Производные пиразола, способ их получения, содержащая их фармацевтическая композиция и промежуточные соединения синтеза - RU2119917C1
Код документа: RU2119917C1
Чертежи



























Описание
Изобретение касается новых производных пиразола, способа их получения и содержащей их фармацевтической композиции.
Многие производные пиразола были описаны в литературе, в частности в патентах EP-A-268554 и DE-A-3910248, в которых заявлены пиразолы, имеющие гербицидные свойства, EP-A-430186 и JP-A-03031840, в которых защищены соединения, применимые в фотографии, и EP-A-418845, в котором заявлены пиразолы, имеющие противовоспалительную, обезболивающую и антитромбозную активность.
В настоящее время было обнаружено, что пиразолы, являющиеся предметом изобретения, имеют высокое сродство к рецептору каннабиноидов и поэтому особенно интересны в терапевтической области, где известно применение каннабиса для терапевтических целей. Соединение Δ9-тетрагидроканнабинол, или Δ9-THC, является основным активным составляющим, экстрагированным из Cannabis sativa (Tuner, 1985; In Marijuana 1984, Ed. Harvey, DY, IRL Press, Oxford).
Эффект каннабиноидов обусловлен взаимодействием со специфическими рецепторами высокого сродства на центральном (Devane et al., Molecular Pharmacology, 1988, 34, 605 - 613) и периферическом уровнях (Nye et al., The Journal of Pharmacology and Experimental Therapeutics, 1985, 234, 784 - 791; Kaminski et al., 1992, Molecular Pharmacology, 42, 736 - 742).
Характеристика этого рецептора
стала
возможной в
результате разработки специфических синтетических лигандов, таких как CP 55, 940, агониста аналога Δ9-THC.
Терапевтические показания каннабиноидов
касаются
различных
областей, таких как иммунная система, центральная нервная система, сердечно-сосудистая система или индокринная система (Hollister, Pharmacological Reviews, 1986, 38, 1 - 20 et
Renv and
Sinha, Progress
in Drug Research, 1991, 36, 71 - 114).
В частности, соединения, имеющие сродство к рецепторам каннабиноидов, имеют применение в качестве иммуномодуляторов, психотропных средств, при заболеваниях тимуса, рвоте, миорелаксации, различных невропатических состояниях, мнезических заболеваниях, дискинезиях, мигрени, астме, эпилепсии, глаукоме, а также при противораковой химиотерапии, ишемии и удушьи, ортостатической гипотонии и сердечной недостаточности.
Более конкретно настоящее изобретение касается соединений формулы I

где g2-g6 и w2-w6 - одинаковы или различны и являются независимо водородом, атомом хлора или брома, C1-C3-алкилом, C1-C3-алкокси, трифторметилом, нитрогруппой;
g4 - при необходимости, фенильной группой;
R4 - водород или C1-C3-алкил;
X - либо прямая связь, либо -(CH2)nN(R3)-группа, где R3 - водород или C1-C3 -алкил, а n - нуль или единица;
R означает
-NR1R2-группу, где R1 и R2 - независимо друг от друга означают C1-C6 -алкил;
неароматический карбоциклический радикал C3-C15, при необходимости замещенный, амино C1-C4 алкильную группу, где аминогруппа при необходимости дизамещена C1-C3-алкилом; циклоалкил C1-C3-алкил, где циклоалкил имеет 3 - 12 атомов углерода; фенил, незамещенный или замещенный одним или несколькими атомами галогена, C1-C5-алкилом или C1-C5 -алкоксигруппой; фенил C1-C3-алкил; дифенил C1-C3-алкил; нафтил; антраценил, гетероциклический насыщенный радикал 5 - 8-членный, незамещенный или замещенный C1-C3-алкилом, гидроксилом или бензилом, 1-адамантилметил, ароматический гетероцикл, незамещенный или замещенный одним или несколькими атомами галогена, C1-C5-алкилом или C1-C5 - алкоксигруппой; C1-C3-алкил, замещенный ароматическим гетероциклом, незамещенным или замещенным одним или несколькими атомами галогена, C1-C5-алкилом или C1-C5-алкоксирадикалом или R1 - водород, а R2 - как определено выше, или же R1 и R2 образуют с атомом азота, с которым они связаны, насыщенный 5 - 8-членный гетероциклический радикал, причем указанный гетероциклический радикал не является морфолином, если w2, w3, w4, w5, w6, g2, g3, g4, g5 и g6 являются все водородом;
группу R2, определенную выше, если X означает (CH2)n N(R3);
группу R5, если X - прямая связь, причем R5 - C1-C3-алкил, C3-C12-циклоалкил, незамещенный или замещенный C1-C5-алкилом, фенил C1 -C3-алкил, незамещенный или замещенный галогеном или C1-C5-алкилом, циклоалкил C1-C3 -алкил, где циклоалкил имеет 3-12 атомов углерода, незамещенный или замещенный C1-C5-алкилом; 2-норборнилметил; или одной из их возможных солей.
Неароматические карбоциклические радикалы C3-C15 включают моно- или полициклические радикалы, конденсированные или мостиковые насыщенные или ненасыщенные, при необходимости терпеновые. Эти радикалы при необходимости моно- или полизамещенные, причем указанный(ые) заместитель(и) не являются замещенной карбонильной группой. Преимущественно моноциклические радикалы замещены по крайней мере одной группой, выбранной из C1-C5 - алкильного, C1-C5- алкоксирадикалов, галогенов или гидроксирадикалов, разумеется, если терпены или терпеновые радикалы, например борнил, метил или мантенил, алкильные группы терпена не рассматриваются как заместители.
Моноциклические радикалы включают циклоалкилы, например циклопропил, циклопентил, циклогексил, циклогептил, циклооктил, циклододецил, незамещенные или замещенные по крайней мере одной C1-C5- алкильной группой, C1-C5- алкоксигруппой, галогеном или гидроксигруппой.
Конденсированные мостиковые или спирановые ди- или трициклические радикалы включают, например, радикалы норборнила, борнила, изоборнила, норадамантила, адамантила, спиро (5,5) ундеканила, причем указанные радикалы незамещены или замещены C1-C5-алкилом.
Под насыщенным гетероциклическим радикалом 5 - 8-членным подразумевают гетероциклический неароматический моно-, ди- или трициклический конденсированный или мостиковый радикал, причем гетероатом S, O или N, или гетероциклический неароматический моноциклический радикал, содержащий атом азота или атом кислорода или серы, причем указанные радикалы, например тетрагидрофуранил, тетрагидротиофуранил, тропил, морфолинил, тиоморфолинил, пиперидинил, пиперазинил, пирролидинил, хинуклидинил, причем предпочтительны радикалы 1-пирролидинила, 1-пиперидинила, 1-гексагидроазепинила, 4-морфолинила и 4-тиоморфолинила.
Ароматические гетероциклы могут быть моно- или дициклическими, как например пиролил, пиридил, индолил, хинолинил, тиазолил, изоиндазолил, причем эти ароматические гетероциклы незамещены или замещены, например, галогенами C1-C5-алкилами, C1-C5 алкоксирадикалами. Предпочтительны ароматические гетероциклы - пиридил, пиррол, индол, 2-индолильный и 3-индолильный радикалы.
В приведенной выше формуле I предпочтительно по крайней мере один из заместителей w2, w3, w4, w5, w6, g2, g3, g4, g5 и g6 не является водородом.
В приведенной выше формуле I, если R является NR1R2-группой,
предпочтительно:
- R1 - водород или алкильная
группа C1-C6, а R2 - как определено выше для (I); или
- R1 и
R2
являются каждый алкильной группой C1-C6, или
циклоалкильной группой C3-C6; или
- R1 - водород или алкильная группа C1-C6, а R2 - циклоалкил C1-C3
алкильная группа, где циклоалкил имеет 3-12 атомов углерода, неароматический карбоциклический радикал C3
-C15,
незамещенный или замещенный, как указано выше, фенил, незамещенный
или замещенный одним или несколькими галогенами, C1-C3-алкилом или C1-C3 алкоксигруппой,
фенил C1-C3-алкил или C1-C3-алкил, замещенный 2- или 3-индолилом.
Особенно предпочтительно, когда в формуле I R является NR1 R2-группой, R1 - водород или алкил C1 -C6, а R2 - карбоциклический неароматический радикал C3-C15, циклоалкил C1 -C3-алкил, где циклоалкил C3-C6, 2- или 3-индолил C1-C3-алкил.
Предпочтительными алкильными группами являются метильная, этильная, пропильная и изопропильная группы.
В формуле I, приведенной выше, R является предпочтительно NR1R2-группой, предпочтительно выбранной из приведенных ниже радикалов (1) - (74).
Если R1 и R2 с атомом азота, с которым они связаны, являются насыщенным гетероциклическим радикалом, он предпочтительно 5-, 6- или 7-членный и может содержать другой гетероатом, в частности кислород или серу, например пирролидин, пиперидин, гексагидроазепин, морфолин или тиоморфолин, с уточненными выше ограничениями.
Радикалы, которыми является R, как определено выше для формулы I,
предпочтительно являются радикалами, выбранными из:
(1) пропиламино
(2)
бутиламино
(3)
изопропиламино
(4) дипентиламино
(5) 2-(N,
N'-диэтиламино)этиламино
(6) бензиламино
(7) 2-фенилэтиламино
(8) 3-фенилпропиламино
(9) 3,
3-дифенилпропиламино
(10) фениламино
(11)
3-хлорфениламино
(12) 4-метилфениламино
(13) циклопропиламино
(14) циклопентиламино
(15)
циклогексиламино
(16) циклогептиламино
(17)
циклооктиламино
(18) циклододециламино
(19) 2-метилциклогексиламино
(20) 3-метилциклогексиламино
(21)
ЦИС 4-метилциклогексиламино
(22) Транс
4-метилциклогексиламино
(23) ЦИС 4-терциобутилциклогексиламино
(24) транс 4-терциобутилциклогексиламино
(25)
4-гидроксициклогексиламино
(26)
2-метоксициклогексиламино
(27) 4-этилциклогексиламино
(28) 2,6-диметилциклогексиламино
(29) N-метилциклогексиламино
(30)
N,
N-дициклогексиламино
(31)
эндо-2-норборниламино (или эндо-бицикло[2.2.1]гептан-2-амино)
(32) экзо-2-норборниламино (или экзо-бицикло[2.2.1]гептан-2-амино)
(33)
1-адамантиламино
(34)
2-адамантиламино
(35) 1-норадамантиламино
(36) (IR)-борниламино
(37) (IR)-изоборниламино
(38) спиро[5.5]ундеканиламино
(39)
циклогексилметиламино
(40) 1-адамантилметиламино
(41) 2-(тетрагидрофуранил)метиламино
(42) 2-(N-метил-2-пирролил)этиламино
(43) 2-(2-пиридинил)этиламино
(44) (2-индолил)метиламино
(45) N-метил(2-индолил)метиламино
(46) 2-(3-индолил)этиламино
(47) N-метил 2-(3-индолил) этиламино
(48)
4-(N-бензилпиперидинил)амино
(49)
3-квинуклидиламино
(50) экзо бицикло[3.2.1]октан-2-амино
(51) бицикло[2.2.2]октан-2-амино
(52)
хлоро-3-бицикло[3.2.1]окт-3-эн-2-амино
(53)
бицикло[2.2.2]окт-2-эн-5-амино
(54) экзо бицикло[3.2.1]октан-3-амино
(55) эндо бицикло[3.2.1]октан-3-амино
(56) эндо
окса-7-бицикло[2.2.1]гептан-2-амино
(57) экзо
окса-7-бицикло[2.2.1]гептан-2-амино
(58) эндо-трицикло[5.2.1.02,6] декан-8-мино
(59) N-этил-1-адамантиламино
(60) трицикло[2.2.1.02,6
]гептан-3-амино
(61) бицикло[3.3.1]нонан-9-амино
(62) эндо-триметил-1,3,3-бицикло[2.2.1]гептан-2-амино (или фенхиламино)
(63) (1R,
2S - эндо) (+) бицикло[2.2.1]
гептан-2-амино
(64)
(1R, 2R - экзо) (-) бицикло[2.2.1] гептан-2-амино
(65) (1S, 2S - эндо) (-) бицикло[2.2.1] гептан-2-амино
(66) (1S, 2S
- экзо) (+) бицикло[2.2.1]
гептан-2-амино
(67)
1-пиперидиниламино
(68) 1-пирролидиниламино
(69) 1-гексагидроазепиниламино
(70) 4-морфолиниламино
(71)
4-тиоморфолиниламино
(72) N-метил экзо
бицикло[2.2.1] гептан-2-амино
(73) N-этил экзо бицикло[2.2.1] гептан-2-амино
(74) N-пропил экзо бицикло[2.2.1] гептан-2-амино
Из продуктов вышеуказанной
формулы I
предпочтительны те, которые отвечают формуле Ia

