Противоинфекционное, противовоспалительное и противоопухолевое лекарственное средство - RU2118532C1
Код документа: RU2118532C1
Чертежи







Описание
Область техники.
Изобретение относится к области медицины, к биологически активным веществам, получаемым химическим путем, конкретно - к производным акридона и моносахаридов, и предназначено для применения в качестве противоинфекционного, противовоспалительного и противоопухолевого средства широкого спектра биологического действия.
Предшествующий уровень техники.
Основу современной противоинфекционной химиотерапии составляют два типа лекарственных средств [ 1]:
- средство для прямого избирательного подавления узловых стадий
репродукции микроорганизмов, развития опухолей, в основном представляющие собой природные, полу- или полностью синтетические соединения (антибиотики, сульфаниламиды, нитрофураны, цитостатики и др.) [2,
3,4,5];
- антимикробные средства широкого спектра действия, одновременно подавляющие воспроизведение многих видов микроорганизмов и новообразований, но косвенным путем, через воздействие на
иммунную систему больного, в связи с чем их называют иммуномодуляторами. При этом детали строения генома, ферментов и мембран чужеродных агентов не имеют принципиального значения.
Это, например, первый из достоверно выявленных стимуляторов иммунитета 1-(-)-2,3,5,6-тетрагидро-6-фенил-имидазо-(2,1)-тиазола гидрохлорид, первоначально используемый в медицинской практике в качестве антигельминтного средства, под названием декарис (левамизол).
Как показали исследования, данное соединение обладает исключительно широким иммуномодулирующим действием и с успехом используется в настоящее время в качестве вспомогательного лекарственного средства при ряде иммунодефицитов [ 6].
Известен также ряд высокомолекулярных иммуностимуляторов на основе полисахаридов и нуклеиновых кислот, например полигуаниловая и полиинозиловая кислоты [7].
Необходимо также указать 2,7-бис[2-(диэтиламино)-этокси]-флюорен-9-он дигидрохлорид (тилорон), который проявляет иммуностимулирующее действие при лечении ряда экспериментальных инфекций (модель - белые мыши) [8,9].
Дальнейшие исследования позволили выявить интерферониндуцирующую активность (один из видов стимуляции иммунитета) у целого ряда гетероциклических низкомолекулярных соединений, например у 3-амино-7-(диметиламино)-2-метил-5-хлор-фенотиазина [10].
Представляют значительный интерес попытки ряда авторов получить иммуномодуляторы на основе моносахаридов с аминокислотными заместителями [11], теоретическое обоснование [12].
Второй из представленных типов лекарственных средств (иммуномодуляторы) с нашей точки зрения значительно более интересен, так как использование таких химических и биологических соединений, во-первых, не вызывает привыкания к ним микроорганизмов (что в значительной степени присуще средствам первого типа, обусловливая кратковременность эффективного их использования в медицинской практике 1,5 - 3 года) и, во-вторых, основывается на мобилизации резервных иммунных сил организма больного.
Традиционно иммуномодуляторы разделяют на две основных группы: а) высокомолекулярные (полисахариды, полинуклеотиды и др.), их молекулярная масса превышает 1500, б) низкомолекулярные (некоторые аномальные моносахариды, нуклеозиды и другие гетероциклы), их молекулярная масса обычно не превышает 1000.
Наиболее перспективными иммуномодуляторами считаются низкомолекулярные, поскольку они, как правило, не вызывают аллергических побочных реакций.
К настоящему времени исследовано большое число низкомолекулярных иммуномодуляторов в качестве потенциальных фармакологических средств для лечения ряда вирусных, бактериальных и грибковых инфекций, а также выявления их опосредованного цититоксического и противовоспалительного действия.
Основные недостатки исследованных препаратов сводятся к следующему:
- высокая токсичность и
наличие кумулятивного эффекта при многократном использовании;
- высокая активность в отношении только одной из групп микроорганизмов, что делает их малоэффективными при смешанных инфекциях;
- широкий спектр фармакологической активности при невысокой эффективности делает их пригодными в качестве вспомогательных лекарственных средств для усиления действия основных (например,
антибиотиков).
- низкая стабильность при длительном хранении в растворах, что является причиной неоднозначности результатов фармакологических испытаний.
Несмотря на перечисленные недостатки в последние годы проведены многочисленные исследования, позволившие наметить ряд интересных подходов к решению проблемы по созданию эффективных низкомолекулярных иммуномодуляторов, способных найти применение в медицине в качестве лекарственных средств.
1) В первую очередь, следует отметить производные акридона: N-метилен-карбокси-9-акридон (акридонуксусная кислота, СМА).
В виде натриевой соли СМА известен под названиями камедон (неовир). Препарат показал высокую активность при лечении ряда заболеваний, вызванных как ДНК-, так и РНК-содержащими вирусами, например вирусов герпеса HSV 1 и HSV 2, лихорадки долины Рифт, лесов Семлики, венесуэльского энцефалита лошадей и др.
К сожалению, высокая активность препарата наблюдается только при лечении ряда вирусных заболеваний, в отношении большинства бактериальных, грибковых и смешанных инфекций препарат не эффективен [13, 14, 15].
2) Представляют также значительный интерес некоторые монозамещенные эфиры моносахаридов: 3-O-(N,N-диметиамино-н-пропил)-1,2:5,6-ди-O-изопропилиден- α,D- глюкофуранозы гидрохлорид (SM-1213), а также некоторые 6-O-алкиламинопроизводные D-галактипиранозы, описанные в патентах США, Великобритании, Германии, Японии [16-21].
Эти соединения имеют относительно широкий спектр биологического действия (достоверно установлено: противовоспалительное, противоопухолевое и противовирусное действие), но недостаточную эффективность.
Это требует использования высоких доз таких препаратов для лечения заболеваний, что крайне нежелательно из-за появления побочных эффектов, связанных с токсическим действием [22].
3). Следует отметить соединение: хлоргидрат N,N-диметиламиноэтилового эфира N-метиленкарбокси-9-акридона [23], которое представляет интерес в качестве попытки объединить близкие по структуре фрагменты препаратов SM-1213 и СМА, представленных выше (N,N-диметиламиноэтил-радикала и акридонового цикла). Это позволило получить препарат, принципиально пригодный для перорального использования, хотя и с несколько сниженными иммуномодулирующими свойствами.
4) Интересная попытка модернизации СМА проведена авторами патента России [Л-24] . В результате проведенной работы, удалось получить соль СМА с природным аминосахаром (D-глюкамином). От натриевой соли СМА полученное соединение отличается более мягкими интерферониндуцирующими свойствами. Кроме того, по утверждению создателей препарат обладает противовирусной, в том числе анти-ВИЧ, антипаразитарной, антипромоторной и радиопротекторной активностями.
К сожалению оказалось, что парентеральная лекарственная форма этого препарата отличается низкой стабильностью в растворах, что, по-видимому, связано с инверсией D-глюкамина в водных растворах, переходящего в пять близких по строению аминомоносахаридов.
Приведенный краткий обзор современного состояния проблемы по созданию иммуномодулирующих средств показывает, что в целом она еще далеко не решена. Каждый из перечисленных выше препаратов имеет ограничения при его практическом использовании.
Раскрытие изобретения.
Сущностью настоящего изобретения является создание лекарственного средства, совмещающего полезные качества акридонуксусной кислоты и монозамещенных эфиров моносахаридов (высокая эффективность первых, при широте спектра фармакологической активности вторых).
Наиболее близки к
заявляемому лекарственному средству - монозамещенные эфиры моносахаридов, имеющее общую формулу
S-O-Y,
где
S - моносахарид (пентозы, гексозы, гептозы);
Y
- органический радикал общей формулы

