Карбоциклические и гетероциклические конденсированные соединения хинолинкарбоновой кислоты, пригодные в качестве иммунодепрессантов - RU2133740C1
Код документа: RU2133740C1
Чертежи










Описание
Это изобретение касается карбоциклических и гетероциклических конденсированных соединений хинолинкарбоновой кислоты, фармацевтических композиций, включающих такие соединения, и способов применения таких соединений для лечения и/или предупреждения отторжения пересаженных органов, реакции "трансплантант против хозяина", аутоиммунных заболеваний и хронических воспалительных заболеваний, включая, но не ограничиваясь, псориаз и ревматоидный артрит, у млекопитающих.
Предпосылки создания изобретения
2-фенил-4-хинолинкарбоновые кислоты открыты как подавляющие опухоли соединения U. S. Patent N 4680299, выданные Hesson 14 июля 1987 г. Применение этих соединений для лечения заболеваний кожи и
эпителия запатентовано U.S. Patent N 4861784, выданным Ackerman et al. 29 августа 1989 г. Применение этих соединений в качестве иммунодепрессантов или иммуномодуляторов открыто U. S. Patent N 4968701,
выданным Ackerman et al. 6 ноября 1990 г. U.S.Patent N 5204329, выданный Ackerman et al., описывает использование этих соединений в сочетании со вторичным иммунодепрессантом для лечения отторжения при
трансплантации и других болезненных состояниях.
Дополнительные примеры раскрывают пригодность 2-фенил-4-хинолинкарбоновых кислот для лечения артрита и для стимуляции иммунодепрессии U.S.Patent N 4847381, выданным Sutherland et al. 11 июля 1989 г., и U.S.Patent N 4968702, выданным Poletto et al. 6 ноября 1990 г.
В химической литературе известны
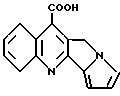
бенз[с]акридин-7-карбоновые кислоты и 5,6-дигидробенз[с] акридин-7-карбоновые кислоты. Их обычно синтезируют по реакции Пфитзингера взаимодействием соответствующего изатина с соответствующим 3,
4-дигидро-1(2Н)-нафталеноном. U. S. Patent N 4918077, выданный Behrens 17 апреля 1990 г., обсуждает множество ссылок, касающихся этих соединений, и также открывает подавляющие опухоли 3-фенил-5,
6- дигидробенз[с]акридин-7-карбоновые кислоты формулы

где R1 - inter alia, COOH, COONa или COOK; R2 и R3 - независимо H, F, Cl, Br, I, метил, этил, CF3, алкилтио, алкилсульфонил или алкилсульфонил: и R4 и R5 независимо являются H или взятые вместе являются S. U.S. Patent N 5135934, выданный Behrens et al 4 августа 1992 г., открывает применение этих соединений как иммунодепрессантов и противовоспалительных агентов.
Синтез конденсированных хинолинкарбоновых кислот формулы:

где n равняется 1-4 и R=H или метил, описан Huisgen et al [Ann. Chem. (1957) 610: 57] и Scholn et al. [Rocz. Chem. (1964) 38: 425; Chem. Abstr. 61, 1827g] . Noelting et al [Chem. Ber. (1911) 44: 2585] также описывал синтез этих соединений, а также соединения, где R - это H и где (CH2)n замещен S. Это же соединение, где R есть метил или (CH2)n замешен S. синтезировал Buu-Hoi [J. Chem. Soc. С (1966) 47] как промежуточный продукт потенциальных канцерогенов.
Синтез 4,5- дигидрофуро[2,3-с]акридин-6-карбоновых кислот формулы:

где R = метил, фенил или 4-бромфенил, описан Takagi et al. [Chem. Pharm. Bull. (1971) 19: 1218 и Chem. Pharm. Bull. (1972) 20: 2051].
Buu-Hoi et al. [J. Chem, Soc. (1958) 2418] описывали синтез соединения, показанного ниже, как промежуточного соединения потенциальных канцерогенов.

Cagniant et al. [Bull. Soc. Chim. Fr. (1969) 991 и Bull. Soc. Chim. Fr. (1970) 322] описывали синтез других аналогов тиенакридина формулы:

где R есть H или метил и n равняется 1 или 2.
Соединение, представленное ниже, описано Braunholtz et al. [J. Chem. Soc. (1962) 4346] как промежуточный продукт реакции
Бензофуро[3,2-b]хинолины формулы:

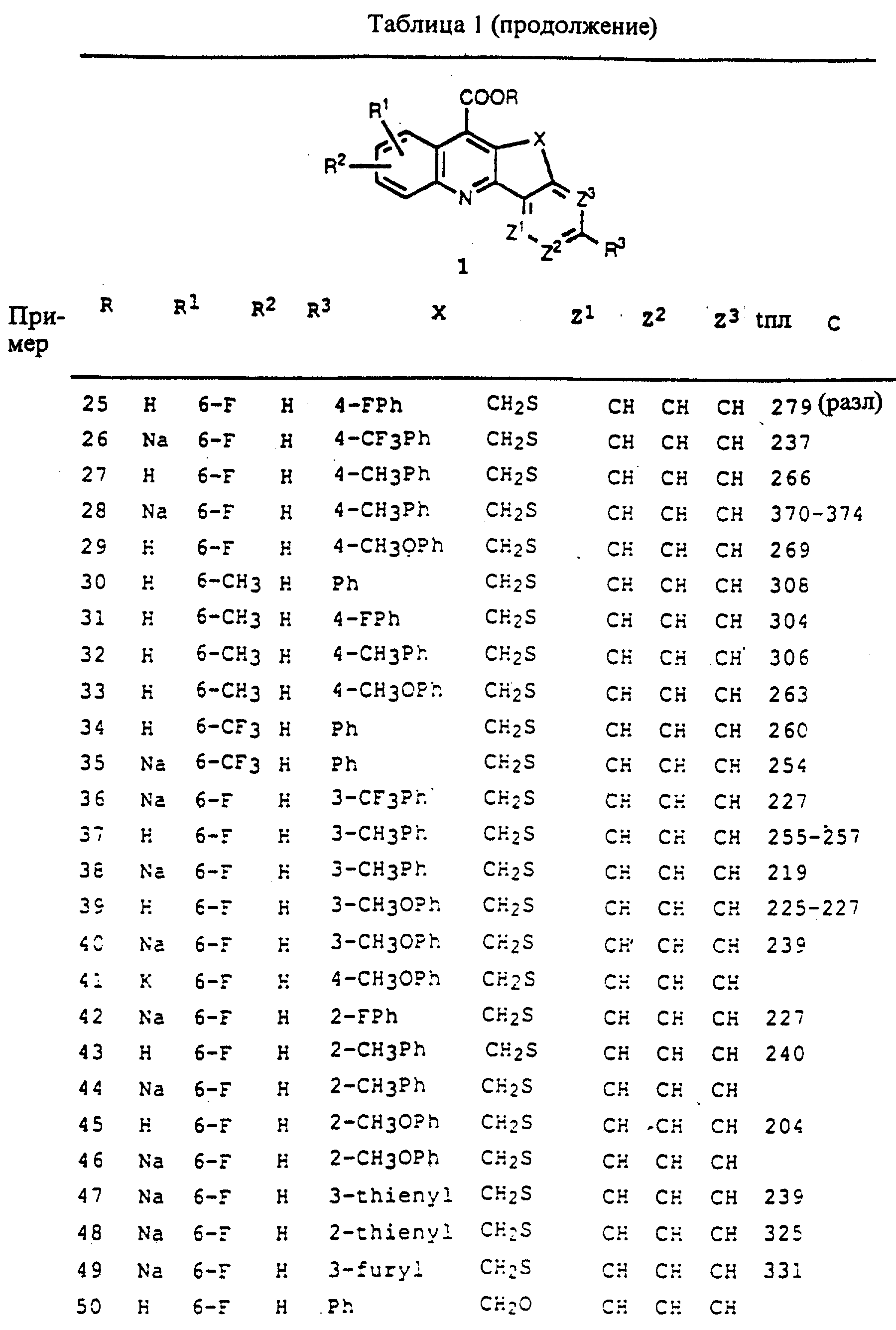
где R - низший алкил или карбоксил, открыты Jpn. Kokai Tokkyo Koho 63 295 579, выданный Kinoshita et al. 1 декабря 1988 г. [Chem. Abstr. Ill, 187618] с применением для лечения остеопороза. Аналогичные карбоновые кислоты, замещенные одиночным атомом галогена, а также соответствующими N-оксидами, синтезированы Yamaguchi et al. [J. Heterocyclic Chem. (1989) 26: 285] как промежуточные соединения возможных канцерогенов, мутагенов и противоопухолевых веществ. Дополнительные соединения такой же формулы, где R= COOH, где на бензофурановом кольце есть метоксизаместитель и где О заменили на S, также синтезировали Buu-Hoi et al. [Israel J. Chem. (1963) 1: 369; Chem. Abstr. 60, 11998fl.
Degutis et el. [Khim. Geterotsiki. Soedin. (1986) 1375; Chem. Abstr. 107, 39658] и Ezerskaite et
al. [Izv. Khim. (1989) 22: 113; Chem. Abstr. 112, 66492 Izv. Khim. (1989) 22: 232; Chem. Abstr. 113, 58979] синтезировали индолхинолинкарбоновые кислоты формулы:

где R1 и R2 являются H или алкилом и R3=H или бром, и открыли пригодность как сенсибилизаторов для электрофотографии.
Holt et al. [J. Chem. Soc. (1947) 607] также обсуждают синтез такого типа соединения.
Fravolini et al. [Ann. Chim. (1968) 58: 1155: Chem. Abstr. 70. 47334] синтезировали фторированные бензотиопиранозинолиновые карбоновые кислоты, представленные ниже.

Hoi et al. [Youji Huaxue (1991) II: 615; Chem. Abstr. 116, 128709] описали синтез соединений, представленных ниже, где R есть H или галоген.

Wang et al. [Gaodeng Xuexiao Huaxue Xuebao (1991) 12: 59; Chem. Abstr. 115, 49457 и Zhongguo Yui ao Gongye Zashi (1991) 22: 103; Chem. Abstr. 115, 183138] открыли синтез и противовоспалительную активностью 7-карбоксиизохромано[4,3-b]хинолинов формулы
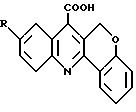
где R = H, галоген или метил. Соединение такой формулы, но без R группы и с О, замещенным S, синтезировали Braun et al. [Chem. Ber. (1929) 62: 2416] и Kiang et al. [J. Chem. Soc. (1951) 1909]. О применении последнего соединения не сообщалось.
Roussel et al. [L. Chem. Soc. (1965) 5458] описывали синтез производных
дибензонафтиридина формулы:
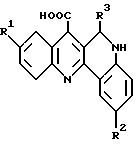
где R1= H, метил или хлор, R2=H, метокси или хлор и R3=H или метил, как потенциальных канцерогенов.
Производные хинолинонафтиридина, показанные ниже (R=H, ацетил) описаны Settimo wt al. [J. Heterocyclic Chem. (1979) 16: 169].

О синтезе похожих соединений, показанных выше, без метильного заместителя и где R1= метил или фенил, сообщили Braunholtz et al. [J. Chem. Soc. (1995) 381] и Mann [J. Chem. Soc. (1949) 2816], соответственно.
Производные
бензохинолиноазепина формулы

где R=метил или толилсульфонил были синтезированы Braunholtz et al. [J. Chem. Soc. (1958) 3377] и Proctor et al. [J. Chem. Soc. Perkin Trans. 1 (1978) 862] . Другие соединения этой формулы, но с NR замещенной на S, были синтезированы Huckle et al. [J.Chem.Soc. С (1971) 2252].
Литературные ссылки, касающиеся конденсированных хинолинкарбоновых кислот или их производных настоящего изобретения или их использования в лечении и/или предупреждении иммунологических нарушений, отсутствуют.
В настоящее время циклоспорин A, иммунодепрессант, применяемый в сочетании с дополнительными лекарственными препаратами, такими как азатиоприн и кортикостероиды, является преобладающим лекарственным препаратом, применяемым для предотвращения отторжения пересаженных органов. Другие иммунодепрессанты, такие как азатиоприн, кортикостероиды (такие как преднизон), ОКТЗ, FK506, микофенолоксилота или ее 2-(N- морфолино)этиловый эфир, 15-деоксиспергуалин, рапамицин, мизорибин, мисопростол и антитела к рецептору интерлейкина-2, использовали или предполагали, что они будут пригодны, в лечении и/или предотвращении отторжения пересаженных органов.
Применение любого из этих известных иммуносупрессорных соединений либо по отдельности, либо в сочетании с другими соединениями связано с высокой частотой побочных эффектов, таких как нефротоксичность и/или гепатотоксичность. Таким образом, в настоящее время есть необходимость в усовершенствованных лекарственных препаратах для лечения рака и лечения и/или предупреждения отторжения пересаженных органов, реакции "трансплантант против хозяина", аутоиммунных заболеваний и хронических воспалительных заболеваний, включая, но не ограничиваясь, псориаз и ревматоидный артрит.
В настоящее время обнаружено, что описываемые здесь карбоциклические и гетероциклические конденсированные соединения хинолинкарбоновой кислоты пригодны для лечения и/или предотвращения отторжения пересаженных органов, реакции "трансплантант против хозяина", аутоиммунных заболеваний и хронических воспалительных заболеваний, включая, но не ограничиваясь заболеваниями псориаз и ревматоидный артрит, у млекопитающих.
Карбоциклические и гетероциклические конденсированные соединения хинолинкарбоновой кислоты этого изобретения могут применяться по отдельности или в сочетании с одним или более дополнительными известными иммунодепрессантами, такими как, циклоспорин A (CSA и CsA) и его аналоги, FK506 (или FK-506) и его аналоги, кортикостероиды, азатиоприн (AZA), микофенолокислота и ее 2-(N-морфолино)этиловый эфир, микофенолат мофетил, рапамицин, 15-деоксипергуалин, мизорибин, лефлуномид, ОКТЗ, антитела к рецептору интерлейкина-2, мисопростол, метотрексат, циклофосфамид и антилимфоцитарные/тимоцитарные сыворотки, уменьшая таким образом требуемую дозу и связанные с действием иммунодепрессантов нежелательные побочные эффекты.
Краткое описание изобретения
Это изобретение касается
карбоциклических и гетероциклических конденсированных соединений хинолинкарбоновой кислоты формул 1-4, описанных ниже, фармацевтических композиций, включающих такие соединения, и способов применения
таких соединений для лечения и/или предупреждения отторжения пересаженных органов, реакции "трансплантант против хозяина", аутоиммунных заболеваний и хронических воспалительных заболеваний, включая,
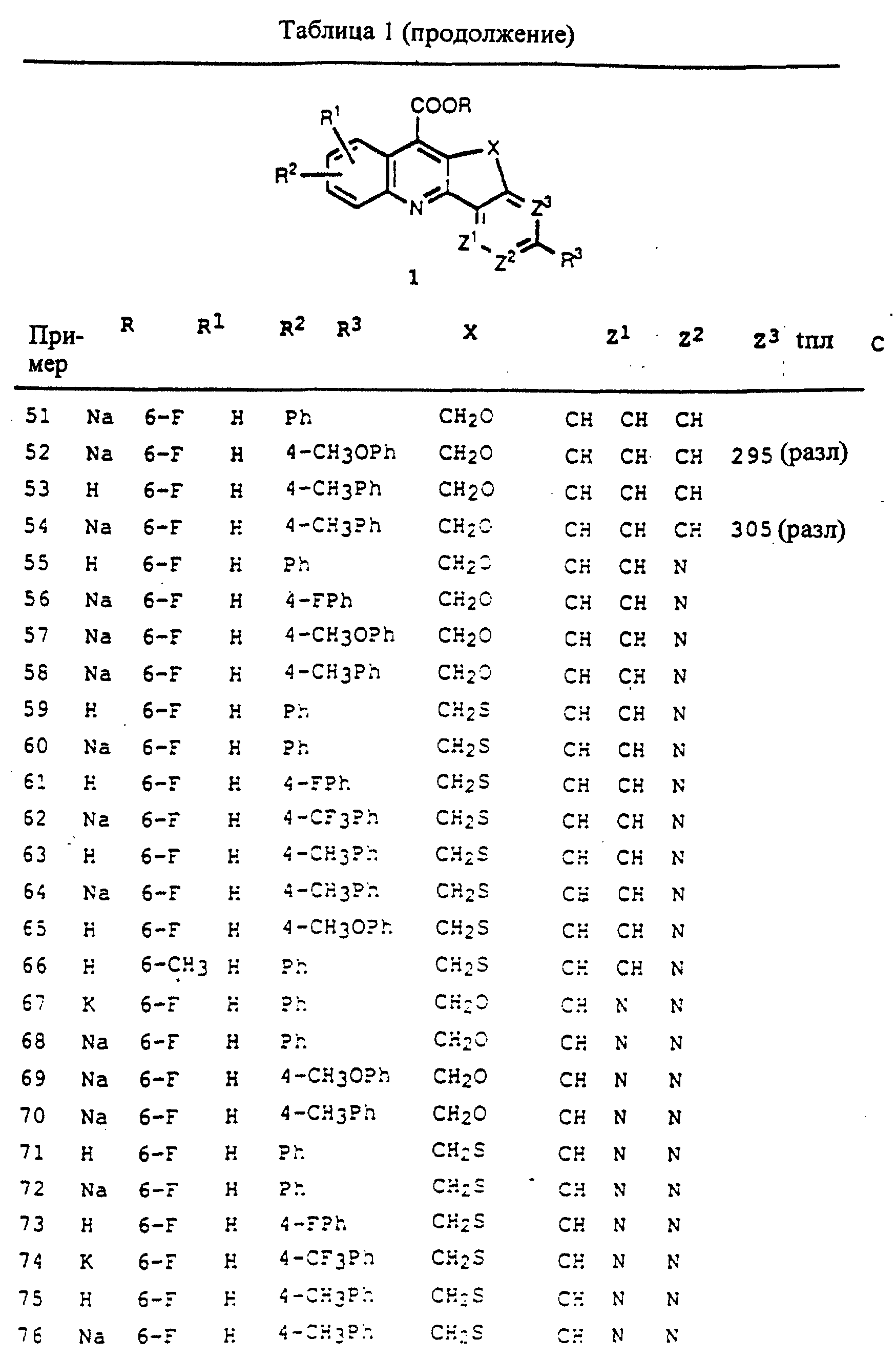
но не ограничиваясь заболеваниями псориаз и ревматоидный артрит, у млекопитающих. Соединения формул 1-4 настоящего изобретения также полезны для лечения опухолей у млекопитающий.
Обнаружено, что описанные здесь соединения формул 1-4 являются иммунодепрессантами или иммуномодуляторами, которые пригодны для лечения и/или предотвращения отторжения пересаженных органов у млекопитающих, реакции "трансплантант против хозяина" и аутоиммунных заболеваний, включая, но не ограничиваясь заболеваниями системная красная волчанка, ревматоидный артрит, псориаз, рассеянный склероз и миастения gravis. Соединения формул 1-4 настоящего изобретения также пригодны для лечения и/или предупреждения у млекопитающих хронических воспалительных заболеваний, включая, но не ограничиваясь, болезнь Крона, воспалительное заболевание кишечника, ревматоидный артрит и первичный билиарный цирроз печени. Соединения формул 1-4 настоящего изобретения также пригодны для лечения опухолей у млекопитающих, в том числе лейкемии и твердых опухолей, включая опухоли молочной железы, толстой кишки и легких.
Настоящее изобретение также предусматривает фармацевтические композиции, включающие соединения формул 1-4 и фармацевтически приемлемый носитель.
Также предусмотренными настоящим изобретением являются методы лечения и/или предотвращения отторжения пересаженных органов, реакции "трансплантант против хозяина", аутоиммунных заболеваний или хронических воспалительных заболеваний, включающие введение терапевтически эффективных количеств соединения формул 1-4 настоящего изобретения млекопитающим, нуждающимся в таком лечении и/или профилактике.
Настоящее изобретение также предусматривает методы лечения и/или предупреждения отторжения пересаженных органов, реакции "трансплантант против хозяина", псориаза, аутоиммунных заболеваний и хронических воспалительных заболеваний у млекопитающих, включающие введение млекопитающим для лечения желаемого вышеупомянутого заболевания терапевтически эффективного количества сочетания: соединения формул 1-4, которое описано ниже и по меньшей мере, одного дополнительного иммунодепрессанта. Такой дополнительный иммунодепрессант может быть выбран из группы, включающей, но не ограниченной, циклоспорин A (CSA и CsA) и его аналоги, FK506 и его аналоги, кортикостероиды, азатиоприн (AZA), микофенолокислота и ее 2-(N-морфолино)этиловый эфир, микофенолят мофетил, рапамицин, 15-деоксипергуалин, мизорибин, лефлуномид, ОКТЗ, антитела к рецептору инлерлейкина-2, мисопростол, метотрексат, циклофосфамид и антилимфоцитарные/тимоцитарные сыворотки.
Соединения формул 1-4 настоящего изобретения можно также вводить в сочетании с нестероидными противовоспалительными агентами, выбранными из группы, включающей, но не ограниченной, аспирин, ибупрофен, напроксин, индометацин, диклофенак, сулиндак, пироксикам, этодолак, кетопрофен, меклофентамат, супрофен и толметин, для лечения и/или предупреждения отторжения пересаженных органов, реакции "трансплантант против хозяина", псориаза, аутоиммунных заболеваний и хронических воспалительных заболеваний у млекопитающих.
Соединения формул 1-4 настоящего изобретения можно также вводить в сочетании с другими подавляющими опухоль агентами, включая, но не ограничиваясь, 5-фторурацил, для лечения опухолей у млекопитающих.
Подробное описание изобретения
Это изобретение касается карбоциклических и гетероциклических конденсированных соединений
хинолинкарбоновой кислоты формул 1-4, описанных ниже, фармацевтических композиций, включающих такие соединения, и способов применения таких соединений для лечения и/или предупреждения иммунологических
расстройств, в том числе отторжения пересаженных органов, реакции "трансплантант против хозяина", аутоиммунных заболеваний и хронических воспалительных заболеваний, включая, но не ограничиваясь,
псориаз и ревматоидный артрит, у млекопитающих. Соединения формул 1-4 настоящего изобретения также полезны для лечения опухолей у млекопитающих.
Обнаружено, что описываемые здесь соединения формул 1-4 являются иммунодепрессантами или иммуномодуляторами, которые пригодны для лечения и/или предотвращения отторжения пересаженных органов у млекопитающих, реакции "трансплантант против хозяина" и аутоиммунных заболеваний, включая, но не ограничиваясь, системную красную волчанку, ревматоидный артрит, псориаз, рассеянный склероз и миастению gravis. Соединения формул 1-4 настоящего изобретения также пригодны для лечения и/или предупреждения у млекопитающих хронических воспалительных заболеваний, включая, но не ограничиваясь, болезнь Крона, воспалительное заболевание кишечника, ревматоидный артрит, остеоартрит, псориаз и первичный билиарный цирроз печени. Соединения формул 1-4 настоящего изобретения также пригодны для лечения опухолей у млекопитающих, в том числе лейкемии и твердых опухолей, включая опухоли молочной железы, толстой кишки и легких.
Карбоциклические и гетероциклические конденсированные соединения хинолинкарбоновой кислоты
настоящего изобретения выбираются из соединений формул 1, 2, 3 или 4 (1-4):