где w2-w6, g2 -g6, R4; R1 и R2 определены выше для формулы I.
Среди соединений формулы Ia наиболее
предпочтительны те соединения, которые
отвечают общей
формуле Ia'

где w2-w6, g2-g6, R4 имеют значения, указанные для соединения формулы I;
R1 - водород или C1-C6-алкил;
R2 - неароматический карбоциклический радикал C3 -C15, или насыщенный гетероциклический радикал 5 - 8-членный, выбранный из 1-пирролидинила, 1-пиперидинила, 1-гексагидроазепинила, 4-морфолинила и 4-тиоморфолинила и их соли.
Среди продуктов формулы I те, которые отвечают приведенным ниже формулам Ia, Ib, Ic, Id, Ie и If, где по крайней мере один из заместителей w2-w6, g2-g6 не является водородом, R1 - водород или алкил C1-C6, R2 - как определено выше, R3 - водород или алкильная группа C1-C3, R4 - водород или метил, а R5 - циклоалкил C1-C3-алкил, причем циклоалкил C3-C6, или фенил C1-C3-алкил, незамещенный или замещенный в ароматическом цикле метильной группой или атомом фтора или хлора, и возможные их соли, являются особенно предпочтительными.
В этих
последних особенно предпочтительных
продуктах,
- если R1 является алкилом C1-C6, предпочтительны метильная,
этильная, пропильная и изопропильная группы;
- если R3 является
алкилом C1-C3, предпочтительна метильная группа;
- группы R2 являются
предпочтительно неароматическими карбоциклическими
радикалами C3-C15, незамещенными или замещенными алкилом C1-C4, в частности метилом, этилом, пропилом,
изопропилом или т-бутилом, или двумя или тремя
метильными группами, например метил-,
этил-, или т-бутилциклогексильным радикалом, или диметил- или триметилциклогексильным радикалом; циклоалкил C1-C3 алкиловыми радикалами,
где циклоалкил - C3-C6, алкильными радикалами C1-C3, замещенными 2- или 3-индолильной группой; 2- и
3-индолильными радикалами и радикалами 1-пирролидинила,
1-пиперидинила, 1-гексагидроазепинила,
4-морфолинила, 4-тиоморфолинина;
- группы R5 являются предпочтительно радикалами
циклогексилметила, циклогексилэтила, бензила,
4-метилбензила и фенэтила.
Из
продуктов вышеуказанной формулы I предпочтительны продукты, представленные формулой i

где R4, X и R - как определено выше для формулы I,
и их соли, особенно, если R4 водород или метильная группа, или если R4 - водород или метил, а X - прямая связь.
Особенно предпочтительны соединения формулы i, где R4 - водород или метил, X - прямая связь, а R - NR1R2-группа, где R1 - водород или метильная группа, а R2 - неароматический карбоциклический радикал C3-C15 или насыщенный гетероциклический радикал 5-8-членный, выбранный из 1-пирролидинила, 1-пиперидинила, 1-гексагидроазепинила, 4-морфолинила и 4-тиоморфолина и их солей.
Также предпочтительны соединения формулы i, где R4 - водород или метил, X - -(CH2 )n-N(R3)-, а R - NR1R2, при этом n равняется нулю или единице, R1 - водород, R3 - водород или метильная группа, а R2 - фенил, незамещенный или замещенный одним или двумя атомами галогена, C1-C5 алкильная или C1-C5 алкоксигруппа или неароматический карбоциклический радикал в C3-C15, и их соли.
Из соединений формулы I также
предпочтительны соединения, отвечающие формуле
ii

где X и R - как определено выше для формулы I, и w4 - метильная или метоксигруппа и их соли, в частности те соединения формулы ii, где w4 - метильная или метоксигруппа, X - прямая связь, а R является NR1R2-группой, где R1 - водород или метильная группа, а R2 - неароматический карбоциклический радикал C3-C15, и их соли.
Предпочтительный подкласс включает соединения формулы ii, где w4 - метильная или метоксигруппа, X - является -(CH2)n-N(R3)-группой, где n равен нулю или единице, R3 - водород или метильная группа, а R является -NR1R2-группой, где R1 - водород, а R2 - фенил, незамещенный или замещенный одним или двумя атомами галогена, C1-C5 алкильная или C1-C5 алкоксигруппа или неароматический карбоциклический радикал C3-C15 или их соли.
Другими интересными соединениями настоящего изобретения являются соединения формулы I, где w2-w6, g2-g6, R4 и X - как определено выше для формулы I, а R является -NR1 R2-группой, где R1 - водород или C1-C6 алкильная группа, а R2 - 2- или 3-индолил C1 -C3 алкильная группа или 2- или 3-индолильная группа и их соли.
Из последних
продуктов особенно интересны продукты формулы iii

где X - как определено выше для формулы I, а R является NR1R2-группой, где R1 - водород или C1-C6-алкил, а R2 - 2- или 3-индолил C1-C3 алкильная группа или 2- или 3-индолильная группа, и, либо w2 - водород, а w4 - метильная или метоксигруппа, либо w2 и w4 - атом хлора, и их соли.
Из продуктов,
включенных в приведенную выше формулу I, также интересны продукты формулы
iv

где X и R - как определено выше для формулы I, а g4 - атом брома, метил или трифторметил, и их соли.
В предпочтительных продуктах формулы iv два атома хлора находятся в позиции 2,3; 2,4; 2, 5 или 3,4; и в этих предпочтительных продуктах формулы iv наиболее предпочтительны те продукты, где X является прямой связью, а R является -NR1R2-группой, где R1 - водород или алкил в C1-C6, а R2 - неароматический карбоциклический радикал, содержащий 3-15 атомов углерода.
Возможные соли соединений настоящего изобретения, в частности соединений указанных выше формул I, Ia', i, ii, iii, iv и приведенных ниже Ia, Ib, Ic, Id, Ie, If, включают как соединения с неорганическими или органическими кислотами, которые позволяют производить разделение или соответствующую кристаллизацию продуктов, такие как пикриновая или щавелевая кислоты, так и те, которые образуют фармацевтически приемлемые соли, такие как хлоргидрат, бромгидрат, сульфат, гидрогенсульфат, дигидрогенсульат, метансульфонат, метилсульфат, малеат, фумарат, 2-нафталенсульфонат, гликонат, цитрат, изетионат.
В соответствии с
другим своим аспектом настоящее изобретение
касается способа получения соединений формулы I, отличающегося
тем, что производное 3-пиразолкарбоновой кислоты формулы

