил)-1Н-индазола
На Фиг.22 показана реакционная схема синтеза соединения 11d-1, имеющего общую Формулу VIII. Раствор соединения 9d-1, полученного согласно Примеру 21, в смеси вода/ацетонитрил (1:1) обрабатывали NaIO4 и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Затем реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Полученный остаток распределяли между водой и CH2Cl2. Органический слой отделяли, фильтровали через бумагу 1PS, концентрировали в вакууме и очищали на картридже с силикагелем SepPak, элюируя градиентом гексан/Et2O (4:1, 2:1 и 1:1) и получая соединение 11d-1.
Пример 23
Получение 1-бензолсульфонил-5-(4-фторфенилсульфанил)-1Н-индазола (13d)
На Фиг.23 показана реакционная схема получения соединения 13d, имеющего общую Формулу VII.
Стадия А: Раствор 5-йодиндазола (соединения 1d) в пиридине обрабатывали бензолсульфонилхлоридом при комнатной температуре в атмосфере азота в течение 18 часов. Реакционную смесь концентрировали в вакууме, остаток переносили в CH2Cl2 и 1 н. HCl. Органический слой отделяли, фильтровали через фильтровальную бумагу 1PS и концентрировали в вакууме. Полученный остаток очищали на колонке Biotage, элюируя смесью гексан/Et2O (5:1) и получая соединение 12d.
Стадия В: Смесь соединения 12d, 5 н. КОН, порошка меди и 4-фтортиофенола в растворе воды и ДМФА нагревали с обратным холодильником в течение примерно 18 часов. Затем смесь охлаждали до комнатной температуры, подкисляли 1 н. HCl и экстрагировали CH2Cl2. Органический слой фильтровали через бумагу 1PS, концентрировали в вакууме, полученный остаток очищали на картридже с силикагелем SepPak, элюируя смесью гексан/Et2O (4:1) и получая соединение 13d.
Пример 24
Получение 3-хлор-6-феноксибензо[d]изоксазола (8е-1)
На Фиг.24 показана реакционная схема синтеза соединений 8е, имеющих общую Формулу V. В этом примере описан синтез соединения 8е-1, где Ar2 представляет собой фенил.
Стадия А: Раствор 4-фтор-2-гидроксибензойной кислоты (соединение 1е) в МеОН медленно обрабатывали концентрированной H2SO4 и затем нагревали с обратным холодильником в течение 12 дней. Затем реакционную смесь концентрировали в вакууме до желтого масла и масло переносили в СН2Cl2. Органический слой промывали насыщенным водным NaHCO3, рассолом и водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая 12,7 г соединения 2е в виде масла янтарного цвета.
Стадия В: Раствор соединения 2е, К2СО3 и бензилхлорида в ДМФА (200 мл) нагревали при 95°С в течение 18 часов. Смесь фильтровали и фильтрат концентрировали в вакууме до желтого масла. Масло очищали на колонке Biotage, элюируя смесью гексан/EtOAc (7:2) и получая 19,4 г соединения 3е в виде прозрачного масла.
Стадия С: Раствор соединения 3е в ДМСО (2 мл) обрабатывали К2СО3 с последующим добавлением Ar2ОН при комнатной температуре в атмосфере азота. В этом примере Ar2ОН представлял собой фенол. Смесь нагревали при 90°С в течение 3 дней в атмосфере азота. Медленно добавляли воду (1 мл) и продукт экстрагировали EtOAc. Водный слой отделяли и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали в вакууме до темного масла. Масло очищали на колонке Biotage, элюируя смесью гексан/Et2О (6:1) и получая соединение 4е-1 в виде прозрачного масла.
Стадия D: 1,0 М раствор соединения 4е-1 в МеОН (30 мл) продували азотом и обрабатывали 20% Pd(OH)2/C (15 мас.% равно 297 мг). Через реакционную смесь пропускали дополнительное количество азота и затем перемешивали при комнатной температуре в течение 2 дней в атмосфере водорода. Катализатор отфильтровывали и промывали МеОН. Фильтрат упаривали в вакууме до прозрачного масла, которое очищали на колонке Biotage, элюируя смесью 5% Et2О/гексан и получая соединение 5е-1 в виде прозрачного масла.
Стадия Е: 3 М NaOH (9 мл) добавляли к раствору NH2OH·HCl в воде (14 мл) с последующим добавлением раствора соединения 5е-1 в диоксане (10 мл). Мутную смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере азота. Полученную прозрачную смесь охлаждали на ледяной бане, подкисляли 2 М HCl и экстрагировали EtOAc. Объединенные органические слои промывали рассолом, фильтровали через бумагу 1PS и упаривали в вакууме, получая 235 мг бежевого твердого вещества. Это твердое вещество растирали в смеси гексан/EtOAc (4:1) и полученное белое твердое вещество (соединение 6е-1) собирали фильтрацией.
Стадия F: Раствор карбонилдиимидазола в ТГФ добавляли к раствору соединения 6е-1 в ТГФ, который нагревали с обратным холодильником и это нагревание продолжали в течение 18 часов. Затем смесь концентрировали в вакууме, разбавляли водой, подкисляли 1 н. HCl и экстрагировали CH2Cl2. Органический слой фильтровали через бумагу 1PS и упаривали в вакууме, получая соединение 7е-1 в виде бледно-желтого твердого вещества или пены.
Стадия G: Суспензию соединения 7е-1 в POCl3 обрабатывали триэтиламином при комнатной температуре и смесь нагревали при 110°С в течение 6 часов. Смесь охлаждали до комнатной температуры и выливали в стакан, содержащий ледяную воду. Продукт экстрагировали CH2Cl2, фильтровали через бумагу 1PS и упаривали в вакууме, получая 10 мг соединения 8е-1 в виде масла янтарного цвета.
Пример 25
Получение 3,6-дифеноксибензо[d]изоксазола (9е-1)
На Фиг.25 показана реакционная схема синтеза соединений 9е, имеющих общую Формулу V. В этом примере описан синтез соединения 9е-1, где Ar1 представляет собой фенил и Ar2 представляет собой фенил. Раствор соединения 8е-1, полученного согласно Примеру 24, в ДМФА (1 мл) добавляли к смеси NaH и фенола (1 мл) в ДМФА. Реакционную смесь нагревали при 110°С в течение 18 часов. Растворитель выпаривали в вакууме и остаток распределяли между 1 н. HCl и СН2Cl2. Органический слой отделяли и фильтровали через бумагу 1PS. Выпаривание растворителя давало коричневое масло, которое очищали на картридже с силикагелем Sep Pak, элюируя смесью гексан/Et2O (4:1) и получая соединение 9е-1 в виде прозрачного масла, которое затвердевало с образованием длинных белых игл.
Пример 26
Получение (4-метоксифенил)-(6-феноксибензо[d]изоксазол-3-ил)амина (10е-1)
На Фиг.26 показана реакционная схема синтеза соединений 10е, имеющих общую Формулу V. В этом примере описан синтез соединения 10е-1, где Ar1 представляет собой 4-метоксифенил и Ar2 представляет собой фенил. Раствор Ar1NH2 в ТГФ охлаждали до -78°С и обрабатывали н-бутиллитием в атмосфере азота. В этом примере Ar1NH2 представлял собой анилин. После перемешивания при -78°С в течение 20 минут добавляли раствор соединения 8е-1, полученного согласно Примеру 25, в ТГФ в атмосфере азота. Смесь медленно нагревали до комнатной температуры, затем гасили водным насыщенным NH4Cl и экстрагировали NH4Cl. Органический слой промывали 1 н. HCl и водой, фильтровали через бумагу 1PS, концентрировали в вакууме и очищали на картридже Sep Pak, элюируя смесью гексан/Et2О (4:1) и получая соединение 10е-1 в виде желтого масла.
Пример 27
Получение оксима (2,4-дифторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (7f-1)
На Фиг.27 показана реакционная схема синтеза соединений 4f, имеющих общую формулу X, и соединений 7f, имеющих общую Формулу XII. В этом примере описан синтез соединения 7f-1, где R1 представляет собой изобутил, R2 представляет собой Н, и Ar представляет собой 2,4-дифторфенил.
Стадия А: Тетрафторборат аммония (20,97 г, 200 ммоль) растворяли в водном растворе уксусной кислоты (500 мл АсОН/250 мл воды) и охлаждали до 0°С. Последовательно добавляли 4-бром-2-метиланилин (соединение 1f, 18,61 г, 100 ммоль) и 42 мл концентрированной водной HCl (36% (мас./мас.), 12 н., 500 ммоль). Смесь перемешивали в течение 20 минут при 0°С и затем добавляли NaNO2 (7,59 г, 110 ммоль). Реакцию перемешивали в течение 1 часа при 0°С и нагревали до комнатной температуры. После 16 часов при комнатной температуре смесь концентрировали при пониженном давлении, остаток подвергали азеотропной перегонке с толуолом и сушили под высоким вакуумом. Твердое вещество суспендировали в 500 мл CHCl3 и добавляли КОАс (12,76 г, 130 ммоль) и 18-краун-6 (7,93 г, 30 ммоль). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре. Смесь промывали водой, сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 30 г 5-бром-1Н-индазола (2f) в виде желто-коричневого твердого вещества. Неочищенное вещество использовали без дальнейшей очистки.
Стадия В: Неочищенное соединение 2f (100 ммоль) растворяли в 250 мл ДМФА. Добавляли К2СО3 (20,7 г, 150 ммоль) и 1-бром-2-метилпропан (16,3 мл, 150 ммоль). Смесь нагревали до 120°С в атмосфере азота в течение 16 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли воду (200 мл) и CH2Cl2 (200 мл) и интенсивно перемешивали в течение 30 минут. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая примерно 30 г неочищенного вещества. Неочищенное вещество очищали посредством хроматографии (эфир/гексаны: от 1:9 до 1:4), получая 12,87 г соединения 3f-1 в виде темно-красного масла с выходом 50,8% для стадий А и В. МС ИЭР (+), обнаружено: m/z 253 и 255 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.93 (s, 1Н), 7.87 (m, 1Н), 7.43 (m, 1Н), 7.29 (m, 1Н), 7.29 (m, 1Н), 4.15 (m, 2Н), 2.33 (m, 1Н), 0.92 (m, 6Н).
Стадия С: Соединение 3f-1 (121,0 мг, 0,478 ммоль) растворяли в 2 мл эфира и охлаждали до -78°С. К этому раствору добавляли трет-BuLi (1,70 М в пентане, 0,59 мл, 1,004 ммоль). Реакционную смесь дополнительно перемешивали в течение часа при -78°С. Добавляли 2,6-дифторбензальдегид (58 мкл, 0,526 ммоль) при -78°С, охлаждающую баню удаляли и реакционную смесь медленно нагревали до комнатной температуры. Реакцию гасили 10 мл воды. Слои разделяли и водный слой несколько раз экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит, концентрировали при пониженном давлении и очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая соединение 4f-1 в виде бледно-желтого кристаллического твердого вещества (104,5 мг, выход 69,1%). МС ИЭР (+), обнаружено: m/z 317 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.96 (s, 1Н), 7.73 (s, 1Н), 7.56 (m, 1Н), 7.40~7.35 (m, 2Н), 6.91 (m, 2Н), 6.78 (m, 1Н), 6.22 (m, 1Н), 4.15 (m, 2Н), 2.39~2.26 (m, 2Н, перекрывается с -ОН), 0.92 (m, 6Н).
Стадия D: Соединение 4f-1 (316,3 мг, 1,00 ммоль), триацетоксипериодинан (445,3 мг, 1,05 ммоль) и 10 мл CH2Cl2 перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь гасили 10 мл насыщенного раствора К2СО3 и слои разделяли. Водный слой экстрагировали СН2Cl2, объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией, используя смесь эфир/гексаны (1:2) и получая 237,6 мг соединения 5f-1 в виде вязкого светло-коричневого масла (выход 75,6%). МС ИЭР (+), обнаружено: m/z 315 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.16 (s, 1Н), 8.11 (s, 1Н), 7.99 (m, 1Н), 7.60 (m, 1Н), 7.47 (m, 1Н), 7.03 (m, 1Н), 6.94 (m, 1Н), 4.21 (m, 2Н), 2.37 (m, 1Н), 0.95 (m, 6Н).
Стадия Е: Смесь соединения 5f-1 (96,7 мг, 0,308 ммоль), гидроксиламин-HCl (соединение 6f-1; 213,8 мг, 3,076 ммоль) и 5 мл пиридина перемешивали при комнатной температуре в течение 65 часов. Избыток пиридина удаляли при пониженном давлении. Остаток растворяли в 20 мл CH2Cl2. Белое твердое вещество выпадало в осадок, смесь переносили в делительную воронку и промывали 1 н. HCl. Органический слой сушили над безводным MgSO4, фильтровали через целит, концентрировали при пониженном давлении и очищали хроматографией, используя смесь эфир/гексаны (1:2) и получая 66,5 мг соединения 7f-1 в виде бледно-желтого пенистого твердого вещества (выход 65,5%), которое представляло собой смесь изомеров (1:4). МС ИЭР (+), обнаружено: m/z 330 (М+1).
Пример 28
Получение О-этилоксима (2,4-дифторфенил)-1-изобутил-1Н-индазол-5-ил)метанона (7f-3)
В этом примере описан синтез соединения 7f-3, имеющего общую Формулу XII, как показано на Фиг.27, где R1 представляет собой изобутил, R2представляет собой этил и Ar представляет собой 2,4-дифторфенил. Соединение 5f, где R1 представляет собой изобутил и Ar представляет собой 2,4-дифторфенил, получали в соответствии со стадиями A-D Примера 27. Смесь соединения 5f (43,3 мг, 0,138 ммоль), хлористоводородной соли О-этилгидроксиламина (53,8 мг, 0,551 ммоль) и 2 мл сухого пиридина перемешивали при комнатной температуре. Смесь перемешивали в течение 90 часов при комнатной температуре. Избыток пиридина удаляли при пониженном давлении. К остатку добавляли 2 мл воды и 2 мл CH2Cl2. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl (20 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:4) и получая 21,2 мг соединения 7f-3 в виде масла (выход 43,1%), которое представляло собой смесь изомеров (1:9).
Пример 29
Получение трет-бутилового эфира {2-[(2,4-дифторфенил)-(1-изобутил-1Н-индазол-5-ил)метиленаминоокси]этил}карбаминовой кислоты (7f-5)
В этом примере описан синтез соединения 7f-5, имеющего общую Формулу XII, как показано на Фиг.27, где R1 представляет собой изобутил, R2представляет собой
CH2CH2NHBoc и Ar представляет собой 2,4-дифторфенил. Соединение 5f, где R1 представляет собой изобутил, и Ar представляет собой 2,4-дифторфенил, получали в соответствии со стадиями A-D Примера 27. Смесь соединения 5f (50 мг, 0,159 ммоль), трет-бутилового эфира (2-аминооксиэтил)карбаминовой кислоты, полученного, как описано в Примере 30 (112 мг, 0,636 ммоль), пиридина (1,5 мл) и капли 6 н. HCl-MeOH (смесь концентрированной HCl и МеОН (1:1 по объему)) перемешивали при комнатной температуре в течение 64 часов. Избыток пиридина удаляли при пониженном давлении, остаток очищали хроматографией, используя смесь эфир/гексаны (1:2) и получая соединение 7f-5 с выходом 63,9%.
Пример 30
Получение трет-бутилового эфира (2-аминооксиэтил)карбаминовой кислоты
Реакционная схема синтеза трет-бутилового эфира (2-аминооксиэтил)карбаминовой кислоты показана на Фиг.29.
Стадия А: Смесь трет-бутилового эфира (2-бромэтил)карбаминовой кислоты (2,77 г, 12,39 ммоль), N-гидроксифталимида (2,02 г, 12,39 ммоль), ТЭА (5,18 мл, 37,16 ммоль) и 25 мл ДМФА перемешивали при комнатной температуре в течение 64 часов. Смесь разбавляли 100 мл воды. Белое твердое вещество выпадало в осадок и его собирали фильтрацией. Твердое вещество растворяли в СН2Cl2 (50 мл) и этот раствор промывали 1 н. HCl (20 мл), насыщенным NaHCO3 (20 мл), водой (20 мл) и рассолом (20 мл). Раствор сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 0,842 г белого твердого вещества (выход 22%).
Стадия В: трет-Бутиловый эфир [2-(1,3-диоксо-1,3-дигидроизоиндол-2-илокси)этил]карбаминовой кислоты (0,842 г, 2,749 ммоль) растворяли в 20 мл CH2Cl2 и добавляли метилгидразин (150 мкл, 2,776 ммоль) при комнатной температуре. Как только добавляли метилгидразин, формировался белый осадок. Реакционную смесь перемешивали при комнатной температуре в течение 72 часов. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении, получая 0,496 г вязкого масла (выход 102%). Неочищенное вещество использовали без дальнейшей очистки.
Пример 31
Получение оксима (4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (7f-2)
В этом примере описан синтез соединения 7f-2, имеющего общую Формулу XII, как показано на Фиг.27, где R1 представляет собой изобутил, R2представляет собой Н и Ar представляет собой 4-фторфенил.
Стадии А и В: Соединение 3f получали, как описано в стадиях А и В Примера 27.
Стадия С: Соединение 3f-2 (616,3 мг, 2,436 ммоль) растворяли в 20 мл эфира и охлаждали до -78°С. К раствору по каплям добавляли трет-BuLi (1,70 М в пентане, 2,94 мл). После добавления трет-BuLi смесь перемешивали в течение 30 минут при -78°С. По каплям добавляли 4-фторбензальдегид (290 мкл, 2,678 ммоль) при -78°С. Смесь медленно нагревали до комнатной температуры. Реакцию гасили CH2Cl2, объединенные экстракты промывали рассолом (20 мл), сушили над безводным MgSO4, фильтровали через целит и концентрировали, получая 750 мг соединения 4f-2 в виде желто-коричневого твердого вещества. Твердое вещество очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 554 мг соединения 4f-2 в виде светло-коричневого твердого вещества (выход 76,3%).
Стадия D: Соединение 4f-2 (100,6 мг, 0,337 ммоль) растворяли в 10 мл CH2Cl2 и к раствору добавляли реагент Десс-Мартина (Dess Martin periodinane) (триацетоксипериодинан; 150,2 мг, 0,354 ммоль). После 25 минут при комнатной температуре смесь становилась мутной. Реакционную смесь перемешивали в течение еще 30 минут при комнатной температуре и переносили в делительную воронку. Смесь разбавляли 30 мл CH2Cl2 и промывали насыщенным NaHCO3. Желтое нерастворимое твердое вещество, которое образовывалось между органическим и водным слоями, удаляли. Органический слой сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая соединение 5f-2 в виде масла с выходом 85,4%.
Стадия Е: Смесь соединения 5f-2 (41,6 мг, 0,140 ммоль) и гидрохлорида гидроксиламина (20,0 мг, 0,281 ммоль) в 1 мл пиридина перемешивали в течение ночи при комнатной температуре. Через один день исследование методом ВЭЖХ показало примерно 50%-ную конверсию. Добавляли еще 5 эквивалентов NH2OH-HCl и реакцию перемешивали в течение 72 часов. Избыток пиридина удаляли при пониженном давлении, остаток очищали хроматографией, используя смесь эфир/гексаны (1:2) и получая 31,4 мг соединения 7f-2 (выход 71,8%) в виде смеси изомеров (1:2). МС ИЭР (+), обнаружено: m/z 312 (М+1).
Пример 32
Получение О-этилоксима (4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (7f-4)
В этом примере описан синтез соединения 7f-4, имеющего общую Формулу XII, как показано на Фиг.27, где R1 представляет собой изобутил, R2представляет собой этил и Ar представляет собой 4-фторфенил.
Стадии A-D: Соединение 5f-2 получали, как описано в стадиях A-D Примера 31.
Стадия Е: Смесь соединения 5f-2 (51,2 мг, 0,173 ммоль), О-этилгидроксиламин-HCl (67,4 мг, 0,691 ммоль) и 2 мл сухого пиридина перемешивали при комнатной температуре. Смесь перемешивали в течение 90 часов при комнатной температуре. Избыток пиридина удаляли при пониженном давлении. К остатку добавляли 2 мл воды и 2 мл CH2Cl2. Слои разделяли и водный слой экстрагировали СН2Cl2. Объединенные экстракты промывали 1 н. HCl (20 мл), сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:4) и получая 47,1 мг соединения 7f-4 в виде масла (выход 80,3%), которое представляло собой смесь изомеров (1:2). МС ИЭР (+), обнаружено: m/z 340 (М+1).
Пример 33
Получение трет-бутилового эфира {2-[(4-фторфенил)-(1-изобутил-1H-индазол-5-ил)метиленаминоокси]этил}карбаминовой кислоты (7f-6)
В этом примере описан синтез соединения 7f-6, имеющего общую Формулу XII, как показано на Фиг.27, где R1 представляет собой изобутил, R2представляет собой
CH2CH2NHBoc и Ar представляет собой 4-фторфенил.
Стадии A-D: Соединение 5f-2 получали, как описано в стадиях A-D Примера 31.
Стадия Е: Смесь соединения 5f-2, трет-бутилового эфира (2-аминооксиэтил)карбаминовой кислоты, полученного, как описано в Примере 30 (120 мг, 0,675 ммоль), пиридина (1,5 мл) и капли 6 н. HCl/MeOH (смесь концентрированной HCl и МеОН (1:1 по объему)) перемешивали при комнатной температуре в течение 39 часов. Избыток пиридина удаляли при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 65,6 мг (выход 85,5%) соединения 7f-6 в виде бледно-желтого масла.1Н-ЯМР - анализ показал, что соединение 7f-6 представляло собой смесь изомеров в соотношении 1:1,8.
Пример 34
Получение О-бензилоксима (2,4-дифторфенил)-(1-изобутил-1H-индазол-5-ил)метанона (7f-7)
Синтез соединения 7f-7, имеющего общую Формулу XII, показан на Фиг.27.
Стадия А: Соединение 5f получали, как описано в Примере 27.
Стадия В: Соединение 5f (76,9 мг, 0,244 ммоль) растворяли в 2 мл пиридина и добавляли гидрохлорид О-бензилгидроксиламина (0,195 г, 1,22 ммоль). Смесь перемешивали при комнатной температуре в течение 2 дней и концентрировали при пониженном давлении. Остаток суспендировали в CH2Cl2, суспензию фильтровали через ватную пробку и очищали хроматографией, используя смесь эфир/гексаны (1:4) и получая 0,069 г соединения 7f-7 в виде смеси Е- и Z-изомеров (1:4, выход 67,2%). МС ИЭР (+), обнаружено: m/z 420 (М+Н).
Пример 35
Получение О-(2-аминоэтил)оксима (2,4-дифторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (7f-8)
Синтез соединения 7f-8, имеющего общую Формулу XII, показан на Фиг.27.
Стадия А: Соединение 7f-5 получали, как описано в Примере 29.
Стадия В: Соединение 7f-5 (32,3 мг, 0,0656 ммоль) растворяли в 2 мл смеси СН2Cl2:ТФУ (1:1) и эту смесь перемешивали в течение 0,5 часа при комнатной температуре. Всю смесь концентрировали при пониженном давлении и сушили под высоким вакуумом в течение ночи. Остаток растворяли в 5 мл CH2Cl2 и промывали насыщенным К2СО3. Органический слой сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 18,6 мг соединения 7f-8 в виде масла (выход 76,1%). МС ИЭР (+), обнаружено: m/z 373 (М+Н).
Пример 36
Получение Q-метилоксима (4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (7f-9)
Синтез соединения 7f-9, имеющего общую Формулу XII, показан на Фиг.27.
Стадия А: Соединение 5f-2 получали, как описано в Примере 31.
Стадия В: Соединение 5f-2 растворяли в трет-бутиловом эфире этил)карбаминовой кислоты (120 мг, 0,675 ммоль), пиридине (1,5 мл) и добавляли одну каплю в 2 мл пиридина и MeONH2-HCl. Смесь перемешивали при комнатной температуре в течение 2 дней и концентрировали при пониженном давлении. Остаток суспендировали в CH2Cl2, суспензию фильтровали через ватную пробку и очищали хроматографией, используя смесь эфир/гексаны (1:4) и получая 33,5 мг фракции 1, 1,0 мг фракции 2 и 17,7 мг смешанной фракции, составляющих в итоге 52,2 мг соединения 7f-9 (выход 58%). МС ИЭР (+), обнаружено: m/z 344 (М+Н).
Пример 37
Получение О-(2-аминоэтил)оксима (4-фторфенил)-(1-изобутил-1H-индазол-5-ил)метанона (7f-10)
Синтез соединения 7f-10, имеющего общую Формулу XII, показан на Фиг.27.
Стадия А: Соединение 7f-6 получали, как описано в Примере 33.
Стадия В: Соединение 7f-6 (50,5 мг, 0,107 ммоль) растворяли в 4 мл СН2Cl2, к раствору добавляли трифторуксусную кислоту (4 мл). После 0,5 часа при комнатной температуре смесь концентрировали при пониженном давлении и сушили под высоким вакуумом в течение ночи. Масло растворяли в 10 мл CH2Cl2 и промывали насыщенным раствором К2СО3. Органический слой сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 34,9 мг соединения 7f-10 в виде масла, содержащего смесь изомеров (1:2, выход 88,6%). МС ИЭР (+), обнаружено: m/z 355 (М+Н).
Пример 38
Получение О-метилоксима (2,4-дифторфенил)-(1-метил-1H-индазол-5-ил)метанона (7f-11)
Синтез соединения 7f-11, имеющего общую Формулу XII, показан на Фиг.28.
Стадия А: Соединение 9f-1 получали, как описано в Примере 74.
Стадия В: Соединение 9f-1 (622 мг, 2,409 ммоль), К2СО3 (499 мг, 1,50 экв.) и ДМФА (10 мл) помещали в пробирку Шленка (Schlenk). Добавляли йодметан (225 мкл, 1,50 экв.) и пробирку герметично закрывали. Пробирку нагревали до 100°С. После 23 часов при 100°С смесь охлаждали до комнатной температуры и вскрывали пробирку. Смесь переносили в круглодонную колбу и концентрировали при пониженном давлении. Остаток гасили водой и CH2Cl2, слои разделяли. Водный слой экстрагировали CH2Cl2. Объединенные органические экстракты сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 176 мг соединения 5f-13 в виде желтого твердого вещества (выход 26,9%). МС ИЭР (+), обнаружено: m/z 273 (М+Н).
Стадия С: Соединение 5f-13 (0,040 г, 0,147 ммоль) и хлористоводородную соль метоксиламина (0,123 г, 1,47 ммоль) помещали в реакционный сосуд на 5 мл и добавляли 1 мл пиридина. Реакционный сосуд герметично закрывали и нагревали до 50°С. Через 18 часов избыток пиридина удаляли при пониженном давлении и к остатку добавляли воду. Водную смесь экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl и насыщенным NaHCO3, сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 0,033 г соединения 7f-11 (выход 74,6%) в виде вязкого масла, содержащего смесь изомеров (1:9).
МС ИЭР (+), обнаружено: m/z 302 (М+Н).
Пример 39
Получение оксима (2,4-дифторфенил)-[1-(2,2,2-трифторэтил)-1Н-индазол-5-ил]метанона (7f-12)
Синтез соединения 7f-12, имеющего общую Формулу XII, показан на Фиг.28.
Стадия А: Соединение 5f-11 получали, как описано в Примере 74.
Стадия В: Соединение 5f-11, гидроксиламин-HCl (0,051 г, 0,735 ммоль) и 1 мл пиридина помещали в сосуд и эту смесь нагревали до 50°С. Через 14,5 часов пиридин удаляли при пониженном давлении, остаток разбавляли CH2Cl2 и водой. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl и насыщенным NaHCO3, сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 22,9 мг (выход 87,7%) соединения 7f-12 в виде белой пены, содержащей смесь изомеров (1:4).
МС ИЭР (+), обнаружено: m/z 356 (М+Н).
Пример 40
Получение О-метилоксима (2,4-дифторфенил)-[1-(2,2,2-трифторэтил)-1Н-индазол-5-ил]метанона (7f-13)
Синтез соединения 7f-13, имеющего общую Формулу XII, показан на Фиг.28.
Стадия А: Соединение 5f-11 получали, как описано в Примере 74.
Стадия В: Соединение 5f-11 (0,023 г, 0,067 ммоль), гидроксиламин-HCl (0,056 г, 0,676 ммоль) и 1 мл пиридина помещали в сосуд и эту смесь нагревали до 50°С. Через 14,5 часов пиридин удаляли при пониженном давлении и остаток разбавляли CH2Cl2 и водой. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl и насыщенным NaHCO3, сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 19,6 мг соединения 7f-13 (выход 78,5%) в виде белой пены, содержащей смесь изомеров (1:4). МС ИЭР (+), обнаружено: m/z 370 (М+Н).
Пример 41
Получение оксима (2,4-дифторфенил)-(1-метансульфонил-1Н-индазол-5-ил)метанона (7f-14)
Синтез соединения 7f-14, имеющего общую Формулу XII, показан на Фиг.28.
Стадия А: Соединение 9f-1 получали, как описано в Примере 74.
Стадия В: Соединение 9f-1 (258 мг, 1,00 ммоль) растворяли в 5 мл пиридина и добавляли метансульфонилхлорид (81 мкл, 1,05 ммоль). Через 15 часов избыток пиридина удаляли при пониженном давлении и к остатку добавляли воду. Водную смесь экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl и насыщенным NaHCOз, сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 238,1 мг соединения 5f-14 в виде белого твердого вещества (общий выход 70,8%).1Н-ЯМР (400 МГц, CDCl3) δ 8.38 (s, 1H), 8.23 (s, 1H), 8.18 (d, 1H), 8.07 (d, 1H), 7.66 (q, 1H), 7.06 (t, 1H), 6.95 (t, 1H), 3.36 (s, 3Н).
Стадия С: Соединение 5f-14 (0,060 г, 0,177 ммоль), гидроксиламин-HCl (0,123 г, 1,77 ммоль) и 1 мл пиридина помещали в сосуд и смесь нагревали до 50°С. Через 26 часов избыток пиридина удаляли при пониженном давлении и остаток разбавляли CH2Cl2 и водой. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl и насыщенным NaHCO3, сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (2:1). Соединение растворяли в смеси МеОН-СН2Cl2 и наносили на колонку, получая 37,4 мг соединения 7f-14 (выход 60,0%) в виде белого порошка, содержащего смесь изомеров (1:2). МС ИЭР (+), обнаружено: m/z 352 (М+Н).
Пример 42
Получение О-метилоксима (2,4-дифторфенил)-(1-метансульфонил-1Н-индазол-5-ил)метанона (7f-15)
Синтез соединения 7f-15, имеющего общую Формулу XII, показан на Фиг.28.
Стадия А: Соединение 5f-14 получали, как описано в Примере 41.
Стадия В: Соединение 5f-14 (0,060 г, 0,250 ммоль), метоксиламин-HCl (0,209 г, 2,50 ммоль) и 1 мл пиридина помещали в сосуд, и эту смесь нагревали до 50°С. Через 26,5 часов избыток пиридина удаляли при пониженном давлении и остаток разбавляли CH2Cl2 и водой. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl и насыщенным NaHCO3, сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 44,8 мг соединения 7f-15 в виде белого твердого вещества, содержащего смесь изомеров (1:4, выход 49%). МС ИЭР (+), обнаружено: m/z 366 (М+Н).
Пример 43
Получение О-метилоксима (2,4-дифторфенил)-(1Н-индазол-5-ил)метанона (7f-16)
Синтез соединения 7f-16, имеющего общую Формулу XII, показан на Фиг.28.
Стадия А: Соединение 9f-1 получали, как описано в Примере 74.
Стадия В: Соединение 9f-1 и хлористоводородную соль метоксиламина помещали в 5 мл реакционный сосуд и добавляли 1 мл пиридина. Реакционный сосуд герметично закрывали и нагревали до 50°С. Через 18 часов избыток пиридина удаляли при пониженном давлении и к остатку добавляли воду (10 мл). Водную смесь экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl (20 мл) и насыщенным NaHCO3 (20 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 33,0 мг соединения 7f-16 (выход 74,6%) в виде вязкого масла, содержащего смесь изомеров (1:4). МС ИЭР (+), обнаружено: m/z 288 (М+Н).
Пример 44
Получение оксима (1-аллил-1Н-индазол-5-ил)-(2,4-дифторфенил)метанона (7f-17)
Синтез соединения 7f-17, имеющего общую Формулу XII, показан на Фиг.28.
Стадия А: Соединение 9f-1 получали, как описано в Примере 74.
Стадия В: Соединение 9f-1 (0,516 г, 2,00 ммоль), К2СО3 (0,0415 г, 3,00 ммоль), ДМФА (10 мл) и аллилбромид (0,363, 3,00 ммоль) добавляли в пробирку Шленка. Пробирку герметично закрывали и нагревали до 100°С. Через 19 часов надосадочный раствор декантировали и соль промывали ДМФА (5 мл × 3). Объединенный надосадочный раствор концентрировали при пониженном давлении. Остаток растворяли в СН2Cl2 и промывали водой. Водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 142,1 мг соединения 5f-12 (выход 23,8%). МС ИЭР (+), обнаружено: m/z 299 (М+Н).
Стадия С: Соединение 5f-12 (0,027 г, 0,090 ммоль), гидроксиламин-HCl (0,063 г, 0,90 ммоль) и пиридин (1 мл) помещали в реакционный сосуд и эту смесь нагревали до 50°С. Через 21,5 часа реакционную смесь переносили в делительную воронку и добавляли воду (10 мл). Смесь экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl (20 мл) и насыщенным NaHCO3, сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 23,1 мг (выход 81,6%) соединения 7f-17 в виде пенистого твердого вещества, содержащего смесь изомеров (1:3). МС ИЭР (+), обнаружено: m/z 356 (М+Н).
Пример 45
Получение О-метилоксима (1-аллил-1Н-индазол-5-ил)-2,4-дифторфенил)метанона (7f-18)
Синтез соединения 7f-18, имеющего общую Формулу XII, показан на Фиг.28.
Стадия А: Соединение 5f-12 получали, как описано в Примере 44.
Стадия В: Соединение 5f-12 (0,027 г, 0,090 ммоль), метоксиламин-HCl (0,063 г, 0,90 ммоль) и пиридин (1 мл) помещали в реакционный сосуд и эту смесь нагревали до 50°С. Через 21,5 часа реакционную смесь переносили в делительную воронку и добавляли воду (10 мл). Смесь экстрагировали CH2Cl2. Объединенные экстракты промывали 1 н. HCl и насыщенным NaHCO3, сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 24,7 мг соединения 7f-18 (выход 83,1%) в виде масла, содержащего смесь изомеров (1:3). МС ИЭР (+), обнаружено: m/z 328 (М+Н).
Примеры 46-61 описывают синтез амидного соединения по этому изобретению, имеющего общую Формулу XIII. На Фиг.30А-30С показана реакционная схема синтеза соединений, имеющих общую структуру 11g.
Пример 46
Получение амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-1)
Стадия А: 1-Фтор-3-метилбензол (соединение 1g; 18,7 г, 170 ммоль) помещали в трехгорлую колбу объемом 500 мл и охлаждали до -78°С. Затем медленно с помощью шприца добавляли раствор трет-бутоксида калия (11,0 г, 170 ммоль) в ТГФ. Через 10 минут к реакции медленно добавляли трет-BuLi (19,0 г, 170 ммоль) в пентане с помощью канюли в атмосфере азота. После перемешивания в течение 2,5 часов реакцию гасили большим количеством измельченного сухого льда, убирали баню с температурой -78°С и вручную перемешивали металлическим шпателем до тех пор, пока темно-коричневое вещество не превратится в очень светлую желтую суспензию. После 20 минут ручного перемешивания добавляли примерно 500 мл воды и эту реакционную смесь перемешивали. Затем реакционную смесь промывали Et2O и затем подкисляли 6 н. HCl до рН менее 3 и экстрагировали Et2O. Органический слой промывали рассолом, сушили над MgSO4, фильтровали и концентрировали с получением 10 мг соединения 2g (выход 45%).1Н-ЯМР (400 МГц, CDCl3) δ 7.90 (t, 1H), 7.04 (d, 1H), 6.97 (d, 1H), 2.39 (s, 3Н).
Стадия В: Соединение 2g (8,0 г, 52 ммоль) добавляли в 500 мл колбу и охлаждали до температуры солевой бани со льдом. Добавляли H2SO4 (150 мл) и смесь перемешивали. Затем в реакционную смесь в течение 10 минут прикапывали смесь свежеприготовленных H2SO4 (6,11 г, 62,3 ммоль) и HNO3(5,2 г, 83 ммоль). После 3 часов при 0°С реакция завершалась, и смесь добавляли к 1500 мл смеси лед/ледяная вода и перемешивали в течение 1 часа. Реакцию фильтровали, промывали несколько раз холодной водой и сушили под высоким вакуумом, получая 8 г соединения 3g (выход 80%).1Н-ЯМР (400 МГц, CDCl3) δ 8.74 (d, 1H), 7.20 (d, 1H), 2.69 (s, 3Н).
Стадия С: Соединение 3g (8 г, 40,0 ммоль) растворяли в МеОН и медленно добавляли H2SO4 (20,0 г, 201 ммоль). Реакционную смесь нагревали до 65°С в течение 20 часов. Реакционную смесь концентрировали, разбавляли льдом и водой, обрабатывали ультразвуком, фильтровали, промывали несколько раз холодной водой и сушили под высоким вакуумом в течение 2 дней. Неочищенное вещество (соединение 4g) использовали непосредственно на следующей стадии.1Н-ЯМР (400 МГц, CDCl3) δ 8.66 (d, 1H), 7.01 (d, 1H), 3.95 (s, 3Н), 2.68 (s, 3Н).
Стадия D: Соединение 4g (5,4 г, 41 ммоль) добавляли к ТГФ и охлаждали до 0°С. Добавляли 4-фторфенол (5,1 г, 45 ммоль). Затем порциями добавляли NaH (60% в масле, 1,8 г, 45 ммоль). Через 1 час реакционную смесь нагревали до комнатной температуры и перемешивали в течение более 2 часов. Реакционную смесь концентрировали и гасили большим избытком 0,5 н. Na2CO3до рН 7.0. Реакционную смесь обрабатывали ультразвуком в течение 30 минут, фильтровали и дополнительно промывали буфером и Н2О. Реакционную смесь сушили под высоким вакуумом в течение 1 часа, затем добавляли к ТГФ и MgSO4 для сушки, фильтровали и упаривали с получением примерно 8 г соединения 5g (выход 75%).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.66 (d, 1H), 7.01 (d, 1Н), 3.95 (s, 3Н), 2.68 (s, 3Н).
Стадия Е: Соединение 5g (10,0 г, 33,0 ммоль) и цинк (11,0 г, 164 ммоль) добавляли к метанолу и перемешивали. Медленно добавляли уксусную кислоту (4,0 г, 66 ммоль). Реакционную смесь перемешивали в течение ночи, обрабатывали ультразвуком и пропускали через целит.Раствор концентрировали с получением примерно 14 г соединения 6g и побочных продуктов цинка. На следующей стадии использовали неочищенное вещество.
Стадия F: Соединение 6g (9,0 г, 33,0 ммоль), тетрафторборат аммония (6,0 г, 65 ммоль) и HCl (17,0 г, 163 ммоль) добавляли к 200 мл АсОН/Н2О (2:1) и обрабатывали ультразвуком. Вещество соскабливали со стенок круглодонной колбы и добавляли NaNO2 (2,7 г, 3 ммоль). Реакцию обрабатывали ультразвуком в течение 10 минут, вращая темно-коричневую смесь, пока не образовывался новый осадок (соль продукта). Реакционную смесь оставляли перемешиваться в течение 4 часов. Реакционную смесь концентрировали в быстром вакууме при 65°С, затем переносили в толуол и упаривали досуха. Неочищенное вещество (соединение 7g) непосредственно использовали на следующей стадии без какой-либо обработки.