и их фармацевтически приемлемых солей, где
R1 и R2 независимо выбираются из H, F, Cl, Br CF3 или алкила из 1-4 атомов углерода;
R3 выбирается из: фенил, фенокси, фенилтио, фенилсульфинил, фенил-N(R4)-, фурил, тиенил, пиридил, тиазолил или оксазолил, где упомянутые фенил, фенокси, фенилтио, фенилсульфинил, фенил-N(R4)-, фурил, тиенил, пиридил, тиазолил или оксазолил замещены по 0-2 группами, независимо выбранными из: F, Cl, Br, CF3, алкил из 1-4 атомов углерода, алкокси из 1-4 атомов углерода, алкилтио из 1-4 атомов углерода или алкилсульфинил из 1-4 атомов углерода;
R4 выбирают из H, алкил из 1-4 атомов углерода или ацил из 1-4 атомов углерода; X выбирается из -Y-, -CH2 Y-, -YCH2, -CH2CH2Y-, -YCH2CH2-, -CH2YCH2-, CR5= C(R6)-, -CR6=N- или -N=C(R6 )- (первый атом перечисленных X прикрепляется к хинолиновому кольцу); где каждая метиленовая группа у X может факультативно замещаться одной или двумя группами, независимо выбранными из алкилов с 1-4 атомами углерода;
Y - это -CH2- (указанная -CH2-группа замещается одной или двумя алкильными группами из 1-4 атомов углерода), -О-, -S- или -N(R7)-;
R5 и R6 являются независимо H или алкилом из 1-4 атомов углерода;
R7 выбирается из H, алкила из 1-4 атомов углерода или ацила из 1-4 атомов углерода;
где в соединениях формулы 1 и формулы 2:
Z1, Z2 и Z3 являются независимо CR8 или N, при условии, что, по меньшей мере, один из Z1, Z2 и Z3 не является азотом;
R8 независимо выбирается из H, F, Cl, Br, CF3 или алкила из 1-4 атомов углерода:
где в соединении формулы 3:
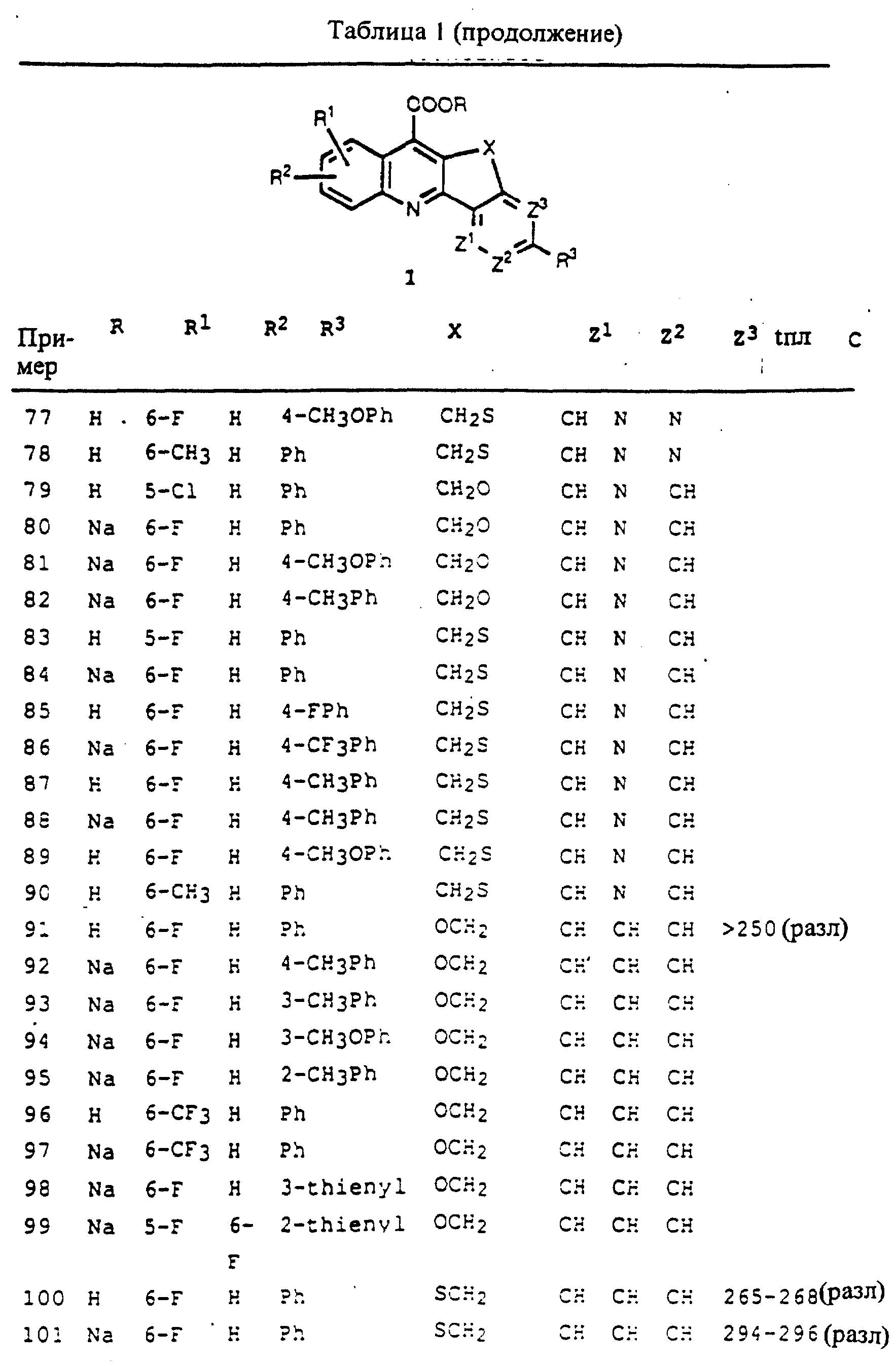
один из Q1 и Q2 является S или NR9, а другой является CR10 или N, при условии, что кольцо, содержащее Q1 и Q2, содержит дополнительную двойную связь, расположенную таким образом, что образуется ароматическое кольцо;
R9 выбирается из H или алкила из 1-4 атомов углерода;
R10 выбирается из H, F, Cl, Br, CF3 или алкила из 1-4 атомов углерода;
где в соединении формулы 4:
один из Q3 и Q4 - это N, а другой - C, при условии, что кольцо формулы 4, содержащее Q3 и Q4, содержит дополнительную двойную связь, расположенную таким образом, что образуется ароматическое пиррольное кольцо; и
R11 - H, F, Cl, Br, CF3 или алкил из 1-4 атомов углерода;
при условии, что:
1) X не является NR7;
2) в соединениях формулы 1, когда X= CH2CH2 и каждый из Z1, Z2 и Z3=CH, то R3 не является незамещенным фенилом;
3) в соединениях формулы 3 или формулы 4, где Q4=N, X должен образовывать мостик из, по меньшей мере, двух атомов, и атом X, присоединенный к кольцу, содержащему Q1 и Q2 или Q3 и Q4, должен быть углеродом;
4) в соединениях формулы 4, где Q3=N, X должен включать либо -CH2CH2-, либо -CH2CH2CH2-, с факультативным замещением каждой метиленовой группы, как описано выше, и
5) соединения формулы 4, где Q4=N, R3 не должен быть фенокси, фенилтио, фенилсульфинил или фенил-N(R4 )-.
Предпочтительными карбоциклическими и гетероциклическими конденсированными соединениями хинолинкарбоновой кислоты настоящего изобретения являются те из описанных выше соединений формул 1-4, где R1 и R2 замещены в положениях 5 и/или 6 на хинолиновое кольцо.
Предпочтительными карбоциклическими и гетероциклическими конденсированными
соединениями хинолинкарбоновой кислоты настоящего изобретения являются те описанные выше соединения формул 1-4, где:
R1 = 6-F или 6-CF3; и/или
R2 = H;
и/или
R3= фенил, тиенил или фурил; упомянутые фенил, тиенил или фурил замещены по 0-2 группами, независимо выбранными из: F, Cl, Br, CF3, алкила из 1-4 атомов углерода;
алкокси из 1-4 атомов углерода, алкилтио из 1-4 атомов углерода или алкилсульфинил из 1-4 атомов углерода: и/или
X = -CH2CH2, -CH2S-, -SCH2-,
-CH2O- или -OCH2-; и/или
где, в соединениях формулы 1 или формулы 2 Z1 есть CH; и/или
где, в соединениях формулы 3 Q1 есть S и Q2 есть CH; и/или
где, в соединениях формулы 4, Q3 есть C и Q4 есть N.
Более предпочтительными карбоциклическими и гетероциклическими
конденсированными соединениями хинолинкарбоновой кислоты формул 1-4 настоящего изобретения являются те описанные выше предпочтительные соединения, у которых:
R3 означает фенил,
2-фторфенил, 3-метоксифенил, 3-метилфенил, 3-трифторметилфенил или тиенил.
Особенно предпочтительными соединениями формул 1-4 настоящего изобретения являются соединения, выбранные из
следующих, или фармацевтически приемлемой соли:
а) соединения формулы 1, где R1 есть 6-F, R2 есть H; R3 = фенил, Z1, Z2 и Z3
все являются CH и X есть CH2S или натриевая соль;
б) соединения формулы 1, где R1 есть CF3, R2 есть H, R3 = фенил, Z1, Z2 и Z3 - все являются CH и X есть CH2S или натриевая соль;
в) соединения формулы 1, где R1 - 6-F, R2-H, R3 = 2-фторфенил, Z1, Z2 и Z3 - все являются CH и X - это CH2S или натриевая соль;
г) соединения формулы 1, где R1 - 6-F, R2-H, R3 =
3-метоксифенил, Z1, Z2 и Z3 - все являются CH и X - это CH2S или натриевая соль;
д) соединения формулы 1, где R1 - есть 6-F, R2 - есть H, R3 = фенил, Z1, Z2 и Z3 - все являются CH и X - это CH2O или натриевая соль;
е) соединения формулы 1, где R1
- есть 6-F, R2 - есть H, R3 = 3-метилфенил, Z1, Z2 и Z3 - все являются CH и X - есть SCH2 или натриевая соль;
ж) соединения
формулы 1, где R1 - есть 6-F, R2 - есть H, R3 = фенил, Z1 и Z2 - CH, Z3 - N и X- есть CH2CH2 или натриевая соль;
з) соединения формулы 1, где R1 - есть 6-F, R2 - есть H, R2 = фенил, Z1, Z2 и Z3 - все CH и X - есть OCH2 или
натриевая соль;
и) соединения формулы 1, где R1 - есть 6-F, R2 - есть H, R3 = фенил, Z1 и Z2- есть CH, Z3 - N и X- есть
SCH2 или натриевая соль;
к) соединения формулы 1, где R1 - есть 6-F, R1 - есть H, R3 = 2- фторфенил, Z1, Z2 и Z3
- все CH и X - есть CH2CH2 или натриевая соль;
л) соединения формулы 1, где R1 - есть 6-F, R2 - есть H, R3 = 3-трифторметилфенил, Z1, Z2 и Z3 - все CH и X - есть CH2CH2 или натриевая соль;
м) соединения формулы 1, где R1 - есть 6-F, R2- есть H, R3 = 3-метоксифенил, Z1, Z2 и Z3 - все CH и X - есть CH2CH2 или натриевая соль.
Настоящее изобретение также обеспечивает фармацевтические композиции, включающие соединения формул 1-4 и фармацевтически приемлемый носитель.
Также предусмотренными настоящим изобретением являются способы лечения и/или предупреждения отторжения пересаженных органов, реакции "трансплантант против хозяина", аутоиммунных заболеваний или хронических воспалительных заболеваний, включающие введение нуждающимся в таком лечении и/или профилактике млекопитающим терапевтически эффективного количества соединения формул 1-4 настоящего изобретения.
Соединения настоящего изобретения также подходят для лечения кожных и слизисто-эпителиальных заболеваний у млекопитающих, таких как псориаз (во всех его формах), лишай (в том числе, красный плоский лишай), хроническая экзема, ихтиоз, питириаз и хроническая крапивница. Фармацевтические композиции, включающие соединения формул 1-4, приготовленные для местного введения, особенно полезны для лечения таких кожных и слизисто-эпителиальных заболеваний.
Настоящее изобретение также предусматривает методы лечения и/или предупреждения иммунологических заболеваний, в том числе, отторжения пересаженных органов, реакции "трансплантант против хозяина", псориаза, аутоиммунных заболеваний и хронических воспалительных заболеваний у млекопитающих, включающие введение млекопитающим терапевтически эффективного количества для лечения желаемого вышеупомянутого заболевания сочетания: 1) соединения формул 1-4, которое описано ниже и 2) по меньшей мере, одного дополнительного иммунодепрессанта. Такой дополнительный иммунодепрессант может быть выбран из группы, включающей, но не ограниченной, циклоспорин A (CSA и CsA) и его аналоги, FK506 (или FK-506) и его аналоги, кортикостероиды, азатиоприн (AZA), микофенолокислота и ее 2-(N-морфолино)этиловый эфир, микофеноляат мофетил, рапамицин, 15- деоксипергуалин, мизорибин, лефлуномид, ОКТЗ, антитела к рецептору инлерлейкина-2, мисопростол, метотрексат, циклофосфамид и антилимфоцитарные/тимоцитарные сыворотки.
Соединения формул 1-4 настоящего изобретения можно также вводить в сочетании с нестероидными противовоспалительными агентами, выбранными из группы, включающей, но не ограниченной, аспирин, ибупрофен, напроксин, индометацин, диклофенак, сулиндак, пироксикам, этодолак, кетопрофен, меклофентамат, супрофен и толметин, для лечения и/или предупреждения отторжения пересаженных органов, реакции "трансплантант против хозяина", псориаза, аутоиммунных заболеваний и хронических воспалительных заболеваний у млекопитающих.
Соединения формул 1-4 настоящего изобретения можно также вводить в сочетании с другими подавляющими опухоль агентами, включая, но не ограничиваясь, 5-фторурацил, для лечения опухолей у млекопитающих.
Соединения формул 1-4 настоящего изобретения являются потенциальными ингибиторами дигидрооротатдегидрогеназы, четвертого фермента в биосинтезе пиримидинов.
Современное рекомендованное лечение предупреждения отторжения пересаженных органов и родственных заболеваний, в том числе, реакции "трансплантант против хозяина", обычно включает лечение больного циклоспорином А и дополнительное лечение кортикостероидами и другими иммунодепрессантами (Jacobs and Elgin, "Cyclosporin A, Current Status, Including the Cape Town Experience" in Immune Modulation Agents and Their Mechanisms, ISBN 0- 8247-7178-8. 1984. pp. 191-228: Transplantation and Clinical Immunology, Volume XX Combined Immunosuppressive Therapy in Transplantation ISBN 0-444-81068-4, 1989). С точки зрения наблюдаемого при лечении серьезного токсического эффекта циклоспорина A (нефротоксичность) и AZA (гепатотоксичность) существует потребность в усовершенствованных лекарственных препаратах для того, чтобы заменить или использовать в сочетании с циклоспорином A или AZA. Представленные результаты показывают, что соединения формул 1-4 настоящего изобретения будут полезны как самостоятельные лекарственные средства, так и как средства, которые нужно использовать в сочетании с другими соединениями, такими как циклоспорин A, применяемыми в настоящее время в этих лечебных схемах.
Соединения формул 1-4 настоящего изобретения имеют уникальный механизм действия (подавление дигидрооротатдегидрогеназы), отличающийся от действия других доступных иммунодепрессантов. Соединения формул 1-4 будут полезны тем, что позволят вводить пониженные дозы других иммунодепрессантов (таких как CsA и AZA), используемых в сочетании с ними, что приведет к уменьшению нежелательных эффектов от действия этих средств.
Выделение FK506 описывается в European Patent Application N 240773, опубликованном 10/14/87, и химический синтез FK506 описан Jones et al. (1989) (J. Am. Chem. Soc. 111:1157-1 159).
Получение азатиоприна описывается в U.S. Patent 3 056 785, выданным Burroughs Wellcome. Азатиоприн доступен как lmurank, для которого информация о продукте, в том числе дозы и способ введения, дана в Physicians' Desk Reference 44th Edition, 1990, pp. 777 - 778.
Получение циклоспорина A описывается в U.S. Patent 4 117 118 выданным Sandoz. Циклоспорин А доступен как Sandimmune R, для которого информация о продукте, включая дозу и информацию, дана в Physicians' Desk Reference 44th Edition, 1990, p. 1950-1952.
Получение преднизона описывается U.S. Patent 2 807 216 и 3 134 718, выданным Schering. Преднизон продается несколькими производителями, как и другие кортикостероиды (см. Physicians' Desk Reference, выше).
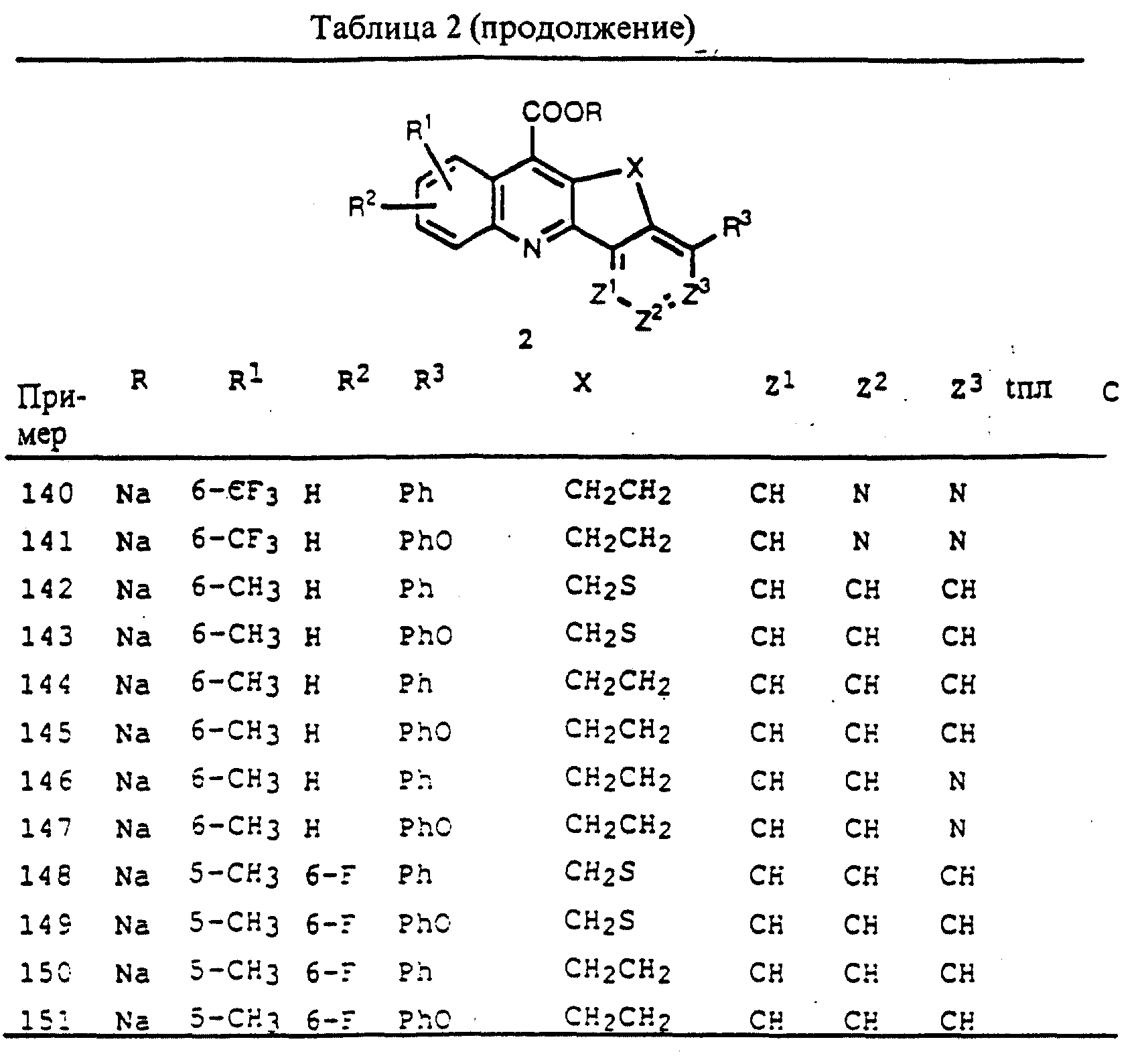
Мышиные моноклональные антитела к человеческому ТЗ антигену (здесь обозначено как ОКТЗ) доступны в виде Orthoclone OKT®3, для которого информация о продукте, включая дозу, способ введения и ссылки на методы получения, дана в Physicians' Desk Reference, 1990, p. 1553 - 1554.
Получение микофенолокислоты описывается в английских патентах 1157099, 1157100 и 1158387, выданных ICI.
15-деоксипергуалин является производным спергуалина, обнаруженного в культуральном фильтрате бактериального штамма BGM162-aFZ по сообщению в Ochiai et al. Prolongation of rat heart allograft survival by 15-deoxyspergualin, J. Antibiot. (Tokyo) 1987; 40: 249.
Мизорибин описывается в U.S. Patent 3 888 843, выданным Toyo Jozo.
Мизопростол, аналог простагландина (PGEI) описывается U.S. Patent 3 965 143, закрепленным за Searle, и U.S. Patent 4 132 738, выданным Miles.
Рапамицин описывается U.S. Patent 4 650 803, 4 316 885, 4885 171, 3993 749 и 3 929 992, все выданы Ayerst.
Антитела к белку рецептора IL-2 описываются U.S. Patent 4 578 335 и 4 845 198 (Immunex) и USSN 7/341 361 и US Patent 4 892 827, выданный Pastan et al.
Также обеспеченными данным изобретением являются способы лечения рака у млекопитающих, в том числе, лейкемии, лимфомы и твердых опухолей, таких как опухоли поджелудочной и молочной желез, легких, грудной клетки, толстой кишки, эпителиальные опухоли и меланомы, включающие введение имеющим такую опухоль млекопитающим терапевтически эффективного, подавляющего опухоль количества соединения формул 1-4, описанного выше.
Соединение формул 1-4 настоящего изобретения могут вводиться в сочетании со вторым иммунодепрессантом для того, чтобы таким образом уменьшить дозы каждого лекарства, требуемые для достижения желаемого терапевтического действия. Таким образом, комбинированное лечение настоящего изобретения позволяет использовать пониженные дозы каждого компонента для уменьшения отрицательных токсических воздействий каждого компонента. Пониженная доза доводит до минимума возможные побочные эффекты препаратов, обеспечивая повышенный коэффициент безопасности по сравнению с коэффициентом безопасности каждого из компонентов, используемых по отдельности.
"Терапевтически эффективное количество" означает то количество соединения формул 1-4, которое при самостоятельном введении или в сочетании с дополнительным имунодепрессантом в клетки или млекопитающим, является эффективным для предупреждения или улучшения болезненного состояния или прогрессирования болезни.
"Введенное в сочетании" или "сочетание", относящееся к компоненту (1) или компоненту (2) настоящего изобретения, означает, что компоненты вводятся в клетку или млекопитающим, которые подвергаются лечению, одновременно. Вводимый в сочетании каждый компонент может быть введен в то же время или последовательно в любом порядке в различные моменты времени. Так, компонент (1) и компонент (2) могут вводиться раздельно, но достаточно близко по времени введения, с тем, чтобы обеспечить желаемый терапевтический эффект.
Это изобретение также включает фармацевтические наборы, содержащие или состоящие по существу из: фармацевтической композиции, включающей соединение формул 1-4 или соединение формул 1-4 вместе с фармацевтической композицией, содержащей, по меньшей мере, еще один иммунодепрессант. Это изобретение также предусматривает методы использования таких фармацевтических наборов для лечения отторжения пересаженных органов, реакции "трансплантант против хозяина", псориаза и аутоиммунных заболеваний, включая, но не ограничиваясь, ревматоидный артрит, системную красную волчанку, рассеянный склероз, миастению gravis, инсулинозависимый диабет, а также хронических воспалительных заболеваний, включая, но не ограничиваясь, болезнь Крона и первичный билиарный цирроз, у млекопитающих.
Это изобретение также включает комбинированные продукты, содержащие фармацевтические композиции, включающие соединение формул 1-4 в медицинском сочетании или в форме однократной дозы со вторым иммунодепрессантом, фармацевтические наборы, содержащие эти комбинированные продукты, и способы применения этих комбинированных продуктов для лечения отторжения пересаженных органов, реакции "трансплантант против хозяина", псориаза и аутоиммунных заболеваний, включая, но не ограничиваясь, ревматоидный артрит, системную красную волчанку, рассеянный склероз, миастению gravis, инсулинозависимый диабет, а также хронических воспалительных заболеваний, включая, но не ограничиваясь, болезнь Крона и первичный билиарный цирроз, у млекопитающих.
Описанные здесь соединения формул 1-4 могут иметь асимметричные центры. В настоящее изобретение входят все хиральные, диастереометрические и рацемические формы. Для этого соединения подразумеваются все хиральные, диастереометрические и рацемические формы, если отдельно не указана стереохимия соединения. В настоящем изобретении рассматриваются все стабильные геометрические изомеры, существующие у описываемых здесь соединений, и подразумеваются все геометрические структурные изомеры, если не указан специально специфический изомер.
Если какая-либо переменная встречается более одного раза в каком-либо структурном компоненте или в формуле 1, 2, 3 или 4 или любой другой представленной здесь формуле, это определение в каждом случае независимо от определения в каждом другом случае. Так, например, если показано, что группа замещается 0-3R8, то указанная группа может факультативно замещаться до трех R8, и R8 в каждом случае выбирается независимо из определенного списка возможных R8. Также, сочетание заместителей и/или переменных допустимо только тогда, когда такое сочетание приводит к стабильным соединениям.
Подразумевается, что используемое здесь определение "ацил из 1-4 атомов углерода" включает алкильную группу из 1-3 атомов углерода, присоединенную через карбонильный мостик, т.е. -C(=O)-(C1-C3)алкил.
Подразумевается, что используемое здесь определение "алкил" включает как разветвленные, так и неразветвленные насыщенные алифатические углеводородные группы, имеющие указанное количество атомов углерода; "алкокси" представляет собой алкильную группу из указанного количества атомов углерода, присоединенную через кислородный мостик; "алкилтио" означает алкильную группу из указанного количества атомов углерода, присоединенную через серный мостик; "алкилсульфинил" означает алкильную группу из указанного количества атомов углерода, присоединенную через сульфинил.
Используемый здесь термин "замещенный" означает, что какой-либо один или более одного атомов водорода у указанного атома замещены по выбору из указанной группы, при условии, что не превышена указанная нормальная валентность атома, и что замещение приводит к стабильному соединению.
Под "стабильным соединением" или "стабильной структурой" здесь подразумевается соединение, достаточно прочное для того, чтобы выдержать выделение до желаемой степени очистки из реакционной смеси и технологию превращения в эффективный лекарственный препарат.
Используемый здесь термин "фармацевтически приемлемые соли" относится к производным открытых соединений, которые модифицируются получением солей кислот или оснований. Примеры включают, но не ограничиваются, соли неорганических или органических кислот основных остатков, таких как амины, и щелочных или органических солей кислых остатков, таких как карбоновые кислоты.
Фармацевтически приемлемые соли соединений этого изобретения могут быть получены взаимодействием этих соединений в форме свободной кислоты или основания со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе или в их смеси; в целом, предпочтительна неводная среда типа эфира, этилацетата, этанола, изопропанола или ацетонитрила. Список соответствующих солей находится в Remington's Pharmaceutical Sciences 17th ed., Mack Publishing Company, Easton, PA, 1985, p. 14, 18 сообщение которого таким образом оформляется ссылкой.
Сообщение всех помещенных здесь ссылок, таким образом, включается здесь в виде ссылок полностью.
Синтез
Соединения настоящего изобретения можно синтезировать, используя описанные ниже методы, а также известные в органической химии методы или их вариации,
которые известны опытным химикам. Предпочтительные методы включают, но не ограничиваются тем, что описано ниже. Все ссылки, помещенные здесь, оформляются полностью в виде ссылок.
В представленном ниже обсуждении на схемах и в сопровождающем тексте представлены соединения формулы 1 и описаны исходные материалы и промежуточные соединения, соответствующие получению соединений формулы 1. Однако, следует понимать, что обсуждаемые методы не ограничиваются получением соединений формулы 1. а, конечно, могут быть использованы также для получения соединений формул 2, 3 и 4.
Соединения формул 1, 2, 3 и 4 могут быть получены по реакции, показанной на схеме 1 (которая представляет эту реакцию для получения соединения формулы 1). Соответственно замещенный изатин формулы 5 конденсируют с соответственно замещенным циклическим кетоном формулы 6. Эта конденсация, обычно называемая конденсацией Пфитзенгера, хорошо известна в химической литературе (например, см. Buu-Hoi et al. J. Org. Chem. (1953), 18: 1209, и Jones, Quinolines Part 1, 1977, Wiley, pp. 197 - 207). Обычно реакцию проводят в соответствующем растворителе, таком как этанол или смесь этанола и воды, в присутствии соответствующего основания, такого как гидроксид натрия или гидроксид калия, при температуре в интервале от 5oC до температуры кипения растворителя, предпочтительно при температуре кипения растворителя. Закисление реакционной смеси неорганической кислотой, такой как соляная кислота, или органической кислотой, такой как уксусная кислота, дает соединения формул 1, 2, 3 или 4. Кроме того, соединения формулы 1, 2, 3 или 4 можно непосредственно выделить из реакционной смеси в виде соли, такой как натриевая соль или калиевая соль.

