Способ получения производных азепина или их солей - SU422149A3
Код документа: SU422149A3
Описание
1
Изобретение относится к области получения новых производных ряда азепипа, которые могут найти применение в фармацевтической промышленности.
Используя известную реакцию алкилирования аминов применительно к соединениям азепинового ряда, получают новые производные азепина общей формулы I
где RI и R2 означают, водород, алкильные группы с числом атомов углерода не более 4 или аллильную группу;
Xi - водород, хлор или трифторметильная группа;
Ха - хлор, если Xi означает водород, или водород, если Xi означает хлор или трифторметильную группу.
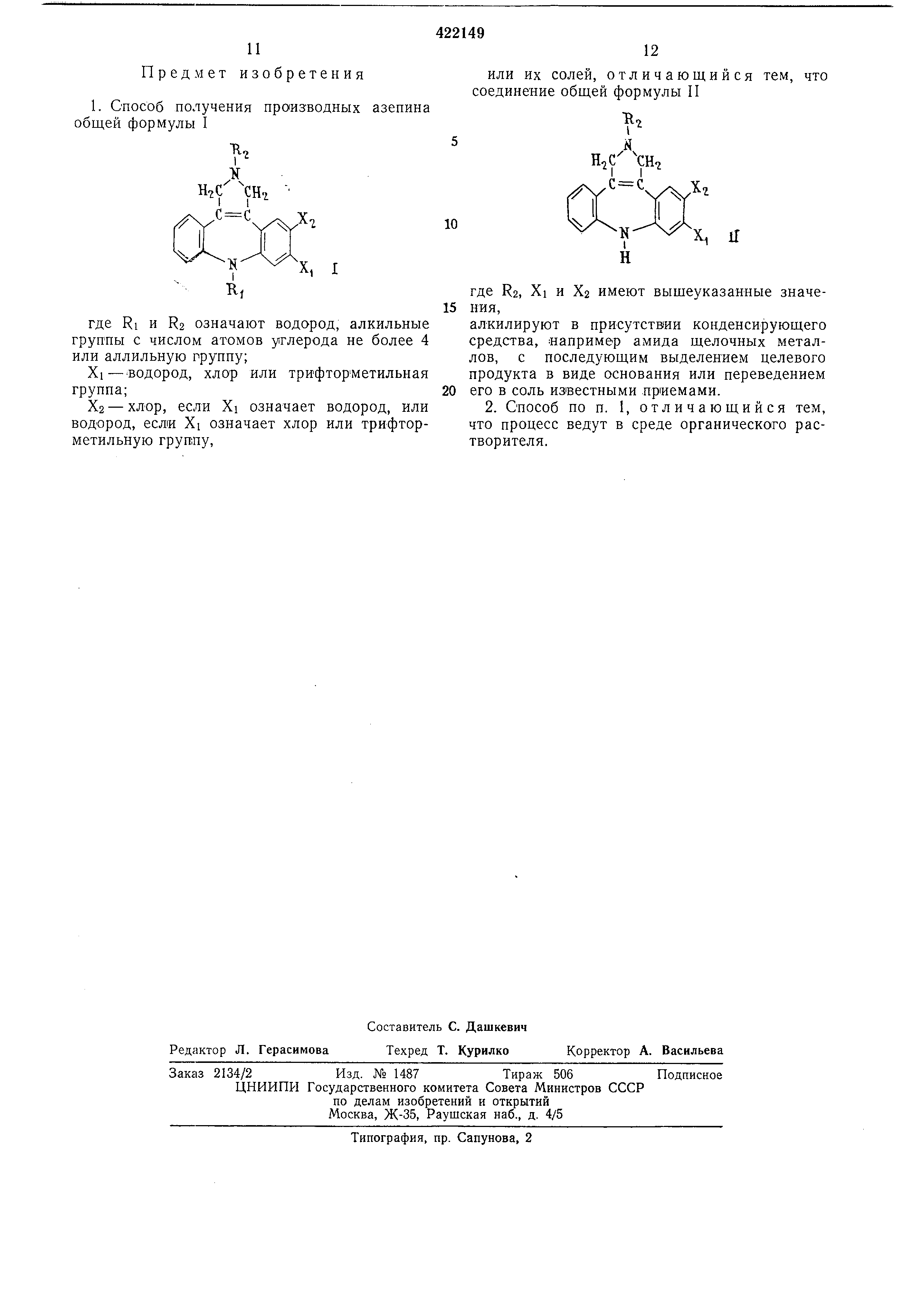
Предлагаемый способ заключается в том, что соединения общей формулы П
S Н.С Но
X.
