Способ получения ценикривирока и родственных аналогов - RU2725888C2
Код документа: RU2725888C2
Чертежи

Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
[1] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №62/096286, поданной 23 декабря 2014 года и озаглавленной ʺСПОСОБ ПОЛУЧЕНИЯ ЦЕНИКРИВИРОКА И РОДСТВЕННЫХ АНАЛОГОВʺ, содержание которой полностью включено в настоящую заявку посредством ссылки для любых целей.
ОБЛАСТЬ ТЕХНИКИ
[2] Настоящее изобретение относится к способам для синтеза соединений, обладающих свойствами антагонистов CCR5 и/или CCR2, или их солей.
УРОВЕНЬ ТЕХНИКИ
[3] Известно, что ценикривирок (CVC) ингибирует рецепторы CCR5 и CCR2 и предотвращает проникновение в клетку человека вируса, такого как вирус ВИЧ (HIV) (патент США №8183273). Также синтез CVC ранее был раскрыт в заявке на патент США №10/506955 и международной публикации WO 2001017947.

[4] В настоящем изобретении предложен перспективный для промышленности способ получения CVC, солей CVC или родственных аналогов посредством оптимизированного способа образования амидных связей с аминосодержащим производным сульфоксида с получением продукта высокой степени чистоты.
[5] В результате применения традиционных способов синтеза CVC, солей CVC и родственных аналогов получают нежелательные примеси. Таким образом, существует потребность в CVC высокой чистоты и способах его получения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[6] В настоящем изобретении предложен технологический способ получения Соединения I, рацемической или оптически чистой формы CVC, и образования его соли метансульфоновой кислоты (Соединение I-MsOH). Согласно некоторым вариантам реализации изобретения Соединение I и Соединение I-MsOH являются рацемическими. Согласно другим вариантам реализации изобретения Соединение I и Соединение I-MsOH содержит оптически активный сульфоксид, такой как (S)-изомер, обозначенный как (S)-Соединение I-MsOH.

[7] Согласно некоторым вариантам реализации изобретения Соединение I-MsOH получают путем добавления метансульфоновой кислоты (MsOH) к Соединению I.
[8] Согласно некоторым вариантам реализации изобретения Соединение I получают путем проведения реакции между Соединением II и Соединением III:

[9] где R1 выбран из группы, состоящей из H, OH, Cl, Br, OR2, OCOR2 и NHR2; и
[10] где R2 выбран из группы, состоящей из H, алкила, замещенного алкила, арила и замещенного арила.
[11] Согласно некоторым вариантам реализации изобретения Соединение I получают путем проведения реакции между Соединением II, где R1=OH (Соединение II-OH), и Соединением III.

[12] Согласно некоторым вариантам реализации изобретения Соединение II-OH получают путем проведения реакции между Соединением IV и Соединением V:

[13] где R3представляет собой Ar1 или OR5; R4 представляет собой Ar2 или OR6; и R5, и R6независимо выбраны из группы, состоящей из H, алкила и замещенного алкила; или R5и R6совместно образуют необязательно замещенный алкил или необязательно замещенный арил; Ar1 и Ar2 независимо представляют собой арил или замещенный арил.
[14] Согласно некоторым вариантам реализации изобретения как R3,так и R4 представляют собой OMe для Соединения V, что обозначено как Соединение V-OMe.
[15] Согласно некоторым вариантам реализации изобретения Соединение V получают из Соединения VI.

[16] В некоторых вариантах реализации настоящего изобретения представлен технологический способ снижения количества примесей, представленных Соединениями I-MsOH-A, I-MsOH-B, I-MsOH-C, I-MsOH-D, I-MsOH-E, (R)-I-MsOH, VII, VIII, IX, и мезилатными сложными эфирами, полученными из MsOH.





[17] Настоящее изобретение включает способ получения 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфоната (Соединение I-MsOH). Например, синтез Соединения I-MsOH включает образование диметил (4-(2-бутоксиэтокси)фенил)бороната (Соединение V), которое затем используется для образования 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновой кислоты (Соединение II-OH) высокой чистоты.
[18] Согласно некоторым вариантам реализации изобретения Соединение V получают путем a) активирования магния в тетрагидрофуране (ТГФ) при нагревании, b) инициирования образования реактива Гриньяра путем добавления части 1-бром-4-(2-бутоксиэтокси)бензола (Соединение VI) к смеси, полученной на стадии a), при нагревании, c) продолжения медленного добавления оставшегося Соединения VI при нагревании, d) охлаждения смеси, полученной на стадии c), до приблизительно -25°C и медленного добавления триметоксиборана, и e) перемешивания смеси, полученной на стадии d) при приблизительно -25°C в течение приблизительно 1 часа и затем нагревания реакционной смеси до приблизительно 20°C в течение приблизительно 1 часа.
[19] Согласно некоторым вариантам реализации изобретения молярное соотношение используемых Соединения VI и триметоксиборана составляет приблизительно 1:1.
[20] Согласно некоторым вариантам реализации изобретения на стадиях b) и/или c) используют Соединение VI без примесей. Согласно другим вариантам реализации изобретения на стадии c) требуется перемешивание реакционной смеси при приблизительно 55°C в течение от приблизительно 3 часов до приблизительно 5 часов.
[21] Соединение V, синтезированное как описано в настоящей заявке, согласно одному из вариантов реализации затем применяется для получения Соединения II-OH. Согласно некоторым вариантам реализации изобретения Соединение II-OH получают путем a) образования двухфазной смеси путем добавления основного водного раствора к раствору Соединения V, b) добавления катализатора и лиганда к смеси, полученной на стадии a), c) добавления 8-бром-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновой кислоты (Соединение IV) к смеси, полученной на стадии b), и нагревания реакционной смеси, и d) окисления смеси, полученной на стадии c). Основание, применяемое на стадии a), согласно некоторым вариантам реализации изобретения, выбрано из группы, состоящей из фосфата калия, карбоната калия, ацетата калия, фторида калия, гидроксида калия, трет-бутоксида калия, карбоната натрия, фосфата натрия, гидроксида натрия, трет-бутоксида натрия, бикарбоната натрия, карбоната цезия, фторида цезия и их комбинации. Согласно некоторым вариантам реализации изобретения катализатор, используемый на стадии b), выбран из группы, состоящей из ацетата палладия, тетракис(трифенилфосфин) палладия, три(дибензилиденацетон)дипалладия, хлорида палладия, ацетилацетоната палладия и их комбинации. Согласно некоторым вариантам реализации изобретения лиганд, используемый на стадии b), выбран из группы, состоящей из три(o-толил)фосфина, трифенилфосфина, три(т-бутил)фосфина, трициклогексилфосфина, пиридина, бипиридина, 2,2'-бис(дифенилфосфин)-1,1'-бинафтила и их комбинации. Согласно другому варианту реализации изобретения каталитическая система стадии b) включает ацетат палладия и три(o-толил)фосфин.
[22] Согласно некоторым вариантам реализации изобретения соотношение катализатора к лиганду составляет приблизительно 1:2. Согласно другим вариантам реализации изобретения количество катализатора, применяемого на стадии b), составляет от приблизительно 0,001 эквивалента (эквив.) до приблизительно 2,500 эквив. по отношению к Соединению IV. Согласно дополнительному варианту реализации изобретения катализатор применяют в количестве от приблизительно 0,001 эквив. до приблизительно 0,005 эквив. по отношению к Соединению IV. Согласно некоторым вариантам реализации изобретения после стадии a) и до стадии d) или во время любой из стадий от a) до d) в реакционную смесь барботируют азот.
[23] Согласно некоторым вариантам реализации изобретения для образования Соединения II-OH Соединение V применяют в количестве от приблизительно 1,5 эквив. до приблизительно 2,2 эквив. по отношению к Соединению IV. Согласно другому варианту реализации изобретения нагревание на стадии c) поддерживают при≤65°C в течение от приблизительно 2 часов до приблизительно 6 часов и стабильно высокой конверсии по Соединению II-OH.
[24] Согласно другим вариантам реализации во время стадии очистки после стадии d), к реакционной смеси, содержащей Соединение II-OH, добавляют активированный уголь с целитом (Celite®) или без него. Согласно другому варианту реализации смесь, содержащую активированный уголь и/или целит и Соединение II-OH, перемешивают и затем подвергают фильтрованию. Согласно одному варианту реализации изобретения соотношение активированного угля к целиту составляет приблизительно 1:2.
[25] Согласно другому варианту реализации во время стадии очистки после стадии d), к реакционной смеси, содержащей Соединение II-OH, добавляют целит, перемешивают и затем подвергают фильтрованию. Согласно одному варианту реализации изобретения во время стадии очистки после стадии d) реакционную смесь подвергают фильтрованию для удаления любых твердых частиц.
[26] Согласно некоторым вариантам реализации изобретения очистка Соединения II-OH включает рекристаллизацию с помощью антирастворителя и/или горячую рекристаллизацию. Согласно некоторым вариантам реализации изобретения антирастворитель, применяемый для рекристаллизации с помощью антирастворителя для получения неочищенного вещества, представляет собой гептаны. Согласно другим вариантам реализации, горячая рекристаллизация включает стадии: i) растворения неочищенного вещества, полученного при рекристаллизации с помощью антирастворителя, в апротонном полярном растворителе и короткоцепочечном спирте при приблизительно 70°C, ii) снижения температуры смеси, полученной на стадии i), до приблизительно 20°C в течение периода времени от приблизительно 3 часов до приблизительно 7 часов, и iii) перемешивания смеси, полученной на стадии ii), при приблизительно 20°C в течение от приблизительно 2 часов до приблизительно 6 часов. Согласно одному варианту реализации изобретения указанный апротонный растворитель представляет собой этилацетат. Согласно другому варианту реализации указанный короткоцепочечный спирт представляет собой изопропанол.
[27] Согласно некоторым вариантам реализации изобретения представленный способ получения Соединения II-OH обеспечивает получение Соединения II-OH со степенью чистоты приблизительно >97,5%. Согласно другому варианту реализации представленный способ получения Соединения II-OH обеспечивает получение Соединения II-OH со степенью чистоты приблизительно > 98,0%. Согласно некоторым вариантам реализации изобретения представленный способ получения Соединения II-OH обеспечивает получение Соединения II-OH со степенью чистоты приблизительно > 99,0%.
[28] Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 4,4'-бис(2-бутоксиэтокси)бифенил (Соединение VII) в количестве приблизительно≤0,10%.
[29] Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту) (Соединение VIII) в количестве приблизительно≤0,20%. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту) (Соединение VIII) в количестве приблизительно≤0,10%. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту) (Соединение VIII) в количестве приблизительно≤0,05%.
[30] Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение IX) в количестве приблизительно≤0,50%. Согласно другому варианту реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение IX) в количестве приблизительно≤0,25%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение IX) в количестве приблизительно≤0,15%.
