Производное п-фенилендиамина как регулятор калиевых каналов, способ его получения и медицинского применения - RU2762562C1
Код документа: RU2762562C1
Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области биомедицины. Более конкретно, настоящее изобретение относится к производным n-диаминобензола, способу их получения и применения в медицине. Такие соединения регулируют калиевые ионные каналы и полезны при лечении и профилактике заболеваний и состояний, на которые влияет активность калиевых каналов.
УРОВЕНЬ ТЕХНИКИ
KCNQ, переименованные позже в каналы Kv 7, являются членами потенциал-зависимого семейства неинактивирующих калиевых каналов. Известно, что семейство KCNQ включает 5 генов. Они известны как KCNQ 1-5 в соответствии с порядком их обнаружения, все они кодируют субъединицы калиевых каналов. Простетические группы KCNQ (KCNQ 1-5), кодируемые генами, регулируют экспрессию, биофизические и фармакологические свойства каналов KCNQ. Мутации генов KCNQ 1-4 могут снижать ток ионов калия. Каналы KCNQ не только участвуют в регулировании многих важных физиологических функций организма, но также играют важную роль в возникновении определенных заболеваний. Среди них мутации 4 генов связаны с различными генетическими заболеваниями. KCNQ 1 экспрессируется в сердце и внутреннем ухе, мутации его гена вызывают синдром L-QT и врожденную глухоту (синдром Джервелла - Ланге-Нильсена). Кроме того, с этим геном может быть связан диабет. Четыре из пяти членов семейства KCNQ (KCNQ 2-5) экспрессируются в нервной системе. Среди них KCNQ 2 и KCNQ 3 широко экспрессируются в неокортексе и гипоталамусе. Их генетические мутации могут вызывать семейные доброкачественные неонатальные судороги (BFNC). Мутации гена KCNQ 2 связаны с чрезмерной возбудимостью периферических нервов. Гетеромультимерный ионный канал, состоящий из KCNQ 2 и KCNQ 3, является молекулярной основой калиевого тока М-типа в нервной системе. Кроме того, калиевый ток М-типа тесно связан с поддержанием устойчивости мембранного потенциала и возбудимости клетки. KCNQ 5 широко экспрессируется в центральной и периферической нервной системе, а также участвует в формировании каналов М-типа. Исследования показывают, что KCNQ 4 ограничен волосковыми клетками внутреннего уха и слуховыми нервами, а генетические мутации в нем могут вызывать неврологическую глухоту. KCNQ являются не только важными определяющими факторами возбудимости миокарда и мембран нервных клеток, но также широко экспрессируются в других гладких мышцах. KCNQ 4 и KCNQ 5 экспрессируются во всем желудочно-кишечном тракте и являются основным регулятором активности гладкой мускулатуры пищеварительного тракта. KCNQ также избирательно распределяются по мышечным клеткам артериовенозных сосудов (Chin J Nerv Merit Dis, 2011, 37, 124-126).
Ретигабин - препарат для лечения эпилепсии. Он был одобрен для продажи в Великобритании, Германии и Дании. Исследования подтвердили, что влияние ретигабина связано с потенциал-управляемыми калиевыми каналами (KCNQ), при этом его основной механизм действует на канал KCNQ2/3 и модулирует калиевый ток М-типа.
Сообщалось, что KCNQ2 и KCNQ3 усиливаются в моделях невропатической боли (Wickenden et al., Sccoety for Neuroscience Abstracts, 2002, 454, 7). Высказана гипотеза, что модуляторы калиевых каналов являются эффективными и в случае невропатической боли, и в случае эпилепсии (Schroder et al., Neuropharmacology, 2001, 40, 888-898).
Исследования показали, что ретигабин эффективен в животных моделях невропатической боли (Blackburn-Munro, European Journal of Pharmacology, 2003, 460, 109 116). Это показывает, что вещества, открывающие калиевые каналы, могут быть использованы для лечения болезненных состояний, включая невропатическую боль.
Поэтому очень важно разрабатывать новые и эффективные вещества, открывающие калиевые каналы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Цель настоящего изобретения - создать новый тип производных п-диаминобензола, которые можно использовать как вещества, открывающие калиевые ионные каналы.
Цель настоящего изобретения - обеспечить способ получения указанного выше соединения.
Целью настоящего изобретения также является обеспечение применения вышеназванных соединений в качестве веществ, открывающих калиевые ионные каналы, для лечения боли, нарушений мозгового кровообращения, эпилепсии и других заболеваний.
Настоящее изобретение также обеспечивает способ предотвращения или лечения заболеваний, связанных с калиевыми каналами, который включает прием соединения или фармацевтической композиции по настоящему изобретению нуждающимся в этом лицом.
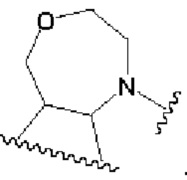
В первом аспекте изобретения предлагается соединение, представленное формулой А, или его фармацевтически приемлемая соль:

в которой:
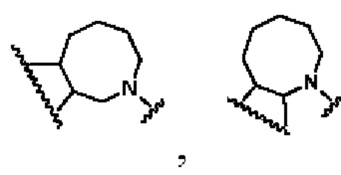
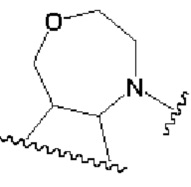
кольцо В представляет собой


кольцо В представляет собой

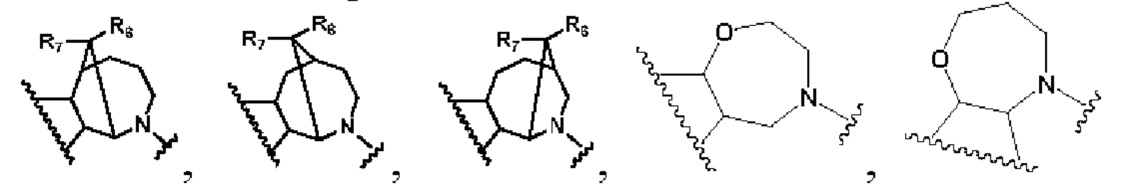
R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1, R2, R3 и R4 представляют собой независимо атом водорода, галогена, нитро-, циано-, алкильную C1-6, алкоксильную C1-6, галогенированную алкильную C1-6, галогенированную алкоксильную C1-6, циклоалкильную С3-6, алкиламинную C1-6 группы;
Y представляет собой N или СН-группу;
Z представляет собой О или (СН2)n, n - это целое число от 1 до 6;
R5 представляет собой алкильную C1-6, циклоалкильную С3-6, циклоалкенильную С3-6, алкенильную С2-6 или алкинильную С2-6 группу, в которой алкильная C1-6, циклоалкильная С3-6, циклоалкенильная С3-6, алкенильная С2-6 или алкинильная С2-6 группа дополнительно замещена одним или несколькими заместителями, выбранными из галогенной, нитро-, циано-, аминной или гидроксильной групп.
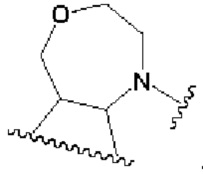
В другом предпочтительном воплощении изобретения кольцо В представляет собой
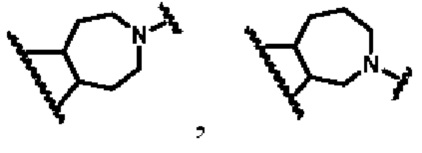


R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1, R2, R3 и R4 каждый представляют собой независимо атом водорода, галогена, нитро-, циано-, алкильную C1-6, алкоксильную С1-6, галогенированную алкильную С1-6, галогенированную алкоксильную C1-6, циклоалкильную С3-6, алканаминную С1-6 группы;
Y представляет собой N или СН-группу;
Z представляет собой О или (СН2)n, n - это целое число от 1 до 6;
R5 представляет собой алкильную С1-6, циклоалкильную С3-6, циклоалкенильную С3-6, алкенильную С2-6 или алкинильную С2-6 группу, в которой алкильная С1-6, циклоалкильная С3-6, циклоалкенильная С3-6, алкенильная С2-6 или алкинильная С2-6 группа дополнительно замещена одним или несколькими заместителями, выбранными из галогенной, нитро-, циано-, аминной или гидроксильной групп.
В другом предпочтительном воплощении изобретения кольцо В представляет собой


кольцо В представляет собой
R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1, R2, R3 и R4 каждый представляют собой независимо атом водорода, галогена, нитро-, циано-, алкильную C1-6, алкоксильную С1-6, галогенированную алкильную С1-6, галогенированную алкоксильную С1-6, циклоалкильную С3-6, алканаминную С1-6 группы;
Y представляет собой N или СН-группу;
Z представляет собой О или (СН2)n, n - это целое число от 1 до 6;
R5 представляет собой алкильную С1-6, циклоалкильную С3-6, циклоалкенильную С3-6, алкенильную С2-6 или алкинильную С2-6 группу, в которой алкильная С1-6, циклоалкильная С3-6, циклоалкенильная С3-6, алкенильная С2-6 или алкинильная С2-6 группа дополнительно замещена одним или несколькими заместителями, выбранными из галогенной, нитро-, циано-, аминной или гидроксильной групп.
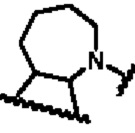
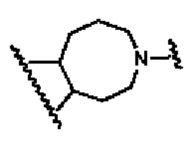
В другом предпочтительном воплощении изобретения В представляет собой
R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1, R2, R3 и R4 представляют собой независимо атом водорода, галогена, нитро-, циано-, алкильную C1-6, алкоксильную С1-6, галогенированную алкильную С1-6, галогенированную алкоксильную С1-6, циклоалкильную С3-6, алкиламинную C1-6 группы;
Y представляет собой N или СН-группу;
Z представляет собой О или (СН2)n, n - это целое число от 1 до 6;
R5 представляет собой алкильную C1-6, циклоалкильную С3-6, циклоалкенильную С3-6, алкенильную С2-6 или алкинильную С2-6 группу, в которой алкильная C1-6, циклоалкильная С3-6, циклоалкенильная С3-6, алкенильная С2-6 или алкинильная С2-6 группа дополнительно замещена одним или несколькими заместителями, выбранными из галогенной, нитро-, циано-, аминной или гидроксильной групп.
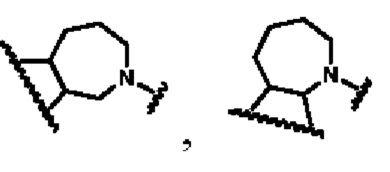
В другом предпочтительном воплощении изобретения кольцо В представляет собой
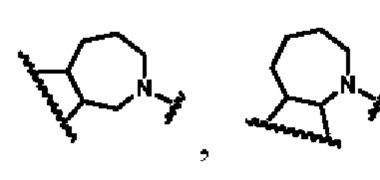
R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1, R2, R3 и R4 каждый представляют собой независимо атом водорода, галогена, нитро-, циано-, алкильную C1-6, алкоксильную C1-6, галогенированную алкильную C1-6, галогенированную алкоксильную C1-6, циклоалкильную С3-6, алканаминную C1-6 группы;
Y представляет собой СН-группу;
Z представляет собой СН2-группу;
R5 представляет собой алкильную C1-6 группу.
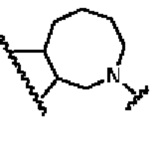
В другом предпочтительном воплощении изобретения кольцо В представляет собой
R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1, R2, R3 и R4 каждый представляют собой независимо атом водорода, галогена, нитро-, циано-, алкильную C1-6, алкоксильную C1-6, галогенированную алкильную C1-6, галогенированную алкоксильную C1-6, циклоалкильную С3-6, алканаминную C1-6 группы;
Y представляет собой СН-группу;
Z представляет собой СН2-группу;
R5 представляет собой алкильную C1-6 группу.
В другом предпочтительном воплощении изобретения кольцо В представляет собой
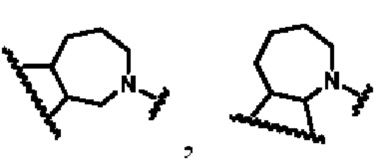
R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1 представляет собой атом водорода, галогена, алкильную С1-6, алкоксильную С1-6, галогенированную алкильную C1-6, галогенированную алкоксильную C1-6, циклоалкильную С3-6 или алкиламинную С1-6 группу;
R2представляет собой атом водорода;
R3 и R4 каждый независимо представляет собой алкильную С1-6 группу;
Y представляет собой СН-группу;
Z представляет собой СН2-группу;
R5 представляет собой алкильную C1-6 группу.
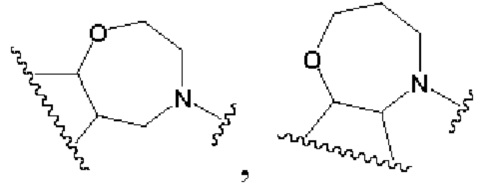
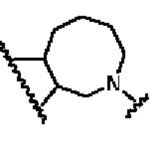
В другом предпочтительном воплощении изобретения кольцо В представляет собой

R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1 представляет собой атом водорода, галогена, алкильную C1-6, алкоксильную С1-6, галогенированную алкильную С1-6, галогенированную алкоксильную С1-6, циклоалкильную С3-6 или алкиламинную С1-6 группу;
R2представляет собой атом водорода;
R3 и R4 каждый независимо представляет собой алкильную C1-6 группу;
Y представляет собой СН-группу;
Z представляет собой СН2-группу;
R5 представляет собой алкильную C1-6 группу.
В другом предпочтительном воплощении изобретения кольцо В представляет собой
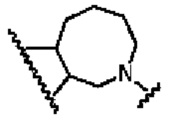
R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1 представляет собой атом водорода, галогена или алкильную C1-6 группу;
R2представляет собой атом водорода;
R3 и R4 каждый независимо представляет собой алкильную C1-6 группу;
Y представляет собой СН-группу;
Z представляет собой СН2-группу;
R5 представляет собой алкильную C1-6 группу.
В другом предпочтительном воплощении изобретения кольцо В представляет собой

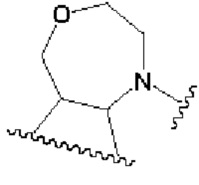
R1 представляет собой заместитель в кольце А;
R2представляет собой заместитель в кольце В;
R3 и R4 представляют собой заместители в шестичленном кольце;
R1 представляет собой атом водорода, галогена или алкильную С1-6 группу;
R2 представляет собой атом водорода;
R3 и R4 каждый независимо представляет собой алкильную С1-6 группу;
Y представляет собой СН-группу;
Z представляет собой СН2-группу;
R5 представляет собой алкильную С1-6 группу.
В другом предпочтительном воплощении изобретения R1 представляет собой атом водорода, галогена или алкильную С1-6 группу.
В другом предпочтительном воплощении изобретения R2представляет собой атом водорода.
В другом предпочтительном воплощении изобретения R3 представляет собой алкильную C1-6 группу, предпочтительно метильную.
В другом предпочтительном воплощении изобретения R4 представляет собой алкильную C1-6 группу, предпочтительно метильную.
В другом предпочтительном воплощении изобретения R3 и R4 являются заместителями в положениях, отличных от положения Y в шестичленном кольце.
В другом предпочтительном воплощении изобретения Y представляет собой СН-группу.
В другом предпочтительном воплощении изобретения Z представляет собой СН2-группу.
В другом предпочтительном воплощении изобретения R5 представляет собой алкильную C1-6 группу, предпочтительно изобутильную.
В другом предпочтительном воплощении изобретения насыщенное или ненасыщенное гетероциклическое соединение, содержащее от 1 до 2 гетероатомов, выбираемых из N, S или О, выбирают из группы, состоящей из пиридина, пиразина, пиридазина, пиримидина, тиофена, фурана, пиррола, тиазола и оксазола.
В другом предпочтительном воплощении изобретения насыщенное или ненасыщенное гетероциклическое соединение, содержащее от 1 до 2 гетероатомов, выбираемых из N, S или О, представляет собой тиофеновое кольцо.
В другом предпочтительном воплощении изобретения соединение выбирают из группы, состоящей из:

В другом предпочтительном воплощении изобретения соединение выбирают из группы, состоящей из:

Во втором аспекте изобретения предлагается фармацевтическая композиция, содержащая одну или несколько фармацевтически приемлемых носителей или разбавителей, и соединение в соответствии с первым аспектом настоящего изобретения или его фармацевтически приемлемую соль.
В третьем аспекте настоящее изобретение обеспечивает применение соединения согласно первому аспекту настоящего изобретения, его фармацевтически приемлемой соли или фармацевтической композиции согласно второму аспекту настоящего изобретения для приготовления лекарственного средства, регулирующего (например, положительная или отрицательная регуляция) ток ионов в калиевых каналах у млекопитающих.
В третьем аспекте настоящее изобретение обеспечивает применение соединения в соответствии с первым аспектом настоящего изобретения или его фармацевтически приемлемой соли или фармацевтической композиции в соответствии со вторым аспектом настоящего изобретения для приготовления лекарственного средства, предназначенного для предотвращения, лечения или подавления нарушения или состояния, вызванного аномальным (например, повышенным или пониженным) потоком ионов в калиевом канале.
В другом предпочтительном воплощении изобретения заболевание или состояние выбирают из группы, состоящей из заболевания или состояния центральной нервной системы, боли, нарушения мозгового кровообращения, нейродегенеративного состояния и нейрональной гипервозбудимости.
В другом предпочтительном воплощении изобретения нарушение или состояние центральной нервной системы представляет собой симптомы эпилептического припадка; и/или
тип боли, включающий в себя воспалительную боль, невропатическую боль, состояния мигрени, аллодинию, гипералгезическую боль, фантомную боль и боль, связанную с онкологическим заболеванием; и/или
нейродегенеративное нарушение, включающее в себя болезнь Альцгеймера, болезнь Хантингтона, рассеянный склероз, амиотрофический латеральный склероз, СПИД-индуцированную энцефалопатию и болезнь Крейтцфельда-Якоба, болезнь Паркинсона, нейродегенерацию, вызванную травмой, другую энцефалопатию, связанную с инфекцией и вызванную вирусом краснухи, герпеса, боррелией и неизвестным возбудителем; и/или
нейрональную гипервозбудимость - состояние при отмене лекарств или отравлении.
В другом предпочтительном воплощении изобретения симптомы эпилептического припадка включают в себя судороги, эпилепсию или эпилептическое состояние.
В другом предпочтительном воплощении изобретения невропатическая боль представляет собой невропатическую боль, связанную с диабетической невропатией или мигренью.
Настоящее изобретение обеспечивает способ приготовления соединения, представляемого формулой А или его фармацевтически приемлемой солью, не ограничиваемый следующими способами:
Способ 1:
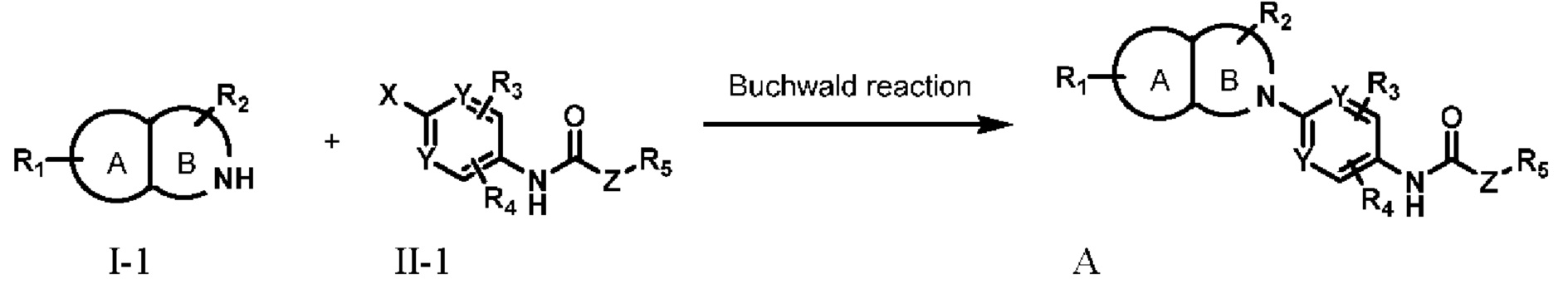
Соединения I-1 и II-1 аминируют по Бухвальду с получением соединения А;
В способе 1 X представляет собой Cl, Br, I, трифлат (OTf) или В(ОН)2; А, В, R1, R2, R3 , R4 , R5, Y, и Z определены в соответствии с формулой А.
Способ 2:
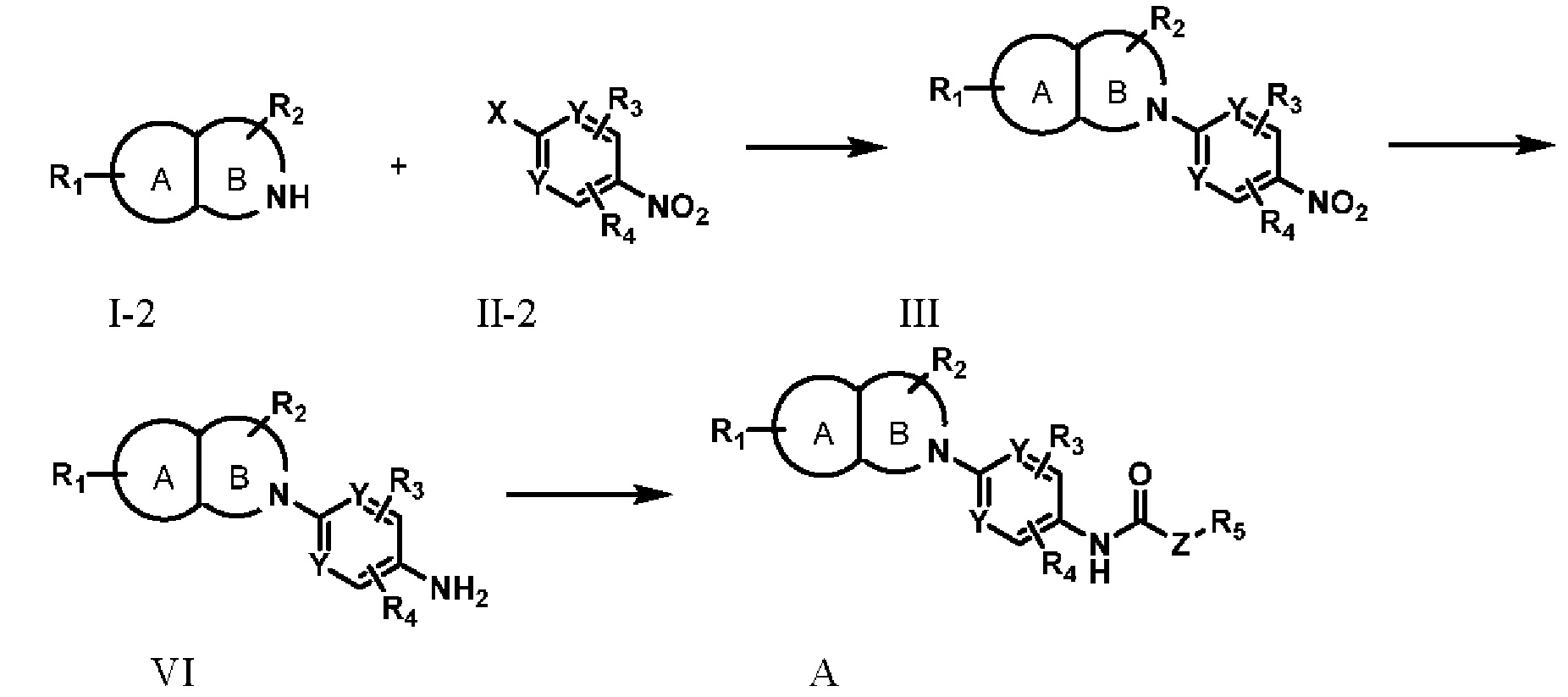
Соединения I-2 и II-2 подвергают реакции нуклеофильного замещения с получением соединения III;
Соединение III подвергают реакции нитровосстановления с получением соединения VI;
Соединение VI в дальнейшем алкилируют с получением соединения А;
В способе 2, X представляет собой F или Cl; А, В, R1, R2, R3, R4, R5, Y и Z соответствуют определениям в формуле А.
Следует понимать, что в пределах объема настоящего изобретения вышеупомянутые его технические признаки и технические признаки, конкретно описанные ниже (например, в примерах), могут сочетаться друг с другом для образования нового или предпочтительного технического решения, которые не будут подробно описаны здесь один за другим.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
За счет обширных и интенсивных исследований авторы изобретения неожиданно впервые открыли класс соединений n-диаминобензола с новой структурой в качестве веществ, открывающих калиевые каналы. Соединения по настоящему изобретению обладают отличной активностью по открыванию KCNQ2/3 и могут использоваться для лечения боли, эпилепсии, инсульта и других заболеваний. На основании вышеизложенного открытия было осуществлено настоящее изобретение.
ТЕРМИНЫ
Если не указано иное, слово «или», упоминаемое в настоящем документе, имеет то же значение, что и «и/или» (относится к «или» и «и»).
Если не указано иное, во всех соединениях по настоящему изобретению каждый хиральный атом углерода (хиральный центр) может необязательно иметь R-конфигурацию, S-конфигурацию или их сочетание.
Используемый здесь термин «алкильный», как в отношении самостоятельного заместителя, так и в составе другого заместителя, относится к линейной (неразветвленной) цепи только из атомов углерода или к разветвленной насыщенной углеводородной группе или к группам, образованным сочетанием линейной цепи и ответвлений. Когда для алкильной группы указано ограниченное количество атомов углерода рядом с ней (например, алкильная группа C1-6), это означает, что алкильная группа содержит от 1 до 6 атомов углерода, включая, например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную или подобные группы.
В контексте настоящего изобретения, будучи отдельным заместителем или компонентом других заместителей, термин «алкоксильная С1-6 группа» относится к С1-6 алкил-О-группе, включая, например, метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси или подобные группы.
В контексте настоящего изобретения, будучи отдельным заместителем или компонентом других заместителей, термин «циклоалкильная С3-6 группа» относится к циклической алкильной группе, содержащей от 3 до 6 атомов углерода, включая, например, циклопропильную, циклобутильную, циклопентильную, циклогексильную или подобные группы.
В контексте настоящего изобретения, будучи отдельным заместителем или компонентом других заместителей, термин «циклоалкенильная С3-6 группа» относится к циклической алкенильной группе, содержащей от 3 до 6 атомов углерода, включая, например, циклобутенильную, циклопентенильную, циклогексенильную или подобные группы.
В контексте настоящего изобретения, будучи отдельным заместителем или компонентом других заместителей, термин «алкенильная С2-6 группа» относится к линейной или разветвленной алкенильной группе, содержащей от 3 до 6 атомов углерода, которая может содержать одну или более алкенильных групп, включая, например, винильную, пропенильную, бутенильную или подобные группы.
В контексте настоящего изобретения, будучи отдельным заместителем или компонентом других заместителей, термин «алкинильная С2-6 группа» относится к разветвленной или линейной алкинильной группе, содержащей от 2 до 6 атомов углерода, которая может содержать одну или более алкинильных групп, включая, например, этинильную, пропинильную, бутинильную или подобные группы.
В контексте настоящего изобретения термин «галоген» относится к фтору, хлору, брому или йоду.
В контексте настоящего изобретения термин «галогенированный» означает фторированный, хлорированный, бромированный или йодированный.
Активный компонент
Соединение по настоящему изобретению относится к соединению, представленному общей формулой А, или его стереоизомеру, или оптическому изомеру, или его фармацевтически приемлемой соли.
«Фармацевтически приемлемая соль» относится к соли, которая образована соединением по настоящему изобретению и фармацевтически приемлемой неорганической и органической кислотой, в котором предпочтительная неорганическая кислота включает (но не ограничивается перечисленным): соляную кислоту, бромистоводородную кислоту, фосфорную кислоту, азотную кислоту, серную кислоту, трифторуксусную кислоту (TFA); предпочтительные органические кислоты включают (но не ограничиваются перечисленным): муравьиную кислоту, уксусную кислоту, пропионовую кислоту, янтарную кислоту, нафталинсульфоновую кислоту (1,5), азиатиновую кислоту, щавелевую кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, валериановую кислоту, диэтилуксусную кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, пимелиновую кислоту, адипиновую кислоту, малеиновую кислоту, яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, β-пиридинкарбоновую кислоту, изоникотиновую кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту и аминокислоты.
Термин «стереоизомер», или «оптический изомер», означает, что хиральный атом углерода, входящий в соединение по настоящему изобретению, может быть в конфигурации R или S или в их сочетании.
Фармацевтическая композиция и способ приема
Поскольку соединение по настоящему изобретению обладает превосходной активностью для открывания KCNQ2/3, соединение по настоящему изобретению и фармацевтическая композиция, содержащая соединение по настоящему изобретению в качестве основного активного ингредиента, могут использоваться для лечения, предотвращения и облегчения заболеваний, связанных с калиевыми каналами. В соответствии с предшествующим уровнем техники, соединения по настоящему изобретению можно использовать для лечения следующих заболеваний (но не ограничиваясь ими): эпилепсии, воспалительной боли, невропатической боли, мигрени, бессонницы, нейродегенеративных заболеваний, тревожных расстройств, инсульта, злоупотребления кокаином, абстинентного синдрома при отказе от никотина, абстинентного синдрома при отказе от алкоголя или шума в ушах и т.д.
Фармацевтическая композиция при реализации настоящего изобретения содержит безопасное и эффективное количество соединения по настоящему изобретению и фармакологически приемлемое вспомогательное вещество или носитель.
«Безопасное и эффективное количество» относится к такому количеству соединения, которого достаточно для существенного улучшения состояния без появления серьезных побочных эффектов. Обычно фармацевтическая композиция содержит от 1 до 2000 мг соединения по настоящему изобретению/компонента, более предпочтительно от 5 до 200 мг соединения по настоящему изобретению/компоненту. Предпочтительно «одна доза» представляет собой капсулу или таблетку.
«Фармацевтически приемлемый носитель» относится к одному или нескольким совместимым твердым или жидким наполнителям или гелеобразным веществам, которые подходят для использования людьми и которые должны иметь достаточную чистоту и достаточно низкую токсичность. «Совместимый» означает, что каждый компонент такой композиции может быть смешан с соединением по настоящему изобретению, а также друг с другом без существенного снижения эффективности этих соединений. Фармацевтически приемлемые носители включают целлюлозу и ее производные (например, карбоксиметилцеллюлозу натрия, этилцеллюлозу натрия, ацетат целлюлозы и т.д.), желатин, тальк, твердые смазочные вещества (например, стеариновую кислоту, стеарат магния), сульфат кальция, растительные масла (например, соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), многоатомные спирты (например, пропиленгликоль, глицерин, маннит, сорбит и т.д.), эмульгаторы (например, Tween®), смачивающие вещества (например, натрий лаурилсульфат), красители, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, апирогенную воду и т.д.
Способ приема соединения или фармацевтической композиции по настоящему изобретению практически не ограничен. Репрезентативные методы приема включают (но не ограничиваются перечисленным) пероральный, ректальный, парентеральный (внутривенный, внутримышечный или подкожный) и местное введение.
Твердые лекарственные формы для перорального приема включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых лекарственных формах активное соединение смешивают как минимум с одним обычным инертным вспомогательным веществом (или носителем), таким как цитрат натрия или дикальцийфосфат, или смешивают со следующими компонентами: (а) наполнители или вещества для улучшения совместимости, такие как крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (б) связующие вещества, такие как гидроксиметилцеллюлоза, альгинат, желатин, поливинил пирролидон, сахароза и аравийская камедь; (в) увлажнители, такие как глицерин; (г) разрыхлители, такие как агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые сложные силикаты и карбонат натрия; (д) агенты, замедляющие растворение, такие как парафин; (е) ускорители абсорбции, такие как четвертичное аммониевое соединение; (ж) смачивающие агенты, такие как цетиловый спирт и глицерилмоностеарат; (з) адсорбент, такой как каолин; и (и) смазывающее вещество, такое как тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия или их смеси. В капсулах, таблетках и пилюлях лекарственные формы могут также содержать буферные агенты.
Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть приготовлены с покрытиями и оболочками, такими как растворимые в кишечнике покрытия и другие материалы, известные в этой области. Они могут содержать опалесцирующие вещества; высвобождение активного соединения или соединения в такой композиции может осуществляться с задержкой в отделе пищеварительного тракта. Примерами герметизирующих компонентов, которые можно использовать, являются полимерные материалы и воск. При необходимости активное соединение также может быть в микрокапсулированной форме с одним или несколькими из вышеупомянутых наполнителей.
Жидкие лекарственные формы для перорального приема включают в себя фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. В дополнение к активному соединению жидкая лекарственная форма может содержать инертные разбавители, обычно используемые в этой области, такие как вода или другие растворители, сжижающие агенты и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид и масла, особенно хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло или смеси этих веществ.
Кроме этих инертных разбавителей, композиции могут содержать адъювантные вещества, такие как смачиватели, эмульгаторы и суспендирующие вещества, подсластители, ароматизаторы и вкусовые добавки.
В дополнение к активному соединению суспензия может содержать суспендирующие вещества, например, этоксилированный изооктадеканол, полиоксиэтиленсорбит и дегидратированный сложный эфир сорбитана, микрокристаллическая целлюлоза, метилат алюминия, агар или их смесь.
Композиции для парентеральной инъекции могут включать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки, которые можно повторно растворять в стерильные растворы или дисперсии для инъекций. Подходящие водные и неводные носители, разбавители, растворители или наполнители включают в себя воду, этанол, многоатомные спирты и любые их подходящие смеси. Лекарственные формы соединений по настоящему изобретению для местного применения включают мази, порошки, пластыри, пропелленты и средства для ингаляции. Активный компонент смешивают в стерильных условиях с физиологически приемлемым носителем и любыми консервантами, буферами или пропеллентами, которые могут потребоваться при необходимости.
Соединения по настоящему изобретению можно принимать самостоятельно или в сочетании с другими фармацевтически приемлемыми соединениями.
При использовании фармацевтической композиции безопасное и эффективное количество соединения по настоящему изобретению применяют для млекопитающего, нуждающегося в лечении (например, для человека). При этом дозировка во время приема представляет собой фармацевтически эффективную дозу, для людей с массой тела 60 кг суточная доза обычно составляет от 1 до 2000 мг, предпочтительно от 5 до 500 мг. Конечно, при выборе конкретных доз следует учитывать и такие факторы, как путь приема, здоровье пациента и т.д., находящиеся в компетенции квалифицированного врача.
Основные преимущества настоящего изобретения включают в себя:
Настоящее изобретение представляет собой разновидность соединения с новой структурой в качестве вещества для открывания калиевых каналов. Соединения по настоящему изобретению обладают превосходной активностью по открыванию калиевых каналов, а также высокой степенью безопасности.
Ожидается, что соединения по настоящему изобретению будут использованы при лечении и профилактике заболеваний и состояний, на которые влияют калиевые каналы.
Соединения по настоящему изобретению обладают более высокой активностью по открыванию калиевых каналов, лучшими фармакокинетическими свойствами, способностью к улучшению мозгового кровотока и большей безопасностью.
Настоящее изобретение будет проиллюстрировано ниже на конкретных примерах. Следует понимать, что эти примеры используются только для иллюстрации настоящего изобретения, а не для ограничения его объема. Методики проведения экспериментов без каких-либо особых условий, описанных в следующих примерах, осуществлялись, как правило, в обычных условиях или в соответствии с рекомендациями производителя. Если не указано иное, процентные доли и части представляют собой массовые процентные доли и части.
Материалы и реагенты, используемые для проведения экспериментов в следующих примерах, могут быть получены из коммерческих источников, если не указано иное.
Пример 1: Получение соединения 03026

Шаг один: 3-(3-((трет-бутоксикарбонил) амино)-1-пропинил)-2-метилтиофенкарбоксилат (соединение 2)
(1,1'-бис(дифенилфосфин)ферроцен)палладия дихлорид (0,42 г, 0,57 ммоль) и йодид меди (0,217 г, 1,14 моль) добавляли в раствор 3-бром-2-тиофенметилформата (2,5 г, 11,3 ммоль), N-трет-бутоксикарбониламинопропина (2,1 г, 13,6 ммоль) и диизопропилэтиламина (3 мл) в ацетонитриле (30 мл). Полученную смесь кипятили с обратным холодильником при перемешивании в течение ночи в защитной атмосфере азота. Реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Фильтрат концентрировали для удаления растворителя, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 2 (2,0 г, выход: 59,5%).
Масс-спектрометрия с электрораспылительной ионизацией MS (ESI): Рассчитано для C14H17NO4S 295; Найдено 318 [М+Н]+.
Шаг два: 3-(3-((трет-бутоксикарбонил)амино)-пропил)-2-метилтиофенкарбоксилат (соединение 3)
10% палладиевый катализатор на углеродной основе (0,2 г) добавляли в раствор 3-(3-((трет-бутоксикарбонил)амино)-1-пропинил)-2-метилтиофенкарбоксилата (2,0 г, 6,8 ммоль) в тетрагидрофуране (30 мл). Реакционную смесь перемешивали в течение ночи в атмосфере водорода 4 атм, фильтровали и концентрировали с получением светло-желтого маслянистого соединения 3 (1,8 г, выход: 90,3%).
MS (ESI): Рассчитано для C14H21NO4S 299; Найдено 322 [M+Na]+.
Шаг три: 3-(3-аминопропил)-2-тиофенметилформат (соединение 4)
Трифторуксусную кислоту (4,6 г, 40 ммоль) медленно добавляли по каплям в раствор 3-(3-((трет-бутоксикарбонил)амино)-пропил)-2-тиофенметилформата (1,8 г, 6,1 ммоль) в дихлорметане (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем непосредственно концентрировали с получением желтого маслянистого сырого соединения 4 (1,2 г, выход: 96,5%).
MS (ESI): Рассчитано для C9H13NO2S 199; Найдено 200 [М+Н]+.
Шаг четыре: 4,5,6,7-тетрагидро-8Н-тиено[2,3-с]азепин-8-он (соединение 5)
При комнатной температуре добавляли метилат натрия (1,1 г) в раствор (3-(3-аминопропил)-2-метилтиофенкарбоксилата (1,2 г, 6,0 ммоль) в метаноле (40 мл). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Реакционный раствор непосредственно концентрировали, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 5 (800 мг, выход: 79,8%).
MS (ESI): Рассчитано для C8H9NOS 167; Найдено 168 [М+Н]+.
Шаг пять: 5,6,7,8-тетрагидро-4Н-тиено[2,3-с]азепин (соединение 6) При комнатной температуре медленно добавляли тетрагидроалюминат лития (410 мг, 10,8 ммоль) в раствор 4,5,6,7-тетрагидро-8Н-тиено[2,3-с]азепин-8-она (600 мг, 3,6 ммоль) в тетрагидрофуране (10 мг). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Реакцию останавливали водой и метанолом, смесь фильтровали и концентрировали, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 6 (450 мг, выход: 81,7%).
MS (ESI): Рассчитано для C8H11NS 153; Найдено 154 [М+Н]+.
Шаг шесть: N-(2,6-диметил-4-(4,5,6,8-тетрагидро-7Н-тиено[2,3-с]азепин-7-ил)фенил)-3,3-диметилбутанамид(соединение 03026)
Смешанный реакционный раствор, содержащий три(дибензилиденацетон)дипалладий (20 мг), трициклогексилфосфин (0,3 мл 10% раствор), 5,6,7,8-тетрагидро-4Н-тиено[2,3-с]азепин (100 мг, 0,65 ммоль), N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (386 мг, 1,3 ммоль), трет-бутоксид калия (218 мг, 1,95 ммоль) и диметилсульфоксид (20 мл), выдерживали в микроволновом реакторе при температуре 150°С в течение 2 часов. Полученную смесь разбавляли водой (25 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом тонкослойной хроматографии с получением соединения 03026 в виде белого твердого вещества (6,35 мг, выход: 2,6%).
Н ЯМР (400 МГц, CD3OD): δ 7,01 (дублет, J=4,4 Гц, 1Н), 6,74 (дублет, J=4,4 Гц, 1Н), 6,63 (синглет, 2Н), 4,65 (синглет, 2Н), 3,94-3,85 (мультиплет, 2Н), 2,94-2,85 (мультиплет, 2Н), 2,27 (синглет, 2Н), 2,14 (синглет, 6Н), 1,87-1,78 (мультиплет, 2Н), 1,13 (синглет, 9Н). MS (ESI): Рассчитано для C22H30N2OS 370; Найдено 371 [М+Н]+. Высокоэффективная жидкостная хроматография (ВЭЖХ): 93,3% (214 нм) / 94,2% (254 нм).
Пример 2: Получение соединения 03027
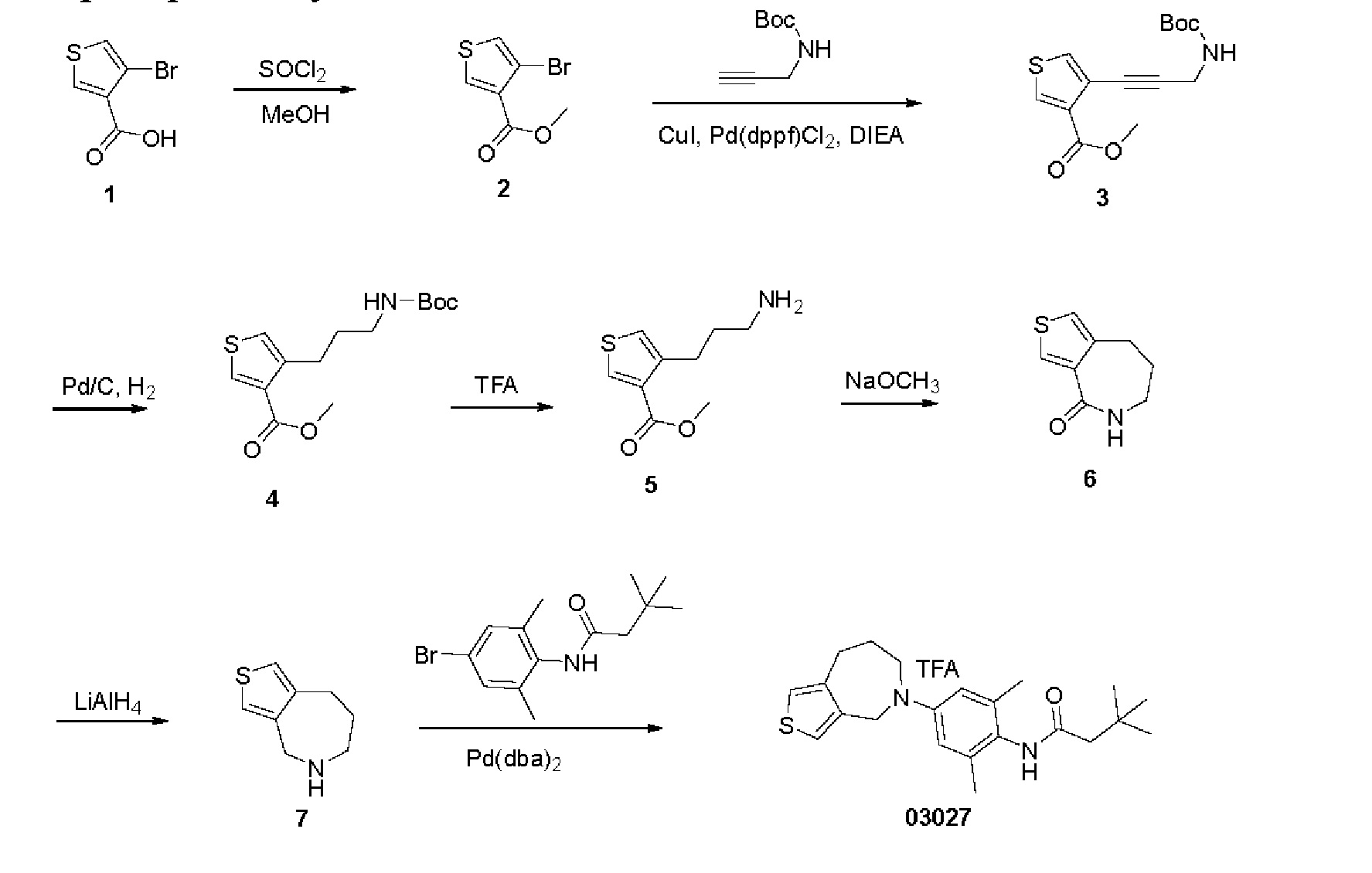
Шаг один: 4-бром-3-тиофенметилформат (соединение 2)
При охлаждении льдом медленно добавляли тионилхлорид (2 мл) в раствор 4-бром-3-тиофенмуравьиной кислоты (2,0 г, 9,7 ммоль) в метаноле (50 мл). Затем реакционную смесь нагревали до 60 градусов и выдерживали в течение 3 часов. Смесь концентрировали под вакуумом с получением желтого маслянистого соединения 2 (2,2 г, выход: 100%).
MS (ESI): Рассчитано для C6H5BrO2S 220; Найдено 221 [М+Н]+.
Шаг два: 4-(3-((трет-бутоксикарбонил)амино)-1-пропинил)-3-тиофенметилформат (соединение 3)
(1,1'-бис(дифенилфосфин)ферроцен) палладия дихлорид (0,36 г, 0,5 ммоль) и йодид меди (0,19 г, 1,0 моль) добавляли в раствор 4-бром-3-тиофенметилформата (2,2 г, 10 ммоль), N-трет-бутоксикарбониламинопропина (1,55 г, 10 ммоль) и диизопропилэтиламина (1,93 г, 15 ммоль) в ацетонитриле (50 мл). Полученную смесь кипятили с обратным холодильником при перемешивании в течение ночи в защитной атмосфере азота. Реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Фильтрат концентрировали для удаления растворителя, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 3 (0,88 г, выход: 29,8%).
MS (ESI): Рассчитано для C14H17NO4S 295; Найдено 318 [М+Н]+.
Шаг три: 4-(3-((трет-бутоксикарбонил)амино)-пропил)-3-тиофенметилформат (соединение 4)
10% палладиевый катализатор на углеродной основе (0,34 г) добавляли в раствор 4-(3-((трет-бутоксикарбонил)амино)-1-пропинил)-3-метилтиофенкарбоксилата (0,88 г, 2,95 ммоль) в метаноле (10 мл). Реакционную смесь перемешивали в течение ночи в атмосфере водорода 4 атм, фильтровали и концентрировали с получением светло-желтого маслянистого соединения 4 (0,76 г, выход: 86,1%).
MS (ESI): Рассчитано для C14H21NO4S 299; Найдено 322 [M+Na]+.
Шаг четыре: 4-(3-аминопропил)-3-тиофенметилформат (соединение 5)
Трифторуксусную кислоту (4,6 г, 40 ммоль) медленно добавляли по каплям в раствор 4-(3-((трет-бутоксикарбонил)амино)-пропил)-3-тиофенметилформата (0,76 г, 2,54 ммоль) в дихлорметане (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем непосредственно концентрировали с получением желтого маслянистого сырого соединения 5 (0,6 г, выход: 100%).
MS (ESI): Рассчитано для C9H13NO2S 199; Найдено 200 [М+Н]+.
Шаг пять: 5,6,7,8-тетрагидро-4Н-тиено[3,4-с]азепин-4-он (соединение 6)
При комнатной температуре добавляли метилат натрия (488 мг) в раствор (4-(3-аминопропил)-3-метилтиофенкарбоксилата (0,6 г, 3,0 ммоль) в метаноле (30 мл). Реакционную смесь кипятили с обратным холодильником в течение 2 часов. Реакционный раствор непосредственно концентрировали, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 6 (0,25 g, выход: 49,9%).
MS (ESI): Рассчитано для C8H9NOS 167; Найдено 168 [М+Н]+.
Шаг шесть: 5,6,7,8-тетрагидро-4Н-тиено[3,4-с]азепин (соединение 7) При комнатной температуре медленно добавляли порциями тетрагидроалюминат лития (284 мг, 7,5 ммоль) в раствор 5,6,7,8-тетрагидро-8Н-тиено[3,4-с]азепин-4-она (250 мг, 1,5 ммоль) в тетрагидрофуране (20 мг). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Смесь быстро охлаждали с помощью 15% NaOH (1 мл) и фильтровали с сульфатом магния. Реакционный раствор непосредственно концентрировали, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 7 (190 мг, выход: 82,8%).
MS (ESI): Рассчитано для C8H11NS 153; Найдено 154 [М+Н]
Шаг семь:
N-(2,6-диметил-4-(4,6,7,8-тетрагидро-5Н-тиено[3,4-с]азепин-5-ил)фенил)-3,3-диметилбутанамид (соединение 03027)
Смешанный реакционный раствор три(дибензилиденацетон)дипалладия (5 мг, 5 мкмоль), трициклогексилфосфина (0,1 мл 10% раствор),
5,6,7,8-тетрагидро-4Н-тиено[3,4-с]азепина (20 мг, 0,13 ммоль),
N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамида (78 мг, 0,26 ммоль), трет-бутоксида калия (44 мг, 0,39 ммоль) и диметилсульфоксида (2 мл) выдерживали в СВЧ-реакторе при температуре 150°С в течение 1 часа. Полученную смесь разбавляли водой (25 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали на хроматографической пластинке с получением белого твердого 03027 трифторацетата (7,7 мг, выход: 16%).
Н ЯМР (400 МГц, CD3OD): δ 7,23 (дублет, J=2,8 Гц, 1H), 6,88 (дублет, J=2,8 Гц, 1H), 6,58 (синглет, 2Н), 4,55 (синглет, 2Н), 3,84 (триплет, J=4,4 Гц, 2Н), 2,94-2,88 (мультиплет, 2Н), 2,27 (синглет, 2Н), 2,14 (синглет, 6Н), 1,82-1,75 (мультиплет, 2Н), 1,12 (синглет, 9Н).
MS (ESI): Рассчитано для C22H30N2OS 370; Найдено 371 [М+Н]+. ВЭЖХ: 98,9% (214 нм) / 99,6% (254 нм).
Пример 3: Получение соединения 03028