N
Чч
, И
где R2, Xi и Х2 имеют указанное для формулы I значение,
алкилируют предпочтительно в присутствии растворителя и основного средства конденсации .
Реакция взаимодействия проходит в присутствии или без инертного органического растворителя . Пригодными инертными растворителями являются, например, углеводороды - бензол, толуол, ксилол, кумол или тетралин, эфироподобные л идкости - диоксан, алканоны - ацетон или метилэтилкетон, амиды карбоновой кислоты - диметилформамид, амиды фосфорной кислоты - триамид гексаметилфосфорной кислоты, или сульфоксиды - днметил
или диэтилсульфоксид. в качестве основных средств конденсации пригодны, например, щелочные металлы - натрий, калий или литий, гидроокиси щелочных металлов - гидроокись натрия или калия, карбонаты щелочных металлов - карбонат калия, амиды щелочных металлов-амид натрия, калия или лития, гидриды щелочных металлов - гидрид натрия или лития, алканолаты щелочных металлов - метилат натрия, этилат натрия или трет бутилат натрия, или алкильные и ариллитиевые соединения - бутил- или фениллитий. Реакционная температура предпочтительно составляет от О до 100°С.
Полученные соединения могут быть переведены в соли с помощью неорганических или органических кислот. Как адсорбент применяют силикагель фирмы Мер с размером зерен 0,05-2 мм.
Пример 1. 4,33 г (0,014 моль) 2-этил-6хлор-1 ,2,3,8-тетрагидродибенз - {,f) - нирроло- (3,4-)-азепина растворяют, в 50 мл триамида гексаметилфосфорной кислоты, а к полученно му раствору добавляют 3,70 мл (0,028 моль) 30%-ной суспензии амида натрия в толуоле. Темно-зеленый раствор в течение 30 мин размещивают в ванне цри 50°С, охлаждают и в вакууме освобождают от образовавщегося газа. Затем, размещивая в течение 5 мин вкапывают раствор из 0,95 мл (0,015 моль) метилйодида в 5 мл триамида гексаметилфосфорной кислоты и продолжают размещивать в течение 15 мин. Реакционный раствор коричнево-красной окраски наливают на лед, разбавляют больщим количеством воды и устанавливают щелочное рИ посредством концентрированного раствора натриевой щелочи . Выделяется сырое основание, которое экстрагируют этиловым уксусным эфиром, раствор этилового уксусного эфира промывают водой и экстрагируют 2 н. соляной кислотой. Солянокислый экстракт обрабатывают активированным углем, нейтрализуют и устанавливают щелочное рН концентрированным раствором едкого кали и экстрагируют выделивщееся основание эфиром. Эфирный экстракт промывают водой, сущат над карбонатом калия и выпаривают в вакууме. Получают 3,46 г сырого 2-этил-6-хлор-8-метил-1,2,3,8-тетрагидродибенз- (b,f) -пирроло- (3,4-d) -азепин.
3 г (0,0096 моль) сырого основания растворяют в эфире. Эфирный раствор встряхивают с 2 к. эфирной соляной кислотой, выделивщийся желтый гидрохлорид отсасывают и сущат в вакууме. Он плавится при 169-171°С и содержит один эквивалент кристаллизационной воды.
Необходимый в качестве исходного продукта 2-этил-6-хлор-1,2,3,8-тетрагидродибенз-(&,)пирроло- (3,4-й)-азепин можно получить следующим путем.
А. 20 г (0,088 моль) мелкоизмельченного 3-хлор-5Н-дибенз-(Ь,/)-азепина в 600 мл 48%яой бромистоводородной кислоты в течение 90 мин кипятят с обратным холодильником.