Кроме того, соединения формул 1, 2, 3 и 4 могут быть получены двухэтапной реакционной последовательностью, как показано на схеме 2 (которая представляет эту последовательность реакций для получения соединения формулы 1). Эта альтернативная двухэтапная реакционная последовательность предпочтительна в том случае, когда кетон формулы 6 нестабилен в условиях реакции, представленной на схеме 1, или не взаимодействует при этих условиях. В этой альтернативной реакционной последовательности соответственно замещенный изатин формулы 5 реагирует с соответственно замещенным циклическим кетоном формулы 6 в подходящем растворителе, таком как этанол, в присутствии соответствующего основания, такого как диэтиламин или пирролидин, при температуре от 5oC до температуры кипения растворителя для того, чтобы получить промежуточный продукт альдольной конденсации формулы 7. Затем продукт формулы 7 взаимодействует далее в соответствующем растворителе, таком как диметиловый эфир этиленгликоля или диоксане, в присутствии неорганической кислоты, такой как водная соляная кислота, или органической кислоты, такой как метансульфокислота, в количестве, составляющем примерно 25 - 100% объема растворителя. Температура реакции находится в интервале от 5oC до точки кипения растворителя. Удаление растворителя и промывка водой дает соединения формул 1, 2, 3 или 4.

Кроме того, соединение формул 1, 2, 3 и 4, где R3 - фенил, фурил, тиенил, пиридил, тиазолил или оксазолил (где указанные фенил, фурил, тиенил, пиридил, тиазолил или оксазолил факультативно замещены) могут быть получены в соответствии с реакционной последовательностью, показанной на схеме 3 (которая представляет реакционную последовательность для получения соединений формулы 1). По этой последовательности кетон формулы 8 (где заместитель R3 кетона формулы 6 заменили соответствующим заместителем, таким как бром) конденсируют с изатином формулы 5, используя конденсацию Пфитзенгера, аналогичную показанной на схеме 1 или двухэтапной реакционной последовательности, аналогичной той, что показана на схеме 2, получая соединение формулы 9. Затем это соединение соединяется с арилборной или гетероарилборной кислотой R3-B(OH)2 по стандартной реакции соединения, такой как описана Miyara etal. [Synth. Comm. (1981) 11: 513], обычно называемой соединением Сузуки (Suzuki Coupling), давая соединение формул 1, 2, 3 или 4. Использование этой реакционной последовательности требует, чтобы R1 и R2 не были Br.

Некоторые заместители у соединений формул 5 и/или 6 могут быть несовместимы с условиями реакций получения соединений формул 1, 2, 3 или 4. В этих случаях заместители можно заменить подходящими защищенными формами желаемых заместителей или подходящими предшественниками желаемых заместителей. После выделения продукта реакционных последовательностей, показанных на схемах 1, 2 или 3, желаемые заместители можно получить соответствующим снятием защиты или другими приемами для получения соединения формул 1, 2, 3 или 4. Случаи, где это может быть необходимо, должны быть очевидны квалифицированному химику, также как и методы, которые нужно использовать для превращения в требуемое состояние.
Соединение формул 1, 2,3 и 4, где X - есть CR5=CR6, CR6=N или N=CR6, могут быть получены из соответствующих соединений, где X=CHR5=CHR6, CHR6NH или NHCHR6 соответственно хорошо известными методами. Примерами являются обработка 2,3-дихлоро-5,6-дициано-1,4-бензохиноном в растворителе, таком как бензол или толуол, как описано Pataki et al. [J. Org. Chem. (1987) 52: 2226], и обработка палладием или активированным углем, как описано Garrett et al. [Tetrahedron Lett., (1969) 191].
Соединения формул 1, 2, 3 или 4, которые содержат ацильные группы в качестве R4 и/или R7, могут быть получены из соответствующего соединения, где R4 и/или R4 являются H, используя хорошо известные методы, такие как ацилирование хлорангидридом или ангидридом.
Изатины формулы 5, используемые в качестве исходных соединений для получения соединений формул 1, 2, 3 или 4, являются либо покупными, либо могут быть получены способами, описанными Papp и по данным здесь ссылкам [Adv. Heterocyclic Chem. (1975) 18: 4].
Кетоны формулы 6 (которые можно использовать для получения соединений формулы 1. и для которых в последующих обсуждениях подразумевается, что они представляют собой кетоны, которые могут быть использованы для получения соединений формул 2, 3 и 4) могут быть получены, используя многообразие стандартных методов, хорошо известных в литературе. Здесь описываются некоторые применимые методы, но эти методы являются только иллюстративными примерами и не ограничивают настоящее изобретение.
В случаях соответствующей реакционной способности, очевидной квалифицированному химику, кетоны формулы 6 могут быть получены реакцией ацилирования Фриделя-Крафтса, показанной на схеме 4. Соответствующий предшественник карбоновой кислоты формулы 10 может либо непосредственно циклизоваться до 6, при использовании кислого катализатора, такого как серная кислота, метансульфокислота или полифосфорная кислота, выборочно в подходящем растворителе, либо кислоту можно сначала активировать превращением в соответствующий хлорангидрид или ангидрид кислоты, используя стандартные методы, после чего ее можно циклизовать, используя в качестве катализатора кислоту Льюиса, такую как хлорид алюминия или трифторид бора, в соответствующем растворителе. Реакция ацилирования Фриделя-Крафтса хорошо известна в химической литературе и широко освещена, например, Gore [Chapter 31 in Olah, Friedel-Crafts and Related Reactions, vol. 3, 1964, Interscience Publishers]. Этот метод будет ограничен случаями, когда кольцо, на котором происходит ацилирование, достаточно реакционноспособно в условиях ацилирования Фриделя-Крафтса, такое как, например, бензольное или тиофеновое кольцо. Карбоновые кислоты, в свою очередь, могут быть получены, используя реакции и способы, известные в химической литературе.