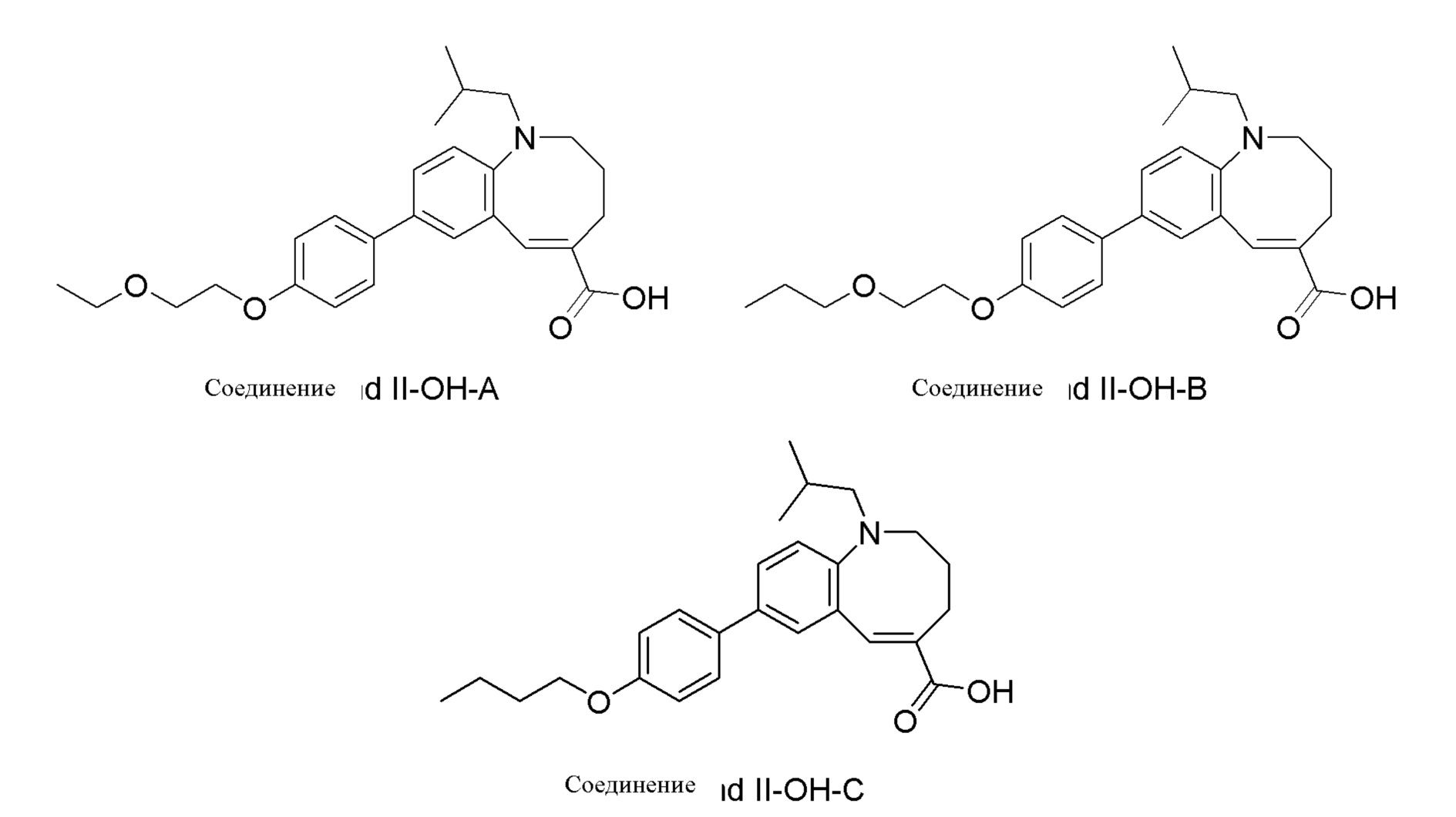
[31] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-(2-этоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-A) в количестве приблизительно≤0,20% от Соединения II-OH. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-(2-этоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-A) в количестве приблизительно≤0,10% от Соединения II-OH. Согласно другому варианту реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-(2-этоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-A) в количестве приблизительно≤0,05% от Соединения II-OH.
[32] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 1-изобутил-8-(4-(2-пропоксиэтокси)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-B) в количестве приблизительно≤0,20% от Соединения II-OH. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения II-OH получают 1-изобутил-8-(4-(2-пропоксиэтокси)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-B) в количестве приблизительно≤0,10% от Соединения II-OH. Согласно другому варианту реализации изобретения в результате описанного синтеза Соединения II-OH получают 1-изобутил-8-(4-(2-пропоксиэтокси)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-B) в количестве приблизительно≤0,05% от Соединения II-OH.
[33] Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-бутоксифенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-C) в количестве приблизительно≤0,50% от Соединения II-OH. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-бутоксифенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-C) в количестве приблизительно≤0,25% от Соединения II-OH. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения II-OH получают 8-(4-бутоксифенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH-C) в количестве приблизительно≤0,10% от Соединения II-OH.
[34] Дополнительно в настоящем изобретении описан способ получения 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфоната (Соединение I-MsOH). Описанный способ получения Соединения I-MsOH включает a) проведение реакции между Соединением II и 4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилином (Соединение III) в присутствии основания с получением 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамида (Соединение I), b) гашение стадии a) водным раствором, c) добавление метансульфоновой кислоты и d) кристаллизацию Соединения I-MsOH. Согласно некоторым вариантам реализации изобретения R1 в Соединении II выбран из группы, состоящей из H, OH, Cl, Br, OR2, OCOR2 и NHR2, а R2 в Соединении II выбран из группы, состоящей из H, алкила, замещенного алкила, арила и замещенного арила.
[35] Согласно некоторым вариантам реализации изобретения R1 в Соединении II представляет собой Cl. Согласно одному варианту реализации изобретения синтез Соединения II включает стадии i) растворения Соединения II-OH в растворителе и ii) добавления к смеси, полученной на стадии i), хлорирующего реагента. Согласно некоторым вариантам реализации изобретения указанный хлорирующий реагент выбран из группы, состоящей из тионилхлорида, трихлорида фосфора, пентахлорида фосфора, оксихлорида фосфора, оксалил хлорида, фосгена и их комбинации. Согласно одному варианту реализации изобретения указанный хлорирующий реагент представляет собой тионилхлорид. Согласно некоторым вариантам реализации изобретения количество применяемого хлорирующего реагента составляет от приблизительно 1,0 эквив. до приблизительно 1,2 эквив. по отношению к Соединению II-OH.
[36] Согласно некоторым вариантам реализации изобретения на стадии a) синтеза Соединения I-MsOH в качестве растворителя применяют дихлорметан. Согласно другим вариантам реализации изобретения на стадии a) синтеза Соединения I-MsOH в качестве основания применяют пиридин. Согласно другим вариантам реализации изобретения на стадии a) синтеза Соединения I-MsOH в качестве Соединения III применяют оптически чистое (S)-Соединение III.
[37] Согласно некоторым вариантам реализации изобретения количество используемого Соединения III составляет от приблизительно 1,0 эквив. до приблизительно 1,2 эквив. по отношению к Соединению II-OH. Согласно некоторым вариантам реализации изобретения количество метансульфоновой кислоты составляет от приблизительно 0,97 эквив. до приблизительно 1,02 эквив. по отношению к Соединению II-OH. Согласно другим вариантам реализации изобретения соотношение метансульфоновой кислоты и Соединения II-OH составляет приблизительно 1:1.
[38] Согласно некоторым вариантам реализации изобретения на стадии b) синтеза Соединения I-MsOH лимонную кислоту применяют в виде водного раствора. Согласно другим вариантам реализации изобретения стадия b) синтеза Соединения I-MsOH дополнительно включает экстрагирование Соединения I и высушивание экстрагированного раствора молекулярными ситами 3 Å.
[39] Согласно некоторым вариантам реализации изобретения для затравки стадии кристаллизации d) синтеза Соединения I-MsOH применяют образец Соединения I-MsOH без примесей. Затравленный кристаллизующий раствор стадии d) согласно некоторым вариантам реализации изобретения дополнительно подвергают стадиям перемешивания при 0°C для осуществления кристаллизации, сбора образовавшихся кристаллов и промывки собранных кристаллов охлажденным этилацетатом. Согласно одному варианту реализации изобретения образовавшиеся кристаллы собирают посредством фильтрования.
[40] Согласно другим вариантам реализации, требуется дополнительная очистка с использованием после стадии d) горячей рекристаллизации. Горячая рекристаллизация Соединения I-MsOH включает i) растворение неочищенных кристаллов Соединения I-MsOH, полученных на стадии d), в ацетонитриле при приблизительно 70°C, ii) снижение температуры смеси, полученной на стадии i) до диапазона от приблизительно 50°C до приблизительно 55°C в течение более 1 часа, iii) затравливание стадии ii) Соединением I-MsOH, iv) перемешивание при от приблизительно 50°C до приблизительно 55°C в течение приблизительно 6 часов, v) снижение температуры смеси, полученной на стадии iii) до приблизительно 20°C, vi) перемешивание при приблизительно 20°C в течение приблизительно 8 часов, vii) сбор кристаллов посредством фильтрования и viii) промывку кристаллов холодным ацетонитрилом.
[41] Согласно некоторым вариантам реализации изобретения в результате описанного способа синтеза Соединения I-MsOH получают Соединение I-MsOH или его энантиомер, стереоизомер или их комбинации со степенью чистоты приблизительно >96,0%. Согласно другому варианту реализации в результате описанного способа синтеза Соединения I-MsOH получают Соединение I-MsOH или его энантиомер, стереоизомер или их комбинации со степенью чистоты приблизительно >97,0%. Согласно одному варианту реализации изобретения в результате описанного способа синтеза Соединения I-MsOH получают Соединение I-MsOH или его энантиомер, стереоизомер или их комбинации со степенью чистоты приблизительно >98,0%. Согласно некоторым вариантам реализации изобретения в результате описанного способа синтеза Соединения I-MsOH получают Соединение I-MsOH или его энантиомер, стереоизомер или их комбинации со степенью чистоты приблизительно >98,5%.
[42] Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH) в количестве приблизительно≤1,0%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH) в количестве приблизительно≤0,80% или приблизительно≤0,50%. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение II-OH) в количестве приблизительно≤0,25%.
[43] Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту) (Соединение VIII) в количестве приблизительно≤0,20%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту) (Соединение VIII) в количестве приблизительно≤0,10%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту) (Соединение VIII) в количестве приблизительно≤0,05%.
[44] Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение IX) в количестве приблизительно≤0,20%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение IX) в количестве приблизительно≤0,10%. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновую кислоту (Соединение IX) в количестве приблизительно≤0,05%.
[45] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид) диметансульфонат (Соединение I-MsOH-G) в количестве приблизительно≤0,40%. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид) диметансульфонат (Соединение I-MsOH-G) в количестве приблизительно≤0,30%. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид) диметансульфонат (Соединение I-MsOH-G) в количестве приблизительно≤0,20%. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид) диметансульфонат (Соединение I-MsOH-G) в количестве приблизительно≤0,15%. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8,8'-(4-(2-бутоксиэтокси)-1,3-фенилен)бис(1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид) диметансульфонат (Соединение I-MsOH-G) в количестве приблизительно≤0,10%.
[46] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилин (Соединение III) или его энантиомер, стереоизомер или их комбинации в количестве приблизительно≤0,25%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилин (Соединение III) или его энантиомер, стереоизомер или их комбинации в количестве приблизительно≤0,15%. Согласно другому варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилин (Соединение III) или его энантиомер, стереоизомер или их комбинации в количестве приблизительно≤0,10%.
[47] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилин (Соединение III) или его энантиомер, стереоизомер или их комбинации в количестве приблизительно≤2000 м.д. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилин (Соединение III) или его энантиомер, стереоизомер или их комбинации в количестве приблизительно≤1750 м.д. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилин (Соединение III) или его энантиомер, стереоизомер или их комбинации в количестве приблизительно≤1500 м.д. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилин (Соединение III) или его энантиомер, стереоизомер или их комбинации в количестве приблизительно≤1250 м.д.
[48] Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают (S)-4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)анилин ((S)-Соединение III) в количестве приблизительно≤1500 м.д.
[49] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-этоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-A) в количестве приблизительно≤0,25%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-этоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-A) в количестве приблизительно≤0,15%. Согласно другому варианту реализации в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-этоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-A) в количестве приблизительно≤0,10%.
[50] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 1-изобутил-8-(4-(2-пропоксиэтокси)фенил)-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-B) в количестве приблизительно≤0,25%. Согласно другим вариантам реализации, в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 1-изобутил-8-(4-(2-пропоксиэтокси)фенил)-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-B) в количестве приблизительно≤0,15%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 1-изобутил-8-(4-(2-пропоксиэтокси)фенил)-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-B) в количестве приблизительно≤0,10%.
[51] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-бутоксифенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-C) в количестве приблизительно≤0,40%. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-бутоксифенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-C) в количестве приблизительно≤0,30%. Согласно другому варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-бутоксифенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-C) в количестве приблизительно≤0,20%.
[52] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфонил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-D) в количестве приблизительно≤2,0%. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфонил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-D) в количестве приблизительно≤1,0%. Согласно другому варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфонил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-D) в количестве приблизительно≤0,50%.
[53] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)тио)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-E) в количестве приблизительно≤0,40%. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)тио)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-E) в количестве приблизительно≤0,30%. Согласно другому варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)тио)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-E) в количестве приблизительно≤0,20%.
[54] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-F) в количестве приблизительно≤0,40%. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-F) в количестве приблизительно≤0,30%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-F) в количестве приблизительно≤0,20%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают 8-(4-(2-бутоксиэтокси)фенил)-1-бутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат (Соединение I-MsOH-F) в количестве приблизительно≤0,15%.