где w2-w6, g2-g6, R4 - как определено выше для формулы I, или одну из его активных форм, сложных эфиров или хлорангидридов кислот, обрабатывают:
либо амином формулы HNR1R2, где R1 и R2 - как определено выше для формулы I, для получения амидов формулы

где w2-w6, g2-g6, R4, R1 и R2 - как определено выше для формулы I,
либо при необходимости первичным амином R3NH2, где R3 - как определено выше для формулы I, для получения промежуточных амидов формулы V

где w2 -w6, g2 -g6, R4 и R3 - как определено выше для формулы I, для получения путем восстановления с помощью гидрида металла промежуточных аминов формулы VI

где w2-w6, g2-g6, R4 и R3 - как определено выше для формулы I, которые переводят в амид или карбамид формулы


где w2-w6, g2-g6, R5, R3 и R4 - как определено выше для формулы I, путем реакции соответственно с хлорангидридом кислоты формулы R2COCl или изоцианатом формулы R2-N=C=O, где R2 - как определено для формулы I,
либо производным азида-дифенилфосфорила в основной среде с последующей кислотной обработкой для получения промежуточного амина формулы

где w2 -w6, g2-g6, R4 - как определено для формулы I, который подвергается воздействию хлорангидрида кислоты R2COCl или изоцианата R2-N=C=O для получения соответственно амидов или карбамидов формулы


где w2-w6, g2-g6, R4 - как определено для формулы I, а R3 - водород, причем те же соединения, где R3 не является водородом, приготавливаются на основе описанного выше первичного амина формулы VII, преобразованного во вторичный амин формулы

где w2-w6, g2-g6, R4 - как определено для формулы I, а R3 означает C1-C5 -алкил, который затем подвергают воздействию хлорангидрида кислоты R2COCl или изоцианата R2-N=C=O для получения амидов и карбамидов формул Id и Ie, как определено выше, где R3 не является водородом,
либо металлоорганическим реактивом, содержащим двухвалентный марганец, R5MnX1, где R5 - как определено для формулы I, а X - галоген, для получения кетоновых производных формулы

полученные таким образом соединения затем при необходимости преобразуются в одну из их солей.
По предпочтительному методу пиразолы формулы I могут быть синтезированы из соответствующих сложных эфиров путем преобразования функции сложного эфира в амид, карбамид или кетон, через кислоту и гидрохлорид кислоты.
Указанные сложные эфиры синтезированы по методике, описанной в Berichte, 1887, 20, 2185.
Схема 1 реакции получения соединений формулы I через их сложные метиловые или этиловые эфиры (Alk=CH3 или C2H5) представлена ниже.
Схема 1

Первый этап (a) состоит из получения соли щелочного металла производного ацетофенона формулы IV, где R4, g2-g6 - как определено выше для формулы I, в которую затем добавляют эквимолярное количество диэтилоксалата (этап b) для получения сложного кетоэфира формулы III.
В случае, когда R4 = H, щелочным металлом и предпочтительно является натрий, а соль сложного кетоэфира формулы III (Alk=CH3) получают по методике, описанной в Bull. Soc. Chim. Fr., 1947, 14, 1098, используя метилат натрия в метаноле для осуществления этапа a).
В случае, когда R4 = CH3, щелочным металлом предпочтительно является литий, а соль сложного кетоэфира формулы III (Alk=C2H5) получают по методике, описанной в J. Heterocyclic. Chem. 1989, 26, 1389, используя литийную соль гексаметилдизилазана в этиловом эфире для осуществления этапа a).
Полученные таким образом соли щелочного металла III и избыток производного гидразина нагревают до температуры флегмы уксусной кислоты (этап c). Путем осаждения в ледяной воде получают сложные эфиры 3-пиразола формулы IIa.
Эти сложные эфиры (IIa) затем преобразуют в их кислоты формулы IIb воздействием щелочного агента, например гидроксида калия, затем подкислением (этап d).
На приведенной выше схеме 1 сложные эфиры
формулы IIa, где w2
и w4 - атом хлора, w3, w5 и w6 - водород, g4 - атом хлора, g2, g3, g5 и g6 - водород, а Alk
является C1-C5-алкилом,
и соответствующие кислоты формулы IIb являются новыми "ключевыми" промежуточными веществами для получения соединений формулы i и
особо предпочтительными
и, таким образом, представляют следующий аспект
изобретения, эти соединения отвечают формулам II'a или II'b

Если X представлен прямой связью, амиды изобретения формулы Ia

где w2-w6, g2-g6, R1, R2 и R4 - определены для формулы I, получают из функционального производного кислоты формулы IIb, предпочтительно хлорида, по обычным методикам с целью их замещения амином формулы HNR1R2, полученным по обычным методикам, для получения соединений формулы Ia в соответствии с изобретением.
Если X - (CH2)n-N(R3)-группа, где n и R3 - определены для формулы I, амиды и карбамиды изобретения формулы Ib
и Ic


где w2-w6, g2-g6, R2, R3 и R4 определены для формулы I, получают из сложного эфира (IIa), описанного выше, по следующей схеме 2:

Переход от сложного эфира (IIa) к промежуточному амиду (V) может осуществляться, например, через хлорангидрид соответствующей кислоты путем его реакции с амином R3NH2 в алканоле, например в этаноле.
Восстановление амида (V) в амин (VI) осуществляется затем посредством гидрида металла, например гидрида лития и алюминия или предпочтительно комплексом ВНЗ-ТГФ в растворе в ТГФ, нагретом до температуры флегмы. Амин (VI) затем преобразуют в амид (Ib) или в карбамид (Ic) в соответствии с изобретением традиционными методами, например соответственно реакцией с хлорангидридом кислоты R2COCl или с изоцианатом R2-N=C=O.
Амиды и карбамиды изобретения формулы Id и
Ie


где w2-w6, g2-g6, R2, R3 и R4 - как определено для формулы I, получают из 3-пирразолкарбоновых кислот, предварительно полученных по следующей схеме 3:
Схема 3

Кислоты (IIb) трансформируются в соответствующие амины (VII) путем реакции Curtius, используя, например, азид дифенилфосфорила в основной среде с последующей обработкой сильной кислотой, например соляной кислотой или трифторуксусной кислотой, как описано в Synthesis, 1990, 295. Амины (VII) трансформируются в амиды (Id) или в карбамид (Ie) в соответствии с изобретением обычными методами, например реакцией с хлорангидридом кислоты R2COCl в случае (Id) при R3 = H или с изоцианатом R2-N=C=O в случае (Ie) при R3 = H.
Альтернативно карбамиды (Ie) при R3 = H могут быть получены путем обратной реакции: кислоты (IIb) трансформируются в соответствующие изоцианаты (VIIc), как описано в J. Org. Chem. 1961, 26, 3511, по приведенной ниже схеме 4.
Реакция изоцианатов (VIIc) с амином R2NH2 дает непосредственно карбамиды
формулы
Ie

Для получения соединений формул Id и Ie, где R3 не является водородом, первичные амиды (VII) предварительно трансформируются во вторичные амины (VIIb) последовательным рядом реакций, например реакцией с хлорангидридом кислоты R'3COCl (с R'3 = C1-C2-алкил) с последующим восстановлением амида (VIIa), полученного, например, путем реакции с ВНЗ в ТГФ. В случае, если R3 является метилом, предпочтительно применяют реакцию аминов (VII) третбутил дикарбонатом, (BOC)2O или со смесью муравьиной кислоты и уксусного ангидрида, что дает соответственно карбамат (VIIa, Z=OtBu) или формамид (VIIa, Z= H), продукты, которые затем восстанавливаются, например, с помощью LiAlH4 для получения аминов (VIIb, R3 = CH3).
Вторичные амины (VIIb) затем трансформируются в амиды (Id) или в карбамиды (Ie) в соответствии с изобретением, как описано выше.
Кетоновые производные изобретения по формуле If

(If, X - прямая связь, а R = R5)
где w2-w6, g2-g6, R4 и R5 - как определено для формулы I, получают предпочтительно из 3-пиразолкарбоновых кислот (IIb), описанных выше, по следующей схеме 5:

Кислоты (IIb) трансформируются в хлорангидрид кислот классическими методами; последние затем трансформируются в кетоновые производные (If) в соответствии изобретением путем реакции с соответствующим органическим реактивом, содержащим двухвалентный магний, R5MnX1, где R5 - как определено для формулы I, а X1 - галоген, предпочтительно атом хлора, например по методу, описанному в Tetrahedron Letters, 1989, 30, 7369.
Альтернативно кетоновые производные (If) могут быть
получены из кислот (IIb) через нитрилы (IIc) по следующей схеме 6:
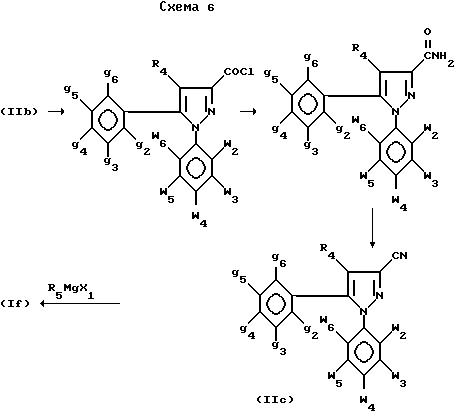
Преобразование (IIb) в (IIc) осуществляется классическим методом, например преобразованием в хлорангидрид кислоты с последующим аминированием (NH3 - ТГФ-вода) и дегидратацией полученного амида, например обработкой CH3SO2Cl в пиридине, как описано в J. Am. Chem Soc. 1955, 77, 1701.
Полученные таким образом нитрилы (IIc) затем обрабатываются маталлоорганическими реактивами, предпочтительно магнийорганическими соединениями формулы R5 MgX1, для получения после кислотной обработки кетоновых производных (If).
Полученные таким образом соединения формулы I выделяют в форме свободного основания или при необходимости в виде соли по классическим технологиям.
Если соединение формулы I получено в форме свободного основания, солеобразование осуществляется обработкой выбранной кислотой в органическом растворителе. Обработкой свободного основания, растворенного, например, в спирте, например изопропаноле, с раствором выбранной кислоты в том же растворителе получают соответствующую соль, которая изолируется по классическим технологиям. Таким образом, получают, например, хлоргидрат, бромгидрат, сульфат, гидрогенсульфат, дигидрогенфосфат, метансульфонат, оксалат, малеат, фумарат, 2-нафталинсульфонат.
В конце реакции соединения формулы I при необходимости могут быть изолированы в форме одной из их солей, например хлоргидрата или оксолата, в этом случае, если необходимо, свободное основание может быть получено путем нейтрализации названной соли неорганическим или органическим основанием, например гидроксидом натрия или триэтиламином, или карбонатом, или бикарбонатом щелочи, например карбонатом или бикарбонатом натрия или калия.
Амины формулы HNR1 R2 либо коммерчески доступны, либо описаны в литературе, либо получают по известным методам согласно главе "Приготовления", описанной ниже.
Среди предпочтительных аминов
можно назвать приведенные ниже:
1) бицикло[3.2.1] октан-2-иламин,
полученный по H. Maskill et al., J. Chem. Soc. Perkin II, 1984, 119;

2) бицикло[2.2.2] октан-2-иламин, полученный по R. Seka et al., Ber. 1942, 1379;

3) эндо- и экзо-бицикло[3.2.1]октан-3-иламин, полученный по H. Maskill et al., J. Chem. Soc. Perkin II, 1984, 1369;

4) эндо- и экзо-окса-7-бицикло[2.2.1]гептан-2-иламин, полученные по W. L. Nelson et al., J. Heterocyclic Chem., 1972, 9, 561;

5) эндо-трицикло[5.2.1.02,6] декан-8-амин, полученный по G. Buchbauer et al., Arch. Pharm., 1990, 323, 367;

6) эндо-триметил-1,3,3-бицикло[2.2.1] гептан-2-иламин, полученный по Ingersoll et al., J. Am. Chem. Soc., 1951, 73, 3360;

7) 3 - метилциклогексиламин, полученный по Smith et al., J. Org. Chem., 1952, 17, 294;

8) 2, 6-диметилциклогексиламин, полученный по Cornubert et al., Bull. Soc. Chim. Fr., 1945, 12, 367;

9) 2-метоксициклогексиламин, полученный по Noyce et al., J. Am. Soc., 1954, 76, 768;

10) 4-этилциклогексиламин, полученный по A. Shirahata et al., Biochem. Pharmacol., 1991, 83, 205;

11) бицикло[2.2.2]окт-2-эн-5-амин, полученный по H.L. Goering et al., J. Am. Chem. Soc., 1961, 83, 1391;
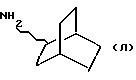
12) N-этил-1-адамантиламин, полученный по V. L. Narayanan et al., J. Med. Chem., 1972, 15, 443;

13) трицикло[2.2.1.02,6] гептан-3-иламин, полученный по G. Miller et al. , Chem. Ber., 1965, 98, 1097;

14) N-метил-экзо-бицикло[2.2.1] гептан-2-иламин, полученный по W. G. Kobalka et al., Synth. Commun., 1991, 20, 231;