Стадия G: Соединение 7g (11,0 г, 31 ммоль), ацетат калия (5,2 г, 53 ммоль) и 18-краун-6 (0,1 экв.) добавляли к хлороформу и обрабатывали ультразвуком в течение 10 минут.Реакция протекала в течение ночи при комнатной температуре. Колонку помещали в фильтровальную колбу объемом 1000 мл, состоящую из примерно 2 дюймов (5 см) силикагеля, 2 дюймов (5 см) целита, расположенного сверху силикагеля, листа фильтровальной бумаги сверху целита и 0,5 дюйма (1,3 см) песка сверху фильтровальной бумаги. Колонку промывали CHCl3. Неочищенное вещество наносили на колонку непосредственно в CHCl3, колонку элюировали CHCl3 до тех пор, пока не выделили большое количество желтого вещества. Затем продукт элюировали из колонки этилацетатом и этилацетатные элюаты объединяли и концентрировали с получением примерно 7 г соединения 8g (выход 95%). МС ИЭР (+), обнаружено: m/z 287 (М+Н).
Стадия Н: Соединение 8g (0,250 г, 0,87 ммоль) добавляли к сухому ДМФА и к этой смеси добавляли изобутилбромид (0,15 мл, 1,2 ммоль) и К2СО3(0,5 г, 3,6 ммоль). Затем эту реакционную смесь помещали в сосуд, покрытый мембраной, и перемешивали при 95°С в течение ночи. Вещество очищали колоночной хроматографией, используя смесь диэтиловый эфир/гексаны (1:1) и получая 0,1 г соединения 9g-1 (выход 33%). МС ИЭР (+), обнаружено: m/z 343 (М+Н).
Стадия I: Соединение 9g-1 (0,100 г, 0,292 ммоль) помещали в смесь 1 н. LiOH/ТГФ (1:1) и перемешивали при 55°С. Через 4 часа ТГФ выпаривали и добавляли 1 н. HCl. Реакционную смесь обрабатывали ультразвуком и фильтровали, выделяя примерно 0,075 г соединения 10g (выход 78%) в виде чистого вещества. МС ИЭР (+), обнаружено: m/z 329 (М+Н).
Стадия J: Раствор соединения 10g (20 мг, 0,061 ммоль) в ТГФ (1 мл) обрабатывали КДИ (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали 0,5 М NH4 в диоксане (0,11 мл, 0,67 ммоль). Еще через 18 часов растворитель оставляли медленно испаряться, и смесь очищали на картридже Sep Pak, элюируя CH2Cl2 - 5% MeOH/CH2Cl2 и получая 2,2 мг соединения 11g-1 в виде масла с выходом 12%.1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.01 (s, 1H), 7.99 (s, 1H), 7.73 (s, 1Н), 7.57 (s, 1Н), 7.26 (s, 1Н), 7.20 (m, 2Н), 7.05 (m, 2Н), 4.27 (d, 2H), 2.24 (m, 1H), 0.86 (d, 6H).
Пример 47
Получение [5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-ил]морфолин-4-илметанона (11g-2)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали морфолином (1 экв.). Еще через 18 часов растворитель оставляли медленно испаряться, и остаток очищали на картридже Sep Pak, элюируя градиентом: от 100% CH2Cl2 до MeOH/CH2Cl2, и получая соединение 11g-2 в виде масла с выходом 93%.
Пример 48
Получение [5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-ил]-4-метилпиперазин-1-ил)метанона (11g-3)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали 1-метилпиперазином (1 экв.). Еще через 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Рак, элюируя градиентом: от 100% CH2Cl2 до 5% MeOH/CH2Cl2, и получая соединение 11g-3 в виде масла с выходом 95%.
Пример 49
Получение (1-бензилпиперидин-4-ил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-4)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали 1-бензилпиперидин-4-иламином (1 экв.). Еще через 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Рак, элюируя градиентом: от 100% CH2Cl2 до 5% MeOH/CH2Cl2, и получая соединение 11g-4 в виде масла с выходом 97%.
Пример 50
Получение (2-бензиламиноэтил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-5)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали N1-бензилэтан-1,2-диамином (1 экв.). Еще через 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Рак, элюируя градиентом: от 100% CH2Cl2 до MeOH/CH2Cl2, и получая соединение 11g-5 в виде масла с выходом 100%.
Пример 51
Получение (2-пиперидинилэтил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-6)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали 2-пиперидин-1-илэтиламином (1 экв.). Еще через 18 часов растворитель оставляли медленно испаряться и остаток очищали на картридже Sep Рак, элюируя градиентом: от 100% CH2Cl2 до 5% MeOH/CH2Cl2, и получая соединение 11g-6 в виде масла с выходом 100%.
Пример 52
Получение (2-пирролидин-1-илэтил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-7)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали 2-пирролидин-1-илэтиламином (1 экв.). Еще через 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Рак, элюируя градиентом: от 100% CH2Cl2 до 5% MeOH/CH2Cl2, и получая соединение 11g-7 в виде масла с выходом 63%.
Пример 53
Получение (3-морфолин-4-илпропил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-8)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали 3-морфолин-4-илпропиламином (1 экв.). Еще через 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Pak, элюируя градиентом: от 100% CH2Cl2 до 5% MeOH/CH2Cl2, и получая соединение 11g-8 в виде масла с выходом 70%.
Пример 54
Получение (3-диметиламинопропил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-9)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали N-1-диметилпропан-1,3-диамином (1 экв.). После дополнительных 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Pak, элюируя градиентом: от 100% CH2Cl2до 5% MeOH/CH2Cl2, и получая соединение 11g-9 в виде масла с выходом 44%.
Пример 55
Получение (2-диметиламиноэтил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-10)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали N1-диметилэтан-1,2-диамином (1 экв.). После дополнительных 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Pak, элюируя градиентом: от 100% CH2Cl2 до 5% МеОН/СН2Cl2, и получая соединение 11g-10 в виде масла с выходом 58%.
Пример 56
Получение метил-(1-метилпиперидин-4-ил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-11)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали метил(1-метилпиперидин-4-ил)амином (1 экв.). После дополнительных 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Pak, элюируя градиентом: от 100% CH2Cl2 до 5% MeOH/CH2Cl2, и получая соединение 11g-11 в виде масла с выходом 3%.
Пример 57
Получение [3-(метилфениламино)пропил]амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (11g-12)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали N1-метил-N1-фенилпропан-1,3-диамином (1 экв.). После дополнительных 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Pak, элюируя градиентом: от 100% CH2Cl2до 5% МеОН/СН2Cl2, и получая соединение 11g-12 в виде масла с выходом 78%.
Пример 58
Получение трет-бутилового эфира 3-{[5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}пирролидин-1-карбоновой кислоты (11g-13)
Раствор 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (соединения 10g, полученного, как описано в Примере 46) в ТГФ обрабатывали карбонилдиимидазолом (1,2 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов реакционную смесь обрабатывали трет-бутиловым эфиром 3-аминопирролидин-1-карбоновой кислоты (1 экв.). После дополнительных 18 часов растворитель оставляли медленно испаряться, остаток очищали на картридже Sep Pak, элюируя градиентом: от 100% CH2Cl2 до 5%
МеОН/СН2Cl2, и получая соединение 11g-13 в виде масла с выходом 94%.
Пример 59
Получение (2-диметиламиноэтил)амида 5-(4-фторфенокси)-1-(2,2,2-трифторэтил)-1Н-индазол-6-карбоновой кислоты (11g-14)
Стадия А: Соединение 8g получали, как описано в Примере 46.
Стадия В: Соединение 8g, 2-бром-1,1,1-трифторэтан, К2СО3 и ДМФА объединяли и реакционную смесь перемешивали в течение ночи при 75°С. Добавляли 2 дополнительных эквивалента 2-бром-1,1,1-трифторэтана и реакционную смесь перемешивали при 90°С. Добавляли еще несколько эквивалентов 2-бром-1,1,1-трифторэтана и реакционную смесь перемешивали при 50°С в течение 72 часов. Реакционную смесь концентрировали, переносили в толуол и очищали колоночной хроматографией (элюируя смесью гексан/Et2О), получая 80 мг соединения 9g-2 (выход 24%). МС ИЭР (+), обнаружено: m/z 369 (М+Н).
Стадия С: Соединение 9g-2 (0,075 г, 0,20 ммоль) помещали в смесь 1 н. LiOH/ТГФ (1:1) и перемешивали в течение 18 часов при комнатной температуре. ТГФ выпаривали и 1 н. HCl добавляли к реакционной смеси, которую затем обрабатывали ультразвуком и фильтровали, выделяя примерно 0,070 г соединения 10g-2 (выход 97%) в виде чистого вещества. МС ИЭР (+), обнаружено: m/z 355 (М+Н).
Стадия D: Соединение 10g-2 (0,03 г, 0,847 ммоль), бензотриазол-1,3-диол (0,022 г, 0,25 ммоль) и (3-диметиламинопропил)этилкарбодиимид (0,011 г, 0,10 ммоль) добавляли к дихлорэтану и перемешивали в течение 5 минут. Затем добавляли N1-диметилэтан-1,2-диамин (0,019 г, 0,10 ммоль) и реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали, переносили в дихлорметан, сушили под высоким вакуумом и очищали обращенно-фазовой ВЭЖХ согласно способу С (см. ниже), получая 25 мг соединения 11g-14 (выход 56%) в виде соли ТФУ.1Н-ЯМР (400 МГц, CDCl3) δ 8.45 (s, 1H), 8.10 (s, 1H), 7.90 (s, 1H), 7.12 (m, 4H), 5.02 (q, 2H), 3.93 (br, 2H), 3.34 (br, 6H), 2.72 (s, 6H).
Пример 60
Получение (2-диметиламиноэтил)амида 5-(4-фторфенокси)-1-метил-1Н-индазол-6-карбоновой кислоты (11g-15)
Стадия А: Соединение 8g получали, как описано в Примере 46.
Стадия В: Соединение 8g, йодметан и К2СО3 добавляли к ДМФА и нагревали примерно до 75°С. Через 48 часов реакционную смесь фильтровали для удаления
К2СО3, концентрировали, переносили в толуол и очищали колоночной хроматографией (элюируя смесью Et2О/гексан (1:1)), получая 70 мг соединения 9g-3 (выход 36,7%). МС ИЭР (+), обнаружено: m/z 301 (М+Н).
Стадия С: Соединение 9g-3 (0,075 г, 0,25 ммоль) помещали в смесь 1 н. LiOH/ТГФ (1:1) и перемешивали в течение 18 часов при комнатной температуре. ТГФ выпаривали и 1 н. HCl добавляли к реакционной смеси, которую затем обрабатывали ультразвуком и фильтровали, получая примерно 0,060 г соединения 10g-3 (выход 84%) в виде чистого вещества. МС ИЭР (+), обнаружено: m/z 287 (М+Н).
Стадия D: Соединение 10g-3 (0,030 г, 0,105 ммоль), бензотриазол-1,3-диол (0,028 г, 0,31 ммоль) и (3-диметиламинопропил)этилкарбодиимид (0,019 г, 0,13 ммоль) добавляли к дихлорэтану и перемешивали в течение 5 минут.
Затем добавляли N1-диметилэтан-1,2-диамин (0,024 г, 0,13 ммоль) и реакционную смесь перемешивали в течение 3 часов. Реакционную смесь затем концентрировали, переносили в дихлорэтан, сушили под высоким вакуумом и очищали обращенно-фазовой ВЭЖХ согласно способу С Примера 86, получая 25 мг соединения 11g-15 (выход 52%) в виде соли ТФУ.1Н-ЯМР (400 МГц, CDCl3) δ 8.44 (br, 1H), 8.21 (s, 1H), 7.85 (s, 1H), 7.05 (m, 4H), 4.15 (s, 3Н), 3.90 (br, 2H), 3.30 (br, 2H), 2.92 (s, 6H).
Пример 61
Получение (2-диметиламиноэтил)амида 5-(4-фторфенокси)-1Н-индазол-6-карбоновой кислоты (11g-16)
Стадия А: Соединение 8g получали, как описано в Примере 46.
Стадия В: Соединение 8g перемешивали в ТГФ, добавляли один объемный эквивалент 1 н. LiOH и реакционную смесь перемешивали при 60°С в течение 6 часов. Реакционную смесь концентрировали, гасили 1 н. HCl, охлаждали, обрабатывали ультразвуком, фильтровали и сушили, получая 0,40 г соединения 10g-4 (84% чистого вещества). МС ИЭР (+), обнаружено: m/z 287 (М+Н).
Стадия С: Соединение 10g-4 (0,030 г, 0,110 ммоль), бензотриазол-1,3-диол (0,029 г, 0,33 ммоль) и (3-диметиламинопропил)этилкарбодиимид (0,020 г, 0,13 ммоль) добавляли к дихлорэтану и перемешивали в течение 5 минут. Затем добавляли N1-диметилэтан-1,2-диамин (0,025 г, 0,13 ммоль) и реакционную смесь перемешивали в течение 3 часов. Реакционную смесь упаривали, переносили в дихлорэтан, сушили под высоким вакуумом и очищали обращенно-фазовой ВЭЖХ согласно способу В Примера 86, получая 25 мг соединения 11g-16 (выход 51%) в виде соли ТФУ.1Н-ЯМР (400 МГц, CDCl3) δ 8.45 (br, 1H), 8.22 (s, 1H), 7.91 (s, 1H), 7.09 (s, 1H), 7.06 (m, 3Н), 3.85 (br, 2Н), 3.20 (br, 2H), 2.90 (s, 6H).
Примеры 62-67 описывают синтез спиртовых соединений, имеющих общую Формулу IX. На Фиг.31 показана реакционная схема синтеза общего соединения 4f.
Пример 62
Получение (2,4-дифторфенил)-(1-изобутил-1Н-индазол-5-ил)метанола (4f-1)
В этом примере описан синтез соединения 4f-1, как показано на Фиг.31, где R1 представляет собой изобутил и Ar представляет собой 2,4-дифторфенил.
Стадия А: Тетрафторборат аммония (20,97 г, 200 ммоль) растворяли в водном растворе уксусной кислоты (500 мл АсОН/250 мл воды) и охлаждали до 0°С. Последовательно добавляли 2-метил-4-броманилин (соединение 1f; 18,61 г, 100 ммоль) и 42 мл концентрированной водной HCl (36% (мас./мас.), 12 н., 500 ммоль). Смесь перемешивали в течение 20 минут при 0°С и добавляли NaNO2(7,59 г, 110 ммоль). Реакционную смесь перемешивали в течение 1 часа при 0°С и нагревали до комнатной температуры. После 16 часов при комнатной температуре смесь концентрировали при пониженном давлении, остаток подвергали азеотропной перегонке с толуолом и сушили под высоким вакуумом. Твердое вещество суспендировали в 500 мл CHCl3 и добавляли КОАс (12,76 г, 130 ммоль) и 18-краун-6 (7,93 г, 30 ммоль). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре. Смесь промывали водой, сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 30 г 5-бром-1H-индазола (соединения 2f) в виде желто-коричневого твердого вещества. Неочищенное вещество использовали без дальнейшей очистки.
Стадия В: Неочищенный 5-бром-1Н-индазол (соединение 2f; 100 ммоль) растворяли в 250 мл ДМФА. Добавляли К2СО3 (20,7 г, 150 ммоль) и 1-бром-2-метилпропан (16,3 мл, 150 ммоль). Смесь нагревали до 120°С в атмосфере азота в течение 16 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли воду (200 мл) и CH2Cl2 (200 мл) и интенсивно перемешивали в течение 30 минут. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая примерно 30 г неочищенного вещества. Неочищенное вещество очищали посредством хроматографии (эфир/гексаны: от 1:9 до 1:4), получая 12,870 г соединения 3f в виде темно-красного масла с выходом 50,8% на стадиях А и В. МС ИЭР (+), обнаружено: m/z 253 и 255 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.93 (s, 1Н), 7.87 (m, 1Н), 7.43 (m, 1Н), 7.29 (m, 1Н), 7.29 (m, 1Н), 4.15 (m, 2Н), 2.33 (m, 1Н), 0.92 (m, 6Н).
Стадия С: Соединение 3f (121,0 мг, 0,478 ммоль) растворяли в 2 мл эфира и охлаждали до -78°С. К раствору добавляли трет-BuLi (1,70 М в пентане, 0,59 мл, 1,004 ммоль). Реакционную смесь перемешивали в течение еще часа при -78°С. Добавляли 2,4-дифторбензальдегид (58 мкл, 0,526 ммоль) при -78°С, охлаждающую баню удаляли, реакционная смесь медленно нагревалась до комнатной температуры. Реакцию гасили 10 мл воды. Слои разделяли, и водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит, концентрировали при пониженном давлении и очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 104,5 мг соединения 4f-1 (выход 69,1%) в виде бледно-желтого кристаллического твердого вещества. МС ИЭР (+), обнаружено: m/z 317 (М+1).1Н-ЯМР (400 МГц, CDCI3) δ 7.96 (s, 1H), 7.73 (s, 1H), 7.56 (m, 1H), 7.40~7.35 (m, 2Н), 6.91 (m, 2Н), 6.78 (m, 1Н), 6.22 (m, 1Н), 4.15 (m, 2Н), 2.39~2.26 (m, 2Н, перекрывается с -ОН), 0.92 (m, 6H).
Пример 63
Получение (4-хлор-2-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанола (4f-7)
В этом примере описан синтез соединения 4f-7, как показано на Фиг.31, где R1 представляет собой изобутил и Ar представляет собой 4-хлор-2-фторфенил.
Стадии А-В: 5-Бром-1-изобутил-1Н-индазол (соединение 3f) получали, как описано в Примере 62, стадии А-В.
Стадия С: Соединение 3f (132 мг, 0,521 ммоль) в 1 мл эфира охлаждали до -78°С. К раствору добавляли трет-BuLi (1,70 М в пентане, 0,64 мл, 1,10 ммоль). После 1 часа при -78°С добавляли раствор 4-хлор-2-фторбензальдегида (86,8 мг, 0,548 ммоль) в 1 мл эфира и смесь медленно нагревали до комнатной температуры. Смесь гасили водой (5 мл) и слои разделяли. Водный слой экстрагировали СН2Cl2, объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией, используя смесь эфир/гексаны (1:2) и получая 43,7 мг соединения 4f-7 в виде бледно-желтого твердого вещества (выход 25,2%). МС ИЭР (+), обнаружено: m/z 333 и 335 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.96 (s, 1H), 7.72 (s, 1H), 7.56 (m, 1Н), 7.39~7.35 (m, 2Н), 7.18 (m, 1Н), 7.05 (m, 1Н), 6.21 (m, 1Н), 4.15 (m, 2Н), 2.37~2.27 (m, 2Н, перекрывается с -ОН), 0.91 (m, 6H).
Пример 64
Получение (2-хлор-4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанола (4f-8)
В этом примере описан синтез соединения 4f-8, как показано на Фиг.31, где R1 представляет собой изобутил и Ar представляет собой 2-хлор-4-фторфенил.
Стадии А-В: 5-Бром-1-изобутил-1Н-индазол (соединение 3f) получали, как описано в Примере 62, стадии А-В.
Стадия С: Раствор соединения 3f (116,2 мг, 0,459 ммоль) в 1 мл эфира охлаждали до -78°С. К раствору при -78°С добавляли трет-BuLi (1,70 М в пентане, 0,57 мл). После 1 часа при -78°С добавляли раствор 2-хлор-4-фторбензальдегида (76,4 мг, 0,482 ммоль) в 1 мл эфира и смесь медленно нагревали до комнатной температуры. Смесь гасили водой (5 мл) и слои разделяли. Водный слой экстрагировали CH2Cl2, объединенные экстракты сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией, используя смесь эфир/гексаны (1:2) и получая 47,6 мг соединения 4f-8 в виде бледно-желтого твердого вещества (выход 31,2%). МС ИЭР (+), обнаружено: m/z 333 и 335 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.96 (s, 1H), 7.72~7.66 (m, 2H), 7.39~7.34 (m, 2Н), 7.13~7.03 (m, 2Н), 6.29 (m, 1Н), 4.15 (m, 2Н), 2.38~2.27 (m, 2Н, перекрывается с -ОН), 0.92 (m, 6H).
Пример 65
Получение (4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанола (4f-2)
В этом примере описан синтез соединения 4f-2, как показано на Фиг.31, где R1 представляет собой изобутил и Ar представляет собой 4-фторфенил.
Стадии А-В: 5-Бром-1-изобутил-1Н-индазол (соединение 3f) получали, как описано в Примере 62, стадии А-В.
Стадия С: Соединение 3f (1,49 г, 5,89 ммоль) растворяли в 50 мл эфира и раствор охлаждали до -78°С. К раствору по каплям добавляли трет-BuLi (1,70 М в пентане, 7,01 мл, 12,07 ммоль). При добавлении трет-BuLi образовывалось коричневое твердое вещество и смесь превращалась в суспензию. После того, как добавление трет-BuLi завершалось, смесь перемешивали еще в течение 30 минут при -78°С. Добавляли по каплям 4-фторбензальдегид (700 мкл, 6,475 ммоль) при -78°С, после чего охлаждающую баню удаляли и реакционную смесь медленно нагревали до комнатной температуры. Реакцию гасили 20 мл воды и слои разделяли. Водный слой экстрагировали CH2Cl2, объединенные экстракты промывали рассолом (20 мл), сушили над MgSO4, фильтровали через целит и концентрировали, получая 1,70 г желто-коричневого твердого вещества. Затем твердое вещество очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 1,233 г соединения 4f-2 в виде светло-коричневого твердого вещества (выход 70,2%). МС ИЭР (+), обнаружено: m/z 299 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.97 (s, 1H), 7.72 (s, 1Н), 7.40~7.31 (m, 4Н), 7.07~7.00 (m, 2Н), 5.96 (m, 1Н), 4.15 (m, 2Н), 2.38~2.27 (m, 2Н, перекрывается с -ОН), 0.92 (m, 6H).
Пример 66
Получение (2,4-дихлорфенил)-(1-изобутил-1Н-индазол-5-ил)метанола (4f-9)
В этом примере описан синтез соединения 4f-9, как показано на Фиг.31, где R1 представляет собой изобутил и Ar представляет собой 2,4-дихлорфенил.
Стадии А-В: 5-Бром-1-изобутил-1Н-индазол (соединение 3f) получали, как описано в Примере 62, стадии А-В.
Стадия С: Соединение 3f (106,8 мг, 0,422 ммоль) растворяли в 2 мл эфира. Раствор охлаждали до -78°С и перемешивали в течение 15 минут.К этой смеси медленно добавляли трет-BuLi (1,70 М в пентане, 0,52 мл, 0,886 ммоль). Смесь превращалась в красную суспензию, и ее перемешивали еще в течение часа при -78°С. 2,4-Дихлорбензальдегид (81,2 мг, 0,464 ммоль) растворяли в 1 мл эфира и раствор переносили в суспензию с помощью иглы с двумя концами. Охлаждающую баню удаляли, позволяя реакционной смеси медленно нагреваться до комнатной температуры. Реакцию гасили 10 мл воды и слои разделяли. Водный слой экстрагировали СН2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит, концентрировали при пониженном давлении и очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая соединение 4f-9 в виде желтой пены (99,6 мг, выход 67,6%). МС ИЭР (+), обнаружено: m/z 349 и 351 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.96 (s, 1H), 7.70 (s, 1H), 7.68 (m, 1H), 7.38~7.36 (m, 3Н), 7.33 (m, 1H), 6.27 (m, 1H), 4.15 (m, 2H), 2.39 (m, 1H, -ОН), 2.37~2.26 (m, 1Н), 0.92 (m, 6Н).
Пример 67
Получение (1-изобутил-1Н-индазол-5-ил)-О-толилметанола (4f-10)
В этом примере описан синтез соединения 4f-10, как показано на Фиг.31, где R1 представляет собой изобутил и Ar представляет собой 2-метилфенил.
Стадии А-В: 5-Бром-1-изобутил-1Н-индазол (соединение 3f) получали, как описано в Примере 62, стадии А-В.
Стадия С: Соединение 3f (123,3 мг, 0,487 ммоль) растворяли в 2 мл эфира. Раствор охлаждали до -78°С и перемешивали в течение 15 минут. К смеси медленно добавляли трет-BuLi (1,70 М в пентане, 0,62 мл, 1,023 ммоль). Смесь превращалась в красную суспензию и ее перемешивали еще в течение часа при -78°С. Добавляли О-толуальдегид (62 мкл, 0,536 ммоль) при -78°С. Охлаждающую баню удаляли, позволяя реакционной смеси медленно нагреваться до комнатной температуры. Реакцию гасили 10 мл воды, слои разделяли и водный слой экстрагировали СН2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит, концентрировали при пониженном давлении и очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая соединение 4f-10 в виде очень вязкого бледно-желтого масла (96,4 мг, выход 67,2%). МС ИЭР (+), обнаружено: m/z 295 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.94 (s, 1Н), 7.64~7.61 (m, 2Н), 7.38~7.33 (m, 2Н), 7.29, (m, 1Н), 7.23 (m, 1Н), 7.17~7.13 (m, 1Н), 6.13 (m, 1Н), 4.15 (m, 2Н), 2.32 (m, 1Н), 2.24 (s, 3Н), 2.18 (m, 1Н, -ОН), 0.91 (m, 6Н).
Примеры 68-75 описывают синтез соединения общей Формулы X. На Фиг.32 и 33 показана реакционная схема синтеза соединений, имеющих общую структуру 5f.
Пример 68
Получение (2,4-дифторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (5f-1)
В этом примере описан синтез соединения 5f-1, как показано на Фиг.32, где R1 представляет собой изобутил и Ar представляет собой 2,4-дифторфенил.
Стадия А: Тетрафторборат аммония (20,97 г, 200 ммоль) растворяли в водном растворе уксусной кислоты (500 мл АсОН/250 мл воды) и охлаждали до 0°С. Последовательно добавляли 2-метил-4-броманилин (18,61 г, 100 ммоль) и 42 мл концентрированной водной HCl (36% (мас./мас.), 12 н., 500 ммоль). Смесь перемешивали в течение 20 минут при 0°С и добавляли NaNO2 (7,59 г, 110 ммоль). Реакционную смесь перемешивали в течение 1 часа при 0°С и нагревали до комнатной температуры. После 16 часов при комнатной температуре эту смесь концентрировали при пониженном давлении, остаток подвергали азеотропной перегонке с толуолом и сушили под высоким вакуумом. Твердое вещество суспендировали в 500 мл CHCl3 и добавляли КОАс (12,76 г, 130 ммоль) и 18-краун-6 (7,93 г, 30 ммоль). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре. Смесь промывали водой, сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 30 г 5-бром-1Н-индазола (соединения 2f) в виде желто-коричневого твердого вещества. Неочищенное вещество использовали без дальнейшей очистки.
Стадия В: Неочищенный 5-бром-1Н-индазол (соединение 2f; 100 ммоль) растворяли в 250 мл ДМФА. Добавляли К2СО3 (20,7 г, 150 ммоль) и 1-бром-2-метилпропан (16,3 мл, 150 ммоль). Смесь нагревали до 120°С в атмосфере азота в течение 16 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли воду (200 мл) и CH2Cl2 (200 мл) и интенсивно перемешивали в течение 30 минут. Слои разделяли и водный слой экстрагировали СН2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая примерно 30 г неочищенного вещества. Неочищенное вещество очищали посредством хроматографии (эфир/гексаны: от 1:9 до 1:4), получая 12,870 г соединения 3f в виде темно-красного масла с выходом 50,8% на стадиях А и В. МС ИЭР (+), обнаружено: m/z 253 и 255 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.93 (s, 1H), 7.87 (m, 1H), 7.43 (m, 1H), 7.29 (m, 1H), 7.29 (m, 1H), 4.15 (m, 2H), 2.33 (m, 1H), 0.92 (m, 6H).
Стадия С: Соединение 3f (121,0 мг, 0,478 ммоль) растворяли в 2 мл эфира и охлаждали до -78°С. К раствору добавляли трет-BuLi (1,70 М в пентане, 0,59 мл, 1,004 ммоль). Реакционную смесь перемешивали еще в течение часа при -78°С. Добавляли 2,4-дифторбензальдегид (58 мкл, 0,526 ммоль) при -78°С, охлаждающую баню удаляли и реакционную смесь медленно нагревали до комнатной температуры. Реакцию гасили 10 мл воды. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит, концентрировали при пониженном давлении и очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая соединение 4f-1 в виде бледно-желтого кристаллического твердого вещества (104,5 мг, выход 69,1%). МС ИЭР (+), обнаружено: m/z 317 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.96 (s, 1H), 7.73 (s, 1Н), 7.56 (m, 1Н), 7.40~7.35 (m, 2Н), 6.91 (m, 2Н), 6.78 (m, 1Н), 6.22 (m, 1Н), 4.15 (m, 2Н), 2.39~2.26 (m, 2Н, перекрывается с -ОН), 0.92 (m, 6H).
Стадия D: Соединение 4f-1 (316,3 мг, 1,00 ммоль), "Реагент Десс-Мартина" (триацетоксипериодинан; 445,3 мг, 1,05 ммоль) и 10 мл CH2Cl2перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь гасили 10 мл насыщенного раствора К2СО3 и слои разделяли. Водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией, используя смесь эфир/гексаны (1:2) и получая 237,6 мг соединения 5f-1 в виде вязкого светло-коричневого масла (выход 75,6%). МС ИЭР (+), обнаружено: m/z 315 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.16 (s, 1Н), 8.11 (s, 1H), 7.99 (m, 1H), 7.60 (m, 1H), 7.47 (m, 1Н), 7.03 (m, 1Н), 6.94 (m, 1Н), 4.21 (m, 2Н), 2.37 (m, 1Н), 0.95 (m, 6Н).
Пример 69
Получение (4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (5f-2)
В этом примере описан синтез соединения 5f-2, как показано на Фиг.32, где R1 представляет собой изобутил и Ar представляет собой 4-фторфенил.
Стадии А-С: (4-Фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанол (соединение
4f-2) получали, как описано в Примере 27, стадии А-С, за исключением того, что вместо 2,4-дифторбензальдегида использовали 4-фторбензальдегид.
Стадия D: Смесь (4-фторфенил)-(1-изобутил-1H-индазол-5-ил)метанола (соединения 4f-2; 745,9 мг, 2,50 ммоль), Реагента Десс-Мартина (триацетоксипериодинана; 1,166 г, 2,75 ммоль) и 50 мл CH2Cl2 перемешивали при комнатной температуре в течение 2 часов. Реакцию гасили 20 мл насыщенного раствора К2СО3. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:4) и получая 599 мг соединения 5f-2 в виде светло-коричневого твердого вещества (выход 80,9%). МС ИЭР (+), обнаружено: m/z 297 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.17 (s, 1H), 8.11 (s, 1Н), 7.94 (m, 1Н), 7.87 (m, 1Н), 7.85 (m, 1Н), 7.49 (m, 1Н), 7.22~7.16 (m, 2Н), 4.23 (m, 2Н), 2.38 (m, 1Н), 0.96 (m, 6Н).
Пример 70
Получение (2,4-дихлорфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (5f-9)
В этом примере описан синтез соединения 5f-9, как показано на Фиг.32, где R1 представляет собой изобутил и Ar представляет собой 2,4-дихлорфенил.
Стадии А-С: (2,4-Дихлорфенил)-(1-изобутил-1Н-индазол-5-ил)метанол (соединение 4f-9) получали, как описано в Примере 27, стадии А-С, за исключением того, что вместо 2,4-дифторбензальдегида использовали 2,4-дихлорбензальдегид.
Стадия D: Смесь соединения 4f-9, Реагента Десс-Мартина (триацетоксипериодинана; 20 мг, 0,046 ммоль) и 1 мл CH2Cl2 перемешивали при комнатной температуре в течение 2 часов. Смесь наносили на систему Biotage и элюировали смесью эфир/гексаны (1:2), получая 12,9 мг соединения 5f-9 (выход 85%). МС ИЭР (+), обнаружено: m/z 347 и 349 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.09 (s, 1Н), 8.06 (m, 1Н), 7.53 (m, 1Н), 7.47 (m, 1Н), 7.41~7.34 (m, 2Н), 4.21 (m, 2Н), 2.36 (m, 1Н), 0.95 (m, 6Н).
Пример 71
Получение (1-изобутил-1Н-индазол-5-ил)-О-толилметанона (5f-10)
В этом примере описан синтез соединения 5f-10, как показано на Фиг.32, где R1 представляет собой изобутил и Ar представляет собой 2-метилфенил.
Стадии А-С: (1-Изобутил-1Н-индазол-5-ил)-O-толилметанол (соединение 4f-10) получали, как описано в Примере 27, стадии А-С, за исключением того, что вместо 2,4-дифторбензальдегида использовали О-толуиловый альдегид.
Стадия D: Соединение 4f-10 (21 мг, 0,070 ммоль), Реагент Десс-Мартина (триацетоксипериодинан; 31 мг, 0,0735 ммоль) и 1 мл CH2Cl2 перемешивали при комнатной температуре в течение 2 часов. Смесь наносили на систему Biotage и элюировали смесью эфир/гексаны (1:2), получая 18,7 мг соединения 5f-10 (выход 91,4%). МС ИЭР (+), обнаружено: m/z 293 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.07 (s, 1Н), 8.06 (s, 1H), 8.04 (m, 1H), 7.46 (m, 1H), 7.41 (m, 1Н), 7.35~7.30 (m, 2Н), 7.30~7.25 (m, 1H), 4.21 (m, 2H), 2.36 (m, 1H), 2.33 (s, 3Н), 0.95 (m, 6H).
Пример 72
Получение (2-хлор-4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (5f-8)
В этом примере описан синтез соединения 5f-8, как показано на Фиг.32, где R1 представляет собой изобутил и Ar представляет собой 2-хлор-4-фторфенил.
Стадии А-С: (2-Хлор-4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанол (соединение 4f-8) получали, как описано в Примере 27, стадии А-С, за исключением того, что вместо 2,4-дифторбензальдегида использовали 2-хлор-4-фторбензальдегид.
Стадия D: (2-Хлор-4-фторфенил)-(1-изобутил-1Н-индазол-5-ил)-метанол (соединение 4f-8; 16,2 мг, 0,0487 ммоль), "Реагент Десс-Мартина" (триацетоксипериодинан; 21,7 мг, 0,0511 ммоль) и 1 мл СН2Cl2 перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь наносили на систему Biotage и элюировали смесью эфир/гексаны (1:2), получая 13,0 мг соединения 5f-8 в виде масла (выход 80,7%). МС ИЭР (+) m/z.
Пример 73
Получение (4-хлор-2-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанона (5f-7)
В этом примере описан синтез соединения 5f-7, как показано на Фиг.32, где R1 представляет собой изобутил и Ar представляет собой 4-хлор-2-фторфенил.
Стадии А-С: (4-Хлор-2-фторфенил-(1-изобутил-1Н-индазол-5-ил)метанол (соединение 4f-7) получали, как описано в Примере 27, стадии А-С, за исключением того, что вместо 2,4-дифторбензальдегида использовали 4-хлор-2-фторфенилбензальдегид.
Стадия D: (4-Хлор-2-фторфенил)-(1-изобутил-1Н-индазол-5-ил)метанол (соединение 4f-7; 20,4 мг, 0,0613 ммоль), Реагент Десс-Мартина (триацетоксипериодинан; 27,3 мг, 0,0644 ммоль) и 1 мл CH2Cl2 перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь наносили на систему Biotage. Элюция смесью эфир/гексаны (1:2) давала 12,0 мг соединения 5f-7 в виде твердого вещества (выход 59,2%). МС ИЭР (+) m/z.1Н-ЯМР (400 МГц, CDCl3) δ 8.15 (s, 1H), 8.11 (s, 1H), 7.99 (m, 1H), 7.53 (m, 1H), 7.47 (m, 1H), 7.30 (m, 1H), 7.24 (m, 1H), 4.21 (m, 1H), 2.37 (m, 1H), 0.95 (m, 6H).
Пример 74
Получение (2,4-дифторфенил)-[1-(2,2,2-трифторэтил)-1Н-индазол-5-ил]метанона (5f-11)
Стадия А: 5-Броминдазол (соединение 2f; 9,852 г, 50,0 ммоль) растворяли в 150 мл эфира и раствор охлаждали до -78°С. Медленно добавляли трет-BuLi (1,70 М раствор в пентане, 88,2 мл, 150 ммоль) при -78°С. После 0,5 часа при -78°С реакцию гасили 2,4-дифторбензальдегидом (10,9 мл, 100,0 ммоль) и медленно нагревали до комнатной температуры. Смесь перемешивали в течение 72 часов при комнатной температуре в атмосфере азота и гасили 100 мл воды. Слои разделяли и водный слой экстрагировали CH2Cl2 (6×50 мл). Объединенные органические экстракты промывали насыщенным раствором NaCl (100 мл), сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая желтое твердое вещество. Реакционную смесь очищали посредством хроматографии, элюируя смесью 5% МеОН в CH2Cl2. Во время обработки образца для хроматографии было обнаружено, что целевые фракции обладают слабой растворимостью в СН2Cl2. Смешанные фракции объединяли и концентрировали при пониженном давлении. Полученное в результате масло обрабатывали CH2Cl2 (примерно 50 мл), и образовывалось твердое вещество. Твердое вещество собирали фильтрацией.1Н-ЯМР спектры для "флэш" и отфильтрованного образца были одинаковы. Так как образцы обладали плохой растворимостью в CHCl3, то к1Н-ЯМР образцам добавляли две капли ДМСО-d6, получали 6,034 г 8f-1 в виде бледно-желтого твердого вещества (выход 46,4%). МС ИЭР (+), обнаружено: m/z 261 (М+Н).
Стадия В: Соединение 8f-1 (4,954 г, 19,04 ммоль) суспендировали в 150 мл CH2Cl2 и порциями добавляли реагент Десс-Мартина (9,156 г, 1,10 экв.) при комнатной температуре. После 3 часов при комнатной температуре смесь концентрировали при пониженном давлении, наносили на Samplet и элюировали 2% МеОН в CH2Cl2, получая твердое вещество. Твердое вещество суспендировали в 300 мл CH2Cl2 и 100 мл насыщенного раствора К2СО3 и интенсивно перемешивали в течение 2 часов. Смесь фильтровали и фильтрат экстрагировали CH2Cl2 (3×100 мл). К водному слою добавляли насыщенный раствор NaCl и слой экстрагировали CH2Cl2 (3×100 мл). Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 9f-1 в виде светло-коричневого твердого вещества (3,407 г, выход 69,3%). МС ИЭР (+), обнаружено: m/z 259 (М+Н).
Стадия С: Соединение 9f-1 (0,258 г, 1,0 ммоль), К2СО3 (0,207, 1,5 ммоль) и ДМФА (5 мл) помещали в небольшую пробирку Шленка, предназначенную для многократной герметизации. Из пробирки откачивали воздух и предварительно охлаждали ее в бане с сухим льдом (без ацетона). Шприц и трифторэтилбромид (0,244 г, 1.5 ммоль) также предварительно охлаждали в бане с сухим льдом. Пробирку открывали и вводили трифторэтилбромид, пока вся система была холодной. Пробирку герметично закрывали и нагревали до 100°С. Через 18 часов избыток ДМФА удаляли при пониженном давлении. Остаток обрабатывали водой (20 мл) и CH2Cl2 (20 мл). Слои разделяли и водный слой экстрагировали CH2Cl2 (4×10 мл). Объединенные экстракты промывали рассолом, сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 64,7 мг соединения 5f-11 (выход 19%). МС ИЭР (+), обнаружено: m/z 341 (М+Н).1Н-ЯМР (400 МГц, CDCl3) δ 8.20 (s, 2H), 8.05 (d, 2H), 7.62 (q, 1H), 7.52 (d, 1H), 7.04, (t, 1Н), 6.95 (t, 1H), 5.00 (q, 2H).