Шаг один: 2-бром-3-тиофенметилформат (соединение 2)
При охлаждении льдом медленно добавляли хлорангидрид щавелевой кислоты (2,5 мл, 29,5 ммоль) в раствор 2-бром-3-тиофенмуравьиной кислоты (5,0 г, 24,1 ммоль) в дихлорметане (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем ее концентрировали для удаления растворителя, а остаток растворяли в метаноле (50 мл) и кипятили с обратным холодильником в течение 4 часов. Смесь концентрировали под вакуумом с получением желтого маслянистого соединения 2 (5,2 г, выход: 93,6%).
MS (ESI): Рассчитано для C6H5BrO2S 220; Найдено 221 [М+Н]+.
Шаг два: 2-(3-((трет-бутоксикарбонил) амино)-1-пропинил)-3-тиофенметилформат (соединение 3)
(1,1'-бис(дифенилфосфин)ферроцен)палладия дихлорид (0,50 г, 0,67 ммоль) и йодид меди (0,13 г, 0,67 моль) добавляли в раствор 2-бром-3-тиофенметилформата (3,0 г, 13,5 ммоль), N-трет-бутоксикарбониламинопропина (2,5 г, 16,3 ммоль) и диизопропилэтиламина (5 мл) в ацетонитриле (30 мл). Полученную смесь кипятили с обратным холодильником при перемешивании в течение ночи в защитной атмосфере азота. Реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Фильтрат концентрировали для удаления растворителя, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 3 (3,4 г, выход: 84,9%).
MS (ESI): Рассчитано для C14H17NO4S 295; Найдено 296 [М+Н]+.
Шаг три: 2-(3-((трет-бутоксикарбонил)амино)-пропил)-3-тиофенметилформат (соединение 4)
10% палладиевый катализатор на углеродной основе (0,34 г) добавляли в раствор 2-(3-((трет-бутоксикарбонил)амино)-1-пропинил-3-метилтиофенкарбоксилата (3,4 г, 11,5 ммоль) в тетрагидрофуране (30 мл). Реакционную смесь перемешивали в течение ночи в атмосфере водорода 4 атм, фильтровали и концентрировали с получением светло-желтого маслянистого соединения 4 (3,4 г, выход: 98,6%).
MS (ESI): Рассчитано для C14H21NO4S 299; Найдено 200 [М-99]+.
Шаг четыре: 2-(3-аминопропил)-3-тиофенметилформат (соединение 5)
Трифторуксусную кислоту (9,1 г, 80 ммоль) медленно добавляли по каплям в раствор 2-(3-((трет-бутоксикарбонил)амино)-пропил)-3-тиофенметилформата (3,4 г, 11,4 ммоль) в дихлорметане (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем непосредственно концентрировали с получением желтого маслянистого сырого соединения 5 (2,2 г, выход: 96,5%).
MS (ESI): Рассч. для C9H13NO2S 199; Найдено 200 [М+Н]+.
Шаг пять: 5,6,7,8-тетрагидро-4Н-тиено[3,2-с]азепин-4-он (соединение 6)
При комнатной температуре добавляли метилат натрия (2,0 г) в раствор (2-(3-аминопропил)-3-метилтиофенкарбоксилата (2,2 г, 11,0 ммоль) в метаноле (40 мл). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Реакционный раствор непосредственно концентрировали, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 6 (1,4 g, выход: 75,8%).
MS (ESI): Рассчитано для C8H9NOS 167; Найдено 168 [М+Н]+.
Шаг шесть: 5,6,7,8-тетрагидро-4Н-тиено[3,2-с]азепин (Соединение 7)
При комнатной температуре медленно добавляли порциями тетрагидроалюминиат лития (956 мг, 25,2 ммоль) в раствор 5,6,7,8-тетрагидро-4Н-тиено[3,2-с]азепин-4-она (1,4 g, 8,4 ммоль) в тетрагидрофуране (50 мг). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Смесь быстро охлаждали с помощью 15% NaOH (1 мл) и фильтровали с сульфатом магния. Фильтрат концентрировали и очищали на колонке с силикагелем с получением желтого маслянистого соединения 6 (800 мг, выход: 62,5%).
MS (ESI): Рассчитано для C8H11NS 153; Найдено 154 [М+Н]+.
Шаг семь: N-(2,6-диметил-4-(4,6,7,8-тетрагидро-5Н-тиено[3,2-с]азепин-5-ил)фенил)-3,3-диметилбутанамид (соединение 03028)
Смешанный реакционный раствор три(дибензилиденацетон)дипалладия (31 мг, 0,034 ммоль), трициклогексилфосфина (0,1 мл 10% раствор), соединения 7 (52 мг, 0,34 ммоль), N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамида (200 мг, 0,68 ммоль), трет-бутоксида калия (76 мг, 0,68 ммоль) и диметилсульфоксида (5 мл) реагировал в микроволновом реакторе при температуре 150°С в течение 2 часов. Полученную смесь разбавляли водой (25 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом хроматографии с получением белого твердого соединения 03028 (12,7 мг, выход: 7,7%).
Н ЯМР (400 МГц, CD3OD):δ5 7,18 (синглет, 2Н), 7,12 (дублет, J=5,2 Гц, 1H), 7,00 (дублет, J=5,2 Гц, 1H), 4,80 (синглет, 2Н), 4,00-3,93 (мультиплет, 2Н), 3,15-3,11 (мультиплет, 2Н), 2,31 (синглет, 2Н), 2,25 (синглет, 6Н), 2,16-2,08 (мультиплет, 2Н), 1,12 (синглет, 9Н). MS (ESI): Рассчитано для C22H30N2OS 370; Найдено 371 [М+Н]+. ВЭЖХ: 98,9% (214 нм) / 99,3% (254 нм).
Пример 4: Получение соединения 03029

Шаг один: 3-(3-((трет-бутоксикарбонил) амино)-1-пропинил)-2-пиридинметилформат (соединение 2)
(1,1'-бис(дифенилфосфин)ферроцен)палладия дихлорид (0,90 г, 1,0 ммоль) и йодид меди (0,2 г, 1,5 моль) добавляли в раствор 3-бром-2-пиридинметилформата (2,5 г, 11,6 ммоль), N-трет-бутоксикарбониламинопропина (2,0 г, 12,9 ммоль) и диизопропилэтиламина (5 мл) в ацетонитриле (30 мл). Полученную смесь кипятили с обратным холодильником в течение 6 часов в защитной атмосфере азота. Реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Фильтрат концентрировали для удаления растворителя, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 2 (1,4 г, выход: 41,7%).
Н ЯМР (400 МГц, CDCl3): δ 8,64 (дублет, J=3,6 Гц, 1H), 7,88 (дублет, J=8,0 Гц, 1H), 7,47-7,38 (мультиплет, 1H), 4,86 (синглет, 1H), 4,23 (дублет, J=2,8 Гц, 2Н), 4,01 (синглет, 3Н), 1,48 (синглет, 9Н).
Шаг два: 3-(3-((трет-бутоксикарбонил)амино)-пропинил)-2-пиридинметилформат (соединение 3)
10% палладиевый катализатор на углеродной основе (0,14 г) добавляли в раствор 3-(3-((трет-бутоксикарбонил)амино)-1-пропинил)-2-пиридинметилформата (1,4 г, 4,8 ммоль) в тетрагидрофуране (30 мл). Реакционную смесь перемешивали в течение ночи в атмосфере водорода 4 атм, фильтровали и концентрировали с получением светло-желтого маслянистого соединения 3 (1,4 г, выход: 98,6%).
MS (ESI): Рассчитано для C15H22N2O4 294; Найдено 295 [М+Н]+, 317 [М+Na]+.
Шаг три: 3-(3-аминопропил)-2-пиридинметилформат (соединение 4)
Трифторуксусную кислоту (4,5 г, 40 ммоль) медленно добавляли по каплям в раствор 3-(3-((трет-бутоксикарбонил)амино)-пропил)-2-пиридинметилформата (1,4 г, 4,7 ммоль) в дихлорметане (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем непосредственно концентрировали с получением желтого маслянистого сырого соединения 4 (1,0 г), которое использовали на следующем этапе без последующей очистки.
MS (ESI): Рассчитано для C10H14N2O2 194; Найдено 195 [М+Н]+.
Шаг четыре: 5,6,7,8-тетрагидро-9Н-пиридо[2,3-с]азепин-9-он (соединение 5)
При комнатной температуре добавляли метилат натрия (1,0 г) в раствор (3-(3-аминопропил)-2-пиридометилформата (1,0 г) в метаноле (20 мл). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Реакционный раствор непосредственно концентрировали, остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 5 (500 мг, выход: 59,8%).
MS (ESI): Рассчитано для C9H10N2O 162; Найдено 163 [М+Н]+.
Шаг пять: 6,7,8,9-тетрагидро-5Н-пиридо[2,3-с]азепин (соединение 6)
При комнатной температуре раствор борана в тетрагидрофуране (1,0 моль/л, 20 мл) медленно добавляли в раствор 5,6,7,8-тетрагидро-9Н-пиридо[2,3-с]азепин-9-она (500 мг, 3,1 ммоль) в тетрагидрофуране (5 мл). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Смесь разбавляли метанолом (10 мл) и концентрировали. Остаток очищали на колонке с силикагелем с получением желтого маслянистого соединения 6 (100 мг, выход: 21,9%).
MS (ESI): Рассчитано для C9H12N2 148; Найдено 149 [М+Н]+.
Шаг шесть: N-(2,6-диметил-4-(5,6,7,9-тетрагидро-8Н-пиридо[2,3-с]азепин-8-ил)фенил)-3,3-диметилбутанамид (соединение 03029)
Смешанный реакционный раствор три(дибензилиденацетон)дипалладия (31 мг, 0,034 ммоль), трициклогексилфосфина (0,1 мл 10% раствор), 6,7,8,9-тетрагидро-5Н-пиридо[2,3-с]азепина (52 мг, 0,34 ммоль), N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамида (200 мг, 0,68 ммоль), трет-бутоксида калия (76 мг, 0,67 ммоль) и диметилсульфоксида (5 мл) выдерживали в микроволновом реакторе при температуре 150°С в течение 2 часов. Полученную смесь разбавляли водой (25 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом тонкослойной хроматографии с получением белого твердого соединения 03029 (6,8 мг, выход: 5,3%).
Н ЯМР (400 МГц, CD3OD): δ 8,27 (дублет, J=5,2 Гц, 1Н), 7,61 (дублет, J=7,2 Гц, 1Н), 7,45-7,20 (двойной дублет, J=5,2 Гц, J=7,2 Гц, 1Н)), 6,63 (синглет, 2Н), 4,78 (синглет, 2Н), 3,90 (триплет, J=4,8 Гц, 2Н), 3,07-3,01 (мультиплет, 2Н), 2,26 (синглет, 2Н), 2,12 (синглет, 6Н), 1,94-1,87 (мультиплет, 2Н), 1,11 (синглет, 9Н). MS (ESI): Рассчитано для C23H31N3O 365; Найдено 366 [М+Н]+. ВЭЖХ: 97,2% (214 нм) / 99,4% (254 нм).
Пример 5: Получение соединения 03033

Шаг один: 5,6,7,8-тетрагидро-4H-циклогептатриено[b]тиофен-4-он (соединение 2)
Пятиокись фосфора (1,5 г, 10,9 ммоль) и молекулярные сита (2 г) добавляли в раствор 5-(тиофен-2-ил)пентановой кислоты (1,00 г, 5,40 ммоль) в толуоле (20 мл). Реакционную смесь нагревали до 130°С и выдерживали в течение 2 часов в защитной атмосфере азота. Затем ее охлаждали до комнатной температуры, фильтровали, промывали насыщенным раствором бикарбоната натрия (30 мл), высушивали, концентрировали под вакуумом для удаления растворителя и очищали методом колоночной хроматографии с получением желтого маслянистого соединения 2 (0,42 г, выход: 46,6%).
MS (ESI): Рассчитано для C9H10OS 166; Найдено 167 [М+Н]+.
Шаг два: 6,7,8,9-тетрагидротиено[3,2-с]азоцин-4(5H)-он (соединение 3)
Азид натрия (501 мг) добавляли в раствор соединения 2 (0,64 г, 3,86 ммоль) в концентрированной соляной кислоте (20 мл). Смесь перемешивали при комнатной температуре в течение 16 часов, выливали в ледяную воду, корректировали рН до значения 7 с помощью карбоната калия, экстрагировали этилацетатом, высушивали над безводным сульфатом натрия. Органическую фазу концентрировали под вакуумом и очищали методом колоночной хроматографии с получением грязно-белого твердого соединения 3 (400,0 мг, выход: 57,25%).
MS (ESI): Рассчитано для C9H11NOS 181; Найдено 182 [М+Н]+.
Шаг три: 4,5,6,7,8,9-гексагидротиено[3,2-с]азоцин (соединение 4)
Алюмогидрид лития (420 мг, 11,05 ммоль) добавляли в раствор соединения 3 (400 мг, 2,21 ммоль) в тетрагидрофуране (40 мл). Смесь нагревали до температуры 80°С и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, добавляли воду (2 мл) и 10% гидроксид натрия (1 мл) для остановки реакции и фильтровали. Фильтрат концентрировали, остаток очищали методом колоночной хроматографии с получением желтого маслянистого соединения 4 (150 мг, выход: 40,90%).
MS (ESI): Рассчитано для C9H13NS 167; Найдено 168 [М+Н]+.
Шаг четыре: N-(2,6-диметил-4-(6,7,8,9-тетрагидротиено[3,2-с]азоцин-5(4H)-ил)фенил)-3,3-диметилбутанамид (соединение 03033)
Добавляли соединение 5 (178 мг, 0,60 ммоль), Pd2(dba)3 (10 мг, 0,034 ммоль), раствор три-трет-бутилфосфон-гексана (1 моль/л, 0,2 мл) и трет-бутоксид калия (100 мг, 0,9 ммоль) в раствор соединения 4 (50 мг, 0,30 ммоль) в ДМСО (2 мл). Смесь выдерживали в СВЧ-реакторе при температуре 150°С в течение 1 часа. Смесь охлаждали до комнатной температуры, реакцию останавливали водой, экстрагировали этилацетатом, высушивали над безводным сульфатом натрия, концентрировали под вакуумом. Остаток очищали методом колоночной хроматографии с получением грязно-белого твердого соединения 03030 (14,78 мг), выход: 12,8%).
MS (ESI): Рассчитано для C23H32N2OS 384; Найдено 385 [М+Н]+.
Н ЯМР (400 МГц, CD3OD): δ 7,10 (дублет, J=5,2 Гц, 1Н), 6,90(дублет, J=5,2 Гц, 1H), 6,44 (синглет, 2Н), 4,55 (синглет, 2Н), 3,56-3,48 (мультиплет, 2Н), 2,82-2,74 (мультиплет, 2Н), 2,29 (синглет, 2Н), 2,15 (синглет, 6Н), 1,80-1,70 (мультиплет, 4Н), 1,14 (синглет, 9Н).
Пример 6: Получение соединения 03034