Затем реакционную смесь охлаждают льдом, причем выпадает часть образовавщегося 3хлор-9-метил-акридингидробромида . Охлаждая льдом к полученной суспензии по порциям добавляют 450 мл концентрированного аммиачного раствора и экстрагируют смесь эфиром. Эфирный раствор промывают водой и экстрагируют 300 мл 1 н. серной кислоты. Если сырой продукт выпадает как сульфат,
то он после добавки воды онять растворяется. Эфирный раствор трижды промывают водой и соединяют промывную воду с кислым экстрактом . Водный, кислый раствор обрабатывают активированным углем, фильтруют и в
светло-желтом фильтрате концентрированным аммиаком устанавливают щелочное рН. Выделивщееся основание поглощают в эфире. Эфирный раствор сущат над сульфатом магния , фильтруют и выпаривают в вакууме. Остаток поглощают в горячем гексане, гексановый раствор чистят активным углем, фильтруют и сгущают раствор. Полученный З-хлор-9метилакридин с т. пл. 117-118°С выкристаллизовывается .
Б. 22,7 г (0,100 моль) полученного по пункту А производного акридина растворяют нагреванием в 120 мл 2 н. серной кислоты. Раствор охлаждают, разбавляют его 120 мл ледяной воды, помещают его в ванну, охлаждаемую льдом с солью и при 9°С, добавляют 29 мл (0,512 моль) охлажденного ацетальдегида . Температура смеси повыщается до 15°С. Размещивая, охлаждают до 8°С и одновременно вкапывают охлажденный до 4°С раствор из 144 г (0,520 моль) ферросульфатагептагидрата в 480 мл воды, а также 60 мл
(0,450) моль) охлажденного до 2°С 75%-кого
т;7ег-бутилгидропероксида.
Во время вкапывания реакционный раствор
сильно перемещивают и скорость вкапывания устанавливают таким образом, чтобы температура в реакционном сосуде составляла 10- 13°С. После добавки половины обоих реактивов на стенке сосуда образуется корка, которую удаляют. По окончании прикапывания продолжают размещивать в течение 15 мин, причем внутренняя температура реакционного сосуда снижается до 3°С. Полученную, коричневую суспензию экстрагируют хлористым
метиленом, экстракт промывают водой, сущат над сульфатом натрия и выпаривают в вакууме . Остаток растворяют в 72 мл теплого абсолютного бензола, охлаждают раствор, фильтруют его через колонну 52 г силикагеля, промывают 320 мл абсолютного бензола и выпаривают элюат бензола. Остаток перекристаллизовывают из эфира-гексана, вследствие чего получается чистый метил-(З-хлор-9метилакридан-9-ил )-кетон, который плавится
нри 116--118°С.
В. 27,4 г (0,100 моль) полученного по пункту Б кетона растворяют в 500 мл метанола. Раствор охлаждают до 10°С и к нему, размещивая в ледяной ванне в течение 10 мин, порциями добавляют 19,1 г (0,500 моль) боргидрида натрия. Перемешивают еще в течение 1 час при 5°С и концентрируют реакционный раствор в вакууме до веса 70 г. Вследствие добавки 6,6 мл воды и некоторых затравочных кристаллов после охлаждения в ледяной ванне начинается кристаллизация . Продолжают далее охлаждать льдом, медленно прибавляют 100 мл воды и в течение 1 час оставляют стоять при 0°С. Затем кристаллы фильтруют на нутче, промывают их водой до нейтрального состояния и сушат их в вакууме над гидроокисью калия. Получают 27,4 г сырого З-хлор-а-9-диметил9-акриданметанола .
Г. В охлажденную до комнатной температуры смесь из 200 мл концентрированной серной кислоты и воды (10:1), сильно размешивая в течение 0,25 час, вносят 27,4 г (0,100 моль) полученного по пункту В гидроксисоединения .
Гид|роксисоединение постепенно растворяется , причем раствор нагревается до 30°С. Через 90 мин после начала добавки образуется прозрачный раствор, который перемешивают в течение дальнейших 45 мин. Затем раствор выливают на смесь из 800 г льда, 1 л воды и 500 мл хлористого метилена. Отделяют органическую фазу и экстрагируют водную фазу хлористым метиленом. Соединенные растворы хлористого метилена промывают водой, сушат над сульфатом натрия и выпаривают в вакууме. Остаток растворяют в абсолютном бензоле и бензольный раствор добавляют к 55 г силикагеля. Адсорбент отфильтровывают на нутче, промывают абсолютным бензолуксусноэтиловЫМ эфиром (10:1) и фильтрат выпаривают в ва кууме. Остаток выкристаллизовывают из эфира-гексана, вследствие чего получают чистый 3-хлор- 0,11 -диметил-5Н-дибенз- (&,f) азенин с т. пл. 137-139°С.