Амины R3NH2 коммерчески доступны или их получают по известным методикам.
Хлорангидриды кислот R2COCl коммерчески доступны или их получают из соответствующих кислот по известным методикам.
Изоцианаты R2-N= C= O также коммерчески доступны или их получают из соответствующих аминов (реакция в фосгене) или соответствующих кислот (перегруппировка Curtius) по известным методикам.
Соединения изобретения стали предметом биохимических исследований.
Соединения формулы I и их соли показали хорошее сродство ин виво к рецепторам каннабиноидов в опытах, осуществленных в экспериментальных условиях, описанных Devane et al., Molecular Pharmacology, 1988, 34, 605-613.
Соединения изобретения имеют также сродство к рецепторам каннабиноидов в препаратах изолированных органов, электрически стимулированных. Эти опыты были осуществлены на подвздошной кишке морской свинки и семявыносящем протоке мыши по Roselt et al. , Acta Physiological, Scandinavia, 1975, 94, 142-144 и по Nicolau et al., Arch. Int. Pharmacodyn., 1978, 236, 131-136.
Соединения изобретения вводят главным образом в единицах дозировки.
Названные единицы дозировки предпочтительно указываются в фармацевтических композициях, где действующее начало смешивается с фармацевтическим экципиентом.
Так, по другому из его аспектов настоящее изобретение касается фармацевтических композиций, включающих в качестве действующего начала соединение формулы I или одну из его фармацевтически приемлемых солей.
Соединения формулы I, описанные выше, и их фармацевтически приемлемые соли могут быть использованы в дневных дозах от 0,01 до 100 мг на 1 кг живого веса млекопитающего, предпочтительно в дневных дозах от 0,1 до 50 мг/кг. Для человека доза может варьироваться предпочтительно от 0,5 до 4000 мг в день, в частности от 2,5 до 1000 мг, в зависимости от возраста пациента или типа лечения: профилактического или целебного.
В фармацевтических композициях настоящего изобретения для орального, подъязычного, подкожного, внутримышечного, внутривенного, трансдермического, локального или ректального введения действующее начало вводят в единичных формах, в смеси с классическими фармацевтическими носителями, для животных и людей. Соответствующие унитарные формы введения выпускаются в форме для орального введения, например таблетки, в желатиновой оболочке, порошки, гранулы, растворы или суспензии для орального применения; в форме для подъязычного и защечного введения, в форме для подкожного, внутримышечного, внутривенного, внутриносового или внутриглазного введения и в форме для ректального введения.
В фармацевтических композициях настоящего изобретения действующее начало, как правило, назначается в единицах дозировки от 0,5 до 1000 мг, предпочтительно 1-2000 мг названного действующего начала на единицу дозировки.
При изготовлении твердых композиций в форме таблеток основное действующее начало смешивают с фармацевтическим носителем, например желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик и аналогичные. Таблетки можно покрыть оболочкой из сахарозы или другими соответствующими веществами, или же обработать их таким образом, чтобы они имели пролонгированное или замедленное действие и чтобы они непрерывно высвобождали определенное количество действующего начала.
Препарат в виде желатиновых капсул получают, смешивая действующее начало с разбавителем и выливая полученную смесь в мягкие или твердые желатиновые оболочки.
Препарат в форме сиропа или эликсира может содержать действующее начало вместе с веществом для подслащивания, предпочтительно малокалорийным, метилпарабена и пропилпарабена в качестве антисептика, а также в качестве агента, придающего вкус, и соответствующего красителя.
Растворимые в воде порошки и гранулы могут содержать действующее начало в смеси с диспергаторами или смачивателями, или агентами образования суспензии, как поливинилпирролидон, а также с веществами для подслащивания или корректирования вкуса.
Для ректального введения, применяют свечи, которые изготавливаются со связующими, набухающими при ректальной температуре, например с маслом какао или полиэтиленгликолями.
Для парентерального, внутриносового или внутриглазного назначения используют водные суспензии, изотонические соленые растворы или стерильные растворы для впрыскивания, которые содержат фармакологически совместимые диспергаторы и/или смачиватели, например пропиленгликоль или бутиленгликоль.
Для ингаляции используют аэрозоль, содержащий, например, триолеат сорбитана или олеиновую кислоту, а также трихлорфторметан, дисхлортетрафторэтан или любой другой биологически совместимый рабочий газ.
Действующее начало может также назначаться в форме микрокапсул, при необходимости с одним или несколькими носителями или присадками.
Действующее начало может также быть представлено в форме комплекса с циклодекстрином, например α,β или γ циклодекстрин, 2-гидроксипропил-β-циклодекстрин или метил-β-циклодекстрин.
Указанные таким образом композиции на основе соединения формулы I могут быть использованы для лечения иммуномодуляции, мигрени, астмы, эпилепсии, глаукомы, болезни Паркинсона, дискинезии, невропатических состояний, мнезических заболеваний и заболеваний тимуса, рвоты, ишемии, удушья, артостатической гипертонии или сердечной недостаточности.
Следующие примеры иллюстрируют изобретения, не ограничивая его.
Точки плавления или разложения продуктов (T.пл.) были измерены в капиллярных трубках с помощью аппарата Tottoli.
Энантиомерный избыток оптически активных аминов (Э.И.) был определен по ядерному магнитному резонансу 19F после реакции с хлорангидридом кислоты Mosher S(+) по J.Org. Chem., 1969, 34, 2543.
Вращательная способность, [α], была измерена в эталоне при c = 1.
Приготовления.
A. Амины NHR1R2.
1. (1R, 2S - эндо) (+) бицикло [2.2.1] гептан-2-иламин.
(1R, 2S - эндо) (-) бицикло [2.2.1] гептан-2-карбоновую кислоту получают по Tetrahedron Letters, 1985, 26, 3095.
Реакцией Curtius, осуществленной по J.Org.Chem, 1961, 26, 3511, она затем трансформируется в соответствующий амин (1R, 2S - эндо) (+).
[α] = +13,40 (c = 1, EtOH).
Э.И. > 95% δ (CF3) = 6,67 ppm по отношению к CF3CO2 H.
2. (1R,2R - экзо) (-) бицикло [2.2.1] гептан-2-иламин.
(1R, 2S - эндо) (-) бицикло [2.2.1] гептан-2-карбоновую кислоту, полученную в предыдущем примере, преобразуют в ее изомер (1R, 2R - экзо) (-) по J. Am.Chem.Soc., 1983, 105, 950, затем преобразуют, как описано в предыдущем примере, в соответствующий амин (1R, 2R - экзо) (-).
[α] = 17,70 (c=1, EtOH).
Э.И. > 94% (определен, как описано выше, δ(CF3) 6,81 ppm).
3. (1S, 2R - эндо) (-) бицикло [2.2.1] гептан-2-иламин.
(1S, 2R - эндо) (+) бицикло [2.2.1] гептан-2-карбоновую кислоту получают по Tetrahedron Letters 1989, 30, 5595, затем трансформируют, как описано выше, в соответствующий амин (1S, 2R - эндо) (-).
Э.И. > 95% (определен, как описано выше, δ (CF3) = 6,62 ppm).
4. (1S, 2S - экзо) (+) бицикло [2.2.1] гептан-2-иламин.
(1S, 2R - эндо) (+) кислоту, полученную в предыдущем примере, преобразуют в ее изомер (1S, 2S - экзо) (+) по J. Am. Chem. Soc., 1983, 105, 950, затем изомер преобразуют в соответствующий амин (1S, 2S - экзо) (+), как описано выше.
Э.И. > 94% (определен, как описано выше, δ (CF3) = 6,91 ppm).
5. экзо-3-хлорбицикло[3.2.1]окт- 3-энил-2-амин.
К раствору 6,1 г экзо-3-хлоро-2-азидбицикло[3.2.1]окт-3-эн, полученному по J. Chem. Soc.Perkin. Trans II, 1984, 119, в 600 мл этанола и 60 мл CHCl3 добавляют 0,4 г PtO2 и гидрогенизируют в аппарате Parr при 4 бар и при комнатной температуре до исчезновения азидной функции. После фильтрации на целите реакционная смесь выпаривается, а остаток кристаллизуется в смеси этанол/ CHCl3. Получают 0,49 г хлоргидрата желаемого амина. Т.пл. > 240oC.
6. N-этил-экзо-бицикло[2.2.1]гептан-2-иламин.
6.1. N-ацетил-экзо-бицикло[2.2.1]гептан-2-иламин.
Раствор 3,50 мл ацетилхлорида в 10 мл CH2 Cl2 добавляют покапельно к раствору 5,00 г экзо-бицикло[2.2.1]гептан-2-иламина и 6,90 мл триэтиламина в 50 мл CH2Cl2, охлажденного до 0oC. После 16 часов перемешивания при комнатной температуре смесь выливают в 100 мл ледяной воды, а органическую фазу отделяют и промывают 5%-ным раствором HCl, затем водой, затем насыщенным раствором NaCl. После высушивания на MgSO4 и выпаривания растворителей получают 5,80 г ожидаемого ацетамида. Т.пл. = 128oC.
6.2. N-этил-экзо-бицикло[2.2.1]гептан-2-иламин.
Раствор 5,10 г предыдущего производного в 30 мл ТГФ, добавляют покапельно в суспензию 2,18 г LiAlH4 в 30 мл ТГФ охлажденную до 0oC, затем смесь нагревают до температуры флегмы в течение 8 часов. Смесь гидролизуют при 0oC в 2,2 мл воды, затем 2,2 мл 15%-ного раствора NaOH, затем 7,5 мл воды. После 15 минут перемешивания осадок фильтруют и промывают в ТГФ, фильтрат выпаривают, затем вводят в 50 мл этилового эфира. Этот эфирный раствор экстрагируют 5%-ной HCl, полученная водная фаза нейтрализуется 30%-ным NaOH, затем экстрагируется этиловым эфиром. После промывания насыщенным раствором NaCl, сушки на MgSO4 и выпаривания получают 3,82 г бледно-желтой жидкости. Растворением в этиловом эфире и обработкой раствором HCl в газовой фазе в безводном этиловом эфире получают белый осадок, который фильтруют, промывают этиловым эфиром и высушивают в вакууме. Таким образом, получают 4,16 г хлоргидрата ожидаемого амина. Т.пл. = 145oC (разложение).
7. N-(H-пропил)-экзо-бицикло[2.2.1]гептан-2-иламин.
7.1. N-пропионил-экзо-бицикло[2.2.1]гептан-2-иламин.
Этот амид получают таким же способом, что и аналог N-ацетил, описанный в предыдущем примере 6, используя пропионилхлорид вместо ацетилхлорида.
7.2.
N-(п-пропил)-экзо-бицикло[2.2.1]гептан-2-иламин
Этот амин получают исходя из
полученного выше амида тем же способом, что и N-этиловый аналог, описанный в предыдущем примере. Образуют соль с
помощью HCl/Et2O в смеси Et2
O/iPr2O и получают
хлоргидрат желаемого амина. Т.пл. = 230oC (разложение).
8. Бицикло[3.3.1]нонан-9-иламин.
8.1. Бицикло[3.3.1]нонан-9-оноксим.
Раствор 1,83 г хлоргидрата гидроксиламина и 2,95 г ацетата натрия в 22 мл воды добавляют к раствору 2,43 г бицикло[3.3.1]нонан-9-она в 9 мл метанола, смесь нагревают до температуры флегмы в течение 24 часов. После охлаждения смесь экстрагируют этиловым эфиром, органические фазы промывают насыщенным раствором NaCl, затем 5%-ным раствором Na2CO3, затем водой, высушиваются на MgSO4 и выпаривают. Получают 3,00 г оксима. Т.пл. = 151oC.
8.2. Бицикло[3.3.1]нонан-9-иламин.
К раствору 1,00 г оксима в 250 мл этанола и 4 мл CHCl3 добавляют 0,20 г PtO2 и гидрогенизируют в аппарате Parr при 6 бар и комнатной температуре в течение 18 часов. После фильтрации на целите сольвенты выпаривают, а остаток кристаллизуют в смеси этанол/гептан. Получают 0,55 г хлоргидрата желаемого амина. Т.пл. > 240oC.
Пример 1. N-(2-адамантил)-1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол- 3-карбоксамид.
(I) : w2, w4 = Cl, R4 = H;
g4 = Cl;

A) Натриевая соль 4-хлорбензоилпирувата метила.
12 г натрия растворяют в 250 мл безводного метанола. Затем добавляют смесь 64,6 мл 4-хлорацетофенона и 67,1 мл диэтилоксалата в 600 мл метанола, поддерживая температуру ниже 10oC. Реакционная смесь затем перемешивается при комнатной температуре в течение 3 часов, затем добавляют 1 л сухого эфира. Снова перемешивают в течение 20 минут, фильтруют, промывают осадок эфиром и высушивают в вакууме для получения 74,6 г желаемой натриевой соли.
B) Метиловый эфир 1-(2,4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-карбоновой кислоты.
Суспензию 26,3 г полученной на этапе A натриевой соли и 23,5 г хлоргидрата 2, 4-дихлорфенилгидразина в 250 мл уксусной кислоты нагревают до температуры флегмы в течение 4 часов. После охлаждения смесь выливают на 250 г льда, полученные кристаллы фильтруют, промывают водой и высушивают в вакууме для получения 26,3 г эфира. Т.пл. = 167oC.
C) 1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-карбоновая кислота.
Раствор 3,70 г KOH в 35 мл воды добавляют к раствору 10,0 г полученного на этапе B эфира в 35 мл метанола.
Смесь нагревают до температуры флегмы в течение 4 часов, охлаждают до комнатной температуры и выливают в 100 мл воды, затем нейтрализуют 5%-ным раствором HCl. Полученные кристаллы фильтруют, промывают водой, затем пентаном и высушивают в вакууме. Получают 9,50 г кислоты. Т.пл. = 185o C.
D) Хлорангидрид 1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-карбоновой кислоты.
5,8 мл тионилхлорида добавляют к суспензии 9,50 г полученной на предыдущей стадии кислоты в 100 мл толуола, смесь нагревают до температуры флегмы в течение 3 часов. Раствор затем выпаривают, затем остаток извлекают 50 мл толуола, а раствор вторично выпаривают (процесс повторяют два раза).
Получают 8,28 г хлорангидрида кислоты.
E) N-(2-адамантил)-1-(2,
4-дихлорфенил)-5-(4-хлорфенил)-1H- пиразол-3-карбоксамид
Раствор 0,50 г
полученного на этапе D хлорангидрида
кислоты в 10 мл CH2Cl2 добавляют покапельно в раствор 0,30
г хлоргидрата 2-адамантанамина и 0,41 мл триэтиламина в 10 мл CH2Cl2, охлажденный до 0oC. Смесь затем перемешивают при комнатной температуре в течение 16 часов,
затем выливают в ледяную воду. Смесь экстрагируют CH2Cl2, а
органическая фаза промывается
последовательно 5%-ным раствором HCl, водой, 5%-ным раствором Na2CO3, затем насыщенным раствором NaCl. После высушивания на MgSO4 и
выпаривания растворителя
неочищенный продукт кристаллизуют в бензоле при высокой температуре для получения 0,32 г
белых кристаллов. Т.пл. = 203oC.
Пример 2. N-(транс-4-гидороксициклогексил)-1-(2, 4-дихлорфенил)-5-(4- хлорфенил)-1H-пиразол-3-карбоксамид.
(I): w2, w4 = Cl; g4 = Cl, R4 = H;

