Новые производные бензимидазола и их применение вкачестве лекарственных препаратов - RU2294326C2
Код документа: RU2294326C2
Описание
Объектом настоящего изобретения являются новые производные бензимидазола (амино- и тиобензимидазолы). Указанные продукты являются агонистами GnRH (гормон, высвобождающий гонадотропин или гонадолиберин). Данное изобретение также относится к фармацевтическим композициям, содержащим указанные продукты, и к их применению при получении лекарственного препарата.
GnRH (гонадолиберин), также называемый LHRH (люлиберин), представляет собой гипоталамический декапептид (pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2), который регулирует репродуктивную систему позвоночных. Он высвобождается в капилляры гипоталамо-гипофизарной портальной системы медиального возвышения и воронки гипоталамуса. С помощью этой сети он достигает передней доли гипофиза и по вторичной капиллярной сети достигает свои клетки-мишени. GnRH действует на мембранном уровне клеток-мишеней через рецепторы с семью трансмембранными фрагментами, связанными с фосфолипазой C, что через G белки приводит к увеличению выхода внутриклеточного кальция. Его действие вызывает биосинтез и высвобождение гонадотропных гормонов FSH (фолликуло-стимулирующий гормон) и LH (лютеинизирующий гормон). Доказано, что агонисты и антагонисты GnRH эффективны для лечения эндометриоза, фибромы, синдрома поликистозных яичников, рака молочных желез, яичника и эндометрия, гонадотропной гипофизарной десенсибилизации при медикаментозной стимуляции яичников для лечения бесплодия у женщин; при лечении доброкачественной простатической гиперплазии и рака простаты; и при лечении преждевременной половой зрелости.
В настоящее время используемыми антагонистами GnRH являются пептидные соединения, которые, как правило, должны вводится внутривенным или подкожным путем из-за своей слабой пероральной биодоступности. Непептидные антагонисты GnRH, обладающие преимуществом перорального введения, являются целью многих научно-исследовательских работ. Например, непептидные соединения, антагонисты GnRH, были описаны в J. Med. Chem, 41, 4190-4195 (1998) и Bioorg. Med. Chem. Lett, 11, 2597-2602 (2001).
Настоящее изобретение относится к новому классу сильных непептидных антагонистов GnRH.
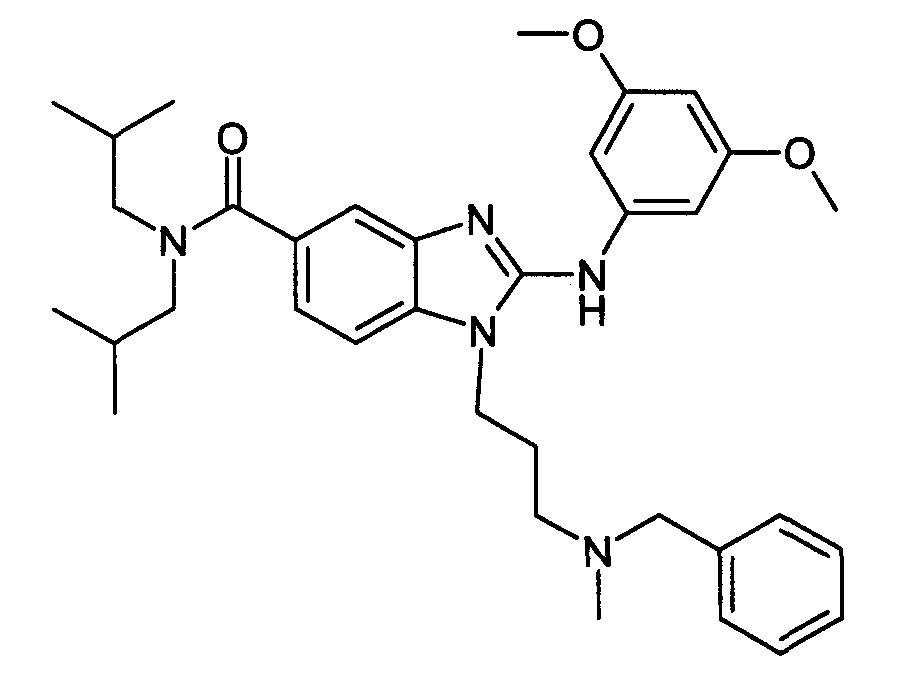
Таким образом, объектом изобретения является соединение общей формулы (I)
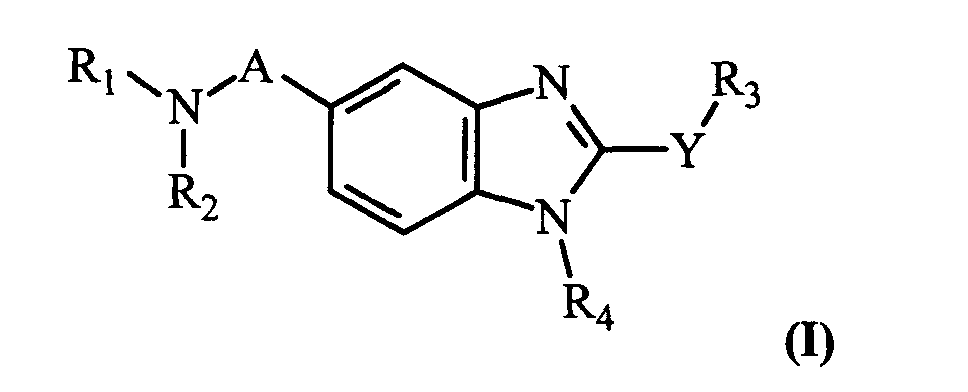
в рацемической, энантиомерной форме или в виде любого сочетания этих форм, где:
A представляет собой -CH2- или -C(O)-;
Y представляет собой -S- или -NH-;
R1 и R2 представляют собой, независимо, атом водорода, (C1-C8)алкил, (C5-C9)бициклоалкил, необязательно замещенный одним или несколькими одинаковыми или различными (C1-C6)алкильными радикалами, или радикал формулы -(CH2)n-X, где X представляет собой амино, (C1-C6)алкиламино, ди((C1 -C6)алкил)амино, (C3-C7)циклоалкил, адамантил, гетероциклоалкил, арил, арилкарбонил или гетероарил, или радикал формулы
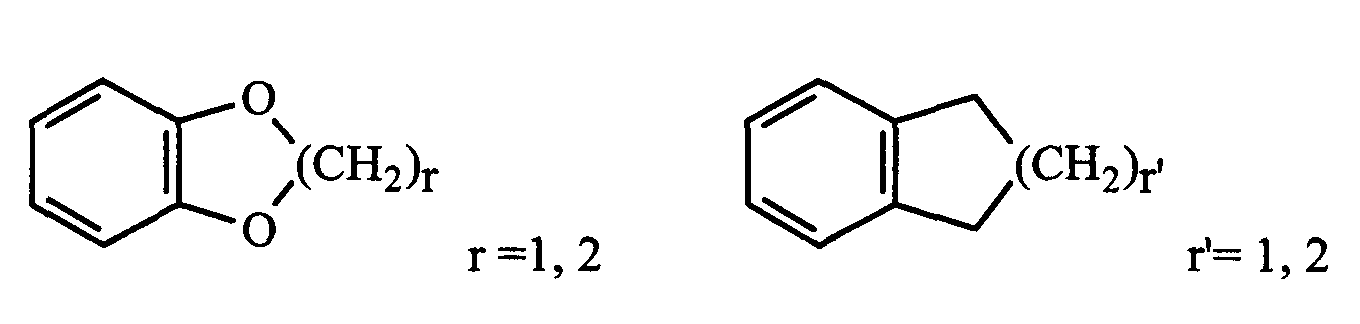
где (C3-C7)циклоалкильный, гетероциклоалкильный, арильный и гетероарильный радикалы необязательно замещены одним или несколькими одинаковыми или различными заместителями, выбранными из: -(CH2)n'-X'-Y', галогена, оксо, нитро, циано, амино, (C1-C6 )алкиламино и ди((C1-C8)алкил)амино, гидрокси, N3;
X' представляет собой -O-, -S-, -C(O)-, -C(O)-O-, -NH-C(O)-, -NH-SO2- или ковалентную связь;
Y' представляет собой (C1-C6)алкильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными галоген радикалами; гетероарил или арил или гетероциклоалкил, необязательно замещенный одним или несколькими одинаковыми или различными заместителями, выбранными из: (C1-C6)алкила, (C1-C6 )алкокси, галогена, нитро, циано, амино, CF3, OCF3, гидрокси, N3, (C1-C6)алкиламино и ди((C1-C6)алкил)амино;
n представляет собой целое число от 0 до 6 и n' представляет собой целое число от 0 до 2;
или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил, гетеробициклоалкил или радикал формулы:
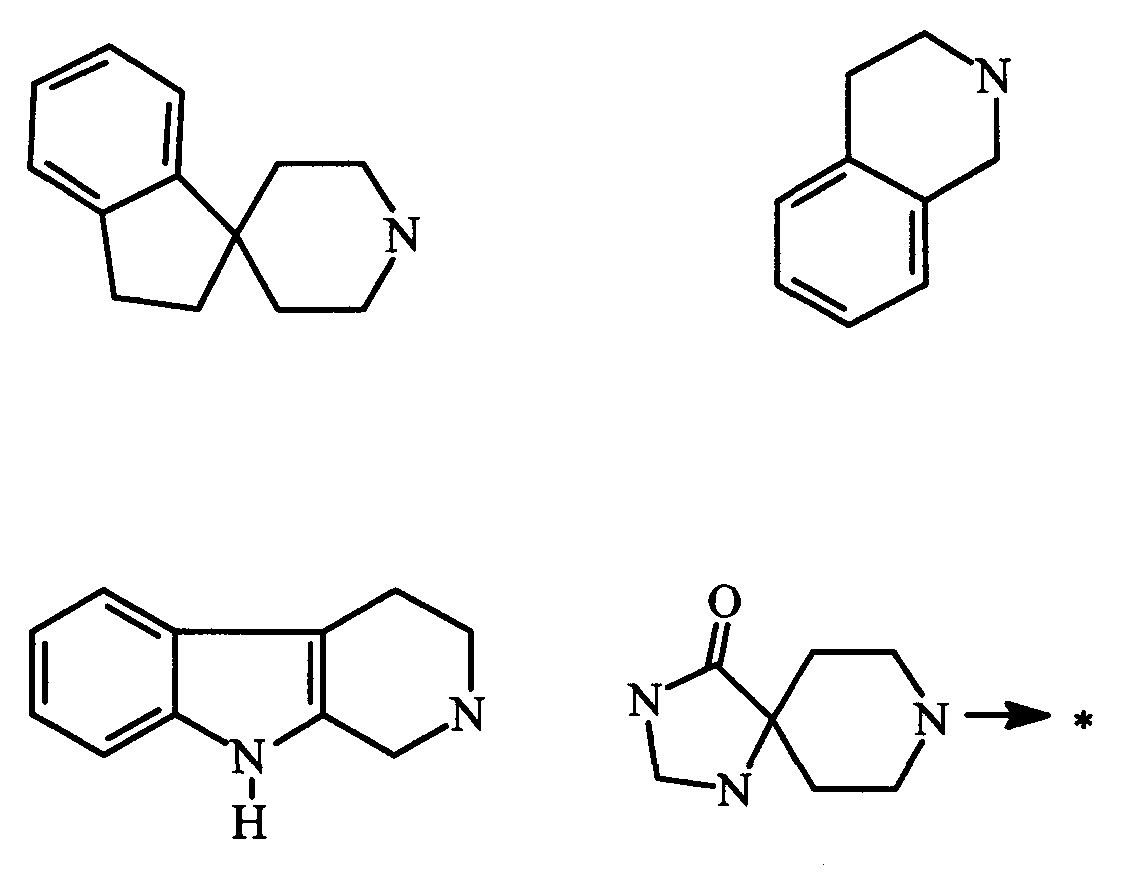
где радикал, образованный взятыми вместе R1 и R2, необязательно замещен одним или несколькими одинаковыми или различными заместителями, выбранными из:
-(CH2)n″-X″-Y″, оксо, гидрокси, галоген, нитро, циано;
X″ представляет собой -O-, -C(O)-, -C(O)-O- или ковалентную связь;
Y″ представляет собой (C1-C6)алкил, амино, (C1-C6)алкиламино, ди((C1-C6)алкил)амино, (C3-C7)циклоалкил, гетероциклоалкил, арилалкил радикал или арильный или гетероарильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными заместителями выбранными из: (C1-C6)алкила, (C1-C6)алкокси, (C1-C6)алкилкарбонила, галогена, гидрокси, нитро, циано, CF3, OCF3, амино, (C1-C6 )алкиламино и ди((C1-C6)алкил)амино); или радикал формулы
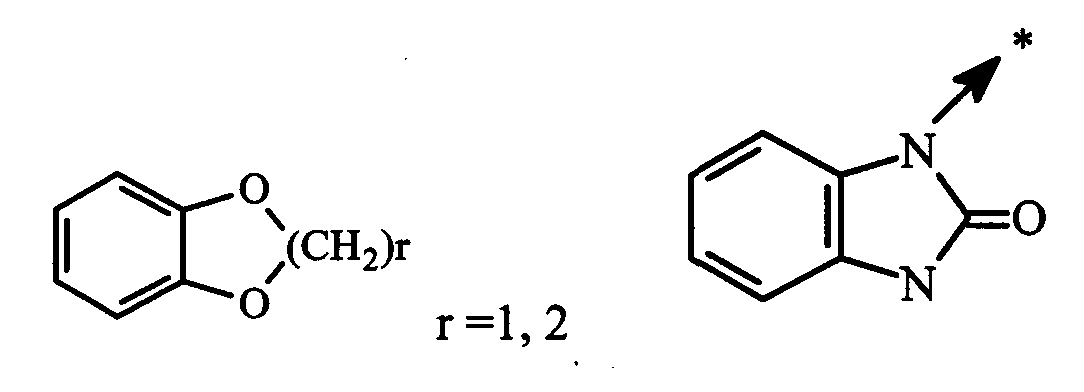
n″ представляет собой целое число от 0 до 4;
R3 представляет собой -(CH2)p-W3-(CH2)p'-Z3.
W3 представляет собой ковалентную связь, -CH(OH)- или -C(O)-;
Z3 представляет собой (C1-C6)алкил, адамантил, арильный радикал, гетероарил или радикал формулы
где арильный радикал необязательно замещен одним или несколькими одинаковыми или различными заместителями, выбранными из: -(CH2)p″-V3-Y3, галогена, нитро, циано, N3, гидрокси;
V3 представляет собой -O-, -S-, -C(O)-, -C(O)-O-, -SO2- или ковалентную связь;
Y3 представляет собой (C1-C6)алкильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными галоген радикалами, амино, (C1-C6)алкиламино, ди((C1-C6)алкил)амино, фенилкарбонилметила, гетероциклоалкила или арильного радикалов;
p, p' и p" представляют собой, независимо, целое число от 0 до 4;
R4 представляет собой радикал формулы-(CH2)s-R"4.
R"4 представляет собой гетероциклоалкил, содержащий, по крайней мере, один атом азота и необязательно замещенный (C1-C6)алкилом или аралкилом; гетероарил, содержащий, по крайней мере, один атом азота и необязательно замещенный (C1-C6)алкилом; или радикал формулы -NW4W'4.
W4 представляет собой атом водорода, (C1-C8)алкил или (C3-C7)циклоалкил;
W'4 представляет собой радикал формулы -(CH2)S'-Q4-Z4;
Q4 представляет собой ковалентную связь, -CH2-CH(OH)-[CH2]t-[O]t'[CH2]t″- или -C(O)-O-;
t, t' и t" представляют собой, независимо, 0 или 1;
Z4 представляет собой атом водорода, (C1-C8)алкил, необязательно замещенный одним или несколькими одинаковыми или различными заместителями, выбранными из: (C1-C6)алкокси, (C1-C6)алкилтио, (C1-C6)алкилдитио и гидрокси; (C2-C6)алкенил; (C2-C6)алкинил; (C3 -C7)циклоалкил, необязательно замещенный одним или несколькими одинаковыми или различными заместителями, выбранными из: (C1-C6)алкила, (C1-C6 )алкоксикарбонила и (C1-C6)гидроксиалкила; циклогексен; адамантил; гетероарил; арил, необязательно замещенный одним или несколькими одинаковыми или различными радикалами, выбранными из формулы -(CH2)q"-V4-Y4, гидрокси, галоген, нитро, циано;
V4 представляет собой -O-, -S-, -NH-C(O)- или ковалентную связь;
Y4 представляет собой (C1-C6)алкильный радикал, необязательно замещенный ди((C1-C6)алкил)амино или одним или несколькими одинаковыми или различными галоген радикалами; амино; (C1-C6)алкиламино; ди((C1-C6)алкил)амино; аралкил; гетероциклоалкил радикалы;
q" представляет собой целое число от 0 до 4;
или Z4 представляет собой радикал формулы
s и s' представляют собой, независимо, целое число от 0 до 6;
или фармацевтически приемлемую соль последнего.
В определениях, указанных выше, выражение галоген представляет собой фтор, хлор, бром или йод, предпочтительно хлор, фтор или бром радикал. Выражение алкил (если не указано иного) предпочтительно представляет собой прямой или разветвленный алкильный радикал, содержащий от 1 до 6 атомов углерода, такой как радикалы метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил, пентил или амил, изопентил, неопентил, 2,2-диметилпропил, гексил, изогексил или 1,2,2-триметилпропил. Термин (C1-C8)алкил обозначает прямой или разветвленный алкильный радикал, содержащий от 1 до 8 атомов углерода, такой как радикалы, содержащие от 1 до 6 атомов углерода, как указано выше, но также гептил, октил, 1,1,2,2-тетраметилпропил, 1,1,3,3-тетраметилбутил. Термин алкилкарбонил предпочтительно обозначает радикалы, где алкильный радикал является таким, как указано выше, такой как, например, метилкарбонил и этилкарбонил. Термин гидроксиалкил обозначает радикалы, где алкильный радикал является таким, как указано выше, такой как, например, гидроксиметил, гидроксиэтил.
Под алкенилом, если не указано иного, понимают прямой или разветвленный алкильный радикал, содержащий от 1 до 6 атомов углерода и содержащий, по крайней мере, один ненасыщенную (двойную) связь, такой как, например, винил, аллил, пропенил, бутенил или пентенил. Под алкинилом, если не указано иного, понимают прямой или разветвленный алкильный радикал, содержащий от 1 до 6 атомов углерода и содержащей, по крайней мере, одну двойную ненасыщенную (тройную) связь, такой как, например, радикал этинил, пропаргил, бутинил или пентинил.
Термин алкокси обозначает радикалы, где алкильный радикал такой, как указано выше, такие как, например, радикалы метокси, этокси, пропилокси или изопропилокси, а также прямой, вторичный или третичный бутокси, пентилокси. Термин алкокси-карбонил предпочтительно обозначает радикалы, где алкокси радикал такой, как указано выше, такие как, например, метоксикарбонил, этоксикарбонил. Термин алкилтио обозначает радикалы, где алкильный радикал такой, как указано выше, такие как, например, метилтио, этилтио. Термин алкилдитио предпочтительно обозначает радикалы, где алкильный радикал такой, как указано выше, такие как, например, метилдитио (CH3-S-S-), этилдитио или пропилдитио.
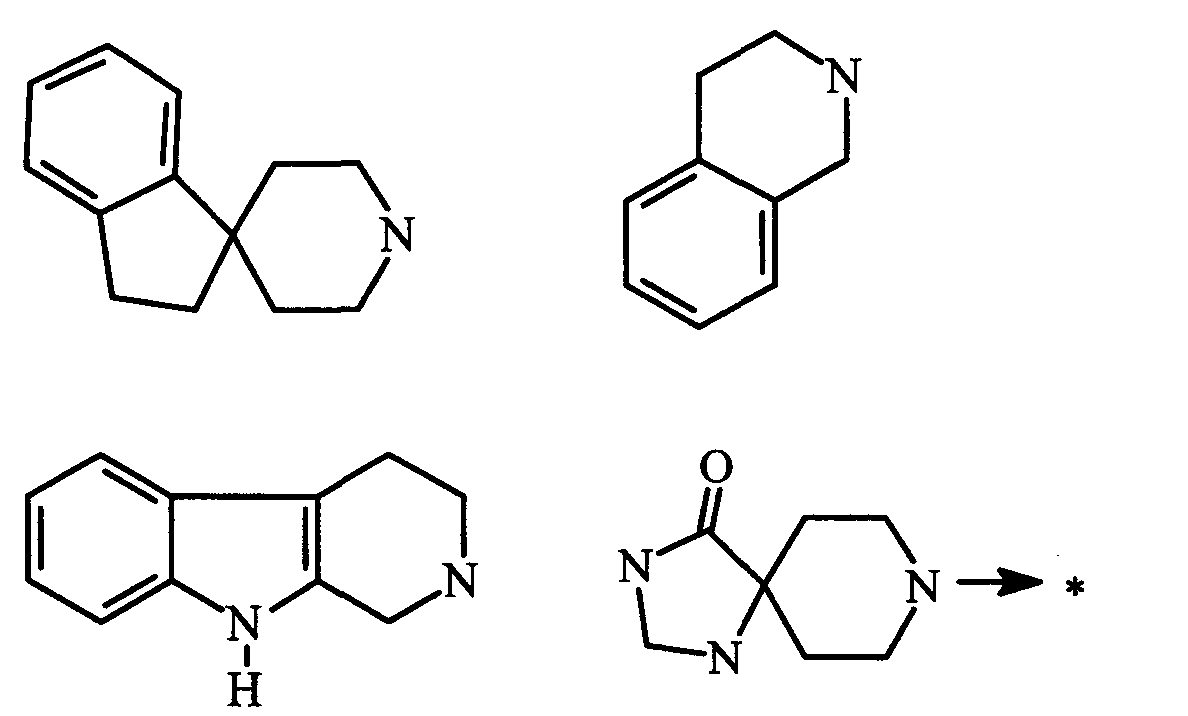
Термин (C3-C7)циклоалкил обозначает насыщенную углеродную моноциклическую систему, содержащую от 3 до 7 атомов углерода, и предпочтительно представляет собой циклопропильное, циклобутильное, циклопентильное, циклогексильное или циклогептильное кольца. Выражение гетероциклоалкил обозначает конденсированную моноциклическую или бициклическую насыщенную систему, содержащую от 2 до 7 атомов углерода и, по крайней мере, один гетероатом. Этот радикал может содержать несколько одинаковых или различных гетероатомов. Предпочтительно гетероатомы выбраны из кислорода, серы или азота. В качестве примера гетероциклоалкила можно упомянуть кольца, содержащие, по крайней мере, один атом азота, такие как пирролидин, имидазолидин, пирразолидин, изотиазолидин, тиазолидин, изоксазолидин, оксазолидин, пиперидин, пиперазин, азепан, диазепан, морфолин, декагидроизохинолин, а также кольца, не содержащие атом азота, такие как тетрагидрофуран или тетрагидротиофен.
Термин (C5-C9)бициклоалкил обозначает неконденсированную насыщенную углеводородную бициклическую систему, содержащую от 5 до 9 атомов углерода, такую как бициклогептан, такую как, например, бицикло[2,2,1]гептан или бициклооктан, такой как, например бицикло[2,2,2]октан или бицикло[3,2,1]октан. Термин гетеробициклоалкил обозначает неконденсированную насыщенную углеводородную бициклическую систему, содержащую от 5 до 8 атомов углерода и, по крайней мере, один гетероатом, выбранный из азота, кислорода и серы. В качестве примера гетеробициклоалкила можно упомянуть азабициклогептан и азабициклооктан, такой как 7-азабицикло[2,2,1]гептан, 2-азабицикло[2,2,2]октан или 6-азабицикло[3,2,1]октан.
Выражение арил представляет собой ароматический радикал, состоящий из конденсированного кольца или колец, такой как, например, фенильный, нафтильный или флуоренильный радикал. Выражение гетероарил обозначает ароматический радикал, состоящий из конденсированного кольца или колец, где, по крайней мере, одно кольцо содержит один или несколько одинаковых или различных гетероатомов, выбранных из серы, азота или кислорода. В качестве примера гетероарильного радикал можно упомянуть радикалы, содержащие, по крайней мере, один атом азота, такие как пирролил, имидазолил, пиразолил, изотиазолил, тиазолил, изоксазолил, оксазолил, триазолил, тиадиазолил, пиридил, пиразинил, пиримидил, хинолил, изохинолил, хиноксалинил, индолил, бензоксадиазоил, карбазолил, а также радикалы, не содержащие атом азота, такие как тиенил, бензотиенил, фурил, бензофурил или пиранил.
Термин аралкил (арилалкил) предпочтительно обозначает радикалы, в которых арильный и алкильный радикал такой, как указано выше; в качестве примера могут быть упомянуты арилалкил, бензил, фенэтил, фенилпропил и фенилбутил. Термин арил-карбонил предпочтительно обозначает радикалы, в которых арильный радикал такой, как указано выше, такие как, например, фенилкарбонил.
Термины алкиламино и диалкиламино предпочтительно обозначают радикалы, в которых алкильные радикалы такие, как указано выше, такие как, например, метиламино, этиламино, диметиламино, диэтиламино или (метил)(этил)амино.
Также в настоящей заявке (CH2)i радикал (i является целым числом, которое может представлять n, n', n″, p, p', p″, s, s', s" и q″, как определено выше) представляет собой прямую или разветвленную углеводородную цепь из i атомов углерода.
Объектом данного изобретения также является соединение общей формулы (I')
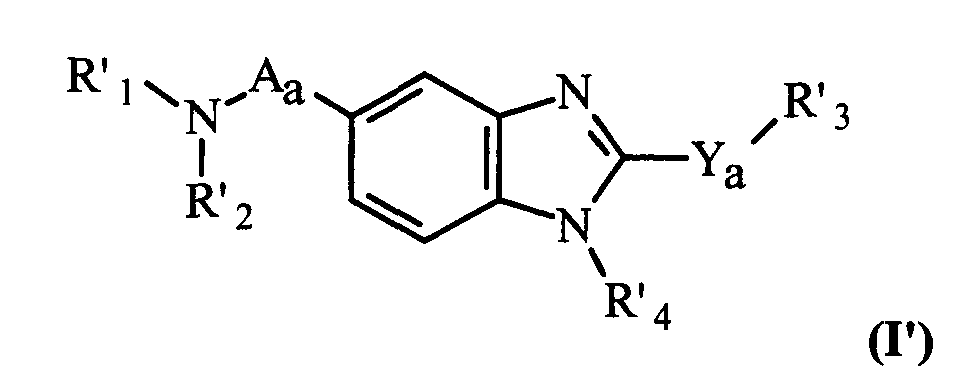
в рацемической, энантиомерной форме или все их сочетания этих форм и где:
Aa представляет собой -CH2- или -C(O)-;
Ya представляет собой -S- или -NH-;
R'1 и R'2 независимо представляют собой атом водорода, (C1-C8)алкильный, (C5-C9)бициклоалкильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными (C1-C6)алкильными радикалами, или радикал формулы -(CH2)n-X, в котором
X представляет собой амино, (C1-C6)алкиламино, ди((C1-C6)алкил)амино, (C3-C7)циклоалкил, адамантил, гетероциклоалкил, арил, арилкарбонил или гетероарил, или радикал формулы
(C3-C7)циклоалкильные, гетероциклоалкильные, арильные и гетероарильные радикалы, необязательно замещенные одним или несколькими одинаковыми или различными заместителями, выбранными из: -(CH2)n'-X'-Y', галогена, оксо, нитро, циано, амино, (C1-C6)алкиламино и ди((C1-C8)алкил)амино, гидрокси, N3;
X' представляет собой -O-, -S-, -C(O)-, -C(O)-O-, -NH-C(O)-, -NH-SO2- или ковалентную связь;
Y' представляет собой (C1-C6)алкильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными галогенами; гетероарильный, или арильный, или гетероциклоалкильный радикалы, необязательно замещенные одним или несколькими одинаковыми или различными заместителями, выбранными из: (C1-C6)алкила, (C1-С6)алкокси, галогена, нитро, циано, амино, CF3, OCF3, гидрокси, N3, (C1-C6)алкиламино и ди((C1-C8)алкил)амино;
n представляет собой целое число от 0 до 6, и n' представляет собой целое число от 0 до 2;
или R'1 и R'2 вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил, гетеробициклоалкил или радикал формулы:
радикал, который совместно образуют R'1 и R'2, необязательно замещен одним или несколькими одинаковыми или различными заместителями, выбранными из:
-(CH2)n″-X″-Y″, оксо, гидрокси, галогена, нитро, циано;
X″ представляет собой -O-, -C(O)-, -C(O)-O- или ковалентную связь;
Y″ представляет собой (C1-C6)алкил, амино, (C1-C6)алкиламино, ди((C1-C6)алкил)амино, (C3-C7)циклоалкильный, гетероциклоалкильный, арилалкильный, или арильный или гетероарильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными заместителями, выбранными из: (C1-C6)алкила, (C1-C6)алкокси, (C1-C6)алкилкарбонила, галогена, гидрокси, нитро, циано, CF3, OCF3, амино, (C1-C6)алкиламино и ди((C1-C6)алкил)амино); или радикал формулы
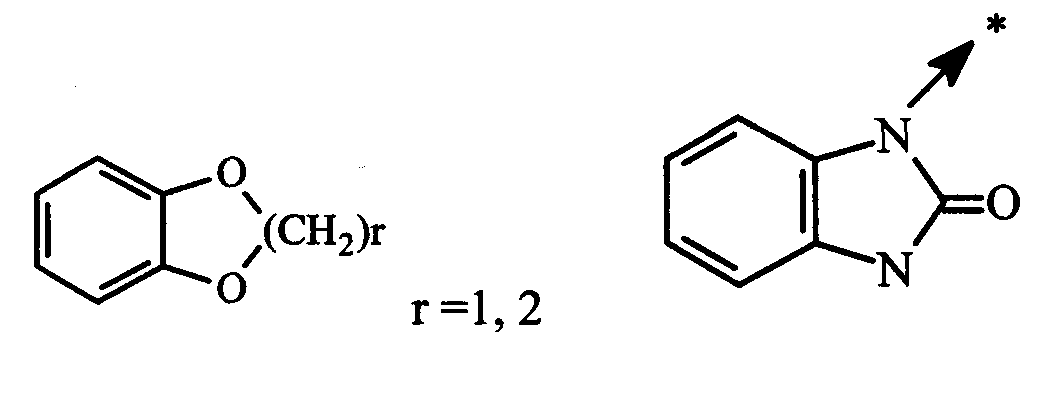
n″ представляет собой целое число от 0 до 4;
R'3 представляет собой -(CH2)p-W3-(CH2)p'-Z3.
W3 представляет собой ковалентную связь, -CH(OH)- или -C(O)-;
Z3 представляет собой (C1-C6)алкильный, адамантильный, арильный, гетероарильный радикал или радикал формулы
арильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными заместителями, выбранными из: -(CH2)p″-V3 -Y3, галогена, нитро, циано, N3, гидрокси;
V3 представляет собой -O-, -S-, -C(O)-, -C(O)-O-, -SO2 - или ковалентную связь;
Y3 представляет собой (C1-C6)алкильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными галоген радикалами, амино, (C1-C6)алкиламино, ди((C1-C6)алкил)амино, фенилкарбонилметильными, гетероциклоалкильными или арильными радикалами;
p, p' и p″ независимо представляют собой целое число от 0 до 4;
R'4 представляет собой радикал формулы-(CH2)s-R″4.
R"4 представляет собой гетероциклоалкил, содержащий, по крайней мере, один атом азота и, необязательно замещенный (C1-C6)алкилом или аралкилом; гетероарил, содержащий, по крайней мере, один атом азота и, необязательно замещенный (C1-C6)алкилом; или радикал формулы -NW4W'4.
W4 представляет собой атом водорода, (C1-C8)алкил или (C3-C7)циклоалкил;
W'4 представляет собой радикал формулы -(CH2)s'-Q4-Z4;
Q4 представляет собой ковалентную связь, -CH2-CH(OH)-[CH2]r[O]t'-[CH2]t″- или -C(O)-O-;
t, t' и t″ независимо равны 0 или 1;
Z4 представляет собой атом водорода, (C1-C8)алкил, (C3-C7)циклоалкил; гетероарил; арил, необязательно замещенный одним или несколькими одинаковыми или различными радикалами, выбранными из формулы -(CH2)q″ -V4-Y4, гидрокси, галогена, нитро, циано;
V4 представляет собой -О-, -S-, -NH-C(O)- или ковалентную связь;
Y4 представляет собой (C1-C6)алкильный радикал, необязательно замещенный ди((C1-C6)алкил)амино или одним или несколькими одинаковыми или различными галогенами; амино; (C1-C6)алкиламино; ди((C1-C6)алкил)амино; аралкильный; гетероциклоалкильный радикалы;
q″ равно целому числу от 0 до 4;
или Z4 представляет собой радикал формулы
s и s' независимо представляют собой целое число от 0 до 6;
или фармацевтически приемлемая соль последнего.
Более конкретным объектом настоящего изобретения является соединение формулы I или I', как определено выше, или фармацевтически приемлемая соль последнего, где А представляет собой -C(O)-.
Еще более конкретным объектом настоящего изобретения является соединение формулы I, как определено выше, или фармацевтически приемлемая соль последнего, где
циклоалкил, представленный X, представляет собой циклогексил или циклогептил,
гетероциклоалкил, представленный X, выбран из: пиперидина, пирролидина, тиазолидина, морфолина и тетрагидротиофена;
арил, представленный X, представляет собой фенильный, нафтильный или флуоренильный радикал;
арил арилкарбонильного радикала, представленный X, представляет собой фенильный радикал;
гетероарил, представленный X, выбран из: пиридина, имидазола, тиофена, индола, карбазола и изохинолина;
гетероарил, представленный Y', выбран из оксазола и имидазола;
арил, представленный Y', представляет фенильный радикал;
гетероциклоалкил, представленный Y', представляет собой пиперазин;
гетероциклоалкил, который совместно с атомом азота, к которому они присоединены, образуют R1 и R2, выбран из: пиперидина, пиперазина, диазепана, тиазолидина и морфолина;
циклоалкил, представленный Y″, представляет собой циклопентил или циклогексил;
гетероциклоалкил, представленный Y″, выбран из: пиперидина, пирролидина и морфолина;
арилалкил и арил, представленный Y″, представляют собой соответственно бензильный радикал и фенильный радикал;
гетероарил, представленный Y″, выбран из: пиридина, пиразина, фурана и тиофена.
Более конкретным объектом настоящего изобретения также является соединение формулы I, как определено выше, или фармацевтически приемлемая соль последнего, где
арил, представленный Z3, представляет собой фенильный или нафтильный радикал;
гетероарил, представленный Z3, выбран из бензо[b]тиофена и бензо[b]фурана;
гетероциклоалкил и арил, представленный Y3, представляют собой соответственно пирролидиновый и фенильный радикалы.
Более конкретным объектом настоящего изобретения также является соединение формулы I, как определено выше, или фармацевтически приемлемая соль последнего, где
гетероциклоалкил, представленный R"4, выбран из: пиперазина, пиперидина, морфолина и пирролидина;
аралкил, необязательно замещенный гетероциклоалкилом, представленный R"4, представляет собой бензильный радикал;
гетероарил, представленный R″4, представляет собой имидазол;
(C3-C7)циклоалкил, представленный Z4, представляет собой циклопропил, циклобутил, циклопентил, циклогексил или циклогептил;
гетероарил, представленный Z4, выбран из: пиридина, тиофена, индола и фурана;
арил, представленный Z4, представляет собой фенил или нафтил;
аралкил, представленный Y4, представляет собой бензил;
гетероциклоалкил, который представляет Y4, представляет собой пирролидин;
аралкил, замещенный на гетероциклоалкиле, который совместно образуют W4 и W'4, представляет собой бензильный радикал.
Предпочтительно объектом данного изобретения является соединение формулы I, как определено выше, или фармацевтически приемлемая соль последнего, где А представляет собой -C(O)- и R1 и R2 независимо представляют собой атом водорода, ((C1-C8)алкильный радикал или радикал формулы -(CH2)n-X, где
X представляет собой, амино, ди(алкил)амино, адаментил, циклогексил, циклогептил, пиперидин, морфолин, пирролидин, фенил, пиридин, имидазол, тиофен, индол, карбазол, необязательно замещенный (C1-C6)алкилом, или радикал формулы
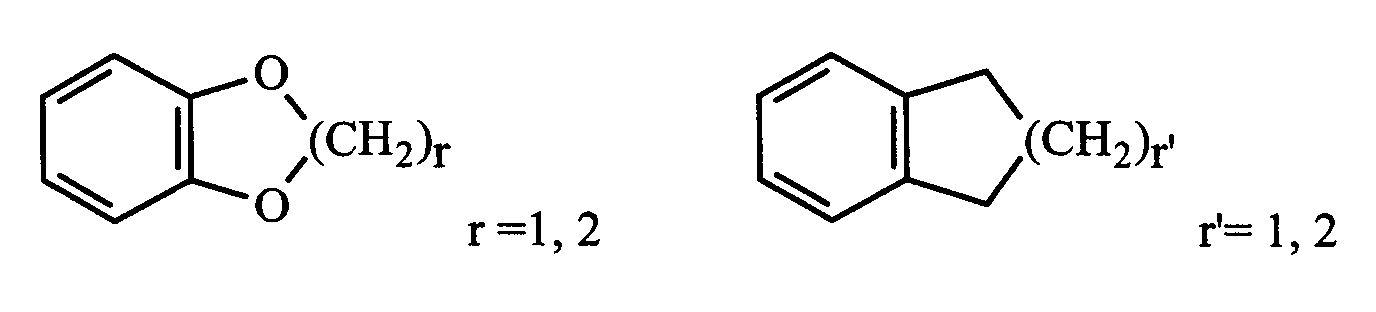
пиперидиновые, пирролидиновые и фенильные радикалы, необязательно замещены одним или несколькими одинаковыми или различными заместителями, выбранными из: -(CH2)n'-X'-Y', галогена, оксо, амино и ди((C1-C8 )алкил)амино;
X' представляет собой -O-, -S-, -C(O)-O-, -NH-C(O)-, -NH-SO2- или ковалентную связь;
Y' представляет собой (C1-C6)алкил, оксазол, фенильный радикал, необязательно замещенный (C1-C4)алкилом, или пиперазин, необязательно замещенный (C1-C4)алкилом;
или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют пиперидин, пиперазин и диазепан, тиазолидин, морфолин или циклический радикал формулы:
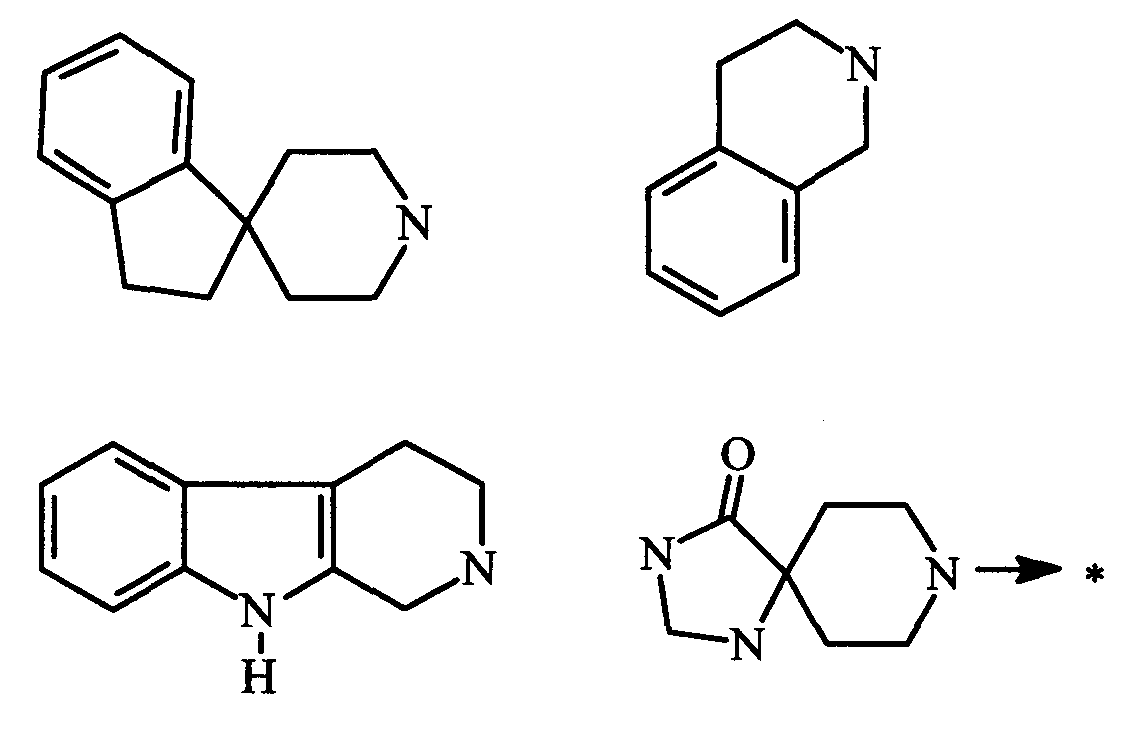
радикал, который совместно образуют R1 и R2, необязательно замещен одним или несколькими одинаковыми или различными заместителями, выбранными из:
-(CH2)n″-X″-Y″;
X″ представляет собой -C(O)-, -C(O)-O- или ковалентную связь;
Y″ представляет собой (C1-C6)алкил; ди(алкил)амино, циклопентильный, циклогексильный, пиперидиновый, пирролидиновый, морфолиновый, бензильный, пиридиновый, пиразиновый, фурановый, тиофеновый или фенильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными заместителями, выбранными из (C1-C6)алкила, (C1-C6)алкокси, (C1-C6)алкилкарбонила и галогена; или Y″ представляет собой радикал формулы
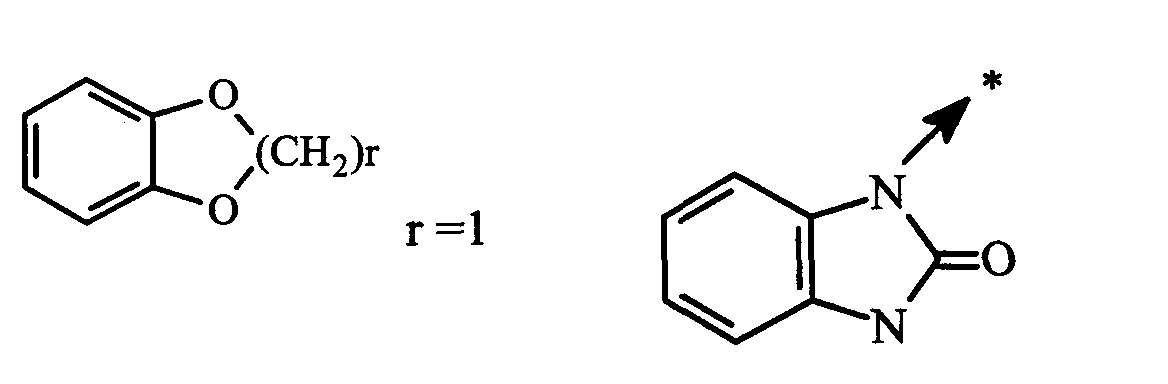
Предпочтительно объектом данного изобретения является соединение формулы I, как определено выше, или фармацевтически приемлемая соль последнего, где А представляет собой -C(O)- и R3 представляет собой -(CH2)p-W3-(CH2)p'-Z3.
W3 представляет собой ковалентную связь, -CH(OH)- или
-C(O)-;
Z3 представляет собой (C1-C6)алкильный, фенильный, нафтильный, бензо[b]тиофеновый, бензо[b]фуранильный радикал или радикал формулы
радикал фенил, необязательно замещен одним или несколькими одинаковыми или различными заместителями, выбранными из: -(CH2)p″-V3-Y3, галогена, нитро, циано;
V3 представляет собой -O-, -S-, -C(O)-, -C(O)-O-, -SO2- или ковалентную связь;
Y3 представляет собой (C1-C6)алкильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными галогенами; амино; ди((C1-C6)алкил)амино; фенилкарбонилметильный; пирролидиновый или фенильный радикалы;
p, p' и p″ независимо равны целому числу от 0 до 2.
Предпочтительно объектом данного изобретения является соединение формулы I', как определено выше, или фармацевтически приемлемая соль последнего, где Aa представляет собой -C(O)- и радикалы R'1, R'2, R'3 и R'4 определены так же как радикалы R1, R2, R3 и R4 соответственно, как определено выше.
Предпочтительно объектом данного изобретения является соединение формулы I, как определено выше, или фармацевтически приемлемая соль последнего, где А представляет собой -C(O)-, и R4 представляет собой радикал формулы-(CH2)s-R″4
R"4 представляет собой пиперидиновое кольцо, необязательно замещенное бензилом, пиперазин, необязательно замещенный бензилом, или радикал формулы -NW4W'4.
W4 представляет собой атом водорода или (C1-C8)алкил;
W'4 представляет собой радикал формулы -(CH2)S'-Q4-Z4;
Q4 представляет собой ковалентную связь, -CH2-CH(OH)-,-CH2-CH(OH)-CH2-O-, -CH2-CH(OH)-CH2-, -CH2-CH(OH)-CH2-O-CH2- или -С(О)-О-;
Z4 представляет собой атом водорода, (C1-C8)алкил, необязательно замещенный (C1-C6)алкокси, (C1-C6)алкилтио, (C1-C6 )алкилдитио или одним или двумя гидрокси; (C2-C6)алкенил; (C2-C6)алкинил; циклопропильные радикалы, необязательно замещенные алкоксикарбонилом; циклобутил, циклопентил, необязательно замещенный гидроксиалкилом; циклогексил, необязательно замещенный одним или несколькими алкилами; циклогептил, циклогексен, адамантил, пиридин, тиофен, индол, фуран, нафтил; фенильные радикалы, необязательно замещенные одним или несколькими одинаковыми или различными радикалами, выбранными из: -(CH2)q″-X4-Y4, гидрокси, галоген и циано;
X4 представляет собой -O- или ковалентную связь;
Y4 представляет собой (C1-C6)алкил, ди((C1 -C6)алкил)амино или пирролидиновый радикал.
Весьма предпочтительно, объектом данного изобретения также является соединение формулы I, как определено выше, где А представляет собой -C(O)-, Y представляет собой -NH- и
R1 и R2 независимо представляют собой (C1-C8)алкильный радикал;
R3 представляет собой -(CH2)p-W3-(CH2)p'-Z3.
W3 представляет собой ковалентную связь; Z3 представляет собой фенильный радикал, замещенный одним или несколькими одинаковыми или различными заместителями, выбранными из: -(CH2)P″-V3-Y3 и галогена; V3 представляет собой -O- или -S-; и Y3 представляет собой (C1-C6)алкильный радикал; p, p' и p″ равны 0;
R4 представляет собой радикал формулы -(CH2)s-R″4
R"4 представляет собой радикал формулы -NW4W'4.
W4 представляет собой атом водорода или (C1-C8)алкил;
W'4 представляет собой радикал формулы -(CH2)S'-Q4-Z4;
Q4 представляет собой ковалентную связь;
Z4 представляет собой атом водорода, (C1-C8)алкил, необязательно замещенный гидрокси, (C3-C7)циклоалкил, гетероарил, арил, необязательно замещенный одним или несколькими одинаковыми или различными радикалами, выбранными из -(CH2)q″ -V4-Y4;
V4 представляет собой -О- или ковалентную связь;
Y4 представляет собой (C1-C6)алкил или ди((C1-C6)алкил)амино радикал;
q″ равно 0; s равно целому числу от 2 до 4, и s' равно целому числу от 1 до 2.
И весьма предпочтительно (C3-C7)циклоалкил выбран из циклопентила и циклогексила, гетероарил представляет собой пиридин и арил представляет собой фенил; или фармацевтически приемлемая соль последнего.
Предпочтительно объектом данного изобретения является соединение формулы I', как указано выше, или фармацевтически приемлемая соль последнего, где Aa представляет собой -C(O)-, Ya, -NH-, радикалы R'1, R'2 и R'3 определены так же, как радикалы R1, R2 и R3 соответственно, как определено выше, и R'4 представляет собой радикал формулы -(CH2)s-R″4.
R"4 представляет собой радикал формулы -NW4 W'4.
W4 представляет собой атом водорода или (C1-C8)алкил;
W'4 представляет собой радикал формулы -(CH2 )s'-Q4-Z4;
Q4 представляет собой ковалентную связь;
Z4 представляет собой атом водорода, (C1-C8)алкил, (C3-C7)циклоалкил, гетероарил, арил, необязательно замещенный одним или несколькими одинаковыми или различными радикалами, выбранными из формулы -(CH2 )q"-V4-Y4;
V4 представляет собой -O- или ковалентную связь;
Y4 представляет собой (C1-C6 )алкильный или ди((C1-C6)алкил)амино радикал;
q″ равно 0; s равно целому числу от 2 до 4, и s' равно целому числу от 1 до 2.
И весьма предпочтительно (C3-C7)циклоалкил выбран из циклопентила и циклогексила, гетероарил представляет собой пиридин и арилфенил; или фармацевтически приемлемая соль последнего.
Весьма предпочтительно объектом данного изобретения также является соединение формулы I, как определено выше, где А представляет собой -C(O)-, Y представляет собой атом серы и
R1 и R2 независимо представляют собой (C1-C8)алкильный радикал;
R3 представляет собой -(CH2)p -W3-(CH2)p'-Z3.
W3 представляет собой ковалентную связь или -C(O)-; Z3 представляет собой фенильный радикал, замещенный одним или несколькими одинаковыми или различными заместителями, выбранными из: -(CH2)p″-V3-Y3 и галогена; V3 представляет собой -O- или ковалентную связь; и Y3 представляет собой (C1-C6)алкил или ди((C1-C6)алкил)амино радикал; p равно 1, и p' и p″ равны 0;
R4 представляет собой радикал формулы-(CH2)s-R″4.
R″4 представляет собой радикал формулы -NW4 W'4.
W4 представляет собой атом водорода или (C1-C8)алкил;
W'4 представляет собой радикал формулы -(CH2 )s'-Q4-Z4;
Q4 представляет собой ковалентную связь;
Z4 представляет собой атом водорода, (C1-C8)алкил, гетероарил, арил;
s равно целому числу от 2 до 4, и s' равно целому числу от 1 до 2.
И весьма предпочтительно гетероарил представляет собой пиридин и арилфенил; или фармацевтически приемлемая соль последнего.
Предпочтительно объектом изобретения является соединение формулы I', как определено выше, или фармацевтически приемлемая соль последнего, где Aa представляет собой -C(O)-, Ya представляет собой атом серы, и радикалы R'1, R'2, R'3 и R'4 определены так же как, радикалы R1, R2, R3 и R4 соответственно, как определено выше, если А представляет собой -C(O)- и Y представляет собой атом серы.
Предпочтительно объектом данного изобретения также является соединение формулы I, как определено выше, где А представляет собой -CH2-, Y представляет собой -NH-, и R1 и R2 независимо представляют собой ((C1-C6)алкильный радикал; R3 представляет собой фенил, замещенный одним или несколькими одинаковыми или различными (C1 -C6)алкокси заместителями; R4 представляет собой радикал формулы -(CH2)s-R″4; R"4 представляет собой радикал формулы -NW4W'4; W4 представляет собой (C1-C8)алкил; W4 представляет собой радикал формулы -(CH2)S'-Q4-Z4; Q4 представляет собой ковалентную связь и Z4 представляет собой пиридин; или фармацевтически приемлемая соль последнего.
Предпочтительно объектом данного изобретения является соединение формулы I', как определено выше, или фармацевтически приемлемая соль последнего, где Aa представляет собой -CH2-, Ya представляет собой -NH-, и радикалы R'1, R'2, R'3 и R'4 определены так же, как радикалы R1, R2, R3 и R4 соответственно, как определено выше, если А представляет собой -CH2-, и Y представляет собой -NH-.
В настоящей заявке символ -> * соответствует месту присоединения радикала. Если место присоединения на радикале не указано, то это обозначает, что присоединение происходит на любом доступном месте этого радикала.
В соответствии с определениями различных групп A, Y, R1, R2, R3 и R4 соединения по данному изобретению могут быть получены в жидкой фазе в соответствии с различными способами А-H, описанными далее.
A. Получение в соответствии co схемой реакции A:
Соединения формулы I по данному изобретению, где Y представляет собой -NH-, и А представляет собой -C(O)-, могут быть получены в соответствии со следующей схемой A:

Как показано на схеме A, 4-фтор-3-нитробензойная кислота (1) может быть соединена с первичным или вторичным амином в присутствии связывающего агента, такого как диизопропилкарбодиимид, дициклогексилкарбодиимид, в присутствии или в отсутствие 1-гидроксибензотриазола (HOBt), в инертном органическом растворителе, таком как метиленхлорид, тетрагидрофуран или диметилформамид, при комнатной температуре в течение 3-24 часов с получением соответствующего амида (2). Обработка фторированного производного (2) первичным амином в присутствии неорганического основания, такого как карбонат цезия или калия, в инертном органическом растворителе, таком как диметилформамид или ацетонитрил, при температуре 20-70°C в течение 2-16 часов приводит к образованию производного (3). Нитрогруппу соединения (3) восстанавливали обработкой дигидратом хлорида олова в инертном растворителе, таком как этилацетат или диметилформамид, при температуре 60-80° C в течение 3-5 часов, или с помощью каталитического восстановления в присутствии 10%-ного палладия на углероде в инертном растворителе, таком как метанол, этанол, этилацетат или смесь этих растворителей, при температуре 18-25°C в течение 2-8 часов с получением дианилина (4). Затем производное (4) обрабатывали изотиоцианатом в присутствии связывающего агента с подложкой из смолы или без нее, такого как диизопропилкарбодиимид или дициклогексилкарбодиимид, или N-метилциклогексилкарбодиимид N-метилполистирольная смола, в инертном растворителе, таком как тетрагидрофуран, метиленхлорид или хлороформ, при температуре 20-70°C в течение 2-72 часов с получением производного (5). Альтернативно производное (4) может быть обработано изотиоцианатом в инертном растворителе, таком как тетрагидрофуран, метиленхлорид или хлороформ, затем полученная тиомочевина может быть обработана метилйодидом в полярном растворителе, таком как этанол, в течение 3-24 часов при температуре 20-70°C с получением (5).
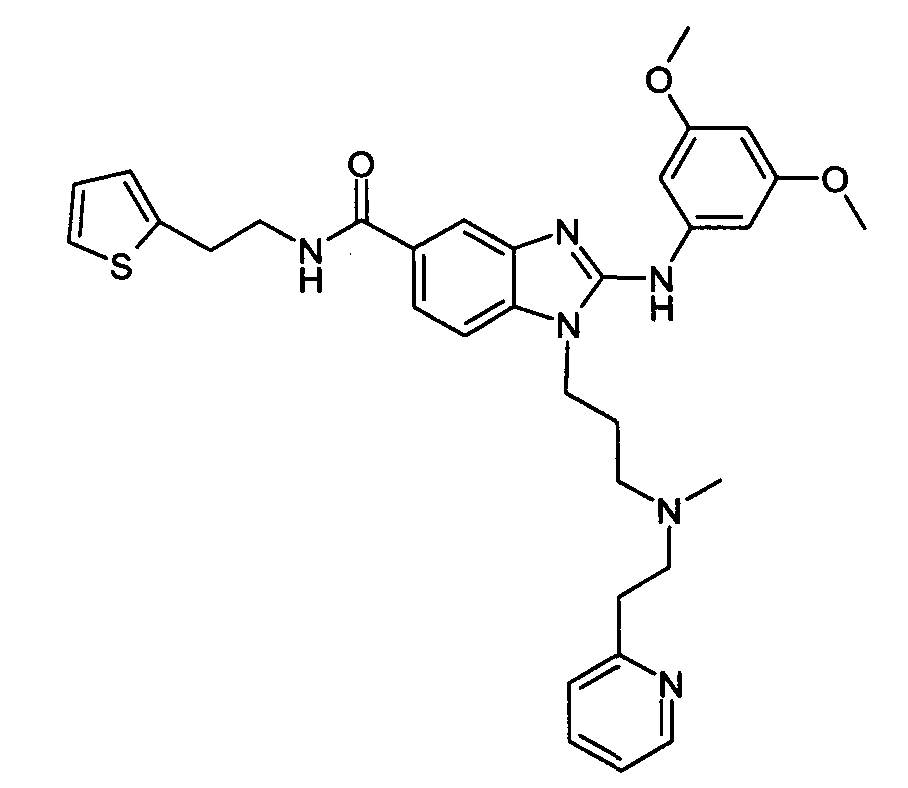
Пример A1: гидрохлорид N,N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-[(3,4,5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамида
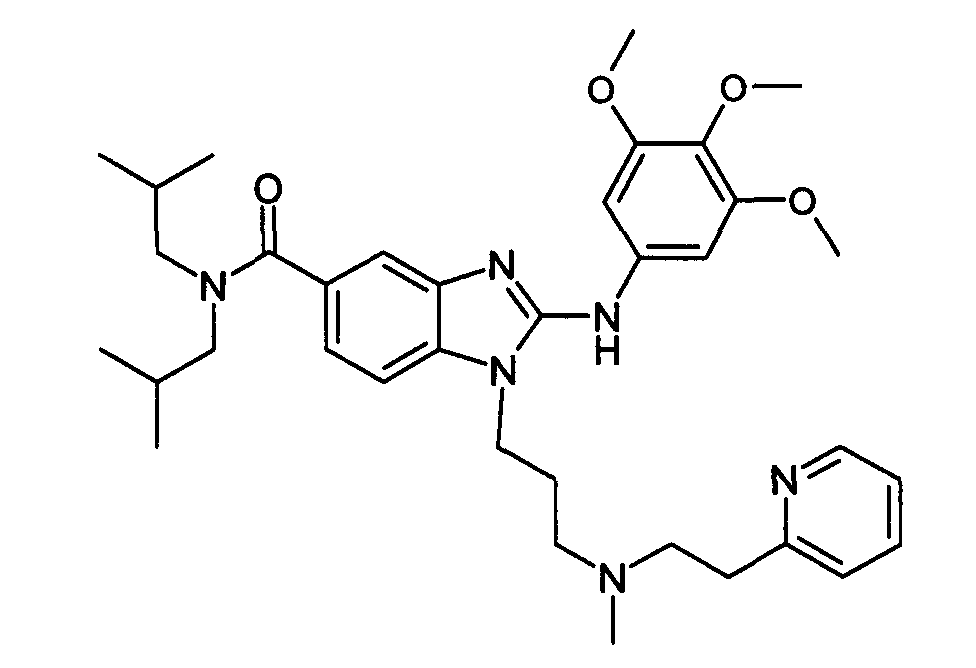
Стадия 1: 4-фтор-N,N-диизобутил-3-нитробензамид
Диизопропилкарбодиимид (13,8 мл, 1,2 экв.) добавляли к 4-фтор-3-нитробензойной кислоте (15 г, 1 экв.) в растворе ТГФ (150 мл). Смесь перемешивали в течение 3 часов при температуре приблизительно 20° C, затем добавляли диизобутиламин (12,9 мл, 1 экв.). После перемешивания в течение 15 часов при температуре приблизительно 20°C реакционную смесь упаривали при пониженном давлении при 40°C. Остаток обрабатывали дихлорметаном (200 мл) и водой (70 мл). После декантации и экстракции объединенные органическом фазы промывали водным солевым раствором, затем сушили над Na2SO4, затем упаривали при пониженном давлении при температуре 40°C. Очистка соединения с помощью флэш хроматографии на силикагеле (элюент: гептан/этилацетат 8:2) давала желаемое соединению в виде желтого твердого продукта (13,8 г; 63%-ный выход).
MS/LC: вычисленный MW = 296,3; m/z = 297,2 (MH+) - точка плавления равна 47°C.
Стадия 2: N,N-диизобутил-4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)-3-нитробензамид
Смесь 4-фтор-N,N-диизобутил-3-нитробензамида (2,07 г, 1 экв.), N-(2-пиридин-2-илэтил)пропан-1,3-диамина (1,6 г, 1,2 экв.) и карбоната цезия (4,5 г, 2 экв.) в ацетонитриле (70 мл) кипятили с обратным холодильником в течение 3 часов, затем концентрировали при пониженном давлении при 40°C. Осадок помещали в дихлорметан (100 мл) и воду (40 мл). После декантации и экстракции объединенные органические фазы промывали водным солевым раствором, сушили над Na2SO4, затем упаривали при пониженном давлении при 40°C. Очищение осадка флэш-хроматографией на силикагеле (элюент : дихлорметан 100 до дихлорметан/метанол 8:2) давало ожидаемое соединение в виде желтого масла (3,1 г; 92%-ный выход).
MS/LC: вычисленный MW = 496,6; m/z = 470,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
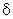
Стадия 3: 3-амино-N,N-диизобутил-4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)бензамид
N,N-диизобутил-4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)-3-нитробензамид (2,9 г) в растворе в смеси этилацетата/этанола (100 мл) и 10% палладий на угле (290 мг) добавляли при автоклавировании. После перемешивания в течение 7 часов в атмосфере водорода (3 бара), катализатор удаляли фильтрацией на целите и фильтрат концентрировали при пониженном давлении при 40°C с получением ожидаемого соединения в виде масла (2,5 г, 92%-ный выход).