Д. 10,0 г (0,39 моль) полученного по пункту Г производного азепина в течение 10 мин в 100 мл ацетангидрида кипятят с обратным холодильником. Затем отгонягот избыточный ацетангидрид под вакуумом при 80°С, поглошают оставшееся красное масло в абсолютном бензоле и хроматографируют раствор на колонне 120 г силикагеля. Колонну промывают 300 мл абсолютного бензола и элюируют абсолютным этиловым бензолуксусным эфиром (10:1). Путем выпаривания элюата в вакууме получают 11,3 г 3хлоп-5-апетил-10 ,11 - диметил - 5Н - дибенз (6,f)-aзeпин, который применяют как сырой .
Е. 11.3 г (0,038 моль) полученного по пункту Д соединения оастворяют в 100 мл ,четыреххлористого углеоода. К паствору добавляют 13.7 г (0.077 моль) N-бромсукнинИМида , освещают полученную суспензию лампами 200 ВТ и кипятят ее в течение 1 час с обратным холодильником. Реакционную смесь охлаждают, фильтруют и фильтрат выпаривают в вакууме. Остаток поглошают в
бензоле, отфильтровывают от малорастворившегося сукцинимида и выпаривают фильтрат в вакууме. Остаток растворяют в эфире, эфирный раствор обрабатывают активным углем, фильтруют и выпаривают в вакууме. Получают 18,0 г аморфного 3-хлор-5-ацетил-10,11бисбромметил-5Н-дибепз- (b.f) -азепина.
Ж. 17,4 г (0,036 моль) полученного по пункту Е соединения растворяют в 350 мл абсолютного бензола. Этот раствор охлаждают и в течение 20 мин размешивают и прикапывают 110 мл (0,51 моль) 21%-ного этиламинового раствора в бензоле. Из реакционного раствора выпадает этиламингидробромид . Продолжают размешивать реакционную смесь еше в течение 10 мин и фильтруют ее очиш.енной диатомовой землей. Избыточный этиламин в фильтрате выпаривают в вакууме и оставшийся бензольный раствор экстрагируют 1 н. соляной кислотой. Солянокислый экстракт обрабатывают активным углем, фильтруют и устанавливают его щелочное рН посредством концентрированного раствора едкого кали. Сырое основание
выпадает масло. Его экстрагируют эфиром , эфирный экстракт промывают водой до нейтрального состояния, сушат над карбонатом калия и выпаривают в вакууме. Получают 10,9 г сырого 2-этил-6-хлор-8-ацетил1 ,2,3,8 - тетрагидродибенз - (fe,f) - пирроло (3,4-)-азепин.
И. 10,9 г (0,032 моль) полученного по пункту Ж соединения растворяют в 50 мл абсолютного этанола. К раствору добавляют
40 мл 20%-ного этанольного раствора едкого кали, кипятят смесь в течение 3 час с обратным холодильником и охлал-сдают. 6,16 г оранжевого, кристаллического продукта реакции отсасывают. К фильтрату добавляют 1 г порошкообразной гидроокиси калия , кипятят смесь, из которой отгоняют 70 МЛ этанола, в течение 1 час и охлаждают ее. Выпадает вторая фракния 1,53 г сырого реакционного продукта, которую отсасывают . Маточный раствор разбавляют водой , извлекают эфиром и эфирный раствор экстрагируют 2 н. соляной кислотой. Солянокислый экстракт обрабатывают активным углем, фильтруют, и в фильтрате при помоши концентрированного раствора едкого кали устанавливают тцелочное рП. Вьпелившееся основание поглощают в эфире, отделяют органическую фазу, сушат ее над капбонатом калия и выпаривают се в вакууме.
Остаток, который выкристаллизовывают из небольшого количества эфира, дает третью фракцию 1,25 г продукта реакции. Соединенные продукты кристаллизации попекриста,члизовывают из бензола, получают 2-этил-6-хлор1 ,2,3,8 - тетрагидродибенз - (h.f - пирроло (3.4-с)-азепиц: т. пл. 193-195°С.