A) транс-4-триметилсилилоксициклогексиламин.
Раствор 1,85 мл хлортриметилсилана в 10 мл CH2Cl2 добавляют покапельно, а в раствор 2,0 г хлоргидрата транс-4-гидроксициклогексиламина и 4,05 мл триэтиламина в 20 мл CH2Cl2, охлажденный до 0oC. После 16 часов перемешивания при комнатной температуре смесь гидролизуют в воде и экстрагируют. Органическую фазу промывают последовательно водой, 5%-ным раствором Na2CO3 и насыщенным NaCl. После высушивания на MgSO4 и выпаривания растворителей получают 1,43 г амина (бесцветная жидкость).
B) N-(транс-4-гидроксициклогексил)-1-(2, 4-дихлорфенил)-5-(4-хлорфенил)- 1H-пиразол-3-карбоксамид.
Раствор 0,60 г приготовленного по примеру 1D хлорангидрида кислоты в 10 мл CH2Cl2 добавляют покапельно в раствор 0,35 г транс-4-триметилсилилоксициклогексиламина и 0,32 мл триэтиламина в 10 мл CH2Cl2, охлажденный до 0oC. После 16 часов перемешивания при комнатной температуре смесь выливают в 30 мл ледяной воды и экстрагируют CH2Cl2. Органическую фазу промывают последовательно 5%-ной HCl и насыщенным раствором NaCl, затем высушивают на MgSO4 и выпаривают. Неочищенный продукт растворяют в 15 мл ТГФ добавляют к раствору 15 мл 5%-ной HCl и перемешивают в течение 1 часа. Смесь экстрагируется эфиром и промывается водой, затем высушивается на MgSO4 и выпаривается для получения после кристаллизации в CH3OH 0,20 г ожидаемого пиразола. Т.пл. = 209oC.
Действуя в соответствии с примером 1, описанным выше, из, например, производных кислот или сложных эфиров, описанных ниже в табл. A, получают соединения, описанные в табл. I-XII, приведенных ниже.
Пример 153. N-(2-адамантил)-1-(2, 4-дихлорфенил)-4-метил-5-(4-хлорфенил)-1H-пиразол-3-карбоксамид.
(I): g4 = Cl; w2, w4 = Cl; R4 = CH3;

A) Литиевая соль, 2,4-диоксо-(4-хлорфенил) бутаноата этила 60 мл раствора соли лития гексаметилдизилазана 1,0 M в ТГФ вводят в 240 мл безводного эфира. Смесь охлаждают до - 78oC и вводят покапельно раствор 10,12 г - 4-хлорпропиофенона в 50 мл эфира. После 30 минут перемешивания при -78oC быстро вводят в раствор 9,16 мл диэтилоксана в 50 мл эфира, затем повышают температуру и перемешивают 5 часов при комнатной температуре. Образующийся бледно-желтый осадок фильтруют, промывают в эфире и высушивают в вакууме. Получают 6,32 г желаемой соли.
B) Сложный этиловый эфир 1-(2, 4-дихлорфенил)-4-метил-5-(4-хлорфенил)-1H-пиразол-3-карбоновой кислоты.
Сложный эфир получают тем же способом, что и в примере 1B), из полученной выше литиевой соли, и очищают путем перекристаллизации в изопропиловом эфире. Т.пл. = 105o C.
C) Соединение 153.
Амид получают из полученного на этапе B эфира тем же способом, что в примере 1C, 1D и 1E, путем превращения сложного эфира в хлорангидрид кислоты, проведения реакции последнего с 2-адамантанамином и очистки перекристаллизацией в изопропиловом эфире. Т.пл. = 190o C.
Действуя по описанному выше примеру 153, получают амиды, описанные в табл. XIII, приведенной ниже.
Пример 159. N-[1-(пара-толил)-5-(4-хлорфенил)-1H-пиразол-3-илметил]-N-метилцикло -гексилкарбоксамид.
(I):
g4 = Cl; w4 = CH3; R4 = H;

A) N-метил-1-(п-толил)-5-(4-хлорфенил)-1H-пиразол-3-карбоксамид.
0,50 г хлорангидрида 1-(4-метилфенил)-5-(4-хлорфенил)-3-пиразол-карбоновой кислоты, растворенных в 5 мл CH2 Cl2, добавляют покапельно в 100 мл 33%-ного раствора метиламина в этаноле. После 2 часов перемешивания при комнатной температуре концентрируют в вакууме, извлекают остаток с помощью смеси 5% Na2CO3 + AcOEt, осветляют, промывают органическую фазу насыщенным раствором NaCl, высушивают на MgSO4 и выпаривают растворители. Остаток снова извлекают изопропиловым эфиром, полученные кристаллы фильтруют и высушивают в вакууме. Получают 0,44 г ожидаемого амида. Т.пл. = 138oC.
B) N-метил-[1-(п-толил)-5-(4-хлорфенил)-1H-пиразол-3-ил]метиламин.
8,76 г полученного выше амида, растворенного в 25 мл безводного ТГФ, добавляют покапельно при температуре в пределах от 0 до 5oC в 75 мл раствора 1,0 M ВНЗ в ТГФ. После повышения температуры до комнатной нагревают реакционную смесь до температуры флегмы в течение 16 часов, затем для охлаждения выливают в ледяную ванну 18 мл HCl 6N, перемешивают в течение 1 часа и 30 мин при комнатной температуре, затем отгоняют ТГФ и концентрируют в вакууме. Реакционную смесь подщелачивают NaOH в таблетках до pH 9 - 10; экстрагируют этилацетатом, высушивают на MgSO4 выпаривают растворители и очищают полученный сырой продукт путем хроматографии на силикагеле (300 г). Элюант: CH2Cl2/CH3OH 97/3 (об./об.). Получают 6,0 г амина. Т.пл. = 85oC.
C) Соединение 159.
К раствору 0,46 г полученного на предыдущем этапе амина в 10 мл CH2Cl2 прибавляют последовательно 0,62 мл триэтиламина, затем раствор 0,23 г хлорангидрида циклогексановой кислоты в 5 мл CH2Cl2.
После 15 минут перемешивания при комнатной температуре реакционную смесь концентрируют в вакууме и остаток извлекают с помощью 30 мл воды и экстрагируют этилацетатом. Органическую фазу промывают последовательно 5% Na2CO3, водой, затем насыщенным раствором NaCl, высушивают на MgSO4, затем растворители выпаривают. Неочищенный продукт очищают хроматографией на силикагеле (25 г). Элюант: толуол/AcOEt 70/30 (об. /об.). Фракции чистого продукта концентрируют в вакууме, а остаток перекристаллизовывают изопропиловым эфиром. Получают 0,38 г амида. Т. пл. = 124oC.
Действуя по описанному выше примеру 159, получают амиды, описанные ниже в таблицах XIV и XV.
Пример 172. N-[1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-илметил] -N'- (4-хлорфенил) мочевина.
(I): g4= Cl; w2, w4 =
Cl; R4 = H;

A) 1-(2,4-дихлофенил)-5-(4-хлорфенил)-1H-пиразол-3-карбоксамид.
Этот амид получают способом, описанным в примере 159 A, путем реакции хлорангидрида кислоты, описанной в примере 1D, с насыщенным раствором аммиака в этаноле. Т.пл. = 178oC.
B) [1, (2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил]метиламин.
Этот амин получают способом, описанным в примере 159B, путем восстановления полученного выше амида с помощью ВН3 в ТГФ.
C) Соединение 172.
0,20 г 4-хлорфенилизоцианата добавляют к 0,45 г полученного выше амина, растворенного в 10 мл толуола, и реакционную смесь перемешивают при комнатной температуре в течение 16 часов.
Растворитель выпаривают, а остаток извлекают 20 мл этилацетата, промывают водой, затем высушивают на MgSO4, а растворители выпаривают. Остаток очищают хроматографией на силикагеле (20 г). Элюант: толуол/AcOEt 60/40 (об. /об). Концентрирование фракций чистого продукта дает остаток, который перекристаллизовывают в смеси изопропанол/изопропиловый эфир. Получают 0,18 г ожидаемого карбамида. Т.пл. = 172oC.
Действуя по описанному выше примеру 172, получают карбамиды, описанные ниже в табл. XVI.
Пример 181. N-[1-(2,4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил] циклогексил-карбоксамид.
(I): g4 = Cl; w2, w4 = Cl; R4 = H;