где
R1 - органический радикал с количеством углеродных атомов от 1 до 7;
R2 и R3 - атом водорода, галогена, тио-, гидроксигруппы, моновалентный органический радикал количеством углеродных атомов от 1 до 7;
описанные в патенте США 4017608 [Л 16], опубликован 12.04.77.
Они выбраны нами в качестве прототипа по наибольшему структурному сходству и основному (базовому) элементу химического соединения.
Как уже было отмечено выше, при всех положительных свойствах прототипа (широта спектра фармакологической активности) оно имеет недостаточно высокую эффективность и требует использования высоких доз препарата и длительного курса лечения, что исключает возможность применения этих соединений при тяжелой быстроразвивающейся патологии.
Задача изобретения - получение нового лекарственного средства, обеспечивающего уменьшение перечисленных выше недостатков, т.е. повышение активности против широкого круга заболеваний (противовирусное, противоопухолевое и противовоспалительное действие), другими словами, уменьшение необходимых доз препарата и курса лечения, а также расширение области применения за счет появления выраженной противобактериальной и противогрибковой активности, получение препарата, обладающего низкой токсичностью, высоким индексом терапевтического действия и стабильного при длительном хранении в растворах.
Поставленная задача решается получением новых химических соединений - солей акридонуксусных кислот и монозамещенных эфиров моносахаридов общей формулы

где
A =

или

X и Z = -OH,-H, -CH2OH или группа

W, W1- -H, алкил, бензил-, (Z и W, X и W), (Z и W, OW1 и W) или Z и W вместе образуют группу

R1, R2 = -H, галоген, алкил-, карбокси-, оксиалкил- или оксиарилгруппы, n = 1 - 4, R3, R4 = -H, алкил или R3 и R4 вместе с атомом азота образуют морфолиновый, пиперидиновый или пиридиновый гетероцикл.
Лучшая реализация заявляемого средства, по мнению авторов
А) - при использовании в качестве аниона: N - акридонуксусной кислоты, а в качестве катиона: 1,2:5,
6-ди-O-изопропилиден 3-O (NN-диметиламино-н-пропил) α,D - глюкофуранозы, образуя соединение формулы

Это соединение представляет собой частный случай общей формулы (1)
Брутто формула - C32H42N2O9
Молекулярная масса 598, 94
Химическое название: 1,2:5,6-ди-O-изопропилиден-3-O-(N,N-диметиламино-н-пропил)- α,D -глюкофуранозы 10'-метиленкарбоксилат-9'-акридон.
Б) при использовании в
качестве аниона 3-хлор-10-метиленкарбоксилат-9-акридон, а в качестве катиона - 1,2:3,4-ди- O-изопропилиден 6-O (N,N-диметиламиноэтил) α,D -галактопиранозу,
образуя соединение
формулы