Шаг один: (N-(2,6-диметил-6-(1-трет-бутилацетил)анилин))-(5,6,7,8-тетрагидро-4H)-тиено [3,2]азаантрацен (СВ03034)
Добавляли соединение 2 (484 мг, 1,63 ммоль), Pd2(dba)3 (149 мг, 0,16 ммоль), раствор три-трет-бутилфосфон-гексана (66 мг, 0,33 ммоль) и трет-бутоксид калия (365 мг, 3,26 ммоль) в раствор соединения 1 (250 мг, 1,63 ммоль) в ДМСО (5 мл). Смесь выдерживали в микроволновом реакторе при температуре 150°С в течение 0,5 часа. Затем смесь охлаждали до комнатной температуры, реакцию останавливали водой, экстрагировали этилацетатом, высушивали над безводным сульфатом натрия и очищали методом колоночной хроматографии с получением грязно-белого твердого соединения 03034 (49,12 мг), выход: 8,12%).
MS (ESI): Рассчитано для C22H30N2OS 370; Найдено 371 [М+Н]+.
Н ЯМР (400 МГц, CDCl3): δ 6,98 (дублет, J=4,0 Гц, 1H), 6,84 (дублет, J=8,0 Гц, 1H), 6,47-6,49 (мультиплет, 3Н), 3,68 (триплет, J=4,0 Гц, 2Н), 3,77 (триплет, J=6,0 Гц, 2Н), 2,29 (синглет, 2Н), 2,16 (синглет, 6Н), 1,89-1,91 (мультиплет, 2Н), 1,74-1,68 (мультиплет, 2Н), 1,16 (синглет, 9Н).
Пример 7: Получение соединения 03039
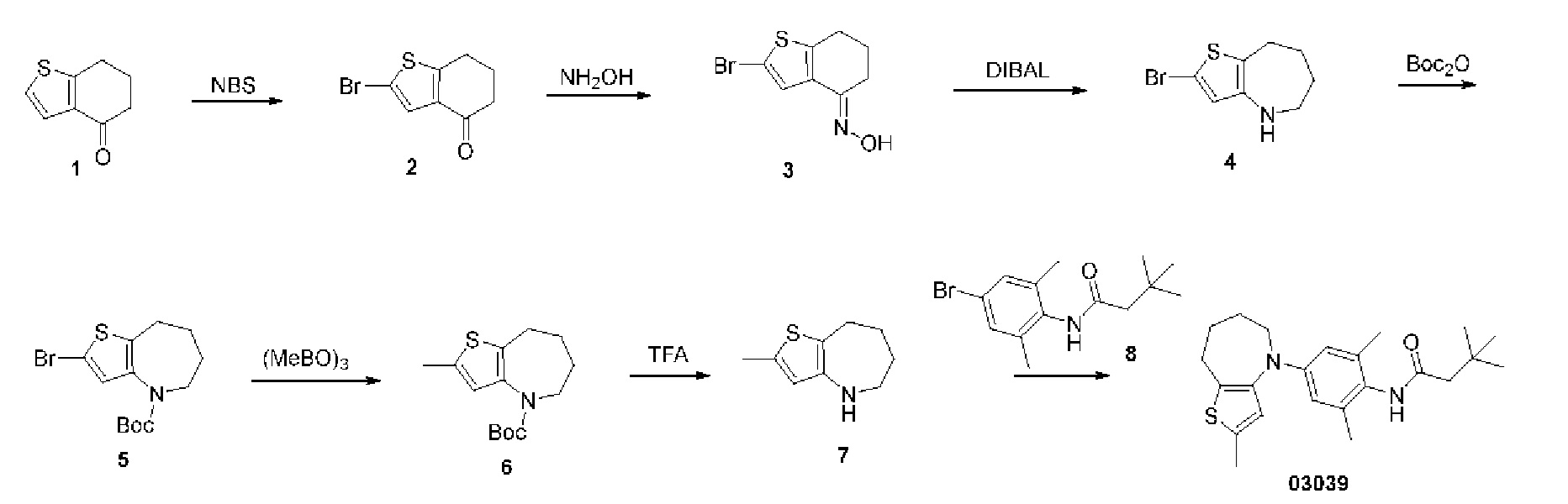
Шаг один: 2-бром-6,7-дигидробензо[b]тиофен-4(5H)-он (соединение 2)
При охлаждении льдом порциями добавляли NBS (266 мг, 1,5 ммоль) в раствор 6,7-дигидробензо[b]тиофен-4(5H)-она (152 мг, 1 ммоль) в диметилформамиде (5 мл). Реакционную смесь выдерживали при комнатной температуре в течение 12 часов. Реакцию в смеси останавливали добавлением воды (10 мл) при 0°С, проводили экстракцию этилацетатом, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного маслянистого соединения 2 (200 мг, выход: 86,0%).
MS (ESI): Рассчитано для C8H7BrOS 230; Найдено 231 [М+Н]+.
Шаг два: 2-бром-6,7-дигидробензо[b]тиофен-4(5H)кетоксим (соединение 3)
Добавляли порциями ацетат натрия (5,10 г, 63 ммоль) и гидроксиламина гидрохлорид (4,30 г, 63 ммоль) в раствор 2-бром-6,7-дигидробензо[b]тиофен-4(5H)-она (4,72 г, 21 ммоль) в этаноле (100 мл) и воде (20 мл). Реакционную смесь выдерживали при температуре 80°С в течение 3 часов и охлаждали до комнатной температуры, фильтровали и концентрировали с получением коричневого твердого вещества 3 (3,80 г, выход: 75,0%).
MS (ESI): Рассчитано для C8H7BrNOS 245; Найдено 246 [М+Н]+.
Шаг три: 2-бром-5,6,7,8-тетрагидро-4H-тиено[3,2-b]азепин (соединение 4)
В бане с ледяной водой в раствор 2-бром-6,7-дигидробензо[b]тиофен-4(5H)кетоксима (300 мг, 1,22 ммоль) в дихлорметане (10 мл) добавляли DIBAL-H (62 мг, 0,46 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, реакцию останавливали водой, смесь экстрагировали этилацетатом, органическую фазу высушивали над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали методом колоночной хроматографии с получением бесцветного маслянистого соединения 4 (100 мг), выход: 35,0%).
MS (ESI): Рассчитано для C8H10BrNS 231; Найдено 232 [М+Н]+.
Шаг четыре: Сложный трет-бутиловый эфир 2-бром-5,6,7,8-тетрагидро-4H-тиено[3,2-b]азепин-4-карбоновой кислоты (соединение 5)
DMAP (223 мг, 1,7 ммоль) и ангидрид трет-бутоксикарбонила (3,77 г, 17 ммоль) добавляли в раствор соединения 4 (2 г, 8,7 ммоль) в тетрагидрофуране (15 мл). Реакционную смесь кипятили с обратным холодильником в течение 6 часов. Растворитель высушивали центрифугированием, разбавляли водой (10 мл) и этилацетатом. Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого соединения 5 (14 г, выход: 48,9%).
MS (ESI): Рассчитано для C13H18BrNO2S 331 Найдено 276 [М-56+Н]+.
Шаг пять: Сложный трет-бутиловый эфир 2-метил-5,6,7,8-тетрагидро-4H-тиено[3,2-b]азепин-4-карбоновой кислоты (соединение 6)
Триметиловый эфир борной кислоты (69 мг, 0,542 ммоль), карбонат калия (125 мг, 0,90 ммоль) и тетракис(трифенилфосфин)палладий (52 мг, 0,045 ммоль) добавляли в раствор соединения 5 (150 мг, 0,452 ммоль) в диметилформамиде (5 мл). Смесь нагревали до температуры 120°С и перемешивали в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали. Остаток отделяли и очищали на колонке с силикагелем с получением желтого маслянистого соединения 6 (112 мг, выход: 93,2%).
MS (ESI): Рассчитано для C14H21NO2S 267; Найдено 212 [М-56+Н]+.
Шаг шесть: 2-метил-5,6,7,8-тетрагидро-4H-тиено[3,2-b]азепин (соединение 7)
Трифторуксусную кислоту (2 мл) добавляли в раствор соединения 6 (112 мг, 0,419 ммоль) в ДХМ (5 мл). Смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли, корректировали значение рН до нейтрального, экстрагировали этилацетатом, концентрировали. Остаток отделяли и очищали в колонке с силикагелем с получением белого твердого соединения 7 (40 мг, выход: 57,1%).
Шаг семь: N-(2,6-диметил-4-(2-метил-5,6,7,8-тетрагидро-4H-тиено[3,2-b]азепин-4-ил)фенил)-3,3-диметилбутирамид (соединение 03039)
Добавляли N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (250 мг, 0,84 ммоль), трет-бутоксид калия (130 мг, 0,84 ммоль), трис(дибензилиденацетон)дипалладий (39 мг, 0,042 ммоль) и три-трет-бутилфосфин (0,3 мг, 0,084 ммоль) в раствор соединения 7 (70 мг, 0,42 ммоль) в ДМСО (2 мл). Реакционную смесь нагревали до температуры 150°С и выдерживали в микроволновом реакторе в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали. Остаток отделяли и очищали на колонке с силикагелем с получением белого твердого соединения 03039 (10 мг, выход: 6,2%).
MS (ESI): Рассчитано для C23H32N2OS 384; Найдено 385 [М+Н]+.
1H ЯМР (400 МГц, CD3CN) δ 7,44 (синглет, 1H), 6,53 (синглет, 1Н), 6,47 (синглет, 2Н), 3,70-3,64 (мультиплет, 2Н), 2,69-2,66 (мультиплет, 2Н), 2,39 (синглет, 3Н), 2,23 (синглет, 2Н), 2,10 (синглет, 6Н), 1,87-1,81 (мультиплет, 2Н), 1,68-1,62 (мультиплет, 2Н), 1,12 (синглет, 9Н).
Пример 8: Получение соединения 03041
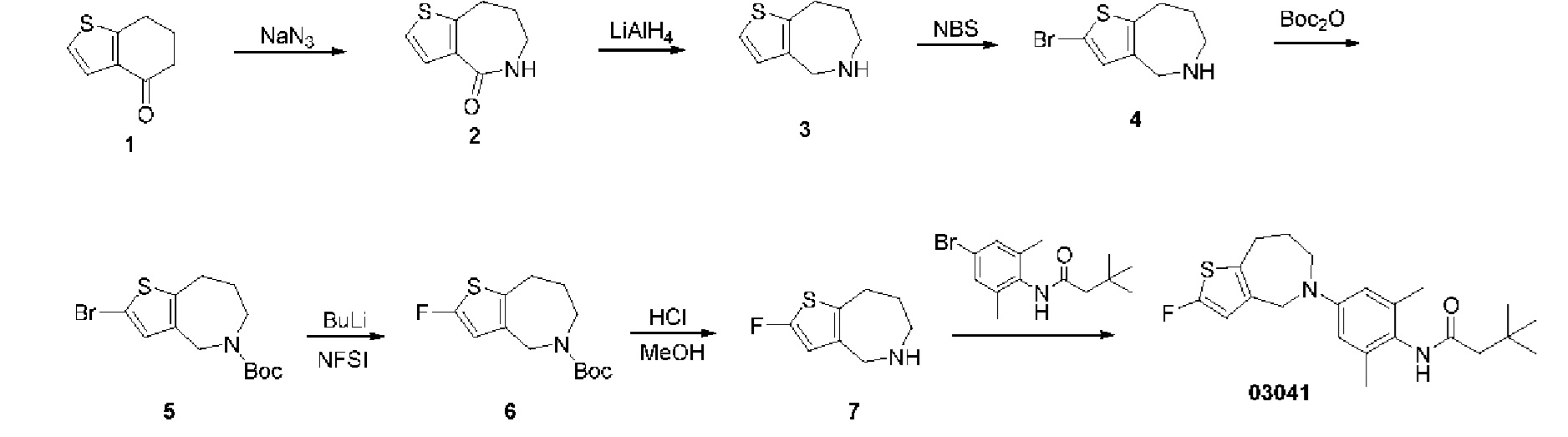
Шаг один: 5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепин-4-он (соединение 2)
При охлаждении льдом порциями добавляли азид натрия (15 г, 250 ммоль) в раствор 6,7-дигидробензо[b]тиофен-4(5H)она (15 г, 98 ммоль) в соляной кислоте (100 мл). Реакционную смесь выдерживали при комнатной температуре в течение ночи. Добавляли кубики льда, корректировали значение рН выше 7 с помощью насыщенного раствора карбоната калия. Смесь экстрагировали дихлорметаном, высушивали над безводным сульфатом натрия, фильтровали, концентрировали с получением белого твердого соединения 2 (13 г) методом колоночной хроматографии.
MS (ESI): Рассчитано для C8H9NOS 167; Найдено 168 [М+Н]+.
Шаг два: 5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепин (соединение 3)
При охлаждении льдом медленно добавляли соединение 2 (13 г, 78 ммоль) в суспензию алюмогидрида лития (8,9 г, 230 ммоль) в тетрагидрофуране (200 мл). Смесь нагревали до температуры 70°С и перемешивали в течение 1 часа. Затем медленно добавляли воду (9 мл), водный раствор гидроксида натрия (15%, 9 мл) и воду (27 мл), добавляли избыток безводного сульфата магния, смесь фильтровали, концентрировали. Остаток подвергали колоночной хроматографии с получением соединения 3 (9,0 г, выход: 75%).
MS (ESI): Рассчитано для C8H11NS 153; Найдено 154 [М+Н]+.
Шаг три: 2-бром-5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепин (соединение 4)
Добавляли соляную кислоту (2,3 г, 62 ммоль) в раствор 5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепина (8,0 г, 50 ммоль) в тетрагидрофуране (100 мл). Смесь перемешивали при комнатной температуре в течение 30 минут. После концентрирования для удаления излишка соляной кислоты последовательно добавляли уксусную кислоту (10 мл) и N-бромимид янтарной кислоты (12,0 г, 68 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, добавляли насыщенный раствор бикарбоната натрия для корректировки значения рН выше 7. На следующем этапе реакцию проводили без очистки.
MS (ESI): Рассчитано для C8H10BrNS 231; Найдено 232 [М+Н]+.
Шаг четыре: трет-бутил-2-бром-4,6,7,8-тетрагидро-5H-тиено[3,2-с]азепин-5-карбоксилат (соединение 5)
Ангидрид трет-бутоксикарбонила (19 г, 87 ммоль) добавляли в раствор соединения 4 (8 г, 35 ммоль) в тетрагидрофуране (200 мл). Смесь выдерживали при комнатной температуре в течение 2 часов, концентрировали и подвергли колоночной хроматографии с силикагелем с получением сырого белого твердого соединения 5(15 г).
MS (ESI): Рассчитано для C13H18BrNO2S 331; Найдено 276 [М-56+1]+.
Шаг пять: трет-бутил-2-фтор-4,6,7,8-тетрагидро-5H-тиено[3,2-с]азепин-5-карбоксилат (соединение 6)
При охлаждении сухим льдом медленно добавляли н-бутиллитий (2,6 мл, 6,24 ммоль) в раствор соединения 5 (1,0 г, 3,02 ммоль) в тетрагидрофуране (20 мл), затем смесь перемешивали при температуре -78°С в течение 1 часа. Затем медленно добавляли N-фтор-бис-бензолсульфонамид (1,91 г, 6,05 ммоль). Реакционную смесь медленно охлаждали до комнатной температуры и перемешивали в течение ночи. После добавления ледяной воды смесь экстрагировали, высушивали и концентрировали. Остаток подвергали колоночной хроматографии с получением сырого соединения 6 (800 мг).
MS (ESI): Рассчитано для C13H18FNO2S 271; Найдено 216 [М-56+1]+.
Шаг шесть: 2-фтор-5,6,7,8-тетрагидро-4Н-тиено[3,2-с]азепин (соединение 7)
Добавляли соляную кислоту (1,87 мл, 7,49 ммоль) в раствор соединения 6 (800 мг, 2,95 ммоль) в 1,4-диоксане (20 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, концентрировали для удаления избытка соляной кислоты; добавляли насыщенный водный раствор бикарбоната натрия для корректировки рН до значения выше 7. Смесь экстрагировали, высушивали и концентрировали, остаток подвергали очистке колоночной хроматографией с получением сырого соединения 7 (1,0 г). MS (ESI):
Рассчитано для C8H10FNS 171; Найдено 172 [М+Н]+.
Шаг семь: N-(4-(2-фтор-4,6,7,8-тетрагидро-5Н-тиено[3,2-с]азепин-5-ил)-2,6-диметилфенил)-3,3-диметилбутанамид (03041)
Промежуточный N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (1,9 г, 6,4 ммоль) и трет-бутоксид натрия (2,25 г, 20 ммоль) добавляли в раствор соединения 7 (1,0 г, 5,8 ммоль) в трет-бутаноле (100 мл). После трехкратного обмена азота добавляли метансульфоновой кислоты (2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)палладий(II) (830 мг, 990 ммоль). Реакционную смесь нагревали до 95°С в атмосфере азота и перемешивали в течение ночи, фильтровали, концентрировали для удаления растворителя. Остаток подвергали колоночной хроматографии с получением сырого соединения 03041, которое затем очищали препаративной хроматографии с получением соединения 03041 (57 мг, выход: 2,5%).
MS (ESI): Рассчитано для C22H29FN2OS 388; Найдено 389 [М+Н]+.
1H ЯМР (400 МГц, CDCl3): δ 6,54 (синглет, 2Н), 6,35 (синглет, 1Н), 4,36 (синглет, 2Н), 3,79-3,74 (мультиплет, 2Н), 2,84-2,78 (мультиплет, 2Н), 2,29 (синглет, 2Н), 2,19 (синглет, 6Н), 2,01-1,96 (мультиплет, 2Н), 1,15 (синглет, 9Н).
Пример 9: Получение соединения 03042
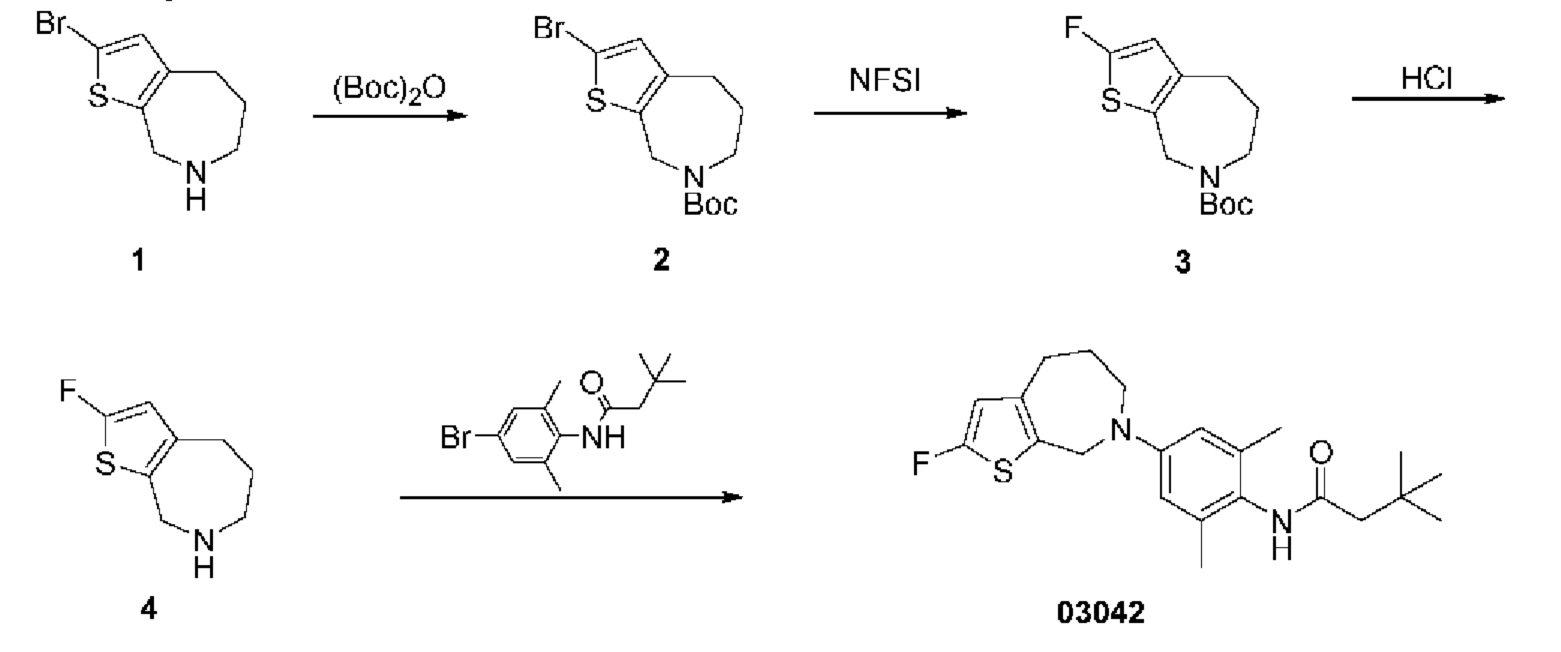
Шаг один: трет-бутил-2-бром-4,5,6,8-тетрагидро-7H-тиено[2,3-с]азепин-7-карбоксилат (соединение 2)
При комнатной температуре добавляли ди-трет-бутилдикарбонат (569 мг, 2,61 ммоль) в раствор 2-бром-5,6,7,8-тетрагидро-4H-тиено[2,3-с]азепина (300 мг, 1,30 ммоль) в тетрагидрофуране (10 мл). Реакционную смесь кипятили с обратным холодильником в течение 16 часов. Реакционную смесь концентрировали для удаления растворителя, а остаток очищали методом колоночной хроматографии с получением соединения 2 (230 мг, выход: 53,5%).
MS (ESI): Рассчитано для C13H18BrNO2S 331; Найдено 276 [М-56]+.
Шаг два: трет-бутил-2-фтор-4,5,6,8-тетрагидро-7H-тиено[2,3-с]азепин-7-карбоксилат (соединение 3)
Соединение 2 (1,2 г, 3,63 ммоль) растворяли в безводном тетрагидрофуране (20 мл), температуру снижали до -78°С в атмосфере азота, затем по каплям добавляли н-бутиллитий (2,4 моль/л, 3,0 мл). Температуру реакции поддерживали при -78°С в течение получаса; затем в реакционную смесь по каплям добавляли раствор N-фтор-бисбензолсулъфонамида (2,29 г, 7,26 ммоль) в тетрагидрофуране. После добавления смесь медленно нагревали до комнатной температуры и выдерживали при ней в течение 16 часов. Реакцию останавливали насыщенным раствором хлорида аммония, смесь экстрагировали этилацетатом, высушивали над безводным сульфатом натрия, фильтровали, концентрировали для удаления растворителя. Остаток очищали методом колоночной хроматографии с получением соединения 3 (400 мг), выход: 40,7%).
MS (ESI): Рассчитано для C13H18FNO2S 271; Найдено 216 [M-56+H]+.
Шаг три: 2-фтор-5,6,7,8-тетрагидро-4H-тиено[2,3-с]азепин (соединение 4)
К соединению 3 добавляли раствор соляной кислоты в метаноле (4 моль/л, 40 мл). Смесь перемешивали при комнатной температуре в течение шестнадцати часов. После концентрирования с целью удаления растворителя добавляли бикарбонат натрия для корректировки рН до значения 8. Остаток очищали колоночной хроматографией с получением соединения 4 (410 мг, выход: 64,9%).
MS (ESI): Рассчитано для C8H10FNS 171; Найдено 172 [М+Н]+.
Шаг четыре: N-(4-(2-фтор-4,5,6,8-тетрагидро-7H-тиено[2,3-с]азепин-7-ил)-2,6-диметилфенил)-3,3-диметилбутанамид (соединение 03042)
N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (712 мг, 2,40 ммоль), трет-бутоксид натрия (921 мг, 9,59 ммоль), метансульфоновой кислоты (2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-связанный бифенил)(2-амино-1,Г-бифенил-2-ил)палладий (II) (201 мг, 0,24 ммоль) добавляли в раствор соединения 4 (410 мг, 2,40 ммоль) в трет-бутаноле (20 мг) в атмосфере азота. Смесь выдерживали при температуре 90°С в течение шестнадцати часов. После охлаждения смеси до комнатной температуры и фильтрации фильтрат концентрировали под вакуумом для удаления растворителя. Остаток очищали колоночной хроматографией с получением сырого продукта, который затем отделяли препаративной хроматографией с получением соединения 03042 (32,7 мг, выход: 3,5%).
MS (ESI): Рассчитано для C22H29FN2OS 388; Найдено 389 [М+Н]+.
Н ЯМР (400 МГц, CD3OD): δ 6,75 (синглет, 2Н), 6,31 (синглет, 1H), 4,59 (синглет, 2Н), 3,89-3,86 (мультиплет, 2Н), 2,82-2,79 (мультиплет, 2Н), 2,29 (синглет, 2Н), 2,19 (синглет, 6Н), 1,91-1,86 (мультиплет, 2Н), 1,13 (синглет, 9Н).
Пример 10: Получение соединение 03043