Пример 75
Получение (1-аллил-1H-индазол-5-ил)-(2,4-дифторфенил)метанона (5f-12)
Стадия А: Соединение 9f-1 получали, как описано в Примере 74.
Стадия В: В пробирку Шленка помещали соединение 9f-1 (0,516 г, 1,0 ммоль), К2СО3 (0,415 г, 1,5 ммоль), ДМФА (10 мл) и аллилбромид (0,363 г, 1,5 ммоль). Пробирку герметично закрывали и нагревали до 100°С. После 19 часов надосадочный раствор декантировали и соль промывали ДМФА (5 мл × 3). Объединенный надосадочный раствор концентрировали при пониженном давлении. Остаток растворяли в CH2Cl2 (20 мл) и промывали водой. Водный слой экстрагировали CH2Cl2 (10 мл × 2). Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 142,1 мг соединения 5f-12 (выход 23,8%). МС ИЭР (+), обнаружено: m/z 299 (М+Н).1Н-ЯМР (400 МГц, CDCl3) δ 8.17 (s, 1H), 8.12 (s, 1Н), 7.98 (d, 1H), 7.60 (m, 1H), 7.48, (d, 1H), 7.04 (td, 1H), 6.95 (td, 1H), 6.05 (m, 1Н), 5.28 (d, 1H), 5.17 (d, 1H), 5.06 (dt, 2H).
Примеры 76-79 описывают синтез анилиновых соединений общей Формулы XI. На Фиг.34 показана реакционная схема синтеза соединений, имеющих общую структуру 2h.
Пример 76
Получение (2,4-дифторфенил)-(1-изобутил-1Н-индазол-5-ил)амина (2h-1)
В этом примере описан синтез соединения 2h-1, как показано на Фиг.34, где R1 представляет собой изобутил и Ar представляет собой 2,4-дифторфенил.
Стадия А: Тетрафторборат аммония (20,97 г, 200 ммоль) растворяли в водном растворе уксусной кислоты (500 мл АсОН/250 мл воды) и охлаждали до 0°С. Последовательно добавляли 2-метил-4-броманилин (18,61 г, 100 ммоль) и 42 мл концентрированной водной HCI (36% (мас./мас.), 12 н., 500 ммоль). Смесь перемешивали в течение 20 минут при 0°С и добавляли NaNO2 (7,59 г, 110 ммоль). Реакционную смесь перемешивали в течение 1 часа при 0°С и нагревали до комнатной температуры. После 16 часов при комнатной температуре смесь концентрировали при пониженном давлении, остаток подвергали азеотропной перегонке с толуолом и сушили под высоким вакуумом. Твердое вещество суспендировали в 500 мл CHCl3 и добавляли КОАс (12,76 г, 130 ммоль) и 18-краун-6 (7,93 г, 30 ммоль). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре. Смесь промывали водой, сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 30 г 5-бром-1Н-индазола (соединения 2f) в виде желто-коричневого твердого вещества. Неочищенное вещество использовали без дальнейшей очистки.
Стадия В: Неочищенный 5-бром-1H-индазол (соединение 2f; 100 ммоль) растворяли в 250 мл ДМФА. Добавляли К2СО3 (20,7 г, 150 ммоль) и 1-бром-2-метилпропан (16,3 мл, 150 ммоль). Смесь нагревали до 120°С в атмосфере азота в течение 16 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли воду (200 мл) и СН2Cl2 (200 мл) и интенсивно перемешивали в течение 30 минут. Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая примерно 30 г неочищенного вещества. Неочищенное вещество очищали посредством хроматографии (эфир/гексаны от 1:9 до 1:4), получая 12,870 г соединения 3f в виде темно-красного масла с выходом 50,8% на стадиях А и В. МС ИЭР (+), обнаружено: m/z 253 и 255 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.93 (s, 1H), 7.87 (m, 1H), 7.43 (m, 1H), 7.29 (m, 1H), 7.29 (m, 1H), 4.15 (m, 2H), 2.33 (m, 1H), 0.92 (m, 6H).
Стадия С: Соединение 3f (2,53 г, 10,0 ммоль) растворяли в 50 мл эфира и раствор охлаждали до -78°С. Добавляли по каплям 12,4 мл трет-BuLi (1,7 М, 21,0 ммоль) и смесь перемешивали еще 30 минут при -78°С. Реакцию гасили В(ОМе)3 (2,4 мл, 21,0 ммоль) и смесь медленно нагревали до комнатной температуры. Через 15 минут реакцию гасили 6 н. HCl (10 мл, 60 ммоль). Реакционную смесь переносили в делительную воронку и добавляли воду (100 мл) и CH2Cl2 (100 мл). Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты сушили над безводным MgSO4, фильтровали через целит и концентрировали при пониженном давлении и очищали посредством хроматографии: эфир/гексаны (2:1) до 5% МеОН в CH2Cl2, получая соединение 1h в виде бледно-желтого твердого вещества (1,41 г, выход 64,7%). МС ИЭР (+), обнаружено: m/z 219 (М+1).
Стадия D: Соединение 1h (109 мг, 0,50 ммоль), ацетат меди (II) (50,3 мг, 0,10 ммоль), миристиновую кислоту (46 мг, 0,20 ммоль) и 2 мл сухого толуола помещали в колбу. Добавляли 2,6-лутидин (58 мкл, 0,50 ммоль) и смесь перемешивали в течение нескольких минут. Добавляли 2,4-дифторанилин (0,75 ммоль, 76 мкл) и смесь интенсивно перемешивали в атмосфере воздуха в течение 90 часов. Смесь разбавляли 10 мл эфира, фильтровали через целит и концентрировали при пониженном давлении, получая вязкое темно-зеленое масло. Неочищенное вещество очищали хроматографией, используя смесь эфир/гексаны (1:4) и получая 59 мг соединения 2h-1 в виде желто-коричневого масла (выход 39%). МС ИЭР (+), обнаружено: m/z 302 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.90 (s, 1Н), 7.39 (m, 1Н), 7.36 (m, 1Н), 7.16 (m, 1Н), 7.07 (m, 1Н), 6.89 (m, 1H), 6.75 (m, 1H), 5.59 (br s, 1H, NH), 4.16 (m, 2H), 2.35 (m, 1H), 0.95 (m, 6H).
Пример 77
Получение (4-фторфенил)-(1 -изобутил-1H-индазол-5-ил)амина (2h-2)
В этом примере описан синтез соединения 2h-2, как показано на Фиг.34, где R1 представляет собой изобутил и Ar представляет собой 4-фторфенил.
Стадии А-С: Соединение 1h получали, как описано в Примере 76, стадии А-С.
Стадия D: Соединение 1h (109 мг, 0,50 ммоль), ацетат меди (II) (25,2 мг, 0,05 ммоль), миристиновую кислоту (23 мг, 0,10 ммоль) и 2 мл сухого толуола помещали в колбу. К смеси добавляли 2,6-лутидин (58 мкл, 0,50 ммоль, 1,0 экв.) и перемешивали ее в течение нескольких минут. Добавляли 4-фторанилин (71 мкл, 0,75 ммоль, 1,5 экв.) и эту смесь интенсивно перемешивали на воздухе (в условиях воздушного окисления для медного катализатора) в течение 21 часа. Смесь разбавляли 10 мл эфира, фильтровали через целит и концентрировали при пониженном давлении, получая очень вязкое темно-зеленое масло. Неочищенное вещество очищали хроматографией, используя смесь эфир/гексаны (1:1) и получая 41 мг соединения 2h-2 (выход 28,9%) в виде желто-коричневого масла. МС ИЭР (+), обнаружено: m/z 284 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.87 (s, 1H), 7.34 (s, 1H), 7.33 (m, 1Н), 7.13 (m, 1Н), 6.98~6.91 (m, 4Н), 4.15 (m, 2Н), 2.35 (m, 1Н), 0.94 (6Н).
Пример 78
Получение (2,4-дихлорфенил)-(1-изобутил-1Н-индазол-5-ил)амина (2h-9)
В этом примере описан синтез соединения 2h-9, как показано на Фиг.34, где R1 представляет собой изобутил и Ar представляет собой 2,4-дихлорфенил.
Стадии А-С: Соединение 1h получали, как описано в Примере 76, стадии А-С.
Стадия D: Соединение 1h (109 мг, 0,50 ммоль), ацетат меди (II) (50,3 мг, 0,10 ммоль), миристиновую кислоту (46 мг, 0,20 ммоль) и 2 мл сухого толуола помещали в колбу. К смеси добавляли 2,6-лутидин (58 мкл, 0,50 ммоль, 1,0 экв.) и перемешивали ее в течение двух минут. Добавляли 2,4-дихлоранилин (122 мг, 0,75 ммоль, 1,5 экв.) и смесь интенсивно перемешивали на воздухе (в условиях воздушного окисления для медного катализатора) в течение 90 часов. Смесь разбавляли 10 мл эфира, фильтровали через целит и концентрировали при пониженном давлении, получая очень вязкое темно-зеленое масло. Неочищенное вещество очищали хроматографией, используя смесь эфир/гексаны (1:4) и получая 59 мг соединения 2h-9 в виде желто-коричневого масла (выход 35%). МС ИЭР (+), обнаружено: m/z 334 и 336 (М+1).
Пример 79
Получение (1-изобутил-1Н-индазол-5-ил)-О-толиламина (2h-10)
В этом примере описан синтез соединения 2h-10, как показано на Фиг.34, где R1 представляет собой изобутил и Ar представляет собой 2-метилфенил.
Стадии А-С: Соединение 1h получали, как описано в Примере 76, стадии А-С.
Стадия D: Соединение 1h (109 мг, 0,50 ммоль), ацетат меди (II) (50,3 мг, 0,10 ммоль), миристиновую кислоту (46 мг, 0,20 ммоль) и 2 мл сухого толуола помещали в колбу. К смеси добавляли 2,6-лутидин (58 мкл, 0,50 ммоль, 1,0 экв.) и перемешивали ее в течение двух минут. Добавляли 80 мкл О-толуидина (0,75 ммоль, 1,5 экв.) и смесь интенсивно перемешивали на воздухе (в условиях воздушного окисления для медного катализатора) в течение 90 часов. Смесь разбавляли 10 мл эфира, фильтровали через целит и концентрировали при пониженном давлении, получая очень вязкое темно-зеленое масло. Неочищенное вещество очищали хроматографией, используя смесь эфир/гексаны (1:4) и получая 77 мг соединения 2h-10 в виде желто-коричневого масла (выход 55%). МС ИЭР (+), обнаружено: m/z 280 (М+1).
Примеры 80-82 описывают синтез аминокислотных соединений общей Формулы XV. На Фиг.35 показана реакционная схема синтеза соединений, имеющих общую структуру 1j.
Пример 80
Получение метилового эфира 4-амино-2-{[5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}масляной кислоты (1j-2)
Стадия А: Соединение 10g-1 получали, как описано в Примере 46.
Стадия В: Раствор соединения 10g-1 (50 мг, 0,15 ммоль) в ТГФ (0,5 мл) обрабатывали КДИ (1,1 экв.) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 часов добавляли метиловый эфир 2-амино-4-трет-бутоксикарбониламиномасляной кислоты (36 мг, 0,165 ммоль) с последующим добавлением N,N-диизопропилэтиламина (29 мг, 0,225 ммоль). После перемешивания в течение 18 часов реакционную смесь концентрировали, остаток переносили в СН2Cl2 и промывали 1 н. HCl. Органический слой фильтровали через бумагу 1PS и очищали на картридже Sep Pak, элюируя смесью CH2Cl2/Et2O (10:1). Целевые фракции концентрировали, получая 72 мг соединения 1j-1 в виде бежевой пены (выход 99%).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.65 (br, 1H), 8.10 (s, 1H), 7.9 (s, 1H), 7.28 (1Н, s), 4.21 (d, 2Н), 4.42 (m, 1Н), 3.6 (s, 3Н), 2.95 (m, 2Н).
Стадия С: Раствор соединения 1j-1 (72 мг, 0,13 ммоль) в CH2Cl2 (0,2 мл) обрабатывали ТФУ (0,1 мл) при комнатной температуре. Через 18 часов растворитель концентрировали и совместно выпаривали с эфиром, получая 70 мг (выход 98%) соединения 1j-2 в виде масла янтарного цвета.1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.85 (br, 1H), 8.01 (s, 1H), 7.98 (s, 1H), 7.70 (br, 2H), 4.60 (m, 1Н), 4.22 (d, 2Н), 3.80 (s, 3Н), 2.85 (m, 2Н).
Пример 81
Получение метилового эфира 4-амино-2-{[5-(4-фторфенокси)-1-(2,2,2-трифторэтил)-1Н-индазол-6-карбонил]амино}масляной кислоты (1j-4)
Стадия А: Соединение 10g-2 получали, как описано в Примере 59.
Стадия В: Соединение 10g-2 (0,026 г, 0,073 ммоль), бензотриазол-1,3-диол (0,013 г, 0,088 ммоль) и (3-диметиламинопропил)этилкарбодиимид (0,017 г, 0,088 ммоль) добавляли к дихлорэтану и смешивали в течение 10 минут. Затем добавляли гетерогенную смесь хлористоводородной соли метилового эфира 2-амино-4-трет-бутоксикарбониламиномасляной кислоты (0,039 г, 0,147 ммоль) и триэтиламина (0,030, 0,29 ммоль) в дихлорэтане. Реакционную смесь перемешивали в течение 3 часов, концентрировали и очищали обращенно-фазовой ВЭЖХ согласно способу А Примера 86, получая примерно 30 мг чистого соединения 1j-3 (выход 71,9%). МС ИЭР (+), обнаружено: m/z 569 (М+Н).
Стадия С: Соединение 1j-3 (0,0012 г, 0,024 ммоль) добавляли к смеси СН2Cl2/ТФУ (1:1) в течение 1,5 часов, затем концентрировали, получая 2,3 мг (выход 100%) соединения 1j-4.1Н-ЯМР (400 МГц, CDCl3) δ 9.21 (br, 1H), 8.40 (br, 1Н), 8.04 (br, 1H), 7.44 (br, 1H), 7.18 (s, 1H), 7.03 (m, 3Н), 5.05 (m, 2H), 4.80 (br, 1Н), 3.75 (s, 3Н), 3.36 (br, 1H), 2.97 (br, 1H), 2.51 (br, 1H), 1.92 (br, 1H).
Пример 82
Получение метилового эфира 4-амино-2-{[5-(4-фторфенокси)-1-метил-1H-индазол-6-карбонил]амино}масляной кислоты (1j-6)
Стадия А: Соединение 10g-3 получали, как описано в Примере 60.
Стадия В: Соединение 10g-3 (0,026 г, 0,090 ммоль), бензотриазол-1,3-диол (0,017 г, 0,11 ммоль) и (3-диметиламинопропил)этилкарбодиимид (0,021 г, 0,017 ммоль) добавляли к дихлорэтану и смешивали в течение 10 минут. Затем добавляли гетерогенную смесь хлористоводородной соли метилового эфира 2-амино-4-трет-бутоксикарбониламиномасляной кислоты (0,05 г, 0,20 ммоль) и триэтиламина (0,037, 0,36 ммоль) в дихлорэтане. Реакционную смесь перемешивали в течение 3 часов и затем очищали обращенно-фазовой ВЭЖХ согласно способу А Примера 86, получая 30 мг (выход 66%) соединения 1j-5 в виде чистого вещества. МС ИЭР (+), обнаружено: m/z 501 (М+Н).
Стадия С: Соединение 1j-5 (0,0012 г, 0,024 ммоль) добавляли к смеси СН2Cl2/ТФУ (1:1) в течение 1,5 часов, затем концентрировали, получая 1,2 мг соединения 1j-6 (выход 100%).1Н-ЯМР (400 МГц, CDCl3) δ 9.10 (br, 1H), 8.32 (br, 1Н), 8.05 (br, 1H), 7.90 (s, 1H), 7.05 (s, 1H), 7.05 (m, 3Н), 4.75 (br, 1H), 4.14 (s, 3Н) 3.65 (s, 3Н), 3.30 (br, 1H), 2.92 (br, 1H), 2.51 (br, 1H), 1.82 (br, 1H).
Примеры 83-85 описывают синтез соединения Формулы XVI, как показано на Фиг.36.
Пример 83
Получение [5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-ил]метил-(2-диметиламиноэтил)амина (1k-1)
Соединение 11g-10 (0,05 г, 0,12 ммоль), полученное, как описано в Примере 55, обрабатывали 6 эквивалентами ВН3 в ТГФ (1 М раствор) и перемешивали при 65°С в течение 6 часов и затем при комнатной температуре в течение 14 часов. Растворитель удаляли путем выпаривания и остаток очищали препаративной ТСХ с использованием смеси гексан/этилацетат (1:1) и 5% триэтиламина, получая 0,014 г продукта (выход 30%). Обнаружено: МН+ 385.
Пример 84
Получение соединения 1k-2
Соединение 1k-1, полученное, как описано в Примере 83, обрабатывали избытком метансульфонилхлорида и триэтиламина в THE при комнатной температуре в течение 4 часов и затем концентрировали, получая 0,010 г 1k-2. Обнаружено: МН+ 427.
Пример 85
Получение соединения 1k-3
Соединение 1k-1, полученное, как описано в Примере 83, обрабатывали избытком уксусного ангидрида и триэтиламином в THE при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали, остаток очищали препаративной ТСХ с использованием смеси гексан/этилацетат (1:1) и 5% триэтиламина, получая 0,005 г (выход 50%). Обнаружено: МН+ 463.
Пример 86
Условия для препаративной обращенно-сразовой ВЭЖХ
Способ А:
Колонка: YMC ODS-AQ, внутр. диам. 250×20 мм, s-10/20 мкм, 12 нм. Растворитель А: Н2О с 0,1% ТФУ. Растворитель В: ацетонитрил с 0,05% ТФУ. Сбор запускался при помощи масс-спектрометра.
Способ В:
Колонка: YMC ODS-AQ, внутр. диам. 250×20 мм, s-10/20 мкм, 12 нм. Растворитель А: Н2О с 0,1% ТФУ. Растворитель В: ацетонитрил с 0,05% ТФУ. Сбор запускался при помощи масс-спектрометра.
Способ С:
Колонка: YMC ODS-AQ, внутр. диам. 250×20 мм, s-10/20 мкм, 12 нм. Растворитель А: Н2О с 0,1% ТФУ. Растворитель В: ацетонитрил с 0,05% ТФУ. Сбор запускался при помощи масс-спектрометра.
Пример 87
Получение соединения 1m-1
Синтез соединения 1m-1 показан на Фиг.37.
Стадия А: Соединение 1j-7 (0,07 г, 0,13 ммоль), полученное способом, аналогичным способу, описанному для соединения 1j-3, обрабатывали борогидридом натрия (10 экв., 0,049 г, 1,3 ммоль) в смеси МеОН/ТГФ (1:1) и нагревали до 60°С в течение 3 часов. Реакционную смесь концентрировали и затем совместно упаривали с МеОН, получая соединение 1l-1.
Стадия В: Соединение 1l-1 помещали в смесь МеОН/4 М HCl в диоксане (1:1) на 1,5 часа и затем реакционную смесь концентрировали. Остаток переносили в хлороформ, промывали 0,6 М раствором Na2CO3 (pH 7,0) и водным насыщенным NaCl и сушили над MgSO4. После фильтрации фильтрат упаривали, получая соединение 1m-1 (с чистотой 99%) в виде свободного основания.1Н-ЯМР (400 МГц), CDCl3: δ 8.39 (d, 1H), 8.34 (s, 1H), 7.90 (s, 1Н), 7.24 (s, 1Н), 6.98 (М, 4Н), 4.27 (m, 1Н), 4.20 (d, 2Н), 3.64 (m, 2Н), 2.65 (m, 1Н), 2.39 (m, 1Н), 2.37 (m, 1Н), 2.18 (m, 1Н), 1.59 (m, 1Н), 0.93 (d, 6H).
Примеры 88-109 описывают синтез анилиновых соединений общей Формулы XVII.
Пример 88
Получение 1-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-3-[2-(1-метил-1Н-индазол-5-илокси)пиридин-3-илметил]мочевины (6n)
Реакционная схема синтеза соединения 6n показана на Фиг.38.
Стадия А: 2-(1-Метил-1Н-индазол-5-илокси)никотинонитрил (3n). 1-Метил-1Н-индазол-5-ол (1n) (синтезированный, как описано в Примере 94) (0,10 г, 0,68 ммоль) и 2-хлорникотинонитрил (2n) (0,11 г, 0,81 ммоль) суспендировали в ДМСО (2 мл). Реакционную смесь нагревали до 110°С в течение 18 часов. Реакционную смесь разбавляли водой и экстрагировали в EtOAc. Объединенные органические вещества сушили Na2SO4 и концентрировали при пониженном давлении, получая неочищенный продукт. Очистка колоночной флэш-хроматографией (20-100% EtOAc/гексаны) давала целевой продукт (3n), (0,152 г, выход 90%).1Н-ЯМР (400 МГц, CD3OD) δ 8.27-8.25 (m, 1Н), 8.23-8.20 (m, 1Н), 8.01 (s, 1Н), 7.62 (d, J=8.6 Гц, 1Н), 7.57 (d, J=2.3 Гц, 1Н), 7.28-7.26 (m, 1Н), 7.23-7.20 (m, 1Н), 4.10 (s, 3Н). МС ИЭР (+), обнаружено: m/z 251 (М+Н).
Стадия В: С-[2-(1-Метил-1Н-индазол-5-илокси)пиридин-3-ил]метиламин (4n). 2-(1-Метил-1Н-индазол-5-илокси)никотинонитрил (3n) (0,132 г, 0,528 ммоль) суспендировали в МеОН (6 мл). Добавляли Pd(OH)2 (0,060 мг, 0,427 ммоль) в атмосфере азота с последующим добавлением концентрированной водной HCl (0,6 мл). Систему продували газообразным Н2 и реакционную смесь перемешивали при комнатной температуре в течение 3 часов в атмосфере Н2 (г). Реакционную смесь фильтровали через наполнитель из целита, промывали МеОН. Органические вещества концентрировали при пониженном давлении, получая неочищенный продукт, который очищали колоночной флэш-хроматографией (MeOH/Et3N/EtOAc), получая целевой продукт (4n) (0,047 г, выход 35%).1Н-ЯМР (400 МГц, CD3OD) δ 7.96 (s, 1H), 7.90 (d, J=4.7 Гц, 1Н), 7.82 (d, J=7.0 Гц, 1Н), 7.56 (d, J=9.4 Гц, 1Н), 7.46 (d, J=2.3 Гц, 1Н), 7.23-7.21 (m, 1H), 7.09-7.06 (m, 1H), 4.07 (s, 3Н), 3.95 (s, 2H).
Стадия С: 1-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-3-[2-(1-метил-1Н-индазол-5-илокси)пиридин-3-илметил]мочевина (6n). С-[2-(1-Метил-1Н- индазол-5-илокси)пиридин-3-ил]метиламин (4n) (0,017 г, 0,067 ммоль) и 2,2,2-трихлорэтиловый эфир (5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)карбаминовой кислоты (5n) (0,035 г, 0,087 ммоль) помещали в 10 мл реакционный сосуд и растворяли в ДМФА (5 мл). К реакционной смеси добавляли ДИЭА (диизопропилэтиламин) (0,058 мл, 0,334 ммоль) и систему нагревали до 80°С в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении, получая неочищенный продукт.Неочищенное вещество очищали колоночной флэш-хроматографией с использованием картриджа Sep Pak (10 г, 35 см3) с диоксидом кремния (50% EtOAc/гексаны), получая целевой продукт (6n) (0,034 г, выход 100%).1Н-ЯМР (400 МГц, ДМСО) δ 8.32 (s, 1Н), 8.00 (s, 1Н), 7.94 (d, J=4.7 Гц, 1Н), 7.65 (d, J=8.6 Гц, 1Н), 7.60 (d, J=7.8 Гц, 1Н), 7.46 (s, 1Н), 7.36 (d, J=7.8 Гц, 2Н), 7.29 (d, J=7.8 Гц, 2Н), 7.19-7.16 (m, 1Н), 7.07-7.03 (m, 2Н), 6.26 (s, 1Н), 4.37 (d, J=6.3 Гц, 2Н), 4.06 (s, 3Н), 2.36 (s, 3H), 1.25 (s, 9H). МС ИЭР (+), обнаружено: m/z 510 (М+Н).
Пример 89
Получение 2-(4-{2-[2-(1-циклобутилметил-1Н-индазол-5-илокси)-5-фторфенил]ацетил}пиперазин-1-ил)-N-изопропилацетамида (12р)
Реакционная схема синтеза соединения 12р показана на Фиг.39.
Стадия А: 1-Аллилокси-4-фторбензол (3р). К раствору 4-фторфенола (1р) (30 г, 268,0 ммоль) в ацетоне (250 мл) добавляли безводный К2СО3 (65 г, 468,3 ммоль), а затем 3-бромпропен (2р) (28 мл, 321,1 ммоль). Полученную смесь нагревали с обратным холодильником в течение 16 часов, охлаждали до комнатной температуры и затем вливали в ледяную воду (500 мл). Водный слой экстрагировали эфиром (3×250 мл), объединенные органические слои промывали 2 М NaOH (2×150 мл) и сушили над смесью безводных К2СО3 и Na2SO4. Растворитель удаляли под вакуумом, получая целевой продукт (3р) (40,4 г, 99%) в виде светло-желтого масла.1Н-ЯМР (400 МГц, CDCl3) δ 7.01-6.92 (m, 2H), 6.89-6.82 (m, 2H), 6.10-6.82 (m, 1H), 5.44-5.41 (m, 1H), 5.39-5.37 (m, 1H), 4.51-4.448 (m, 2H).
Стадия В: 2-Аллил-4-фторфенол (4р). Промежуточное соединение (3р) (14,7 г, 96,6 ммоль) нагревали до 210°С в течение 7 часов, охлаждали до комнатной температуры и выдерживали в течение ночи. Ход реакции контролировали с помощью тонкослойной хроматографии. В ТСХ анализе обнаружили одно новое пятно (Rf ~0,65 в смеси гексан/этилацетат, 7:3). ВЭЖХ неочищенной смеси давала основной пик с временем удерживания 2,07 минут и минорный пик при 2,36 мин. Основной неочищенный продукт (4р) подтверждали в качестве целевого продукта и направляли его сразу на следующую стадию без очистки.1Н-ЯМР (400 МГц, CDCl3) δ 6.88-6.78 (m, 2H), 6.78-6.72 (m, 1H), 6.05-5.93 (m, 1H), 5.21-5.13 (m, 2H), 4.8 (br s, OH), 3.38 (d, J=6.26 Гц, 2Н).
Стадия С: 2-Аллил-4-фторфениловый эфир уксусной кислоты (5р). К неочищенному (4р) добавляли уксусный ангидрид (36,5 мл, 386,4 ммоль) и пиридин (37,5 мл, 463,7 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 18 часов и на следующий день контролировали с помощью ВЭЖХ (реакция, по-видимому, была в основном завершена). Затем смесь вливали в холодную смесь H2O/Et2O, водный слой экстрагировали Et2O (2×), объединенные органические слои последовательно промывали 10%-ным раствором HCl (3×), насыщенным NaHCO3 (2×), Н2О (2×) и рассолом и затем сушили над безводным Na2SO4. Чистоту неочищенного продукта после концентрирования проверяли с помощью тонкослойной хроматографии (гексан/этилацетат, 7:3) и ВЭЖХ. Масс-ион не обнаружили. Неочищенный продукт (5р) направляли сразу на следующую стадию без дальнейшей очистки.1Н-ЯМР (400 МГц, CDCl3) δ 7.04-6.91 (m, 3H), 6.09-5.65 (m, 1Н), 5.19-5.06 (m, 2Н), 3.27 (d, J=6.26 Гц, 2Н), 2.30 (s, CH3).
Стадия D: (2-Ацетокси-5-фторфенил)уксусная кислота (6р-2). К раствору (5р) (10 г, 51,5 ммоль) в 100 мл смеси CCl4/ацетонитрил (1:1) добавляли раствор метапериодата натрия (NaIO4, 33,6 г, 154,5 ммоль) в 500 мл Н2О. После перемешивания в течение нескольких минут добавляли гидрат трихлорида рутения (0,93 г, 4,12 ммоль). Темную смесь перемешивали при комнатной температуре в течение 2 часов и добавляли ДХМ (600 мл). Слои разделяли, водную фазу экстрагировали ДХМ (3×), объединенные органические слои промывали Н2О и сушили над Na2SO4. Фильтрация через целит 545 и упаривание давали смесь альдегида (6р-1) и кислоты (6р-2) (9,1 г, 83%) в виде коричневого масла, которое направляли на следующую стадию без очистки.
Стадия Е: (2-Ацетокси-5-фторфенил)уксусная кислота (7р). Раствор хлорита натрия (52,16 г, 576,7 ммоль) и дигидрофосфата натрия (44,5 г, 371 ммоль) в 225 мл Н2О добавляли к раствору кислоты (6р-2) и альдегида (6р-1) в 100 мл изо-PrOH при 0°С. Полученный раствор перемешивали при 0°С в течение 3 часов, разбавляли эфиром и затем разделяли слои. Органическую фазу промывали Н2О, 10%-ным тиосульфатом натрия (2×), Н2О и рассолом и сушили над Na2SO4. После упаривания до небольшого объема добавляли несколько капель гексана. Постепенно образовывались кристаллы, которые собирали фильтрацией и промывали холодной смесью эфир/гексан, получая целевое соединение (7р) (3,95 г, выход выделенного продукта 36%).1Н-ЯМР (400 МГц, CDCl3) δ 7.12-6.98 (m, 3Н), 3.57 (s, 2H), 2.29 (s, CH3). MC (ХИАД-) (химическая ионизация при атмосферном давлении), обнаружено: m/z 422,7 (2М-Н).
Стадия F: (5-Фтор-2-гидроксифенил)уксусная кислота (8р). Соединение (7р) (3,5 г, 16,5 ммоль) растворяли в 65 мл МеОН и добавляли 7 мл гидроксида аммония (49,5 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и затем анализировали с помощью ТСХ (ДХМ/МеОН/АсОН (9:1:0,15)), ВЭЖХ и МС. Исходного вещества не обнаружили. Вещество концентрировали досуха, получая целевой продукт (8р), который непосредственно направляли на следующую стадию. МС (ХИАД-), обнаружено: m/z 168,9 (М-Н), 338,7 (2М-Н).
Стадия G: [2-(1-Циклобутилметил-1Н-индазол-5-илокси)-5-фторфенил]уксусная кислота (10р). К раствору (8р) (2,8 г, 16,5 ммоль) в 6 мл NMP (N-метилпирролидона) добавляли карбонат цезия (24,2 г, 74,24 ммоль), и реакционная смесь отверждалась. Добавляли еще 12 мл NMP и карбоната цезия (6,29 г, 19,3 ммоль) и реакционную смесь продували азотом. После интенсивного перемешивания добавляли соединение (9р) (5,25 г, 19,8 ммоль) и 2,2,6,6-тетраметилгептан-3,5-дион (90,86 мл, 4,12 ммоль). Реакционную смесь дегазировали и продували азотом. Добавляли хлорид меди (I) (0,82 г, 8,24 ммоль), реакционную смесь дегазировали, продували азотом и нагревали до 140°С. После перемешивания в течение 18 часов реакционную смесь охлаждали до комнатной температуры (примерно 23°С), разбавляли Et2O и фильтровали. Собранные твердые вещества промывали несколько раз эфиром, растворяли в Н2О, подкисляли 6 н. HCl и экстрагировали ДХМ (4×). Объединенные органические слои промывали Н2О и рассолом и сушили над Na2SO4. После концентрирования остаток очищали нормально-фазовой хроматографией, используя смесь гексан/EtOAc/АсОН (9:1:0,15), получая целевой продукт (10р) (1,01 г, выход выделенного продукта 17%).1Н-ЯМР (400 МГц, CDCl3) δ 7.84 (s, 1Н), 7.36 (d, J=8.62 Гц, 1Н), 7.14 (d, J=2.35 Гц, 1Н), 7.11 (dd, J=8.61, 2.35 Гц, 1Н), 7.05 (dd, J=8.61, 3.13 Гц, 1Н), 6.92 (ddd, J=8.61, 8.61, 3.13 Гц, 1H), 6.79 (dd, J=8.61, 4.70 Гц, 1H), 4.35 (d, J=7.04 Гц, 2H), 3.73 (s, 2H), 2.93-2.82 (m, 1H), 2.06-1.97 (m, 2H), 1.94-1.76 (m, 4H). MC ИЭР (+), обнаружено: m/z 355 (М+Н).
Стадия Н: 2-(4-{2-[2-(1-Циклобутилметил-1Н-индазол-5-илокси)-5-фторфенил]ацетил}пиперазин-1-ил)-N-изопропилацетамид (12р). Соединение (10р) (0,087 г, 0,247 ммоль) растворяли в CHCI3 (1,6 мл), смешивали с EDCI (1-этил-3,3-бис-(метиламино)пропилкарбодиимидом) (0,072 г, 0,372 ммоль) и перемешивали при комнатной температуре в течение 30 минут. Добавляли N-изопропил-2-пиперазин-1-илацетамид (12р) (0,069 г, 0,372 ммоль), а затем дополнительно 0,8 мл CHCl3. Полученный раствор перемешивали при комнатной температуре в течение 18 часов. Добавляли PS-изоцианат (0,850 г, 1,6 ммоль/г) и реакционную смесь встряхивали в течение 1 часа. После фильтрации фильтрат промывали Н2О (2×), сушили над Na2SO4 и концентрировали. Остаток очищали посредством хроматографии (Sep Pak, 10 г) (ДХМ, EtOAc), получая целевой продукт (12р) (0,1 г, 77%).1Н-ЯМР (400 МГц, CDCl3) δ 7.87 (s, 1Н), 7.40 (d, J=8.61 Гц, 1Н), 7.13-7.06 (m, 3Н), 6.91 (ddd, J=8.61, 8.61, 3.13 Гц, 1Н), 6.83-6.72 (m, 2Н), 4.38 (d, J=7.04 Гц, 2Н), 4.15-4.02 (m, 1Н), 3.74 (s, 2Н), 3.67-3.60 (m, 2Н), 3.55-3.49 (m, 2Н), 2.99-2.87 (m, 1Н), 2.91 (s, 2Н), 2.44-2.33 (m, 4Н), 2.10-2.00 (m, 2Н), 1.97-1.79 (m, 4Н), 1.16 (s, СН3), 1.15 (s, СН3). МС (ХИАД+), обнаружено: m/z 522,2 (М+Н).
Пример 90
Получение 2-[2-(1-изобутил-1Н-индазол-5-илокси)фенил]-N-(4-морфолин-4-илфенил)ацетамида (16р)
Реакционная схема синтеза соединения 16р показана на Фиг.40.
Стадия А: 5-Бром-1-изобутил-1Н-индазол (14р): К раствору 5-броминдазола в ДМФА добавляли К2СО3. Смесь нагревали до 105°С. После исчезновения 5-броминдазола реакционную смесь вливали в ДХМ/рассол. Два слоя разделяли, водный слой экстрагировали ДХМ (2×) и анализировали ТСХ. Объединенные органические вещества промывали Н2О (2×) и рассолом и сушили над Na2SO4. После фильтрации фильтрат концентрировали, полученный остаток очищали хроматографией, используя смесь гексан/EtOAc (9,5:0,5) и получая целевой продукт (14р).
Стадия В: [2-(1-Изобутил-1Н-индазол-5-илокси)фенил]уксусная кислота (15р). К дегазированной суспензии 2-гидроксибензойной кислоты (2,4 г, 15,8 ммоль) и Cs2CO3 (7,72 г, 23,7 ммоль) в NMP (13 мл) добавляли 2,2,6,6-тетраметилгептан-3,5-дион (0,41 мл, 1,97 ммоль) и соединение 14р (2,0 г, 7,90 ммоль), а затем небольшое количество NMP для промывки. Полученную смесь снова дегазировали азотом, затем добавляли CuCl (0,39 г, 3,95 ммоль) и реакционную смесь снова дегазировали. Смесь нагревали до 140-150°С. После перемешивания в течение 22 часов реакционную смесь вливали в эфир/Н2О. Два слоя разделяли и водный слой (рН ~11) промывали эфиром. Водный слой подкисляли до рН 7 и экстрагировали эфиром (4×), объединенные органические слои сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении. Осадок формировался постепенно в небольшом объеме смешанного растворителя эфир/гексан/ДХМ и его собирали фильтрацией, получая целевое соединение (15р) (0,93 г, выход выделенного продукта 36%).1Н-ЯМР (400 МГц, CDCl3) δ 7.87 (br s, 1H), 7.38-7.28 (m, 2H), 7.25-7.17 (m, 2Н), 7.13 (d, J=9.39 Гц, 1Н), 7.10-7.03 (m, 1H), 6.79 (d, J=8.61 Гц, 1Н), 4.15 (br s, 2H), 3.79 (s, 2H), 2.40-2.27 (m, 1H), 0.93 (s, 3Н), 0.92 (s, 3Н). МС (ХИАД+), обнаружено: m/z 325 (М+Н), МС (ХИАД-), обнаружено: m/z 322,8 (М-Н) и 646,8 (2М-Н).
Стадия С: 2-[2-(1-Изобутил-1Н-индазол-5-илокси)фенил]-N-(4-морфолин-4-илфенил)ацетамид (16р). К раствору (15р) (0,04 г, 0,123 ммоль), PyBOP (0,135 г, 0,26 ммоль) и ДИЭА (0,02 мл, 0,12 ммоль) в CHCl3 (2 мл) добавляли 4-морфолин-4-илфениламин (0,044 г, 0,247 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, обрабатывали смолой АР-трисамин (0,25 г, 2,49 ммоль/г) и растворитель окончательно удаляли при пониженном давлении после фильтрации из смолы. Полученный остаток очищали посредством хроматографии (Sep Pak, 10 г) с эфиром, получая целевой продукт (16р) (0,024 г, 40%).1Н-ЯМР (400 МГц, CDCl3) δ 7.90 (s, 1Н), 7.50 (br s, 1Н), 7.45 (dd, J=7.83, 1.57 Гц, 1Н), 7.39 (d, J=9.39 Гц, 2Н), 7.31-7.26 (m, 3Н), 7.23 (dd, J=7.83, 1.57 Гц, 1Н), 7.15-7.09 (m, 2Н), 6.86-6.79 (m, 3Н), 4.17 (d, J=7.04 Гц, 2Н), 3.86-3.82 (m, 4Н), 3.80 (s, 2H), 3.11-3.06 (m, 4Н), 2.41-2.29 (m, 1Н), 0.95 (s, CH3), 0.94
(s, CH3). МС (ХИАД+), обнаружено: m/z 485,2 (М+Н).
Пример 91
Получение 1-[5-циклопропил-2-(4-трифторметилфенил)-2H-пиразол-3-ил]-3-[5-фтор-2-(1-метил-1Н-индазол-5-иламино)бензил]мочевины (9q-1) и 1-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-3-[2-(1-циклобутилметил-1Н-индазол-5-иламино)-5-фторбензил1мочевины (9q-2)
Реакционная схема синтеза соединений 9q-1 и 9q-2 показана на Фиг.41А и В.