[55] Согласно другому варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают мезилатные сложные эфиры, полученные из MsOH, в количестве приблизительно≤1,0%. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают мезилатные сложные эфиры, полученные из MsOH, в количестве приблизительно≤0,50%. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают мезилатные сложные эфиры, полученные из MsOH, в количестве приблизительно≤0,25%.
[56] Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают мезилатные сложные эфиры, полученные из MsOH, в количестве приблизительно≤20 м.д. Согласно другим вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают мезилатные сложные эфиры, полученные из MsOH, в количестве приблизительно≤10 м.д. Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций получают мезилатные сложные эфиры, полученные из MsOH, в количестве приблизительно≤5 м.д. Согласно некоторым вариантам реализации изобретения Соединение I-MsOH или его энантиомер, стереоизомер или их комбинации содержит 10 м.д. мезилатного сложного эфира на 150 мг.


[57] Согласно одному варианту реализации изобретения в результате описанного синтеза Соединения I-MsOH получают (S)-Соединение I-MsOH. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза получают (S)-Соединение I-MsOH с чистотой более 96% или более 98,5%.
[58] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза (S)-Соединения I-MsOH получают (R)-8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат ((R)-Соединение I-MsOH) в количестве приблизительно≤1,00%. Согласно другому варианту реализации в результате описанного синтеза (S)-Соединения I-MsOH получают (R)-8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат ((R)-Соединение I-MsOH) в количестве приблизительно≤0,50%. Согласно одному варианту реализации изобретения в результате описанного синтеза (S)-Соединения I-MsOH получают (R)-8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфонат ((R)-Соединение I-MsOH) в количестве приблизительно≤0,25%.
[59] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза Соединения I-MsOH или его энантиомера, стереоизомера или их комбинаций полученное содержание воды составляет 5,0 мас.% или менее, или 2,0 мас.%, или менее.
[60] Согласно некоторым вариантам реализации изобретения в результате описанного синтеза (S)-Соединения I-MsOH получают≤3,0% примесей, включая (R)-Соединение I-MsOH, но не включая (S)-Соединение III. Согласно одному варианту реализации изобретения в результате описанного синтеза (S)-Соединения I-MsOH получают≤2,5% примесей, включая (R)-Соединение I-MsOH, но не включая (S)-Соединение III. Согласно другому варианту реализации в результате описанного синтеза (S)-Соединения I-MsOH получают≤2,3% примесей, включая (R)-Соединение I-MsOH, но не включая (S)-Соединение III. Согласно некоторым вариантам реализации изобретения в результате описанного синтеза (S)-Соединения I-MsOH получают≤2,0% примесей, включая (R)-Соединение I-MsOH, но не включая (S)-Соединение III.
ПОДРОБНОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[61] На ФИГ 1. представлен протонный ЯМР (ядерная магнитно-резонансная спектроскопия) спектр (S)-Соединения II-OH.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
[62] Хотя предполагается, что следующие термины хорошо известны среднему специалисту в области техники, следующие определения представлены для облегчения понимания объекта настоящего изобретения.
[63] Термины «a» или «an» относится к одному или более описанному объекту; например, «галоген» относится к одному или более галогенам, или к по меньшей мере одному галогену. Как таковые, термины «a» (или «an»), «один или более» и «по меньшей мере один» в настоящей заявке являются взаимозаменяемыми. Кроме того, указание на «алкильную группу» с помощью неопределенного артикля «a» или «an» не исключает возможности присутствия более, чем одной алкильной группы, если в контексте явным образом не требуется, чтобы алкильная группа была одна и только одна.
[64] Используемый в настоящем описании и формуле изобретения глагол «содержать» и формы его спряжения в неограничивающем смысле означают, что объекты, следующие за указанным словом, включены в перечень, но не означают, что из него исключены объекты, не указанные конкретно.
[65] Используемая в настоящей заявке фраза «алкильная группа» относится к углеводородам с прямой цепью, разветвленной цепью или к циклическим углеводородам, содержащим от 1 до приблизительно 10 атомов углерода. Неограничивающие примеры алкильной группы включают C1-C10 алкильную группу,такую как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, гексил, гептил, октил, нонил, децил и тому подобные.
[66] Используемая в настоящей заявке фраза «арильная группа» относится к ароматической группе, содержащей от 6 до 14 атомов углерода. Неограничивающие примеры арильной группы включают фенил, нафтил, антрил, флуоренил и тому подобные.
[67] Используемая в настоящей заявке фраза «заместитель (заместители)» в необязательно замещенной алкильной группе и необязательно замещенной арильной группе включает атом галогена (например, фтор, хлор, бром, йод и тому подобные), нитрогруппу, циано группу, необязательно замещенную гидроксильную группу (например, гидроксильная группа, C1-C4 алкокси и тому подобные), необязательно замещенную тиольную группу (например, тиол, C1-C4 алкилтио и тому подобные), необязательно замещенную аминогруппу (например, амино, моно-C1-C4 алкиламино, ди-C1-C4 алкиламино, 5- или 6-членная циклическая аминогруппа, такая как пирролидин, пиперазин, пиперидин, морфолин, тиоморфолин, пиррол и имидазол, и тому подобные), необязательно этерифицированную или амидированную карбоксильную группу (например, карбоксил, C1-C4 алкоксикарбонил, карбамоил, моно-C1-C4 алкилкарбамоил, ди-C1-C4 алкилкарбамоил и тому подобные), необязательно галогензамещенную C1-C4 алкоксигруппу (например, метокси, этокси, пропокси, бутокси, трифторметокси, трифторэтокси и тому подобные), необязательно галогензамещенную C1-C4 алкокси-C1-C4 алкокси группу (например, метоксиметокси, метоксиэтокси, этоксиэтокси, трифторметоксиэтокси, трифторэтоксиэтокси и тому подобные), формильную группу, C2-C4 алканоильную группу (например, ацетил, пропионил и тому подобные) и C1-C4 алкилсульфонильную группу (например, метансульфонил, этансульфонил и тому подобные).
[68] Используемая в настоящей заявке фраза «короткоцепочечный спирт» относится к спирту, содержащему 1-8 атомов углерода. Неограничивающие примеры короткоцепочечного спирта включают метанол, этанол, пропанол, изопропанол, бутанол, пентанол, гексанол, гептанол, октанол и тому подобные.
[69] Используемая в настоящей заявке фраза «апротонный растворитель» относится к органическому растворителю или смеси органических растворителей, который не подвергается депротонированию в присутствии сильного основного реагента. Неограничивающие примеры апротонных растворителей включают эфиры, диметилформамид (DMF), диметилацетамид (DMAC), 1,3-диметил-3,4,5,6- тетрагидро-2(1H)-пиримидинон (DMPU), 1,3-диметил-2-имидазолидинон (DMI), N-метилпирролидинон (NMP), формамид, N-метилацетамид, N-метилформамид, ацетонитрил, диметил сульфоксид, пропионитрил, этилформиат, метилацетат, гексахлорацетон, ацетон, этил метил кетон, этилацетат, сульфолан, N,N- диметилпропионамид, тетраметилмочевину, нитрометан, нитробензол или гексаметилфосфорамид, диэтоксиметан, тетрагидрофуран, 1,3-диоксан, 1,4-диоксан, фуран, диэтиловый эфир, тетрагидропиран, диизопропиловый эфир, дибутиловый эфир, этиленгликоль диметиловый эфир, этиленгликоль диэтиловый эфир, диэтиленгликоль диметиловый эфир, диэтиленгликоль диэтиловый эфир, триэтиленгликоль диметиловый эфир, анизол, т-бутил метиловый эфир и тому подобные.
[70] Используемая в настоящей заявке фраза «протонный растворитель» относится к растворителю или смеси растворителей, способному функционировать в качестве кислоты для протонирования любых непрореагировавших сильноосновных промежуточных соединений реакции. Неограничивающие примеры протонных растворителей включают воду, метанол, этанол, 2-нитроэтанол, 2-фторэтанол, 2,2,2-трифторэтанол, этиленгликоль, 1-пропанол, 2-пропанол, 2-метоксиэтанол, 1-бутанол, 2-бутанол, и-бутиловый спирт, т-бутиловый спирт, 2-этоксиэтанол, диэтиленгликоль, 1-, 2-, или 3- пентанол, неопентиловый спирт, т-пентиловый спирт, диэтиленгликоль монометиловый эфир, диэтиленгликоль моноэтиловый эфир, циклогексанол, бензиловый спирт, фенол, глицерин и тому подобные.
[71] Используемая в настоящей заявке фраза «часть (части)» в случае описания объема жидкости относится к приблизительной оценке коэффициента объема к соединению, веществу или жидкости, к которой относится, или о которой говорилось ранее. Например, 50 частей воды по отношению к Соединению A означают, что используют объем воды, приблизительно в 50 раз превышающий объем Соединения A.
[72] Используемый в настоящей заявке символ «≤» означает «не более, чем» или «равный или меньший, чем»; « < » означает «менее, чем»; «≥» означает «не менее, чем» или «равный или больший, чем»; и « > » означает «более, чем». Кроме того, численные показатели, используемые в настоящей заявке применительно к чистоте или содержанию примесей, включают не только конкретное число, но также приблизительный диапазон значений поблизости указанного числового значения. Например, фраза «степень чистоты 99,0%» означает степень чистоты приблизительно 99,0%.
Способ получения Соединения V
[73] Соединение V согласно некоторым вариантам реализации изобретения представляет собой бороновые кислоты, бороновые сложные эфиры, пинаколбораны, димеры бороновой кислоты, триммеры бороновой кислоты, их смеси или тому подобные. Традиционно из уровня техники следует понимать, что Соединение V может быть представлено в виде различных производных бороновых кислот.
[74] Согласно некоторым вариантам реализации изобретения диметил (4-(2-бутоксиэтокси)фенил)боронат (Соединение V-OMe) получают путем образования 1-бром-4-(2-бутоксиэтокси)бензола (Соединение VI) посредством реакции Гриньяра и последующей реакцией с триметоксибораном.
[75] Было обнаружено, что при крупномасштабном производстве инициирование реакции Гриньяра затруднено. Предшествующий способ включал разбавленный раствор Соединения VI, приблизительно 50-70 частей тетрагидрофурана (ТГФ) по отношению к Соединению VI. Инициирование проходило очень медленно в разбавленном растворе Соединения VI с изопропилмагний хлоридом (iPrMgCl), и происходило после продолжительного нагревания с обратным холодильником и добавления увеличенного количества Соединения VI с получением концентрации приблизительно 25 частей ТГФ по отношению к Соединению VI. В дополнение к сложностям в инициировании реакции Гриньяра, было обнаружено, что применение iPrMgCl отрицательно влияло на последующие стадии (более низкая конверсия стадии сочетания Сузуки; см. раздел Способы получения Соединения II-OH).
[76] Для преодоления трудностей инициации реакции Гриньяра согласно некоторым вариантам реализации изобретения необходимо провести стадию активации магниевой стружки при нагревании и перемешивании перед образованием реактива Гриньяра. Согласно некоторым вариантам реализации изобретения магниевые стружки перемешивают в течение 1 часа в приблизительно 9 частях эфирного растворителя, такого как ТГФ. Затем количество растворителя может быть снижено до 3 частей методом дистилляции.
[77] Трудности инициирования реакции Гриньяра согласно некоторым вариантам реализации изобретения решаются путем применения Соединения VI без примесей для получения более концентрированного раствора, чем в предшествующих способах. Согласно некоторым вариантам реализации изобретения приблизительно 20% общего количества Соединения VI, не содержащего примесей, добавляют в раствор активированной магниевой стружки в течение периода времени по меньшей мере 15 минут, в то время как экзотерма контролируется таким образом, что температура реакции поддерживается ниже точки кипения растворителя. Полученный раствор нагревают в точке кипения растворителя или в ближайшем диапазоне в течение от приблизительно 1 часа до приблизительно 4 часов. Затем реакционную смесь охлаждают на приблизительно 10°C и разбавляют тем же растворителем, который использовали ранее (5 частей). Указанная описанная стадия инициации реакции Гриньяра согласно некоторым вариантам реализации изобретения приводит к полному отсутствию iPrMgCl.