Это соединение представляет собой частный случай общей формулы (2)
Брутто формула C31H39N2O9Cl,
Молекулярная масса 619,11.
Химическое название: 1,2:3,4-ди-O-изопропилиден-6 - O-(N, N-диметиламиноэтил)- α,D -галактопиранозы-3'-хлор-10'-метиленкарбоксилат-9'-акридон.
Сущность изобретения поясняется ниже, в том числе:
- результатами всестороннего
исследования терапевтических характеристик заявляемого лекарственного средства;
- общей технологией получения заявляемых соединений;
- 6 примерами изготовления различных конкретных
препаратов;
- 7 таблицами сравнительных испытаний заявляемого средства, где табл. 1 - сравнительная характеристика видов активностей заявляемых соединений и исходных компонентов; табл. 2
- результаты излучения заявляемых препаратов на модели экспериментальной оспенной инфекции в сравнении с прототипом, камедоном и рибамидилом; табл. 3 - результаты излучения заявляемых препаратов на
модели экспериментальной лихорадки долины Рифт в сравнении с прототипом, натрия 3-хлор-N-карбоксиметил-9-акридоном и рибамидилом; табл. 4 - влияние введения заявляемых препаратов на показатели,
характеризующие противогрибковую активность макрофагов в отношении Candida albicans; табл. 5 - сравнительное изучение антибактериальной активности заявляемых соединений, прототипа и акридонового
составляющего; табл. 6 - влияние заявляемых препаратов на рост солидного рака Эрлиха в сравнении с прототипом и акридоновым компонентом; табл. 7 - влияние заявляемых препаратов на рост лейкоза Р-388 в
сравнении с прототипом и акридоновым компонентом.
Заявители провели синтезы всех вышеуказанных соединений. В общем виде он проводился следующим способом. Эквимолекулярные количества обоих компонентов смешиваются в безводном ацетоне, смесь кипятят с обратным холодильником в течение 20 - 40 мин, предохраняя от попадания влаги. Полученный прозрачный раствор упаривают в вакууме до объема, необходимого для выпадения осадка соли. Выпавший осадок отделяют фильтрацией, высушивают и затем перекристаллизовывают из безводного метанола. Перекристаллизованный осадок высушивают в вакууме при температуре не выше 35oC. Какой либо существенной разницы в технологическом процессе получения различных солей формулы (1)) и (2) не выявлено. Как правило, полученные соединения представляют собой крупные гигроскопические кристаллы светло-желтого цвета, хорошо растворимые в воде, диметилацетамиде, диметилсульфоксиде, глицерине в горячем этаноле, мало растворимы в эфире и хлороформе, практически не растворимы в углеводородах.
Синтезированные соединения полностью охарактеризованы методами элементного анализа, тонкослойной хроматографии в ряде систем растворителей, и измерениями УФ-, ИК- и ЯМР-(H1 и C13)-спектрометрии.
Все полученные соли хорошо растворимы в воде и являются удобными для изготовления всех типов лекарственных форм.
Растворы полученных лекарственных форм имеют значения pH, близкие к физиологическим, стабильны при хранении в растворах в течение 1,5 - 2,0 лет (срок наблюдения), 20oC, без доступа света.
В результате изучения заявляемых химических соединений в острых опытах определены пороги токсических действий препаратов на организм как по интегральному показателю - гибели животных, так и по воздействию на основные, жизненно важные системы: кровь, СС и ЦНС. Показано, что эти препараты не вызывают гемолиза эритроцитов. Внутримышечное и внутривенное применение их в дозах 10 - 100 мг/кг не оказывают отрицательного воздействия на гемодинамику, деятельность сердца и ЦНС. В дозах 300 мг/кг ни одно из заявляемых соединений не вызывало гибели животных и проявления токсического действия у мышей и крыс.
Эти препараты не обладают выраженными кумулятивными свойствами.
Внутривенное введение препаратов крысам в возрастающих дозах в хронических экспериментах не приводило к накоплению токсического эффекта (коэффициент кумуляции больше 5).
При внутримышечном введении соединения быстро всасываются в кровь, достигая максимальных концентраций через 20 - 50 мин.
Период полувыведения их (Т 50%) составляет, как правило, 100 - 130 мин, элиминируются из организма, главным образом, за счет почечной экскреции. Через 24 ч после внутривенных инъекций в дозах 10 - 100 мг/кг они практически полностью выводятся и в крови либо совсем не определяются, либо присутствуют в следовых количествах.
Вещества A и B вводили ежедневно в течение 15 и 30 дней внутримышечно и внутривенно кроликам и собакам в дозах 10 и 50 мг/кг. Это не сказалось на состоянии животных, их массе тела, картине крови, функции печени, почек, поджелудочной железы, деятельности сердца и не сопровождалось выраженными патоморфологическими изменениями внутренних органов. Препараты A и B при всех способах введения не оказывали местнораздражеющего и пирогенного действий.
LD 50 синтезированных соединений были в 2 - 2,5 раза ниже, чем сумма LD 50 составляющих их компонентов, взятых в отдельности.