MS/LC: вычисленный MW = 439,6; m/z = 440,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 4: гидрохлорид N,N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-[(3,4,5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамида
3,4,5-триметоксифенилизотиоцианат (27 мг, 1,2 экв.) и N-метилциклогексилкарбодиимид-N-метил-полистирольную смолу (приобретена у Novabiochem; загрузка 1,69 ммоль/г, 236 мг, 4 экв.) последовательно добавляли к раствору 3-амино-N,N-диизобутил-4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)бензамида (48 мг, 1 экв.) в тетрагидрофуране (2 мл). Смесь кипятили с обратным холодильником в течение 18 часов, затем охлаждали до комнатной температуры и добавляли аминометил-полистирольную смолу (приобретена у Novabiochem, 2 экв.). После перемешивания в течение 4 часов при комнатной температуре, смесь фильтровали на фритте, и фильтрат концентрировали при пониженном давлении при 40°C. Полученный осадок растворяли в этиловом эфире и по каплям добавляли раствор 1н. HCl в этиловом эфире с получением ожидаемого соединения в виде гидрохлоридной соли (80 мг, 89%-ный выход).
MS/LC: вычисленный MW = 630,8; m/z = 631,4 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Пример A2: гидрохлорид 1-{3-[бензил(метил)амино]пропил}-2-[(3,5-диметоксифенил)амино]-N,N-диизобутил-1H-бензимидазол-5-карбоксамида
Стадия 1: бензамид 3-амино-4-({3-[бензил(метил)амино]пропил}амино)-N,N-диизобутила
К раствору 4-({3-[бензил(метил)амино]пропил}амино)-N,N-диизобутил-3-нитробензамида (1,44 г, получен в соответствии со способом, описанным для примера A1) в этилацетате (40 мл) добавляли дигидрат хлорида олова (3,58 г, 5 экв.). Смесь кипятили с обратным холодильником в течение 7 часов, затем охлаждали до температуры приблизительно 20°C и выливали в насыщенный раствор NaHCO3. После декантации и экстракции этилацетатом органические фазы объединяли, промывали водным солевым раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении при 40°C. Очистка с помощью флэш-хроматографии на силикагеле (элюент дихлорметан/метанол 95:5) давала соединение в виде пены (1,06 г, 78%-ный выход).
MS/LC: вычисленный MW = 424,3; m/z = 425,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 2: гидрохлорид 1-{3-[бензил(метил)амино]пропил}-2-[(3, 5-диметоксифенил)амино]-N,N-диизобутил-1H-бензимидазол-5-карбоксамида
3,4-Диметоксифенилизотиоцианат (35 мг, 1,2 экв.) и N-метилциклогексилкарбодиимид-N-метилполистирольную смолу (приобретена у Novabiochem; загрузка 1,69 ммоль/г, 355 мг, 4 экв.) последовательно добавляли к раствору 3-амино-4-({3-[бензил(метил)амино]пропил}амино)-N,N-диизобутилбензамида (65 мг, 1 экв.) в тетрагидрофуране (2 мл). Смесь кипятили с обратным холодильником в течение 18 часов, затем охлаждали до комнатной температуры и добавяли аминометил-полистирольную смолу (приобретена у Novabiochem, 2 экв.). После перемешивания в течение 4 часов при комнатной температуре смесь фильтровали на фритте и фильтрат концентрировали при пониженном давлении при 40°C. Полученный осадок растворяли в этиловом эфире и по каплям добавляли 1н. раствор HCl в этиловом эфире с получением ожидаемого соединение в виде гидрохлоридной соли (81 мг, 92%-ный выход).
MS/LC: вычисленный MW = 585, 3; m/z = 586,5 (MH+).
ЯМР(1H, 400 МГц, DMSO-d6):
Нижеследующие соединения получали в соответствии co схемой реакции А и тем же способом, описанным для синтеза N,N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-[(3,4, 5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамида или 1-{3-[бензил(метил)амино]пропил}-2-[(3,5-диметоксифенил)амино]-N,N-диизобутил-1H-бензимидазол-5-карбоксамида:
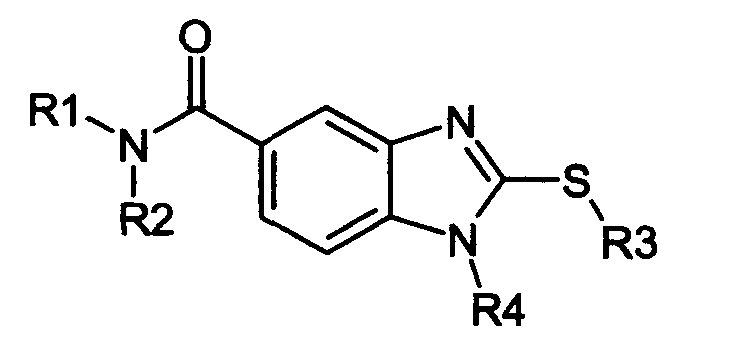
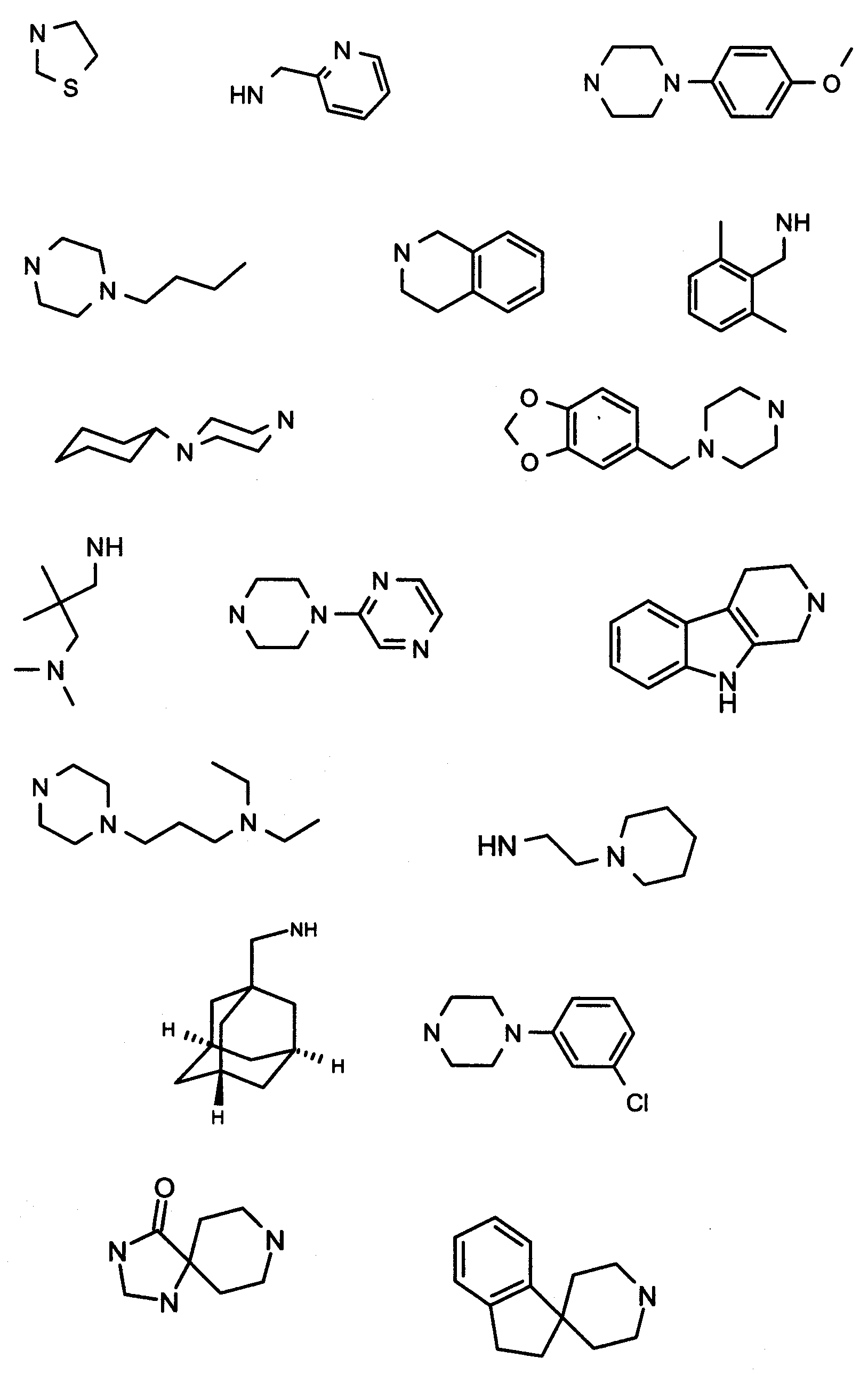
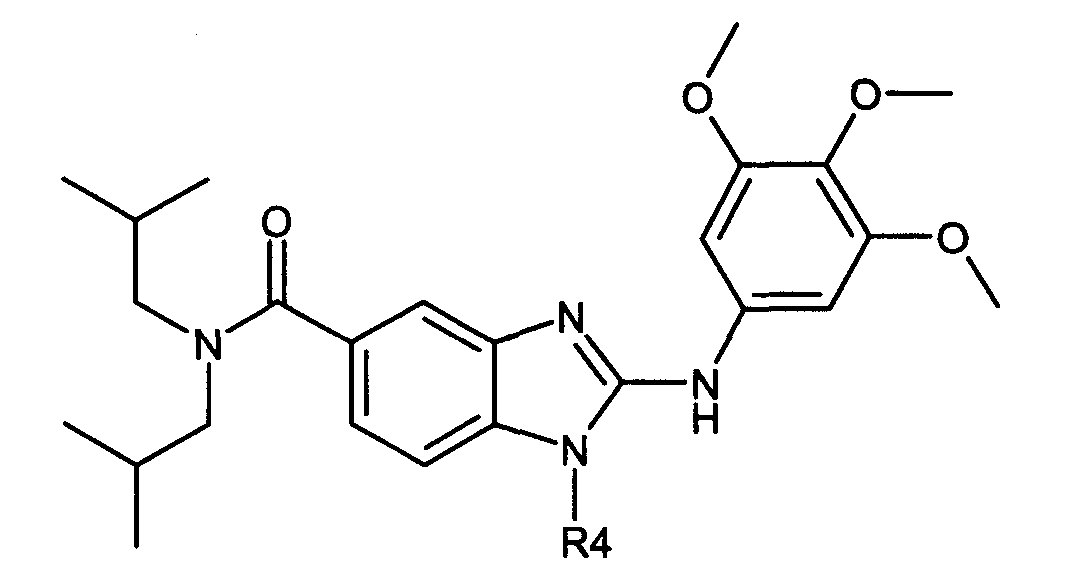
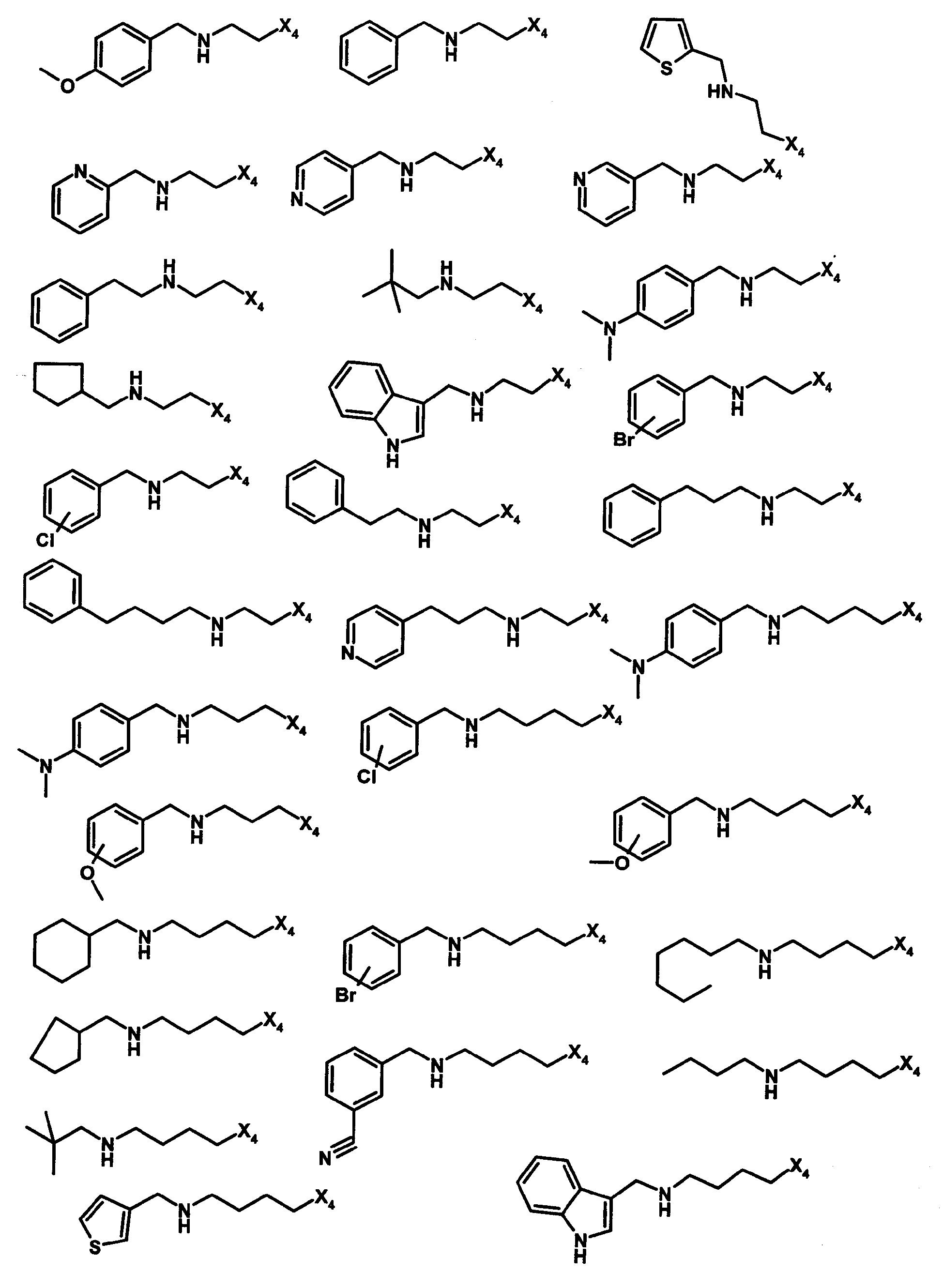
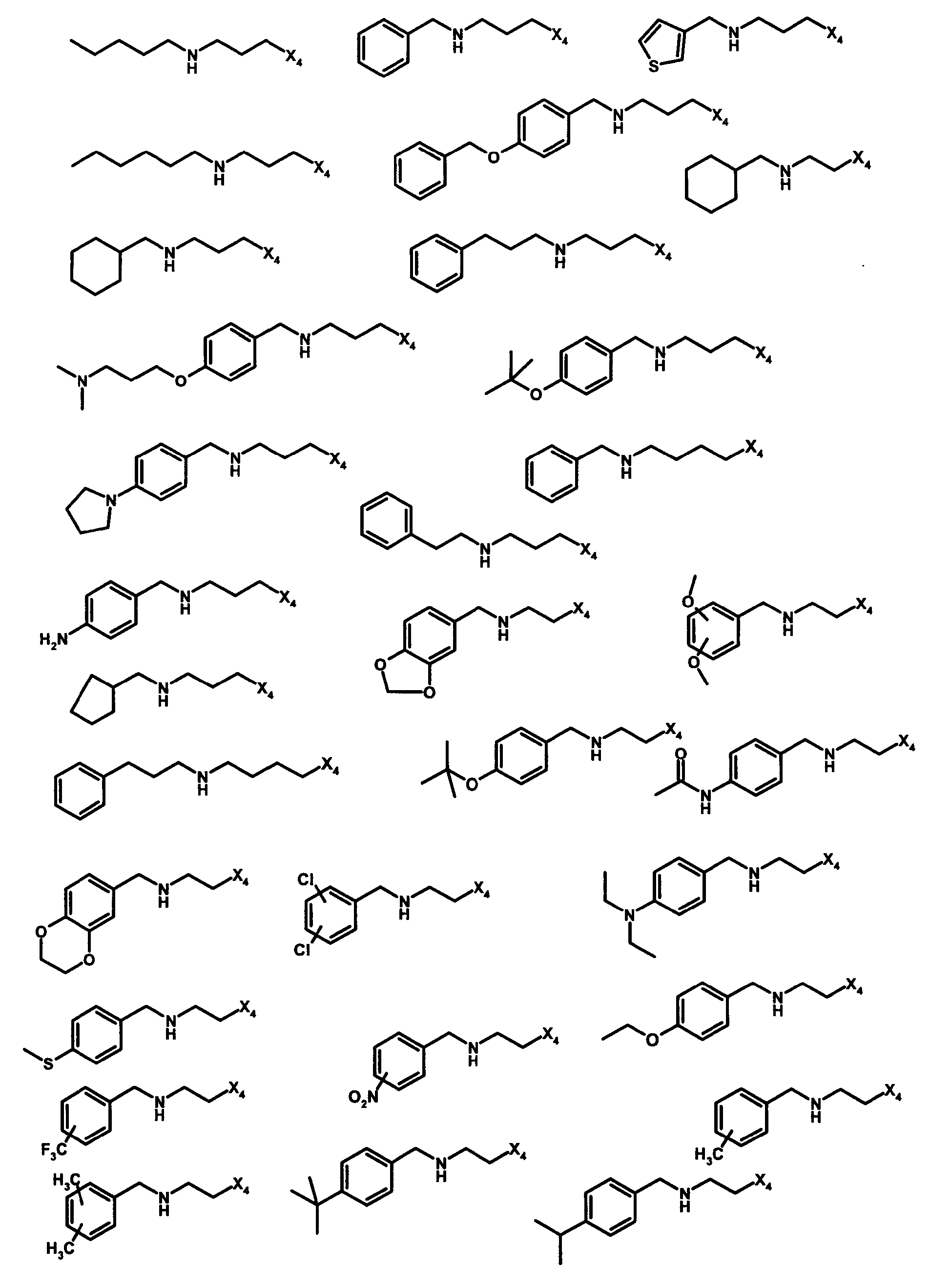
В вышеуказанной формуле R1,R2,N являются одним из нижеприведенных радикалов:
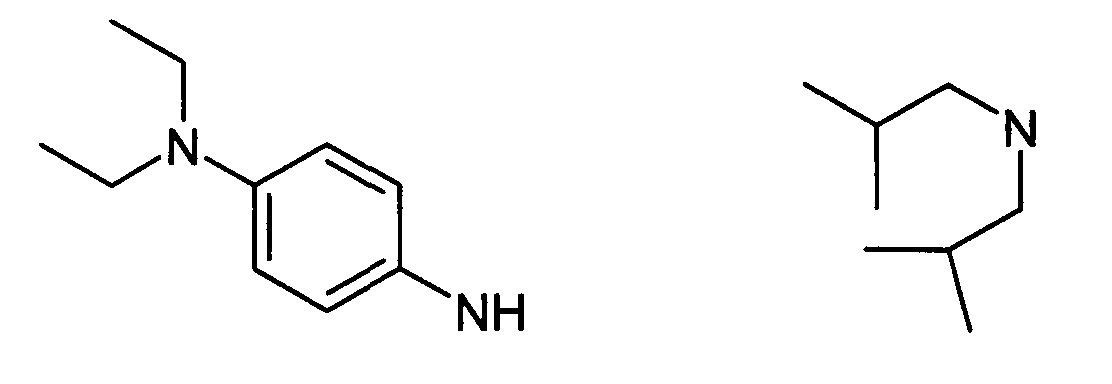
R3 является одним из нижеприведенных радикалов
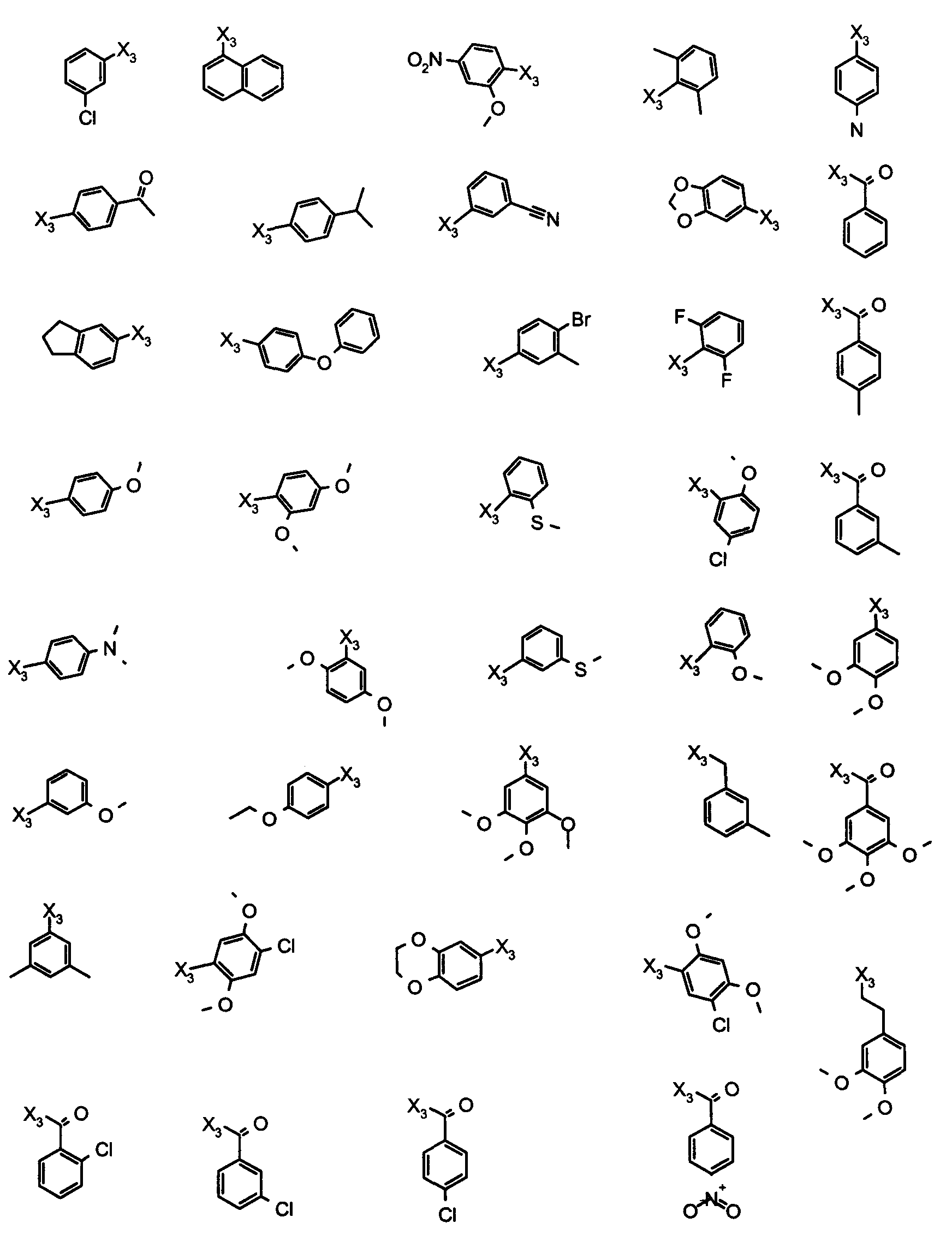
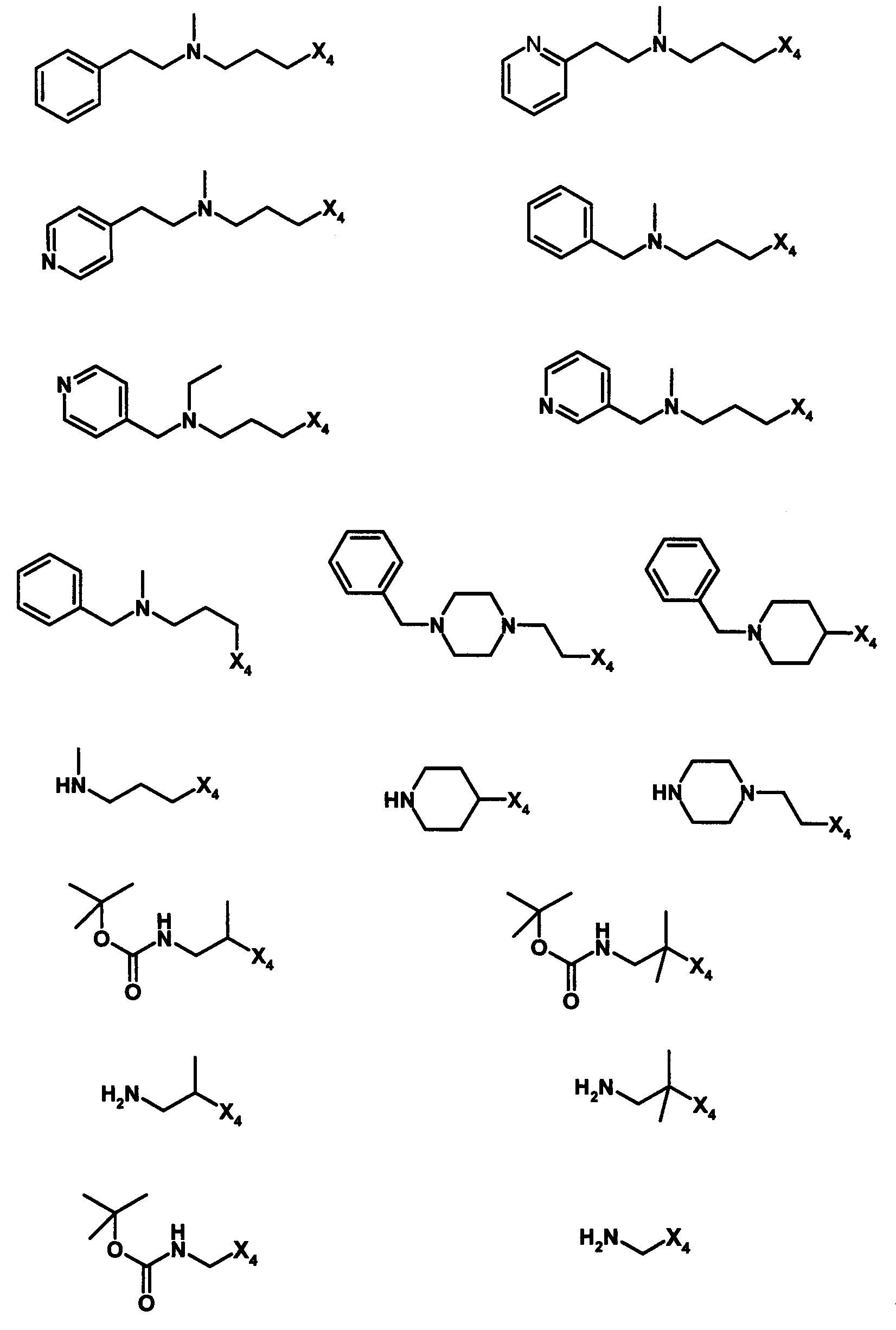
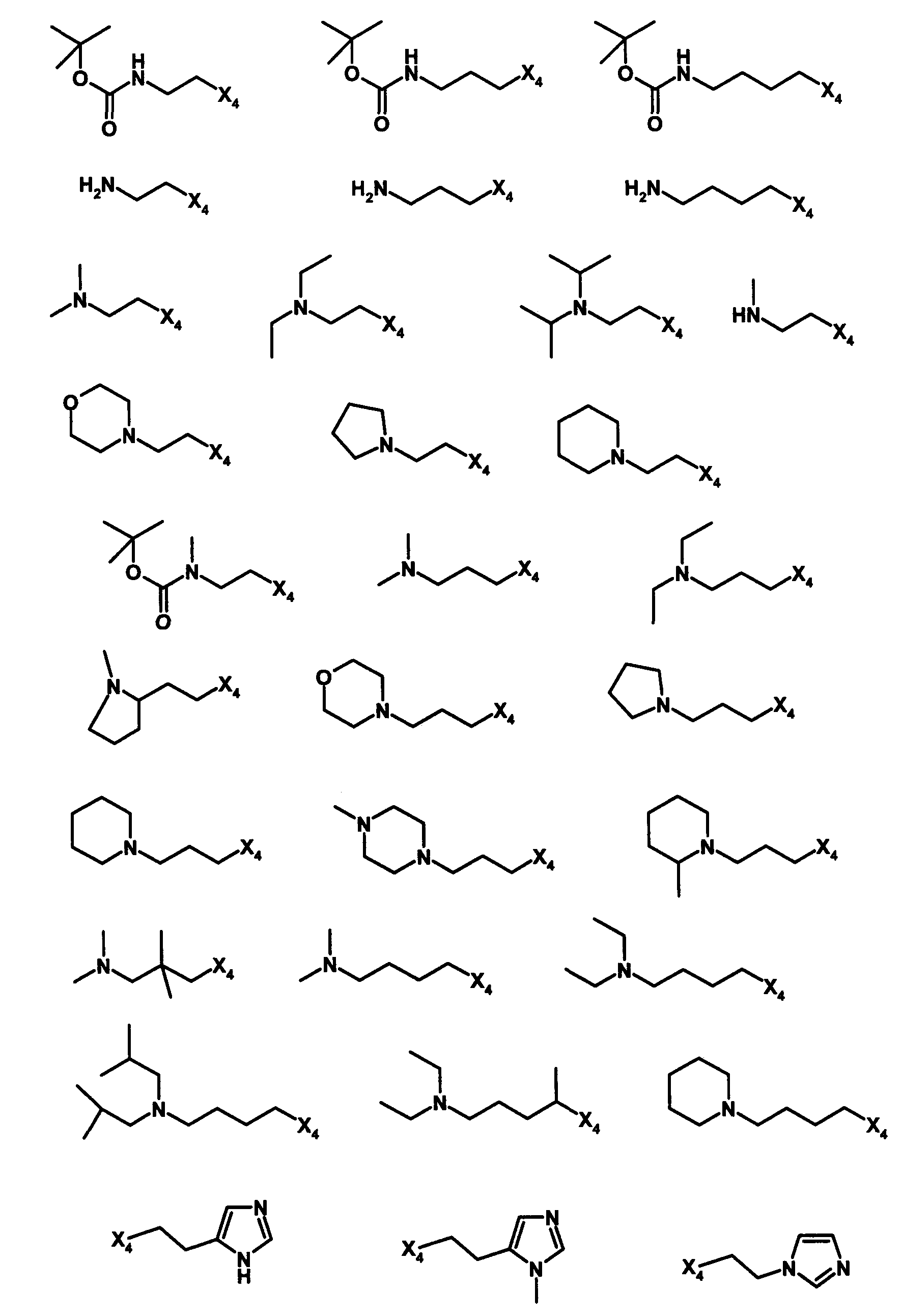
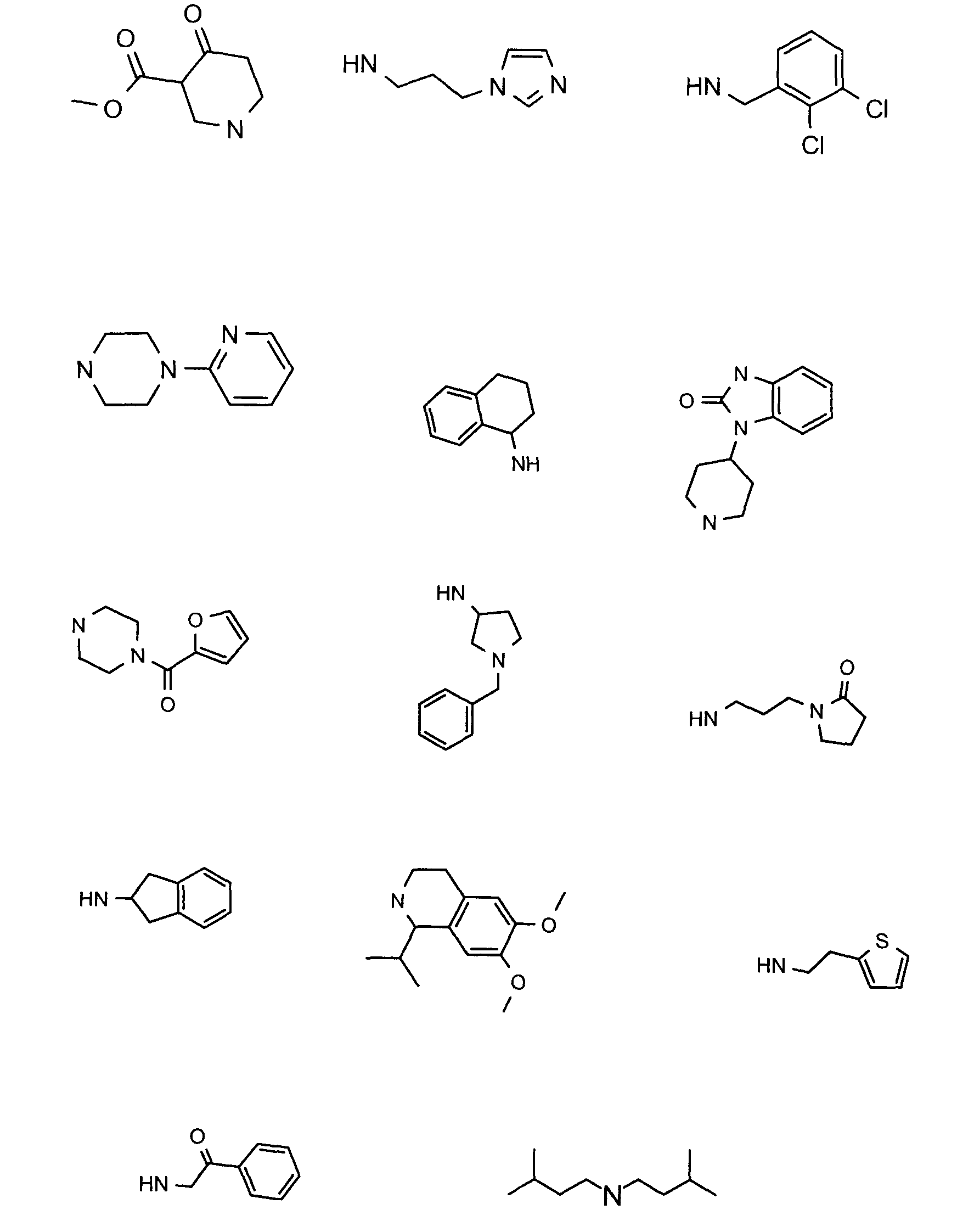
и R4 является одним из нижеприведенных радикалов (если R4 представляет собой радикал, содержащий на конце вторичный амин, например пропиламинометил, то соединения получали путем каталитического гидрирования в присутствии палладия на угле соответствующих N-бензильных производных; и если R4 представляет собой радикал, содержащий на конце первичный амин, например этиламино, то соединения получали обработкой соответствующего производного, защищенного третбутоксикарбонильной группой, кислотой).
B. Получение в соответствии co схемой реакции B:
Соединения формулы I по данному изобретению, где Y представляет собой -S-, и А представляет собой -C(O)-, могут быть получены в соответствии со следующей схемой B:
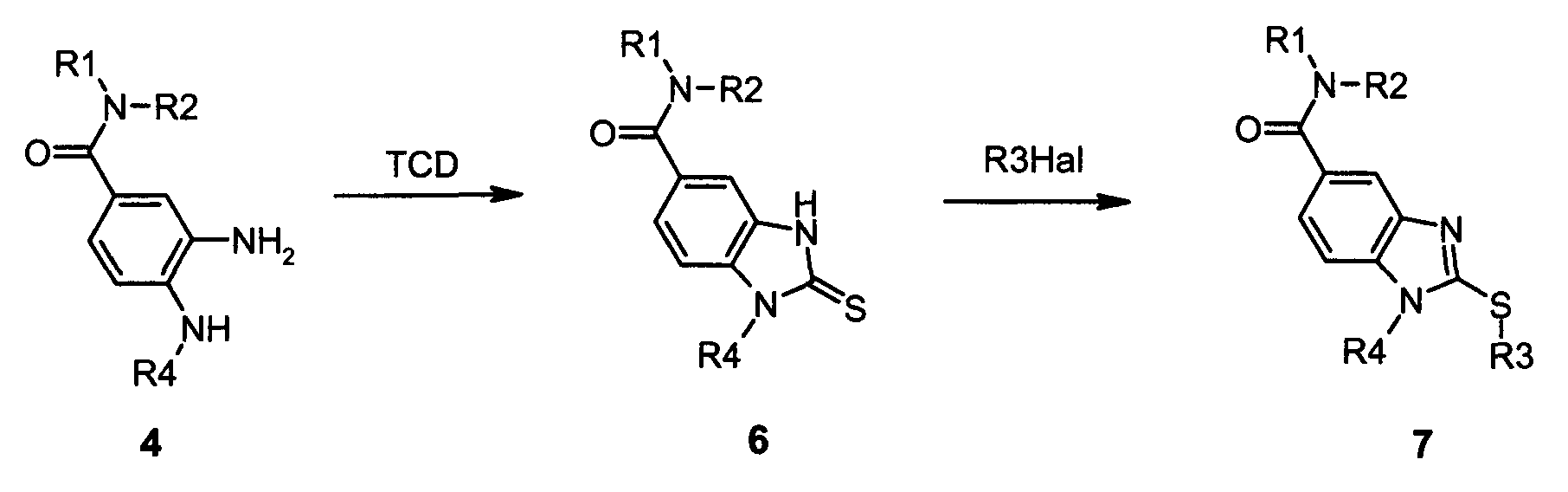
Как показано на схеме B, дианилин (4) может быть обработан тиокарбонилдиимидазолом (TCD) или тиофосгеном в инертном органическом растворителе, таком как тетрагидрофуран, метиленхлорид или хлороформ, при комнатной температуре в течение 2-17 часов с получением производного (6). Затем соединение (6) алкилировали взаимодействием галогенпроизводного, такого как алкил или бензил йодида, с бромидом или хлоридом или бромкетоном в присутствии третичного основания, такого как триэтиламин или диизопропилэтиламин, или в присутствии третичного основания на полимерной подложке, такого как морфолинометил полистирольная смола, в инертном органическом растворителе, таком как тетрагидрофуран, хлороформ или метиленхлорид, при температуре 20-70°C в течение 3-24 часов. Полученный тиобензимидазол (7) может быть выделен либо с помощью флэш-хроматографии на силикагеле либо добавлением к реакционной смеси нуклеофильного агента на полимерной подложке, такого как, например, аминометилполистирольная смола, и электрофильного агента на полимерной подложке, такого как, например, 4-бромметилфеноксиметилполистирольная смола, с последующей фильтрацией и упариванием фильтрата.
Пример B1: N, N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-{[2-оксо-2-(3,4,5-триметоксифенил)этил]тио}-1H-бензимидазол-5-карбоксамид
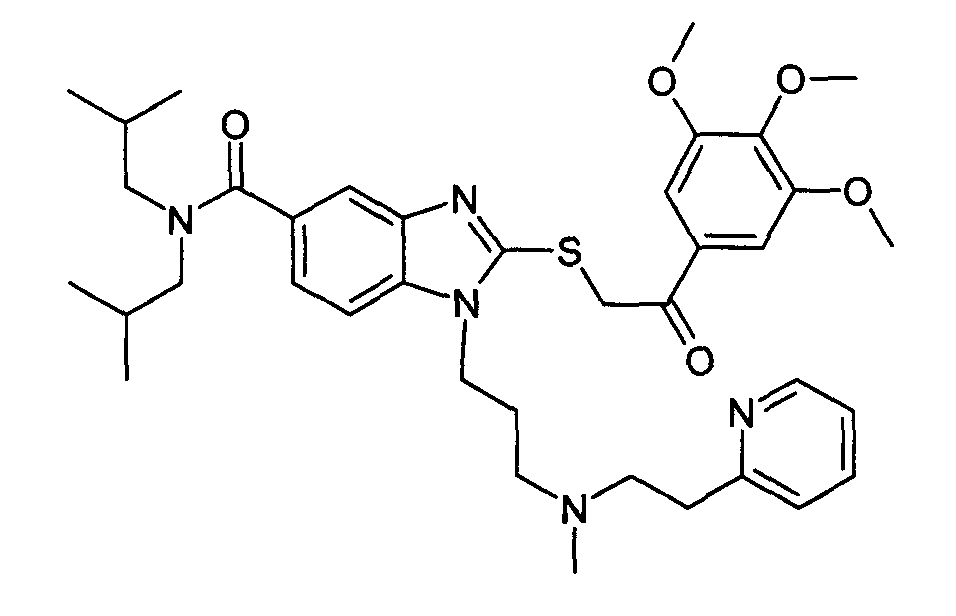
Стадия 1: N,N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-тиоксо-2, 3-дигидро-1H-бензимидазол-5-карбоксамид
Смесь 3-амино-N,N-диизобутил-4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)бензамида (1,52 г, 1 экв.) и тиокарбонилдиимидазола (0,74 г, 1,2 экв.) в ТГФ (30 мл) перемешивали при приблизительно 20°C в течение 15 часов. После концентрирования при пониженном давлении при 40°C полученный осадок помещали в дихлорметан (80 мл) и воду (30 мл). После декантирования и экстрагирования объединенные органические фазы промывали водным солевым раствором, сушили над Na2SO4, затем упаривали при пониженном давлении при 40°C. Очищение осадка с помощью флэш-хроматографии на силикагеле (элюент: от 100% дихлорметана до дихлорметана/метанола 8:2) давало ожидаемое соединение в виде светло-бежевой пены (1,2 г; 72%-ный выход).
MS/LC: вычисленный MW = 481,7; m/z = 482,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 2: N, N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-{[2-оксо-2-(3,4,5-триметоксифенил)этил]тио}-1H-бензимидазол-5-карбоксамид
К раствору N, N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-тиоксо-2,3-дигидро-1H-бензимидазол-5-карбоксамида в тетрагидрофуране последовательно добавляли морфолинометилполистирольную смолу (приобретена у Novabiochem, 2 экв.) и 2-бром-1-(3,4,5-триметоксифенил)этанон. Смесь перемешивали в течение 15 часов при приблизительно 20°C, затем добавляли тетрагидрофуран, аминометилполистирольную смолу (2 экв. приобретена у Novabiochem) и 4-бромметилфеноксиметилполистирольную смолу (3 экв. приобретена у Novabiochem). После перемешивания в течение 6 часов, смесь фильтровали на фрите. Фильтрат затем досуха концентрировали при пониженном давлении при 40°C с получением ожидаемого соединение.
MS/LC: вычисленный MW = 689,9; m/z = 690,5 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Нижеследующие соединения получали в соответствии co схемой реакции в и тем же способом, описанным для синтеза N, N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-[(3,4,5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамида:
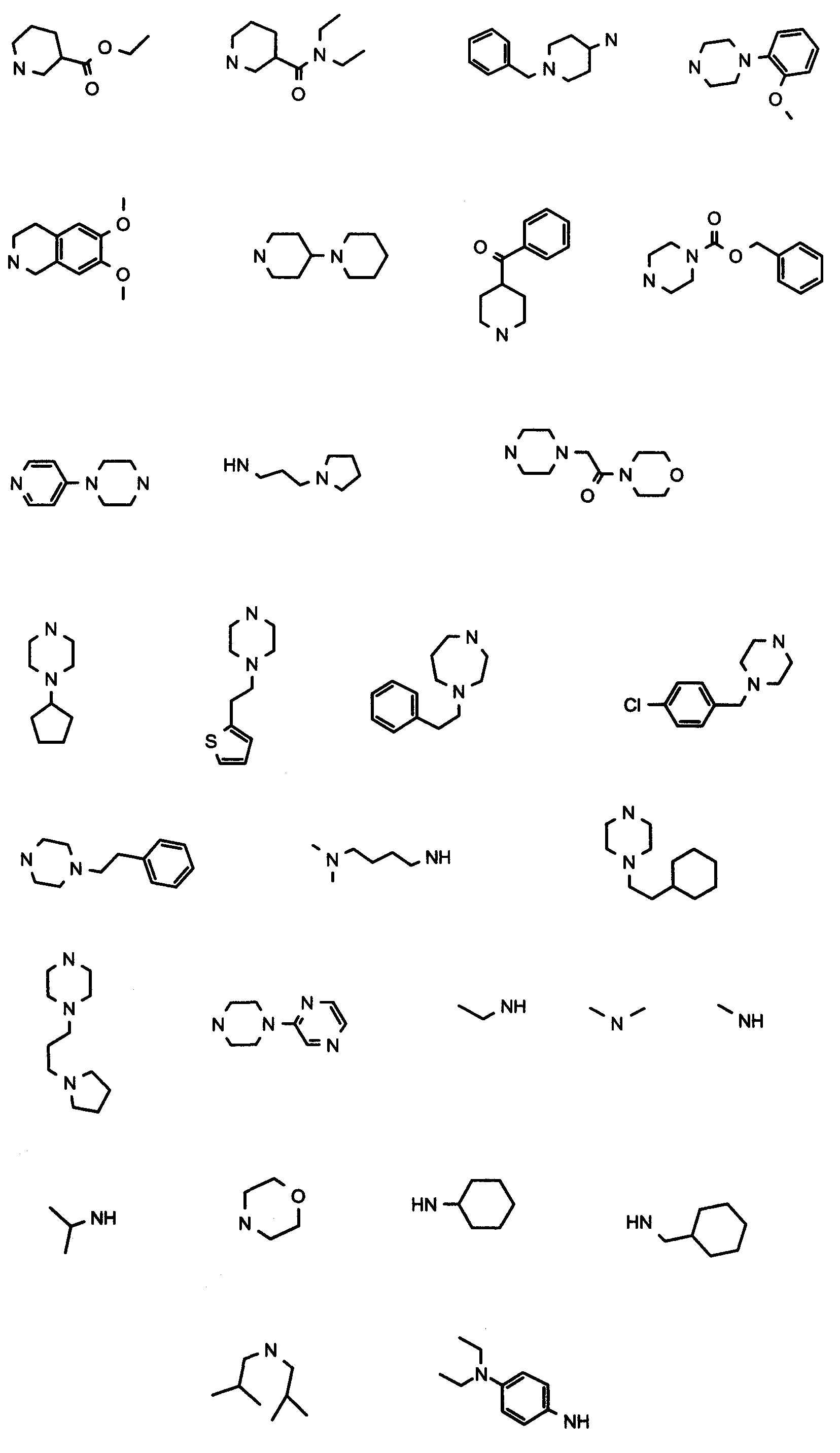
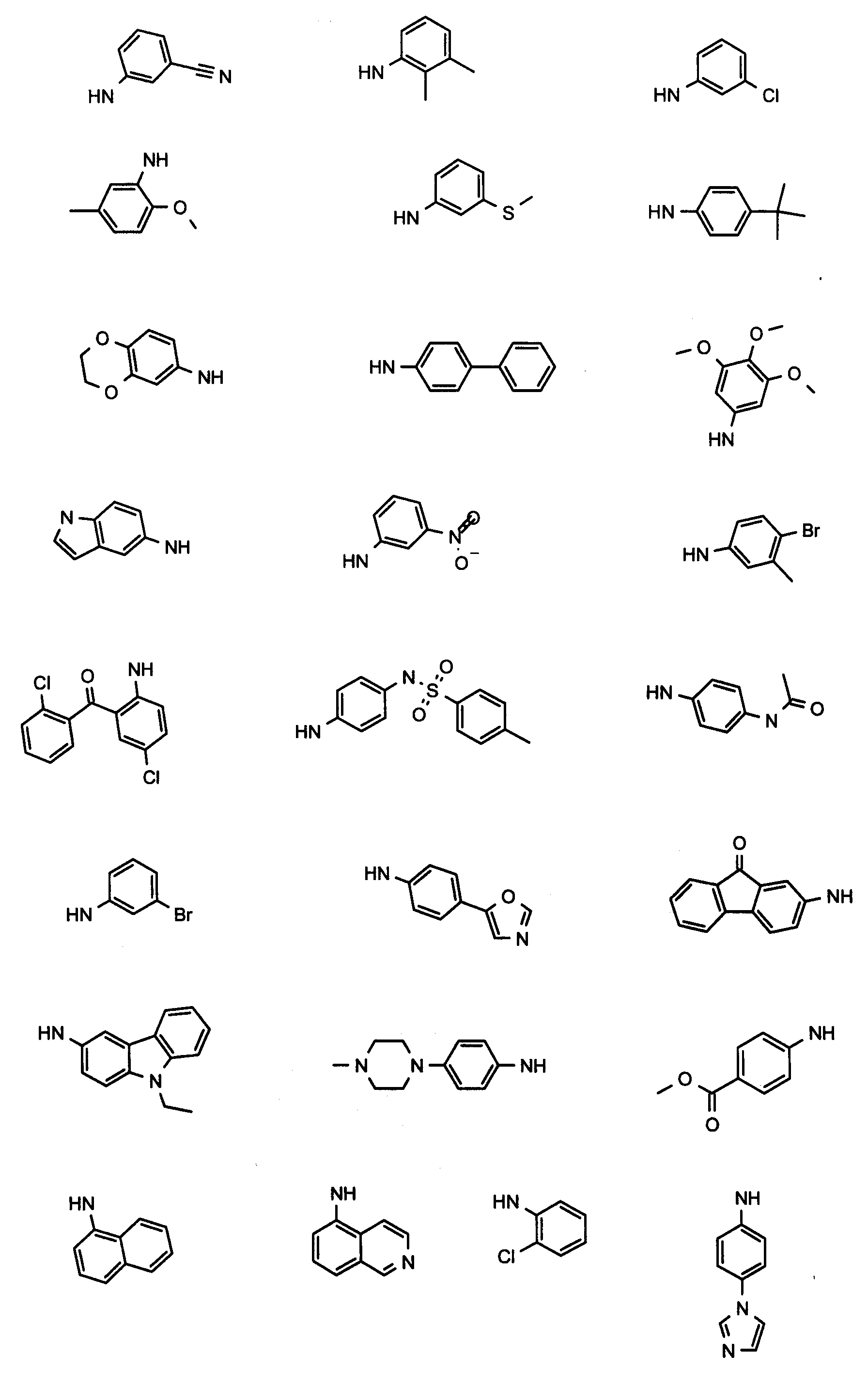
В вышеуказанной формуле R1,R2,N является одним из нижеприведенных радикалов:
R3 является одним из нижеприведенных радикалов
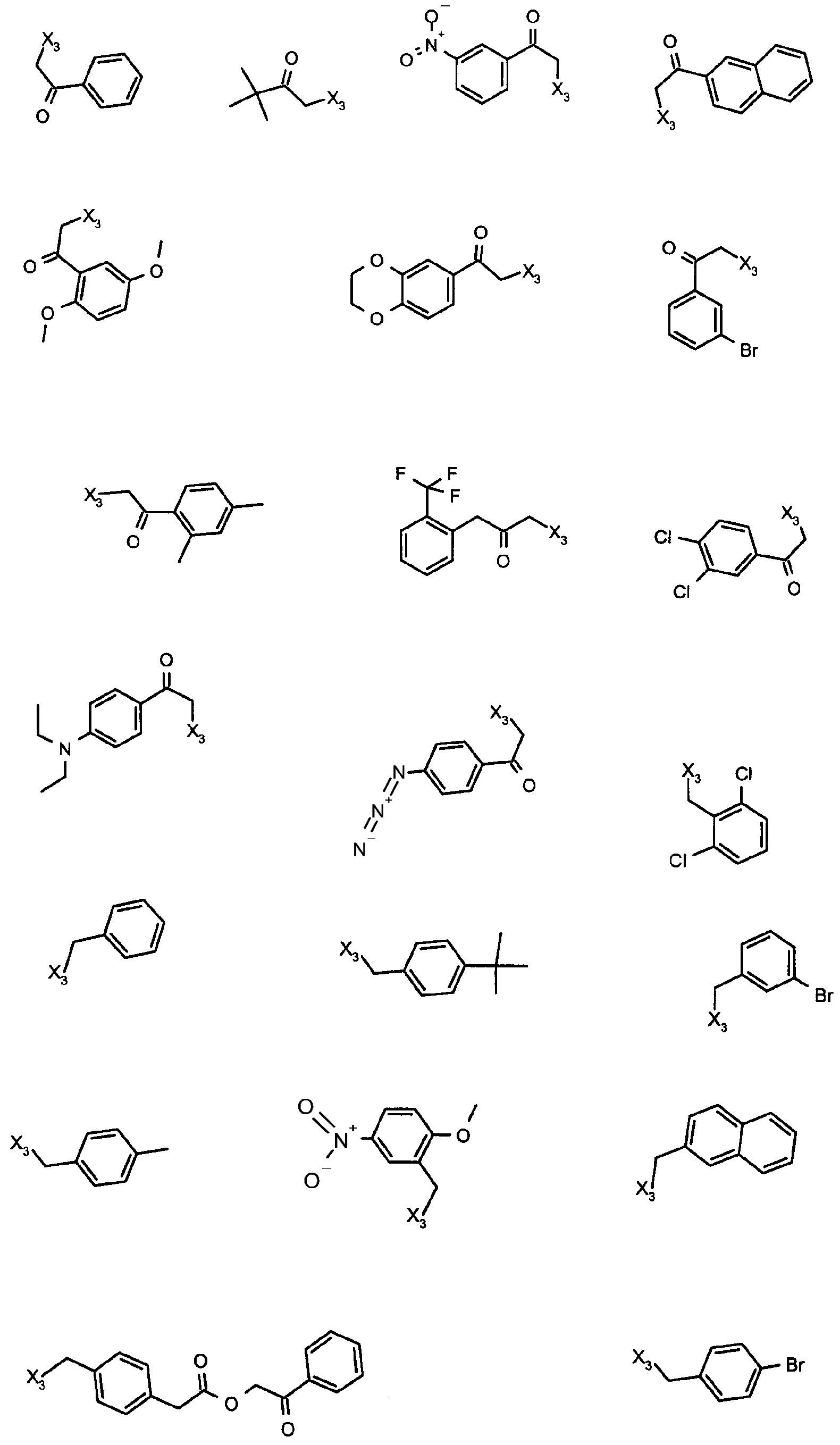
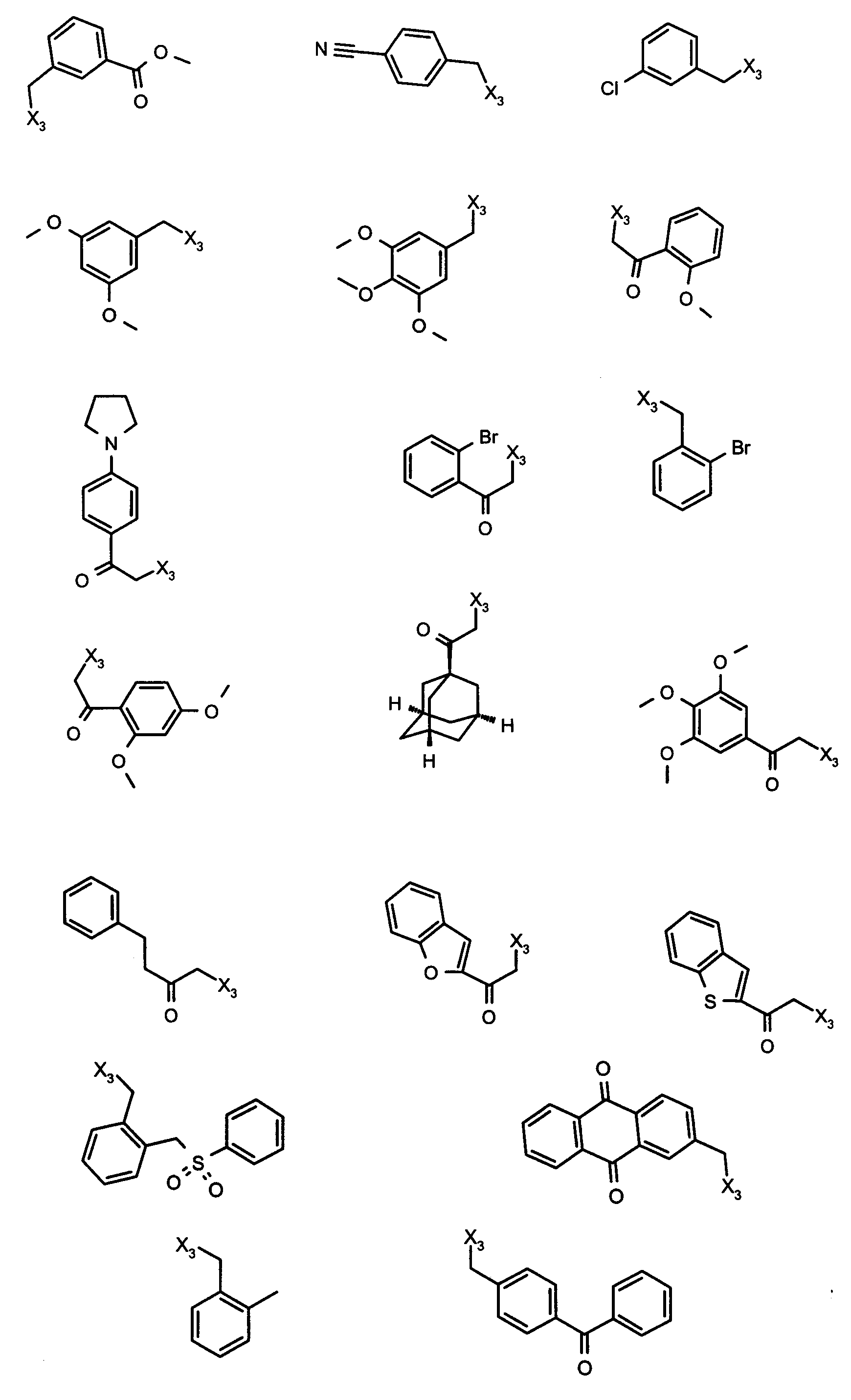
и R4 является одним из нижеприведенных радикалов
C. Получение в соответствии co схемой реакции C:
Соединения формулы I по настоящему изобретению, где Y представляет собой -NH-, и А представляет собой -C(O)-, могут быть получены в соответствии со следующей схемой C:
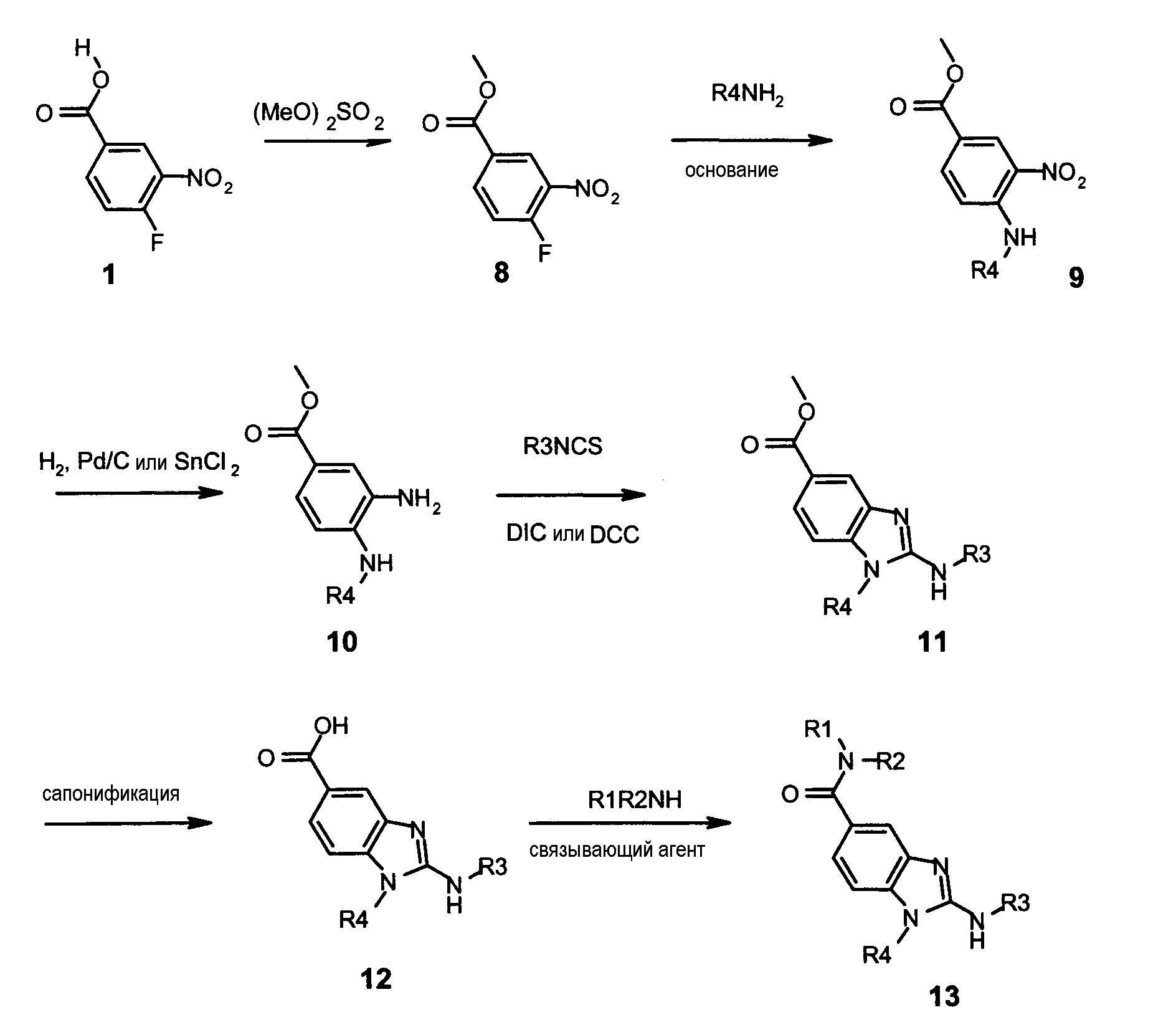
Как показано на схеме C, 4-фтор-3-нитробензойная кислота может быть преобразована в метиловый эфир (8) путем образования карбоксилатной соли, используя неорганическое основание, такое как дигидрат гидроксида лития или карбонат цезия, при комнатной температуре в течение от 30 минут до 2 часов, в инертном органическом растворителе, таком как тетрагидрофуран, с последующим добавлением диметилсульфата при комнатной температуре и перемешивании при кипячении с обратным холодильником в течение 5-15 часов. Фторированное производное (8) может быть обработано первичным амином в присутствии неорганического основания, такого как карбонат цезия или калий, в инертном органическом растворителе, таком как диметилформамид или ацетонитрил, при температуре 20-70°C в течение 2-16 часов с получением производного (9). Нитрогруппу соединения (9) восстанавливали обработкой дигидратом хлорида олова в инертном растворителе, таком как этилацетат или диметилформамид, при температуре 60-80°C в течение 3-15 часов, или с помощью каталитического восстановления в присутствии 10%-ного палладия на углероде в инертном растворителе, таком как метанол, этанол, этилацетат или смесь этих растворителей, при температуре 18-25°C в течение 2-8 часов с получением дианилина (10). Затем производное (10) обрабатывали изотиоцианатом в присутствии связывающего агента, такого как диизопропилкарбодиимид или дициклогексилкарбодиимид, в инертном растворителе, таком как тетрагидрофуран, метиленхлорид или хлороформ, при температуре 20-70°C в течение 2-72 часов с получением производного (11). Альтернативно производное (10) может быть обработано изотиоцианатом в инертном растворителе, таком как тетрагидрофуран, метиленхлорид или хлороформ, затем полученная тиомочевина может быть обработана с метил йодидом в полярном растворителе, таком как этанол, в течение 3-24 часов при температуре 20-70°C с получением (11). Метиловый эфир (11) может затем быть сапонирован в присутствии неорганического основания, такого как дигидрат гидроксида лития, в смеси полярных растворителей, таких как вода и тетрагидрофуран, при температуре 20-70°C в течение 3-17 часов. Полученная кислота (12) может быть соединена с первичным или вторичным амином в присутствии связывающего агента, такого как диизопропилкарбодиимид, дициклогексилкарбодиимид или карбонилдиимидазол, с или без 1-гидроксибензотриазола (HOBt) в инертном органическом растворителе, таком как метиленхлорид, тетрагидрофуран или диметилформамид, при комнатной температуре в течение 3-24 часов. Соответствующий амид (13) может быть выделен либо с помощью флэш-хроматографии на силикагеле, либо добавлением к реакционной смеси нуклеофильного реагента на полимерной подложке, такого как, например, аминометилполистирольная смола, и электрофильного агента на полимерной подложке, такого как, например, метилизотиоцианатполистирольная смола, с последующей фильтрацией и упариванием фильтрата.