[78] Согласно некоторым вариантам реализации изобретения к горячему инициированному раствору Гриньяра, который дополнительно разбавляют, медленно добавляют оставшееся Соединение VI, не содержащее примесей, в течение периода времени от приблизительно 30 минут до приблизительно 1 часа. Добавление Соединения VI является экзотермическим, и реакционную смесь во время добавления тщательно поддерживают при температуре, значительно более низкой, чем точка кипения. Согласно некоторым вариантам реализации изобретения полученную смесь перемешивают и нагревают до температуры ниже точки кипения растворителя, например, приблизительно 55°C для ТГФ, в течение от приблизительно 3 часов до приблизительно 4 часов. Согласно некоторым вариантам реализации изобретения время нагревания может быть увеличено до того момента, когда анализ методом высокоэффективной жидкостной хроматографии (ВЭЖХ) указывает на остаток менее 1% Соединения VI. Было замечено, что увеличенное время нагревания не оказывает положительного эффекта на выход последующей стадии или предотвращение образования основных примесей.
[79] Предшествующий способ получения Соединения V-OMe включал охлаждение смеси Гриньяра до приблизительно -15°C и добавление раствора триметоксиборана в ТГФ. Авторы настоящего изобретения обнаружили, что указанный температурный диапазон не является оптимальным и приводит к меньшему выходу и большему количеству примесей. Также было обнаружено, что реакция чувствительна к скорости добавления триметоксиборана.
[80] Принимая во внимание вышеуказанные заключения, согласно некоторым вариантам реализации изобретения смесь Гриньяра (по завершении образования) охлаждают до приблизительно -25°C и порционно добавляют триметоксиборан, не содержащий примесей, в течение более 2 часов. Реакционную смесь перемешивают при приблизительно -25°C в течение от приблизительно 1 часа до приблизительно 2 часов по завершении добавления триметоксиборана, а затем нагревают до приблизительно 20°C и перемешивают в течение от приблизительно 1 часа до приблизительно 2 часов с получением Соединения V-OMe. Согласно некоторым вариантам реализации изобретения триметоксиборан, не содержащий примесей, перед добавлением к смеси Гриньяра охлаждают.
[81] Согласно некоторым вариантам реализации изобретения соотношение магниевой стружки, Соединения VI и триметоксиборана составляет приблизительно 1,08:1:1.
[82] Согласно некоторым вариантам реализации изобретения для получения Соединения V используют безводные растворители. Согласно другим вариантам реализации изобретения реакцию синтеза Соединения V проводят под атмосферным давлением азота или аргона и из реакционных емкостей и оборудования перед использованием удаляют влагу.
[83] Согласно некоторым вариантам реализации изобретения как Соединение VI, так и триметоксиборан применяют в качестве раствора без примесей для уменьшения использования реактора.
[84] Было замечено, что нет необходимости в фильтровании неочищенного Соединения V-OMe для удаления избыточного магния и солей магния, поскольку оно не влияет на последующую стадию в части предотвращения образования основных примесей.
Способ получения Соединения II-OH
[85] Согласно некоторым вариантам реализации изобретения Соединение II-OH получают путем проведения реакции между Соединением IV и Соединением V. Согласно другим вариантам реализации изобретения Соединение II-OH получают способом с использованием переходных металлов в качестве катализаторов, например, путем реакции сочетания Сузуки между Соединением IV и Соединением V. Согласно одному варианту реализации изобретения используемое количество Соединения V составляет от приблизительно 1 эквивалента (эквив.) до приблизительно 3 эквив. по отношению к Соединению IV. Согласно другим вариантам реализации изобретения используемое количество Соединения V составляет приблизительно 2 эквив. по отношению к Соединению IV.
[86] Предшествующий способ получения Соединения II-OH также включал реакцию сочетания Сузуки, где к реакционной смеси, содержащей Соединение V, добавляли ацетат палладия в качестве катализатора (Pd(OAc)2) и трифенилфосфин в качестве лиганда (PPh3) перед добавлением водного раствора основания (вода и основание в виде твердого вещества). В результате такого способа синтеза получали Соединение II-OH со средним выходом от приблизительно 55% до приблизительно 64% со степенью чистоты от приблизительно 92% до приблизительно 99%.
[87] Было обнаружено, что согласно некоторым вариантам реализации изобретения добавление водного раствора основания с образованием двухфазной смеси перед добавлением палладиевого (Pd) катализатора и лиганда создают благоприятный эффект для снижения примеси Соединения VII, возникающий в связи с гомосопряжением Соединения V. Согласно некоторым вариантам реализации изобретения раствор основания в приблизительно 6,5 частях воды добавляют к реакционной смеси, содержащей Соединение V, полученное, как описано выше. Согласно другим вариантам реализации изобретения основание может быть выбрано из группы, состоящей из карбонатов щелочных металлов (карбонат калия, карбонат натрия, карбонат цезия и тому подобные), гидрокарбонатов щелочных металлов (бикарбонат калия, бикарбонат натрия и тому подобные), ацетатов щелочных металлов (ацетат калия, ацетат натрия и тому подобные), фосфатов щелочных металлов (фосфат калия, фосфат натрия и тому подобные), фторидов щелочных металлов (фторид калия, фторид цезия и тому подобные), алкоксидов щелочных металлов (трет-бутоксид калия, трет-бутоксид натрия и тому подобные), гидроксидов щелочных металлов (гидроксид калия, гидроксид натрия и тому подобные), и органических оснований, таких как алкиламины (триэтиламин, диизопропиламин, диизопропилэтиламин и тому подобные), пиридиноны (пиридин, диметламинопиридин и тому подобные), циклические амины (морфолин, 4-метилморфолин и тому подобные) и их комбинации. Согласно одному варианту реализации изобретения основание представляет собой карбонат калия (K2CO3). Согласно некоторым вариантам реализации изобретения эквивалент основания составляет от приблизительно 1 эквив. до приблизительно 8 эквив. по отношению к Соединению IV.
[88] Добавление водного раствора основания согласно некоторым вариантам реализации изобретения проводят в течение периода времени, составляющего от по меньшей мере 30 минут до по меньшей мере 1 часа. Было обнаружено, что медленное введение раствора основания имеет решающее значение для выхода реакции сочетания Сузуки. Не связываясь конкретной теорией, это, по-видимому, связано с предотвращением образования соли при образовании двухфазной смеси.
[89] Предыдущие пути синтеза реакции сочетания Сузуки вызывали сложности, связанные с конверсией реакции, проводимой в промышленных масштабах. Было обнаружено, что продувка двухфазной реакционной смеси азотом (N2) путем барботирования N2 непосредственно в реакционную смесь в течение примерно 1 часа для удаления воздуха, такого как кислород, обеспечивала желаемую конверсию реакций. Этот процесс известен как дегазация. Дегазация реакционной смеси также оказалась эффективной для снижения количества примесей Соединения VII на стадии сочетания Сузуки.
[90] Согласно некоторым вариантам реализации изобретения к дегазированной двухфазной реакционной смеси, содержащей Соединение V, добавляют Pd-катализатор и лиганд. В предшествующем способе синтеза использовалась каталитическая система тетракис(трифенилфосфин) палладий (Pd(PPh3)4), полученная путем добавления Pd(OAc)2 и PPh3. Выход реакции Сузуки с использованием каталитической системы Pd(PPh3)4не был оптимальным, как показано посредством среднего выхода Соединения II-OH (от приблизительно 55% до приблизительно 64%).
[91] Дальнейшую оптимизацию каталитической системы проводили для улучшения выхода и снижения примесей Соединения VIII. Как описано в Примере 1 и Таблице 1, оптимизация каталитической системы Pd(PPh3)4показала достижение хорошей конверсии только при значительном увеличении загрузки катализатора (от приблизительно 2 мол.% до приблизительно 10 мол.%, Таблица 1, запись 6) или в случае, когда реакционную смесь нагревали с обратным холодильником в течение значительно большего времени (приблизительно 27 часов, запись 5). Также было отмечено, что при высокой загрузке катализатора количество примеси Соединения VIII было значительно ниже (0,04%, запись 6); однако, высокая загрузка катализатора мешала кристаллизации продукта. Кроме того, снижение температуры реакции сочетания Сузуки показало, что не удалось предотвратить образование примеси Соединения VIII.
[92] Далее, как показано в Примере 2 и Таблице 2, рассматривались различные каталитические системы. Было показано, что определяющим для конверсии реакции является удаление фосфиновых лигандов (Таблица 2, запись 1). Авторы настоящего изобретения обнаружили, что каталитическая система Pd(OAc)2/P(o-tol)3увеличивала выход реакции (приблизительно 80-85%) и степень чистоты продукта (>99%) по сравнению с предшествующей каталитической системой Pd(PPh3)4. Кроме того, с использованием новооткрытой каталитической системы Pd(OAc)2/P(o-tol)3, загрузка катализатора может быть значительно минимизирована от приблизительно 2 мол.% до приблизительно 0,25 мол.%. Было также отмечено, что с использованием обнаруженной каталитической системы дегазация реакционной смеси не влияла на скорость конверсии, степень чистоты продукта или количество Соединения VIII. Исследование оптимизации катализатора согласно Примерам 1-2 указывает на то, что количество примеси Соединения VIII достаточно мало коррелирует с условиями реакции сочетания Сузуки.
[93] Согласно некоторым вариантам реализации изобретения Pd-катализатор и лиганды добавляют к двухфазной реакционной смеси, содержащей Соединение V. Согласно некоторым вариантам реализации изобретения Pd-катализатор может представлять собой частицы Pd(0) или частицы Pd(II). Неограничивающие примеры Pd-катализатора включают теракис(трифенилфосфин) палладий (Pd(PPh3)4), три(дибензилиденацетон) дипалладий, бис(три-т-бутилфосфин) палладий, бис[1,2-бис(дифенилфосфин)этан] палладий, бис(трициклогексилфосфин) палладий, ацетат палладия (Pd(OAc)2), хлорид палладия (PdCl2), дихлорбис(трифенилфосфин) палладий, ацетилацетонат палладия, бромид палладия, йодид палладия, цианид палладия, гидроксид палладия, нитрат палладия, хлорид гидрат тетраамина палладия (II), динитродиамин палладий, ди-μ-хлорбис(η-аллил) палладий, дихлорбис(бензонитрил) палладий, дихлорбис(ацетонитрил) палладий, пропиона палладия, [1,1′-бис(дифенилфосфин)ферроцен] палладий (II) хлорид, тетракис(три-o-толилфосфин) палладий, тетракис(три-т-бутилфосфин) палладий, бис(1,2-бис(дифенилфосфин)этан) палладий, бис(1,1′-бис(дифенилфосфин)ферроцен) палладий, тетракис(триэтилфосфит) палладий и их комбинацию.
[94] Согласно некоторым вариантам реализации изобретения лиганд выбран из группы, состоящей из фосфиновых лигандов (тритолилфосфин, трифенилфосфин, триметилфосфин, триэтилфосфин, триметилфосфит, триэтилфосфит, три-n-бутилфосфит, три-трет-бутилфосфин, ди-трет-бутилметилфосфин и т.д.), азот-содержащих лигандов (пиридин, бипиридин и т.д.), NHC лигандов (содержащие N-гетероциклические карбоны) (N,N′-бис(2,6-диизопропилфенил)имидазол-2-илиден и т.д.), и их комбинаций.
[95] Согласно некоторым вариантам реализации изобретения система Pd-катализатор/лиганд представляет собой Pd(OAc)2/P(o-tol)3. Согласно другим вариантам реализации, Pd-катализатор и лиганд добавляют при непрерывной дегазации реакционной смеси.
[96] Согласно некоторым вариантам реализации изобретения количество используемого Pd-катализатора составляет от приблизительно 0,001 мол.% до приблизительно 10,0 мол.% по отношению к Соединению IV. Согласно одному варианту реализации изобретения количество используемого Pd-катализатора составляет от приблизительно 0,05 мол.% до приблизительно 0,25 мол.% по отношению к Соединению IV.
[97] Согласно некоторым вариантам реализации изобретения соотношение лиганда к Pd-катализатору составляет от приблизительно 1:1 до приблизительно 3:1. Согласно некоторым вариантам реализации изобретения соотношение лиганда к Pd-катализатору составляет приблизительно 2:1.
[98] Согласно некоторым вариантам реализации изобретения Соединение IV добавляют к двухфазной смеси, содержащей Соединение V и систему Pd-катализатор/лиганд. Согласно одному варианту реализации изобретения Соединение IV добавляют при непрерывной дегазации реакционной смеси.