Кетоны формулы 6 могут также быть получены, используя реакционную последовательность, показанную на Схеме 5. Соответствующее исходное соединении формулы 11, где G1 и G2 являются подходящими функциональными группами, такими как сложные эфиры карбоновой кислоты, нитрилы или карбоновые кислоты, может циклизоваться в условиях, при которых заместители G1 и G2 дают промежуточный продукт формулы 12, который в дальнейшем может превращаться в кетон формулы 6. В том случае, где G1 и G2 являются сложными эфирами карбоновых кислот, циклизация обычно известна под названием конденсация Дикмана. В случае, где G1 и G2 - это нитрилы, циклизация обычно известна под названием конденсация Торпа-Циглера. Эти реакции известны в химической литературе (см., например, Schafer et al., [Org. Reactions (1967) 15: 1]), и могут проводиться в соответствующем растворителе при использовании основного катализатора, такого как метилат натрия, этилат натрия или t-бутилат калия. В случае, где G1 и G2 являются карбоновыми кислотами, реакция может проводиться обработкой уксусным ангидридом и уксусной кислотой в соответствии с методом Normant- Chefnay et al. [Compt. Rend. Acad. Sci (C) (1068) 267: 547] . Циклизованный промежуточный продукт формулы 12 может в дальнейшем взаимодействовать при соответствующих условиях для отщепления группы G2, давая желаемый кетон формулы 6. Такие условия известны в химической литературе и обычно предполагают катализируемый кислотой или основанием гидролиз нитрила или сложного эфира соответствующей карбоновой кислоты, которую затем декарбоксилируют, используя известные методы для получения соединения формулы 6. В случае, где G1 и G2 являются карбоновыми кислотами, промежуточное соединение не выделяется, а подвергается декарбоксилированию в реакционной смеси, непосредственно давая соединение формулы 6.

Кетоны формулы 6, где X - есть CH2O или CH2S, могут также быть получены последовательностью реакций, показанной на схеме 6. Соответствующий метилкетон формулы 13, где Y - есть О или S, может быть превращен в соединение формулы 14, где волнистая линия изображает либо одинарную, либо двойную связь и где либо R является алкильной группой, такой как метил или этил, либо NR2 является циклической аминогруппой, такой как пирролидин, пиперидин или морфолин. В случае, где волнистая линия представляет одинарную связь, реакция называется реакцией Манниха и хорошо известна в химической литературе (см. , например, Blicke [Org. Reactions (1942) 1:303 и Cox et al [Synthesis (1989) 709] ). В случае, где волнистая линия является двойной связь, это взаимодействие может достигаться, например, конденсацией 13 с соединением, таким как N, N-диметилформамид диметилацеталь, по сообщению Lin et al. [J. Heterocyclic Chem. (1977) 14: 345]. В некоторых случаях, соединение формулы 14 может спонтанно циклизоваться, давая желаемое соединение формулы 15 (которое эквивалентно кетонам формулы 6, где X - это CH2O или CH2S), тогда как в других случаях циклизация может вызываться обработкой соответствующим реагентом, таким как кислотный или основной катализатор. В случае, где волнистая линия 14 - это двойная связь, кетоновое кольцо 15 будет содержать двойную связь, которая может быть восстановлена, используя традиционные методы, известные в литературе, для получения желаемого соединения формулы 15.

В случае, где R3 - фенил, фурил, тиенил, пиридил, тиазолил или оксазолил (где указанные фенил, фурил, тиенил, пиридил, тиазолил или оксазолил выборочно замещены), кетоны формулы 6 могут быть также синтезированы из подходящего предшественника кетона 16, где G - это бром или гидрокси, реакцией соединения с арилборной кислотой или гетероарилборной кислотой, как показано на схеме 7. В случае, где G - это бром, реакция соединения может проводиться, используя соединение Сузуки, упомянутое ранее, согласно схеме 3. Кроме того, в случае, где G -это гидрокси, гидроксигруппа может активироваться превращением в соответствующий трифторметансульфонат, используя традиционные методы и технологии. Полученный трифторметансульфонат может быть соединением с арилборной кислотой или гетероарилборной кислотой, используя, например, метод Oh-e et al. pSynlett (1990) 221].