Как показали опыты, указанные вещества не обладают эмбриотоксическим и тератогенным действием при введении в течение всего периода беременности крысам в дозах, в 10 и 100 раз превышающие терапевтические.
Все полученные результаты подтверждают, что заявляемые химические соединения обладают низкой токсичностью, не вызывают выраженных побочных эффектов, хорошо переносятся больными, имеют высокий терапевтический индекс 50 - 100 и более, что позволяет использовать данные вещества в широком интервале доз.
Неожиданно оказалось, что полученные соединения в опытах на животных и культуре тканей проявили ярковыраженную противобактериальную и противогрибковую виды активностей, не обнаруживаемые как у производных акридонуксусной кислоты, так и у монозамещенных эфиров моносахаридов, что позволяет использовать эти вещества в качестве потенциальных универсальных лекарственных средств при смешанных инфекциях.
Также было выявлено усиление противовирусных свойств: индекс защиты (ИЗ) животных, инфицированных 10 - 100 LD 50 как РНК-, так и ДНК- геномными вирусами, составлял 70 - 85% как при лечебно-профилактической, так и при лечебной схеме введения этих препаратов.
Исследование противовоспалительной активности синтезированных соединений, проведенное на модели заживления полнослойных кожных ран и серотонинового отека (эксудативная фаза воспаления), показало высокую эффективность этих препаратов.
Исследование противоопухолевой активности на модели перевиваемых опухолей (солидные опухоли: рак Эрлиха, меланома B-16 и асцитный лейкоз P-388) позволяет констатировать наличие у данных соединений противоопухолевой активности. Отсутствие дозазависимого ответа на введение заявляемых препаратов заставляет предполагать, что они не являются прямыми цитостатическими агентами, а эффективно воздействуют на замедление роста опухолей через иммунную систему организма.
Сравнительная характеристика видов активностей заявляемых соединений и исходных соединений приведена в табл. 1.
Примеры конкретных способов
получения заявляемых соединений приведены ниже:
Пример 1. 20 г 3-O-(N,N-диметиламино-н-пропил)- 1,2:5,6- ди -O-изопропилиден- α,D -глюкофуранозы, представляющей собой прозрачную вязкую
гидрофобную жидкость, смешивают с 14,5 г N-акридонуксусной кислоты в 150 мл безводного ацетона. Смесь нагревают до кипения и кипятят с обратным холодильником в течение 30 мин. За это время осадок
N-акридонуксусной кислоты полностью растворяется, смесь становится гомогенной. Раствор концентрируют в вакууме до общего объема 95 мл, охлаждают до +5oC, выдерживают при этой температуре в
течение 7 ч. Выпавший осадок фильтруют и высушивают. Полученный светло-желтый крупнокристаллический осадок перекристаллизовывают из безводного метанола (1/3), отделяют от раствора фильтрацией и
высушивают в вакууме (5 мм рт.ст.) при температуре не более 32oC. Получают 29 г (выход - 84%) конечного продукта.
Химическое название: 3-O-(N, N-диметиламино-н-пропил)-1,2:5, 6-ди -O-изопропилиден α,D -глюкофуранозы N-метиленкарбоксилат-9'-акридон.
Брутто формула - C32H42N2O9.
Чистота (хроматографически) - 98,5%.
Молекулярная масса 598,76.
Элементный анализ:
вычислено - C - 64,19 H 7,08 N 4,71,
найдено - C - 64,26 H 7,15 N 4,63.
Данные ЯМР - (H 1); DMSO (d 6):
1,1 - 1,5 м.д. - 4 группы -CH3 в диоксолановых циклах дают 3 синглета: (1.25 м.д.; s(6H)); (1.31 м.д.; s(3H)); (1,40 м.д.; s (3H)).
1,5 - 3,7 м.д. - область сигналов протонов N,N-диметиламино-пропил- заместителя: -N(CH3)2 (2,47 м. д. ; s(6H)); -CH2-N (2,78 м.д.; t(2H); -CH2- (1,78 м.д.; q(2H)); -CH2-O- (3,3,-3,7 перекр. m).
3,5 - 6,0 м.д. - область сигналов протонов глюкофуранозы: H1-H6 (3,5 - 4,5 м.д.; m; H2 (4,62 м.д.; d(1H)); H1 (5,83 м.д. d(1H));
4,98 м. д. - область сигналов протонов
N-метиленкарбоксизаместителя: (s(2H)).
7,2 - 8,5 м.д. - область сигналов ароматических протонов акридона (7,35 м.д.; t(2H)); (7,7 м.д.; d(2H)); (7,83 м.д.; t(2H)); (8,4 м.д.; d(2H)).
Пример 2. 25 г 6-O-(N,N-диметиламиноэтил-1,2:3,4-ди -O-изопропилиден- α,D -галактопиранозы, представляющей собой прозрачную вязкую гидрофобную жидкость, смешивают с 21.6
г 3-хлор-N-акридонуксусной кислоты в 170 мл безводного ацетона. Смесь кипятят с обратным холодильником в течение 40 мин. За это время осадок 3-хлор-N-акридонуксусной кислоты полностью растворяется,
раствор становится прозрачным. Реакционную массу упаривают в вакууме до общего объема 80 мл, охлаждают до 0oC, выдерживают при этой температуре в течение 2 ч. Образовавшийся осадок
фильтруют и высушивают. Полученный крупнокристаллический осадок желто-зеленого цвета перекристаллизовывают из безводного метанола (1/3), отделяют от раствора фильтрацией и высушивают в вакууме (5 мм
рт.ст.), при температуре не более 25oC. Получают 41,5 г (выход - 89%) конечного продукта:
Химическое название: 6-O-(N,N-диметиламиноэтил)-1,2:3,4-ди-O-изопропилиден- α,D
-галактопиранозы 3'-хлор-N-метиленкарбоксилат-9'-акридон.