Пример C1: пропан-1-он 1-(2-[(3,5-диметоксифенил)амино]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-ил)-3-тиен-2-ила
Стадия 1: метил 4-фтор-3-нитробезоат
Моногидрат гидроксида лития (4,5 г, 1 экв.) маленькими порциями добавляли к раствору 4-фтор-3-нитробензойной кислоты (20 г, 1 экв.) в тетрагидрофуране (100 мл). После перемешивания в течение 1 часа при приблизительно 20°C к желтому осадку по каплям добавляли диметилсульфат (10,2 мл). Реакционную смесь затем кипятили с обратным холодильником в течение 8 часов, затем концентрировали при пониженном давлении при 40°C. Осадок растворяли в дихлорметане и воде, насыщенной Na2CO3. После декантирования и экстракции объединенные органическом фазы промывали водном солевым раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении при 40°C. Полученный желтый твердый продукт перекристаллизовывали смесью диэтилового эфира/петролейного эфира с получением ожидаемого соединения в виде светло-желтого порошка (16,7 г, 78%-ный выход). Точка плавления = 59°C.
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 2: метил 4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)-3-нитро безоат
Смесь метил 4-фтор-3-нитробезоата (5,08 г, 1 экв.), N-(2-пиридин-2-илэтил)пропан-1,3-диамина (5,4 г, 1,2 экв.) и карбоната калия (7,0 г, 2 экв.) в ацетонитриле (180 мл) кипятили с обратным холодильником в течение 3 часов, затем концентрировали при пониженном давлении при 40°C. осадок is помещали в дихлорметан (150 мл) и воду (60 мл). После декантирования и экстракции объединенные органическом фазы промывали водным солевым раствором, сушили над Na2SO4, затем упаривали при пониженном давлении при 40°C. Очистка соединения с помощью флэш-хроматографии на силикагеле (элюент: от дихлорметана до дихлорметана/метанола 9:1) давала ожидаемое соединение в виде оранжевого масла (9,2 г; 97%-ный выход).
MS/LC: вычисленный MW = 372,4; m/z = 373,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 3: метил 3-амино-4-({3-[метил(2-пиридин-2-илметил)амино]пропил}амино) безоат
Метил 4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)-3-нитробезоат (9,1 г) в растворе в смеси этилацетата/метанола и 10% палладий на угле (910 мг) добавляли при автоклавировании. После перемешивания в течение 4 часов в атмосфере водорода (3 бара), катализатор удаляли фильтрацией на силикагеле и фильтрат концентрировали при пониженном давлении при 40°C с получением ожидаемого соединения в виде масла (8,2 г, 98%-ный выход).
MS/LC: вычисленный MW = 342,4; m/z = 343,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 4: метил-2-[(3, 5-диметоксифенил)амино]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоксилат
3,5-диметоксифенилизотиоцианат (571 мг, 1 экв.) и диизопропилкарбодиимид (1,35 мл, 4 экв.) последовательно добавляли к раствору метил 3-амино-4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)безоата (1,0 г, 1 экв.) в тетрагидрофуране (10 мл). смесь кипятят с обратным холодильником в течение 18 часов, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении при 40°C, осадок помещали в этилацетат (100 мл) и воду (40 мл). После декантирования и экстракции объединенные органическом фазы промывали водным солевым раствором, сушили над Na2SO4, затем упаривали при пониженном давлении при 40°C. Очистка осадока с помощью флэш-хроматографии на силикагеле (элюент: дихлорметан/метанол от 99:1 до 98:2) давала ожидаемое соединение в виде бежевой пены (1,12 г; 76%-ный выход).
MS/LC: вычисленный MW = 503,6; m/z = 504,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 5: 2-[(3, 5-диметоксифенил)амино]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоновая кислота
Гидроксид лития (0,350 г, 4 экв.) добавляли к раствору метил-2-[(3, 5-диметоксифенил)амино]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоксилата (1,05 г, 1 экв.) в смеси тетрагидрофурана (10 мл) и воды (5 мл). Смесь перемешивали при 65°C в течение 18 часов, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении при 40° C. К осадку добавляли этилацетат и воду. Смесь подкисляли, добавляя уксусную кислоту до pH 5. После декантирования и экстракции объединенные органические фазы сушили над сульфатом натрий и концентрировали при пониженном давлении. Очистка с помощью флэш-хроматографии на силикагеле (элюент: дихлорметан/этанол 95/5-70/30) давала ожидаемое соединение в виде белой пены (0,93 г, 91%-ный выход).
MS/LC: вычисленный MW = 489,6; m/z = 490,1 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 6: 1-(2-[(3, 5-диметоксифенил)амино]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-ил)-3-тиен-2-илпропан-1-он
К раствору 2-[(3, 5-диметоксифенил)амино]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоновой кислоты (24 мг, 1 экв.) в смеси диметилформамида (0,2 мл) и тетрагидрофурана (0,4 мл) добавляли карбонилдиимидазол (10,5 мг, 1,3 экв.) в растворе с хлороформом (0,2 мл). Смесь перемешивали в течение 15 часов при приблизительно 20°C, затем добавляли тиофен-2-этиламин (13 мг, 2 экв.) в растворе с тетрагидрофураном (0,1 мл). После перемешивания в течение 15 часов при приблизительно 20°C к смеси растворенной в дихлорметане, добавляли аминометил полистирольную смолу (2 экв.), TBD-метил полистирольную смолу (2 экв.) и метилизотиоцианат полистирольную смолу (4 экв.). После перемешивания в течение 6 часов при приблизительно 20 C смесь фильтровали и фильтрат концентрировали при пониженном давлении при 40°C с получением ожидаемого соединения в виде масла (27 мг, 90%-ный выход).
MS/LC: вычисленный MW = 598,8; m/z = 599,2 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Нижеследующие соединения получали в соответствии co схемой реакции С и тем же способом, описанным для синтеза 1-(2-[(3, 5-диметоксифенил)амино]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-ил)-3-тиен-2-илпропан-1-она:
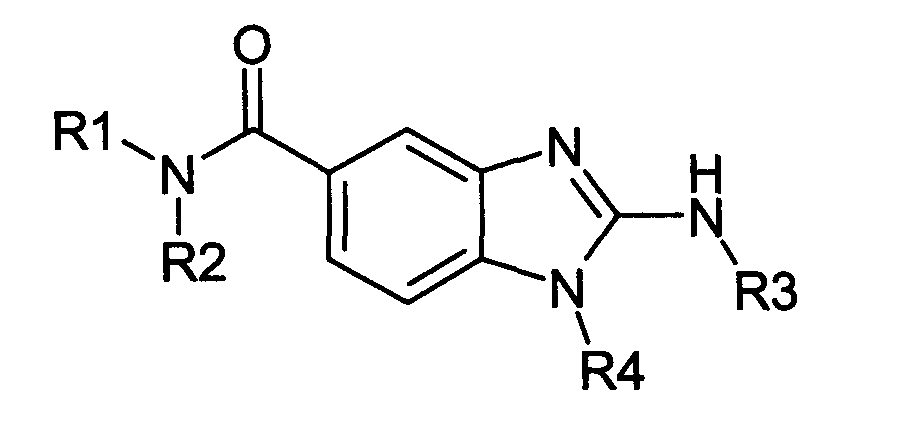
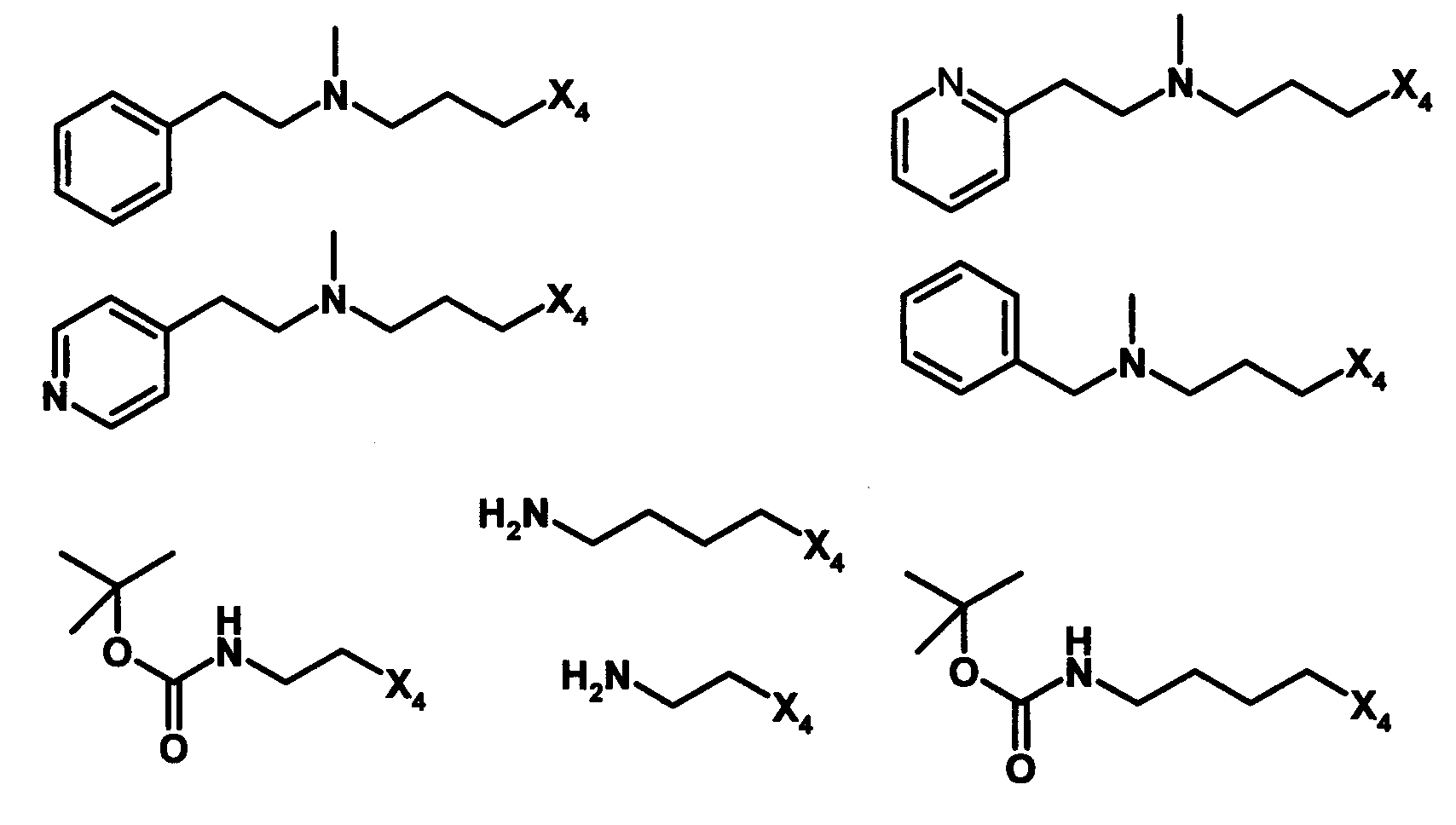
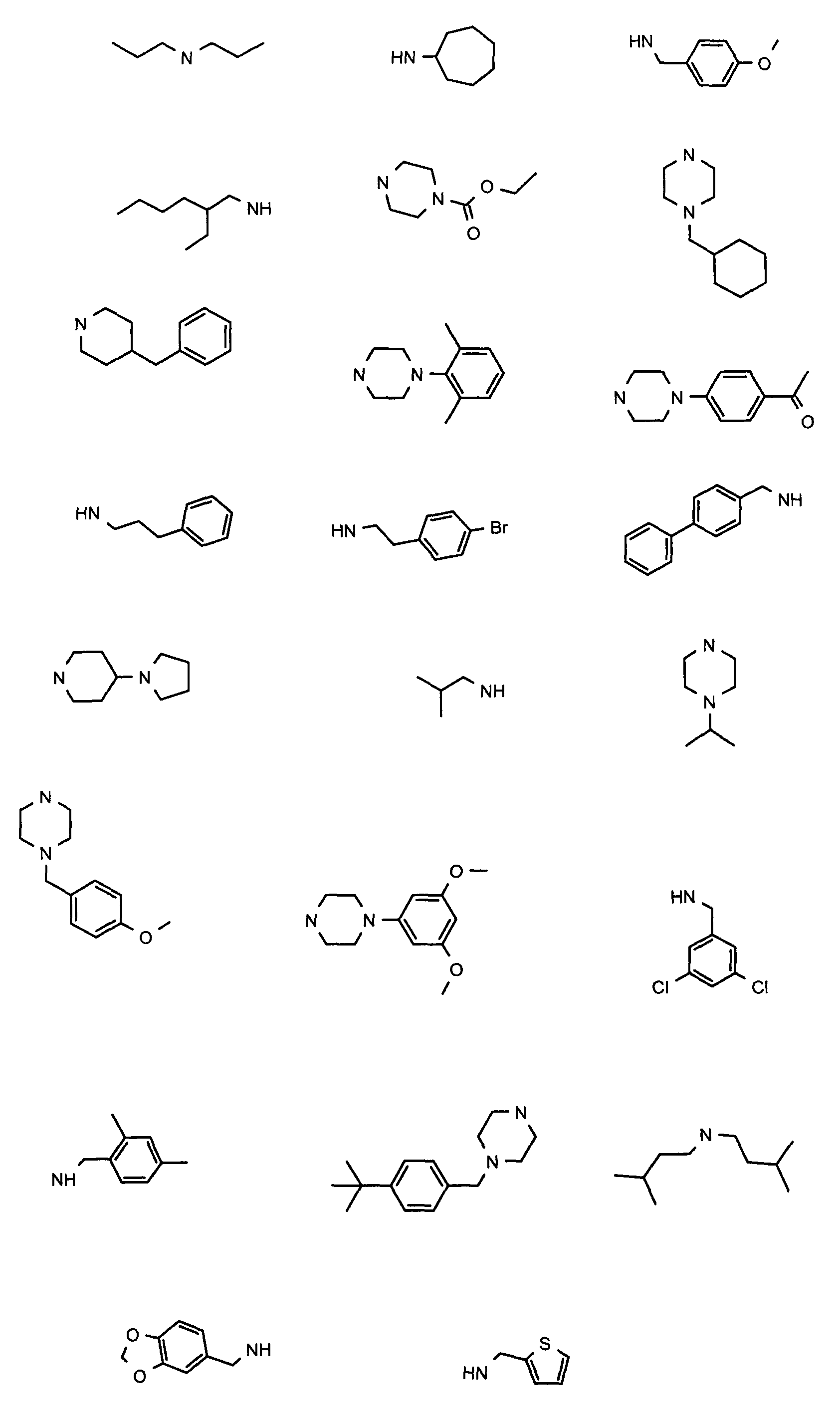
В вышеуказанной формуле, R1,R2,N представляет собой один из нижеприведенных радикалов:
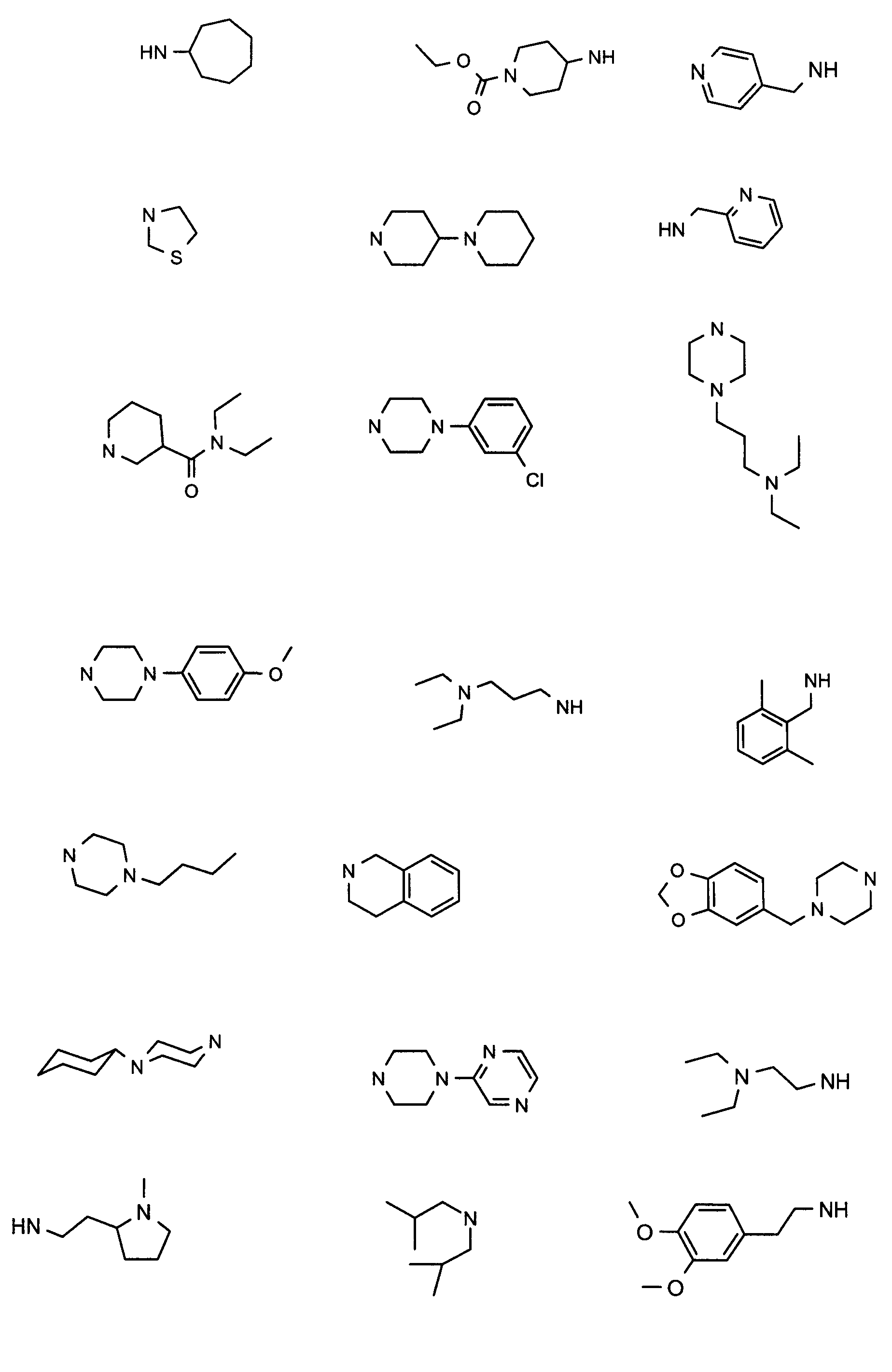
R3 является одним из нижеприведенных радикалов:
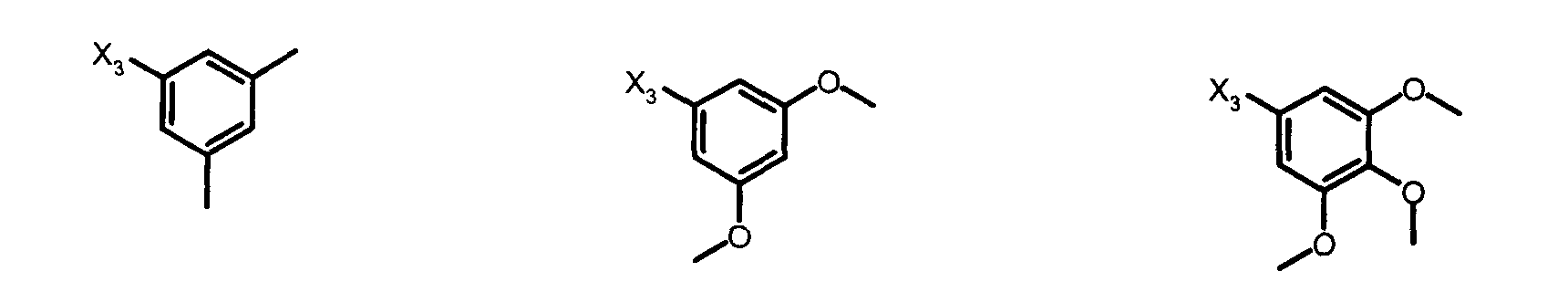
и R4 является одним из нижеприведенных радикалов:
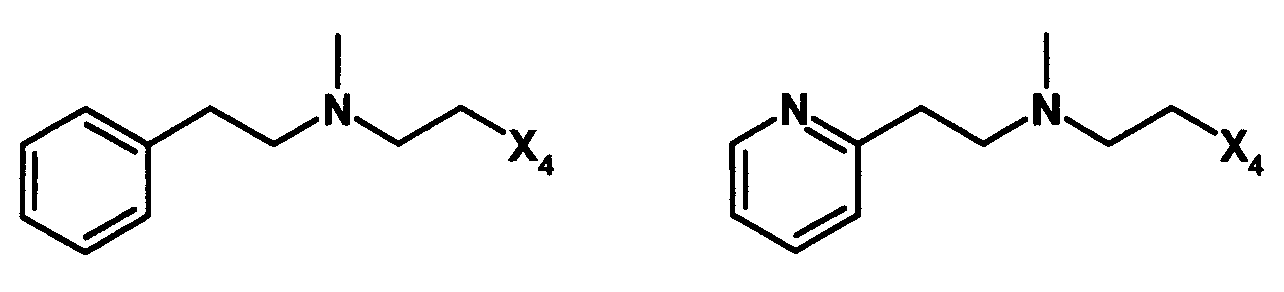
D. Получение в соответствии co схемой реакции D:
Соединения формулы I в соответствии с данным изобретением, где Y представляет собой -S-, и А представляет собой -C(O)-, могут быть получены в соответствии со следующей схемой D:
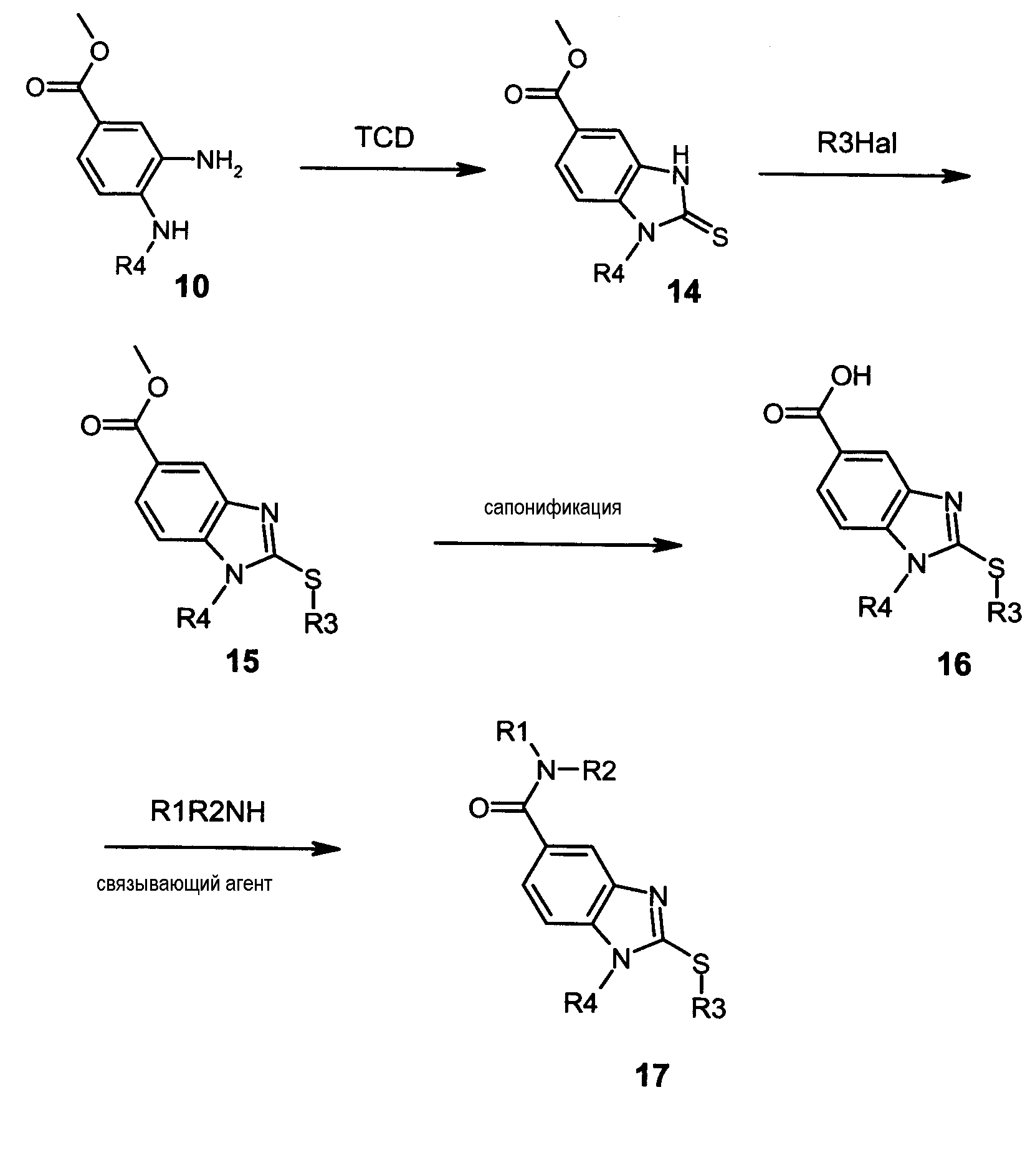
Как показано на схеме D, дианилин (10) может быть обработан тиокарбонилдиимидазолом (TCD) или тиофосгеном в инертном органическом растворителе, таком как тетрагидрофуран, при комнатной температуре в течение 2-17 часов с получением производного (14). Соединение (14) затем алкилировали путем взаимодействия с галогенированным производным, таким как алкил или бензил йодид, бромид или хлорид или бромкетон, в присутствии третичного основания, такого как триэтиламин или диизопропилэтиламин, в инертном органическом растворителе, таком как тетрагидрофуран, хлороформ или метиленхлорид, при температуре 20-70°C в течение 3-24 часов с получением тиобензимидазольного производного (15). Затем метиловый эфир (15) может быть сапонирован в присутствии неорганического основания, такого как моногидрат гидроксида лития, в смеси полярных растворителей, таких как вода и тетрагидрофуран, при температуре 20-70°C в течение 3-17 часов. Полученная кислота (16) может быть соединена с первичным или вторичным амином в присутствии связывающего агента, такого как диизопропилкарбодиимид, дициклогексилкарбодиимид или карбонилдиимидазол, с или без 1-гидроксибензотриазола (HOBt) в инертном органическом растворителе, таком как метиленхлорид, тетрагидрофуран или диметилформамид, при комнатной температуре в течение 3-24 часов. Соответствующий амид (17) может быть выделен либо с помощью флэш-хроматографии на силикагеле либо добавлением к реакционной смеси нуклеофильного агента на полимерной подложке, такого как, например, аминометилполистирольная смола, и электрофильного агента на полимерной подложке, такого как, например, метилизотиоцианатполистирольная смола, с последующим фильтрованием и упариванием фильтрата.
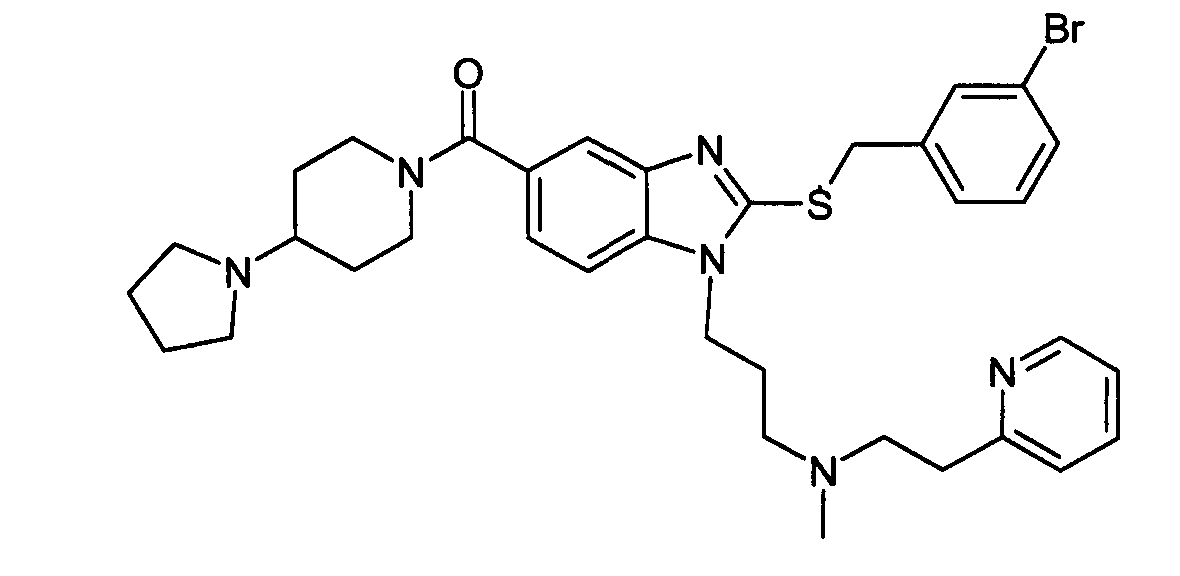
Пример D1: 3-(2-[(3-бромбензил)сульфанил]-5-{[4-(1-пирролидинил)-1-пиперидинил]карбонил}-1H-бензимидазол-l-ил)-N-метил-N-[2-(2-пиридинил)этил]-1-пропанамин
Стадия 1: метил 1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-тиоксо-2, 3-дигидро-1H-бензимидазол-5-карбоксилат
Смесь метил 3-амино-4-({3-[метил(2-пиридин-2-илэтил)амино]пропил}амино)безоата (4,09 г, 1 экв.) и тиокарбонилдиимидазола (2,77 г, 1,3 экв.) в тетрагидрофуране (100 мл) перемешивали при приблизительно 20°C в течение 15 часов. После концентрирования при пониженном давлении при 40°C, полученный осадок помещали в дихлорметан (150 мл) и воду (50 мл). После декантирования и экстракции объединенные органические фазы промывали водным солевым раствором, сушили над Na2SO4, затем упаривали при пониженном давлении при 40°C. Очистка с помощью флэш-хроматографии на силикагеле (элюент: от 100% дихлорметан до дихлорметана/метанола 9:1) давала ожидаемое соединение в виде пены (3,94 г; 85%-ный выход).
MS/LC: вычисленный MW = 384,5; m/z = 385,2 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 2: метил 2-[(3-бромбензил)тио]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоксилат
Триэтиламин (0,82 мл, 1,6 экв.) и 3-бромбензилбромид (0,97 г, 1 экв.) последовательно добавляли к раствору метил 1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-тиоксо-2,3-дигидро-1H-бензимидазол-5-карбоксилата (1,5 г) в тетрагидрофуране (30 мл). Смесь перемешивали в течение 15 часов при приблизительно 20°C, затем концентрировали при пониженном давлении при 40°C. Полученный осадок растворяли в этилацетате и воде. После декантирования и экстракции органическую фазу промывали водным солевым раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении при 40°C. Очистка с помощью флэш-хроматографии на силикагеле (элюент: дихлорметан/метанол от 95/5 до 90/10) давала ожидаемое соединение в виде бесцветного масла (1,5 г; 70%-ный выход).
MS/LC: вычисленный MW = 553,5; m/z = 553,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 3: 2-[(3-бромбензил)тио]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоновая кислота
Гидроксид лития (0,315 г, 3 экв.) добавляли к раствору метил 2-[(3-бромбензил)тио]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоксилата (1,03 г, 1 экв.) в смеси тетрагидрофурана (10 мл) и воды (5 мл). Смесь кипятили с обратным холодильником в течение 18 часов, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении при 40° C. К осадку добавляли этилацетат и воду. Смесь подкисляли, добавляя уксусную кислоту, до pH 5. После декантирования и экстракции объединенные органические фазы сушили над сульфатом натрия и концентрировали при пониженном давлении. Очистка с помощью флэш-хроматографии на силикагеле (элюент: дихлорметан/метанол от 95/5 до 80/20) давала ожидаемое соединение в виде пены (0,85 г, 85%-ный выход).
MS/LC: вычисленный MW = 539,5; m/z = 539,2 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 4: 3-(2-[(3-бромбензил)сульфанил]-5-{[4-(1-пирролидинил)-1-пиперидинил]карбонил}-1H-бензимидазол-1-ил)-N-метил-N-[2-(2-пиридинил)этил]-1-пропанамин
К раствору 2-[(3-бромбензил)тио]-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоновой кислоты (27 мг, 1 экв.) в смеси диметилформамида (0,2 мл) и тетрагидрофурана (0,4 мл) добавляли карбонилдиимидазол (10,5 мг, 1,3 экв.) в растворе с хлороформом (0,2 мл). Смесь перемешивали в течение 15 часов при приблизительно 20°C, затем добавляли 4-(1-пирролидинил)пиперидин (15 мг, 2 экв.). После перемешивания в течение 15 часов при приблизительно 20°C к смеси разбавленной в дихлорметане, добавляли аминометил полистирольную смолу (2 экв. приобретена у Novabiochem), TBD-метил полистирольную смолу (2 экв. приобретена у Novabiochem) и метилизотиоцианат полистирольную смолу (4 экв. приобретена у Novabiochem). После перемешивания в течение 6 часов при приблизительно 20°C смесь фильтровали, и фильтрат концентрировали при пониженном давлении при 40°C с получением ожидаемого соединения в виде масла (28 мг, 84%-ный выход).
MS/LC: вычисленный MW = 675,7; m/z = 674,2 (MH+)
ЯМР(1H, 400 МГц, CDCl3):
Нижеследующие соединения получали в соответствии co схемой реакции D и тем же способом, описанным для синтеза 3-(2-[(3-бромбензил)сульфанил]-5-{[4-(1-пирролидинил)-1-пиперидинил]карбонил}-1H-бензимидазол-1-ил)-N-метил-N-[2-(2-пиридинил)этил]-1-пропанамин:
В вышеприведенной формуле, R1,R2,N представляет собой один из нижеприведенных радикалов:
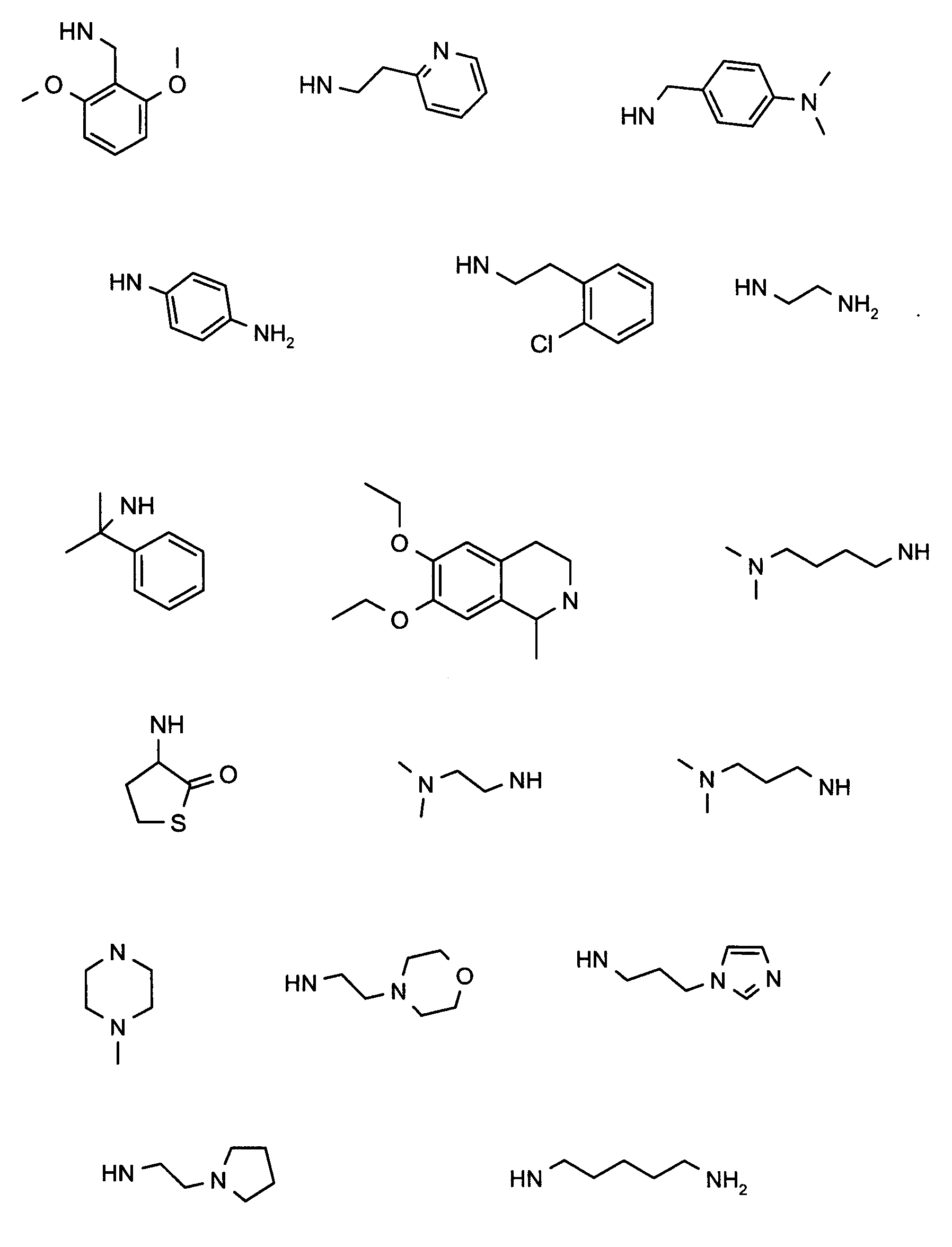
R3 является одним из нижеприведенных радикалов:
и R4 является одним из нижеприведенных радикалов:
E. Получение в соответствии co схемой реакции E:
Соединения формулы I по данному изобретению, где А представляет собой -(CH2)-, могут быть получены из соединений, где А представляет собой -C(O)-, в соответствии со следующей схемой E:
Как показано на схеме E, амид (18), полученный в соответствии с реакционными схемами А или B, может быть восстановлен до соответствующего амина (19), используя боран или алюмогидрид лития в апротонном растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температуре от 0 до 70°C, в течение 1-6 часов.
Пример E1: 5-[(диизобутиламино)метил]-1-(3-{метил[2-(2-пиридинил)этил]амино}пропил)-N-(3,4, 5-триметоксифенил)-1H-бензимидазол-2-амин
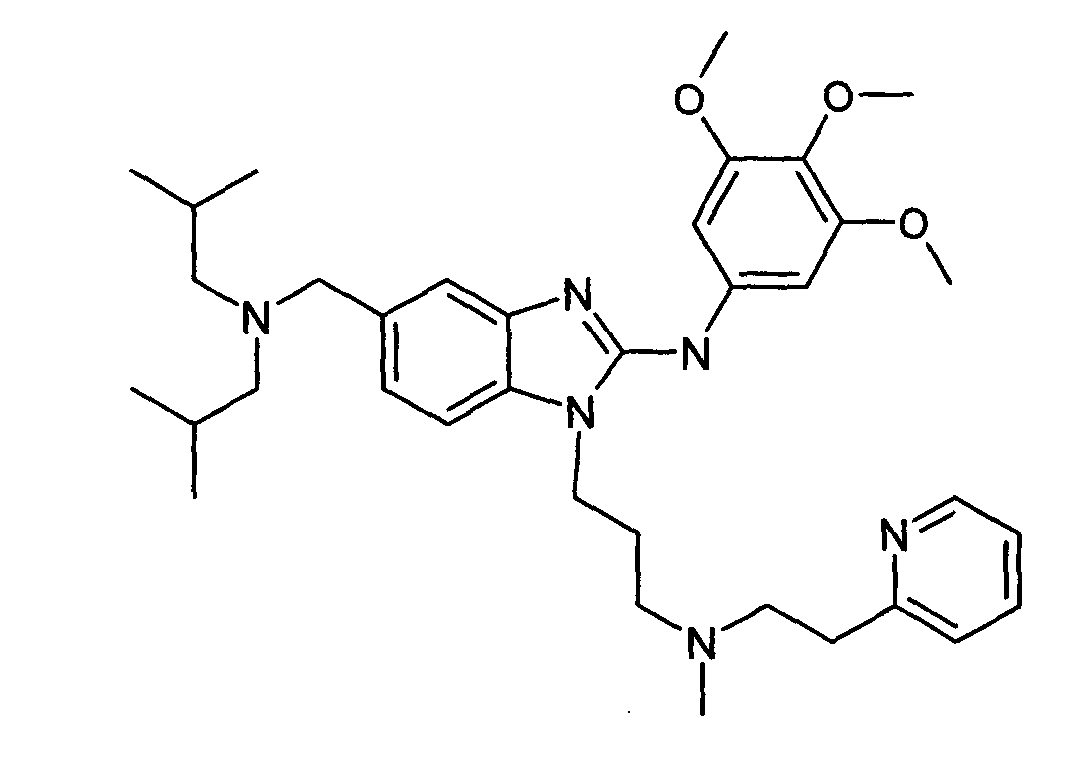
Молярный раствор алюмогидрида лития в тетрагидрофуране (0,83 мл, 5 экв.) добавляли по каплям к раствору N,N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-[(3,4,5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамида (105 мг, 1 экв., получен в соответствии с примером A1), охлажденному до 0°С в тетрагидрофуране (3 мл). После перемешивания в течение 15 минут при 0°C, смесь нагревали при 60°C в течение 3 часов, затем охлаждали до 0°C и гидролизовали. После добавления этилацетата, декантирования и экстракции объединенные органические фазы промывали водным солевым раствором, затем сушили над сульфатом натрия и концентрировали при пониженном давлении. Очистка с помощью флэш-хроматографии на силикагеле (элюент: от 100% дихлорметана до дихлорметана/метанола 9:1) давала ожидаемое соединение в виде пены (63 мг, 62%-ный выход).
MS/LC: вычисленный MW = 616,8; m/z = 617,4 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
F. Получение в соответствии co схемой реакции F:
Соединения формулы I по данному изобретению, где Y представляет собой -S- и -NH-, и А представляет собой -CH2 -, могут быть получены в соответствии со следующей схемой F:
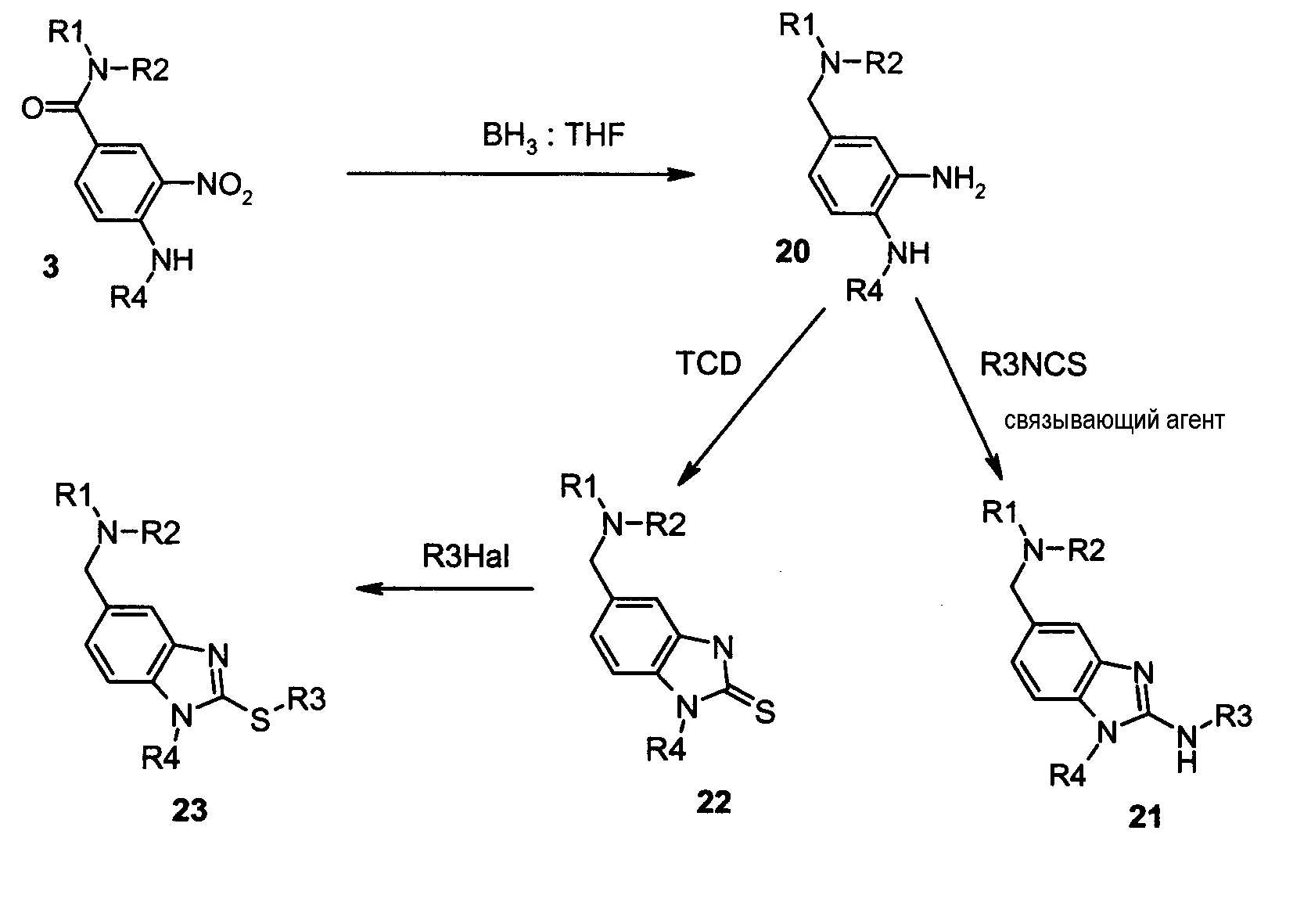
Как показано на схеме F, производное (3) может быть восстановлено до соединения (20), используя боран в апротонном растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температуре от 0 до 70°C в течение 18-24 часов. Затем дианилин (20) может быть обработан изотиоцианатом в присутствии связующего агента без или на полимерной подложке, такого как диизопропилкарбодиимид или дициклогексилкарбодиимид, или N-метилциклогексилкарбодиимид N-метил полистирольная смола, в инертном растворителе, таком как тетрагидрофуран, метиленхлорид или хлороформ, при температуре 20-70° C в течение 2-72 часов с получением производного (21). Альтернативно производное (4) может быть обработано изотиоцианатом в инертном растворителе, таком как тетрагидрофуран, метиленхлорид или хлороформ, затем полученная тиомочевина может быть обработана метилйодидом в полярном растворителе, таком как этанол, в течение 3-24 часов при температуре 20-70°C с целью получения соединения (21).
Как также показано на схеме реакции B и в примере B1, дианилин (20) может быть обработан тиокарбонилдиимидазолом (TCD) или тиофосгеном в инертном органическом растворителе, таком как тетрагидрофуран, метиленхлорид или хлороформ, при комнатной температуре в течение 2-17 часов с получением производного (22). Соединение (22) затем алкилировали путем взаимодействия с галогенированным производным, таким как алкил- или бензилйодид, бромид, или хлорид, или бромкетон, в присутствии третичного основания, такого как триэтиламин или диизопропилэтиламин, или в присутствии третичного основания по полимерной подложке, такого как морфолинометилполистирольная смола, в инертном органическом растворителе, таком как тетрагидрофуран, хлороформ или метиленхлорид, при температуре 20-70°C в течение 3-24 часов. Полученное тиобензимидазольное производное (23) может быть выделено либо на силикагеле, либо добавлением к реакционной смеси нуклеофильного агента на полимерной подложке, такого как, например, аминометилполистирольная смола, и электрофильного агента на полимерной подложке, такого как, например, 4-бромметилфеноксиметилполистирольная смола, с последующей фильтрацией и упариванием фильтрата.
Пример F1: 5-[(диизобутиламино)метил]-1-(3-{метил[2-(2-пиридинил)этиламино}пропил)-N-(3,4, 5-триметоксифенил)-1H-бензимидазол-2-амин
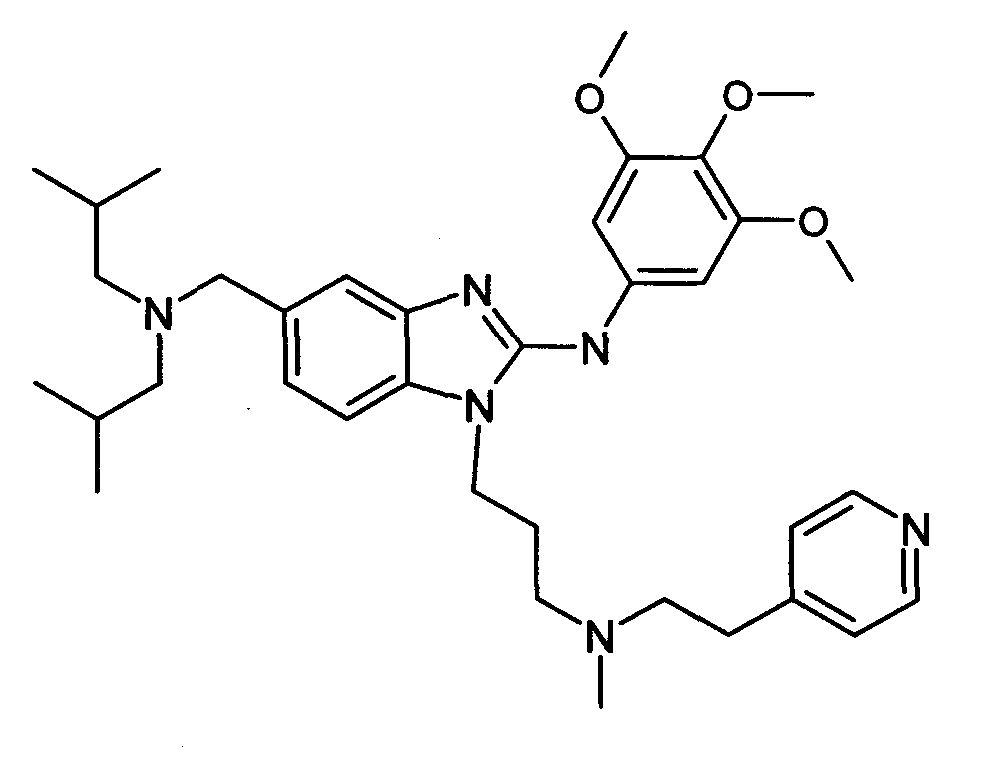
Стадия 1: 4-[(диизобутиламино)метил]-N-(3-{метил[2-(4-пиридинил)этил]амино}пропил)-1,2-бензолдиамин
Молярный раствор комплекса бор-тетрагидрофуран (6,25 мл, 15 экв.) по каплям добавляли к раствору N,N-диизобутил-4-({3-[метил(2-пиридин-4-илэтил)амино]пропил}амино)-3-нитробензамида (200 мг, 1 экв.) в тетрагидрофуране (3 мл), охлажденному до 0°C. Смесь кипятили с обратным холодильником в течение 20 часов, затем охлаждали до 0°C и гидролизовали 6н. водным раствором хлористоводородной кислоты (12 мл). Через 1 час 30 минут смесь охлаждали до 0°С с обратным холодильником и с помощью 6 н. водного раствора соды доводили до щелочной pH. После добавления этилацетата декантирования и экстракции органические фазы объединяли, а затем промывали водным солевым раствором, сушили над сульфатом натрия и упаривали при пониженном давлении. Очистка с помощью флэш-хроматографии на силикагеле (элюент: от 100% дихлорметана до дихлорметана/метанола 8:2) давала ожидаемое соединение в виде масла (92 мг, 51%-ный выход).
MS/LC: вычисленный MW = 425,6; m/z = 426,4 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 2: 5-[(диизобутиламино)метил]-1-(3-{метил[2-(2-пиридинил)этил]амино}пропил)-N-(3,4,5-триметоксифенил)-1H-бензимидазол-2-амин
К раствору 4-[(диизобутиламино)метил]-N-(3-{метил[2-(4-пиридинил)этил]амино}пропил)-l,2-бензолдиамина (90 мг, 1 экв.) в тетрагидрофуране (2 мл) последовательно добавляли 3,4,5-триметоксифенилизотиоцианат (57 мг, 1,2 экв.) и N-метилциклогексилкарбодиимид-N-метил полистирольную смолу (приобретена у Novabiochem; загрузка 1,69 ммоль/г, 501 мг, 4 экв.). Смесь кипятили с обратным холодильником в течение 18 часов, затем охлаждали до комнатной температуры и добавляли аминометилполистирольную смолу (приобретена у Novabiochem, 2 экв.). После перемешивания в течение 4 часов при комнатной температуре, смесь фильтровали на фритте и фильтрат концентрировали при пониженном давлении при 40°C. Очистка с помощью флэш-хроматографии на силикагеле (элюент: от 100% дихлорметана до дихлорметана/метанола 9:1) давала ожидаемое соединение в виде бежевой пены (92 мг, 83%-ный выход).
MS/LC: вычисленный MW = 616,8; m/z = 617,4 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
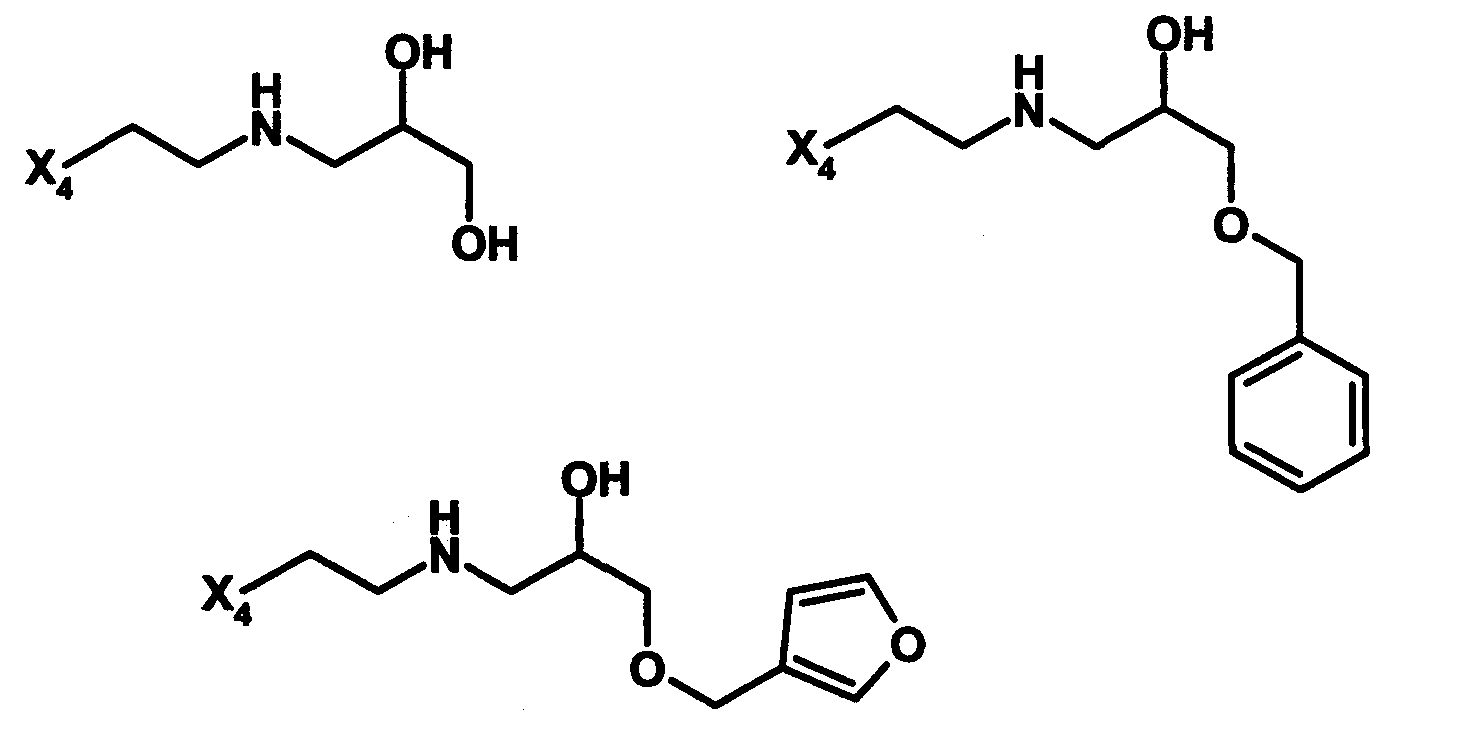
G. Получение в соответствии co схемой реакции G:
Соединения формулы I по данному изобретению, где А представляет собой -C(O)- и R4 представляет собой -NW4W'4, могут быть получены в соответствии со следующей схемой G:
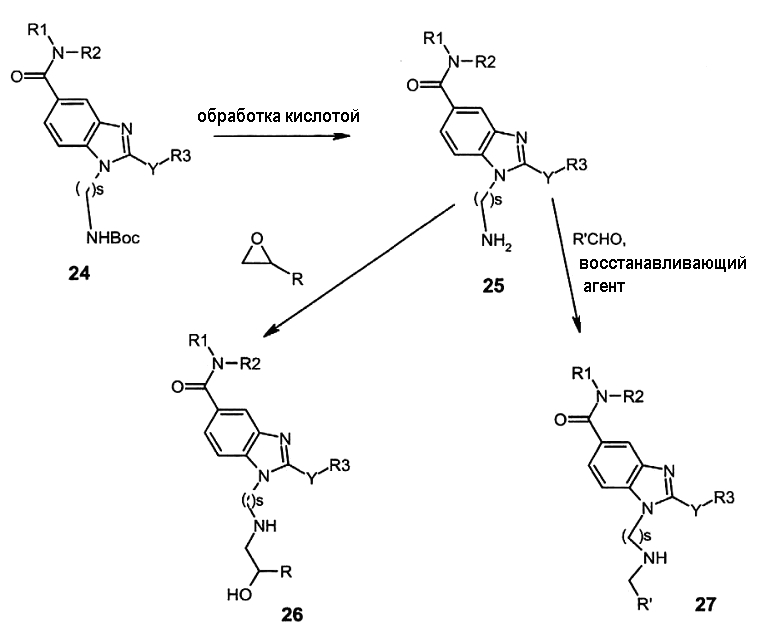
Как представлено на схеме G, бензимидазольное производное (24), полученное в соответствии с реакционными схемами A, B, C или D, может быть обработано органической или неорганической кислотой, такой как трифторуксусная кислота или хлористый водород (водная или газообразная форма), в апротонном растворителе, таком как дихлорметан или этилацетат, при температуре 0-20°C в течение 0,5-5 часов с получением амина (25). Амин (25) затем может быть обработан эпоксидом в протонном или апротонном полярном растворителе, таком как метанол, этанол или ацетонитрил, в присутствии или в отсутствие перхлората лития или трифлат иттербия при температуре 20-80°C в течение 4-48 часов с получением соединения (26). Амин (25) может также взаимодействовать с альдегидом в протонном или апротонном растворителе, таком как дихлорметан, тетрагидрофуран или метанол, в течение 1-15 часов при температуре 0-50°C. Полученный имин затем восстанавливали in situ с помощью восстанавливающего агента без или на полимерной подложке, предпочтительно с помощью натрий триацетоксиборгидрида на полимерной подложке, натрия цианоборгидрида или боргидрида, в присутствии или в отсутствие кислоты, такой как уксусная кислота, при температуре от 20 до 50°C в течение 0,2 до 5 часов с получением соединения (27).