Стадия А: 2-Азидо-5-фторбензонитрил (1q). Смесь NaN3 (1,17 г, 1,8 ммоль) и дифторбензонитрила (0,5 г, 3,6 ммоль) в ДМФА (60 мл) нагревали при 100°С в течение 30 минут. Затем смесь разбавляли водой (300 мл) и эфиром (300 мл). Органический слой трижды промывали водой и рассолом. Органический слой сушили (MgSO4) и концентрировали. Неочищенный продукт очищали колоночной флэш-хроматографией, используя в качестве элюента смесь эфир:гексан (1:5) и получая целевой продукт (1q) в виде белых кристаллов (0,3 г, выход выделенного продукта 53%).1Н-ЯМР (400 МГц, CDCl3) δ 7.38-7.31 (m, 2H), 7.27-7.18 (m, 1H).
Стадия В: 2-Амино-5-фторбензонитрил (2q). К раствору CoBr2 (15 мг, 0,068 ммоль) в этаноле (3 мл) добавляли 2,2′-дипиридил (10 мг, 0,068 ммоль) при комнатной температуре с последующим добавлением NaBH4 (40 мг, 1,02 ммоль). Реакционную смесь охлаждали до -10°С, а затем по каплям добавляли промежуточное соединение (2q) (0,22 г, 1,36 ммоль) в этаноле (1 мл) в течение 10 минут. Реакционную смесь перемешивали в течение 15 минут и затем гасили уксусной кислотой и метанолом при -10°С. Остаток затем растворяли в этилацетате и промывали насыщенным бикарбонатом натрия, рассолом, сушили (MgSO4) и растворители удаляли при пониженном давлении. Неочищенный продукт очищали колоночной флэш-хроматографией, используя в качестве элюента смесь эфир:гексан (1:2) и получая соединение (2q) в виде белых кристаллов (0,16 г, выход выделенного продукта 87%).1Н-ЯМР (400 МГц, CDCl3) δ 7.12-7.08 (m, 2Н), 6.7 (dd, J=10.4, 4.8 Гц, 1Н), 4.3 (or s, 2H).
Стадия С: трет-Бутиловый эфир (2-циано-4-фторфенил)-бис-(карбаминовой кислоты) (3q). К раствору (2q) (33 мг, 0,24 ммоль) в ТГФ (3 мл) добавляли Вос2О (200 мг, 0,72 ммоль) и DMAP (диметиламинопиридин) (5,9 мг, 0,048 ммоль) при комнатной температуре. Реакционную смесь нагревали с обратным холодильником в течение 2,5 часов, затем охлаждали до комнатной температуры и растворитель выпаривали при пониженном давлении. Неочищенный продукт очищали колоночной флэш-хроматографией, используя в качестве элюента смесь эфир:гексан (1:3) и получая продукт (3q) в виде белых кристаллов (0,08 г, выход выделенного продукта 98%).1Н-ЯМР (400 МГц, CDCl3) δ 7.4-7.26 (m, 3H), 1.45 (s, 18H).
Стадия D: трет-Бутиловый эфир (2-аминометил-4-фторфенил)-бис-(карбаминовой кислоты) (4q). К раствору (3q) (1 г, 2,97 ммоль) в этаноле (30 мл) добавляли CoBr2 (27 мг, 0,12 ммоль), 2,2′-дипиридил (57 мг, 0,36 ммоль) при комнатной температуре с последующим добавлением NaBH4 (350 мг, 9,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и гасили уксусной кислотой и метанолом при 0°С. Затем остаток растворяли в этилацетате и промывали насыщенным бикарбонатом натрия, рассолом, сушили (MgSO4) и растворители удаляли при пониженном давлении. Неочищенный продукт (4q) использовали непосредственно на следующей стадии (1 г, выход выделенного продукта 100%).
Стадия Е: Хлористоводородная соль 2-аминометил-4-фторфениламина (5с). Неочищенное промежуточное соединение (4q) (0,95 г, 2,8 ммоль) растворяли в МеОН/ДХМ (15 мл). Затем добавляли 4 н. HCl (10,5 мл, 42,0 ммоль) в диоксане и перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении и остаток (5q) направляли на следующую стадию без дополнительной очистки или идентификации.
Стадия F: трет-Бутиловый эфир (2-амино-5-фторбензил)карбаминовой кислоты (6q). Раствор Вос-ангидрида (0,49 г, 2,5 ммоль) в диоксане (5 мл) по каплям добавляли к охлажденному на ледяной бане раствору (5q) (2,8 ммоль, 1 экв.) в 5,7 мл 1 М NaHCO3 (5,63 ммоль) и диоксане (11,2 мл) (1:2). Реакционную смесь оставляли нагреваться до комнатной температуры и продолжали перемешивание при комнатной температуре в течение 18 часов. На следующий день смесь разбавляли Et2O и промывали рассолом. Слои разделяли. Водный слой (рассол) экстрагировали Et2O (3×), объединенные органические слои экстрагировали 10%-ным KHSO4 (3×), промывали Н2О и рассолом и сушили над Na2SO4. Полученный после концентрирования неочищенный продукт очищали посредством хроматографии (Sep Pak) с использованием гексана и смеси гексан/EtOAc (9:1), получая продукт (6q) (0,34 г, выход выделенного продукта 50%).1Н-ЯМР (400 МГц, CD3OD) δ 6.85-6.75 (m, 2Н), 6.6 (dd, J=7.8, 4.7 Гц, 1Н), 4.82 (br s, NH), 4.21 (d, J=6.2 Гц, 2H), 4.06 (br s, NH2), 1.45 (s, 9H). ЖХ-МС ИЭР (+), обнаружено: m/z 241 (M+H).
Стадия G: трет-Бутиловый эфир [5-фтор-2-(1-циклобутилметил-1Н-индазол-5-иламино)бензил]карбаминовой кислоты (7q-2). В колбу, содержащую бороновую кислоту (0,175 г, 0,76 ммоль), амин (6q) (0,22 г, 0,915 ммоль), Cu(ОАс)2 (0,135 г, 0,76 ммоль) и 4Å сита (0,2 г) в ДХМ, медленно добавляли Et3N (0,52 мл, 3,7 ммоль). Смесь перемешивали при комнатной температуре в течение 3 дней. К реакционной смеси добавляли ДХМ и фильтровали. Фильтрат промывали Н2О (2×), рассолом и сушили над Na2SO4. После концентрирования остаток очищали посредством хроматографии (Sep Pak; 10 г), используя смесь гексан/Et2О (3:1). Фракции, содержащие продукт, объединяли, получая (7q-2) (0,12 г, выход 37%).1Н-ЯМР (400 МГц, CDCl3) δ 7.82 (s, 1Н), 7.34 (d, J=8.6 Гц, 1Н), 7.24 (br s, 1Н), 7.13 (d, J=8.6 Гц, 1Н), 6.94-6.84 (m, 3Н), 5.02 (br s, NH), 4.35 (d, J=7.8 Гц, 2Н), 4.29 (d, J=6.2 Гц, 2Н), 2.98-2.85 (m, 1H), 2.10-1.98 (m, 2H), 1.95-1.79 (m, 4H), 1.44 (s, 9H). MC (ХИАД+), обнаружено: m/z 425 (M+H).
Стадия Н: (2-Аминометил-4-фторфенил)-(1-циклобутилметил-1Н-индазол-5-ил)амин (8q-2). Промежуточное соединение (7q-2) (0,076 г, 0,18 ммоль) растворяли в ДХМ/изо-PrOH (5 мл, 1:1), добавляли 0,5 мл HCl (1,97 ммоль) в диоксане и реакционную смесь перемешивали в течение 3 дней. Растворитель выпаривали, получая продукт (8q-2), который направляли на следующую стадию. ЖХ-МС ИЭР (+), обнаружено: m/z 308 (М-NH2). Как (7q-1), так и (8q-1) направляли на конечную стадию, используя протокол для аналогичных 7q-2 и 8q-2.
Стадия I: 1-[5-Циклопропил-2-(4-трифторметилфенил)-2Н-пиразол-3-ил]-3-[5-фтор-2-(1-метил-1Н-индазол-5-иламино)бензил]мочевина (9q-1). Раствор (8q-1) (0,15 г, 0,54 ммоль) в ДМФА (4,5 мл) обрабатывали при комнатной температуре карбаматом 10q (0,26 г, 0,6 ммоль), а затем ДИЭА (0,35 мл, 2,0 ммоль). Смесь нагревали при 80°С в течение 18 часов и затем растворитель выпаривали при пониженном давлении. Остаток переносили в ДХМ и промывали 1 н. HCl. Органический слой фильтровали через бумагу 1PS и упаривали досуха. Затем неочищенный продукт очищали с помощью ВЭЖХ, получая продукт (9q-1) (0,027 г, выход выделенного продукта 9%).1Н-ЯМР (500 МГц, CDCl3) δ 7.88 (s, 1Н), 7.59 (d, J=7.96 Гц, 2Н), 7.54 (d, J=7.43 Гц, 2Н), 7.57 (br s, NH), 7.37 (d, J=8.49 Гц, 1Н), 7.22-7.16 (m, 2H), 7.15 (s, 1H), 6.99-6.91 (m, 2Н), 6.21 (s, 1Н), 4.34 (s, 2Н), 4.07 (s, 3Н), 2.01-1.93 (m, 1Н), 1.14-0.98 (m, 2Н), 0.90-0,83 (m, 2Н). МС (ХИАД+), обнаружено: m/z 564 (М+Н).
Стадия J: 1-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-3-[2-(1-циклобутилметил-1Н-индазол-5-иламино)-5-фторбензил]мочевина (9q-2). Соединение (8q-2) (0,18 ммоль) растворяли в ДМФА (2,5 мл), добавляли карбамат 10q (0,08 г, 0,20 ммоль), а затем ДИЭА (0,1 мл, 0,57 ммоль). Реакционную смесь нагревали до 80°С в течение 18 часов. Растворитель выпаривали при пониженном давлении, остаток переносили в ДХМ и промывали 1 н. HCl. Органический слой фильтровали через бумагу 1PS и упаривали при пониженном давлении до масла. Затем неочищенный продукт очищали с помощью ВЭЖХ, получая продукт (9q-2) (0,045 г, выход выделенного продукта 44%).1Н-ЯМР (400 МГц, CDCl3) δ 7.81 (s, 1H), 7.7 (br s, NH), 7.32 (d, J=9.39 Гц, 1H), 7.18-7.06 (m, 7H), 6.94-6.85 (m, 2H), 6.50 (s, 1H), 4.37-4.30 (m, 6H), 2.96-2.81 (m, 1H), 2.27 (s, 3H), 2.09-1.96 (m, 2H), 1.94-1.76 (m, 4H), 1.34 (s, 9H). МС (ХИАД+), обнаружено: m/z 580 (M+H).
Пример 92
Получение 1-(5-трет-бутил-2-п-хлорфенил-2Н-пиразол-3-ил)-3-[2-(1-метил-1Н-индазо-5-илсульфанил)-5-фторбензил]мочевины (6r-2)
Реакционная схема синтеза соединения 6r-2 показана на Фиг.42.
Стадия А: 5-Бром-1Н-индазол (1r). 4-Бром-2-метиланилин (20 г, 107 ммоль), тетрафторборат аммония (23 г, 215 ммоль) и концентрированную HCl (45 мл, 537 ммоль) добавляли к АсОН/Н2О (350 мл, 2:1) и обрабатывали ультразвуком. Затем медленно добавляли NaNO2 (8,9 г, 129 ммоль) и реакционную смесь обрабатывали ультразвуком в течение еще 10 минут (она становилась темно-коричневой, и сразу образовывался осадок). Реакционную смесь оставляли перемешиваться в течение ночи. На следующий день исходное вещество не было обнаружено. Смесь упаривали в быстром вакууме при 65°С и затем подвергали азеотропной перегонке с толуолом досуха. Вещество использовали непосредственно на следующей стадии без дополнительной очистки. Упомянутое выше неочищенное вещество, ацетат калия (42 г, 428 ммоль) и 18-краун-6 (2,8 г, 11 ммоль) добавляли к хлороформу (300 мл) и обрабатывали ультразвуком в течение 10 минут. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Вещество пропускали через фильтровальную воронку со смесью силикагель/целит/песок и неоднократно промывали CHCl3 (вещество не было собрано). Затем колонку промывали EtOAc, получая оранжевое вещество, которое собирали, объединяли и упаривали, получая примерно 16 г вещества. Затем неочищенный продукт подвергали флэш-хроматографии на силикагеле с использованием ДХМ:МеОН (5%) в качестве элюента и сушили под высоким вакуумом в течение ночи, получая целевой продукт (1 г) (8 г, 50% выход).1Н-ЯМР (400 МГц, CDCl3) δ 11.9 (br s, 1H), 8.05 (s, 1H), 7.9 (s, 1H), 7.46 (d, J=8.8 Гц, 1Н), 7.39 (d, J=8.8 Гц, 1Н). МС (ИЭР) m/z 197,1 (М+Н)+.
Стадия В: 5-Бром-1-метил-1Н-индазол (2 г). 5-Бром-1Н-индазол (1 г) (10 г, 51 ммоль) в ТГФ медленно добавляли к холодному раствору NaH (2,2 г, 60 мас.% в масле, 56 ммоль) в ТГФ в атмосфере азота. Через 15 минут к темному раствору добавляли йодметан (10,8 г, 76 ммоль) при 0°С. Через 2 часа смесь вливали в 1 н. HCl (30 мл), экстрагировали EtOAc (2×50 мл), объединенные экстракты промывали рассолом (50 мл), сушили над Na2SO4) фильтровали и концентрировали. Колоночная хроматография (силикагель): гексан:EtOAc (10-40%) давала 8,2 г конечного продукта (2r).1Н-ЯМР (400 МГц, CDCl3) δ 7.9 (s, 1Н), 7.84 (s, 1Н), 7.43 (d, J=8.8 Гц, 1Н), 7.24 (d, J=8.8 Гц, 1Н), 4.04 (s, 3Н). МС (ИЭР) m/z 213 (М+Н)+.
Стадия С: 5-(Триизопропилсилилсульфанил)-1-метил-1Н-индазол (3r). КН (1,3 г, 30 мас.%, 9,8 ммоль) промывали ТГФ и затем суспендировали в ТГФ (10 мл) при 5°С. В течение 15 минут добавляли триизопропилсилилтиол (1,8 г, 9,3 ммоль) с интенсивным выделением водорода. Смесь перемешивали при 5°С в течение часа и затем при 25°С в течение 1 часа. Этот раствор добавляли к раствору 1-метил-5-броминдазола (2 г) (2 г, 9,5 ммоль) и (Ph3P)4Pd (1,1 г, 0,93 ммоль) в ТГФ (15 мл). Желтую суспензию перемешивали в течение 1 часа при 70°С. После охлаждения добавляли эфир, раствор промывали рассолом, сушили (Na2SO4) и концентрировали. Остаток подвергали хроматографии (силикагель, 3% EtOAc в гексане), получая 5-(триизопропилсульфанил)-1-метил-1Н-индазол (3r) (1,8 г, 59%).1Н-ЯМР (400 МГц, CDCl3) δ 7.89 (s, 1Н), 7.86 (s, 1Н), 7.48 (d, J=8.8 Гц, 1Н), 7.25 (d, J=8.4 Гц, 1Н), 4.05 (s, 3Н), 1.28-1.19 (m, 3Н), 1.08 (d, J=7.6 Гц, 18Н).
Стадия D: 2-(1-Метил-1Н-индазол-5-илсульфанил)-5-фторбензонитрил (4r). Соединение (3r) (0,65 г, 2 ммоль), карбонат калия (0,34 г, 2,4 ммоль), CsF (0,46 г, 3 ммоль), 2,5-дифторбензонитрил (0,56 г, 4,1 ммоль) и ДМФА (5 мл) помещали в 60 мл реакционный сосуд и этот сосуд герметично закрывали. Смесь нагревали до 100°С в течение 16 часов. Избыток ДМФА удаляли при пониженном давлении. Это вещество переносили в ДХМ (50 мл) и промывали водой (20 мл). Водный слой экстрагировали ДХМ (3×). Объединенные органические слои промывали рассолом (2×) и сушили над MgSO4, фильтровали через наполнитель целит/силикагель и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле с гексан/EtOAc (20%), получая конечный продукт в виде вязкой жидкости (4 г) (0,43 г, выход выделенного продукта 75%).1Н-ЯМР (400 МГц, CDCl3) δ 7.99 (s, 1Н), 7.97 (s, 1Н), 7.46 (dd, J=16.8, 8.8 Гц, 2Н), 7.35-7.32 (m, 1H), 7.14-7.07 (m, 1Н), 7.05-7.01 (m, 1Н), 4.1 (s, 3Н). МС (ИЭР) m/z 284,2 (М+Н)+.
Стадия Е: 2-(1-Метил-1Н-индазол-5-илсульфанил)-5-фторбензиламин (5r). Раствор соединения (4r) (0,43 г, 1,5 ммоль) в МеОН (30 мл) продували азотом и обрабатывали катализатором Pd(OH)2/C (15 мас.%, 280 мг, 0,3 ммоль), а затем концентрированной HCl (0,38 мл, 4,6 ммоль). После еще одной продувки азотом в верхней части колбы размещали баллон, заполненный водородом. После перемешивания при комнатной температуре в течение 18 часов анализ методом ЖХ показал отсутствие исходного вещества. Затем добавляли К2СО3 (0,5 г). Катализатор фильтровали через наполнитель силикагель/целит/песок и промывали смесью CHCl3/Et3N, растворитель удаляли при пониженном давлении. Полученную бледно-желтую пену (5r) (0,43 г, выход выделенного продукта 87%) хранили в атмосфере азота. МС (ИЭР) m/z 287,9 (М+Н)+.
Стадия F: 1-(5-Циклопропил-2-п-хлорфенил-2Н-пиразол-3-ил)-3-[2-(1-метил-1Н-индазо-5-илсульфанил)-5-фторбензил]мочевина (6r-1). Раствор соединения (5r) (70 мг, 0,21 ммоль) в ДМФА (1 мл) обрабатывали соответствующим карбаматом (97 мг, 0,24 ммоль), а затем ДИЭА (70 мкл, 0,54 ммоль). Смесь нагревали при 80°С в течение 18 часов в атмосфере азота. Затем неочищенный продукт очищали препаративной тонкослойной хроматографией, используя смесь гексан/EtOAc (1:1) в качестве элюента (Rf равно 0,6) и получая конечный продукт (6r-1) (80 мг, выход выделенного продукта 68%).1Н-ЯМР (400 МГц, CDCl3) δ 7.85 (s, 1H), 7.57 (s, 1H), 7.34-7.26 (m, 5Н), 7.2-7.14 (m, 2Н), 6.97 (dd, J=9.2, 2.8 Гц, 1Н), 6.92-6.84 (m, 2H), 5.99 (s, 1H), 5.7 (t, J=6.0 Гц, 1Н), 4.4 (d, J=5.6 Гц, 2Н), 1.89 (m, 1H), 0.95-0.9 (m, 2H), 0.75-0.71 (m, 2Н). МС (ИЭР) m/z 547,1 (М+Н)+.
Стадия G: 1-(5-трет-Бутил-2-п-хлорфенил-2Н-пиразол-3-ил)-3-[2-(1-метил-1Н-индазо-5-илсульфанил)-5-фторбензил]мочевина (6r-2). Раствор амина (5r) (70 мг, 0,21 ммоль) в ДМФА (1 мл) обрабатывали соответствующим карбаматом (100 мг, 0,24 ммоль), а затем ДИЭА (70 мкл, 0,54 ммоль). Смесь нагревали при 80°С в течение 18 часов в атмосфере азота. Затем неочищенный продукт очищали препаративной тонкослойной хроматографией, используя смесь гексан/EtOAc (1:1) в качестве элюента (Rf равно 0,6) и получая конечный продукт (6r-2) (80 мг, выход выделенного продукта 66%).1Н-ЯМР (400 МГц, CDCl3) δ 7.86 (s, 1Н), 7.58 (s, 1Н), 7.39-7.26 (m, 5Н) 7.21-7.14 (m, 2Н), 6.98 (dd, J=9.2, 2.4 Гц, 1Н), 6.88 (d(t), J=8.4, 2.4 Гц, 1Н), 6.68 (s, 1H), 6.24 (s, 1H), 5.64 (t, J=6.0 Гц, 1Н), 4.42(d, J=6.4 Гц, 2Н), 4.02 (s, 3H), 1.3 (s, 9H). МС (ИЭР) m/z 563,1 (М+Н)+.
Пример 93
Получение 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{2-[1-(3-изопропиламинопропил)-1Н-индазол-5-иламино]бензил}мочевины (8s-2)
Реакционная схема синтеза соединения 8s-2 показана на Фиг.43А-В.
Стадия А: 1-Аллил-5-бром-1Н-индазол (1s). 5-Броминдазол (Bioorg. Med. Chem. Lett., 11: 1153-1156 (2001)) (3,94 г, 20,0 ммоль), аллилбромид (2,6 мл, 30 ммоль) и карбонат калия (4,15 г, 30,0 ммоль) нагревали в ДМФА (25 мл) при 100°С в течение 18 часов. Реакционную смесь охлаждали, фильтровали через целит и твердые вещества промывали EtOAc. Раствор концентрировали почти досуха и затем распределяли между EtOAc и водой. Органическую фазу промывали NaHCOз, сушили (MgSO4), концентрировали и очищали колоночной хроматографией (силикагель, 7% EtOAc/гексаны), получая N1 изомер (быстро элюируемый) 1-аллил-5-бром-1Н-индазола (1s) (1,7 г, выход 36%).1Н-ЯМР (400 МГц, CDCl3) δ 7.95 (s, 1Н), 7.88 (d, J=2.3 Гц, 1Н), 7.44 (dd, J=8.6, 1.6 Гц, 1Н), 7.29 (d, J=8.6 Гц, 1Н), 6.06-5.97 (m, 1Н), 5.24 (dd, J=10.2, 1.6 Гц, 1Н), 5.12 (d, J=16.4 Гц, 1Н), 5.01-5.00 (m, 2H).
МС ИЭР (+), обнаружено: m/z 237, 239 (М+Н, Br образец). ВЭЖХ (5-95%) 2,98 мин.
Стадия В: 3-(5-Броминдазол-1-ил)пропан-1-ол (2s). 1-Аллил-5-бром-1Н-индазол (1s) (0,50 г, 2,1 ммоль) растворяли в 2 мл ТГФ и охлаждали до 0°С. Затем медленно через шприц, в атмосфере азота и при перемешивании добавляли раствор 9-BBN в ТГФ (0,5 М раствор, 8,9 мл, 4,4 ммоль). Реакционную смесь нагревали до комнатной температуры в течение 6,5 часов. Затем к этому раствору медленно добавляли раствор водной Н2О2 (30 мас.%-ный раствор; 1,4 мл) в 1 н. NaOH (14 мл, 14 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, что приводило к образованию белого осадка. Реакционную смесь разбавляли Н2О и Et2O. Слои разделяли и органическую фазу промывали рассолом. Водные фазы экстрагировали один раз Et2O. Органические фазы объединяли, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенное вещество (2s) направляли на следующую стадию без идентификации.
Стадия С: 5-Бром-1-[3-(трет-бутилдифенилсиланилокси)пропил]-1Н-индазол (3s). Неочищенный 3-(5-броминдазол-1-ил)пропан-1-ол (2s) (2,1 ммоль) и имидазол (0,22 г, 3,2 ммоль) растворяли в 10 мл CH2Cl2. К раствору добавляли трет-бутилдифенилсилилхлорид (0,58 г, 2,1 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Добавляли дополнительные количества имидазола (0,07 г, 1,0 ммоль) и трет-бутилдифенилсилилхлорида (0,16 г, 0,63 ммоль), реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли Et2O и последовательно промывали 3%-ной водной HCl и рассолом. Водные фазы экстрагировали один раз Et2O. Органические фазы объединяли, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали на силикагеле с Et2O/гексан (1:6), получая (3s) (1,0 г, выход на 2 стадиях 96%) в виде бесцветного масла.1Н-ЯМР (400 МГц, CDCl3) δ 7.92 (s, 1Н), 7.86 (s, 1Н), 7.61 (d, J=7.8 Гц, 4Н), 7.44-7.42 (m, 3Н), 7.36-7.33 (m, 5Н), 4.53 (t, J=10.2 Гц, 2H), 3.63 (t, J=9.0 Гц, 2H), 2.16-10 (m, 2H), 1.08 (s, 9H). ВЭЖХ (5-95%) 4,72 мин.
Стадия D: 1-[3-(трет-Бутилдифенилсиланилокси)пропил]-1Н-индазол-5-бороновая кислота (4s). 5-Бром-1-[3-(трет-бутилдифенилсиланилокси)пропил]-1Н-индазол (3s) (200 мг, 0,41 ммоль) растворяли в 4,0 мл ТГФ и охлаждали до -78°С. Медленно добавляли раствор н-бутиллития в смеси гексанов (2,5 М, 0,17 мл). Желтый раствор перемешивали в течение 30 минут. Добавляли триметилборат (130 мг, 1,2 ммоль), реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. Реакцию гасили 10 мл 0,3%-ной водной HCl и полученную смесь перемешивали в течение 30 минут. Реакционную смесь разбавляли Et2O и слои разделяли. Органическую фазу промывали рассолом. Водные фазы экстрагировали один раз Et2O. Органические фазы объединяли, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт частично очищали на силикагеле с 3% MeOH/CH2Cl2, получая (4s) (97 мг, 52%). МС ИЭР (+) m/z 459 (М+Н)+. ВЭЖХ (5-95%) 3,74 мин. Эту смесь направляли на следующую стадию без дополнительной очистки.
Стадия Е: 1-(2-Аминобензил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)мочевина (5s). 5-трет-Бутил-2-метил-2Н-пиразол-3-иламин (10s) (4,8 г, 31 ммоль) и карбонилдиимидазол (4,6 г, 32 ммоль) частично растворяли в ДХЭ (100 мл) и нагревали при 70°С в течение 2 часов. Реакционную смесь охлаждали и добавляли 2-аминометилфениламин (9s) (4,2 г, 34 ммоль), реакционную смесь перемешивали в течение 14 часов. Реакционную смесь концентрировали для удаления растворителя и затем распределяли между EtOAc и 0,5 н. HCl (60 мл). Органическую фазу промывали NH4Cl и водой и сушили (MgSO4). Раствор концентрировали и перекристаллизовывали из EtOAc (200 мл), получая целевой продукт (5s) (4,6 г, выход 49%).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.31 (s, 1Н), 6.98 (d, J=1.6 Гц, 1Н), 6.96 (dt, J=7.8, 1.6 Гц, 1Н), 6.63 (t, J=6.3 Гц, 1Н), 6.59 (d, J=7.0 Гц, 1Н), 6.48 (t, J=6.3 Гц, 1Н), 5.93 (s, 1H), 5.07 (s, 2Н), 4.12 (d, J=6.3 Гц, 2Н), 3.51 (s, 3H), 1.16 (s, 9H).
Стадия F: 1-(2-{1-[3-(трет-Бутилдифенилсиланилокси)пропил]-1Н-индазол-5-иламино}бензил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)мочевина (6s). Бороновую кислоту 4 (240 мг, 0,52 ммоль), 1-(2-аминобензил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)мочевину (5s) (170 мг, 0,58 ммоль), ацетат меди (II) (90 мг, 0,52 ммоль) и 240 мг 4Å молекулярных сит суспендировали в 10 мл СН2Cl2. Добавляли триэтиламин (0,36 мл, 2,6 ммоль) и смесь перемешивали при комнатной температуре в течение ночи, подвергая воздействию воздуха. Добавляли еще 3 мл CH2Cl2, смесь фильтровали через целит и летучие вещества удаляли в вакууме. Неочищенный продукт очищали на силикагеле с 2-4% МеОН/СН2Cl2, получая (6s) (170 мг, выход 45%) в виде коричневой смолы.
МС ИЭР (+), обнаружено: m/z 714 (М+Н)+. ВЭЖХ (5-95%) 4,32 мин.
Стадия G: 1-(5-трет-Бутил-2-метил-2Н-пиразол-3-ил)-3-{2-[1-(3-гидроксипропил)-1Н-индазол-5-иламино]бензил}мочевина (7s). 1-(2-{1-[3-(трет-Бутилдифенилсиланилокси)пропил]-1Н-индазол-5-иламино}бензил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)мочевину (6s) (40 мг, 0,056 ммоль) растворяли в 0,5 мл ТГФ и обрабатывали TBAF (1,0 М раствор в ТГФ, 0,11 мл, 0,11 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли дополнительное количество TBAF (0,3 мл, 0,3 ммоль) и реакционную смесь перемешивали в течение еще 2 часов. Реакционную смесь разбавляли CH2Cl2 и промывали Н2О. Водную фазу экстрагировали один раз CH2Cl2. Органические фазы объединяли, сушили (MgSO4), фильтровали и концентрировали в вакууме. Продукт очищали на силикагеле с 5% МеОН/СН2Cl2, получая целевое соединение (7s) (8 мг, 30%, чистота примерно 90%, как обнаружено1Н-ЯМР и ВЭЖХ).1Н-ЯМР (400 МГц, CDCl3) δ 7.81 (s, 1Н), 7.36-7.31 (m, 2Н), 7.21-7.12 (m, 4Н), 6.85 (s, 1H), 6.81 (t, J=10.6 Гц, 1Н), 5.93 (s, 1Н), 5.41-5.38 (m, 1Н), 4.49 (t, J=9.0 Гц, 2Н), 4.43 (d, J=6.3 Гц, 2Н), 3.57 (t, J=8.2 Гц, 3Н), 3.52 (s, 3Н), 2.13-2.07 (m, 2Н), 1.25 (s, 9H). МС (ХИАД) m/z 476 (М+Н)+. ВЭЖХ (5-95%) 2,79 мин.
Стадия Н: 1-(5-трет-Бутил-2-метил-2Н-пиразол-3-ил)-3-{2-[1-(3-диметиламинопропил)-1Н-индазол-5-иламино]бензил}мочевина (8s-1). Ангидрид метансульфокислоты (12 мг, 0,070 ммоль) добавляли к раствору спирта (7s) (24 мг, 0,050 ммоль) и диизопропилэтиламина (20 мг, 0,15 ммоль) при комнатной температуре. Раствор перемешивали в течение 1 часа. Добавляли диметиламин (2,0 М в ТГФ, 0,25 мл, 0,50 ммоль) и реакционную смесь перемешивали в течение ночи. Добавляли дополнительное количество диметиламина (2,0 М в ТГФ, 0,25 мл, 0,50 ммоль) и реакционную смесь перемешивали в течение еще 2 дней. Затем смесь распределяли между CHCl3и водой. Водную фазу экстрагировали один раз CHCl3. Органические фазы объединяли, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали на силикагеле, используя смесь 5% МеОН/СН2Cl2, содержащую 1% Et3N, и получая целевое соединение (8s-1) (11 мг, выход 43%) в виде темной пены.1Н-ЯМР (400 МГц, CDCl3) δ 7.81 (s, 1H), 7.42 (s, 1Н), 7.35-7.33 (m, 2Н), 7.19-7.11 (m, 4Н), 6.80-6.75 (m, 2Н), 5.94 (s, 1H), 5.55 (m, 1Н), 4.43 (d, J=6.6 Гц, 2Н), 4.38 (t, J=10.5 Гц, 2Н), 3.57 (s, 3Н), 2.24 (t, J=10.5 Гц, 2Н), 2.19 (s, 6Н), 2.08-2.01 (m, 2Н), 1.24 (s, 9H). МС ИЭР (+), обнаружено: m/z 503 (М+Н)+. ВЭЖХ (5-95%) 2,59 мин.
Стадия I: 1-(5-трет-Бутил-2-метил-2Н-пиразол-3-ил)-3-{2-[1-(3-изопропиламинопропил)-1Н-индазол-5-иламино]бензил}мочевина (8s-2). Ангидрид метансульфокислоты (18 мг, 0,11 ммоль) добавляли к раствору спирта (7s) (36 мг, 0,076 ммоль) и диизопропилэтиламина (29 мг, 0,23 ммоль) при комнатной температуре. Раствор перемешивали в течение 1 часа. Добавляли изопропиламин (0,13 мл, 1,50 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 60 часов. Летучие вещества удаляли в вакууме. Неочищенный продукт очищали на силикагеле, используя смесь 5% МеОН/СН2Cl2, содержащую 1% Et3N, а затем на силикагеле С18 с CH3CN/H2O, получая целевое соединение (8s-2) (8 мг, выход 20%).1Н-ЯМР (400 МГц, MeOD) δ 7.88 (s, 1Н), 7.51 (d, J=8.6 Гц, 1Н), 7.31-7.24 (m, 3Н), 7.17-7.16 (m, 2Н), 6.93-6.89 (m, 1Н), 6.05 (s, 1Н), 4.52 (t, J=10.2 Гц, 2Н), 4.41 (s, 2Н), 3.57 (s, 3Н), 3.35-3.30 (m, 1Н), 3.03 (t, J=11.7 Гц, 2Н), 2.29-2.22 (m, 2Н), 1.29 (d, J=6.3 Гц, 6Н), 1.26 (s, 9H). МС ИЭР (+), обнаружено: m/z 517 (М+Н)+. ВЭЖХ (5-95%) 2,61 мин.
Пример 94
Получение 1-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]мочевины (7t-1) и 1-[5-трет-бутил-2-(4-хлорфенил)-2Н-пиразол-3-ил]-3-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]мочевины (7t-2)
Реакционная схема синтеза соединения 7t-2 показана на Фиг.44.
Стадия А: 5-Метокси-1-метил-1Н-индазол (2t). Раствор 5-метоксииндазола (1t) (5 г, 33,75 ммоль; Tet Lett., 43 (15): 2695 (2002)) в ДМФА (200 мл) обрабатывали карбонатом калия (6,06 г, 43,87 ммоль) при комнатной температуре. После перемешивания в течение 15 минут добавляли метилйодид (2,33 мл, 37,12 ммоль). Полученную смесь нагревали при 110°С в течение 18 часов. Анализ методом ЖХ показал незначительные остатки исходного вещества. Добавляли дополнительное количество метилйодида (2,33 мл) и перемешивание продолжали в течение еще 18 часов. Анализ методом ЖХ показал наличие смеси N1 и N2 алкилированных изомеров (1:2). Растворитель выпаривали в вакууме, остаток переносили в ДХМ и промывали 1 н. HCl. Органический слой фильтровали через бумагу 1PS, упаривали в вакууме и очищали на Biotage, элюируя смесью гексан/Et2О (4:3, 3:1). Объединенные целевые фракции (N1 изомер) упаривали в вакууме, получая целевой продукт (2t) в виде желтого масла (2,57 г; 47%).1Н-ЯМР (400 МГц, CDCl3) δ 7.38 (d, J=7.8 Гц, 1Н), 7.17 (dd, J=7.8, 1.6 Гц, 1Н), 7.13 (d, J=1.6 Гц, 1Н), 5.19-5.18 (m, 1H), 4.51-4.44 (m, 1H), 4.43-4.36 (m, 1H), 2.53-2.45 (m, 1H), 2.36-2.30 (m, 1H). МС ИЭР (+), обнаружено: m/z 163 (М+Н).
Стадия В: 1-Метил-1Н-индазол-5-ол (3t). К раствору (2t) (0,99 г, 6,1 ммоль) в толуоле (30 мл) добавляли AlCl3 (2,44 г, 18,3 ммоль) при комнатной температуре, после чего образовывалась смесь пурпурного цвета. После нагревания с обратным холодильником в течение 20 минут образовывалась смесь оливкового цвета. Смесь нагревали с обратным холодильником в течение 2 часов, оставляли охлаждаться до комнатной температуры и вливали в баню с ледяной водой. Нерастворимые твердые вещества собирали фильтрацией (398 мг). Фильтрат экстрагировали ДХМ, фильтровали через бумагу 1PS, упаривали в вакууме и очищали на Biotage, элюируя смесью Et2O/ДХМ ((1:9), затем (3:7)) и окончательно смесью ДХМ/Et2O (1:1). Фракции продукта упаривали в вакууме, получая соединение (3t) в виде коричневой пены (122 мг). Общий объединенный выход составлял 520 мг (58%).1Н-ЯМР (400 МГц, CDCl3) δ 7.38 (d, J=7.8 Гц, 1Н), 7.17 (dd, J=7.8, 1.6 Гц, 1Н), 7.13 (d, J=1.6 Гц, 1Н), 5.19-5.18 (m, 1H), 4.51-4.44 (m, 1H), 4.43-4.36 (m, 1H), 2.53-2.45 (m, 1Н), 2.36-2.30 (m, 1Н). МС ИЭР (+), обнаружено: m/z 149 (М+Н).
Стадия С: 5-Фтор-2-(1-метил-1Н-индазол-5-илокси)бензонитрил (4t). Раствор (3t) (0,70 г, 4,74 ммоль) в ДМФА (50 мл) охлаждали до 0°С и обрабатывали 60 мас.%-ным гидридом натрия (0,28 г, 7,11 ммоль). После перемешивания при этой температуре в течение 20 минут добавляли арилфторид (0,79 г, 5,69 ммоль) при 0°С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Затем смесь охлаждали до 0°С и обрабатывали водой (50 мл), экстрагировали Et2O (3×150 мл), объединенные органические слои промывали водой (2×20 мл), рассолом (2×20 мл), сушили над MgSO4 и упаривали в вакууме до масла. Это вещество очищали колоночной флэш-хроматографией (EtOAc/гексан равно 2:3; наносили в теплой смеси толуола и ДМФА). Целевые фракции упаривали в вакууме и подвергали азеотропной перегонке с толуолом. Получали соединение (4t) в виде белых кристаллов (1,09 г; выход выделенного продукта 86%).1Н-ЯМР (400 МГц, CDCl3) δ 7.38 (d, J=7.8 Гц, 1Н), 7.17 (dd, J=7.8, 1.6 Гц, 1Н), 7.13 (d, J=1.6 Гц, 1Н), 5.19-5.18 (m, 1H), 4.51-4.44 (m, 1H), 4.43-4.36 (m, 1H), 2.53-2.45 (m, 1H), 2.36-2.30 (m, 1H). МС ИЭР (+), обнаружено: m/z 268 (М+Н).
Стадия D: Гидрохлорид 5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензиламина (5t). Раствор (4t) (0,32 г, 1,22 ммоль) в МеОН (20 мл) продували азотом и обрабатывали катализатором 20% Pd(OH)2/C (15 мас.%, 180 мг), а затем концентрированной HCl (0,3 мл, 3,6 ммоль). После дополнительной продувки азотом в верхней части колбы размещали баллон, заполненный водородом. После перемешивания при комнатной температуре в течение 18 часов анализ методом ЖХ показал отсутствие исходного вещества. Катализатор отфильтровывали через наполнитель силикагель/целит/песок и промывали МеОН. Растворитель выпаривали в вакууме и остаток совместно упаривали с эфиром. Полученную бледно-желтую пену (5t) хранили в атмосфере N2 (0,34 г; выход выделенного продукта 91%).1Н-ЯМР (400 МГц, CDCl3) δ 7.38 (d, J=7.8 Гц, 1Н), 7.17 (dd, J=7.8, 1.6 Гц, 1Н), 7.13 (d, J=1.6 Гц, 1Н), 5.19-5.18 (m, 1H), 4.51-4.44 (m, 1H), 4.43-4.36 (m, 1H), 2.53-2.45 (m, 1H), 2.36-2.30 (m, 1H). МС ИЭР (+), обнаружено: m/z 272 (М+Н).