A) N-(третбутоксикарбонил)-[1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H- пиразол-3-ил] амин.
К раствору 2,05 мл азида дифенилфосфорила в 40 мл безводного третбутанола добавляют 3,25 г 1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-3-пиразолкарбоновой кислоты, полученной по примеру 1C, затем 1,32 мл триэтиламина и реакционную смесь нагревают до температуры флегмы в азоте в течение 12 часов. После охлаждения, реакционную смесь обрабатывают насыщенным раствором NaHCO3 и экстрагируют этилацетатом. После промывания водой, затем насыщенным раствором NaCl, высушивания на MgSO4 и выпаривания растворителей, неочищенный продукт очищается хроматографией на силикагеле 70-230 меш. Элюант: CH3OH/CH2Cl 1/99 (об/об). Получают 1,09 г желаемого продукта.
B) Хлоргидрат 1-(2,4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3- ил-аммония.
1,09 г полученного выше продукта растворяют в 20 мл насыщенного раствора HCl в EtOH, разведенного до 50%, реакционную смесь нагревают до температуры флегмы в течение 2 часов. Растворитель затем выпаривают, а остаток истирают в этилацетате во флегме, затем фильтруют и высушивают в вакууме. Получают 0,55 г хлоргидрата.
C) Соединение 181.
Раствор 0,11 мл хлорангидрида циклогексанкарбоновой кислоты в 2 мл CH2Cl2 добавляют покапельно в раствор 0,20 г полученного выше хлоргидрата и 0,19 мл триэтиламина в 5 мл CH2Cl2. После 24 часов перемешивания при комнатной температуре смесь промывают последовательно 5%-ным раствором HCl, водой, 5%-ным раствором Na2CO3, затем насыщенным раствором NaCl, высушивают на MgSO4, затем растворители выпаривают. Неочищенный продукт кристаллизуется в iPr2O. Получают 0,12 г желаемого амида. Т.пл = 213oC.
Пример 182. N-метил-N-[1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил] адамантил-1-карбоксамид.
(I): w2 = w4 = Cl; g4 = Cl; R4 = H;

A) N-[1-(2,4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил]формамид.
В смесь 4 мл муравьиной кислоты и 0,5 мл уксусного ангидрида, охлажденную в ледяной ванне, добавляют небольшими порциями 0,50 г 1-(2,4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-иламина, полученного в предыдущем примере. После 30 мин перемешивания растворители выпариваются в вакууме, а осадок извлекают изопропиловым эфиром. Полученное белое твердое вещество фильтруют, промывают в изопропиловом эфире и высушивают в вакууме. Получают 0,49 г ожидаемого формамида. Т.пл. = 181oC.
B) N-метил-[1-(2,4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил]амин.
Раствор 1,15 г полученного в предыдущем примере формамида в 10 мл безводного ТГФ добавляют покапельно при комнатной температуре в суспензию 0,24 г L: AlH4 в 40 мл безводного ТГФ. Смесь затем нагревают во флегме в течение 20 минут, охлаждают до 0oC и гидролизуют 0,24 мл воды, затем 0,24 мл 15% NaOH, затем 0,72 мл воды. После 20 минут перемешивания при комнатной температуре фильтруют, промывают в ТГФ, а фильтрат выпаривают досуха. Остаток извлекают изопропиловым эфиром, фильтруют и высушивают в вакууме. Получают 1,02 г ожидаемого амида. Т.пл.=157oC.
C) Соединение 182.
Действуя, как в примере 181C, путем реакции полученного выше амина с хлоридом адамантан-1-карбоновой кислоты получают ожидаемый амид, который очищается хроматографией на силиконовой колонке. Элюант: AcOEt/толуол 7:93. Т.пл. = 65oC.
Аналогично примеру 182 получают соединения, указанные в табл. XVII.
Пример 187. N-метил-[1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил] -N'-(4-хлорфенил) мочевина.
(I): w2 = w4 = Cl; g4 = Cl; R4 = H;

К суспензии 0,40 г 1-(2,4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-иламина, полученного нейтрализацией хлоргидрата, полученного в примере 181B, в 15 мл толуола добавляют 225 мг 4-хлорфенилизоцианата и нагревают смесь до 40oC в течение 1 часа, затем проводят реакцию при комнатной температуре в течение 16 часов. Полученный осадок фильтруют, промывают толуолом и высушивают в вакууме. Получают 0,46 г ожижаемого карбамида. Т.пл. = 215oC.
Пример 188. N-[1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил]-N'- (4-адамантил) мочевина.
(I): w2 = w4 = Cl; g4 = Cl; R4 = H

A) К раствору 10,0 г хлорангидрида кислоты, полученного по примеру 1D, в 320 мл ацетона, охлажденному до 0oC, добавляют раствор 2,54 г азида натрия в 10 мл воды. После 1 часа перемешивания при 0oC, полученный осадок фильтруют и промывают ацетоном, затем высушивают в вакууме. Получают 9,86 г ожидаемого ацилазида.
B) N-[1-(2,4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил] - N'-(1-адамантил) мочевина.
Раствор 1,00 г ацилазида, полученного выше, в 5 мл толуола нагревают до температуры флегмы в течение 30 минут. После установления температуры добавляют к полученному таким образом раствору изоцианата 0,39 г 1-адамантанамида и перемешивают смесь в течение 1 ч 30 мин. Полученный осадок фильтруют, промывают толуолом, затем изопропиловым эфиром, затем очищают тритированием в смеси ацетон/метанол. После высушивания в вакууме, получают 0,48 г ожидаемого карбамида. Т.пл. = 244oC.
Аналогично получают соединения, указанные в табл. XVIII.
Пример 191. 1-Циклогексилметил-[1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол -3-ил]-кетон.
(I): w2 = w4 = Cl; g4 = Cl; R4 = H;

2,5 мл раствора 0,625 M MnLi2Cl2 в ТГФ (Tetrahedron 1989, 45, 4163) охлаждают до 0oC и добавляют к нему покапельно 3,12 мл раствора 0,50 М бромида метилциклогексилмагния в ТГФ, затем реакционная смесь перемешивается при 0oC в течение 2 часов. Реакционная смесь охлаждается до -10oC, а раствор 0,50 г хлорангидрида кислоты, полученного по примеру 1D, в 8 мл ТГФ добавляют покапельно. Смесь перемешивают при комнатной температуре в течение 5 часов, затем гидролизуют насыщенным раствором NH4Cl, экстрагируют эфиром, промывают водой, затем насыщенным раствором NaCl. После высушивания на MgSO4 и выпаривания растворителей, сырой продукт очищают хроматографией на силикагеле 230-400 меш. Элюант: AcOEt/гексан 5/95 (об./об.). Таким образом, получают 0,09 г ожидаемого кетона. Т.пл. = 118oC.
Пример 192. 1-[1-(2, 4-дихлорфенил)-5-(4-хлорфенил)-1H-пиразол-3-ил] -2-(метил-4-фенил)-1-этанон.
(I): w2 = w4 = Cl; g4 = Cl; R4
= H;

A) 1-(2,4-дихлорфенил)-3-циано-5-(4-хлорфенил)-пиразол.
Раствор 0,70 г 1-(2.4-дихлорфенил)-5-(4-хлорфенил)-1H- пиразол-3-карбоксамида, полученного по примеру 172A, и 0,74 мл хлорида мезила в 6 мл пиридина нагревают до 50oC в течение 8 часов. Растворитель выпаривают в вакууме, а остаток растворяют в 20 мл CH2Cl2. Промывают последовательно 5%-ным раствором HCl, затем водой, затем насыщенным раствором NaCl, высушивают на MgSO4, затем выпаривают растворитель. Остаток кристаллизуют в изопропиловом эфире. Получают 0,66 г ожидаемого нитрила. Т.пл.= 123oC.
C) Соединение 192.
6, 3 мл раствора 1,0 M хлорида метил-4-бензилмагния в этиловом эфире добавляют покапельно в раствор 0,73 г полученного выше нитрила в 20 мл этилированного эфира. После 2 часов реакции при комнатной температуре смесь гидролизуют в 50 мл 5%-ной соляной кислоты, а полученная в результате биофазная смесь перемешивается в течение 30 минут. Образовавшийся розовый осадок фильтруется, промывается водой и этилированным эфиром, затем растворяется в 100 мл CH2Cl2 и перемешивается 30 минут в присутствии приблизительно 10 г влажной двуокиси кремния. Двуокись кремния затем фильтруется, фильтрат выпаривается, а остаток кристаллизуется в смеси CH2Cl2/iPr2O. Получают 0,37 г ожидаемого кетона. Т.пл. = 175oC.
Аналогично описанному выше получают соединения, указанные в табл. XIX.
Реферат
Изобретение относится к производным пиразола общей формулы I, где g2, g3 и g6 водород; g4 - атом хлора или брома, С1-С3-алкил, трифторметил, или фенил; g5 - водород или атом хлора; w2, w3, w5 и w6 - водород или атом хлора; w4 - водород, атом хлора, С1-С3-алкил, С1-С3-алкокси или нитро; Х - прямая связь или группа -(CH2)nN(R3)-, где R3 - водород или С1-С3-алкил; n равно 0 или 1; R4 - водород или С1-С3-алкил и, когда Х означает прямую связь, R представляет собой группу -NR1R2, где R1 - водород, С1-С6-алкил или циклогексил, а R2 - С1-С6-алкил, неароматический карбоциклический радикал С3-С15, возможно замещенный гидроксильной группой, одним или несколькими С1-С5-алкилами, С1-С5 алкоксигруппой или галогеном; группу амино С1-С4-алкил, в которой амино возможно двузамещен С1-С3-алкилом, циклогексил С1 -С3-алкил; фенил, незамещенный или замещенный галогеном, или С1-С5-алкилом; фенил С1-С3 -алкил, дифенил С1-С3-алкил, насыщенный гетероциклический радикал, выбранный из пирролидинила, пиперидила, гексагидроазепина, морфолинила, хинуклидинила и оксабициклогептинила, незамещенного или замещенного С1-С3-алкилом или бензилом; 1-адамантанилметил; С1-С3-алкил, замещенный ароматическим гетероциклом, выбранным из пирролила, пиридила или индолила, незамещенного или замещенного С1-С5-алкилом, или R1 и R2 образуют с атомом азота, с которым они связаны, пирролидинил, пиперидил или морфолинил; или группу R5, представляющую фенил С1-С3-алкил, незамещенный или замещенный С1-С5-алкилом; циклогексил С1-С3 -алкил, или 2-норборнилметил; когда Х представляет собой группу -(CH2)n N(R3)-, то R представляет группу R2а, которая представляет собой неароматический карбоциклический радикал С3-С15; фенил, замещенный галогеном; фенил С1-С3-алкил, возможно замещенный галогеном; индолил, возможно замещенный С1 -С5 алкоксигруппой; антраценил, или группу NHR2b, в которой R2b - циклогексил, адамантил, фенил, незамещенный или замещенный одним или двумя атомами галогена, С1-С5-алкилом или С1-С5 алкоксигруппой или их кислотно-аддитивным солям. Соединения формулы I получают взаимодействием соответствующих производных 3-пиразолкарбоновой кислоты с амином формулы HNR1R2, где R1 и R2 определены выше, либо с первичным амином R3NH2, где R3 имеет значения, определенные выше, полученный промежуточный амид восстанавливают для получения соответствующего промежуточного аминометильного производного, которое подвергают взаимодействию с хлорангидридом кислоты R2аCOCl, где R2а определен выше, либо с изоцианом R2bN=C=O, где R2b определен выше. Целевые соединения также получают взаимодействием соответствующих производных 3-пиразолкарбоновой кислоты с производным дифенилфосфорилазида с получением промежуточного амина, который при необходимости отрабатывают алкилирующим агентом для получения вторичного амина и полученные промежуточные амины подвергают взаимодействию с хлорангидридом кислот R2аCOCl или с изоцианатом R2bN=C=O, где R2а и R2b определены выше. Целевые кетоновые производные пиразола получают взаимодействием производных 3-пиразолкарбоновой кислоты с соединением формулы R5MnX1, где R5 определен выше, Х1- галоген. Соединения формулы I проявляют сродство к рецепторам каннабиноидов. Предложена также фармацевтическая композиция, обладающая вышеуказанной активностью, содержащая в качестве активного начала 0,5 - 1000 мг соединения формулы I на единицу дозировки. Изобретение также относится и к промежуточным производным 3-пиразолкарбоновой кислоты формулы II, где R' - водород или С1-С5-алкил. 7 с. и 12 з.п. ф-лы, 20 табл.