Соединения 27, где s = 3, может быть также получено в соответствии со следующей схемой G':
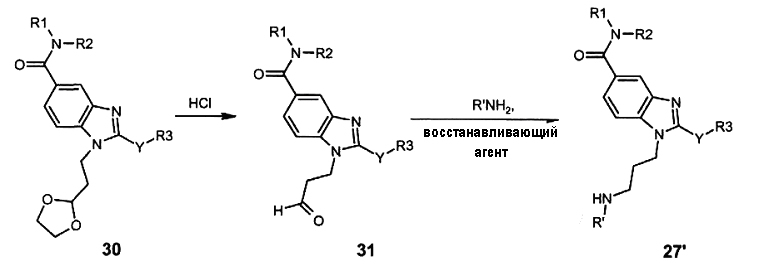
Как показано на схеме G', производное (30), полученное в соответствии с реакционными схемами A, B, C или D, может быть обработано либо органической кислотой, такой как пиридин тозилат или паратолуолсульфоновая кислота, в апротонном растворителе, таком как ацетон, в присутствии воды, при температуре 20-70°C в течение 2-12 часов, либо неорганической кислотой, такой как водный хлористый водород в апротонном растворителе, таком как тетрагидрофуран. при температуре 0-20°C в течение 6-18 часов с получением соединения (31). Затем альдегид (31) может быть обработан амином в протонном или апротонном растворителе, таком как дихлорметан, тетрагидрофуран или метанол, в течение 1-18 часов при температуре 20°C. Полученный имин затем восстанавливали in situ с помощью восстанавливающего агента, предпочтительно с помощью натрия триацетоксиборгидрида или натрия цианоборгидрида, в присутствии или в отсутствие кислоты, такой как уксусная кислота, при температуре 20-50°C в течение 0,2-6 часов с получением соединения (27').
Пример G1: 1-{2-[(циклогексилметил)амино]этил}-N,N-диизобутил-2-[(3,4, 5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамид
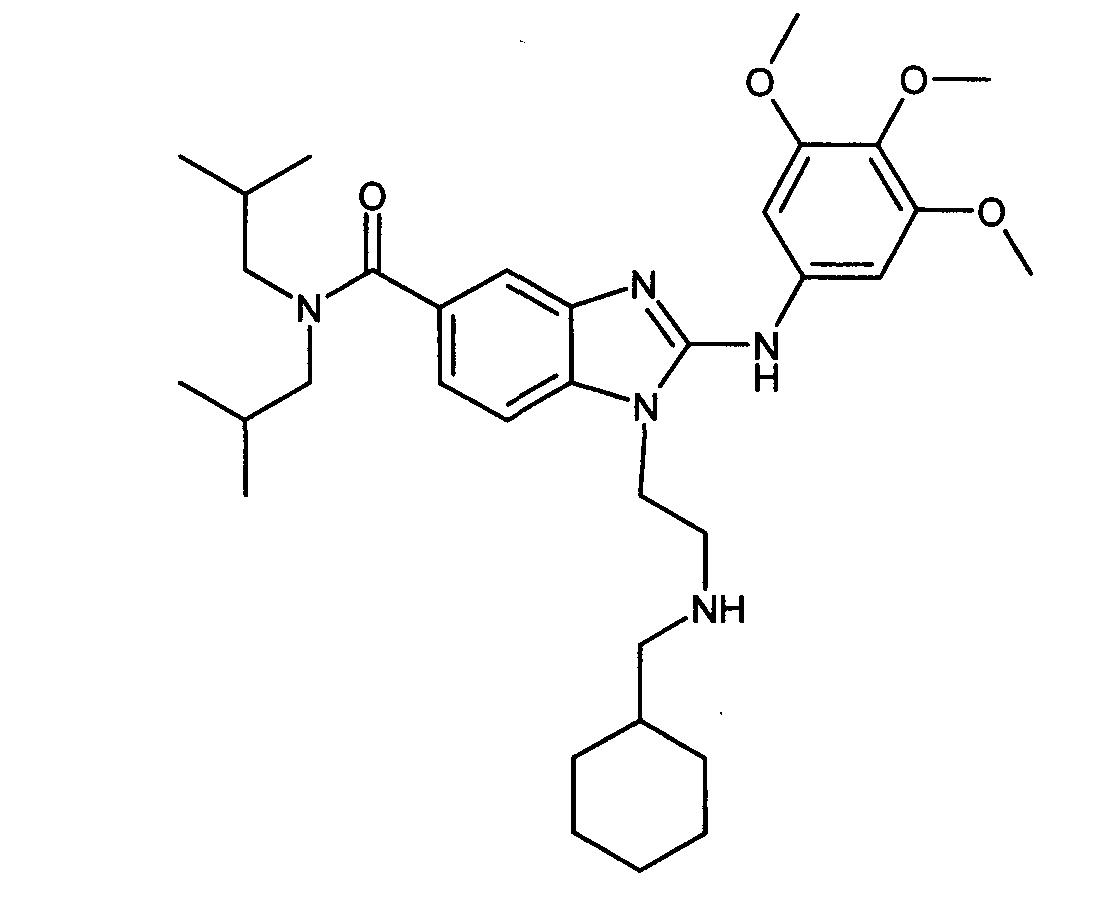
Стадия 1: гидрохлорид 1-(2-аминоэтил)-N,N-диизобутил-2-[(3,4,5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамида
Поток сухого HCl пропускали через раствор трет-бутил 2-{5-[(диизобутиламино)карбонил]-2-[(3,4,5-триметоксифенил)амино]-1H-бензимидазол-1-ил}этилкарбамата (2,56 г, получен в соответствии со способом, описанным в примере A1, схема реакции A) в этилацетате (100 мл), (100 % этилацетат) охлаждали до 0°C до полного исчезновения исходного продукта, определяемого с помощью ТСХ. Полученную смесь затем упаривали при пониженном давлении. Полученный твердый продукт растирали в порошок с диэтилэфиром и фильтровали с получением ожидаемого соединение в виде белых кристаллов (2,25 г, 97%-ный выход).
MS/LC: вычисленный MW = 497,6; m/z = 498,3 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Стадия 2: 1-{2-[(циклогексилметил)амино]этил}-N,N-диизобутил-2-[(3,4,5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамид
Раствор 1-(2-аминоэтил)-N,N-диизобутил-2-[(3,4, 5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамид (30 мг, 1 экв.) и циклогексанкарбоксальдегида (5 мг, 0,8 экв.) в метаноле (0,7 мл) перемешивали при температуре приблизительно 20°C в течение 4 часов. Добавляли боргидридный полимер (48 мг, 2,5 ммоль/г, Amberlite ®, IRA-400), и смесь перемешивали в течение 18 часов, затем добавляли дихлорметан (0.5 мл) и бензилоксибензальдегидный полимер Wang (37 мг, 3,22 ммоль/г, Novabiochem). После перемешивания в течение ночи смесь фильтровали и фильтрат упаривали при пониженном давлении с получением ожидаемого соединение в виде бежевой пены (18 мг, 65%).
MS/LC: вычисленный MW = 593,8; m/z = 594,4 (MH+)
ЯМР(1H, 400 МГц, CDCl3):
Нижеследующие соединения получали в соответствии co схемой реакции G и тем же способом, описанным для синтеза 1-{2-[(циклогексилметил)амино]этил}-N,N-диизобутил-2-[(3,4, 5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамида (также проводили окончательную очистку с помощью флэш-хроматографии на силикагеле):
В вышеуказанной формуле R4 представляет собой один из нижеприведенных радикалов:
Пример G2: 1-{2-[(1-гидрокси-2-фенилэтил)амино]этил}- N,N-диизобутил-2-[(3,4,5-триметоксифенил)амино]-1Н-бензимидазол-5-карбоксамид
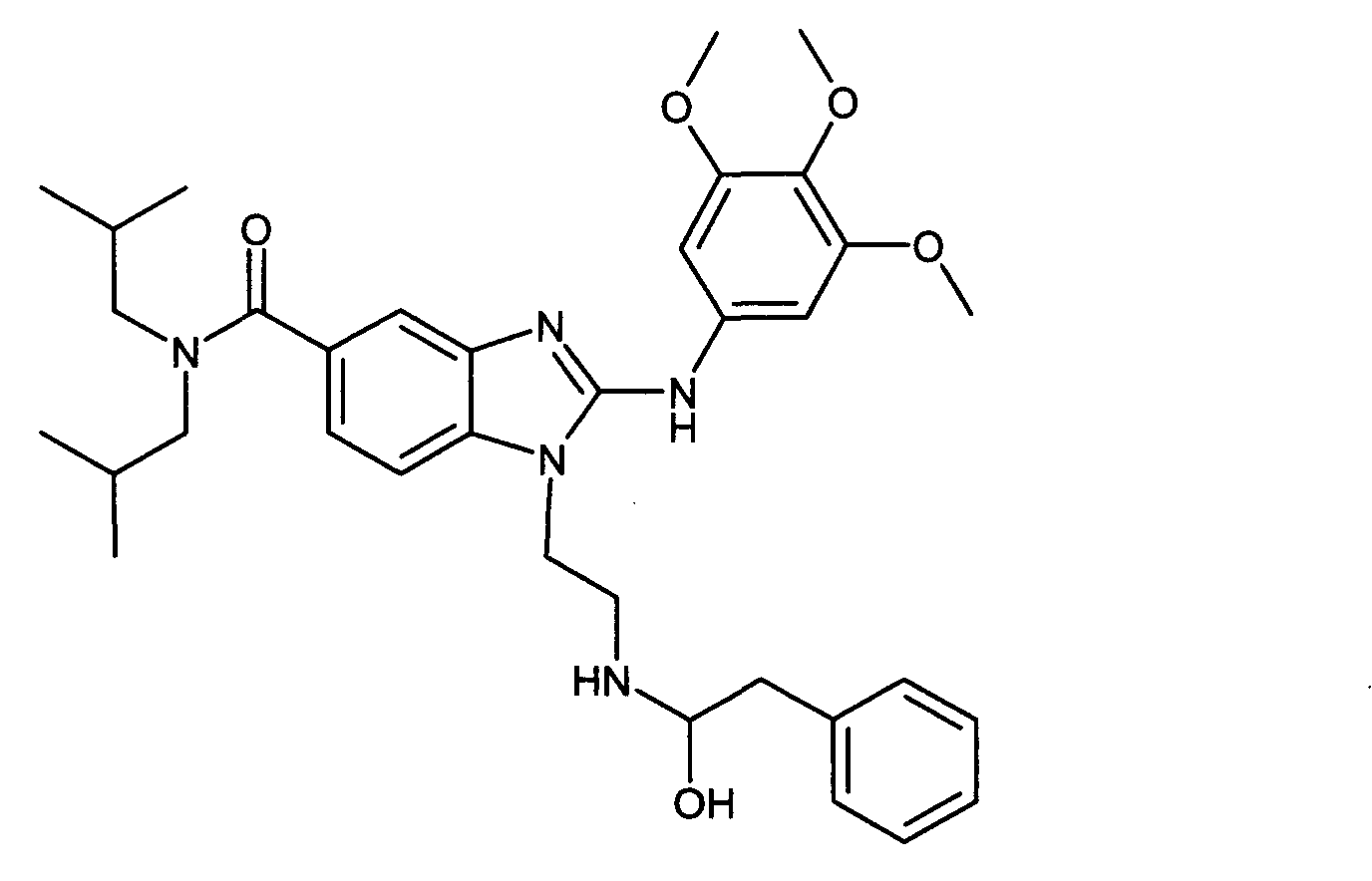
К раствору 2,3-эпоксипропилбензола (7 мг, 1 экв.) в ацетонитриле (0,5 мл) добавляли перхлорад лития (16 мг, 3 экв.), а через 5 минут 1-(2-аминоэтил)-N,N-диизобутил-2-[(3,4,5-триметоксифенил)амино]-1Н-бензимидазол-5-карбоксамид (25 мг, 1 экв.) при температуре приблизительно 20 С. Смесь кипятили с обратным холодильником в течение 24 часов, затем охлаждали до температуры окружающей среды и добавляли воду, насыщенную гидрокарбонатом и дихлорметаном. После декантирования и экстракции органические фазы объединяли и промывали водным солевым раствором, а затем сушили над сульфатом магния и упаривали при пониженном давлении при 40 С. Очистка полученного масла с помощью флэш-хроматографии на силикагеле (от 100% дихлорметана до дихлорметана/метанола 80:20) давала ожидаемое соединение в виде масла (31 мг, 55% выход).
MS/LC: вычисленный MW = 631,8; m/z = 632,4 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Нижеследующие соединения получали в соответствии co схемой реакции G и тем же способом, описанным для синтеза 1-{2-[(1-гидрокси-2-фенилэтил)амино]этил}-N,N-диизобутил-2-[(3,4,5-триметоксифенил)амино]-1H-бензимидазол-5-карбоксамид:
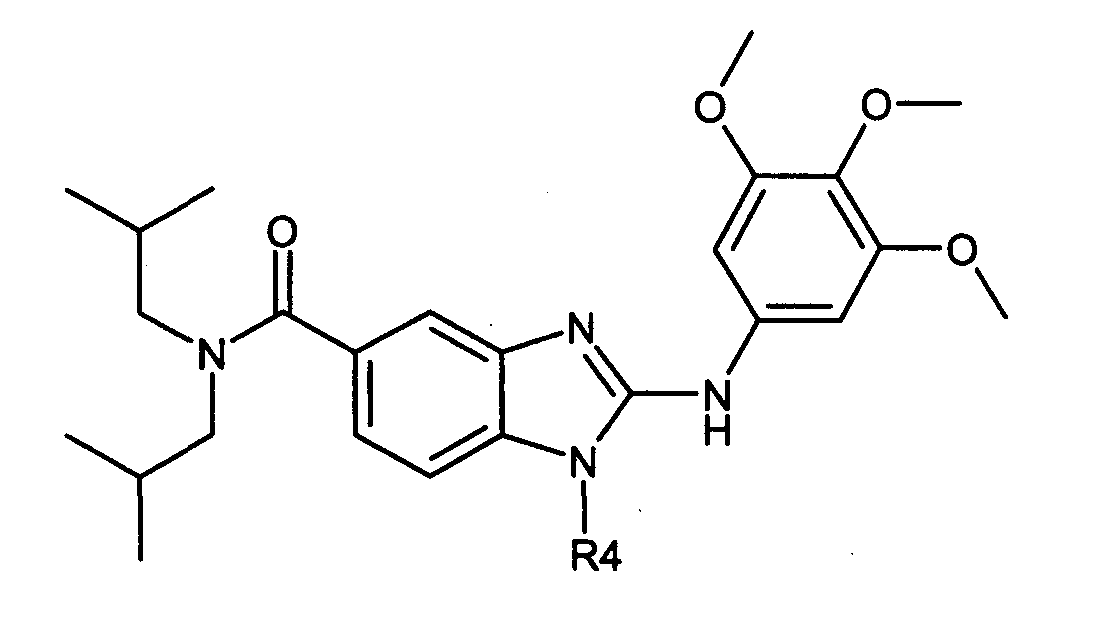
в вышеуказанной формуле R4 представляет собой один из нижеприведенных радикалов:
H. Получение в соответствии co схемой реакции H:
Соединения формулы I по данному изобретению, где А представляет собой -C(O)-, Y представляет собой -S-, и R3 представляет собой -(CH2)p-CH(OH)-(CH2)p''-Z3, могут быть получены в соответствии со следующей схемой H:
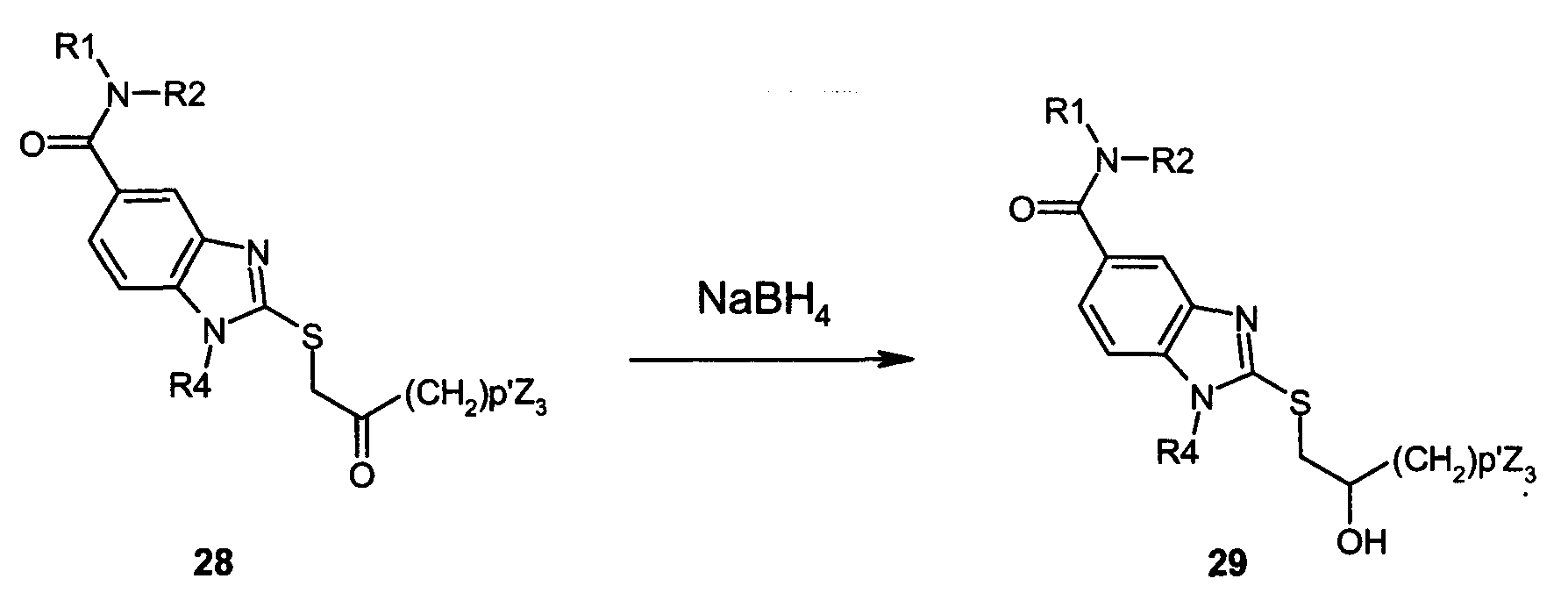
Как представлено на схеме H, тиобензимидазольное производное (28), полученное в соответствии с реакционными схемами B или D, может быть обработано восстанавливающим агентом, таким как боргидрид натрия, в протонном растворителе, таком как метанол, при температуре 0-20°C в течение от 0,2 часа до 1 часа с получением соответствующего спирта (29).
Пример H1: 2-{[2-гидрокси-2-(3,4, 5-триметоксифенил)этил]тио}-N,N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-1H-бензимидазол-5-карбоксамид
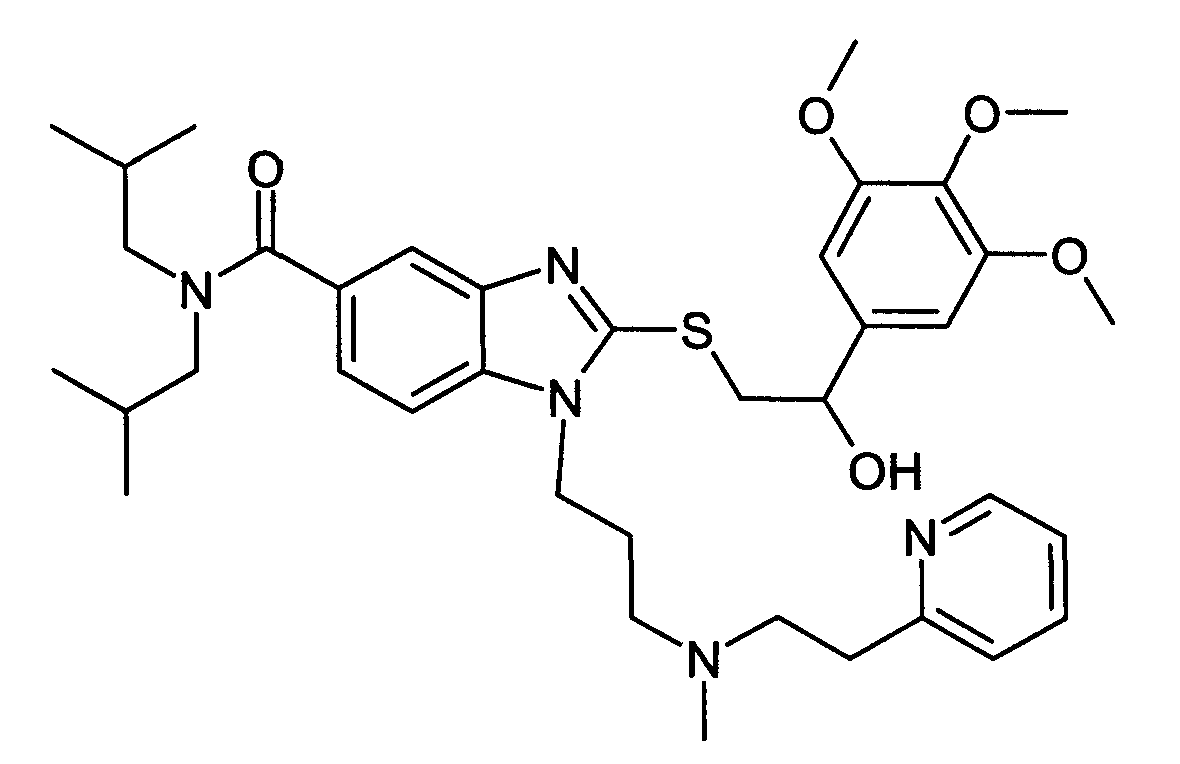
Боргидрид натрия (8 мг, 2 экв.) добавляли при 0°C к раствору N,N-диизобутил-1-{3-[метил(2-пиридин-2-илэтил)амино]пропил}-2-{[2-оксо-2-(3,4, 5-триметоксифенил)этил]тио}-1H-бензимидазол-5-карбоксамида (69 мг, 1 экв.) в метаноле (2 мл). После перемешивания в течение 10 минут при 0°C смесь доводили до температуры приблизительно 20°C и перемешивали при этой температуре в течение 30 минут. Затем смесь концентрировали при пониженном давлении при 40°C, затем добавляли воду, насыщенную хлоридом аммония и дихлорметаном. После декантирования и экстракции органические фазы объединяли и промывали водным солевым раствором, сушили над сульфатом натрия и упаривали при пониженном давлении при 40 C. Очистка полученного масла с помощью флэш-хроматографии на силикагеле (от 100% дихлорметана до дихлорметана/метанола 80:20) давала ожидаемое соединение в виде масла (61 мг, 88%-ный выход).
MS/LC: вычисленный MW = 691,9; m/z = 692,4 (MH+)
ЯМР(1H, 400 МГц, DMSO-d6):
Получение агентов синтеза
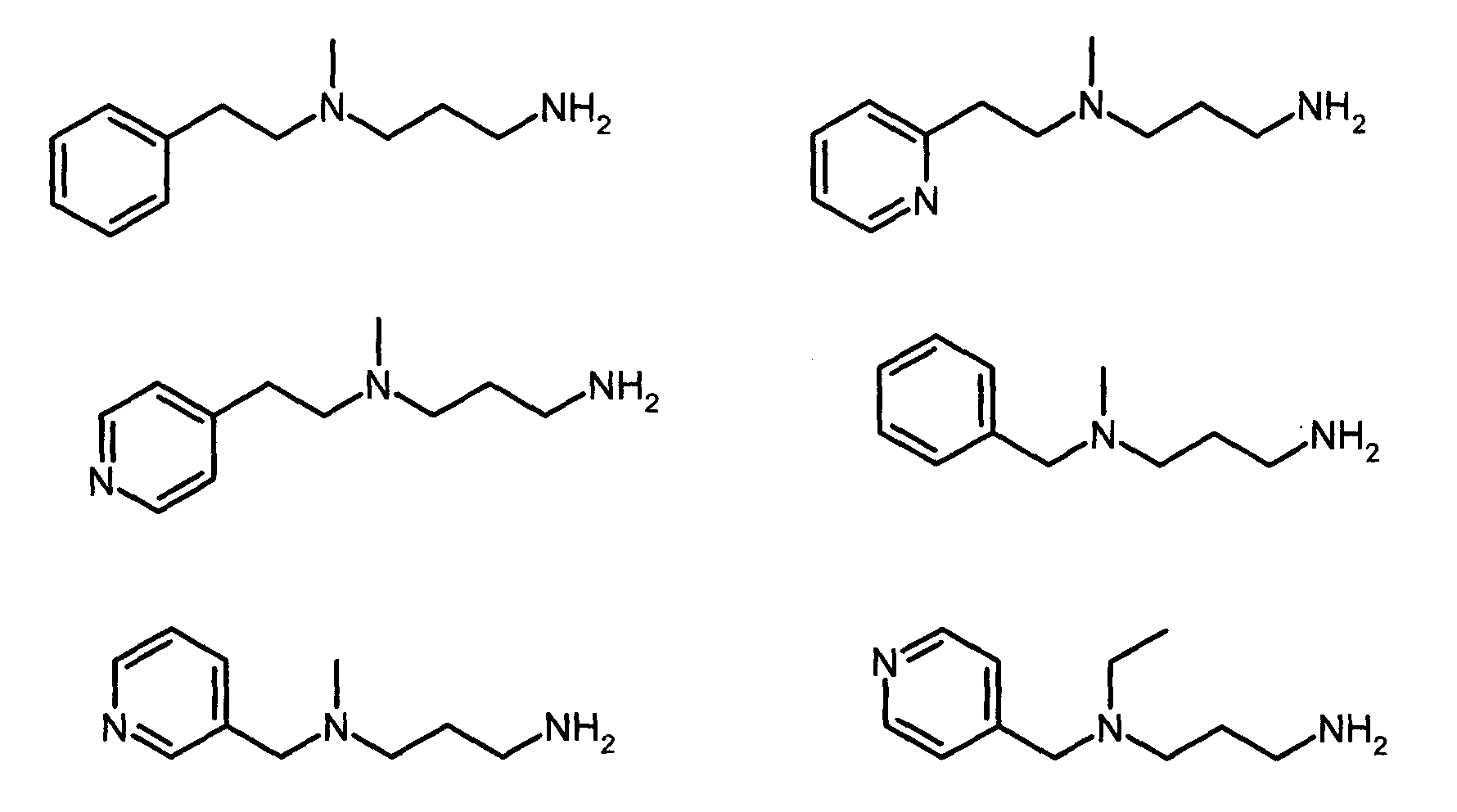
N-(2-пиридин-2-ил этил)пропан-1,3-диамин
К раствору, охлажденному до приблизительно 4°C, 2-[2-(метиламино)этил]пиридина (19,5 мл, 1 экв.) в метаноле (200 мл) медленно добавляли акрилонитрил (10,1 мл, 1,1 экв.). Реакционную среду затем перемешивали в течение 3 часов при приблизительно 20°C, затем концентрировали при пониженном давлении при 40°C с получением 3-[(2-пиридин-2-илэтил)амино]пропаннитрила в виде желтого масла (25,6 г, 96%-ный выход).
Раствор этого масла (15,3 г) в метаноле, насыщенном аммонием, (250 мл) затем гидрировали в присутствии никеля Ренея (1,5 г) при приблизительно 20°C в течение 15 часов. Реакционную смесь затем фильтровали через целит. Фильтрат концентрировали при пониженном давлении при приблизительно 40°C с получением ожидаемого соединения в виде зеленоватого масла (15,5 г, 97%-ный выход).
Нижеследующие соединения получали тем же способом, описанным для синтеза N-(2-пиридин-2-илэтил)пропан-1,3-диамина:
2-бром-1-(3,4,5-триметоксифенил)этанон
Гидробромид пербромида пиридина на полимерной подложке (23, г, 1 экв.) добавляли к раствору 3,4,5-триметокси-ацетофенона (10 г, 1 экв.) в метаноле (150 мл). После перемешивания в течение 3 часов при приблизительно 20°C смесь фильтровали и фильтрат концентрировали при пониженном давлении. Очистка полученного осадка с помощью флэш-хроматографии на силикагеле (элюент: гептан/этилацетат 8/2, затем 7/3) давала ожидаемое соединение в виде белого порошка (8,2 г, 60%-ный выход). Точка плавления = 66°C.
3,4,5-триметоксибензоил изотиоцианат
Тиоцианат калия добавляли к раствору 3,4,5-триметоксибензоилхлорида (2,3 г) в ацетонитриле (40 мл). После перемешивания в течение 15 минут при приблизительно 20°C, смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением ожидаемого соединение в виде бежевого порошка (2,4 г, 96%-ный выход). Точка плавления = 101°C.
Соединения I (или I') по настоящему изобретению обладают эффективными фармакологическими свойствами. Таким образом, было обнаружено, что соединения I (или I') по настоящему изобретению обладают антагонизмом в отношении GnRH (гонадолиберин).
Таким образом, соединения по настоящему изобретению могут иметь широкое терапевтическое применение. Они могут эффективно использоваться у женщин при лечении эндометриоза, фибромы, синдрома поликистозных яичников, рака молочных желез, яичника и эндометрия, гонадотропной гипофизарной десенсибилизации при медикаментозной стимуляции яичников для лечения бесплодия; у мужчин при лечении доброкачественной простатической гиперплазии и рака простаты; и при лечении преждевременной половой зрелости у мужчин и женщин. Примеры фармакологического использования соединений по данному изобретению можно найти далее, в экспериментальной части.