Стадия Е: 1-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]мочевина (7t-1). Раствор (5t) (70 мг, 0,23 ммоль) в ДМФА (1 мл) обрабатывали соответствующим карбаматом (6t-1) (100 мг, 0,25 ммоль), а затем ДИЭА (99 мкл, 0,57 ммоль). Смесь нагревали при 80°С в течение 18 часов, продувая азотом. Растворитель выпаривали в вакууме, остаток переносили в ДХМ и промывали 1 н. HCl. Органический слой фильтровали через бумагу 1PS и упаривали в вакууме до масла, которое очищали на картридже с силикагелем Sep Pak, элюируя смесью ДХМ/Et2O (10:1). Целевые фракции упаривали в вакууме, получая целевое соединение (7t-1) в виде бледно-желтого масла (80 мг; выход выделенного продукта 67%).1Н-ЯМР (400 МГц, CDCl3) δ 7.38 (d, J=7.8 Гц, 1Н), 7.17 (dd, J=7.8, 1.6 Гц, 1Н), 7.13 (d, J=1.6 Гц, 1Н), 5.19-5.18 (m, 1H), 4.51-4.44 (m, 1H), 4.43-4.36 (m, 1H), 2.53-2.45 (m, 1H), 2.36-2.30 (m, 1H). МС ИЭР (+), обнаружено: m/z 527 (М+Н).
Стадия F: 1-[5-трет-Бутил-2-(4-хлорфенил)-2Н-пиразол-3-ил]-3-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]мочевина (7t-2). Раствор (5t) (74 мг, 0,57 ммоль) в ДМФА (1 мл) обрабатывали соответствующим карбаматом (6t-2) (110 мг, 0,25 ммоль), а затем ДИЭА (99 мл, 0,57 ммоль). Смесь нагревали при 80°С в течение 18 часов, продувая азотом. Растворитель выпаривали в вакууме, остаток переносили в ДХМ и промывали 1 н. HCl. Органический слой фильтровали через бумагу 1PS и упаривали в вакууме до масла, которое очищали на картридже с силикагелем Sep Pak, элюируя смесью ДХМ/Et2O (10:1). Целевые фракции упаривали в вакууме, получая целевое соединение (7t-2) в виде бледно-желтого масла (80 мг; выход выделенного продукта 64%).1Н-ЯМР (400 МГц, CDCl3) δ 7.38 (d, J=7.8 Гц, 1Н), 7.17 (dd, J=7.8, 1.6 Гц, 1Н), 7.13 (d, J=1.6 Гц, 1Н), 5.19-5.18 (m, 1H), 4.51-4.44 (m, 1H), 4.43-4.36 (m, 1H), 2.53-2.45 (m, 1Н), 2.36-2.30 (m, 1Н). МС ИЭР (+), обнаружено: m/z 547 (М+Н).
Аналогичным способом были синтезированы следующие соединения.
Пример 95
Получение 2-(1-циклобутилметил-1Н-индазол)-5-фторбензиламида циклопропанкарбоновой кислоты (9t)
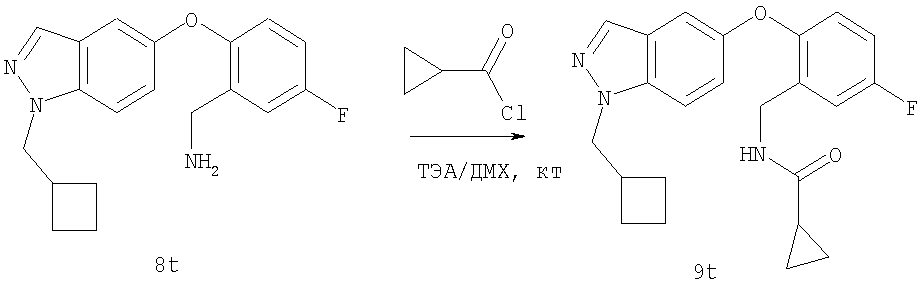
Раствор (8t) (20 мг, 0,06 ммоль; полученного способом, описанным в Примере 94, стадии A-D, за исключением обработки концентрированной HCl) в ДХМ (0,5 мл) обрабатывали основанием (13 мкл, 0,09 ммоль), а затем циклопропанкарбонилхлоридом (6 мкл, 0,07 ммоль) при комнатной температуре и продувке азотом. Смесь перемешивали при комнатной температуре в течение 18 часов и затем очищали на картридже с силикагелем Sep Pak, элюируя смесью ДХМ-Et2О (10:1). Целевые фракции упаривали в вакууме, получая продукт (9t) в виде бледно-желтого масла (10,2 мг; выход выделенного продукта 42%). МС ИЭР (+), обнаружено: m/z 394 (М+Н).
Пример 96
Получение N-[5-фтор-2-(1-изобутил-1H-индазол-5-илокси)бензил]-3-трифторметилбензамида (11t)

Раствор соединения (10t) (14 мг, 0,05 ммоль; полученного способом, описанным в Примере 94, стадии A-D, за исключением обработки концентрированной HCl) в ДХМ (0,5 мл) обрабатывали основанием (11 мкл, 0,06 ммоль), а затем 3-трифторметилбензоилхлоридом (12 мг, 0,075 ммоль) при комнатной температуре и продувке азотом. Смесь перемешивали при комнатной температуре в течение 18 часов и затем очищали на картридже с силикагелем Sep Pak, элюруя смесью ДХМ-Et2O (10:1). Целевые фракции упаривали в вакууме, получая продукт (11t) в виде желтого масла (16,6 мг; выход выделенного продукта 53%).1Н-ЯМР (400 МГц, CDCl3) δ 7.38 (d, J=7.8 Гц, 1Н), 7.17 (dd, J=7.8, 1.6 Гц, 1Н), 7.13 (d, J=1.6 Гц, 1Н), 5.19-5.18 (m, 1H), 4.51-4.44 (m, 1H), 4.43-4.36 (m, 1H), 2.53-2.45 (m, 1H), 2.36-2.30 (m, 1H). МС ИЭР (+), обнаружено: m/z 468 (М+Н).
Пример 97
Получение N-[2-(1-изобутил-1H-индазол-5-илокси)бензил]-2-(3-трифторметилфенил)ацетамида (14t)
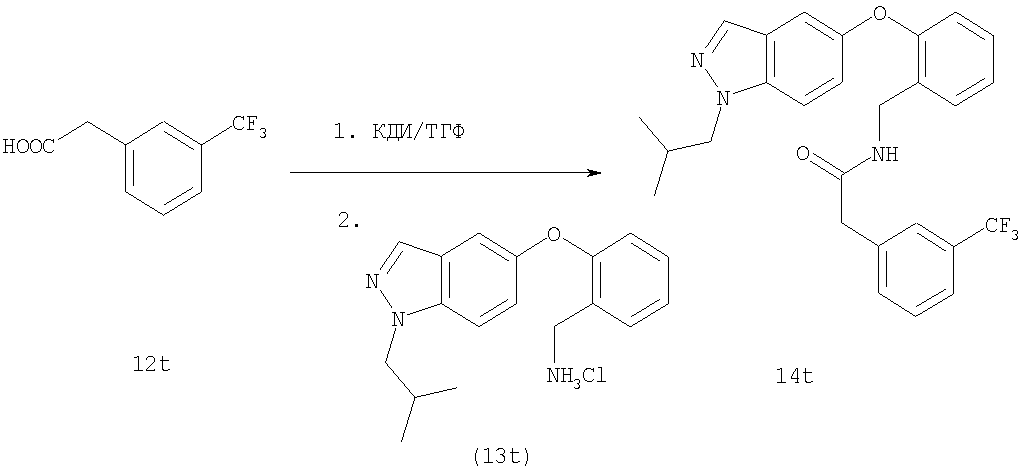
Получение N-[2-(1-изобутил-1H-индазол-5-илокси)бензил]-2-(3-трифторметилфенил)ацетамида (14t)
Раствор (3-трифторметилфенил)уксусной кислоты (12t) (10 мг, 0,051 ммоль) в ТГФ (0,5 мл) при комнатной температуре обрабатывали 1,1-карбонилдиимидазолом (КДИ, 9 мг, 0,055 ммоль). После перемешивания при комнатной температуре и продувке азотом в течение 18 часов добавляли бензиламин (13t) (17 мг, 0,05 ммоль; полученный способом, описанным в Примере 94, стадии A-D) при комнатной температуре. Перемешивание продолжали в течение еще 18 часов. Растворитель выпаривали в вакууме, остаток переносили в ДХМ и очищали на картридже с силикагелем Sep Pak, элюируя смесью ДХМ-Et2О (10:1). Целевые фракции упаривали в вакууме, получая продукт (14t) в виде бледно-желтого масла (10 мг; выход выделенного продукта 42%). МС ИЭР (+), обнаружено: m/z 482 (М+Н).
Пример 98
Получение 5-фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензиламида 5-трет-бутил-1-пиридин-2-ил-1Н-пиразол-4-карбоновой кислоты (16t)

Раствор 5-трет-бутил-1-пиридин-2-ил-1Н-пиразол-4-карбоновой кислоты (15t) (12 мг, 0,05 ммоль) в ТГФ (0,5 мл) обрабатывали 1,1-карбонилдиимидазолом (КДИ, 9 мг, 0,05 ммоль) при комнатной температуре. После перемешивания при комнатной температуре и продувке азотом в течение 18 часов добавляли бензиламин (13t) (15 мг, 0,05 ммоль; полученный способом, описанным в Примере 94, стадии A-D) при комнатной температуре. Перемешивание продолжали в течение еще 18 часов. Растворитель выпаривали в вакууме, остаток переносили в ДХМ и очищали на картридже с силикагелем Sep Pak, элюируя МеОН (2-10%) в ДХМ. Целевые фракции упаривали в вакууме, получая продукт (16t) в виде желтого масла (5,2 мг; выход выделенного продукта 23%). МС ИЭР (+), обнаружено: m/z 541 (М+Н).
Пример 99
Получение 2-циклопропил-N-[5-фтор-2-(1-изобутил-1H-индазол-5-илокси)бензил]ацетамида (18t)
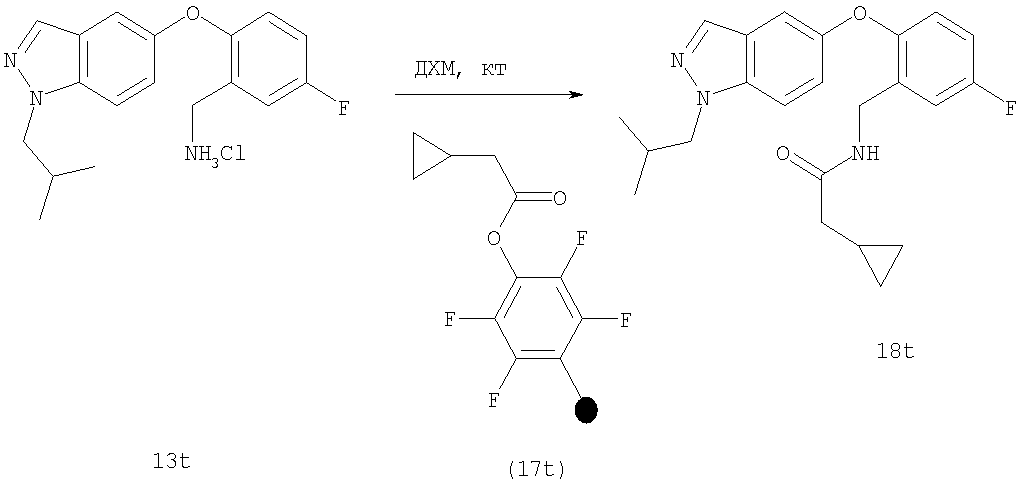
Раствор соединения (13t) (15 мг, 0,05 ммоль; полученного способом, описанным в Примере 94, стадии A-D) в ДХМ (0,5 мл) добавляли к соответствующему TFP (тетрафторфениловому) сложному эфиру (17t) (1 ммоль/г) при комнатной температуре. Смесь встряхивали в течение 18 часов. Смолу промывали ДХМ. Объединенные фильтраты концентрировали в вакууме и очищали на картридже с силикагелем Sep Pak, элюируя смесью ДХМ-Et2O (10:1). Целевые фракции упаривали в вакууме, получая продукт (18t) в виде желтого масла (12,6 мг; выход выделенного продукта 66%). МС ИЭР (+), обнаружено: m/z 400 (М+Н).
Пример 100
Получение 3-хлор-N-[5-фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензил]бензамида (20t)

Раствор (13t) (15 мг, 0,05 ммоль; полученного способом, описанным в Примере 94, стадии A-D) в ДХМ (0,5 мл) добавляли к соответствующему TFP сложному эфиру (19t) (1 ммоль/г) при комнатной температуре. Смесь встряхивали в течение 18 часов. Смолу промывали ДХМ. Объединенные фильтраты концентрировали в вакууме и очищали на картридже с силикагелем Sep Pak, элюируя смесью ДХМ-Et2O (10:1). Целевые фракции упаривали в вакууме, получая продукт (20t) в виде желтого масла (14,4 мг; выход выделенного продукта 66%). МС ИЭР (+), обнаружено: m/z 452 (М+Н).
Пример 101
Получение N-[5-фтор-2-(1-изобутил-1H-индазол-5-илокси)бензил]-4-трифторметилбензолсульфонамида (21t)

Раствор соединения (13t) (15 мг, 0,04 ммоль; полученного способом, описанным в Примере 94, стадии A-D) в пиридине (0,5 мл) обрабатывали 4-трифторметилбензолсульфонилхлоридом (13 мг, 0,05 ммоль) при комнатной температуре и продувке азотом. Смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель выпаривали в вакууме, переносили в ДХМ и промывали 1 н. HCI. Органический слой фильтровали через бумагу 1PS, концентрировали в вакууме и очищали на картридже с силикагелем Sep Pak, элюируя смесью ДХМ-Et2O (10:1). Целевые фракции упаривали в вакууме, получая продукт (21t) в виде желтого масла (15,6 мг; выход выделенного продукта 70%). МС ИЭР (+), обнаружено: m/z 522 (М+Н).
Пример 102
Получение N-[5-фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензил]метансульфонамида (22t)

Раствор соединения (13t) (15 мг, 0,043 ммоль; полученного способом, описанным в Примере 94, стадии A-D) в пиридине (0,5 мл) обрабатывали метансульфонилхлоридом (4 мкл, 0,05 ммоль) при комнатной температуре и продувке азотом. Смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель выпаривали в вакууме, переносили в ДХМ и промывали 1 н. HCl. Органический слой фильтровали через бумагу 1PS, концентрировали в вакууме и очищали на картридже с силикагелем Sep Pak, элюируя смесью ДХМ-Et2O (10:1). Целевые фракции упаривали в вакууме до желтого масла (22t) (13,1 мг; выход выделенного продукта 78%). МС ИЭР (+), обнаружено: m/z 392 (М+Н).
Пример 103
Получение 5-фтор-2-(1Н-индазол-5-илокси)бензонитрила (26t)
Реакционная схема синтеза соединения 26t показана на Фиг.45.
Стадия А: 5-Фтор-2-гидроксибензонитрил (23t). 2,5-Дифторбензонитрил (14,9 г, 107 ммоль) и бензиловый спирт (11,1 мл, 11,6 г, 107 ммоль) растворяли в ДМФА (330 мл) и охлаждали до 0°С. К этому раствору добавляли гидрид натрия (60% в масле, 6,40 г, 161 ммоль) при 0°С и реакционную смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение 1 часа при комнатной температуре реакционный раствор охлаждали до 0°С и к этому раствору постепенно добавляли воду (330 мл). Смесь переносили в делительную воронку и три раза экстрагировали 500 мл Et2O. Объединенный органический слой дважды промывали 100 мл воды, один раз рассолом, затем сушили над MgSO4. После фильтрации раствор концентрировали при пониженном давлении с получением неочищенного бледно-желтого твердого вещества. Неочищенное твердое вещество растворяли в МеОН (500 мл). К раствору добавляли 10% палладия на активированном угле в атмосфере азота. Газообразный азот заменяли газообразным водородом, реакционную смесь перемешивали при комнатной температуре (если в течение 30 минут реакция не происходила, то палладий на угле отфильтровывали и реакцию проводили снова). После перемешивания в течение 2 часов палладий на угле отфильтровывали и промывали МеОН. Раствор концентрировали при пониженном давлении с получением бледно-желтого твердого вещества. Твердое вещество перекристаллизовывали из горячего толуола (100 мл), добавляя гексан (10 мл) с последующим охлаждением до 0°С. Полученные белые иглы промывали смесью толуола и гексана (1:1, 7,23 г, выход 49%). Маточный раствор концентрировали и очищали на силикагеле с Et2О/гексан (2:3-1:1), получая целевое соединение (23t) (6,94 г, выход 47%). Общий выход на 2 стадиях 14,2 г (96%).1Н-ЯМР (400 МГц, d6-ДМСО) δ 11.09 (s, 1Н), 7.58 (dd, J=8.4, 3.2 Гц, 1Н), 7.40 (td, J=8.6, 3.2 Гц, 1Н), 7.03 (dd, J=9.2, 4.4 Гц, 1Н) млн-1.
Стадия В: 5-Фтор-2-(3-метил-4-нитрофенокси)бензонитрил (24t). Промежуточное соединение (23t) (10,0 г, 72,9 ммоль), 4-фтор-2-метил-1-нитробензол (13,6 г, 87,5 ммоль) и карбонат калия (11,1 г, 80,2 ммоль) растворяли в диметилацетамиде (ДМА, 400 мл) и затем смесь нагревали до 100°С при интенсивном перемешивании. После перемешивания в течение 16 часов реакционную смесь охлаждали до комнатной температуры, а затем к этой смеси добавляли 400 мл воды. Смесь трижды экстрагировали 500 мл Et2O. Объединенный органический слой трижды промывали 100 мл воды и один раз рассолом. Затем раствор сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученное оранжевое неочищенное твердое вещество промывали 100 мл теплого (примерно 50°С) гексана и дополнительно 400 мл гексана (не теплого), получая бледно-желтое твердое вещество (16,4 г). Оранжевый фильтрат концентрировали и остаток очищали на силикагеле с Et2О/гексан (1:4-1:3), получая слегка оранжевое твердое вещество, которое промывали смесью Et2O и гексана (1:3) (1,2 г). Объединяли две порции бледно-желтого твердого вещества, получая соединение (24t) (17,6 г, выход 89%).
1Н ЯМР (400 МГц, CDCl3) δ 8.08 (d, J=8.6 Гц, 1Н), 7.43 (dd, J=7.6, 3.1 Гц, 1Н), 7.35 (ddd, J=9.7, 3.1 Гц, 1Н), 7.09 (dd, J=9.5, 4.7 Гц, 1Н), 6.95-6.87 (m, 2H), 2.62 (s, 3Н) млн-1.
Стадия С: 2-(4-Амино-3-метилфенокси)-5-фторбензонитрил (25t). Промежуточное соединение (24t) (14,3 г, ммоль) и цинковую пыль (17,2 г, 263 ммоль) суспендировали в смешанном растворителе МеОН/ТГФ (1:1, 125 мл) и добавляли насыщенный NH4Cl (125 мл). Реакционная смесь становилась теплой. Происходило явное изменение в цинковой суспензии. Реакция завершалась через 10 минут. Реакционную смесь фильтровали через наполнитель из диоксида кремния и разбавляли EtOAc и насыщенным NaHCO3. Слои разделяли, объединенные органические вещества сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенный остаток (25t) был достаточно чистым для спектра ЯМР.1Н ЯМР (400 МГц, d6-ДМСО) δ 7.83 (dd, J=8.5, 3.1 Гц, 1Н), 7.47 (td, J=8.9, 3.1 Гц, 1Н), 6.83-6.77 (m, 2H), 6.74 (dd, J=8.6, 2.3 Гц, 1Н), 6.66 (d, J=8.6 Гц, 1Н), 4.88 (s, 1H), 2.06 (s, 3H) млн-1.
Стадия D: 5-Фтор-2-(1Н-индазол-5-илокси)бензонитрил (26t). Промежуточное соединение (25t) (13,2 г, 54,5 ммоль) и NH4BF4 растворяли в 550 мл смешанного растворителя АсОН и воды (2:1) и охлаждали до 0°С. К раствору при 0°С добавляли концентрированную HCl (12 н., 23 мл, 272 ммоль) и NaNO2 (4,14 г, 59,9 ммоль), затем смесь оставляли нагреваться до комнатной температуры и перемешивали. После 3 часов перемешивания раствор концентрировали при пониженном давлении и 4 раза подвергали азеотропной перегонке с толуолом досуха, получая бледно-желтое неочищенное твердое вещество. Твердое вещество растворяли в 600 мл EtOAc и к этому раствору добавляли КОАс, затем смесь перемешивали при комнатной температуре. Через 30 минут желтый раствор становился оранжевым, и его дополнительно перемешивали. После перемешивания в течение ночи оранжевую суспензию фильтровали и несколько раз промывали EtOAc до общего объема 1000 мл. Органический раствор переносили в делительную воронку и промывали насыщенным NaHCO3 и рассолом, сушили над MgSO4, фильтровали и упаривали. Полученное неочищенное оранжевое твердое вещество очищали на силикагеле с EtOAc /гексан (1:2-1:1), получая оранжевое твердое вещество (13,2 г), которое промывали толуолом. Толуольный раствор оранжевого цвета концентрировали и снова промывали толуолом. Повторение такой же толуольной промывки давало соединение (26t) в виде твердого соединения слегка оранжевого цвета (11,7 г, выход на 2 стадиях 84%).1Н-ЯМР (400 МГц, CDCl3) δ 12.12 (s, 1Н), 8.03 (s, 1Н), 7.58 (d, J=9.4 Гц, 1Н), 7.40 (d, J=2.3 Гц, 1Н), 7.37 (dd, J=7.9, 3.1 Гц, 1Н), 7.18 (ddd, J=9.4, 7.9, 3.1 Гц, 1Н), 7.13 (dd, J=8.5, 2.3 Гц, 1Н), 6.80 (dd, J=9.3, 3.9 Гц, 1Н) млн-1.
Пример 104
Получение 3-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-1-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]-1-метилмочевины (28t)
Реакционная схема синтеза соединения 28t показана на Фиг.46.
Стадия А: Гидрохлорид 5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензиламина (5t). Промежуточное соединение (4t) (1,13 г, 4,23 ммоль; полученное, как описано в Примере 94, стадии А-С) растворяли в метаноле (50 мл) и к раствору добавляли 20%-ный гидроксид палладия на активированном угле (0,50 г, 0,71 ммоль) в атмосфере азота. После добавления концентрированной HCl (12 н., 5,0 мл, 60 ммоль) смесь перемешивали в атмосфере водорода в течение ночи (18 часов). Смесь фильтровали и гидроксид палладия промывали МеОН. После упаривания неочищенный остаток подвергали азеотропной перегонке со смесью толуола и EtOH (досуха), получая соединение (5t) в виде белого порошка (1,29 г, выход 99%).1Н-ЯМР (400 МГц, CDCl3) δ 8.72 (br, 3Н), 8.01 (s, 1Н), 7.73 (d, J=8.6 Гц, 1Н), 7.57 (dd, J=9.4, 2.3 Гц, 1Н), 7.38 (s, 1Н), 7.19 (d, J=3.1 Гц, 1Н), 7.04 (td, J=74.8, 3.1 Гц, 1Н), 7.02 (dd, J=195.1,4.7 Гц, 1Н), 4.07 (s, 3H) млн-1.
Стадия В: трет-Бутиловый эфир [5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]карбаминовой кислоты (26t). Промежуточное соединение (5t) (134 мг, 0,43 ммоль) растворяли в СН2Cl2 (5 мл) и к раствору добавляли диизопропилэтиламин (151 мкл, 112 мг, 0,87 ммоль) и ди-трет-бутилдикарбонат (94,7 мг, 0,43 ммоль). После перемешивания в течение ночи (12 часов) реакционную смесь разбавляли этилацетатом (100 мл), промывали 0,2 н. HCl (5 мл), насыщенным NaHCO3 (5 мл) и рассолом, сушили над MgSO4 и концентрировали при пониженном давлении, получая желтое неочищенное масло, которое очищали на силикагеле с EtOAc/гексан (1:2), получая продукт (26t) в виде бесцветного масла (150 мг, выход 93%).1Н-ЯМР (400 МГц, CDCl3) δ 7.85 (s, 1Н), 7.35 (d, J=9.4 Гц, 1Н), 7.15-7.08 (m, 3Н), 6.87 (t, J=9.4 Гц, 1Н), 6.75 (dd, J=8.7, 4.7 Гц, 1Н), 5.09 (br, 1Н), 4.35 (d, J=6.3 Гц, 2Н), 4.06 (s, 3Н), 1.42 (s, 9Н) млн-1.
Стадия С: трет-Бутиловый эфир [5-фторо-2-(1-метил-1Н-индазол-5-илокси)бензил]метилкарбаминовой кислоты (27t). Промежуточное соединение (26t) (50 мг, 0,135 ммоль) растворяли в ДМФА (2 мл) и охлаждали до 0°С. К раствору при 0°С добавляли гидрид натрия (60%, 8,1 мг, 0,20 ммоль) и метилйодид (42 мкл, 95,5 мг, 0,67 ммоль), затем смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение 1 часа смесь вливали в насыщенный раствор NH4Cl и трижды экстрагировали 30 мл эфира. Объединенный органический слой дважды промывали 5 мл воды и один раз рассолом, сушили над MgSO4 и упаривали при пониженном давлении. Бледно-желтый остаток очищали на силикагеле с EtOAc/гексан (1:2), получая соединение (27t) в виде бесцветного масла (52 мг, количественный выход).1Н-ЯМР (400 МГц, CDCl3) δ 7.83 (s, 1H), 7.35 (s, 0,4H), 7.33 (s, 0,6H), 7.11 (s, 0,6H), 7.08 (s, 1,4Н), 6.98 (d, J=8.6 Гц, 1Н), 6.92-6.81 (m, 1), 6.81-6.73 (m, 1Н), 4.49 (s, 0,8Н), 4.45 (s, 1,2Н), 4.04 (s, 3H), 2.89 (s, 1.8H), 2.85 (s, 1,2H), 1.45 (s, 3,6H), 1.41 (s, 5,4Н) млн-1.
Стадия D: 3-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-1-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]-1 -метилмочевина (28t).
Промежуточное соединение (27t) (52 мг, 0,135 ммоль) растворяли в CH2Cl2 (2 мл) и к раствору при комнатной температуре добавляли ТФУ (1 мл). После перемешивания в течение 1 часа реакционный раствор упаривали при пониженном давлении. Остаток разбавляли EtOAc (50 мл) и нейтрализовали насыщенным NaHCO3, промывали рассолом, сушили над MgSO4 и упаривали при пониженном давлении. Неочищенное бледно-желтое масло растворяли в ДМА (2 мл). К раствору добавляли карбамат (6t-1) и диизопропилэтиламин и нагревали до 80°С в запаянной пробирке. После перемешивания в течение 16 часов реакционную смесь разбавляли Et2O (50 мл) и трижды промывали 5 мл воды и один раз рассолом, сушили над MgSO4 и упаривали при пониженном давлении. Неочищенное масло очищали колоночной флэш-хроматографией (этилацетат/гексан, 1:1), получая соединение (28t) в виде белой пены (63,8 мг, выход на 2 стадиях 87%).1Н-ЯМР (400 МГц, CDCl3) δ 7.83 (s, 1H), 7.32 (d, J=9.4 Гц, 1Н), 7.22 (d, J=7.8 Гц, 2Н), 7.13 (d, J=7.8 Гц, 2Н), 7.02 (s, 1H), 6.98 (d, J=8.6 Гц, 1Н), 6.89 (t, J=8.6 Гц, 1Н), 6.72 (dd, J=7.7, 4.7 Гц, 1Н), 6.62 (s, 1H), 6.39 (s, 1H), 4.51 (s, 2H), 4.05 (s, 3H), 2.94 (s, 2H), 2.32 (s, 3H), 1.30 (s, 9H) млн-1.
Пример 105
Получение 1-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]-1-метилмочевины (32t)
Реакционная схема синтеза соединения 32t показана на Фиг.47.
Стадия А: [5-Фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]-(4-метоксибензил)амин (29t). Промежуточное соединение (5t) (136 мг, 0,442 ммоль; полученное, как описано в Примере 106, стадия А) растворяли в EtOAc (100 мл) и нейтрализовали насыщенным NaHCO3, затем промывали рассолом, сушили над MgSO4 и упаривали при пониженном давлении, получая свободный амин. Свободный амин растворяли в 1,2-дихлорэтане (5 мл) и к этому раствору при комнатной температуре добавляли п-анисовый альдегид. После перемешивания в течение 2 часов раствор упаривали при пониженном давлении. Остаток растворяли в МеОН (5 мл) и охлаждали до 0°С. К раствору добавляли борогидрид натрия при 0°С. После перемешивания в течение 40 минут при 0°С реакционную смесь гасили несколькими каплями уксусной кислоты при 0°С, затем реакционную смесь упаривали при пониженном давлении. Остаток разбавляли EtOAc (50 мл) и нейтрализовали насыщенным NaHCO3 и промывали рассолом, сушили над MgSO4 и упаривали, получая неочищенное масло, которое очищали на силикагеле с EtOAc/гексан (1:1) с 1% Et3N, получая соединение (29t) в виде бесцветного масла (139 мг, выход 80%).1Н-ЯМР (400 МГц, CDCl3) δ 7.85 (s, 1Н), 7.34 (d, J=9.4 Гц, 1Н), 7.28 (d, J=7.0 Гц, 1Н), 7.23-7.15 (m, 2Н), 7.13-7.06 (m, 2Н), 6.92-6.85 (m, 2Н), 6.84-6.78 (m, 2Н), 4.61 (s, 1H), 4.06 (s, 3H), 3.82 (s, 2H), 3.77 (s, 3H), 3.72 (s, 2H) млн-1.
Стадия В: 3-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-1-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]-1-(4-метоксибензил)мочевина (30t). Промежуточное соединение (29t) (135 мг, 0,354 ммоль), трихлорэтилкарбамат (6t-1) (154 мг, 0,379 ммоль) и диизопропиламин (120 мкл, 89 мг, 0,69 ммоль) растворяли в ДМА (5 мл) и нагревали до 80°С. После перемешивания при 80°С в течение 12 часов реакционную смесь охлаждали до комнатной температуры, разбавляли эфиром (50 мл) и трижды промывали 5 мл воды и один раз рассолом, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученное неочищенное масло очищали на силикагеле с EtOAc/гексан (2:3), получая соединение (30t) в виде бесцветного масла (180 мг, выход 83%).1Н-ЯМР (400 МГц, CDCl3) δ 7.82 (s, 1H), 7.07-6.95 (m, 8Н), 6.94-6.89 (m, 1Н), 6.89-6.84 (m, 1Н), 7.29 (d, J=9.0 Гц, 1Н), 6.75 (d, J=8.6 Гц, 2Н), 6.69 (dd, J=8.9, 4.7 Гц, 1Н), 6.60 (s, 1H), 6.41 (s, 1H), 4.57 (s, 2H), 4.44 (s, 2H), 4.04 (s, 3Н), 3.78 (s, 1H), 3.77 (s, 3Н), 2.31 (s, 3Н), 1.30 (s, 9H) млн-1.
Стадия С:
1-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]-3-(4-метоксибензил)-1-метилмочевина (31t). Промежуточное соединение (30t) (150 мг, 0,23 ммоль) растворяли в ДМФА (2 мл) и охлаждали до 0°С. К раствору при 0°С добавляли гидрид натрия (60% в масле, 14 мг, 0,36 ммоль) и метилйодид (73,8 мкл, 168 мг, 1,19 ммоль) и реакционную смесь перемешивали при 0°С в течение 1 часа. Реакционную смесь гасили путем добавления 5 мл воды при 0°С и затем трижды экстрагировали 50 мл Et2O. Объединенный органический слой дважды промывали 5 мл воды и один раз рассолом, сушили над MgSO4, фильтровали и упаривали при пониженном давлении. Остаток очищали на силикагеле с EtOAc/гексан (1:2), получая соединение (31t) в виде бледно-желтого аморфного вещества (134 мг, выход 87%).1Н-ЯМР (400 МГц, CDCl3) δ 7.82 (s, 1H), 7.37 (d, J=8.6 Гц, 2Н), 7.28 (d, J=9.4 Гц, 1Н), 7.15-7.02 (m, 2H), 7.00-6.81 (m, 6H), 6.80-6.60 (m, 3Н), 5.85 (s, 1H), 4.21 (s, 2H), 4.20 (s, 2H), 4.05 (s, 3Н), 3.76 (s, 3Н), 2.98 (s, 3Н), 2.29 (s, 3Н), 1.23 (s, 9H) млн-1.
Стадия D: 1-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1Н-индазол-5-илокси)бензил]-1-метил мочевина (32t). Промежуточное соединение (31t) (85 мг, 0,131 ммоль) растворяли в 5 мл раствора 2%-ного (об./об.) анизола в трифторуксусной кислоте и перемешивали при комнатной температуре в течение 1,5 часов. После упаривания неочищенный остаток растворяли в EtOAc (80 мл) и нейтрализовали насыщенным NaHCO3, промывали рассолом, сушили над MgSO4, фильтровали и упаривали при пониженном давлении. Полученное неочищенное масло очищали на силикагеле с EtOAc/гексан (2:3), получая соединение (32t) в виде белого аморфного вещества (68,6 мг, выход 99%).1Н-ЯМР (400 МГц, CDCl3) δ 7.84 (s, 1Н), 7.32 (d, J=9.4 Гц, 1Н), 7.25 (d, J=7.8 Гц, 2Н), 7.13 (d, J=7.8 Гц, 2Н), 7.07 (t, J=10.2 Гц, 1Н), 7.20-6.96 (m, 1Н), 6.93 (td, J=8.6, 3.1 Гц, 1Н), 6.85 (td, J=8.6, 3.1 Гц, 1Н), 6.68 (dd, J=8.6, 4.7 Гц, 1Н), 6.11 (s, 1H), 5.31 (t, J=6.3 Гц, 0,8 Н), 5.17 (t, J=6.3 Гц, 0,2Н), 4.48-4.26 (m, 2Н), 4.05 (s, 3Н), 3.00 (s, 3Н), 2.33 (s, 2,4Н), 2.32 (s, 0.6H), 1.36 (s, 1,8H), 1.29 (s, 7,2H) млн-1.
Пример 106
Получение 1-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1Н-пиразоло[3,4-с]пиридин-5-илокси)бензил]мочевины (4u)
Реакционная схема синтеза соединения 4u показана на Фиг.48.
Стадия А: 5-Фтор-2-гидроксибензонитрил (23t). 2,5-Дифторбензонитрил (14,9 г, 107 ммоль) и бензиловый спирт (11,1 мл, 11,6 г, 107 ммоль) растворяли в ДМФА (330 мл) и охлаждали до 0°С. К раствору при 0°С добавляли гидрид натрия (60% в масле, 6,40 г, 161 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение 1 часа при комнатной температуре реакционный раствор охлаждали до 0°С и к раствору постепенно добавляли воду (330 мл). Смесь переносили в делительную воронку и трижды экстрагировали 500 мл Et2O. Объединенный органический слой дважды промывали 100 мл воды, один раз рассолом, затем сушили над MgSO4. После фильтрации раствор концентрировали при пониженном давлении, получая неочищенное бледно-желтое твердое вещество. Неочищенное твердое вещество растворяли в МеОН (500 мл). В атмосфере азота к раствору добавляли 10% палладия на активированном угле. Газообразный азот заменяли газообразным водородом и реакционную смесь перемешивали при комнатной температуре (если в течение 30 минут реакция не происходила, то Pd/C отфильтровывали и реакцию проводили снова). После перемешивания в течение 2 часов палладий на угле отфильтровывали и промывали МеОН. Раствор концентрировали при пониженном давлении, получая бледно-желтое твердое вещество. Твердое вещество перекристаллизовывали из горячего толуола (100 мл), добавляя гексан (10 мл) с последующим охлаждением до 0°С. Полученные белые иглы промывали смесью толуола и гексана (1:1, 7,23 г, выход 49%). Маточный раствор концентрировали и очищали на силикагеле с Et2О/гексан (2:3-1:1), получая целевое соединение (23t) (6,94 г, выход 47%). Общий выход на 2 стадиях 14,2 г (96%).1Н-ЯМР (400 МГц, d6-ДМСО) δ 11.09 (s, 1H), 7.58 (dd, J=8.4, 3.2 Гц, 1Н), 7.40 (td, J=8.6, 3.2 Гц, 1Н), 7.03 (dd, J=9.2, 4.4 Гц, 1Н) млн-1.
Стадия В: 5-Фтор-2-(4-метил-5-аминопиридин-2-илокси)бензонитрил (1u). Промежуточное соединение (23t) (1,78 г, 13,0 ммоль), 2-хлор-4-метил-5-нитропиридин (2,31 г, 13,0 ммоль) и карбонат калия (1,80 г, 13,0 ммоль) растворяли в ДМФА (120 мл). Когда смесь нагревали до 60°С, бесцветный раствор в течение 10 минут становился синим. После перемешивания в течение 16 часов при 60°С реакционную смесь оставляли охлаждаться до комнатной температуры и затем разбавляли 100 мл Et2O. Неорганический осадок удаляли фильтрацией и промывали Et2O. Объединенный органический раствор (всего 600 мл) переносили в делительную воронку и трижды промывали 60 мл воды и один раз рассолом. Раствор сушили над MgSO4 и затем концентрировали при пониженном давлении, получая неочищенное коричневое твердое вещество. Неочищенное твердое вещество растворяли в МеОН (240 мл). К раствору в атмосфере азота добавляли 10% палладия на активированном угле. Газообразный азот заменяли газообразным водородом и реакционную смесь перемешивали при комнатной температуре (если в течение 30 минут реакция не происходила, то Pd/C отфильтровывали и реакцию проводили снова). После перемешивания в течение 1,5 часов палладий на угле отфильтровывали и промывали МеОН. Раствор концентрировали при пониженном давлении, получая бледно-желтое твердое вещество. Неочищенное соединение очищали на силикагеле с EtOAc/гексан (1:1-3:2), получая (1u) в виде белого твердого вещества (2,21 г, выход на 2 стадиях 70%).1Н-ЯМР (400 МГц, CDCl3) δ 7.57 (s, 1H), 7.33 (ddd, J=7.8, 3.1, 1.6 Гц, 1Н), 7.29-7.22 (m, 1Н), 7.23 (s, ОН), 7.15 (ddd, J=9.3, 4.7, 1.6 Гц, 1Н), 6.82 (s, 1Н), 2.22 (s, ЗН)млн-1.
Стадия С: 5-Фтор-2-(1Н-пиразоло[3,4-с]пиридин-5-илокси)бензонитрил (2u): Промежуточное соединение (1u) (0,25 г, 1,03 ммоль) и NH4BF4 (0,22 г, 2,06 ммоль) растворяли в 10 мл смешанного растворителя АсОН/вода (2:1) и охлаждали до 0°С, затем к раствору при 0°С добавляли концентрированную HCl (0,43 мл, 5,16 ммоль) и NaNO2 (0,078 г, 1,13 ммоль). После добавления нитрата натрия цвет раствора сразу же становился желтым. Затем реакционную смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение 2 часов растворитель удаляли при пониженном давлении и трижды подвергали азеотропной перегонке с толуолом для удаления воды, получая бледно-желтое неочищенное твердое вещество. Твердое вещество растворяли в 10 мл EtOAc и к раствору добавляли КОАс. Через 30 минут цвет суспензии становился темно-оранжевым и затем ее перемешивали еще в течение часа. Белую неорганическую соль удаляли фильтрацией и промывали EtOAc. Объединенный фильтрат разбавляли EtOAc до общего объема 100 мл, переносили в делительную воронку, промывали насыщенным NaHCO3 (10 мл) и рассолом, сушили над MgSO4, фильтровали и упаривали, получая твердое вещество темно-оранжевого цвета. Неочищенное твердое вещество очищали на силикагеле с EtOAc/гексан (2:3), получая твердое вещество оранжево-желтого цвета (2u) (0,23 г, выход на 2 стадиях 88%).1Н-ЯМР (400 МГц, CDCl3) δ 8.65 (s, 1H), 8.15 (s, 1H), 7.30-7.34 (m, 2H), 7.32-7.22 (m, 2Н), 7.12 (dd, J=9.4, 4.7 Гц, 1Н) млн-1.