Формула

в которой g2, g3 и g6 - водород;
g4 - атом хлора или брома, (C1 - C3)алкил, трифторметил или фенил;
g5 - водород или атом хлора;
w2, w3, w5 и w6 - водород или атом хлора;
w4 - водород, атом хлора, (C1 - C3)алкил, (C1 - C3)алкокси или нитро;
X означает прямую связь или группу -(CH2)n - N(R3)-, в которой R3 - водород или (C1 - C3)алкил, а n равно 0 или единице;
R4 - водород иди (C1 - C3)алкил,
и когда X означает прямую связь, R представляет собой группу -NR1R2, в которой R1 - водород, (C1 - C3 )алкил или циклогексил, а R2 - (C1 - C6)алкил, неароматический карбоциклический радикал C3 - C15, возможно замещенный гидроксильный группой, одним или несколькими (C1 - C5)алкилами, (C1 - C5)алкоксигруппой или галогеном; группу амино (C1 - C4)алкил, в которой амино возможно двузамещен (C1 - C3)алкилом; циклогексил (C1 - C3)алкил; фенил, незамещенный или замещенный галогеном или (C1 - C5)алкилом; фенил (C1 - C3)алкил; дифенил (C1 - C3)алкил; насыщенный гетероциклический радикал, выбранный из пирролидинила, пиперидила, гексагидроазепинила, морфолинила, хинуклидинила и оксабициклогептанила, незамещенного или замещенного (C1 - C3)алкилом или бензилом; 1-адамантилметил; (C1 - C3)алкил, замещенный ароматическим гетероциклом, выбранным из пирролила, пиридила или индолила, незамещенного или замещенного (C1 - C5)алкилом; или R1 и R2 образуют с атомом азота, с которым они связаны, пирролидинил, пиперидил или морфолинил; или группу R5, представляющую собой фенил (C1 - C3)алкил, незамещенный или замещенный (C1 - C5)алкилом, циклогексил (C1 - C3)алкил или 2-норборнилметил; когда X представляет собой группу -(CH2)nN(R3)-, то R представляет собой группу R2a, которая представляет собой неароматический карбоциклический радикал C3 - C15; фенил, замещенный галогеном; фенил (C1 - C3)алкил, возможно замещенный галогеном; индолил, возможно замещенный (C1 - C5) алкоксигруппой; антраценил; или группу -NHR2b, в которой R2b - циклогексил; адамантил; фенил, незамещенный или замещенный одним или двумя атомами галогена, (C1 - C5) алкилом или (C1 - C5) алкоксигруппой; или их кислотно-аддитивные соли.

в которой w2, w3, w4, w5, w6, g2, g3, g4, g5, g6 и R4 имеют значения, определенные в п.1;
R1 - водород или (C1 - C6)алкил, а R2 - карбоциклический неароматический радикал C3 - C15 или насыщенный гетероциклический радикал, выбранный из 1-пирролидинила, 1-пиперидинила, 1-гексагидроазепинила и 4-морфолинила;
или их кислотно-аддитивные соли.

в которой R4, X и R имеют значения, определенные в п.1,
или их кислотно-аддитивные соли.

в которой X и R имеют значения, определенные в п.1; W4 - метильная или метоксильная группа, или их кислотно-аддитивные соли.

в которой X и R имеют значения, определенные в п.1; g4 - атом брома или метильная или трифторметильная группа, или их кислотно-аддитивные соли.

в которой g2 - g6, w2 - w6, R1, R2 и R4 имеют значения, определенные в п. 1, или их кислотно-аддитивных солей, отличающийся тем, что производное 3-пиразолкарбоновой кислоты общей формулы (IIa)

в которой g2 - g6, w2 - w6 и R4 имеют значения, определенные в п.1, или его сложный эфир, или ангидрид или хлорангидрид, подвергают взаимодействию с амином формулы HNR1R2, в которой R1 и R2 имеют значения, определенные в п.1 формулы, с последующим выделением целевого соединения в виде свободного основания или в виде кислотно-аддитивной соли.

в которой g2 - g6, w2 - w6, R3, R4 и R имеют значения, определенные в п. 1, или их кислотно-аддитивных солей, отличающийся тем, что производное 3-пиразолкарбоновой кислоты общей формулы (IIa)

в которой g2 - g6, w2 - w6 и R4 имеют значения, определенные в п.1, или его сложный эфир, ангидрид или хлорангидрид, подвергают взаимодействию с первичным амином R3NH2, где R3 имеет значения, определенные в п.1, полученный при этом промежуточный амид формулы (V)

в которой g2 - g6, w2 - w6, R3 и R4 имеют значения, определенные в п.1, подвергают восстановлению гидридом металла с получением промежуточного амина формулы (VI)

в которой g2 - g6, w2 - w6, R3 и R4 имеют значения, определенные в п.1, который подвергают взаимодействию с хлорангидридом кислоты формулы R2aCOCl, в которой R2a имеет значения, определенные в п.1, или с изоцианатом формулы R2bN= C= O, в которой R2b имеет значения, определенные в п.1, с последующим выделением полученных целевых амидов формулы (Ib), в которой R - определенная выше группа R2a, или полученных целевых карбамидов формулы (Ib), в которой R - группа NHR2b, определенная в п.1, в свободном виде или в виде кислотно-аддитивных солей.

в которой g2 - g6, w2 - w6 и R4 имеют значения, определенные в п.1, R3 - водород или (C1 - C3)алкил и R - группа R2a или группа -NHR2b, определенные в п. 1, или их кислотно-аддитивных солей, отличающийся тем, что производное 3-пиразолкарбоновой кислоты формулы (IIa)

в которой g2 - g6, w2 - w6 и R4 имеют значения, определенные в п.1, или его сложный эфир, ангидрид или хлорангидрид обрабатывают производным дифенилфосфорилазида в основной среде с последующей обработкой кислотой в спиртовой среде с получением промежуточного амина формулы (VII)

в которой g2 - g6, w2 - w6 и R4 имеют определенные выше значения,
с последующим при необходимости взаимодействием амина формулы (VII) с алкилирующим агентом с получением вторичного амина формулы (VIIb):

в которой g2 - g6, w2 - w6 и R4 имеют значения, определенные выше, и R'3 - (C1 - C2) алкил,
и полученный амин формулы (VII) или формулы (VIIb) подвергают взаимодействию с хлорангидридом кислоты формулы R2aCOCl, в которой R2a имеет указанные выше значения, или с изоцианатом формулы R2bN=C=O с последующим выделением целевого амида формулы (Id), или целевого карбамида формулы (Ie):


в которой g2 - g6, w2 - w6, R2a, R2, R3 и R4 имеют значения, определенные в п.1,
в свободном виде или в виде кислотно-аддитивной соли.

в которой g2 - g6, w2 - w6, R4 и R5 имеют значения, определенные в п.1,
или их кислотно-аддитивных солей, отличающийся тем, что производное 3-пиразолкарбоновой кислоты формулы (IIa)

в которой g2 - g6, w2 - w6 и R4 имеют значения, определенные в п.1,
или его сложный эфир, или ангидрид, или хлорангидрид обрабатывают органическим соединением двухвалентного марганца формулы R5МnX1, в которой R5 имеет указанные выше значения, а X1 - галоген, с последующим выделением целевого соединения в свободном виде или в виде кислотно-аддитивной соли.

в которой R4 имеет значения, определенные в п.1, а R1 - водород или (C1 - C5) алкил.
Комментарии