Объектом данного изобретения также являются продукты указанной выше формулы I (или I'), используемые в качестве лекарственных средств, а также соли добавления фармацевтически приемлемых неорганических или органических кислот указанных продуктов формулы I (или I'), а также фармацевтические композиции, содержащие в качестве активного ингредиента, по крайней мере, одно лекарственное средство, указанное выше, в сочетании с фармацевтически приемлемым носителем.
Фармацевтическая композиция может находиться в твердой форме, например, в виде порошков, гранул, таблеток, желатиновых капсул или суппозиториев. Подходящими твердыми носителями могут быть, например, фосфат кальция, стеарат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, поливинилпирролидин и воск.
Фармацевтические композиции, содержащие соединение по данному изобретению, также могут находиться в жидкой форме, например в виде растворов, эмульсий, суспензий или сиропов. Подходящими жидкими носителями могут быть, например, вода, органические растворители, такие как глицерин или гликоли, а также их смеси, в различных соотношениях, в воде с добавлением фармацевтически приемлемых масел или жиров. Стерильные жидкие композиции могут использоваться для внутримышечных, внутрибрюшинных или подкожных инъекций, и стерильные композиции также могут вводиться внутривенно.
Значения всех специальных и научных терминов, использованных в настоящем документе, хорошо понятны специалисту в данной области. Более того, все патенты (или патентные заявки), а также другие библиографические ссылки приведены в качестве ссылки.
Экспериментальная часть:
Соединения по данному изобретению, полученные в соответствии со способами примеров A, B, C, D, E, F, G и H, описанными ранее, перечислены в нижеследующей таблице.
Соединения описаны с помощью их времени задержки (вр.задерж.) и их молекулярным пиком, определенным с помощью масс спектрометрии (МН+).
Для масс спектрометрии использовался унифицированный квадрупольный масс-спектрометр (Micromass, Platform model), оснащенный источником электрораспыления, с разрешением 0,8 Да с 50% долиной. Калибровку осуществляли ежемесячно в области масс 80-1000 Да, используя калибровочную смесь йодида натрия и йодида рубидия в растворе смеси изопропанола/воды (1/1 об./об.).
Для жидкостной хроматографии использовали системы Waters, включая линейный дегазатор, кватернарный носос Waters 600, аппликатор образцов на плашку Gilson 233 и ультрафиолетовый детектор Waters 996 PDA.
Использовали следующие условия элюирования:
Элюент А вода + 0,04% трифторуксусная кислота
В ацетонитрил
Скорость потока: 1 мл/мин
Инъекция: 10 мкл
Колонка 3 мкм 75×4,6 мм в.д. Uptisphere ODS
Эти примеры представлены в качестве иллюстрации вышеуказанных способов и не в коей мере не предназначены для ограничения области данного изобретения.
В каждом примере радикалы R1, R2, R3 и R4, радикалы X1, X2, X3 и X4 представляют собой соответственно остальную часть соединения общей формулы (I).
Примеры 1-253, 254-255 и 256-538 иллюстрируют соответственно соединения I, где А представляют собой -C(O)-, и Y представляют собой -S-, А представляет собой -CH2-, и Y представляет собой -NH-, и А представляют собой -C(O)- и Y представляют собой -NH-.
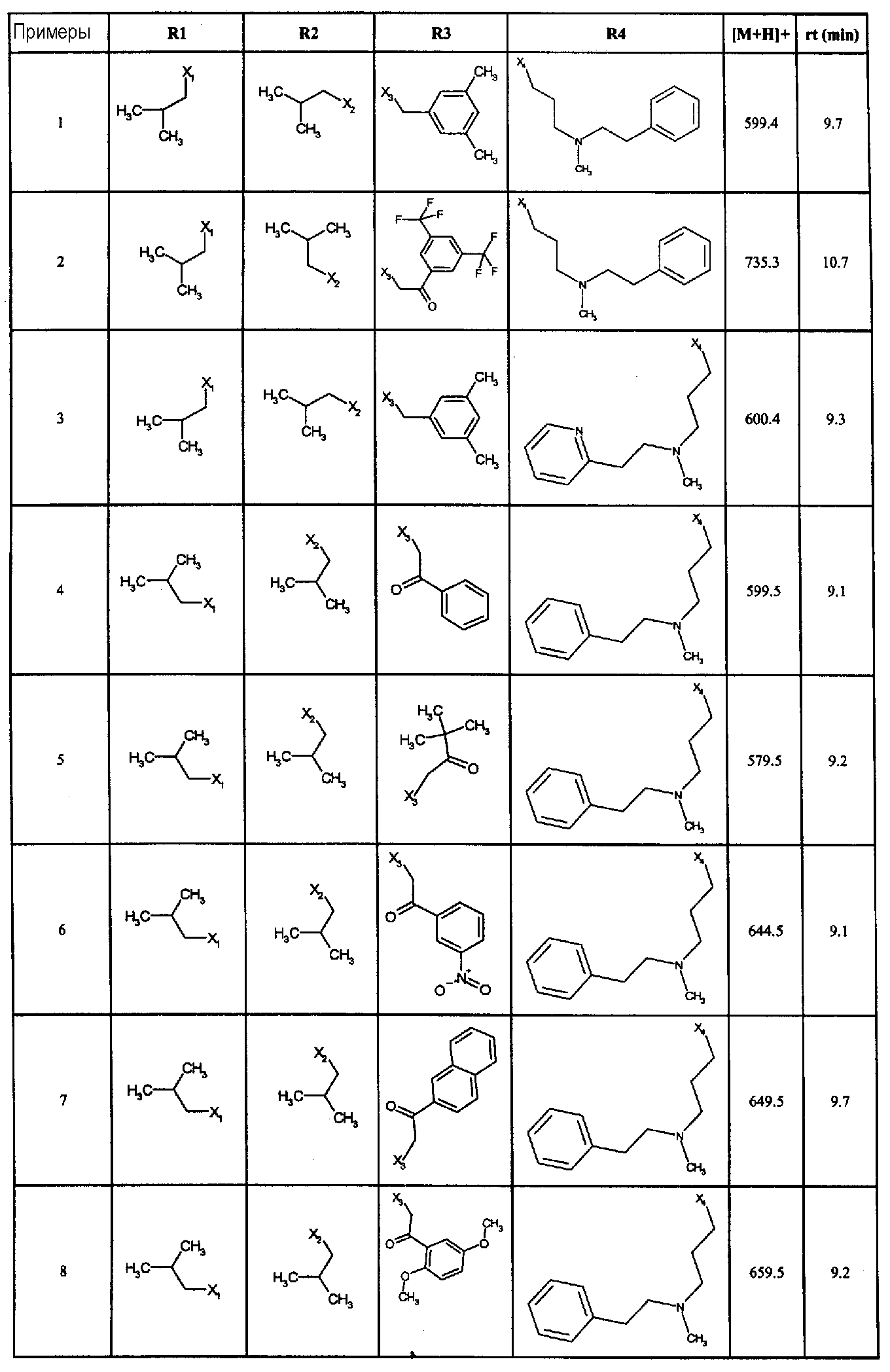
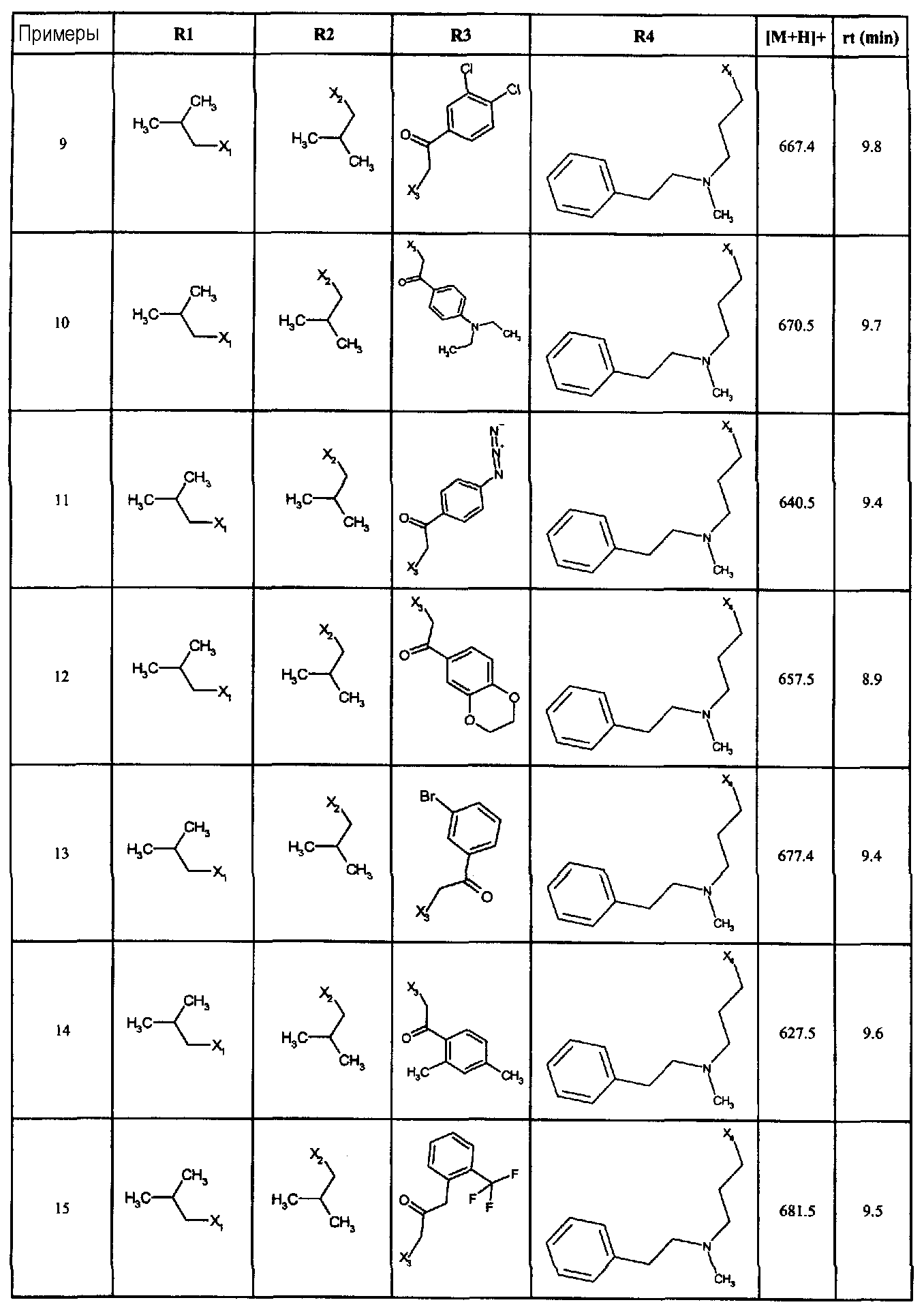
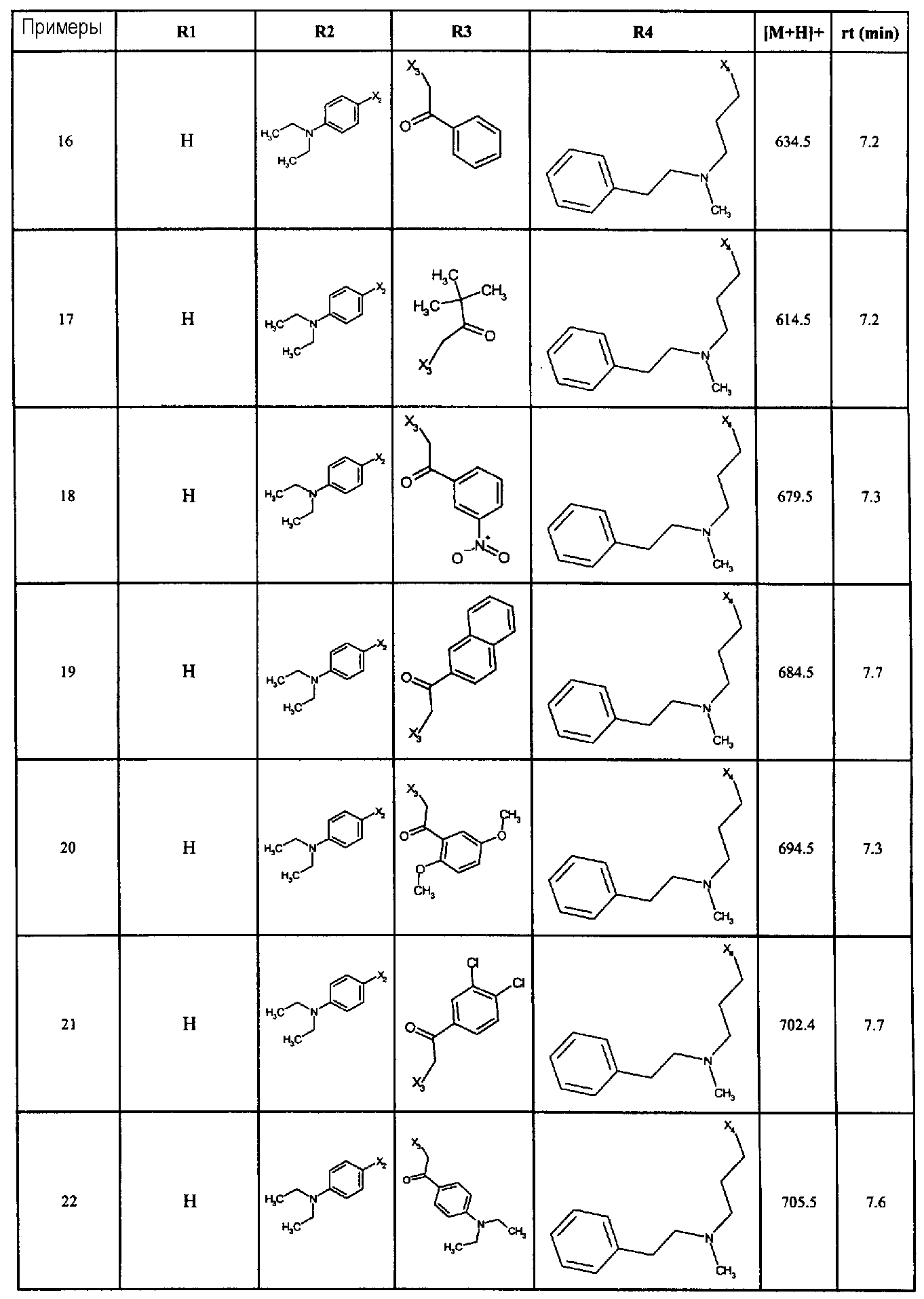
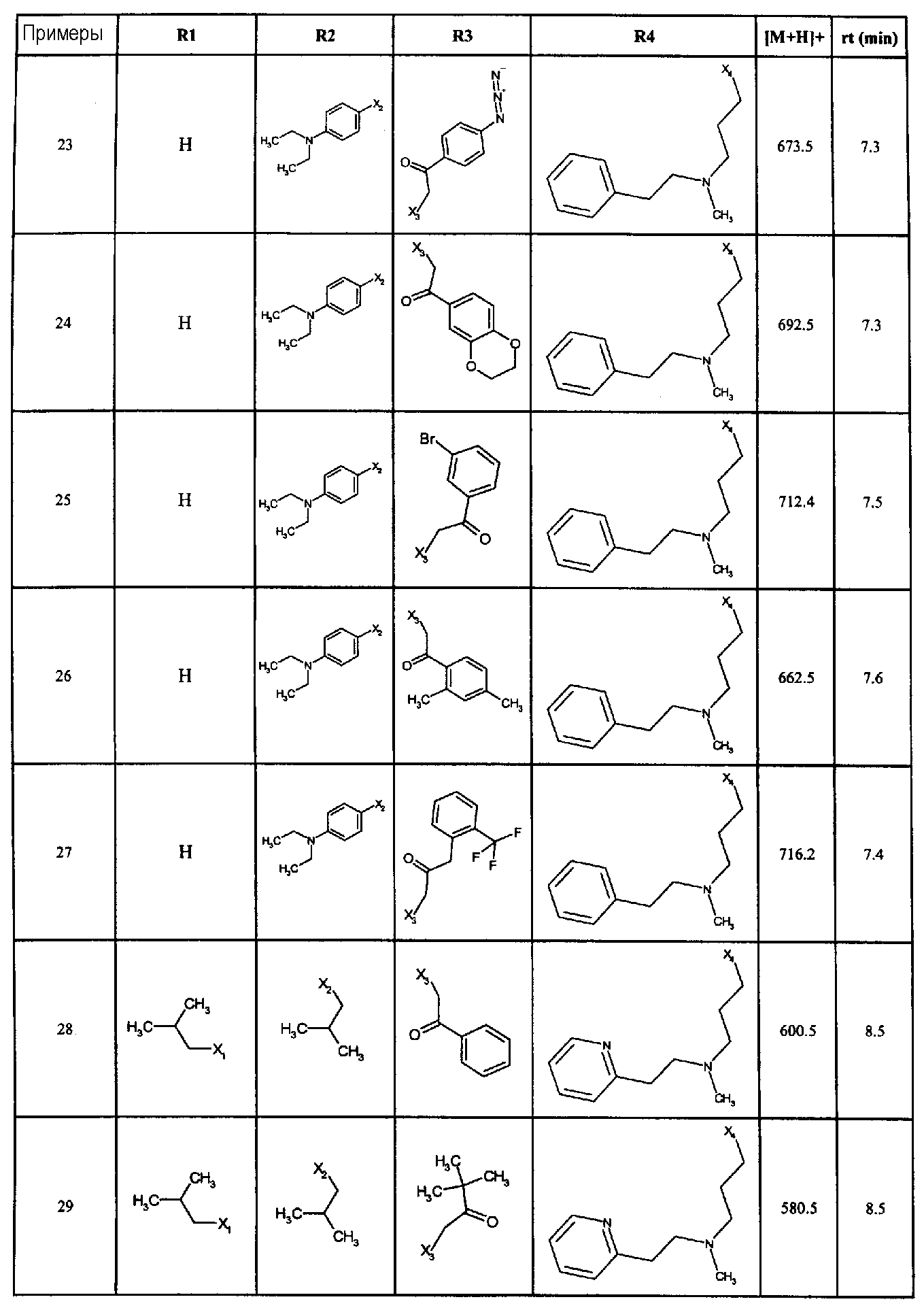
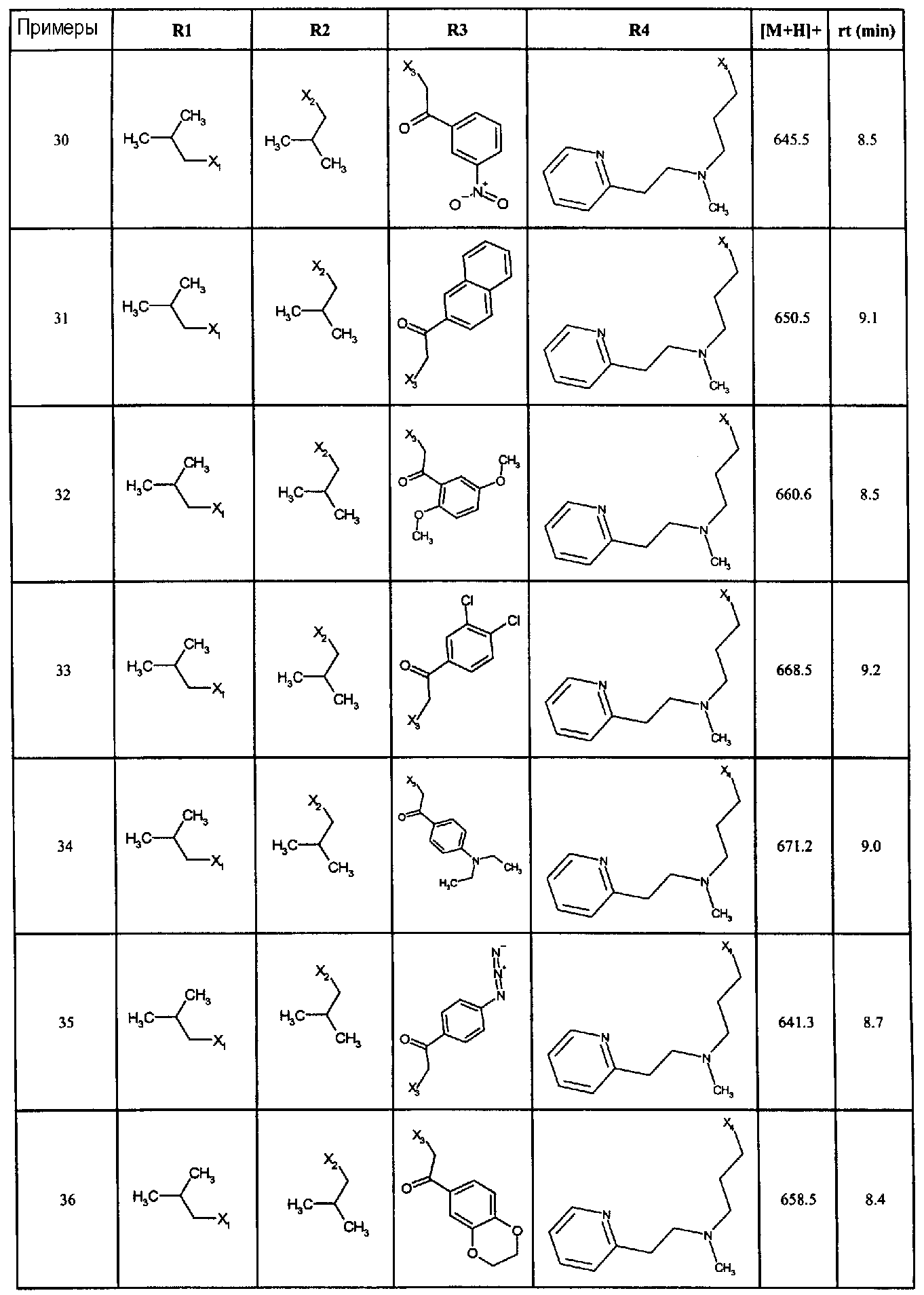
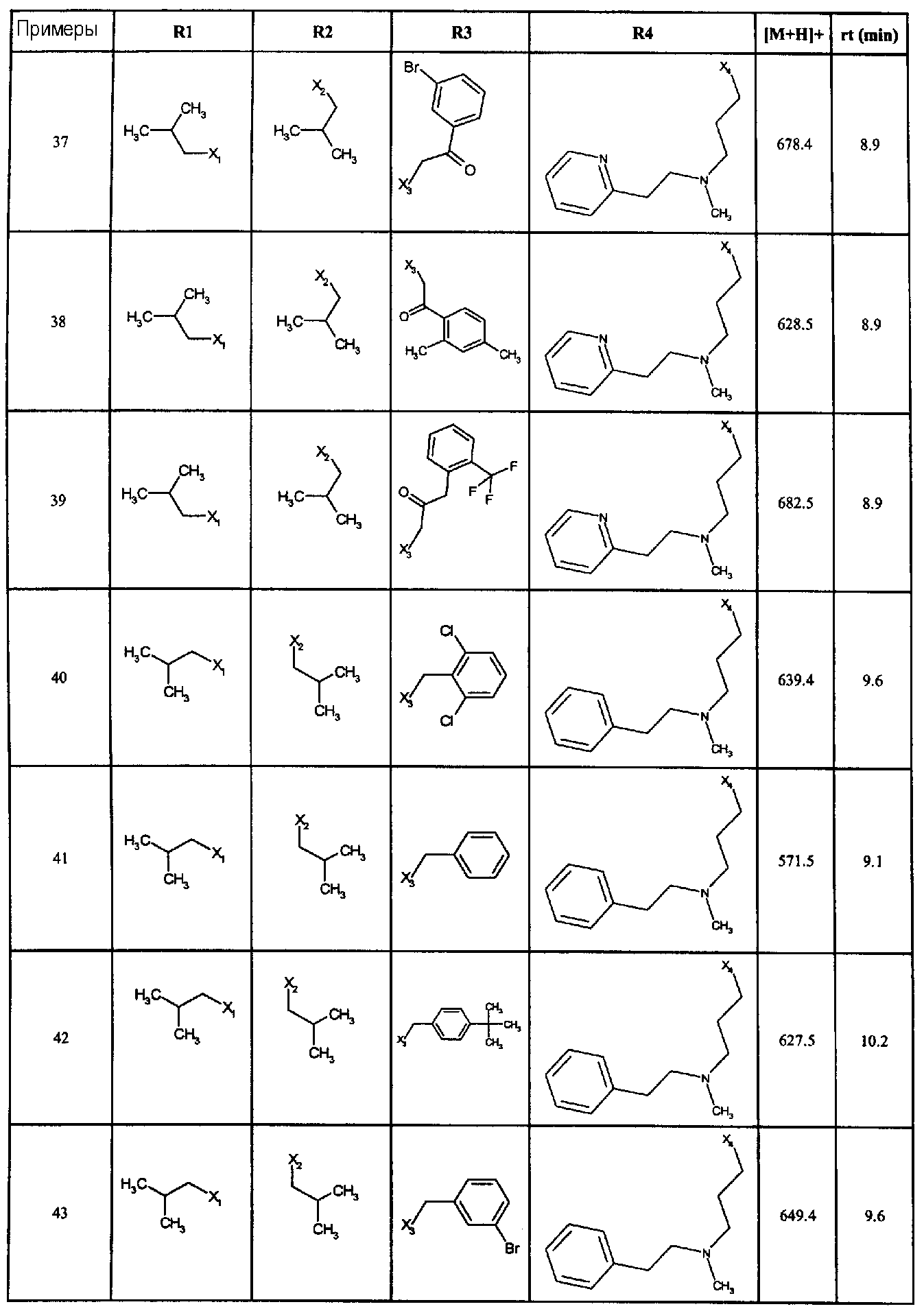
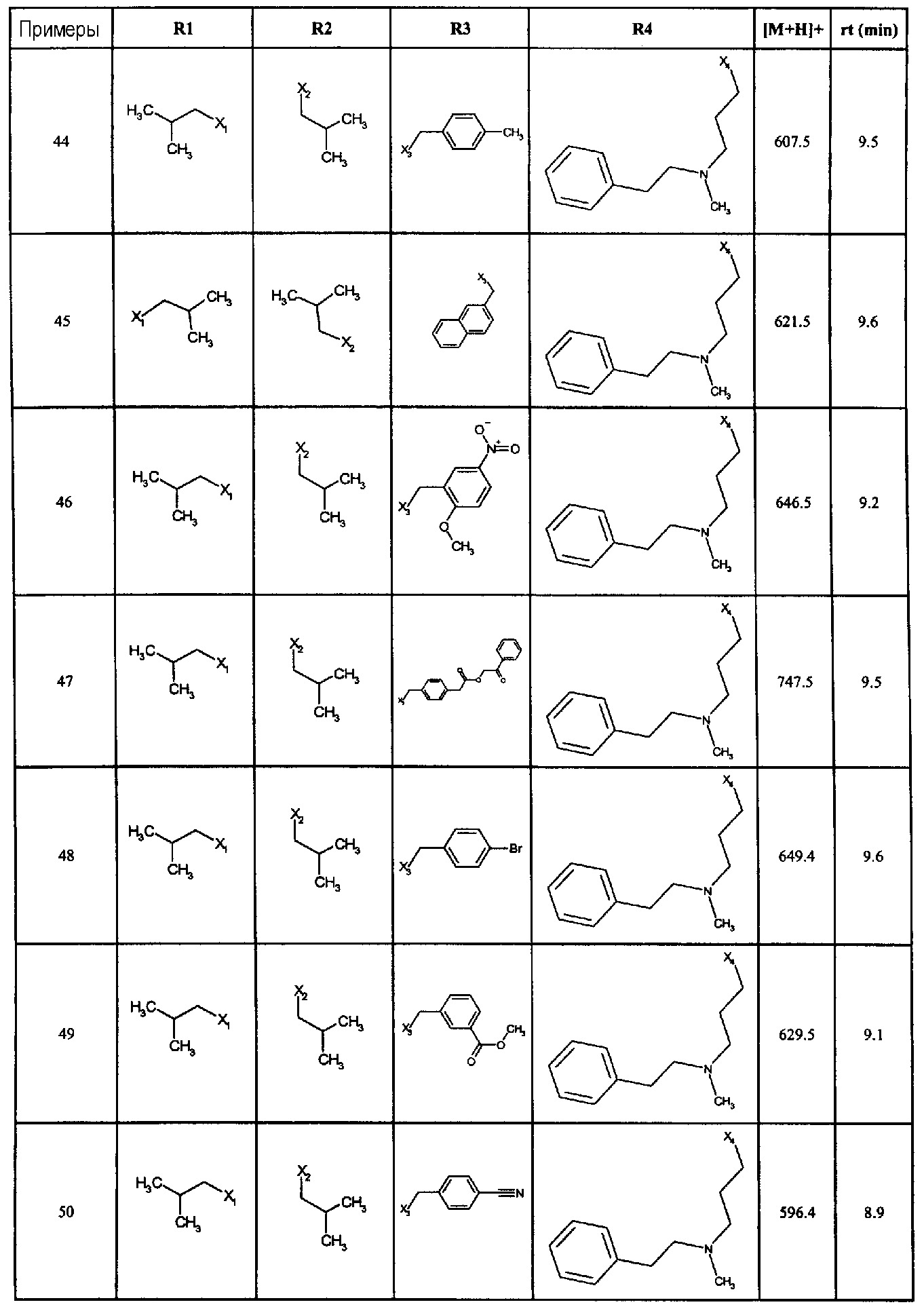
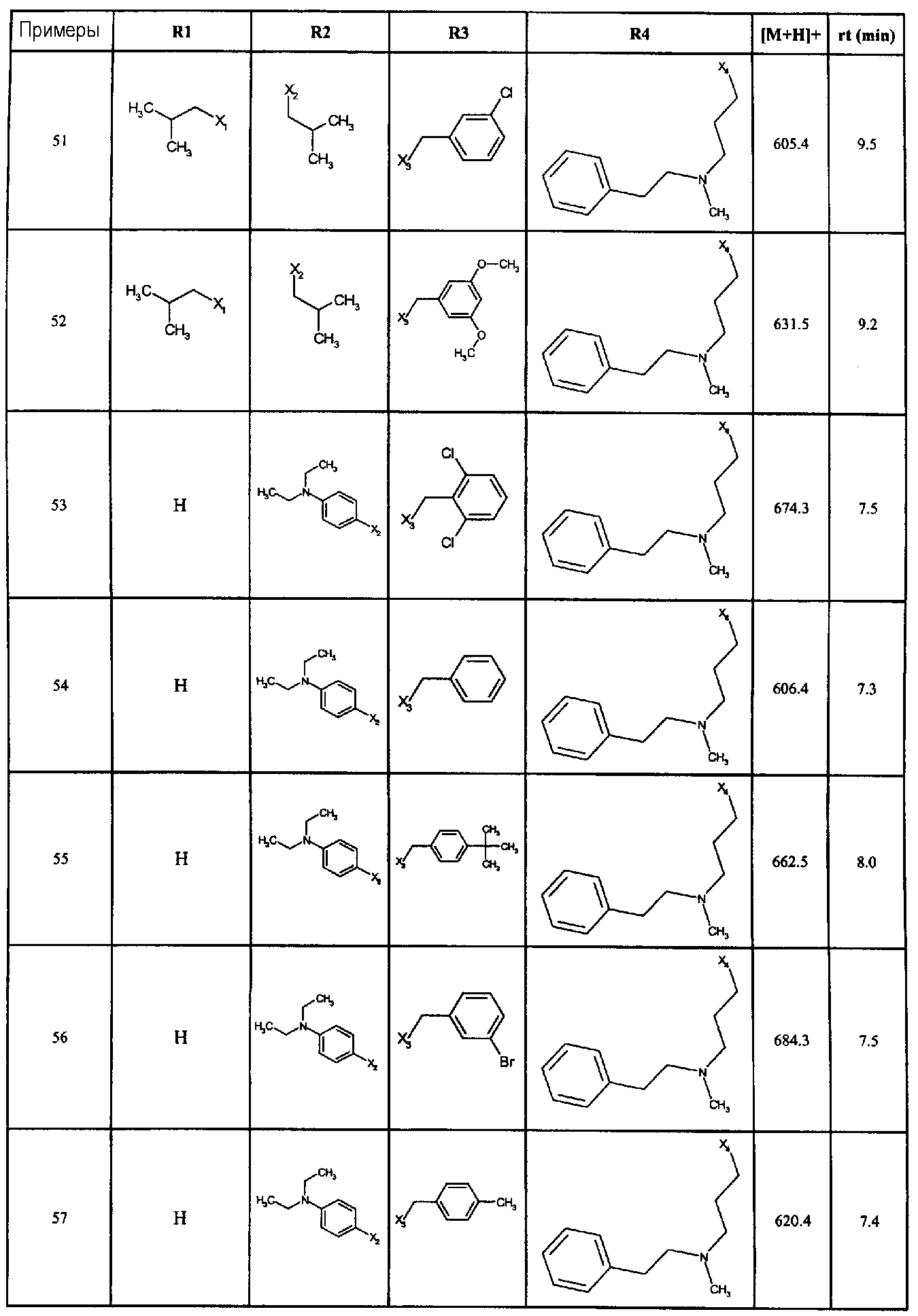
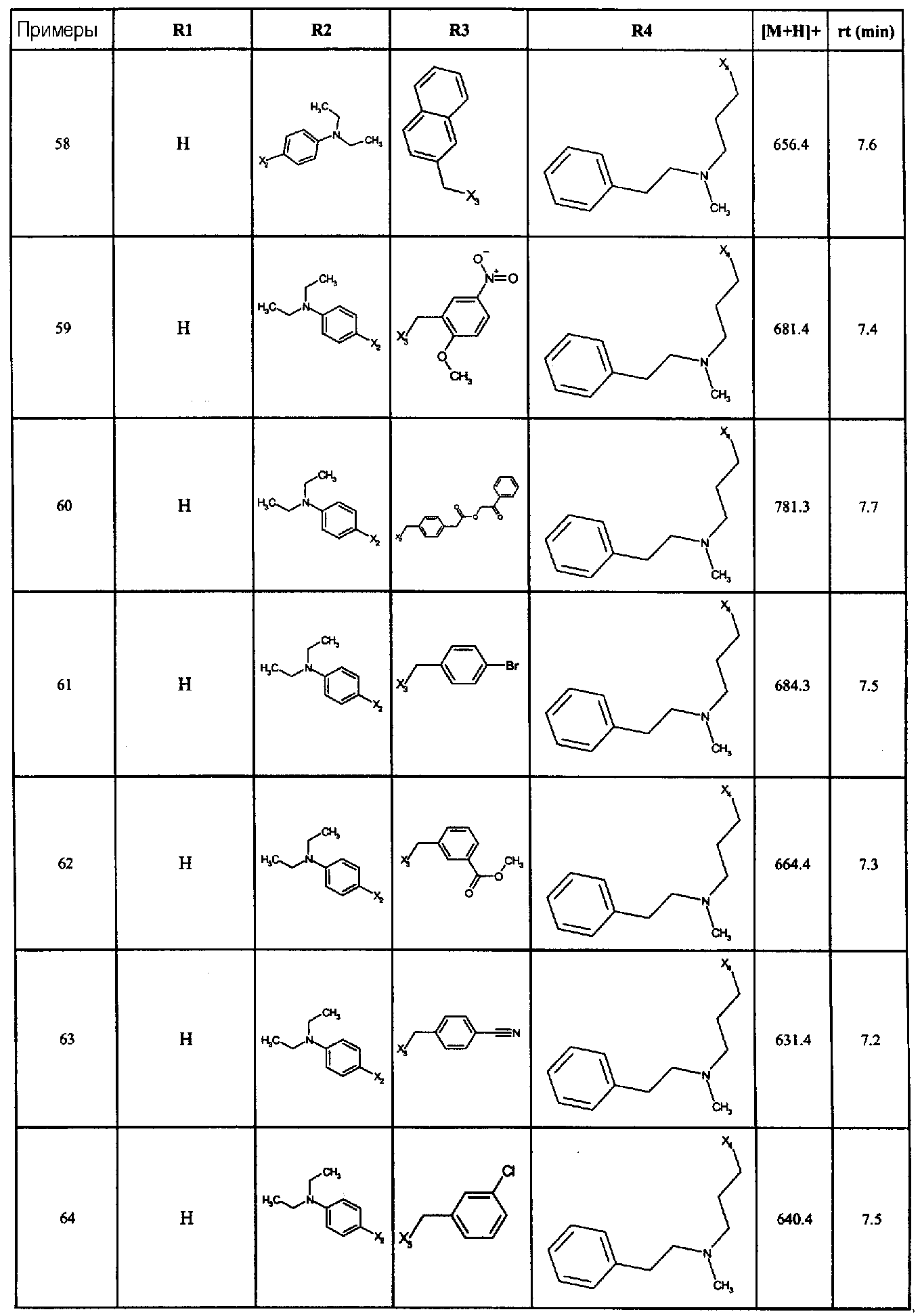
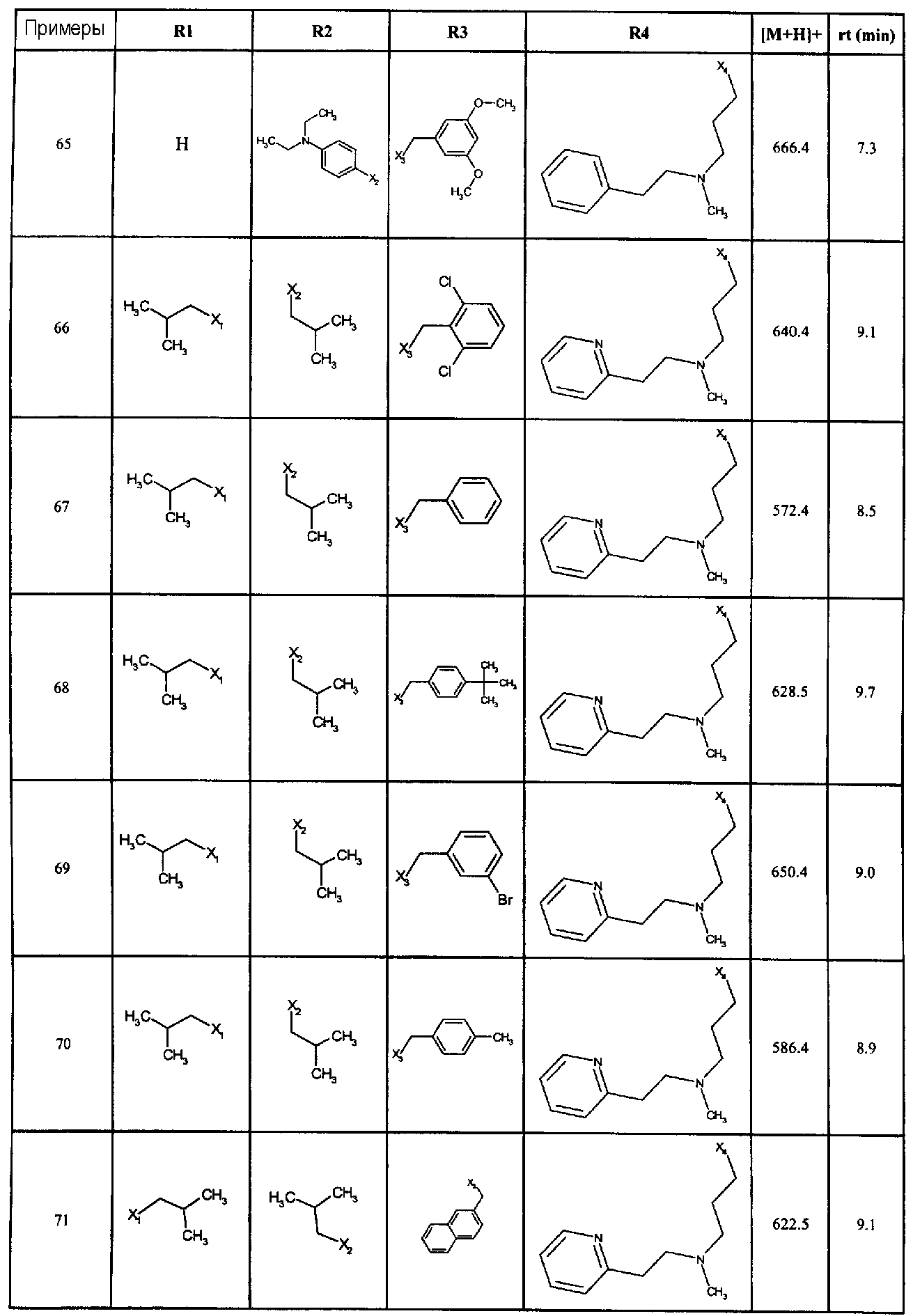
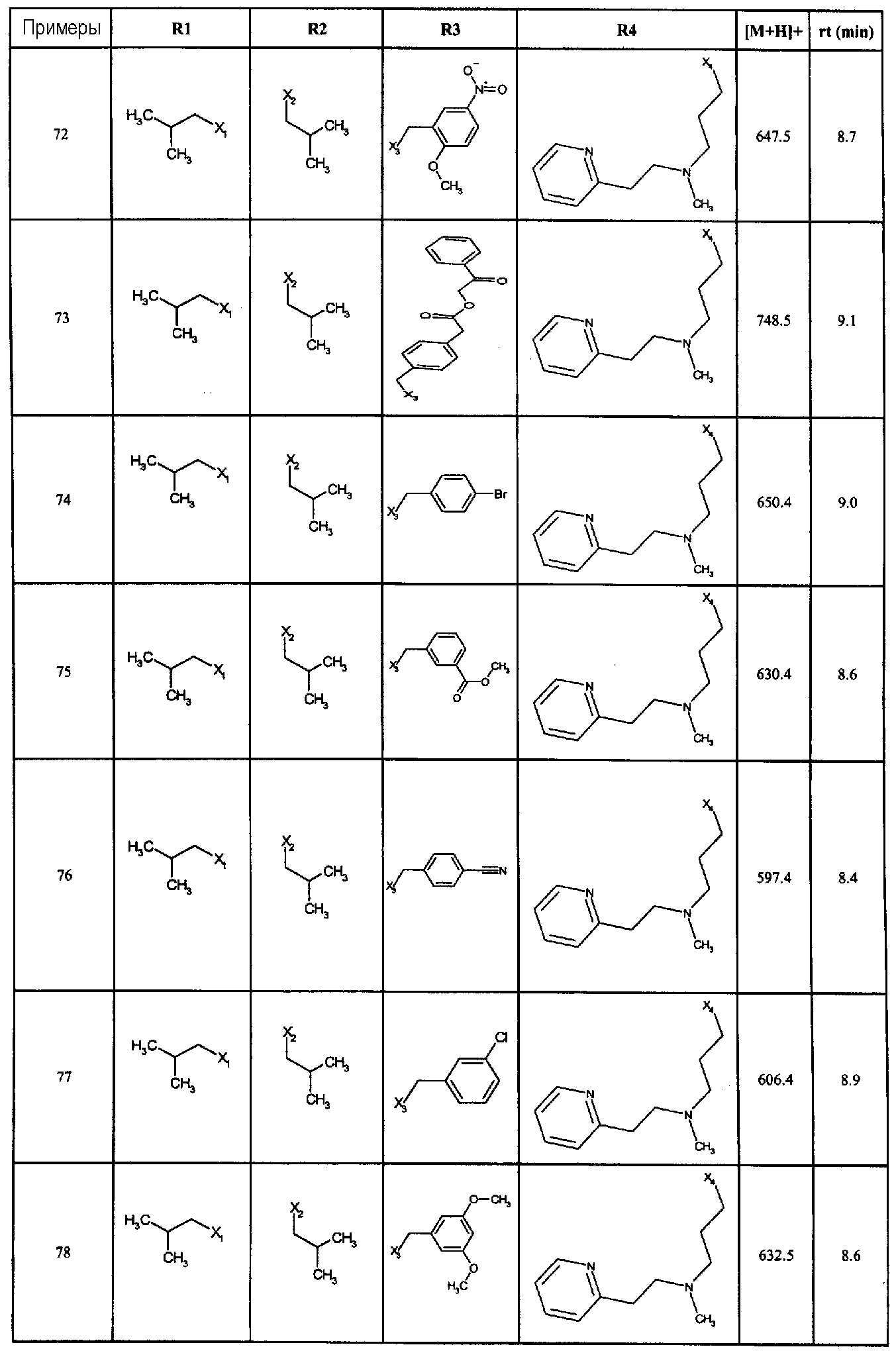
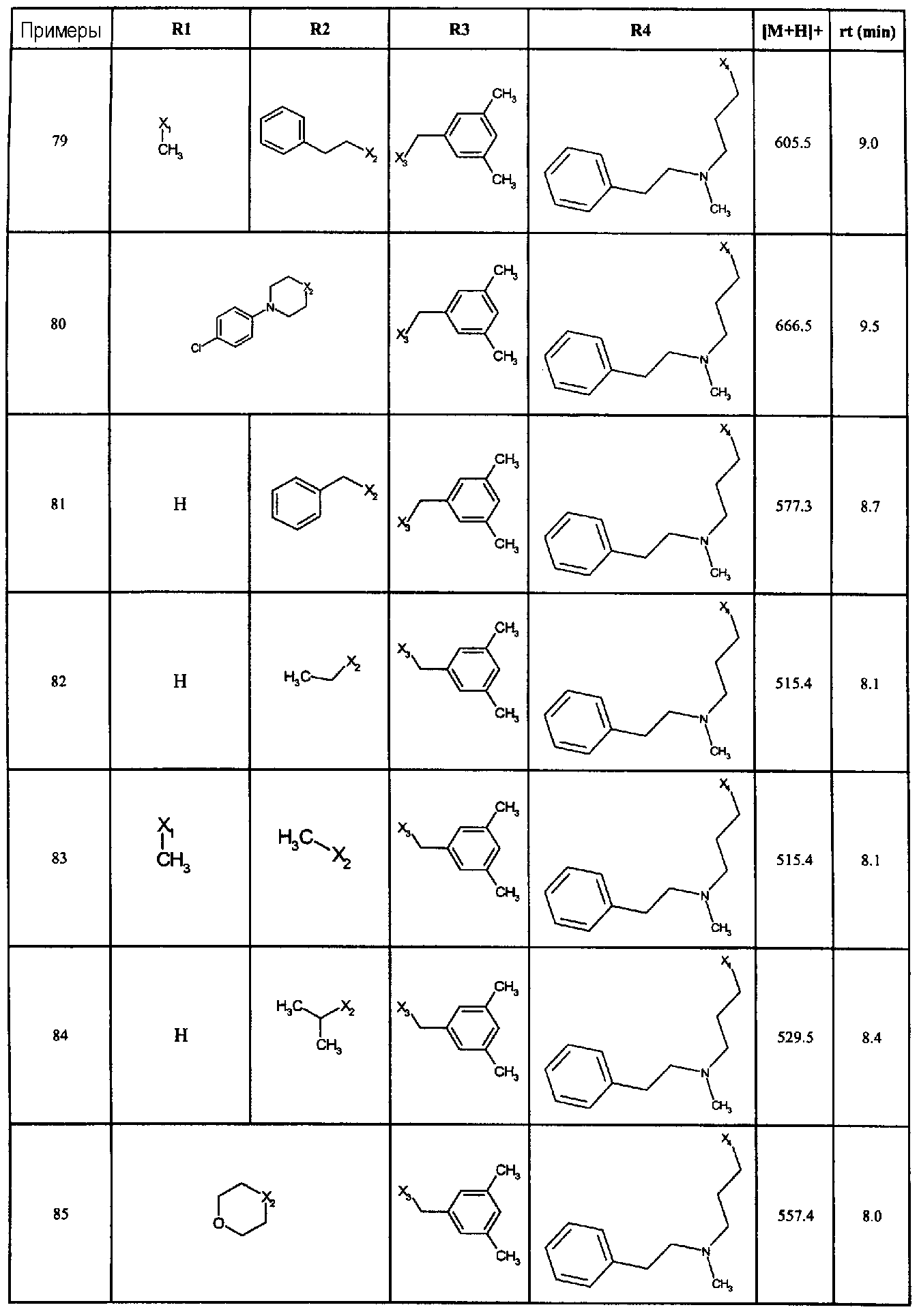
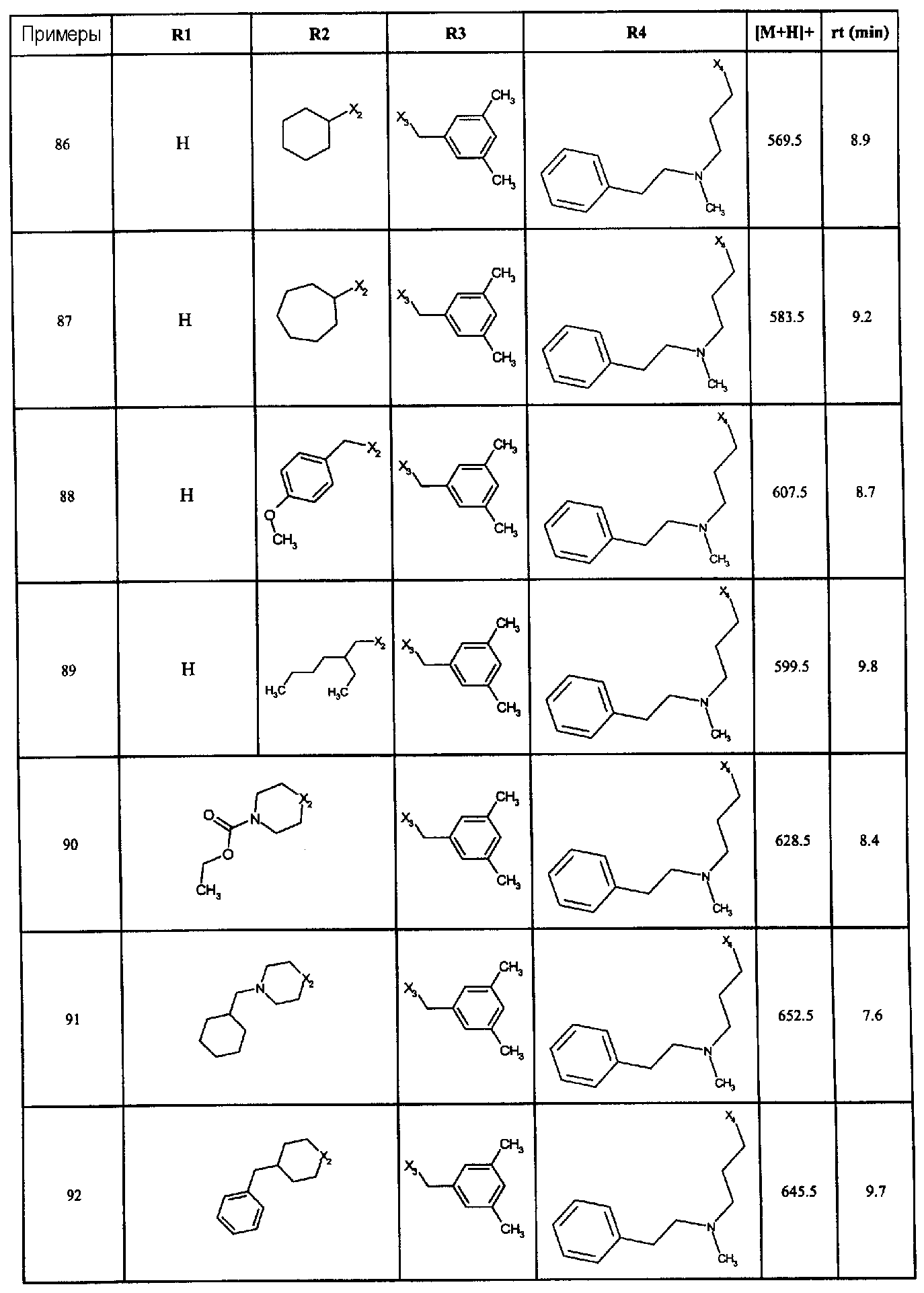
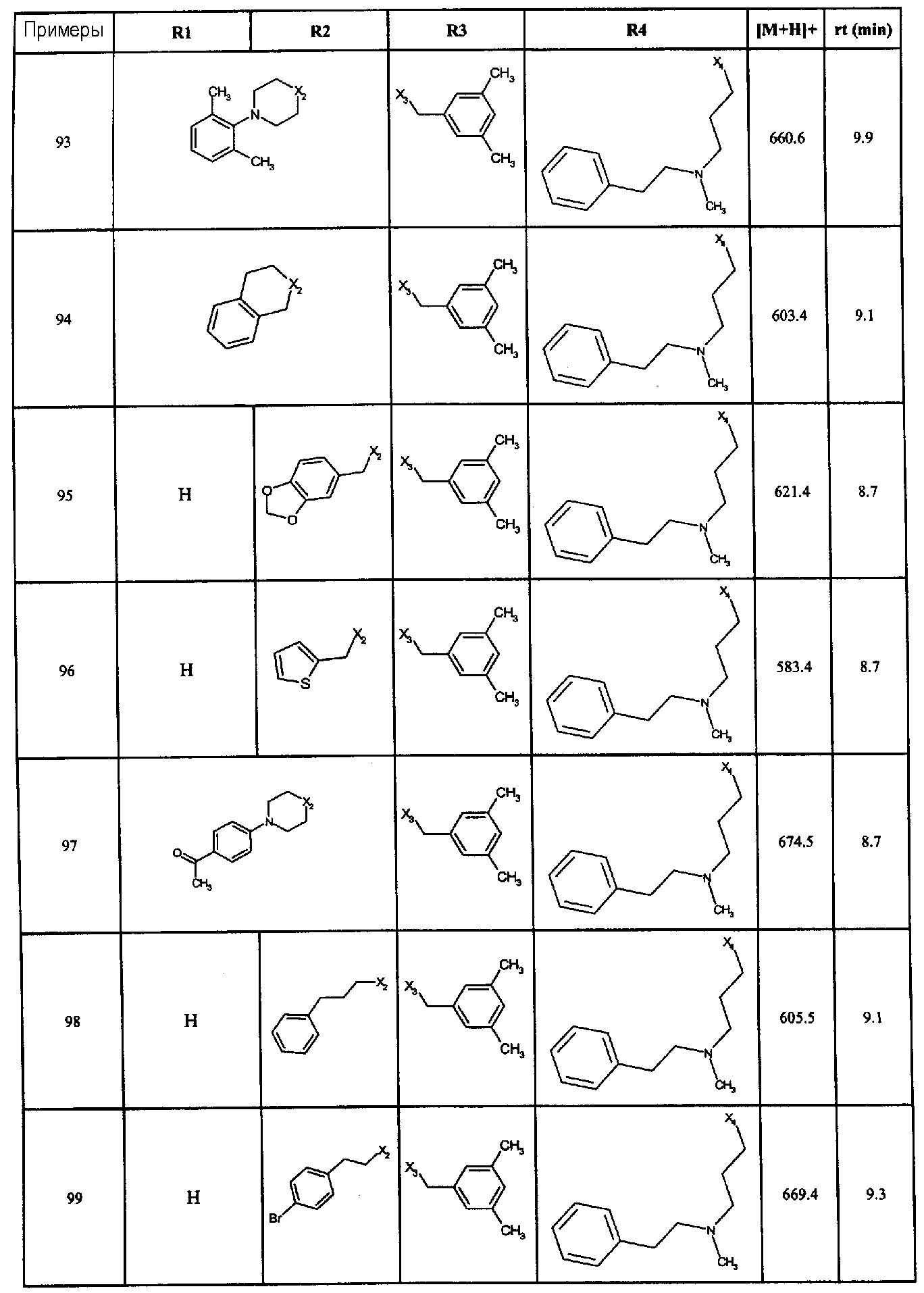
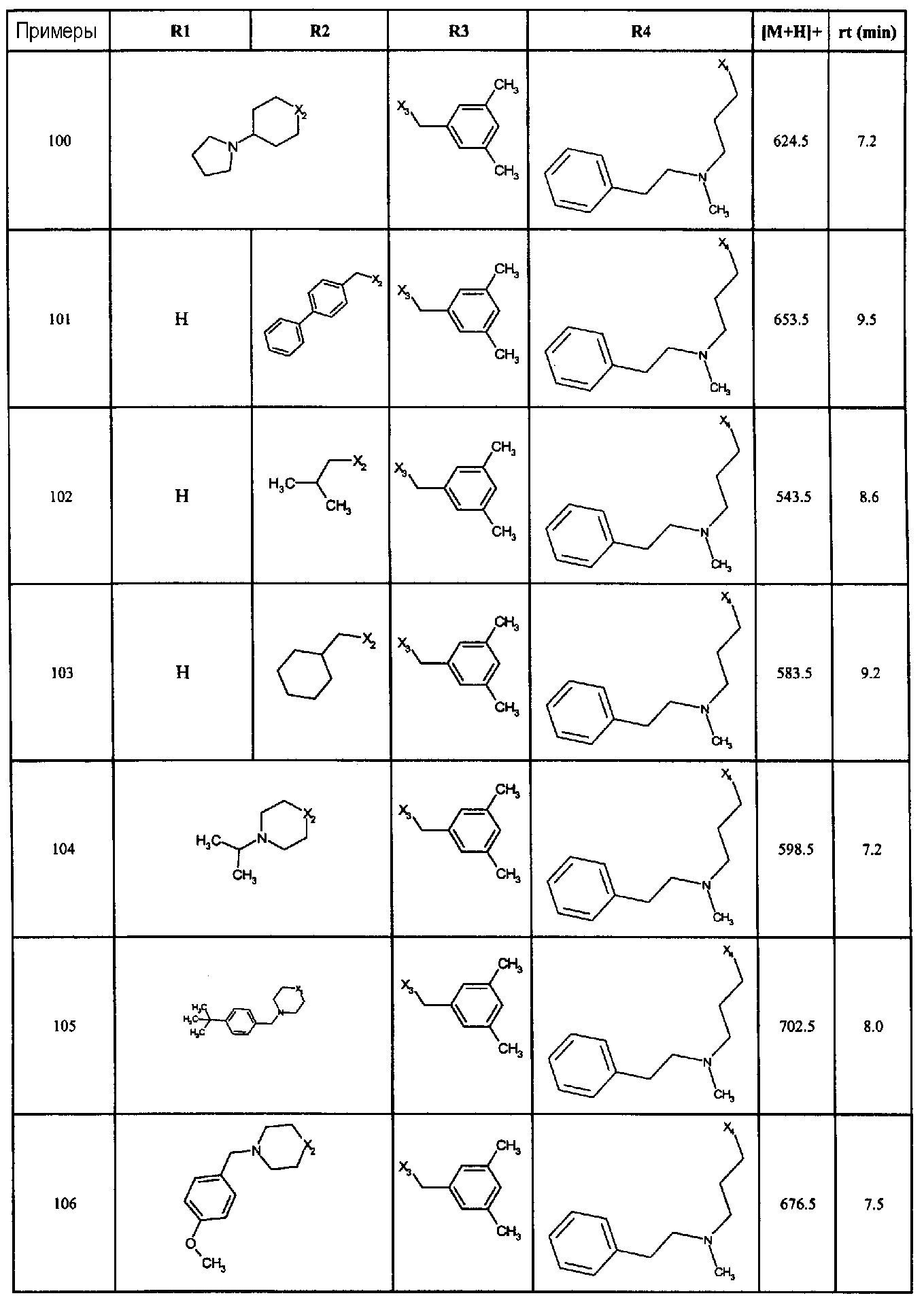
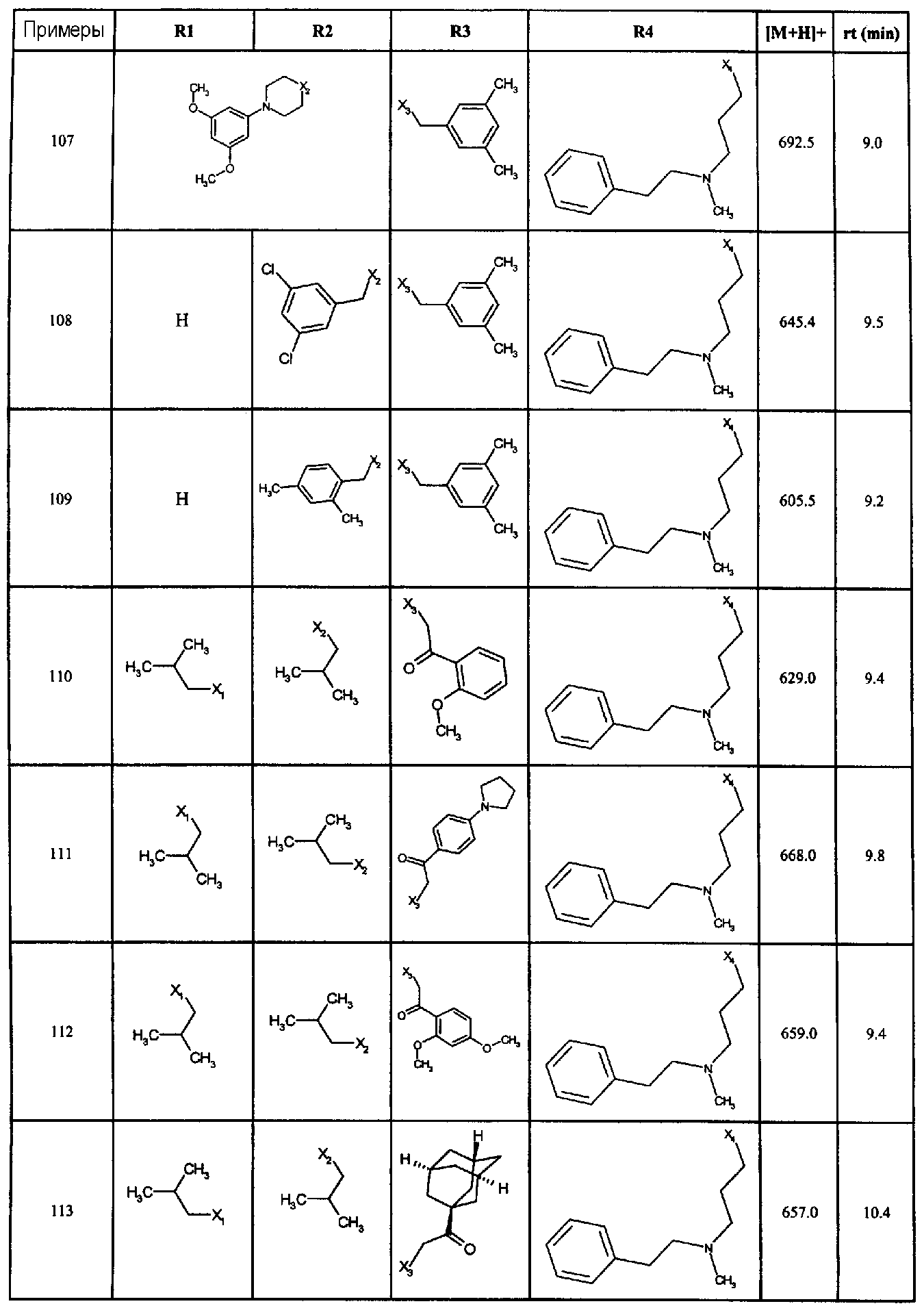
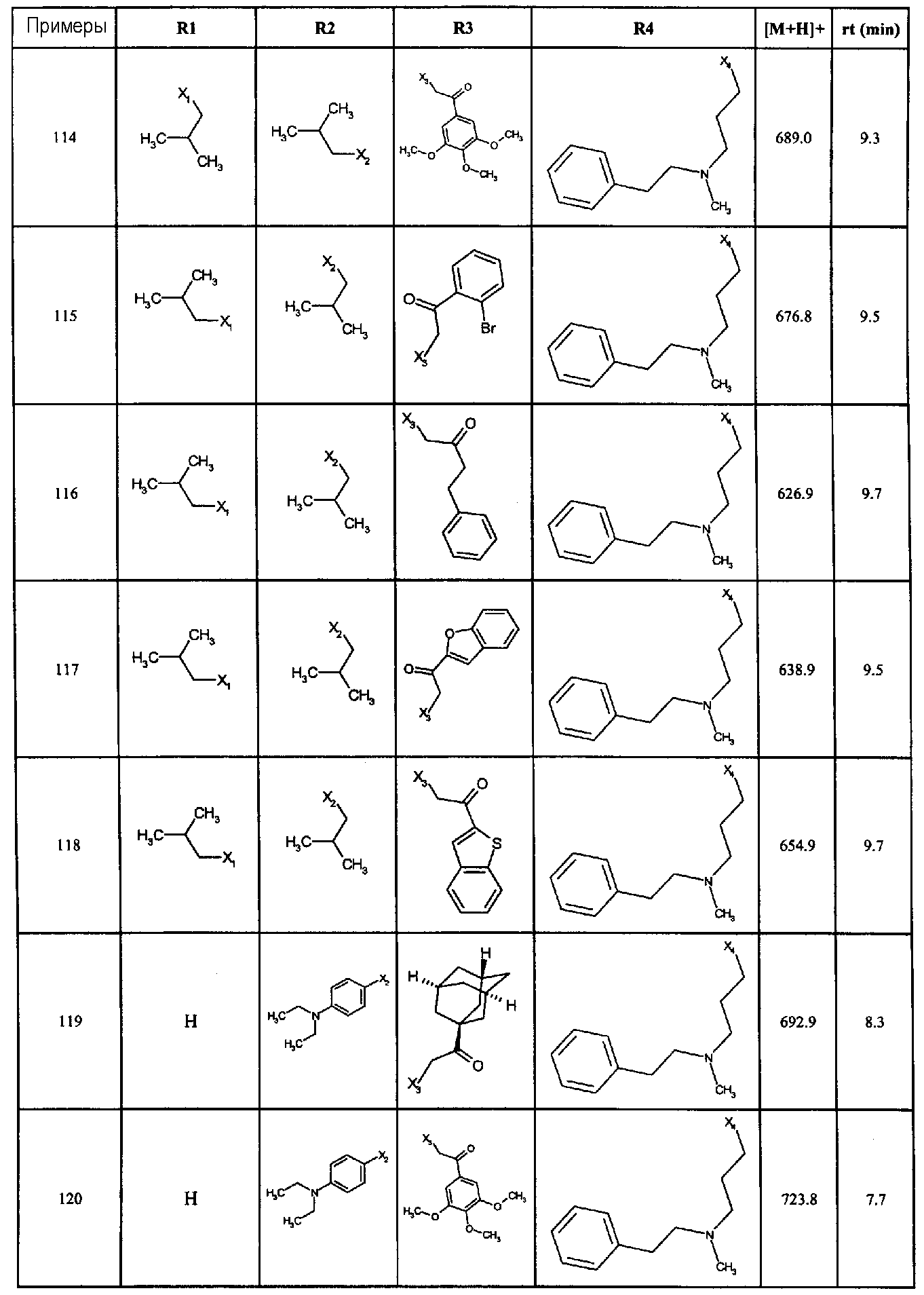
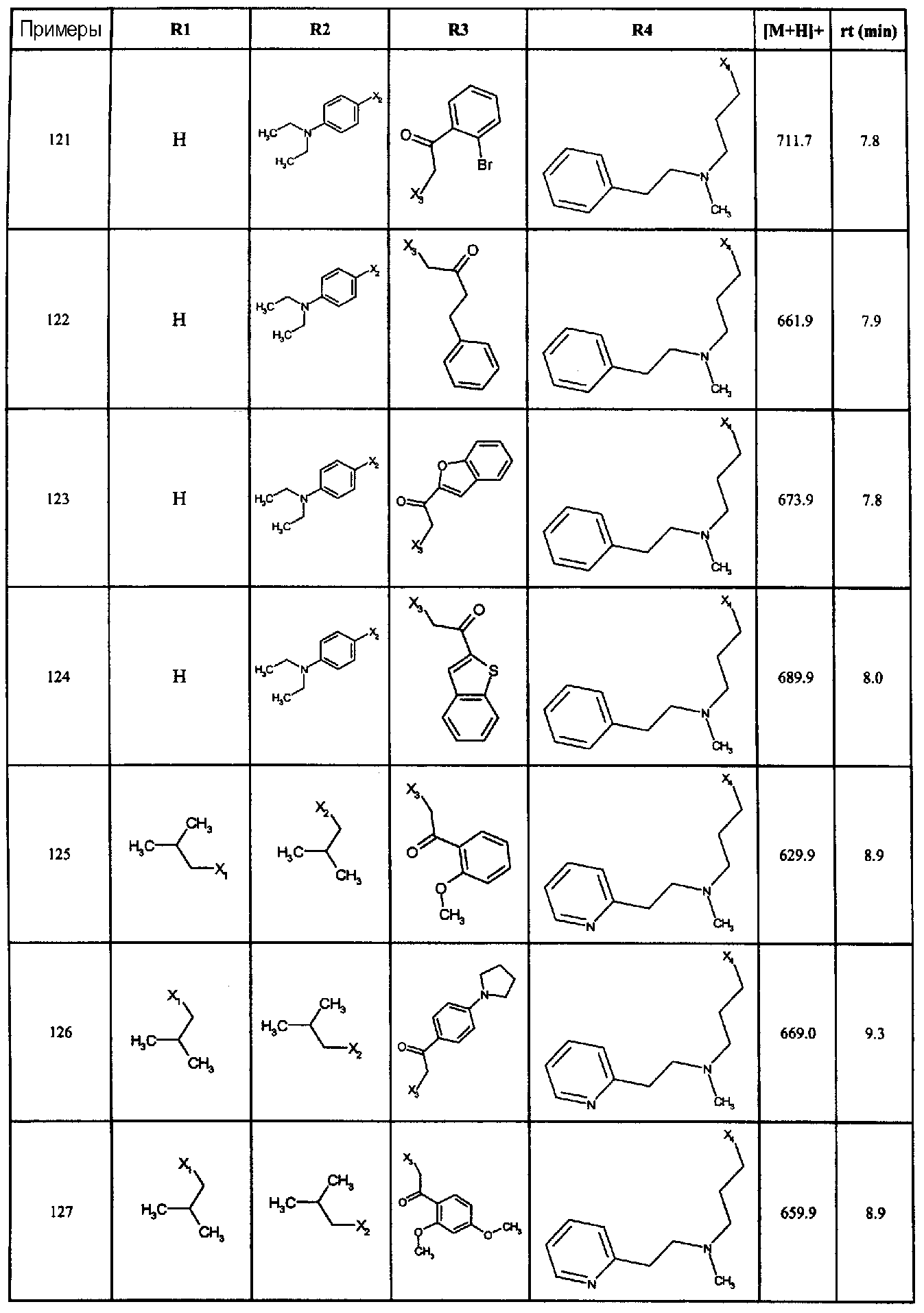
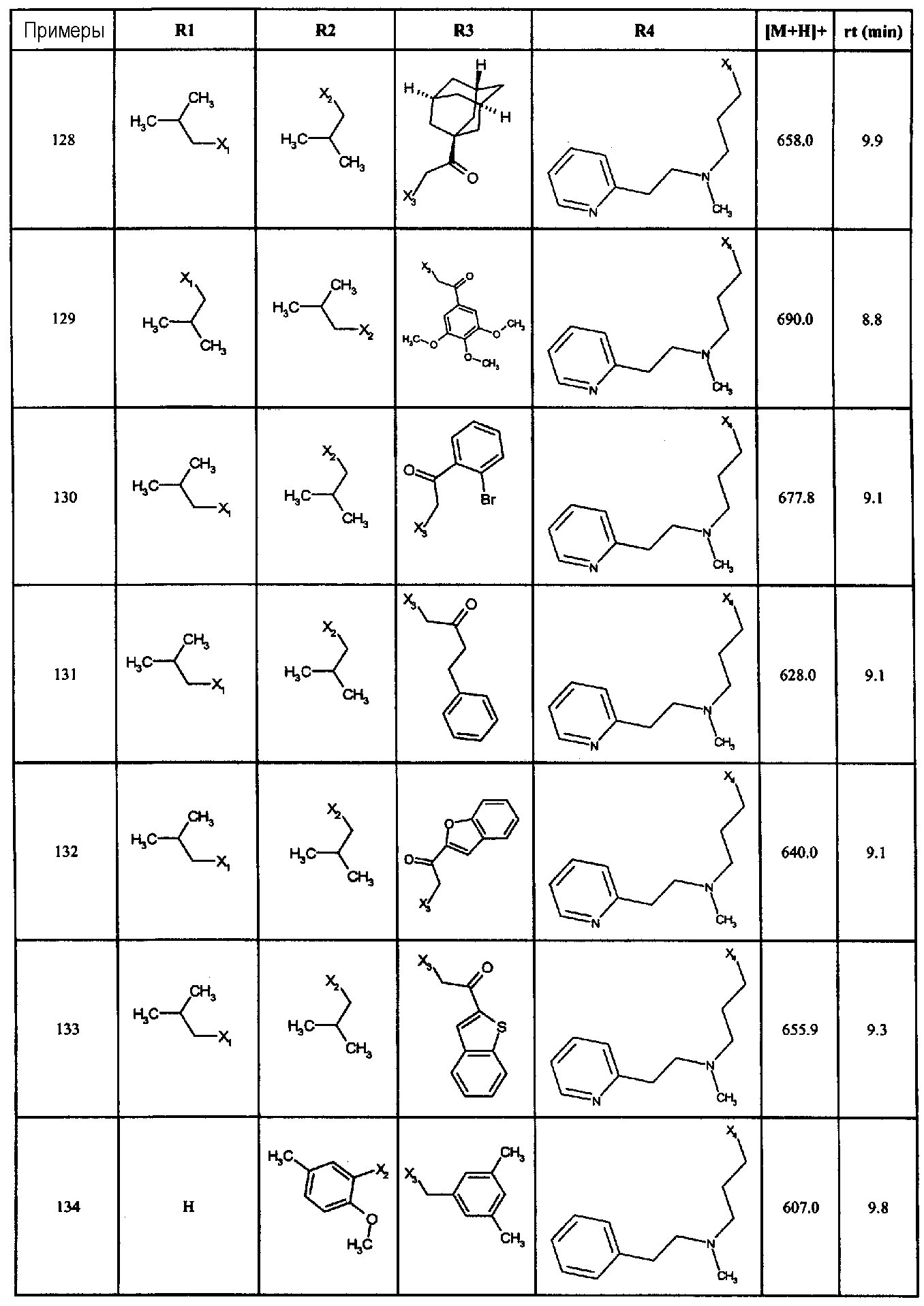
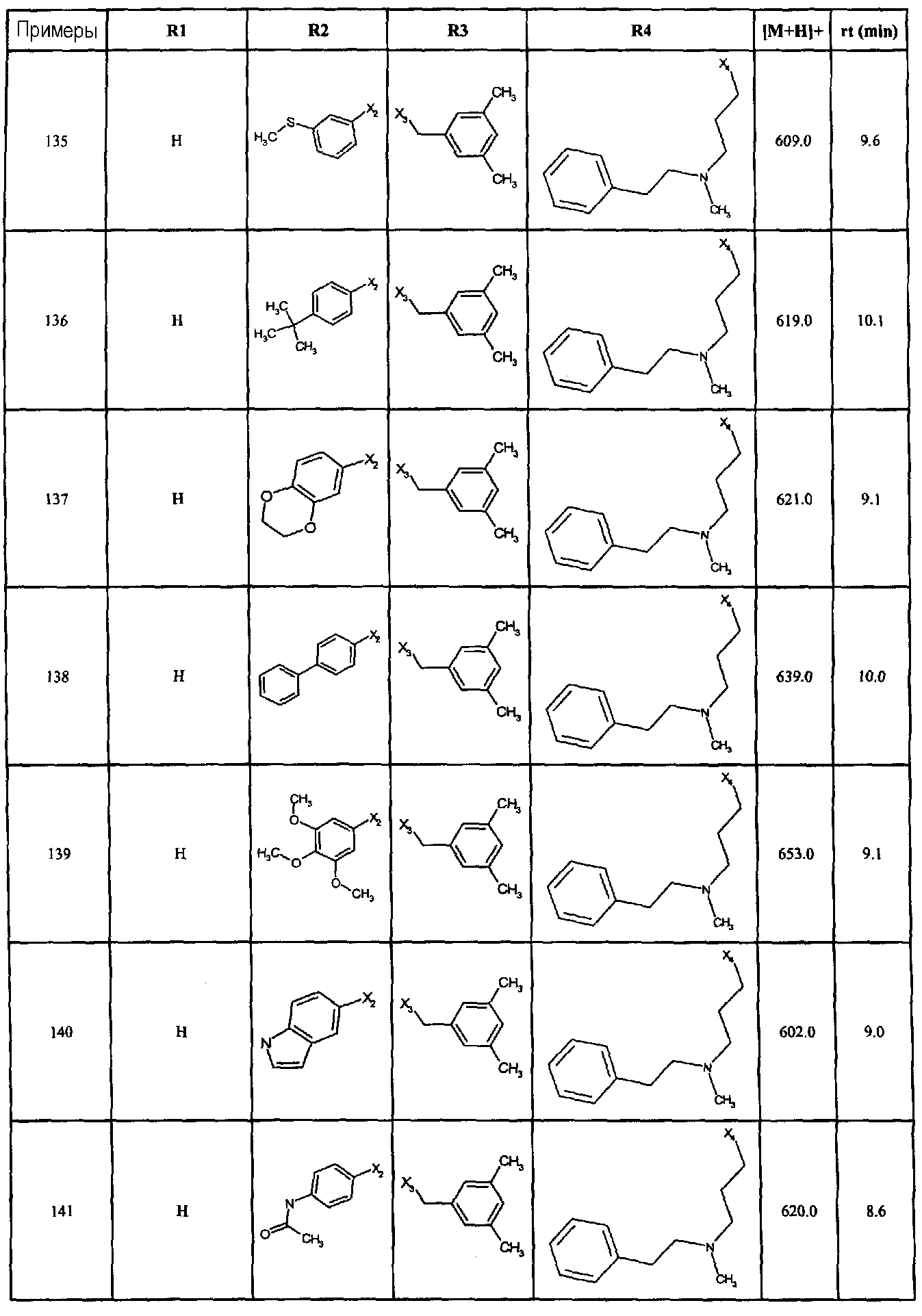
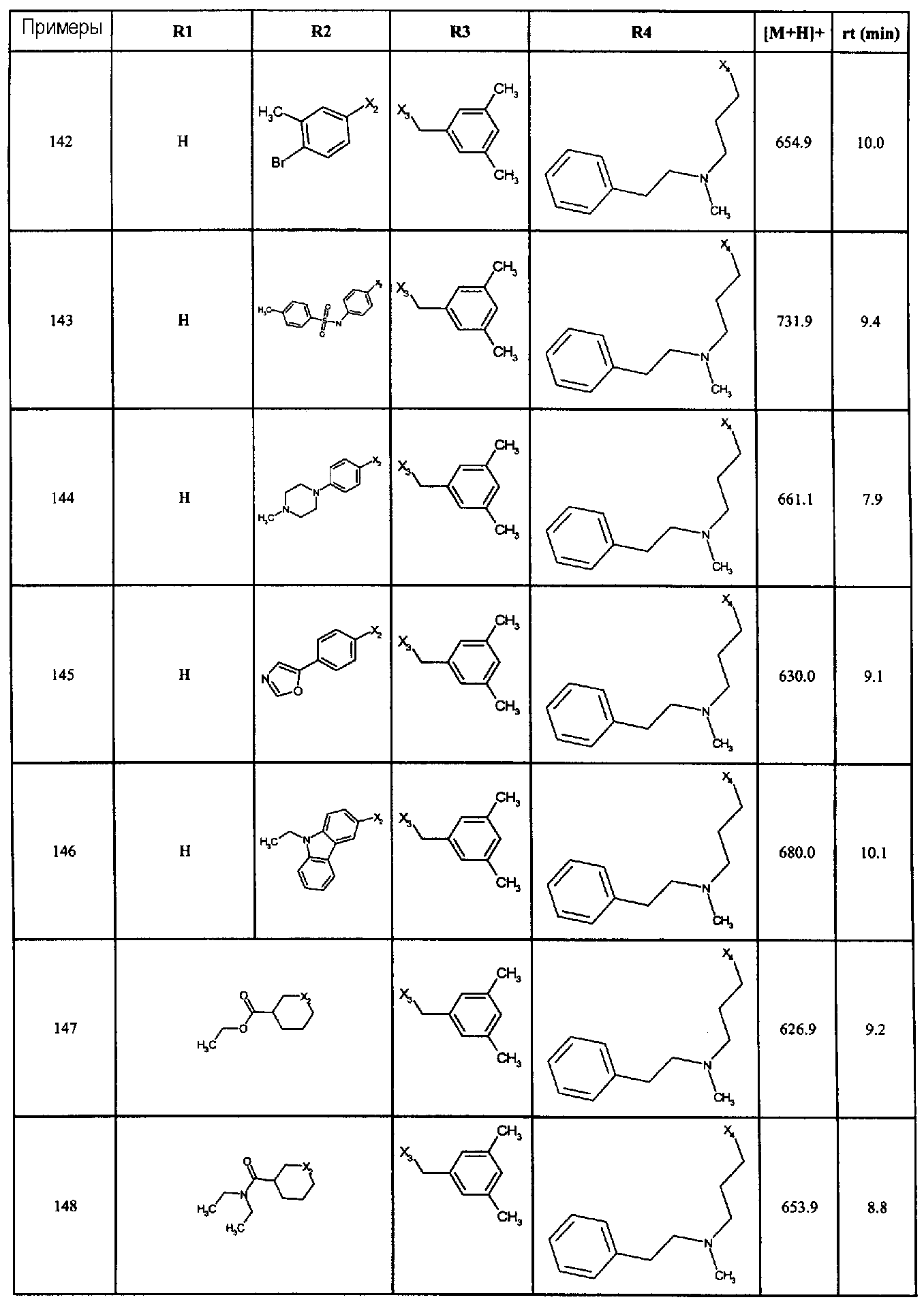
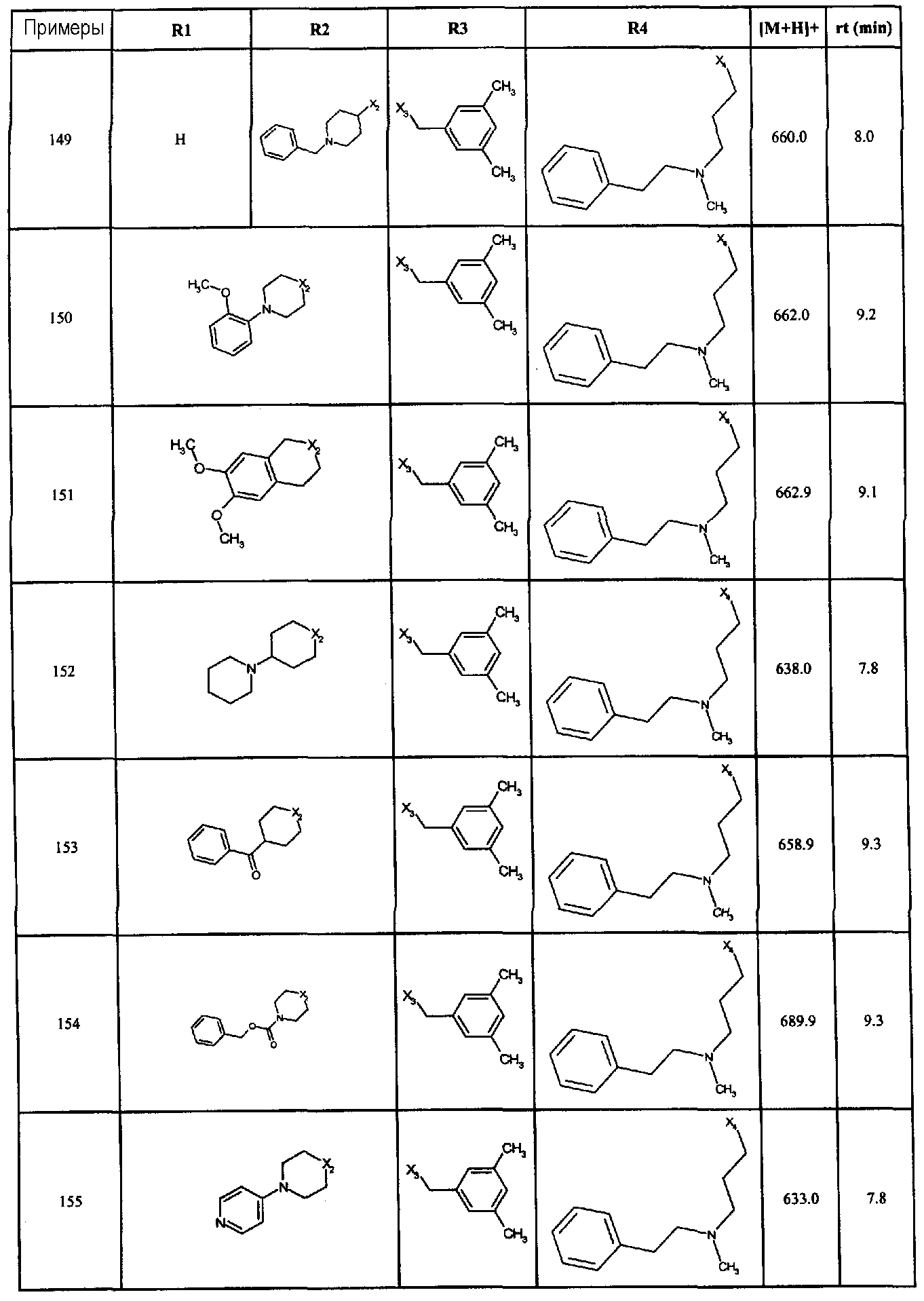
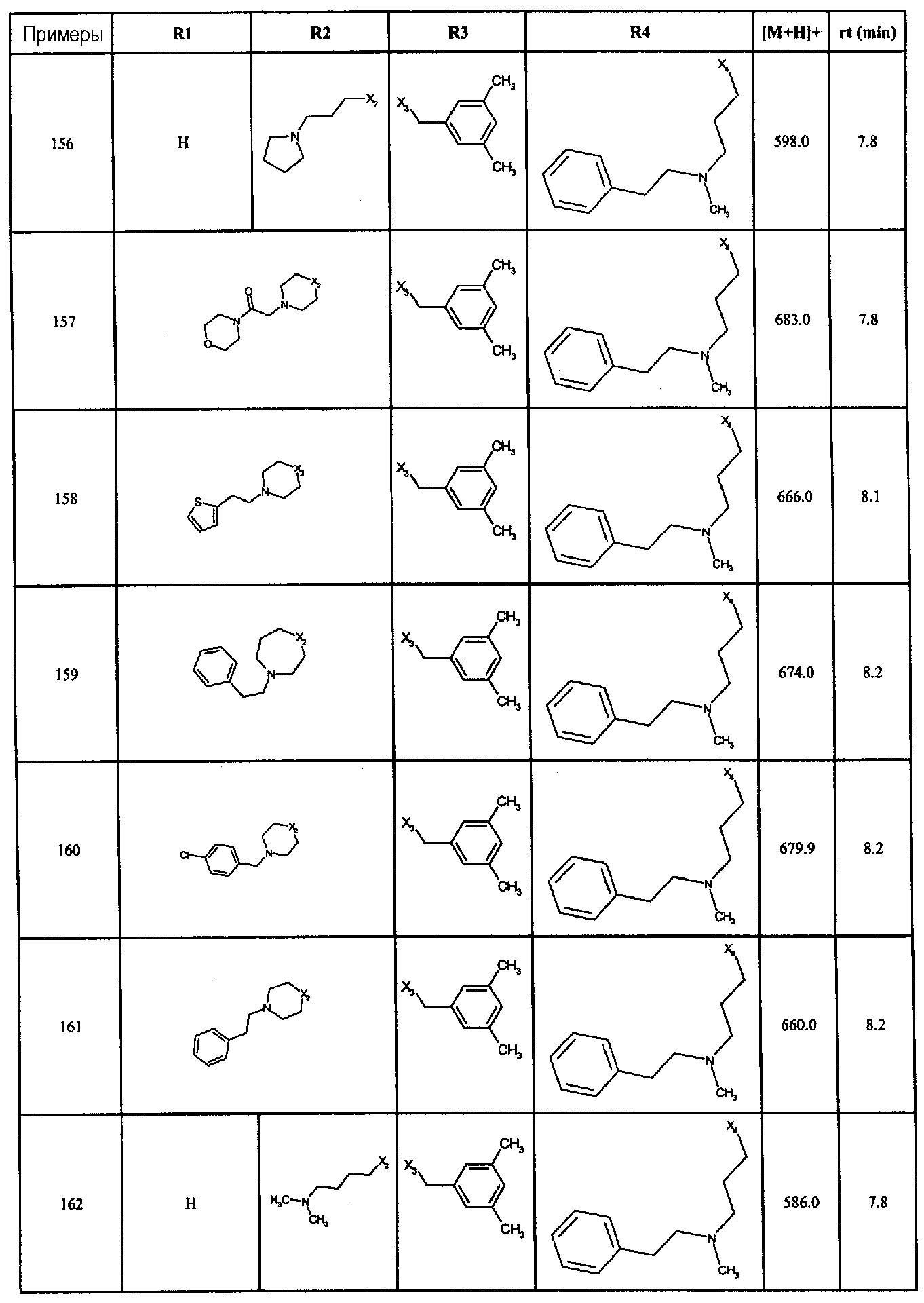
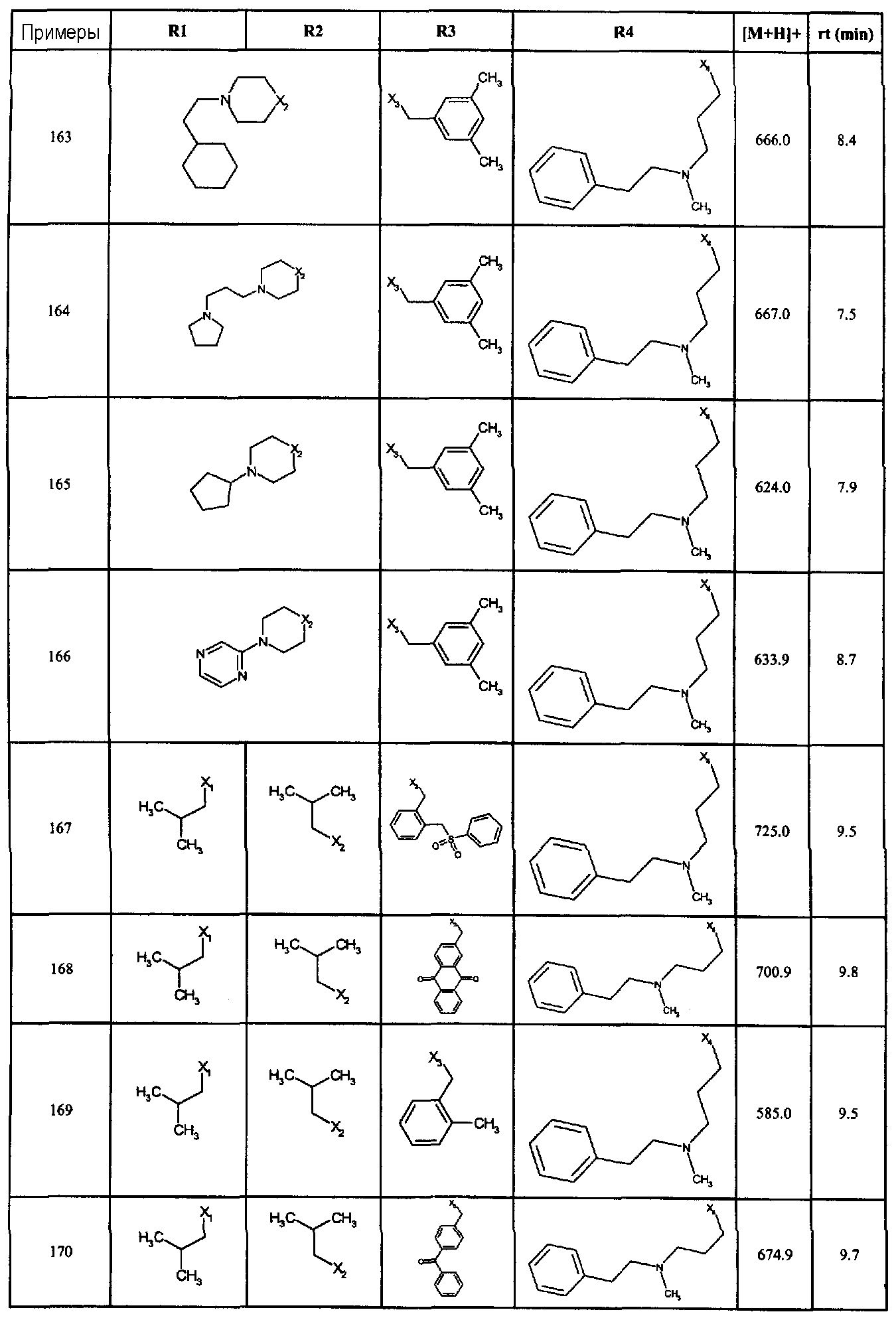
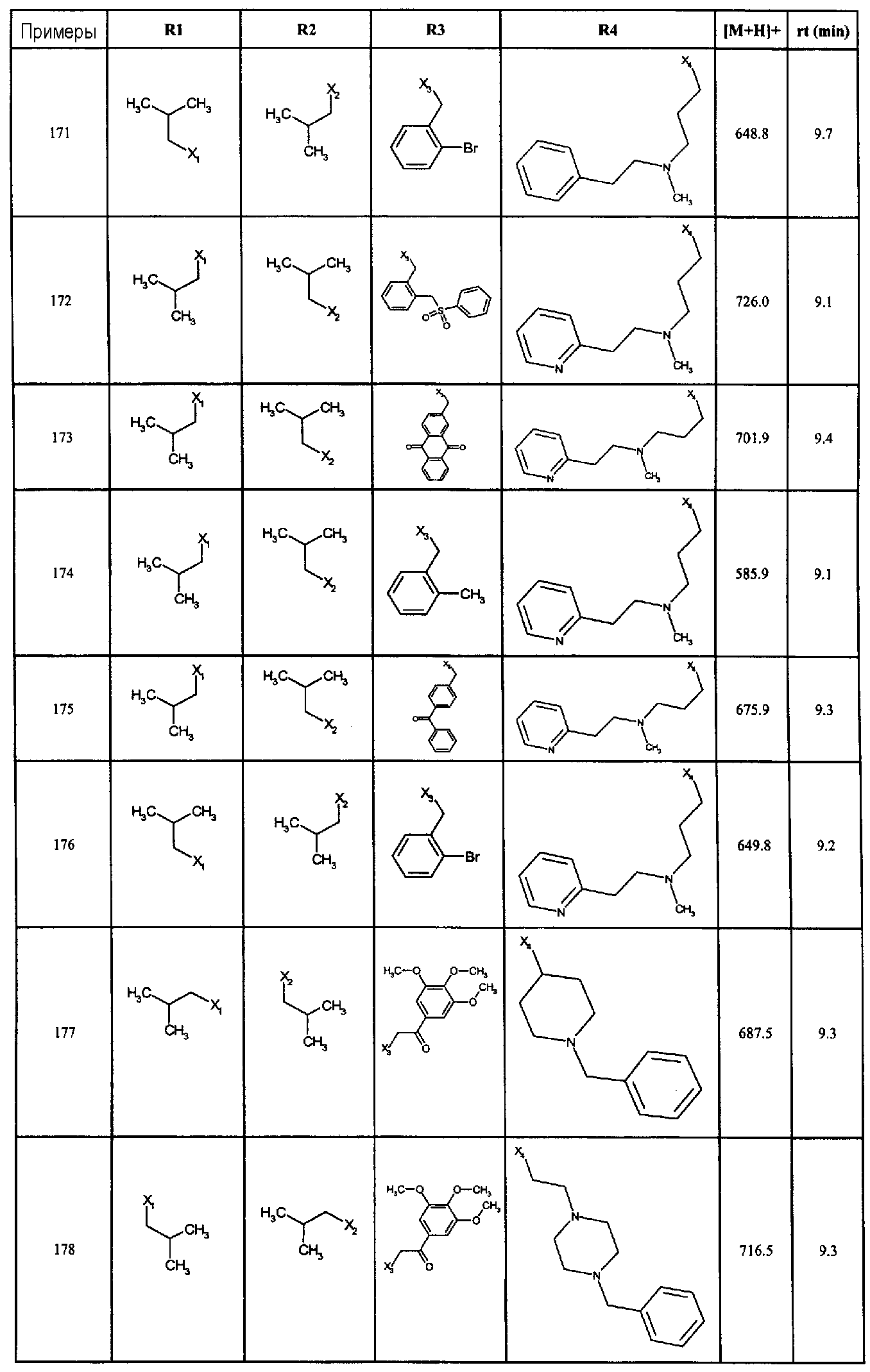