[99] Согласно некоторым вариантам реализации изобретения после добавления Соединения IV реакционную смесь нагревают в течение от приблизительно 2 часов до приблизительно 5 часов и затем охлаждают до температуры окружающей среды. Согласно некоторым вариантам реализации изобретения реакционную смесь нагревают не более чем до 65°C. Было замечено, что при повышении температуры выше 65°C Pd-катализатор теряет активность. Например, реакция Сузуки, проводимая при температуре 90°C, не доходит до завершения. Согласно одному варианту реализации изобретения реакционную смесь нагревали до тех пор, пока не получали результаты анализа ВЭЖХ, указывающие на то, что оставшееся количество Соединения IV составляет ≤2% и на образование Соединения II-OH.
[100] Как только согласно результатам ВЭЖХ реакцию считали завершенной, согласно некоторым вариантам реализации изобретения реакционную смесь охлаждали до температуры окружающей среды и pH реакционной смеси доводили до от приблизительно 2,0 до приблизительно 3,0 используя водные растворы кислоты. Согласно некоторым вариантам реализации используют соляную кислоту (HCl).
[101] Согласно некоторым вариантам реализации изобретения Соединение V представляет собой Соединение V-OMe.
Очистка Соединения II-OH
[102] Согласно предшествующему способу очистки Соединения II-OH требовалось две горячих рекристаллизации и две обработки активированным углем. В описанном способе очистки, согласно некоторым вариантам реализации изобретения, требуется одна обработка активированным углем, одна рекристаллизация с помощью антирастворителя и/или одна горячая рекристаллизация.
[103] Окисленную двухфазную реакционную смесь, содержащую неочищенное Соединение II-OH, согласно некоторым вариантам реализации изобретения разделяют на водный слой и органический слой. Согласно некоторым вариантам реализации изобретения полученный водный слой экстрагируют органическим растворителем. Согласно одному варианту реализации изобретения водный слой экстрагируют толуолом (приблизительно 10 частей).
[104] Объем комбинированных органических слоев согласно некоторым вариантам реализации изобретения снижают до приблизительно 6,5 частей. Согласно некоторым вариантам реализации изобретения объем комбинированного органического слоя снижают методом дистилляции. Полученный уменьшенный органический слой согласно некоторым вариантам реализации изобретения обрабатывают активированным углем. Согласно другим вариантам реализации полученный уменьшенный органический слой обрабатывают активированным углем и целитом. Согласно одному варианту реализации изобретения соотношение активированного угля к целиту составляет приблизительно 1:2 по массе. Согласно некоторым вариантам реализации реакционную смесь, содержащую активированный уголь, перемешивают в течение от приблизительно 1 часа до приблизительно 5 часов при температуре окружающей среды. После этого согласно другим вариантам реализации изобретения активированный уголь отфильтровывают и объем реакционной смеси снижают до приблизительно 3 частей. Согласно одному варианту реализации изобретения объем снижают методом дистилляции.
[105] Согласно некоторым вариантам реализации изобретения для очистки Соединения II-OH применяют рекристаллизацию с помощью антирастворителя. К уменьшенной неочищенной смеси добавляют полярные растворители, такие как изопропанол и этилацетат, и концентрируют до маслянистого состояния. Согласно одному варианту реализации изобретения в течение периода времени, большего, чем 1 час к неочищеной масляной смеси порционно добавляют неполярный антирастворитель. Полученную суспензию перемешивают в течение от приблизительно 1 часа до приблизительно 8 часов. Согласно некоторым вариантам реализации изобретения осажденные кристаллы затем собирают путем фильтрования. Согласно некоторым вариантам реализации изобретения исходный раствор не рециркулируют для удаления остаточных кристаллов из реакционных емкостей; вместо этого могут дополнительно проводить множественную промывку растворителем, используя свежеприготовленный растворитель.
[106] Согласно некоторым вариантам реализации изобретения антирастворитель представляет собой гептаны. Согласно другим вариантам реализации изобретения полярный растворитель представляет собой изопропанол или смесь изопропанола и этилацетата. Согласно некоторым вариантам реализации изобретения продукт осаждают без добавления антирастворителя.
[107] Согласно некоторым вариантам реализации изобретения для очистки Соединения II-OH применяют горячую рекристаллизацию. Неочищенное вещество, содержащее Соединение II-OH или неочищенные кристаллы Соединения II-OH растворяют в полярных растворителях, таких как изопропанол и этилацетат при повышенной температуре. Температуру раствора медленно понижают до температуры окружающей среды и перемешивают до завершения рекристаллизации, а затем кристаллы собирают путем фильтрования.
[108] Согласно некоторым вариантам реализации изобретения используемый полярный растворитель представляет собой изопропанол или смесь изопропанола и этилацетата. Согласно некоторым вариантам реализации изобретения неочищенное Соединение II-OH растворяют в смеси изопропанола и этилацетата в соотношении приблизительно 9:1 при приблизительно 70°C. Согласно другим вариантам реализации изобретения температуру горячего раствора снижают на приблизительно 10°C приблизительно каждый час до достижения температуры окружающей среды. Согласно некоторым вариантам реализации изобретения, как только растворитель охлажден до температуры окружающей среды, раствор перемешивают в течение от приблизительно 2 часов до приблизительно 6 часов. Полученные кристаллы согласно некоторым вариантам реализации изобретения собирают путем фильтрования. Согласно некоторым вариантам реализации изобретения исходный раствор не рециркулируют для удаления остаточных кристаллов из реакционных емкостей; вместо этого могут дополнительно проводить множественную промывку растворителем, используя свежеприготовленный растворитель.
[109] Исследование растворителя для рекристаллизации выявило, что при проведении горячей рекристаллизации только в изопропаноле выход Соединения II-OH был высоким (90-93%) при снижении примесей Соединения VIII на приблизительно 50-60%. При проведении рекристаллизации только в этилацетате выход Соединения II-OH был ниже (70-75%), чем при использовании системы с изопропанолом, однако снижение примеси Соединения VIII было выше (на 80-83%). При проведении рекристаллизации в смеси изопропанола и этилацетата получали как высокий выход Соединения II-OH (90-92%), так и эффективное снижение примесей Соединения VIII (на 75-80%).
[110] Согласно некоторым вариантам реализации изобретения используется как рекристаллизация с помощью антирастворителя, так и горячая рекристаллизция. Согласно некоторым вариантам реализации изобретения комбинирование рекристаллизации с помощью растворителя и горячей рекристаллизации позволяет значительно уменьшить примеси Соединений VIII и IX. Согласно некоторым вариантам реализации изобретения стадии рекристаллизации могут повторяться до достижения желаемой чистоты. Согласно другим вариантам реализации, в результате способа получения Соединения II-OH, описанного в настоящей заявке, чистота Соединения II-OH составляет >97,5% с≤0,20% Соединения VII, с≤0,20% Соединения VIII и с≤0,50% Соединения IX. Согласно некоторым вариантам реализации изобретения в результате способа получения Соединения II-OH, описанного в настоящей заявке, чистота Соединения II-OH составляет >97,5% с≤0,10% Соединения VII, с≤0,10% Соединения VIII и с≤0,25% Соединения IX. Согласно одному варианту реализации изобретения в результате способа получения Соединения II-OH, описанного в настоящей заявке, чистота Соединения II-OH составляет >97,5% с≤0,05% Соединения VII, с≤0,05% Соединения VIII и с≤0,15% Соединения IX.
[111] Согласно одному варианту реализации изобретения в результате способа получения Соединения II-OH, описанного в настоящей заявке, чистота Соединения II-OH составляет >97,5% с≤0,20% Соединения II-OH-A, с≤0,20% Соединения II-OH-B, и с≤0,50% Соединения II-OH-C. Согласно другому варианту реализации в результате способа получения Соединения II-OH, описанного в настоящей заявке, чистота Соединения II-OH составляет >97,5% с≤0,10% Соединения II-OH-A, с≤0,10% Соединения II-OH-B и с≤0,25% Соединения II-OH-C. Согласно некоторым вариантам реализации изобретения в результате способа получения Соединения II-OH, описанного в настоящей заявке, чистота Соединения II-OH составляет >97,5% с≤0,05% Соединения II-OH-A, с≤0,05% Соединения II-OH-B и с≤0,15% Соединения II-OH-C.
[112] Согласно одному варианту реализации изобретения в результате способа получения Соединения II-OH, описанного в настоящей заявке, чистота Соединения II-OH составляет >98,0%. Согласно одному варианту реализации изобретения в результате способа получения Соединения II-OH, описанного в настоящей заявке, чистота Соединения II-OH составляет >99,0%.
Получение Соединения I
[113] Сложности, связанные с предшествующим способом получения Соединения I и затем Соединения I-MsOH, были связаны с присутствием Соединения II-OH (исходного вещества) в конечном продукте. Было обнаружено, что образование Соединения II-OH зависит от нескольких стадий или условий реакции. Прежде всего, образование хлорангидридного Соединения II-Cl (Соединение II, где R1=Cl). Во-вторых, выбор растворителя для реакции, влияющий на количество получаемого Соединения II-OH. В-третьих, относится к стадии образования соли. Заявленный способ, описанный в настоящей заявке, позволяет решить указанные проблемы и описывает протоколы, позволяющие значительно снизить образование Соединения II-OH.
[114] Согласно некоторым вариантам реализации изобретения Соединение I получают путем проведения реакции между Соединением II и Соединением III. Согласно некоторым вариантам реализации изобретения Соединение II-OH реагирует с хлорирующим реагентом с образованием Соединения II-Cl. Согласно некоторым вариантам реализации изобретения Соединение II-Cl реагирует с Соединением III с образованием Соединения I.
[115] Согласно некоторым вариантам реализации изобретения Соединение II-OH растворяют в растворителе и добавляют хлорирующий реагент для получения Соединения II-Cl. Согласно некоторым вариантам реализации изобретения используемый растворитель включает, но не ограничивается указанными, тетрагидрофуран (ТГФ), диметилформамид (DMF), диэтиловый эфир и метилен хлорид (DCM). Согласно одному варианту реализации изобретения указанный растворитель представляет собой метилен хлорид.
[116] В предшествующем способе ТГФ использовали в качестве растворителя для образования хлорангидрида с добавлением DMF. Было обнаружено, что образование Cоединения II-OH может быть сведено к минимуму, в случае если DCM используется в качестве растворителя для образования хлорангидрида.
[117] Перед добавлением хлорирующего реагента раствор, содержащий Соединение II-OH, охлаждают ниже температуры окружающей среды. Согласно некоторым вариантам реализации изобретения раствор, содержащий соединение II-OH, охлаждают до от приблизительно 10°C до приблизительно 15°C. Согласно некоторым вариантам реализации изобретения хлорирующий реагент добавляют в течение более чем от приблизительно 10 минут до приблизительно 30 минут, в то время как температура раствора поддерживается ниже температуры окружающей среды. Согласно некоторым вариантам реализации изобретения смесь поддерживают при температуре от приблизительно 10°C до приблизительно 15°C и перемешивают в течение от примерно 2 часов до примерно 4 часов, затем охлаждают примерно до 0°C или ниже. Согласно одному варианту реализации изобретения реакционную смесь перемешивают до тех пор, пока анализ ВЭЖХ не показал наличие≤3,0% Соединения II-OH.
[118] Неограничивающие примеры хлорирующих реагентов включают тионил хлорид, трихлорид фосфора, пентахлорид фосфора, оксихлорид фосфора, оксалил хлорид, фосген и тому подобные или их комбинации. Согласно одному варианту реализации изобретения хлорирующий реагент представляет собой тионил хлорид. Согласно другому варианту реализации хлорирующий реагент используют в количестве от приблизительно 1,0 эквив. до приблизительно 2,0 эквив. по отношению к Соединению II-OH. Согласно одному варианту реализации изобретения хлорирующий реагент используют в количестве от приблизительно 1,0 эквивалента до приблизительно 1,1 эквив. по отношению к Соединению II-OH. Согласно другому варианту реализации соотношение хлорирующего агента и Соединения II-OH составляет приблизительно 1:1.