Стадия D: 5-Фтор-2-(1-метил-1Н-пиразоло[3,4-с]пиридин-5-илокси)бензонитрил (3u). Промежуточное соединение (2u) (0,23 г) растворяли в ДМФА (9 мл) и охлаждали до 0°С. К раствору при 0°С добавляли гидрид натрия (60% в масле, 0,054 г, 1,36 ммоль) и метилйодид (0,28 мл, 642 мг, 4,52 ммоль), реакционную смесь затем перемешивали в течение часа при 0°С. Смесь гасили 10 мл воды и трижды экстрагировали 30 мл эфира. Объединенный органический слой дважды промывали 5 мл воды и один раз рассолом, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный маслянистый остаток очищали на силикагеле с EtOAc/гексан (1:1), получая бесцветное масло (3u) (0,124 г, выход 51%).1Н ЯМР (400 МГц, CDCl3) δ 8.56 (s, 1H), 8.02 (s, 1H), 7.40-7.32 (m, 2H), 7.31-7.23 (m, 2H), 7.11-7.05 (m, 1H), 4.17 (s, 3H) млн-1.
Стадия Е:
1-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1Н-пиразоло[3,4-с]пиридин-5-илокси)бензил]мочевина (4u). Промежуточное соединение (3u) растворяли в 10 мл МеОН и к раствору затем добавляли концентрированную HCl (12 н., 1,0 мл, 12 ммоль) и 10% палладия на активированном угле. Смесь перемешивали в атмосфере водорода. После перемешивания в течение 24 часов палладий на активированном угле отфильтровывали и фильтрат концентрировали при пониженном давлении. Полученный остаток дважды подвергали азеотропной перегонке со смесью толуола и этанола (досуха), получая неочищенную хлористоводородную соль амина. Неочищенную соль растворяли в 5 мл ДМФА. К раствору добавляли диизопропилэтиламин и трихлорэтилкарбамат и нагревали до 80°С. После перемешивания в течение 16 часов ДМФА выпаривали при пониженном давлении, остаток разбавляли 50 мл EtOAc и промывали насыщенным NaHCO3 и рассолом, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученное неочищенное масло очищали препаративной ТСХ с EtOAc/гексан (2:1), а затем со 100%-ным CH2Cl2, получая бесцветное масло (4u) (9,0 мг, выход 4%).1Н-ЯМР (400 МГц, CDCl3) δ 8.23 (s, 1Н), 7.90 (s, 1Н), 7.24 (s, 1Н), 7.20 (d, J=8.2 Гц, 2Н), 7.08 (d, J=8.6 Гц, 2Н), 7.06-7.00 (m, 2Н), 6.91 (td, J=8.3, 3.1 Гц, 1Н), 6.82 (dd, J=8.9, 4.7 Гц, 1Н), 6.46 (br, 1Н), 6.18 (s, 1Н), 5.84 (t, J=8.6 Гц, 1Н), 4.36 (s, 1H), 4.34 (s, 2H), 4.05 (s, 3H), 2.28 (s, 3H), 1.28 (s, 9H) млн-1.
Пример 107
Получение 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензил]мочевины (7v) и 1-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензил]мочевины (8v)
Реакционная схема синтеза соединений 7v и 8v показана на Фиг.49.
Стадия А: 5-Фтор-2-(3-метилбензо[d]изоксазол-6-илокси)бензонитрил (3v). 3-Метилбензо[d]изоксазол-6-ол (1v) (ссылка на синтез: Indian J. Chem. Sect. В; 19: 571-575 (1980)) (1,58 г, 10,6 ммоль) и 2,5-дифторбензонитрил (2v) (1,47 г, 10,6 ммоль) объединяли с карбонатом калия (1,46 г, 10,6 ммоль) в 15 мл сухого ДМА. Реакционную смесь нагревали в 100°С бане в течение 6 часов. Реакционную смесь охлаждали и разбавляли EtOAc (200 мл) и трижды промывали водой, NH4Cl, NaHCO3 и рассолом. Органический раствор сушили над MgSO4 и концентрировали. Неочищенный остаток очищали колоночной хроматографией (диоксид кремния, 70-80% СН2Cl2/гексан), получая соединение (3v) (1,65 г, выход 58%).1Н-ЯМР (400 МГц, CDCl3) δ 7.63 (d, J=8.6 Гц, 1Н), 7.44-7.41 (m, 1H), 7.33-7.26 (m, 1H), 7.10 -7.06 (m, 2H), 7.03 (dd, J=8.2, 4.7 Гц, 1Н), 2.59 (s, 3Н). МС ИЭР (+), обнаружено: m/z 269 (М+Н).
Стадия В: 2-(3-Бромметилбензо[d]изоксазол-6-илокси)-5-фторбензонитрил (4v). 5-Фтор-2-(3-метилбензо[d]изоксазол-6-илокси)бензонитрил (3v), (0,87 г, 3,2 ммоль), N-бромсукцинимид (0,82 г, 4,6 ммоль), пероксид бензоила (0,12 г, 0,49 ммоль) и о-дихлорбензол (6 мл) объединяли в 15 мл пробирке для реакций под давлением. Смесь перемешивали и нагревали в 150°С бане. Через 2,5 часа реакционную смесь охлаждали и остаток сразу же очищали колоночной хроматографией (диоксид кремния, 70% СН2Cl2/гексаны), получая соединение (4v) (0,29 г, выход 26%) и восстановленное исходное вещество (3v) (0,57 г, 65%).1Н-ЯМР (400 МГц, CDCl3) δ 7.84 (d, J=7.0 Гц, 1Н), 7.45-7.43 (m, 1Н), 7.35-7.31 (m, 1Н), 7.16-7.13 (m, 2Н), 7.08-7.05 (m, 1Н), 4.72 (s, 2Н).
Стадия С: 5-Фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензонитрил (5v). 2-(3-Бромметилбензо[d]изоксазол-6-илокси)-5-фторбензонитрил (4v) (0,295 г, 0,850 ммоль) растворяли в дихлорметане и по каплям добавляли пирролидин (0,18 г, 2,5 ммоль). Через 2 часа реакционную смесь концентрировали и распределяли между NaHCO3 (15 мл) и EtOAc (30 мл). Водный слой снова экстрагировали EtOAc, органические фракции объединяли, промывали рассолом и сушили (MgSO4), получая соединение (5v) (0,27 г, 94%), которое использовали без дополнительной очистки.1Н-ЯМР (400 МГц, CDCl3) δ 7.90 (d, J=8.6 Гц, 1Н), 7.42 (d, J=6.3 Гц, 1Н), 7.30-7.26 (m, 1H), 7.11 (s, 1Н), 7.07-7.02 (m, 2Н), 4.02 (s, 2H), 2.62 (s, 4H), 1.82 (s, 4H). МС ИЭР (+), обнаружено: m/z 338 (М+Н).
Стадия D: 5-Фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензиламин (6v). 5-Фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензонитрил (5v) (0,20 г, 0,59 ммоль) растворяли в ТГФ (4 мл) и охлаждали до 0°С. Медленно добавляли раствор алюмогидрида лития в ТГФ (1,0 мл, 1,0 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры. Через 1 час реакционную смесь охлаждали до 0°С и добавляли дополнительную порцию LAH в ТГФ (0,9 мл, 0,9 ммоль). Реакционную смесь оставляли перемешиваться в течение 20 минут. Затем реакцию гасили, добавляя порциями декагидрат сульфата натрия при 0°С до прекращения выделения газа. Добавляли ТГФ, смесь фильтровали через целит и элюировали EtOAc. Раствор концентрировали, получая 5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензиламин (6v) (0,15 г), который на следующей стадии использовали без дополнительной очистки. МС (ХИАД+), обнаружено: m/z 342 (М+Н).
Стадия Е: 1-(5-трет-Бутил-2-метил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензил]мочевина (7v). 5-Фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензиламин (6v) (50 мг, 0,15 ммоль), 2,2,2-трифторэтиловый эфир (5-трет-бутил-2-метил-2Н-пиразол-3-ил)карбаминовой кислоты (а) (68 мг, 0,21 ммоль) и диизопропилэтиламин (0,038 мл, 0,22 ммоль) объединяли в ДМФА (1,5 мл) и нагревали при 85°С в течение 3 часов. Реакционную смесь охлаждали, концентрировали в вакууме и очищали колоночной хроматографией (диоксид кремния, 4% MeOH/CH2Cl2/0,5% Et3N), получая 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензил]мочевину (7v) (30 мг, 39%).1Н-ЯМР (400 МГц, CDCl3) δ 7.79 (d, J=8.6 Гц, 1Н), 7.16 (d, J=8.6 Гц, 1Н), 7.03-6.80 (m, 4H), 6.11 (s, 1H), 5.93 (s, 1H), 5.25 (t, J=8.6 Гц, 1Н), 4.38 (d, J=6.3 Гц, 2Н), 3.99 (s, 2H), 3.67 (s, 3Н), 2.61 (s, 4Н), 1.81 (s, 4H), 1.26 (s, 9H). МС (ХИАД+), обнаружено: m/z 521 (М+Н). ВЭЖХ (5-95%) 2,61 мин.
Стадия F: 1-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензил]мочевина (8v). 5-Фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензиламин (6v) (50 мг, 0,15 ммоль), 2,2,2-трифторэтиловый эфир (5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)карбаминовой кислоты (b) (75 мг, 0,18 ммоль) и диизопропилэтиламин (0,038 мл, 0,22 ммоль) объединяли в ДМФА (1,5 мл) и нагревали при 85°С в течение 3 часов. Реакционную смесь охлаждали, концентрировали в вакууме и очищали колоночной хроматографией (диоксид кремния, 40-60% EtOAc/гексаны/0,25% Et3N), получая 1-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)бензил]мочевину (8v) (30 мг, выход 34%).1Н-ЯМР (400 МГц, CDCl3) δ 7.78 (d, J=8.6 Гц, 1Н), 7.31 (d, J=7.0 Гц, 2Н), 7.20 (d, J=7.8 Гц, 2Н), 7.02-6.88 (m, 5Н), 6.17 (s, 1Н), 6.02 (s, 1Н), 5.30 (t, J=8.6 Гц, 1Н), 4.36 (d, J=5.5 Гц, 2Н), 3.98 (s, 2H), 2.60 (s, 4H), 2.36 (s, 3Н), 1.80 (s, 4H), 1.32 (s, 9H). МС ИЭР (+), обнаружено: m/z 597 (М+Н). ВЭЖХ (5-95%) 2,93 мин.
Пример 108
Получение 1-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-метилбензо[d]изоксазол-6-илокси)бензил]мочевины (10v)
Реакционная схема синтеза соединения 10v показана на Фиг.50.
Стадия А: 5-Фтор-2-(3-метилбензо[d]изоксазол-6-илокси)бензиламин (9v). 5-Фтор-2-(3-метилбензо[d]изоксазол-6-илокси)бензонитрил (3v) (84 мг, 0,31 ммоль; полученный, как описано в Примере 108) растворяли в ТГФ (3 мл) и охлаждали до 0°С. Медленно добавляли раствор алюмогидрида лития в ТГФ (0,35 мл, 0,35 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры. Через 1 час реакцию гасили, добавляя порциями декагидрат сульфата натрия при 0°С до прекращения выделения газа. Добавляли ТГФ и смесь фильтровали через целит и элюировали EtOAc. Раствор концентрировали, получая неочищенный продукт (9v) (66 мг), который использовали без дополнительной очистки.
Стадия В: 1-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-метилбензо[d]изоксазол-6-илокси)бензил]мочевина (10v). 5-Фтор-2-(3-метилбензо[d]изоксазол-6-илокси)бензиламин (9v) (60 мг, 0,22 ммоль), 2,2,2- трифторэтиловый эфир (5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)карбаминовой кислоты (12v) (89 мг, 0,22 ммоль) и диизопропилэтиламин (0,058 мл, 0,33 ммоль) объединяли в ДМФА (2 мл) и нагревали при 75°С в течение 3 часов. Реакционную смесь охлаждали, концентрировали в вакууме и очищали колоночной хроматографией (диоксид кремния, 30-40% EtOAc/гексаны), получая соединение (10v) (90 мг, выход 77%).1Н-ЯМР (400 МГц, CDCl3) δ 7.51 (d, J=8.6 Гц, 1Н), 7.30 (d, J=8.6 Гц, 2Н), 7.20 (d, J=7.8 Гц, 2Н), 7.02-6.88 (m, 5Н), 6.15 (s, 1Н), 5.99 (s, 1Н), 5.28 (t, J=7.6 Гц, 1Н), 4.36 (d, J=5.5 Гц, 2Н), 2.53 (s, 3H), 2.36 (s, 3H), 1.31 (s, 9H). МС ИЭР (+), обнаружено: m/z 528 (М+Н). ВЭЖХ (5-95%) 3,49 мин.
Пример 109
Получение 5-бром-1-циклопропилметил-1Н-индазола

К раствору 5-броминдазола (15 г, 76,1 ммоль) и (бромметил)циклопропана (8,12 мл, 83,7 ммоль) в 75 мл ДМФА добавляли К2СО3 (16 г, 114,0 ммоль). Смесь нагревали до 105°С. Через 24 часа все еще наблюдалось исходное вещество. Добавляли дополнительное количество (бромметил)циклопропана (5,7 мл, 57,0 ммоль) и реакционную смесь нагревали до 105°С в течение еще 24 часов. Снова обнаруживали 5-броминдазол, поэтому добавляли дополнительное количество (бромметил)циклопропана (4 мл, 38 ммоль) и реакционную смесь нагревали при 95°С в течение еще 48 часов. После исчезновения 5-броминдазола реакционную смесь вливали в смесь ДХМ/рассол. Два слоя разделяли, водный слой экстрагировали ДХМ (2×) и анализировали с помощью ТСХ. В водном слое не обнаружили никакого продукта. Объединенные органические вещества промывали Н2О (2×) и рассолом и сушили над Na2SO4. После фильтрации фильтрат концентрировали, полученный остаток очищали хроматографией, используя смесь гексан/EtOAc (9,5:0,5) и получая 5-бром-1-циклопропилметил-1Н-индазол (8,74 г, выход выделенного продукта 45%).1Н-ЯМР (400 МГц, CDCl3) δ 7.93 (s, 1Н), 7.86 (d, J=1.57 Гц, 1Н), 7.43 (dd, J=8.61, 1.57 Гц, 1Н), 7.31 (d, J=8.61 Гц, 1Н), 4.24 (d, J=6.26 Гц, 2Н), 1.37-1.26 (m, 1Н), 0.62-0.55 (m, 2Н), 0.43-0.37 (m, 2Н). МС (ХИАД+), обнаружено: m/z 251/253 (М/М+2Н, 1:1).
Пример 110
Получение 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (17d)
Реакционная схема синтеза соединения 17d согласно этому Примеру показана на Фиг.51.
Стадия А: 1,2-Дибром-4-метил-5-нитробензол. 3,4-Дибромтолуол (108,11 мл, 800 ммоль) добавляли по каплям в течение 4 часов к азотной кислоте (90%, 280 мл, 6000 ммоль), которую охлаждали до 0°С в атмосфере азота при механическом перемешивании. Во время добавления внутреннюю температуру смеси поддерживали ниже 10°С, а после окончания добавления смесь перемешивали в течение 1 часа при 0°С. К смеси по каплям добавляли воду (840 мл), поддерживая внутреннюю температуре ниже 10°С. Неочищенный продукт собирали фильтрацией и промывали водой (5×500 мл) для удаления избытка азотной кислоты. Твердые вещества сушили под высоким вакуумом и очищали перекристаллизацией из этанола (800 мл), получая 180,9 г целевого продукта (выход 77%) в виде твердого вещества.1Н-ЯМР (400 МГц, CDCl3) δ 8.24 (s, 1H), 7.64 (s, 1H), 2.55 (s, 3Н).
Стадия В: 1-Бром-2-(2,4-дифторфенокси)-4-метил-5-нитробензол. Смесь 1,2-дибром-4-метил-5-нитробензола (84,3 г, 286 ммоль), 2,4-дифторфенола (37,2 г, 286 ммоль) и К2СО3 (43,5 г, 315 ммоль) нагревали до 100°С в течение 45 часов. Реакционную смесь охлаждали до комнатной температуры и затем хранили в холодильнике при 5°С в течение ночи. Всю реакционную смесь за один раз вливали в 1200 мл ледяной воды. Полученное влажное твердое вещество собирали, частично измельчали и перемешивали в 900 мл Н2О в течение 45 минут. Твердое вещество собирали фильтрацией и промывали 700 мл воды (порциями). Полученное твердое вещество сушили под высоким вакуумом в течение ночи, получая 93,5 г коричневого твердого вещества (выход 95%).1Н-ЯМР (400 МГц, CDCl3) δ 8.38 (s, 1H), 7.18 (m, 1H), 7.03 (m, 1Н), 6.97 (m, 1Н), 6.52 (s, 1H), 2.50 (s, 3Н).
Стадия С: 5-Бром-4-(2,4-дифторфенокси)-2-метилфениламин. 1-Бром-2-(2,4-дифторфенокси)-4-метил-5-нитробензол (87,0 г, 253 ммоль) растворяли в ТГФ (300 мл) и разбавляли МеОН (900 мл). Добавляли цинковую пыль (82,7 г, 126 моль) и медленно добавляли 1 л насыщенного NH4Cl, чтобы температура реакции никогда не превышала 42°С. Реакционную смесь интенсивно механически перемешивали в течение 16 часов. Реакционную смесь фильтровали через целит и осадок на фильтре промывали этилацетатом. Затем фильтрат концентрировали с помощью 1,2 л насыщенного NH4OAc. После удаления ТГФ/МеОН твердые вещества собирали и промывали водой. Затем твердые вещества перемешивали в 1 л воды в течение 30 минут, собирали фильтрацией и промывали тремя порциями воды (1 л). Полученное твердое вещество сушили под высоким вакуумом в течение 48 часов, получая 64 г целевого продукта (выход 81%). МС (ИЭР+), обнаружено: m/z 314, 316 (М+1, Br образец).1Н-ЯМР (400 МГц, CDCl3) δ 6.92 (m, 1H), 6.91 (s, 1H), 6.75 (m, 2Н), 6.70 (s, 1H), 3.57 (br. s, 2H), 2.08 (s, 3Н).
Стадия D: 6-Бром-5-(2,4-дифторфенокси)-1Н-индазол (14d).
Тетрафторборат 5-бром-4-(2,4-дифторфенокси)-2-метилбензолдиазония. 5-Бром-4-(2,4-дифторфенокси)-2-метилфениламин (30,0 г, 96 ммоль) растворяли в смеси АсОН/Н2О (2:1, 960 мл). Добавляли NH4BF4 (20,0 г, 191 ммоль) и смесь охлаждали до 3°С (примерно 30 минут). Затем за один раз добавляли концентрированную HCl (40 мл), смесь нагревали до 6°С. Смесь охлаждали до 2°С и затем добавляли NaNO2 (7,25 г, 105 ммоль). Реакционную смесь перемешивали в ледяной бане в течение 5 минут и затем оставляли перемешиваться в течение 1 часа при комнатной температуре. Смесь концентрировали при пониженном давлении и остаток подвергали азеотропной перегонке с толуолом (3×400 мл). Неочищенное вещество (тетрафторборат 5-бром-4-(2,4-дифторфенокси)-2-метилбензолдиазония) использовали на следующей реакционной стадии без дополнительной очистки.
6-Бром-5-(2,4-дифторфенокси)-1Н-индазол. Неочищенный тетрафторборат 5-бром-4-(2,4-дифторфенокси)-2-метилбензолдиазония суспендировали в этилацетате (650 мл) и обрабатывали 10 эквивалентами КОАс. Смесь интенсивно перемешивали при комнатной температуре в течение 1,5 часов и затем фильтровали и разбавляли этилацетатом до общего объема 1 л. Смесь промывали смесью насыщенный NaHCO3/рассол (800 мл, 1:1). Водную фазу экстрагировали этилацетатом (400 мл). Органические вещества объединяли, сушили (MgSO4) и концентрировали до коричневого твердого вещества (31 г, выход 99%).1Н-ЯМР (400 МГц, CDCl3) δ 10.55 (br. s, 1H), 7.98 (s, 1Н), 7.84 (s, 1Н), 7.20 (s, 1Н), 6.99 (m, 1Н), 6.94 (m, 1Н), 6.84 (m, 1Н).
Стадия Е: 6-Бром-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол (15d). 6-Бром-5-(2,4-дифторфенокси)-1Н-индазол (60,0 г, 185 ммоль) растворяли в ДМФА и обрабатывали К2СО3 (76,5 г, 554 ммоль) и изобутилбромидом (126,4 г, 923 ммоль). Смесь перемешивали и нагревали до 80°С в течение 16 часов. Добавляли еще 15 г
К2СО3 и смесь интенсивно перемешивали в течение еще 24 часов. Затем реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и растворяли в эфире (1 л). Смесь промывали смесью рассол/вода (1:5, 2×600 мл). Водные фазы экстрагировали эфиром (300 мл), объединенные органические вещества сушили (MgSO4) и концентрировали при пониженном давлении. Неочищенный продукт подвергали хроматографии на Biotage Flash 75 двумя порциями (примерно по 35 г каждая), элюируя смесью 5% этилацетата в гексанах. Объединенные очищенные продукты давали 30,1 г целевого продукта в виде твердого вещества (выход 43%). МС (ИЭР+), обнаружено: m/z 381, 383 (М+1, Br образец).1Н-ЯМР (400 МГц, CDCl3) δ 7.86 (s, 1Н), 7.72 (s, 1Н), 7.16 (s, 1Н), 6.98 (m, 1Н), 6.92 (m, 1Н), 6.82 (m, 1Н), 4.12 (d, 2H), 2.34 (m, 1Н), 0.94 (d, 6H).
Стадия F: 5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-карбонитрил (16d). 6-Бром-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол (31,2 г, 82 ммоль) и Cu(I)CN (13,9 г, 156 ммоль) растворяли в ДМА и дегазировали азотом под вакуумом. Реакционную смесь нагревали до 150°С в течение 16 часов. Смесь охлаждали до комнатной температуры и разбавляли этилацетатом перед двойной промывкой 7 М NH4OH. Органические слои промывали рассолом и дегазировали азотом, после чего сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенный продукт подвергали хроматографии, элюируя смесью 10% этилацетата в гексанах, получая 25,1 г продукта (выход 95%).
Стадия G: 5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновая кислота (17d). 5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-карбонитрил (25,1 г, 77 ммоль) суспендировали в этаноле (620 мл) и КОН (2,5 М, 310 мл) и нагревали до температуры дефлегмации в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении для удаления этанола. Полученный водный раствор разбавляли водой и промывали эфиром. Водный слой подкисляли концентрированной HCl до рН 1 и несколько раз экстрагировали этилацетатом. Органические слои объединяли и концентрировали при пониженном давлении, получая 25,5 г продукта (выход 96%).1Н-ЯМР (400 МГц, CDCl3) δ 8.37 (s, 1H), 7.91 (s, 1Н), 7.20 (m, 1Н), 7.07 (s, 1Н), 7.04 (m, 1Н), 6.95 (m, 1Н), 4.24 (d, 2H), 2.36 (m, 1Н), 0.94 (d, 6H).
Пример 111
Получение {(3-[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-илокси]пропил}диметиламина (20d)
Реакционная схема синтеза соединения 20d согласно этому Примеру показана на Фиг.52.
Стадия А: 5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-ол (15d; полученный согласно Примеру 110, стадии А-Е). 6-Бром-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол (1,36 г, 3,6 ммоль) растворяли в сухом эфире (17,5 мл) и охлаждали до -78°С. Добавляли по каплям н-бутиллитий (1,7 мл 2,5 М раствора в гексане) в течение 10 минут и смесь перемешивали в течение 30 минут. Добавляли по каплям триметилборат (6 мл, 53,5 ммоль) в течение 10 минут, реакционную смесь оставляли нагреваться до комнатной температуры и перемешиваться в течение 18 часов. Смесь охлаждали до -10°С и добавляли 2 н. NaOH (3,6 мл) и воду (3 мл), а затем Н2О2 (3,6 мл) и 2 н. NaOH (3,6 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, пока образовывался белый осадок. Добавляли воду (3 мл), 4 н. NaOH (4 мл) и Н2О2 (1 мл), смесь перемешивали в течение еще 20 минут. Затем смесь разбавляли эфиром и слои разделяли. Водный слой промывали эфиром, подкисляли и затем экстрагировали эфиром (2×). Объединенные эфирные экстракты промывали водой и рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении, получая 0,465 г продукта (выход 41%).
Стадия В: 6-(3-Хлорпропокси)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол (19d). К раствору 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-ола (0,015 г, 0,05 ммоль) в ДМФА (1 мл) добавляли Cs2CO3. Реакционную смесь перемешивали в течение 30 минут, после чего добавляли 1-бром-3-хлорпропан (0,01 г, 0,08 ммоль) и смесь затем нагревали до 80°С в течение 25 часов. После охлаждения смеси до комнатной температуры реакционную смесь разбавляли водой и эфиром, слои разделяли. Водный слой экстрагировали эфиром и объединенные органические слои промывали 1 н. NaOH, водой и рассолом. Неочищенную смесь сушили над Na2SO4 и концентрировали при пониженном давлении, получая продукт с небольшим количеством примесей. Неочищенную смесь использовали на следующей реакционной стадии без дополнительной очистки.
Стадия С: {3-[5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-илокси]пропил}диметиламин (20d). К раствору 2 н. диметиламина в ТГФ (1,4 мл) добавляли 6-(3-хлорпропокси)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол (0,019 г, 0,05 ммоль). Смесь перемешивали при комнатной температуре в течение 62 часов и затем нагревали до 45°С в течение 3 часов. Растворитель удаляли при пониженном давлении и остаток распределяли между дихлорметаном и 0,1 н. NaOH. Слои разделяли и водный слой экстрагировали дихлорметаном (3×). Объединенные органические слои промывали водой и рассолом и затем сушили над Na2SO4. Смесь концентрировали при пониженном давлении, получая неочищенную смесь, которую очищали обращенно-фазовой ВЭЖХ. Целевую фракцию концентрировали до соли ТФУ (7 мг, выход 37%). МС (ИЭР+), обнаружено: m/z 404 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.88 (s, 1H), 7.34 (s, 1H), 6.97 (m, 1H), 6.82 (s, 1Н), 6.78 (m, 2Н), 4.14 (m, 2Н), 4.11 (d, 2Н), 2.98 (m, 2Н), 2.76 (s, 6H), 2.35 (m, 1Н), 2.22 (m, 2Н), 0.95 (d, 6H).
Пример 112
Получение 5-(2,4-дифторфенокси)-1-изобутил-6-(пиперидин-4-илметокси)-1Н-индазола (21d)
К раствору 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-ола (0,06 г, 0,19 ммоль) и трет-бутилового эфира 4-(толуол-4-сульфонилоксиметил)пиперидин-1-карбоновой кислоты (0,08 г, 0,20 ммоль) в ДМФА (3 мл) добавляли NaI (0,014 г, 0,009 ммоль) и К2СО3 (0,08 г, 0,56 ммоль). Реакционную смесь нагревали до 70°С в течение 20 часов. Смесь разбавляли эфиром и водой, слои разделяли. Водный слой экстрагировали эфиром (2×), объединенные органические слои промывали водой и рассолом и сушили над Na2SO4. Неочищенную смесь концентрировали при пониженном давлении и очищали обращенно-фазовой ВЭЖХ. Целевую фракцию концентрировали до 0,037 г, которые затем обрабатывали раствором HCl (4 н. в диоксане) в течение 7 часов. Растворитель удаляли при пониженном давлении и конечный продукт сушили под высоким вакуумом, получая 0,031 г твердого вещества (выход 37%). МС (ХИАД+), обнаружено: m/z 416 (М+1).
Пример 113
Получение 5-(2,4-дифторфенокси)-1-изобутил-6-(3-пиперазин-1-илпропокси)-1Н-индазола (22d)
К раствору 6-(3-хлорпропокси)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазола (0,025 г, 0,063 ммоль) и NaI (0,019 г, 0,13 ммоль) в ДМА (0,5 мл) и ТГФ (5 мл) добавляли трет-бутиловый эфир пиперазин-1-карбоновой кислоты (0,059 г, 0,32 ммоль). Реакционную смесь нагревали в запаянном реакционном сосуде до 65°С в течение 20 часов. Смесь концентрировали при пониженном давлении, остаток распределяли между водой, рассолом и эфиром. Слои разделяли и водный слой экстрагировали эфиром (2×). Объединенные органические слои промывали водой и рассолом и сушили над Na2SO4. Неочищенную смесь концентрировали при пониженном давлении и обрабатывали раствором HCl (4 н. в диоксане) в течение 3 часов. Полученную смесь концентрировали и очищали обращенно-фазовой ВЭЖХ, получая 0,025 г соли ТФУ (выход 51%). МС (ХИАД+), обнаружено: m/z 445 (М+1).1Н-ЯМР (400 МГц, CD3OD) δ 7.88 (s, 1Н), 7.32 (s, 1Н), 7.17 (s, 1Н), 7.14 (m, 1Н), 6.89 (m, 2Н), 4.23 (t, 2Н), 4.18 (d, 2Н), 3.52 (m, 4Н), 3.41 (m, 4Н), 3.14 (t, 2Н), 2.31 (m, 1Н), 2.19 (m, 2Н), 0.92 (d, 6H).
Пример 114
Получение 5-(2,4-дифторфенокси)-1-изобутил-6-(морфолин-2-илметокси)-1Н-индазола (23d)
Стадия А: трет-Бутиловый эфир 2-гидроксиметилморфолин-4-карбоновой кислоты. К раствору (4-бензилморфолин-2-ил)метанола (0,66 г, 3,18 ммоль, Synth. Comm. 1980, 10, 59-73) в МеОН (20 мл) добавляли Вос-ангидрид (0,83 г, 3,82 ммоль), а затем Pd/C (0,66 г, 6,20 ммоль). Смесь перемешивали в атмосфере водорода в течение 60 часов. Катализатор удаляли фильтрацией и фильтрат концентрировали при пониженном давлении, получая продукт в виде бесцветного масла (0,69 г, выход 99%).
Стадия В: трет-Бутиловый эфир 2-бромметилморфолин-4-карбоновой кислоты. К охлажденному (0°С) раствору трет-бутилового эфира 2-гидроксиметилморфолин-4-карбоновой кислоты (1,04 г, 4,79 ммоль, см. стадию 1) в дихлорметане (20 мл) добавляли CBr4 (1,98 г, 5,98 ммоль). После перемешивания смеси в течение 10 минут порциями добавляли трифенилфосфин (2,20 г, 8,38 ммоль). Реакционную смесь перемешивали при 0°С в течение 6 часов, затем оставляли нагреваться до комнатной температуры и перемешивали в течение 60 часов. Смесь концентрировали при пониженном давлении и затем разбавляли эфиром. Неочищенную смесь фильтровали, фильтрат концентрировали, получая неочищенный продукт, который подвергали хроматографии на Biotage, элюируя дихлорметаном. Целевые фракции объединяли и концентрировали с получением 0,50 г продукта (выход 37%).
Стадия С: 5-(2,4-Дифторфенокси)-1-изобутил-6-(морфолин-2-илметокси)-1Н-индазол. К раствору 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-ола (0,035 г, 0,11 ммоль) в ДМА (3,5 мл) добавляли Cs2CO3 (0,11 г, 0,33 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, после чего добавляли трет-бутиловый эфир 2-бромметилморфолин-4-карбоновой кислоты (0,062 г, 0,22 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 14 часов. Реакционную смесь разбавляли эфиром и водой, слои разделяли. Водный слой экстрагировали эфиром (3×), объединенные органические слои промывали водой и рассолом и сушили над Na2SO4. Неочищенную смесь концентрировали при пониженном давлении и очищали обращенно-фазовой ВЭЖХ, получая целевой продукт. МС (ХИАД+), обнаружено: m/z 418 (М+1).1Н-ЯМР (400 МГц, CD3OD) δ 7.90 (s, 1Н). 7.38 (s, 1H), 7.21 (s, 1H), 7.11 (m, 1H), 6.84 (m, 2H), 4.22 (m, 2H), 4.18 (d, 2H), 4.05 (m, 2H), 3.31 (m, 3Н), 3.03 (m, 2H), 2.31 (m, 1H), 0.92 (d, 6H).
Пример 115
Получение 1-[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-илокси]-3-пирролидин-1-илпропан-2-ола (24d)
Стадия А: 5-(2,4-Дифторфенокси)-1-изобутил-6-оксиранилметокси-1Н-индазол. К раствору 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-ола (0,15 г, 0,47 ммоль) в ДМА (5 мл) добавляли Cs2CO3 (0,46 г, 1,41 ммоль). После перемешивания смеси в течение 3 часов добавляли 2-бромметилоксиран (0,13 г, 0,94 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь разбавляли эфиром и водой, слои разделяли. Водный слой экстрагировали эфиром (3×), объединенные органические слои промывали водой и рассолом и сушили над Na2SO4. Неочищенный продукт концентрировали при пониженном давлении и использовали на следующей реакционной стадии без дополнительной очистки (0,135 г, выход 77%).
Стадия В: 1-[5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-илокси]-3-пирролидин-1-илпропан-2-ол. К раствору 5-(2,4-дифторфенокси)-1-изобутил-6-оксиранилметокси-1Н-индазола (0,035 г, 0,093 ммоль, стадия 1) в МеОН (3 мл) добавляли пирролидин (0,007 г, 0,093 ммоль). Смесь перемешивали при комнатной температуре в течение 36 часов и затем концентрировали при пониженном давлении. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ, получая конечный продукт в виде соли ТФУ (0,037 г, выход 59%). МС (ХИАД+), обнаружено: m/z 446 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.92 (s, 1Н), 7.33 (s, 1Н), 6.96 (m, 1Н), 6.88 (s, 1Н), 6.79 (m, 2Н), 4.34 (m, 1Н), 4.25 (m, 1Н), 4.14 (d, 2Н), 3.95 (m, 1Н), 3.82 (m, 2Н), 3.17 (m, 1Н), 3.08 (m, 1Н), 2.88 (m, 1Н), 2.78 (m, 1Н), 2.33 (m, 1Н), 2.12 (m, 4Н), 0.94 (d, 6H).
Пример 116
Получение (3-диметиламинопропил)амида 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфоновой кислоты (26d)
Реакционная схема синтеза соединения 26d согласно этому Примеру показана на Фиг.53.
Стадия А: 5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-сульфонилхлорид (25d). К охлажденному (-78°С) раствору 6-бром-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазола (15d; полученного, как описано в Примере 110, стадии А-Е) (2,0 г, 5,2 ммоль) в ТГФ (50 мл) в атмосфере N2добавляли по каплям н-бутиллитий (1,5 мл 2,5 М раствора). Полученный раствор перемешивали при -78°С в течение 5 минут и затем переносили при помощи канюли в охлажденную (-78°С) суспензию SO2 (0,34 г, 5,25 ммоль) в ТГФ (5 мл). Смесь перемешивали при -78°С в течение 2 часов, затем разбавляли эфиром (20 мл) и перемешивали при комнатной температуре в течение 1 часа. Суспензию концентрировали при пониженном давлении и остаток перемешивали в ледяной бане с насыщенным NaHCO3 (15 мл) и NCS (0,77 г, 5,8 ммоль) в течение 45 минут. Реакционную смесь экстрагировали этилацетатом (3×), объединенные органические слои промывали рассолом и сушили над Na2SO4. Раствор концентрировали при пониженном давлении, получая жидкость с высокой вязкостью, которую использовали на следующей реакционной стадии без дополнительной очистки.
Стадия В: (3-Диметиламинопропил)амид 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфоновой кислоты (26d). К охлажденному (0°С) раствору 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфонилхлорида (0,20 г, 0,50 ммоль) в дихлорметане в атмосфере N2 добавляли порциями 3-(диметиламино)пропиламин (0,05 г, 0,50 ммоль) и триэтиламин (0,15 г, 1,5 ммоль). Реакционную смесь перемешивали в течение 4 часов, затем разбавляли дихлорметаном (20 мл), промывали водой, насыщенным NaHCO3 и рассолом и затем сушили над MgSO4. Смесь концентрировали при пониженном давлении и подвергали препаративной хроматографии на ТСХ пластинках, элюируя смесью дихлорметан/МеОН/Et3N (95:4:1) и получая 93 мг конечного продукта (выход 40%). МС (ХИАД-), обнаружено: m/z 466.1Н-ЯМР (400 МГц, CDCl3) δ 8.15 (s, 1Н), 7.91 (s, 1Н), 7.20 (m, 1Н), 7.09 (s, 1Н), 7.00 (m, 1Н), 6.90 (m, 1H), 4.22 (d, 2H), 3.12 (t, 2H), 2.35 (m, 3Н), 2.13 (s, 6H), 1.70 (m, 2H), 0.94 (d, 6H).
Пример 117
Получение (S)-метил-2-(5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфонамидо)-4-(диметиламино)бутаноата (27d)
Получали, как описано в Примере 116, стадии А и В, заменяя 3-(диметиламино)пропиламин дигидрохлоридом метилового эфира 2-амино-4-диметиламиномасляной кислоты. Неочищенный продукт подвергали препаративной хроматографии на ТСХ пластинках, элюируя смесью гексаны/этилацетат/Et3N (50:50:5) и получая конечный продукт с выходом 37%. MeOH/Et3N (95:4:1) с получением 93 мг конечного продукта (выход 40%). МС (ХИАД-), обнаружено: m/z 523 (М-1).1Н-ЯМР (400 МГц, CDCl3) δ 8.08 (s, 1H), 7.90 (s, 1H), 7.23 (m, 1H), 7.08 (s, 1H), 7.00 (m, 1H), 6.89 (m, 1H), 4.32 (t, 1H), 4.21 (d, 2Н), 3.45 (s, 3Н), 2.37 (m, 3Н), 2.15 (s, 6Н), 2.05 (m, 1Н), 1.90 (m, 1Н), 0.92 (dd, 6Н).
Пример 118
Получение [2-(1-метилпирролидин-2-ил)этил]амида 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфоновой кислоты (28d)
Получали, как описано в Примере 116, стадии А и В, заменяя 3-(диметиламино)пропиламин 2-(1-метилпирролидин-2-ил)этиламином. Неочищенный продукт подвергали препаративной хроматографии на ТСХ пластинках, элюируя смесью гексаны/этилацетат/Et3N (1:1:0,1) и получая конечный продукт с выходом 24%. МС (ХИАД+), обнаружено: m/z 493 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.14 (s, 1H), 7.93 (s, 1H), 7.19 (m, 1H), 7.12 (s, 1H), 7.00 (m, 1H), 6.90 (m, 1H), 4.23 (d, 2H), 3.12 (m, 3Н), 2.44 (m, 1H), 2.36 (m, 1H), 2.34 (s, 3Н), 2.24 (m, 1H), 1.92 (m, 1H), 1.77 (m, 4H), 1.51 (m, 1H), 0.94 (d, 6H).
Пример 119
Получение (2-диметиламиноэтил)амида 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфоновой кислоты (29d)
Получали, как описано в Примере 116, стадии А и В, заменяя 3-(диметиламино)пропиламин диметиламиноэтиламином. Неочищенный продукт подвергали препаративной хроматографии на ТСХ пластинках, элюируя смесью гексаны/этилацетат/Et3N (1:1:0,1), с получением конечного продукта. МС (ХИАД+), обнаружено: m/z 453 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.15 (s, 1Н), 7.92 (s, 1Н), 7.18 (m, 1Н), 7.12 (s, 1Н), 7.00 (m, 1Н), 6.89 (т, 1Н), 4.23 (d, 2Н), 3.07 (t, 2Н), 2.41 (t, 2Н), 2.36 (т, 1Н), 2.15 (s, 6H), 0.94 (d, 6H).