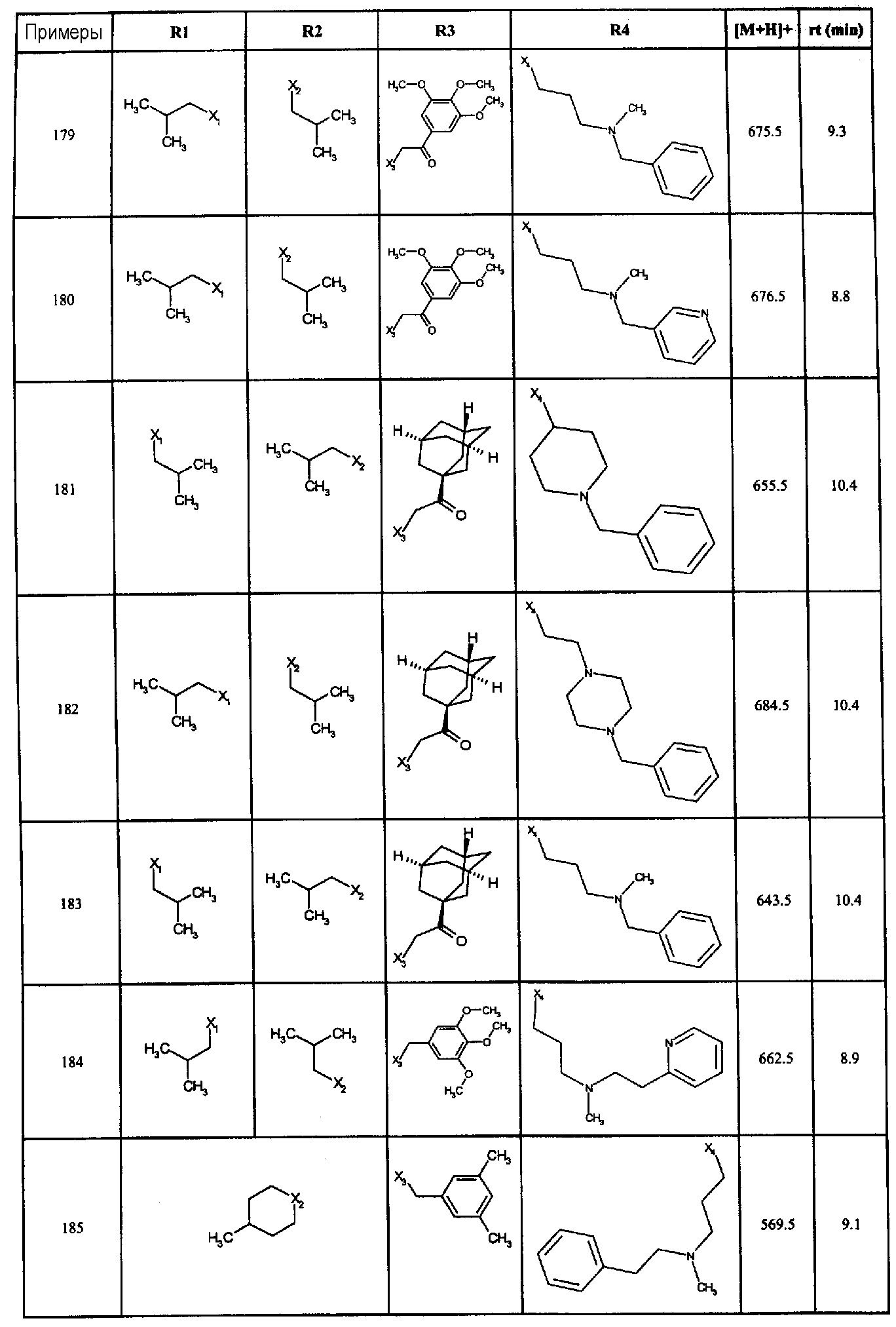
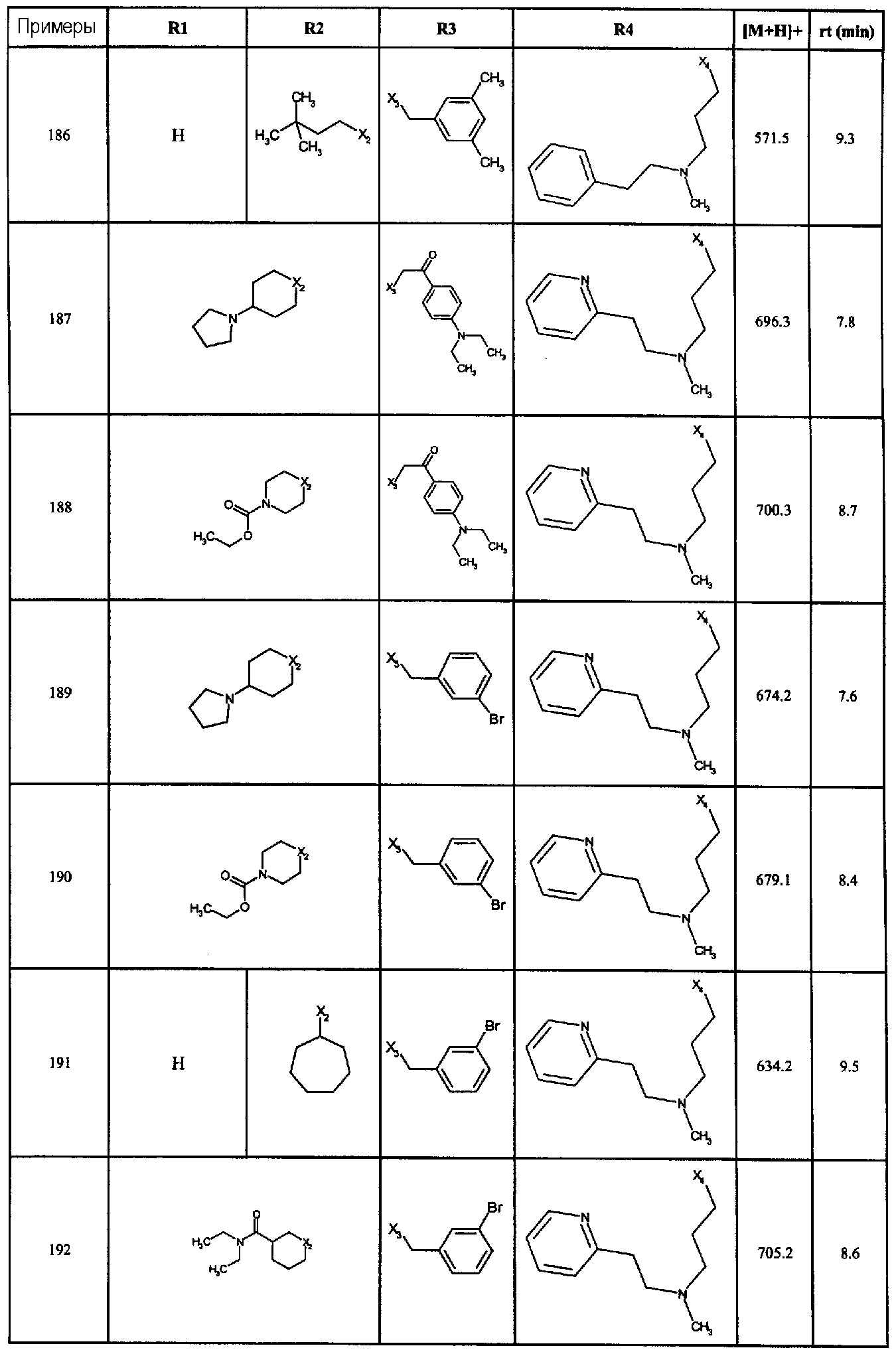
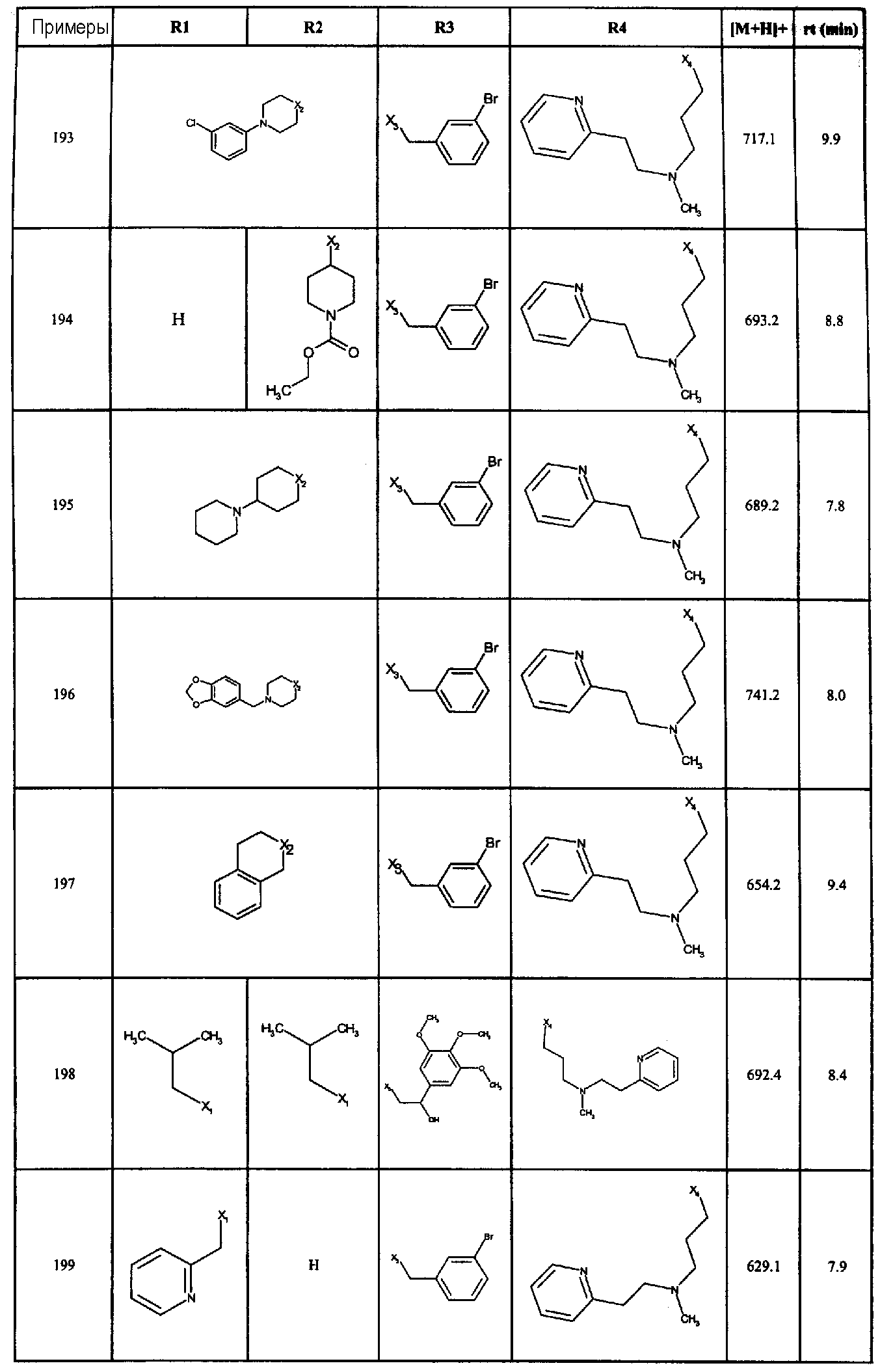
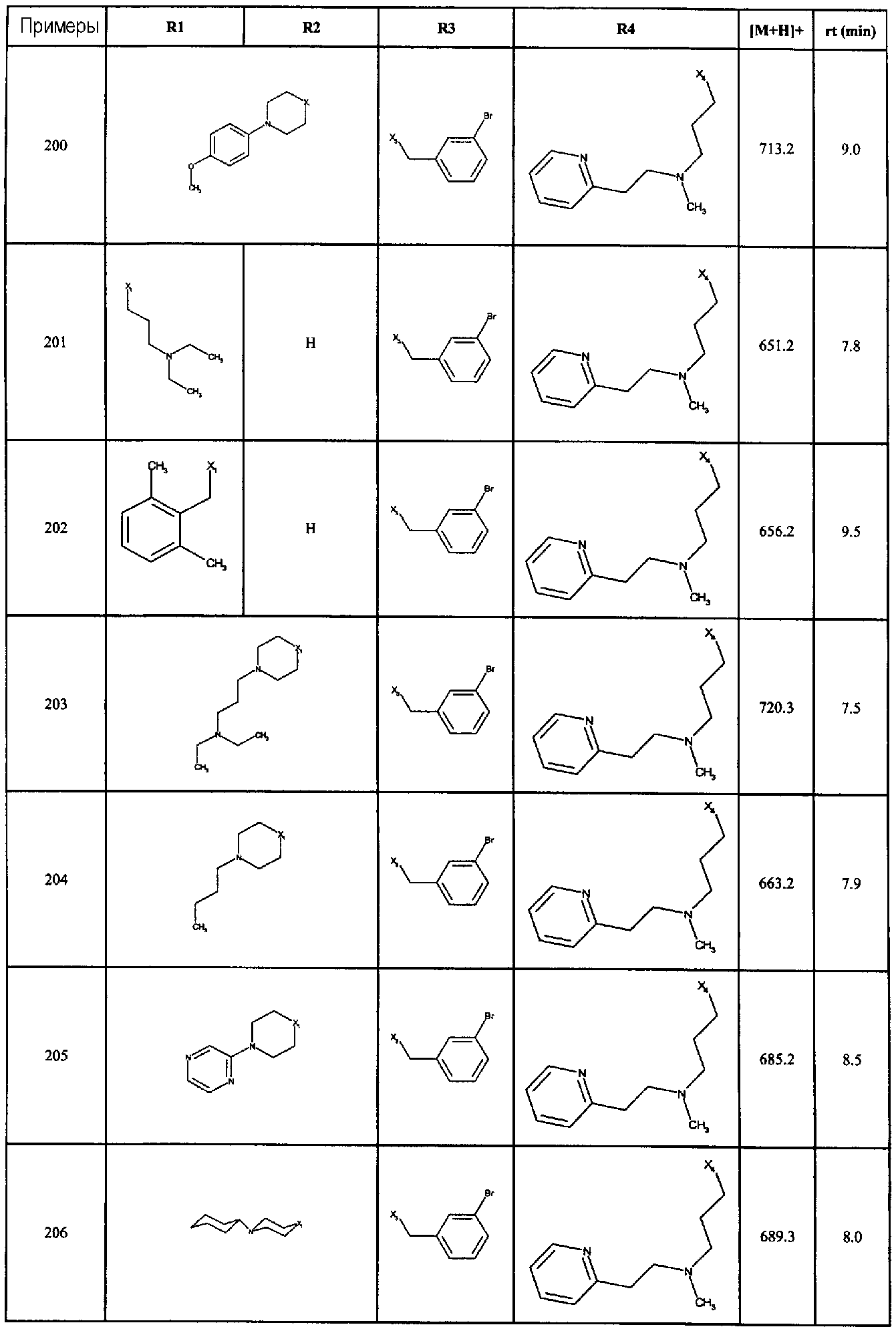
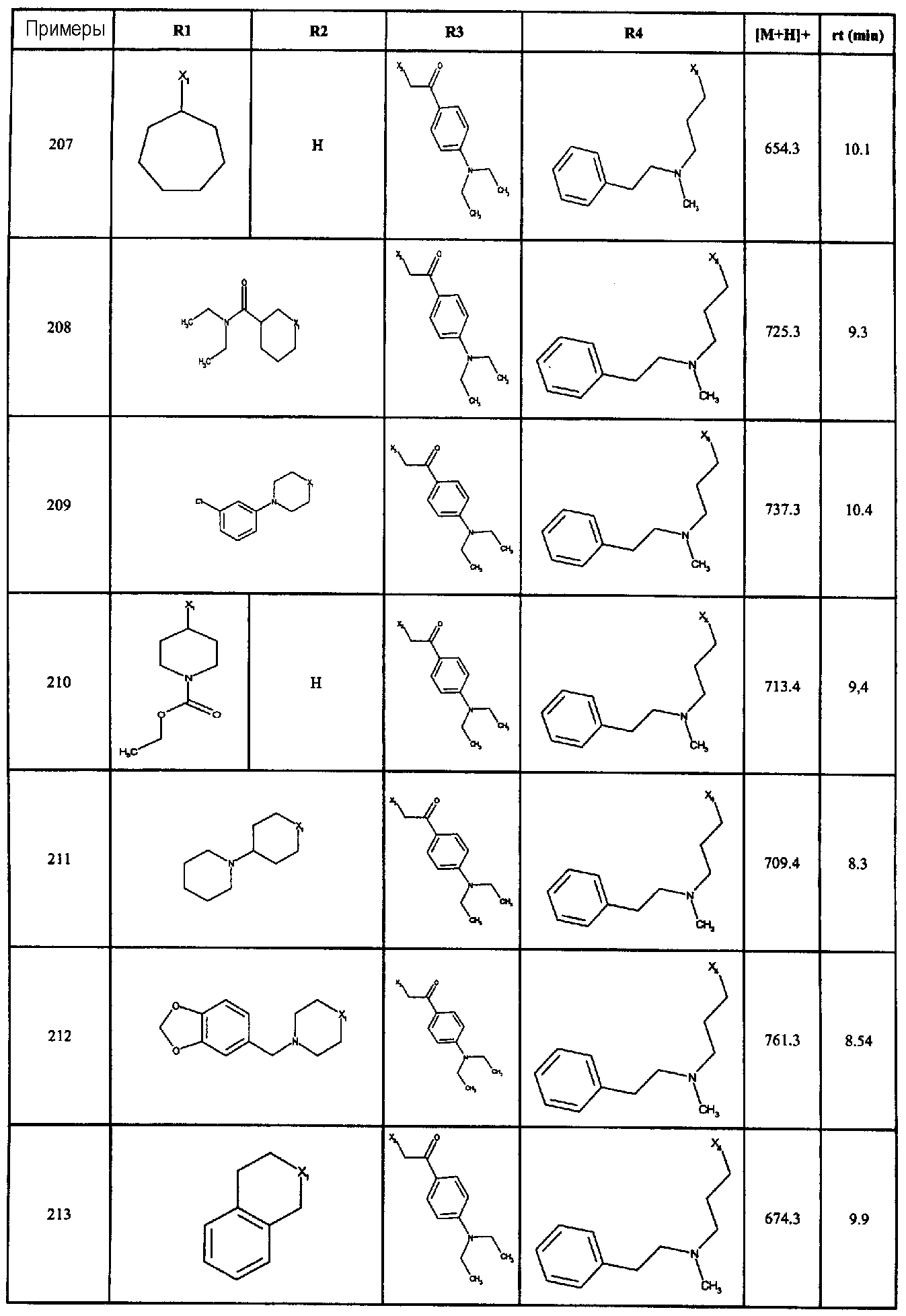
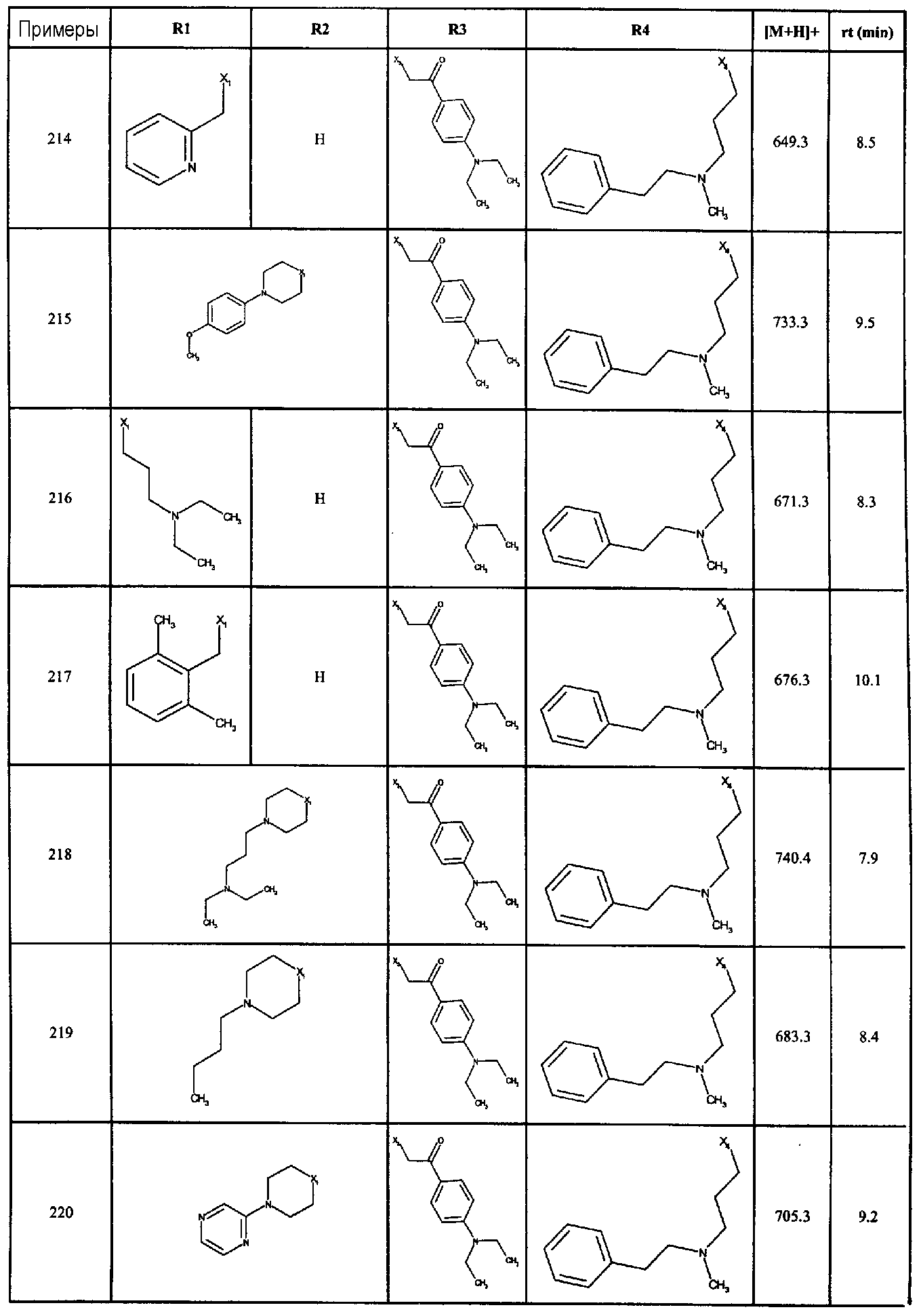
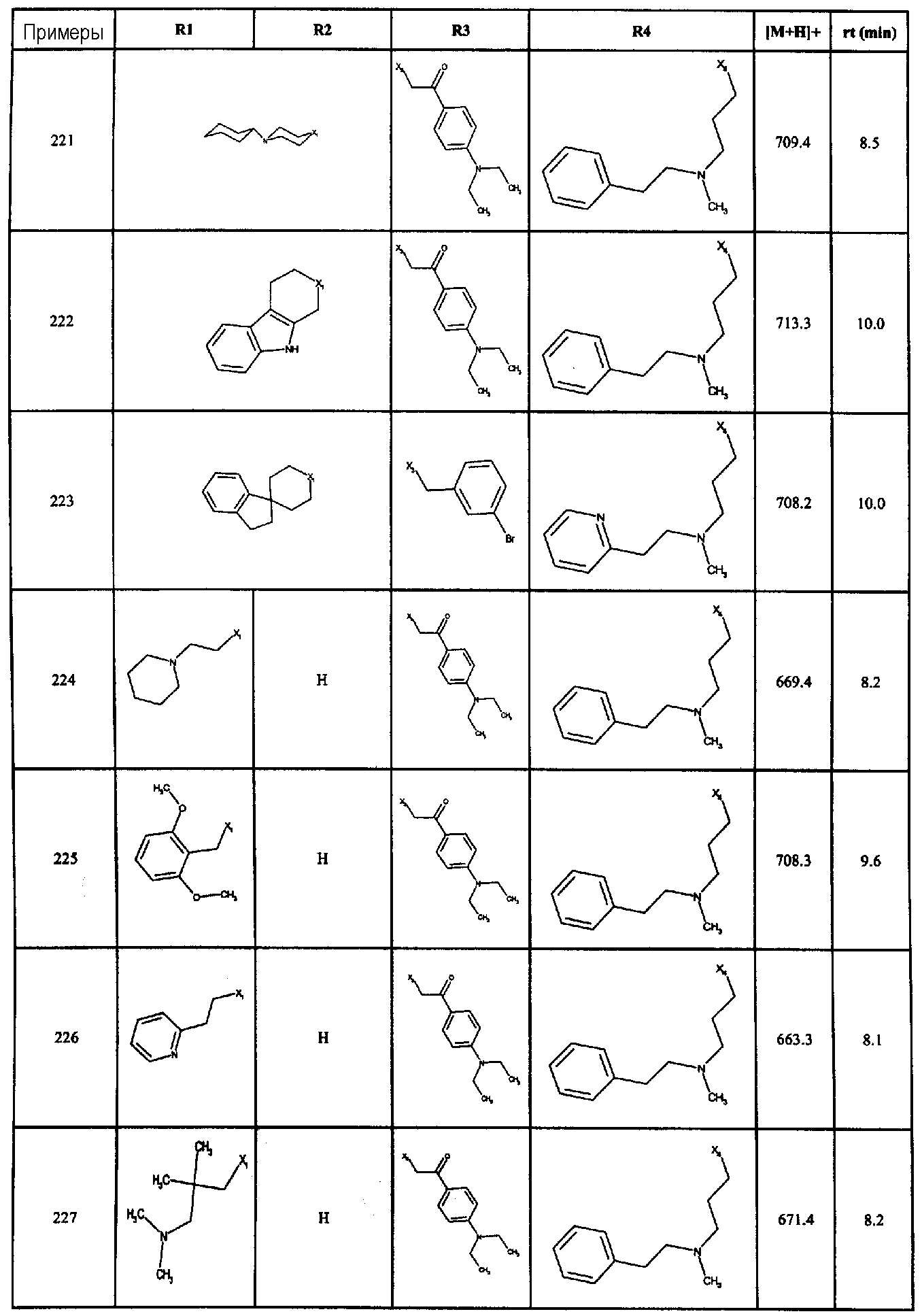
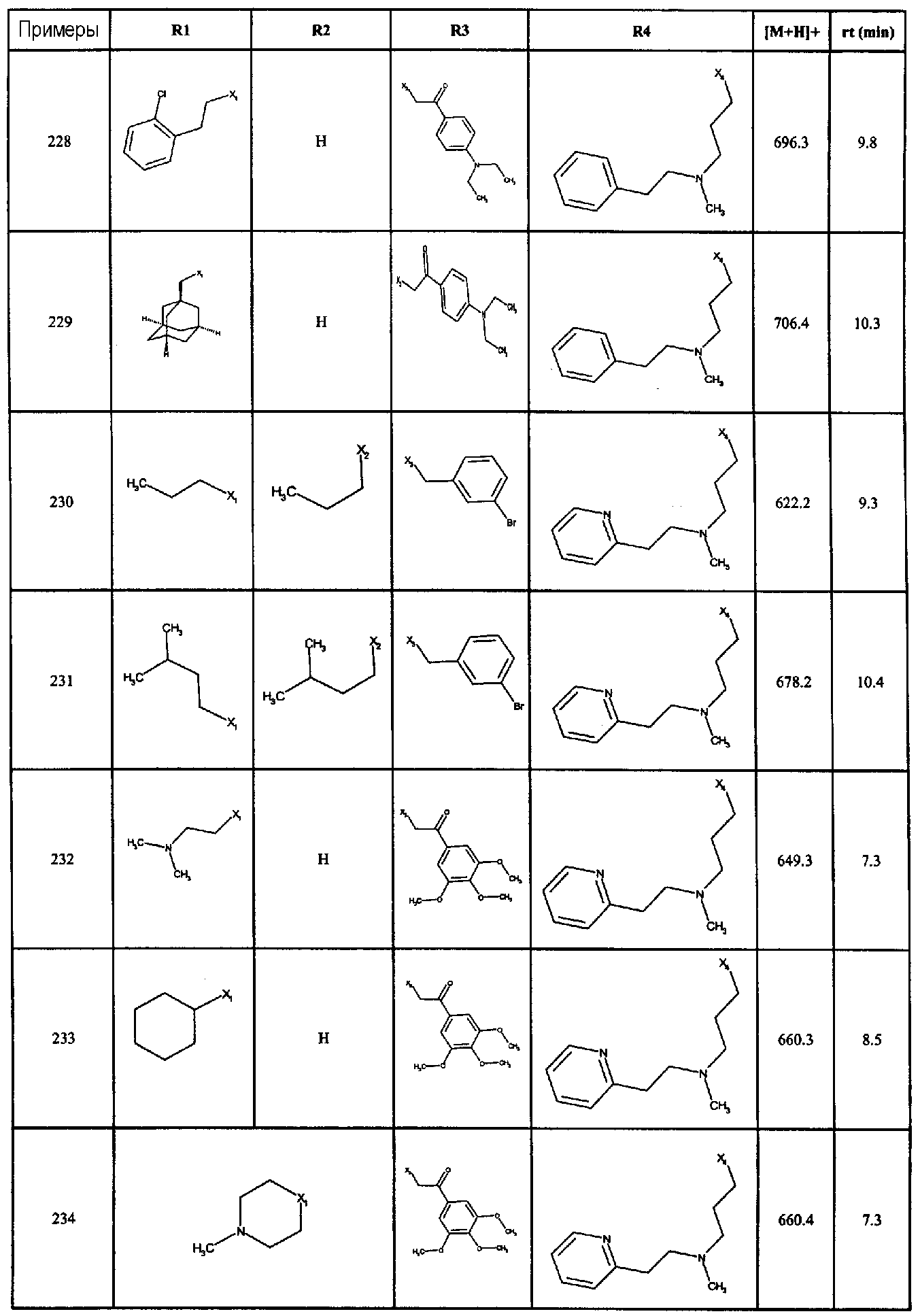
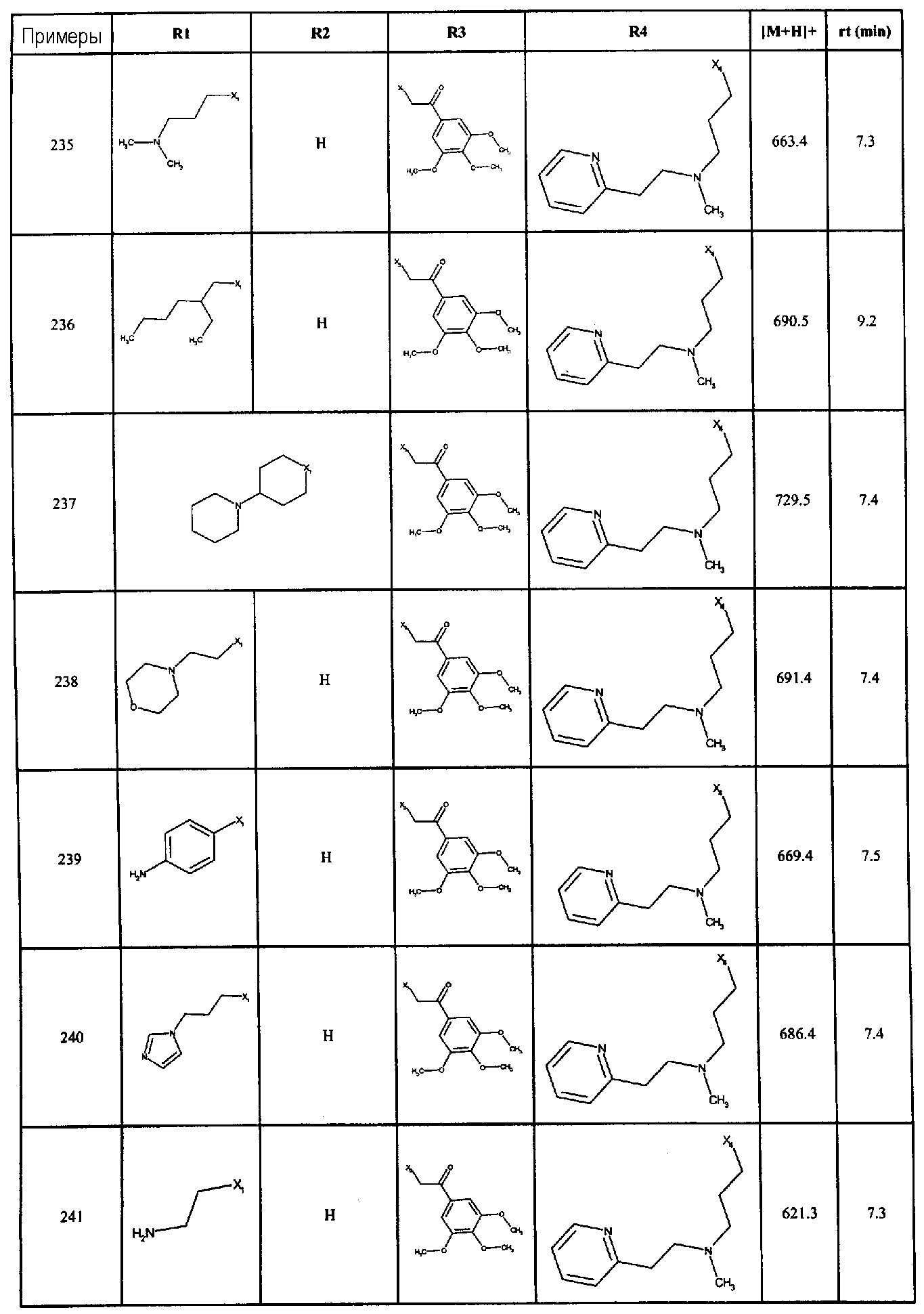
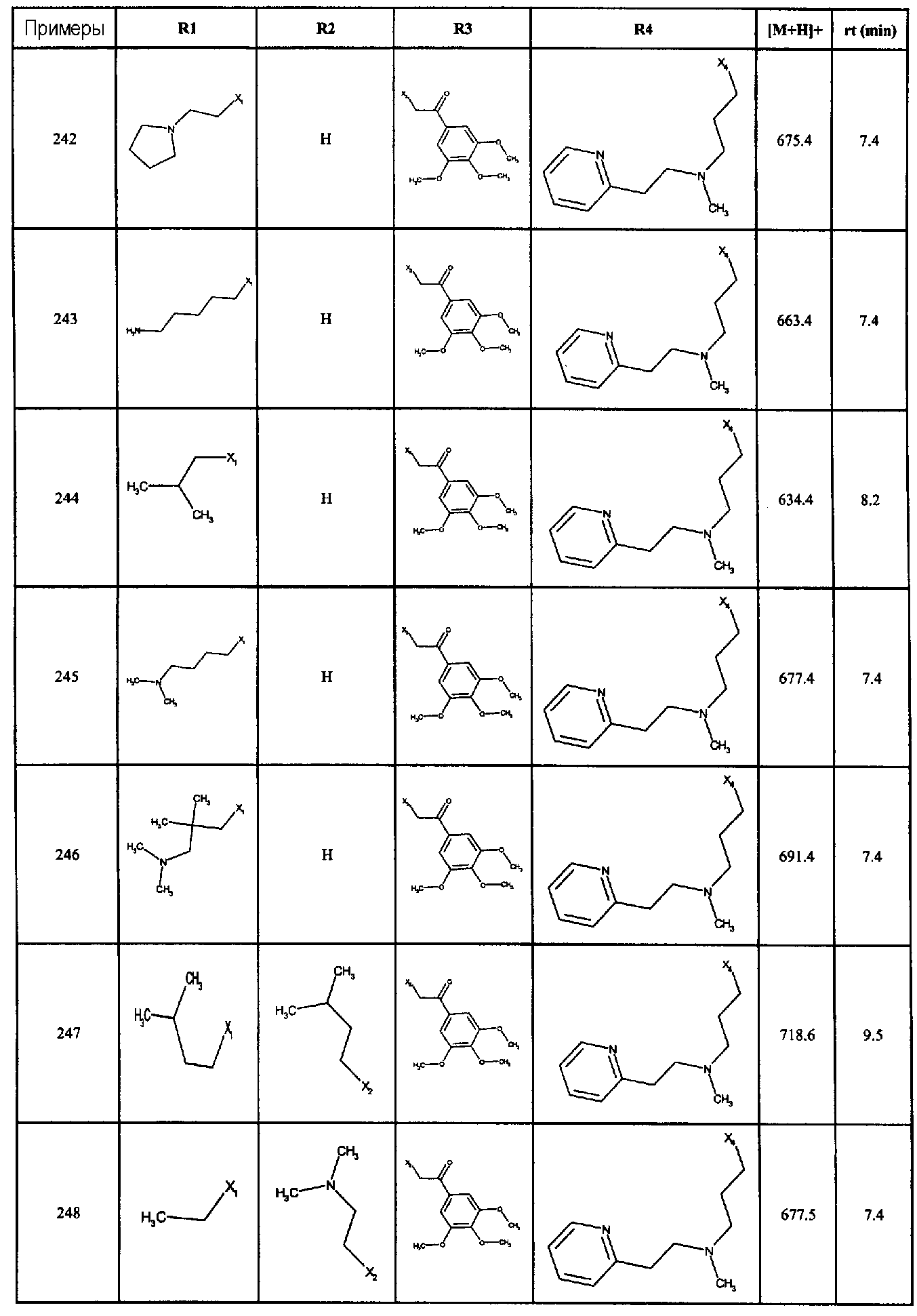
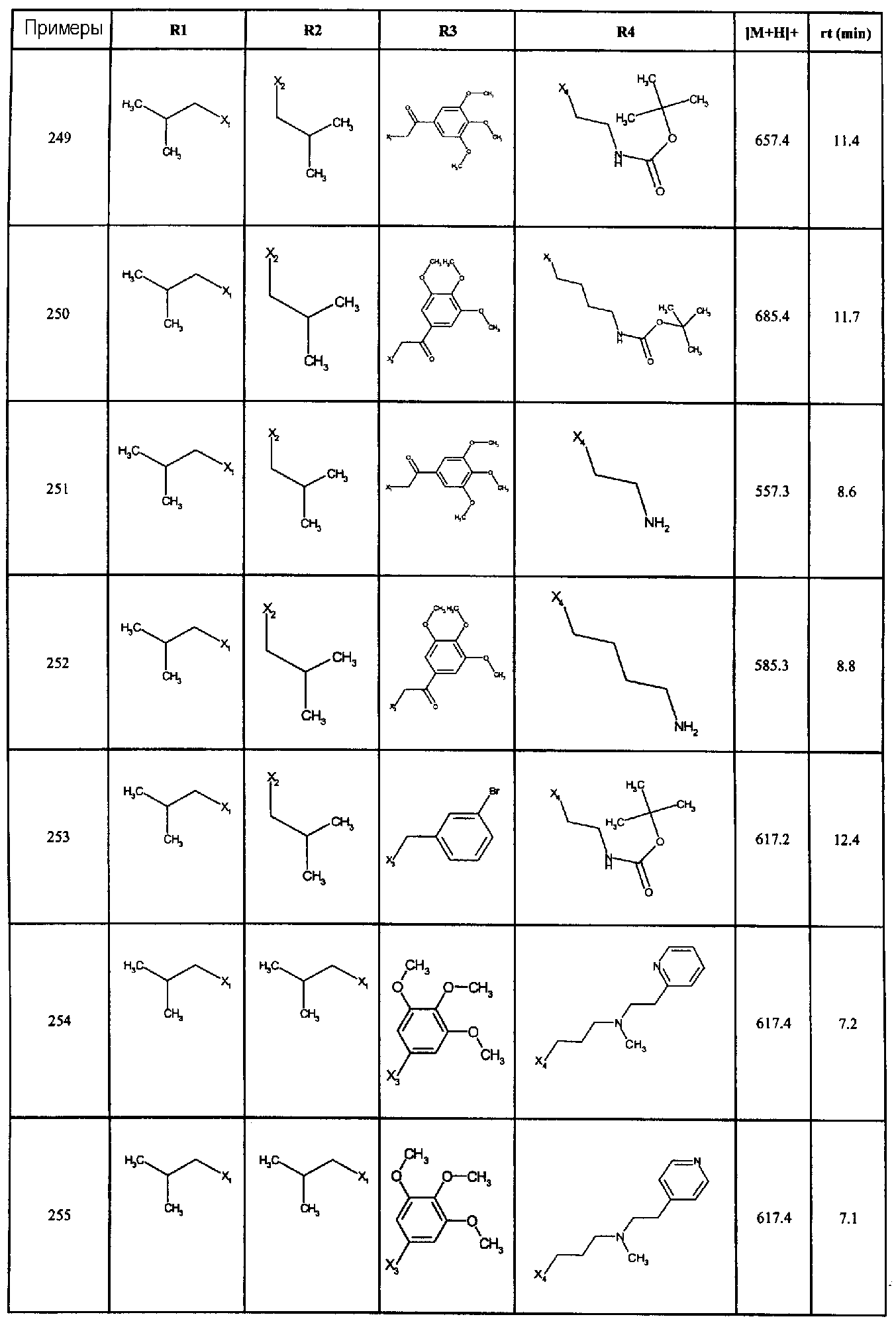
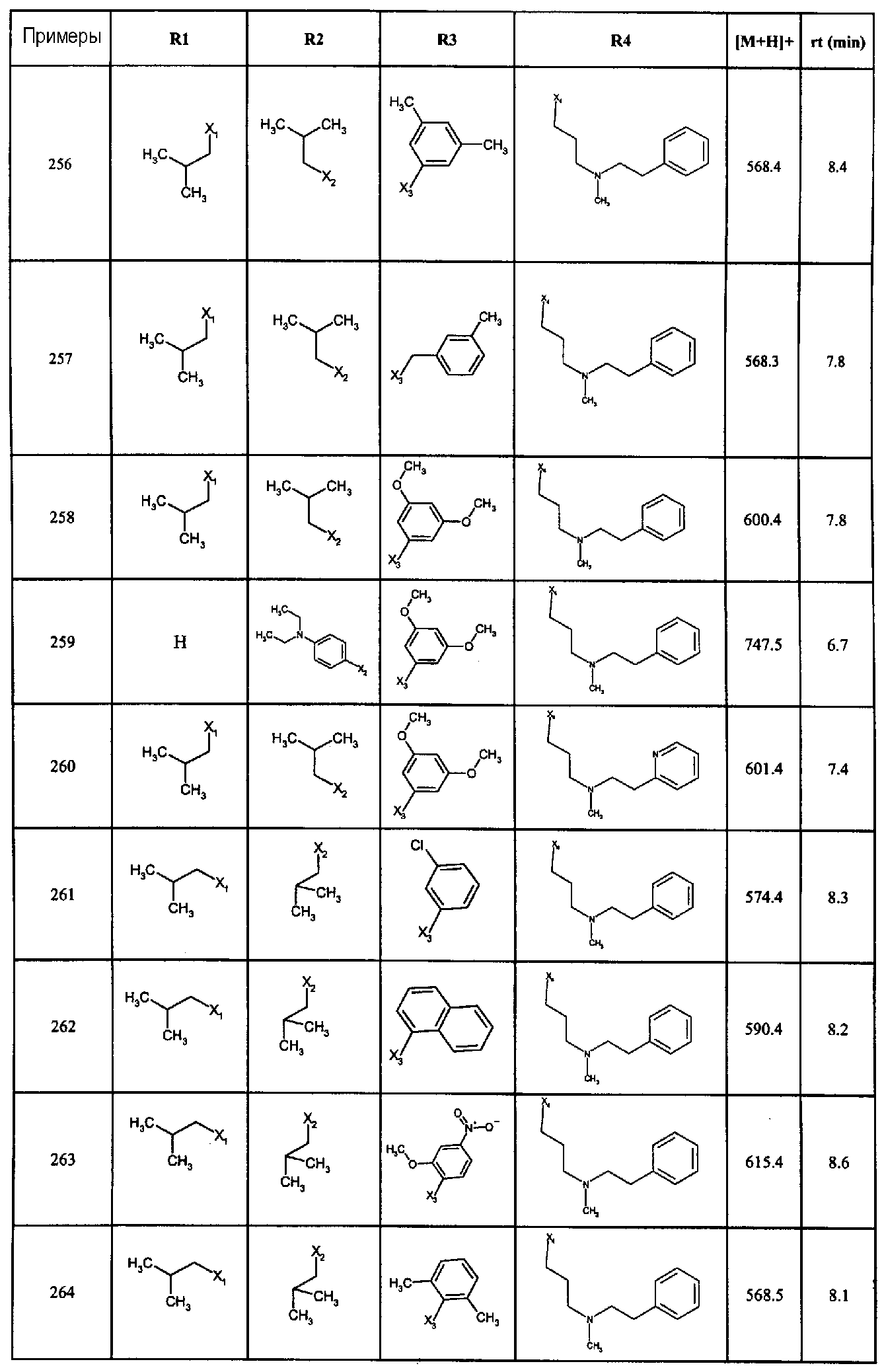
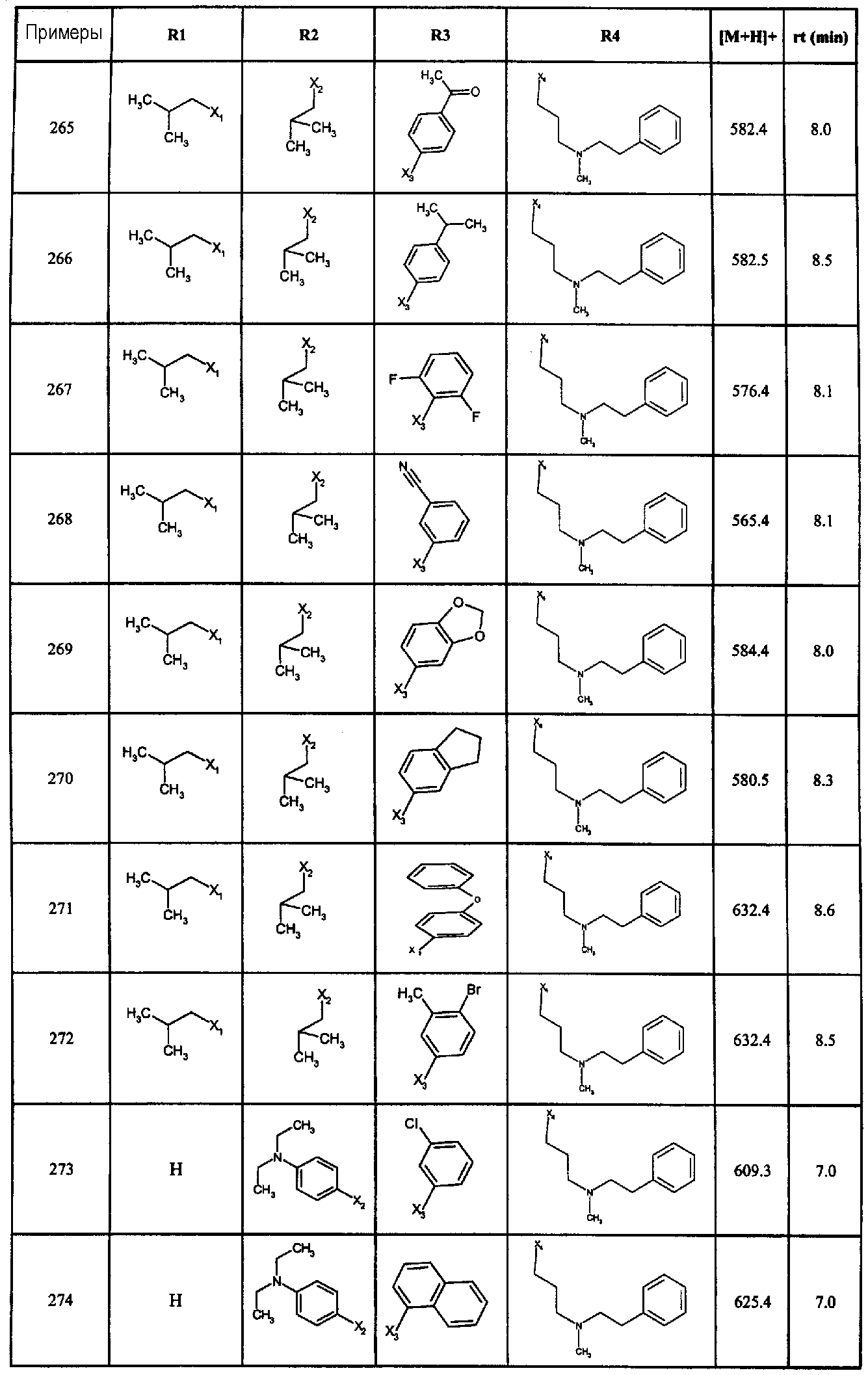
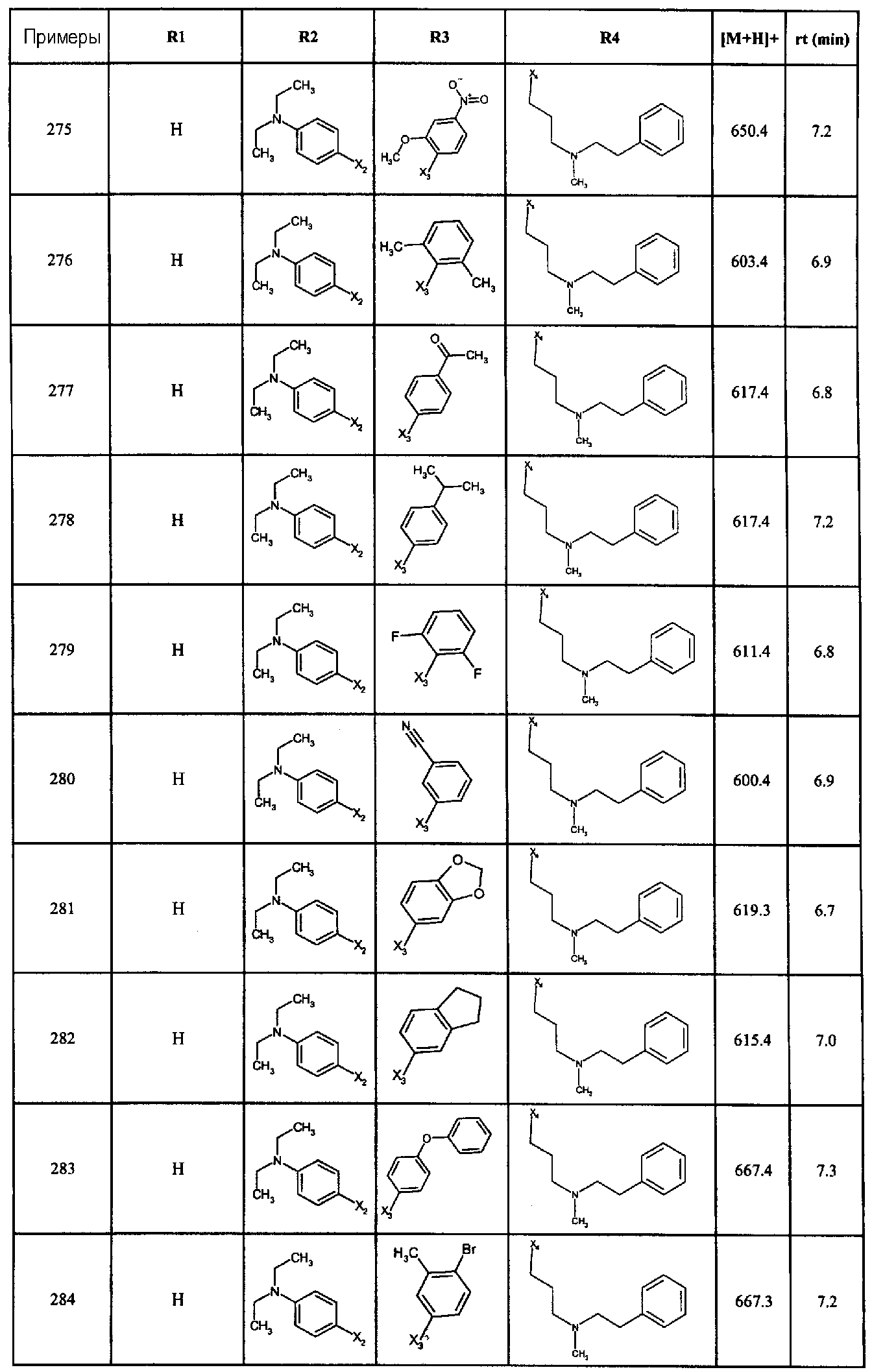
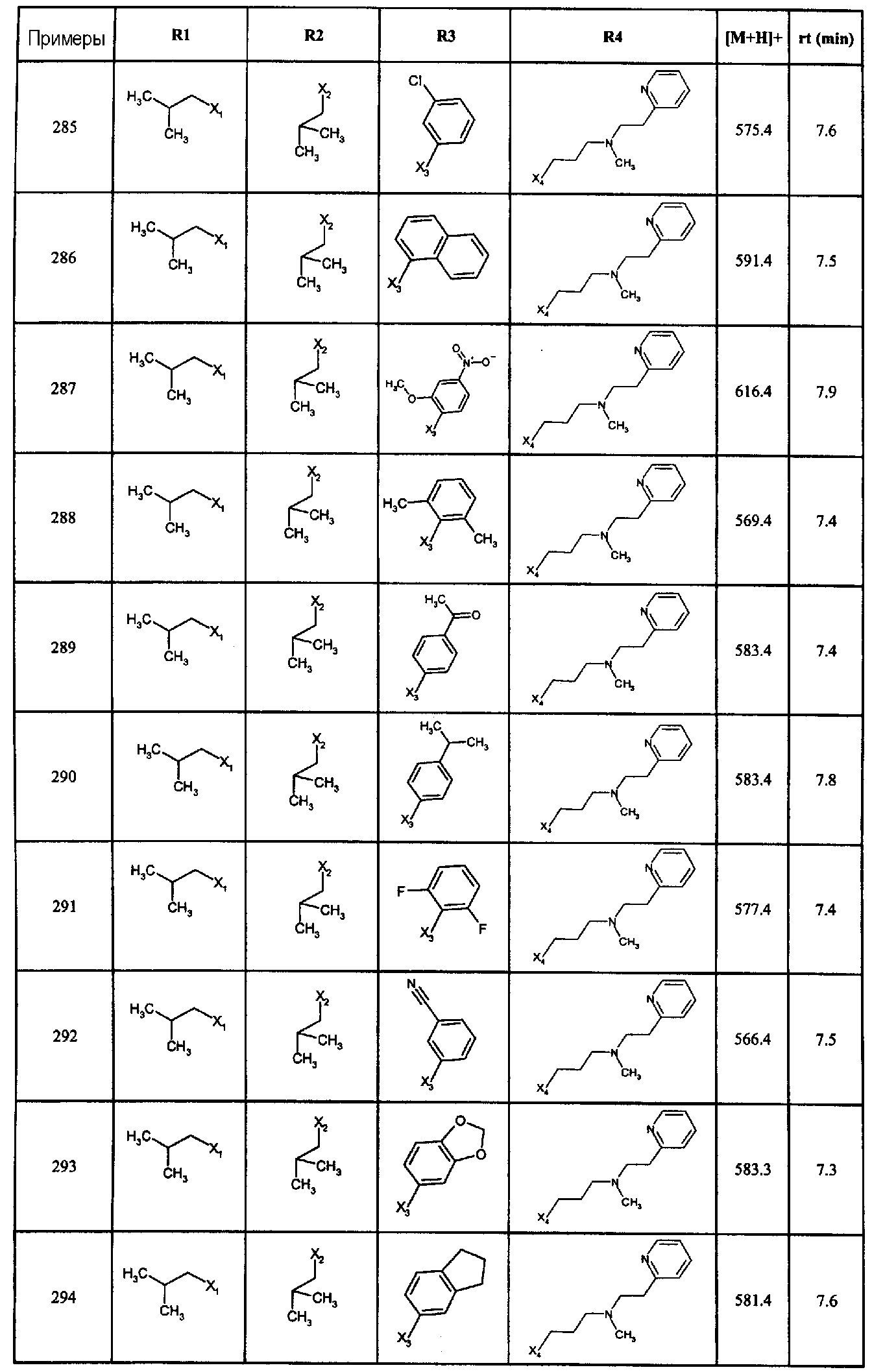
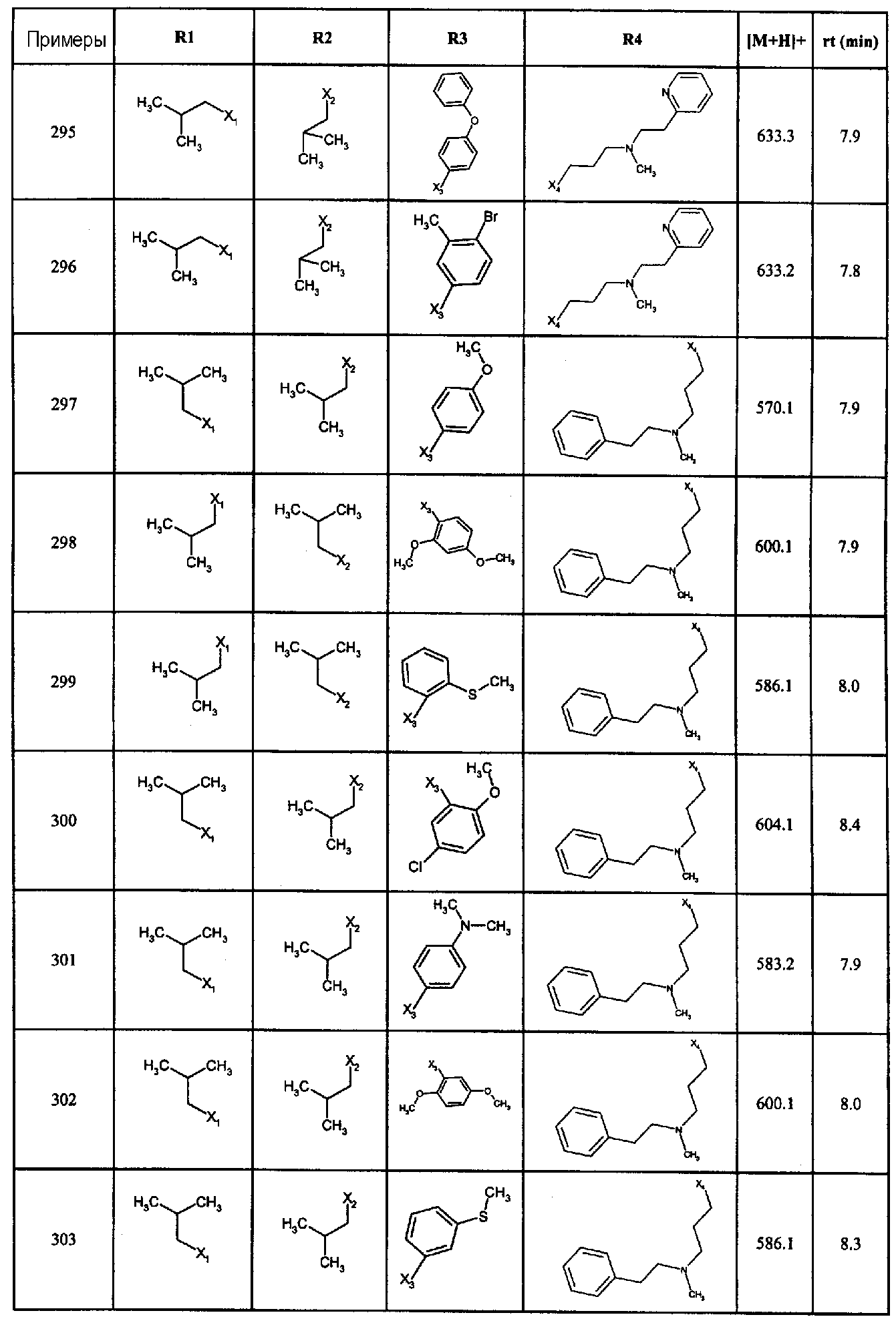
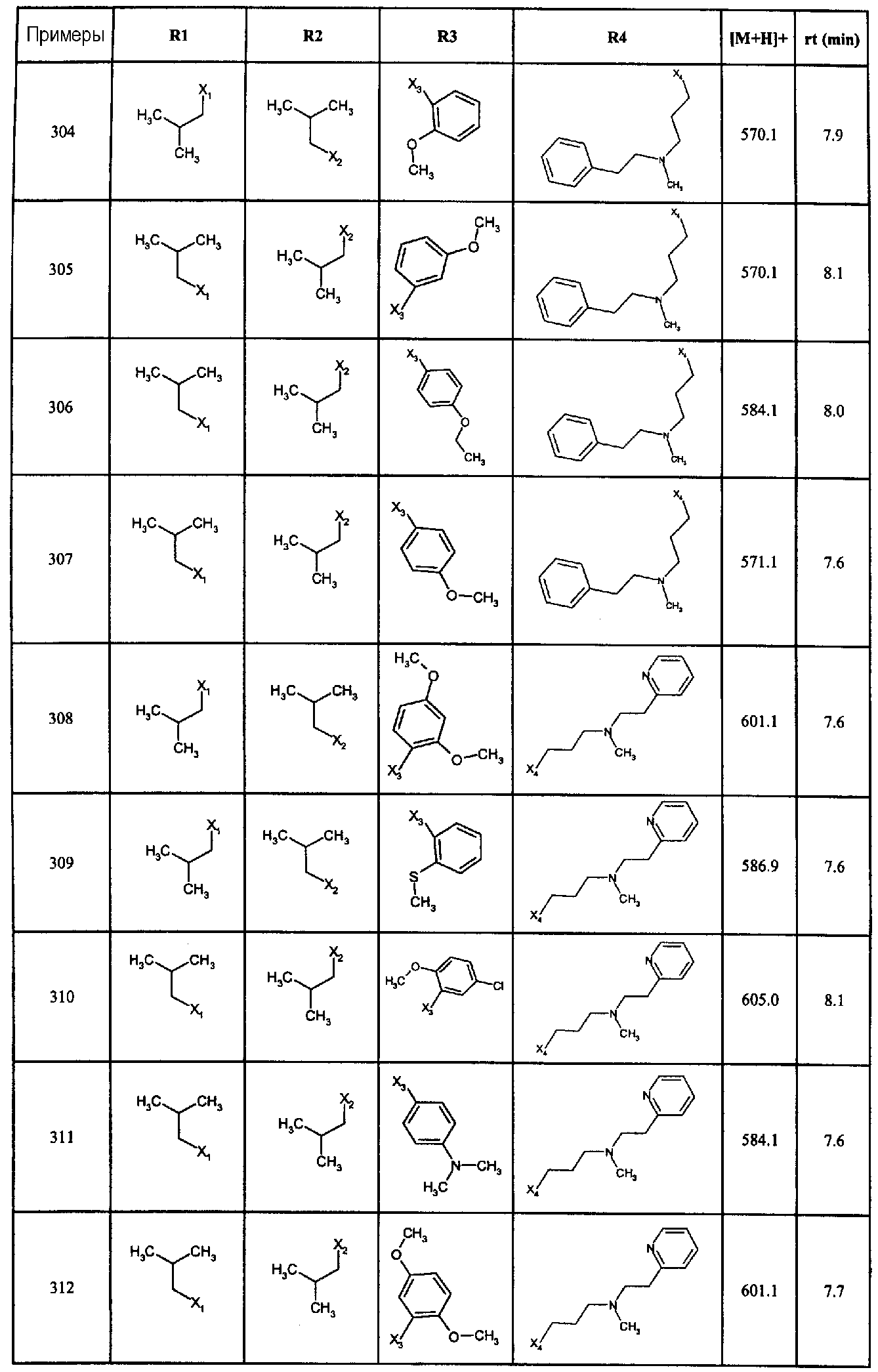
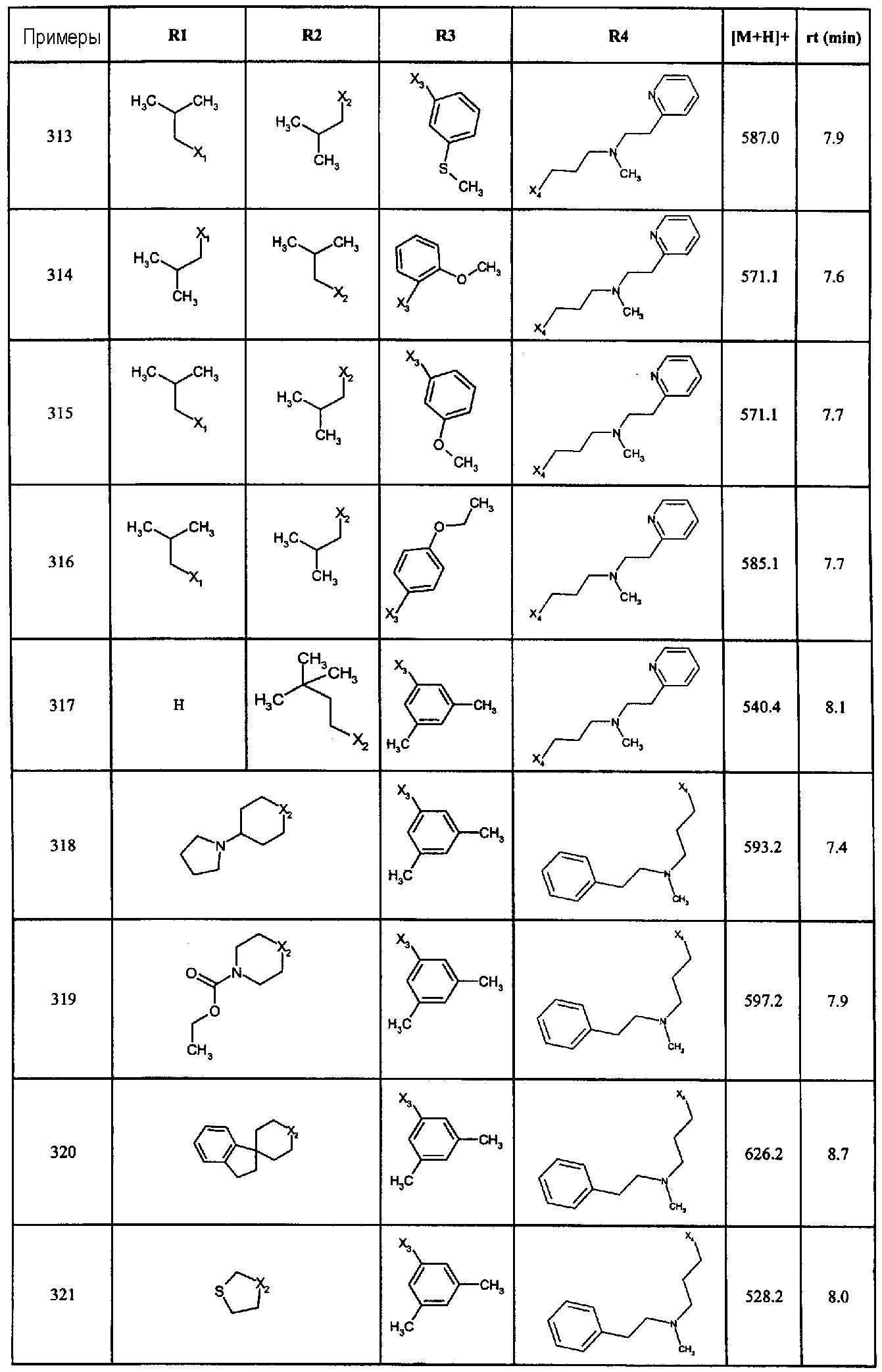
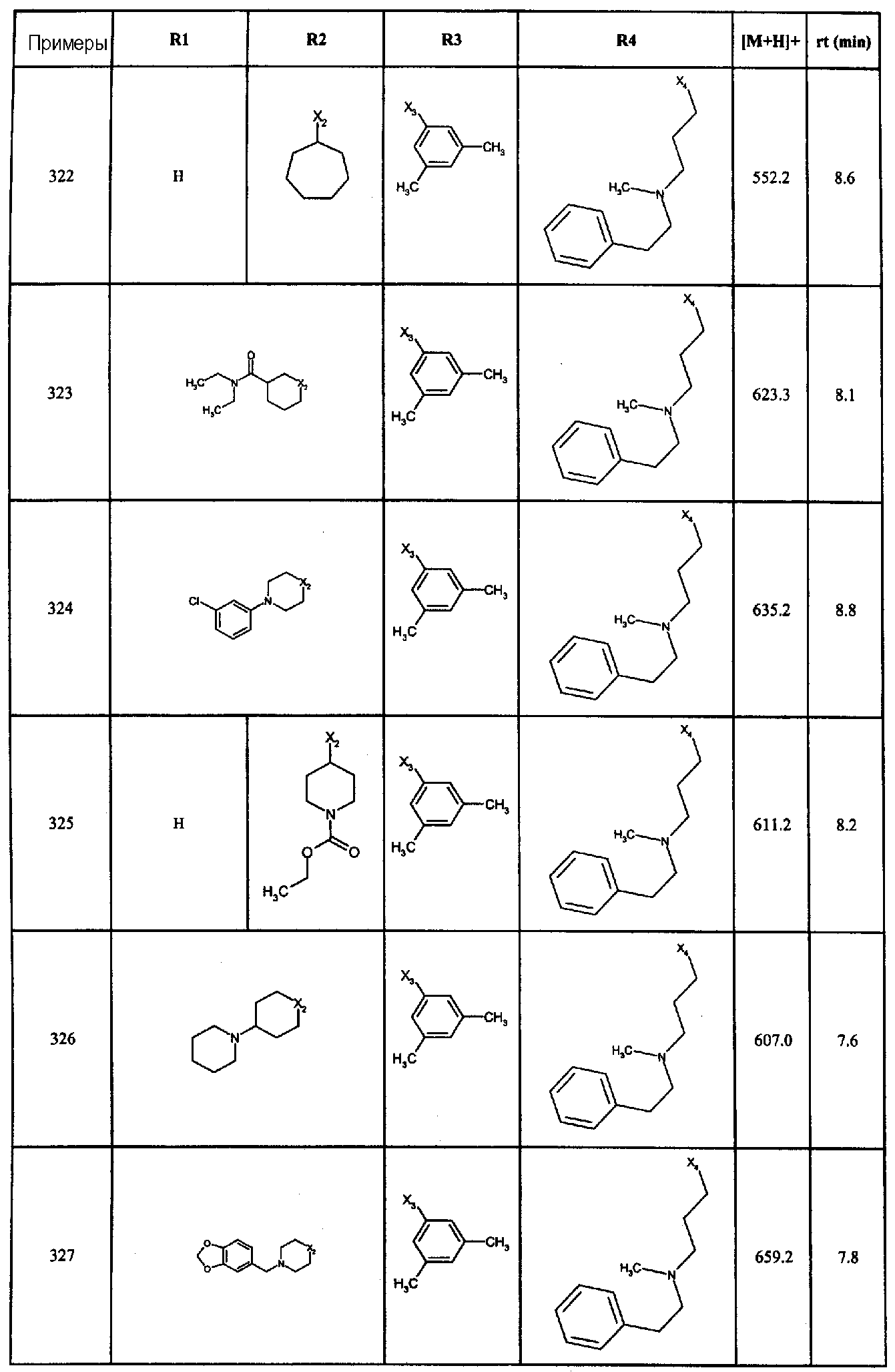
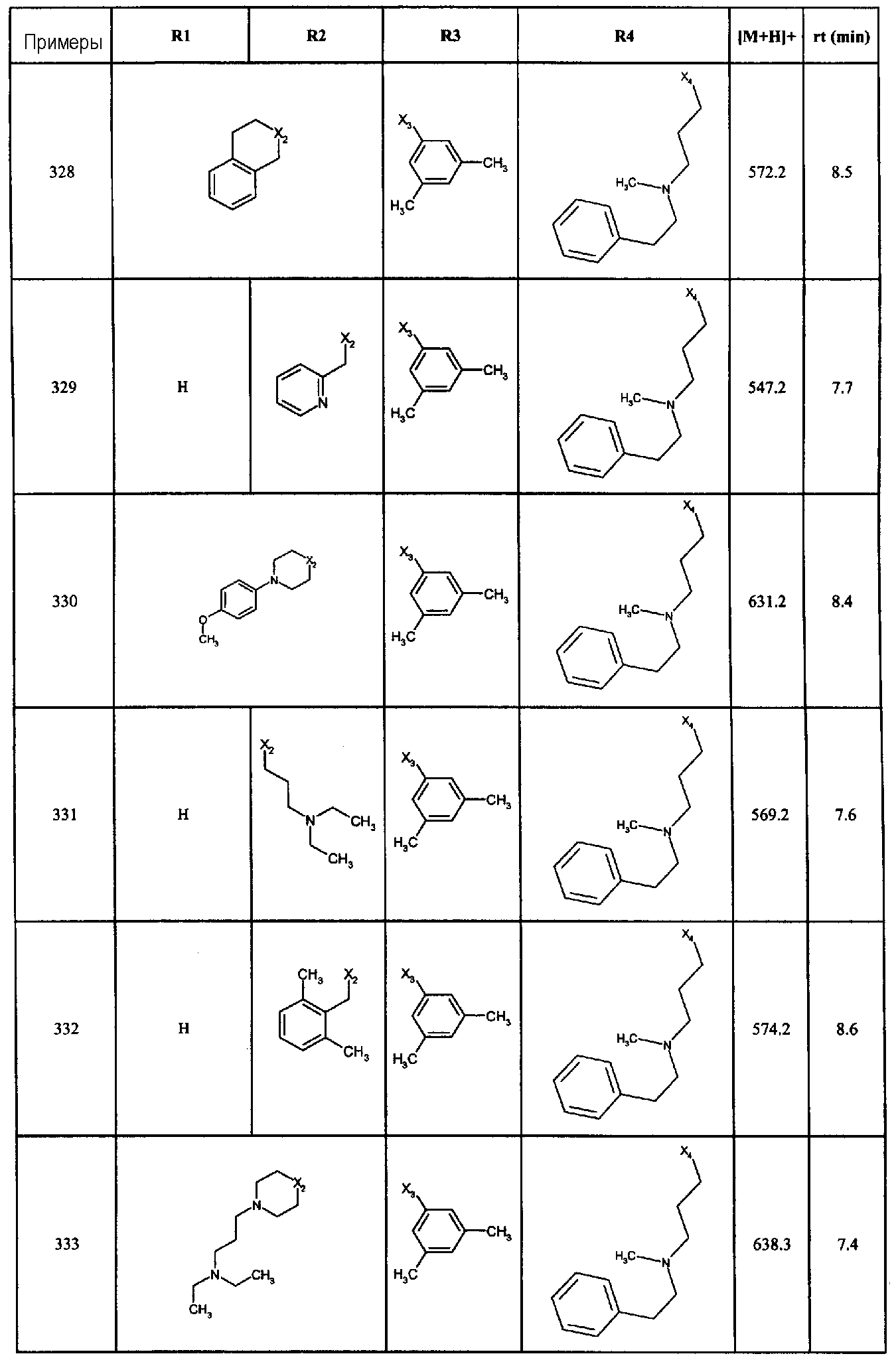
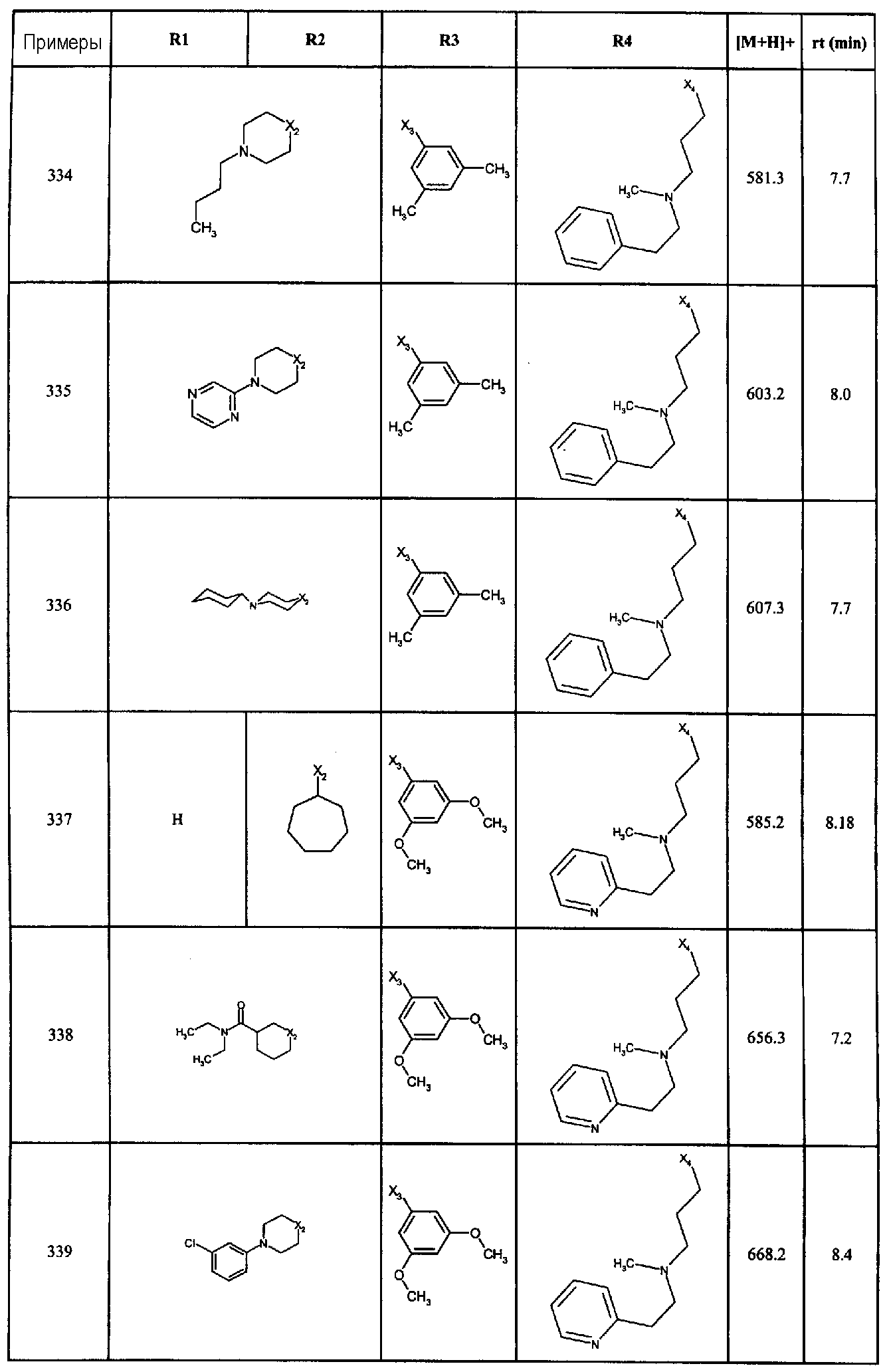
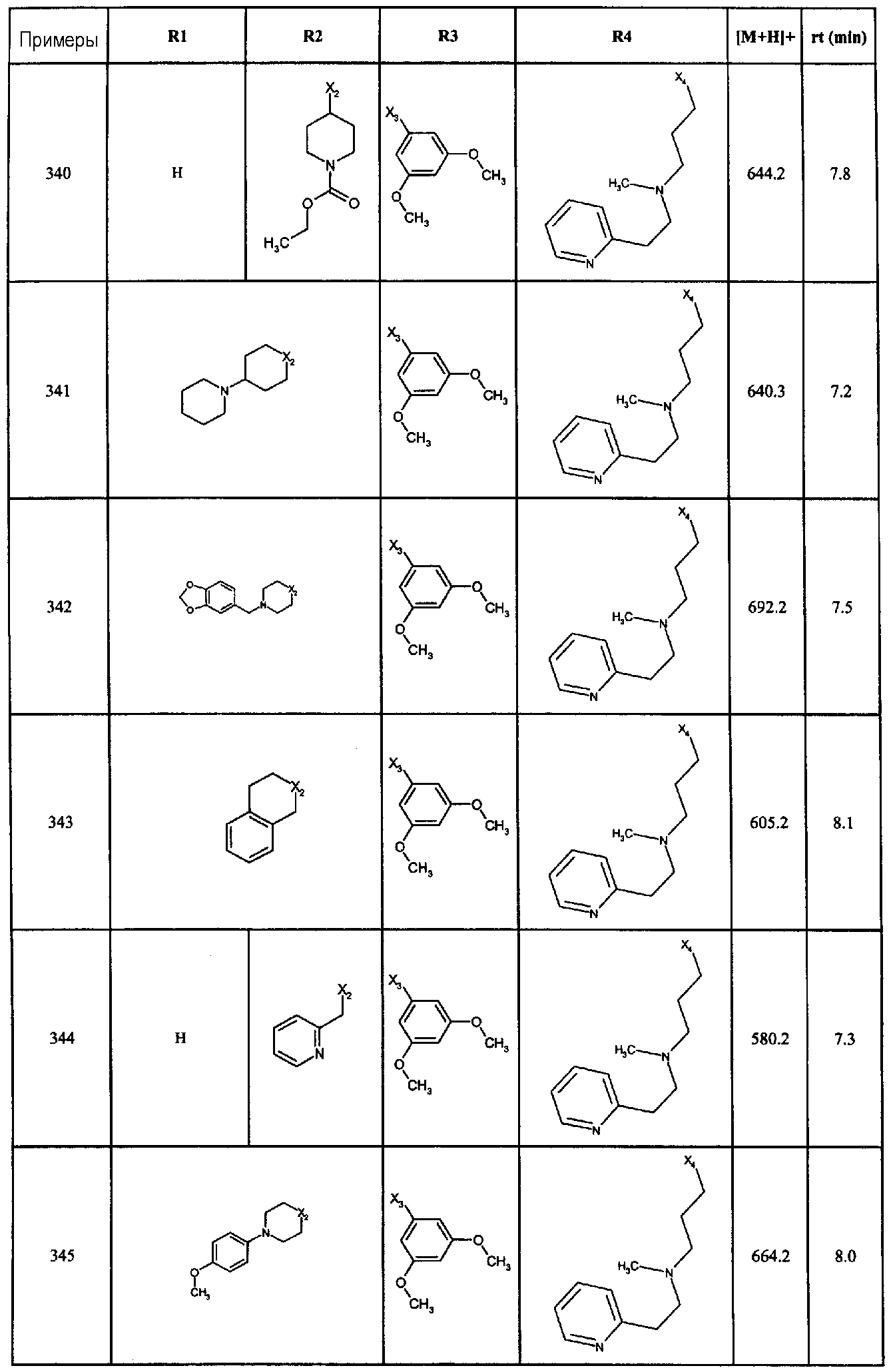
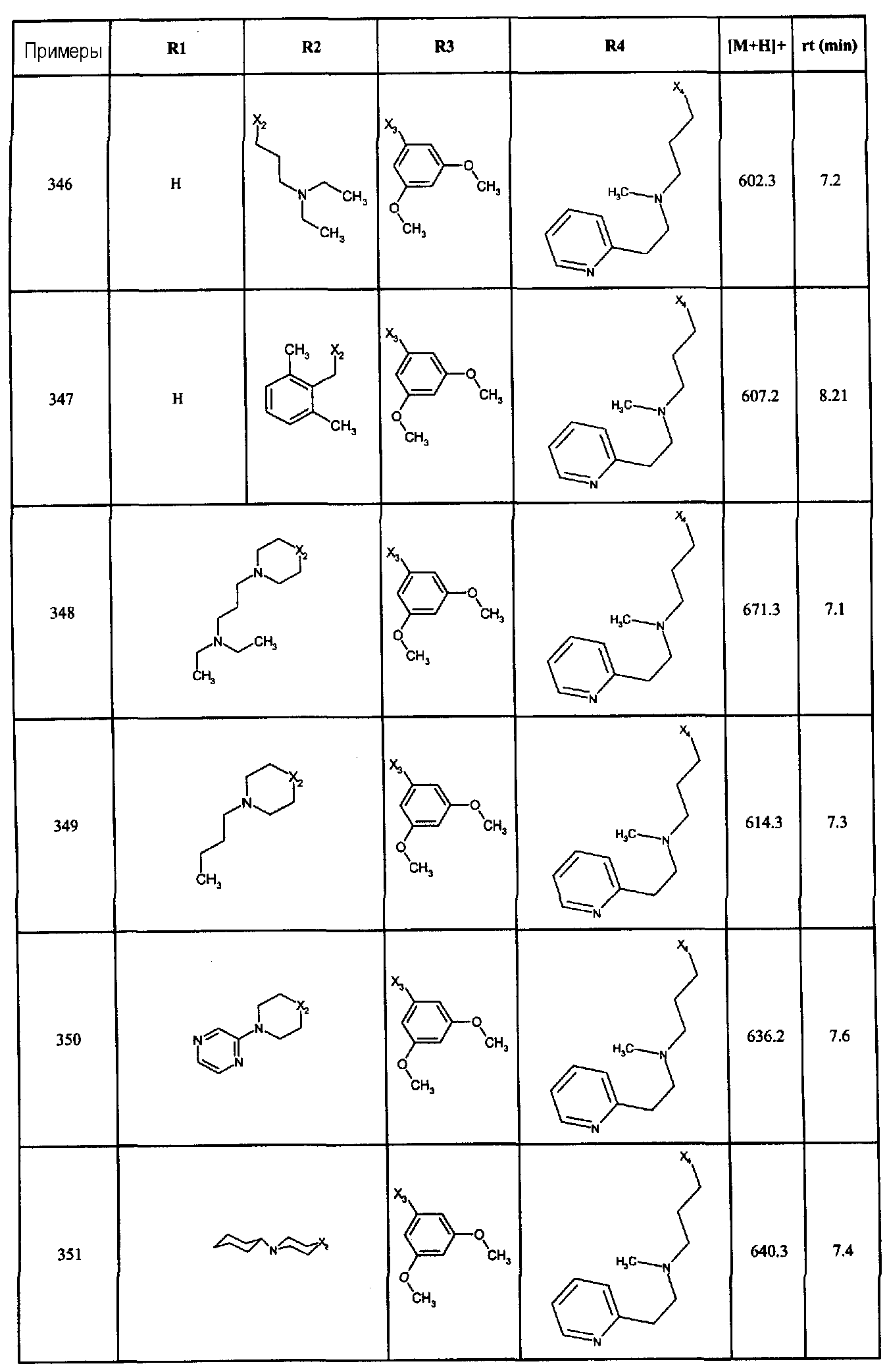
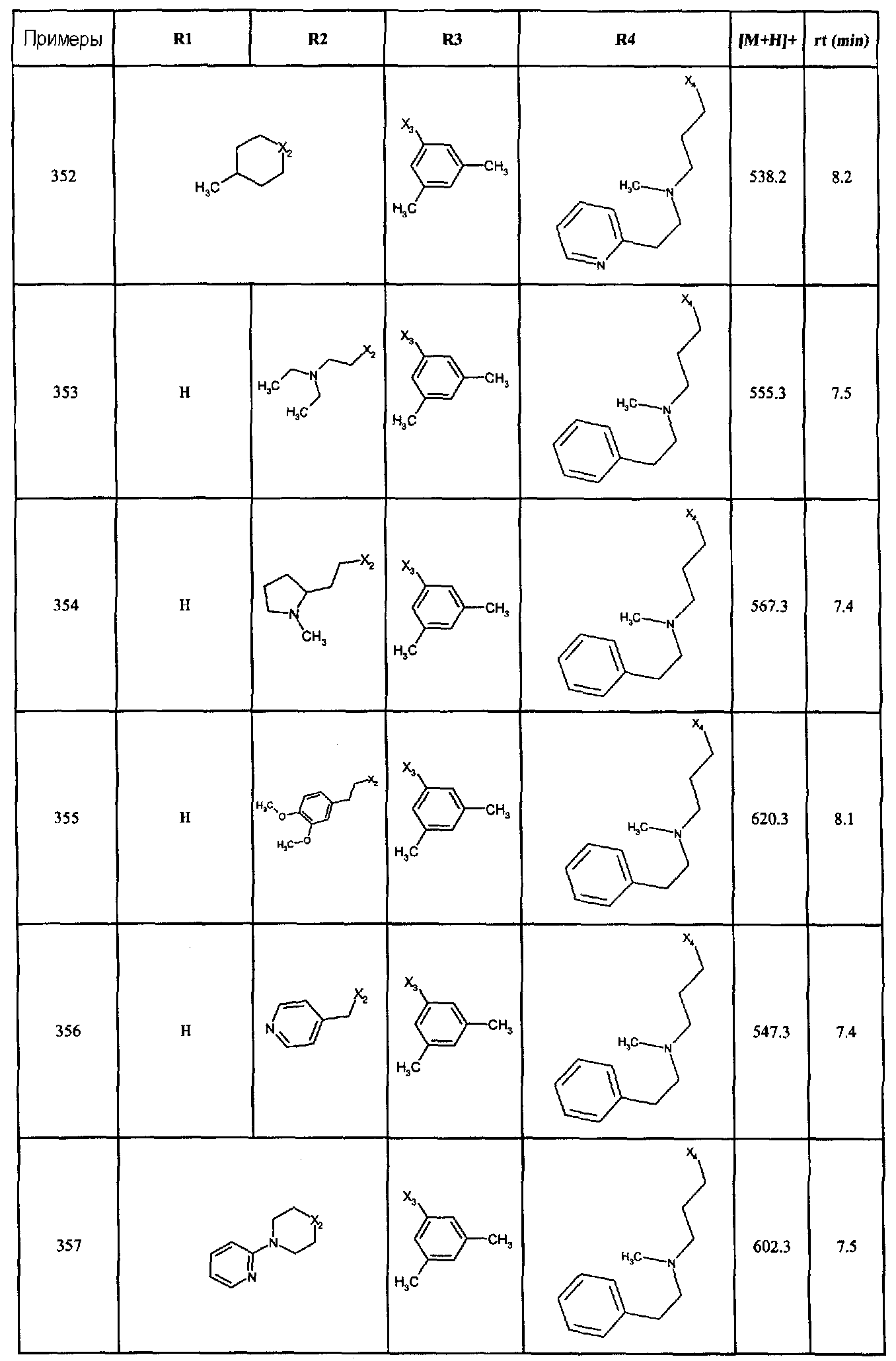
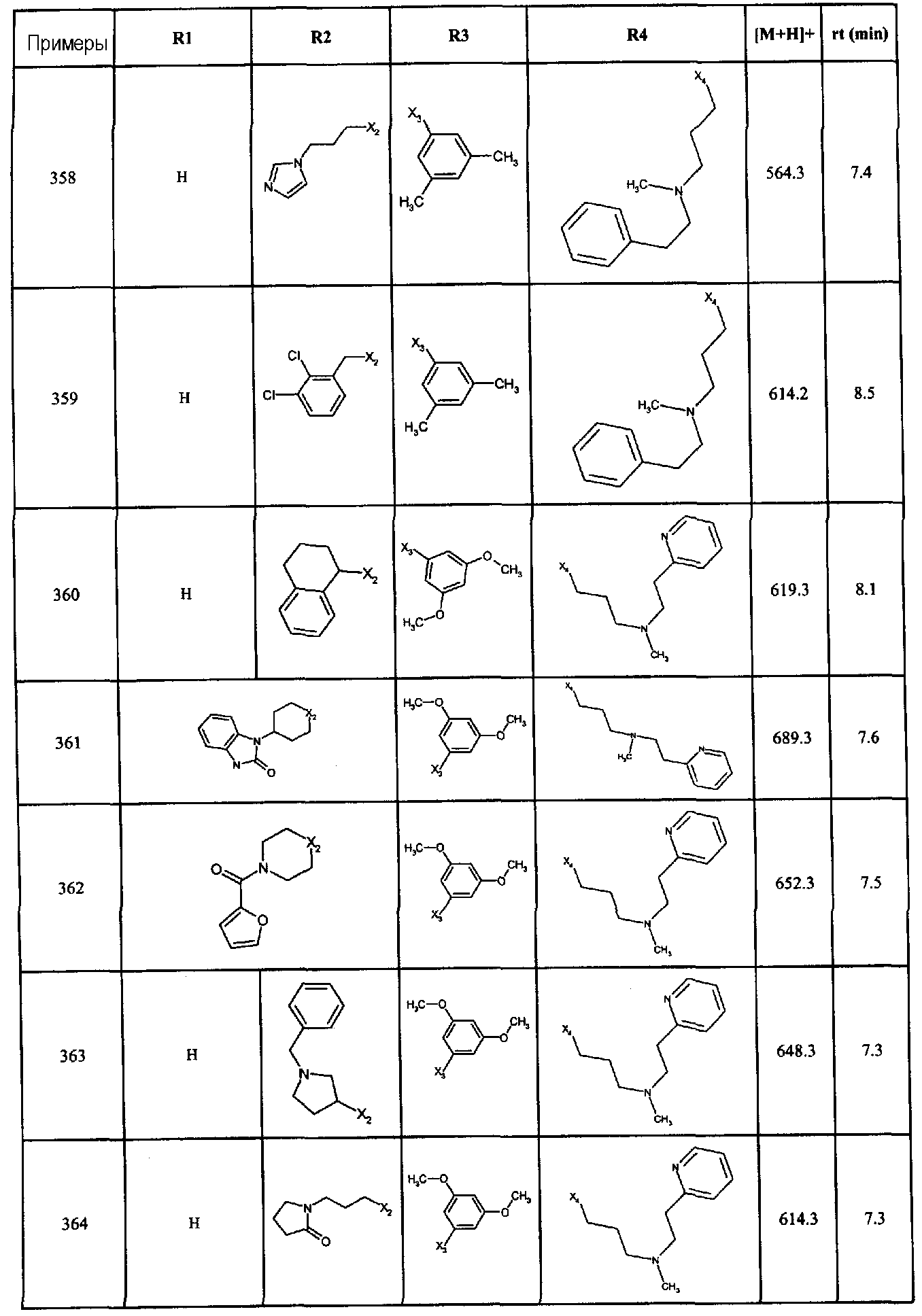
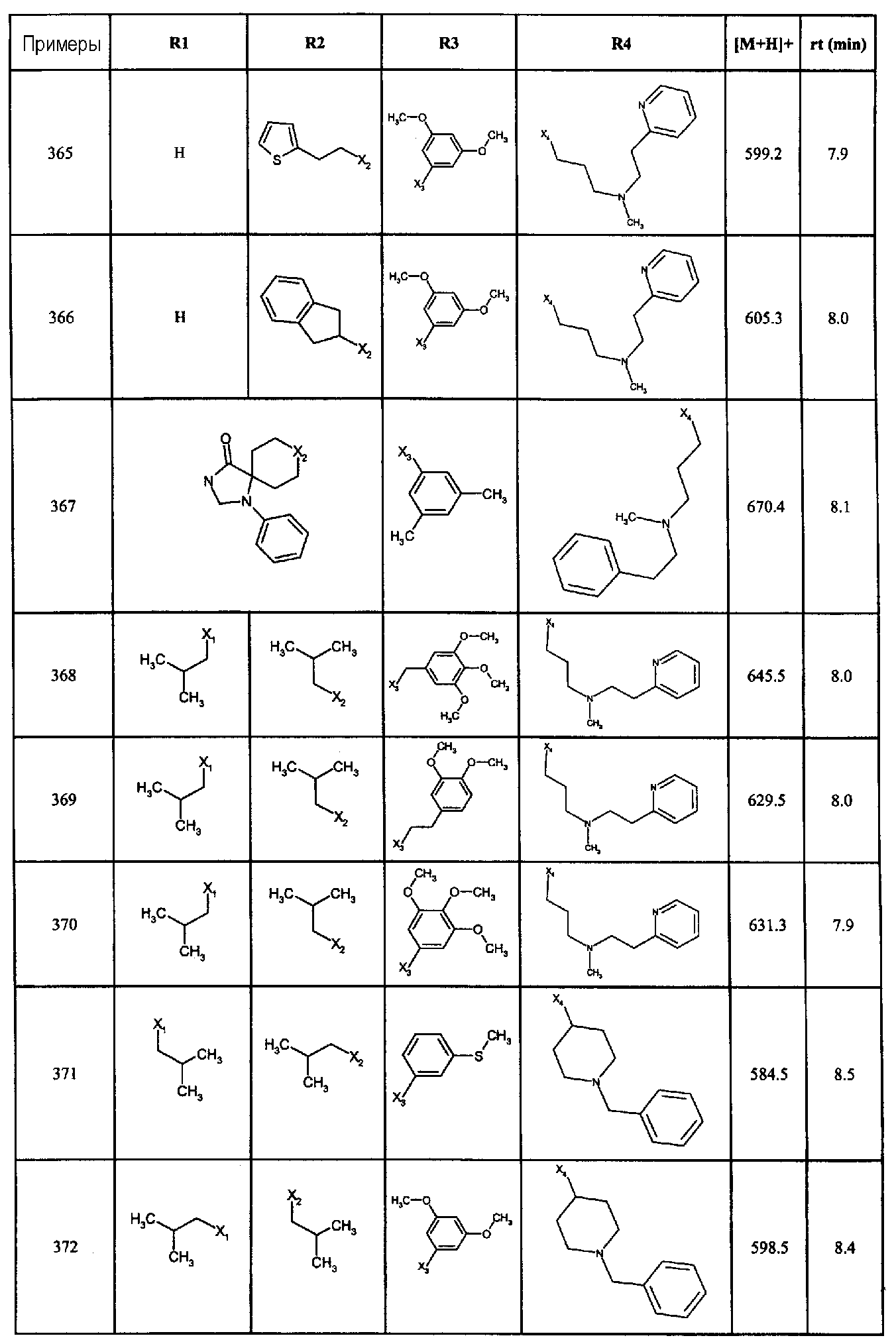
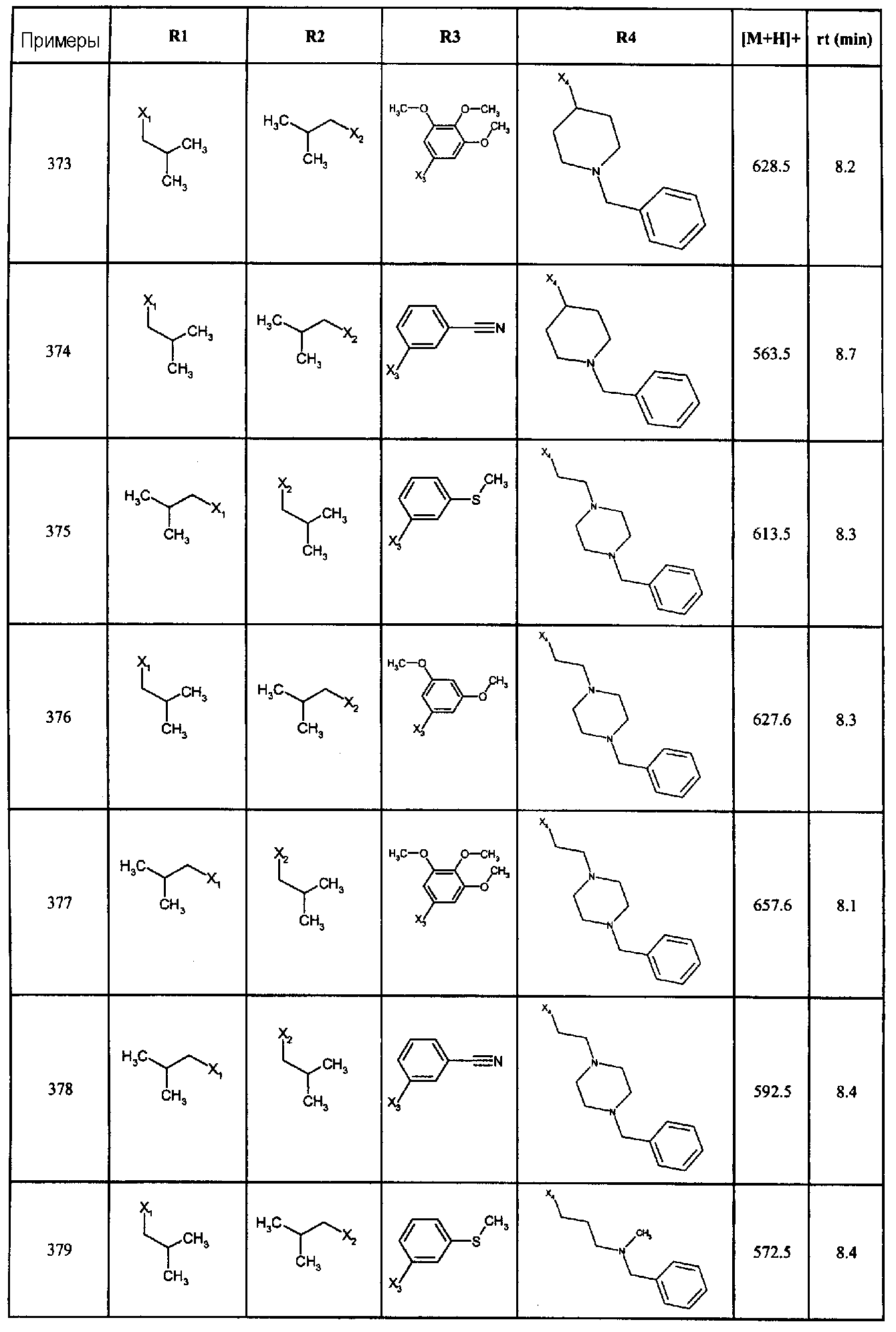
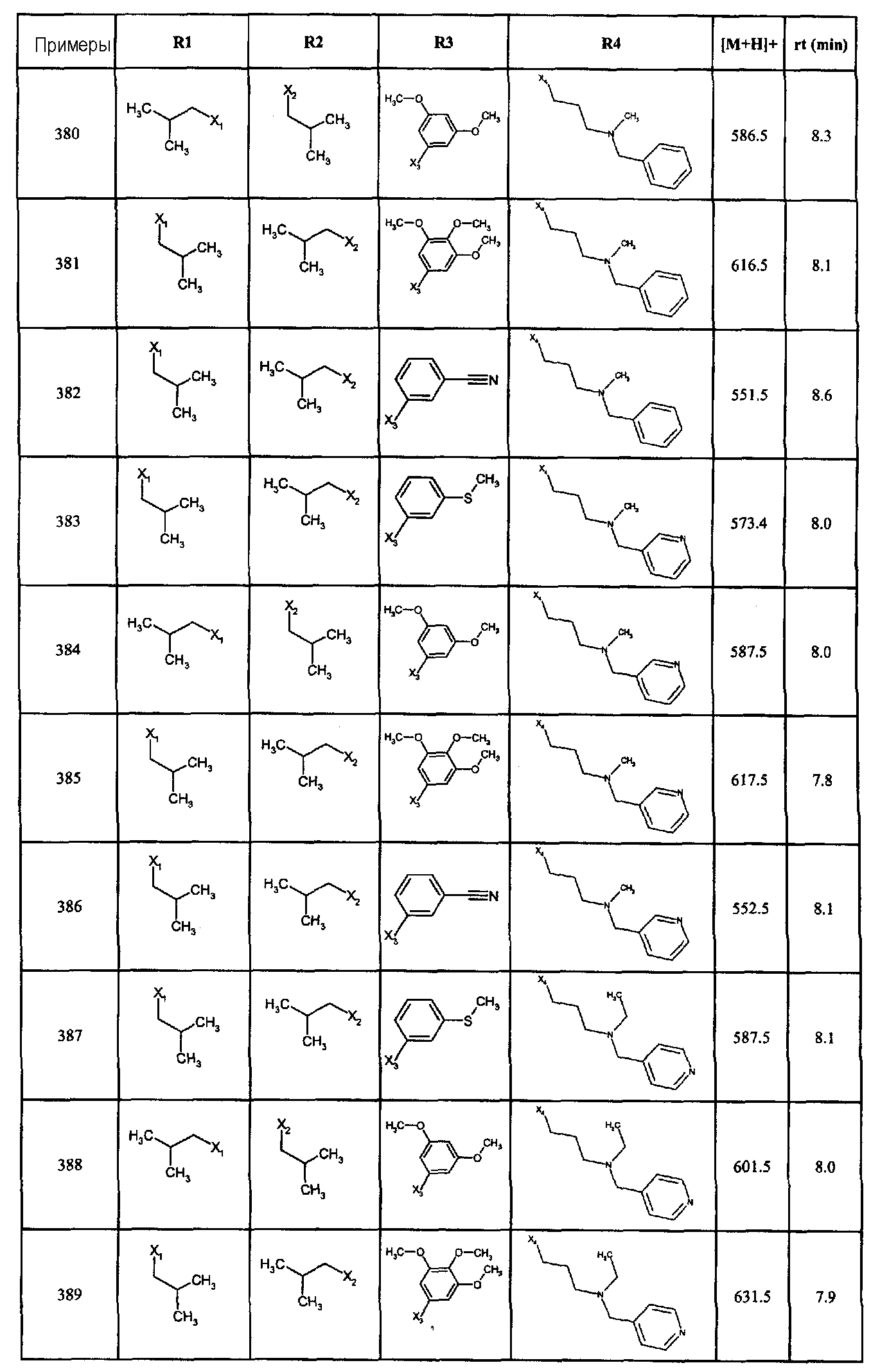
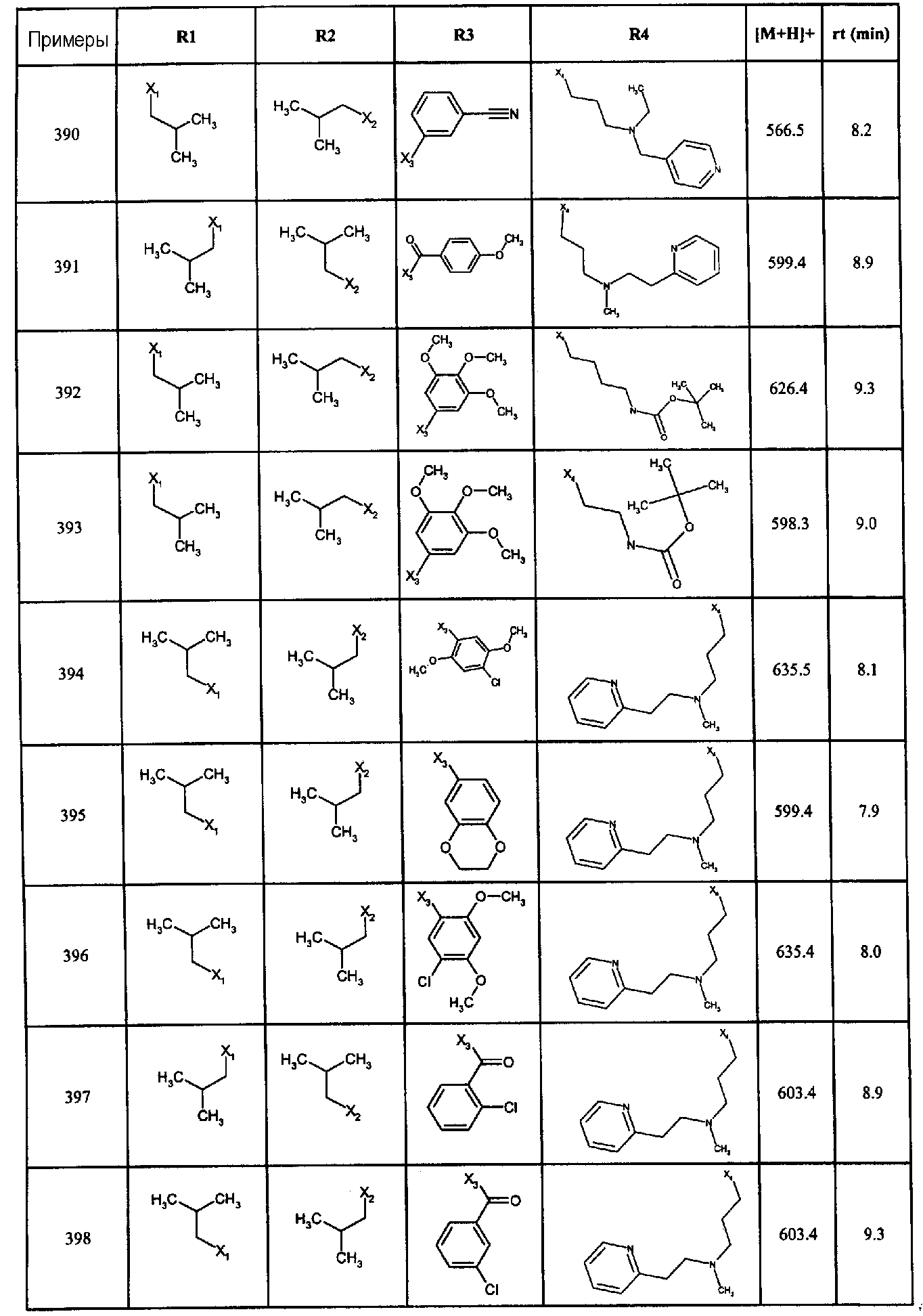
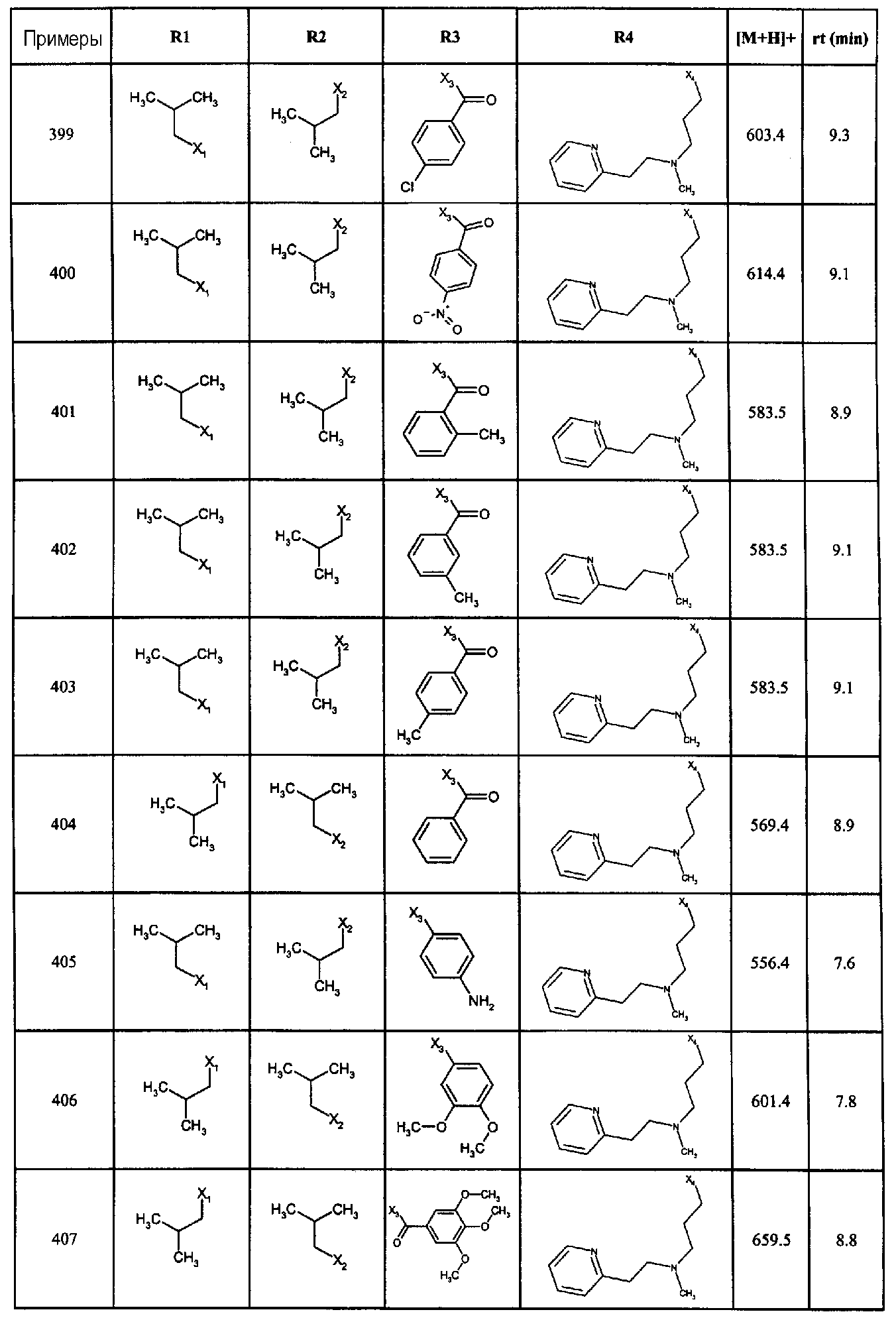
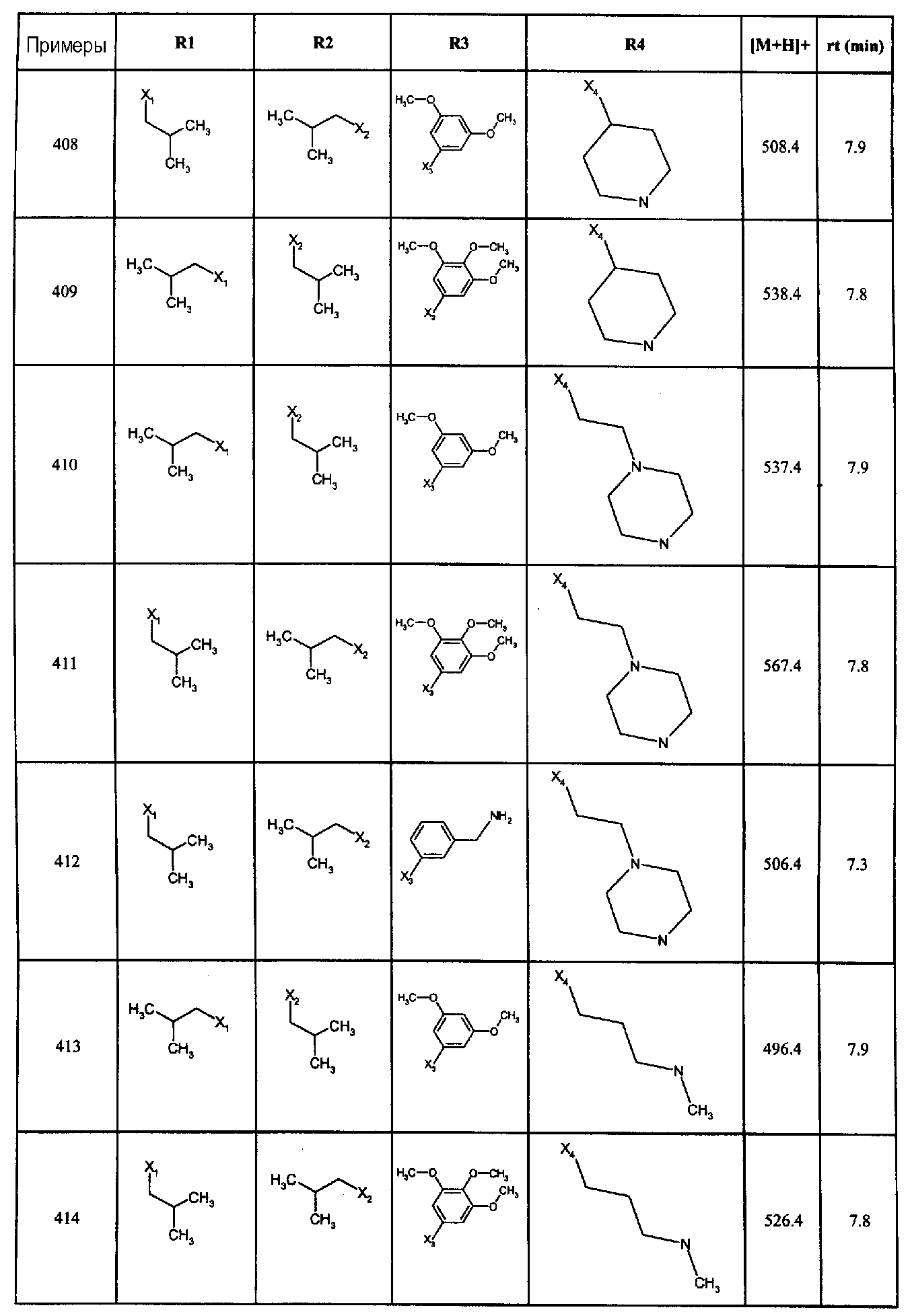
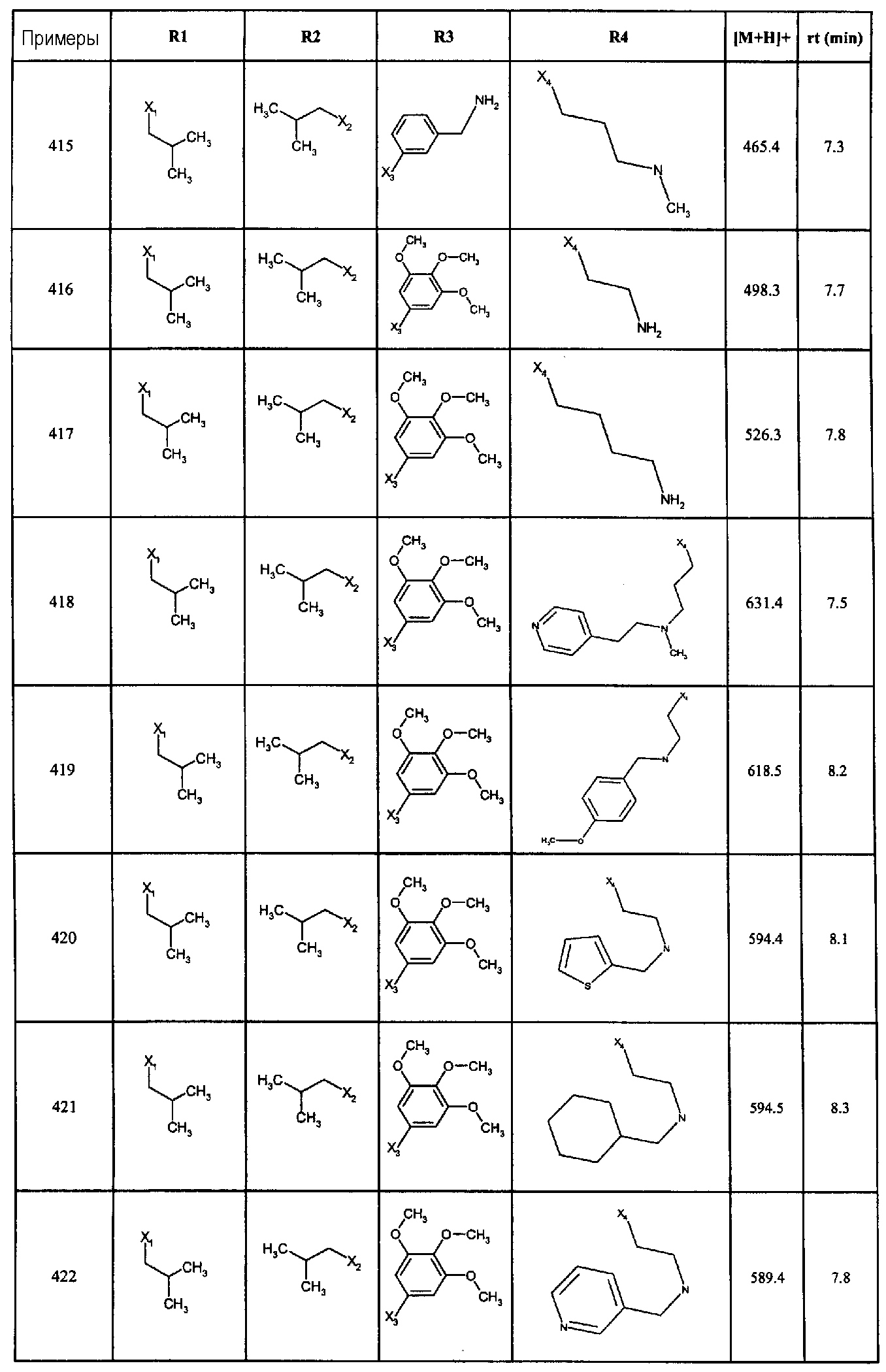
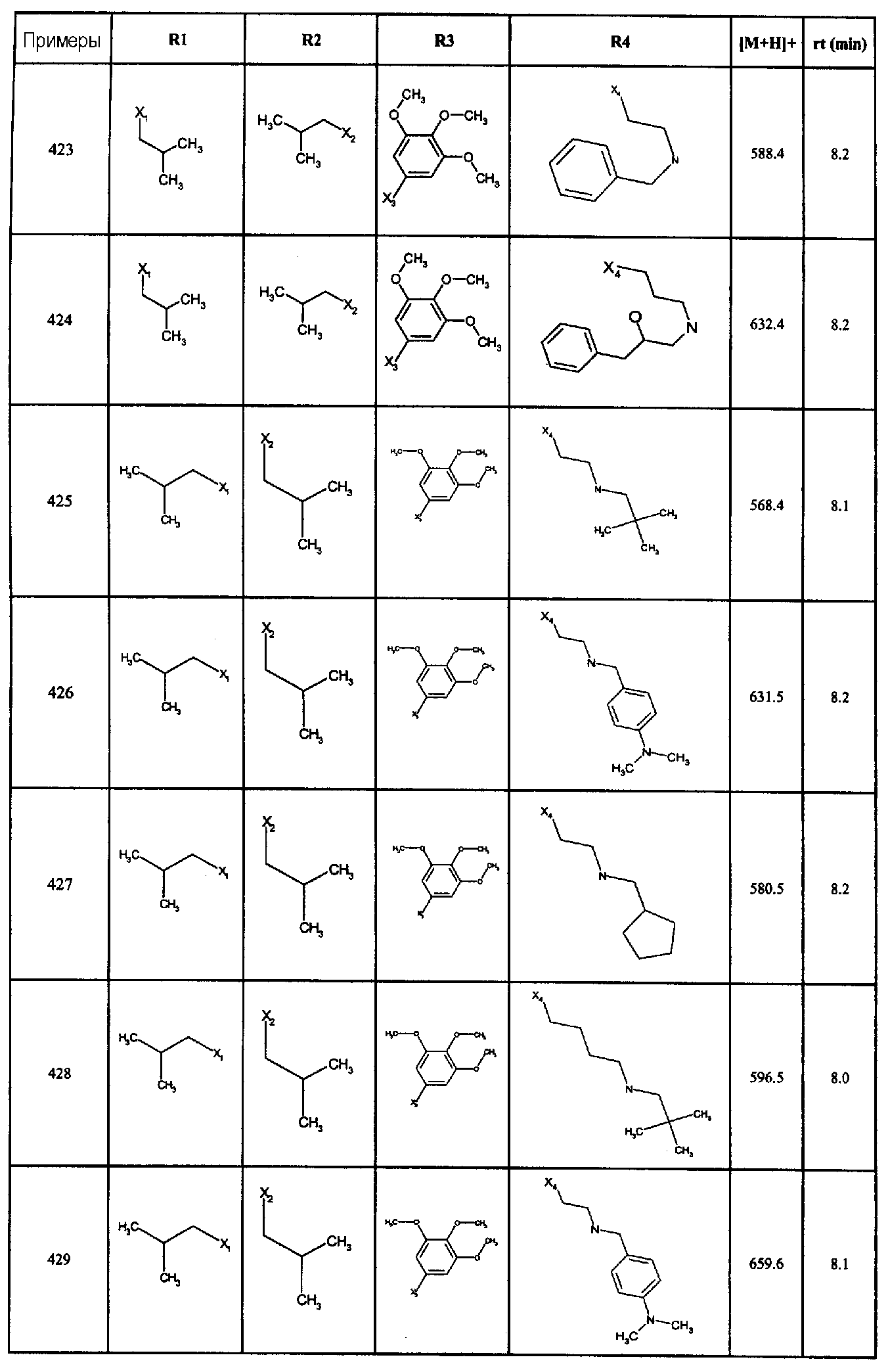
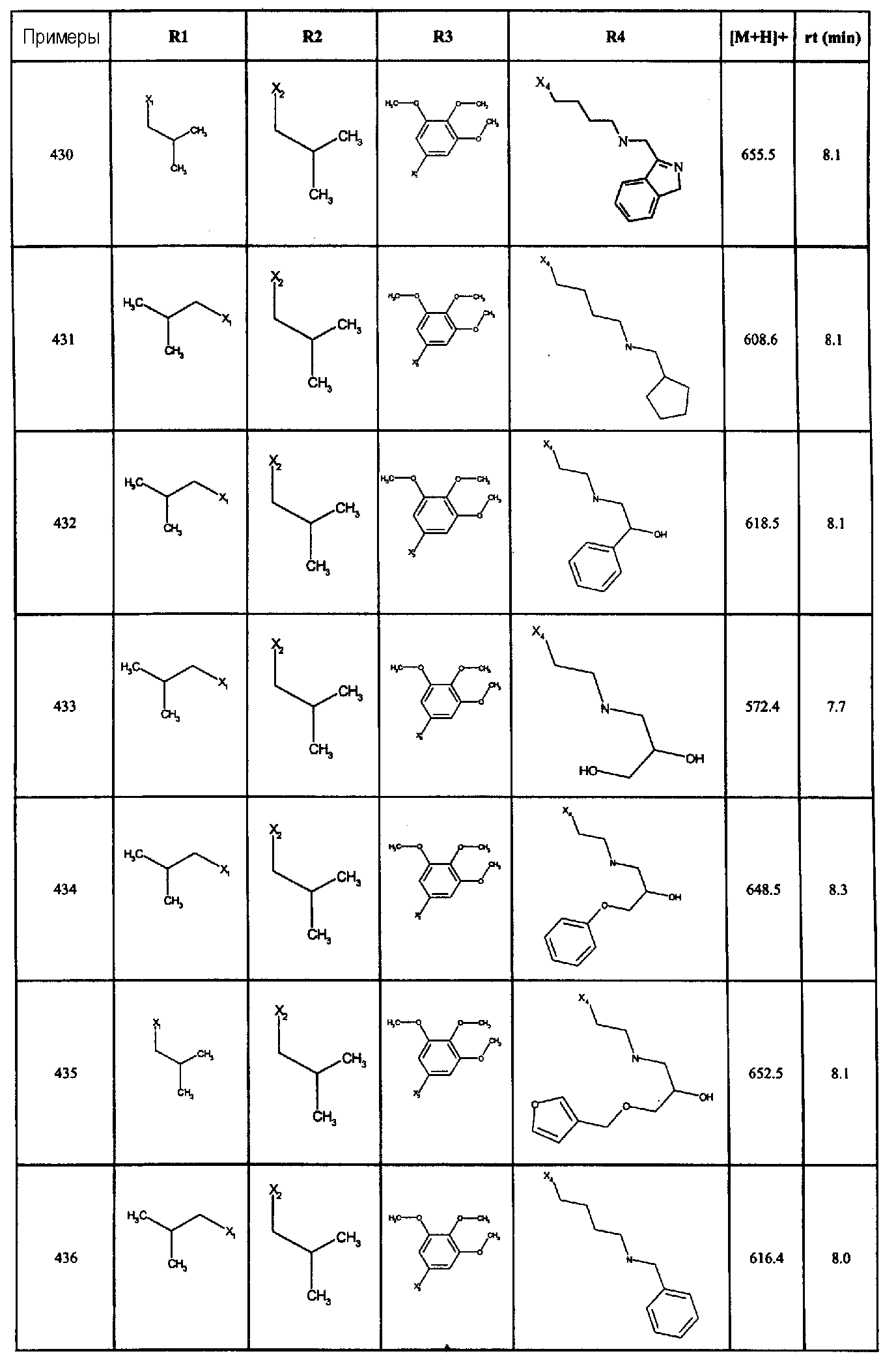

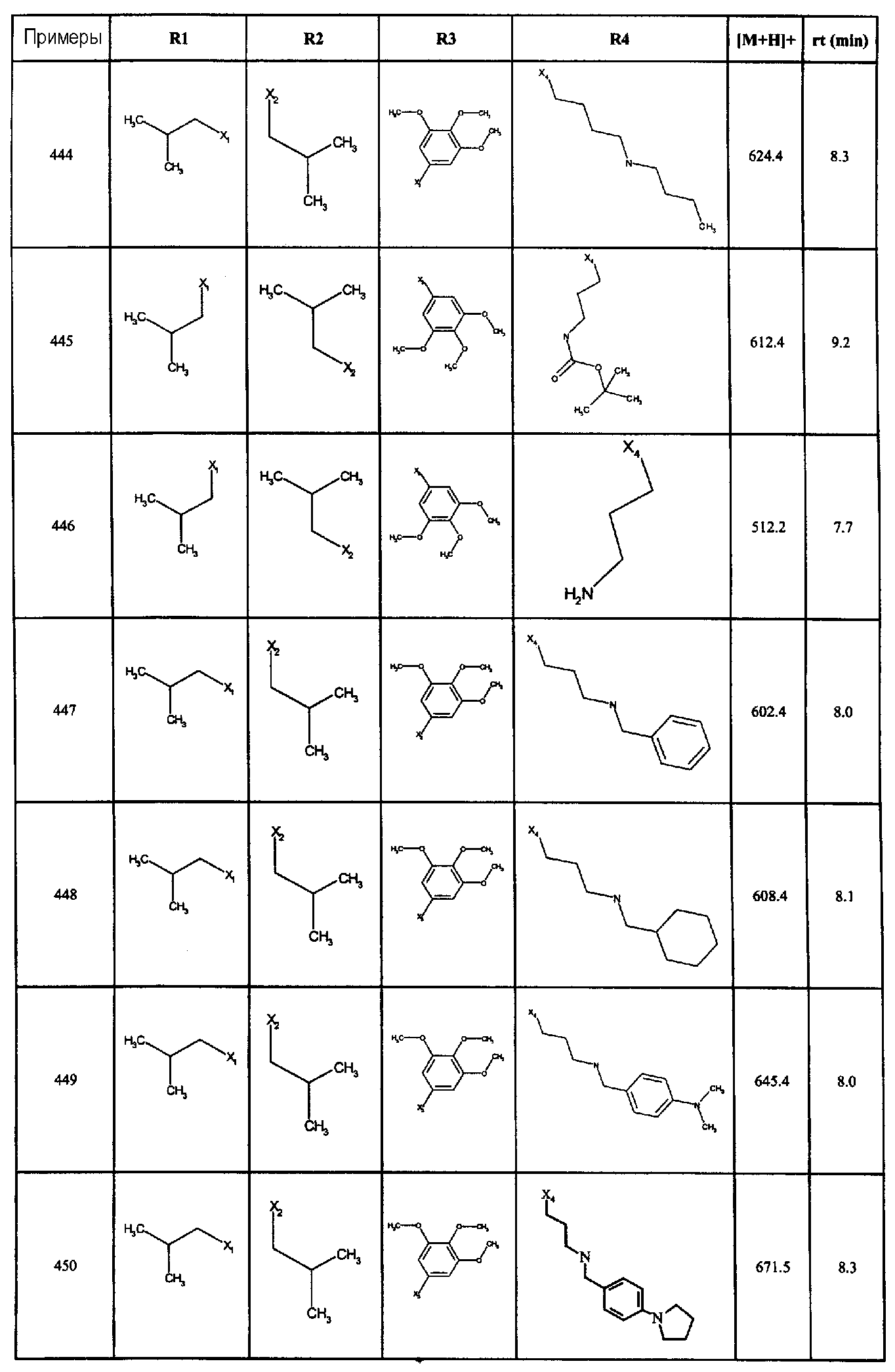
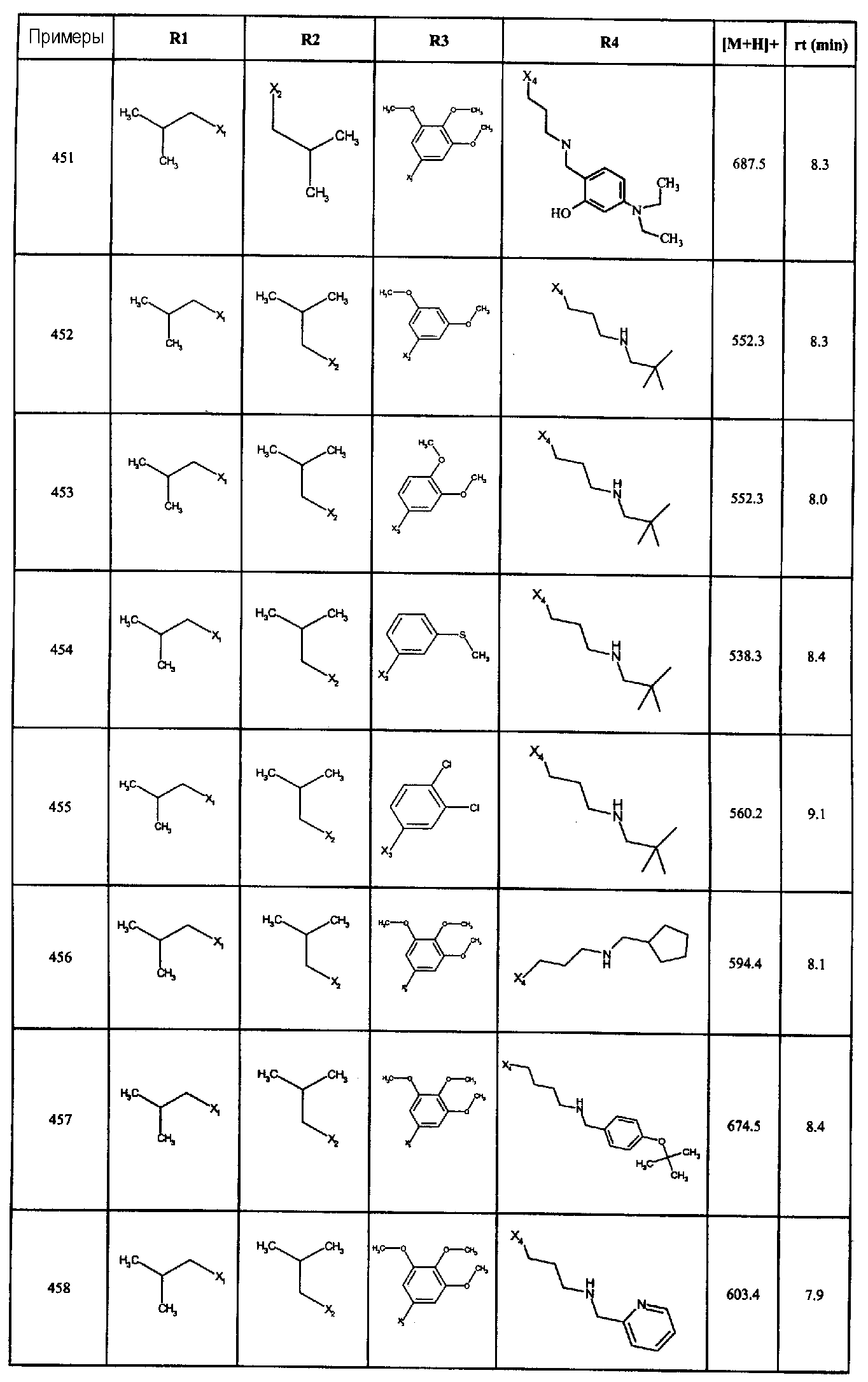
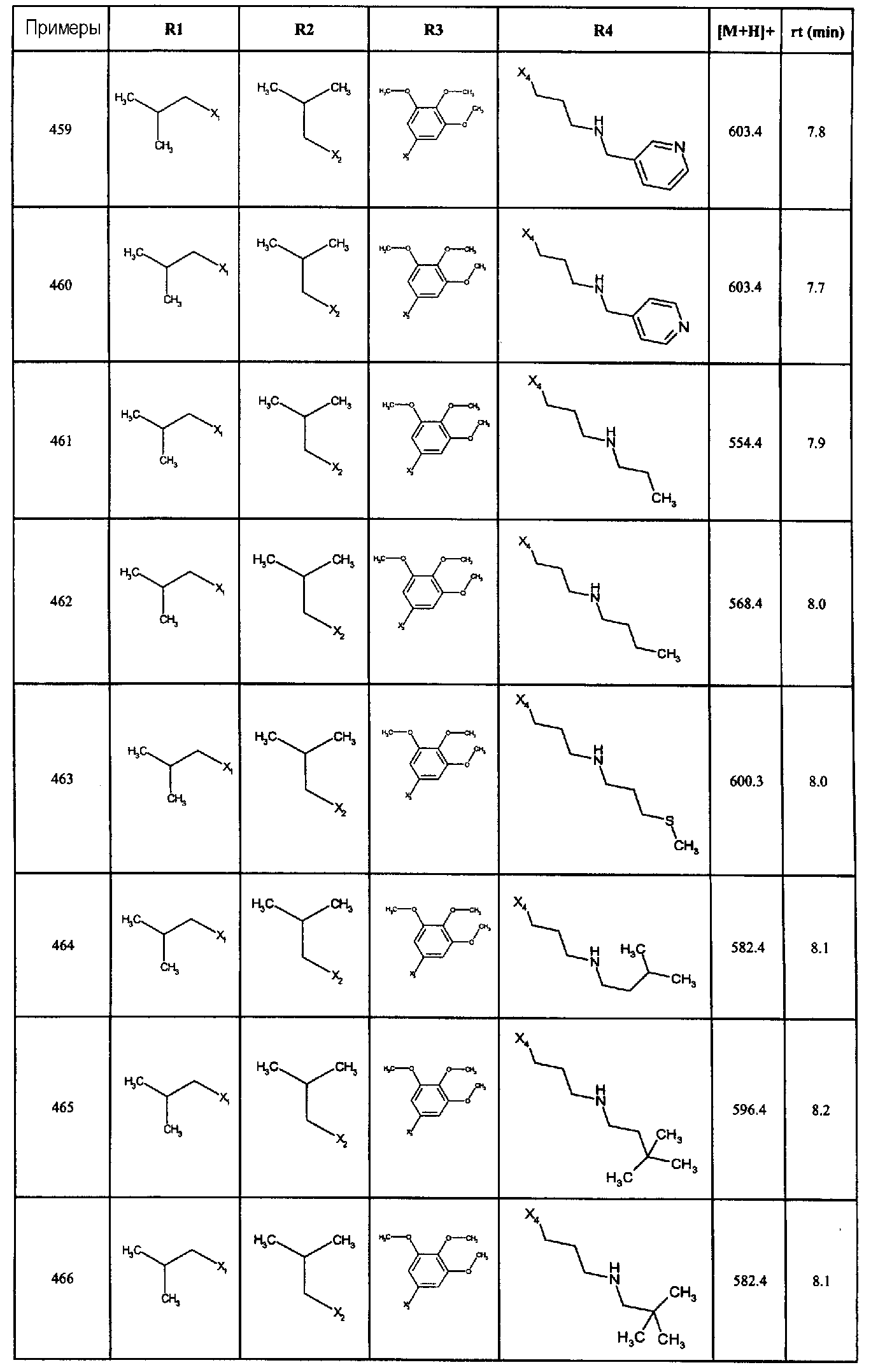
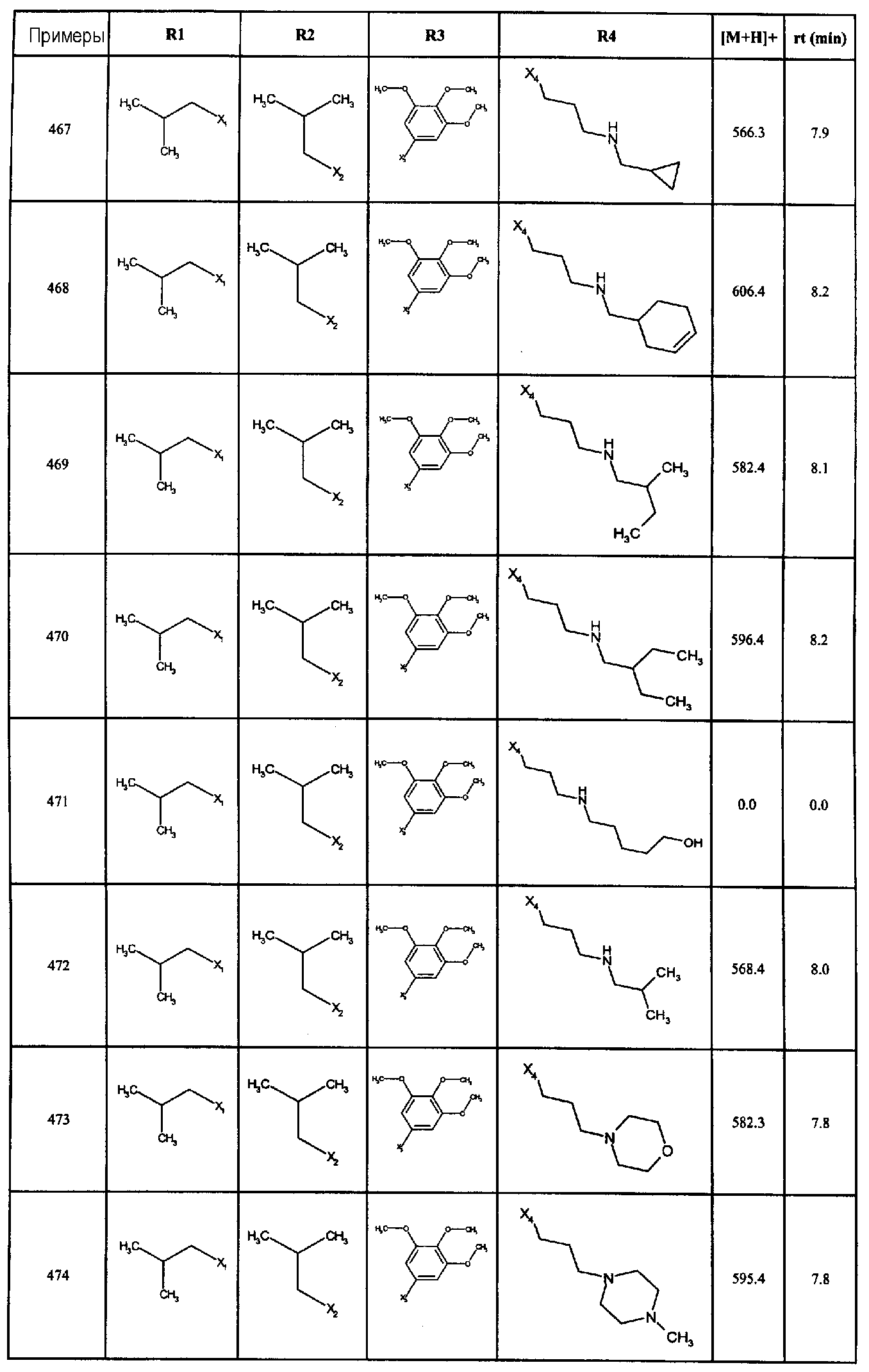
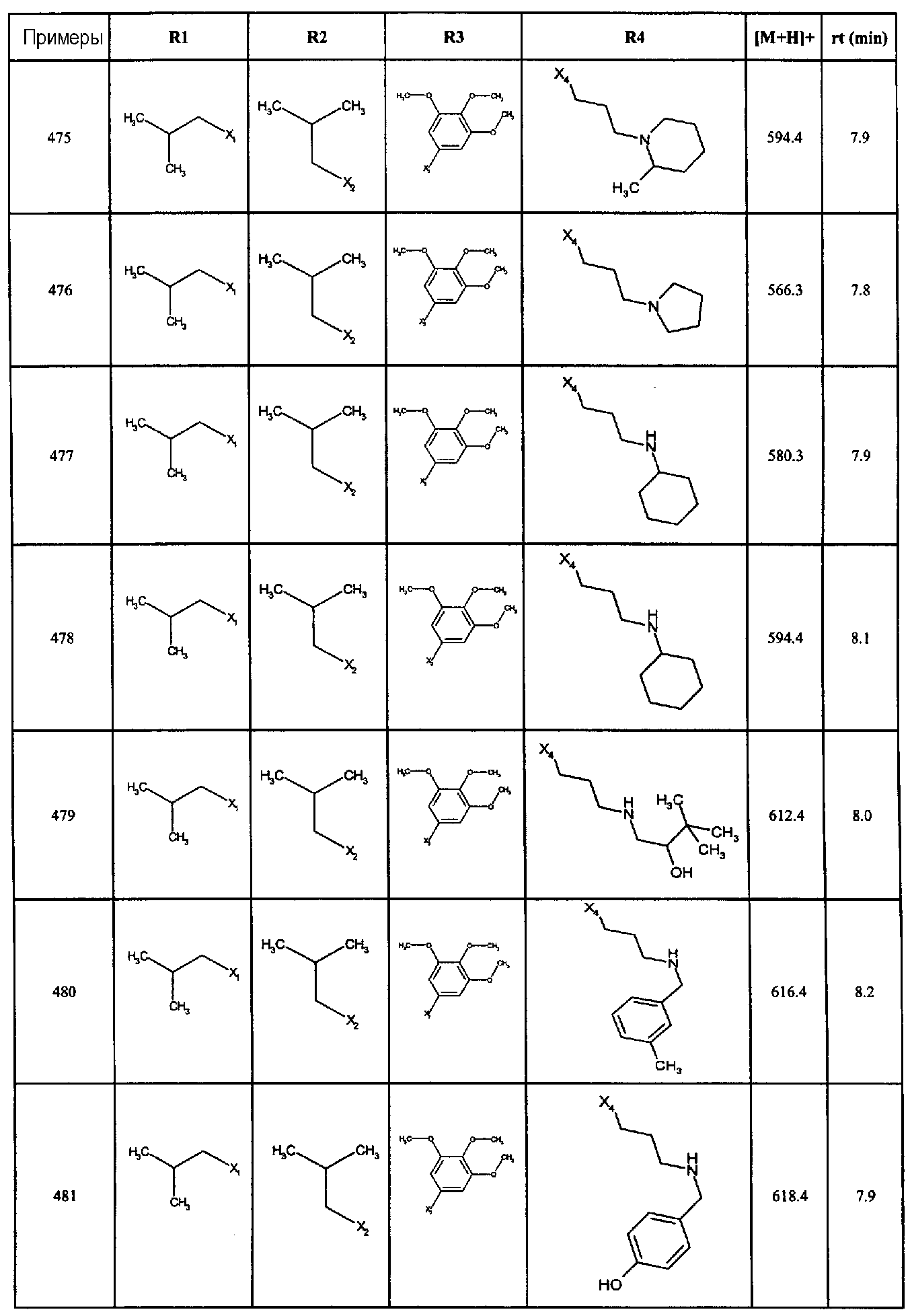
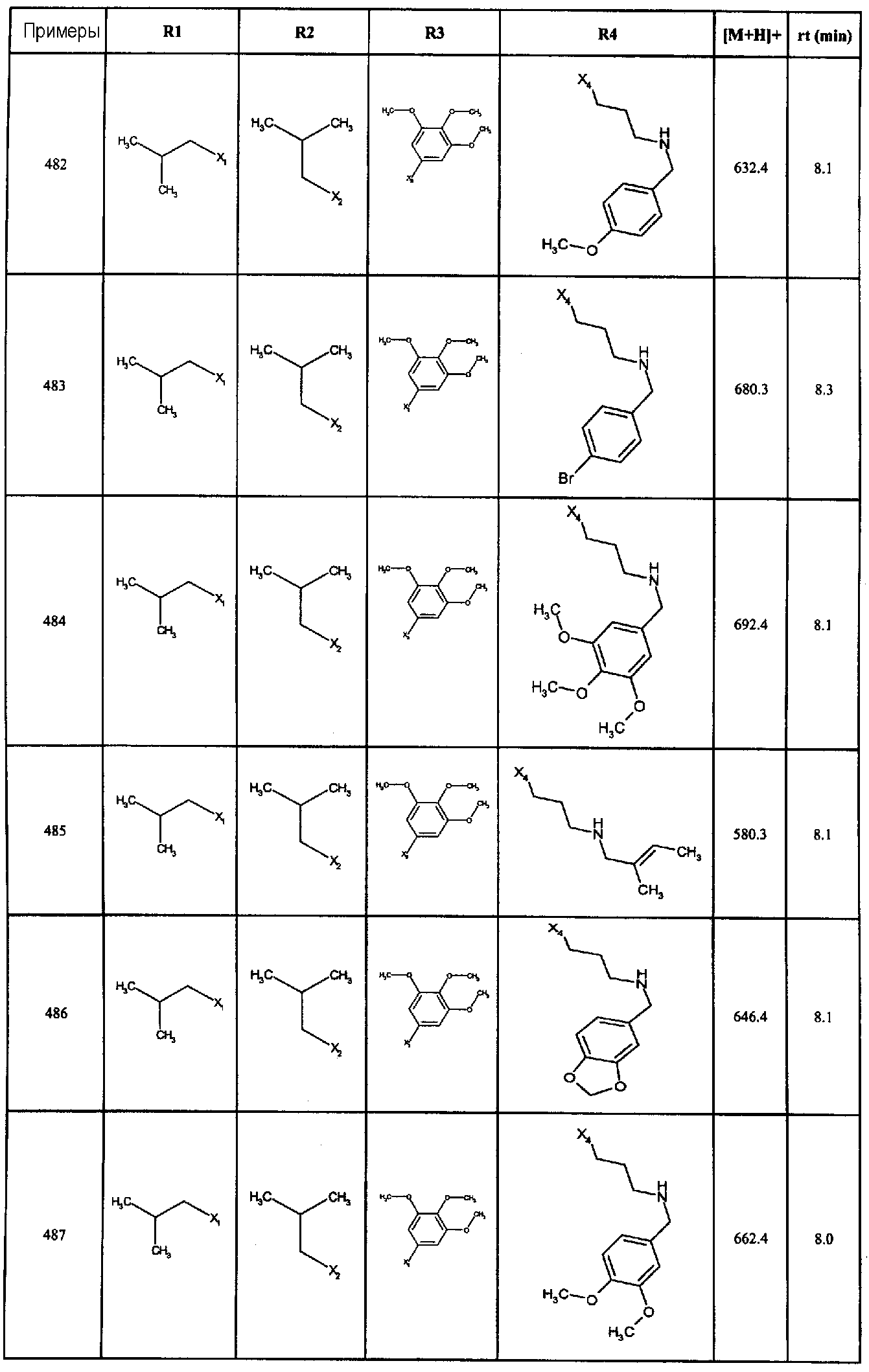

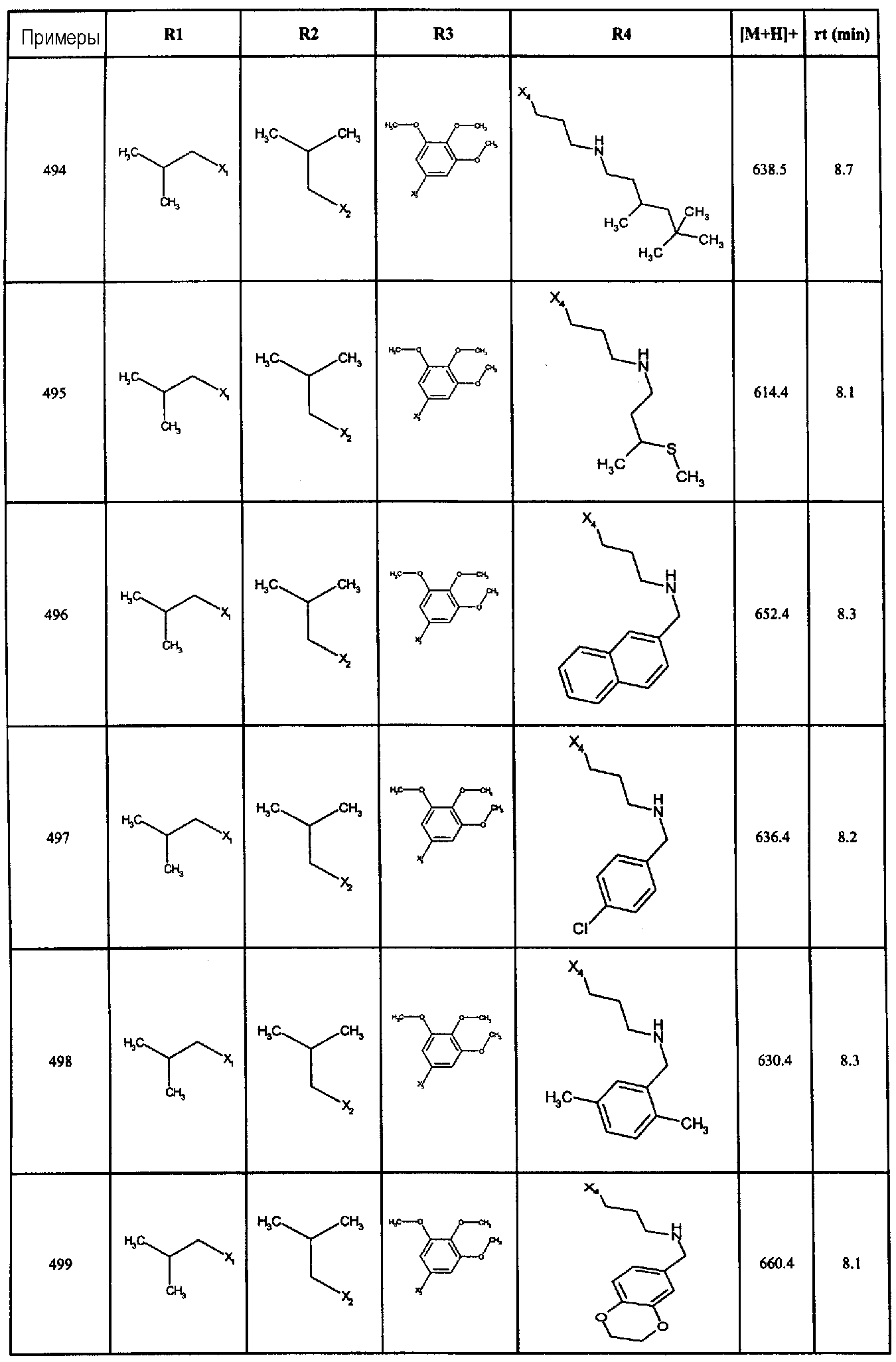

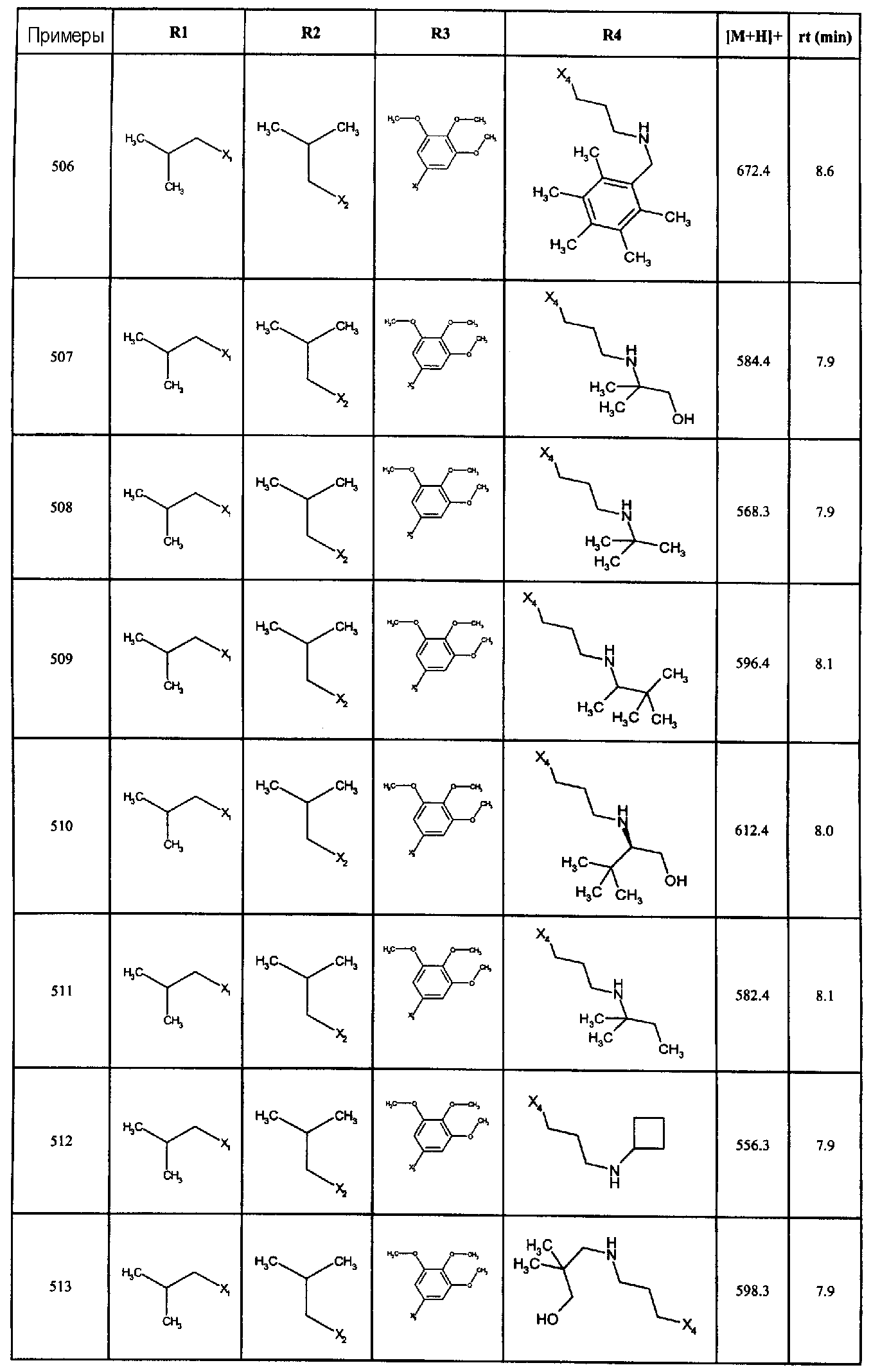
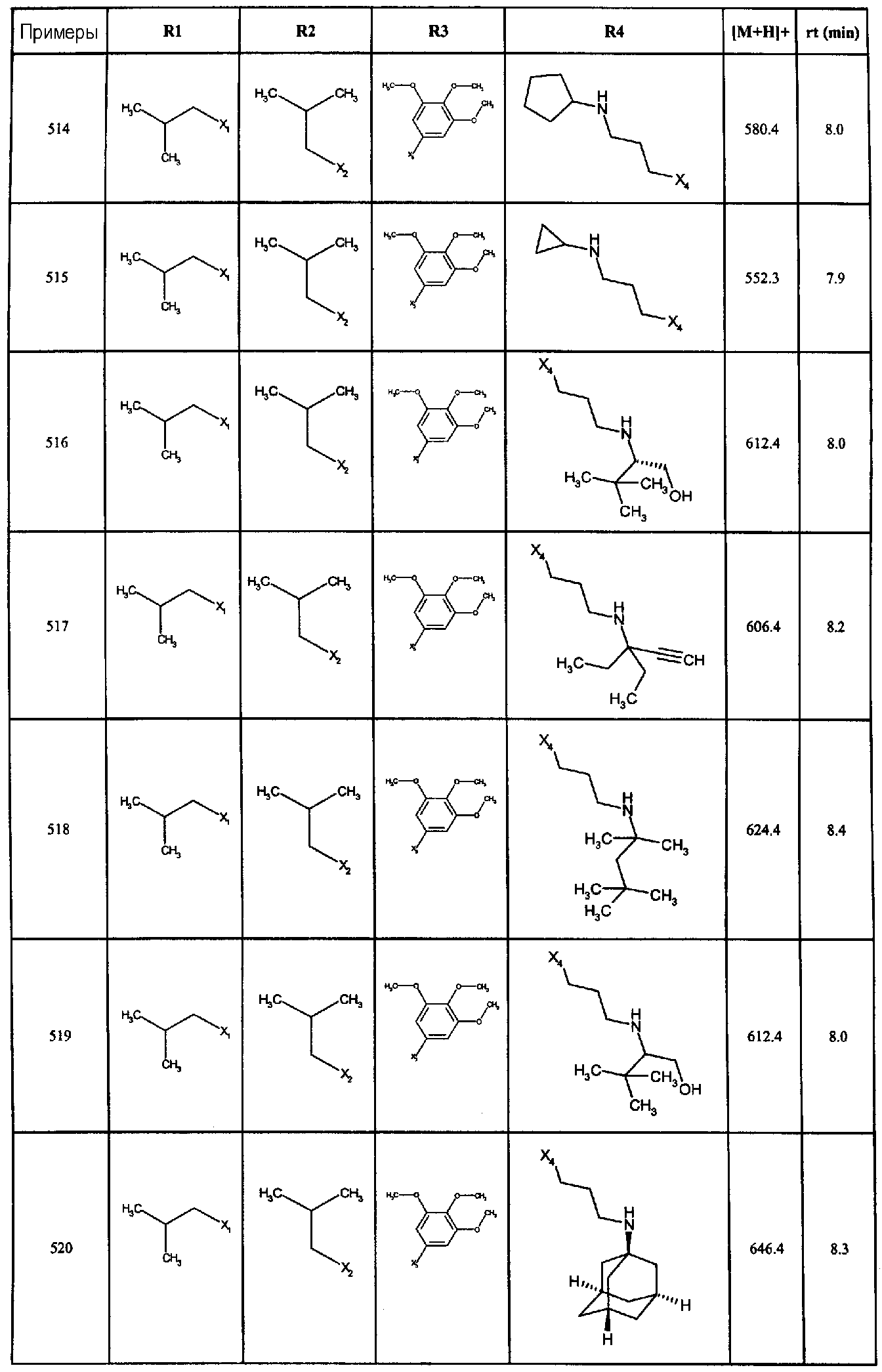
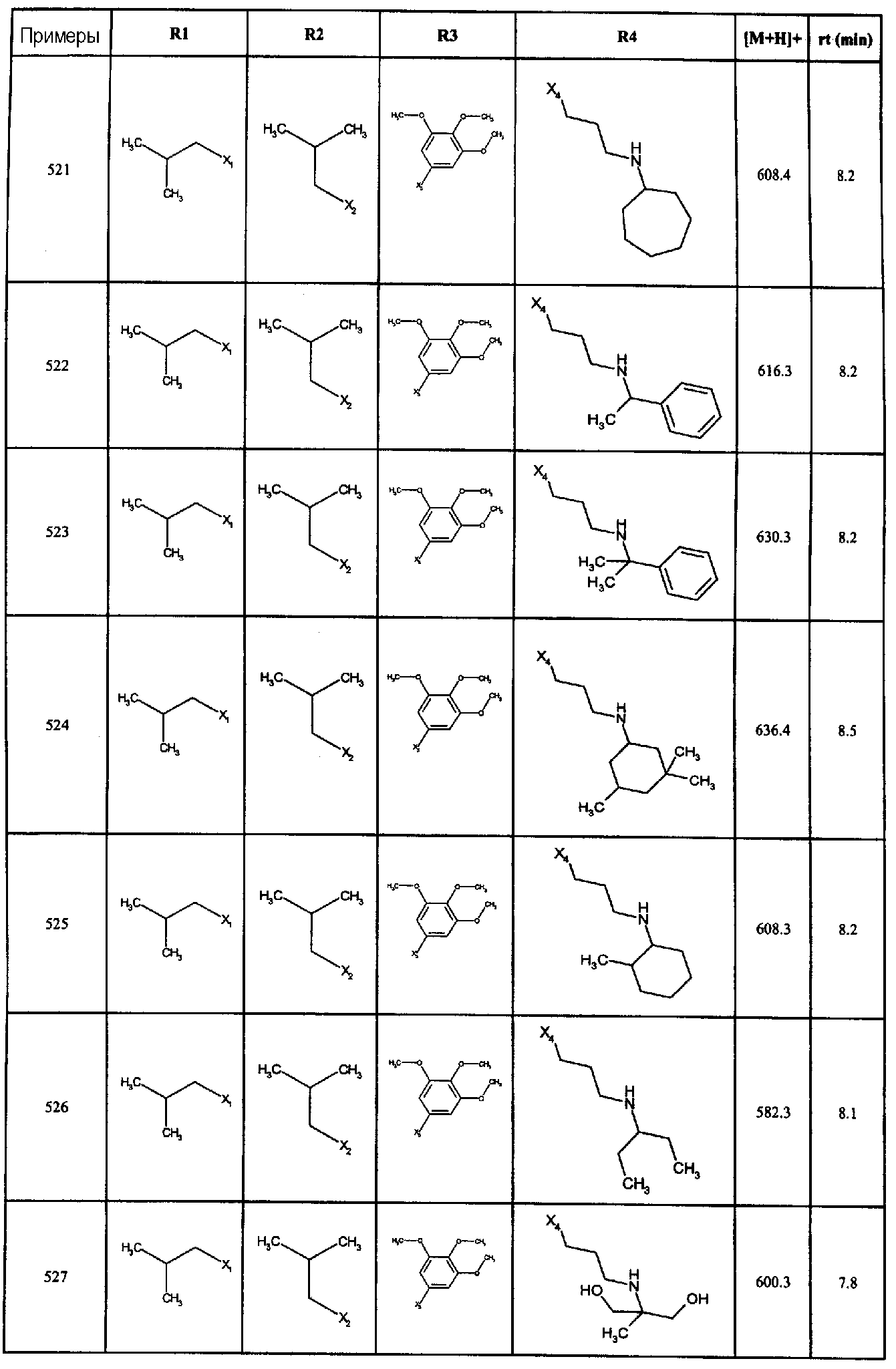
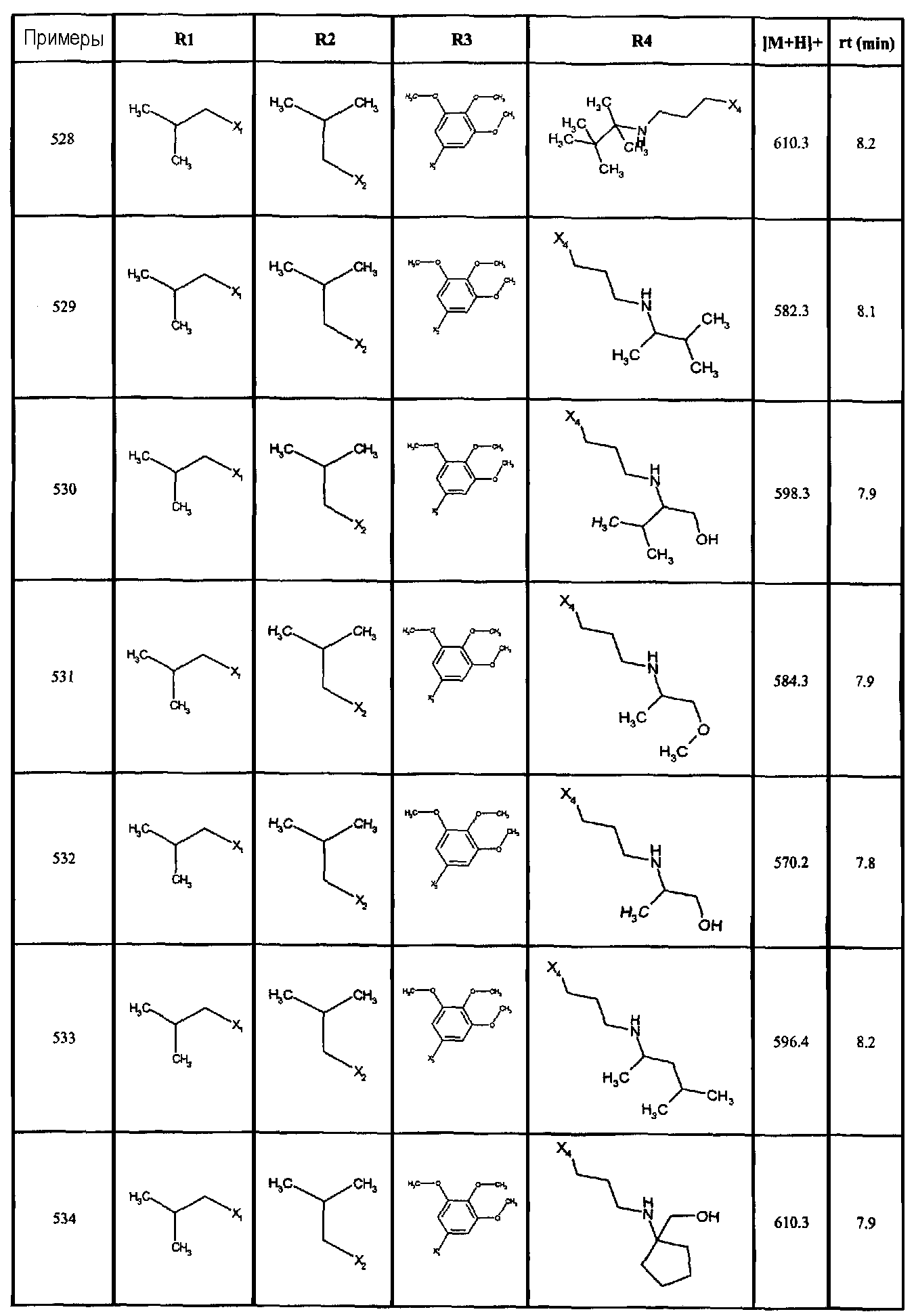
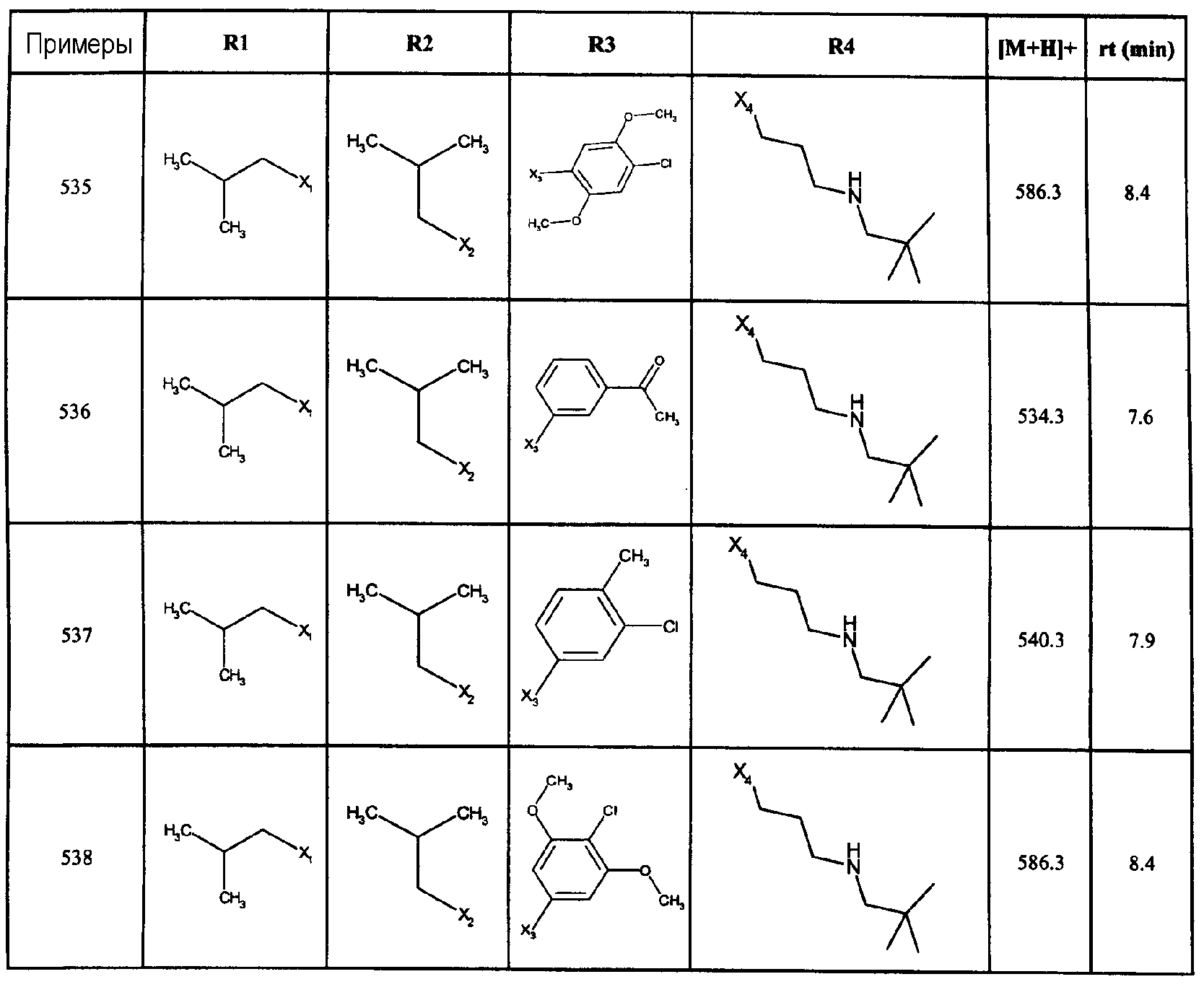
Фармакологическое исследование
Антагоническая активность GnRH соединений по данному изобретению измеряли в соответствии со следующими протоколами:
Получение стабильной линии, трансфецированной рецептором LHRH человека
кДНК рецептора LHRH человека клонировали в сайт экспрессирующего вектора млекопитающего pCDNA3,1 (InVitrogen Lie). Эту плазмидную конструкцию трансфецировали, используя Effectene в соответствии с рекомендациями изготовителя (Qiagen), в линию клеток, полученных из эмбриональной почки человека, HEK-293 (ATCC) и проводили селекцию на среде DMEM, содержащей 0,5 мг/мл генетицина. Клетки, содержащие вектор экспрессии рецептора LHRH, затем клонировали путем лимитированных разведений, затем мультиплицировали в культуре. Эти клонированные клетки затем исследовали на экспрессию рецептора LHRH человека с помощью тестов на конкурентное ингибирование связи и проводя измерение инозитол фосфатов.
Клеточная культура и получение мембран
Клетки HEK-293, стабильно экспрессирующие рецептор LHRH человека, как описано выше, культурировали в среде DMEM, содержащей 10% фетальную сыворотку теленка и дополненной 0,4 мг/мл генетицина (G418, Sigma Chemical Company). Клетки отделяли от культуральной среды с помощью 0,5 мМ EDTA и центрифугировали при 500 g в течение 10 минут при 4°C. Клеточный осадок промывали 50 мМ Tris, pH 7,4, и дважды центрифугировали при 500 g в течение 10 минут. В завершении клетки лизировали ультразвуком, затем центрифугировали при 39000 g в течение 10 минут при 4°C. Осадок ресуспендировали в 50 мМ Tris 50, pH 7,4 и центрифугировали при 50000 g в течение 10 минут при 4°C для получения осадка мембран, который затем разделяли на несколько аликвот и хранили при -80°C перед использованием.
Исследование аффинности в отношении рецептора LHRH человека
Аффинность соединения по данному изобретению в отношении рецептора LHRH человека определяли путем измерения ингибирования связывания [125I-Tyr5]-DTrp6-LHRH на клетках человека, трансфецированных кДНК рецептора LHRH человека.
Исследование конкурентного ингибирования связывания [125I-Tyr5]-DTrp6-LHRH проводили в 96-лучных полипропиленовых плашках, дублируя их. Мембраны клеток HEK-293, стабильно экспрессирующие рецептор LHRH человека (20 мкг белка/лунку), инкубировали в присутствии [125I-Tyr5]-DTrp6-LHRH (0,2 нМ) в течение 60 минут при 4°C в среде, содержащей 50 мМ Tris/HCl, pH 7,4, бактитрацин 0,1 мг/мл, BSA 0,1% (1 мг/мл).
Связанные [125I-Tyr5]-DTrp6-LHRH отделяли от свободных [125I-Tyr5]-DTrp6 -LHRH путем фильтрации через фильровальные пластины из стеклянных волок GF/C (Unifilter, Packard), пропитанные 0,1% полиэтиленимином, используя FilterMate 96 (Packard). Затем фильтры промывали 50 мМ буфером Tris/HCl при 4°C в течение 4 секунд и с помощью сцинциляционного счетчика (Packard, Topcount) подсчитывали радиоактивность.