Пример 2. К 1.84 г (0,0065 моль) -метил-5-хлор-1 ,2,3,8-тетрагидро - дибенз - {h,f}пирроло- (3.4-с/)-азепина, растворенных в
50 мл триамида гексаметилфосфорной кислоты , добавляют 1,67 мл (0,0125 моль) 30%-ной суспензии амида натрия в толуоле. Реакционную смесь в течение 30 мин перемешивают нри 30°С, охлаждают ее и в вакууме освобождают ее от образовавшегося газа. Затем, охлаждая водой и размешивая прикапывают 0,43 мл (0,007 моль) метилйодида, растворенного в 5 мл триамида гексаметилфосфорной кислоты. Затем реакционную смесь размешивают еше 15 мин, повторно добавляют то же самое количество суспензии амида натрия, освобождают смесь в вакууме от образовавшегося газа, повторяют также добавку того же количества метилйодида и еще раз перемешивают в течение 15 мин. Затем смесь наливают на лед и экстрагируют эфиром. Водный слой из-за образования четвертичных солей остается желто окрашенным. Эфирный раствор экстрагируют 2 н. соляной кислотой, в солянокислом экстракте устанавливают щелочное рН концентрированным раствором едкого калия, и выделившееся основание экстрагируют эфиром. Эфирный раствор сушат над карбонатом калия и выпаривают его. Остаток хроматографируют над колонной, которую изготовляют из 50 г силикагеля и абсолютного бензола и сырой продукт элюируют содержащим 1 % этанола хлороформочм. Элюат выпаривают в вакууме и остаток перекристаллизовывают из изопропанола, причем получают 103 мг желтого 2,8-диметил-5-хлор1 ,2,3,8 - тетрагидродибенз - (6,f) - пирроло (3,4-сг)-азеп-ина с т. пл. 178-190°С. Употребленный в качестве исходного продукта 2-метил-5-хлор-1,2,3,8-тетрагидродибенз (6,f)-пиppoлo-(3,4-d)-aзeпин получают следующим образом. А. 22,8 г (0,100 моль) 2-хлор-9-метилакридина в 35 мл 2 н. соляной кислоты и 65 мл ледяной БОТЫ подвергают взаимодействию с 7,2 мл (0,128 моль) ацетальдегида, 15 мл (0,114 моль) 75%-ного трет-бутилгидропероксила и 36 г (0,130 моль) ферросульфата - гептагидрата. в водном растворе 120 мл воды, причем получают 13,20 г метил-(2-хлор-9-метилакрияан-9-ил )-кетона с т. пл. 134-135°С (из эфира-гексана); выход 51% от теоретического из расчета на 21,8 г прореагировавшего исхотного продукта. Б. 3,11 г (0,0114 моль) полученного по пункту А кетона растворяют в 40 мл метанола. К раствору добавляют 0,50 г (0,013 моль) боргилрипа натрия и в течение 1 час размешивают при комнатной температуре. Реакционную смесь ОСТОРОЖНО выпаривают в вакууме и остаток поглоп1ают в 100 мл хлористого метилена. К раствору хлористого метилена прибавляют немного безводного сульфата магния, фильтруют и выпаривают фильтрат в вакууме. Получают 3,18 г сырого 2хлор-а-9-диметил-9-акриданметанола и его перерабатывают как сырой продукт. Если полученное соединение не применяется сейчас же, его следует хранить при 0°С. В. 15,1 г (0,055 моль) полученного по пункту В гидроксисоединения размешивают в 300 мл концентрированной серной кислоты-воды (10:3) при комнатной температуре, пока не образуется раствор. Затем реакционную смесь цродолжают размешивать еще 30 мин при той же температуре, и, размешивая ее вносят в смесь из 700 мл 50%-ного раствора гидроокиси калия « 2 кг льда. Полученную суспензию разбавляют водой, чтобы выделившийся сульфат калия растворился бы и экстрагируют раствор эфиром. Эфирный раствор промывают водой, сушат над сульфатом магния и вьгааривают в вакууме. Перекристаллизовывают остаток из эфира-гексана и получают 11,47 г 2-хлор-10,11-диметил-5Н-дибенз- (6,/)-азепина с т. пл. 137-138°С. Г. Аналогично цримеру 1, Д, 13,11 г (0,051 моль) полученного по пункту 2, В соединения в течение 15 мин кипятят с обратным холодильником с 100 Мл ацетангидрида и полученный сырой продукт чистят над силикагелем . Получают 14,66 г 2-хлор-5-ацетил10 ,11-диметил-5Н-дибенз-(6,/)-азепин, который применяют в качестве сырого продукта. Д. 13,50 г (0,045 моль) полученного по пункту Г сырого продукта аналогично примеру 1,Е подвергают взаимодействию с 17,7 г (0,091 моль) N-бромсукцинимида. Полученный сырой продукт очищают над колонной 150 г силикагеля. В качестве элюата сначала применяют абсолютный бензол, который элюирует побочный продукт, и затем смесь из абсолютного этилового бензолуксусного эфира ( 10 : 1). Элюат этилового бензолуксусного эфира выпаривают в вакууме, в результате получают 20,74 г чистого желтоватого, аморфного 2-хлор-5-ацетил-10,11-бисбромметил-5Н-ди6eH3- (&,f)-a3enHHa. Е. Размешивая 100 мл (0,32 моль) 10%-ного метиламинового раствора в бензоле в течение 10 мин вкапывают в 19,0 г (0,040 моль) полученного по пункту Д соединения, которое растворяют в 200 мл абсолютного бензола и охлаждают ледяной ванной. Скорость прикапывания регулируют таким образом, чтобы температура реакции составляла 20°С. Затем размешивают еще 30 мин, выделившуюся соль фильтруют на нутче и вынаривают фильтрат в вакууме. Остаток поглощают в эфире и эфирный раствор экстрагируют 1 н. соляной кислотой. В солянокислом экстракте устанавливают щелочное рН посредством концентрированного раствора едкого кали и экстрагируют выделившееся сырое основание эфиром. Эфирный раствор сушат над карбонатом калия и выпаривают в вакууме. Получ-ают аморфный, желтоватый 2-метил-5-хлор8-ацетил-1 ,2,3,8-тетрагидродибенз - (b,f) - пирроло- (3,4-г)-азепин. Продукт является чувствительным к воздуху и его следует хранить в холоде под азотом . Ж. 6,63 г (0,020 моль) полученного по пункту Е соединения растворяют в 50 мл
9
абсолютного этанола. К этому раствору добавляют 4,60 г гидроокиси калия, кипятят смесь под азотом и с обратным холодильником в течение 5 час и затем охлаждают ее до 0°С. Выделившиеся оранжевые кристаллы (4,61 г) отсасывают, промывают небольшим количеством холодного как лед этанола и сушат. Фильтрат выпаривают в вакууме , остаток растворяют в эфире, эфирный раствор экстрагируют 2 н. соляной кислотой и в солянокислом экстракте устанавливают ш,елочное рН концентрированным раствором едкого «али. Выделившееся свободное основание поглошают в хлористом метилене, сушат раствор хлористого метилена над карбонатом калия и выпаривают его. Кристаллический остаток 0,66 г соединяют с. первой фракцией кристаллов и смесь растворяют в бензоле, обрабатывают активным углем, фильтруют и сгущают. Получают 5,03 г 2-метил5-хлор-1 ,2,3,8-тетрагидродибенз - (6,f) - пирроло- (3,4-с)-азепин с т. пл. 210-212°С.
Пример 3. 4,50 г (0,0152 моль) 2-этил5-хлор-1 ,2,3,8-тетрагидродибенз - (6,/)-пирроло- (3,4-й)-азепина растворяют в 100 мл тиамида гексаметилфосфорной кислоты и под азотом добавляют 6,25 мл суспензии 0,0322 моль амида натрия в толуоле. Смесь размешивают в течение 30 мин при 50°С, и полученный аммиак удаляют обезгаживанием . Охлаждая льдом, в течение 5 мин вкапывают раствор 2,94 мл н-пропилйодида в 5 мл триамида гексаметилфосфорной кислоты, и смесь продолжают размешивать еш;е в течение 15 мин. После добавления 500 мл воды смесь трижды экстрагируют по 250 мл этилового уксусного эфира. Соединенные экстракты трижды промывают, каждый раз в 300 мл воды, высушивают над сульфатом магния и сгушают в вакууме. Остаюшееся красное масло растворяют в хлороформе и хроматографируют над колонной силикагеля (фирмы Мерк, 0,05-0,2 мм). Элюированием хлороформом и добавляя 1 % метанола и выпаривая хлороформ получают желтое масло, которое кристаллизуется при стоянии и представляет собой 2-этил-5-8-н-пропил-1.2,3,8-тетрагидродибенз- (Ь.П-пирроло - (3,4-d) .- азепин , т. пл. 90-92°С.