Примеры получения кетонов формулы 6, являющихся исходными соединениями, и родственных кетонов, которые могут быть использованы для синтеза соединений формул 1, 2, 3 и 4, даны ниже.
Все температуры плавления нескорректированы. Все реакции проводились в атмосфере азота, за исключением случаев, где указаны другие условия. Все коммерческие реактивы использовались как общепринято. Хроматография проводилась на силикагеле 60 Merck (230 - 400 mesh). Хроматографические элюенты даются как объемные отношения. Органическую фазу экстрактов растворитель/растворитель обычно выпаривали над сульфатом магния, если не оговаривается другое. Растворители обычно удаляли выпариванием при пониженном давлении на роторном испарителе, если не оговаривается другое, Позиции пиков1H ЯМР спектров даны как части на миллион (δ) по полю по внутреннему стандарту тетраметилсилану. Сокращения для1H ЯМР спектров следующие: s - синглет, d - дублет, m - мультиплет, dd - дублет дублетов, dm - дублет мультиплетов. Масс-спектры получали, используя химическую ионизацию аммиаком в качестве газа реагента.
Получение 6-(4-метилфенил)-1 -тетралона
а. 6-метокситетралон (50 г, 280 ммоль) суспендировали в ледяной уксусной кислоте (250 мл). Добавляли бромоводородную кислоту (47%, 500 мл) и смесь нагревали с обратным холодильником 6 ч, охлаждали до
комнатной температуры и выливали на 500 г измельченного льда. Через 1 ч осадок собирали и перекристаллизовывали из этанола, получая 32 г 6- гидрокситетралона. Концентрирование маточного раствора
давало дополнительно 9 г продукта, t пл. 149 - 152oC;1H ЯМР (CDCl3) δ 7,80 (d, 1H), 6,78 (d, 1H), 6,68 (s, 1H), 2,90 (t, 2H), 2,05 (m, 2H).
б. Порцию продукта части а (9 г, 55.5 ммоль) растворяли в диметоксиэтане (100 мл). Добавляли растертый карбонат калия (16, 88 г, 122 ммоль) и нагревали реакционную смесь с обратным холодильником до комнатной температуры. Добавляли N-фенилтрифторметансульфонимид (19,85 г, 55,5 ммоль) и нагревали смесь 0,5 ч с обратным холодильником, затем охлаждали до комнатной температуры. Растворитель декантировали с твердого остатка, который дополнительно промывали диметоксиэтаном. Промывки добавляли к декантированному раствору и объединенные растворители выпаривали, получая масло. Очистка колоночной хроматографией на силикагеле при использовании в качестве элюента гексана/этилацетат 5:1 давала 15,7 г (96%) 6-трифторметансульфонокси-1-тетралона в виде не совсем белого твердого вещества;1H ЯМР (CDCl3) δ 8,15 (d, 1H), 7,25 - 7,15 (m, 2H), 3,02 (t, 2H), 2,65 (t, 2H), 2,18 (m, 2H).
в. Порцию продукта части б (8,0 г, 27,2 ммоль), 4-метилфенилборную кислоту (4,07 г, 29,9 ммоль) и растертый трехосновный фосфат калия (8,65 г, 40,7 ммоль) суспендировали в безводном диоксане. Реакционную смесь дегазировали, пробулькивая азот через суспензию в течение 10 мин. Добавляли тетракис(трифенилфосфин)палладий (943 мг, 0,8 ммоль) и смесь нагревали 5 ч с обратным холодильником, охлаждали до комнатной температуры, разбавляли диэтиловым эфиром, фильтровали и выпаривали. Очистка хроматографией на силикагеле при использовании в качестве элюента дихлорметана давала 2,2 г (34%) 6-(4-метилфенил)-1- тетралона в виде не совсем белого твердого остатка;1H ЯМР (CDCl3) δ 8,05 (d, 1H), 7,58 - 7.25 (m, 6H), 3.05 (t, 2H), 2,64 (t, 2H), 2,40 (s, 3H): масс-спектр (химическая ионизация метаном) m/z 265 (М+C2H5), 237 (М+H).
Получение 2-фенил-7,8-дигидрохинолин-5(6H)-она
3-Аминоциклогексенон (5,55 г, 50 ммоль) и 3-диметиламинопропиофенона гидрохлорид (10,65 г, 50
ммоль) суспендировали в ледяной уксусной кислоте (15 мл) и нагревали с обратным холодильником 1 ч. Смесь охлаждали до комнатной температуры и осторожно выливали в 20% водный раствор карбоната натрия.
После прекращения выделения газа остаток экстрагировали хлороформом, высушивали над безводным карбонатом натрия, фильтровали и выпаривали. Очистка хроматографией на силикагеле при использовании в
качестве элюента хлороформа/дихлорметана 1:1 давала 2,95 г (26%) 2-фенил-7,8- дигидрохинолин-5(6H)-она в виде не совсем белого твердого остатка: tпл 128-130oC;1H ЯМР (CDCl3) δ 8,32 (d, 1H), 8,05 (dd, 2H), 7,66 (d, 2H), 7,55-7,42 (m, 3H), 3,21 (t, 2H), 2,72 (t, 2H), 2,22 (m, 2H).
Получение 2-фенил-7,8-дигидро-1,3-хиназолин-5(6H)-она
а. 1,3-циклогександион (11,2 г, 100 ммоль) суспендировали в диметилформамиде диметилацетале (28 мл) и смесь нагревали с обратным холодильником 1 ч. Смесь охлаждали до комнатной температуры и
растворитель выпаривали, получая оранжевый твердый остаток, который перекристаллизовывали из этилацетата для получения 12 г (72%) 2- диметиламинометилен-1,3- циклогександиона в виде светло-оранжевых
игольчатых кристаллов, tпл 116-118oC;1H ЯМР (CDCl3) δ 8,05 (s, 1H), 3,40 (s, 3H), 3,19 (s, 3H), 2,26 (t, 4H), 1,95 (m, 1H).
б. Натрий (1,09 г, 47 ммоль) осторожно маленькими порциями добавляли к безводному этанолу в течение 15 мин и смесь перемешивали до полного растворения натрия. Добавляли бензамидина гидрохлорид (7,80 г, 49 ммоль), вслед за этим растворяли порцию вещества из части а. (8,31 г, 50 ммоль). Смесь нагревали с обратным холодильником 1 ч и охлаждали до комнатной температуры. Растворитель выпаривали и осадок обрабатывали водой (100 мл), продукт фильтровали. Продукт перекристаллизовывали из этилацетата, получая 5,2 г (42%) 2-фенил-7,8-дигидро-1,3- хиназолин-5(6H)-она в виде светло-желтых кубиков, tпл 122- 124oC;1H ЯМР (CDCl3) δ 9,25 (s, 1H), 8,55 (dd, 2H), 7,58-7,48 (m, 3H), 3,18 (t, 2H), 2,74 (t, 2H), 2,24 (m, 2H), масс-спектр m/z 225 (M+H).
Получение
7-фенилтиохромана-4-она
а. 3-бромтиофенол (10 г, 52,9 ммоль) растворяли в акрилонитриле (11,28 г, 213 ммоль). Добавляли Тритон Б (1 мл). При добавлении Тритона Б смесь разогревалась и
начинала кипеть без дополнительного нагрева. Смеси давали остыть до комнатной температуры, выливали в холодный водный раствор гидроксида натрия (2%, 200 мл) и экстрагировали диэтиловым эфиром.
Органический слой высушивали, фильтровали и выпаривали, получая 12 г (93%) 3-[(3- бромфенил)тио] пропионитрила в виде коричневатого масла;1H ЯМР (CDCl3) δ 7,54 (s, 1H), 7,
42 (d, 1H), 7,32 (d, 1H), 7,25-7,15 (m, 2H), 3,15 (t, 2H), 2,62 (t, 2H); масс-спектр m/z 261,259 (M+NH4).
б. Продукт части а (12,0 г, 50 ммоль) растворяли в ледяной уксусной кислоте (10 мл). Добавляли концентрированную соляную кислоту (250 мл) и реакционную смесь нагревали с обратным холодильником 4 ч. Смесь охлаждали до комнатной температуры и выливали на 250 г измельченного льда. После растворения льда осадок фильтровали и растворяли в насыщенном растворе бикарбоната натрия (250 мл) и раствор промывали этилацетатом. Водную фазу осторожно закисляли концентрированной соляной кислотой и экстрагировали этилацетатом. Органическую фазу высушивали, фильтровали и выпаривали, получая 7,4 г (54%) 3-[(3-бромофенил)тио] пропановой кислоты в виде белого порошка, tпл 87-88oC;1H ЯМР (DMSO-d6) δ 7,52 (s, 1H), 7,40-7,25 (m, 3H), 3,18 (t, 2H), 2,53 (t, 2H); масс-спектр m/z 280, 278 (M+NH4), 262, 260 (m+H).
в. Порцию продукта части б (2,61 г, 10 ммоль) растирали, добавляли к концентрированной серной кислоте (8 мл) и смесь перемешивали 3 ч при комнатной температуре. Смесь выливали на лед. После таяния льда смесь экстрагировали этилацетатом. Органический экстракт осторожно промывали насыщенным водным раствором бикарбоната натрия и рассолом. Продукт перекристаллизовывали из бензол/гексана, получая 1,6 г (66%) 7-бромтиохроман-4она в виде белого порошка, tпл 55-57oC:1H ЯМР (CDCl3) δ 7,94 (d, 1H), 7,45 (d, 1H), 7,30 (dd, 1H), 3,25 (t, 2H), 2,97 (t, 2H); масс-спектр m/z 262, 260 (M+NH4), 245, 243 (M+H).
г. Продукт части в (15,00 г, 61,47 ммоль), фенилборную кислоту и бромид тетрабутиламмония (0,99 г, 3,07 ммоль) растворяли в толуоле (200 мл), водном растворе карбоната натрия (2М, 60 мл) и этаноле (30 мл). Смесь дегазировали, пропуская струю азота через интенсивно перемешиваемую смесь в течение 0,5 ч. Добавляли тетракис(трифенилфосфин)палладия (2,14 г, 1,8 ммоль) и смесь нагревали ночь (около 16 ч) с обратным холодильником. Смесь охлаждали до комнатной температуры, фазы разделяли. Водную фазу экстрагировали этилацетатом (2х50 мл). Объединенные органические фазы высушивали, фильтровали через пробку силикагеля и выпаривали до образования белого твердого остатка, который перекристаллизовывали из бензо/гексана, получая 10,9 (74%) 7-фенилтиохроман-4-она в виде не совсем белого порошка, tпл 85-87oC;1H ЯМР (CDCl3) δ/ 8,17 (d, 1H), 7,60-7,36 (m, 7H), 3, 25 (t, 2H), 2,98 (t, 2H); масс-спектр m/z 258 (M+NH4), 241 (M+H).
Получение 7-(4-метилфенил)хроман-4-она
а. 3-Бромфенол (100 r, 580 ммоль) и триэтиламин (104 мл,
750 ммоль) растворяли в дихлорметане (500 мл) и раствор охлаждали в ледяной бане. Добавляли по каплям ацетилхлорид (49,7 г, 630 ммоль). Через 1 ч охлаждающую баню убирали и смесь перемешивали ночь
(около 16 ч) при комнатной температуре. Смесь промывали соляной кислотой (1 н, 250 мл), водой (250 мл), 5% гидрокарбонатом натрия (250 мл) и рассолом (250 мл). Органический слой высушивали и упаривали,
получая 124 г (99%) 3-бромфенилацетата в виде светло-коричневой жидкости,1H ЯМР (CDCl3) δ 7,36 (d, 1H), 7,32-7,20 (m, 2H), 7,05 (d, 1H), 2,28 (s, 3H).
б. Продукт части а. (60 г, 280 ммоль) добавляли к хлориду алюминия (120 г, 890 ммоль) и помещали в масляную баню на 3 ч при 160oC. Смеси давали остыть до комнатной температуры и выливали на 300 г смесь лед/соляная кислоты (1 н). После расплавления льда смесь экстрагировали этилацетатом. Органический слой высушивали, фильтровали через подушку из силикагеля и выпаривали до образования светло-желтого масла, которое затвердевало под вакуумом, давая 53 г (88%) 4-бром-2-гидроксиацетофенона,1H ЯМР (CDCl3) δ 12,32 (s, 1H), 7.58 (d, 1H), 7,18 (s, 1H), 7,05 (d, 1H), 3,60 (s, 3H).
в. Продукт части б (53 г, 250 ммоль) растворяли в диметилацетале диметилформамиде (98 мл) и нагревали с обратным холодильником в атмосфере азота 1 ч в течение которого формировался осадок. Смесь охлаждали до комнатной температуры, осторожно добавляли ледяную кислоту (100 мл) и смесь нагревали с обратным холодильником 1 ч. Анализ аликвот реакционной смеси тонкослойной хроматографией и1H ЯМР показал, что полностью реакция не прошла, поэтому добавляли еще уксусной кислоты (100 мл) и нагревали смесь с обратным холодильником дополнительно 2 ч. Затем оставили смесь на ночь (около 16 ч) при комнатной температуре. Осадок отфильтровывали, высушивали и перекристаллизовывали из этанола, получая 12 ч (68%) 7- бромхромен-4-она. Концентрирование маточного раствора давало дополнительно 12 г продукта, tпл 154-155oC;1H ЯМР (CDCl3) δ 8,05 (d, 1H), 7,82 (d, 1H), 7,62 (s, 1H), 7,55 (d, 1H), 6,35 (d, 1H); масс-спектр m/z 244, 242 (M+NH4), 225, 227 (M+H).
г. Продукт части в (4,5 г, 20 ммоль), 4-метилфенилборную кислоту (3,26 г, 24 ммоль) и бромид тетрабутиламмония (3,22 мг, 1 ммоль) растворяли в смеси толуола (40 мл), этилового спирта (10 мл) и 2М водного раствора карбоната натрия и дегазировали, пропуская струю азота через интенсивно перемешиваемую смесь в течение 0,5 ч. Добавляли тетракис(трифенилфосфни)палладия (139 мг, 0,12 ммоль) и нагревали смесь с обратным холодильником ночь (около 16 ч). Смесь охлаждали до комнатной температуры и фильтровали через целит, слои разделяли и проводили экстракцию водного слоя этилацетатом. Объединенные органические слои фильтровали через подушку силикагеля и выпаривали, tпл 159-160oC;1H ЯМР (CDCl3) δ 8,35 (d, 1H), 7,95 (d, 1H), 7,72 (m, 1H), 7,65 (d, 2H), 7,38 (d, 2H), 6,42 (d, 1H), 2,50 (s, 3H); масс-спектр m/z 237 (M+H).
д. Продукт части г (1,80 г, 7,62 ммоль) растворяли в безводном тетрагидрофуране (100 мл) и охлаждали в бане сухой лед/ацетон. По каплям добавляли трибутил-2- боргидрид калия (1,0 М в тетрагидрофуране; 7,6 мл, 7.6 ммоль) и смесь перемешивали 1 ч при температуре охлаждающей бани. Реакционную смесь выливали в 20%-ный водный раствор одноосновного фосфата калия и экстрагировали этилацетатом. Органический слой высушивали, фильтровали и выпаривали. Очистка колоночной хроматографией на силикагеле с 1: 1 хлороформ/дихлорметаном давала 1,46 г (81%) 7-(4-метилфенил)хроман-4- она в виде не совсем белого твердого остатка,1H ЯМР (CDCl3) δ 7,85 (d, 1H), 7,52 (d, 2H), 7,35-7,18 (m, 4H), 4,58 (t, 2H), 2,82 (t, 2H), 2,40 (s, 3H).
Получение 7-фенилизохроман-4-она
а. К смеси
4-бром-2-метилбензойной кислоты (25,0 г, 116 ммоль) и карбоната калия (20,0 г, 145 ммоль) в безводном диметилформамиде (75 мл) добавляли метилйодид (10,0 мл, 160 ммоль) при комнатной температуре в
атмосфере азота и перемешивали 18 ч. Смесь выливали в воду и экстрагировали этилацетатом (3х50мл). Органические фазы объединяли, промывали раствором сульфата меди (50 мл) и рассолом (50 мл),
высушивали и выпаривали. Очистка колоночной хроматографией на силикагеле с 9:1 гексан/этилацетатом в качестве элюента давала 21,0 г (79%) метил 4-бром-2-метилбензоата в виде прозрачной жидкости,1H ЯМР (CDCl3) δ 7,85 (d, 1H), 7,42-7,47 (m, 2H), 3,95 (s, 3H), 2,64 (s, 3H); IR (острый) 1726 см-1; масс-спектры m/z 197,199 (М+H-ОCH3).
б. Смесь продукта части а (20,0 г, 87 ммоль), N-бромсукцинимида (15,54 г, 87 ммоль) и пероксида бензоила (0,21 г, 1 ммоль%) в тетрахлориде углерода (75 мл) кипятили с обратным холодильником в атмосфере азота 3 ч. Раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали, получая оранжевое масло, которое при стоянии образовывало кристаллы. Белый твердый остаток собирали фильтрацией под вакуумом и перекристаллизовывали из гексана, получая 15,23 г (56%) метил 2-бромметил-4-бромбензоата в виде белого порошка, tпл 78-79oC;1H ЯМР (CDCl3) δ 7, 84 (d, 1H), 7,63 (s, 1H), 7,50 (dd, 1H), 4,89 (s, 2H), 3,94 (s, 3H); IR (KBr) 1722 см-1; масс-спектр m/z 325 (М+H); Анал. Рассч. для C9H8Br2O2: C 35,10, H 2, 62, Br 51.89; Найдено: C 35,09, H 2,63, Br 51,67.
в. Гидрид натрия (1,43 г, 60% в масле, 36 ммоль) суспендировали в безводном тетрагидрофуране (10 мл) в атмосфере азота. При комнатной температуре добавляли раствор продукта части б. (10,0 г, 32 ммоль) и метилгликолята (2,76 мл, 36 ммоль) в безводном тетрагидрофуране (40 мл) и смесь нагревали с обратным холодильником 3 ч. После охлаждения до комнатной температуры раствор гасили метанолом (2 мл), выливали в воду (200 мл) и экстрагировали этилацетатом (50 мл). Органическую фазу промывали рассолом, высушивали и выпаривали, получая белый остаток. Очистка колоночной хроматографией на силикагеле с 4: 1 гексан/этилацетатом в качестве элюента давала 7,10 г (69%) метил 2-[(2-метокси-2- оксоэтокси)метил]-4-бромбензоата в виде белого твердого остатка, tпл 60-62oC;1H ЯМР (CDCl3) δ 7,93 (s, 1H), 7,83 (d, 1H), 7,48 (dd, 1H), 5,01 (s, 2H), 4,25 (s, 2H), 3,88 (s, 3H), 3,79 (s, 3H); IR (KBr) 1758, 1720 см-1; масс-спектр m/z 334 (M+NH4); Анал. Рассч. для C12H13BrO5: C 45,45, H 4,13, Br 25,20; Найдено: C 45.42, H 4.03, Br 25, 17.
г. Смесь продукта части в (6,77 г, 21 ммоль) и гидроксида натрия (50% водной раствор, избыток) в 50% водной этаноле (200 мл) нагревали с обратным холодильником 5 ч. Раствор охлаждали до комнатной температуры, выливали в воду (400 мл) и подкисляли концентрированной соляной кислотой для получения белого осадка, который собирали фильтрацией и высушивали на воздухе, получая 6,04 г (98%) 2-[(2-гидрокси-2-оксоэтокси)метил]-4-бромбензойной кислоты в виде белого твердого остатка, tпл 194-196oC;1H ЯМР (DMSO-d6) 7,84 (m. 2H), 7.62 (d, 1H), 4.90 (s, 2H), 4,19 (s, 2H); ИК-спектр (KBr) 1704 см-1; масс-спектр m/z 230 (M-CH2COOH); Анал. Рассч. для C10H9O5Br: C 41,55, H 3,14, Br 27,64: Найдено: C 41,33, H 3,08, Br 27,42.
д. Смесь продукта части г. (5,48 г, 19 ммоль) и ацетата калия (3,0 г, 3 ммоль) в уксусном ангидриде (10 мл) нагревали с обратным холодильником в атмосфере азота 18 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Осторожно добавляли водный раствор гидроксида натрия (1 M, 100 мл) и водный раствор экстрагировали диэтиловым эфиром (3 х 50 мл). Органические экстракты объединяли, высушивали и выпаривали. Остаток собирали водным раствором гидроксида натрия (1 н, 100 мл) и перемешивали 5 мин при комнатной температуре. Смесь выливали в воду (400 мл) и твердый остаток собирали фильтрацией. Очистка колоночной хроматографией на силикагеле с 5:1 гексан/этилацетатом в качестве элюента давала 1,15 г (27%) 7-бромизохроман-4-она в виде белого твердого остатка, tпл 144-146oC;1H ЯМР (CDCl3) δ 7,91 (d, 1H), 7,57 (dd, 1H), 7,41 (s, 1H), 4,86 (s, 2H), 4,36 (s, 2H); ИК-спектр (KBr) 1688 см-1; масс-спектр m/z 226 (М+H); Анал. Рассч. для C9H2O2Br: C 47,61, H 3,11, Br 35,19; Найдено: C 47,65, H 3,10, Br 35,04.
e. Азот пробулькивали через смесь продукта д. (0,611 г, 27 ммоль) и фенилборной кислоты (0,36 г, 29 ммоль) 5:2 толуол/этаноле (30 мл) 4 ч. После добавления карбоната натрия (0,57 г, 5,38 ммоль) добавляли бромид тетрабутиламмония (0,049 г, 0,13 ммоль) и тетракис(трифенилфосфин)палладия (0,012 г, 0,04 ммоль). Раствор нагревали с обратным холодильником 18 ч, охлаждали до комнатной температуры и выпаривали. Остаток собирали водой (50 мл) и экстрагировали этилацетатом (3 х 20 мл). Остаток собирали водой (50 мл) и экстрагировали этилацетатом (3 х 20 мл). Органические фазы объединяли, промывали рассолом, высушивали и выпаривали. Твердый остаток перекристаллизовывали из гексана, получая 0,429 г (70%) 7- фенилизохроман-4-она в виде золотистого твердого остатка, tпл 127- 128oC;1H ЯМР (CDCl3) 8,12 (d, 1H), 7,60-7,66 (m, 3H), 7,39-7,51 (m, 4H), 4,96 (s, 2H), 4,41 (s, 2H); ИК-спектр (KBr) 1688 см-1; масс- спектр m/z= 225 (М+H): Анал. Рассч. для C15H12O2: C 80,34, H 5,39; Найдено: C 80,03, H 5,45.
Получение 7-фенилизотиохроман-4-она
а. К раствору
3-бромбензилбромида (23,75 г, 90 ммоль) и тиогликолиевой кислоты (11,42 г, 120 ммоль) в этаноле (150 мл) добавляли гидроксид калия (14,24 г, 250 ммоль) в воде (50 мл). Реакционную смесь нагревали с
обратным холодильником 3,5 ч, охлаждали и выпаривали этанол. Остаток гасили водой (100 мл) и экстрагировали смесь эфиром (50 мл). Водный слой подкисляли концентрированной соляной кислотой до pH 1,
экстрагировали этилацетатом (2 х 100 мл) и высушивали органическую фазу. Выпаривание растворителя и последующая промывка остатка гексаном давали 3-бромфенилметилтиоуксусную кислоту в виде белого
твердого остатка (92%), tпл 90-91oC;1H ЯМР (CDCl3) δ 3,10 (s, 2H), 3,82 (s, 2H), 7,20-7,29 (m, 2H), 7,39-7,42 (dd, 1H), 7,52 (d, 1H); ИК-спектр (KBr) 1706
см-1; масс-спектр m/z=280 (M+NH4); Анал. Рассч. для C9H9O2SBr: C 41,39, H 3,47, S 12,18; Найдено: C 41,43, H 3,49, S 12,10.
б. К
холодному (0oC) раствору продукта части а. (22,00 г, 80 ммоль) в метиленхлориде (200 мл) добавляли по каплям оксалилхлорид (11,81 г, 90 ммоль) и затем 4 капли диметилформамида. Реакционную
смесь интенсивно перемешивали при 0oC и затем 5 ч при комнатной температуре. Смесь концентрировали до желтого масла, которое перерастворяли в свежем метиленхлориде (100 мл). Раствор
добавляли по каплям при 0oC к суспензии хлорида алюминия (11,30 г, 80 ммоль) в метиленхлориде (150 мл). Смесь нагревали до комнатной температуры и перемешивали 48 ч, затем выливали в
измельченный лед (150 г). Органический слой отделяли и промывали водой (50 мл), насыщенным бикарбонатом натрия (50 мл) и рассолом (50 мл), высушивали и выпаривали до темно-коричневого остатка. Очистка