Брутто формула -C31H39N2O9Cl.
Чистота (хроматографически) - 98%.
Молекулярная масса 619,11.
Элементный анализ:
вычислено - C - 60,14 H 6,35 N 4,52 Cl 5,73,
найдено - C - 60,05 H 6,41 N 4,63 Cl 5,70.
Данные ЯМР - (H 1); DMSO (d 6):
1,15 - 1,5 м.д. - 4 группы -CH3 в диоксолановых циклах дают 3 синглета: (1.23 м.д.; s(6H)); (1.37 м.д.; s(3H)); (1.48 м.д.; s(3H));
2,3 - 3,
7 м.д. - область сигналов протонов N,N-диметиламинэтил-заместителя: -N(CH3)2 (2,5 м.д.; s(6H)); -CH2-N и -CH2-O-(2,7 - 3,5 перекр. m).
3,5 - 6,0 м.д. - область сигналов протонов
галактопиранозы: H2-H6 (3,5 - 4,8 м.д.; m); H1 (5,71 м.д.; d(1H));
5.0 м. д. - область сигналов протонов N-метиленкарбоксизаместителя: (s(2H)).
7,2 - 8,6 м.д. - область сигналов ароматических протонов акридона.
Пример 3. 22 г 3-O-(1'-этил-2'-(N-морфолинил)-1,2-O-изопропилиден- α,D - глюкофуранозы, представляющей собой прозрачную вязкую жидкость,
смешивают с 19,5 г 3-хлор-6-метил-N-акридонуксусной кислоты в 250 мл безводного ацетона. Смесь нагревают до кипения и кипятят с обратным холодильником в течение 20 мин. Полученный прозрачный раствор
упаривают в вакууме до общего объема 100 мл, охлаждают до 0oC, выдерживают при этой температуре в течение 4 ч. Образовавшийся осадок фильтруют, высушивают и перекристаллизовывают из
безводного метанола (1/3), отделяют от раствора фильтрацией и высушивают в вакууме (5 мм рт. ст. ), при температуре не более 25oC. Получают 36,7 г (выход - 88,4%) конечного продукта:
Химическое название: 3-O-(1'-этил-2'-(N-морфолинил)-1,2-O-изопропилиден- α,D - глюкофуранозы 3''-хлор-6''-метил-N-метиленкарбоксилат-9''-акридон.
Брутто формула -C31H39N2O10Cl.
Чистота (хроматографически) - >98%.
Молекулярная масса 635,17.
Элементный анализ:
вычислено - C - 58,62 H 6,20 N 4,41 Cl 5,58
найдено - C - 58,73 H 6,21 N 4,37 Cl 5,62
Данные ЯМР - (H 1); DMSO (d 6):
1,2 - 1,5 м.д. - 2 группы -CH3 в диоксолановом цикле
дают 2 синглета, интенсивностью в 3 протона каждый.
1,5 - 3,9 м.д. - область сигналов протонов N-этилморфолирилзаместителя: -CH2-N- и -CH2-N-CH2-морфолина (2,7 - 3,1 м.д.; m(6H)); -CH2-O- и -CH2-N-CH2-морфолина (3,6 - 3,9 перекр. m).
3,5 - 6,0 м.д. - область сигналов протонов глюкофуранозы: H3-H6 (3,5 - 4,3 м.д.; m); H2 (4,7 м.д.; d(1H)); H1 (5,8 м.д. d(1H));
4.99 м. д. - область сигналов протонов N-метиленкарбоксизаместителя: (s(2H)).
7,0 - 8,5 м.д. - область сигналов ароматических протонов акридона.
Пример 4. 15 г 3-O-(N,N-диэтиламино-(2'-изобутил)-1,2-O-изопропилиден-D- аллофуранозы, представляющей собой прозрачную желтоватого цвета вязкую жидкость смешивают с 12,8 г 2'-карбокси-N-метиленкарбоксилат-9'-акридона кислоты в 100 мл безводного ацетона. Смесь нагревают до кипения и кипятят с обратным холодильником в течение 20 мин. Далее поступают аналогично способу, приведенному в примере 2. Получают 21 г (выход - 75,5%) конечного продукта.
Химическое название: 3-O-(N,N-диэтиламино-(2'-изобутил)-1,2-O-изопропилиден- α,D - глюкофуранозы 2''-карбокси-N-метиленкарбоксилат-9''-акридон.
Брутто формула -C33H44N2O11.
Чистота (хроматографически) - 98%.
Молекулярная масса 644,83.
Элементный анализ:
вычислено - C - 61,47 H 6,88 N 4,36,
найдено - C - 61,35 H 6,91 N 4,42.
Пример 5. 30 г 1-O-этил-5-O-(1'-бутил-4'-(N-пиридинил)-D-рибофуранозид смешивают с 25,5 г 2-оксиэтил-7-карбоксиэтил-N-акридонуксусной кислоты в 150 мл безводного ацетона с обратным холодильником в течение 40 мин. Далее поступают аналогично способу, приведенному в примере 2. Получают 43,5 г (выход - 78%) конечного продукта.
Химическое название: 1-O-этил-5-O-(1'-бутил-4'-(N-пиридинил)-D-рибофуранозида 2''-оксиэтил-7''-карбоксиэтил N-метиленкарбоксилат-9-акридон.
Брутто формула -C36H45N2O11.
Чистота (хроматографически) - 97%.
Молекулярная масса 681,83.
Элементный анализ:
вычислено - C - 63,41 H 6,67 N 4,11,
найдено - C - 63,49 H 6,58 N 4,12.
Пример 6. 75 г 5-O-бензил-3-O-(1'-этил-2'-(N-пиперидинил)-1,2-O-изопропилиден- α,D -ксилофуранозы смешивают с 74,2 г 2-(1'-этил-2'-бензилокси)-N-акридонуксусной кислоты в 350 мл безводного ацетона. После кипячения в течение 20 мин раствор упаривают в вакууме до объема 180 мл. Далее поступают аналогично способу, приведенному в примере 2. Получают 122 г (выход - 82%) конечного продукта.
Химическое название: 5-O-бензил-3-O-(1'-этил-2'-(N-пиперидинил)-1,2-O-изопропилиден- α,D -ксилофуранозы 2''-(1'''-этил-2'''-бензилокси) N-метиленкарбоксилат-9''-акридон.
Брутто формула -C46H54N2O9.
Чистота (хроматографически) - 97%.
Молекулярная масса 779,02.
Элементный анализ:
вычислено - C - 70,92 H 7,00 N 3,60,
найдено - C - 70,47 H 7,05 N 3,66.
Промышленная применимость.
Изучение противовирусной активности заявляемых препаратов проводилось на экспериментальной ортопокс вирусной инфекции, вызванной у хлопковых крыс (массой 50 - 70 г), интрназальным введением вируса вакцины (штамм Л-ИВП) с инфекционной активностью 1,6•10(9) ООЕ/мл.