4,85 г основания растворяют в 100 мл хлористого метилена и добавляют 1,90 мл 19.5%ного раствора хлористого водорода в этаноле. После десятиминутной выдержки раствоо в вакууме выпаривают досуха и остаток с 40 мл этилового уксусного эфира размешивают в течение 2 час. Кристаллы отсасывают и сушат в вакууме; они представляют собой гидрохлорид основания, т. пл. 212-217°С.
10
Употребленный в качестве исходного продукта 2-этил-5-хлор-1,2,3,8-тетрагидрод ибенз (6,/)-пирроло-(3,4-й)-азепин получают следующим образом.
А. 14,6 г (0,0307 моль) полученного согласно примеру 2 Е соединения растворяют в 200 мл бензола и при охлаждении в азотной атмосфере, размешивая, по каплям добавляют 93 мл 21%-ного раствора этиламина в
бензоле. Работая согласно примеру 1 Ж нолучают 2-этил-5-хлор-8-ацетил-1,2,3,8-тетрагидродибенз- (b,f) -пирроло- (3,4-d) -азепин, который
хранят под азотом в виде бесцветной пены.
Б. 5,0 г (0,01475 моль) полученного соединения с 3,28 г гидроокиси калия в 50 мл этанола в течение 2,5 час кипятят с обратным холодильником под азотом. После охлаждения отсасывают выделившиеся кристаллы . К маточному раствору еще раз добавляют то же самое количество гидроокиси калия и этанола и омыляют тем же образом , причем получают дальнейшее количество кристаллов. Соединенные крнсталлизаты перекристаллизовывают из бензола, причем получают чистый 2-этил-5-хлор-1,2,3,8тетрагидродибенз- (&,)-пирроло - (3,4-rf) - азепин в виде желтых кристаллов; т. пл. 202- 204°С.
Пример 4. 5,0 г (0,0169 моль) полуценного согласно примеру 1 И соединения обменно разлагают согласно примеру 3 с нпропилйодидом . После переработки получают сырой продукт, который как раствор в хлороформе очищают над колонной с десятикратным количеством силикагеля (фирмы Мерк, размер зерен 0,05-0,2 мм). С целью удаления побочных продуктов, колонну затем промывают хлороформом и затем элюиРЗЮТ хлороформом, содерл-сашим 1 % метанола . После выпаривания растворителя и перекристаллизации остатка из гексана получают чистый 2-этил-6-хлор-8-я-пропил-1,2,3,8тетрагидродибенз- (6,f)-пирроло - (3,4-rf) - азецин в виде желтых пластинок, т. пл. 127-
129°С.
Из 3,65 г этого основания аналогично примеру 3 получают гидрохлорид в виде желтых кристаллов; т. пл. 235-238°С. Пример 5. Соответственно примеру 3, из
4,135 г (0,01395 моль), полученного согласно примеру 1 И соединения и аллилбромида, получают 2-этил-6-хлор-8-аллил-1,2,3,8-тетрагидpoдибeнз- (&,f)-пирроло-(3.4-rf)-азепин в виде красноватого вязкого масла, которое после
очистки над силикагелем дает чистое основание в виде светло-желтого масла.
Аналогично примеру 3 из 3,60 г оспования получают гидрохлорид в виде желтых кристаллов; т. пл.208-211°С.
И
Предмет изобретения
1. Способ получения производных азепина бщей формулы I
|. .
н,с сн, -
ОС
У
ЧА
X, I
R,
где RI и R2 означают водород, алкильные группы с числом атомов углерода не более 4 или аллильную грунну;
Xi-водород, хлор или трифторметильная группа;
Хз - хлор, если Xi означает водород, или водород, есл1И Xi означает хлор или трифторметильную груплу.
12
или их солей, отличающийся тем, что соединение общей формулы II
И
где Ra, Xi и Х2 имеют выщеуказанные значения ,
алкилируют в присутствии конденсирующего средства, «апример амида щелочных металлов , с последующим выделением целевого продукта в виде основания или переведением его в соль известными приемами.
2. Способ по п. I, отличающийся тем, что процесс ведут в среде органического растворителя .





Реферат
Формула
Комментарии