хроматографией на силикагеле с 20:1 гексан/этилацетатом в качестве элюента давала 7-бромизотиохроман-4-он (88%) в виде темно-коричневого твердого остатка, tпл 81oC;1H ЯМР
(CDCl3) 3,54 (s, 2H), 3,88 (s, 2H), 7,26 (s, 1H), 7,50 (d, 1H), 7,95 (d, IH); ИК-спектр (KBr) 1676 см-1; масс- спектр m/z 245 (М+H); Анал. Рассч. для C9H7
BrOS: C 44,46, H 2,90, S 13,19: Найдено: C 44,28, H 2,77, S 13,09,
в. 7-фенилизотиохроман-4он получали с выходом 62% способом, используемым в части д. получения 7-фенилизохроман-4-она и
перекристаллизовывали из бензологексанов, tпл 87-89oC;1H ЯМР (CDCl3) δ 3,59 (s, 2H), 3,99 (s, 2H), 7,38-7,47 (m, 4H), 7,49-7,60 (m, 3H), 8,17 (d, 1H);
ИК-спектр (KBr) 1674 см-1; масс-спектр m/z 241 (100); Анал. Рассч. для Cl5H12O5: C 74,97, H 5,03, S 13,34; Найдено: C 75,06, H 5,00, S 13,20.
Получение 7-фенил-8-азаизотиохроман-4-она
К раствору этил 2-метил-6-фенилникотината (приготовленному по Spath et al. [Monatsh. Chem. (1928) 49: 265]) (1,00 г, 4,10 ммоль) в
тетрахлориде углерода (10 мл) добавляли N-бромсукцинимид (0,81 г, 4,60 ммоль) и пероксид бензоила (0,05 г). Реакционную смесь нагревали 18 ч с обратным холодильником, охлаждали и фильтровали. Фильтрат
концентрировали и перерастворяли в безводном тетрагидрофуране (10 мл). Добавляли метилтиогликолят (0,37 мл, 4,10 ммоль), а затем гидрид натрия (80%, 0,12 г, 4,1 ммоль). Реакционной смеси давали
перемешаться при комнатной температуре в течение 3 ч. Добавляли еще гидрид натрия (80%, 0,14 г, 4,6 ммоль) и раствор нагревали с обратным холодильником при слабом кипении 18 ч. Реакционную смесь
охлаждали до 0oC и гасили водой (20 мл). Смесь промывали этилацетатом (50 мл). Водный слой подкисляли до pH 5 уксусной кислотой и экстрагировали продукт этилацетатом (3 х 40 мл).
Органический экстракт промывали рассолом (10 мл), высушивали и выпаривали до получения оранжевого масла. Хроматография на силикагеле с использованием в качестве элюента 6:1 гексан/этилацетата давала
смесь из нециклизованного соединения в виде желтого масла (20%) и метил 7-фенил-8-азаизотиохроман-4-он-3- карбоксилата (23%) в виде желтого твердого остатка, tпл 78-81oC. Это вещество
нагревали с обратным холодильником в 3 н. водной соляной кислоте (30 мл) 12 ч, затем охлаждали и доводили pH до щелочного значения. Экстракция этилацетатом (4 х 50 мл) после высушивания и выпаривания
tпл 97-99oC;1H ЯМР (CDCl3) δ 3,58 (s, 2H), 4,14 (s, 2H), 7,48 (m. 4H), 7,78 (d, 1H), 8,04-8,07 (m, 2H), 8,40 (d, 1H); ИК-спектр (KBr) 1680 см-1;
масс-спектр m/z 242 (М+H); Анал. Рассч. для C14H11NOS: C 69,68, H 4,54, N 5,80; Найдено: C 69,39, H 4,58, N 5,69.
Соединения настоящего изобретения и их получения далее могут быть поняты по следующим характерным примерам, которые не являются ограничением данного изобретения. Все температуры плавления нескорректированы. Все реакции проводились в атмосфере азота, за исключением случаев, где указаны другие условия. Все продажные реактивы использовались как общепринято. Хроматография проводилась на силикагеле 60 Merck (230 - 400 меш.). Хроматографические элюенты даются как объемные отношения. Органическую фазу экстрактов растворитель/растворитель обычно выпаривали над сульфатом магния, если не оговаривается другое. Растворители обычно удаляли выпариванием при пониженном давлении на роторном испарителе, если не оговаривается другое. Позиции пиков1H ЯМР спектров даны как части на миллион (δ) по полю по внутреннему стандарту тетраметилсилану. Сокращения для1H ЯМР спектров следующие: s - синглет, d - дублет, m - мультиплет, dd - дублет дублетов, dm -дублет мультиплетов. Масс-спектры получали, используя химическую ионизацию аммиаком в качестве газа реагента.
Пример 1
Получение соединения формулы 1, где R1 есть 6-F, R2 = H, R3 = 4-метилфенил, X = CH2CH2 и Z1, Z2 и Z3 все являются CH.
5-фторизатин (1,40 г, 8,48 ммоль) и 6-(4-метилфенил)-1-тетралон (2,0 г, 8,47 ммоль) суспендировали в безводном этаноле. Добавляли гидроксид калия (4,92 г, 84,8 ммоль), растворенный в воде (10 мл), и смесь нагревали с обратным холодильником ночь (около 16 ч). Смесь охлаждали до комнатной температуры и выпаривали для удаления растворителя. Остаток суспендировали в воде (50 мл) и промывали эфиром (5 х 20 мл). Водный слой подкисляли 1 н. соляной кислотой. Получающийся осадок собирали и растирали в порошок с небольшим количеством этилацетата, получая 1,17 г (36%) названного соединения, tпл 305oC (разложение);1H ЯМР (DMSO-d6) 8,55 (d, 1H), 8,28 (m, 1H), 7,85-7,68 (m, 5H), 7,60 (d, 1H), 7,38 (d, 2H), 3,18 (m, 4H), 2,40 (s, 3H); масс-спектры 384 (M+H); масс-спектры высокого разрешения рассчитано 383,1340, найдено 384,1404.
Пример 2
Получение натриевой соли соединения формулы 1, где R1 есть 6-F, R2 = H, R3 = 4-метилфенил, X
= CH2CH2 и Z1, Z2 и Z3 все являются CH.
Продукт пример 1 (1,17 г, 3,05 ммоль) суспендировали в этаноле (50 мл) и нагревали до кипения с обратным холодильником. Добавляли по каплям водный раствор гидроксида натрия (1 н. , 3,05 мл, 3,05 ммоль) в течение времени растворения вещества. Смесь нагревали с обратным холодильником 1ч, охлаждали до комнатной температуры, фильтровали и выпаривали, получая названное соединение в виде светло-рыжего порошка, tпл 342oC (разложение);1H ЯМР (DMSO-d6) δ 8,42 (d, 1H), 8,02 (m, 1H), 7,75-7,50 (m, 6H), 7,30 (d, 2H), 3,15-2,95 (m, 4H), 2,18 (s, 3H).
Пример 3
Получение натриевой соли соединения формулы 1, где R1 есть 6-F, R2 = H, R3 = фенил, X = CH2CH2 и Z1 и Z2 = CH и Z3 = N.
2-фенил-7, 8-дигидрохинолин-5(6H)-он (3,25 г, 14,6 ммоль) и 5-фторизатин (2,40 г, 14,6 ммоль) суспендировали в этаноле (50 мл). К реакционной смеси добавляли растворенный в воде (10 мл) гидроксид натрия (3,50 г, 87.5 ммоль), затем смесь нагревали с обратным холодильником 16 ч. Смесь охлаждали до комнатной температуры и выпаривали растворитель. Остаток разбавляли водой (100 мл) и диэтиловым эфиром (100 мл) и перемешивали 0,5 ч. Водный слой промывали диэтиловым эфиром (3 х 100 мл), отделяли и разбавляли равным объемом насыщенного раствора хлорида натрия. Образовывался осадок, который собирали спустя 1 ч, промывали небольшим количеством воды (10 мл), затем растирали в порошок в ацетоном (50 мл), фильтровали и высушивали, получая 3,3 г (58%) названного соединения в виде не совсем белого порошка, tпл >350oC (разложение);1H ЯМР (DMSO-d6) δ 8,75 (d, 1H), 8,19 (d, 2H), 8,10-7,98 (m, 2H), 7,70-7,42 (m, 5H), 3,28-3,14 (m, 4H); масс-спектры 410 (M+NH4), 393 (М+H); масс-спектры высокого разрешения рассч. 371,1195, найдено 371,1180. Анал. рассч. для C23H14FN2O2Na(H2O): C 67,32, H 3,93, N 6,83: Найдено: C 67,43, H 3,70, N 6,78.
Пример 4
Получение натриевой соли соединения формулы 1, где R1 есть 6-F, R2 = H, R3 = фенил, X = CH2CH2 и Z1 = CH, Z2 и Z3=N.
5-фторизатин (3,30 г, 20 ммоль) и 2-фенил-7,8-дигидро-1,3- хиназолин-5(6H)-он (4,48 г, 20 ммоль) суспендировали в этаноле (100 мл). Растворяли в воде (20 мл) гидроксид натрия (4,80 г, 120 ммоль) и добавляли к реакционной смеси, которую затем нагревали с обратным холодильником ночь (около 16 ч). Смесь охлаждали до комнатной температуры и растворитель выпаривали. Остаток разбавляли водой (250 мл) и фильтровали. Твердый остаток промывали небольшим количеством воды, ацетоном (100 мл) и высушивали, получая 6,50 г названного соединения в виде не совсем белого порошка, tпл >400oC (разложение);1H ЯМР (DMSO-d6) δ 9,70 (s, 1H), 8,58 (m, 2H), 8,14 (m, 1H), 7,78-7,60 (m, 5H), 3,28 (dm, 4H); Анал. рассч. для C22H13FN3O2Na(0,5 H2O): C 65,67, H 3,51, N 10,44; Найдено: C 65,83, H 3,44, N 10, 32.
Пример 22
Получение соединения формулы 1. где R1 есть 6-F, R2 =H, R3=фенил, X= CH2S и Z1, Z2 и Z3 = CH.
5-фторизатин (497 мг, 3,01 ммоль) и 7-фенилтиохроман-4-он (723 мг, 3,01 ммоль) суспендировали в этаноле (10 мл). Добавляли растворенный в воде (1 мл) гидроксид калия (1, 01 г, 18 ммоль) и смесь нагревали с обратным холодильником ночь (около 16 ч). Растворитель выпаривали, остаток разбавляли водой (10 мл) и промывали диэтиловым эфиром (2 х 10 мл). Водный слой подкисляли водной соляной кислотой (1 н. ) до pH 3 и давали постоять в течение 1 ч. Осадок собирали и высушивали с выходом 889 мг (76%) названного соединения. Перекристаллизация из метилизобутилкетона давала светло-желтый порошок, tпл >280oC (разложение);1H ЯМР (DMSO-d6) δ 8,56 (d, 1H), 8,19 (m, 1H), 7,60-7,40 (m, 9H), 4,30 (s, 2H); масс-спектры 388 (M+H); масс-спектры высокого разрешения рассч. 387,0729. найдено 387,0721; Анал. рассч. для C23H14FNO2S: C 71,30, H 3,64, N 3,62; Найдено: C 70,84, H 3,53, N 3.51.
Пример 23
Получение натриевой соли соединения формулы 1, где R1 есть 6-F, R2 = H, R3 = фенил, X = CH2S и Z1, Z2
и Z3 = CH.
Продукт примера 22 (3,0 г, 7,74 ммоль) суспендировали в этаноле (50 мл) и нагревали с обратным холодильником до кипения. Добавляли по каплям водный гидроксид натрия (1 н., 7,75 мл. 7,75 ммоль) до растворения вещества. Реакционной смеси давали остыть до комнатной температуры, фильтровали и выпаривали с выходом названного соединения в виде не совсем белого порошка, tпл >230-235oC (разложение);1H ЯМР (DMSO-d6) δ 8,56 (d, 1H), 8,10 (m, 1H), 7,62-7,38 (m, 9H), 4,30 (s, 2H).
Пример 53
Получение соединения формулы 1, где R1 есть 6-F, R2 = H, R3 = 4-метилфенил, X = CH2O и Z1, Z2 и Z3 = CH.
5-фторизатин (1,00 г, 5.9 ммоль) и 7-(4-метилфенил)хроман-4-он (1.46 г, 6,1 ммоль) суспендировали в этаноле (30 мл). Добавляли диэтиламин (445 мг, 6,1 ммоль) и перемешивали реакционную смесь ночь (около 16 ч), в течение которой формировался осадок. Осадок отфильтровывали, высушивали и растворяли в диметоксиэтане (20 мл). После добавления воды (10 мл) добавляли метансульфокислоту (10 мл) и смесь нагревали с обратным холодильником ночь (около 16 ч). Смесь охлаждали до комнатной температуры и выливали в смесь льда и воды (20 мл) и оставляли на 1 ч. Собирали осадок и перемешивали ночь с этилацетатом (50 мл). Твердый остаток отфильтровывали, получая 105 г названного соединения в виде светло-оранжевого порошка,1H ЯМР(DMSO-d6) 8,44(d,m, 1H), 8,24 (m, 1H), 7,90-7, 30 (m, 8H), 5,60 (s, 2H), 2,42 (s, 3H).
Пример 54
Получение натриевой соли соединения формулы 1, где R1 - ее 6-F, R2-H, R3 = 4-метилфенил, X
- CH2O и Z1, Z2 и Z3 = CH.
Продукт примера 53 (100 мг, 0,25 ммоль) суспендировали в этаноле (10 мл) и нагревали с обратным холодильником до кипения. Добавляли по каплям водный раствор гидроксида натрия (1 н., 0,26 мл, 0,26 ммоль), во время добавления которого вещество растворялось. Смесь нагревали с обратным холодильником еще 0,5 ч, охлаждали до комнатной температуры, фильтровали и выпаривали, получая названное соединение в виде светло-рыжего остатка, tпл 305oC (разложение);1H ЯМР (DMSO-d6) δ 8,44 (d,m, 1H), 8,10 (m, 1H), 7,95-7,35 (m, 8H), 5,44 (s, 2H), 2,42 (s, 3H).
Пример 91
Получение соединения формулы 1, где R1 - ее 6-F, R2-H,
R3-фенил, X = OCH2 и Z1, Z2 и Z3 = CH.
Смесь 7-фенилизохроман-4-она (0,412 г, 1,84 ммоль), 5- фторизатина (0,303 г, 1,84 ммоль) и диэтиламина (0,19 мл, 1,84 ммоль) перемешивали в этаноле (10 мл) при комнатной температуре в атмосфере азота 4 ч. Растворитель удаляли, получая коричневатый остаток. Остаток собирали 1, 2-диметоксиэтаном (10 мл) и добавляли 50% водный раствор метансульфокислоты (10 мл). Смесь нагревали с обратным холодильником 18 ч, охлаждали до комнатной температуры и выливали в воду (25 мл). Водный раствор защелачивали 1 н. гидроксидом натрия и экстрагировали диэтиловым эфиром (3 х 25 мл). Водный раствор закисляли концентрированный соляной кислотой и осадок собирали фильтрацией, получая 62,5 мг (17%) названного соединения в виде желтого твердого остатка, tпл >250oC (разложение);1H ЯМР (DMSO-d6) δ 8,42 (d, 1H), 8,16 (dd, 1H), 7,88 (d, 1H), 7,77 (t, 3H), 7,41-7,65 (m, 5H), 5,53 (s, 2H); масс-спектры 372 (M+H); масс-спектры высокого разрешения рассч. 372,1036, найдено 372,1027.
Пример 100
Получение соединения формулы
1, где R1 - есть 6-F, R2 = H, R3 = фенил, X = SCH2 и Z1, Z2 и Z3 = CH.
Гидроксид натрия (0,49 г, 12,5 ммоль) растворяли в воде (0,5 мл) и добавляли к раствору 5-фторизатина (0,34 г, 2,08 ммоль) в этаноле (15 мл). Смесь осторожно кипятили с обратным холодильником 0,5 ч. Затем к вышеназванному раствору порциями добавляли 7-фенилизотиохроман-4-он и смесь нагревали с обратным холодильником 24 ч. Этанол выпаривали и остаток разбавляли водой (75 мл). Непрореагировавшие исходные вещества экстрагировали эфиром (2 х 50 мл) и отбрасывали. Водный слой обрабатывали рассолом (75 мл). Сформировавшийся осадок отфильтровывали, промывали 2-бутаноном (20 мл) и высушивали под вакуумом. Твердый остаток растворяли в воде (60 мл) и осторожно подкисляли уксусной кислотой до pH 5. Осадок отфильтровывали и высушивали под вакуумом, получая названное соединение в виде желтых кристаллов (52%), tпл > 265-268oC (разложение);1H ЯМР (DMSO-d6) δ 4,18 (s, 2H), 7,45 (d, 1H), 7,50-7,55 (ds, 2H), 7,59-7,63 (dd, 1H), 7,73-7,76 (dt, 1H), 7,78-7,84 (m, 4H), 8,21 (dd, 1H), 8,47 (d, 1H); масс-спектры 388 (М+H).
Пример 101
Получение натриевой соли соединения формулы 1, где R1 - есть 6-F, R2 = H, R3 = фенил, X
= SCH2 и Z1, Z2 и Z3 = CH.
К раствору соединения пример 100 (0,30 г, 0,78 ммоль) в этаноле (4 мл) добавляли 1 н. водный раствор гидроксида натрия (0,77 мл). Смесь осторожно нагревали 1 ч и выпаривали этанол. Остаток промывали 2-бутаноном (10 мл), фильтровали и высушивали под вакуумом, получая названное соединения (87%), tпл > 294-296oC (разложение);1H ЯМР (DMSO-d6) δ 4,00 (s, 2H), 7,40 (m, 1H), 7,54 (m, 3H), 7,72 (s, 1H), 7,75-7,78 (dm, 4H), 8,04 (dd, 1H), 8,46 (d, 1H); масс-спектры 388 (М+H, свободная кислота); масс-спектры высокого разрешения рассч. для C23H14FNO2S (свободная кислота) 388.0807, найдено 388,0817.
Пример 102
Получение натриевой соли соединения формулы 1, где R1 - есть 6-F, R2 - H, R3 = фенил, X = SCH2, Z1 и Z1 = CH и Z3 =
N.
Названное соединение получали из 5-фторизатина и 7-фенил-8- азаизотиохроман-4-она с выходом 53%, используя способ, описанный в примере 100, tпл 210oC (разложение);1H ЯМР (DMSO-d6) δ 4,28 (s, 2H), 7,50-7,58 (d, 3H), 7,72- 7,69 (m, 2H), 8,13-8,22 (dm, 4H), 8,71 (d, 1H); масс-спектры 389 (М+H, свободная кислота).
В Таблицах 1, 2, 3, и 4 (см. в конце описания) представлены соединения примеров 1-4, 22,23, 53, 54, 91 и 100-102 и другие соединения, которые были получены, используя процедуры примеров 1-4, 22, 23, 53, 54, 91 и 100-102, а также другие соединения, которые могут быть получены этими способами. В этих таблицах буква d после температуры плавления указывает, что при этой температуре или указанном интервале температур происходит разложение соединения.
Применение
Реакция смешанной культуры лимфоцитов человека
Тест реакция смешанной культуры лимфоцитов человека, описанный
ниже, может быть использован для демонстрации того, что соединения этого изобретения обладают способностью подавлять или тормозить ответ иммунных клеток. Реакция смешанной культуры лимфоцитов
используется для определения совместимости при трансплантации между донором (трансплантант) и реципиентом (Park and Good, p. 71, in Tissue Typing and Organ Transplantation (Yunis et al.). 1973.
Academic Press Inc., NY). Реакции смешанной культуры лимфоцитов человека является иммунным ответом in vitro. Подавление иммунного ответа реакции смешанной культуры лимфоцитов человека является
стандартным иммунологическим методом, который, как считается, указывает на иммуноподавляющую активность in vivo. В частности, активность в реакции смешанной культуры лимфоцитов показывает, что
соединения данного изобретения были бы эффективны в предупреждении отторжения пересаженных органов и реакции "трансплантант против хозяина". Смешанные лимфоциты человека также используются в качестве
модельной системы иммунных ответов, в том числе, иммунных ответов Т-клеток. Эти иммунные ответы Т-клеток связаны с патологическими состояниями, ассоциированными с аутоиммунными заболеваниями,
хроническими воспалительными заболеваниями, реакцией "трансплантант против хозяина" и отторжением органов при трансплантации. Таким образом, в свете представленных в заявке результатов, ожидается, что
соединения формул 1-4 настоящего изобретения будут эффективными в лечении этих заболеваний.
Кровь получали венепункцией от двух неродственных доноров. Используя метод Leuco Prep (Becton-Dickinson), из этих образцов выделяли одноядерные клетки периферической крови (ОКПК). ОКПК дважды промывали фосфатно-солевым буфером (без кальция и магния) и проводили посев выделенных клеток при соответствующей концентрации в среде (RPMI 1640) с добавлением 10% сыворотки AB человека и 50 мкл/мл гентамицина. Клетки донора A (2•105) инкубировали с клетками донора B (2•105) в 96- луночных микропланшетах со сферическим дном с или без соединения при 37oC, 5% CO2 в течение 6 дней. За 18 ч до сбора клеток из планшетов во все лунки добавляли 1 мкКи тритированного тимидина. На шестой день клетки из планшетов промывали и определяли включение тритированного тимидина с помощью сцинцилляционного счетчика.
В таблице 5 ( см. в конце описания) представлены результаты для характерных соединений настоящего изобретения. В таблице ++ показывает, что значение IC50 меньше 5•10-8 М и + показывает, что значение IC50 находится в интервале от 5•10-8 М до 1•10-5 М.
Результаты тестирования в реакции пролиферации смешанной культуры лимфоцитов человека показывают, что соединения настоящего изобретения тормозят или подавляют иммунный ответ in vitro (т.е., обладают потенциальной иммуносупрессорной активностью) и будут эффективны в лечении и/или предупреждении отторжения пересаженного органа, реакции "трансплантант против хозяина", псориаза, ревматоидного артрита, аутоиммунных заболеваний и хронических воспалительных заболеваний, которые все включают иммунные ответы Т-лимфоцитов. Также, антипролиферативная активность соединений настоящего изобретения указывает на способность этих соединений подавлять рост опухоли.
Контактная чувствительность к ДНФБ у мышей
Контактную чувствительность к ДНФБ широко изучали и охарактеризовали у мышей для того, чтобы выявить
регуляторные механизмы, вовлеченные в клеточные ответы (Claman et al., Immunol. Rev. 50: 105, 1980; Young and Young, "Cutaneous Models of Inflammation for evaluation of Topical and Systemic
Pharmacological Agents" in Pharmacological Methods in Control of Inflammation" (Chang and Lewis, Eds.), Alan R. Liss, Inc., New York, pp. 215-231, 1989). Это антиген- специфичный опосредованный
Т-клетками воспалительный ответ, который представляет реакции гиперчувствительности замедленного типа, показанные как у человека, так и у других млекопитающих. Животная модель контактной
чувствительности используется в текущей работе многих лабораторий фармакологического скрининга противовоспалительных средств и средств лечения аутоиммунных заболеваний.
Самок balb/c
мышей (примерно 20 г, Charles River) сенсибилизировали на выбритом животе 25 мкл 0,5% 2,4-динитрофторбензола (ДНФБ) {Eastman Kodak Co. ] в растворителе ацетон/оливковое масло 4:1 в день 0 и 1. Мышам
вводили в ухо по 20 мкл 0,2% ДНФБ в растворителе ацетон/оливковое масло 4:1 на 5 день. Одинаковый сегмент уха был измерен непосредственно перед введением и через 24 ч инженерным микрометром. Набухание
уха выражали как разницу в толщине уха до и после введения в единицах 10-4 дюйма ±SEM. Процент подавления рассчитывали как:

Соединения (приготовленные в 0,25% Methocel®) вводили перорально. Контрольные животные получали только растворитель (0,25%; Methocel®). Негативный контроль не сенсибилизировали в день 0 и 1, но делали ушное введение на 5 день. Обычно использовали по 10 мышей в группе.
В таблице 6 (см. в конце описания) показаны результаты теста на контактную чувствительность для характерных соединений настоящего изобретения. В этой таблице + означает более чем 30% подавление контрольного набухания уха.
Результаты представленных выше биологических тестов показывают, что карбоциклические и гетероциклические конденсированные соединения хинолинкарбоновой кислоты формул 1-4 настоящего изобретения обладают способностью к подавлению или торможения контактной чувствительности к 2,4-динитрофторбензолу (ДНФБ) у мышей.
Контактная чувствительность к ДНФБ - это форма гиперчувствительности замедленного типа, которую широко изучают для того, чтобы понять регуляцию иммунологических процессов (Claman et al., выше). Эта реакция опосредуется Т-лимфоцитами, которые становятся чувствительными к антигену, отвечая пролиферацией и превращением в зрелые эффекторные клетки (Claman et al., выше). Эта опосредованная клетками иммунная реакция (опосредованный Т-клетками иммунитет) является центральной при многих болезненных состояниях, таких как отторжение органа при трансплантации и реакция "трансплантант против хозяина" (Benacerrafand Unanue (1979), Textbook of Immunology, Williams&Wilkins Co. , Eisen (1980), Immunology, An Introduction to Molecular and Cellular Principles of the Immune Responses, Harper&Row, Inc.; Loveland and McKenzie (1982), Immunology, 46: 313- 320; Gallin et al. (1988), Imflammation, Basic Principles and Clinical Correlates, Raven Press).
Модель контактной чувствительности, используемая в этом исследовании, является модельной системой реакций замедленной гиперчувствительности, которые связаны с патологическими состояниями, связанными с отторжением пересаженных органов, реакцией "трансплантант против хозяина", рассеянным склерозом, миастенией gravis, системной красной волчанкой, ревматоидным артритом и другими хроническими воспалительными заболеваниями и аутоиммунными заболеваниями, для которых Т-клетки являются основными в создании иммунного или аутоиммунного ответа.
Модель контактной чувствительность - это широко используемая модель реакции замедленной гиперчувствительности, в том числе клеточных реакций (опосредованных Т-клетками ответов), которые связаны с отторжением пересаженных органов и реакцией "трансплантант против хозяина", а также болезненных состояний, связанных с псориазом, ревматоидным артритом, аутоиммунными заболеваниями и хроническими воспалительными болезненными состояниями. Поскольку известно, что эти заболевания вовлекают Т-лимфоциты, предполагается, что заявленные иммунодепрессанты формул 1-4 будут эффективными в лечении этих заболеваний.
Представленные результаты показывают, что соединения этого изобретения обладают как иммуномодулирующей, так и противовоспалительной эффективностью.
Вызванный адъювантом артрит
Вызванный адъювантом артрит у крыс представляет
собой системное воспалительное заболевание с изменениями кости и хряща, подобными наблюдаемым при ревматоидном артрите, но в ускоренном промежутке времени (Pearson, Arthr. Rheum. 7$ 80, 1964).
Активность тестируемых соединений в модели вызванного адъювантом артрита является показателем противовоспалительной активности для лечения хронических воспалительных заболеваний, таких как
ревматоидный артрит, псориаз и воспалительное заболевание кишечника.
Самцам крыс Lewis (Charles River) весом 160 - 210 г подкожно вводят 0,1 мл полного адъюванта Фрейнда, содержащего 5 мг М. бутирикум/мл парафинового масла (Difco Laboratories) в подошву правой задней лапы. Контрольным животным вводят парафиновое масло. В каждое группе обычно используют по 10 крыс. Соединения готовят в 0,25 Methocel® (Dow Chemical Со.) с одной каплей Tween® 80 на 10 мл Methocel®. Животные получают дозу каждый день, начиная с момента инъекции в лапу до 18 дня. Через день записывается вес животных, начиная со дня инъекций в лапу. На 18 день животных взвешивают и измеряют объем задней лапы, в которую делали инъекции, используя плетизмограф Ugo Basile Volume Differential Plethismometer.
Гиперпролиферация, вызванная ТФА
Тест на гиперпролиферацию, вызванную ТФА, описанный ниже, может быть использован
для того, чтобы установить, обладают ли соединения настоящего изобретения способностью подавлять гиперплазию кожи, вызванную повторным приложением тетрадеканоилфорболацета (ТФА) на уши мыши (Marks et
al., Cancer Res. , 36: 2636, 1976). Известно, что ТФА вызывает изменения на мышиной коже, которые имитируют многие воспалительные и эпителиальные изменения, имеющие место при заболеваниях человека,
таких как псориаз.
Самцам мышей CF-I (Charles River, вес 20-25 г) вводят перорально тестируемое соединение, приготовленное в 0,25 Methocel® (Dow Chemical Co.) за 1 ч до аппликации с 1 мкг ТФА (в ацетоне) на правое ухо и только с ацетоном на левое ухо. Эта обработка повторяется раз в день 4 последующих дня. На 5 день животным внутрибрюшинно вводится 2 мг/кг винбластин сульфата для остановки деления клеток в метафазе. Четыре дня спустя животных забивают и удаляют уши для гистологического исследования. Гистологические срезы затем исследовали под световым микроскопом и считали количество метафаз на миллиметр базальной мембраны. В группе обычно используют по 10 мышей.
Результаты теста могут быть использованы для демонстрации того, что описываемые здесь соединения эффективно подавляют митотическую активность, связанную с гиперплазией кожи, вызванной ТФА, у мышей, эффективны в лечении кожных и слизисто-эпителиальных заболеваний у человека, таких как, псориаз (во всех его формах), лишай, хроническая экзема, ихтиоз, питириаз и хроническая крапивница.
Противораковая активность
Характерные соединения
данного изобретения могут тестироваться в различных преклинических животных моделях опухолей, описанных ниже, на противораковую активность, которая является показателем клинической пригодности.
Противораковая активность может быть также проверена in vitro методом подавления роста клеток, описанного ниже.
Активность, подавляющая рост in vitro
Реактивы для культуры
ткани покупают у GIBCO (Grand Island, NY). 5-(диметилтиазол-2-ил)-2,5-дифенилтетразолия бромид (МТТ) покупали у Sigma Chemical Company (St. Louis, МО). Все соединения готовят в виде исходного раствора
2 мг/мл на ДМСО. МТТ готовят в виде исходного раствора 1 мг/мл на фосфатно-солевом буфере Дульбекко (ФСБ). Все растворы хранятся замороженными в темноте при -20oC.
Клон A раковых клеток толстой кишки человека выделяли из гетерогенной линии DLD-1 опухоли толстой кишки и культивировали, как ранее было описано (Dexter et al. Cancer Research 1979, 39, 1020; Dexter et al. Am. J. Med. 1981, 71, 949). L1210 лейкозные клетки мыши культивируются в среде RPMI-L как описано (Chen et al. Cancer Research 1986, 46, 5014). Все клеточные линии инкубируют при 37oC во влажной атмосфере 5% CO2 + 95% воздуха.
Экспоненциально растущие L1210 клетки (1•103) или клетки клона A (8•102) в 0,1 мл среды высевают
на 96-луночные микропланшеты в день 0. В день 1, к клеткам на микропланшетах добавляют аликвоты по 0,1 мл среды, содержащей повышающиеся концентрации тестируемых соединений. После инкубации в течение
42 ч при 37oC в инкубаторе с влажной атмосферой планшеты, содержащие клетки L 1210, коротко центрифугируют и отбирали по 100 мкл ростовой среды. Культуры клеток инкубируют с 50 мкл МТТ 4 ч
при 37oC. Получающийся пурпурный осадок формазан солюбилизируют 200 мкл 0,04 н. HCl в изопропаноле. Поглощение промеряют на спектрофотометре для сканирования лунок Titertek Multiskan MCC
(Flow Laboratories) при длине волны тестирования 570 нм и длине волны сравнения 630 нм. Значения 1050 определяют компьютерной программой, заполняющей данными уравнение
Y = ((Am - Ao)/(1 + (
X/IC50)n)) + Ao
где Am - поглощение контрольных клеток; Ao - поглощение клеток в присутствии наивысшей концентрации соединения; Y - наблюдаемое поглощение; X
- концентрация соединения; IC50 - доза соединения, подавляющая число удвоений популяции клеток наполовину по сравнению с числом удвоений популяции контрольных клеток, и n равняется наклону
прямого участка кривой.
Животные модели опухолей
В описываемых ниже животных моделях опухолей противоопухолевая активность соединений данного изобретения может оцениваться по
одному или более из описанных ниже параметров.
В методе определения подавления роста опухоли эффективность тестируемых соединений определяется по степени подавления роста опухоли у
леченых мышей по сравнению с контрольными животными, получавшими растворитель. Вес опухоли рассчитывается по измерениям кронциркулем с помощью формулы для вытянутого эллипсоида (мг веса опухоли =
(длина•ширина2)/2). Прирост опухоли определяется для каждой группы, в которой проводилось лечение, и контрольной группы, получавшей растворитель, путем вычитания первоначального веса
опухоли из конечного веса опухоли после окончания эксперимента. Результаты обсчитываются по формуле:

В опытах на выживание противоопухолевая активность рассчитывается по формуле:

В методе определения задержки роста опухоли значение Т/С рассчитывается как среднее время (в днях), необходимое для достижения опухолью предопределенного размера (750 мг) в опыте, деленное на среднее время, необходимое для достижения опухолью того же размера у контрольной группы

В некоторых случаях задержка роста опухоли выражается в "днях", рассчитанных вычитанием среднего времени в днях для достижения предопределенного веса опухоли в контрольной группе из среднего времени для достижения того же предопределенного веса в опытной группе.
В экспериментах на мышиной модели лейкемии, активность тестируемых соединений может также выражаться как % увеличения
продолжительности жизни хозяина (% ILS) согласно формуле:
где СПЖО - средняя продолжительность жизни в опыте, СПЖК - средняя продолжительность жизни в контроле.
Для подкожно растущих опухолей, гибель опухолевых клеток рассчитывается следующим образом:

где T-C - задержка роста опухоли и TD - время удвоения опухоли в днях. В моделях опухоли, описанных ниже, в группе используется обычно по 5 - 10 мышей.
В16 Меланома: Опухолевая линия В 16 спонтанно появляется на коже в основании уха у C57BL мышей (NIH Publication N. 84-2635, February 1984, In Vivo Cancer Models). Опухолевая линия подкожно (п/к) культивируется последовательным пассированием у самок C57BL мышей. Для тестирования самкам B6C3FI мышей весом 18-22 г в день 0 внутрибрюшинно инокулируют 0,25 мл 1-5 взвеси опухоли. Мышей произвольно распределяют на группы. В контроле и опыте в качестве растворителя используется 0,25 Methocel® /2% Tween® 80. Тестируемые соединения и контрольный растворитель вводятся внутрибрюшинно раз в день в течение 9 последующих дней, начиная со дня 1.
Р388 лейкемия: Опухолевая линия Р388 появляется из лимфолейкоза у самок DBA/2 мышей после окраски кожи 3-метил-холантреном (NIH Publication N 84-2635, February 1984, In Vivo Cancer Models). Опухолевая линия культивируется последовательным пассированием DBA/2 мышей. Для тестирования самкам CDFI мышей весом 18-22 г в день 0 инокулируют в/б 1•106 жизнеспособных Р388 клеток, собранных из асцитов DBA/2 мышей. Мышей произвольно делят на группы. Контрольный растворитель и тестируемые соединения вводятся в/б или в/в раз в день 5 или 9 последующих дней, начиная с 1 дня (способ введения и количество дней определяются заранее). В контроле и опыте в качестве растворителя использовали 0,25% Methocel® /2% Tween® 80.
L1210 лейкемия: Опухолевая L1210 линия первоначально в 1948 г была химически индуцирована в селезенке и лимфатических узлах DBA мышей путем окрашивания кожи метилхолантреном в этиловом эфире (NIH Publication N 84-2635, February 1984, In Vivo Cancer Models). Опухолевая линия культивируется последовательным пассированием у самок DBA/2 мышей. Для тестирования на день 0 самкам CDFI мышей весом 18-22 г инокулируется в/б 1•105 L1210 клеток (0,1 мл/мышь), собранных из асцитов DBA/2 мышей. Мышей произвольно делят на группы. Контрольный растворитель и тестируемые соединения вводятся в/б или в/в раз в день 5 или 9 последующих дней, начиная с 1 дня (способ введения и количество дней определяются заранее). В контроле и опыте в качестве растворителя использовали 0,25% Methocel® /2% Tween® 80.
Аденокарцинома протоков поджелудочной железы (Panc02 и Panc03): Опухолевая линия аденокарциномы протоков поджелудочной железы возникла из опухоли, вызванной имплантацией нити, несущей 3-метилхолантрен, в ткань поджелудочной железы мыши (Corbett et al., Cancer Research (1984) 44: 717-726). Фрагменты опухоли имплантируются подкожно билатерально троакаром и тестируемые соединения вводятся в/в, п/к или п/о, начиная с 1-3 дня с момента имплантации, по плану 1 или 2 раза в день.
Аденокарцинома молочной железы мыши 16/С (Маm 16 / С): Аденокарциному молочной железы мыши 16/С первоначально выделяли и культивировали трансплантацией метастатических очагов легких (Corbett et al., Cancer Research (1978) 62: 1471-1488). Фрагменты опухоли имплантируются п/к билатерально троакаром и 1-3 дня спустя начинают лечение тестируемыми соединениями. Соединения вводятся 1 или 2 раза в день. Опухоли измеряются кронциркулем один или два раза в неделю. Мышей забивают, когда вес опухоли в контрольной группе превышает средний вес 1500 мг.
Аденокарцинома молочной железы мыши 17 (Маm 17): Эта опухолевая линия хранится в хранилище глубокого холода Developmental Therapeutics Program, поддерживаемой Biological Testing Branch, Frederick MD (Mucci-LoRusso et al. , Investigational New Drugs (1990) 8(3): 253-261). Опыты по химиотерапии на самках C3H мышей, несущих Маm 17 опухоли, проводятся также, как описано выше для опухоли Мат 16/С.
Подкожно-имплантированная карцинома толстой кишки 38 (Colon 38) и карцинома толстой кишки 51 Colon 51): Эти опухолевые линии возникли из химически индуцированной опухоли толстой кишки у C57BL/6 мыши, индуцированной повторными п/к инъекциями 1,2-диметилгидразина (Corbett et al., Cancer Research (1975) 35: 2434-2439). Куски опухоли имплантируются билатерально п/к троакаром, и через 1-3 дня после имплантации начинается лечение тестируемым соединением. Соединения вводятся 1 или 2 раза в день. Опухоли измеряются кронциркулем раз или два раза в неделю до тех пор, пока опухоли у контрольной группы не превысят средний вес 1500 мг.
Модели in vivo ксенотрансплантанта опухоли человека (MX-1, LX- 1, СX-1 и DLD-2)
MX-1 карциному молочной железы человека, LX-1 карциному легких человека и СX-1 и DLD-2 карциному толстой кишки
человека первоначально получали из удаленных хирургическим путем первичной опухоли груди, опухоли легких и карциномы толстой кишки, соответственно. Линии опухолей человека культивируются
последовательным пассированием у бестимусных мышей. МX-1 карцинома молочной железы человека - установленная опухоль, используемая NCI. Модели опухолей МX-1 и DLD-2 полностью охарактеризованы.
Подопытным мышами могут быть неродственно скрещенные Swiss мыши и BALB/c мыши, несущие nude (nu/nu) ген. На день 0 30-60 кусочков опухоли имплантируются подкожно билатерально троакаром самкам мышей (20-25 г). Кусочки опухолей готовят из свежих опухолей, растущих подкожно у перевиваемых мышей. Прощупываемые опухоли весом примерно 50 мг появляются у мышей в течение 7-10 дней после инокуляции. Мышей делят на группы и тестируемые соединения и контрольный растворитель вводится внутривенно (в/в) раз в день на 3, 5, 7 и 12-16 дни.
Обмеры опухоли и вес опухоли записываются раз или два раза в неделю. Мыши забиваются, когда опухоли достигают среднего 1500 мг (примерно день 20).
Эффективность тестируемых соединений определяется как процент подавления роста опухоли. В моделях рака in vivo использовали критерии активности Национального Института Рака (NIH Publication N 84-2635, February 1984: In vivo Cancer Models). Фактические регрессии опухоли (IR - неполная регрессия; FR - полная регрессия) показывают превосходную - выдающуюся активность. Подавление роста опухоли >90% рассматривается как хорошее-превосходное и подавление 58-89% рассматривается как умеренное-хорошее. Соединения, показывающие <58% подавления роста, рассматриваются как неактивные.
Дозировка и технология приготовления
лекарственного средства
Соединения настоящего изобретения могут вводиться для лечения или предупреждения отторжения пересаженных органов, реакции "трансплантант против хозяина", псориаза,
аутоиммунных заболеваний и хронических воспалительных заболеваний, любыми способами, которые создают контакт активного компонента с местом его действия в теле млекопитающего. Соединения настоящего
изобретения могут также применяться для лечения кожных и слизисто-эпителиальных заболеваний, таких как псориаз (во всех его формах), лишай, хроническая экзема, ихтиоз, питириаз и хроническая
крапивница. Они могут вводиться любыми традиционными способами, доступными для применения, в сочетании с данным фармацевтическим препаратом, либо как индивидуальные лекарственные препараты, либо в
комбинации лекарственных препаратов. Они могут вводиться отдельно, но обычно вводятся с фармацевтическим носителем, выбранным на основе избранного способа введения и стандартной фармацевтической
деятельности.
Соответствующие фармацевтические носители и методы приготовления лекарственных форм описываются в Remington's Pharmaceutical Sciences, Mack Publishig Company, стандартном справочнике данной области.
Вводимая доза будет варьировать в зависимости от известных факторов, принимаемых во внимание практикующими опытными врачами, таких как фармакодинамические характеристики отдельных соединений и способ их применения и путь введения, возраст, состояние здоровья, вес реципиента, природа и степень выраженности симптомов, вид одновременного лечения, частота лечения и желаемый эффект. Согласно общепринятой практике, ежедневная доза активного ингредиента может составлять около 0,1-100 мг на кг веса тела. Обычно 0,5-50, и предпочтительно, 1-25 мг на кг в день, даваемые в виде небольших доз 1-6 раз в день или в виде постоянно высвобождаемой лекарство форме, являются эффективными для достижения желаемых результатов.
Фармацевтические композиции (лекарственные формы), подходящие для внутреннего введения, содержат от примерно 1 мг до примерно 500 мг активного компонента не единицу. В этих фармацевтических композициях активный компонент будет обычно присутствовать в количестве примерно 0,5-95% по весу в расчете на общий вес композиции.
Активный компонент может вводиться орально в твердых лекарственных формах, таких как капсулы, таблетки и порошки, или в жидких лекарственных формах, таких как эликсиры, сиропы и суспензии. Также он может быть введен парентерально в стерильных жидких лекарственных формах. Также он может быть введен ингаляцией в форме аэрозоля для носа и легочного ингалятора. Он может также применяться местно в виде мази, крема, геля, пасты, примочек, раствора, аэрозоля, липосом или наклеек. Лекарственные формы, используемые для введения активного компонента, обычно содержат соответствующие носители, разбавители, консерванты или другие наполнители, описанные в Remington's Pharmaceutical Sciences, Mack Publishig Company, стандартном справочнике данной области.
Желатиновые капсулы содержат активный компонент и растертые в порошок носители, такие как лактоза, сахароза, маннитол, крахмал, производные целлюлозы, стеарат магния, стеариновая кислота и т.п. Подобные разбавители могут быть использованы для производства таблеток и порошков. Как таблетки, так и капсулы могут быть изготовлены в виде лекарственных форм с длительным высвобождением активного компонента для обеспечения постоянного высвобождения лекарственного средства в течение нескольких часов. Таблетки могут покрываться сахаром или пленкой для маскировки какого-либо неприятного вкуса и защиты таблетки от воздействия воздуха или эндеросолюбильной оболочкой для выборочного распада в пищеварительном тракте.
Жидкие лекарственные формы для орального введения могут содержать красители и корригенты для повышения их восприятия больным.
В общем, соответствующее масло, вода, физиологический раствор, водный раствор декстрозы (глюкозы) и подобные растворы Сахаров и гликоли, такие как пропиленгликоль или полиэтиленгликоли, являются подходящими носителями для парентеральных растворов. Растворы для парентерального введения содержат активный компонент, соответствующее стабилизирующее соединение и, в случае необходимости, буферные вещества. Подходящими стабилизирующими соединениями являются противоокислительные соединения, такие как бисульфит натрия, сульфит натрия или аскорбиновая кислота, либо по отдельности, либо в сочетании. Применяются также лимонная кислота и ее соли и ЭДТА-натриевая соль. Кроме того, растворы для парентерального введения могут содержать консерванты, такие как бензалконий хлорид, метил- или пропилпарабен и хлорбутанол.
Мази, кремы, гели и пасты могут включать разбавители, такие как воски, парафины, крахмал, полиэтиленгликоль, силиконы, бентониты, кремниевую кислоту, растительные и животные жиры, тальк и оксид цинка или смеси этих или других разбавителей. Растворы и эмульсии местного применения могут, например, содержать обычные разбавители (исключая растворители, имеющие молекулярную массу меньше 200, кроме случаев присутствия поверхностно-активного соединения), такие как растворители, растворяющие соединение, и эмульгаторы; специфическими примерами являются вода, этанол, 2-пропанол, этилкарбонат, бензиловый спирт, пропиленгликоль, масла, глицерин и сложные эфиры карбоновых кислот и сорбитола или их смеси. Композиции лекарственных форм местного применения могут также содержать консерванты или противоокислительные соединения. Соединения настоящего изобретения в лекарственных формах местного применения могут использоваться в сочетании со стероидными лекарственными веществами, особенно стероидами местного применения, такими как Synalar (фторциналона ацетонид), Lidex (фторциналон), Westcort (гидрокортизона валерат), Valisone (бетаметазона валеат) и Diprasone (бетаметазона дипропионат).
Порошки и аэрозоли могут содержать обычные разбавители, такие как лактозы, тальк, кремниевая кислота, гидроксид алюминия, силикат кальция и полиамидные порошки или смеси этих веществ. Аэрозоли могут содержать обычные пропелленты. Липосомы могут быть приготовлены из таких веществ, как животные или растительные жиры, которые будут образовывать липидные бислои, в которые может включаться активный компонент.
Наклейки могут быть изготовлены из матрикса, такого как полиакриламид, и полупроницаемой мембраны из соответствующей полимера для контроля скорости поступления соединения к коже.
Примеры полезных фармацевтических композиций (лекарственных форм) для введения соединений этого изобретения приводятся ниже.
Капсулы. Капсулы могут быть приготовлены заполнением стандартных двойных твердых желатиновых капсул 50 мг порошкообразного активного компонента, 175 мг лактозы, 24 мг талька и 6 мг стеарата магния каждая.
Мягкие желатиновые капсулы. Готовится смесь активного компонента в соевом масле и вводится с помощью нагнетательного поршневого насоса в желатин для формирования желатиновых капсул, содержащих 50 мг активного компонента. Капсулы промываются петролейным эфиром и высушиваются.
Таблетки. Таблетки могут быть приготовлены традиционными методами так, чтобы стандартная доза содержала 50 мг активного компонента, 6 мг стеарата магния, 70 мг микрокристаллической целлюлозы, 11 мг кукурузного крахмала и 225 мг лактозы. Для улучшения вкусовых качеств и задержки всасывания могут наноситься соответствующие покрытия.
Суспензии. Водная суспензия для орального введения готовится таким образом, чтобы каждые 5 мл содержали 25 мг точно разведенного активного компонента. 200 мг натриевой карбоксиметилцеллюлозы, 5 мг бензоата натрия, 1,0 г раствора сорбитола, фармакопея США, и 0,025 мг ванилина.
Формы для инъекций. Парентеральные композиции, подходящие для введения инъекциями, готовятся перемешиванием 1,5% по весу активного компонента в 10% по объему пропиленгликоле и воде. Раствор стерилизуют общепринятыми способами.
Носовые спреи. Водный раствор готовится таким образом, чтобы каждый 1 мл содержал 10 мг активного компонента, 1,8 мг метилпарабена, 0,2 мг пропилпарабена и 10 мг метилцеллюлозы. Раствор разливается в 1 мл ампулы.
Легочная ингаляция. Гомогенная смесь активного компонента в полисорбате 80 готовится таким образом, чтобы конечная концентрация активного компонента в упаковке составляла 10 мг/на упаковку и конечная концентрация полисорбата 80 в упаковке составляла 1% по весу. Смесь разливают по баллонам, на баллоне закрепляют вентили и под давлением добавляют требуемое количество дихлортетрафторэтана.
Мазь. Активный компонент добавляют при 70oС к смеси 48% по весу белого вазелина, 10 % вазелинового масла, 8 % моностеарата глицерина, 3% изопропил миристата и 20% ланолина. После тщательного перемешивания добавляют теплый раствор метил- и пропилпарабенов в воде, содержащий ацетон бисульфит натрия, так, чтобы конечная концентрация каждого парабена составляла 0,15 %, воды 8% и ацетонбисульфита натрия 0,5%. Смесь перемешивают до тех пор, пока она не примет комнатную температуру.
Технология приготовления лекарственных форм местного применения
Мазь для местного применения может быть также приготовлена
при 70oC добавлением активного компонента к смеси 48% по весу белого вазелина, 10% вазелинового масла, 8% моностеарата глицерина, 3% изопропилмиристата и 20% ланолина. После тщательного
перемешивания добавляют теплый раствор метил- и пропилпарабенов в воде, содержащий ацетон бисульфит натрия, так, чтобы конечная концентрация каждого парабена составляла 0,15%, воды 8% и
ацетонбисульфита натрия 0 - 5%. Смесь перемешивают до тех пор, пока она не примет комнатную температуру.
Крем для местного применения готовится при 75oC добавлением активного компонента к смеси 1% лаурилсульфата натрия, 12% пропиленгликоля, 25% стеаринового спирта, 25% белого вазелина и 37% воды. Смесь перемешивается до загустения.
Гель для местного применения готовится при 70oC добавлением активного компонента к смеси 0,75% карбопола 940 (поликарбопола), 46,25% воды, 3% эмульгатора гидроксилированного ланолина, 50% этанола и, необязательно, 1-2% диизопропаноламина. Смесь перемешивается до охлаждения ее до комнатной температуры.
Фармацевтические наборы, включающие терапевтически эффективное количество фармацевтической композиции, содержащей соединение формул 1-4 в одном или более стерильных контейнерах, также входят в это изобретение. Стерилизация контейнера может проводиться традиционными способами стерилизации, известными специалистам. Такие наборы далее могут включать, при желании, один или более компонентов различных стандартных фармацевтических наборов, таких как, например, один или более фармацевтически приемлемый носитель, дополнительные флаконы для смешивания компонентов и т.д., что будет очевидно специалисту. В набор могут также входить инструкции, либо в виде вкладыша, либо в виде наклейки, указывающие количество лекарственного средства, которое следует принимать, указания на способ применения и/или указание для смешивания компонентов.
Соединения формул 1-4 (компонент 1) могут вводиться в сочетании со вторым иммунодепрессантом (компонент 2), как комбинированное лечение, любыми традиционными способами, доступными для применения в сочетании с данными лекарственными препаратами, либо в виде отдельных доз, вводимых одновременно или последовательно, либо в физическом сочетании лекарственного соединения каждого компонента в однократной или комбинированной дозе.
Такой второй иммунодепрессант (компонент 2) может быть выбран из группы, состоящей из, но не ограниченной, циклоспорина A, азатиоприна, кортикостероиды, такие как преднизон, ОКТЗ, FK506, микофенолокислота или ее 2-(N- морфолино)этиловый эфир, 15-деоксиспергуалин, рапамицин, мизорибин, мисопростол и антитела к рецептору интерлейкина-2 (ИЛ-2). Комбинированное лечение может проводиться для лечения иммуномодуляторных расстройств и воспалительных заболеваний и, особенно, для предупреждения или лечения отторжения органов при трансплантации, реакции "трансплантант против хозяина", псориаз, ревматоидный артрит, аутоиммунных заболеваний и хронических воспалительных заболеваний и связанных расстройств, любыми средствами, которые создают контакт активного компонента(ов) с участком действия вещества в теле млекопитающих.
Как обсуждалось выше, вводимая доза в комбинированном лечении будет изменяться в зависимости от использования и известных факторов, таких как, фармакодинамические характеристики отдельных соединений и способ их применения и путь введения, возраст, состояние здоровья, вес реципиента, природа и степень выраженности симптомов, вид одновременного лечения, частота лечения и желаемый эффект. Согласно общепринятой практике, ежедневная доза активного ингредиента может составлять около 0,001-1000 мг на кг веса тела. Обычно в дозе от 0,1 до 100 мг/кг в день, даваемые в виде небольших доз 1-6 раз в день или в виде постоянно высвобождаемой лекарство форме, являются эффективными для достижения желаемых результатов.
Активные компоненты в комбинированном лечении могут вводиться в виде различных лекарственных форм, необязательно с фармацевтическим носителем и различными путями, что обсуждалось выше для лечения одним препаратом.
Компонент 1 и компонент 2 изобретения могут быть в одном лекарственном препарате, в виде однократной дозы (т.е., объединены вместе в одной капсуле, таблетке, порошке или жидкости и т.д.) как комбинированный продукт. Если компоненты 1 и 2 не входят в один препарат, соединение формул 1-4 (компонент 1) может вводиться одновременно со вторым иммунодепрессантом (компонент 2) или в любом порядке; например, компонент 1 этого изобретения может вводиться первым, а вслед за ним - компонент 2 или они могут вводиться в обратном порядке. Если соединения не вводятся одновременно, предпочтительно, чтобы введение компонента 1 и 2 этого изобретения производилось в пределах 1 ч. Предпочтительно, чтобы путь введения компонента 1 и 2 настоящего изобретения был оральным. Термины оральный агент, оральный ингибитор, оральное соединение или подобные, используемые здесь, обозначают соединения, которые могут быть введены через рот. Несмотря на то, что предпочтителен один и тот же путь введения для компонента 1 и компонента 2 изобретения (т.е., например, оба вводятся орально) или одна лекарственной формы, при желании, они могут вводиться различными путями (т.е., например, один компонент комбинированного продукта может вводиться орально и другой компонент может быть введен внутривенно) или в разных лекарственных формах.
Вводимая доза будет варьировать в зависимости от известных факторов, принимаемых во внимание практикующими опытными врачами, таких как фармакодинамические характеристики отдельных соединений и способ их применения и путь введения, возраст, состояние здоровья, вес реципиента, природа и степень выраженности симптомов, вид одновременного лечения, частота лечения и желаемый эффект.
Подходящие дозы компонентов 1 и 2 этого изобретения будут легко установлены опытными практикующими врачами на основе настоящего открытия. В соответствии с существующими рекомендациями, обычно ежедневная доза может составлять от примерно 100 мкг до примерно 1 г каждого компонента. В соответствии с существующими рекомендациями, если соединение компонента 1 и компонента 2 вводятся в сочетании, количество каждого вводимого компонента может быть снижено на 70-80% относительно обычной дозы компонента при его использовании в качестве единственного средства.
Комбинированные продукты этого изобретения могут быть составлены таким образом, что несмотря на объединение активных компонентов в одной единице дозы, физический контакт между активными компонентами будет минимальным. Для уменьшения возможности контакта, например, при оральном введении продукта, один активный компонент может быть покрыт энтеросолюбильной оболочкой. Покрытие энтеросолюбильной оболочкой одного из активных компонентов, возможно, не только сводит к минимум контакт между скомбинированными активными компонентами, но также, вероятно, контролирует выделение одного из компонентов в пищеварительном тракте таким образом, что один из этих компонентов не высвобождается в желудке, а предпочтительно высвобождается в тонком кишечнике. Другое воплощение этого изобретения, где желательно оральное введение препаратов, предусматривает комбинированный продукт, в котором один из активных компонентов покрыт обеспечивающим длительное высвобождение материалом, который обеспечивает длительное высвобождение на протяжении пищеварительного тракта и также служит для уменьшения физического контакта между скомбинированными активными компонентами. Более того, длительно высвобождающийся компонент может быть дополнительно покрыт энтеросолюбильной оболочкой для того, чтобы высвобождение этого компонента происходило только в тонком кишечнике. Еще одно достижение включает технологию приготовления комбинированного продукта, в котором один компонент покрыт полимером, обеспечивающим длительное высвобождение, и/или энтеросолюбильным полимером и второй компонент также имеет оболочку из полимерного материала, такой как гидроксиметилцеллюлоза с низкой вязкостью или другие подходящие известные материалы, для дополнительного разделения активных компонентов. Полимерное покрытие служит для того, чтобы создавать дополнительный барьер для взаимодействия с другим компонентом.
Лекарственными формами комбинированных продуктов настоящего изобретения, где один из активных компонентов покрыт энтеросолюбильной оболочкой, могут быть таблетки, такие, что компонент, покрытый энтеросолюбильной оболочкой, и второй активный ингредиент смешиваются вместе и затем спрессовываются в таблетке или такие, что покрытый энтеросолюбильной оболочкой компонент прессуется в виде одного слоя таблетки и другой активный компонент прессуется в еще один слой. В некоторых случаях для дополнительного разделения двух слоев может иметь место слой из индифферентного вещества (плацебо-слой), такой, чтобы плацебо-слой располагался между слоями активных компонентов.
Кроме того, лекарственные формы настоящего изобретения могут быть в форме капсул, где один из активных компонентов спрессован в таблетке, или в форме множества микротаблеток, частиц, гранул или не перилов, которые затем покрываются энтеросолюбильной оболочкой. Эти покрытые энтеросолюбильными оболочками микротаблетки, частицы, гранулы или неперилы затем помещаются в капсулу или сжимаются в капсулу вместе с гранулированным другим активным компонентов.
Эти, а также и другие способы сведения к минимуму контакта между компонентами комбинированных продуктов настоящего изобретения, которые вводятся как одна лекарственная форма или вводятся разными лекарственными формами, но одновременно или последовательно таким же образом, будут очевидны специалистам, опираясь на настоящее изобретение.
Характерные соединения изобретения, пригодные в лечении отторжения пересаженных органов, реакции "трансплантант против хозяина", псориаза, аутоиммунных заболеваний и воспалительный заболеваний представлены ниже в таблицах.
Реферат
Карбоциклические и гетероциклические конденсированные соединения хинолинкарбоновой кислоты общих формул 1-4 или их фармацевтически приемлемая соль соединения, где R1, R2 -Н, F, Cl, Br, CF3 или С1-4 алкил; R3 - незамещенный фенил, фенокси, фенилтио, фенилсульфинил, фенил-N(R4)-, фурил, тиенил, пиридил, тиазолил или оксазолил, или замещенные 1-2 F, Cl, Br, CF3 или С1-4 алкилом, алкокси, алкилтио или алкилсульфинилом (значения других радикалов см. в п.1 ф-лы изобретения). Соединения формулы 1-4 являются иммунодепрессантами или иммуномодуляторами, которые пригодны для лечения или предотвращения отторжения пересаженных органов у млекопитающих. 7 с. и 6 з. п. ф-лы, 5 табл.
Формула



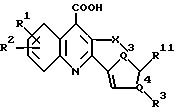
или их фармацевтически приемлемые соли соединений, где R1 и R2 независимо выбираются из H, F, Cl, Br, CF3 или алкила с 1 - 4 атомами углерода;
R3 означает незамещенные фенил, фенокси, фенилтио, фенилсульфинил, фенил-N(R4)-, фурил, тиенил, пиридил, тиазолил или оксазолил или замещенные 1 - 2 группами, независимо выбранными из F, Cl, Br, CF3, алкил с 1 - 4 атомами углерода, алкокси с 1 - 4 атомами углерода, алкилтио с 1 - 4 атомами углерода или алкилсульфинила с 1 - 4 атомами углерода;
R4 означает H, алкил с 1 - 4 атомами углерода или ацил с 1 - 4 атомами углерода;
X означает -Y-, -CH2Y-, -YCH2-, -CH2CH2Y-, -YCH2CH2-, -CH2YCH2-, CR5 = C(R6)-, -CR6= N- или -N=C(R6)-, при этом первый атом углерода из перечисленных значений X прикрепляется к хинолиновому кольцу и каждая метиленовая группа в значениях X может быть независимо замещена одной или двумя группами, независимо выбранными из алкилов с 1 - 4 атомами углерода;
Y означает -CH2-, причем указанная -CH2- группа может быть замещена одной или двумя алкильными группами, содержащими 1 - 4 атомов углерода, -O-, -S-, или -N(R7)-;
R5 и R6 являются независимо H или алкилом с 1 - 4 атомами углерода;
R7 означает H, алкил с 1 - 4 атомами углерода или ацил с 1 - 4 атомами углерода;
и где в соединениях формулы 1 и формулы 2
Z1, Z2 и Z3 являются независимо CR8 или N при условии, что по меньшей мере один из Z1, Z2 и Z3 не является азотом;
R8 независимо выбирается из H, F, Cl, Br, CF3 или алкила с 1 - 4 атомами углерода;
где в соединении формулы 3
один из Q1 и Q2 является S или NR9, а другой является CR10 или N, при условии, что кольцо, содержащее Q1 и Q2, содержит дополнительную двойную связь, расположенную так, что образуется ароматическое кольцо;
R9 выбирается из H или алкила с 1 - 4 атомами углерода;
R10 выбирается из H, F, Cl, Br, CF3 или алкила с 1 - 4 атомами углерода;
и где в соединении формулы 4
один из Q3 и Q4 есть N, а другой C, при условии, что кольцо формулы 4, содержащее Q3 и Q4, содержит дополнительную двойную связь, расположенную так, что образуется ароматическое пиррольное кольцо; и
R11 есть H, F, Cl, Br, CF3 или алкила с 1 - 4 атомами углерода;
при условии, что:
X не является NR7;
в соединениях формулы 1, когда X означает CH2CH2 и каждый из Z1, Z2 и Z3 означает CH, то R3 не является незамещенным фенилом;
в соединениях формулы 3 или формулы 4, где Q4 означает N, X должен образовывать мостик из по меньшей мере двух атомов, и атом X, присоединенный к кольцу, содержащему Q1 и Q2 или Q3 и Q4, должен быть углеродом;
в соединениях формулы 4, где Q3 есть N, X должен включать либо -CH2 CH2-, либо -CH2CH2CH2-, с факультативным замещением каждой метиленовой группы, как описано выше, и
в соединениях формулы 4, где Q4 означает N, R3 не должен быть фенокси, фенилтио, фенилсульфинил или фенил-N(R4)-.
соединение формулы 1, где R1 есть 6-F, R2 есть H, R3 - фенил, Z1, Z2 и Z3 все являются CH и X есть CH2S, или его натриевая соль;
соединение формулы 1, где R1 - 6-F, R2 - H, R3 - фенил, Z1, Z2 и Z3 все являются CH и X есть CH2S, или его натриевая соль;
соединение формулы 1, где R1 - 6-F, R2 - H, R3 - 2-фторфенил, Z1, Z2 и Z3 все являются CH и X есть CH2S, или его натриевая соль;
соединение формулы 1, где R1 - 6-F, R2 - H, R3 - 3-метоксифенил, Z1, Z2 и Z3 все являются CH и X есть CH2S, или его натриевая соль;
соединение формулы 1, где R1 есть 6-F, R2 есть H; R3 - фенил, Z1, Z2 и Z3 все являются CH и X есть CH2O; или его натриевая соль;
соединение формулы 1, где R1 есть 6-F, R2 есть H, R3 - 3-метилфенил, Z1, Z2 и Z3 все являются CH и X есть SCH2, или его натриевая соль;
соединение формулы 1, где R1 есть 6-F, R2 есть H, R3 - фенил, Z1 и Z2 - CH, Z3 - N и X есть CH2CH2, или его натриевая соль;
соединение формулы 1, где R1 есть 6-F, R2 есть H, R3 - фенил, Z1, Z2 и Z3 все являются CH и X есть OCH2, или его натриевая соль;
соединение формулы 1, где R1 есть 6-F, R2 есть H, R3 - фенил, Z1, Z2 есть CH, Z3 - N и X есть SCH2, или его натриевая соль;
соединение формулы 1, где R1 есть 6-F, R2 есть H, R3 - 2-фторфенил, Z1, Z2 и Z3 все являются CH и X есть CH2CH2, или его натриевая соль;
соединение формулы 1, где R1 есть 6-F, R2 есть H, R3 - 3-трифторметилфенил, Z1, Z2 и Z3 все являются CH и X есть CH2CH2, или его натриевая соль;
соединение формулы 1, где R1 есть 6-F, R2 есть H; R3 - 3-метоксифенил, Z1, Z2 и Z3 означает CH и X - CH2CH2, или его натриевая соль.
Комментарии