Для стандартизации условий опыта заражение во всех случаях проводили постоянной дозой вируса, равной 10 LD 50.
Препараты вводили в дозе 50 мг/кг крысам за 4 ч до и на вторые сутки после заражения лабораторных животных, применяя внутримышечный и внутрибрюшинный методы введения.
Эксперименты считались достоверными только при 100% гибели животных в контроле. В каждом опыте в качестве рефененс-препарата использовали рибамидил (виразол), который давали крысам перорально в дозе 100 мг/кг по стандартной лечебно-профилактической схеме.
Результаты изучения приведены в табл. 2.
Из данных табл. 2 следует сделать выводы, что заявляемые соединения проявляют высокую противовирусную активность, особенно выраженную при внутримышечном введении препарата (ИЗ=80%), и являются перспективными противовирусными препаратами, значительно превосходящими прототип, камедон и рибамидил (референс-препарат).
Эффективность заявляемых препаратов при экспериментальной лихорадке долины Рифт исследовалась на белых беспородных мышах массой 10 - 12 г при подкожном заражении и на белых крысах массой 35 - 50 г, инфицированных врутрибрюшинно. Для воспроизводства инфекции использовали вирулентный штамм вируса лихорадки долины Рифт, пассированный в многослойных культурах клеток GMK. Оценку эффективности проводили на мышах, инфицированных дозами вируса 25 - 50 БОЕ, и крысах, зараженных дозой вируса 250 БОЕ. Препараты вводили внутрибрюшинно по схеме экстренной профилактики (через 2 ч после заражения и затем 1 раз в сутки в течение 3 дней). Эксперименты считались достоверными только при 100% гибели животных в контроле. В опыте в качестве референс-препарата использовали рибамидил (виразол), который вводили животным в дозе 100 мг/кг по той же схеме.
Результаты изучения заявляемых препаратов на модели экспериментальной лихорадки долины Рифт приведены в табл. 3.
Из данных табл. 3 следует, что заявляемые соединения проявляют исключительно высокую противовирусную активность на модели экспериментальной лихорадки долины Рифт, значительно превосходящую активности исходных компонентов.
Противогрибковую активность заявляемых препаратов 3-O-(N,N-диметиламино-н-пропил)-1,2: 5,6-ди -O-изопропилиден- α,D -глюкофуранозы N-метиленкарбоксилат-9''-акридон (1 в табл. 4) и 3-O-(N, N-диэтиламино-(2'-изобутил)-1,2-O-изопропилиден- α,D -аллофуранозы 2''-карбокси-N-метиленкарбоксилат-9''-акридон (2 в табл. 4) изучали на белых беспородных мышах. Препараты вводили внутрибрюшинно в дозах: 25, 50 и 75 мг/кг массы тела животного. В качестве препаратов сравнения в опытах использовали 3-O-(N,N-диэтиламино-(2'-изобутил)-1,2-O-изопропилиден- α,D -аллофуранозу (3 в табл. 4), являющуюся прототипом, и 2'-карбокси-N-метиленкарбоксилат-9-акридон (4 в табл. 4), компонент, входящий в соединение (2) в дозах 25 и 75 мг/кг. Взвесь клеток C. albicans (штамм N 13, выращенных на среде Сабуро при 37oC в течение 2 суток), приготовленных по стандарту 50 млн., через 1 ч вводили по 1 мл суспензии мышам, получившим испытуемые препараты.
У животных через 7 ч после введения C.albicans экстирпировали сальники и готовили пленочные препараты, которые окрашивали с помощью PAS-реакции. Затем подсчитывали число дрожжевых клеток с ростовыми трубочками в камере Горяева.
Результаты исследований представлены в табл. 4. Как видно из данных табл. 4, размножение возбудителя в пленках сальника животных, получавших препараты (1) и (2), резко снижалось по сравнению с контролем. Наиболее выраженное снижение вегетации C. albicans отмечено через 7 ч после внутрибрюшинного введения в дозах 25 и 50 мг/кг, что свидетельствует о противогрибковой эффективности заявляемых соединений, тогда как соединение (3), являющееся прототипом, не проявило достоверных противогрибковых свойств. Соединение (4) также оказалось неактивным.
Антибактериальное действие заявляемых препаратов изучено на модели хронического стафилококкового сепсиса, вызванного внутривенным введением золотистого стафилококка штамм 5 а. Мышам вводили внутримышечно препараты в разовых дозах 25 и 50 мг/кг по лечебной (1 раз в сутки в течение 5 дней) и по лечебно-профилактической схеме: за 2 суток до заражения, и продолжали в течение 5 суток после заражения.
По истечении срока терапии, животных забивали, иссекали почки, гомогенизировали и производили высев на агар Чистовича для учета количества клеток St. aureus. Через 72 ч инкубации при 37oC определяли количество выросших колоний стафилококка. Результаты сравнительного изучения антибактериальной активности заявляемых соединений приведены в табл. 5.
Уровень обсемененности почечной ткани мышей, получавших заявляемые соединения, снизился более, в 10000 - 100000 раз по сравнению с контролем, у прототипа также обнаружена слабо выраженная противобактериальная активность, значительно меньшая, чем у заявляемых соединений, у акридонового составляющего активность не выявлена.
Влияние заявляемых синтезированных
соединений:
1. 3-O-(N, N-диметиламино-н-пропил)-1,2:5,6-ди -O-изопропилиден- α,D -глюкофуранозы-N-карбоксиметилакридон,
2.
1-O-этил-5-O-(1'-бутил-4'-(N-пиридинил)-D-рибофуранозида 2''-оксиэтил-7''карбоксиэтил N-метиленкарбоксилат-9-акридон в сравнении с
3. 1-O-этил-5-O-(1'-бутил-4'-(N-пиридинил)-D-рибофуранозой
(прототип),
4. 2-оксиэтил-7-карбоксиэтил N-метиленкарбоксилат-9-акридоном (акридоновым компонентом вещества (2) на рост перевиваемых опухолей изучали на моделях:
1. Солидный рак
Эрлиха (РЭ) - (табл. 6).
2. Асцитный лейкоз Р-388 (табл. 7).
Солидную опухоль (РЭ) перевивали в подушечку задней лапы мышей-самцов весом 18 - 20 г линии SHR и C57B1, соответственно в количестве 10(6) клеток РЭ. Лейкоз Р-388 перевивали внутрибрюшинно мышам-самцам линии DBA в количестве 106 клеток. Начиная с 3-го дня прививки РЭ (ко времени формирования опухоли и ее стромальных элементов) и со 2-го дня после прививки Р-388, мышам контрольных групп ежедневно внутрибрюшинно вводили по 0,1 мл физраствора, а животным подопытных групп вводили препарат в дозах 20, 100 и 250 мг/кг.
Как следует из представленных результатов (табл. 6 и 7), заявляемые соединения обладают ярковыраженной способностью к торможению роста перевиваемых опухолей и по своей активности значительно превосходит прототип и акридоновый компонент. Результаты, приведенные в табл. 2 - 7, убедительно подтверждают достижение поставленной фармакологической цели - усиление фармакологической активности и расширение спектра показаний нового препарата.
Из вышеизложенного следует, что получены новые химические соединения - соли монозамещенных эфиров моносахаридов и производных акридонуксусной кислоты, являющиеся эффективными терапевтическими препаратами, при этом с широким спектром биологической активности, в том числе активностью неизвестной для соединений прототипа - антимикотической.
Приведенные способы синтеза данных соединений легко осуществимы в промышленных масштабах, не требуют дефицитного сырья и нестандартного оборудования. Учитывая все это, начата разработка на их основе лекарственных форм для парентерального, перорального и наружного использования.
Таким образом, можно утверждать, что предложенное решение ново, неочевидно и промышленно применимо, т.е. удовлетворяет требованиям, предъявляемым к изобретениям.
Литература
1. Chemotherapy and
Immunity. Ed. G. Pulverer, J. Jeljaszewicz, Gustav Fisher Verlag, 1985, 245 с.
2. Регистр лекарственных средств России (РЛС). Под ред. Крылова Ю.Ф. -М. : Инфармхим, 1993, 1006 с.
3. Бердникова Т. Ф., Ломакина Н.Н. Новые антибиотики группы полициклических пептидов. Антибиотики и медицинская биотехнология, 1986, N 11, с. 814 - 820.
4. Коган Э.З., Синицин Н.И. и др. Линкомицин и его лекарственные формы. - Антибиотики и медицинская биотехнология, 1985, N 10, с. 783 - 788.
5. Яковлев В. П. Антибактериальная терапия в неинфекционной клинике: новые беталактамы, монобактамы и хинолоны. Фармакология: химиотерапевтические средства, т. 20, ВИНИТИ, 1992, 203 с.
6. Schneiden H. Levamisole - a general pharmacological perspective. Int. J. Immunopharmacological, 1981, v. 3, p. 9 - 13.
7. Gordon G., Minks M.A. The interferone renaissance: molecular aspects of induction and action. Microbiol. Rev., 1981, v. 45, p. 244.
8. De Clercq E., Merigan T.S.J. infect. Dis. 1971, v. 123., p. 190 -199.
9. F. Smejkal, D. Zelena, J.Krepelka, I Vankurova Derivatives of benzo (c)-fluorene; Acta virol., 1984, v. 29, N 11 - 18, p. 11.
10. Diederich J., Lodemann E. Arch. ges. Virusforsch. 1973. Bd 59., s. 172 - 173.)
11. E.P. N
653434, МКИ C 07 H 15/12, опубл. 17.04.95.
12. Малайцев В.В. и др. Бюл. экспер. биол. 1985, N 8, с. 221 - 223.
13. Патент США N 3681360, опубл. 09.04.71.
14. Taylor J.L., Grossberg S.E. Tex. Rep. Biol. Med. 1981, 82 v. 41 p. 158 - 163.
15. Szulc B., Piasecki E. Arch. Immun. Ther. Exp., 1988, 36, N 5, p. 537 - 545.
16. Патент США N 4017608, МКИ A 61 K 31/70, НКИ 424/180, 536/129, опубл. 12.04 1977 - прототип.
17. Патент Великобритании 1497409, кл. A 61 K 31/33, опубл. 12.01.1978.
18. Патент Японии 62-149694, кл. C 07 H 15/12, A 61 K 31/70.
19. Заявка Германии 2455026, кл. A 61 K, опубл. 03.07.1975.
20. Заявка Германии 2612717, кл. С 07 Н 15/04, опубл. 14.10.1976.
21. Патент США 4735934, кл. A 61 K 31/70, опубл. 05.04.1988.
22. Nishikava Y. , Yoshimoto K., Chem. Pharm. Bul., 1987, -35, N 7, p. 2894 - 2899.
23. Патент Польши 139805, кл. C 07 D 219/16, опубл. 31.07.1987.
24. Заявка России N 93017260/04 от 1.04.93.
Реферат
Изобретение предназначено для использования в медицинской практике. Задача изобретения - снижение доз препарата и
длительности курса лечения, расширение области применения за счет выраженной противобактериальной и противогрибковой активности, помимо указанных выше (противовоспалительной и противоопухолевой).
Заявлено средство, представляющее собой соль акридонуксусной кислоты и монозамещенного эфира моносахарида общей формулы