[119] В отдельной реакционной емкости Соединение III растворяют в растворителе с основанием. К раствору Соединения III и основанию медленно добавляют раствор Соединения II-Cl. Согласно некоторым вариантам реализации изобретения растворитель, используемый для растворения Соединения III, может представлять собой тетрагидрофуран, диметилформамид, диэтиловый эфир, метилен хлорид и их смеси. Согласно одному варианту реализации изобретения растворитель представляет собой метилен хлорид. Согласно некоторым вариантам реализации изобретения реакционную смесь охлаждают до приблизительно 0°C перед добавлением Соединения III. Согласно одному варианту реализации изобретения реакционную смесь выдерживают при 0°C в течение от приблизительно 3 часов до приблизительно 7 часов после добавления Соединения III до те х пор, пока анализ ВЭЖХ не покажет присутствие≤0,5% Соединения II-Cl. Согласно другому варианту реализации Соединение III используют в количестве от приблизительно 1,0 эквив. до приблизительно 1,2 эквив. по отношению к Соединению II-OH.
[120] Согласно некоторым вариантам реализации изобретения основание используют в количестве от приблизительно 1 эквив. до приблизительно 4 эквив. Неограничивающие примеры основания включают карбонаты щелочных металлов (карбонат калия, карбонат натрия, карбонат цезия и тому подобные), гидрокарбонаты щелочных металлов (бикарбонат калия, бикарбонат натрия и тому подобные), ацетаты щелочных металлов (ацетат калия, ацетат натрия и тому подобные), фосфаты щелочных металлов (фосфат калия, фосфат натрия и тому подобные), фториды щелочных металлов (фторид калия, фторид цезия и тому подобные), алкоксиды щелочных металлов (трет-бутоксид калия, трет-бутоксид натрия и тому подобные), гидроксиды щелочных металлов (гидроксид калия, гидроксид натрия и тому подобные), и органические основания, такие как алкиламины (триэтиламин, диизопропиламин, диизопропилэтиламин и тому подобные), пиридины (пиридин, диметиламинопиридин и тому подобные), циклические амины (морфолин, 4-метилморфолин и тому подобные), и их комбинации. Согласно одному варианту реализации изобретения основание представляет собой пиридин. Согласно некоторым вариантам реализации изобретения пиридин вступает в реакцию с Соединением II-Cl с образованием соли пиридин-HCl и для предотвращения агрегации соли может потребоваться энергичное перемешивание.
[121] Согласно одному варианту реализации изобретения после указания на превращение Соединения II-Cl в Соединение I реакционную смесь подкисляли. Согласно некоторым вариантам реализации изобретения для подкисления реакционной смеси, содержащей неочищенное Соединение I, использовали раствор лимонной кислоты. Согласно одному варианту реализации изобретения лимонную кислоту использовали в количестве от приблизительно 1,5 эквив. до приблизительно 2,0 эквив. в приблизительно 10 частях воды по отношению к Соединению II-OH и добавляли ее более чем от приблизительно 30 минут до приблизительно 1 часа. Согласно одному варианту реализации изобретения охлажденный водный раствор лимонной кислоты добавляли к охлажденной реакционной смеси при сохранении внутренней температуры приблизительно 0°C.
[122] Согласно некоторым вариантам реализации изобретения летучий растворитель удаляли для получения объема приблизительно 13 частей. Согласно другим вариантам реализации изобретения к уменьшенной реакционной смеси добавляли различные растворители (приблизительно 5 частей) и объем снижали снова с получением общего объема приблизительно 13 частей. Согласно некоторым вариантам реализации изобретения применяют полярный растворитель, такой как этилацетат. Согласно другому варианту реализации изобретения растворитель удаляли при пониженном давлении.
[123] Уменьшенную реакционную смесь, в которой преобладает водный кислотный слой, согласно некоторым вариантам реализации изобретения экстрагируют полярным растворителем, таким как этилацетат в количестве приблизительно 10 частей. Согласно некоторым вариантам реализации изобретения органический слой, содержащий желаемый продукт, Соединение I, промывают водным раствором несколько раз, например, раствором бикарбоната натрия и солевым раствором.
[124] Исследовали стабильность Соединения I во время обработки. Было показано, что Соединение I не показало во время исследования особенной чувствительности к свету, и применение прозрачной реакционной емкости или амбровой реакционной емкости не показало усиленного гидролиза Соединения I до Соединения II-OH. Кроме того, Соединение I во время обработки исследовали при различных pH и температуре; однако, не было выявлено корреляции с повышенным гидролизом Соединения I до Соединения II-OH. Хотя существует вероятность, что Соединение I может гидролизоваться в Соединение II-OH во время обработки, амидная связь в этих условиях довольно стабильна.
[125] Было обнаружено, что содержание воды в органическом слое, полученном в результате экстракционной обработки, оказывает влияние на общий выход образования соли Соединения I (Соединение I-MsOH). Согласно некоторым вариантам реализации изобретения присутствие воды во время образования соли усиливало гидролиз Соединения I в Соединение II-OH, поэтому сушка в жестких условиях является идеальной. Согласно некоторым вариантам реализации изобретения органический слой, содержащий Соединение I, высушивают с помощью 3 Å порошковых молекулярных сит. Согласно некоторым вариантам реализации изобретения полученную суспензию перемешивают в течение от приблизительно 15 часов до приблизительно 30 часов при температуре окружающей среды перед удалением молекулярных сит методом фильтрования. Отфильтрованные молекулярные сита промывают полярным растворителем, таким как этилацетат. Согласно некоторым вариантам реализации изобретения оставшееся содержание воды определяют с помощью титрования. Согласно некоторым вариантам реализации изобретения стадию сушки с использованием молекулярных сит могут повторять до тех пор, пока остаток воды не составит≤2,5%.
[126] Сразу после высушивания органического слоя, содержащего Соединение I, и определения того факта, что в нем по существу отсутствует вода, согласно некоторым вариантам реализации изобретения растворитель удаляют с получением общего объема приблизительно 3 части. Согласно некоторым вариантам реализации изобретения растворитель удаляют методом дистилляции. Согласно другому варианту реализации раствор анализируют с помощью ВЭЖХ до или после снижения количества растворителя для расчета количества присутствующего Соединения I.
Получение Соединения I-MsOH
[127] К концентрированному неочищенному раствору Соединения I согласно некоторым вариантам реализации изобретения добавляют приблизительно 4 части растворителя. Согласно одному варианту реализации изобретения применяемый растворитель представляет собой ацетонитрил. К раствору, содержащему Соединение I, добавляют метансульфоновую кислоту (MsOH). Согласно некоторым вариантам реализации изобретения MsOH добавляют в виде одной части. Согласно другим вариантам реализации, MsOH применяют в количестве от приблизительно 0,9 эквив. до приблизительно 1,5 эквив. по отношению к Соединению I, как определено с помощью анализа ВЭЖХ. Согласно одному варианту реализации изобретения MsOH применяют в количестве от приблизительно 0,97 эквив. до приблизительно 1,02 эквив.
[128] Согласно некоторым вариантам реализации изобретения MsOH смывают в раствор, содержащий Соединение I и MsOH с дополнительным растворителем, таким как ацетонитрил или этилацетат. Реакционную смесь перемешивают при температуре окружающей среды в течение от приблизительно 30 минут до приблизительно 1 часа. Было обнаружено, что избыток MsOH отрицательно влияет на образование Соединения II-OH посредством гидролиза амидной связи Соединения I, поэтому точный анализ соединения I имеет решающее значение для определения точного количества присутствующего соединения I и точного количества MsOH для достижения стехиометрического соотношения 1: 1 при образовании соли. Согласно одному варианту реализации изобретения Соединение I и MsOH используют в соотношении 1:1 для снижения гидролиза амидной связи.
[129] Согласно одному варианту реализации изобретения растворитель, используемый на стадии превращения Соединения I в Соединение I-MsOH, не содержит спиртовых растворителей. Было обнаружено, что остаточные уровни спиртовых растворителей (например, метанола, этанола и тому подобных) в реакционной смеси приводят к нежелательным примесям Соединения I-MsOH в виде мезилатных эфиров. Такие полученные мезилатные эфиры являются известными мутагенами.
[130] Согласно некоторым вариантам реализации изобретения перед кристаллизацией реакционную смесь промывают солевым раствором и высушивают с помощью 3 Å молекулярных сит. В некоторых случаях было определено, что присутствие небольшого количества воды в реакционной смеси способно предотвратить кристаллизацию и/или привести к более низкому выходу Соединения I-MsOH. Не связываясь какой-либо теорией, более низкий выход в системах с более высоким содержанием воды обусловлен большей скоростью гидролиза для получения Соединения II-OH, которое в исходном растворе при более высокой концентрации было обнаружено в исследовании с более высоким содержанием воды.
[131] Для кристаллизации Соединения I-MsOH из реакционной смеси согласно некоторым вариантам реализации изобретения в качестве затравки используют чистую пробу Соединения I-MsOH. Раствор, с затравкой или без нее, согласно некоторым вариантам реализации изобретения перемешивают при температуре окружающей среды в течение от приблизительно 6 часов до приблизительно 10 часов. Кроме того, согласно некоторым вариантам реализации изобретения раствор перемешивают при приблизительно 0°C в течение от приблизительно 6 часов до приблизительно 10 часов. Осажденные кристаллы согласно некоторым вариантам реализации изобретения собирают путем фильтрования. Согласно некоторым вариантам реализации изобретения кристаллы промывают холодным растворителем, таким как этилацетат, с получением неочищенного Соединения I-MsOH.
[132] Неочищенные кристаллы Соединения I-MsOH согласно некоторым вариантам реализации изобретения дополнительно очищают с использованием методики перекристаллизации. Согласно некоторым вариантам реализации изобретения кристаллы Соединения I-MsOH растворяют в растворителях (приблизительно 10 частей) при повышенной температуре. Согласно другим вариантам реализации, кристаллы соединения I-MsOH растворяют в ацетонитриле при приблизительно 70°C. Горячий раствор Соединения I-MsOH медленно охлаждают до от приблизительно 50°C до приблизительно 55°C в течение периода времени, большего приблизительно 1 часа. Согласно некоторым вариантам реализации изобретения раствор I-MsOH затравляют пробой Соединения I-MsOH без примесей при от приблизительно 50°C до приблизительно 55°C. Раствор, с затравкой или без нее, перемешивают при от приблизительно 50°C до приблизительно 55°C в течение от приблизительно 4 часов до приблизительно 8 часов согласно некоторым вариантам реализации. Горячий раствор согласно некоторым вариантам реализации изобретения охлаждают до температуры окружающей среды в течение более чем приблизительно 1 часа и перемешивают при температуре окружающей среды в течение от приблизительно 6 часов до приблизительно 10 часов. Согласно одному варианту реализации изобретения горячая рекристаллизация Соединения I-MsOH из ацетонитрила снижает количество примесей, включая мезилатные сложные эфиры.
[133] Осажденные кристаллы Соединения I-MsOH согласно некоторым вариантам реализации изобретения собирают путем фильтрования. Согласно другим вариантам реализации отфильтрованные кристаллы Соединения I-MsOH промывают ацетонитрилом. Согласно одному варианту реализации изобретения отфильтрованные кристаллы Соединения I-MsOH промывают холодным ацетонитрилом. Степень чистоты кристаллов анализируют путем титрования и ВЭЖХ. В случае необходимости горячую рекристаллизацию можно повторять до достижения желательной степени чистоты. Согласно некоторым вариантам реализации изобретения отфильтрованные кристаллы Соединения I-MsOH высушивают при пониженном давлении. Согласно другим вариантам реализации высушенные кристаллы дополнительно измельчают порошковой мельницей и струйной мельницей или т.п.
[134] Исследования кристаллов Соединения I-MsOH под микроскопом выявили, что поверхность кристаллов со временем стала маслянистой, что считается результатом гидролиза на поверхности кристаллов. Было обнаружено, что ацетонитрил представляет собой растворитель, в котором Соединение II-OH растворимо лучше, чем Соединение I-MsOH. Таким образом, после рекристаллизации целесообразно промыть отфильтрованные кристаллы ацетонитрилом. В связи с тем, что Соединение I-MsOH также до некоторой степени растворимо в ацетонитриле, согласно некоторым вариантам реализации изобретения для промывки кристаллов следует использовать холодный ацетонитрил, а объем и частоту промывки следует ограничить до приблизительно двух раз от приблизительно 2 объемных частей до приблизительно 3 объемных частей.
[135] Гидролиз Соединения I или Соединения I-MsOH чувствителен в присутствии воды или кислоты. Согласно некоторым вариантам реализации изобретения реакционная смесь по существу не должна содержать воды до и во время стадий очистки Соединения I-MsOH. Согласно другим вариантам реализации изобретения реакционная смесь по существу не должна содержать водной кислоты до и во время стадий очистки Соединения I-MsOH. Согласно некоторым вариантам реализации изобретения стадии образования соли и очистки Соединения I-MsOH должны проводиться при осторожном перемешивании.