Шаг один: 2-хлор-6,7-дигидробензо[b]тиофен-4(5H)-он (соединение 2)
Добавляли N-хлорсукцинимид (32 г, 0,24 моль) в раствор 6,7-дигидробензо[b]тиофен-4(5H)она (25 г, 0,16 моль) в уксусной кислоте (50 мл), реакционную смесь нагревали до 50°С и выдерживали в течение ночи. Смесь охлаждали до комнатной температуры и концентрировали. Добавляли воду и этилацетат для экстракции, экстракт высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток подвергали колоночной хроматографии с получением соединения 2 (19 г, выход: 62%).
MS (ESI): Рассчитано для C8H7ClOS 186; Найдено 187 [М+Н]+.
Шаг два: 2-хлор-5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепин-4-он (соединение 3)
При охлаждении льдом медленно добавляли азид натрия (10 г, 0,15 ммоль) в раствор соединения 2 (14 г, 0,07 ммоль) в соляной кислоте (100 мл). Реакционную смесь выдерживали при комнатной температуре в течение ночи. Добавляли соответствующее количество льда и насыщенного карбоната калия для корректировки рН выше 7, смесь экстрагировали, высушивали, фильтровали и концентрировали. Остаток подвергали колоночной хроматографии с получением соединения 3 (6 г, выход: 40%).
MS (ESI): Рассчитано для C8H8ClNOS 201; Найдено 202 [М+Н]+.
Шаг три: 2-хлор-5,6,7,8-тетрагидро-4H-тиено[3,2-c]азепин (соединение 4)
При охлаждении льдом медленно добавляли соединение 3 (6 г, 29,8 ммоль) в суспензию алюмогидрида лития (5,6 г, 147 ммоль) в тетрагидрофуране (50 мл). Смесь перемешивали при температуре 70°С в течение 1 часа. Затем медленно последовательно добавляли воду (5,6 мл), водный раствор гидроксида натрия (15%, 5,6 мл) и воду (16,8 мл), избыток безводного сульфата магния. Смесь фильтровали, концентрировали. Остаток подвергали колоночной хроматографии с получением соединения 4 (3 г, выход: 53,7%).
MS (ESI): Рассчитано для C8H10ClNS 187; Найдено 188 [М+Н]+.
Шаг четыре: N-(4-(2-хлор-4,6,7,8-тетрагидро-5H-тиено[3,2-с]азепин-5-ил)-2,6-диметилфенил)-3,3-диметилбутанамид (соединение 03043)
Промежуточный N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (3,6 г, 12 ммоль) и трет-бутоксид натрия (3,5 г, 36 ммоль) добавляли в раствор соединения 4 (1,7 г, 9 ммоль) в трет-бутаноле (50 мл). После трехкратного замещения азота добавляли метансульфоновой кислоты (2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)п алладий(II) (1,29 г, 1,53 ммоль). Реакционную смесь нагревали до температуры 85°С в защитной атмосфере азота и выдерживали в течение ночи, затем фильтровали и концентрировали для удаления растворителя. Остаток подвергали колоночной хроматографии с получением сырого соединения 03043, который затем очищали препаративной хроматографией с получением соединения 03043 (600 мг, выход: 16,7%).
MS (ESI): Рассчитано для C22H29CIN2 OS 404; Найдено 405[M+H]+.
1H ЯМР (400 МГц, CD3OD): δ 6,92 (синглет, 1Н), 6,55 (синглет, 2Н), 4,46 (синглет, 2Н), 3,88-3,80 (мультиплет, 2Н), 2,92-2,84 (мультиплет, 2Н), 2,28 (синглет, 2Н), 2,15 (синглет, 6Н), 1,88 (синглет, 2Н), 1,13 (синглет, 9Н).
Пример 11: Получение соединения 03044

Шаг один: Метил 3-(3-((трет-бутоксикарбонил)амино)проп-1-ин-1-ил)-тиофен-2-карбоксилат (соединение 2)
N-трет-бутоксикарбониламинопропин (8,4 г, 54 ммоль), йодид меди (0,86 г, 4,5 ммоль), Pd(dppf)Cl2 (1,7 г, 2,3 ммоль) и DIEA (8,8 г, 68 ммоль) добавляли в указанном порядке в раствор метилового эфира 3-бромтиофен-2-карбоновой кислоты (10,0 г, 45 ммоль) в ацетонитриле (100 мл). Реакционную смесь нагревали до температуры 80°С в защитной атмосфере азота и перемешивали в течение 16 часов. Затем ее охлаждали до комнатной температуры, концентрировали под вакуумом для удаления растворителя и очищали методом колоночной хроматографии с получением желтого маслянистого соединения 3 (6,0 г, выход: 45,2%).
MS (ESI): Рассчитано для C14H17NO4S 295; Найдено 318 [+]+.
Шаг два: Метил 3-(3-((трет-бутоксикарбонил)амино)пропил)тиофен-2-карбоксилат (соединение 3)
Палладиевый катализатор на углеродной основе (600 мг) добавляли в раствор соединения 2 (6,0 г, 0,02 моль) в метаноле (40 мл). Смесь перемешивали при комнатной температуре в атмосфере водорода 0,4 МПа в течение 16 часов, фильтровали на нутч-фильтре. Фильтрат концентрировали и очищали колоночной хроматографией с получением желтого маслянистого соединения 3 (6,0 г, выход: 100%).
MS (ESI): Рассчитано для C14H21NO4S 299; Найдено 322 [M+Na]+.
Шаг три: Метил-3-(3-аминопропил)тиофен-2-карбоксилат (соединение 4)
2 моль/л раствор трифторуксусной кислоты в дихлорметане (50 мл) добавляли в соединение 3 (6,0 г, 0,02 моль). Смесь перемешивали при комнатной температуре в течение 2 часов. Фильтрат концентрировали с получением желтого маслянистого соединения 4 (4,0 г, выход: 100%).
MS (ESI): Рассчитано для C9H13NO2S 199; Найдено 200 [М+Н]+.
Шаг четыре: 4,5,6,7-тетрагидро-8H-тиено[2,3-с]азепин-8-он (соединение 5)
Добавляли метилат натрия (3,2 г, 0,06 моль) в раствор соединения 4 (4,0 г, 0,02 моль) в метаноле (50 мл). Смесь нагревали до температуры 70°С и перемешивали в течение 5 часов. Смесь охлаждали до комнатной температуры, фильтровали. Фильтрат концентрировали и очищали колоночной хроматографией с получением соединения 5 в виде желтого масла (2,8 г, выход: 83,8%).
MS (ESI): Рассчитано для C8H9NOS 167; Найдено 168 [М+Н]+.
Шаг пять: 5,6,7,8-тетрагидро-4H-тиено[2,3-с]азепин (соединение 6)
Алюмогидрид лития (1,9 г, 0,05 моль) добавляли в раствор соединения 5 (2,8 г, 17 ммоль) в тетрагидрофуране (50 мл) на ледяной бане. Смесь непрерывно перемешивали в течение 0,5 часа, затем нагревали до температуры 80°С и выдерживали в течение 2 часов. Смесь охлаждали до комнатной температуры, останавливали реакцию, растворитель концентрировали под вакуумом и очищали колоночной хроматографией с получением соединения 6 в виде грязно-белого твердого вещества (2,18 г, выход: 83,8%). MS (ESI):
Рассчитано для C8H11NS 153; Найдено 154 [М+Н]+.
Шаг шесть: 2-хлор-5,6,7,8-тетрагидро-4H-тиено[2,3-с]азепин (соединение 7)
Концентрированную соляную кислоту (1 мл) добавляли в раствор соединения 6 (2,0 г, 0,013 ммоль) в тетрагидрофуране (10 мл). Смесь перемешивали при комнатной температуре в течение 10 минут и концентрировали для удаления растворителя. Остаток растворяли в тетрагидрофуране (50 мл) и порциями добавляли уксусную кислоту (15 мл) и NCS (1,58 г, 0,012 моль). Смесь выдерживали при комнатной температуре в течение 2 часов. Смесь концентрировали под вакуумом и очищали колоночной хроматографией с получением соединения 7 в виде грязно-белого твердого вещества (2,1 г, выход: 86,4%).
MS (ESI): Рассчитано для C8H10ClNS 187; Найдено 188 [М+Н]+.
Шаг семь: N-(4-(2-хлор-4,5,6,8-тетрагидро-7H-тиено[2,3-с]азепин-7-ил)-2,6-диметилфенил)-3,3-диметилбутанамид (соединение 03044)
Ruphos-Pd-G3 (940 мг, 11 ммоль), трет-бутоксид натрия (4,3 г, 45 ммоль) и соединение 8 (4,67 г, 15 ммоль) добавляли в раствор соединения 7 (2,1 г, 11 ммоль) в трет-бутаноле (40 мл). Смесь нагревали до температуры 90°С в атмосфере азота и перемешивали в течение ночи. Смесь охлаждали до комнатной температуры, фильтровали, промывали этилацетатом и концентрировали. Остаток очищали методом флэш-хроматографии с получением бледно-желтого соединения 03044 (540 мг, выход: 12,2%).
MS (ESI): Рассчитано для C22H29CIN2 OS 404; Найдено 405 [M+H]+.
Н ЯМР (400 МГц, CD3OD): δ 8,75 (синглет, 1H), 6,81 (синглет, 1H), 6,53 (синглет, 2Н), 4,57 (синглет, 2Н), 3,84-3,75 (мультиплет, 2Н), 2,82-2,73 (мультиплет, 2Н), 2,16 (синглет, 2Н), 2,04 (синглет, 6Н), 1,75-1,65 (мультиплет, 2Н), 1,04 (синглет, 9Н).
Пример 12: Получение соединения 03045

Шаг один: 5,6,7,8-тетрагидро-4/7-тиено[3,2-с]азепин-4-он (соединение 2)
При охлаждении льдом порциями добавляли азид натрия (15 г, 250 ммоль) в раствор 6,7-дигидробензо[b]тиофен-4(5H)она (15 г, 98 ммоль) в соляной кислоте (100 мл). Реакционную смесь выдерживали при комнатной температуре в течение ночи. Добавляли кубики льда, корректировали значение рН выше 7 с помощью насыщенного раствора карбоната калия. Смесь экстрагировали дихлорметаном, высушивали над безводным сульфатом натрия, фильтровали, концентрировали и подвергали очистке методом колоночной хроматографии с силикагелем с получением сырого белого твердого соединения 2 (13 г).
MS (ESI): Рассчитано для C8H9NOS 167; Найдено 168 [М+Н]+.
Шаг два: 5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепин (соединение 3)
При охлаждении льдом медленно добавляли соединение 2 (13 г, 78 ммоль) в суспензию алюмогидрида лития (8,9 г, 230 ммоль) в тетрагидрофуране (200 мл). Смесь нагревали до температуры 70°С и перемешивали в течение 1 часа. Затем медленно последовательно добавляли воду (9 мл), водный раствор гидроксида натрия (15%, 9 мл) и воду (27 мл). Затем добавляли избыток безводного сульфата магния, смесь фильтровали, концентрировали. Остаток подвергали колоночной хроматографии с получением соединения 3 (9 г, выход: 75%).
MS (ESI): Рассчитано для C8H11NS 153; Найдено 154 [М+Н]+.
Шаг три: 2-бром-5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепин (соединение 4)
Добавляли соляную кислоту (2,3 г, 62 ммоль) в раствор 5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепина (8 г, 50 ммоль) в тетрагидрофуране (100 мл). Смесь перемешивали при комнатной температуре в течение 30 минут. После концентрирования для удаления излишка соляной кислоты последовательно добавляли уксусную кислоту (10 мл) и N-бромимид янтарной кислоты (12 г, 68 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, добавляли насыщенный раствор бикарбоната натрия для корректировки значения рН выше 7. На следующем этапе реакцию проводили без очистки.
MS (ESI): Рассчитано для C8H10BrNS 230.9; Найдено 231.9 [М+Н]+.
Шаг четыре: трет-бутил-2-бром-4,6,7,8-тетрагидро-5H-тиено[3,2-с]азепин-5-карбоксилат (соединение 5)
Ангидрид трет-бутоксикарбонила (19 г, 87 ммоль) добавляли в раствор соединения 4 (8 г, 35 ммоль) в тетрагидрофуране (200 мл). Смесь выдерживали при комнатной температуре в течение 2 часов, концентрировали и подвергли колоночной хроматографии с получением сырого белого твердого соединения 5 (15 г).
MS (ESI): Рассчитано для C13H18BrNO2S 331; Найдено 276 [М-56+1]+.
Шаг пять: трет-бутил-2-метил-4,6,7,8-тетрагидро-5H-тиено[3,2-с]азепин-5-карбоксилат (соединение 6)
Добавляли карбонат калия (2,5 г, 18 ммоль) в раствор соединения 5 (2 г, 6 ммоль) в 1,4-диоксане (100 мл), замещали азот три раза. Затем добавляли 2,4,6-триметил-1,3,5,2,4,6-триоксатриборинан (3 г, 24 ммоль) и тетракис(трифенилфосфин)палладий (2 г, 1,8 ммоль). Реакционную смесь выдерживали в течение ночи при температуре 120°С, фильтровали, концентрировали для удаления растворителя, а остаток очищали методом колоночной хроматографии с получением сырого соединения 6 (400 мг).
MS (ESI): Рассчитано для C14H21NO2S 267; Найдено 212 [М-56+1]+.
Шаг шесть: 2-метил-5,6,7,8-тетрагидро-4H-тиено[3,2-с]азепин (соединение 7)
Добавляли соляную кислоту (1,87 мл, 7,49 ммоль) в раствор соединения 6 (400 мг, 1,49 ммоль) в метаноле (20 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, концентрировали для удаления избытка соляной кислоты; добавляли насыщенный водный раствор бикарбоната натрия для корректировки значения рН выше 7. Смесь экстрагировали, высушивали и концентрировали, остаток подвергали очистке колоночной хроматографией с получением сырого соединения 7 (270 мг).
MS (ESI): Рассчитано для C9H13NS 167; Найдено 168 [М+Н]+.
Шаг семь: N-(2,6-диметил-4-(2-метил-4,6,7,8-тетрагидро-5H-тиено[3,2-с]азепин-5-ил)фенил)-3,3-диметилбутанамид (соединение 03045)
Промежуточный N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (528 мг, 1,78 ммоль) и трет-бутоксид натрия (620 мг, 6,5 ммоль) добавляли в раствор соединения 7 (270 мг, 1,62 ммоль) в трет-бутаноле (30 мл), замещали азот три раза. Затем добавляли метансульфоновой кислоты (2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)п алладий(II) (229 мг, 0,27 ммоль), реакционную смесь нагревали до температуры 95°С в защитной атмосфере азота и выдерживали всю ночь, фильтровали и концентрировали для удаления растворителя. Остаток подвергали колоночной хроматографии с получением сырого соединения 03045, который затем очищали препаративной хроматографии с получением соединения 03045 (15 мг, выход: 2,4%).
MS (ESI): Рассчитано для C23H32N2OS 384; Найдено 385 [М+Н]+.
1H ЯМР (400 МГц, MeOD):δ 7,04 (синглет, 2Н), 6,69 (синглет, 1H), 4,65 (синглет, 2Н), 3,96-3,92 (мультиплет, 2Н), 3,04-3,00 (мультиплет, 2Н), 2,39 (синглет, 3Н), 2,33 (синглет, 2Н), 2,25 (синглет, 6Н), 2,07 (синглет, 2Н), 1,15 (синглет, 9Н).
Пример 13: Получение соединения 03046