Пример 120
Получение (S)-метил-2-(5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоксамидо)-4-(диметиламино)бутаноата (30d)
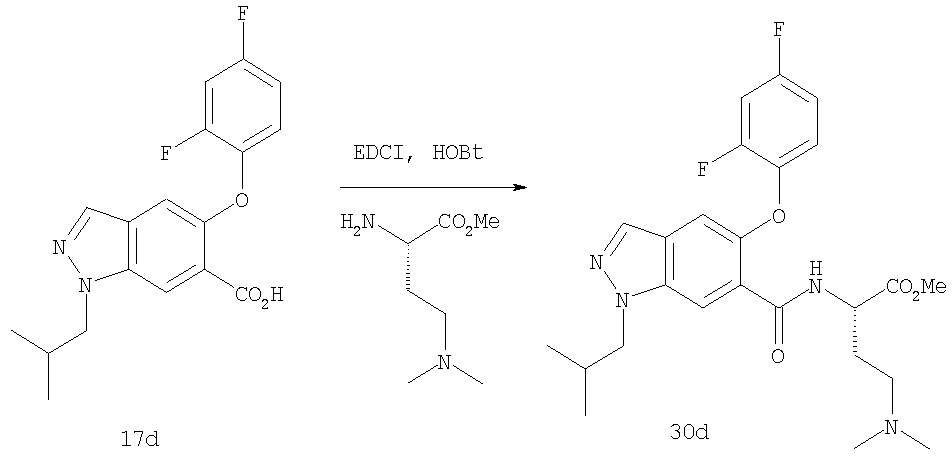
Стадия А: (S)-2-(трет-Бутоксикарбонил)-4-гидроксимасляная кислота. К раствору L-гомосерина (49,9 г, 419 ммоль) в 1 н. NaOH (460 мл) и EtOH (400 мл) добавляли раствор Вос-ангидрида (100,6 г, 461 ммоль) в ТГФ (400 мл) в течение 15 минут. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем смесь промывали эфиром (3×500 мл), подкисляли 1 н. HCl до рН 2 и экстрагировали этилацетатом (6×250 мл). Объединенные органические экстракты промывали рассолом (2×250 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 72,6 г белого твердого вещества (выход 79%).
Стадия В: Комплекс (S)-2-(трет-бутоксикарбонил)-4-гидроксимасляной кислоты и дициклогексиламина. К раствору (S)-2-(трет-бутоксикарбонил)-4-гидроксимасляной кислоты (72,6 г, 331 ммоль) в EtOH (500 мл) добавляли по каплям дициклогексиламин (73 мл, 364 ммоль). Смесь перемешивали в течение 2 часов при комнатной температуре и затем концентрировали при пониженном давлении. Белое твердое вещество сушили под высоким вакуумом и затем растирали с эфиром (1000 мл). Тонкий белый порошок собирали фильтрацией, промывали эфиром и сушили под высоким вакуумом (125,6 г, выход 95%).
Стадия С: Метиловый эфир (S)-2-трет-бутоксикарбониламино-4-гидроксимасляной кислоты. К суспензии комплекса (S)-2-(трет-бутоксикарбонил)-4-гидроксимасляной кислоты и дициклогексиламина (110 г, 275 ммоль) в ДМФА (900 мл) добавляли йодметан (20,5 мл, 330 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Прозрачный раствор концентрировали при пониженном давлении и подвергали азеотропной перегонке с толуолом (5×200 мл). Остаток разбавляли водой (500 мл) и этилацетатом (500 мл) и перемешивали в течение 2 часов, после чего слои разделяли. Водный слой экстрагировали этилацетатом (9×250 мл). Объединенные экстракты промывали рассолом (250 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая желтое масло. Неочищенное масло подвергали хроматографии на диоксиде кремния, элюируя смесью эфир/гексаны (3:1) и получая 53 г бесцветного масла (выход 83%).
Стадия D: Метиловый эфир (S)-4-бром-2-трет-бутоксикарбониламиномасляной кислоты. К охлажденному (0°С) раствору метилового эфира (S)-2-трет-бутоксикарбониламино-4-гидроксимасляной кислоты (28,7 г, 123 ммоль) в дихлорметане (500 мл) добавляли CBr4 (51,0 г, 154 ммоль). Смесь перемешивали в течение 5 минут, после чего порциями добавляли трифенилфосфин (48,41 г, 185 ммоль). Смесь продолжали перемешивать при 0°С в течение 1 часа и затем оставляли нагреваться до комнатной температуры. Растворитель удаляли при пониженном давлении и затем разбавляли эфиром (500 мл) и перемешивали в течение 30 минут. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя смесью эфир/гексаны (1:2) и получая 27,5 г белого твердого вещества (выход 76%).
Стадия Е: Метиловый эфир (S)-2-трет-бутоксикарбониламино-4-диметиламиномасляной кислоты. К раствору метилового эфира (S)-4-бром-2-трет-бутоксикарбониламиномасляной кислоты (27,5 г, 93 ммоль) в ТГФ (100 мл) в сосуде для реакций под давлением добавляли триэтиламин (26 мл) и диметиламин (93 мл 2,0 М раствора в ТГФ). Реакционный сосуд герметично закрывали и нагревали до 60°С в течение 16 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении и затем растворяли в дихлорметане (500 мл). Раствор промывали водой (3×200 мл) и рассолом (200 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток сушили под высоким вакуумом, получая 23,4 г желтого масла (выход 97%).
Стадия F: Дигидрохлорид (S)-метил-2-амино-4-(диметиламино)бутаноата. К охлажденному (0°С) раствору метилового эфира (S)-2-трет-бутоксикарбониламино-4-диметиламиномасляной кислоты (23,4 г, 90 ммоль) в диоксане (100 мл) по каплям добавляли HCl (225 мл, 4 М раствор в диоксане). Смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Твердое вещество фильтровали, промывали эфиром (3×100 мл) и сушили под высоким вакуумом, получая 20,2 г продукта (выход 96%).
Стадия G: (S)-Метил-2-(5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоксамидо)-4-(диметиламино)бутаноат (30d). 5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновую кислоту (17d; полученную согласно Примеру 110) (0,396 г, 1,14 ммоль) перемешивали с HOBt (N-гидроксибензотриазол) (0,192 г, 1,26 ммоль) и EDCI (0,241 г, 1,26 ммоль) в дихлорэтане (2 мл) в течение 10 минут при комнатной температуре. Затем эту смесь добавляли к суспензии дигидрохлорида (S)-метил-2-амино-4-(диметиламино)бутаноата (0,280 г, 1,20 ммоль) и триэтиламина (1 мл, 6,9 ммоль) в дихлорметане (6 мл). Реакционную смесь перемешивали в течение 3 часов и затем концентрировали при пониженном давлении. Остаток разбавляли хлороформом (50 мл) и промывали 1 н. HCl (2×25 мл), насыщенным К2СО3 (2×50 мл), водой (25 мл), рассолом (25 мл) и сушили над MgSO4. Отфильтрованный раствор концентрировали при пониженном давлении, получая желтое масло. Масло подвергали хроматографии, элюируя 5% МеОН в дихлорметане, и получали вязкое бесцветное масло, которое после сушки под высоким вакуумом затвердевало (0,393 г, выход 71%). МС (ИЭР+), обнаружено: m/z 489 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.91 (d, 1H), 8.36 (s, 1H), 7.86 (s, 1H), 7.16 (m, 1Н), 7.03 (m, 1H), 7.00 (s, 1H), 6.93 (m, 1H), 4.88 (m, 1H), 4.21 (d, 2H), 3.74 (s, 3Н), 2.34 (m, 3Н), 2.06 (s, 6H), 2.01 (m, 2H), 0.92 (d, 6H).
Пример 121
Получение (S)-5-(2,4-дифторфенокси)-N-(4-(диметиламино)-1-гидроксибутан-2-ил)-1-изобутил-1Н-индазол-6-карбоксамида (31d)
Смесь (S)-метил-2-(5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоксамидо)-4-(диметиламино)бутаноата (30d; Пример 120, стадии А-Е) (0,370 г, 0,76 ммоль) и
NaBH4 (0,114 г, 3,04 ммоль) в ТГФ/EtOH (20 мл, 3:2) нагревали до 60°С в течение 7 часов. Реакционную смесь концентрировали при пониженном давлении и разбавляли дихлорметаном. Суспензию подвергали хроматографии на Biotage, элюируя смесью 10% МеОН в дихлорметане с 1% триэтиламина. Получали 0,337 г продукта в виде вязкого масла (выход 97%). МС (ИЭР+), обнаружено: m/z 461 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.34 (d, 1H), 8.31 (s, 1H), 7.86 (s, 1H), 7.13 (m, 1H), 7.02 (s, 1H), 7.00 (m, 1H), 6.90 (m, 1H), 4.33 (m, 1H), 4.22 (d, 2H), 3.64 (m, 2H) 2.37 (m, 1H), 2.17 (s, 6H), 2.10 (m, 1H), 1.82 (m, 1Н), 1.07 (m,2H), 0.93 (d, 6H).
Пример 122
Получение (1-гидроксиметил-3-изопропиламинопропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (32d)
Стадия А: Изопропил-(4-метоксибензил)амин. Смесь 4-метоксибензиламина (1,37 г, 10 ммоль) и ацетона (0,81 мл, 11 ммоль) в сухом дихлорэтане (20 мл) перемешивали при комнатной температуре в течение 30 минут. К раствору добавляли триацетоксиборогидрид натрия (3,18 г, 15 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 17 часов. Реакционную смесь гасили 1 н. NaOH (50 мл) и слои разделяли. Водный слой экстрагировали дихлорметаном (2×20 мл). Объединенные экстракты промывали водой (20 мл), рассолом (20 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя смесью 10% МеОН в дихлорметане с 1% триэтиламина и получая 1,53 г масла (выход 85%).
Стадия В: (S)-Метил-4-бром-2-(трет-бутоксикарбонил)бутаноат. К охлажденному (0°С) раствору (S)-метил-2-амино-4-бромбутаноата (1,80 г, 6,5 ммоль) в ТГФ (20 мл) добавляли триэтиламин (4,53 г, 32,5 ммоль) и Вос-ангидрид (1,49 г, 6,83 ммоль, раствор в 20 мл ТГФ). Смесь перемешивали при 0°С в течение 30 минут, затем нагревали до комнатной температуры и перемешивали в течение 18 часов. Реакционную смесь гасили 1 н. HCl (50 мл) и слои разделяли. Водный слой экстрагировали эфиром (2×20 мл), объединенные органические экстракты промывали водой (20 мл) и рассолом (20 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая бледно-желтое масло. Масло подвергали хроматографии, элюируя смесью эфир/гексаны (1:2), и получали 1,45 г бесцветного масла, которое под высоким вакуумом затвердевало (выход 75%).
Стадия С: Гидрохлорид (S)-метил-4-((4-метоксибензил)(изопропил)амино)-2-(трет-бутоксикарбонил)бутаноата. Смесь изопропил-(4-метоксибензил)амина (0,111 г, 0,62 ммоль), (S)-метил-4-бром-2-(трет-бутоксикарбонил)бутаноата (0,150 г, 0,51 ммоль) и триэтиламина (0,21 мл, 1,52 ммоль) в ТГФ (5 мл) нагревали до температуры дефлегмации в течение 65 часов. Затем смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя 5% МеОН в дихлорметане и получая 0,026 г бесцветного масла (выход 13%). Затем соединение обрабатывали HCl (1 мл 4 н. раствора в диоксане) при комнатной температуре в течение 3 часов. Смесь концентрировали при пониженном давлении и сушили под высоким вакуумом.
Стадия D: (S)-Метил-4-((4-метоксибензил)(изопропил)амино)-2-(5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоксамидо)бутаноат. 5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновую кислоту (17d; полученную согласно Примеру 110) (0,022 г, 0,064 ммоль) перемешивали с HOBt (0,011 г, 0,070 ммоль) и EDCI (0,014 г, 0,070 ммоль) в дихлорэтане (1 мл) в течение 10 минут при комнатной температуре. Затем к этой смеси добавляли суспензию гидрохлорида (S)-метил-4-((4-метоксибензил)(изопропил)амино)-2-(трет-бутоксикарбонил)бутаноата (0,025 г, 0,067 ммоль) и триэтиламина (0,054 мл, 0,384 ммоль) в дихлорметане (2 мл). Смесь перемешивали при комнатной температуре в течение 16 часов. Раствор фильтровали через целит и концентрировали при пониженном давлении. Неочищенное масло подвергали хроматографии, элюируя 2% МеОН в дихлорметане с 1% триэтиламина и получая 0,035 г вязкого бледно-желтого масла (выход 89%).
Стадия Е: (S)-N-(4-((4-Метоксибензил)(изопропил)амино)-1-гидроксибутан-2-ил)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоксамид. (S)-Метил-4-((4-метоксибензил)(изопропил)амино)-2-(5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоксамидо)бутаноат (0,035 г, 0,057 ммоль) и NaBH4 (0,022 г, 0,57 ммоль) растворяли в смеси ТГФ/МеОН (5 мл, 3:2) и нагревали до 50°С в течение 3 часов. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя 5% МеОН в дихлорметане с 1% триэтиламина, получая 0,020 г геля (выход 59%).
Стадия F: (1-Гидроксиметил-3-изопропиламинопропил)амид (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (32d). К раствору (S)-N-(4-((4-метоксибензил)(изопропил)амино)-1-гидроксибутан-2-ил)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоксамида (0,020 г, 0,033 ммоль) в МеОН (5 мл) добавляли влажный Pd/C (0,020 г, 10 мас.%). Смесь несколько раз продували водородом и затем перемешивали при комнатной температуре в атмосфере Н2 в течение 5 часов. Катализатор удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя смесью 10% МеОН в дихлорметане с 2% триэтиламина и получая 9,4 мг бесцветного геля (выход 60%). МС (ИЭР+), обнаружено: m/z 475 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 8.40 (d, 1Н), 8.31 (s, 1Н), 7.87 (s, 1Н), 7.13 (m, 1Н), 7.03 (s, 1Н), 7.01 (m, 1Н), 6.91 (m, 1Н), 4.33 (m, 1Н), 4.22 (d, 2Н), 3.69 (m, 2Н), 2.69 (m, 2Н), 2.37 (m, 2Н), 1.32 (m, 2Н), 1.09 (d, 3H), 1.01 (d, 3H), 0.93 (d, 6H).
Пример 123
Получение (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}4-диметиламиномасляной кислоты (33d)
Стадия А: 2,5-Диоксопирролидин-1-иловый эфир 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты. 5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновую кислоту (17d; полученную согласно Примеру 110) (25,0 г, 72,2 ммоль), EDCI (18,0 г, 93,8 ммоль) и 1-гидроксипирролидин-2,5-дион (9,97 г, 86,6 ммоль) суспендировали в дихлорметане и перемешивали при комнатной температуре в течение 2 часов. Смесь разбавляли дихлорметаном (500 мл) и промывали насыщенным NH4Cl (2×200 мл), насыщенным NaHCO3 (2×200 мл) и рассолом (200 мл). Органические слои сушили над MgSO4 и концентрировали при пониженном давлении, получая неочищенный продукт в виде желтой пены.
Стадия В: Метиловый эфир (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-диметиламиномасляной кислоты. К раствору 2,5-диоксопирролидин-1-илового эфира 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (32,0 г, 72,2 ммоль) и дигидрохлорида метилового эфира 2-амино-4-диметиламиномасляной кислоты (19,35 г, 83,0 ммоль) в дихлорметане добавляли триэтиламин (35 мл, 253 ммоль) и смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении и разбавляли дихлорметаном (400 мл). Раствор промывали насыщенным NH4Cl (2×200 мл), насыщенным NaHCO3(2×200 мл) и рассолом (200 мл). Органические слои сушили над MgSO4 и концентрировали при пониженном давлении, получая неочищенный продукт.
Стадия С: (S)-2-{[5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-диметиламиномасляная кислота (33d). К раствору метилового эфира (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-диметиламиномасляной кислоты (3,36 г, 6,88 ммоль) в ТГФ (5 мл) добавляли триметилсиланоат калия (1,77 г, 13,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Перед концентрированием смеси при пониженном давлении к ней добавляли раствор HCl (17 мл 4 М раствора в диоксане). Остаток суспендировали в дихлорметане и фильтровали. Фильтрат концентрировали при пониженном давлении, получая 2,23 г продукта (выход 68%). МС (ИЭР+), обнаружено: m/z 475 (М+1).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.81 (d, 1H), 8.02 (s, 1H), 7.99 (s, 1H), 7.48 (m, 1H), 7.25 (m, 1Н), 7.22 (s, 1Н), 7.11 (m, 1Н), 4.50 (m, 1Н), 4.28 (d, 2Н), 3.17 (m, 1Н), 3.04 (m, 1Н), 2.70 (s, 6Н), 2.25 (m, 2Н), 2.13 (m, 1Н), 0.87 (d, 6H).
Пример 124
Получение (1-гидроксиметил-3-пиперидин-1-илпропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (34d)
Стадия А: Метиловый эфир (S)-2-трет-бутоксикарбониламино-4-пиперидин-1-илмасляной кислоты. Метиловый эфир (S)-4-бром-2-трет-бутоксикарбониламиномасляной кислоты (0,10 г, 0,34 ммоль) (полученный, как описано в Примере 120, стадии A-D) и пиперидин (1 мл) нагревали до 50°С в течение 16 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток подвергали азеотропной перегонке с толуолом (3×10 мл), а затем хроматографии, элюируя смесью МеОН/дихлорметан (1:9) и получая 93 мг бесцветного масла (выход 97%).
Стадия В: Дигидрохлорид метилового эфира (S)-2-амино-4-пиперидин-1-илмасляной кислоты. К метиловому эфиру (S)-2-трет-бутоксикарбониламино-4-пиперидин-1-илмасляной кислоты добавляли HCl (0,45 мл 4 М раствора в диоксане) и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении и сушили под высоким вакуумом в течение 16 часов, получая продукт с выходом 46%.
Стадия С: Метиловый эфир (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-пиперидин-1-ил-масляной кислоты. К раствору 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (0,050 г, 0,14 ммоль), дигидрохлорида метилового эфира (S)-2-амино-4-пиперидин-1-илмасляной кислоты (0,043 г, 0,16 ммоль), EDCI (0,033 г, 0,17 ммоль) и HOBt (0,023 г, 0,17 ммоль) в дихлорметане по каплям добавляли ДИЭА (0,093 г, 0,72 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока ВЭЖХ-анализ не показал, что исходное вещество израсходовалось, и затем разбавляли дихлорметаном и промывали насыщенным NaHCO3. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии, получая 0,051 г продукта (выход 67%).
Стадия D: (1-Гидроксиметил-3-пиперидин-1-илпропил)амид (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (34d). Борогидрид натрия (0,012 г, 0,31 ммоль) порциями добавляли к нагретому (50°С) раствору метилового эфира (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-пиперидин-1-илмасляной кислоты (0,022 г, 0,042 ммоль) в МеОН. Реакционную смесь перемешивали при 50°С до тех пор, пока ВЭЖХ-анализ не показал, что исходное вещество израсходовалось. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли этилацетатом и 1 н. HCl. Органические вещества экстрагировали 1 н. HCl до тех пор, пока органический слой содержал продукт, который определяли ВЭЖХ-анализом. Водный раствор подщелачивали NaOH до рН 14 и затем несколько раз экстрагировали дихлорметаном. Объединенные органические вещества сушили над MgSO4 и концентрировали при пониженном давлении, получая 9,1 мг масла (выход 44%). МС (ХИАД+), обнаружено: m/z 501 (М+1).
Пример 125
Получение (3-диметиламино-1-диметилкарбамоилпропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (35d)
К раствору (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-диметиламиномасляной кислоты (33d; Пример 123) (0,100 г, 0,21 ммоль), диметиламина (0,095 г, 2,11 ммоль), EDCI (0,053 г, 0,27 ммоль), HOBt (0,037 г, 0,27 ммоль), растворенному в дихлорметане, по каплям добавляли ДИЭА (0,082 г, 0,63 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока исходное вещество не было полностью израсходовано, что определяли ВЭЖХ-анализом. Затем реакционную смесь разбавляли дихлорметаном и промывали насыщенным NaHCO3, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии на флэш-колонке Isolute SPE Si (5 г), элюируя градиентом ТЭА/дихлорметан (100 мл, 0,3:99,7), ТЭА/МеОН/дихлорметан (100 мл, 0,3:0,5:99,2), ТЭА/МеОН/дихлорметан (100 мл, 0,3:2,5:97,2), ТЭА/МеОН/дихлорметан (100 мл, 0,3:5:94,7). Конечный продукт получали с выходом 86%. МС (ИЭР+), обнаружено: m/z 502 (М+1).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.63 (d, 1Н), 8.01 (s, 1Н), 7.96 (s, 1Н), 7.48 (m, 1Н), 7.23 (m, 1Н), 7.20 (s, 1Н), 7.09 (m, 1Н), 4.98 (m, 1Н), 4.26 (d, 2H), 3.07 (s, 3H), 2.84 (s, 3H), 2.23 (m, 2Н), 2.13 (m, 1Н), 2.03 (s, 6Н), 1.80 (m, 1Н), 1.65 (m, 1Н), 0.86 (d, 6H).
Пример 126
Получение (3-диметиламино-1-метилкарбамоилпропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (36d)
Получали в соответствии с методикой, описанной в Примере 125, заменяя диметиламин метиламином. Продукт получали с выходом 78%. МС (ИЭР+), обнаружено: m/z 488 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 9.05 (d, 1H), 8.29 (s, 1Н), 7.86 (s, 1Н), 7.44 (m, 1Н), 7.19 (m, 1Н), 7.02 (m, 1Н), 7.01 (s, 1H), 6.93 (m, 1Н), 4.78 (m, 1Н), 4.21 (d, 2Н), 2.81 (d, 3Н), 2.50 (m, 1Н), 2.42 (m, 1Н), 2.36 (m, 1Н), 2.23 (s, 6Н), 2.15 (m, 1Н), 1.88 (m, 1Н), 0.92 (d, 6H).
Пример 127
Получение (1-карбамоил-3-диметиламинопропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (37d)
Получали в соответствии с методикой, описанной в Примере 125, заменяя диметиламин аммиаком. Продукт получали с выходом 70%. МС (ХИАД+), обнаружено: m/z 474 (М+1).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.59 (d, 1Н), 8.07 (s, 1Н), 8.01 (s, 1Н), 7.50 (m, 1Н), 7.39 (s, 1Н), 7.27 (m, 1Н), 7.19 (s, 1H), 7.11 (m, 2Н), 4.44 (m, 1Н), 4.27 (d, 2Н), 2.24 (m, 2Н), 2.15 (m, 1Н), 2.00 (s, 6H), 1.88 (m, 1Н), 1.71 (m, 1Н), 0.86 (d, 6H).
Пример 128
Получение [1-(2-диметиламиноэтил)-2-гидрокси-2-метилпропил]амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (38d)
К охлажденному (0°С) раствору метилового эфира (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-диметиламиномасляной кислоты (Пример 123, стадии А-В) (0,112 г, 0,229 ммоль) в ТГФ (2 мл) по каплям добавляли метилмагнийбромид (2,00 мл 1,4 М раствора). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешиваться в течение 16 часов в атмосфере N2. Смесь распределяли между этилацетатом и насыщенным NH4Cl. Слои разделяли и водный слой дважды экстрагировали этилацетатом. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток подвергали хроматографии на флэш-колонке Isolute SPE Si (5 г), элюируя градиентом: ТЭА/СН2Cl2 (100 мл, 0,3:99,7), ТЭА/МеОН/СН2Cl2 (100 мл, 0,3:0,5:99,2), ТЭА/МеОН/СН2Cl2 (100 мл, 0,3:2,5:97,2), ТЭА/МеОН/СН2Cl2 (100 мл, 0,3:5:94,7). Конечный продукт получали с выходом 41%. МС (ХИАД+), обнаружено: m/z 489 (М+1).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.00 (s, 1Н), 7.97 (d, 1Н), 7.89 (s, 1Н), 7.48 (m, 1Н), 7.23 (m, 1Н), 7.18 (s, 1Н), 7.10 (m, 1Н), 4.58 (m, 1Н), 4.27 (d, 2Н), 3.88 (m, 1Н), 2.23 (m, 2Н), 2.07 (s, 6Н), 1.88 (m, 1Н), 1.43 (m, 1Н), 1.13 (s, 3H), 1.04 (s, 3H), 0.86 (dd, 6H).
Пример 129
Получение {1-гидроксиметил-3-[(2-метоксиэтил)метиламино]-пропил}амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (39d)
Стадия А: Метиловый эфир (S)-2-трет-бутоксикарбониламино-4-йодмасляной кислоты: Смесь метилового эфира (S)-4-бром-2-трет-бутоксикарбониламиномасляной кислоты (1,19 г, 4,0 ммоль) (Пример 120, стадии A-D) и NaI (6,0 г, 40,0 ммоль) в ацетоне (25 мл) нагревали до 70°С в течение 2 часов. Затем смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении и распределяли между водой (10 мл) и эфиром (40 мл). Слои разделяли, органический слой промывали водой (10 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 1,26 г продукта (выход 92%).
Стадия В: Метиловый эфир (S)-2-трет-бутоксикарбониламино-4-[(2-метоксиэтил)метиламино]масляной кислоты. Смесь метилового эфира (S)-2-трет-бутоксикарбониламино-4-йодмасляной кислоты (0,200 г, 0,58 ммоль), (2-метоксиэтил)метиламина (0,062 г, 0,70 ммоль) и триэтиламина (0,41 мл, 2,9 ммоль) в диоксане (1 мл) перемешивали при 70°С в течение 16 часов. Смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении и растворяли в дихлорметане (20 мл). Раствор промывали водой (3×10 мл), рассолом (10 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении. Желто-коричневое масло подвергали хроматографии, элюируя эфиром и получая 0,87 г бледно-желтого масла (выход 49%).
Стадия С: Дигидрохлорид метилового эфира (S)-2-амино-4-[(2-метоксиэтил)метиламино]масляной кислоты. Метиловый эфир (S)-2-трет-бутоксикарбониламино-4-[(2-метоксиэтил)метиламино]масляной кислоты (0,086 г, 0,28 ммоль) обрабатывали HCl (1 мл 4 М раствора в диоксане) и обрабатывали ультразвуком. Смесь концентрировали и сушили под высоким вакуумом, получая продукт.
Стадия D: Метиловый эфир (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-[(2-метоксиэтил)метиламино]масляной кислоты. К раствору 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (полученной, как описано в Примере 110, стадии A-G) (0,094 г, 0,27 ммоль), дигидрохлорида метилового эфира (S)-2-амино-4-[(2-метоксиэтил)метиламино]масляной кислоты (0,079 г, 0,28 ммоль), EDCI (0,057 г, 0,30 ммоль) и HOBt (0,045 г, 0,30 ммоль) в дихлорэтане (2 мл) по каплям добавляли триэтиламин (0,23 мл, 1,62 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов. Смесь концентрировали при пониженном давлении и подвергали хроматографии, элюируя этилацетатом и получая 0,100 г продукта (выход 69%).
Стадия Е: {1-Гидроксиметил-3-[(2-метоксиэтил)метиламино]пропил}амид (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (39d). Метиловый эфир (S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]амино}-4-[(2-метоксиэтил)метиламино]масляной кислоты (0,023 г, 0,043 ммоль) и NaBH4 (0,016 г, 0,43 ммоль) растворяли в ТГФ/МеОН (7 мл, 5:2) и нагревали до 70°С в запаянном сосуде в течение 5 часов. Смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении и подвергали хроматографии, элюируя смесью ТЭА/этилацетат (1:4) и получая 0,007 г продукта в виде вязкого масла (выход 31%). МС (ХИАД+), обнаружено: m/z 505 (М+1).
Пример 130
Получение [3-диметиламино-1-(2-гидроксиэтилкарбамоил)пропил]амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты (40d)
Получали в соответствии с методикой, описанной в Примере 125, заменяя диметиламин 2-аминоэтанолом. Продукт получали с выходом 51%. МС (ХИАД+), обнаружено: m/z 518 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 9.00 (d, 1H), 8.29 (s, 1H), 7.86 (s, 1H), 7.81 (t, 1H), 7.18 (m, 1H), 7.02 (m, 1H), 7.00 (s, 1H), 6.92 (m, 1H), 4.82 (m, 1H), 4.21 (d, 2H), 3.42 (m, 2H), 2.86 (m, 1H), 2.49 (m, 2H), 2.35 (m, 1H), 2.23 (s, 6H), 2.17 (m, 2H), 1.94 (m, 1H), 0.92 (d, 6H).
Пример 131
Получение N′-[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-ил]-N,N-диметилпропан-1,3-диамина (41d)
Смесь 6-бром-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазола (15d; Пример 110, стадии А-Е) (0,030 г, 0,079 ммоль), 3-(диметиламино)пропиламина (0,012 г, 0,118 ммоль), BINAP (0,009 г, 0,016 ммоль), Pd2dba3 (0,007 г, 0,008 ммоль) и NaO-трет-Bu (0,008 г, 0,087 ммоль) в диоксане перемешивали в колбе и трижды продували азотом. Затем реакционную смесь нагревали до 100°С в течение 16 часов. Смесь охлаждали до комнатной температуры и распределяли между этилацетатом (20 мл) и рассолом (20 мл). Водную фазу экстрагировали этилацетатом, объединенные органические слои сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии на диоксиде кремния, элюируя смесью ацетон/эфир (1:1,5) с 0,2% триэтиламина. Продукт снова подвергали хроматографии, элюируя 5% МеОН в дихлорметане и получая 0,022 г продукта в виде бесцветного масла (выход 69%). МС (ИЭР+), обнаружено: m/z 403 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.70 (s, 1Н), 6.95 (m, 3Н), 6.80 (m, 1Н), 6.40 (s, 1H), 5.48 (br. s, 1H), 4.06 (d, 2H), 3.28 (m, 2Н), 2.43 (m, 2Н), 2.35 (m, 1Н), 2.20 (s, 6Н), 1.86 (m, 2Н), 0.94 (d, 6H).
Пример 132
Получение [5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-ил]пиперидин-4-иламина (42d)
Стадия А: трет-Бутиловый эфир 4-[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-иламино]пиперидин-1-карбоновой кислоты.
Получали, как описано в Примере 131, заменяя 3-(диметиламино)пропиламин трет-бутиловым эфиром 4-аминопиперидин-1-карбоновой кислоты. Неочищенный продукт подвергали хроматографии на диоксиде кремния, элюируя смесью эфир/гексаны (1:2) и получая 0,035 г продукта в виде желтого масла (выход 78%).
Стадия В: [5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-ил]пиперидин-4-иламин (42d). К раствору трет-бутилового эфира 4-[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-иламино]пиперидин-1-карбоновой кислоты (0,035 г, 0,070 ммоль) в МеОН (2 мл) добавляли HCl (2 мл 4 М раствора в диоксане). Смесь перемешивали при комнатной температуре в течение 40 минут и затем концентрировали при пониженном давлении. Остаток подвергали азеотропной перегонке с МеОН (2×), получая 0,032 г желтого твердого вещества в виде хлористоводородной соли (выход 97%).1Н-ЯМР (400 МГц, CD3OD) δ 8.06 (s, 1H), 7.26 (m, 1H), 7.20 (m, 1H), 7.04 (m, 1H), 6.93 (s, 1H), 6.89 (s, 1Н), 4.27 (d, 2Н), 3.95 (m, 1Н), 3.52 (m, 2Н), 3.28 (m, 2Н), 2.34 (m, 3Н), 1.83 (m, 2Н), 0.95 (d, 6H).
Пример 133
Получение [5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-ил]пиперидин-3-илметиламина (43d)
Стадия А: трет-Бутиловый эфир 3-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-иламино]метил}пиперидин-1-карбоновой кислоты. Получали в соответствии с методикой, описанной в Примере 131, заменяя 3-(диметиламино)пропиламин трет-бутиловым эфиром 3-аминометилпиперидин-1-карбоновой кислоты. Неочищенный продукт подвергали хроматографии на диоксиде кремния, элюируя смесью эфир/гексаны (1:2) и получая 0,051 г продукта в виде желтой пены (выход 94%).
Стадия В: [5-(2,4-Дифторфенокси)-1-изобутил-1Н-индазол-6-ил]пиперидин-3-илметиламин (43d). К охлажденному (0°С) раствору трет-бутилового эфира 3-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-иламино]метил}пиперидин-1-карбоновой кислоты (0,051 г, 0,099 ммоль) в МеОН (3 мл) добавляли концентрированную HCl (0,18 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Добавляли дополнительно концентрированную HCl (0,4 мл) и смесь перемешивали при комнатной температуре в течение более 24 часов. Смесь концентрировали при пониженном давлении и подвергали азеотропной перегонке с МеОН (3×), получая 0,037 г желтоватого твердого вещества (выход 77%). МС (ИЭР+), обнаружено: m/z 415 (М+1).
Пример 134
Получение 2-(5-{2-[3-(5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)уреидометил]-4-фторфенокси}индазол-1-ил)-N,N-диметилацетамида (47d)
Реакционная схема синтеза соединения 47d по этому изобретению показана на Фиг.54.
Стадия А: 2-[5-(2-Циано-4-фторфенокси)индазол-1-ил]-N,N-диметилацетамид (45d). К раствору 5-фтор-2-(1Н-индазол-5-илокси)бензонитрила (44d) (0,200 г, 0,790 ммоль) в ДМФА (6 мл) добавляли 2-хлор-N,N-диметилацетамид (0,115 г, 0,948 ммоль) и тетрабутиламмония йодид (0,088 г, 0,237 ммоль), а затем К2СО3 (0,164 г, 1,19 ммоль). Смесь нагревали до 110°С в течение 48 часов в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении и растворяли в дихлорметане. Раствор промывали 1 н. HCl, фильтровали и подвергали хроматографии на Biotage, элюируя смесью 5% МеОН в дихлорметане и получая 0,032 г продукта (выход 12%).
Стадия В: 2-[5-(2-Аминометил-4-фторфенокси)индазол-1-ил]-N,N-диметилацетамид (46d). К раствору 2-[5-(2-циано-4-фторфенокси)индазол-1-ил]-N,N-диметилацетамида (0,090 г, 0,266 ммоль) в EtOH (0,5 мл) добавляли CoBr2 (27 мкл, 0,005 ммоль), а затем [2,2′]бипиридинил (81 мкл, 0,015 ммоль). К смеси добавляли NaBH4 (0,030 г, 0,798 ммоль) и перемешивали при комнатной температуре в течение 18 часов. Смесь обрабатывали другой порцией CoBr2, [2,2′]бипиридинила и NaBH4 и перемешивали в течение еще 18 часов. Смесь гасили МеОН, а затем уксусной кислотой и затем концентрировали при пониженном давлении. Белый остаток распределяли между насыщенным NaHCO3 и этилацетатом. Органический слой фильтровали и концентрировали при пониженном давлении, получая 10 мг белого твердого вещества (выход 11%).
Стадия С: 5-трет-Бутил-2-п-толил-2Н-пиразол-3-иламин. Раствор гидрохлорида п-толилгидразина (15,86 г, 100 ммоль) и пивалоилацетонитрила (17,9 г, 143 ммоль) в МеОН (65 мл) нагревали до температуры дефлегмации в течение 18 часов в атмосфере N2. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток растирали с эфиром и собирали фильтрацией. Твердое вещество сушили под высоким вакуумом, получая 26,6 г белого твердого вещества (выход 99%).
Стадия D: 2,2,2-Трихлорэтиловый эфир (5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)карбаминовой кислоты. Охлажденный (0°С) двухфазный раствор 5-трет-бутил-2-п-толил-2Н-пиразол-3-иламина (26,6 г, 100 ммоль) в воде (80 мл) и этилацетате (180 мл) обрабатывали NaOH (10 г, 250 ммоль), а затем трихлорэтилхлорформиатом (29,7 г, 140 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Слои разделяли и органический слой промывали рассолом (100 мл), сушили над MgSO4, фильтровали через целит и концентрировали при пониженном давлении, получая 40,3 г бледно-желтого твердого вещества (выход 99%).
Стадия Е: 2-(5-{2-[3-(5-трет-Бутил-2-п-толил-2Н-пиразол-3-ил)уреидометил]-4-фторфенокси}индазол-1-ил)-N,N-диметилацетамид (47d). К раствору 2-[5-(2-аминометил-4-фторфенокси)индазол-1-ил]-N,N-диметилацетамида (46d) (0,010 г, 0,029 ммоль) и 2,2,2-трихлорэтилового эфира (5-трет-бутил-2-п-толил-2Н-пиразол-3-ил)карбаминовой кислоты (0,013 г, 0,032 ммоль) в ДМФА (1 мл) добавляли ДИЭА (0,01 мл, 0,058 ммоль). Смесь нагревали до 80°С в течение 18 часов в атмосфере N2. Смесь концентрировали при пониженном давлении и растворяли в дихлорметане. Раствор промывали 1 н. HCl, фильтровали и концентрировали при пониженном давлении. Масло подвергали хроматографии, элюируя смесью дихлорметан/эфир (10:1) и затем 5% МеОН в дихлорметане, получая 6,3 мг бледно-желтого масла (выход 36%). МС (ХИАД+), обнаружено: m/z 598 (М+1).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.29 (s, 1Н), 7.97 (s, 1H), 7.58 (d, 1H), 7.36 (d, 2H), 7.28 (d, 2H), 7.19 (d, 1H), 7.12 (d, 1H), 7.07 (m, 2Н), 6.99 (m, 1Н), 6.84 (m, 1Н), 6.24 (s, 1H), 5.40 (s, 2Н), 4.28 (d, 2H), 3.10 (s, 3H), 2.84 (s, 3H), 2.35 (s, 3H), 1.25 (s, 9H).
Пример 135
Получение 1-(5-трет-бутилизоксазол-3-ил)-3-[5-фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензил]мочевины (48d)
Стадия А: 1-Изобутил-5-метокси-1Н-индазол. Раствор 5-метокси-1Н-индазола (5,00 г, 33,7 ммоль) в ДМФА (100 мл) обрабатывали К2СО3 (5,83 г, 42,2 ммоль) и перемешивали при комнатной температуре в течение 15 минут. К этому раствору добавляли 1-бром-2-метилпропан (5,09 г, 37,1 ммоль) и полученную смесь нагревали до 110°С в течение 18 часов. Добавляли еще один эквивалент 1-бром-2-метилпропана и продолжали нагревать смесь в течение еще 48 часов. Смесь концентрировали при пониженном давлении и растворяли в дихлорметане. Раствор промывали 1 н. HCl, фильтровали и концентрировали при пониженном давлении. Остаток подвергали хроматографии на Biotage, элюируя смесью гексаны/эфир (5:1) и получая 2,51 г оранжевого масла (выход 36%).
Стадия В: 1-Изобутил-1Н-индазол-5-ол. К охлажденному (-78°С) раствору 1-изобутил-5-метокси-1Н-индазола (2,57 г, 12,6 ммоль) в дихлорметане (100 мл) добавляли BBr3 (25 мл 1 М раствора в дихлорметане). Смесь перемешивали при -78°С в течение 2 часов и затем нагревали до комнатной температуры и перемешивали в течение 18 часов. Реакционную смесь вливали в ледяную воду и экстрагировали дихлорметаном. Органический экстракт фильтровали и концентрировали при пониженном давлении, получая 2,3 г твердого вещества (выход 96%).