[136] Согласно некоторым вариантам реализации изобретения Соединение III, используемое в реакции получения Соединения I или Соединения I-MsOH является оптически чистым. В этом случае получают оптически чистое Соединение I или оптически чистое Соединение I-MsOH. Согласно одному варианту реализации изобретения Соединение III представляет собой (S)-Соединение III. Согласно другому варианту реализации Соединение I-MsOH представляет собой (S)-Соединение I-MsOH.
[137] Описанный способ получения Соединения II-OH и его последующее применение в описанном способе получения Соединения I-MsOH согласно некоторым вариантам реализации изобретения приводит к получению Соединения I-MsOH высокой чистоты, по существу не содержащему Соединений I-MsOH-A, I-MsOH-B, I-MsOH-C, I-MsOH-D, I-MsOH-E, I-MsOH-F, I-MsOH-G, II-OH, III, VI, VII, VIII, IX и мезилатных эфиров, полученных из MsOH. Согласно некоторым вариантам реализации изобретения Соединение I-MsOH, полученное, например, способом, описанным в настоящей заявке, характеризуется степенью чистоты >96%. Согласно другим вариантам реализации изобретения Соединение I-MsOH, полученное, например, способом, описанным в настоящей заявке, характеризуется степенью чистоты >97%. Согласно одному варианту реализации изобретения Соединение I-MsOH, полученное, например, способом, описанным в настоящей заявке, характеризуется степенью чистоты >98%. Согласно другому варианту реализации Соединение I-MsOH, полученное, например, способом, описанным в настоящей заявке, характеризуется степенью чистоты >99%.
[138] Описанный способ получения Соединения II-OH и его последующее применение в описанном способе получения Соединения I-MsOH согласно некоторым вариантам реализации изобретения приводит к получению (S)-Соединения I-MsOH высокой чистоты, по существу не содержащему (R)-Соединения I-MsOH, R или S изомеров (I-MsOH-A, I-MsOH-B, I-MsOH-C, I-MsOH-D, I-MsOH-E, I-MsOH-F, I-MsOH-G), II-OH, III, VI, VII, VIII, IX и мезилатных эфиров, полученных из MsOH. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, полученное, например, способом, описанным в настоящей заявке, характеризуется степенью чистоты >96%. Согласно другим вариантам реализации, (S)-Соединение I-MsOH, полученное, например, способом, описанным в настоящей заявке, характеризуется степенью чистоты >97%. Согласно одному варианту реализации изобретения (S)-Соединение I-MsOH, полученное, например, способом, описанным в настоящей заявке, характеризуется степенью чистоты >98%. Согласно другому варианту реализации (S)-Соединение I-MsOH, полученное, например, способом, описанным в настоящей заявке, характеризуется степенью чистоты >99%.
[139] Согласно другим вариантам реализации, Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,2% каждой примеси, включая Соединения I-MsOH-A, I-MsOH-B, I-MsOH-C, I-MsOH-F, I-MsOH-G, VII, VIII и IX. Согласно другим вариантам реализации изобретения Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤1,0%,≤0,8%,≤0,6% или≤0,4% каждой примеси, включая I-MsOH-D и Соединение II-OH. Согласно другим вариантам реализации изобретения Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤1500 м.д. Соединения III. Согласно другому варианту реализации Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,3% каждой примеси, включая I-MsOH-C, I-MsOH-E и I-MsOH-F. Согласно некоторым вариантам реализации изобретения Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,002% (20 м.д.) мезилатного эфира, полученного из MsOH. Согласно некоторым вариантам реализации изобретения Соединение I-MsOH содержит≤0,002% (20 м.д.) мезилатного эфира на дозу в 150 мг. Согласно некоторым вариантам реализации изобретения Соединение I-MsOH содержит≤15 м.д. мезилатного сложного эфира на дозу в 150 мг. Согласно одному варианту реализации изобретения Соединение I-MsOH содержит≤0,001% (10 м.д.) мезилатного сложного эфира на дозу в 150 мг.
[140] Согласно другим вариантам реализации, Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,3% каждой из примесей, включая Соединения I-MsOH-A, I-MsOH-B, I-MsOH-C, I-MsOH-F, I-MsOH-G, VII, VIII, и IX. Согласно другим вариантам реализации изобретения Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0.5% каждой из примесей, включая I-MsOH-D и Соединение II-OH. Согласно другим вариантам реализации, Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤1000 м.д. Соединения III. Согласно другому варианту реализации Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,15% каждой из примесей, включая Соединения I-MsOH-C, I-MsOH-E и I-MsOH-F, VII, VIII и IX. Согласно некоторым вариантам реализации изобретения Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,001% (10 м.д.) мезилатного эфира, полученного из MsOH.
[141] Согласно другим вариантам реализации, Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,05% каждой из примесей, включая Соединения I-MsOH-A, I-MsOH-B, I-MsOH-C, I-MsOH-F, I-MsOH-G, VII, VIII и IX. Согласно другим вариантам реализации изобретения Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,30% каждой из примесей, включая Соединение I-MsOH-D и Соединение II-OH. Согласно некоторым вариантам реализации изобретения Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,1% каждой из примесей, включая I-MsOH-A, I-MsOH-B, I-MsOH-C, I-MsOH-F, I-MsOH-G, VII, VIII и IX. Согласно другим вариантам реализации изобретения Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,15% каждой из примесей, включая Соединение I-MsOH-D и Соединение II-OH. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤1,0% (R)-Соединения I-MsOH. Согласно другому варианту реализации (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,5% (R)-Соединения I-MsOH. Согласно одному варианту реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,25% (R)-Соединения I-MsOH. Согласно одному варианту реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,20% (R)-Соединения I-MsOH.
[142] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤5,0 мас.% воды, как определено с помощью Фармакопеи (ФСША) <921>, способ 1C. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤2,5 мас.% воды, как определено с помощью ФСША <921>, способ 1C. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤2,0 мас.% воды, как определено с помощью ФСША <921>, способ 1C. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤1,0 мас.% воды, как определено с помощью ФСША <921>, способ 1C.
[143] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤20 мас.% метансульфоновой кислоты. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤15 мас.% метансульфоновой кислоты. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤13 мас.% метансульфоновой кислоты. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит от приблизительно 5% до приблизительно 15 мас.% метансульфоновой кислоты. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит от приблизительно 11% до приблизительно 13 мас.% метансульфоновой кислоты.
[144] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤500 м.д. ацетонитрила в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤425 м.д. ацетонитрила в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤410 м.д. ацетонитрила в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤350 м.д. ацетонитрила в качестве остаточного растворителя.
[145] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤7500 м.д. этилацетата в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤5000 м.д. этилацетата в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤4000 м.д. этилацетата в качестве остаточного растворителя.
[146] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤300 м.д. пиридина в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤200 м.д. пиридина в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤100 м.д. пиридина в качестве остаточного растворителя.
[147] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤750 м.д. дихлорметана в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤600 м.д. дихлорметана в качестве остаточного растворителя. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤500 м.д. дихлорметана в качестве остаточного растворителя.
[148] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤1,0 м.д. элементных примесей кадмия, как определено с помощью ФСША <232> и/или ≤1,0 м.д. свинца. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤0,5 м.д. элементных примесей кадмия, как определено с помощью ФСША <232> и/или ≤0,5 м.д. свинца. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤0,25 м.д. элементных примесей кадмия, как определено с помощью ФСША <232> и/или ≤0,25 м.д. свинца.
[149] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤2,0 м.д. элементных примесей мышьяка, как определено с помощью ФСША <232>. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤1,5 м.д. элементных примесей мышьяка, как определено с помощью ФСША <232>. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤1,0 м.д. элементных примесей мышьяка, как определено с помощью ФСША <232>.
[150] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤10,0 м.д. элементных примесей ртути, как определено с помощью ФСША <232> и/или ≤10,0 м.д. кобальта. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤5,0 м.д. элементных примесей ртути, как определено с помощью ФСША <232> и/или ≤5,0 м.д. кобальта. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤3,0 м.д. элементных примесей ртути, как определено с помощью ФСША <232> и/или ≤2,5 м.д. кобальта. Согласно одному варианту реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤2,0 м.д. элементных примесей ртути, как определено с помощью ФСША <232>. Согласно одному варианту реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤2,0 м.д. элементных примесей кобальта, как определено с помощью ФСША <232>.
[151] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤20,0 м.д. элементных примесей ванадия, как определено с помощью ФСША <232> и/или ≤20,0 м.д. палладия. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤10,0 м.д. элементных примесей ванадия, как определено с помощью ФСША <232> и/или ≤10,0 м.д. палладия. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤5,0 м.д. элементных примесей ванадия, как определено с помощью ФСША <232> и/или ≤5,0 м.д. палладия.
[152] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤30,0 м.д. элементных примесей никеля, как определено с помощью ФСША <232>. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤20,0 м.д. элементных примесей никеля, как определено с помощью ФСША <232>. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤10,0 м.д. элементных примесей никеля, как определено с помощью ФСША <232>.
[153] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤1500 м.д. элементных примесей хрома, как определено с помощью ФСША <232>. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит≤1250 м.д. элементных примесей хрома, как определено с помощью ФСША <232>. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤1100 м.д. элементных примесей хрома, как определено с помощью ФСША <232>. Согласно одному варианту реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤1000 м.д. элементных примесей хрома, как определено с помощью ФСША <232>.
[154] Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤500 м.д. элементных примесей молибдена, как определено с помощью ФСША <232>. Согласно некоторым вариантам реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤300 м.д. элементных примесей молибдена, как определено с помощью ФСША <232>. Согласно одному варианту реализации изобретения (S)-Соединение I-MsOH, например, полученное согласно описанному способу, содержит ≤250 м.д. элементных примесей молибдена, как определено с помощью ФСША <232>.
ПРИМЕРЫ
[155] Если не указано иное, чистоту соединений определяли с использованием стандартного анализа ВЭЖХ. Например, применяли колонку Capcellpak C18 (Shisedo), имеющую размеры 4,6 см x 150 см, 5 микрон, с PDA детектором 290 нм. Устанавливали температуру колонки 40°C, и две подвижные фазы представляли собой A: 100% 0,05M NH4OAc в воде и B: 100% ацетонитрил. Устанавливали скорость потока при 1,0 мл/мин при времени записи хроматограммы 45-60 минут для одной пробы. Объем вводимой пробы составлял 10 мкл. В другой системе применяли прибор Кларка с PDA детектором 293 нм. Объем вводимой пробы составлял 20 мкл, а время записи хроматограммы составляло 120 минут для одной пробы.
[156] Пример 1: Оптимизация реакции сочетания Сузуки с помощью системы Pd(PPh3)4
[157] В Таблице 1 показаны попытки оптимизации реакции сочетания Сузуки с использованием каталитической системы Pd(PPh3)4 между Соединением IV и Соединением V-OMe. В реакции, представленной в Таблице 1, применяли Соединение IV (5 г, 1 эквив.), Соединение V-OMe (2 эквив.) и основание (6,3 эквив.) в растворителе (соотношение об/мас. по отношению к Соединению IV) и нагревали с обратным холодильником. Такая серия экспериментов показала, что изменение условий реакции приводило к некоторому снижению количества примеси Соединения VIII с использованием системы Pd(PPh3)4, хотя в большинстве случаев наблюдалось замедление или невозможность рекристаллизации продукта.
Таблица 1
[158] Пример 2: Оптимизация реакции сочетания Сузуки с Pd каталитической системой
[159] В Таблице 2 показаны попытки оптимизации реакции сочетания Сузуки с использованием каталитической системы Pd(PPh3)4 между Соединением IV и Соединением V-OMe. В реакции, представленной в Таблице 2, применяли Соединение IV (5 г, 1 эквив.), Соединение V-OMe (2 эквив.) и основание (6,3 эквив.) в растворителе (соотношение об/мас. по отношению к Соединению IV) и нагревали с обратным холодильником. Согласно результатам, представленным в Таблице 2, в системе Pd(OAc)2/P(o-tol)3 применяется значительно меньшее количество катализатора, значительно меньшее количество фосфинового лиганда, и она, как правило, всегда доходит до завершения в течение 2 часов без видимых замедлений даже в отсутствии дегазации. Каталитическая система Pd(OAc)2/P(o-tol)3позволяла получить Соединение II-OH с увеличенным выходом и увеличенной степенью чистоты (>99%) по сравнению с оригинальной каталитической системой Pd(PPh3)4. Кроме того, серия экспериментов также показала, что изменение условий реакции приводило к значительному снижению количества примеси Соединения VIII по сравнению с системами Pd(PPh3)4.