Шаг один: 2-метил-4,5,6,7-тетрагидро-8H-тиено[2,3-с]азепин-8-он (соединение 2)
Карбонат калия (2,5 г, 0,018 моль) добавляли в раствор соединения 1 (1,5 г, 0,006 моль) в 1,4-диоксане (50 мл), азот замещали три раза. Затем добавляли тетракис(трифенилфосфин)палладий (1,42 г, 0,0012 моль) и 2,4,6-триметил-1,3,5,2,4,6-триоксатриборинан (775 мг, 0,006 моль), реакционную смесь выдерживали при температуре 100°С в течение ночи, затем фильтровали, экстрагировали и концентрировали. Остаток подвергали колоночной хроматографии с получением сырого соединения 2 (1,5 г).
MS (ESI): Рассчитано для C9H11NOS 181; Найдено 182 [М+Н]+.
Шаг два: 2-метил-5,6,7,8-тетрагидро-4H-тиено[2,3-с]азепин (соединение 3)
При охлаждении льдом соединение 2 (1,5 г, 0,008 моль) медленно добавляли в суспензию алюмогидрида лития (0,9 г, 0,024 моль) в тетрагидрофуране (50 мл). Смесь перемешивали при температуре 70°С в течение 1 часа, затем последовательно медленно добавляли воду (1 мл), водный раствор гидроксида натрия (15%, 1 мл) и воду (3 мл), затем достаточное количество безводного сульфата магния. Смесь фильтровали, концентрировали, остаток очищали колоночной хроматографией с получением соединения 8 (500 мг, выход: 36,5%).
MS (ESI): Рассчитано для C9H13NS 167; Найдено 168 [М+Н]+.
Шаг три: N-(2,6-диметил-4-(2-метил-4,5,6,8-тетрагидро-7H-тиено[2,3-с]азепин-7-ил)фенил)-3,3-диметилбутанамид (соединение 03046)
Промежуточный N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (897 мг, 3 ммоль) и трет-бутоксид натрия (1,05 г, 10,9 ммоль) добавляли в раствор соединения 3 (500 мг, 2,7 ммоль) в трет-бутаноле (50 мл). После трехкратного замещения азота добавляли метансульфоновой кислоты (2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)п алладий(II) (390 мг, 0,46 ммоль). Реакционную смесь выдерживали в течение ночи при температуре 95°С, фильтровали и концентрировали для удаления растворителя. Остаток подвергали колоночной хроматографии с получением сырого соединения 03046, который затем очищали препаративной хроматографии с получением соединения 03046 (46 мг, выход: 4,3%).
MS (ESI): Рассчитано для C23H32N2OS 384; Найдено 385 [М+Н]+.
1H ЯМР (400 МГц, CD3OD): δ 6,79-6,74 (мультиплет 2Н), 4,68 (синглет, 2Н), 3,93-3,88 (мультиплет, 2Н), 2,84-2,79 (мультиплет, 2Н), 2,29 (синглет, 2Н), 2,17 (синглет, 6Н), 2,11 (синглет, 3Н), 1,95-1,87 (мультиплет, 2Н), 1,13 (синглет, 9Н).
Пример 14: Получение соединения 03049
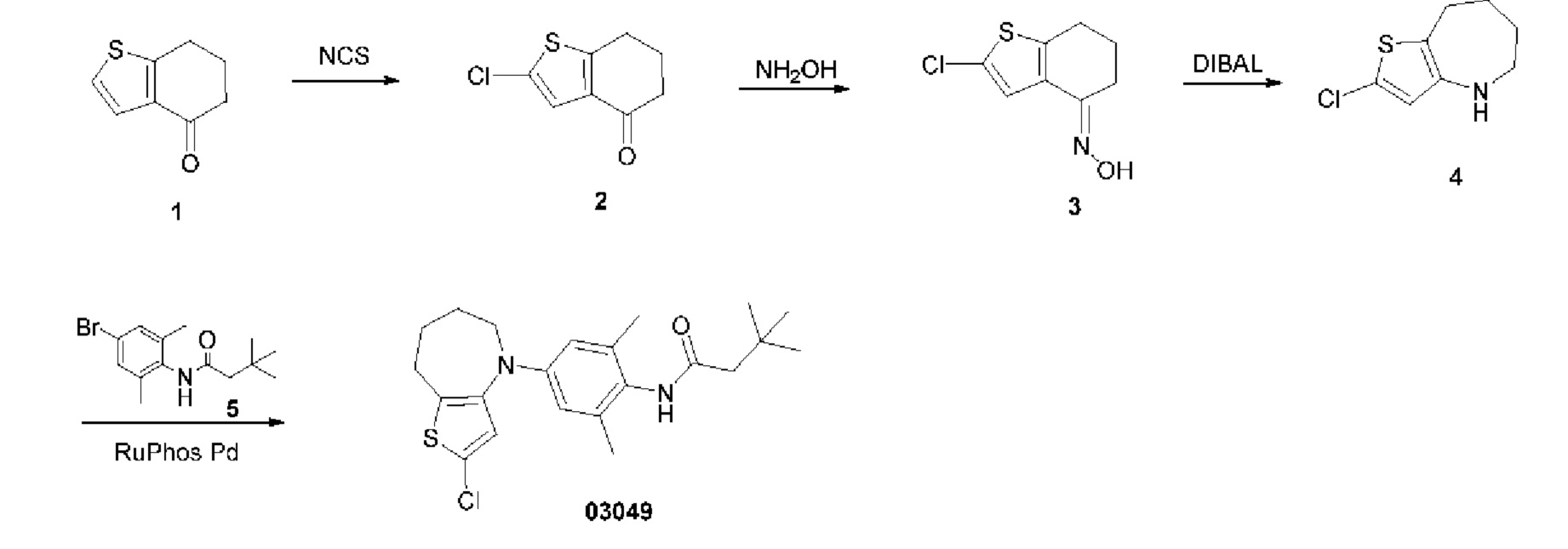
Шаг один: 2-хлор-6,7-дигидробензо[b]тиофен-4(5Я)-он (соединение 2)
Добавляли порциями N-хлорсукцинимид (3,2 г, 23,96 ммоль) в раствор 6,7-дигидробензо[b]тиофен-4(5H)-она (3,0 г, 19,74 моль) в уксусной кислоте (30 мл), реакционную смесь нагревали до 50°С и выдерживали в течение 16 часов. Смесь охлаждали до комнатной температуры, испаряли для удаления большей части растворителя, экстрагировали этилацетатом, осушали над безводным сульфатом натрия, концентрировали и подвергали колоночной хроматографии с получением соединения 2 (3,6 г, сырой продукт, выход: 100%).
MS (ESI): Рассчитано для C8H7ClOS 186; Найдено 187 [М+Н]+.
Шаг два: 2-хлор-6,7-дигидробензо[b]тиофен-4(5H)кетоксим (соединение 3)
Добавляли гидроксиламина гидрохлорид (3,9 г, 56,52 ммоль) и ацетат натрия (4,6 г, 56,52 ммоль) в смешанный раствор соединения 2 (3,6 г, 19,30 ммоль) в этаноле (50 мл) и воде (10 мл), затем кипятили смесь с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, концентрировали для удаления растворителя, а остаток очищали методом колоночной хроматографии с получением сырого соединения 3 (3,9 г выход: 100%).
MS (ESI): Рассчитано для C8H8ClNOS 201; Найдено 202 [М+Н]+.
Шаг три: 2-хлор-5,6,7,8-тетрагидро-4H-тиено[3,2-b]азепин (соединение 4)
В раствор соединения 3 (1,0 г, 4,90 ммоль) в дихлорметане (30 мл) по каплям добавляли раствор диизобутилалюминий гидрида в н-гексане (1 моль/л, 10 мл) на бане с ледяной водой. Смесь медленно нагревали до комнатной температуры в защитной атмосфере азота и перемешивали при комнатной температуре в течение 3 часов. Когда температура понижалась до 0°С, медленно добавляли воду (0,4 мл), водный раствор гидроксида натрия (15%, 0,4 мл) и воду (1,0 мл) в указанной последовательности, затем избыток безводного сульфата магния. Смесь фильтровали, концентрировали, остаток подвергали колоночной хроматографии с получением соединения 4 (340 мг, выход:: 36,6%).
MS (ESI): Рассчитано для C8H10ClNS 187; Найдено 188 [М+Н]+.
Шаг четыре: N-(4-(2-хлор-5,6,7,8-тетрагидро-4H-тиено[3,2-6]азепин-4-ил)-2,6-диметилфенил)-3,3-диметилбутанамид (соединение 03049)
В раствор соединения 5 (500 мг, 2,67 ммоль) в трет-бутаноле (20 мл) добавляли N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (1,19 г, 4,00 ммоль), трет-бутоксид (1,03 г, 10,70 ммоль), метансульфоновой кислоты (2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)палладий(II) (224 мг, 0,27 ммоль) в защитной атмосфере азота. Смесь выдерживали при температуре 90°С в течение шестнадцати часов. После охлаждения смеси до комнатной температуры и фильтрации фильтрат концентрировали под вакуумом для удаления растворителя. Остаток очищали колоночной хроматографией с получением сырого продукта, который затем отделяли препаративной хроматографией с получением соединения 03049 (27,02 мг, выход: 2,5%).
MS (ESI): Рассчитано для C22H29CIN2 OS 404; Найдено 405 [М+Н]+.
Н ЯМР (400 МГц, CD3OD): δ 6,67 (синглет, 1H), 6,50 (синглет, 2Н), 3,71-3,68 (мультиплет, 2Н), 2,71-2,68 (мультиплет, 2Н), 2,30 (синглет, 2Н), 2,15 (синглет, 6Н), 1,90-1,86 (мультиплет, 2Н), 1,72-1,71 (мультиплет, 2Н), 1,15 (синглет, 9Н).
Пример 15: Получение соединения 03058
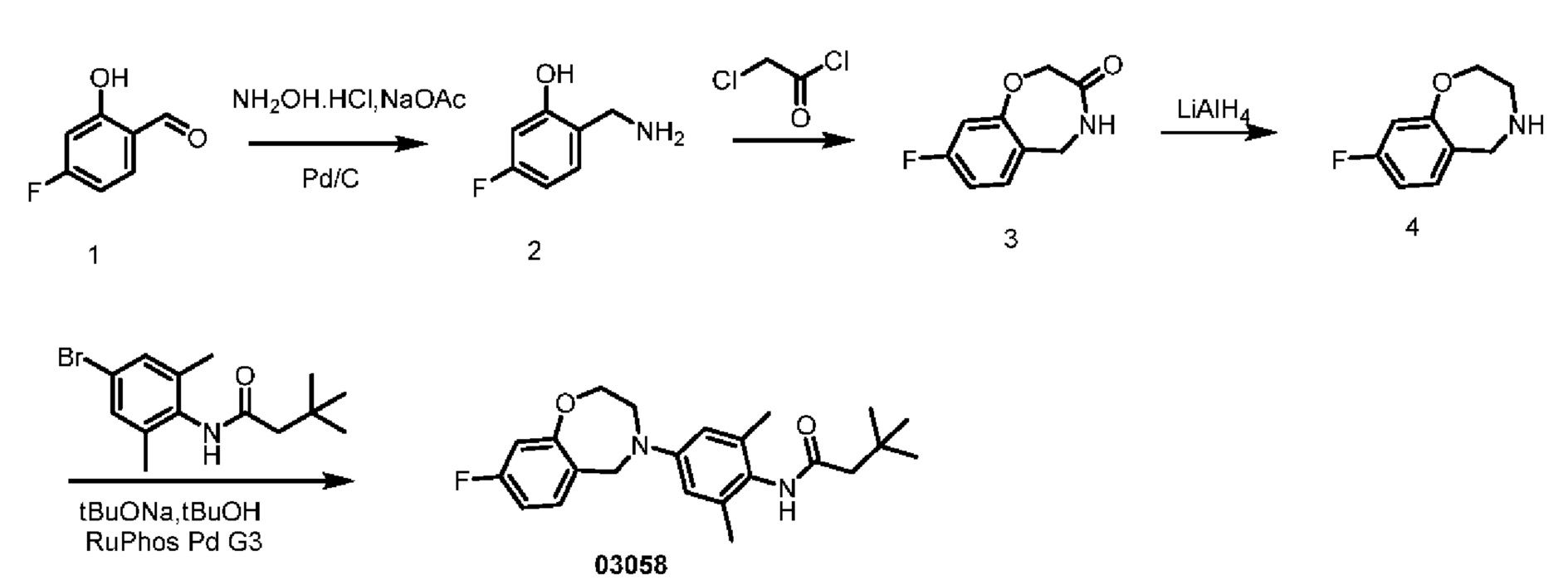
Шаг один: 2-(аминометил)-5-фторфенол (соединение 2)
Добавляли гидроксиламина гидрохлорид (1,5 г, 21,59 ммоль) и ацетат натрия (2,34 г, 28,54 ммоль) в раствор 4-фтор-2-гидроксибензальдегида (1,0 г, 7,14 ммоль) в этаноле (50 мл), затем кипятили смесь с обратным холодильником в течение двух часов. Смесь охлаждали до комнатной температуры, концентрировали для удаления растворителя, экстрагировали этилацетатом, высушивали над безводным сульфатом натрия, фильтровали, концентрировали для удаления растворителя с получением сырого продукта. Сырой продукт растворяли в этаноле (50 мл), добавляли концентрированную соляную кислоту (3,5 мл) и 10% палладиевый катализатор на углеродной основе (300 мг). Смесь перемешивали при комнатной температуре в атмосфере азота под давлением 0,4 МПа в течение 16 часов. рН фильтрата корректировали до значения 8 с помощью бикарбоната натрия. После концентрирования остаток подвергали колоночной хроматографии с получением соединения 2 (1,0 г, выход: 100%).
MS (ESI): Рассчитано для C7H8FNO 141; Найдено 125 [M-NH2]+.
Шаг два: 8-фтор-4,5-дигидробензо[f][1,4]оксазепин-3(2H)он (соединение 3)
В раствор соединения 2 (100 мг, 0,71 ммоль) в ацетонитриле (10 мл) последовательно добавляли хлорацетилхлорид (88 мг, 0,78 ммоль), карбонат калия (294 мг, 2,13 ммоль) и тетрабутиламмонийбромид (23 мг, 0,071 ммоль). Реакционную смесь нагревали до температуры 80°С в защитной атмосфере азота и перемешивали в течение 16 часов. Смесь охлаждали до комнатной температуры, фильтровали на нутч-фильтре, фильтрат концентрировали под вакуумом и очищали колоночной хроматографией с получением соединения 3 (80 мг, выход: 62,5%).
MS (ESI): Рассчитано для C9H8FNO2 181; Найдено 182 [М+Н]+.
Шаг три: 8-фтор-2,3,4,5-тетрагидробензо[f][1,4]оксазепин (соединение 4)
Соединение 3 (800 мг, 2,99 ммоль) добавляли в суспензию алюмогидрида лития (420 мг, 11,05 моль) в тетрагидрофуране (20 мл). Смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере азота. Реакцию в смеси останавливали водой (0,42 мл), 15% раствором гидроксида натрия в воде (0,42 мл) и водой (1,26 мл), высушивали над безводным сульфатом магния и фильтровали на нутч-фильтре. Отфильтрованный осадок промывали дихлорметаном, фильтрат концентрировали и очищали колоночной хроматографией с получением соединения 4 (583 мг, выход: 79,0%).
MS (ESI): Рассчитано для C9H10FNO 167; Найдено 168 [М+Н]+.
Шаг четыре: N-(4-(8-фтор-2,3-дигидробензо[f][1,4]оксазепин-4(5H)-ил)-2,6-диметилфенил)-3,3-диметилбутанамид (соединение 03058)
В раствор соединения 4 (583 мг, 3,49 ммоль) в трет-бутаноле (20 мл) добавляли N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (1,04 г, 3,49 ммоль), трет-бутоксид натрия (1,34 г, 13,97 ммоль), метансульфоновой кислоты (2-дициклогексилфосфин-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)палладий (II) (292 мг, 0,35 ммоль). Смесь выдерживали при температуре 90°С в течение шестнадцати часов. После охлаждения смеси до комнатной температуры и фильтрации фильтрат концентрировали под вакуумом для удаления растворителя. Остаток очищали колоночной хроматографией с получением сырого продукта, который затем отделяли препаративной хроматографией с получением соединения 03058 (78,91 мг, выход: 5,9%).
MS (ESI): Рассчитано для C23H29FN2O2 384; Найдено 385 [М+Н]+.
Н ЯМР (400 МГц, CD3OD): δ 7,41-7,31 (мультиплет, 1H), 6,75-6,61 (мультиплет, 4Н), 4,61 (синглет, 2Н), 4,16-4,15 (мультиплет, 2Н), 3,90-3,88 (мультиплет, 2Н), 2,27 (синглет, 2Н), 2,14 (синглет, 6Н), 1,14 (синглет, 9Н).
Пример 16: Получение соединения 03059
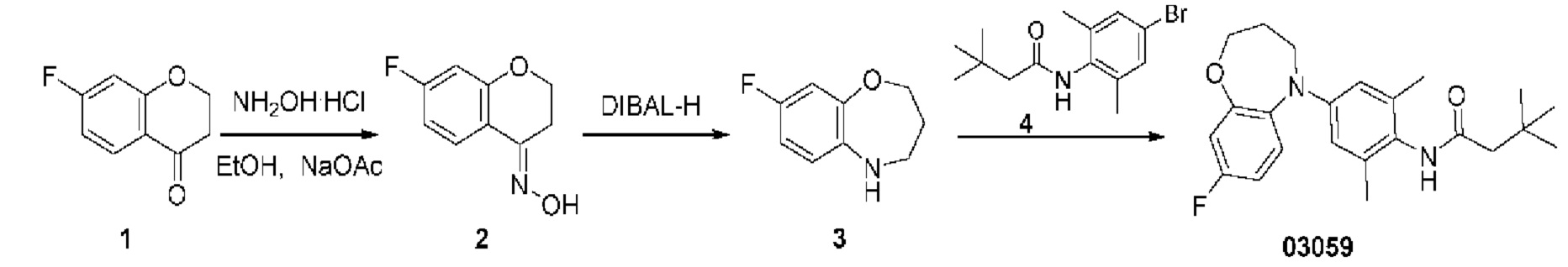
Шаг один: 7-фторхроман-4-оксим (соединение 2)
При комнатной температуре последовательно добавляли ацетат натрия (1,6 г, 19,7 ммоль), гидроксиламин гидрохлорид (1,3 г, 19,7 ммоль) и воду (2 мл) в раствор 7-фторхроман-4-она (1,0 г, 6,5 ммоль) в этаноле (10 мл). Реакционную смесь нагревали до температуры 90°С и выдерживали в течение 2 часов. Смесь охлаждали до комнатной температуры, разбавляли водой, экстрагировали этилацетатом, высушивали над безводным сульфатом натрия, фильтровали, концентрировали и очищали методом флэш-хроматографии с получением твердого соединения 2 (1,10 г), выход: 93,4%).
MS (ESI): Рассчитано для C9H8FNO2 181; Найдено 182 [М+Н]+.
Шаг два: 8-фтор-2,3,4,5-тетрагидробензо[b][1,4]оксазепин (соединение 3)
В ледяной бане медленно добавляли раствор диизобутилалюминий гидрида (1 N, 30,37 мл) в раствор соединения 2 (1,1 г, 6,07 ммоль) в дихлорметане (30 мл). Смесь перемешивали при комнатной температуре в защитной атмосфере азота в течение 5 часов, затем последовательно добавляли воду (1,2 мл), 10% раствор гидроксида натрия (1,2 мл) и воду (3,6 мл), перемешивали при комнатной температуре в течение 30 минут и фильтровали. Отфильтрованный осадок промывали этилацетатом, фильтрат экстрагировали этилацетатом, промывали насыщенным рассолом, высушивали с помощью безводного сульфата натрия, концентрировали и очищали методом флэш-хроматографии с получением соединения 3 (0,9 г, выход: 88,3%).
MS (ESI): Рассчитано для C9H10FNO 167; Найдено 168 [М+Н]+.
Шаг три: N-(4-(8-фтор-3,4-дигидробензо[b][1,4]оксазепин-5(2H)-ил)-2,6-диметилфенил)-3,3-диметилбутанамид (соединение 03059)
Ruphos-Pd-G3 (150,5 мг, 0,18 ммоль), трет-бутоксид натрия (695 мг, 7,16 ммоль) и соединение 4 (586 мг, 1,79 ммоль) добавляли в раствор соединения 3 (300 мг, 1,79 ммоль) в трет-бутаноле (10 мл). Смесь перемешивали при температуре 80°С в течение ночи. Смесь охлаждали до комнатной температуры, фильтровали, промывали этилацетатом и концентрировали. Осадок очищали флэш-хроматографией (этилацетат/петролейный эфир=1 / 5) с получением светло-желтого соединения 03059 (470,08 мг, выход: 78,5%).
MS (ESI): Рассчитано для C23H29FN2O2 384; Найдено 385 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 7,10-7,02 (мультиплет, 1H), 6,75-6,66 (мультиплет, 1H), 6,68-6,60 (мультиплет, 1H), 6,57 (синглет, 2Н), 6,50 (синглет, 1H)), 4,12 (триплет, J=6,0 Гц, 2Н), 3,87 (триплет, J=6,0 Гц, 2Н), 2,29 (синглет, 2Н), 2,17 (синглет, 6Н), 2,17-2,07 (мультиплет, 2Н), 1,16 (синглет, 9Н).
Пример 17: Получение соединения 03063
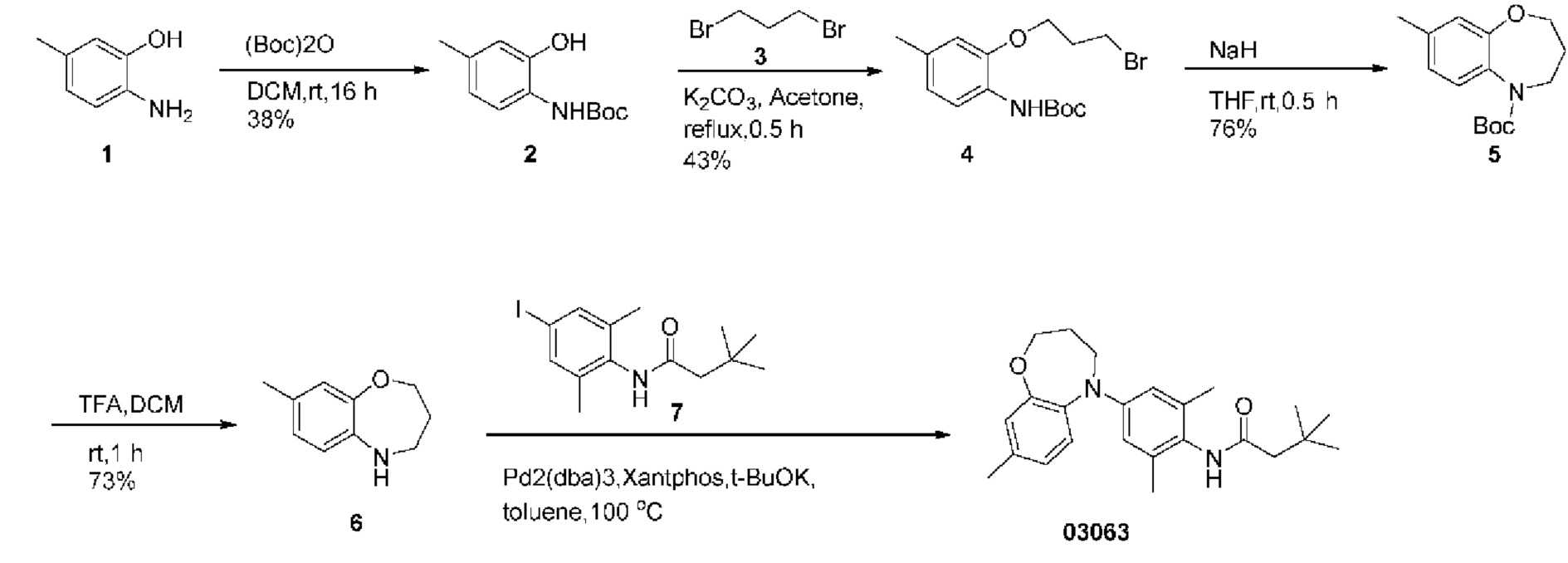
Шаг один: (2-гидрокси-4-метилфенил)третбутилкарбамат (соединение 2) Добавляли ВОС2О (10,63 г, 48,717 ммоль) и триэтиламин (14,79 г, 146,151 ммоль) в раствор 2-амино-5-метилфенола 1 (6 г, 48,717 ммоль) в дихлорметане (60 мл). Смесь перемешивали при комнатной температуре в течение ночи. Смесь промывали водой и насыщенным раствором NaCl, высушивали над Na2SO4, концентрировали и очищали колоночной хроматографией (н-гексан/этилацетат = 10:1) с получением сложного трет-бутилового эфира (2-гидрокси-4-метилфенил)карбамата 2 (4,2 г, 38%).
LCMS: [M+Na]+=246,2
Шаг два: трет-бутил(2-(3-бромпропокси)-4-метилфенил)карбамат (соединение 4)
Добавляли 1,3-дибромпропан (3,1 г, 15,695 ммоль) и K2CO3 (4,96 г, 35,874 ммоль) в раствор трет-бутил(2-гидрокси-4-метилфенил)карбамата 2 (1 г, 4,484 ммоль) в ацетоне (30 мл). Смесь перемешивали при температуре 75°С в течение 0,5 часа. После охлаждения при комнатной температуре смесь разбавляли этилацетатом и фильтровали. Фильтрат концентрировали и очищали колоночной хроматографией (н-гексан/этилацетат = 15:1) с получением трет-бутил(2-(3-бромпропокси)-4-метилфенил)карбамата 4 (660 мг, 43%) в виде бесцветного масла.
LCMS: [M+Na]+=366,1.
Шаг три: Сложный трет-бутиловый эфир 8-метил-3,4-дигидробензо[b][1,4]оксазепин-5(2Н)-карбоновой кислоты (соединение 5) в виде твердого вещества белого цвета.
Добавляли NaH (60%, 307 мг, 7,674 ммоль) в раствор трет-бутил(2-(3-бромпропокси)-4-метилфенил)карбамата 4 (660 мг, 1,918 ммоль) в тетрагидрофуране (20 мл); смесь перемешивали при комнатной температуре в течение получаса. Смесь выливали в ледяную воду, экстрагировали этилацетатом (2×20 мл), промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (н-гексан/этилацетат = 15:1) с получением сложного трет-бутилового эфира 8-метил-3,4-дигидробензо[b][1,4]оксазепин-5(2Н)-карбоновой кислоты 5 (385 мг, 76%) в виде твердого вещества белого цвета.
LCMS: [M+Na]+=286,2
Шаг четыре: 8-метил-2,3,4,5-тетрагидробензо[b][1,4]оксазепин (соединение 6)
Добавляли 4 мл трифторуксусной кислоты в раствор сложного трет-бутилового эфира 8-метил-3,4-дигидробензо[b][1,4]оксазепин-5(2Н)-карбоновой кислоты 5 (385 мг, 1,464 ммоль) в дихлорметане (4 мл); смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали и добавляли 30 мл дихлорметана для растворения осадка. Раствор промывали насыщенным раствором NaHCO3 и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (н-гексан/этилацетат = 7:3) с получением 8-метил-2,3,4,5-тетрагидробензо[b][1,4]оксазепина 6 (175 мг, 73%) в виде коричневого масла.
LCMS: [М+Н]+=164,2.
Шаг пять: N-(2,6-диметил-4-(8-метил-3,4-дигидробензо[b][1,4]оксазепин-5(2Н)-ил)фенил)-3,3-диметилбутанамид (03063)
N-(4-йод-2,6-диметилфенил)-3,3-диметилбутанамид 7 (1,12 г, 3,25 ммоль), Pd2(dba)3 (229 мг, 0,25 ммоль), ксантфос (289 мг, 0,50 ммоль) и t-BuOK (842 мг, 7,50 ммоль) добавляли в раствор 8-метил-2,3,4,5-тетрагидробензо[b][1,4]оксазепина 6 (408 мг, 2,50 ммоль) в толуоле (25 мл). Смесь перемешивали при температуре 100°С в течение 16 часов в атмосфере азота. Смесь фильтровали, фильтрат концентрировали и очищали колоночной хроматографией (дихлорметан/метанол = 30:1) с получением желтого масла. Очищали методом препаративной ВЭЖХ (колонка: Kromasil-C18 100×21,2 мм, 5 мкм подвижная фаза: ACN-H2O (0,05% NH3) градиент: 35-45) с получением целевого соединения 03063 (30,4 мг, 0,08 ммоль, 3%) в виде твердого вещества грязно-белого цвета.
LCMS: [М+Н]+=381,3.
1Н NMR (400 МГц, DMSO) δ 8,83 (синглет, 1H), 7,03-6,74 (мультиплет, 3Н), 6,48 (синглет, 2Н), 3,98 (синглет, 2Н), 3,78 (синглет, 2Н), 2,31-1,97 (мультиплет, 13Н), 1,05 (синглет, 9Н).
Пример 18: Получение соединения 03066