Стадия С: 5-Фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензонитрил. К раствору 1-изобутил-1Н-индазол-5-ола (2,33 г, 12,2 ммоль) и К2СО3 (2,03 г, 14,7 ммоль) в ДМФА (75 мл) добавляли 2,5-дифторбензонитрил (1,87 г, 13,5 ммоль). Смесь нагревали до 110°С в течение 18 часов в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в дихлорметане. Раствор промывали 1 н. HCl, фильтровали и концентрировали при пониженном давлении. Остаток подвергали хроматографии на Biotage, элюируя смесью гексаны/эфир (5:2) и получая 3,05 г бледно-желтого масла (выход 81%).
Стадия D: 5-Фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензиламин. К раствору 5-фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензонитрила (3,05 г, 9,86 ммоль) в МеОН (50 мл), подвергнутому азотной продувке, добавляли концентрированную HCI (1,6 мл) и Pd(OH)2/C (15 мас.%, 0,457 г). Смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере Н2. Катализатор удаляли фильтрацией и раствор концентрировали при пониженном давлении, получая 3,32 г бледно-желтой пены (выход 96%).
Стадия Е: 1-(5-трет-Бутилизоксазол-3-ил)-3-[5-фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензил]мочевина. К охлажденному (0°С) раствору 5-фтор-2-(1-изобутил-1Н-индазол-5-илокси)бензиламина (0,364 г, 1,04 ммоль) и ДИЭА (0,5 мл, 2,08 ммоль) в дихлорметане (10 мл) добавляли трифосген (0,131 г, 0,374 ммоль). Смесь перемешивали при 0°С в течение 1 часа и затем перемешивали при комнатной температуре в течение 18 часов в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении и суспендировали в дихлорметане (10 мл) с получением 0,123 М раствора. 0,4 мл этого раствора (0,015 г, 0,048 ммоль) обрабатывали 5-трет-бутилизоксазол-3-иламином (0,008 г, 0,053 ммоль). Продукт получали с выходом 44%. МС (ХИАД+), обнаружено: m/z 480 (М+1).
Пример 136
Получение 1-(3-трет-бутилизоксазол-5-ил)-3-{5-фтор-2-[1-(2-пиперазин-1-илэтил)-1Н-индазол-5-илокси]бензил)мочевины (49d)
Стадия А: 2-[1-(2,2-Диметоксиэтил)-1Н-индазол-5-илокси]-5-фторбензонитрил. К охлажденному (0°С) раствору 5-фтор-2-(1Н-индазол-5-илокси)бензонитрила (0,100 г, 0,395 ммоль) и 2-бром-1,1-диметоксиэтана (0,114 г, 0,671 ммоль) в ДМФА (4 мл) добавляли NaH (0,024 г, 60%, 0,59 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. К смеси добавляли тетрабутиламмония йодид (0,029 г, 0,079 ммоль) и нагревали ее до 60°С в течение 3 часов. Смесь охлаждали до комнатной температуры, разбавляли водой (4 мл) и экстрагировали эфиром (3×30 мл). Объединенные экстракты промывали водой (2×5 мл) и рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя смесью этилацетат/гексаны (1:2) и получая 0,066 г продукта (выход 49%).
Стадия В: 5-Фтор-2-[1-(2-оксоэтил)-1Н-индазол-5-илокси]бензонитрил. К раствору 2-[1-(2,2-диметоксиэтил)-1Н-индазол-5-илокси]-5-фторбензонитрила (1,42 г, 4,16 ммоль) в дихлорметане (62 мл) порциями в течение 3 часов добавляли йодтриметилсилан (3,33 г, 16,64 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. К смеси добавляли водный NaHCO3 (60 мл) и экстрагировали ее этилацетатом (2×50 мл). Объединенные экстракты промывали Na2S2O4, рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей реакционной стадии без дополнительной очистки.
Стадия С: трет-Бутиловый эфир 4-{2-[5-(2-циано-4-фторфенокси)индазол-1-ил]этил}пиперазин-1-карбоновой кислоты. К раствору 5-фтор-2-[1-(2-оксоэтил)-1Н-индазол-5-илокси]бензонитрила (0,307 г, 1,04 ммоль) и триацетоксиборогидрида (0,66 г, 3,1 ммоль) в дихлорэтане (10 мл) добавляли трет-бутиловый эфир пиперазин-1-карбоновой кислоты (0,65 г, 3,49 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили МеОН (2 мл) и разбавляли этилацетатом (100 мл). Раствор промывали водным NaHCO3, рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя смесью этилацетат/гексаны (2:3, с 1% триэтиламина) и получая 0,29 г продукта (выход 60%).
Стадия D: трет-Бутиловый эфир 4-{2-[5-(2-аминометил-4-фторфенокси)индазол-1-ил]этил}пиперазин-1-карбоновой кислоты. К раствору трет-бутилового эфира 4-{2-[5-(2-циано-4-фторфенокси)индазол-1-ил]этил}пиперазин-1-карбоновой кислоты (0,160 г, 0,344 ммоль), CoBr2 (0,008 г, 0,034 ммоль), [2,2′]бипиридинила (0,016 г, 0,010 ммоль) в EtOH (6 мл) добавляли NaBH4 (0,039 г, 1,0 ммоль). Смесь перемешивали в течение 3 часов при комнатной температуре. Реакцию гасили МеОН (3 мл) и уксусной кислотой (10 капель). Раствор концентрировали при пониженном давлении и разбавляли этилацетатом. Смесь промывали водным NaHCO3, рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенное масло подвергали хроматографии, элюируя смесью 1% триэтиламина в этилацетате, смесью МеОН/СН2Cl2/гексаны (1:15:15, 1% триэтиламина), затем смесью МеОН/СН2Cl2/гексаны (1:10:10, 1% триэтиламина). Чистый продукт получали с выходом 86%.
Стадия Е: 4-Нитрофениловый эфир (3-трет-бутилизоксазол-5-ил)карбаминовой кислоты. Раствор 3-трет-бутилизоксазол-5-иламина (2,50 г, 17,83 ммоль) в дихлорметане обрабатывали пиридином (2 мл, 26,7 ммоль), а затем п-нитрофенилхлорформиатом (3,77 г, 18,73 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь промывали 1 н. HCl, фильтровали и концентрировали при пониженном давлении. Остаток растирали с эфиром и собирали фильтрацией, получая 2,2 г продукта (выход 41%).
Стадия F: трет-Бутиловый эфир 4-[2-(5-{2-[3-(3-трет-бутилизоксазол-5-ил)уреидометил]-4-фторфенокси}индазол-1-ил)этил]пиперазин-1-карбоновой кислоты. Раствор трет-бутилового эфира 4-{2-[5-(2-аминометил-4-фторфенокси)индазол-1-ил]этил}пиперазин-1-карбоновой кислоты (0,050 г, 0,107 ммоль) в дихлорметане (2 мл) обрабатывали 4-нитофениловым эфиром (3-трет-бутилизоксазол-5-ил)карбаминовой кислоты (0,081 г, 0,266 ммоль) и перемешивали при комнатной температуре в течение 24 часов. Смесь разбавляли этилацетатом (60 мл), промывали 1 н. NaOH (5 мл), водой (2×10 мл), рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя смесью ацетон/гексаны (2:3, затем 1:1) и получая 0,056 г продукта (выход 83%).
Стадия G: 1-(3-трет-Бутилизоксазол-5-ил)-3-{5-фтор-2-[1-(2-пиперазин-1-илэтил)-1Н-индазол-5-илокси]бензил}мочевина. трет-Бутиловый эфир 4-[2-(5-{2-[3-(3-трет-бутилизоксазол-5-ил)уреидометил]-4-фторфенокси}индазол-1-ил)этил]пиперазин-1-карбоновой кислоты (0,056 г, 0,088 ммоль) обрабатывали смесью ТФУ/СН2Cl2 (1:1, 2 мл) и перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении и подвергали азеотропной перегонке с толуолом. Остаток разбавляли этилацетатом (40 мл) и промывали 1 н. NaOH и рассолом. Раствор концентрировали при пониженном давлении, получая 0,038 г продукта (выход 80%). МС (ИЭР+), обнаружено: m/z 536 (М+1).1Н-ЯМР (400 МГц, CDCl3) δ 7.81 (s, 1H), 7.35 (d, 1Н), 7.15 (dd, 1H), 7.08 (s, 1H), 7.05 (d, 1H), 6.88 (m, 1H), 6.76 (m, 1Н), 6.28 (m, 1Н), 5.99 (s, 1Н), 4.46 (m, 4Н), 2.86 (m, 2Н), 2.79 (m, 4Н), 2.43 (m, 4Н), 1.26 (s, 9H).
Пример 137
Получение 1-(3-трет-бутилизоксазол-5-ил)-3-{2-[1-(2-диметиламиноэтил)-1Н-индазол-5-илокси]-5-фторбензил}мочевины (50d)
Стадия А: 2-[1-(2-Диметиламиноэтил)-1Н-индазол-5-илокси]-5-фторбензонитрил. Раствор 5-фтор-2-[1-(2-оксоэтил)-1Н-индазол-5-илокси]бензонитрила (0,110 г, 0,372 ммоль) (полученного в соответствии с методикой, описанной в Примере 136, стадии А-В) и триацетоксиборогидрида натрия (0,39 г, 1,9 ммоль) в дихлорэтане (3 мл) обрабатывали диметиламином (0,17 г, 3,7 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Реакцию гасили МеОН (1 мл) и разбавляли этилацетатом (30 мл). Раствор промывали водным NaHCO3, рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя этилацетатом, а затем смесью ацетон/гексаны (2:3 с 1% триэтиламина) и получая 0,115 г продукта (выход 95%).
Стадия В: {2-[5-(2-Аминометил-4-фторфенокси)индазол-1-ил]этил}диметиламин. К охлажденному (0°С) раствору 2-[1-(2-диметиламиноэтил)-1Н-индазол-5-илокси]-5-фторбензонитрила (0,115 г, 0,303 ммоль) в ТГФ (3 мл) добавляли LAH (0,61 мл 1 М раствора в ТГФ). Смесь нагревали до комнатной температуры и перемешивали в течение 1,5 часов. Смесь гасили водой (23 мкл), 3 н. NaOH (23 мкл) и водой (69 мкл). Соли удаляли фильтрацией, и фильтрат концентрировали при пониженном давлении, получая 0,113 г продукта (выход 97%).
Стадия С: 1-(3-трет-Бутилизоксазол-5-ил)-3-{2-[1-(2-диметиламиноэтил)-1Н-индазол-5-илокси]-5-фторбензил}мочевина. Раствор {2-[5-(2-аминометил-4-фторфенокси)индазол-1-ил]этил}диметиламина (0,020 г, 0,061 ммоль) в ДМФА (1 мл) обрабатывали 4-нитрофениловым эфиром (3-трет-бутилизоксазол-5-ил)карбаминовой кислоты (0,020 г, 0,067 ммоль) (полученным согласно Примеру 136, стадия Е). Смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере N2. Реакционную смесь подвергали хроматографии, элюируя смесью дихлорметан/эфир (10:1), а затем 5% МеОН в дихлорметане и получая 5,3 мг бледно-желтого масла (выход 18%). МС (ХИАД+), обнаружено: m/z 495 (М+1).1Н-ЯМР (400 МГц, ДМСО-d6) δ 10.14 (s, 1H), 7.98 (s, 1Н), 7.74 (d, 1H), 7.22 (d, 1H), 7.16 (m, 2H), 7.08 (m, 1Н), 6.88 (m, 2Н), 5.93 (s, 1H), 4.48 (t, 2H), 4.35 (d, 2H), 2.70 (t, 2H), 2.17 (s, 6Н), 1.23 (s, 9H).
Пример 138
Получение 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{5-фтор-2-[1-(2-гидрокси-2-метилпропил)-1Н-индазол-5-илокси]бензил)мочевины (51d)
Стадия А: 5-Фтор-2-[1-(2-гидрокси-2-метилпропил)-1Н-индазол-5-илокси]бензонитрил. Раствор 5-фтор-2-(1Н-индазол-5-илокси)бензонитрила (0,100 г, 0,395 ммоль) в ДМФА (4 мл) обрабатывали NaH (0,022 г, 60%, 0,56 ммоль) и перемешивали в течение 5 минут. Добавляли 2,2-диметилоксиран (0,035 г, 0,48 ммоль) и раствор перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь нагревали до 80°С в течение 1,5 часов. Раствор охлаждали до комнатной температуры, разбавляли эфиром (50 мл), промывали водой (3×5 мл), рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя смесью этилацетат/гексаны (1:1) и получая 0,070 г продукта (выход 47%).
Стадия В: 2-{1-[2-(трет-Бутилдиметилсиланилокси)-2-метилпропил]-1Н-индазол-5-илокси}-5-фторбензонитрил. К охлажденному (0°С) раствору 5-фтор-2-[1-(2-гидрокси-2-метил пропил)-1Н-индазол-5-илокси]бензонитрила (0,070 г, 0,215 ммоль) и 2,6-лутидина (0,030 г, 0,284 ммоль) в дихлорметане (2 мл) добавляли TBSOTf (0,063 г, 0,240 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Смесь разбавляли эфиром (50 мл) и промывали 0,2 н. HCl, NaHCO3 и рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии, элюируя смесью эфир/гексаны (1:3) и получая 0,071 г бледно-желтого масла (выход 94%).
Стадия С: 2-{1-[2-(трет-Бутилдиметилсиланилокси)-2-метилпропил]-1Н-индазол-5-илокси}-5-фторбензиламин. К охлажденному (0°С) раствору 2-{1-[2-(трет-бутилдиметилсиланилокси)-2-метилпропил]-1Н-индазол-5-илокси}-5-фторбензонитрила (0,071 г, 0,162 ммоль) в ТГФ (2 мл) добавляли LAH (0,32 мл 1 М раствора в ТГФ). Раствор нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь охлаждали до 0°С и гасили водой (12 мкл), 3 н. NaOH (12 мкл) и водой (36 мкл). Соли удаляли фильтрацией, фильтрат концентрировали при пониженном давлении и использовали на следующей реакционной стадии без дополнительной очистки.
Стадия D: 2,2,2-Трихлорэтиловый эфир (5-трет-бутил-2-метил-2Н-пиразол-3-ил)карбаминовой кислоты. К охлажденному (10°С) раствору 5-трет-бутил-2-метил-2Н-пиразол-3-иламина (3,75 г, 24,5 ммоль) и NaOH (1,5 г в 20 мл воды) в этилацетате (45 мл) добавляли 2,2,2-трихлорэтилхлорформиат (7,52 г, 35,5 ммоль) в течение 5 минут. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Смесь разбавляли этилацетатом и слои разделяли. Органический слой промывали водой, рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток разбавляли смесью этилацетат/дихлорметан и обрабатывали аминометилдиоксидом кремния (10 г) в течение 1 часа. Смесь фильтровали и фильтрат концентрировали при пониженном давлении, получая продукт.
Стадия Е: 1-(2-{1-[2-(трет-Бутилдиметилсиланилокси)-2-метилпропил]-1Н-индазол-5-илокси}-5-фторбензил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)мочевина. Смесь 2-{1-[2-(трет-бутилдиметилсиланилокси)-2-метилпропил]-1Н-индазол-5-илокси}-5-фторбензиламина (0,072 г, 0,162 ммоль), 2,2,2-трихлорэтилового эфира (5-трет-бутил-2-метил-2Н-пиразол-3-ил)карбаминовой кислоты (0,080 г, 0,24 ммоль) и ДИЭА (0,06 мл, 0,324 ммоль) в ДМА (3 мл) нагревали до 80°С в течение 16 часов. Смесь разбавляли эфиром (60 мл) и промывали водой (3×5 мл), рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии на диоксиде кремния, элюируя смесью этилацетат/гексаны (3:1) и получая 0,084 г продукта (выход 83%).
Стадия F: 1-(5-трет-Бутил-2-метил-2Н-пиразол-3-ил)-3-{5-фтор-2-[1-(2-гидрокси-2-метилпропил)-1Н-индазол-5-илокси]бензил}мочевина. К раствору 1-(2-{1-[2-(трет-бутилдиметилсиланилокси)-2-метилпропил]-1Н-индазол-5-илокси}-5-фторбензил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)мочевины (0,084 г, 0,135 ммоль) в дихлорметане (3 мл) добавляли TBAF (0,70 мл 1 М раствора в ТГФ). Смесь перемешивали при комнатной температуре в течение 5 дней. Реакционную смесь вливали в NH4Cl и экстрагировали этилацетатом (3×30 мл). Объединенные экстракты промывали рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографии на диоксиде кремния, элюируя смесью ацетон/гексаны (2:1) и получая 0,060 г продукта (выход 87%). МС (ИЭР+), обнаружено: m/z 509 (М+1).1Н-ЯМР (400 МГц, ДМСО-d6) δ 8.45 (s, 1H), 7.98 (s, 1Н), 7.74 (d, 1Н), 7.17 (m, 3Н), 7.08 (m, 1Н), 6.85 (m, 2Н), 5.95 (s, 1H), 4.66 (s, 1H), 4.33 (d, 2H), 4.30 (s, 2H), 2.50 (s, 3H), 1.18 (s, 9H), 1.12 (s, 6H).
Приведенное выше описание рассматривается только в качестве иллюстрации принципов изобретения. Более того, поскольку многочисленные модификации и изменения будут очевидны специалистам в данной области техники, то не желательно ограничивать изобретение точным толкованием и способом, как описано выше. Соответственно, могут быть осуществлены все подходящие модификации и эквиваленты, которые находятся в рамках настоящего изобретения, как определено нижеследующей формулой изобретения.
Слова "содержат", "содержащий", "включают в себя", "включающий в себя" и "включает в себя" при использовании в этом описании и в формуле изобретения, предназначены для определения присутствия заявленных признаков, целых чисел, компонентов или стадий, но они не исключают присутствия или добавления одного или более других признаков, целых чисел, компонентов, стадий или групп.
Реферат
Настоящее изобретение может быть применено в медицине и относится к ингибиторам МАР-киназы р38 формулы (I) ! ! где W представляет собой N или О, когда Y представляет собой С, и W представляет собой С, когда Y представляет собой N; U представляет собой СН или N; V представляет собой С-Е или N; X представляет собой О, S, SO, SO2, NH, C=O, -C=NOR1 или CHOR1; В представляет собой Н или NH2; R1, Е и А представляют собой Н или различные алкильные, гетероалкильные, ароматические и гетероароматические заместители. Технический результат - получение новых биологически активных соединений. 6 н. и 42 з.п. ф-лы, 54 ил.
Формула
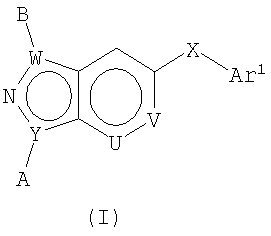
или его фармацевтически приемлемая соль, где
Y представляет собой С, N;
W представляет собой С, N или О, при условии, что W представляет собой N или О, когда Y представляет собой С, и W представляет собой С, когда Y представляет собой N;
U представляет собой СН или N;
V представляет собой С-Е или N;
X представляет собой О, S, SO, SO2, NH, C=O, -C=NOR1 или CHOR1;
В представляет собой Н или NH2, когда W представляет собой С, или В представляет собой Н, когда W представляет собой N, или В отсутствует, когда W представляет собой О;
R1 представляет собой Н, алкил или Zn-Ar1;
Е представляет собой Н, Zn-NR2R3, Zn-(C=O)R4, Zn-(C=O)R5, Zn-OR6, Zn-SO2R4 или R5;
Z представляет собой алкилен, имеющий от 1 до 4 атомов углерода, необязательно замещенный группой, выбранной из ОН или С1-6алкила;
R2 представляет собой Н, алкил, Zn-гетероциклоалкил или Zn-Ar1;
R3 представляет собой Н, алкил, трет-бутоксикарбонил, метилсульфонил или ацетил;
или R2 вместе с R3 и N образуют гетероциклоалкил;
R4 представляет собой природную или неприродную аминокислоту,
NHCH(R8)(CH2)mOR7 или NR2R3;
где n означает 0 или 1;
m означает целое число от 1 до 4;
R5 представляет собой Н, ОН, алкил, С1-6алкокси или Zn-гетероциклоалкил;
R6 представляет собой Н, Zn-гетероциклоалкил или алкил;
R7 представляет собой Н или алкил;
R8 представляет собой Zn-NR2R3;
Ar1 представляет собой арил или гетероарил;
А в случае, когда Y представляет собой С, имеет следующие значения: Н, ОН,
Zn-NR2R3, Zn-NR3(C=O)R3, Zn-SO2R10, Zn-OR10, Zn-(C=O)R9, алкил, алкенил, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1;
А в случае, когда Y представляет собой N, имеет следующие значения: Н, Z-NR2R3, Z-NR3(C=O)R3, Zn-SO2R10, Z-OR10, Zn-(C=O)R9, алкил, алкенил, Zn-циклоалкил, Zn-гетероциклоалкил или Z-Ar1;
R9 представляет собой Н, ОН, C1-6алкоксигруппу или аминогруппу, необязательно замещенную С1-6алкилом или бензилом;
R10 представляет собой Н, алкил или арил;
где алкенил представляет собой С1-6алкенил, который может быть замещен арилом;
где алкил представляет собой С1-6алкил и может быть замещен одной или более чем одной группой, выбранной из галогена, циклоалкила, гетероциклоалкила, -СООН, -COOR (где R представляет собой C1-6алкил), -CONH2, -CONH(С1-6алкил), -CON(С1-6алкил)(С1-6алкил), аминогруппы или гидроксигруппы, где аминогруппа или гидроксигруппа могут быть необязательно замещены одной или более чем одной группой, выбранной из C1-6алкила, бензила или трет-бутоксикарбонила;
где циклоалкил представляет собой циклопропил, циклобутил, циклопентил, циклогексил и циклогептил;
где гетероциклоалкил представляет собой насыщенный 5-8-членный моноцикл, который может быть конденсирован с одним шестичленным ароматическим кольцом и имеет кроме атомов углерода от одного до двух гетероатомов, выбранных из N, О и S, который может быть замещен группами, выбранными из С1-6алкила, бензила, трет-бутоксикарбонила или оксогруппы;
где гетероарил представляет собой ароматический моноцикл из 5-6 членов или бицикл из 9-10 членов, включающий кроме атомов углерода от одного до двух гетероатомов, выбранных из N, О и S, который может быть замещен группами, выбранными из галогена, С1-4алкила, С1-4алкокси, С1-4алкилтио или С1-4алкилсульфонила;
где арил представляет собой 6- или 10-членное ароматическое кольцо, которое может быть замещено группами, выбранными из галогена, С1-4алкила, С1-4алкокси,
С1-4алкилтио или С1-4алкилсульфонила;
при условии, что когда Е представляет собой водород, и соединение формулы (I) представляет собой соединение формулы

тогда А представляет собой ОН, Zn-NR2R3, Zn-NR3(C=O)R3, Zn-SO2R10, Zn-OR10, Zn-(C=O)R9, алкил, аллил, Zn-циклоалкил, Zn-гетероциклоалкил или Z-Ar1;
при условии, что когда Е представляет собой водород, и соединение формулы (I) представляет собой соединение формулы

тогда группа Ar1 представляет собой арил, и А является иным, чем водород;
при условии, что когда Е представляет собой водород, и соединение формулы (I) представляет собой соединение формулы

тогда А является иным, чем водород;
и при условии, что соединение формулы (I) не является 2-(1-метил-1Н-индазол-5-иламино)бензойной кислотой, 5-(2,4-дихлорфенокси)-1Н-индазол-1-уксусной кислотой, 3-метил-6-фенилсульфонилбензизоксазолом или 6-(2-хлор-4-трифторметилфенокси)-3-метилбензизоксазолом.
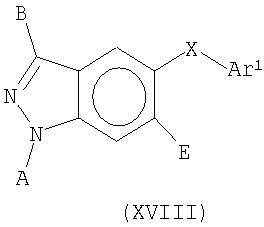
где Е представляет собой Zn-(C=O)R4 или Zn-SO2R4, и Z, n и R4 являются такими, как определено в п.1.

где А, В, X, R2, R3 и Ar1 являются такими, как определено в п.1.
амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
[5-(4-фторфенокси)-1 -изобутил-1H-индазол-6-ил]морфолин-4-ил-метанона;
[5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-ил]-(4-метилпиперазин-1-ил)-метанона;
(1-бензилпиперидин-4-ил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(2-бензиламиноэтил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(2-пиперидин-ил-этил)амида 5-(4-фторфенокси)-1-изобутил-1H-индазол-6-карбоновой кислоты;
(2-пирролидин-1-илэтил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(3-морфолин-4-илпропил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(3-диметиламинопропил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(2-диметиламиноэтил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
метил-(1-метилпиперидин-4-ил)амида 5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
[3-(метилфениламино)-пропил]амида 5-(4-фторфенокси)-1-изобутил-1H-индазол-6-карбоновой кислоты;
трет-бутилового эфира 3-{[5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбонил]-амино}-пирролидин-1-карбоновой кислоты;
(2-диметиламиноэтил)амида 5-(4-фторфенокси)-1-(2,2,2-трифторэтил)-1H-индазол-6-карбоновой кислоты;
(2-диметиламиноэтил)амида 5-(4-фторфенокси)-1-метил-1H-индазол-6-карбоновой кислоты;
(2-диметиламиноэтил)амида 5-(4-фторфенокси)-1H-индазол-6-карбоновой кислоты;
метилового эфира 4-амино-2-{[5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-карбонил]-амино}масляной кислоты;
метилового эфира 4-амино-2-{[5-(4-фторфенокси)-1-(2,2,2-трифторэтил)-1H-индазол-6-карбонил]-амино}масляной кислоты;
метилового эфира 4-амино-2-{[5-(4-фторфенокси)-1-метил-1H-индазол-6-карбонил]-амино} масляной кислоты;
(S)-N-(4-амино-1-гидроксибутан-2-ил)-5-(4-фторфенокси)-1-изобутил-1H-индазол-6-карбоксамида;
(S)-метил-2-(5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоксамидо)-4-(диметиламино)бутаноата;
(S)-5-(2,4-дифторфенокси)-N-(4-(диметиламино)-1-гидроксибутан-2-ил)-1-изобутил-1Н-индазол-6-карбоксамида;
(1-гидроксиметил-3-изопропиламинопропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(S)-2-{[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбонил]-амино}-4-диметиламиномасляной кислоты;
(1-гидроксиметил-3-пиперидин-1-илпропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(3-диметиламино-1-диметилкарбамоилпропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(3-диметиламино-1-метилкарбамоилпропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
(1-карбамоил-3-диметиламинопропил)амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
[1-(2-диметиламиноэтил)-2-гидрокси-2-метилпропил]амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты;
{1-гидроксиметил-3-[(2-метоксиэтил)метиламино]пропил}амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты; и
[3-диметиламино-1-(2-гидроксиэтилкарбамоил)пропил]амида (S)-5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-карбоновой кислоты.
(3-диметиламинопропил)амида 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфоновой кислоты;
(S)-метил-2-(5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфонамидо)-4-(диметиламино)бутаноата;
[2-(1-метилпирролидин-2-ил)-этил]амида 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфоновой кислоты; и
(2-диметиламиноэтил)амида 5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-сульфоновой кислоты.
где Е представляет собой Н, Zn-NR2R3, Zn-(C=O)R5, Zn-OR6 или R5; и Z, n, R2, R3, R5 и R6 являются такими, как определено в п.1.

где А, В, X, R2, R3 и Ar1 являются такими, как определено в п.1.
[5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-ил]метил-(2-диметиламиноэтил)амина;
N-(2-(диметиламино)этил)-N-((5-(4-фторфенокси)-1 -изобутил-1H-индазол-6-ил)метил)метансульфонамида;
N-(2-(диметиламино)этил)-N-((5-(4-фторфенокси)-1-изобутил-1Н-индазол-6-ил)метил)ацетамида;
N′-[5-(2,4-дифторфенокси)-1-изобутил-1H-индазол-6-ил]-N,N-диметилпропан-1,3-диамина;
[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-ил]-пиперидин-4-ил-амина и
[5-(2,4-дифторфенокси)-1-изобутил-1H-индазол-6-ил]-пиперидин-3-илметиламина.
{3-[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-илокси]-пропил}-диметиламина;
5-(2,4-дифторфенокси)-1-изобутил-6-(пиперидин-4-илметокси)-1H-индазола;
5-(2,4-дифторфенокси)-1-изобутил-6-(3-пиперазин-1-ил-пропокси)-1H-индазола;
5-(2,4-дифторфенокси)-1-изобутил-6-(морфолин-2-илметокси)-1Н-индазола и 1-[5-(2,4-дифторфенокси)-1-изобутил-1Н-индазол-6-илокси]-3-пирролидин-1-ил-пропан-2-ола.

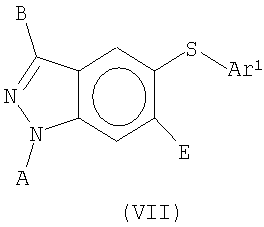



в которой А, В, Е, Ar1 и R1 являются такими, как определено в п.1.
в которой А, В, Е и Ar1 являются такими, как определено в п.1.
в которой А, В, Е и Аг1 являются такими, как определено в п.1.
в которой А, В, Е и Ar1 являются такими, как определено в п.1.
в которой А, В, Е и Ar1 являются такими, как определено в п.1.
в которой А, В, Е, R1 и Ar1 являются такими, как определено в п.1.
в которой А, В, Е и Ar1 являются такими, как определено в п.1.
(2,4-дифторфенил)-(1-изобутил-1H-индазол-5-ил)-амина;
(4-фторфенил)-(1-изобутил-1H-индазол-5-ил)-амина;
(2,4-дихлорфенил)-(1-изобутил-1H-индазол-5-ил)-амина и
(1-изобутил-1H-индазол-5-ил)-O-толил-амина.
в которой А, В, X, Ar1 и Е являются такими, как определено в п.1.
где А, В, X и Ar1 являются такими, как определено в п.1.
где А, В, Е, X и Ar1 являются такими, как определено в п.1.
где А, В, Е, X и Ar1 являются такими, как определено в п.1.
6-(4-фторфенилсульфанил)-3-(4-метоксибензил)-1H)индазола;
[6-(4-фторфенилсульфанил)-1H-индазол-3-ил]метанола;
6-(4-фторфенилсульфанил)-3-метоксиметил-1H-индазола;
6-(4-фторфенокси)-3-метил-1H-индазола;
6-(4-хлорфенокси)-3-метил-1H-индазола;
3-метил-6-(м-толилокси)-1H-индазола;
6-(3-фторфенокси)-3-метил-1H-индазола;
6-(3-хлорфенокси)-3-метил-1H-индазола;
3-метил-6-(3-(метилтио)фенокси)-1H-индазола;
3-метил-6-(3-(метилсульфонил)фенокси)-1H-индазола;
6-(4-фтор-3-метилфенокси)-3-метил-1H-индазола;
3-этил-6-(4-фторфенилсульфанил)-1H-индазола;
N-[6-(4-фторфенокси)-1H-индазол-3-ил]ацетамида;
2-[6-(4-фторфенокси)-1H-индазол-3-ил]изоиндол-1,3-диона и
3-(1,3-дигидроизоиндол-2-ил)-6-(4-фторфенокси)-1H-индазола.
где А, Е, X и Ar1 являются такими, как определено в п.1.
3,6-дифеноксибензо[d]изоксазола и
N-(4-метоксифенил)-6-феноксибензо[d]изоксазол-3-амина.
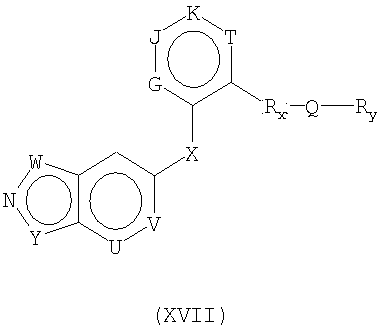
или его фармацевтически приемлемая соль,
где Y представляет собой CR1 или NR2;
W представляет собой CR3 или О, при условии, что W представляет собой О, когда Y представляет собой CR1, и W представляет собой CR3, когда Y представляет собой NR2;
R3 представляет собой Н или NH2;
R1 представляет собой Н, ОН, Zn-NRaRb, Zn-NRb(C=O)Rb, Zn-SO2Rg, Zn-ORg, Zn-(C=O)Re, алкил, алкенил, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1;
R2 представляет собой H, Z-NRaRb, Z-NRb(C=O)Rb, Zn-SO2Rg, Z-ORg, Zn-(C=O)Re, алкил, алкенил, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar1;
Ra представляет собой Н, алкил, Zn-гетероциклоалкил или Zn-Ar1;
Rb представляет собой Н или алкил;
или Ra вместе с Rb и N образуют гетероциклоалкил;
Re представляет собой ОН, Н, С1-6алкоксигруппу или аминогруппу, необязательно замещенную С1-6алкилом или бензилом;
Rg представляет собой Н, С1-6алкил или фенил, где фенил необязательно замещен галогеном или С1-6алкоксигруппой;
Z представляет собой алкилен, имеющий от 1 до 4 атомов углерода, необязательно замещенный группой, выбранной из ОН или С1-6алкила;
n означает 0 или 1;
Ar1 представляет собой арил или гетероарил;
U представляет собой СН или N;
V представляет собой СН или N;
X представляет собой О, S, SO, SO2, NH или С=О;
G, К, J и Т независимо представляют собой N или CRz, при условии, что когда любые из указанных G, К, J и Т представляют собой N, тогда общее количество G, К, J или Т, которые представляют собой N, не превышает 1;
Rz представляет собой Н, F, Cl, Br, CF3, OR6, SR6, низший (С1-С4)алкил CN или
NR6R7;
R6 и R7 независимо представляют собой Н, CF3, низший (С1-С4)алкил, цепь которого необязательно разделена гетероатомами, выбранными из N или О;
Q представляет собой -NR8C(O)NH-, -NHC(O)-, -NR8SO2NH-, -NHSO2-или -CONR8-;
R8 представляет собой Н или низший (С1-С4)алкил;
Rx представляет собой -(СН2)m-;
m означает 1-3;
Ry представляет собой Н, алкил, Zn-циклоалкил, Zn-гетероциклоалкил или Zn-Ar2;
Ar2 представляет собой арил или гетероарил;
где, когда Q представляет собой -CONR8-, тогда Ry в сочетании с R8 дополнительно представляет собой гетероциклоалкил; и
где алкенил представляет собой С1-6алкенил, который может быть замещен арилом;
где алкил представляет собой С1-6алкил и может быть замещен одной или более чем одной группой, выбранной из галогена, циклоалкила, гетероциклоалкила, аминогруппы или гидроксигруппы, где аминогруппа или гидроксигруппа могут быть необязательно замещены одной или более чем одной группой, выбранной из С1-6алкила или бензила;
где циклоалкил представляет собой циклопропил, циклобутил, циклопентил, циклогексил и циклогептил;
где гетероциклоалкил представляет собой насыщенный 5-8-членный моноцикл, который может быть конденсирован с одним шестичленным ароматическим кольцом и имеет кроме атомов углерода от одного до двух гетероатомов, выбранных из N, О и S, который может быть замещен группами, выбранными из С1-6алкила, бензила, трет-бутоксикарбонила или оксогруппы;
где гетероарил представляет собой ароматический моноцикл из 5-6 членов или бицикл из 9-10 членов, включающий кроме атомов углерода от одного до двух гетероатомов, выбранных из N, О и S, который для Ar1 может быть замещен галогеном или С1-4алкоксигруппой, а для Ar2 может иметь 1-3 заместителя, независимо выбранных из F, Cl, Br, CF3, CN, С1-6алкила, Zn-циклоалкила, Zn-гетероциклоалкила или Zn-Ar1;
где арил представляет собой 6- или 10-членное ароматическое кольцо, которое для Ar1 может быть замещено галогеном или С1-4алкоксигруппой, а для Ar2 может иметь 1-3 заместителя, независимо выбранных из F, Cl, Br, CF3, CN, С1-6алкила, Zn-циклоалкила, Zn-гетероциклоалкила или Zn-Ar1.





1-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-3-[2-(1-метил-1Н-индазол-5-илокси)-пиридин-3-илметил]мочевины;
2-(4-{2-[2-(1-циклобутилметил-1Н-индазол-5-илокси)-5-фторфенил]-ацетил}-пиперазин-1-ил)-N-изопропилацетамида;
2-[2-(1-изобутил-1H-индазол-5-илокси)-фенил]-N-(4-морфолин-4-илфенил)-ацетамида;
1-[5-циклопропил-2-(4-трифторметилфенил)-2H-пиразол-3-ил]-3-[5-фтор-2-(1-метил-1H-индазол-5-иламино)бензил]мочевины;
1-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-3-[2-(1-циклобутилметил-1H-индазол-5-иламино)-5-фторбензил]мочевины;
1-(5-трет-бутил-2-n-хлорфенил-2H-пиразол-3-ил)-3-[2-(1-метил-1H-индазо-5-илсульфанил)-5-фторбензил]мочевины;
1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{2-[1-(3-изопропиламино-пропил)-1Н-индазол-5-иламино]-бензил}мочевины;
1-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1H-индазол-5-илокси)-бензил]мочевины;
1-[5-трет-бутил-2-(4-хлор-фенил)-2H-пиразол-3-ил]-3-[5-фтор-2-(1-метил-1H-индазол-5-илокси)-бензил]мочевины;
2-(1-циклобутилметил-1Н-индазол)-5-фторбензиламида циклопропанкарбоновой кислоты;
N-[5-фтор-2-(1-изобутил-1H-индазол-5-илокси)-бензил]-3-трифторметил-бензамида;
N-[2-(1-изобутил-1H-индазол-5-илокси)-бензил]-2-(3-трифторметилфенил)-ацетамида;
5-фтор-2-(1-изобутил-1H-индазол-5-илокси)-бензиламида 5-трет-бутил-1-пиридин-2-ил-1H-пиразол-4-карбоновой кислоты;
2-циклопропил-N-[5-фтор-2-(1-изобутил-1H-индазол-5-илокси)бензил]ацетамида;
3-хлор-N-[5-фтор-2-(1-изобутил-1H-индазол-5-илокси)бензил]бензамида;
N-[5-фтор-2-(1-изобутил-1H-индазол-5-илокси)-бензил]-4-трифторметил-бензолсульфонамида;
N-[5-фтор-2-(1-изобутил-1H-индазол-5-илокси)-бензил]-метансульфонамида;
3-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-1-[5-фтор-2-(1-метил-1H-индазол-5-илокси)-бензил]-1-метилмочевины;
1-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1H-индазол-5-илокси)-бензил]-1-метилмочевины;
1-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(1-метил-1Н-пиразоло[3,4-с]пиридин-5-илокси)-бензил]мочевины;
1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)-бензил]мочевины;
1-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-пирролидин-1-илметилбензо[d]изоксазол-6-илокси)-бензил]мочевины;
1-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-3-[5-фтор-2-(3-метил-бензо[d]изоксазол-6-илокси)-бензил]мочевины;
2-(5-{2-[3-(5-трет-бутил-2-n-толил-2Н-пиразол-3-ил)-уреидометил]-4-фторфенокси}-индазол-1-ил)-N,N-диметилацетамида;
1-(5-трет-бутил-изоксазол-3-ил)-3-[5-фтор-2-(1-изобутил-1Н-индазол-5-илокси)-бензил]мочевины;
1-(3-трет-бутил-изоксазол-5-ил)-3-{5-фтор-2-[1-(2-пиперазин-1-ил-этил)-1Н-индазол-5-илокси]-бензил}мочевины;
1-(3-трет-бутил-изоксазол-5-ил)-3-{2-[1-(2-диметиламиноэтил)-1Н-индазол-5-илокси]-5-фтор-бензил}мочевины; и
1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{5-фтор-2-[1-(2-гидрокси-2-метилпропил)-1Н-индазол-5-илокси]-бензил}мочевины.
Приоритет по пунктам:
Комментарии