Таблица 2
1Показаны результаты двух различных экспериментов.2Соединение II-OH измельчали.
[160] Пример 3: Получение Соединения II-OH
[161] Безводный тетрагидрофуран (ТГФ, 9 частей) добавляли к магнию (0,185 кг, 2,15 эквив.) и раствор перемешивали в течение 1 часа. ТГФ удаляли методом дистилляции до достижения общего объема раствора, составлявшего приблизительно 3 части. К получившемуся раствору добавляли Соединение VI (0,775 кг, 0,4 эквив.), не содержащее примесей, и раствор нагревали до приблизительно 66°C в течение 2 часов. Реакционную смесь охлаждали до приблизительно 55°C и добавляли дополнительный безводный ТГФ (5 частей). К горячему раствору добавляли Соединение VI (1,163 кг, 1,6 эквив.), не содержащее примесей, в течение более чем 1 часа, и смесь перемешивали при приблизительно 55°C в течение приблизительно 4 часов с образованием реактива Гриньяра. После того, как согласно анализу ВЭЖХ было показано, что оставшееся количество Соединения VI составляло менее 1%, реакционную смесь охлаждали приблизительно до -25°C. К охлажденной реакционной смеси порционно добавляли триметоксиборан (0,739 кг, 2,0 эквив.), не содержащий примесей в течение 2 часов. Полученную смесь перемешивали при -25°C в течение 1 часа, а затем нагревали до приблизительно 20°C и перемешивали в течение 1 часа с получением Соединения V-OMe.
[162] К реакционной смеси, содержащей соединение V-OMe, порционно добавляли раствор карбоната калия (3,64 кг, 6,25 эквив.) в воде (25 мл) в течение более 1 часа. Двухфазный раствор дегазировали азотом в течение 1 часа, затем добавляли ацетат палладия (0,002 кг, 0,0025 эквив.) и три-o-толилфосфин (0,0054 кг, 0,0050 эквив.) при продолжающейся дегазации. Затем при продолжающейся дегазации добавляли Соединение IV (1,200 кг, 1,0 эквив.). Полученную в результате реакционную смесь перемешивали при 65°C или ниже в течение 4 часов или до тех пор, пока результаты анализа ВЭЖХ не показывали, что количество оставшегося Соединения IV составляло ≤2%. Как только реакцию считали завершенной, реакционную смесь охлаждали до температуры окружающей среды.
[163] Реакционную смесь подкисляли с помощью водной соляной кислоты до тех пор, пока pH не доводили до приблизительно 2,0-3,0. После подкисления слои разделяли и водный слой экстрагировали толуолом (10 частей). Объединенные органические слои дистиллировали до достижения приблизительного объема 6,5 частей, затем добавляли целит (Celite®) (0,6 мас.%, 0,720 кг) и Draco KBG (0,3 мас.%, 0,360 кг, активированный уголь) и перемешивали в течение 3 часов при приблизительно 20°C. Активированный уголь и целит удаляли путем фильтрования и фильтрат концентрировали при пониженном давлении с получением объема приблизительно 3 части.
[164] К указанному раствору добавляли изопропанол (5 частей) и смесь вновь концентрировали до объема 3 части. К полученному маслу порционно добавляли гептаны (12 частей) в течение более чем 1 часа. Полученную суспензию перемешивал при приблизительно 20°C в течение 6 часов и собирали кристаллы путем фильтрования.
[165] Неочищенные кристаллы, собранные путем фильтрования, затем растворяли в этилацетате (0,4 части) и изопропаноле (3,6 частей) при 70°C. Температуру раствора снижали на 10°C чаждый час до достижения температуры 20°C. Раствор перемешивали при 20°C в течение 4 часов и кристаллы собирали путем фильтрования и промывали гептанами. Соединение II-OH высушивали с получением 0,938 кг твердого вещества желтого цвета (выход 58,5%, степень чистоты 99,42%).
[166] Способ очистки ВЭЖХ:
Колонка: Capcellpak C18, Shisedo, 4,6×150 см, 5 микрон
Детекторная длина волны: PDA 290 нм
Температура колонки: 40°C
Подвижная фаза: A: 100% 0,05M NH40Ac в воде
B: 100% ACN
Скорость потока: 1,0 мл/мин
Время записи хроматограммы: 45 минут.
Объем вводимой пробы: 10 мкл
Таблица градиента:
Соединение VI=8,3 минуты
Соединение V=2,3-2,6 минуты (три компонента в смеси: Соединение V-(OMe)2, Соединение V-(OMe)(Ar1), и Соединение V-(Ar1)(Ar2))
Соединение IV= 3,0 минуты
Соединение II-OH=8,3 минуты; чистота=99,42%.
[167] Пример 4: Синтез соединения I-MsOH
[168] Соединение II-OH (34,7 кг, 1,0 эквив.) растворяли в дихлорметане (5 частей) и охлаждали до приблизительно 10-15°C. Порционно добавляли тионил хлорид без примесей (10,1 кг, 1,10 эквив.) в течение более, чем 10 минут, и смесь перемешивали при приблизительно 10-15°C в течение 3 часов. После того, как анализ ВЭЖХ показал, что остаток Соединения II-OH составлял ≤3%, реакционную смесь охлаждали до 0°C. Раствор (S)-Соединения III (21,2 кг, 1,05 эквив.) и пиридина (21,3 кг, 3,5 эквив.) в дихлорметане (6 частей) отдельно подготавливали и охлаждали до 0°C. К раствору (S)-Соединения III медленно добавляли раствор хлорангидрида при 0°C и перемешивали в течение 5 часов.
[169] По завершении реакции, которое определяли посредством анализа ВЭЖХ, свидетельствующего о том, что количество Соединения II-Cl оставляло≤0,5%, в течение 30 минут добавляли охлажденный раствор лимонной кислоты (27,7 кг, 1,7 эквив.) в воде (10 частей), при сохранении внутренней температуры 0°C. Дихлорметан удаляли при пониженном давлении до общего объема, приблизительно составляющего 13 частей, затем добавляли этилацетат (5 частей) и объем снова снижали под давлением до приблизительно 13 частей. Полученный остаток экстрагировали этилацетатом (10 частей) и органический слой промывали водным раствором бикарбоната натрия (41,7 кг, 6,45 эквив.) в воде (10 частей) и повторяли промывку. Органический слой дополнительно промывали солевым раствором (10 частей).
[170] К полученному в результате органическому слою добавляли 3Å порошкообразные молекулярные сита (100 мас.%, 34,8 кг) и суспензию перемешивали в течение 20 часов, а затем фильтровали. Фильтровальный осадок промывали этилацетатом (2 части). Высушенный органический слой, содержащий Соединение I, анализировали методом ВЭЖХ для определения количества. К раствору добавляли ацетонитрил (4 части), затем добавляли метансульфоновую кислоту (6,9 кг, 1,01 эквив.) в виде одной порции. Этилацетат (1 часть) использовали для переноса всей метансульфоновой кислоты. Смесь перемешивали при 20°C в течение приблизительно 30 минут.
[171] Затем реакционную смесь затравляли (S)-Соединением I-MsOH и смесь перемешивали при 20°C в течение 8 часов. Осажденные кристаллы собирали путем фильтрования и промывали охлажденным этилацетатом (1 часть). Неочищенные кристаллы растворяли в ацетонитриле (10 частей) при 70°C и раствор охлаждали до 50-55°C в течение более чем 1 часа и затравляли (S)-Соединением I-MsOH. Раствор перемешивали при 50-55°C в течение 6 часов, затем охлаждали до 20°C в течение более 1 часа, затем перемешивали в течение 8 часов. Осажденные кристаллы собирали путем фильтрования и дважды промывали охлажденным ацетонитрилом (2,5 части каждый). Кристаллы высушивали с получением 47,72 кг (S)-Соединения I-MsOH в виде твердого вещества ярко-желтого цвета (выход 78%, степень чистоты 99,10%). Высушенные кристаллы затем измельчали порошковой мельницей и струйной мельницей с получением композиции конечного продукта.
[172] Следует понимать, что в приведенном выше описании представлены только типичные иллюстративные варианты реализации изобретения и примеры. Для удобства читателя приведенное выше описание сосредоточено на ограниченном числе типичных примеров всех возможных вариантов реализации изобретения, на примерах, в которых показаны основные положения настоящего изобретения. Авторы настоящего изобретения не пытались в описании исчерпывающе перечислить все возможные варианты или даже комбинации описанных вариантов. То, что указанные альтернативные варианты реализации изобретения могут быть не представлены в конкретных разделах описания, или то, что дополнительные неописанные альтернативные варианты реализации могут быть подходящими для какой-либо его части, не следует рассматривать как ограничение указанных альтернативных вариантов реализации. Для среднего специалиста в области техники будет понятно, что множество таких неописанных вариантов реализации изобретения скорее включают отличия в материалах и методах, чем отличия в применении основных положений настоящего изобретения. Соответственно, не предполагается, что настоящее изобретение ограничено менее чем до объема притязаний согласно следующей формуле изобретения.
[173] Способ очистки ВЭЖХ:
Колонка: Capcellpak C18, Shisedo, 4,6×150 см, 5 микрон
Детекторная длина волны: PDA290 нм
Температура колонки: 40°C
Подвижная фаза: A: 100% 0,05M NH4OAc в воде
B: 100% ACN
Скорость потока: 1,0 мл/мин
Время записи хроматограммы: 60 минут
Объем вводимой пробы: 10 мкл
Таблица градиента:
Соединение II-OH=18,54 мин
Соединение I/Соединение I-MsOH=26,05
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛКИ
Все ссылки, статьи, публикации, патенты, патентные публикации и заявки на патенты, цитированные в настоящей заявке, полностью включены в настоящую заявку посредством ссылки для любых целей. Однако, упоминание в настоящей заявке любой ссылки, статьи, публикации, патента, патентной публикации и заявки на патент не подразумевается и не должно подразумеваться как подтверждение или любая форма вероятности того, что они составляют действующий уровень техники или частично образуют общеизвестные знания любой страны мира.
Реферат
Изобретение относится к способу получения 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновой кислоты (Соединение II-OH), включающий стадии: a) образования двухфазной смеси путем добавления к раствору диметил (4-(2-бутоксиэтокси)фенил)бороната (Соединение V-OMe) водного основного раствора, причем указанный водный основный раствор образован основанием, выбранным из группы, состоящей из фосфата калия, карбоната калия, ацетата калия, фторида калия, гидроксида калия,-бутоксида калия, карбоната натрия, фосфата натрия, гидроксида натрия,-бутоксида натрия, гидрокарбоната натрия, карбоната цезия, фторида цезия и их комбинации; b) добавления катализатора и лиганда к смеси, полученной на стадии a); причем указанный катализатор выбран из группы, состоящей из ацетата палладия, тетракис(трифенилфосфин) палладия, три(дибензилиденацетон)дипалладия, хлорида палладия, ацетилацетоната палладия и их комбинации; причем указанный лиганд выбран из группы, состоящей из три(-толил)фосфина, трифенилфосфина, три(т-бутил)фосфина, трициклогексилфосфина, пиридина, бипиридина, 2,2'-бис(дифенилфосфино)-1,1'-бинафтила и их комбинации; c) добавления 8-бром-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновой кислоты (Соединение IV) к смеси, полученной на стадии b), и нагревания реакционной смеси; и d) окисления смеси, полученной на стадии c); с получением 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновой кислоты (Соединение II-OH), где после стадии a) и до стадии d) в реакционную смесь барботируют азот. Изобретение также относится к способу получения 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоксамид метансульфоната. Технический результат: предложен способ получения 8-(4-(2-бутоксиэтокси)фенил)-1-изобутил-1,2,3,4-тетрагидробензо[b]азоцин-5-карбоновой кислоты (Соединение II-OH), который является исходным соединением в синтезе ценикривирока, с высоким выходом и чистотой более 97,5%. 2 н. и 15 з.п. ф-лы, 2 табл., 4 пр., 1 ил.
Формула




Комментарии