Шаг один: 2-хлор-N-(2-(гидроксиметил)-4-метилфенил)ацетамид (соединение 3)
В 50 мл дихлорметана растворяли 2-амино-5-метилбензиловый спирт 1 (3,0 г, 21,9 ммоль), 2-хлорацетилхлорид (2,7 г, 24 ммоль) и DIEA (5,66 г, 43,8 ммоль), затем смесь перемешивали при комнатной температуре в течение 2 часов. Реакцию в растворе останавливали насыщенным раствором бикарбоната натрия. Органическую фазу отделяли и промывали водой (2×50 мл) и насыщенным раствором бикарбоната натрия (2×50 мл), высушивали над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (н-гексан/этилацетат = 5/1) с получением 2-хлор-N-(2-(гидроксиметил)-4-метилфенил)ацетамида 3 (3,6 г, 77%) в виде твердого вещества желтого цвета.
LCMS: [M+H]+=196,1
Шаг два: 7-метил-1,5-дигидробензо[е][1,4]оксазепин-2(3Н)он (соединение 4)
При температуре 0°С добавляли NaH (600 мг, 25 ммоль) в раствор 2-хлор-N-(2-(гидроксиметил)-4-метилфенил)ацетамида 3 (2,6 г, 12,5 ммоль) в тетрагидрофуране (60 мл), смесь перемешивали при температуре 0°С в течение 1 часа. Затем реакцию останавливали добавлением воды, а затем добавляли этилацетат (2×50 мл). Органическую фазу промывали водой (2×50 мл) и насыщенным раствором бикарбоната натрия (2×50 мл), высушивали над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (н-гексан/этилацетат = 3/1) с получением 7-метил-1,5-дигидробензо[е][1,4]оксазепин-2(3Н)она 4 (2,1 г, 94%) в виде твердого вещества желтого цвета.
LCMS: [М+Н]+=178,1
Шаг три: 7-метил-1,2,3,5-тетрагидробензо[Е][1,4]оксазепин (соединение 5)
Медленно добавляли LiAlH4 (350 мг, 9,15 ммоль) в раствор 7-метил-1,5-дигидробензо[е][1,4]оксазепин-2(3Н)он 4 (810 мг, 4,57 ммоль) в тетрагидрофуране при температуре 0°С, реакционную смесь кипятили с обратным холодильником в течение 1 часа. Раствор охлаждали до 0°С и медленно добавляли тетрагидрофуран и раствор Na2SO4. Смесь перемешивали в течение 10 минут, затем добавляли раствор Na2SO4. Смесь фильтровали, органическую фазу концентрировали и очищали колоночной хроматографией (н-гексан/этилацетат = 5/1) с получением 7-метил-1,2,3,5-тетрагидробензо[е][1,4]оксазепин 5 (600 мг, 80%) в виде твердого вещества желтого цвета.
LCMS: [М+Н]+=164,1
Шаг четыре: N-(2,6-диметил-4-(7-метил-2,3-дигидробензо[е][1,4]оксазепин-1(5Н)-ил)фенил)-3,3-диметил бутанамид (соединение 03066)
Раствор соединения 5 (300 мг, 1,84 ммоль), соединение 6 (762 мг, 2,2 ммоль), Pd2(dba)3 (165 мг, 0,18 ммоль), X-Phos (213 мг, 0,37 ммоль) и Cs2CO3 (1,2 г, 3,68 ммоль) в толуоле (15 мл) перемешивали при температуре 110°С в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры в смесь добавляли 10 мл этилацетата, и полученную смесь промывали насыщенным раствором хлорида натрия (2×20 мл) и высушивали с помощью безводного сульфата натрия. Раствор концентрировали и очищали препаративной ВЭЖХ (0,1% FA) с получением соединения 03066 (55 мг, 8%) в виде твердого вещества белого цвета.
LCMS: [М+Н]+=381,2
1H ЯМР (400 МГц, ДМСО) δ 8,82 (широкий синглет, 1H), 7,20 (синглет, 1H), 7,11 (двойной дублет, J=8,4, 2,0 Гц, 1Н), 7,01 (дублет, J=8,0 Гц, 1Н), 6,40 (синглет, 2Н), 4,42 (синглет, 2Н), 3,74-3,70 (мультиплет, 4Н), 2,30 (синглет, 3Н), 2,16 (синглет, 2Н), 2,01 (синглет, 6Н), 1,04 (синглет, 9Н).
Пример 19: Получение соединения 03060
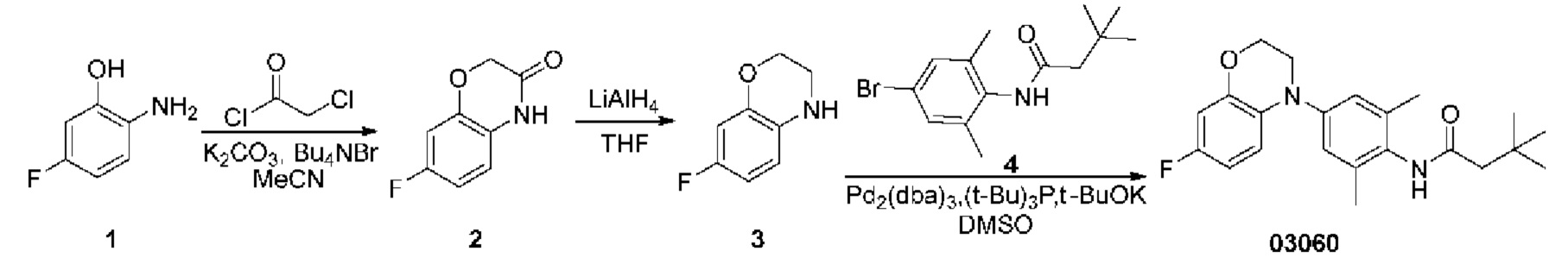
Шаг один: 7-фтор-2H-бензо[1,4]оксазин-3(4H)-он (соединение 2)
Хлорацетилхлорид (490 мг, 4,33 ммоль), карбонат калия (1,63 г, 11,82 ммоль) и тетрабутиламмонийбромид (126 мг, 0,39 ммоль) последовательно добавляли в раствор 2-амино-5-фторфенола (500 мг, 3,94 ммоль) в ацетонитриле (5 мл). Реакционную смесь нагревали до температуры 65°С в защитной атмосфере азота и перемешивали в течение 15 часов. Смесь охлаждали до комнатной температуры, фильтровали на нутч-фильтре, фильтрат концентрировали под вакуумом и очищали колоночной хроматографией с получением желтого маслянистого соединения (500 мг, выход: 76,05%).
MS (ESI): Рассчитано для C8H6FNC2 167; Найдено 168 [М+Н]+.
Шаг два: 7-фтор-3,4-дигидро-2H-бензо[1,4]оксазин (соединение 3)
Соединение 2 (500 мг, 2,99 ммоль) добавляли в суспензию алюмогидрида лития (284 мг, 7,48 ммоль) в тетрагидрофуране (5 мл). Смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере азота. Реакцию в системе останавливали водой и 15% водным раствором гидроксида натрия. Смесь фильтровали, фильтрат экстрагировали этилацетатом, концентрировали и очищали колоночной хроматографией с получением соединения 3 в виде желтого масла (252 мг, выход: 55,01%).
MS (ESI): Рассчитано для C8H8FNO 153; Найдено 154 [М+Н]+.
Шаг три: N-(2,6-диметил-6-(1-трет-бутилацетил)анилин))4-(7-фтор-2,3-дигидро-4H-бензо[1,4]оксазин (соединение 03060)
Добавляли соединение 4 (532 мг, 1,79 ммоль), Pd2(dba)3 (126,6 мг, 0,16 ммоль) и трет-бутоксид натрия (632,4 мг, 6,52 ммоль) в раствор соединения 3 (250 мг, 1,63 ммоль) в трет-бутаноле (5 мл). Смесь выдерживали при температуре 85°С в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали, фильтрат концентрировали под вакуумом. Остаток очищали методом препаративной ВЭЖХ с получением белого твердого соединения 03060 (46,40 мг, выход: 7,67%).
MS (ESI): Рассчитано для C22H27FN2O2 370; Найдено 371 [М+Н]+.
Н ЯМР (400 МГц, CD3Cl3): δ 6,87-6,90 (мультиплет, 3Н), 6,66-6,48 (мультиплет, 3Н), 4,25 (триплет, J=4,0 Гц, 2Н), 3,67 (синглет, 2Н), 2,33 (синглет, 2Н), 2,23 (синглет, 6Н), 1,18 (синглет, 9Н).
Пример 20: Получение соединения 03037
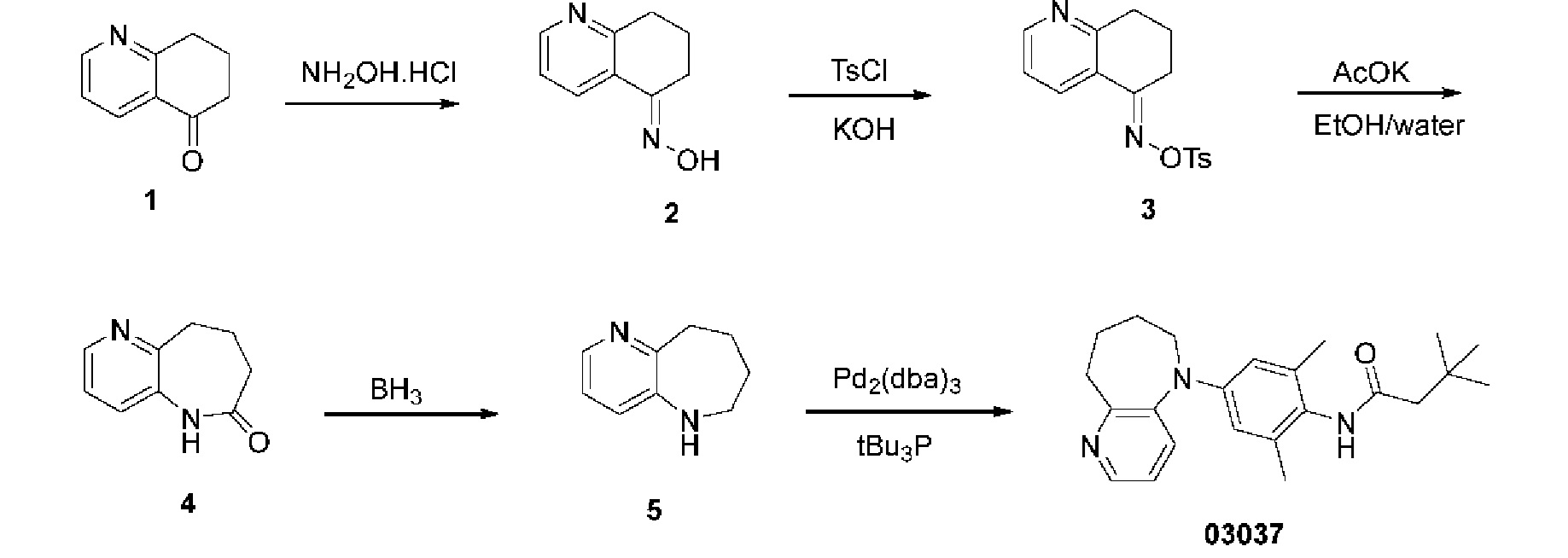
Шаг один: (Е)-7,8-дигидрохинолин-5(6H)-оноксим (соединение 2)
Добавляли гидроксиламина гидрохлорид (1,13 г, 16,29 ммоль) и ацетат натрия (1,34 г, 16,29 ммоль) в смешанный раствор 7,8-дигидрохинолин-5(6H)-она (1,0 г, 6,79 ммоль) в метаноле/воде (40 мл/6 мл), затем смесь кипятили с обратным холодильником в течение двух часов. Смесь охлаждали до комнатной температуры, концентрировали для удаления растворителя, добавляли воду и фильтровали. Отфильтрованный осадок высушивали с получением соединения 2 (1,1 г сырого продукта, выход: 100%).
MS (ESI): Рассчитано для C9H10N2O 162; Найдено 163 [М+Н]+.
Шаг два: (E)-7,8-дигидрохинолин-5(6H)-он-п-толуолсульфонилоксим (соединение 3)
Добавляли гидроксид калия (381 мг, 6,79 ммоль) и воду (10 мл) в раствор соединения 2 (1,10 г, 6,79 ммоль) в ацетоне (25 мл), затем добавляли п-толуолсульфонилхлорид (1,94 г, 10,18 ммоль). Смесь кипятили с обратным холодильником в течение 1 часа. Смесь охлаждали до комнатной температуры, концентрировали для удаления растворителя. Добавляли воду, фильтровали, отфильтрованный осадок высушивали с получением соединения 3 (2,1 г сырого продукта, выход: 100%).
MS (ESI): Рассчитано для C16H16N2O3S 316; Найдено 317 [М+Н]+.
Шаг три: 5,7,8,9-тетрагидро-6H-пиридо[3,2-b]азепин-6-он (соединение 4)
Ацетат калия (1,5 г, 15,28 ммоль) добавляли в смешанный раствор соединения 3 (2,1 г, 6,65 ммоль) в этаноле/воде (20 мл / 40 мл). Смесь кипятили с обратным холодильником в течение шестнадцати часов. Смесь охлаждали до комнатной температуры, концентрировали для удаления растворителя. Добавляли воду и корректировали значение рН до 10 с помощью 5 н гидроксида натрия. Реакционную смесь экстрагировали дихлорметаном, высушивали над безводным сульфатом натрия, фильтровали, концентрировали для удаления растворителя. Сырой продукт подвергали колоночной хроматографии с получением соединения 4 (750 мг, выход: 75,0%).
MS (ESI): Рассчитано для C9H10N2O 162; Найдено 163 [M+H]+.
Шаг четыре: 6,7,8,9-тетрагидро-5H-пиридо[3,2-b]азепин (соединение 5)
В раствор соединения 4 (750 мг, 4,63 ммоль) в тетрагидрофуране (30 мл) по каплям добавляли раствор борана в тетрагидрофуране (1 моль/л, 14 мл). Смесь непрерывно перемешивали в течение 0,5 часа, затем нагревали до температуры 60°С и выдерживали в течение 2 часов. Смесь охлаждали до комнатной температуры, реакцию останавливали метанолом, смесь концентрировали под вакуумом для удаления растворителя. Остаток очищали методом колоночной хроматографии с получением соединения 5 (400 мг выход: 58,0%).
MS (ESI): Рассчитано для C9H12N2 148; Найдено 149 [М+Н]+.
Шаг пять: N-(2,6-диметил-4-(6,7,8,9-тетрагидро-5H-пиридо[3,2-6]азепин-5-ил)фенил)-3,3-диметилбутанамид (соединение 03037)
В атмосфере азота добавляли N-(4-бром-2,6-диметилфенил)-3,3-диметилбутанамид (160 мг, 0,54 ммоль), Pd2(dba)3 (25 мг, 0,034 ммоль), раствор три-трет-бутилфосфина в н-гексане (0,1 мл) и трет-бутоксид калия (61 мг, 0,54 ммоль) в раствор соединения 5 (40 мг, 0,27 ммоль) в ДМСО (2 мл). Смесь выдерживали в микроволновом реакторе при температуре 150°С в течение одного часа. После охлаждения смеси до комнатной температуры реакцию прерывали водой, смесь экстрагировали этилацетатом, высушивали над безводным сульфатом натрия, концентрировали под вакуумом для удаления растворителя. Остаток очищали колоночной хроматографией с получением сырого продукта, который далее очищали препаративной хроматографией с получением соединения 03030 (13,59 мг, выход: 11,0%).
MS (ESI): Рассчитано для C23H31N3O 365; Найдено 366 [М+Н]+.
Н ЯМР (400 МГц, CD3OD): 3 8,29 (синглет, 1H), 7,97-7,93 (мультиплет, 1H), 7,66-7,64 (мультиплет, 1H), 6,85 (синглет, 2Н), 3,90-3,88 (мультиплет, 2Н)), 3,33-3,25 (мультиплет, 2Н), 2,34 (синглет, 2Н), 2,21 (синглет, 6Н), 1,99-1,95 (мультиплет, 4Н), 1,16 (синглет, 9Н).
Определение активности вещества для открывания ионных каналов (обнаружение FDSS/μCELL)
1. Экспериментальный метод:
1.1 Экспериментальный процесс
Подготовка клеток: Клетки CHO-KCNQ2 выращивали в колбе для выращивания культур объемом 175 см2. Когда клетки вырастали до плотности 60-80%, удаляли питательную среду, промывали один раз 7 мл PBS (физиологическим раствором с фосфатным буфером), затем добавляли 3 мл 0,25% трипсина для гидролиза. После завершения гидролиза добавляли 7 мл питательного раствора (90% DMEM/F12 + 10% FBS + 500 мкг/мл G418) для нейтрализации, затем центрифугировали в течение 3 минут со скоростью вращения 800 об/мин. Надосадочную жидкость отсасывали, затем добавляли 5 мл питательной среды для повторного суспендирования, затем подсчитывали количество клеток.
Посев клеток: Регулировали плотность клеток до 3×104/лунка в соответствии с результатами подсчета клеток. После выдержки при комнатной температуре в течение 30 минут клетки помещали в СО2 инкубатор с температурой 37°С и выдерживали в течение ночи на 16-18 часов. Плотность клеток достигала прибл. 80%.
Инкубация флуоресцентного красителя: Среду для культивирования клеток удаляли, добавляли загрузочный буфер 80 мкл/лунка, затем клетки инкубировали в темноте при комнатной температуре в течение 60 минут.
Инкубация соединения: удаляли загрузочный буфер, добавляли приготовленный раствор соединения в количестве 80 мкл/лунка, инкубировали в темноте при комнатной температуре в течение 20 минут.
Сбор флуоресцентных данных: Для записи сигналов использовали прибор FDSS/μCELL для измерения флуоресценции в реальном времени, в котором длина волны возбуждения составляла 480 нм, длина волны излучения составляла 540 нм, а сигналы записывались 1 раз в секунду. После записи базовой линии в течение 10 секунд начинали добавлять симулирующий буфер 20 мкл/лунка, а сигнал непрерывно записывали до конца 180 секунд.
1.2 Приготовление растворов




Вышеуказанный буфер был взят из серийно производимого набора для анализа каналов ионов калия FluxOR от изготовителя Invitrogen, каталожный №F10017, номер партии 913728.
1.3 Подготовка соединения
Готовили маточный раствор соединения в ДМСО 20 ммоль/л. 10 мкл маточного раствора с концентрацией 20 ммоль/л отбирали и добавляли в 20 мкл ДМСО, раствор последовательно разбавляли 3 раза с получением 8 промежуточных концентраций. Затем отбирали среднюю концентрацию соединения в тестовый буфер, разбавление в 200 раз для получения конечной концентрации для анализа. Отбирали 80 мкл и добавляли в испытательный планшет.
Наивысшая тестовая концентрация составляла 100 мкмоль/л, за ней следовали 100; 33,33; 11,11; 3,70; 1,23; 0,41; 0,137; 0,045 мкмоль/л, всего 8 значений концентрации. Каждое значение концентрации повторяют в 3 лунки.
Содержание ДМСО в конечной концентрации для анализа не превышало 0,5%. Концентрация ДМСО не влияла на калиевый канал KCNQ2.
1.4 Анализ данных
Экспериментальные данные были обработаны программами Excel 2007 и GraphPad Prism 5.0, и для расчета эффекта возбуждения было вычислено уровень в 180 секунд Эффект возбуждения соединения рассчитывали по следующей формуле:

1.5 Контроль качества
Условия окружающей среды: Температура около 25°С
Реагенты: Набор для определения FluxORTM (Invitrogen, кат. №F0017)
Экспериментальные данные в протоколе могут соответствовать следующим критериям: коэффициент Z'>0,5
2. Результаты определения: подробности см. в Таблице 1, в которой чем меньше было ЕС50, тем была выше активность соответствующего соединения.
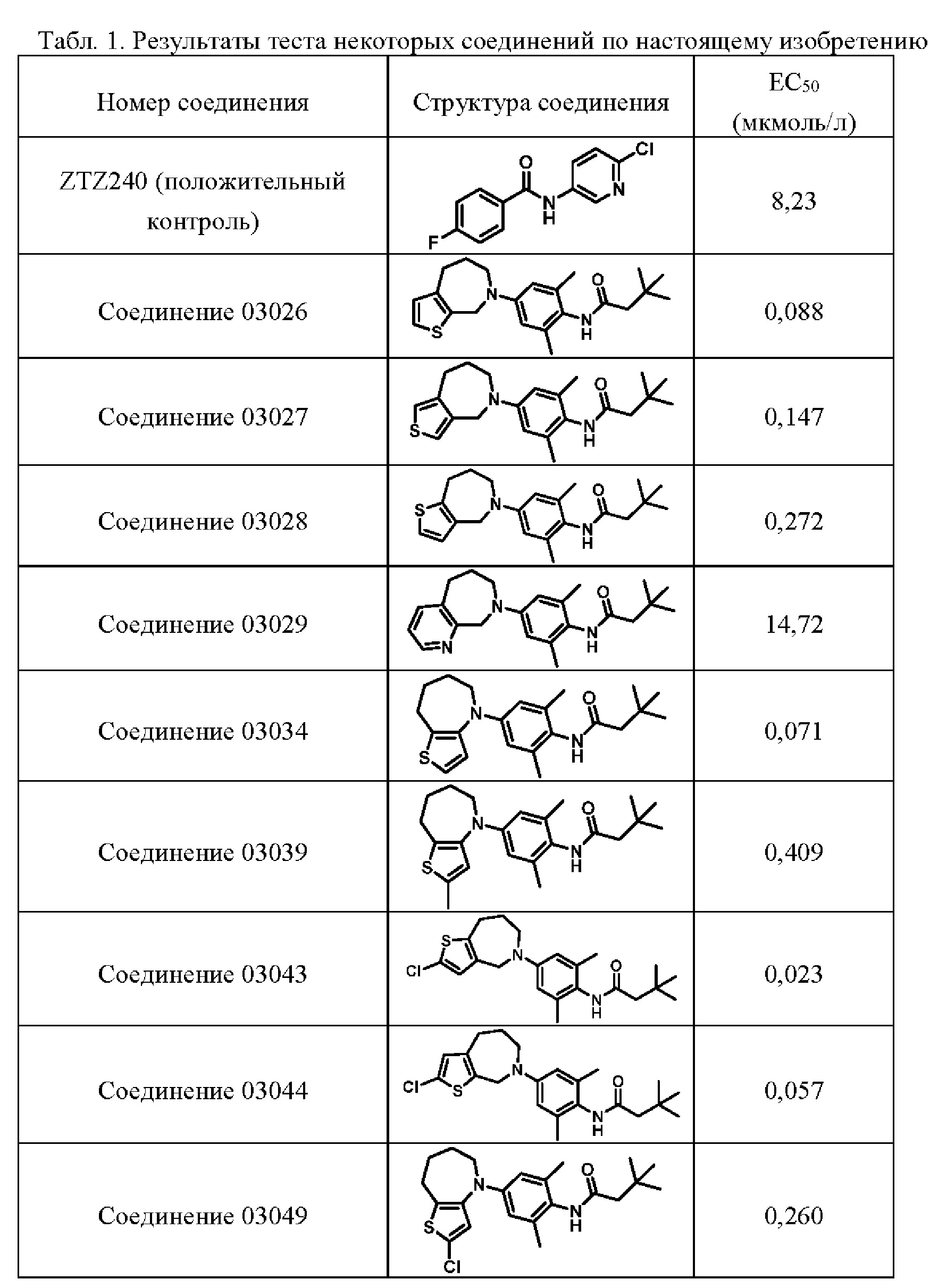

Ссылки на вышеописанные методы испытаний:
(1) Zhaobing Gao et al. Journal of Biological Chemistry. 2010, 285(36): 28322-28332.
(2) Jinfeng Yue et al. Acta Pharmacologica Sinica. 2016, 37:105 110.
Из Таблицы 1 можно увидеть:
1) сравнивая соединение А (структурная формула:
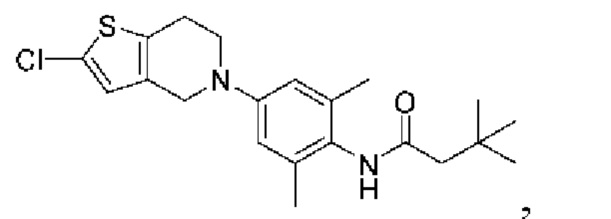
ЕС50=0,065 мкмоль/л) и соединение 03043 (структурная формула:
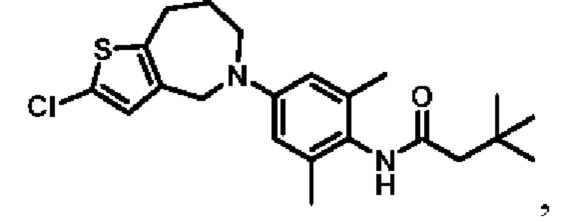
ЕС50=0,023 мкмоль/л), можно видеть, что после преобразования азотсодержащего 6-членного кольца в 7-членное активность полученного соединения значительно увеличилась прибл. в 2,83 раза (=0,065/0,023);
2) сравнивая соединение В (структурная формула:

ЕС50=0,098 мкмоль/л) и соединение 03058 (структурная формула:
ЕС50=0,008 мкмоль/л), можно видеть, что после преобразования азот-содержащего 6-членного гетероцикла в 7-членный гетероцикл, содержащий один атом азота и один атом кислорода, активность полученного соединения значительно увеличилась прибл. в 12,25 раза (=0,098/0,008);
3) сравнивая соединение 03060 (структурная формула:
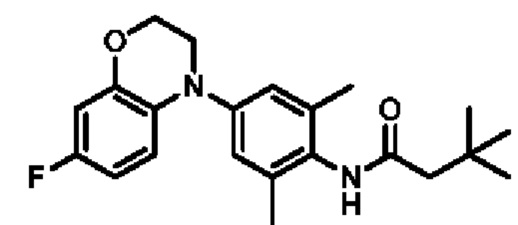
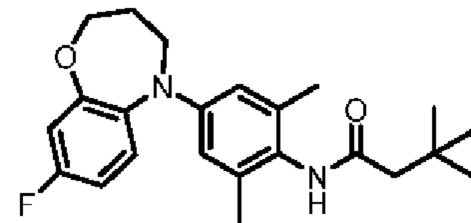
Исследование фармакокинетических свойств соединения 03058
1. Цель исследования: Определение фармакокинетических характеристик соединения 03058 у самцов мышей линии ICR
2. Суть эксперимента
Брали 6 здоровых самцов мышей линии ICR (с массой тела в диапазоне 18 22 грамма), разделяли их на 2 группы (3 мыши/группа и 3 мыши / контрольный момент времени), выдерживали их без приема пищи более 12 часов (только для группы с пероральным приемом) и вводили препарат внутривенно (0,05 мг/кг), перорально (1 мг/кг), в моменты времени 0,083 (только внутривенное введение), 0,25; 0,5; 1; 2; 4; 6 (только пероральный прием). Через 8 часов и 24 часа отбирали кровь пункцией сердца. Минимум 0,3 мл цельной крови переносили в пробирку с антикоагулянтом ЭДТА-К2, и через полтора часа плазму отделяли центрифугированием (6000 об/мин, 8 минут, 4°С) и замораживали при -20°С для использования. (Конфигурация соединения: 5% DMAC + 10% Solutol HS 15 + 85% солевой раствор использовали для приготовления раствора, содержащего концентрацию для внутривенного 0,01 мг/мл и перорального 0,1 мг/мл приема).
Экспериментальные результаты: Согласно полученным данным о концентрации препарата в крови, для расчета фармакокинетических параметров после его введения использовали некомпартментную модель программы WinNonlin® 7.0 (Pharsight, США).

Из представленных в Таблице 2 результатов видно, что соединение 03058 обладает хорошими фармакокинетическими свойствами.
Исследование фармакокинетических свойств соединений 03043 и 03044
1. Цель исследования: Получить фармакокинетические характеристики соединений 03043 и 03044 у самцов крыс линии SD и их проникновение через гематоэнцефалический барьер (ГЭБ)
2. Суть эксперимента
Брали 15 здоровых самцов крыс линии SD (диапазон массы тела 200-250 г) и разделяли на 3 группы. 3 крысы использовали для внутривенного введения в группе 1, 3 крысы -для перорального введения в группе 2, а 9 крыс - для определения доли соединения в церебральной крови после перорального введения в группе 3 (3 крысы в контрольный момент времени). Группы 2 и 3 выдерживали без приема пищи более 12 часов. Внутривенное введение: 1 мг/кг, пероральное 5 мг/кг, в момент времени 0,083 (только группа 1), 0,25 (только группы 1 и 2), 0,5 (только группы 1 и 2), 1,2 (только группы 1 и 2). Через 4 часа, 8 часов и 24 часа (только группа 1 и группа 2) отбирали кровь из яремной вены или пункцией сердца. Минимум 0,3 мл цельной крови отбирали в пробирку с антикоагулянтом ЭДТА-К2. В течение получаса плазму отделяли центрифугированием (6000 об/мин, 8 минут, 4°С) и замораживали при температуре -20°С для последующего использования. Одновременно у животных из группы 3 отбирали мозговую ткань (моменты времени 1 час, 4 и 8 часов, соответственно), промывали нормальным физиологическим раствором, высушивали промоканием впитывающей бумагой, взвешивали и замораживали при температуре -20°С для последующего использования. (Приготовление соединения: 5% DMAC + 10% Solutol HS 15+85% солевой раствор использовали для приготовления раствора, содержащего концентрацию для внутривенного 0,2 мг/мл и перорального 0,3 мг/мл приема).
Экспериментальные результаты: Согласно полученным данным о концентрации препарата в крови, для расчета фармакокинетических параметров после его введения использовали некомпартментную модель программы WinNonlin® 7.0 (Pharsight, США).

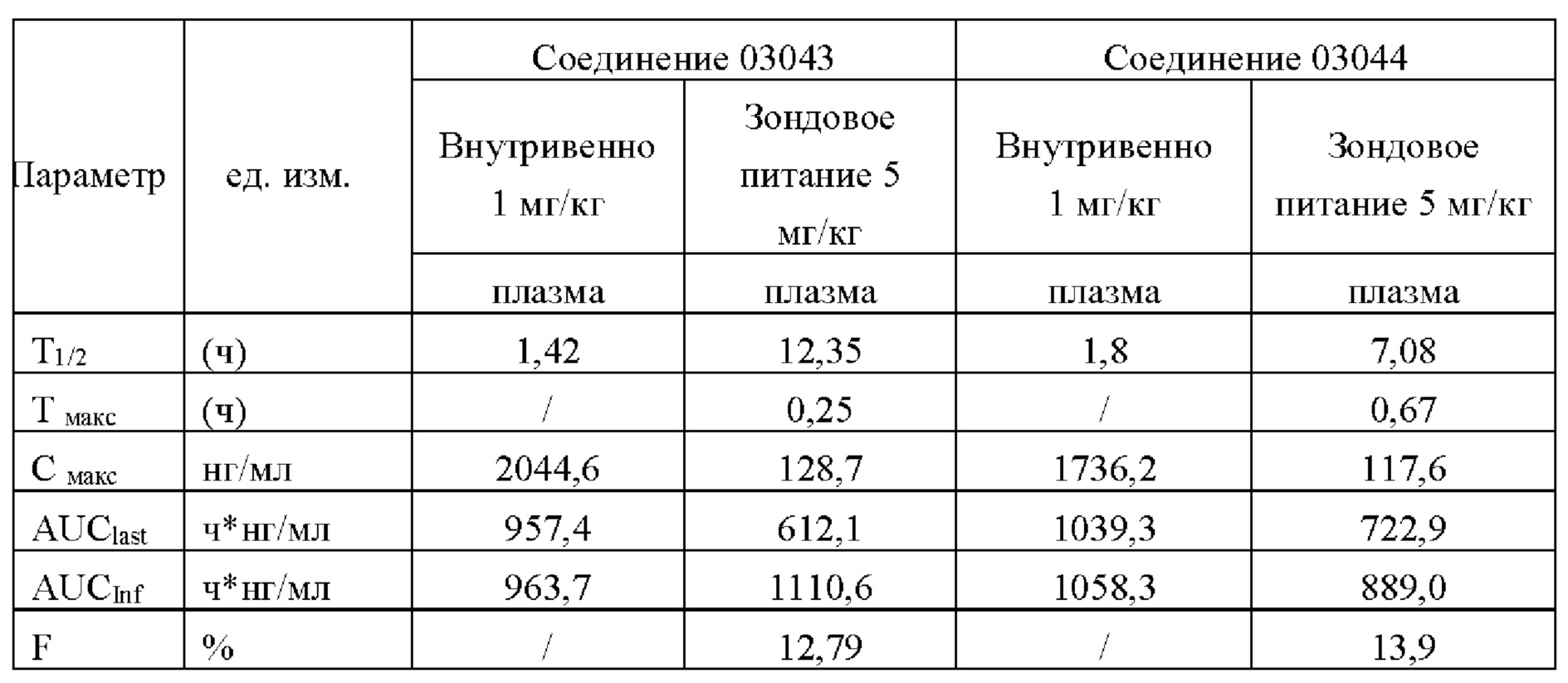
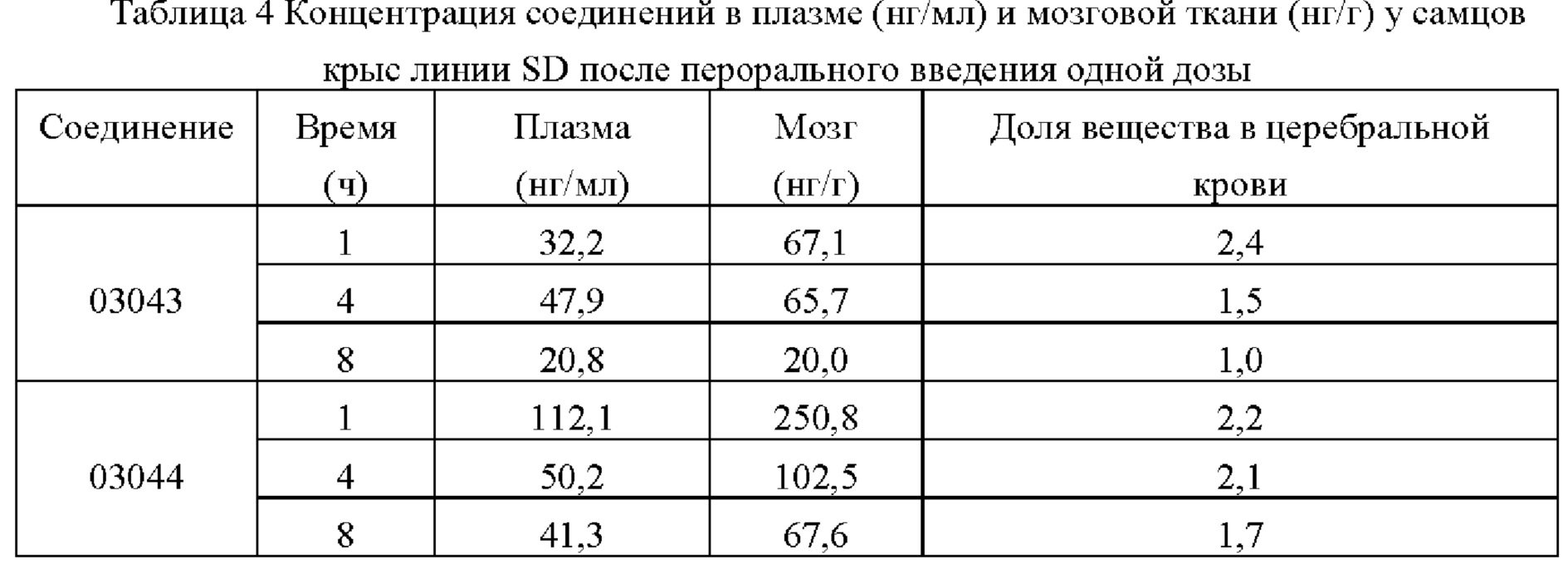
Из представленных в Таблице 3 результатов видно, что соединения 03043 и 03044 обладают хорошими фармакокинетическими свойствами.
Из результатов, представленных в Таблице 4, можно видеть, что при пероральном введении соединений 03043 и 03044 самцам крыс линии SD препараты показывают высокое их содержание в церебральной крови (от 1,0 до 2,4) в каждый контрольный момент времени. В предыдущих исследованиях мы установили, что доля соединения А в церебральной крови (WO 2014/048165 А1) составляла приблизительно 0,5 (содержание в церебральной крови через 2 часа составляла 0,56; через 4 часа - 0,46). Приведенные выше результаты предполагают, что по сравнению с соединением А, содержащим шестичленное кольцо, соединения 03043 и 03044 с семичленным кольцом демонстрируют более высокое содержание в церебральной крови.
Все литературные источники, упомянутые в настоящем изобретении, включены в настоящий документ в виде ссылок, как если бы они были включены при помощи ссылки по отдельности. Кроме того, следует понимать, что после изучения вышеприведенного описания специалистами в данной области техники могут быть внесены многочисленные изменения и модификации, и такие эквиваленты также попадают в объем, определяемый прилагаемой формулой изобретения.
Реферат
Настоящее изобретение относится к соединению, представленному общей формулой А, в которой: когда кольцо В представляет собой
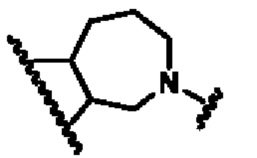


Формула
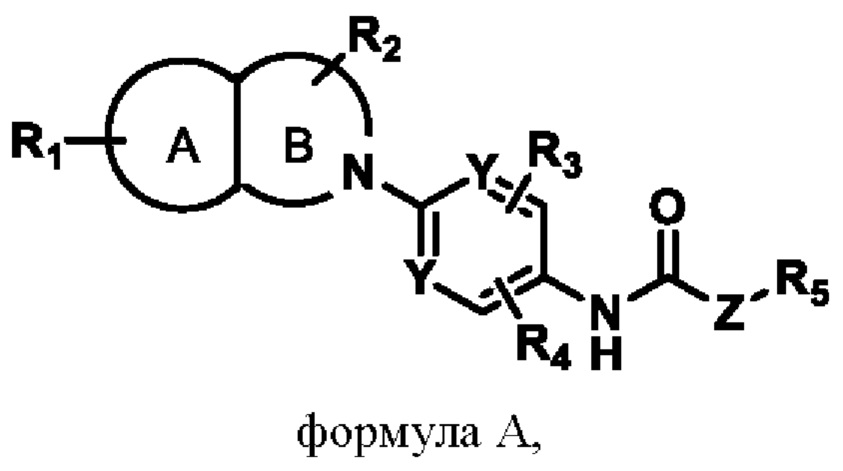
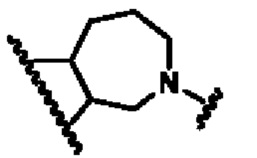
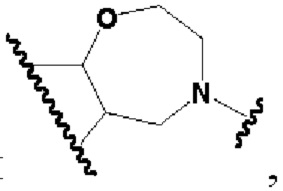

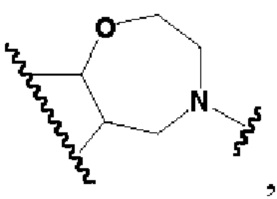

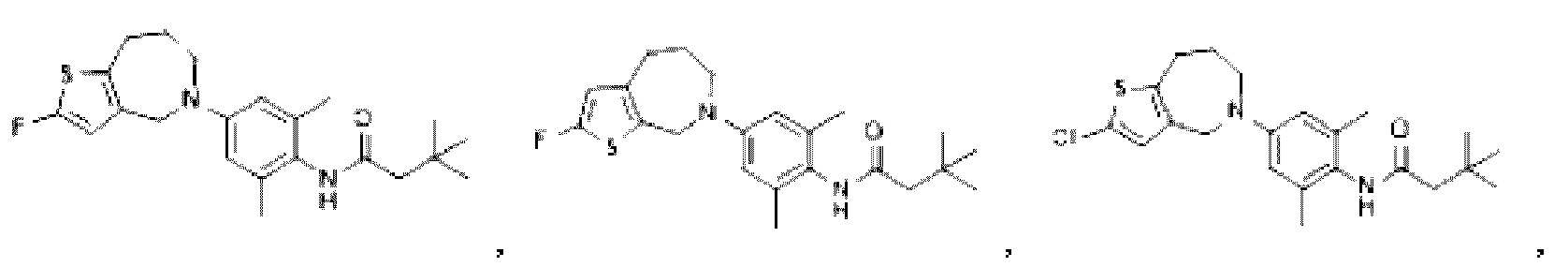
Документы, цитированные в отчёте о поиске
Способ получения производных n-арилпиперазиналканамида
Комментарии