где A =
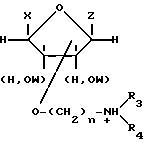
или

X и Z- -OH, -H, -CH2OH или группа

W, W'= -H, алкил-, бензил-, (Z и W, X и W), (Z и W, OW' и W) или Z и W вместе образуют группу

R1, R2- - H, галоген, алкил-, карбокси-, оксиалкил- или оксиарилгруппы, n = 1 - 4, R3, R4- - H, алкил или R3 и R4 вместе с атомом азота образуют морфолиновый, пиперидиновый или пиридиновый гетероцикл. Препарат обладает низкой токсичностью, высоким индексом терапевтического действия и стабилен при длительном хранении в растворах. 2 з.п. ф-лы, 7 табл.
Формула

где А

или
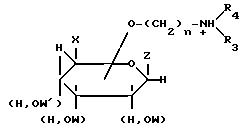
Х и Z = -OH, -H, -CH2 OH или группа

W, W' = -H, алкил-, бензил-, (Z и W, X и W), (Z и W, OW' и W) или Z и W вместе образуют группу

R1, R2 = -H, галоген, алкил-, карбокси-, оксиалкил- или оксиарилгруппы;
n = 1 - 4;
R3, R4 = -H, алкил-, или R3 и R4 вместе с атомом азота образуют морфолиновый, пиперидиновый или пиридиновый гетероцикл.


3. Средство по п.1, представляющее собой соль 3-хлор-10-метиленкарбоксилат-9-акридона и 1,2: 3,4-0-диизопропилиден-6-О-(N, N-диметиламино-этил)-α,D-галактопиранозы общей формулы


Комментарии