Специфическое связывание подсчитывали после вычитания неспецифического связывания (определяли в присутствии 0,1 мкМ DTrp6-LHRH) от общего связывания. Данные, относящиеся к связыванию, получали с помощью анализа нелинейной регрессии и определением величин константы ингибирования (Ki).
Определение параметров агонизма или антагонизма соединения по настоящему изобретению проводили способом, описанным ниже.
Функциональный тест: Ингибирование продукции внутриклеточных инозитол фосфатов
Клетки HEK-293, стабильно экспрессирующие рецептор GnRH человека, культурировали в количестве 200000 клеток на лунку в 24-луночной плашке, покрытой поли-D-лизином (Falcon Biocoat) в среде DMEM, содержащей 10% фетальную сыворотку теленка и 0,4 мг/мл генетицина, в течение 24 часов.
Среду затем заменяли средой DMEM без инозитола, содержащей 10% фетальную сыворотку теленка и 1 мкКю/мл [3H]миоинозитола (Amersham), в течение 16-18 часов при 37°C.
Клетки промывали DMEM без инозитола, содержащей 10 мМ хлорида лития и инкубировали в течение 30 минут при 37°C.
Продукцию инозитол фосфатов стимулировали добавлением 0,5 нМ DTrp6-LHRH в течение 45 минут при 37°C.
Антагонистический эффект соединения измеряли путем одновременного добавления 0,5 нМ DTrp6-LHRH, и исследуемого соединения с различными увеличивающимися концентрациями (пример: от 10-10 M до 10-5 M).
Реакционную среду элиминировали, добавляли 1 мл 0,1 M муравьиной кислоты и инкубировали в течение 5 минут при 4°C.
Плашку затем замораживали при -80°C, затем размораживали при комнатной температуре.
Затем на ионно-обменной смоле (Biorad) отделяли инозитол фосфаты от всех внутриклеточных инозитолов, элюируя 1 M формиатом аммония и 0,1 M муравьиной кислотой.
Количество инозитол фосфатов, покинувших колонку, окончательно измеряли в присутствии сцинцилирующей жидкости.
Результаты
Проведенные в соответствии со вышеописанными протоколами исследования показали, что продукты общей формулы (I), определенной в настоящей заявке, обладают хорошей аффинностью в отношении рецептора LHRH, константа ингибирования Ki на этом рецепторе ниже микромолярной для конкретных примеров соединений.
Реферат
Изобретение относится к новым производным бензимидазола общей формулы
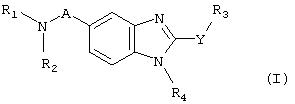
где А представляет собой -СН2- или -С(O)-; Y представляет собой -S- или -NH-; R1 и R2 представляют собой, независимо, атом водорода, (С1-С8)алкил, (С5-С9)бициклоалкил, необязательно замещенный одним или несколькими одинаковыми или различными (С1-С6)алкильными радикалами, или радикал формулы -(СН2)n-Х, где Х представляет собой амино, (С3-С7)циклоалкил, а также другие значения радикалов, указанные в формуле изобретения; R3 представляет собой -(CH2)p-W3-(CH2)p'-Z3; W3 представляет собой ковалентную связь, -СН(O)- или -С(O)-; Z3 представляет собой (С1-С6)алкил, арильный радикал, гетероарил, а также другие значения радикалов, указанные в формуле изобретения; V3 представляет собой -О-, -S-, -C(O)-, -C(O)-O-, -SO2- или ковалентную связь; Y3 представляет собой (С1-С6 )алкильный радикал, необязательно замещенный одним или несколькими одинаковыми или различными галоген радикалами, амино, ди((С1-С6)алкил)амино, фенилкарбонилметила, гетероциклоалкила или арильного радикалов; р и р' представляют собой, независимо, целое число от 0 до 4; R4 представляет собой радикал формулы -(CH2)s-R"4, R"4 представляет собой гетероциклоалкил, содержащий, по крайней мере, один атом азота и необязательно замещенный (С1-С6)алкилом или аралкилом, а также другие значения радикалов, указанные в формуле изобретения. Изобретение относится также к фармацевтической композиции, являющейся антогонистом GnRH, на основе этих соединений. Также рассматривается применение вышеуказанных соединений для получения лекарственного средства. Технический результат - получение новых соединений, фармацевтической композиции и лекарственного средства на их основе в целях лечения таких заболеваний, как: эндометриоз, фиброма, синдром поликистозных яичников, рак молочных желез, яичника и эндометрия, гонадотропная гипофизарная десенсибилизация при медикаментозной стимуляции яичников для лечения бесплодия у женщин.5 н. и 13 з.п. ф-лы, 2 табл.
Формула
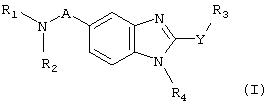
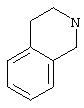
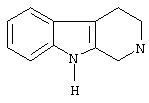
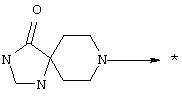
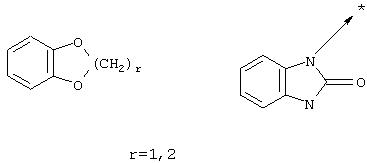
Комментарии